 Shanshan Wang
Shanshan Wang- Department of Microbiology, Immunology and Infectious Diseases, University of Calgary, Calgary, AB, Canada
Gasdermins (GSDMs) are a group of proteins that are cleaved by inflammatory caspases to induce pore formation in the plasma membrane to cause membrane permeabilization and lytic cell death or pyroptosis. All GSDMs share a conserved structure, containing a cytotoxic N-terminal (NT) pore-forming domain and a C-terminal (CT) repressor domain. Entamoeba histolytica (Eh) in contact with macrophages, triggers outside-in signaling to activate inflammatory caspase-4/1 via the noncanonical and canonical pathway to promote cleavage of gasdermin D (GSDMD). Cleavage of GSDMD removes the auto-inhibition that masks the active pore-forming NT domain in the full-length protein by interactions with GSDM-CT. The cleaved NT-GSDMD monomers then oligomerize to form pores in the plasma membrane to facilitate the release of IL-1β and IL-18 with a measured amount of pyroptosis. Pyroptosis is an effective way to counteract intracellular parasites, which exploit replicative niche to avoid killing. To date, most GSDMs have been verified to perform pore-forming activity and GSDMD-induced pyroptosis is rapidly emerging as a mechanism of anti-microbial host defence. Here, we review our comprehensive and current knowledge on the expression, activation, biological functions, and regulation of GSDMD cleavage with emphases on physiological scenario and related dysfunctions of each GSDM member as executioner of cell death, cytokine secretion and inflammation against Eh and other protozoan parasitic infections.
Introduction
The gastrointestinal tract encounters innumerable luminal insults, including those caused by noxious substances, commensal bacteria, and/or pathogens. The enteric protozoan parasite Entamoeba histolytica (Eh) is the causative agent of intestinal amebiasis, which manifests as amebic colitis and amebic liver abscess (1, 2) and is named for its ability to lyse host tissues. Of approximately 10 individuals that are infected, one will develop symptoms such as diarrhea and abdominal pain (3). On rare circumstances, Eh penetrates the intestinal mucosa and cause necrosis that extend to the submucosa and muscularis, causing “flask-shaped ulcer” (3, 4) and enter the bloodstream and disseminate to other extraintestinal sites, most commonly the liver, lung and brain. Collectively, there are approximately 100 million annual cases of amebic dysentery, colitis and liver abscess resulting 11,300 deaths in 2013 (5).
The first line of innate host defence in the gastrointestinal tract is the mucus layer produced by goblet cells (6). In the colon, the epithelial barrier consists of three layers: the single layer of intestinal epithelial cells (IECs) that separates the underlying lamina propria, the mucus bilayer and indigenous microbiota that reside in/on the mucus layer (7). The colonic mucus layer contains two components: a firm sterile inner mucus layer adherent on the apical surface of IECs (8), and a superficial loose outer mucus layer that is heavily colonized by commensal microorganisms (9) (Figure 1A). Colonic MUC2 mucins are the structural component of the mucus gel that are rich in galactose (Gal) and N-acetyl galactosamine (GalNAc) (11). Eh colonizes the gut by binding of the surface Gal/GalNAc adherence lectin (Gal-lectin) adhesin to mucin Gal and GalNAc glycans. Here it also uses the Gal-lectin adhesin to bind bacteria and host cells that it feeds upon (12–15). In disease pathogenesis, Eh cysteine proteases degrades peptides in the poor glycosylated regions of the MUC2 C-terminus (16) to abrogate mucus protective functions allowing the parasite to penetrate through the mucus layer (17) (Figure 1B). In addition to mucin degradation, Eh also evokes hypersecretion (exocytosis) of mucus by engaging Eh cysteine protease-5 (EhCP-A5) RGD motif (arginine-glycine-aspartate) to αvβ3 integrin on goblet cells (18) as a means not only to defend against the parasite but also to deplete intracellular mucin stores.
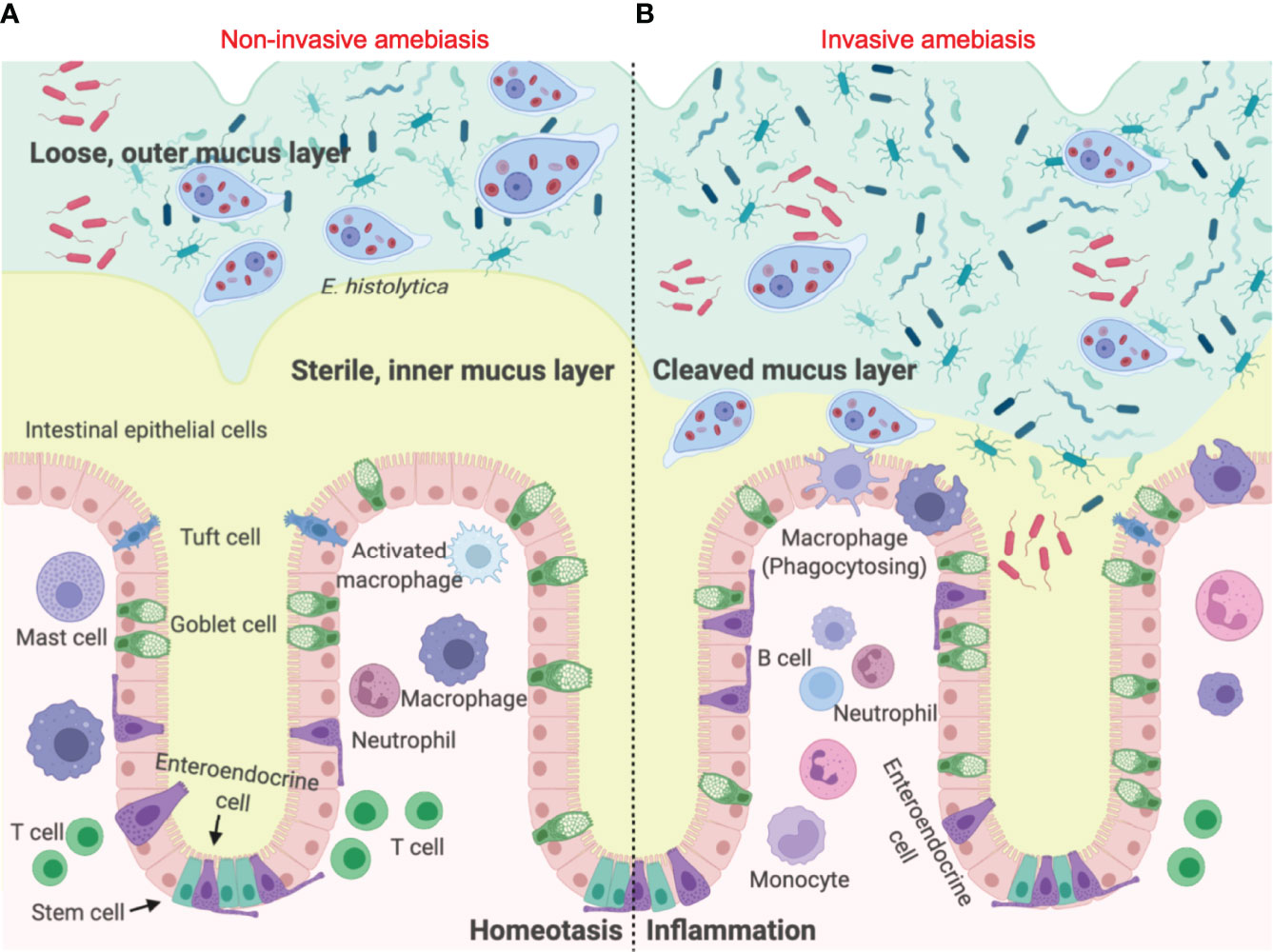
Figure 1 Entamoeba histolytica breaches the mucus layer and results in invasive amebiasis. (A) When Eh infects a healthy individual it colonizes the looser outer mucus layer by binding Gal/GalNAc mucin glycans to the parasite Gal-lectin adhesin (10). Here the parasite feeds on bacteria, cellular debris and sugar from mucus to satisfy its energetic needs, establishing asymptomatic infections. (B) In invasive amebiasis Eh disrupts innate host defences by cleaving mucin glycans with parasite glycosidases followed by the degradation of MUC2 mucin in its C-terminal domain via Eh cysteine proteases to dissolve the protective colonic mucus gel (11). This allows Eh to come in direct contact with and disrupts intestinal epithelial cells where the parasite comes in direct contact with lamina propria macrophages that express high amounts of NLRP3 inflammasomes evoking a prompt pro-inflammatory response dominated by TNF-α, IL-1β and IL-8.
When the mucus barrier is degraded and/or depleted by mucus hypersecretion that exceeds MUC2 biosynthesis, Eh comes in contact with and disrupts the surface epithelium. This allows Eh to interact with resident macrophages in the lamina propria to elicit a robust pro-inflammatory response through activation of the nucleotide-binding oligomerization domain (NOD)-like receptor-pyrin containing 3 (NLRP3) inflammasome, leading to interleukin (IL)-1β and IL-18 release (10) (Figure 2A). The direct sensing of Eh via Gal-lectin induces NLRP3 inflammasome activation, driving the processing and release of IL-1β and an aggressive inflammatory response that are aimed at eradicating Eh and recruiting immune cells to the site of infection (10). The canonical NLRP3 inflammasomes are the cytosolic sensor that target pathogens and various cellular stresses that form multimeric high molecular complexes to mediate recruitment and activation of caspase-1 (24). Until recently, most work on the NLRP3 inflammasome has been focused on the functions of caspase-1 (25–29). However, recent studies using Casp1–/– mice that have a mutation in Casp11, led to the discovery of the noncanonical inflammasome pathway regulated by caspase-11 and identification of a lytic cell death pathway induced by caspase-11 independent of the canonical NLRP3 inflammasome complex (30). Most gram-negative bacteria such as Escherichia coli, Citrobacter rodentium and Vibrio cholerae, activate caspase-11/4-mediated signaling pathway in mouse/human macrophages resulting in maturation and release of IL-1β/IL-18 and pyroptosis (30) (Figure 2B). However, the precise role of caspase-11/4 in macrophage inflammasome signaling and their mechanism of activation against parasitic infections remain to be clarified.
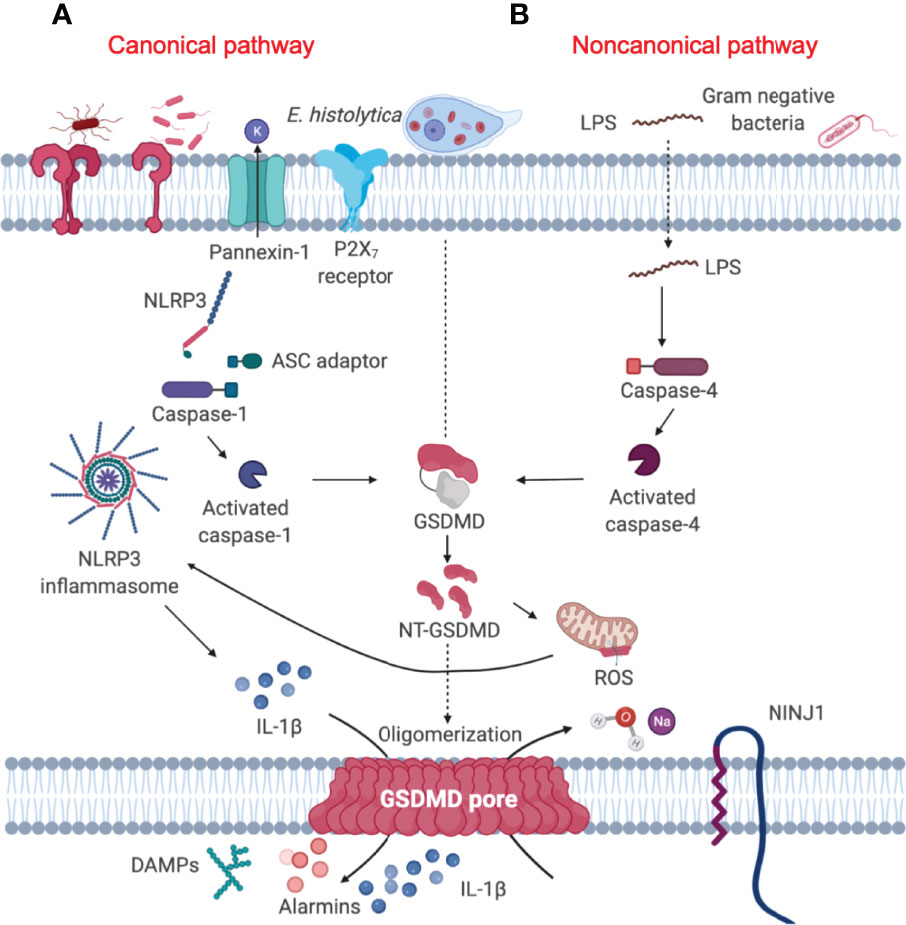
Figure 2 Gasdermin D-mediated pyroptosis is regulated by both canonical and noncanonical signaling pathway. (A) Canonical inflammasome pathway (left) constitute a Nod-like receptor (NLRP3), the adaptor protein ASC and pro-caspase-1, which converts pro-IL-1β into its active form. Activated caspase-1 cleaves GSDMD to unleash the NT-GSDMD, which mediates bioactive IL-1β release and ultimately causes cell pyroptosis (19). (B) Cytosolic LPS released from Gram-negative bacteria (20) and the extracellular protozoan Eh can trigger the activation of caspase-4 (21), leading to activation of the noncanonical signaling pathway (right). Activated caspase-4 in turn, cleaves GSDMD in a similar fashion as canonical caspase-1 to mediate the secretion of mature pro-inflammatory cytokines (22). NINJ1 is a cell-surface protein that contains two transmembrane domains and is a mediator of plasma membrane rupture that can release pro-inflammatory cytokines (23).
Although caspases have different effector functions, all caspases share a common structure: caspases constitute an N-terminal (NT) pro-domain, a central large domain containing an active cysteine residue and a small C-terminal (CT) subunit domain (31). The pro-domains of caspases contain caspase activation and recruitment domains (CARDs) or death effector domains (DEDs). A growing body of literature suggests that the activation of inflammatory caspase requires at least two steps: the catalytic component of two pro-caspases need to undergo dimerization, which is made up of two large and two small subunits (Figure 3). Following this, autocleavage is triggered within the dimerized full protease (32). Auto-proteolytic processing of pro-caspase occurs when the protein is cleaved between the pro-domain and the large subunit as well as between the large subunit and the small subunit, which allows both large/small subunits to re-assemble as an active heterotetramer (31) (Figure 3). Caspases need to be tightly regulated as their hyperactivation can promote auto-inflammatory diseases, while insufficient activation of caspases can lead to susceptibility to infection and sepsis (33). It is still unclear how each specific caspase works, if there is any redundancy or interaction among them and how caspase functions are fine-tuned.
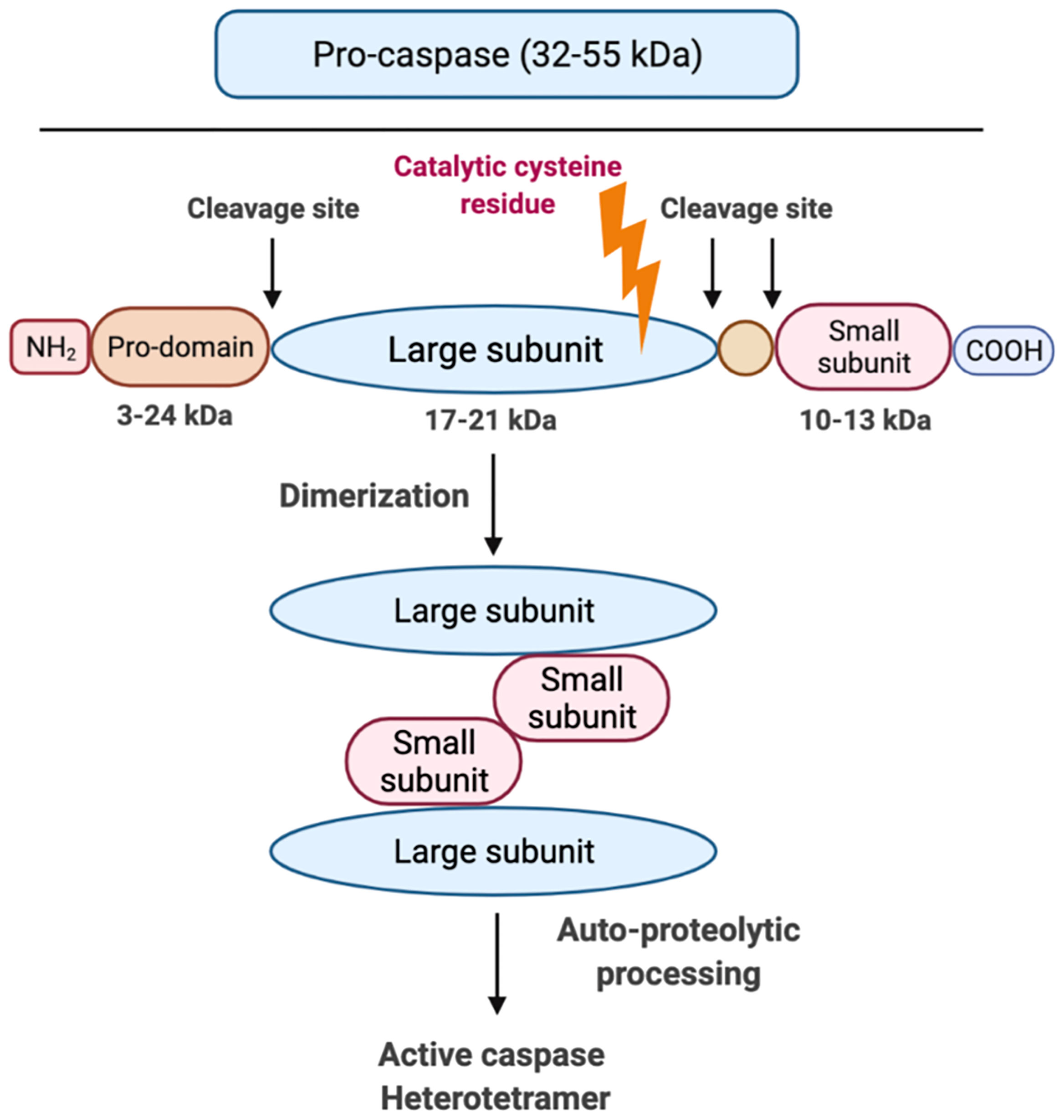
Figure 3 Graphic representation of caspase structure. Caspases share a common structure, consisting of a 3-24 kDa NT pro-domain, a 17-21 kDa central large domain and a 10-13 kDa small CT subunit domain. All caspases consist of an active cysteine residue (bold lightning) in the large domain that specifically cleaves after the aspartate residue in substrates. The activation of inflammatory caspases usually requires two steps: step 1 is dimerization that is induced by proximity and step 2 is auto-processing within the catalytic domain of each caspase monomer.
