 Kelsey Ouyang1,2,3†
Kelsey Ouyang1,2,3† George W. Agak
George W. Agak- 1Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH, United States
- 2Division of Dermatology, Department of Medicine, University of California, Los Angeles (UCLA), Los Angeles, CA, United States
- 3David Geffen School of Medicine at University of California, Los Angeles (UCLA), Los Angeles, CA, United States
- 4Department of Dermatology, Penn State University College of Medicine, Hershey, PA, United States
The role of extracellular traps (ETs) in the innate immune response against pathogens is well established. ETs were first identified in neutrophils and have since been identified in several other immune cells. Although the mechanistic details are not yet fully understood, recent reports have described antigen-specific T cells producing T cell extracellular traps (TETs). Depending on their location within the cutaneous environment, TETs may be beneficial to the host by their ability to limit the spread of pathogens and provide protection against damage to body tissues, and promote early wound healing and degradation of inflammatory mediators, leading to the resolution of inflammatory responses within the skin. However, ETs have also been associated with worse disease outcomes. Here, we consider host-microbe ET interactions by highlighting how cutaneous T cell-derived ETs aid in orchestrating host immune responses against Cutibacterium acnes (C. acnes), a commensal skin bacterium that contributes to skin health, but is also associated with acne vulgaris and surgical infections following joint-replacement procedures. Insights on the role of the skin microbes in regulating T cell ET formation have broad implications not only in novel probiotic design for acne treatment, but also in the treatment for other chronic inflammatory skin disorders and autoimmune diseases.
Introduction
The skin is the largest organ in the body, and it provides an effective physical and immune barrier between the internal and external environment. As the skin is under constant assault from physical, chemical and biological threats, disturbance of this barrier can manifest as inflammatory skin diseases, such as atopic dermatitis and acne vulgaris. Contributing to the healthy skin barrier is a collection of diverse skin microbiota, which blanket the skin’s surface and populate the hair follicles, helping to promote skin homeostasis, immune defense and education of host immune cells (1, 2). Hair follicles penetrate deep into the dermis and contain a diverse population of commensal bacteria in comparison to the skin surface (1, 2). Although the hair follicle is an immune-privileged site, it may serve as an important site for interactions between bacteria and host T cells, particularly in acne, wherein the epithelium of the follicles and sebaceous glands are breached. In the healthy state, little is known about the potential for bi-directional movement of immune cells, including T cells from the dermis through the intact follicular epithelium to contact luminal bacteria. This interaction between immune cells and the diverse skin bacteria may contribute to both skin homeostasis and/or disease.
Human skin contains abundant populations of memory αβT cells, antigen presenting cells (APCs), natural killer cells, γδ T cells, and innate lymphoid cells (3). Effector and memory T cells within the skin coordinate immune responses against microbes. However, inappropriate T cell activation against innocuous or autoantigens can lead to chronic skin inflammatory disorders. In this review, we focus on the immune responses mediated by skin-resident TH17 cells that develop in response to Cutibacterium acnes (C. acnes), a bacterium that has been associated with acne vulgaris. We discuss the antimicrobial capacity of these cells in acne resolution and/or inflammation (4). The etiology and pathogenesis of acne vulgaris is briefly discussed, followed by the CD4+ T cell responses against C. acnes and their ability to form extracellular traps (ETs) in response to microbial threats. On this basis, mechanisms of T cell extracellular trap (TET) formation are proposed and the dual nature of ET formation in acne and select skin diseases is detailed, with a focused discussion on the specific factors that may contribute to host protection versus exacerbation of disease. We highlight new perspectives into how healthy skin commensals are critical to the instruction of our immune system and how T cell-derived–antimicrobial molecules may provide new therapeutic strategies in our overall defense against pathogens. The elucidation of subcellular events leading to TET formation may generate novel therapeutic strategies that target both the innate and adaptive immune system in order to ameliorate skin diseases.
Etiology and Pathogenesis of Acne
Acne vulgaris is a chronic inflammatory disorder of the pilosebaceous unit (PSU) that most commonly occurs during adolescence but may linger into adulthood (5–8). In most cases, this skin disease is characterized with an array of lesions, which consist of comedones, papules, pustules, and nodules that are concentrated on the face, back, or chest. While the course of acne progression may be limited in most patients, the aftereffects can be life-long, with physical scars and psychological impairment, especially in adolescents (9). The pathogenesis of acne involves several factors, and at least four factors have been identified. These key factors include: follicular epidermal hyperproliferation, sebum production, C. acnes (in italics), and inflammation/immune response. Each of these processes are interrelated and can be influenced by both hormones and the immune response (9–15).
Current evidence supports the notion that acne lesions begin with the formation of the microcomedo with subsequent development into clinically detectable lesions and scarring, in the most severe cases. Follicular epidermal hyperproliferation contributes to microcomedo formation. During lesion development, the microcomedo expands with a densely packed layer of keratin, and there is increased sebum production that supports bacteria growth within the PSU. Eventually this ballooning effect causes the walls of the follicles to rupture leading to extrusion of keratin, sebum, and bacteria into the surrounding dermis, triggering a rapid inflammatory response. The predominant cell type present within 24 hours of rupture is the lymphocyte. CD4+ T lymphocytes are present around the ruptured PSU, while CD8+ T cells are found within the perivascular region. One to two days after the comedo is ruptured, neutrophils become the main cell type surrounding the ruptured microcomedo (9–15), further amplifying skin inflammation.
It is generally accepted that acne does not occur without sebum. Sebaceous gland activity increases during puberty in response to androgens (16). The anaerobic and lipophilic microenvironment of the PSU favors the growth of C. acnes over other skin microbes. Sebum, the lipid-rich fluid of sebaceous glands, serves as the primary nutrient source for C. acnes. Breakdown of sebum triglycerides into free fatty acids by C. acnes contributes to the inflammatory response (17, 18).
C. acnes also contributes to skin inflammation through activation of the immune response. C. acnes-induced secretion of proinflammatory cytokines from monocytes involves Toll-like receptor 2 (TLR2) (19), which is expressed on macrophages surrounding the PSU and in the epidermis of inflammatory acne lesions (19, 20). In addition, C. acnes induces IL-1β secretion and inflammasome activation via NLR family pyrin domain containing 3 (NLRP3) and caspase-1 in monocytes and sebocytes (21–23). The antimicrobial peptides, cathelicidin, and histone H4 can also be secreted locally in response to C. acnes. Histone H4 secreted by sebocytes exerts direct microbial killing, while cathelicidin interacts with components of the innate immune system within the acne microenvironment, such as psoriasin and beta-defensins, all of which contribute to the direct killing of C. acnes (24, 25).
CD4+ T Cells in the Skin
In the periphery, naive CD4+ T cells undergo differentiation into distinct T cell lineages upon interaction with APCs and the influence of specific cytokines. These distinct lineages play a major role in mediating immune responses mainly through the secretion of specific cytokines and antimicrobial molecules. The CD4+ T cells perform multiple functions ranging from the activation of innate immune cells, B cells, cytotoxic T cells, as well as other non-immune cells. They also play a critical role in the quelling of immune reactions (26).
Traditionally, the CD4+ T cell subset lineages have been classified based on the cytokines they produce expression of characteristic transcriptional factors (TFs). Based on this classification, CD4+ T cells have been designated into T-helper-1 (TH1), T-helper 2 (TH2), T-helper 17 (TH17), follicular helper T cells (Tfh), induced T-regulatory cells (iTregs), regulatory type 1 cells (Tr1), and T-helper 9 cells (TH9) (27–33). In recent years, the diversity of TH subsets has increased in complexity and the designation of TH subsets beyond TH1, TH2 and TH17 cells remains a subject of intense debate as the traditional classification system fails to account for TH cells that are involved in the induction of various pathologies. Of note, the emergence of novel technologies has enabled the simultaneous measurement of several cytokines at once along with other markers such as TFs, chemokine receptors and integrins at the single cell level, making it impossible to categorize TH cells based on a dominant cytokine or even a family of cytokines (34). The new proposed paradigm reorganizes and expands the TH universe based on the help these cells provide to the actual cell targets, rather than on the transient expression of certain cytokines and TFs (35). The debate on taxonomy to capture the complexity and diversity of TH cells is described elsewhere (35–37).
Functionally, TH17 cells produce IL-17, IL-22 and IL-26 and play an important role in the clearance of extracellular bacteria and fungi from epithelial surfaces (38–40). TH17 cells are an important component of T cell immunity as patients with loss-of-function mutations in genes coding for IL-17, IL-17 receptor and/or the transcriptional factor RORγt have been shown to suffer from recurrent infections of the skin, nails, and mucosal surfaces. IL-22 can cooperate with IL-17 to activate epithelial cells to produce antimicrobial peptides (41–43). Finally, IL-26 is a cationic antimicrobial protein that kills extracellular bacteria by creating pores on the bacterial membrane (39). Like other TH17 cell cytokines, IL-26 is highly expressed in the skin lesions of psoriatic patients (44), colonic lesions in patients with inflammatory bowel disease (45, 46) and in the synovial fluid of individuals with rheumatoid arthritis (47, 48). A risk locus containing IL26 and single-nucleotide polymorphisms within the IL26 gene region have been associated with multiple sclerosis (49), highlighting the fact that IL-26 may be involved in driving TH17 cell–associated inflammatory activity.
IL-17 is a key cytokine involved in the recruitment, activation and migration of neutrophils to sites of inflammation (50, 51). Recruitment of neutrophils not only results in the phagocytosis and clearance of microbes, but can also result in tissue destruction. Similar to neutrophils, TH17 cells may also contribute to host defense and/or tissue damage as a result of uncontrolled TET activation and release. We envisage a scenario where an appropriate and timely decline of TH17 responses within the dermis may be required to minimize tissue injury or damage. In essence, this decline in TH17 response may be achieved by a shift toward production of cytokines such as IL-10 by TH17 cells. It is therefore likely that, in the steady-state, skin resident TH17 cells may have the capacity to adjust their cytokine profiles and secrete a combination of cytokines such as IL-26 and IL-10 that together can help dampen the inflammation and simultaneously time aid in combatting diverse skin pathogens, such as Staphylococcus aureus (S. aureus) and C. acnes, as needed. Thus together, with other innate immune cells, rapid cytokine secretion and TET formation may represent an efficient local host defense for controlling pathogenic microbes on the skin.
The components of both the innate and adaptive immune system have been demonstrated to play a part in the inflammatory responses that are observed during acne pathogenesis. Notably, in an elegant histological study of acne lesions, Norris et al. demonstrated that lymphocytes were the predominant cell types, with CD4+ T cells being among the cells that were detected within the early inflammatory infiltrate surrounding acne lesions (15). Polymorphonuclear neutrophils (PMNs), were increasingly evident at 24 and 72h and were linked to the disruption of the PSU. Follow up studies from our group and others further demonstrated that both TH1 and TH17 associated cytokines such as IFN-γ and IL-17 were prominent in acne lesions (4, 52, 53), and may contribute to either the inflammatory and/or to antimicrobial activity observed in acne. In early lesions, therefore, it is likely that antimicrobial activity may be driven by T cell-derived cytokines, that ultimately lead to bacterial lysis. The release of microbial components upon bacterial lysis may also lead to direct activation of the innate immune response, further enhancing inflammation. Moreover, TH17 cells recruit neutrophils, which in inflamed lesions, infiltrate around hair follicles and phagocytose C. acnes (54). Additionally, neutrophils can also release reactive oxygen species (ROS) and lysosomal enzymes that may further exacerbate acne disease (55–57). The proposed mechanisms by which immune cells interact with each other within an acne lesion are summarized in Figure 1.
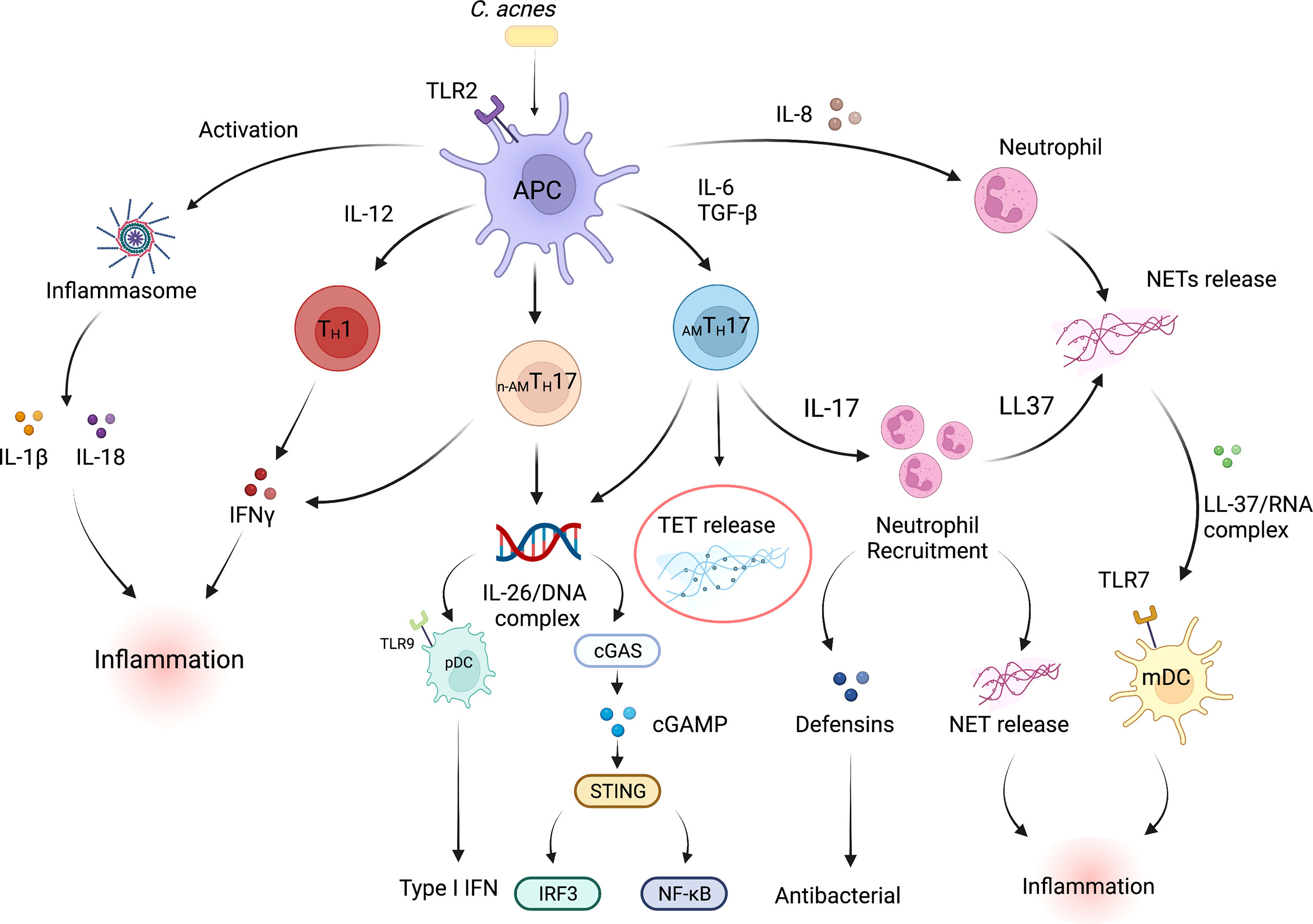
Figure 1 Inflammatory and antimicrobial pathways in acne vulgaris. Multiple interconnected innate and adaptive immune pathways contribute to skin inflammation associated with acne. C. acnes is the pre-dominant organism in the sebaceous region of the skin. Dysbiosis of the skin microbiome, C. acnes colonization or a disrupted homeostasis can upregulate the production of pro-inflammatory cytokines by skin-resident APCs such as dendritic cells, including high levels of IL-1β, IL-6, IL-8, IL-12p70, and TGFβ. Cytokines such as IL-6 and TGFβ induce naive T cells to differentiate into effector TH17 cells, whereas IL-12 drives a TH1 differentiation program. C. acnes strains within the pilosebaceous unit can also influence CD4+ T cell differentiation; C. acnes strains-associated with healthy skin promote the differentiation of naïve T cells into IL-10-producing AMTH17 cells whereas acne-associated strains promote the development of a non-antimicrobial TH17 subpopulation (n-AMTH17). IL-8, IL-17, IL-26, LL-37 and TET production by immune cells within acne lesion promote neutrophil recruitment and increased antimicrobial activity. Expression of defensins can also drive antibacterial action. On the other hand, NET/TET release and formation of LL-37/RNA and/or IL-26/DNA complexes can activate both myeloid and plasmacytoid DCs leading to the production of proinflammatory cytokines that further promote inflammation (39). mtDNA can also activate the cGAS-STING pathway and production of type I IFNs (58, 59). C. acnes can also activate TLR2 receptor expression on macrophages and subsequent secretion of IL-1β and IL-18 secretion in an inflammasome-mediated fashion. IFN-γ release by TH1 cells further drives the inflammatory responses within acne lesions. In addition, proinflammatory conditions exist when IFN-γ-producing TH1 cells are exposed to TNF-α and IL-12. APC, Antigen presenting cell; TGFβ, Transforming growth factor- Beta; pDC, plasmacytoid dendritic cell; IFN-γ, Interferon gamma; TLR, Toll-like receptor; mDC, myeloid dendritic cell; cGAS-STING, cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway. Created with Biorender.com.