Upon sensing multiple pathogen and host-derived cellular insults via pathogen and damage-associated molecular patterns (PAMPs and DAMPs, respectively), canonical NLRP3 sensor triggers oligomerization of the adaptor protein apoptosis-associated speck like protein containing a CARD (ASC) and the effector protein caspase-1 through homotypic interactions, leading to the formation of the multimeric inflammasome complex to regulate cytokine maturation and pyroptosis. Yet, the accurate mechanism and interaction governing the crosstalk between canonical NLRP3 and noncanonical caspase-4/5/11 inflammasomes in response to parasites require further elucidation. It has been suggested that caspase-4 and caspase-5 may have originated from caspase-11 (34) as caspase-11 share approximately 60% amino acid identity to caspase-4 (35). Hence, there is a degree of crosstalk between the inflammatory caspases that determine the fate of the cell, and manage the outcome of host response to infections. Macrophages deficient in caspase-4 have decreased caspase-1 activation and reduced IL-1β secretion (22) when stimulated with ATP and monosodium urate crystals (MSU) (36), suggesting caspase-4 functions similarly to caspase-11, as it might regulate caspase-1 activation. The distinct role of caspase-4 is also implicated in dengue virus infection where it was discovered to be critical in regulating caspase-1 activity and subsequent IL-1β secretion (37). By serendipitous discovery, caspase-11 and the human ortholog caspase-4 are shown to directly bind lipopolysaccharide (LPS) via their CARD domains that undergo oligomerization and activation (38). Though caspases are known to require the assembly of receptor complex for their activation, caspase-11 and caspase-4 appear to occur without the formation of a specific receptor complex. Cytosolic LPS released from gram-negative bacteria is detected by murine caspase-11 or human caspase-4 and 5, leading to activation of the noncanonical inflammasome (Figure 2B). These activated inflammatory caspases in turn, cleave GSDMD to cause cell pyroptosis. Similar to macrophages, caspase-4/1 initiate pyroptotic cell death in epithelial cells infected with S. typhimurium to restrict bacterial growth (39). These studies show that caspase-4 activation is important for inflammatory signaling processes, displaying diverse functions, however, extensive studies are required to decipher caspase-4 and caspase-11 functions in inflammatory response evoked by protozoan parasites.
The Gasdermin Family
The gasdermins (GSDMs) are a family of recently identified intracellular proteins that elicit pyroptosis. The human genome encodes six paralogous members of GSDM family proteins: GSDMA, GSDMB, GSDMC, GSDMD, GSDME/DFNA5 and DFNB59 (also known as pejvakin) (40), and ten in mouse, including GSDMA1, GSDMA2, GSDMA3, GSDMC, GSDMC2, GSDMC3, GSDMC4, GSDMD, GSDME, DFNB59 (40, 41). The name gasdermin comes from the expression in the gastrointestinal tract and epithelium of the skin. All members of the GSDM family excluding DFNB59 are conserved and comprised of highly conserved NT and CT fragments that are separated by a central flexible and viable linker that harbour pore-forming activity (42). Upon cleavage by caspases or granzymes, the NT fragment of GSDMs binds to acidic phospholipids in the inner leaflet of cell membranes to form GSDM pores (42–45).
GSDMA
The human genome encodes a single GSDMA, whereas the mouse genome encodes three paralogs known as GSDMA1, GSDMA2 and GSDMA3 (46–48). Mouse GSDMA1 was the first cloned member of the GSDM family protein and was found exclusively in the upper gastrointestinal tract (46). Human GSDMA is expressed in skin and the gastrointestinal tract, including the epidermis, hair follicles, stomach and esophagus (40, 47, 49, 50) (Table 1), however, the protease that activates GSDMA has not been elucidated, though by conducting protein-protein network (STRING-db) (86) analysis, it predicts caspase-1 (red stippled circle) has a strong relationship with GSDMA (Figure 4A). Some evidence suggested that GSDMA may participate in TGF-β production of gastric epithelial cell apoptosis, and gain-of-function mutations in this protein have been discovered to result in hair loss and keratosis (47, 87, 88). Human GSDMA has three mice orthologs, and the generation of knockout (KO) mice displayed increased susceptibility to invasive infection (89) (Table 2). GSDMA genetic polymorphisms indicates a connection with inflammatory bowel disease (IBD) and asthma in children (103–106). However, the gain-of-function mutations were discovered to impair the interaction between the NT and CT fragments by interfering with autoinhibition, causing constitutive activation and inducing pyroptosis without unleashing autoinhibition (88) (Table 1). Similar to GSDMD-NT, when the NT fragments of GSDMA and GSDMA3 were overexpressed in HEK 293T cells, they enhanced pore formation in the cell plasma membrane and trigger pyroptosis-like features (42). Other studies have shown that the NT of mouse GSDMA3 associates with the chaperone protein, heat shock protein 90 (HSP90) (107), and STRING analysis reveals a potentially functional link between GSDMA and heat shock-related 70 kDa protein 2 (HSPA2) (Figure 4A). Though there are no studies to date depicting a connection between GSDMA and protozoan parasites (Table 1), recent studies conducted on group A Streptococcus pyogenes (GAS) revealed that the GAS cysteine protease SpeB cleaves GSDMA to trigger pyroptosis in keratinocytes (52, 89). Upon SpeB cleavage, GSDMA unleashes its NT that forms pores in the plasma membrane which act as a conduit to release intracellular products simultaneously allowing extracellular SpeB to diffuse into the cell, resulting in more GSDMA cleavage and IL-1β processing. Notably, SpeB processes IL-1β at a different site compared to caspase-1, indicating a caspase-1 independent pathway to release IL-1β. In addition, mouse deficient in Gsdma1 gene displayed blunt immune response to GAS, causing disseminated bacterial infection (52). It would be intriguing to determine if protozoan parasites could trigger the cleavage of GSDMA by activating proteases, and what are the consequences of forming GSDMA pores.

Table 1 Gasdermin family expression patterns and functions in protozoan parasite infection.
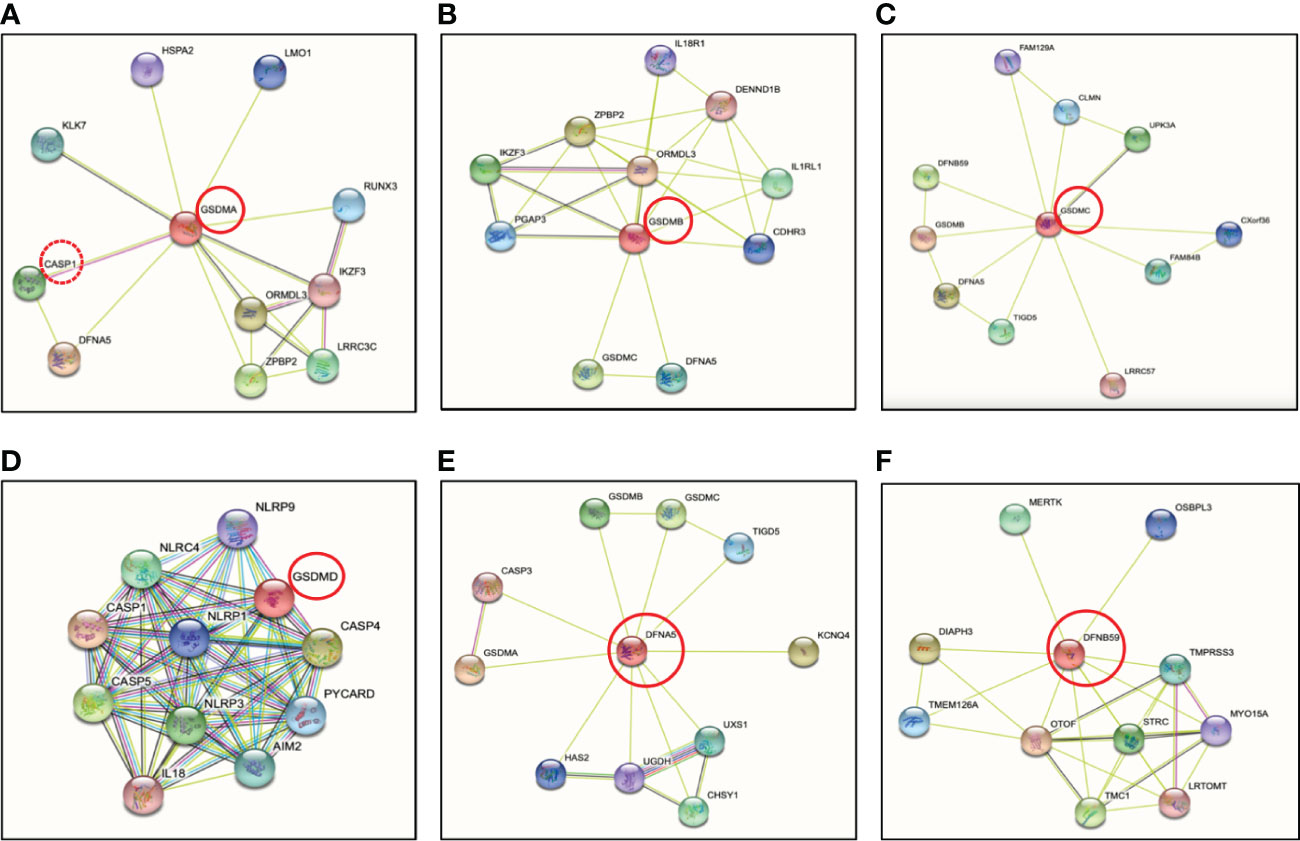
Figure 4 Protein-protein interactions of the human gasdermin family members by STRING analysis. (A) GSDMA is cleaved by caspase-1 with predicted interaction between GSDMA and DFNA5. GSDMA is co-expressed with KLK7, IKZF3 and ORMDL3. (B) GSDMB is co-expressed with ORMDL3 as GSDMA. ORMDL3 is ORM1-like protein 3, a negative regulator of sphingolipid synthesis, which may indirectly regulate endoplasmic reticulum-mediated Ca2+ signaling. (C) The co-expression of GSDMC is UPK3A or uroplakin-3a, is a component of the asymmetric unit membrane (AUM). UPK3A is a highly specialized biomembrane elaborated by terminally differentiated urothelial cells, that play an important role in preventing bacterial adherence. (D) GSDMD is essential effector of NLRP3 inflammasome-dependent caspase-1 activation and IL-1β/IL-18 secretion in response to noncanonical activators, including UVB radiation, cholera enterotoxin subunit B and cytosolic LPS. (E) GSDME or nonsyndromic hearing impairment protein 5 (DFNA5) plays a role in the TP53-regulated cellular response to DNA damage probably by cooperating with TP53, which is known to activate the transcription of numerous tumor suppressors and activators of apoptosis. In addition to pyroptotic activity, GSDME interacts with caspase-3/7 to trigger apoptosis by targeting mitochondria, but is more strongly implicated in tumor suppression. (F) Protein-protein interaction analysis shows no direct connection between DFNB59 and other GSDMs. Otoferlin (OTOF) is a key Ca2+ sensor involved in Ca2+ dependent SNARE-mediated exocytosis. Possible interaction between DFNB59 and OTOF suggests DFNB59 might be involved in vesicle transportation but requires further confirmation.
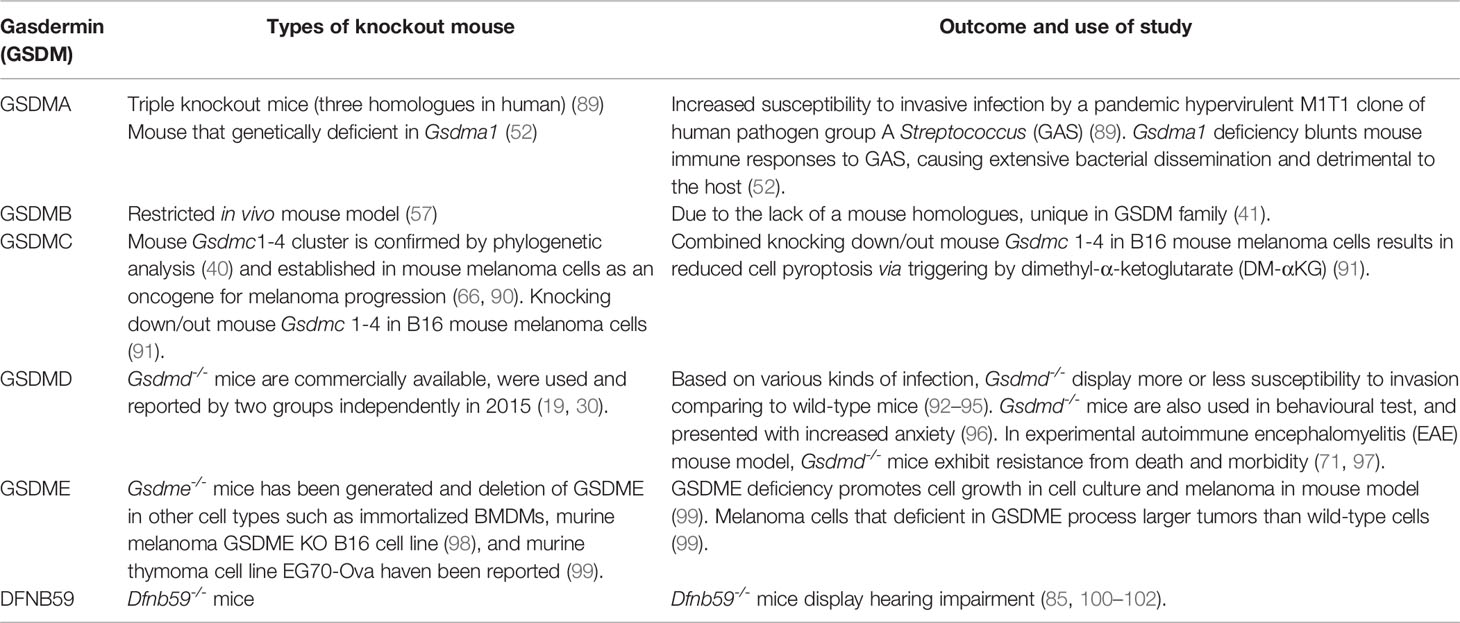
Table 2 Available experimental knockout mouse in gasdermin family members.
GSDMB
At present, GSDMB remains the least studied. GSDMB is the most divergent member among the GSDM family dominantly located in airway epithelium, oesophagus, stomach, liver, small intestine and colon (108, 109). Human GSDMB have at least four isoforms with viable linkers that are cleaved by granzyme A from cytotoxic lymphocytes to induce target cell pyroptosis (55, 110). However, Gsdmb has not been found in rodents and this limits the development of experimental tools to study GSDMB in mouse models (Table 2). The granzyme A-GSDMB pyroptotic axis may also play important roles in immunity to cancers in addition to microbial infections (Table 1), however, no predicted interactions were found between them (Figure 4B). Genome-wide association studies (GWAS) uncovered a strong correlation between GSDMB and susceptibility to inflammatory diseases including Crohn’s disease, asthma, and type I diabetes (111–113). However, the direct relationship between GSDMB and these inflammatory diseases need to be further identified. A single nucleotide polymorphism (SNP) was discovered to associate with upregulated GSDMB expression and increased susceptibility in asthma (56). GSDMB is frequently expressed in cancer cells, consequently, significant correlations were indicated between expression levels of GSDMB and overall survival of patients with bladder carcinoma or skin cutaneous melanoma (55). More recently, a study showed that upregulated of GSDMB in IECs was associated with increased susceptibility of IBD (57) (Table 1). An essential physiological restoration and recovery mechanism to maintain intestinal homeostasis is the resolution of inflammation, and IECs play a major role to undergo proper tissue restitution and repair (114). Specifically, GSDMB was found to be significantly upregulated in IECs from active IBD patients as compared to healthy individuals, suggesting a strong correlation between GSDMB in IECs and IBD progressions (57) (Table 1). Unlike other GSDM members, GSDMB can bind to cell membrane even with the full-length protein (113). Another unique feature about GSDMB is its NT was found to directly interact with caspase-4 CARD protein which may have the capability to upregulate the cleavage of GSDMD mediated by caspase-4 (61). This study discovered that a reduction of GSDMB expression alleviated GSDMD cleavage and programmed pyroptotic cell death (61). GSDMB overexpression facilitated GSDMD cleavage along with elevated lactate dehydrogenase (LDH) release (61), indicating that GSDMB could act as a regulator for GSDMD cleavage in certain circumstances including protozoan parasite infection. In addition, since GSDMB can promote caspase-4 activity by directly binding to the CARD domain of caspase-4, this suggests that GSDMB can play a role in Eh pathogenesis as caspase-4 regulates GSDMD pore-forming activity in response to Eh (22).
GSDMC
GSDMC, also known as melanoma-derived leucine zipper containing extranuclear factor, was named by its initial observation in melanoma metastases in mice (66). The expression of GSDMC is restricted to the esophagus, skin, spleen and vagina (109) (Table 1) and its expression was first used as a marker for melanoma progression (66). Upregulation of GSDMC was detected in human colorectal cancer tissues and was experimentally confirmed with its oncogenic potential (115). Human GSDMC is preferentially expressed in metastatic melanoma cells (66). Mice lack GSDMB but express GSDMC1, 2, 3, 4 (19) (Table 2). Knockdown of mGSDMC1-4 in B16 mouse melanoma was an accidental finding when dimethyl-α-ketoglutarate (DM-αKG) was used to treat HeLa cervical carcinoma cells (Table 2). The treatment of DM-αKG vastly triggered cell death, which presented with distinct morphology alterations in pyroptosis (91). Studies in Chinese Han population revealed that genetic polymorphisms of Gsdmc can alleviate the risk of Lumbar disc herniation, which is a common spinal disease (116). Like other GSDM family proteins, overexpressed GSDMC-NT can drive pore formation and cell pyroptosis in 293T cells (42). To date, no protease has been identified to initiate GSDMC cleavage (66) (Table 1). Based on STRING analysis (86), GSDMC interacts with LRRC57 (Figure 4C), and Synaptosome Associated Protein (SNAP23) was identified as the downstream of LRRC57 in human (117), indicating an interesting relationship between GSDMC and SNAP23. SNAP23 was found to be downregulated in Eh-induced hyperactivated macrophages (22), which may play a major role in repairing GSDMs pores on the plasma membrane. Delineating the interactions between GSDMC and SNAP23 could strengthen our understanding on the balance between cell hyperactivation and cell pyroptosis, and what triggers GSDMs to switch from a pyroptotic role to a non-pyroptotic dominant function. It was recently shown that GSDMC is specifically cleaved by caspase-8 following tumor necrosis factor α (TNF-α) stimulation under hypoxic conditions (65). TNF-α-induced GSDMC cleavage at Asp365, unleashing the GSDMC-NT to form pores and initiate cell pyroptosis in breast cancer cells (65). In Eh-stimulated macrophages, we detected bioactive IL-1β release in CRISPR-Cas9 gene edited GSDMD KO cells, suggesting that perhaps other GSDM pores (22) including GSDMC may have occurred to elicit robust pro-inflammatory responses.
GSDMD
GSDMD is found in the gastrointestinal tract and in sentinel cells of the immune system, macrophages and dendritic cells (118). It was the first GSDM protein identified as the executor of pyroptosis that acts downstream of inflammatory caspases (19, 20). Gsdmd-/- mice has been widely used in research including inflammatory-caspase-associated autoinflammatory conditions and septic shock, and display protection from death as compared to wild-type counterparts (Table 2). By investigating polymorphisms in GSDMD, it was found that polymorphisms can dramatically impact GSDMD functions by affecting sites that are considered as structurally important (119). A prominent portion of GSDMD research has been focused on bacterial exposure, whereas in vitro studies in human and mouse macrophages discovered that Eh activates caspase-4 via noncanonical inflammasome pathway to regulate the cleavage of GSDMD to mediate IL-1β secretion (21), and STRING analysis (86) indicates strong interaction between GSDMD and caspase-4 (Figure 4D). GSDMD is the most well-characterized GSDM protein with its pore-forming NT effector that punches pores in plasma membrane. This discovery has shifted the paradigm of our understanding on how programmed cell death occurs (19). Remarkably, the effector molecule that induced pyroptotic cell death remained unknown until 2015 when GSDMD was discovered as the key effector of pyroptotic cell death and a cleavage target for caspase-1 and -11 (19, 20) (Figure 4D). The gene Gsdmd encodes the 480-residue protein GSDMD, which contains an NT and a CT domain (20, 120). Mechanistic studies demonstrated that GSDM-NT harbours the intrinsic pore-forming and pyroptosis-inducing activity (19, 20, 42). GSDMD are expressed as inactive forms in the resting state, where the CT fragment is held back in check with the NT component to trigger an intramolecular auto-inhibition to inhibit GSDMD cleavage (42, 121). Upon activation by cytosolic danger and infection sensors in macrophages and dendritic cells cleave GSDMD within the linker region, liberating the active NT pore-forming domain to assemble GSDMD pores in the cell membrane (19) (Figure 5). Recently, it was reported that GSDMD-CT not only auto-inhibits GSDMD-NT, but also provides a platform for recruiting inflammatory caspases and GSDMD activation (122). Thus, it is not surprising that mutations within the GSDM-CT region can impair the inhibitory interaction between the CT and NT domains that can trigger spontaneous cleavage of GSDMs. GSDM pores induce disruption in cell membrane integrity that ultimately leads to plasma membrane rupture and osmotic cell lysis, in which pro-inflammatory cytokines are released into the extracellular space (118, 123, 124) (Figure 5). Expression of GSDMD-NT itself can trigger inflammatory cell death, whereas overexpression of GSDMD-CT can inhibit pyroptosis (19). Immune cells release pro-inflammatory molecules into the extracellular space, therefore eliciting inflammatory and immune responses that play a role in innate host responses to parasitic infections. Eh as an extracellular protozoan parasite that induces outside-in signaling in human macrophages to activate caspase-4 similar to Eh-induced caspase-1 activation (22). Importantly, we recently shown that caspase-4 triggered GSDMD cleavage was more efficient in the release of IL-1β independent of the canonical NLRP3 inflammasome pathway while keeping the cells alive (22) (Table 1, see section below on intracellular protozoan parasites). By functional enrichment analysis on protein-protein interaction networks (86), GSDMD is intensively connected with both caspase-4/1 and NLRP3/1 inflammasomes. Humans with mutations in NLRC4 develops an autoinflammatory syndrome presented with acute fever and feature indicative of Macrophage Activation Syndrome (MAS). MAS patients present with elevated serum IL-18 as predicted by STRING analysis (Figure 4D).
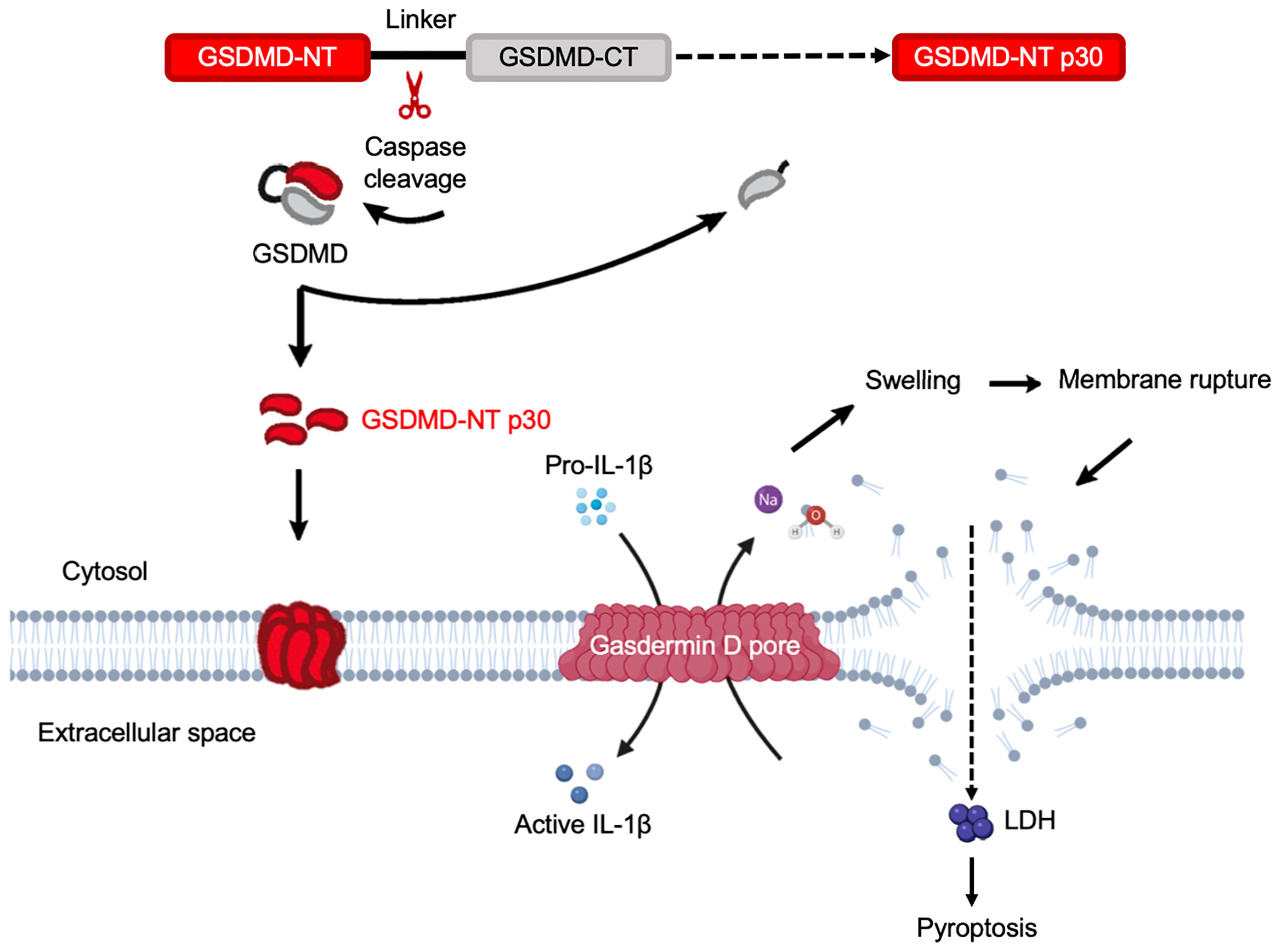
Figure 5 Gasdermin D functions as the executor of cell pyroptosis. Inflammatory caspases are activated by a wide array of inflammasomes upon sensing cytosolic contamination or perturbation. Upon activation, caspases cleave pro-IL-1β, pro-IL-18, and GSDMD. Full length GSDMD is composed of an NT effector domain, also known as pore-forming domain (red) and a CT repressor domain (grey). These two components are connected by a flexible link containing the caspase cleavage sites. The liberated NT pore-forming domain then inserts into the plasma membrane and triggers oligomerization of approximately 16 monomers to form a GSDMD pore. This is a relatively small pore allowing IL-1β to release through it. Simultaneously, sodium gets into the cell, bringing along water that increases cell volume. This can rapidly exceed the volume capacity of the cell, causing a big tear in the plasma membrane that is much larger in size than the GSDMD pore. This membrane-rupture event results in massive release of soluble cytosolic contents, including lactate dehydrogenase (LDH).
A mounting body of evidence has demonstrated that neutrophils are crucial effector cells against Eh and some intracellular protozoan parasites (125–128) (Table 1). Inflammasome activation promotes neutrophil recruitment, which is critical to parasite dissemination and/or exacerbation of disease (129). In contrast to macrophages, GSDMD cleavage in neutrophils is caspase-independent, and is mediated by a neutrophil-specific serine protease, elastase (ELANE) released from cytoplasmic granules into the cytosol during neutrophil death (92). ELANE cleaves GSDMD to perforate the plasma membrane to trigger pyroptosis in neutrophils. GSDMD in neutrophils are linked to azurophilic granules and upon cleavage into GSDMD-NT, it transports to azurophilic granules that results in leakage of ELANE into the cytosol to cleave GSDMD, leading to a secondary cleavage on GSDMD (130). Neutrophil extracellular traps (NETs) catch and damage Eh and play an important role in innate host defense against infection (131). These findings highlight the principal differences between neutrophil and macrophage in GSDMD regulation that implicate neutrophil-specific functions in response to parasites (130).
GSDME (DFNA5)
GSDME is variably expressed in a range of human cells and tissues, including the brain, endometrium, placenta and intestine (132) and was characterized as the causative gene for nonsyndromic hearing loss and as a tumor suppressor (40, 133) (Table 1). GSDME deletion by CRISPR/Cas9 gene editing in various cell types has been generated, for example, murine melanoma cell line B16 and thymoma cell lone EG7-Ova (99) (Table 2). Gsdme-/- mice exhibit healthier and more robust phenotypes after peritoneal injection of cisplatin comparing to wild-type mice (74) (Table 2). Other studies have reported gain-of-function in Gsdme leads to disruption of exon 8 at mRNA level and cause nonsyndromic DFNA5 hearing impairment (77–79, 134–137). It is not clear what the physiological role of GSDME is in these tissues and cells, where inflammation might be harmful (96). GSDME and pejvakin (PJVK) belong to the deafness-associated genes (DFN) and their protein sequences cluster together, distant from the other GSDMs (GSDMA-D). Evolutionarily, GSDME and PJVK are among the oldest GSDM members to date, with resemble sequences found in lower vertebrates and some invertebrates (138, 139). DFNA5 plays a role in the TP-53-regulated cellular response to DNA damage probably by cooperating with TP53 (140). Notably, GSDME was recently shown to be cleaved and activated by caspase-3 with a role in anti-tumor immunity (141, 142) (Table 1), driving chemotherapy drug-induced normal-tissue damage or viral infection-mediated secondary necrosis (74, 75) as confirmed by STRING analysis (86) (Figure 4E, Table 1). A recent study revealed that NLRP3 inflammasome activation causes delayed necrotic cell death by recruiting ASC in caspase-1/11 deficient macrophages (143). Additionally, ASC triggered caspase-8 activation to process GSDME, which is essential for caspase-1 independent necrosis without detectable IL-1β secretion, suggesting an alternative pathway for necrotic cell death regulated by NLRP3 inflammasome upon inhibition on caspase-1 (143). Furthermore, constitutive NLRP3 activation retains the capability to trigger inflammation in mice lacking GSDMD likely following GSDME-regulated pyroptosis (144). Thus, GSDME presents as a potential treatment for inflammatory disorders induced by sustained inflammasome activation (Table 1).
DFNB59
DFNB59 or PJVK is a protein related to hearing impairment or loss in humans and mice (85, 145, 146). Since all GSDM family members share the two-domain structure, excluding DFNB59, it is a more remotely associated GSDM family member with a truncated non-homologous CT fragment (147) with no interaction with other GSDM family members (Figure 4F). DFNB59 mRNA is present in the brain, eyes, ears, heart, liver and intestines (85, 148) (Table 1). Even though GSDME and DFNB59 are associated with nonsyndromic hearing loss and impairment, Dfnb59-/- but not Gsdme-/- mice display hearing dysfunction (85, 100–102) (Table 2). Using Pjvk KO alleles, it was shown that PJVK was essential for regular mechanotransduction in hair cells (149). Missense mutations within the NT of DFNB59 results in impairment of transmitting the auditory signal through auditory neurons (Table 1), present either dysfunctional in cochlear outer hair cells (100) or auditory neuropathy (85). It is still not known if DFNB59 is a pore-forming protein or if it remains physiologically active under homeostasis, since the NT of DFNB59 is extremely short and might fail to trigger autoinhibition.
Gasdermin D-Mediated Cell Pyroptosis
Pyroptosis (in Greek, pyro means fire and ptosis means falling) is a lytic form of regulated cell death that is induced by inflammatory caspase-1, 4, 5, and 11 (mouse) (150, 151), and was first reported as a cell biological phenomenon in macrophages during Shigella flexneri and Salmonella enterica serovar Typhimurium infections, respectively (152–154). GSDMD pores in the plasma membrane act as conduits through which low molecular weight cellular contents are released into the extracellular space to elicit inflammation. However, once the volume exceeds membrane capacity, a membrane rupture event occurs to release soluble cytosolic contents (19). This tear is robust enough to instantly dissipate soluble protein like LDH, while organelles are retained (155). During cell pyroptosis, the damaged membrane forms large ballooning bubbles and dying cells begin to flatten as their cytoplasmic contents are released (156). The rapidity of cell death and morphological alterations are signatures to distinguish pyroptosis and apoptosis (118). It is predicted that after the rupture event, the osmotic pressure achieves a new balance, and stops further volume increases (157). Once the pore-forming fragment is unleashed from the CT repressor domain, it subsequently oligomerizes to form the pyroptotic pores, in which an estimated 16 pore-forming fragment (NT) monomers oligomerize to form pores with a diameter of approximately 10-15 nm of range (42, 43, 45), or around 21 nm (44). These pores mediate the passage of cytokines and diverse cytoplasmic contents and undermine the cellular ionic gradients (Figure 5). The opening of GSDMD pore breaks the normal permeability barrier of the plasma membrane, resulting in membrane disruption, which causes an increase in osmotic pressure causing an influx of water, leading to the cell volume to increase (157). As a consequence, this drives cell swelling and plasma membrane rupture, which eventually lead to cell pyroptosis (Figure 5).
NINJ1 Actively Mediates Plasma Membrane Rupture During Lytic Programmed Cell Death
It was originally thought that plasma rupture occurred after pore formation as a passive cellular event triggered by shear force (158). However, recent studies have shown that plasma membrane rupture is an actively regulated process that is mediated by a specific membrane protein, ninjurin-1 (NINJ1) (23). Deletion of NINJ1 in bone marrow-derived macrophages (BMDMs), inhibited the release of HMGB1, a pro-inflammatory DAMP (159), despite exhibiting normal GSDMD-dependent release of IL-1β, indicating that the rupture of plasma membrane was an abnormal event. Macrophage’s deficiency in NINJ1 displayed significant decreased in LDH release in the absence of GSDMD cleavage (23). IL-1β secretion was unaffected in NINJ1-/- BMDMs, supporting the notion that IL-1β is predominantly secreted through GSDMD pores.
NINJ1 is a 16 kDa cell-surface protein that contains two transmembrane regions with N and C termini exposing to the cytoplasm (Figure 2). With the use of artificial dyes of various sizes, it was revealed that NINJ1 acted downstream of GSDMD pores, and overexpressing NINJ1 in HEK 293T cells enhanced cytotoxicity (160). This finding was paradigm-shifting, as membrane rupture event has been long presumed to passively and spontaneously occur after cell death is triggered. Though much work needs to be done on the interaction between GSDMD and NINJ1, there are many unanswered questions: what drives the oligomerization to induce NINJ1 activation; what role does GSDMD play in regulating NINJ1 activity; what are the functions of the other NINJ family members and do they interact with each other and what is the biochemical activity of the NINJ family. More importantly, the link between NINJ1 activity in plasma membrane rupture and cytotoxicity requires more attention.