This review focuses on the antimicrobial subpopulation of TH17 cells (AMTH17), whose antimicrobial capacity were discovered using C. acnes as a model organism. Moreover, our recent advances in TH17 cell research have reshaped the concept of immunomodulatory CD4+ T cells and have shown that direct antimicrobial activity by AMTH17 subpopulations requires ET formation, which may be critical for pathogen clearance within the cutaneous environment (60).
TH17 Differentiation
TH17 cells express the transcription factor RAR-related orphan receptor-γt (RORγt), and are induced by IL-6, TGFβ and IL-1β. The differentiation of the TH17 lineage has also been shown to be distinct from that of TH1 and TH2 cells. TH17 cells are induced by signal transducer and activator of transcription 3 (STAT3) and RORγt working synergistically with one another (61). The transcription factor, forkhead box P3 (FOXP3), is the negative regulator of RORγt and TH17 programming. FOXP3 maintains tolerance by inducing Regulatory T cells (Tregs) differentiation via STAT6 and downregulating differentiation of TH17 cells (62). However, the TH17/Treg balance is shifted in favor of TH17 when proinflammatory cytokines, such as IL-1β, IL-6 and IL-21, are present. TGFβ is a critical cytokine required for TH17 differentiation. TGFβ not only induces the differentiation of TH17 cells in conjunction with IL-6 in mice and IL-1β in humans, but also independently orchestrates the differentiation of naïve CD4+ T cells into Tregs (63, 64). Exposure of naïve CD4+ helper T cells to TGFβ/IL-1β and IL-6 results in the inhibition of FOXP3 and subsequent RORγt activation, thus initiating TH17 differentiation and programming (63, 65–67). RORγt activation therefore promotes both the expression of IL-17 and IL-23 receptor (68). Interestingly, we have demonstrated that C. acnes stimulates the expression of both RORγt and RORα in peripheral blood mononuclear cells (PBMCs) suggesting that C. acnes contributes to TH17 differentiation (4, 69). In addition, we and others have demonstrated that IL-23, by itself, does not induce the development of TH17 cells from naïve CD4+ helper T cells, but instead IL-23 is important in the maintenance and survival of TH17 cells (70–72). IL-21, which belongs to the IL-2 family of cytokines, further amplifies TH17 cell differentiation and stabilizes the development of TH17 cells in cooperation with Transforming growth factor B (TGFB) in an IL-6-independent manner (61, 73–75).
As highlighted above, TGFβ is critical for TH17 differentiation, but it also influences TH17 biological functions. Different isoforms of TGF have also been shown to induce distinct phenotypic functions in TH17 cells. As an example, in the absence of IL-23, the combination of TGFβ1 and IL-6-induced TH17 cells did not cause experimental autoimmune encephalomyelitis (EAE), but TGFβ3-induced TH17 cells were highly pathogenic (76). TH17 cells, themselves, exhibit high phenotypic and functional plasticity, which suggests that these cells can transdifferentiate into other T cell subsets that are dependent on the inflammatory context (77–79).
In summary, TH17 cell differentiation and regulation is mediated by a tightly controlled complex of cytokine and transcription factors, all of which results in both pathologic and/or non-pathologic immune effector functions.
Diversity of C. acnes Strains and Influence on TH17 Differentiation
Similar to other bacterial species, C. acnes possesses phenotypic and genotypic diversity (70), and the influence of this diversity at the strain level and its connection to either healthy skin or acne is unclear (80). As observed in other human diseases, recent typing of C. acnes strains revealed that not all strains are pathogenic. Certain C. acnes strains were associated with acne, while others were associated with healthy skin (80–84). Strains of C. acnes also show differences in their pathogenic potential and secretome profiles (85–87), as they differ in their ability to induce human β-defensins, influence cell growth, and activate both innate and adaptive components of the immune system (88–92).
We have demonstrated that C. acnes strains differentially modulate the fate of TH17 cells (70). Some healthy-skin associated C. acnes strains induce TH17 subpopulations that produce the anti-inflammatory cytokine IL-10, thus downregulating their pathogenic functions (70, 93). TH17 cells in the gut mucosa have also been shown to transdifferentiate to IL-10-producing regulatory T cells (Tr-1 cells), a process seemingly reliant on aryl hydrocarbon receptor (AhR) and TGFβ (40, 94). The ability of TH17 cells to secrete IL-10 has also been reported following treatment with TNFα inhibitors (95). IL-10 production by TH17 cells has been suggested to be under the regulation of several transcription factors, such as c-Maf and Aiolos (94, 95). In contrast, the majority of acne-associated C. acnes strains induce TH17 subpopulations with a pro-inflammatory phenotype that produce IL-17 and IFN-γ (70, 96).
CD4+ T Cell–Mediated Killing of Target Cells
CD4+ T cell–mediated killing of target cells is well described in the literature, but the extent to which TH1 and TH17 CD4+ T subsets mediate antimicrobial activity is unknown (60, 97). In the case of the TH17 cells, their role in pathologic inflammation and disease is well-defined (73, 98), yet it is unclear what distinguishes the inflammatory TH17 cells elicited by pathogens from those within the PSU or the inner layers of the dermis that are induced by commensals. Two subpopulations of TH17 cells that are functionally different were demonstrated to simultaneously reside within the gut microenvironment during pathogen-induced inflammation. In particular, commensal filamentous bacteria within the gut were shown to induce TH17 cells that were non-inflammatory and homeostatic in nature. In contrast, Citrobacter rodentium, a model pathogen for gastrointestinal human disease research, induced TH17 cells that were highly inflammatory and feature a distinct cytokine profile that likely contributes to pathogenic intestinal pathologies (99). Similar to gut microbes, acne-associated and healthy-associated strains of C. acnes differentially regulate CD4+ T cell responses to induce TH17 cells that secrete either IL-17 and IFN-γ (pro-inflammatory) or IL-17 and IL-10 (anti-inflammatory) respectively (70). We and others revealed that C. acnes induce IL-17 and IFN-γ in CD4+ T cells, and that IL-17-secreting cells are visualized within the perifollicular infiltrates of acne lesions, which is consistent with the fact that TH17 cells may contribute to both the inflammatory and/or antimicrobial responses during acne progression (4, 52, 100).
More recently, while studying the antimicrobial mechanisms involved in TH17 cell–mediated killing of bacteria, we identified a subpopulation of TH17 cells that expressed and secreted multiple antimicrobial proteins. These findings were strengthened by the observation that this antimicrobial subpopulation, termed “AMTH17”, released histone-rich T cell extracellular traps (TETs) in conjunction with other antimicrobial proteins that entangled and killed C. acnes and other bacteria (60). This observation suggested that AMTH17-mediated killing of bacteria is likely to be a general feature that is essential for the homeostatic control of bacterial colonization. By contrast, a separate subpopulation of TH17 cells that were induced by acne-associated strains did not exhibit antibacterial activity (n-AMTH17) against Gram-positive and Gram-negative bacteria.
TH17 and Extracellular Traps
Secretion of extracellular traps loaded with DNA is likely an ancient, conserved function of the innate immune system (101). The discovery and observation that neutrophil extracellular traps (NETs) trapped and killed bacteria changed our collective thinking of the role of these cells in host defense mechanisms, above and beyond the traditional function of microbe phagocytosis cytokine, secretion and antimicrobial peptide release (102). Now, we know that ETs are part of the arsenal of several immune cells including basophils, mast cells, eosinophils, macrophages, and TH17 cells (60, 103–112).
The first observations of lymphocytes extruding DNA were made in 1972. It was difficult to dissect the contribution of extruded DNA in immunity, and it took close to 50 years of intensive investigation for researchers to identify the lymphocyte populations that were capable of releasing ETs. Taking advantage of the fact that NETs are formed in patients with systemic lupus erythematosus (SLE), Rocha Arrieta et al. discovered that both B and T cells had the ability to secrete DNA into the extracellular microenvironment in response to SLE serum and other inflammatory stimulants. These extracellular DNA were not considered as ETs since this study did not evaluate the proteins associated with DNA (113). Focusing on T cells, work by Costanza and colleagues further showed that following activation, murine CD4+ T cells were able to extrude DNA fibers that they termed, “threads” (114). This release of DNA “threads” from CD4+ T cells was also shown to be dependent on mitochondrial ROS (114). However, the responsible T cell subset remained unknown. From the work of our lab, we demonstrated that TH17 cells extrude fibrous DNA threads coated with antimicrobial molecules that trap bacteria. We termed these threads TETs (60).
As previously reported, ETs are released by different immune cells and these ETs entrap not only Gram-positive/negative bacteria and group A streptococci, but also pathogenic fungi (60, 103–111, 115). Importantly, we observed that C. acnes can induce the AMTH17 cells, which upon activation are able to extrude TETs: fibrous DNA structures that are strikingly decorated with histones and antimicrobial molecules. TETs released by AMTH17 cells form interlaced structures in the extracellular space and entangle C. acnes (60). After entrapment, most of the C. acnes were killed (60). However, it is important to highlight that some bacteria species, such as Streptococcus pneumoniae, have developed strategies to evade capture and killing by repelling cationic antimicrobial peptides (CAMPs) found within ETs or by degrading the DNA backbone of ETs with enzymes such as, DNases (115–117). DNase treatment inactivates TETs and renders them ineffective, suggesting that DNA is required for TET structure and function. During acne pathogenesis, disruption of the PSU allows for the entry of C. acnes into the dermis, which initiates an inflammatory response. Using confocal microscopy, we visualized TETs in vivo in biopsy specimens from acne lesions, observing the colocalization of fibrous structures composed of DNA and histone H2B in proximity to CD4+ T cells secreting IL-17 (60). The potential receptors on T cells that mediate interactions with bacteria leading to TET induction are currently unknown (Figure 2).
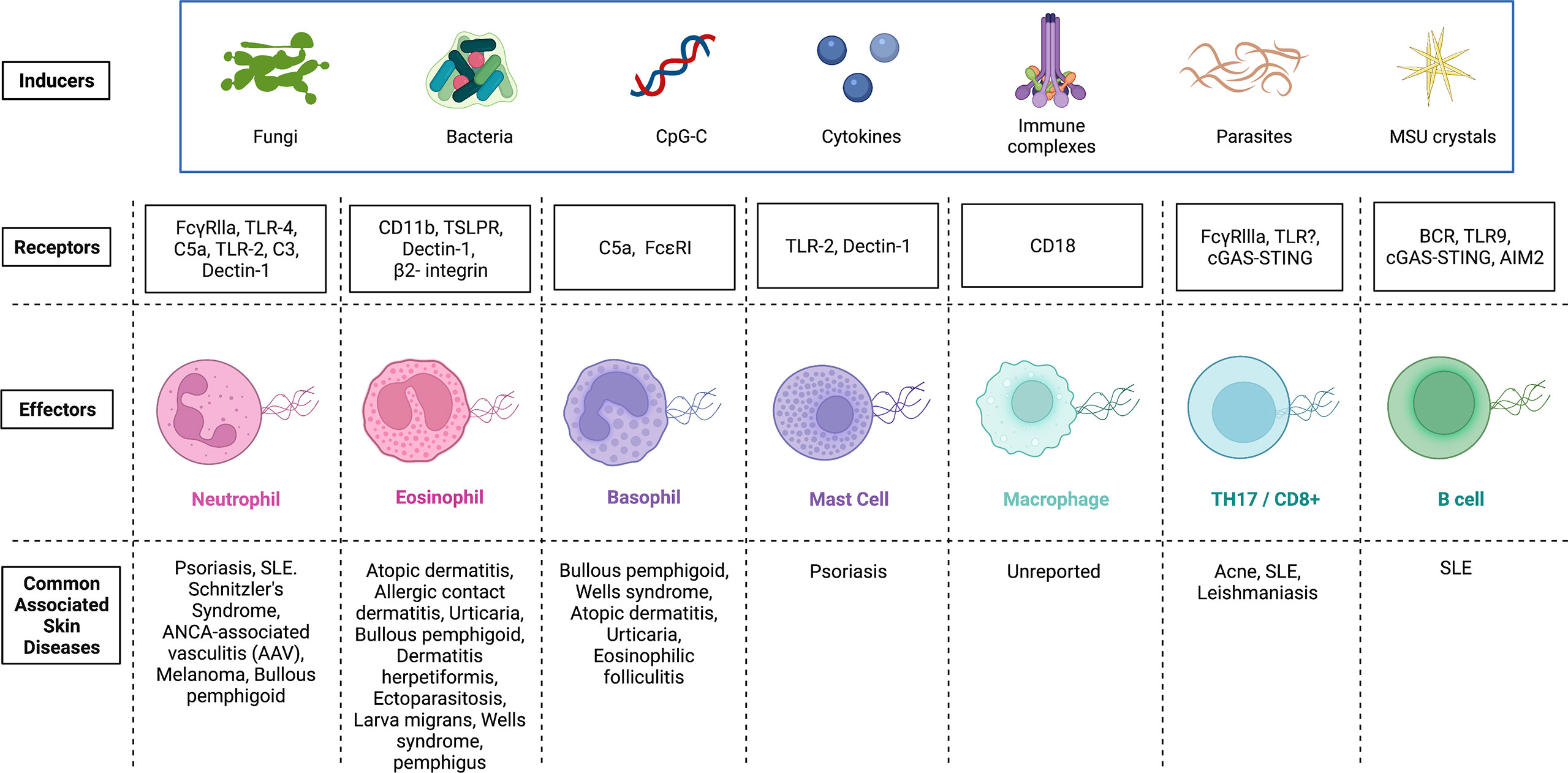
Figure 2 Inducers and potential receptors that activate the release of extracellular traps in immune cells and the associated skin diseases. Extracellular traps were first identified in neutrophils, and later, this phenomenon was detected in several other immune cells. Although there exists some overlap, inducers and receptors vary widely. Activation of immune cells by microbes, CpG-C, cytokines, immune complexes, parasites and MSU crystals can initiate the release of extracellular traps. Receptors on immune cells (Effectors) that have been identified and demonstrated to interact with inducers of extracellular trap formation are shown. Mechanisms by which ET formation are initiated in various effector cells vary and any dysregulation of each cascade can be pathological. Skin diseases associated with dysregulated extracellular trap formation are highlighted. FcγRs, Receptors for the constant region of IgG immunoglobulin; TLR, Toll-like receptor; C5a, Complement component 5a; C3, Complement component 3; TSLPR, Thymic stromal lymphopoietin receptor; FcϵRI, High affinity IgE; BCR, B cell receptor; STING, Stimulator of interferon genes; MSU, Monosodium urate; CpG-C, Synthetic short single-stranded DNA molecules made of cytosine triphosphate deoxynucleotide. Created with Biorender.com.
Histones and Extracellular Traps
Our findings were the first to demonstrate that AMTH17 cells make TETs loaded with histones H2B and H4 and that these AMTH17 cells secrete molecules, which could kill different bacterial strains (60). Through confocal imaging of acne lesions, we were able to detect TETs that were loaded with histones H2B, suggesting that ETs may play an important role in the clearance and elimination of pathogens within the extracellular matrix during acne disease progression (60). In unstimulated AMTH17 cells, we discovered that core histones, such as H2A, H2B, H3 and H4, had similar levels of expression. However, in TETs, H2B and H4 were found in higher concentrations than H2A and H1, and antibodies to histones H2B and H4 led to 60% reduction in TET-mediated bacterial killing (60). Similarly, neutralizing antibodies against core histones H2A and H2B interfered with NET-mediated bacterial killing and elimination of bacteria, further highlighting the importance of histones (102). Since ETs and histones in NETs and TETs can mediate injurious effects, a fine balance is needed between the host defense benefits and adverse effects that can be orchestrated by ETs (60, 118).