In terms of therapy, it is promising to target GSDMD or NINJ1 to limit inflammation rather than to simply block either NLRP3 or IL-1β, because the ultimate goal is to prevent pyroptosis in diseases such as autoinflammatory genetic disorders (97). In addition to this, a group of small molecules that target and inhibit GSDMD has been reported (161, 162). Because robust evidence indicates that NINJ1 as another new therapeutic target to limit inflammation. This could be achieved by either knocking out NINJ1 or with an antagonistic anti-NINJ1 antibody, aimed at inhibiting NINJ1 to reduce inflammation and suppression of plasma membrane rupture (23).
The GSDME/Caspase-3 Signaling Function as a Switch Between Apoptosis and Pyroptosis Against E. histolytica and Other Parasitic Infections
Apoptosis constitutes a programmed non-lytic and non-inflammatory mode of cell death involving apoptotic executioner caspase-3, caspase-6 and caspase-7. Cell membrane integrity is preserved at early stages of apoptosis, and apoptotic cells are efficiently eradicated from healthy tissues by phagocytosis prior to being lysed. Classical apoptosis is a physiological programmed cell suicide in which cells undergo characteristic morphological changes, including membrane blebbing, cell shrinkage, DNA fragmentation, chromatin condensation (141, 142), whereas pyroptotic cells rapidly lose cell membrane integrity, increase in size, and have smaller nuclei by forming GSDM pores in which the damaged membrane forms large ballooning bubbles, which are of distinct morphological changes as apoptosis (156, 163–165) (Figure 6A). It is a well-defined step-wise process for Eh to kiss host cells to death via Gal-lectin contact with upregulation of intracellular Ca2+ levels followed by dephosphorylation of host proteins that activates caspase-3 to promote cell death (166). Blocking Ca2+ channels and/or Ca2+ chelators inhibit Eh killing of host cells (166, 167). Eh-induced apoptosis was found not to be dependent on caspase-8 or caspase-9 (168). Casp-3 KO mice are tolerant to amebiasis and a caspase-3 pharmacological inhibitor decreased Eh cytotoxicity (168, 169). Apoptosis is immunologically silent, thus by inducing apoptosis, Eh can dampen the inflammatory response to escape from being recognized.
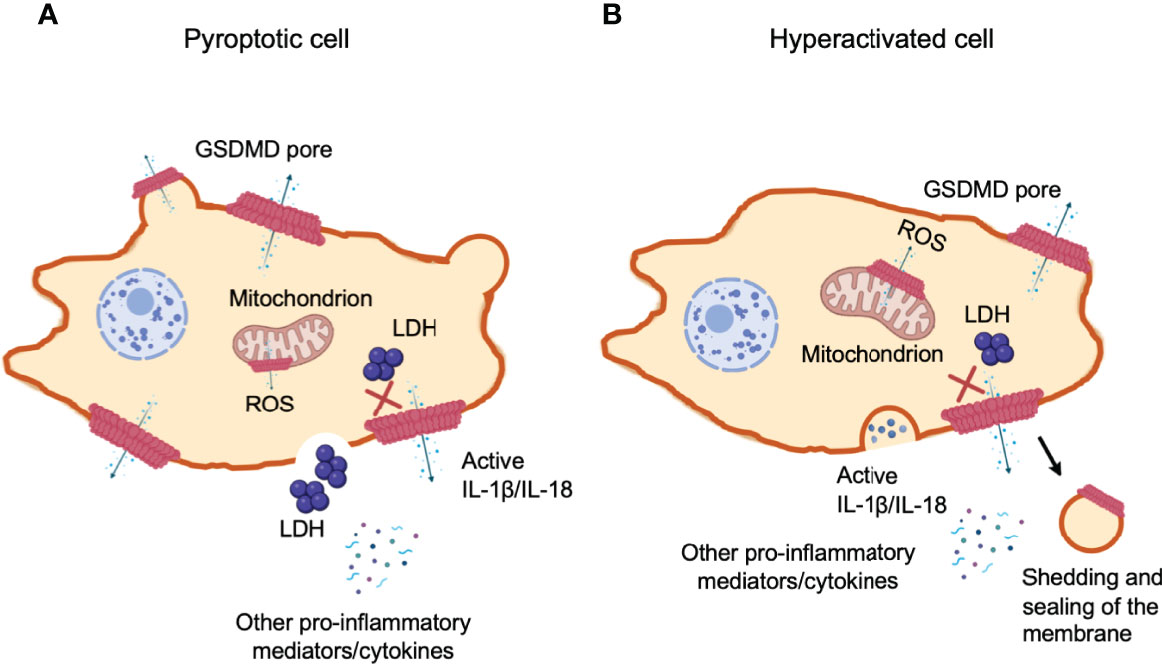
Figure 6 Gasdermin D-mediated cell hyperactivation. Cell pyroptosis and IL-1β secretion can be uncoupled under conditions of cell hyperactivation. This is a stage where phagocytes release IL-1β while remaining intact. The cleaved GSDM-NT fragments can also bind to mitochondrial membranes with higher affinity. NT pore-forming fragments of both GSDMD and GSDME permeabilize the outer membrane of the mitochondria, driving the generation of reactive oxygen species (ROS), loss of transmembrane potential and the release of cytochrome (c). (A) Pyroptotic cells form large balloon-like membrane structures and dying cells appear to flatten as their cytoplasmic contents are released, which is followed by cell swelling and membrane rupture. (B) Hyperactivated and pyroptotic cells can be differentiated by measuring lactate dehydrogenase (LDH) release, as it is too large to exit via GSDMD-NT pores and fully dependent on cell lysis for its release. Hyperactivated cells might have the capability to repair GSDMD pores through shedding of the disrupted membrane to recover damaged area. However, the mechanism to determine if GSDMD pores trigger cell pyroptosis or hyperactivation remains unclear.
Recent studies have significantly advanced our knowledge on the mechanisms and physiology roles of pyroptosis. Although pyroptosis and apoptosis have been considered as two distinct pathways, apoptotic stimuli can trigger pyroptosis under selective scenarios. GSDME is activated during classical apoptosis, following caspase-3 cleavage after Asp270 within the linker region (Table 1), which switches non-inflammatory apoptotic cell death into inflammatory pyroptotic death that express GSDME, alternatively, depletion of GSDME can switch apoptosis to pyroptosis in cells (74, 75). More specifically, activated caspase-3 can cleave GSDME to induce pyroptosis instead of apoptosis (74, 75). When GSDMs are activated by granzymes or caspases, it can also induce apoptosis in certain situations, where the expression of GSDMs is too low to induce pyroptosis, and vice versa, caspase and granzyme-induced apoptosis can be switched to pyroptosis by the expression of GSDMs (164). Another example is the discovery of the apoptotic initiator caspase-8 to interact with the adaptor protein ASC (170, 171). On the other hand, TNF receptor activation activates caspase-8 to induce GSDMD-dependent pyroptosis during pathogenic Yersinia infection (67, 68). Interestingly, activation of the inflammasomes drives the activation of apoptotic caspase-3/7 (172–175). Strikingly, deficiency of GSDMD remarkably postponed and decreased the amount of LPS-induced caspase-3 activation (75). The pore-forming fragment of GSDMD, but not full-length GSDMD, was discovered to cause cytochrome c release and caspase-3 activation (99), indicating that targeting at mitochondrial membrane, releasing cytochrome c and activating caspase-3 is a common mechanism employed by the gasdermin family members to potentiate apoptosis. At the onset of apoptosis, caspase-3 proteolytically cleaves GSDME, which is strongly implicated in tumor suppression (74). By inducing GSDME expression in vitro, it supresses colony formation and tumor cell proliferation in gastric cancer, melanoma and colorectal cancer and reduced metastasis in breast cancer (99, 176–178). Caspase-3 was shown to be involved in Jurkat T cell apoptosis induced by Eh through in vitro studies (168). Eh-induced apoptosis required contact via the Gal lectin but was independent of their cysteine proteases by applying the cell-permeable cysteine proteinase inhibitor, E64c. With the use of caspase-3 inhibitor Ac-DEVD-CHO, it was determined that caspase-3-like activity was necessary for Eh to trigger Jurkat cell apoptosis, since this inhibitor completely blocked Jurkat cell DNA fragmentation.
Transcriptomic analysis of mice brain that were infected with T. gondii compared to uninfected mice using RNA sequencing, showed that inflammasome-regulated pyroptosis was an important host cell death pathway to control infection (128, 179). By performing host-parasite interactome analysis, nine pyroptosis-related differentially expressed transcripts (DETs) were upregulated in infected mice, including casp-4, gsdmd and pycard, while gsdme was downregulated in chronic infection (180). Additionally, IL-1β and IL-18 were upregulated during chronic T. gondii infection, suggesting these inflammatory cytokines are crucial to assist mice to mount an appropriate inflammatory response to prevent latent T. gondii infection (180). During T. gondii infection, infected cells secrete growth factors to inhibit neutrophil apoptosis, by triggering Mcl-1 expression (181). Delayed neutrophil apoptosis may contribute to a robust pro-inflammatory response (181). Leishmania infection reduces caspase-3 activity and postpones spontaneous apoptosis in human neutrophils (182), serving as “trojan horses” to invade macrophages that are phagocytosing apoptotic cells (183). Clearly, more work needs to be done on how GSDM family members are involved as a master switch from apoptosis to pyroptosis, especially with protozoan parasite infections (Table 1).
Gasdermin D Serves as a Gatekeeper to Release IL-1β in Hyperactivated Macrophages in Response to E. histolytica
When Eh contacts macrophage via the Gal-lectin adhesin, surface EhCP-A5 RGD sequence binds α5β1 integrin that triggers Src family kinase phosphorylation and the opening of pannexin-1 channel. Pannexin-1 is a membrane channel for ATP that induces an extracellular burst of ATP, which engages the P2X7 receptor in an autocrine or paracrine fashion to activate the NLRP3 inflammasome and caspase-1 resulting in cell pyroptosis (184). By inducing the outside-in signaling in macrophage, Eh activates caspase-4 via the noncanonical inflammasome pathway to initiate IL-1β secretion similar to caspase-1 by keeping the cells intact (Figure 2) (22). The mechanism of human caspase-4 activation requires live Eh in direct contact with macrophage to trigger outside-in signaling in macrophage to induce the generation of reactive oxygen species (ROS) and K+ efflux (21). Activation of pro-caspases undergoes dimerization or oligomerization with a succeeding cleavage into a small subunit and a large subunit. Upon being activated, it cleaves GSDMD to release the NT pore-forming domain that promotes pore formation to cause Na+ influx, cell swelling that ultimately triggers rupture in the plasma membrane. In response to Eh stimulation, caspase-4 also interacts with caspase-1 in a protein complex that potentiate the cleavage of caspase-1 CARD domains to enhance IL-1β secretion (21). More recently (22), we showed that Eh-induced IL-1β release was not due to massive cell pyroptosis, but rather induce macrophages to reach a stage of “hyperactivation” that caused sustained release of pro-inflammatory cytokines, revealing a significant non-pyroptotic role for GSDMD. This is in marked contrast to nigericin stimulated LPS-primed macrophages that activated caspase-1, resulting in massive cell pyroptosis (22). Mechanistically, Eh triggered caspase-4 activation, GSDMD cleavage and pore formation within 5 min and with IL-1β secretion as early as 30 min in the absence of cell death (22). An important finding was that caspase-4 cleavd GSDMD at the identical amino acid as caspase-1 to maximally regulate IL-1β secretion in response to Eh. Eh-induced IL-1β secretion was independent of pyroptosis as revealed by pharmacologically inhibiting GSDMD pore formation and in CRISPR-Cas9 gene edited GSDMD KO macrophages (22). We theorize that Eh-induced caspase-4/1 activation induced fewer GSDMD pores than NLRP3 inflammasome agonist such as LPS + Nigericin. If fewer GSDMD pores are generated, the cell might respond by initiating compensatory mechanisms to downregulate cell volume allowing for sustained IL-1β secretion while maintaining cell viability, and this may be a unique mechanism in the biology of Eh that could play a crucial role in disease pathogenesis and host defence.
It is controversial whether IL-1β is effectively released via pyroptotic membrane rupture or in the absence of pyroptosis (185–189). With intracellular pathogens, pyroptosis mediated by GSDMD was necessary for IL-1β release from macrophages exposed to inflammasome activators (155). Cell- and liposome-based assays demonstrated that GSDMD pores were required for IL-1β transport across an intact lipid bilayer (42–45). Of note, GSDMD-/- macrophages are still able to release caspase-1-processed bioactive IL-1β via PIP2-mediated plasma membrane binding that is of a much slower rate than GSDMD-dependent release (190). These findings identify a non-pyroptotic function for GSDMD, and raised the possibility that GSDMD pores represent conduits for the secretion of cytosolic cytokines under conditions of cell hyperactivation (191, 192).
Gasdermin D-Regulated Cell Hyperactivation
How can a cell release IL-1β/IL-18 through GSDM pores in the absence of cell lysis? Secretion of the IL-1 family of cytokines was widely considered a caspase triggered pyroptosis-dependent event, as IL-1β and IL-18 lack a protein secretion signal sequence (193). Recently, the concept of “unconventional protein secretion” was advanced to describe GSDMs pore-mediated pro-inflammatory cytokine release (194, 195) and cellular alarmins such as ATP and high-mobility group box 1 (HMGB1) at the same time to augment inflammation in tissues to recruit immune cells against infection and damage. Phagocytes can achieve a state of hyperactivation, which is defined by their ability to secrete the IL-1 family of cytokines while retaining viability, but it remains unclear how IL-1β can be secreted from living cells (196).
When macrophages, dendritic cells and neutrophils survive inflammasome-mediated GSDMD pore-forming activity without membrane rupture and pyroptosis, they retain their ability to produce pro-inflammatory cytokines, termed “hyperactivated” cells (197). The hyperactivated cells and pyroptotic cells can be distinguished by measuring LDH in cell supernatants, which are too large to be secreted through GSDMD-NT pores and are only released upon cell lysis (Figure 6A). Mature IL-1 family cytokines and alarmin molecule HMGB1 are small proteins that are readily released through these pores. Alternatively, if the number of GSDMD pores patched in cell membrane exceeds the recovery capability of the cell, cell volume in turn increases. Once the volume exceeds membrane capacity, the plasma membrane divides from the cytoskeleton in large fluid-filled balloons. These pores disintegrate the cell membrane and are large enough to rapidly lose soluble proteins including LDH, whereas, organelles are retained (155). Shortly after the formation of GSDMD pores, a pyroptosis-associated membrane-rupture event proceeds which releases soluble cytosolic contents. Pyroptotic cells release both LDH and cytokines (196) (Figure 6A). Hyperactivated cells can process and pump out more inflammatory components as compared to their pyroptotic counterparts (141) (Figure 6B). At present, it still remains unclear what determines whether cleaved GSDMD causes pyroptosis or hyperactivation. Recently, researchers have proposed a possible mechanism of cell membrane repair response at the site of lesion, which efficiently recover membrane integrity, allowing cell survival. The battle between how promptly and efficiently the membrane repair response is initiated and how robust and fast the damaged is caused, is presumably dependent on how much and how efficiently GSDMD is cleaved.
Mechanisms of Pyroptotic Membrane Repair Regulation
Cells that have GSDM pores on plasma membrane can be sensed by an instantaneous increase in intracellular Ca2+ to trigger membrane repair by recruiting the endosomal sorting complexes required for transport (ESCRT) machinery to disrupted membrane areas and repair them in ectosomes (198) (Figure 7). Mammalian cells can repair plasma membrane damages when size of the disruption is not large (<100 nm) by a variety of mechanisms (199). Cell membrane that are damaged by GSDM pores likely induce activation of the ubiquitous plasma membrane damage repair response in eukaryotic cells, restoring membrane integrity rapidly to increase cell survival (200). The two major well-defined mechanisms are: Acidic Sphingomyelinase (ASM)-dependent endocytosis of plasma membrane pores and ESCRT-mediated shedding of injured plasma membrane (Figure 7). The ESCRT machinery was initially found in yeast, where it was identified to regulate protein trafficking and intraluminal vesicle formation at the distal side of cell membrane (201). The ESCRT machinery is a multiprotein complex contained a group of subcomplexes, including ESCRT 0, I, II, III, and ESCRT-III was discovered to be essential in membrane repair process (202, 203). The pyroptosis membrane repair involves massive endocytosis of damaged membrane, mostly from lysosomes (204) and exocytosis by promoting lipid raft formation (205, 206) (Figure 7). We have shown that when EhCP-A5 engages αvβ3 integrin on goblet cells, it facilitated outside-in signaling cascade by activating SRC family kinase, resulting in the activation of several kinases including PI3K and PKC (18). The trafficking vesicle marker, myristolated alanine-rich C-kinase substrate (MARCKS), was the target for PKC and has been implicated in docking vesicles for SNARE-mediated membrane fusion. We discovered that the R-SNARE vesicle-associated membrane proteins 8 (VAMP8) expressed on mucin vesicles was critical for SNARE-mediated exocytosis of mucin secretion in response to Eh (18). Thus, SNARE complex in goblet cells regulates mucin exocytosis and degradation of these regulatory pathways aggravates Eh pathogenesis (18). After the membrane is damaged and the uncontrolled entry of Ca2+ occurs, lysosomes are recruited to the site of the lesion and subsequently fuse to the plasma membrane, releasing ASM that hydrolyzes sphingomyelin into ceramide. This in turn, facilitates endosomes formation that internalize the lesion and repair membrane integrity (206–209) (Figure 7). A group evaluated the involvement of ASM in the repairing plasma membrane that caused by Eh. They found that damage to Eh membrane promotes lysosomes migration to the site of impairment where lysosomal ASM is released, forming membrane patches and endosomes to incorporate the lesion area and repair cell membrane, thus this mechanism facilitates amoebic viability (210).
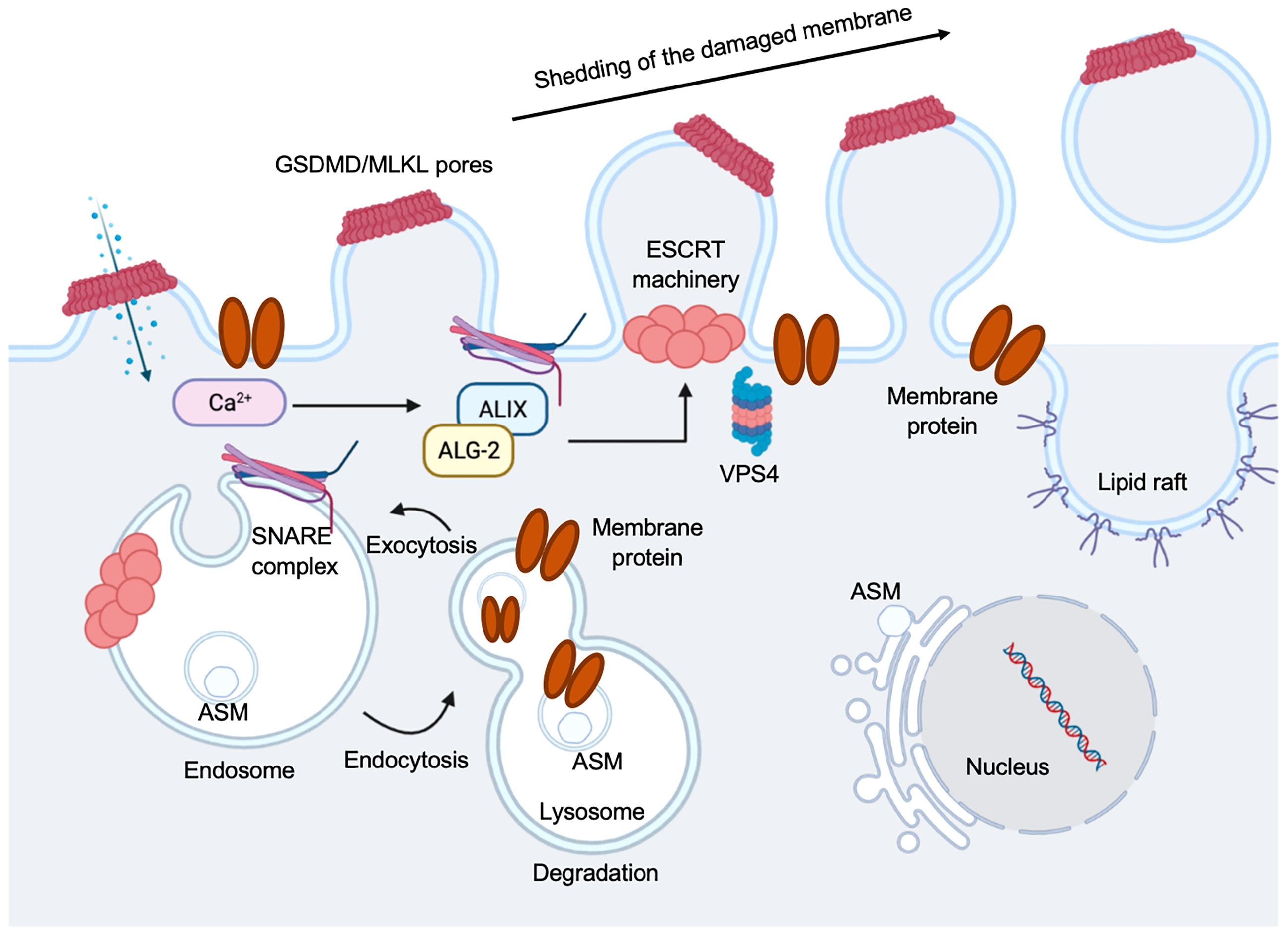
Figure 7 Regulation of pyroptosis membrane repair mechanisms. Pyroptotic pores formed by GSDMD, or necroptotic pores formed by mixed lineage kinase domain-like protein (MLKL), triggers ion exchanges across the plasma membrane. Ca2+ influx occurs through plasma membrane wounds, which in turn binds to apoptosis linked gene 2 (ALG-2) facilitating its recruitment and migration to the damage site. Elevation in the intracellular Ca2+ concentration triggers exocytosis of lysosomes. Lysosomal acid sphingomyelinase (ASM) is transported to the outer leaflet of the membrane. This is followed by the recruitment of ALG-2 interacting protein X (ALIX) and this occurs in an ALG-2 and Ca2+ dependent manner. This initiates ESCRT-III assembly, leading to membrane deformation into the extracellular milieu. A promising candidate is SNARE-mediated exocytosis to facilitate vesicle and plasma membrane fusion events. Vacuolar protein sorting-associated protein 4 (VPS4) leads to membrane repair and shedding of the damaged part of the cell membrane, recycling ESCRT subunits during this process.
Ca2+ influx through GSDMD pores acts as a signal for cells to undergo membrane repair by recruiting the ESCRT machinery to the site of damage or ruptured membrane (198) (Figure 7). Inhibiting the ESCRT-III assembly robustly favors pyroptosis in mouse BMDMs (198), suggesting a crucial anti-inflammatory role for the ESCRT-III machinery to restore cell homeostasis after GSDMD pore formation (Figure 7). Imaging the disruption of the electrochemical gradient using the Ca2+ dye Fluo-8, and loss of membrane integrity using propidium iodide (PI) in mouse BMDMs, showed that the ESCRT system dampens GSDMD pore-forming activity and downregulated cell death and IL-1β secretion by acting downstream of caspase activation and GSDMD oligomerization (198). However, it remains vague whether ESCRTs could serve as a control of caspase activation to regulate GSDMD activity, and if other GSDMs are involved in the process. ESCRT-III was discovered to repair mixed lineage kinase domain-like protein (MLKL)-induced plasma membrane injury in the process of necroptosis, indicating this membrane repair process can increase cell viability (triggering hyperactivation instead of pyroptosis) to combat the pore-forming activity of GSDMD (198) (Figure 7). The mechanisms on if bioactive IL-1β is packaged into micro-vesicles to be released or they are secreted primarily from the GSDMD pores remain to be elucidated. It is still unclear if these membrane repair processes occur in cells with GSDMD-disrupted plasma membranes.
Gasdermin Pores Target Mitochondrial Membrane to Eradicate E. histolytica and Other Protozoan Parasites
GSDM-NT can bind and insert into internal cellular membranes (70, 99, 107, 130), including mitochondria and bacteria to kill the cells (43). However, bacteria are known to survive during pyroptosis (155, 211) so the physiological relevance of bacterial membrane-targeting remains unclear. The NT of GSDMD and GSDME when translocated to mitochondria, permeabilize the outer membrane and destroy their function, resulting in the production of ROS (Figure 2, Figure 6 and Figure 8), loss of mitochondrial membrane potential and the release of cytochrome c, which subsequently activates caspase-3 to augment cell death (99). For example, bacteria that remain viable during pyroptosis but are damaged may drive by the formation of GSDMD pores (43) and ROS (212). ROS and the release of mitochondrial DNA, mitochondria, nuclei, and ASC specks via cell membrane rupture and Ca2+ influx and K+ efflux through GSDM pores can trigger activation of inflammasomes (213). Noncanonical signaling pathway primarily triggers GSDMD cleavage and induces pyroptosis, however, activation of noncanonical signaling pathway can trigger assembly of the canonical NLRP3 inflammasome, possibly via the generation of mitochondrial ROS and K+ efflux, implicating a certain degree of crosstalk between these two pathways (214–216) (Figure 2). The noncanonical caspase-4 activation in response to Eh involves K+ efflux and ROS generation that can play an important role not only in amplifying downstream pro-inflammatory responses (Figure 2), but by interacting with the canonical NLRP3 inflammasome pathway to enhance the cleavage of caspase-1 CARD proteins (21). K+ channels are the most common identified ion transporter, and their critical roles was determined from colon biopsies from human with amebiasis with the demonstration of restrained K+ channel expression. Blocking K+ channels with genetic silencing or pharmacologic inhibitors suppressed caspase-1 activation, IL-1β secretion and cell pyroptosis in macrophages (217). The production of ROS is dispensable in killing Leishmania amazonensis but plays a major role in regulating inflammatory responses by regulating neutrophil infiltration into lesions (218). Activated phagocytes elicit cytotoxic impacts through ROS generation to kill pathogens by oxidative damage in Chagas disease causing Trypanosoma cruzi (219–223).
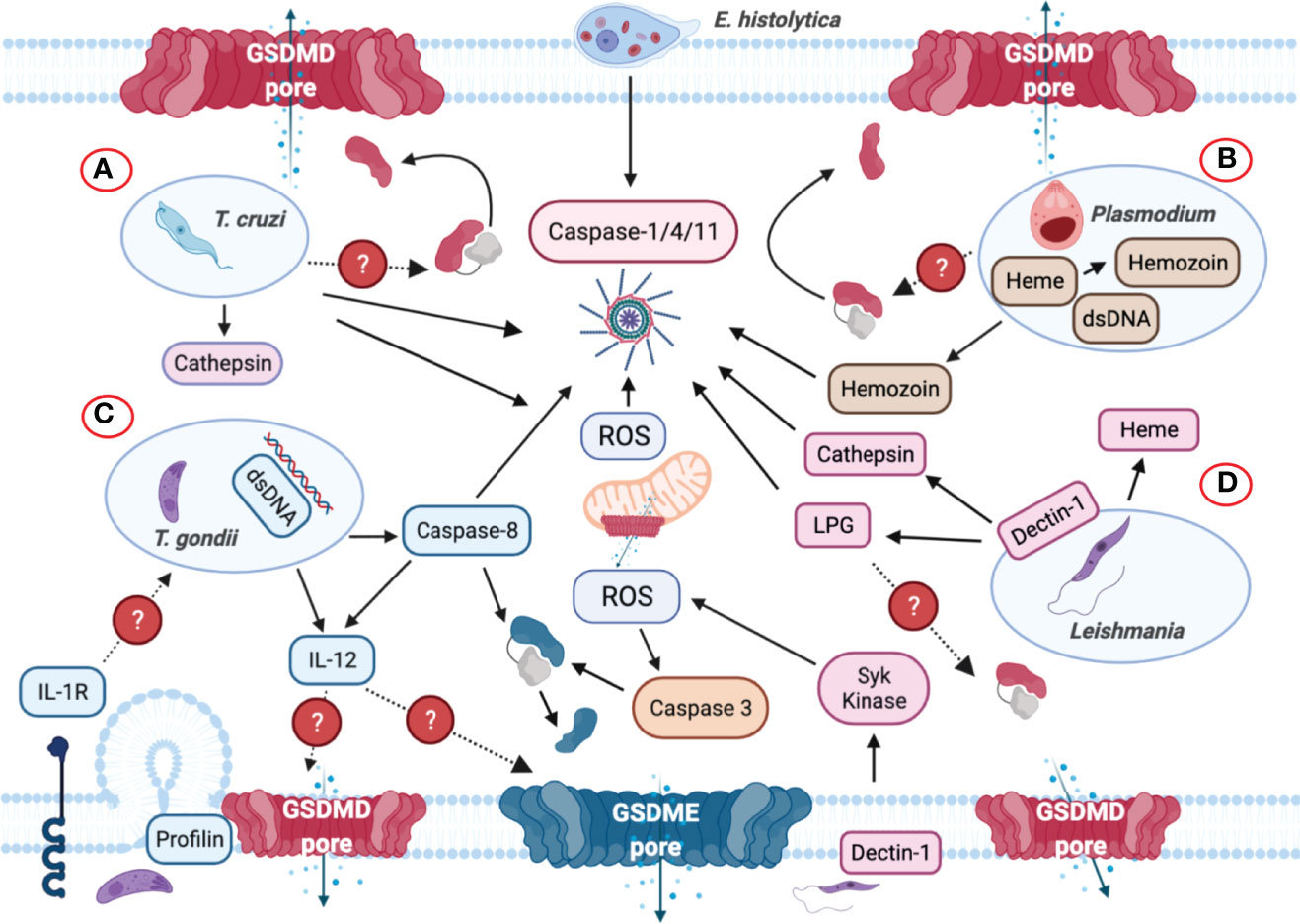
Figure 8 Inflammasome and GSDMs regulation by intracellular protozoan parasites. (A) Trypanosoma cruzi triggers NLRP3 inflammasome activation when the parasite lyses the parasitophorous vacuole to gain access to the cell cytoplasm. Cathepsin and reactive oxygen species (ROS) are involved in NLRP3 inflammasome activation. (B) Plasmodium-derived molecules such as hemozoin and dsDNA are released to the host cell cytosol upon lysosomal rupture and cathepsin release that trigger activation of the NLRP3 inflammasome. dsDNA can trigger NLRP3 inflammasome activation via the generation of ROS and membrane disruption. It is unclear if GSDMD or other GSDMs are involved in inflammasome-mediated IL-1β/IL-18 secretion. (C) Though Toxoplasma gondii is widely considered as a silent parasite, NLRP3 inflammasomes are activated upon infection. The parasite can be recognized by TLR-dependent sensing of T. gondii-derived molecules in murine dendritic cells that induces the production of IL-12. The identification of the parasite profilin as a PAMP mediates the recognition of T. gondii. Subsequently, the production of ROS is also elevated upon sensing this parasite but it remains unknown how GSDMD cleavage occurs during infection. Caspase-8 was discovered to mediate T. gondii control through innate production of IL-12, and IL-12 may be released via GSDMD/GSDME pores. Caspase-8 can induce the cleavage of both GSDMD and GSDME and act as the upstream regulator for caspase-3. (D) Upon phagocytosis by macrophages, Leishmania triggers Dectin-1, a C-type lectin receptor that signals containing the participation of Syk kinase, which in turn produces ROS to promote NLRP3 inflammasome activation. Additionally, Leishmania lipophosphoglycan (LPG) triggers caspase-11 activation, promoting NLRP3 inflammasome assembly via the noncanonical pathway. During this process, mitochondria generate ROS, lose their transmembrane potential and release cytochrome c into the cytosol, which subsequently activate caspase-3 to cleave GSDME or GSDMD, causing pyroptosis instead of apoptosis.
Pyroptosis Defends Against Intracellular Protozoan Parasites
Intracellular protozoan parasites reside in neutrophils, dendritic cells and macrophages. GSDMD-mediated pyroptosis can effectively damage intracellular parasites-residing niche, releasing cytosolic contents to promote pathogen expulsion. This in turn, triggers a strong inflammatory response to eliminate the compromised cell. Removing the intracellular replicative niche is beneficial to the host, however, it raises the interesting question, how does pyroptotic cells or infected macrophages ultimately eradicate pathogens?
Trypanosoma cruzi
Trypanosoma cruzi is an intracellular protozoan parasite that causes Chagas disease, which manifests as fever, fatigue, and headaches during early stage of infection, with potential to progress to heart failure and severe gastrointestinal complications (224–226). During the acute phase of infection, T. cruzi in circulating blood results in fever and swelling around the site of infection (227). Innate and adaptive host immune responses are both indispensable to control T. cruzi infection (228, 229). Upon lysing the vacuolar membrane, T. cruzi gains access to the host cell cytoplasm, where it undergoes replication (Figure 8A). Innate immune sensing by the NLRP3 inflammasome critically contribute to eliminating T. cruzi from healthy tissue (230, 231) (Table 3). NLRP3-/-, caspase-1-/- and Asc-/- mice display more severe parasitemia (231) and lower survival probability (230) compared to wild-type mice. Cathepsin B is essential for NLRP3 activation in response to T. cruzi, and pharmacologically inhibiting cathepsin B abolishes IL-1β release (231) (Figure 8A). Interestingly, NLRP3 deficient macrophages are not only presented with impaired IL-1β secretion but also nitric oxide (NO) release that causes increase in parasite replication (231). NO produced by innate immune cells is trypanocidal to control T. cruzi replication (246). Macrophages serve as first responders by activation of NAD(P)H oxidase (NOX2) and ROS production to eliminate T. cruzi (247) (Figure 8A). NOX2 and ROS promote T cell-mediated adaptive immune response and deficiency in them causes defective splenic activation of cytotoxic T cell immunity against T. cruzi invasion (248). By infecting p47phox-/- macrophages with T. cruzi in vitro, NOX2 and ROS were discovered to regulate cytokinopathy through controlling cytokine gene expression. Cytokinopathy was demonstrated as a molecular mechanism in cardiomyopathy in Chagas’ disease by cardiac gene expression profiling (249). Under situations like trauma, infection, and inflammation, macrophages become activated and secret robust level of inflammatory cytokines including IL-1 and IL-6, triggering the cascade that results in cytokinopathy (250). However, with deficiency of NOX2, CD8+T cell generation and activation were compromised, causing parasite burden, tissue damage and death (248).
Table 3 The signaling and functions of gasdermin proteins defend against intracellular protozoan parasite infection.
GSDMD pore formation from NLRP3 inflammasome activation induces autophagy and may serve as a valuable anti-parasitic mechanism to eliminate T. cruzi from the host (251) (Table 1 and Figure 8A). By studying the interplay between inflammasome and autophagy in T. cruzi-infected mice and peritoneal macrophages, it was demonstrated that autophagy governs trypomastigotes outcome and activity at early stages of invasion by forming the NLRP3-dependent autolysosomes (252). NLRP3 inflammasome is also a requirement for triggering functional autophagy in T. cruzi activated macrophages (252) (Table 3). Autophagy and autophagy associated proteins not only regulate excessive inflammatory responses (232) but also target intracellular pathogens and organelles (253). Thus, deficits in autophagy molecules can result in uncontrolled inflammasome activation and subsequent immunopathology (254, 255). Autophagy disrupts pyroptosis by downregulating GSDMD cleavage (256) (Table 3). A recent study revealed that inhibition of mammalian target of rapamycin (mTOR) signaling upregulated NLRP3 expression, ROS generation and IL-1β release that boosted the efficiency of parasite control in macrophages (257) (Table 3); however, the direct effect and connection between autophagy and rapamycin treatment are unknown (257). Another study reported that rapamycin as an autophagy agonist, prevents GSDMD-mediated pyroptosis after LPS treatment (258). Autophagy was demonstrated to coordinate GSDME-induced pyroptosis by utilizing chloroquine to block autophagy pathway in human melanoma cells following an unclear mechanism (233). It will be of interest to uncover the interplay between autophagy and GSDMD/GSDME to shed light on the role of GSDMs in T. cruzi infection (Table 3).