The antibacterial activity of histones is well documented, with first reports of antibacterial activity of histones and histone-like proteins originating in 1942 (119). Four core histones (H2A, H2B, H3, and H4) form an octamer, around which DNA enfold within nucleosomes (120). Apart from being part of the nucleosome structures, these core histones also play an important role in protecting the host epithelial surfaces from microbial invasion (120). Histones are predominant in the ETs of neutrophils, eosinophils, basophils, macrophages and other innate immune cells (102, 121–123). Pioneering studies by Hirsch et al. demonstrated the antimicrobial effect of arginine-rich histones against bacteria (124). Subsequent studies, including ours, have also shown that lysine-rich histones have bactericidal activity (60, 120). Lysine-rich histones, such as histones H2A and H2B, are highly expressed on the placental epithelial surface and have been suggested to be important in providing fetal protection against in utero microbial infection (125). Additionally, histones H2A and H2B are also effective in the neutralization of endotoxin, a major structural component of Gram-negative bacterial cell walls, and an important virulent factor during microbial colonization (24). From our work, H2B-loaded TETs released by the AMTH17 cells are antimicrobial against C. acnes, E. coli, Pseudomonas aeruginosa, and S. aureus (60). Furthermore, AMTH17 cells also released granulysin. Importantly, histone H2B gene expression is highly correlated with granulysin activity, which was consistent with the observed antimicrobial action. However, how this increased antimicrobial response is activated in vivo is unknown (120). While histone H3 exhibits antibacterial activity against both E. coli and S. aureus, less is known about its antibacterial actions on other bacteria (24). Histone H4 mediates antimicrobial activity through disruption of the cell membrane. Human sebocytes, the cells comprising sebaceous glands, can release H4, which is bactericidal against C. acnes (24). We observed the expression of the arginine-rich histone H4 in AMTH17 cells (60) and the antibacterial activity of H4 could be intensified by free fatty acids found on acne skin (24).
Formation of Extracellular Traps (ETs) by Neutrophils
Immune cells are able to fight pathogens using various modes of action. They can secrete antibacterial proteins stored in granules, phagocytose microbes and then kill them intracellularly by antibacterial proteins or reactive oxygen species, or form and release ETs. The most well-characterized cells to secrete ETs are neutrophils (NETs). In addition to their phagocytic function, neutrophils utilize NETs for extracellular microbial killing (102). Different mechanisms are involved in NET formation. Generally, NET formation is a cell-destructive process, during which disassembly of nuclear material and disintegration of intracellular organelle membranes occur. After rupture of the plasma membrane, release of DNA, granular and cytoplasmic proteins (e.g. MPO and elastase) can facilitate extracellular killing of various pathogens, such as bacteria and fungi. Importantly, microbes and microbial components can induce NET formation (102, 116, 126–131). These inducers include bacteria or their structural components, such as lipoteichoic acid, lipopolysaccharides and bacterial breakdown products (132).
Proposed Mechanisms of ET Release by B and T Lymphocytes
The first reported descriptions of lymphocyte-derived ETs demonstrated that both T and B cells secreted DNA following stimulation with SLE serum and other inflammatory stimuli (113, 133). The treatment with anti-IgM + lipopolysaccharides (LPS), which mimics stimulation of B cells under physiological conditions, initiated the release of DNA into the extracellular milieu, which suggests that ET release by B cells occurs in vivo (113). With regard to B cells, immune complexes can induce FcγRIIB activation independent of BCR-specific antigen binding (134, 135). Immune complexes can similarly mediate BCR cross-linking and induce downstream signaling pathways, promoting the secretion of ETs. DNA produced by ionomycin-stimulated peripheral blood B cells had high molecular weight, with no observed random or internucleosomal fragmentation that is observed in necrosis or apoptosis, respectively (136). Thus, B cell released DNA has similar characteristics to NETs (137, 138). Notably, the overall purity of B cell preparation was 80%, and therefore future work should examine the composition of the DNA extruded into the extracellular environment by B cells. The identification of proteins including histone composition or citrullination status will further help define the B cell-derived ETs (113).
ET formation in CD8+ T lymphocytes has been well characterized (139). Koh et al. demonstrated that upon activation of CD8+ T cells with α-CD3/CD28, these cells released ETs that they termed lymphocyte extracellular traps (LETs). LETs were shown to be morphologically different from CD4+ T cells-induced ETs as they appeared as long thin filaments that connected neighboring cells together. In contrast, CD4+ T cell-derived ETs formed a diffused patterns that appeared as a halo. Importantly, released LETs were loaded with CD107a+ cytotoxic vesicles that could kill distant target cells. The downside of this cytotoxic mechanism was that LETs release by CD8+CD107a+ T cells was associated with increased inflammatory infiltrates and subsequent severe disease in patients with cutaneous and mucosal leishmaniasis, suggesting that LETs could drive disease pathology within leishmania lesions. However, this study did not identify the CD8+ T cell subpopulations that were responsible for LET release (139).
Costanza et al. demonstrated that activated human and mouse CD4+ T cells produce extracellular DNA, which they termed, T helper-released extracellular DNAs or threads (114). However, the responsible T cell subset also remained unknown. Using RNA-seq and functional analysis of CD4+ TH17 cells, we demonstrated that AMTH17 cells, but not the n-AMTH17, TH1 or TH2 cells, released TETs that trap bacteria (60). After entrapment, most of the bacteria were killed (60). Further, T cell-derived DNA strands contained both mitochondrial and nuclear DNA, suggesting that both are involved in the process of TET formation (60, 114). Further exploration to determine whether the formation of TETs (TETosis) resembles vital (involving vesicular exportation or mitochondrial DNA) or a suicidal process is needed. However, it is plausible that several mechanisms contribute to the release of DNA traps by CD4+ T cells. These mechanisms are discussed below.
Vital TETosis
Contrasting prior studies detailing the mechanisms of NETosis as a process that required several hours, we noted that C. acnes-stimulated TET formation in AMTH17 cells occurred within just 30 min (60). These TETs were likely induced by the recognition of bacteria or bacterial products. Using confocal microscopy, we demonstrated that activated AMTH17 formed TETs comprised of DNA decorated with histones, proteases and cytosolic proteins. These proteins ensnared bacteria, as well as provided large concentration of antimicrobial molecules, such as granulysin, that assist in trapping and killing of bacteria. This phenomenon has also been reported in neutrophils, as neutrophils that release NETs remained impermeable to SYTOX Green, suggesting that their structure remained intact. In neutrophils, vital NETosis has clearly been demonstrated by Pilsczek et al. as a process that involves vesicles of DNA budding off from the nuclear envelope, and delivered out of the cells without membrane perforation (140). Likewise, TH17 cells also remain structurally intact after TET formation, although additional studies are required to elucidate the precise mechanisms involved in vital TETosis. However, based on the similarities with NETs, we envisage a process where bacteria induce TET release through an initial blebbing event that is followed by substantive protrusions of the nuclear envelope. This is followed by transient detachment of the blebs/vesicles and transport of the vesicles through the cytoplasm and subsequent release of the DNA-loaded ETs in vitro and in vivo (133, 140–142). As a result, this proposed mechanism preserves the structural integrity of the T cell plasma membranes (Figure 3). It still remains unclear whether a TH17 cell that has ejected (parts of its) DNA is still viable and capable of performing T cell function, such as antigen recognition, immunomodulation, and cytokine secretion.
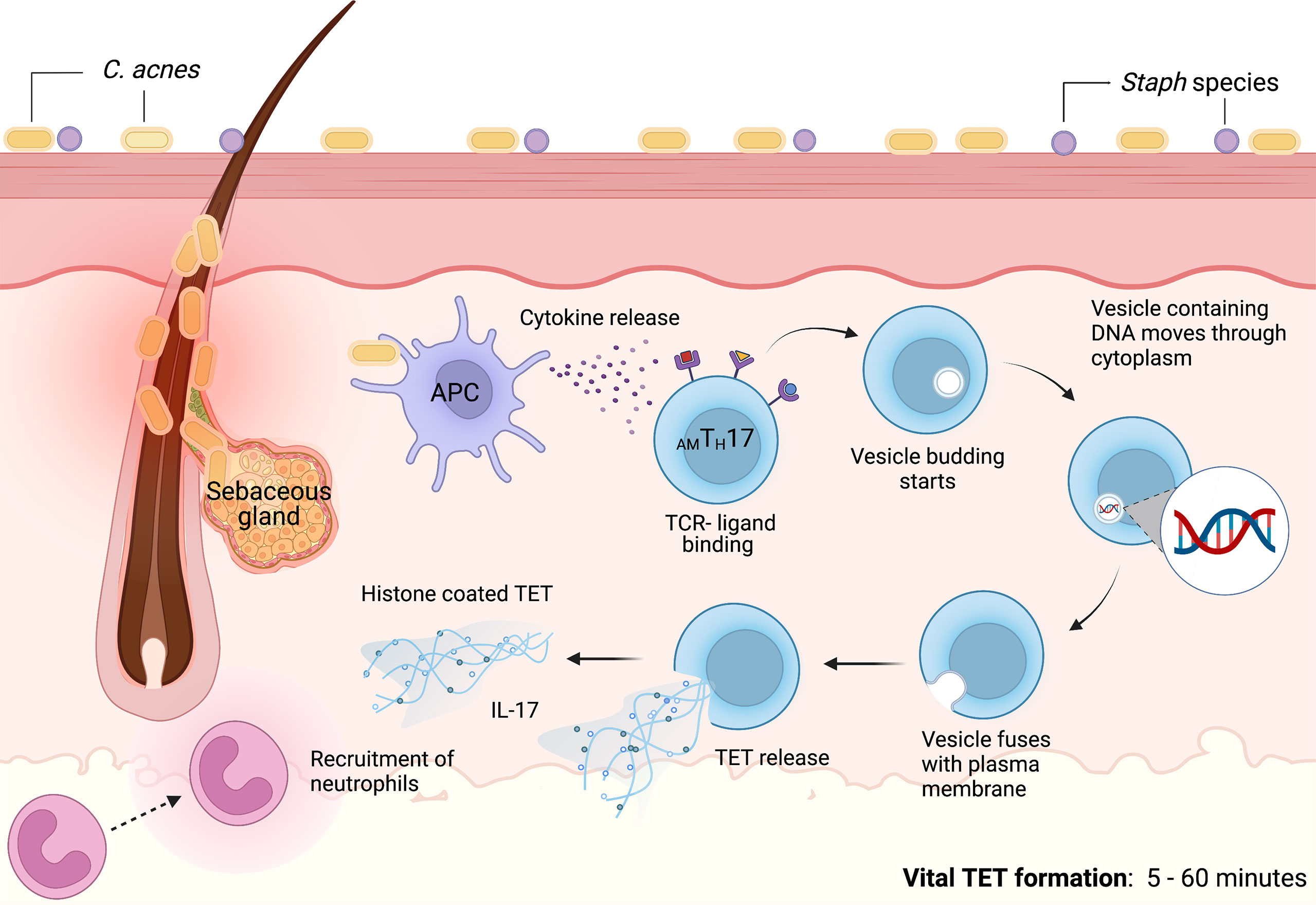
Figure 3 Proposed mechanism of vital TET formation in acne. In the pilosebaceous follicles, C. acnes strains trigger antigen presenting cells to secrete cytokines that induce naïve T cells to differentiate into distinct T cell subsets. Upon induction, AMTH17 cells can further be activated by bacterial ligands, PMA and other stimuli. TCR-ligand binding induce the formation of vesicles containing DNA. Blebbing of T cell nuclear envelope, vesicular exportation and exocytosis of vesicles containing DNA generate extracellular traps while preserving the integrity of the cell. As a result, AMTH17 cells retain conventional functions of viable T cells such as cytokine secretion. In vitro, we have observed that AMTH17 cells release TETs as early as 30 minutes after stimulation with C. acnes. Both the released TETs and IL-17 can recruit neutrophils to inflamed acne lesions. TETs trap C. acnes and are also loaded with antimicrobial molecules, including granulysin and histone H2B, H3 and H4 that kill bacteria. Created with BioRender.com.
Vital TET Formation: Mitochondrial DNA
ETs produced in response to bacterial invasion have been shown to contain nuclear DNA as their critical structural component (143–146). However, other studies have reported ETs are composed of mitochondrial DNA (mtDNA) suggesting that the mitochondria can be involved in the process of vital ET formation and inflammatory response (147) (Figure 4). For example, GM-CSF primed neutrophils, upon stimulation via TLR4 or complement receptor 5a, produced ETs comprising of only mtDNA. In addition, in vivo, NETs containing mtDNA are seen in the serum of individuals post-trauma (148) and after orthopedic surgery (143). mtDNA-facilitated NET formation appears to be ROS-mediated (147). Interestingly, Yousefi et al. found ROS was essential for the release of mtDNA-NETs as mtDNA-NET formation was blocked when a ROS production inhibitor (diphenyleneiodonium) was used (147). However, the exact molecular mechanism of mtDNA-ET release is not fully understood in both neutrophils and lymphocytes. Whether mtDNA is involved in TH17-mediated TET formation remains to be determined.
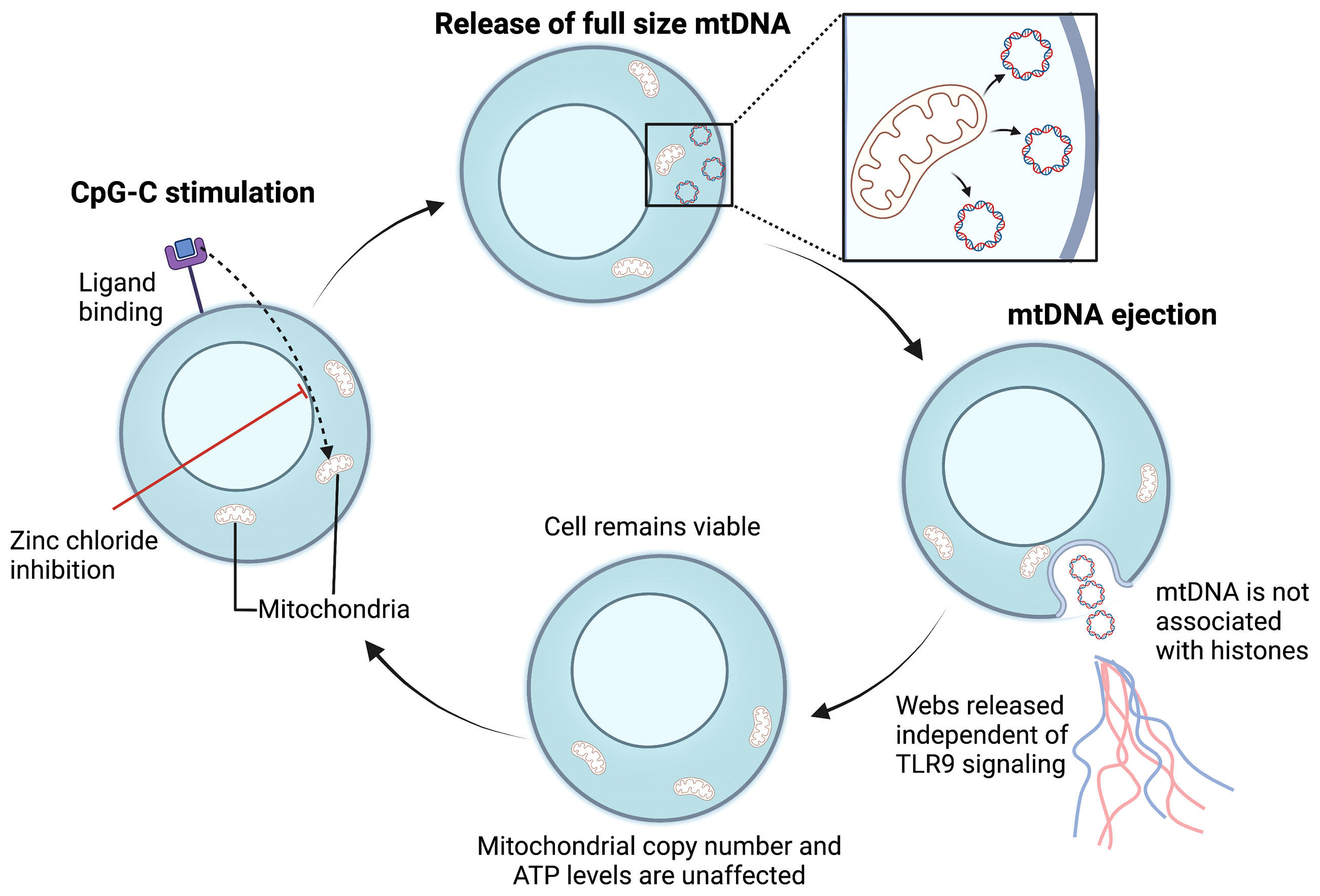
Figure 4 Proposed mechanism for mitochondrial vital TET formation. In B cells, CpG-C activation initiates the ejection of full-sized mtDNA that is free of histones (133). Free mtDNA enters the extracellular space in a web-like configuration, independent of TLR-9 activation. Following mtDNA ejection, the cell membrane is preserved, and the number of mitochondria remain unaffected. The cycle is repeated upon CpG-C re-stimulation and mtDNA release is an active process. Whether bacterial ligands can also induce ETs composed of mtDNA in T cells warrants further investigation. mtDNA, mitochondrial DNA; TLR, Toll-like receptor; ATP, Adenosine triphosphate; CpG-C, Synthetic short single-stranded DNA molecules made of cytosine triphosphate deoxynucleotide. Created with BioRender.com.
mtDNA is a dominant driver of systemic inflammatory response after injury (143, 149, 150). It is recognized as an “alarmin” (151) that can stimulate the innate immune system at physiological concentrations. Free mtDNA from tissue and necrosis following injury or mtDNA-NETs could possibly re-activate neutrophils and other cells within the acne microenvironment through TLR2 and generate further ET production (19, 152). ET formation from neutrophils occurs through activation of TLR4 by another alarmin, more specifically, the high-mobility group box protein 1 (HMGB1) (153).