Plasmodium
Malaria is a parasitic disease caused by the protozoa pathogens of the Plasmodium genus, which accounts for 228 million infected people globally in 2018 (259). Transmission occurs primarily through the bite of an infected female Anopheles mosquitoes, which inoculate the highly infective sporozoite forms into the mammalian host (260, 261). Different species can result in various clinical outcomes. This parasite has a unique life cycle in which sporozoites enter circulation and liver, where they undergo replication and differentiation (262). Upon exiting infected hepatocytes, red blood cells (RBCs) are infected where the parasites transform into trophozoites (263). Excessive activation of inflammatory responses in malaria causes a pro-inflammatory cytokine storm that coincides with severe fever episodes in patients that can be detrimental to the host. The cytokine storm contains IL-1β, TNF-α and IL-12 (262, 264), which effectively can control parasite replication (262). The release of inflammatory effectors is the consequence of hemozoin (265) (Figure 8B). The majority of the liver damage in malaria is an outcome of high oxidative stress accumulated by heme and infiltrated neutrophils regulated by NF-κB (266). When Plasmodium digests hemoglobin it converts free heme into an insoluble crystalline manner named hemozoin. Hemozoin drives inflammation in both sterile and infectious conditions, contributing to the pathogenesis of hemolytic disorders, including sickle cell disease and malaria (267, 268). Since the generation of hemozoin is a critical step for Plasmodium survival, it becomes a promising target for antimalarial drug development (269).
In the parasite vacuole, some Plasmodium-derived specific molecules, including hemozoin-derived from RBCs, parasite double-stranded DNA (dsDNA) and cathepsin are released in the cytoplasm to trigger NLRP3 inflammasome activation (234, 235) (Figure 8B). Sufficient NLRP3 inflammasome activation requires K+ efflux and NADPH oxidase induced by hemozoin in P. berghei ANKA sporozoites, suggesting NLRP3 as an essential player in cerebral malaria in mice, while not controlling parasitemia (270). By dissecting the mechanism of hemolysis, it was found that hemozoin triggered NLRP3 inflammasome activation in LPS-primed macrophages to promote IL-1β release (271). In addition, hemozoin-induced sterile hemolysis was demonstrated to trigger ROS production, tyrosine kinase (Syk) and NADPH oxidase activation that was crucial to inflammasome assembly (271). It is not known if this mechanism is involved in malaria pathogenesis. Conversely, NLRP3 was found to have a toxic impact in malaria, as mice deficiency in inflammasome components had higher survival rate with a lethal dose of P. chabaudi (234).
Caspase-8 was reported to be a primary mediator of systemic inflammatory response in P. chabaudi infected mice and in P. berghei experimental cerebral malaria. Knockout of caspase-8/1/11 or caspase-8/GSDMD resulted in disruption of TNF-α and IL-1β production, uncovering a supplementary and indispensable role for caspase-8 and GSDMD in malaria pathogenesis (236). The role of caspase-8 on IL-1β release needs more investigation as it is was shown to be involved in the cleavage of GSDMD (67). However, none of these roles in malaria pathogenesis has been addressed to date. It is not known whether these cytokines are secreted through GSDMD pores or it is a pyroptosis-dependent event or if other GSDMs might be involved (Figure 8B). A more comprehensive understanding using different Plasmodium species and in different models of disease would be beneficial to dissect the mechanisms on progression and regulation of malaria.
Toxoplasma gondii
Toxoplasma gondii is an obligate intracellular parasite and is the causative agent of toxoplasmosis (272). In most, T. gondii infection remains asymptomatic, whereas devastating diseases can occur in immunosuppressed individuals, causing infections in the brain and other tissues (273). As an apicomplexan, T. gondii has evolved specialized secretory organelles, including the rhoptries (ROPs) and dense granules (GRAs), which are designated to inject effector molecules into the host cell (274). The parasite can also invade and replicate in host cell by secreting ROP effector proteins and building a parasitophorous vacuole (PV) (Figure 8C), with the injection of GRA molecules into the cell cytosol to promote parasite growth (275). Profilin was identified from T. gondii as a PAMP recognized by murine Toll-like receptor (TLR)11/12 (276–278) (Figure 8C). Innate detection of T. gondii induces IL-12 production in both dendritic cell and macrophage to directly sense and recognize parasite-derived molecules (Figure 8C). Ripk3-/-Casp8-/- mice succumbed to invasion with T. gondii that was rescued with exogenous IL-12 (239). Caspase-8 is an essential regulator of cell apoptosis and is indispensable for optimal transcription of inflammatory defense genes such as il12 and il1β (239). Caspase-8 is an upstream regulator of caspase-3 that control cell apoptosis, while preventing RIPK3-MLKL-dependent necroptosis (Figure 8C). By using a small-molecule inhibitor to block TGFβ-activated kinase 1 (TAK1), the activation of caspase-8 was discovered to induce both GSDMD and GSDME cleavage in murine macrophages, causing cell pyroptosis (68). Additionally, caspase-8 was found to initiate NLRP3 inflammasomes in macrophages (240) that might serve as a compensatory pathway when caspase-1 activity is inhibited (170, 237). More work needs to be conducted on how T. gondii regulates caspase-8 activity and if GSDMD/GSDME pores mediates IL-12 release during this process (Figure 8C and Table 1).
T. gondii-induced macrophage death occurs via a guanylate binding protein (GBP) dependent event independent of GSDMD to promote apoptosis (279). GBP1 was found to be targeted in Salmonella-containing vacuoles to facilitate caspase-4 recruitment causing its activation and pyroptosis. These finding suggested an immune role for GBPs as a conduit for not only pyroptosis, but also apoptosis (279). T. gondii stimulation in human monocytes stimulates the Syk-CARD9/MALT-1-NF-κB signaling pathway and activation of the NLRP3 inflammasome to release IL-1β in a cell death- and GSDMD-independent manner (280). In contrast, T. gondii infection in microglia elicits NF-κB signaling to mediate pro-inflammatory cytokine secretion in innate host defence. Other studies have shown that GSDMD dependent IL-1α but not IL-1β release in microglia impaired parasite control and dysregulated immune cell infiltration. Unlike IL-1β, IL-1α functions as an alarmin molecule directly released without processing upon sensing invasive signals, acting as a rapid initiator in inflammatory response (281). IL-1α and IL-1β both process through the same receptor (IL-1R) (282) (Figure 8C). It was concluded that microglia act differently from macrophages as they can release the alarmin IL-1α to promote neuroinflammation and parasite control in T. gondii infection though they are present in the identical microenvironment in the central nervous system (241). Furthermore, IL-1R1 is primarily expressed on blood vasculature in the brain, and the pro-inflammatory response triggers in the brain during chronic T. gondii infection is regulated through IL-1α, but not IL-1β, since brain-resident microglia lack an NF-κB signature compared to monocyte-differentiated macrophages (241). In GSDMD KO there is a robust increase in T. gondii cyst burden six weeks post infection compared to wild-type mice, indicating an inflammasome-mediated neuroinflammation requires GSDMD to control T. gondii in the brain (241). However, studies are still needed to determine what role IL-1R could play in controlling T. gondii infection in the gut and macrophages. Activating the P2X7 receptor with extracellular ATP in T. gondii-stimulated macrophages enhanced parasite clearance by producing ROS and lysosome fusion with parasitophorous vacuole, forming a phagolysosome (238). Extracellular ATP also activated the NLRP3 inflammasome to potentiate IL-1β secretion by recruiting caspase-1. IL-1β released in the extracellular milieu binds to its receptor accelerating T. gondii eradication through mitochondrial ROS production (Figure 8C). T. gondii-mediated activation of the inflammasome results in caspase-1-mediated processing of IL-1α, IL-1β and IL-18 and pyroptosis (Figure 8C).
Leishmania
Leishmaniasis is a vector-borne parasitic inflammatory disease caused by the protozoan parasite of the Leishmania genus. This disease affects millions of people, over 88 countries worldwide, especially prevalent in tropical and subtropical areas of the world (283). Self-healing cutaneous and debilitating visceral leishmaniasis affect approximately 1.5 million people globally (283, 284).
The parasite exists extracellularly as flagellated promastigotes in sand flies (283). Leishmania spp. interact with a range of cells, including neutrophils, monocytes, and macrophages, where the parasites differentiate into amastigotes and replicate inside the parasitophorous vacuole (283) (Figure 8D). Inflammasome activation is a signature event of leishmaniasis and is protective and responsible for the restriction of parasite replication in macrophages (242, 285). All different species of Leishmania can activate the NLRP3 inflammasome via K+ efflux and cathepsins to release IL-1β (242) (Figure 8D). Upon phagocytosis, the parasite turns on the C-type lectin receptor: Dectin-1, which elicits a Syk-dependent signaling pathway that results in ROS generation to contribute to NLRP3 inflammasome activation (243) (Figure 8D). More recently, it was shown that Leishmania lipophosphoglycan (LPG) can trigger the activation of caspase-11 to promote GSDMD pore formation, IL-1β secretion and pyroptotic cell death (244) (Figure 8D).
Receptor interacting protein kinase 1 (RIPK1) is a critical kinase that mediates necroptotic cell death following activation of various cell death receptors and TLRs (245). Human visceral leishmaniasis displayed increased serum levels of heme (Figure 8D), which is generated by hemoglobin catabolism, and is a potent stimulator of necroptotic macrophage (245). By examining the correlation between heme and necroptosis, it was found that heme strongly prevented Leishmania replication in BMDMs, and blocking RIPK1 kinase activity upregulated Leishmania replication without the presence of heme (245). Since GSDMD can be cleaved by RIPK1 (Table 1) and RIPK1 kinase activity is required for IL-1β expression in response to Leishmania (245), it would be of interest to detect if any crosstalk between necroptosis and pyroptosis exists in the presence of Leishmania. In addition, how pyroptosis affects parasite survival and virulence remains to be elucidated. Interestingly, it was reported that the host cell secretory pathway can export Leishmania zinc-metalloprotease GP63 and LPG (286) out of the vacuole, suggesting these virulent factors could act as the upstream regulator for caspase-11 activation. On the contrary, Leishmania can negatively regulate inflammasome activation by using GP63 to block PKC signaling in human monocytes, avoiding ROS generation, inflammasome activation and IL-1β production (287).
Concluding Remarks
A protective role of inflammasome signaling and inflammatory cytokine release has been determined against protozoan parasites, particularly in the acute phase of infection. It is worth noting that in the past several decades new modes of regulated cell death have emerged to involve inflammatory form of necroptosis and pyroptosis, and other inflammatory manners of caspase-independent programmed cell death. Inhibiting pyroptosis ameliorates disease, including septic shock and autoinflammation, conversely, it can be detrimental for infections. The key roles of GSDMs in autoimmune and inflammatory diseases, infection, deafness and cancer are evolving, which reveals possible novel therapeutic avenues (76, 113, 288, 289). However, of note, the non-pyroptotic function of GSDMD has been uncovered in Eh-induced hyperactivated macrophages (22), and in GSDMD deficient LS174T cells (goblet cell line) that displayed vigorous reduction in MUC2 secretion when stimulated with mucus secretagogues such as ATP, histamine and PMA (290), suggesting a significant non-pyroptotic role of GSDMD in regulating cortical actin cytoskeleton disassembly during mucin granule exocytosis (291). By proteomics analysis, we determined SNAP23 is highly involved in membrane trafficking and is downregulated in Eh-induced hyperactivated macrophages (22), implying potential relevance may exist between exocytosis (membrane repair response) and conversion between hyperactivation and pyroptosis in macrophage. Mechanistically, if GSDM pores are effectively repaired by membrane repair pathways, pyroptotic cells might transform into non-pyroptotic or hyperactivated counterparts, whereas if GSDM pores keep releasing inflammatory cytokines and ions, the cell would finally burst. Thus, studying the mechanisms of the membrane repair system might shed light on defining the non-pyroptotic roles of GSDMs in the pathogenesis of protozoan parasites.
Pyroptosis and the connection between pyroptosis and apoptosis need further investigation. Caspase-3 as the common main protease that triggers the cleavage of GSDME (65) is the key determinant of which mode of programmed cell death pathway the cell follows. GSDME can be exploited as a master switch molecule in shifting between apoptosis and pyroptosis. The level of GSDME expression determines the fate of the cell: cells that express adequate GSDME trigger pyroptosis, while cells expressing insufficient GSDME elicit apoptosis (74). Moreover, GSDME cleavage particularly depends on caspase-3 and not caspase-7 to switch TNF-induced apoptosis to pyroptosis in HeLa cells (74). Since GSDME enhances caspase-3/7 activation in apoptotic cell death through targeting the mitochondria to release cytochrome c (99), we predict an inflammatory role of GSDME to trigger pyroptotic cell death is vital in alleviating protozoan parasite infections (Figure 8). Therefore, by targeting caspase-3 or GSDME, it holds much hope to explore the mechanism of development and treatment in inflammatory disorders, and to search new targets and strategies that are feasible for clinical use.
Recent studies have significantly advanced our understanding of the mechanisms and physiological roles of pyroptosis. Overall, there is a remarkable knowledge gap between GSDMs and protozoan parasite infection. More in-depth research needs to be focused on delineating how diverse immune cells regulate GSDMs pore-forming activity differently, how parasites manipulate GSDMs pore-forming activity to prevent being eradicated, what determines whether GSDMD cleavage induces hyperactivation or pyroptosis, how GSDMs pores initiate membrane repair, and what are the differences between extracellular and intracellular parasites in triggering GSDMs pore formation. Given the primary function of GSDMs in combating protozoan parasites, the search for GSDM-regulating pore-forming activity is likely to be extraordinarily developed in the future.
Author Contributions
KC and SW contributed and design the content of the review. SW wrote the first draft of the manuscript and KC and FM edited the text. SW and FM did the recent published study that led to this review. All authors contribute to manuscript revision, read, and approved the submitted version.