A recent study demonstrated a clear link between Severe acute respiratory syndrome associated coronavirus (SARS-CoV)-induced tissue damage in the skin and lungs of COVID-19 patients to the activation of the cyclic GMP-AMP synthase (cGAS- stimulator of interferon genes (STING) pathway and type I IFN signaling (58, 59). SARS-CoV-2 infection caused disruption of mitochondrial homeostasis in lung epithelial and vascular endothelial cells, leading to the accumulation of mtDNA. Both the mtDNA and DNA released from dying cells subsequently activated the cGAS-STING pathway and led to the secretion of type I IFNs and pro-inflammatory cytokines that further promoted hyperinflammation and tissue damage. This study clearly demonstrated that infections that disrupt mitochondrial function can lead to immunopathology. Whether the observed mtDNA was released by the process of vital ET formation is unknown. Additionally, the mechanisms and detrimental responses that might be induced by mtDNA and/or damaged DNA released from dying sebocytes and other cells within a disrupted PSU microenvironment in acne lesions are yet to be elucidated. Therefore, it is reasonable to speculate that mtDNA release by neutrophils, TH17 cells and other immune cells during chronic inflammation and/or after tissue injury could feed into a vicious cycle of immune activation. Such a vicious cycle could exacerbate the chronic inflammatory responses that are observed in acne (Figure 1).
Suicidal TETosis
In neutrophils, suicidal NETosis is frequently initiated by ligand binding to neutrophil TLRs and receptors for IgG-Fc, complement or cytokines (102, 154, 155). Release of NETs was initially proposed to require the complete rupture of the neutrophil, a process that was termed NETosis (137). As such, NETosis is a cell death process, unlike apoptosis and necrosis, that relies on the generation of ROS by reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, via PKC and Raf-MEK-ERK signaling pathways (156–160).
Whether overstimulation of T cells with C. acnes can lead to suicidal/lytic TETosis in T cells within the acne microenvironment is currently unclear. Further investigations into the signaling pathways involved in TETosis are required. As has been observed in neutrophils, we speculate that the mechanisms by which TETs are formed will depend on the stimulus and the duration of the stimulation.
It has also been reported that S100A9-deficient neutrophils that are induced by S. aureus produce more mitochondrial superoxide, resulting in the release of NETs through suicidal NETosis. Increased suicidal NETosis failed to improve neutrophil removal of S. aureus in isolation. Rather, enhanced macrophage trapping and killing of bacteria was mediated by increased phagocytosis and the direct action of antimicrobial peptides (161). It has, therefore, been proposed that accelerated NET formation can be an immune-mediated mechanism that can amplify the antibacterial activity of macrophage (161). However, it is important to highlight that various pathogens such as S. aureus have developed survival mechanisms involving the secretion of nucleases and DNAses that allow them to evade and uncouple cooperation among immune cells through ET degradation (161). Our proposed mechanism of suicidal TETosis is illustrated in Figure 5 and may likely resemble NETosis as previously described (156–160).
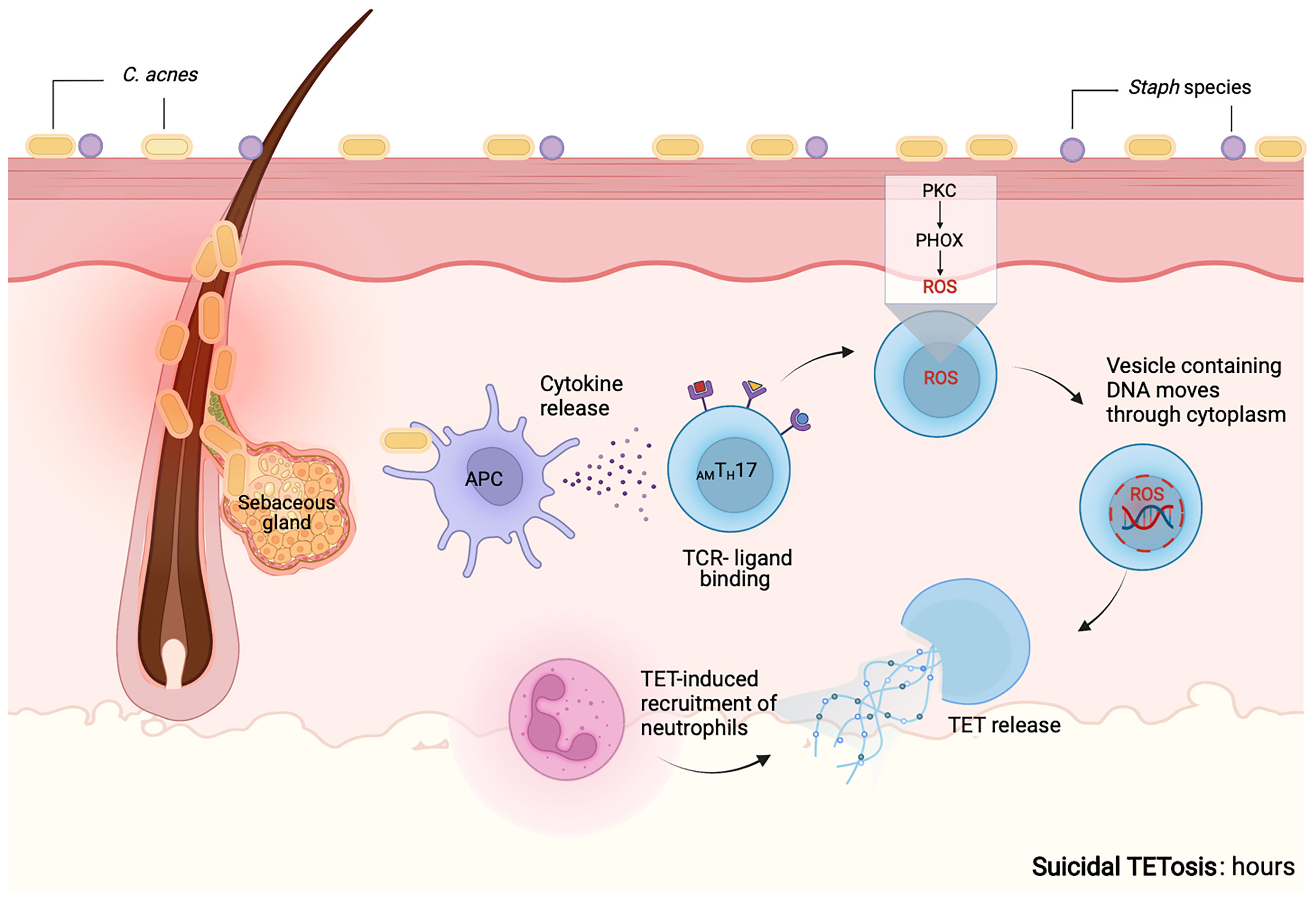
Figure 5 Proposed mechanism for suicidal TETosis. Similar to neutrophils, we propose that upon activation of TLR and receptors for IgG-Fc, complement or cytokine receptors on T cells, calcium is released from the endoplasmic reticulum into the cytoplasm. Higher levels of calcium within the cytoplasm, promote the activity of protein kinase C (PKC). This further induces the assembly of both the membrane-bound and cytosolic subunits of NADPH oxidase (phagocytic oxidase, PHOX) into functional complexes. This is followed by the release of reactive oxygen species (ROS), that causes the granules and the nuclear envelope to rupture. Following rupture both the nuclear and cytoplasmic contents are able to mix. Enzymes associated with the nucleus and the cytoplasm go on to degrade the linker histone H1, further processes the core histones (H2A, H2B, H3 and H4) and amplify the decondensation of chromatin. These events eventually lead to the rupture of the plasma membrane and the TETs are released into the extracellular space (160). The release of TETs can lead to T cell death and the loss of viable T cell functions such as antigen recognition and cytokine release. Enzymes that may mediate the proposed molecular processes are not known. Created with BioRender.com.
The Duality of Extracellular Traps
Accumulation, incomplete removal, and improper localization of ETs can promote inflammation and cellular damage during infections and sterile inflammatory conditions (122). Although TETs are an important component of the host antimicrobial response against extracellular pathogens, we cannot preclude the possibility of TETs contributing to pathology. In psoriasis, NETs promote TH17 induction as part of the pathophysiology of the disease (162). Additionally, correlation of presence of NET-associated DNA with pathology has been implicated in other inflammatory skin diseases (Figure 2). Still, exploration of whether these observations are true, and whether C. acnes–induced TETs are part of the inflammatory cascade observed in acne is currently unclear (102, 163). In addition, it is plausible that the combined activity of CAMPs and histone-loaded TETs can lead to reduction of bacteria load within the PSU of acne patients. Further studies are needed to identify whether TETs are a double-edged sword in acne pathogenesis.
In summary, ETs have been implicated in diseases related to tissue injury: impaired wound healing, autoinflammation, autoimmunity, and tumor metastasis, highlighting their contributory role in numerous dermatological pathologies. Some select pathologies are discussed below.
ETs in Tissue Injury and Wound Healing
Initially, it was proposed that ETs serve a protective function for the host by controlling infections. During pathogen invasion, these ETs can capture and degrade invasive microbes (164). However, additional studies have revealed that dysfunctional release and clearance of ETs can harm the host and contribute to disease (Figure 2). Structural components of ETs (i.e. decondensed DNA skeleton tangled with histones and granules) can function as damage associated molecular patterns (DAMPs), inciting additional tissue and organ injury (165–168). For example, the overabundance of histones decorating DNA filaments of traps promotes platelet aggregation and platelet dependent thrombin generation, resulting in disruption of circulation (159). Furthermore, NETs are associated with enzymes, myeloperoxidase (MPO) and elastase, that activate the immune system. When overexpressed, both enzymes can damage the epithelial barrier of the lung (169). Additional research is needed to elucidate whether TETs are associated with similar peroxidase and protease enzymes and whether these enzymes contribute to TET-induced tissue damage.
Degradation of the DNA backbone of ETs triggers the release of antibacterial proteins and upregulates inflammatory cytokines, such as type I interferon, IL-17, and IL-8 (170–173). While hyperinflammation is important for fighting infections, after injury, hypoinflammation is needed during tissue healing and repair (174). Therefore, overabundance of TETs during repair is counterproductive, unless there is continued need for protecting against inadvertent infections. In the case of acne, overabundance of TETs may be associated with hyperinflammation and tissue damage, contributing to acne scarring. It is plausible that skin conditions associated with aberrant wound healing, such as keloids, may be related to dysregulated TET formation and removal, but evidence on TET formation in keloids is currently lacking.
ETs in Autoinflammation and Autoimmunity
Prior research demonstrates that NET components activate APCs, which present self-antigens to corresponding lymphocytes, leading to autoreactivity. Presence of ETs within the extracellular environment has been observed in various autoimmune diseases, including psoriasis, rheumatoid arthritis, and systemic lupus erythematosus (SLE) (175–177). In psoriasis, NETs, their citrullinated histones and antigens stimulate immunocompetent cells to produce antibodies against cytoplasmic antigens (ANCAs) and antibodies against nuclear antigens (ANAs) (175, 176). These antibodies act against neutrophil-associated enzymes and activate neutrophils to release more NETs (178). Autoantigens released by NETs become the target of successive antibodies perpetuating the development of autoimmune disease. In addition, abnormal NET formation and inefficient degradation can also initiate and/or facilitate autoimmune diseases (144, 169, 175, 176, 179). In SLE, ANCAs and ANAs lead to the generation of immune complexes and chronic activation of plasmacytoid dendritic cells (169). The inappropriate release of intracellular autoantigens during apoptotic cell removal caused continual production of autoantibodies; this cycle is considered the key pathogenic mechanism in SLE (154, 169). By binding DNA and cathelicidin (LL-37) of NETs, autoreactive antibodies form autoantibody-DNA/LL-37 complexes, which can be detected by other immune cells as a signal to “sustain” the autoimmune response (154). A relationship between inadequate NET degradation and SLE development has also been proposed (169). The ability to degrade NETs increased during SLE remission but decreased during relapse (176). Moreover, patients with increased SLE severity had higher NET antibody levels. In SLE, non-degraded NETs activate components of the complement system, such as C1q, which breaks down NETs. The opsonization of NETs and/or inhibition of DNase I activity were hypothesized to be responsible for this process (176). Thus, the continued presence and mis-regulated degradation of NETs contribute to the autoimmunity.
ETs in Cancer
Identification of ETs in blood and solid tumors of animals and humans suggests that traps play a role in cancer. However, there exists conflicting evidence as to whether ETs serve as pro- or anti-tumoral factors (180, 181). For example, studies on NETs have shown that several NET components, such as MPO, proteinases, and histones are cytotoxic, whereas other studies have proposed that NETs promote metastasis (182–184). By increasing the concentration of LPS and upregulating the complement component, C3a, NETs induce more coagulation and tumorigenesis, further feeding into a positive-feedback loop that favors a pro-tumorigenic phenotype (185–187). These potential oncological mechanisms have been described for neutrophils but have yet to be studied in T cells. Given the crucial role of T cells in cancer, exploration of TETs in cancer is an exciting topic of interest.
Murine models demonstrate that ETs contribute to skin cancer and inflammation-mediated skin tumor cell growth (188, 189). NETs contribute to adverse reactions in murine melanoma models, regardless of whether they are spontaneous or immunotherapy-induced (190). In melanoma studies, IL-8 and type I IFNs act as potential therapeutic targets associated with NETs (191–193).
TETs have not been investigated in cutaneous cancers, such as cutaneous T-cell lymphomas (CTCL). For example, there is no evidence regarding ET production in Sézary syndrome (SS), an aggressive and rare leukemic form of CTCL (194). The chemokine CXCL8 is highly expressed in CTCL skin lesions and can act in concert with IL-8 and IL-17 to facilitate the priming and recruitment of neutrophils to the CTCL microenvironment. However, in CTCL patients, there is no evidence demonstrating the release of NETs within the CTCL tumor microenvironment, even though neutrophils isolated from the peripheral blood of these patients are phenotypically active (195). As the disease progresses, other cells such as myeloid-derived suppressor cells become activated and secrete ROS (196, 197). Increased ROS release has been linked to T cell tolerance and unresponsiveness within CTCL skin lesions (198). Additionally, if present within CTCL, neutrophils show compromised functionality, with diminished ingestion and intracellular killing of pathogens, as well as reduced NET production. Moreover, impairment of the host immune response against pathogens increases susceptibility to severe infections, and complications observed in CTCL patients (199, 200).
As the CTCL progresses, the tumor microenvironment is associated with increasing expression of TH2 TFs and cytokines (e.g. GATA-3, IL-4, IL-5, and IL-13) and declining levels of TH1 and TH17 associated TFs and cytokines (e.g. T-bet, RORγt, IL-12, IL-17 and IFN-γ) (201–207). Accordingly, late-stage CTCL is dominated by a TH2 tumor microenvironment and small numbers of TH1 cells and CD8+ T cells (205, 208–212). The increased TH2-bias is believed to be a key process that suppresses cellular immunity and anti-tumor responses in CTCL (213, 214). It has also been observed that different cellular sources of IL-17 may exist within the CTCL microenvironment. However, these IL-17-secreting cells fail to express the characteristic TH17 phenotype, indicating that IL-17 production may originate from dysregulated signaling and cannot be classified as a true TH17 response in SS (201, 215). Understanding how the low numbers of neutrophils, TH1 and TH17 TETs promote or inhibit tumorigenesis in CTCL may inform future development of skin cancer therapies and treatments.
Research Gaps in TETs
Since the discovery of ETs two decades ago, substantial advances in defining their role in immune-mediated antimicrobial activity have been made (216). Processes related to their formation and mechanism of action are being identified. It is now generally accepted that ET formation is not unique to neutrophils as this phenomenon has also been observed in other immune cells (60, 103–112). Furthermore, work from our lab and others demonstrate that adaptive immune cells, namely antigen-specific T cells, extrude ETs suggesting that lymphocyte ETs serve as an important link between cells of the innate and adaptive immune system. Recent findings provide new perspectives on how T cell-derived ETs are involved in antimicrobial responses and influence host immune homeostasis. Though significant progress has been made regarding ETs, the complete role of TETs in host defense and inflammation is still unclear. More work is needed to elucidate the unique role of TETs and how that may inform treatment of ET-associated diseases.
Even though in vitro and in vivo formation of TETs has been observed, huge research gaps exist in our knowledge about how TETs form in general. Various particles and microbial agents can induce the release of histone-coated DNA ETs by T cells, but additional research is needed to identify the exact stimuli and receptors that induce TET formation (60, 113, 133, 139). Defining the distinctive characteristics of these stimuli is critical to advancing our understanding of diseases with TET-related pathophysiology. Equally important, research is needed to understand how ET formation is blocked. For example, the bacteria, Lactobacillus rhamnosus, inhibits NET release suggesting that different microbial species can act as agonists or antagonists of TET formation (217, 218). This brings up the interesting possibility that agents that modulate the microbiome, probiotics or antibiotics, may be beneficial in the management of TET-related diseases. An improved understanding of the role genetics play in TET formation and activity can also guide future therapeutics. Some NET associated diseases, such as psoriasis, have genetic component, so research efforts should pursue putative gene variants that predispose individuals to TET dysfunction. Understanding how host genetics influence TET activity may help us prognosticate individuals who are at risk for pathological conditions linked to extracellular traps.
Dissecting the mechanisms involved in TET induction and release may inform how TET activity can be moderated. With the introduction of novel computational and imaging algorithms that utilize high content screening-cellomics platform, efficient and precise detection of ETs are now possible (219). This high-throughput screening method uses cell membrane specific DNA dyes (permeable and impermeable) in situ to discern the cellular and morphologic characteristics of ET-forming cells. The combination of high-throughput screening with single-cell analysis of morphological changes within the nucleus and chromatin dynamics can provide precise detection of ET-forming cells with attention to specificity while eliminating user bias that is seen with other cell death assays. Furthermore, combining live cell imaging with staining for cell death markers in situ will help identify the specific death pathway and thus, discriminate between NETs, TETs, apoptosis, and necrosis (219).
An improved understanding of the uni-directional and/or bi-directional communication pathways between TETs and immune cells is also needed to fully understand the ultimate cellular response to TETs. We know that NETs do not always work in isolation. NETs can work cooperatively with macrophages in granuloma formation. NETosis by itself does not improve the killing potential of neutrophils against S. aureus but rather it augments macrophage microbial killing (161). Even though TH17 cells are adaptive immune cells,, the extracellular trap timing and response are more characteristic of an innate immune response; thus, TH17 cells may link the two arms of immunity by working cooperatively with other cells. TH17 cells recruit neutrophils and other proinflammatory cells to the infection site, where neutrophils activate and promote differentiation of TH17 cells, suggesting cross-talk between innate and adaptive cells (220, 221). These cooperative mechanisms have strong clinical relevance, as NETs have been connected to tumor progression and metastasis (185). Therefore, more research is needed to understand how TH17 cells coordinate host defense mechanisms with other cells.
Insights into how ET formation acts as a conduit for various immune cells, including neutrophils, T cells and macrophages, to fight microbes cooperatively within the deeper layers of the skin during acne disease is of particular importance. We have shown that AMTH17 cells release TETs that are coated with histones as part of the host antimicrobial defense. The fact that AMTH17 cells release ETs implies that this TH17 subpopulation can act as an important link between the innate and adaptive arms of the immune response, which ensures the efficient capture and destruction of invading microbes. However, there is a need to delineate the specific CD4 + T cell populations that have a capacity to release ETs, as the molecular markers that can be used to identify, purify and isolate TET-forming T cells are yet to be discovered. In the interim, it is possible that TET-forming TH17 have a specific pro-inflammatory signature as TH17 cells differentiated in the presence of IL-23 provoke EAE, but not TH17 induced by IL-6 and TGFβ alone (76). We are still in the beginning of understanding how the cytokine milieu influence the plasticity and heterogeneity of TH17 cells into either pathologic or non-pathologic conditions within different inflammatory settings. Yet, the verification of TH17 cell plasticity in humans and its functional importance implies that during pathological conditions, both anti-inflammatory and antimicrobial therapeutics should be designed to specifically target recruited cells while sparing TH17 subpopulations and other tissue-resident cells that promote skin tissue repair. Teasing apart, the likely, yin and yang activities associated with TET-forming TH17 cells will be of future interest (60).
Many unanswered questions remain when it comes to TETs. Future studies should focus on clarifying which pathologies, including autoimmune diseases, cancer, and chronic inflammation, that are associated with T cell derived ETs. In doing so, we may also begin to identify autoantigens that may be associated with TETs, thereby improving our understanding of the underlying mechanisms contributing to these diseases. Simultaneously, we will gain a greater understanding and appreciation for the critical protective role that TETs serve. As ET and TET research unfolds, therapeutic opportunities to exploit and control the formation, modulation, and regulation of these ETs for the treatment of inflammatory and autoimmune diseases may be on the horizon.
Author Contributions
KO: data curation, formal analyses, data interpretation and manuscript writing; NO: data curation, formal analyses, data interpretation and manuscript writing; AN: manuscript writing/editing; GA: conceptualization, data curation, data analyses and interpretation, funding acquisition, methodology, project administration, resources, supervision, visualization, and manuscript writing/editing and submission. All authors edited the manuscript, gave final approval for publication, and agreed to be accountable for the work. KO and NO contributed equally to this work.
Funding
This work was supported by NIH K01AR071479 (GA) and NIH R01AR081337 (GA).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hall JB, Cong Z, Imamura-Kawasawa Y, Kidd BA, Dudley JT, Thiboutot DM, et al. Isolation and Identification of the Follicular Microbiome: Implications for Acne Research. J Invest Dermatol (2018) 138(9):2033–40. doi: 10.1016/j.jid.2018.02.038
2. Grice EA, Kong HH, Renaud G, Young AC, Program NCS, Bouffard GG, et al. A Diversity Profile of the Human Skin Microbiota. Genome Res (2008) 18(7):1043–50. doi: 10.1101/gr.075549.107
3. Ho AW, Kupper TS. T Cells and the Skin: From Protective Immunity to Inflammatory Skin Disorders. Nat Rev Immunol (2019) 19(8):490–502. doi: 10.1038/s41577-019-0162-3
4. Agak GW, Qin M, Nobe J, Kim MH, Krutzik SR, Tristan GR, et al. Propionibacterium Acnes Induces an IL-17 Response in Acne Vulgaris That Is Regulated by Vitamin A and Vitamin D. J Invest Dermatol (2014) 134(2):366–73. doi: 10.1038/jid.2013.334
5. Marples RR. The Microflora of the Face and Acne Lesions. J Invest Dermatol (1974) 62(3):326–31. doi: 10.1111/1523-1747.ep12724285
6. Chiller K, Selkin BA, Murakawa GJ. Skin Microflora and Bacterial Infections of the Skin. J Investig Dermatol Symp Proc (2001) 6(3):170–4. doi: 10.1046/j.0022-202x.2001.00043.x
7. Bruggemann H, Henne A, Hoster F, Liesegang H, Wiezer A, Strittmatter A, et al. The Complete Genome Sequence of Propionibacterium Acnes, a Commensal of Human Skin. Science (2004) 305(5684):671–3. doi: 10.1126/science.1100330
8. Bruggemann H. Insights in the Pathogenic Potential of Propionibacterium Acnes From its Complete Genome. Semin Cutan Med Surg (2005) 24(2):67–72. doi: 10.1016/j.sder.2005.03.001
9. Goh C, Agak GW, Zaenglein AL, Graber EM, Thiboutot D, Kim J, et al. Fizpatrick’s Dermatology. 9 ed. Kang S, Amagai M, Bruckner AL, Enk AH, Maragolis DJ, McMichael AJ, editors. New York: McGraw Hill (2018).
10. Thiboutot D. Hormones and Acne: Pathophysiology, Clinical Evaluation, and Therapies. Semin Cutan Med Surg (2001) 20(3):144–53. doi: 10.1053/sder.2001.28208
11. Plewig G, Fulton JE, Kligman AM. Cellular Dynamics of Comedo Formation in Acne Vulgaris. Arch Dermatol Forsch (1971) 242(1):12–29. doi: 10.1007/BF00595286
12. Leyden JJ. New Understandings of the Pathogenesis of Acne. J Am Acad Dermatol (1995) 32(5 Pt 3):S15–25. doi: 10.1016/0190-9622(95)90416-6
13. Leyden JJ, McGinley KJ, Mills OH, Kligman AM. Propionibacterium Levels in Patients With and Without Acne Vulgaris. J Invest Dermatol (1975) 65(4):382–4. doi: 10.1111/1523-1747.ep12607634
14. Cunliffe WJ. The Sebaceous Gland and Acne–40 Years on. Dermatology (1998) 196(1):9–15. doi: 10.1159/000017859
15. Norris JF, Cunliffe WJ. A Histological and Immunocytochemical Study of Early Acne Lesions. Br J Dermatol (1988) 118(5):651–9. doi: 10.1111/j.1365-2133.1988.tb02566.x
16. Carmina E, Dreno B, Lucky WA, Agak WG, Dokras A, Kim JJ, et al. Female Adult Acne and Androgen Excess: A Report From the Multidisciplinary Androgen Excess and PCOS Committee. J Endocr Soc (2022) 6(3):bvac003. doi: 10.1210/jendso/bvac003
17. Gribbon EM, Cunliffe WJ, Holland KT. Interaction of Propionibacterium Acnes With Skin Lipids In Vitro. J Gen Microbiol (1993) 139(8):1745–51. doi: 10.1099/00221287-139-8-1745
18. Ro BI, Dawson TL. The Role of Sebaceous Gland Activity and Scalp Microfloral Metabolism in the Etiology of Seborrheic Dermatitis and Dandruff. J Investig Dermatol Symp Proc (2005) 10(3):194–7. doi: 10.1111/j.1087-0024.2005.10104.x
19. Kim J, Ochoa MT, Krutzik SR, Takeuchi O, Uematsu S, Legaspi AJ, et al. Activation of Toll-Like Receptor 2 in Acne Triggers Inflammatory Cytokine Responses. J Immunol (2002) 169(3):1535–41. doi: 10.4049/jimmunol.169.3.1535
20. Jugeau S, Tenaud I, Knol AC, Jarrousse V, Quereux G, Khammari A, et al. Induction of Toll-Like Receptors by Propionibacterium Acnes. Br J Dermatol (2005) 153(6):1105–13. doi: 10.1111/j.1365-2133.2005.06933.x
21. Qin M, Pirouz A, Kim MH, Krutzik SR, Garban HJ, Kim J. Propionibacterium Acnes Induces IL-1beta Secretion via the NLRP3 Inflammasome in Human Monocytes. J Invest Dermatol (2014) 134(2):381–8. doi: 10.1038/jid.2013.309
22. Kistowska M, Gehrke S, Jankovic D, Kerl K, Fettelschoss A, Feldmeyer L, et al. IL-1beta Drives Inflammatory Responses to Propionibacterium Acnes In Vitro and In Vivo. J Invest Dermatol (2014) 134(3):677–85. doi: 10.1038/jid.2013.438
23. Li ZJ, Choi DK, Sohn KC, Seo MS, Lee HE, Lee Y, et al. Propionibacterium Acnes Activates the NLRP3 Inflammasome in Human Sebocytes. J Invest Dermatol (2014) 134(11):2747–56. doi: 10.1038/jid.2014.221
24. Lee DY, Huang CM, Nakatsuji T, Thiboutot D, Kang SA, Monestier M, et al. Histone H4 Is a Major Component of the Antimicrobial Action of Human Sebocytes. J Invest Dermatol (2009) 129(10):2489–96. doi: 10.1038/jid.2009.106
25. Lee DY, Yamasaki K, Rudsil J, Zouboulis CC, Park GT, Yang JM, et al. Sebocytes Express Functional Cathelicidin Antimicrobial Peptides and can Act to Kill Propionibacterium Acnes. J Invest Dermatol (2008) 128(7):1863–6. doi: 10.1038/sj.jid.5701235
26. Luckheeram RV, Zhou R, Verma AD, Xia B. Cd4(+)T Cells: Differentiation and Functions. Clin Dev Immunol (2012) 2012:925135. doi: 10.1155/2012/925135
27. Jadali Z. Th9 Cells as a New Player in Inflammatory Skin Disorders. Iran J Allergy Asthma Immunol (2019) 18(2):120–30. doi: 10.18502/ijaai.v18i2.915
28. Schlapbach C, Gehad A, Yang C, Watanabe R, Guenova E, Teague JE, et al. Human TH9 Cells are Skin-Tropic and Have Autocrine and Paracrine Proinflammatory Capacity. Sci Transl Med (2014) 6(219):219ra8. doi: 10.1126/scitranslmed.3007828
29. Clark RA. Resident Memory T Cells in Human Health and Disease. Sci Transl Med (2015) 7(269):269rv1. doi: 10.1126/scitranslmed.3010641
30. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two Types of Murine Helper T Cell Clone. I. Definition According to Profiles of Lymphokine Activities and Secreted Proteins. J Immunol (1986) 136(7):2348–57.
31. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A Novel Transcription Factor, T-Bet, Directs Th1 Lineage Commitment. Cell (2000) 100(6):655–69. doi: 10.1016/S0092-8674(00)80702-3
32. Zhang DH, Cohn L, Ray P, Bottomly K, Ray A. Transcription Factor GATA-3 is Differentially Expressed in Murine Th1 and Th2 Cells and Controls Th2-Specific Expression of the Interleukin-5 Gene. J Biol Chem (1997) 272(34):21597–603. doi: 10.1074/jbc.272.34.21597
33. Zheng W, Flavell RA. The Transcription Factor GATA-3 is Necessary and Sufficient for Th2 Cytokine Gene Expression in CD4 T Cells. Cell (1997) 89(4):587–96. doi: 10.1016/S0092-8674(00)80240-8
34. Tortola L, Jacobs A, Pohlmeier L, Obermair FJ, Ampenberger F, Bodenmiller B, et al. High-Dimensional T Helper Cell Profiling Reveals a Broad Diversity of Stably Committed Effector States and Uncovers Interlineage Relationships. Immunity (2020) 53(3):597–613.e6. doi: 10.1016/j.immuni.2020.07.001
35. Tuzlak S, Dejean AS, Iannacone M, Quintana FJ, Waisman A, Ginhoux F, et al. Repositioning TH Cell Polarization From Single Cytokines to Complex Help. Nat Immunol (2021) 22(10):1210–7. doi: 10.1038/s41590-021-01009-w
36. Jankovic D, Ciucci T, Coffman RL, Coquet JM, Le Gros G, Mosmann TR, et al. Comment on: Repositioning TH Cell Polarization From Single Cytokines to Complex Help. Nat Immunol (2022) 23(4):501–2. doi: 10.1038/s41590-022-01144-y
37. Tuzlak S, Ginhoux F, Korn T, Becher B. Reply to 'Comment on: Repositioning TH Cell Polarization From Single Cytokines to Complex Help'. Nat Immunol (2022) 23(4):503–4. doi: 10.1038/s41590-022-01142-0
38. Kolls JK. Th17 Cells in Mucosal Immunity and Tissue Inflammation. Semin Immunopathol (2010) 32(1):1–2. doi: 10.1007/s00281-010-0198-8
39. Meller S, Di Domizio J, Voo KS, Friedrich HC, Chamilos G, Ganguly D, et al. T(H)17 Cells Promote Microbial Killing and Innate Immune Sensing of DNA via Interleukin 26. Nat Immunol (2015) 16(9):970–9. doi: 10.1038/ni.3211
40. Bystrom J, Clanchy FIL, Taher TE, Al-Bogami M, Ong VH, Abraham DJ, et al. Functional and Phenotypic Heterogeneity of Th17 Cells in Health and Disease. Eur J Clin Invest (2019) 49(1):e13032. doi: 10.1111/eci.13032
41. Wolk K, Witte E, Witte K, Warszawska K, Sabat R. Biology of Interleukin-22. Semin Immunopathol (2010) 32(1):17–31. doi: 10.1007/s00281-009-0188-x
42. Wolk K, Sabat R. Interleukin-22: A Novel T- and NK-Cell Derived Cytokine That Regulates the Biology of Tissue Cells. Cytokine Growth Factor Rev (2006) 17(5):367–80. doi: 10.1016/j.cytogfr.2006.09.001
43. Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, et al. Chronic Mucocutaneous Candidiasis in Humans With Inborn Errors of Interleukin-17 Immunity. Science (2011) 332(6025):65–8. doi: 10.1126/science.1200439
44. Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, et al. Development, Cytokine Profile and Function of Human Interleukin 17-Producing Helper T Cells. Nat Immunol (2007) 8(9):950–7. doi: 10.1038/ni1497
45. Dambacher J, Beigel F, Zitzmann K, De Toni EN, Goke B, Diepolder HM, et al. The Role of the Novel Th17 Cytokine IL-26 in Intestinal Inflammation. Gut. (2009) 58(9):1207–17. doi: 10.1136/gut.2007.130112
46. Silverberg MS, Cho JH, Rioux JD, McGovern DP, Wu J, Annese V, et al. Ulcerative Colitis-Risk Loci on Chromosomes 1p36 and 12q15 Found by Genome-Wide Association Study. Nat Genet (2009) 41(2):216–20. doi: 10.1038/ng.275
47. Corvaisier M, Delneste Y, Jeanvoine H, Preisser L, Blanchard S, Garo E, et al. IL-26 Is Overexpressed in Rheumatoid Arthritis and Induces Proinflammatory Cytokine Production and Th17 Cell Generation. PLoS Biol (2012) 10(9):e1001395. doi: 10.1371/journal.pbio.1001395
48. Vandenbroeck K, Cunningham S, Goris A, Alloza I, Heggarty S, Graham C, et al. Polymorphisms in the Interferon-Gamma/Interleukin-26 Gene Region Contribute to Sex Bias in Susceptibility to Rheumatoid Arthritis. Arthritis Rheumatol (2003) 48(10):2773–8. doi: 10.1002/art.11236
49. Goris A, Marrosu MG, Vandenbroeck K. Novel Polymorphisms in the IL-10 Related AK155 Gene (Chromosome 12q15). Genes Immun (2001) 2(5):284–6. doi: 10.1038/sj.gene.6363772
50. Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, et al. Requirement of Interleukin 17 Receptor Signaling for Lung CXC Chemokine and Granulocyte Colony-Stimulating Factor Expression, Neutrophil Recruitment, and Host Defense. J Exp Med (2001) 194(4):519–27. doi: 10.1084/jem.194.4.519
51. Maddur MS, Miossec P, Kaveri SV, Bayry J. Th17 Cells: Biology, Pathogenesis of Autoimmune and Inflammatory Diseases, and Therapeutic Strategies. Am J Pathol (2012) 181(1):8–18. doi: 10.1016/j.ajpath.2012.03.044
52. Kistowska M, Meier B, Proust T, Feldmeyer L, Cozzio A, Kuendig T, et al. Propionibacterium Acnes Promotes Th17 and Th17/Th1 Responses in Acne Patients. J Invest Dermatol (2015) 135(1):110–8. doi: 10.1038/jid.2014.290
53. Mouser PE, Baker BS, Seaton ED, Chu AC. Propionibacterium Acnes-Reactive T Helper-1 Cells in the Skin of Patients With Acne Vulgaris. J Invest Dermatol (2003) 121(5):1226–8. doi: 10.1046/j.1523-1747.2003.12550_6.x
54. Toyoda M, Morohashi M. Pathogenesis of Acne. Med Electron Microsc (2001) 34(1):29–40. doi: 10.1007/s007950100002
55. Abdel Fattah NS, Shaheen MA, Ebrahim AA, El Okda ES. Tissue and Blood Superoxide Dismutase Activities and Malondialdehyde Levels in Different Clinical Severities of Acne Vulgaris. Br J Dermatol (2008) 159(5):1086–91. doi: 10.1111/j.1365-2133.2008.08770.x
56. Levell MJ, Cawood ML, Burke B, Cunliffe WJ. Acne is Not Associated With Abnormal Plasma Androgens. Br J Dermatol (1989) 120(5):649–54. doi: 10.1111/j.1365-2133.1989.tb01351.x
57. Chiu A, Chon SY, Kimball AB. The Response of Skin Disease to Stress: Changes in the Severity of Acne Vulgaris as Affected by Examination Stress. Arch Dermatol (2003) 139(7):897–900. doi: 10.1001/archderm.139.7.897
58. Domizio JD, Gulen MF, Saidoune F, Thacker VV, Yatim A, Sharma K, et al. The cGAS-STING Pathway Drives Type I IFN Immunopathology in COVID-19. Nature (2022) 603(7899):145–51. doi: 10.1038/s41586-022-04421-w
59. Andreakos E. STINGing Type I IFN-Mediated Immunopathology in COVID-19. Nat Immunol (2022) 23(4):478–80. doi: 10.1038/s41590-022-01174-6
60. Agak GW, Mouton A, Teles RM, Weston T, Morselli M, Andrade PR, et al. Extracellular Traps Released by Antimicrobial TH17 Cells Contribute to Host Defense. J Clin Invest (2021) 131(2):e141594. doi: 10.1172/JCI141594
61. Yang J, Sundrud MS, Skepner J, Yamagata T. Targeting Th17 Cells in Autoimmune Diseases. Trends Pharmacol Sci (2014) 35(10):493–500. doi: 10.1016/j.tips.2014.07.006
62. Fasching P, Stradner M, Graninger W, Dejaco C, Fessler J. Therapeutic Potential of Targeting the Th17/Treg Axis in Autoimmune Disorders. Molecules (2017) 22(1):134. doi: 10.3390/molecules22010134
63. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the Context of an Inflammatory Cytokine Milieu Supports De Novo Differentiation of IL-17-Producing T Cells. Immunity (2006) 24(2):179–89. doi: 10.1016/j.immuni.2006.01.001
64. Ichiyama K, Yoshida H, Wakabayashi Y, Chinen T, Saeki K, Nakaya M, et al. Foxp3 Inhibits RORgammat-Mediated IL-17a mRNA Transcription Through Direct Interaction With RORgammat. J Biol Chem (2008) 283(25):17003–8. doi: 10.1074/jbc.M801286200
65. Wang M, Tian T, Yu S, He N, Ma D. Th17 and Treg Cells in Bone Related Diseases. Clin Dev Immunol (2013) 2013:203705. doi: 10.1155/2013/203705
66. Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming Growth Factor-Beta Induces Development of the T(H)17 Lineage. Nature (2006) 441(7090):231–4. doi: 10.1038/nature04754
67. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal Developmental Pathways for the Generation of Pathogenic Effector TH17 and Regulatory T Cells. Nature (2006) 441(7090):235–8. doi: 10.1038/nature04753
68. Khan D, Ansar Ahmed S. Regulation of IL-17 in Autoimmune Diseases by Transcriptional Factors and microRNAs. Front Genet (2015) 6:236. doi: 10.3389/fgene.2015.00236
69. Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, et al. T Helper 17 Lineage Differentiation is Programmed by Orphan Nuclear Receptors ROR Alpha and ROR Gamma. Immunity (2008) 28(1):29–39. doi: 10.1016/j.immuni.2007.11.016
70. Agak GW, Kao S, Ouyang K, Qin M, Moon D, Butt A, et al. Phenotype and Antimicrobial Activity of Th17 Cells Induced by Propionibacterium Acnes Strains Associated With Healthy and Acne Skin. J Invest Dermatol (2018) 138(2):316–24. doi: 10.1016/j.jid.2017.07.842
71. Zhu J, Yamane H, Paul WE. Differentiation of Effector CD4 T Cell Populations (*). Annu Rev Immunol (2010) 28:445–89. doi: 10.1146/annurev-immunol-030409-101212
72. Banuelos J, Cao Y, Shin SC, Lu NZ. Immunopathology Alters Th17 Cell Glucocorticoid Sensitivity. Allergy (2017) 72(3):331–41. doi: 10.1111/all.13051
73. Bunte K, Beikler T. Th17 Cells and the IL-23/IL-17 Axis in the Pathogenesis of Periodontitis and Immune-Mediated Inflammatory Diseases. Int J Mol Sci (2019) 20(14):3394. doi: 10.3390/ijms20143394
74. Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, et al. IL-21 Initiates an Alternative Pathway to Induce Proinflammatory T(H)17 Cells. Nature (2007) 448(7152):484–7. doi: 10.1038/nature05970
75. Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, et al. Essential Autocrine Regulation by IL-21 in the Generation of Inflammatory T Cells. Nature (2007) 448(7152):480–3. doi: 10.1038/nature05969
76. Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, et al. Induction and Molecular Signature of Pathogenic TH17 Cells. Nat Immunol (2012) 13(10):991–9. doi: 10.1038/ni.2416
77. Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, et al. Fate Mapping of IL-17-Producing T Cells in Inflammatory Responses. Nat Immunol (2011) 12(3):255–63. doi: 10.1038/ni.1993
78. Veldhoen M. Interleukin 17 Is a Chief Orchestrator of Immunity. Nat Immunol (2017) 18(6):612–21. doi: 10.1038/ni.3742
79. Zenobia C, Hajishengallis G. Basic Biology and Role of Interleukin-17 in Immunity and Inflammation. Periodontol 2000 (2015) 69(1):142–59. doi: 10.1111/prd.12083
80. Fitz-Gibbon S, Tomida S, Chiu BH, Nguyen L, Du C, Liu M, et al. Propionibacterium Acnes Strain Populations in the Human Skin Microbiome Associated With Acne. J Invest Dermatol (2013) 133(9):2152–60. doi: 10.1038/jid.2013.21
81. Lomholt HB, Kilian M. Population Genetic Analysis of Propionibacterium Acnes Identifies a Subpopulation and Epidemic Clones Associated With Acne. PLoS One (2010) 5(8):e12277. doi: 10.1371/journal.pone.0012277
82. McDowell A, Gao A, Barnard E, Fink C, Murray PI, Dowson CG, et al. A Novel Multilocus Sequence Typing Scheme for the Opportunistic Pathogen Propionibacterium Acnes and Characterization of Type I Cell Surface-Associated Antigens. Microbiology (2011) 157(Pt 7):1990–2003. doi: 10.1099/mic.0.049676-0
83. McDowell A, Nagy I, Magyari M, Barnard E, Patrick S. The Opportunistic Pathogen Propionibacterium Acnes: Insights Into Typing, Human Disease, Clonal Diversification and CAMP Factor Evolution. PLoS One (2013) 8(9):e70897. doi: 10.1371/journal.pone.0070897
84. Tomida S, Nguyen L, Chiu BH, Liu J, Sodergren E, Weinstock GM, et al. Pan-Genome and Comparative Genome Analyses of Propionibacterium Acnes Reveal its Genomic Diversity in the Healthy and Diseased Human Skin Microbiome. mBio (2013) 4(3):e00003–13. doi: 10.1128/mBio.00003-13
85. Nakatsuji T, Liu YT, Huang CP, Zouboulis CC, Gallo RL, Huang CM. Vaccination Targeting a Surface Sialidase of P. Acnes: Implication for New Treatment of Acne Vulgaris. PLoS One (2008) 3(2):e1551. doi: 10.1371/journal.pone.0001551
86. Holland C, Mak TN, Zimny-Arndt U, Schmid M, Meyer TF, Jungblut PR, et al. Proteomic Identification of Secreted Proteins of Propionibacterium Acnes. BMC Microbiol (2010) 10:230. doi: 10.1186/1471-2180-10-230
87. Suh DH, Kwon HH. What's New in the Physiopathology of Acne? Br J Dermatol (2015) 172(Suppl 1):13–9. doi: 10.1111/bjd.13634
88. Akaza N, Akamatsu H, Kishi M, Mizutani H, Ishii I, Nakata S, et al. Effects of Propionibacterium Acnes on Various mRNA Expression Levels in Normal Human Epidermal Keratinocytes In Vitro. J Dermatol (2009) 36(4):213–23. doi: 10.1111/j.1346-8138.2009.00626.x
89. Nagy I, Pivarcsi A, Kis K, Koreck A, Bodai L, McDowell A, et al. Propionibacterium Acnes and Lipopolysaccharide Induce the Expression of Antimicrobial Peptides and Proinflammatory Cytokines/Chemokines in Human Sebocytes. Microbes Infect (2006) 8(8):2195–205. doi: 10.1016/j.micinf.2006.04.001
90. Nagy I, Pivarcsi A, Koreck A, Szell M, Urban E, Kemeny L. Distinct Strains of Propionibacterium Acnes Induce Selective Human Beta-Defensin-2 and Interleukin-8 Expression in Human Keratinocytes Through Toll-Like Receptors. J Invest Dermatol (2005) 124(5):931–8. doi: 10.1111/j.0022-202X.2005.23705.x
91. Bojar RA, Holland KT. Acne and Propionibacterium Acnes. Clinics Dermatol (2004) 22(5):375–9. doi: 10.1016/j.clindermatol.2004.03.005
92. Beylot C, Auffret N, Poli F, Claudel JP, Leccia MT, Del Giudice P, et al. Propionibacterium Acnes: An Update on its Role in the Pathogenesis of Acne. J Eur Acad Dermatol Venereol (2014) 28(3):271–8. doi: 10.1111/jdv.12224
93. Guo B. IL-10 Modulates Th17 Pathogenicity During Autoimmune Diseases. J Clin Cell Immunol (2016) 7(2):400. doi: 10.4172/2155-9899.1000400
94. Stockinger B, Omenetti S. The Dichotomous Nature of T Helper 17 Cells. Nat Rev Immunol (2017) 17(9):535–44. doi: 10.1038/nri.2017.50
95. Evans HG, Roostalu U, Walter GJ, Gullick NJ, Frederiksen KS, Roberts CA, et al. TNF-Alpha Blockade Induces IL-10 Expression in Human CD4+ T Cells. Nat Commun (2014) 5:3199. doi: 10.1038/ncomms4199
96. McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al. TGF-Beta and IL-6 Drive the Production of IL-17 and IL-10 by T Cells and Restrain T(H)-17 Cell-Mediated Pathology. Nat Immunol (2007) 8(12):1390–7. doi: 10.1038/ni1539
97. Hahn S, Gehri R, Erb P. Mechanism and Biological Significance of CD4-Mediated Cytotoxicity. Immunol Rev (1995) 146:57–79. doi: 10.1111/j.1600-065X.1995.tb00684.x
98. Kryczek I, Zhao E, Liu Y, Wang Y, Vatan L, Szeliga W, et al. Human TH17 Cells are Long-Lived Effector Memory Cells. Sci Transl Med (2011) 3(104):104ra0. doi: 10.1126/scitranslmed.3002949
99. Omenetti S, Bussi C, Metidji A, Iseppon A, Lee S, Tolaini M, et al. The Intestine Harbors Functionally Distinct Homeostatic Tissue-Resident and Inflammatory Th17 Cells. Immunity (2019) 51(1):77–89.e6. doi: 10.1016/j.immuni.2019.05.004
100. Kelhala HL, Palatsi R, Fyhrquist N, Lehtimaki S, Vayrynen JP, Kallioinen M, et al. IL-17/Th17 Pathway is Activated in Acne Lesions. PLoS One (2014) 9(8):e105238. doi: 10.1371/journal.pone.0105238
101. Zhang X, Soldati T. Of Amoebae and Men: Extracellular DNA Traps as an Ancient Cell-Intrinsic Defense Mechanism. Front Immunol (2016) 7:269. doi: 10.3389/fimmu.2016.00269
102. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil Extracellular Traps Kill Bacteria. Science (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
103. Goldmann O, Medina E. The Expanding World of Extracellular Traps: Not Only Neutrophils But Much More. Front Immunol (2012) 3:420. doi: 10.3389/fimmu.2012.00420
104. Liu P, Wu X, Liao C, Liu X, Du J, Shi H, et al. Escherichia Coli and Candida Albicans Induced Macrophage Extracellular Trap-Like Structures With Limited Microbicidal Activity. PLoS One (2014) 9(2):e90042. doi: 10.1371/journal.pone.0090042
105. Loureiro A, Pais C, Sampaio P. Relevance of Macrophage Extracellular Traps in C. Albicans Killing. Front Immunol (2019) 10:2767. doi: 10.3389/fimmu.2019.02767
106. Mohanan S, Horibata S, McElwee JL, Dannenberg AJ, Coonrod SA. Identification of Macrophage Extracellular Trap-Like Structures in Mammary Gland Adipose Tissue: A Preliminary Study. Front Immunol (2013) 4:67. doi: 10.3389/fimmu.2013.00067
107. Munoz-Caro T, Silva LM, Ritter C, Taubert A, Hermosilla C. Besnoitia Besnoiti Tachyzoites Induce Monocyte Extracellular Trap Formation. Parasitol Res (2014) 113(11):4189–97. doi: 10.1007/s00436-014-4094-3
108. Mukherjee M, Lacy P, Ueki S. Eosinophil Extracellular Traps and Inflammatory Pathologies-Untangling the Web! Front Immunol (2018) 9:2763. doi: 10.3389/fimmu.2018.02763
109. Yousefi S, Morshed M, Amini P, Stojkov D, Simon D, von Gunten S, et al. Basophils Exhibit Antibacterial Activity Through Extracellular Trap Formation. Allergy (2015) 70(9):1184–8. doi: 10.1111/all.12662
110. Wong KW, Jacobs WR Jr. Mycobacterium Tuberculosis Exploits Human Interferon Gamma to Stimulate Macrophage Extracellular Trap Formation and Necrosis. J Infect Dis (2013) 208(1):109–19. doi: 10.1093/infdis/jit097
111. Guimaraes-Costa AB, Nascimento MT, Froment GS, Soares RP, Morgado FN, Conceicao-Silva F, et al. Leishmania Amazonensis Promastigotes Induce and are Killed by Neutrophil Extracellular Traps. Proc Natl Acad Sci U S A (2009) 106(16):6748–53. doi: 10.1073/pnas.0900226106
112. Campillo-Navarro M, Leyva-Paredes K, Donis-Maturano L, Gonzalez-Jimenez M, Paredes-Vivas Y, Cerbulo-Vazquez A, et al. Listeria Monocytogenes Induces Mast Cell Extracellular Traps. Immunobiology (2017) 222(2):432–9. doi: 10.1016/j.imbio.2016.08.006
113. Rocha Arrieta YC, Rojas M, Vasquez G, Lopez J. The Lymphocytes Stimulation Induced DNA Release, A Phenomenon Similar to NETosis. Scand J Immunol (2017) 86(4):229–38. doi: 10.1111/sji.12592
114. Costanza M, Poliani PL, Portararo P, Cappetti B, Musio S, Pagani F, et al. DNA Threads Released by Activated CD4(+) T Lymphocytes Provide Autocrine Costimulation. Proc Natl Acad Sci U S A (2019) 116(18):8985–94. doi: 10.1073/pnas.1822013116
115. Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M, et al. DNase Expression Allows the Pathogen Group A Streptococcus to Escape Killing in Neutrophil Extracellular Traps. Curr Biol (2006) 16(4):396–400. doi: 10.1016/j.cub.2005.12.039
116. Beiter K, Wartha F, Albiger B, Normark S, Zychlinsky A, Henriques-Normark B. An Endonuclease Allows Streptococcus Pneumoniae to Escape From Neutrophil Extracellular Traps. Curr Biol (2006) 16(4):401–7. doi: 10.1016/j.cub.2006.01.056
117. Wartha F, Beiter K, Albiger B, Fernebro J, Zychlinsky A, Normark S, et al. Capsule and D-Alanylated Lipoteichoic Acids Protect Streptococcus Pneumoniae Against Neutrophil Extracellular Traps. Cell Microbiol (2007) 9(5):1162–71. doi: 10.1111/j.1462-5822.2006.00857.x
118. Cheng OZ, Palaniyar N. NET Balancing: A Problem in Inflammatory Lung Diseases. Front Immunol (2013) 4:1. doi: 10.3389/fimmu.2013.00001
119. Miller BF, Abrams R, Dorfman A, Klein M. Antibacterial Properties of Protamine and Histone. Science (1942) 96(2497):428–30. doi: 10.1126/science.96.2497.428
120. Hoeksema M, van Eijk M, Haagsman HP, Hartshorn KL. Histones as Mediators of Host Defense, Inflammation and Thrombosis. Future Microbiol (2016) 11(3):441–53. doi: 10.2217/fmb.15.151
121. Daniel C, Leppkes M, Munoz LE, Schley G, Schett G, Herrmann M. Extracellular DNA Traps in Inflammation, Injury and Healing. Nat Rev Nephrol (2019) 15(9):559–75. doi: 10.1038/s41581-019-0163-2
122. Brinkmann V. Neutrophil Extracellular Traps in the Second Decade. J Innate Immun (2018) 10(5-6):414–21. doi: 10.1159/000489829
123. Brinkmann V, Laube B, Abu Abed U, Goosmann C, Zychlinsky A. Neutrophil Extracellular Traps: How to Generate and Visualize Them. J Vis Exp (2010) 36:1724. doi: 10.3791/1724
124. Hirsch JG. Bactericidal Action of Histone. J Exp Med (1958) 108(6):925–44. doi: 10.1084/jem.108.6.925
125. Kim HS, Cho JH, Park HW, Yoon H, Kim MS, Kim SC. Endotoxin-Neutralizing Antimicrobial Proteins of the Human Placenta. J Immunol (2002) 168(5):2356–64. doi: 10.4049/jimmunol.168.5.2356
126. Juneau RA, Pang B, Weimer KE, Armbruster CE, Swords WE. Nontypeable Haemophilus Influenzae Initiates Formation of Neutrophil Extracellular Traps. Infect Immun (2011) 79(1):431–8. doi: 10.1128/IAI.00660-10
127. Ramos-Kichik V, Mondragon-Flores R, Mondragon-Castelan M, Gonzalez-Pozos S, Muniz-Hernandez S, Rojas-Espinosa O, et al. Neutrophil Extracellular Traps are Induced by Mycobacterium Tuberculosis. Tuberculosis (Edinb) (2009) 89(1):29–37. doi: 10.1016/j.tube.2008.09.009
128. Berends ET, Horswill AR, Haste NM, Monestier M, Nizet V, von Kockritz-Blickwede M. Nuclease Expression by Staphylococcus Aureus Facilitates Escape From Neutrophil Extracellular Traps. J Innate Immun (2010) 2(6):576–86. doi: 10.1159/000319909
129. Delbosc S, Alsac JM, Journe C, Louedec L, Castier Y, Bonnaure-Mallet M, et al. Porphyromonas Gingivalis Participates in Pathogenesis of Human Abdominal Aortic Aneurysm by Neutrophil Activation. Proof concept rats. PLoS One (2011) 6(4):e18679. doi: 10.1371/journal.pone.0018679
130. Marin-Esteban V, Turbica I, Dufour G, Semiramoth N, Gleizes A, Gorges R, et al. Afa/Dr Diffusely Adhering Escherichia Coli Strain C1845 Induces Neutrophil Extracellular Traps That Kill Bacteria and Damage Human Enterocyte-Like Cells. Infect Immun (2012) 80(5):1891–9. doi: 10.1128/IAI.00050-12
131. Young RL, Malcolm KC, Kret JE, Caceres SM, Poch KR, Nichols DP, et al. Neutrophil Extracellular Trap (NET)-Mediated Killing of Pseudomonas Aeruginosa: Evidence of Acquired Resistance Within the CF Airway, Independent of CFTR. PloS One (2011) 6(9):e23637. doi: 10.1371/journal.pone.0023637
132. Neeli I, Khan SN, Radic M. Histone Deimination as a Response to Inflammatory Stimuli in Neutrophils. J Immunol (2008) 180(3):1895–902. doi: 10.4049/jimmunol.180.3.1895
133. Ingelsson B, Soderberg D, Strid T, Soderberg A, Bergh AC, Loitto V, et al. Lymphocytes Eject Interferogenic Mitochondrial DNA Webs in Response to CpG and non-CpG Oligodeoxynucleotides of Class C. Proc Natl Acad Sci U S A (2018) 115(3):E478–E87. doi: 10.1073/pnas.1711950115
134. Pearse RN, Kawabe T, Bolland S, Guinamard R, Kurosaki T, Ravetch JV. SHIP Recruitment Attenuates Fc Gamma RIIB-Induced B Cell Apoptosis. Immunity (1999) 10(6):753–60. doi: 10.1016/S1074-7613(00)80074-6
135. Tzeng SJ, Bolland S, Inabe K, Kurosaki T, Pierce SK. The B Cell Inhibitory Fc Receptor Triggers Apoptosis by a Novel C-Abl Family Kinase-Dependent Pathway. J Biol Chem (2005) 280(42):35247–54. doi: 10.1074/jbc.M505308200
136. Fink SL, Cookson BT. Apoptosis, Pyroptosis, and Necrosis: Mechanistic Description of Dead and Dying Eukaryotic Cells. Infect Immun (2005) 73(4):1907–16. doi: 10.1128/IAI.73.4.1907-1916.2005
137. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel Cell Death Program Leads to Neutrophil Extracellular Traps. J Cell Biol (2007) 176(2):231–41. doi: 10.1083/jcb.200606027
138. Farrera C, Fadeel B. Macrophage Clearance of Neutrophil Extracellular Traps Is a Silent Process. J Immunol (2013) 191(5):2647–56. doi: 10.4049/jimmunol.1300436
139. Koh CC, Wardini AB, Vieira M, Passos LSA, Martinelli PM, Neves EGA, et al. Human CD8+ T Cells Release Extracellular Traps Co-Localized With Cytotoxic Vesicles That Are Associated With Lesion Progression and Severity in Human Leishmaniasis. Front Immunol (2020) 11:594581. doi: 10.3389/fimmu.2020.594581
140. Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A Novel Mechanism of Rapid Nuclear Neutrophil Extracellular Trap Formation in Response to Staphylococcus Aureus. J Immunol (2010) 185(12):7413–25. doi: 10.4049/jimmunol.1000675
141. Byrd AS, O'Brien XM, Johnson CM, Lavigne LM, Reichner JS. An Extracellular Matrix-Based Mechanism of Rapid Neutrophil Extracellular Trap Formation in Response to Candida Albicans. J Immunol (2013) 190(8):4136–48. doi: 10.4049/jimmunol.1202671
142. Yipp BG, Kubes P. NETosis: How Vital Is It? Blood (2013) 122(16):2784–94. doi: 10.1182/blood-2013-04-457671
143. McIlroy DJ, Jarnicki AG, Au GG, Lott N, Smith DW, Hansbro PM, et al. Mitochondrial DNA Neutrophil Extracellular Traps are Formed After Trauma and Subsequent Surgery. J Crit Care (2014) 29(6):1133 e1–5. doi: 10.1016/j.jcrc.2014.07.013
144. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil Extracellular Traps Enriched in Oxidized Mitochondrial DNA are Interferogenic and Contribute to Lupus-Like Disease. Nat Med (2016) 22(2):146–53. doi: 10.1038/nm.4027
145. White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, et al. Apoptotic Caspases Suppress mtDNA-Induced STING-Mediated Type I IFN Production. Cell (2014) 159(7):1549–62. doi: 10.1016/j.cell.2014.11.036
146. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome During Apoptosis. Immunity (2012) 36(3):401–14. doi: 10.1016/j.immuni.2012.01.009
147. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable Neutrophils Release Mitochondrial DNA to Form Neutrophil Extracellular Traps. Cell Death Differ (2009) 16(11):1438–44. doi: 10.1038/cdd.2009.96
148. Itagaki K, Kaczmarek E, Lee YT, Tang IT, Isal B, Adibnia Y, et al. Mitochondrial DNA Released by Trauma Induces Neutrophil Extracellular Traps. PLoS One (2015) 10(3):e0120549. doi: 10.1371/journal.pone.0120549
149. Zhang Q, Itagaki K, Hauser CJ. Mitochondrial DNA is Released by Shock and Activates Neutrophils via P38 Map Kinase. Shock (2010) 34(1):55–9. doi: 10.1097/SHK.0b013e3181cd8c08
150. Simmons JD, Lee YL, Mulekar S, Kuck JL, Brevard SB, Gonzalez RP, et al. Elevated Levels of Plasma Mitochondrial DNA DAMPs Are Linked to Clinical Outcome in Severely Injured Human Subjects. Ann Surg (2013) 258(4):591–6. doi: 10.1097/SLA.0b013e3182a4ea46
151. Harris HE, Raucci A. Alarmin(g) News About Danger: Workshop on Innate Danger Signals and HMGB1. EMBO Rep (2006) 7(8):774–8. doi: 10.1038/sj.embor.7400759
152. Balogh ZJ, McIlroy DJ, Smith DW, Hansbro PM. The Origin and the Role of Mitochondrial DNA in Postinjury Inflammation. J Crit Care (2013) 28(6):1099–100. doi: 10.1016/j.jcrc.2013.08.027
153. Tadie JM, Bae HB, Jiang S, Park DW, Bell CP, Yang H, et al. HMGB1 Promotes Neutrophil Extracellular Trap Formation Through Interactions With Toll-Like Receptor 4. Am J Physiol Lung Cell Mol Physiol (2013) 304(5):L342–9. doi: 10.1152/ajplung.00151.2012
154. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting Neutrophils are Major Inducers of Type I IFN Production in Pediatric Systemic Lupus Erythematosus. Sci Transl Med (2011) 3(73):73ra20. doi: 10.1126/scitranslmed.3001201
155. Munks MW, McKee AS, Macleod MK, Powell RL, Degen JL, Reisdorph NA, et al. Aluminum Adjuvants Elicit Fibrin-Dependent Extracellular Traps In Vivo. Blood (2010) 116(24):5191–9. doi: 10.1182/blood-2010-03-275529
156. Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, et al. Activation of the Raf-MEK-ERK Pathway is Required for Neutrophil Extracellular Trap Formation. Nat Chem Biol (2011) 7(2):75–7. doi: 10.1038/nchembio.496
157. Steinberg BE, Grinstein S. Unconventional Roles of the NADPH Oxidase: Signaling, Ion Homeostasis, and Cell Death. Sci STKE (2007) 2007(379):pe11. doi: 10.1126/stke.3792007pe11
158. Castanheira FVS, Kubes P. Neutrophils and NETs in Modulating Acute and Chronic Inflammation. Blood (2019) 133(20):2178–85. doi: 10.1182/blood-2018-11-844530
159. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr., et al. Extracellular DNA Traps Promote Thrombosis. Proc Natl Acad Sci U S A (2010) 107(36):15880–5. doi: 10.1073/pnas.1005743107
160. Yang H, Biermann MH, Brauner JM, Liu Y, Zhao Y, Herrmann M. New Insights Into Neutrophil Extracellular Traps: Mechanisms of Formation and Role in Inflammation. Front Immunol (2016) 7:302. doi: 10.3389/fimmu.2016.00302
161. Monteith AJ, Miller JM, Maxwell CN, Chazin WJ, Skaar EP. Neutrophil Extracellular Traps Enhance Macrophage Killing of Bacterial Pathogens. Sci Adv (2021) 7(37):eabj2101. doi: 10.1126/sciadv.abj2101
162. Uyemura K, Ohmen JD, Grisso CL, Sieling PA, Wyzykowski R, Reisinger DM, et al. Limited T-Cell Receptor Beta-Chain Diversity of a T-Helper Cell Type 1-Like Response to Mycobacterium Leprae. Infect Immun (1992) 60(11):4542–8. doi: 10.1128/iai.60.11.4542-4548.1992
163. Cho JS, Pietras EM, Garcia NC, Ramos RI, Farzam DM, Monroe HR, et al. IL-17 is Essential for Host Defense Against Cutaneous Staphylococcus Aureus Infection in Mice. J Clin Invest (2010) 120(5):1762–73. doi: 10.1172/JCI40891
164. Petretto A, Bruschi M, Pratesi F, Croia C, Candiano G, Ghiggeri G, et al. Neutrophil Extracellular Traps (NET) Induced by Different Stimuli: A Comparative Proteomic Analysis. PLoS One (2019) 14(7):e0218946. doi: 10.1371/journal.pone.0218946
165. Shi C, Yang L, Braun A, Anders HJ. Extracellular DNA-A Danger Signal Triggering Immunothrombosis. Front Immunol (2020) 11:568513. doi: 10.3389/fimmu.2020.568513
166. Hu Z, Murakami T, Tamura H, Reich J, Kuwahara-Arai K, Iba T, et al. Neutrophil Extracellular Traps Induce IL-1beta Production by Macrophages in Combination With Lipopolysaccharide. Int J Mol Med (2017) 39(3):549–58. doi: 10.3892/ijmm.2017.2870
167. Denning NL, Aziz M, Gurien SD, Wang P. DAMPs and NETs in Sepsis. Front Immunol (2019) 10:2536. doi: 10.3389/fimmu.2019.02536
168. Magna M, Pisetsky DS. The Alarmin Properties of DNA and DNA-Associated Nuclear Proteins. Clin Ther (2016) 38(5):1029–41. doi: 10.1016/j.clinthera.2016.02.029
169. Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of Neutrophil Extracellular Trap Degradation is Associated With Lupus Nephritis. Proc Natl Acad Sci U S A (2010) 107(21):9813–8. doi: 10.1073/pnas.0909927107
170. Thierry AR, Roch B. Neutrophil Extracellular Traps and By-Products Play a Key Role in COVID-19: Pathogenesis, Risk Factors, and Therapy. J Clin Med (2020) 9(9):2942. doi: 10.3390/jcm9092942
171. Mohanty T, Fisher J, Bakochi A, Neumann A, Cardoso JFP, Karlsson CAQ, et al. Neutrophil Extracellular Traps in the Central Nervous System Hinder Bacterial Clearance During Pneumococcal Meningitis. Nat Commun (2019) 10(1):1667. doi: 10.1038/s41467-019-09040-0
172. Domer D, Walther T, Moller S, Behnen M, Laskay T. Neutrophil Extracellular Traps Activate Proinflammatory Functions of Human Neutrophils. Front Immunol (2021) 12:636954. doi: 10.3389/fimmu.2021.636954
173. Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, et al. Mast Cells and Neutrophils Release IL-17 Through Extracellular Trap Formation in Psoriasis. J Immunol (2011) 187(1):490–500. doi: 10.4049/jimmunol.1100123
174. Zhu S, Yu Y, Ren Y, Xu L, Wang H, Ling X, et al. The Emerging Roles of Neutrophil Extracellular Traps in Wound Healing. Cell Death Dis (2021) 12(11):984. doi: 10.1038/s41419-021-04294-3
175. Panda R, Krieger T, Hopf L, Renne T, Haag F, Rober N, et al. Neutrophil Extracellular Traps Contain Selected Antigens of Anti-Neutrophil Cytoplasmic Antibodies. Front Immunol (2017) 8:439. doi: 10.3389/fimmu.2017.00439
176. Leffler J, Martin M, Gullstrand B, Tyden H, Lood C, Truedsson L, et al. Neutrophil Extracellular Traps That are Not Degraded in Systemic Lupus Erythematosus Activate Complement Exacerbating the Disease. J Immunol (2012) 188(7):3522–31. doi: 10.4049/jimmunol.1102404
177. Sur Chowdhury C, Giaglis S, Walker UA, Buser A, Hahn S, Hasler P. Enhanced Neutrophil Extracellular Trap Generation in Rheumatoid Arthritis: Analysis of Underlying Signal Transduction Pathways and Potential Diagnostic Utility. Arthritis Res Ther (2014) 16(3):R122. doi: 10.1186/ar4579
178. Palic D, Ostojic J, Andreasen CB, Roth JA. Fish Cast NETs: Neutrophil Extracellular Traps are Released From Fish Neutrophils. Dev Comp Immunol (2007) 31(8):805–16. doi: 10.1016/j.dci.2006.11.010
179. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs Are a Source of Citrullinated Autoantigens and Stimulate Inflammatory Responses in Rheumatoid Arthritis. Sci Transl Med (2013) 5(178):178ra40. doi: 10.1126/scitranslmed.3005580
180. Homa-Mlak I, Majdan A, Mlak R, Malecka-Massalska T. Metastatic Potential of NET in Neoplastic Disease. Postepy Hig Med Dosw (Online) (2016) 70(0):887–95. doi: 10.5604/17322693.1216275
181. Saffarzadeh M. Neutrophil Extracellular Traps as a Drug Target to Counteract Chronic and Acute Inflammation. Curr Pharm Biotechnol (2018) 19(15):1196–202. doi: 10.2174/1389201020666190111164145
182. Monti M, De Rosa V, Iommelli F, Carriero MV, Terlizzi C, Camerlingo R, et al. Neutrophil Extracellular Traps as an Adhesion Substrate for Different Tumor Cells Expressing RGD-Binding Integrins. Int J Mol Sci (2018) 19(8):2350. doi: 10.3390/ijms19082350
183. Kanamaru R, Ohzawa H, Miyato H, Yamaguchi H, Hosoya Y, Lefor AK, et al. Neutrophil Extracellular Traps Generated by Low Density Neutrophils Obtained From Peritoneal Lavage Fluid Mediate Tumor Cell Growth and Attachment. J Vis Exp (2018) 138:58201. doi: 10.3791/58201
184. Najmeh S, Cools-Lartigue J, Rayes RF, Gowing S, Vourtzoumis P, Bourdeau F, et al. Neutrophil Extracellular Traps Sequester Circulating Tumor Cells via Beta1-Integrin Mediated Interactions. Int J Cancer (2017) 140(10):2321–30. doi: 10.1002/ijc.30635
185. Masucci MT, Minopoli M, Del Vecchio S, Carriero MV. The Emerging Role of Neutrophil Extracellular Traps (NETs) in Tumor Progression and Metastasis. Front Immunol (2020) 11:1749. doi: 10.3389/fimmu.2020.01749
186. Guglietta S, Chiavelli A, Zagato E, Krieg C, Gandini S, Ravenda PS, et al. Coagulation Induced by C3aR-Dependent NETosis Drives Protumorigenic Neutrophils During Small Intestinal Tumorigenesis. Nat Commun (2016) 7:11037. doi: 10.1038/ncomms11037
187. Yuen J, Pluthero FG, Douda DN, Riedl M, Cherry A, Ulanova M, et al. NETosing Neutrophils Activate Complement Both on Their Own NETs and Bacteria via Alternative and Non-Alternative Pathways. Front Immunol (2016) 7:137. doi: 10.3389/fimmu.2016.00137
188. Blenman KRM, Wang J, Cowper S, Bosenberg M. Pathology of Spontaneous and Immunotherapy-Induced Tumor Regression in a Murine Model of Melanoma. Pigment Cell Melanoma Res (2019) 32(3):448–57. doi: 10.1111/pcmr.12769
189. Munir H, Jones JO, Janowitz T, Hoffmann M, Euler M, Martins CP, et al. Stromal-Driven and Amyloid Beta-Dependent Induction of Neutrophil Extracellular Traps Modulates Tumor Growth. Nat Commun (2021) 12(1):683. doi: 10.1038/s41467-021-20982-2
190. Schedel F, Mayer-Hain S, Pappelbaum KI, Metze D, Stock M, Goerge T, et al. Evidence and Impact of Neutrophil Extracellular Traps in Malignant Melanoma. Pigment Cell Melanoma Res (2020) 33(1):63–73. doi: 10.1111/pcmr.12818
191. Shao BZ, Yao Y, Li JP, Chai NL, Linghu EQ. The Role of Neutrophil Extracellular Traps in Cancer. Front Oncol (2021) 11:714357. doi: 10.3389/fonc.2021.714357
192. Shang A, Gu C, Zhou C, Yang Y, Chen C, Zeng B, et al. Exosomal KRAS Mutation Promotes the Formation of Tumor-Associated Neutrophil Extracellular Traps and Causes Deterioration of Colorectal Cancer by Inducing IL-8 Expression. Cell Commun Signal (2020) 18(1):52. doi: 10.1186/s12964-020-0517-1
193. Yang L, Liu L, Zhang R, Hong J, Wang Y, Wang J, et al. IL-8 Mediates a Positive Loop Connecting Increased Neutrophil Extracellular Traps (NETs) and Colorectal Cancer Liver Metastasis. J Cancer (2020) 11(15):4384–96. doi: 10.7150/jca.44215
194. Miyashiro D, Souza BCE, Torrealba MP, Manfrere KCG, Sato MN, Sanches JA. The Role of Tumor Microenvironment in the Pathogenesis of Sezary Syndrome. Int J Mol Sci (2022) 23(2):936. doi: 10.3390/ijms23020936
195. Goddard DS, Yamanaka K, Kupper TS, Jones DA. Activation of Neutrophils in Cutaneous T-Cell Lymphoma. Clin Cancer Res (2005) 11(23):8243–9. doi: 10.1158/1078-0432.CCR-05-1434
196. Geskin LJ, Akilov OE, Kwon S, Schowalter M, Watkins S, Whiteside TL, et al. Therapeutic Reduction of Cell-Mediated Immunosuppression in Mycosis Fungoides and Sezary Syndrome. Cancer Immunol Immunother (2018) 67(3):423–34. doi: 10.1007/s00262-017-2090-z
197. Tadmor T, Attias D, Polliack A. Myeloid-Derived Suppressor Cells–Their Role in Haemato-Oncological Malignancies and Other Cancers and Possible Implications for Therapy. Br J Haematol (2011) 153(5):557–67. doi: 10.1111/j.1365-2141.2011.08678.x
198. Ohl K, Tenbrock K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front Immunol (2018) 9:2499. doi: 10.3389/fimmu.2018.02499
199. Durgin JS, Weiner DM, Wysocka M, Rook AH. The Immunopathogenesis and Immunotherapy of Cutaneous T Cell Lymphoma: Pathways and Targets for Immune Restoration and Tumor Eradication. J Am Acad Dermatol (2021) 84(3):587–95. doi: 10.1016/j.jaad.2020.12.027
200. Fierro MT, Cuffini AM, Novelli M, Banche G, Allizond V, Comessatti A, et al. Functional and Phenotypical Alterations of Polymorphonuclear Cells in Sezary Syndrome Patients. Eur J Dermatol (2011) 21(6):921–9. doi: 10.1684/ejd.2011.1544
201. Krejsgaard T, Lindahl LM, Mongan NP, Wasik MA, Litvinov IV, Iversen L, et al. Malignant Inflammation in Cutaneous T-Cell Lymphoma-a Hostile Takeover. Semin Immunopathol (2017) 39(3):269–82. doi: 10.1007/s00281-016-0594-9
202. Torrealba MP, Manfrere KC, Miyashiro DR, Lima JF, de MOL, Pereira NZ, et al. Chronic Activation Profile of Circulating CD8+ T Cells in Sezary Syndrome. Oncotarget (2018) 9(3):3497–506. doi: 10.18632/oncotarget.23334
203. Saed G, Fivenson DP, Naidu Y, Nickoloff BJ. Mycosis Fungoides Exhibits a Th1-Type Cell-Mediated Cytokine Profile Whereas Sezary Syndrome Expresses a Th2-Type Profile. J Invest Dermatol (1994) 103(1):29–33. doi: 10.1111/1523-1747.ep12388985
204. Hsi AC, Lee SJ, Rosman IS, Carson KR, Kelley A, Viele V, et al. Expression of Helper T Cell Master Regulators in Inflammatory Dermatoses and Primary Cutaneous T-Cell Lymphomas: Diagnostic Implications. J Am Acad Dermatol (2015) 72(1):159–67. doi: 10.1016/j.jaad.2014.09.022
205. Vowels BR, Lessin SR, Cassin M, Jaworsky C, Benoit B, Wolfe JT, et al. Th2 Cytokine mRNA Expression in Skin in Cutaneous T-Cell Lymphoma. J Invest Dermatol (1994) 103(5):669–73. doi: 10.1111/1523-1747.ep12398454
206. Hahtola S, Tuomela S, Elo L, Hakkinen T, Karenko L, Nedoszytko B, et al. Th1 Response and Cytotoxicity Genes are Down-Regulated in Cutaneous T-Cell Lymphoma. Clin Cancer Res (2006) 12(16):4812–21. doi: 10.1158/1078-0432.CCR-06-0532
207. Netchiporouk E, Litvinov IV, Moreau L, Gilbert M, Sasseville D, Duvic M. Deregulation in STAT Signaling Is Important for Cutaneous T-Cell Lymphoma (CTCL) Pathogenesis and Cancer Progression. Cell Cycle (2014) 13(21):3331–5. doi: 10.4161/15384101.2014.965061
208. Abeni D, Frontani M, Sampogna F, Sera F, Bolli S, Corona R, et al. Circulating CD8+ Lymphocytes, White Blood Cells, and Survival in Patients With Mycosis Fungoides. Br J Dermatol (2005) 153(2):324–30. doi: 10.1111/j.1365-2133.2005.06755.x
209. Papadavid E, Economidou J, Psarra A, Kapsimali V, Mantzana V, Antoniou C, et al. The Relevance of Peripheral Blood T-Helper 1 and 2 Cytokine Pattern in the Evaluation of Patients With Mycosis Fungoides and Sezary Syndrome. Br J Dermatol (2003) 148(4):709–18. doi: 10.1046/j.1365-2133.2003.05224.x
210. Geskin LJ, Viragova S, Stolz DB, Fuschiotti P. Interleukin-13 is Overexpressed in Cutaneous T-Cell Lymphoma Cells and Regulates Their Proliferation. Blood (2015) 125(18):2798–805. doi: 10.1182/blood-2014-07-590398
211. Quaglino P, Fava P, Pileri A, Grandi V, Sanlorenzo M, Panasiti V, et al. Phenotypical Markers, Molecular Mutations, and Immune Microenvironment as Targets for New Treatments in Patients With Mycosis Fungoides and/or Sezary Syndrome. J Invest Dermatol (2021) 141(3):484–95. doi: 10.1016/j.jid.2020.07.026
212. Samimi S, Benoit B, Evans K, Wherry EJ, Showe L, Wysocka M, et al. Increased Programmed Death-1 Expression on CD4+ T Cells in Cutaneous T-Cell Lymphoma: Implications for Immune Suppression. Arch Dermatol (2010) 146(12):1382–8. doi: 10.1001/archdermatol.2010.200
213. Kim EJ, Hess S, Richardson SK, Newton S, Showe LC, Benoit BM, et al. Immunopathogenesis and Therapy of Cutaneous T Cell Lymphoma. J Clin Invest (2005) 115(4):798–812. doi: 10.1172/JCI24826
214. Axelrod PI, Lorber B, Vonderheid EC. Infections Complicating Mycosis Fungoides and Sezary Syndrome. JAMA (1992) 267(10):1354–8. doi: 10.1001/jama.267.10.1354
215. Krejsgaard T, Ralfkiaer U, Clasen-Linde E, Eriksen KW, Kopp KL, Bonefeld CM, et al. Malignant Cutaneous T-Cell Lymphoma Cells Express IL-17 Utilizing the Jak3/Stat3 Signaling Pathway. J Invest Dermatol (2011) 131(6):1331–8. doi: 10.1038/jid.2011.27
216. Takei H, Araki A, Watanabe H, Ichinose A, Sendo F. Rapid Killing of Human Neutrophils by the Potent Activator Phorbol 12-Myristate 13-Acetate (PMA) Accompanied by Changes Different From Typical Apoptosis or Necrosis. J Leukoc Biol (1996) 59(2):229–40. doi: 10.1002/jlb.59.2.229
217. Dominguez-Diaz C, Varela-Trinidad GU, Munoz-Sanchez G, Solorzano-Castanedo K, Avila-Arrezola KE, Iniguez-Gutierrez L, et al. To Trap a Pathogen: Neutrophil Extracellular Traps and Their Role in Mucosal Epithelial and Skin Diseases. Cells (2021) 10(6)1469. doi: 10.3390/cells10061469
218. Vong L, Lorentz RJ, Assa A, Glogauer M, Sherman PM. Probiotic Lactobacillus Rhamnosus Inhibits the Formation of Neutrophil Extracellular Traps. J Immunol (2014) 192(4):1870–7. doi: 10.4049/jimmunol.1302286
219. Singhal A, Yadav S, Chandra T, Mulay SR, Gaikwad AN, Kumar S. An Imaging and Computational Algorithm for Efficient Identification and Quantification of Neutrophil Extracellular Traps. Cells (2022) 11(2):191. doi: 10.3390/cells11020191
220. Wu X, Tian J, Wang S. Insight Into Non-Pathogenic Th17 Cells in Autoimmune Diseases. Front Immunol (2018) 9:1112. doi: 10.3389/fimmu.2018.01112
Keywords: T cell extracellular traps, acne, Cutibacterium acnes (C acnes), TH17 cell, skin
Citation: Ouyang K, Oparaugo N, Nelson AM and Agak GW (2022) T Cell Extracellular Traps: Tipping the Balance Between Skin Health and Disease. Front. Immunol. 13:900634. doi: 10.3389/fimmu.2022.900634
Received: 21 March 2022; Accepted: 23 May 2022;
Published: 20 June 2022.
Edited by:
Daniel (Söderberg) Appelgren, Linköping University, SwedenReviewed by:
Massimo Costanza, Fondazione IRCCS Istituto Neurologico C. Besta (IRCCS), ItalyStavros Giaglis, University of Basel, Switzerland
Copyright © 2022 Ouyang, Oparaugo, Nelson and Agak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: George W. Agak, R2FnYWtAbWVkbmV0LnVjbGEuZWR1
†These authors have contributed equally to this work