Funding
This work was funded by a Discovery Grant (RGPIN/04139-2019) from the Natural Sciences and Engineering Research Council of Canada and project grants from the Canadian Institutes of Health Research (PJT-173551, PJT-162284) awarded to KC.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mortimer L, Moreau F, Cornick S, Chadee K. The NLRP3 Inflammasome Is a Pathogen Sensor for Invasive Entamoeba histolytica via Activation of Alpha5beta1 Integrin at the Macrophage-Amebae Intercellular Junction. PLoS Pathog (2015) 11:5:e1004887. doi: 10.1371/journal.ppat.1004887
3. Mortimer L, Chadee K. The Immunopathogenesis of Entamoeba histolytica. Exp Parasitol (2010) 126:3366–80. doi: 10.1016/j.exppara.2010.03.005
4. Ralston KS, Petri WA Jr. Tissue Destruction and Invasion by Entamoeba histolytica. Trends Parasitol (2011) 27:6254–63. doi: 10.1016/j.pt.2011.02.006
5. Mortality GBD, Causes of Death C. Global, Regional, and National Age-Sex Specific All-Cause and Cause-Specific Mortality for 240 Causes of Death, 1990-2013: A Systematic Analysis for the Global Burden of Disease Study 2013. Lancet (2015) 385:9963117–71. doi: 10.1016/S0140-6736(14)61682-2
6. Cornick S, Tawiah A, Chadee K. Roles and Regulation of the Mucus Barrier in the Gut. Tissue Barriers (2015) 3:1–2.e982426. doi: 10.4161/21688370.2014.982426
7. Turner JR. Intestinal Mucosal Barrier Function in Health and Disease. Nat Rev Immunol (2009) 9:11799–809. doi: 10.1038/nri2653
8. Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The Inner of the Two Muc2 Mucin-Dependent Mucus Layers in Colon Is Devoid of Bacteria. Proc Natl Acad Sci USA (2008) 105:3915064–9. doi: 10.1073/pnas.0803124105
9. Johansson ME, Larsson JM, Hansson GC. The Two Mucus Layers of Colon Are Organized by the MUC2 Mucin, Whereas the Outer Layer Is a Legislator of Host-Microbial Interactions. Proc Natl Acad Sci USA (2011) 108 Suppl 1:4659–65. doi: 10.1073/pnas.1006451107
10. Mortimer L, Moreau F, Cornick S, Chadee K. Gal-Lectin-Dependent Contact Activates the Inflammasome by Invasive Entamoeba histolytica. Mucosal Immunol (2014) 7:4829–41. doi: 10.1038/mi.2013.100
11. Chadee K, Petri WA Jr., Innes DJ, Ravdin JI. Rat and Human Colonic Mucins Bind to and Inhibit Adherence Lectin of Entamoeba histolytica. J Clin Invest (1987) 80:51245–54. doi: 10.1172/JCI113199
12. Dodson JM, Lenkowski PW Jr., Eubanks AC, Jackson TF, Napodano J, Lyerly DM, et al. Infection and Immunity Mediated by the Carbohydrate Recognition Domain of the Entamoeba histolytica Gal/GalNAc Lectin. J Infect Dis (1999) 179:2460–6. doi: 10.1086/314610
13. Bracha R, Mirelman D. Adherence and Ingestion of Escherichia Coli Serotype 055 by Trophozoites of Entamoeba histolytica. Infect Immun (1983) 40:3882–7. doi: 10.1128/IAI.40.3.882-887.1983
14. Ravdin JI, Guerrant RL. Role of Adherence in Cytopathogenic Mechanisms of Entamoeba histolytica. Study With Mammalian Tissue Culture Cells and Human Erythrocytes. J Clin Invest (1981) 68:51305–13. doi: 10.1172/jci110377
15. Petri WA Jr., Smith RD, Schlesinger PH, Murphy CF, Ravdin JI. Isolation of the Galactose-Binding Lectin That Mediates the In Vitro Adherence of Entamoeba histolytica. J Clin Invest (1987) 80:51238–44. doi: 10.1172/JCI113198
16. Moncada D, Keller K, Chadee K. Entamoeba histolytica Cysteine Proteinases Disrupt the Polymeric Structure of Colonic Mucin and Alter Its Protective Function. Infect Immun (2003) 71:2838–44. doi: 10.1128/iai.71.2.838-844.2003
17. Lidell ME, Moncada DM, Chadee K, Hansson GC. Entamoeba histolytica Cysteine Proteases Cleave the MUC2 Mucin in its C-Terminal Domain and Dissolve the Protective Colonic Mucus Gel. Proc Natl Acad Sci USA (2006) 103:249298–303. doi: 10.1073/pnas.0600623103
18. Cornick S, Moreau F, Chadee K. Entamoeba histolytica Cysteine Proteinase 5 Evokes Mucin Exocytosis From Colonic Goblet Cells via Alphavbeta3 Integrin. PLoS Pathog (2016) 12(4):e1005579. doi: 10.1371/journal.ppat.1005579
19. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature (2015) 526:7575660–5. doi: 10.1038/nature15514
20. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 Cleaves Gasdermin D for Non-Canonical Inflammasome Signalling. Nature (2015) 526:7575666–71. doi: 10.1038/nature15541
21. Quach J, Moreau F, Sandall C, Chadee K. Entamoeba histolytica-Induced IL-1beta Secretion is Dependent on Caspase-4 and Gasdermin D. Mucosal Immunol (2019) 12:2323–39. doi: 10.1038/s41385-018-0101-9
22. Wang S, Moreau F, Chadee K. The Colonic Pathogen Entamoeba histolytica Activates Caspase-4/1 That Cleaves the Pore-Forming Protein Gasdermin D to Regulate IL-1β Secretion. PLoS Pathog (2022) 18(3):e1010415. doi: 10.1371/journal.ppat.1010415
23. Kayagaki N, Kornfeld OS, Lee BL, Stowe IB, O'Rourke K, Li Q, et al. NINJ1 Mediates Plasma Membrane Rupture During Lytic Cell Death. Nature (2021) 591:7848131–6. doi: 10.1038/s41586-021-03218-7
24. Martinon F, Mayor A, Tschopp J. The Inflammasomes: Guardians of the Body. Annu Rev Immunol (2009) 27:229–65. doi: 10.1146/annurev.immunol.021908.132715
25. Dinarello CA. Interleukin-1 in the Pathogenesis and Treatment of Inflammatory Diseases. Blood (2011) 117:143720–32. doi: 10.1182/blood-2010-07-273417
26. Franchi L, Eigenbrod T, Nunez G. Cutting Edge: TNF-Alpha Mediates Sensitization to ATP and Silica via the NLRP3 Inflammasome in the Absence of Microbial Stimulation. J Immunol (2009) 183:2792–6. doi: 10.4049/jimmunol.0900173
27. Jin C, Flavell RA. Molecular Mechanism of NLRP3 Inflammasome Activation. J Clin Immunol (2010) 30:5628–31. doi: 10.1007/s10875-010-9440-3
28. Schroder K, Tschopp J. The Inflammasomes. Cell (2010) 140:6821–32. doi: 10.1016/j.cell.2010.01.040
29. Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, et al. A Novel Heterodimeric Cysteine Protease Is Required for Interleukin-1 Beta Processing in Monocytes. Nature (1992) 356:6372768–74. doi: 10.1038/356768a0
30. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-Canonical Inflammasome Activation Targets Caspase-11. Nature (2011) 479:7371117–21. doi: 10.1038/nature10558
31. Chowdhury I, Tharakan B, Bhat GK. Caspases - an Update. Comp Biochem Physiol B Biochem Mol Biol (2008) 151:110–27. doi: 10.1016/j.cbpb.2008.05.010
32. Ramirez MLG, Salvesen GS. A Primer on Caspase Mechanisms. Semin Cell Dev Biol (2018) 82:79–85. doi: 10.1016/j.semcdb.2018.01.002
33. Scott AM, Saleh M. The Inflammatory Caspases: Guardians Against Infections and Sepsis. Cell Death Differ (2007) 14:123–31. doi: 10.1038/sj.cdd.4402026
34. Martinon F, Burns K, Tschopp J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-Beta. Mol Cell (2002) 10:2417–26. doi: 10.1016/s1097-2765(02)00599-3
35. Kamens J, Paskind M, Hugunin M, Talanian RV, Allen H, Banach D, et al. Identification and Characterization of ICH-2, a Novel Member of the Interleukin-1 Beta-Converting Enzyme Family of Cysteine Proteases. J Biol Chem (1995) 270:2515250–6. doi: 10.1074/jbc.270.25.15250
36. Sollberger G, Strittmatter GE, Kistowska M, French LE, Beer HD. Caspase-4 is Required for Activation of Inflammasomes. J Immunol (2012) 188:41992–2000. doi: 10.4049/jimmunol.1101620
37. Cheung KT, Sze DM, Chan KH, Leung PH. Involvement of Caspase-4 in IL-1 Beta Production and Pyroptosis in Human Macrophages During Dengue Virus Infection. Immunobiology (2018) 223:4–5356-64. doi: 10.1016/j.imbio.2017.10.044
38. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory Caspases Are Innate Immune Receptors for Intracellular LPS. Nature (2014) 514:7521187–92. doi: 10.1038/nature13683
39. Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C, et al. Noncanonical Inflammasome Activation of Caspase-4/Caspase-11 Mediates Epithelial Defenses Against Enteric Bacterial Pathogens. Cell Host Microbe (2014) 16:2249–56. doi: 10.1016/j.chom.2014.07.002
40. Tamura M, Tanaka S, Fujii T, Aoki A, Komiyama H, Ezawa K, et al. Members of a Novel Gene Family, Gsdm, are Expressed Exclusively in the Epithelium of the Skin and Gastrointestinal Tract in a Highly Tissue-Specific Manner. Genomics (2007) 89:5618–29. doi: 10.1016/j.ygeno.2007.01.003
41. Broz P, Pelegrin P, Shao F. The Gasdermins, a Protein Family Executing Cell Death and Inflammation. Nat Rev Immunol (2020) 20:3143–57. doi: 10.1038/s41577-019-0228-2
42. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-Forming Activity and Structural Autoinhibition of the Gasdermin Family. Nature (2016) 535:7610111–6. doi: 10.1038/nature18590
43. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-Activated Gasdermin D Causes Pyroptosis by Forming Membrane Pores. Nature (2016) 535:7610153–8. doi: 10.1038/nature18629
44. Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD Membrane Pore Formation Constitutes the Mechanism of Pyroptotic Cell Death. EMBO J (2016) 35:161766–78. doi: 10.15252/embj.201694696
45. Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, et al. GsdmD P30 Elicited by Caspase-11 During Pyroptosis Forms Pores in Membranes. Proc Natl Acad Sci U S A (2016) 113:287858–63. doi: 10.1073/pnas.1607769113
46. Saeki N, Kuwahara Y, Sasaki H, Satoh H, Shiroishi T. Gasdermin (Gsdm) Localizing to Mouse Chromosome 11 Is Predominantly Expressed in Upper Gastrointestinal Tract But Significantly Suppressed in Human Gastric Cancer Cells. Mamm Genome (2000) 11:9718–24. doi: 10.1007/s003350010138
47. Runkel F, Marquardt A, Stoeger C, Kochmann E, Simon D, Kohnke B, et al. The Dominant Alopecia Phenotypes Bareskin, Rex-Denuded, and Reduced Coat 2 Are Caused by Mutations in Gasdermin 3. Genomics (2004) 84:5824–35. doi: 10.1016/j.ygeno.2004.07.003
48. Yu Y, Zhang C, Zhou G, Wu S, Qu X, Wei H, et al. Gene Expression Profiling in Human Fetal Liver and Identification of Tissue- and Developmental-Stage-Specific Genes Through Compiled Expression Profiles and Efficient Cloning of Full-Length cDNAs. Genome Res (2001) 11:81392–403. doi: 10.1101/gr.175501
49. Tanaka S, Mizushina Y, Kato Y, Tamura M, Shiroishi T. Functional Conservation of Gsdma Cluster Genes Specifically Duplicated in the Mouse Genome. G3 (Bethesda) (2013) 3:101843–50. doi: 10.1534/g3.113.007393
50. Lunny DP, Weed E, Nolan PM, Marquardt A, Augustin M, Porter RM. Mutations in Gasdermin 3 Cause Aberrant Differentiation of the Hair Follicle and Sebaceous Gland. J Invest Dermatol (2005) 124:3615–21. doi: 10.1111/j.0022-202X.2005.23623.x
51. Kumar S, Rathkolb B, Budde BS, Nurnberg P, de Angelis MH, Aigner B, et al. Gsdma3(I359N) Is a Novel ENU-Induced Mutant Mouse Line for Studying the Function of Gasdermin A3 in the Hair Follicle and Epidermis. J Dermatol Sci (2012) 67:3190–2. doi: 10.1016/j.jdermsci.2012.05.001
52. Deng W, Bai Y, Deng F, Pan Y, Mei S, Zheng Z, et al. Streptococcal Pyrogenic Exotoxin B Cleaves GSDMA and Triggers Pyroptosis. Nature (2022) 602:7897496–502. doi: 10.1038/s41586-021-04384-4
53. Li J, Zhou Y, Yang T, Wang N, Lian X, Yang L. Gsdma3 Is Required for Hair Follicle Differentiation in Mice. Biochem Biophys Res Commun (2010) 403:118–23. doi: 10.1016/j.bbrc.2010.10.094
54. Sato H, Koide T, Masuya H, Wakana S, Sagai T, Umezawa A, et al. A New Mutation Rim3 Resembling Re(den) Is Mapped Close to Retinoic Acid Receptor Alpha (Rara) Gene on Mouse Chromosome 11. Mamm Genome (1998) 9:120–5. doi: 10.1007/s003359900673
55. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme A From Cytotoxic Lymphocytes Cleaves GSDMB to Trigger Pyroptosis in Target Cells. Science (2020) 368:6494. doi: 10.1126/science.aaz7548
56. Das S, Miller M, Beppu AK, Mueller J, McGeough MD, Vuong C, et al. GSDMB Induces an Asthma Phenotype Characterized by Increased Airway Responsiveness and Remodeling Without Lung Inflammation. Proc Natl Acad Sci USA (2016) 113:4613132–7. doi: 10.1073/pnas.1610433113
57. Rana N, Privitera G, Kondolf HC, Bulek K, Lechuga S, De Salvo C, et al. GSDMB Is Increased in IBD and Regulates Epithelial Restitution/Repair Independent of Pyroptosis. Cell (2022) 185:2283–98.e17. doi: 10.1016/j.cell.2021.12.024
58. Hu Y, Jin S, Cheng L, Liu G, Jiang Q. Autoimmune Disease Variants Regulate GSDMB Gene Expression in Human Immune Cells and Whole Blood. Proc Natl Acad Sci USA (2017) 114:38:E7860–E2. doi: 10.1073/pnas.1712127114
59. Verlaan DJ, Berlivet S, Hunninghake GM, Madore AM, Lariviere M, Moussette S, et al. Allele-Specific Chromatin Remodeling in the ZPBP2/GSDMB/ORMDL3 Locus Associated With the Risk of Asthma and Autoimmune Disease. Am J Hum Genet (2009) 85:3377–93. doi: 10.1016/j.ajhg.2009.08.007
60. Kang MJ, Yu HS, Seo JH, Kim HY, Jung YH, Kim YJ, et al. GSDMB/ORMDL3 Variants Contribute to Asthma Susceptibility and Eosinophil-Mediated Bronchial Hyperresponsiveness. Hum Immunol (2012) 73:9954–9. doi: 10.1016/j.humimm.2012.06.009
61. Chen Q, Shi P, Wang Y, Zou D, Wu X, Wang D, et al. GSDMB Promotes non-Canonical Pyroptosis by Enhancing Caspase-4 Activity. J Mol Cell Biol (2019) 11:6496–508. doi: 10.1093/jmcb/mjy056
62. Wei J, Xu Z, Chen X, Wang X, Zeng S, Qian L, et al. Overexpression of GSDMC Is a Prognostic Factor for Predicting a Poor Outcome in Lung Adenocarcinoma. Mol Med Rep (2020) 21:1360–70. doi: 10.3892/mmr.2019.10837
63. Cui YQ, Meng F, Zhan WL, Dai ZT, Liao X. High Expression of GSDMC Is Associated With Poor Survival in Kidney Clear Cell Cancer. BioMed Res Int (2021) 2021:5282894. doi: 10.1155/2021/5282894
64. Xi R, Montague J, Lin X, Lu C, Lei W, Tanaka K, et al. Up-Regulation of Gasdermin C in Mouse Small Intestine Is Associated with Lytic Cell Death in Enterocytes in Worm-Induced Type 2 Immunity. Proc Natl Acad Sci USA (2021) 118:30. doi: 10.1073/pnas.2026307118
65. Hou J, Zhao R, Xia W, Chang CW, You Y, Hsu JM, et al. PD-L1-Mediated Gasdermin C Expression Switches Apoptosis to Pyroptosis in Cancer Cells and Facilitates Tumour Necrosis. Nat Cell Biol (2020) 22:101264–75. doi: 10.1038/s41556-020-0575-z
66. Watabe K, Ito A, Asada H, Endo Y, Kobayashi T, Nakamoto K, et al. Structure, Expression and Chromosome Mapping of MLZE, a Novel Gene Which Is Preferentially Expressed in Metastatic Melanoma Cells. Jpn J Cancer Res (2001) 92:2140–51. doi: 10.1111/j.1349-7006.2001.tb01076.x
67. Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, et al. Pathogen Blockade of TAK1 Triggers Caspase-8-Dependent Cleavage of Gasdermin D and Cell Death. Science (2018) 362:64181064–9. doi: 10.1126/science.aau2818
68. Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, et al. Caspase-8 Induces Cleavage of Gasdermin D to Elicit Pyroptosis During Yersinia Infection. Proc Natl Acad Sci USA (2018) 115:46E10888–E97. doi: 10.1073/pnas.1809548115
69. Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin D Plays a Vital Role in the Generation of Neutrophil Extracellular Traps. Sci Immunol (2018) 3:26. doi: 10.1126/sciimmunol.aar6689
70. Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, et al. Noncanonical Inflammasome Signaling Elicits Gasdermin D-Dependent Neutrophil Extracellular Traps. Sci Immunol (2018) 3:26. doi: 10.1126/sciimmunol.aar6676
71. Li S, Wu Y, Yang D, Wu C, Ma C, Liu X, et al. Gasdermin D in Peripheral Myeloid Cells Drives Neuroinflammation in Experimental Autoimmune Encephalomyelitis. J Exp Med (2019) 216:112562–81. doi: 10.1084/jem.20190377
72. Kanneganti A, Malireddi RKS, Saavedra PHV, Vande Walle L, Van Gorp H, Kambara H, et al. GSDMD Is Critical for Autoinflammatory Pathology in a Mouse Model of Familial Mediterranean Fever. J Exp Med (2018) 215:61519–29. doi: 10.1084/jem.20172060
73. Begum S, Moreau F, Leon Coria A, Chadee K. Entamoeba histolytica Stimulates the Alarmin Molecule HMGB1 from Macrophages to Amplify Innate Host Defenses. Mucosal Immunol (2020) 13:2344–56. doi: 10.1038/s41385-019-0233-6
74. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy Drugs Induce Pyroptosis Through Caspase-3 Cleavage of a Gasdermin. Nature (2017) 547:766199–103. doi: 10.1038/nature22393
75. Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by Caspase-3 During Apoptosis Mediates Progression to Secondary Necrotic/Pyroptotic Cell Death. Nat Commun (2017) 8:14128. doi: 10.1038/ncomms14128
76. Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin E Suppresses Tumour Growth by Activating Anti-Tumour Immunity. Nature (2020) 579:7799415–20. doi: 10.1038/s41586-020-2071-9
77. Bischoff AM, Luijendijk MW, Huygen PL, van Duijnhoven G, De Leenheer EM, Oudesluijs GG, et al. A Novel Mutation Identified in the DFNA5 Gene in a Dutch Family: A Clinical and Genetic Evaluation. Audiol Neurootol (2004) 9:134–46. doi: 10.1159/000074185
78. Cheng J, Han DY, Dai P, Sun HJ, Tao R, Sun Q, et al. A Novel DFNA5 Mutation, IVS8+4 A>G, in the Splice Donor Site of Intron 8 Causes Late-Onset non-Syndromic Hearing Loss in a Chinese Family. Clin Genet (2007) 72:5471–7. doi: 10.1111/j.1399-0004.2007.00889.x
79. Li-Yang MN, Shen XF, Wei QJ, Yao J, Lu YJ, Cao X, et al. IVS8+1 DelG, a Novel Splice Site Mutation Causing DFNA5 Deafness in a Chinese Family. Chin Med J (Engl) (2015) 128:182510–5. doi: 10.4103/0366-6999.164980
80. Yu C, Meng X, Zhang S, Zhao G, Hu L, Kong X. A 3-Nucleotide Deletion in the Polypyrimidine Tract of Intron 7 of the DFNA5 Gene Causes Nonsyndromic Hearing Impairment in a Chinese Family. Genomics (2003) 82:5575–9. doi: 10.1016/s0888-7543(03)00175-7
81. Park HJ, Cho HJ, Baek JI, Ben-Yosef T, Kwon TJ, Griffith AJ, et al. Evidence for a Founder Mutation Causing DFNA5 Hearing Loss in East Asians. J Hum Genet (2010) 55:159–62. doi: 10.1038/jhg.2009.114
82. Nishio A, Noguchi Y, Sato T, Naruse TK, Kimura A, Takagi A, et al. A DFNA5 Mutation Identified in Japanese Families with Autosomal Dominant Hereditary Hearing Loss. Ann Hum Genet (2014) 78:283–91. doi: 10.1111/ahg.12053
83. Jiang S, Gu H, Zhao Y, Sun L. Teleost Gasdermin E is Cleaved by Caspase 1, 3, and 7 and Induces Pyroptosis. J Immunol (2019) 203:51369–82. doi: 10.4049/jimmunol.1900383
84. Forn-Cuni G, Meijer AH, Varela M. Zebrafish in Inflammasome Research. Cells (2019) 8:8. doi: 10.3390/cells8080901
85. Delmaghani S, del Castillo FJ, Michel V, Leibovici M, Aghaie A, Ron U, et al. Mutations in the Gene Encoding Pejvakin, a Newly Identified Protein of the Afferent Auditory Pathway, Cause DFNB59 Auditory Neuropathy. Nat Genet (2006) 38:7770–8. doi: 10.1038/ng1829
86. Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res (2019) 47:D1D607–D13. doi: 10.1093/nar/gky1131
87. Tanaka S, Tamura M, Aoki A, Fujii T, Komiyama H, Sagai T, et al. A New Gsdma3 Mutation Affecting Anagen Phase of First Hair Cycle. Biochem Biophys Res Commun (2007) 359:4902–7. doi: 10.1016/j.bbrc.2007.05.209
88. Shi P, Tang A, Xian L, Hou S, Zou D, Lv Y, et al. Loss of Conserved Gsdma3 Self-Regulation Causes Autophagy and Cell Death. Biochem J (2015) 468:2325–36. doi: 10.1042/BJ20150204
89. LaRock DL, Johnson AF, Wilde S, Sands JS, Monteiro MP, LaRock CN. Group A Streptococcus Induces GSDMA-Dependent Pyroptosis in Keratinocytes. Nature (2022) 605:527–31. doi: 10.1038/s41586-022-04717-x
90. Katoh M, Katoh M. Identification and Characterization of Human DFNA5L, Mouse Dfna5l, and Rat Dfna5l Genes in Silico. Int J Oncol (2004) 25:3765–70. doi: 10.3892/ijo.25.3.765
91. Zhang JY, Zhou B, Sun RY, Ai YL, Cheng K, Li FN, et al. The Metabolite Alpha-KG Induces GSDMC-Dependent Pyroptosis Through Death Receptor 6-Activated Caspase-8. Cell Res (2021) 31:9980–97. doi: 10.1038/s41422-021-00506-9
92. Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, et al. Gasdermin D Exerts Anti-Inflammatory Effects by Promoting Neutrophil Death. Cell Rep (2018) 22:112924–36. doi: 10.1016/j.celrep.2018.02.067
93. Zhu Q, Zheng M, Balakrishnan A, Karki R, Kanneganti TD. Gasdermin D Promotes AIM2 Inflammasome Activation and is Required for Host Protection Against Francisella Novicida. J Immunol (2018) 201:123662–8. doi: 10.4049/jimmunol.1800788
94. Wang J, Deobald K, Re F. Gasdermin D Protects From Melioidosis Through Pyroptosis and Direct Killing of Bacteria. J Immunol (2019) 202:123468–73. doi: 10.4049/jimmunol.1900045
95. Dubois H, Sorgeloos F, Sarvestani ST, Martens L, Saeys Y, Mackenzie JM, et al. Nlrp3 Inflammasome Activation and Gasdermin D-Driven Pyroptosis are Immunopathogenic Upon Gastrointestinal Norovirus Infection. PLoS Pathog (2019) 15:4:e1007709. doi: 10.1371/journal.ppat.1007709
96. Lammert CR, Frost EL, Bellinger CE, Bolte AC, McKee CA, Hurt ME, et al. AIM2 Inflammasome Surveillance of DNA Damage Shapes Neurodevelopment. Nature (2020) 580:7805647–52. doi: 10.1038/s41586-020-2174-3
97. Xiao J, Wang C, Yao JC, Alippe Y, Xu C, Kress D, et al. Gasdermin D Mediates the Pathogenesis of Neonatal-Onset Multisystem Inflammatory Disease in Mice. PLoS Biol (2018) 16:11:e3000047. doi: 10.1371/journal.pbio.3000047
98. Overwijk WW, Restifo NP. B16 as a Mouse Model for Human Melanoma. Curr Protoc Immunol (2001) 39:1–20. doi: 10.1002/0471142735.im2001s39
99. Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin Pores Permeabilize Mitochondria to Augment Caspase-3 Activation During Apoptosis and Inflammasome Activation. Nat Commun (2019) 10:11689. doi: 10.1038/s41467-019-09397-2
100. Schwander M, Sczaniecka A, Grillet N, Bailey JS, Avenarius M, Najmabadi H, et al. A Forward Genetics Screen in Mice Identifies Recessive Deafness Traits and Reveals That Pejvakin is Essential for Outer Hair Cell Function. J Neurosci (2007) 27:92163–75. doi: 10.1523/JNEUROSCI.4975-06.2007
101. Van Laer L, Pfister M, Thys S, Vrijens K, Mueller M, Umans L, et al. Mice Lacking Dfna5 Show a Diverging Number of Cochlear Fourth Row Outer Hair Cells. Neurobiol Dis (2005) 19:3386–99. doi: 10.1016/j.nbd.2005.01.019
102. Harris SL, Kazmierczak M, Pangrsic T, Shah P, Chuchvara N, Barrantes-Freer A, et al. Conditional Deletion of Pejvakin in Adult Outer Hair Cells Causes Progressive Hearing Loss in Mice. Neuroscience (2017) 344:380–93. doi: 10.1016/j.neuroscience.2016.12.055
103. Soderman J, Berglind L, Almer S. Gene Expression-Genotype Analysis Implicates GSDMA, GSDMB, and LRRC3C as Contributors to Inflammatory Bowel Disease Susceptibility. BioMed Res Int (2015) 2015:834805. doi: 10.1155/2015/834805
104. Moreno-Moral A, Bagnati M, Koturan S, Ko JH, Fonseca C, Harmston N, et al. Changes in Macrophage Transcriptome Associate With Systemic Sclerosis and Mediate GSDMA Contribution to Disease Risk. Ann Rheum Dis (2018) 77:4596–601. doi: 10.1136/annrheumdis-2017-212454
105. Terao C, Kawaguchi T, Dieude P, Varga J, Kuwana M, Hudson M, et al. Transethnic Meta-Analysis Identifies GSDMA and PRDM1 as Susceptibility Genes to Systemic Sclerosis. Ann Rheum Dis (2017) 76:61150–8. doi: 10.1136/annrheumdis-2016-210645
106. Yu J, Kang MJ, Kim BJ, Kwon JW, Song YH, Choi WA, et al. Polymorphisms in GSDMA and GSDMB are Associated With Asthma Susceptibility, Atopy and BHR. Pediatr Pulmonol (2011) 46:7701–8. doi: 10.1002/ppul.21424
107. Lin PH, Lin HY, Kuo CC, Yang LT. N-Terminal Functional Domain of Gasdermin A3 Regulates Mitochondrial Homeostasis via Mitochondrial Targeting. J BioMed Sci (2015) 22:44. doi: 10.1186/s12929-015-0152-0
108. Panganiban RA, Sun M, Dahlin A, Park HR, Kan M, Himes BE, et al. A Functional Splice Variant Associated With Decreased Asthma Risk Abolishes the Ability of Gasdermin B to Induce Epithelial Cell Pyroptosis. J Allergy Clin Immunol (2018) 142:51469–78.e2. doi: 10.1016/j.jaci.2017.11.040
109. Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, et al. Analysis of the Human Tissue-Specific Expression by Genome-Wide Integration of Transcriptomics and Antibody-Based Proteomics. Mol Cell Proteomics (2014) 13:2397–406. doi: 10.1074/mcp.M113.035600
110. Voskoboinik I, Whisstock JC, Trapani JA. Perforin and Granzymes: Function, Dysfunction and Human Pathology. Nat Rev Immunol (2015) 15:6388–400. doi: 10.1038/nri3839
111. Saleh NM, Raj SM, Smyth DJ, Wallace C, Howson JM, Bell L, et al. Genetic Association Analyses of Atopic Illness and Proinflammatory Cytokine Genes With Type 1 Diabetes. Diabetes Metab Res Rev (2011) 27:8838–43. doi: 10.1002/dmrr.1259
112. Zhao CN, Fan Y, Huang JJ, Zhang HX, Gao T, Wang C, et al. The Association of GSDMB and ORMDL3 Gene Polymorphisms With Asthma: A Meta-Analysis. Allergy Asthma Immunol Res (2015) 7:2175–85. doi: 10.4168/aair.2015.7.2.175
113. Chao KL, Kulakova L, Herzberg O. Gene Polymorphism Linked to Increased Asthma and IBD Risk Alters Gasdermin-B Structure, a Sulfatide and Phosphoinositide Binding Protein. Proc Natl Acad Sci USA (2017) 114:7E1128–E37. doi: 10.1073/pnas.1616783114
114. Leoni G, Neumann PA, Sumagin R, Denning TL, Nusrat A. Wound Repair: Role of Immune-Epithelial Interactions. Mucosal Immunol (2015) 8:5959–68. doi: 10.1038/mi.2015.63
115. Miguchi M, Hinoi T, Shimomura M, Adachi T, Saito Y, Niitsu H, et al. Gasdermin C is Upregulated by Inactivation of Transforming Growth Factor Beta Receptor Type II in the Presence of Mutated Apc, Promoting Colorectal Cancer Proliferation. PLoS One (2016) 11:11:e0166422. doi: 10.1371/journal.pone.0166422
116. Wu J, Sun Y, Xiong Z, Liu J, Li H, Liu Y, et al. Association of GSDMC Polymorphisms With Lumbar Disc Herniation Among Chinese Han Population. Int J Immunogenet (2020) 47:6546–53. doi: 10.1111/iji.12488
117. Jia W, Qiu K, He M, Song P, Zhou Q, Zhou F, et al. SOAPfuse: An Algorithm for Identifying Fusion Transcripts From Paired-End RNA-Seq Data. Genome Biol (2013) 14:2R12. doi: 10.1186/gb-2013-14-2-r12
118. Liu X, Lieberman J. Knocking 'Em Dead: Pore-Forming Proteins in Immune Defense. Annu Rev Immunol (2020) 38:455–85. doi: 10.1146/annurev-immunol-111319-023800
119. Rathkey JK, Xiao TS, Abbott DW. Human Polymorphisms in GSDMD Alter the Inflammatory Response. J Biol Chem (2020) 295:103228–38. doi: 10.1074/jbc.RA119.010604
120. Man SM, Kanneganti TD. Gasdermin D: The Long-Awaited Executioner of Pyroptosis. Cell Res (2015) 25:111183–4. doi: 10.1038/cr.2015.124
121. Liu Z, Wang C, Yang J, Zhou B, Yang R, Ramachandran R, et al. Crystal Structures of the Full-Length Murine and Human Gasdermin D Reveal Mechanisms of Autoinhibition, Lipid Binding, and Oligomerization. Immunity (2019) 51:143–9.e4. doi: 10.1016/j.immuni.2019.04.017
122. Liu Z, Wang C, Yang J, Chen Y, Zhou B, Abbott DW, et al. Caspase-1 Engages Full-Length Gasdermin D Through Two Distinct Interfaces That Mediate Caspase Recruitment and Substrate Cleavage. Immunity (2020) 53:1106–14.e5. doi: 10.1016/j.immuni.2020.06.007
123. Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci (2017) 42:4245–54. doi: 10.1016/j.tibs.2016.10.004
124. Liu X, Lieberman J. A Mechanistic Understanding of Pyroptosis: The Fiery Death Triggered by Invasive Infection. Adv Immunol (2017) 135:81–117. doi: 10.1016/bs.ai.2017.02.002
125. Seydel KB, Zhang T, Stanley SL Jr. Neutrophils Play a Critical Role in Early Resistance to Amebic Liver Abscesses in Severe Combined Immunodeficient Mice. Infect Immun (1997) 65:93951–3. doi: 10.1128/iai.65.9.3951-3953.1997
126. Velazquez C, Shibayama-Salas M, Aguirre-Garcia J, Tsutsumi V, Calderon J. Role of Neutrophils in Innate Resistance to Entamoeba histolytica Liver Infection in Mice. Parasite Immunol (1998) 20:6255–62. doi: 10.1046/j.1365-3024.1998.00128.x
127. Hartley MA, Drexler S, Ronet C, Beverley SM, Fasel N. The Immunological, Environmental, and Phylogenetic Perpetrators of Metastatic Leishmaniasis. Trends Parasitol (2014) 30:8412–22. doi: 10.1016/j.pt.2014.05.006
128. Zamboni DS, Lima-Junior DS. Inflammasomes in Host Response to Protozoan Parasites. Immunol Rev (2015) 265:1156–71. doi: 10.1111/imr.12291
129. Dey R, Joshi AB, Oliveira F, Pereira L, Guimaraes-Costa AB, Serafim TD, et al. Gut Microbes Egested During Bites of Infected Sand Flies Augment Severity of Leishmaniasis via Inflammasome-Derived IL-1beta. Cell Host Microbe (2018) 23:1134–43.e6. doi: 10.1016/j.chom.2017.12.002
130. Karmakar M, Minns M, Greenberg EN, Diaz-Aponte J, Pestonjamasp K, Johnson JL, et al. N-GSDMD Trafficking to Neutrophil Organelles Facilitates IL-1beta Release Independently of Plasma Membrane Pores and Pyroptosis. Nat Commun (2020) 11:12212. doi: 10.1038/s41467-020-16043-9
131. Avila EE, Salaiza N, Pulido J, Rodriguez MC, Diaz-Godinez C, Laclette JP, et al. Entamoeba histolytica Trophozoites and Lipopeptidophosphoglycan Trigger Human Neutrophil Extracellular Traps. PLoS One (2016) 11:7:e0158979. doi: 10.1371/journal.pone.0158979
132. van der Meer JW, Vogels MT, Netea MG, Kullberg BJ. Proinflammatory Cytokines and Treatment of Disease. Ann N Y Acad Sci (1998) 856:243–51. doi: 10.1111/j.1749-6632.1998.tb08331.x
133. Van Laer L, Huizing EH, Verstreken M, van Zuijlen D, Wauters JG, Bossuyt PJ, et al. Nonsyndromic Hearing Impairment is Associated With a Mutation in DFNA5. Nat Genet (1998) 20:2194–7. doi: 10.1038/2503
134. Chai Y, Chen D, Wang X, Wu H, Yang T. A Novel Splice Site Mutation in DFNA5 Causes Late-Onset Progressive non-Syndromic Hearing Loss in a Chinese Family. Int J Pediatr Otorhinolaryngol (2014) 78:81265–8. doi: 10.1016/j.ijporl.2014.05.007
135. Chen S, Dong C, Wang Q, Zhong Z, Qi Y, Ke X, et al. Targeted Next-Generation Sequencing Successfully Detects Causative Genes in Chinese Patients With Hereditary Hearing Loss. Genet Test Mol Biomarkers (2016) 20:11660–5. doi: 10.1089/gtmb.2016.0051
136. Wang H, Guan J, Guan L, Yang J, Wu K, Lin Q, et al. Further Evidence for "Gain-of-Function" Mechanism of DFNA5 Related Hearing Loss. Sci Rep (2018) 8:18424. doi: 10.1038/s41598-018-26554-7
137. Yuan Y, Li Q, Su Y, Lin Q, Gao X, Liu H, et al. Comprehensive Genetic Testing of Chinese SNHL Patients and Variants Interpretation Using ACMG Guidelines and Ethnically Matched Normal Controls. Eur J Hum Genet (2020) 28:2231–43. doi: 10.1038/s41431-019-0510-6
138. Kersey PJ, Allen JE, Allot A, Barba M, Boddu S, Bolt BJ, et al. Ensembl Genomes 2018: An Integrated Omics Infrastructure for Non-Vertebrate Species. Nucleic Acids Res (2018) 46:D1D802–D8. doi: 10.1093/nar/gkx1011
139. Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, et al. Ensembl 2018. Nucleic Acids Res (2018) 46:D1D754–D61. doi: 10.1093/nar/gkx1098
140. Op de Beeck K, Van Camp G, Thys S, Cools N, Callebaut I, Vrijens K, et al. The DFNA5 Gene, Responsible for Hearing Loss and Involved in Cancer, Encodes a Novel Apoptosis-Inducing Protein. Eur J Hum Genet (2011) 19:9965–73. doi: 10.1038/ejhg.2011.63
141. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ (2018) 25:3486–541. doi: 10.1038/s41418-017-0012-4
142. Nagata S, Tanaka M. Programmed Cell Death and the Immune System. Nat Rev Immunol (2017) 17:5333–40. doi: 10.1038/nri.2016.153
143. Aizawa E, Karasawa T, Watanabe S, Komada T, Kimura H, Kamata R, et al. GSDME-Dependent Incomplete Pyroptosis Permits Selective IL-1alpha Release Under Caspase-1 Inhibition. iScience (2020) 23:5101070. doi: 10.1016/j.isci.2020.101070
144. Wang C, Yang T, Xiao J, Xu C, Alippe Y, Sun K, et al. NLRP3 Inflammasome Activation Triggers Gasdermin D-Independent Inflammation. Sci Immunol (2021) 6:64:eabj3859. doi: 10.1126/sciimmunol.abj3859
145. Collin RW, Kalay E, Oostrik J, Caylan R, Wollnik B, Arslan S, et al. Involvement of DFNB59 Mutations in Autosomal Recessive Nonsyndromic Hearing Impairment. Hum Mutat (2007) 28:7718–23. doi: 10.1002/humu.20510
146. Mujtaba G, Bukhari I, Fatima A, Naz S. A P.C343S Missense Mutation in PJVK Causes Progressive Hearing Loss. Gene (2012) 504:198–01. doi: 10.1016/j.gene.2012.05.013
147. Zheng Z, Deng W, Lou X, Bai Y, Wang J, Zeng H, et al. Gasdermins: Pore-Forming Activities and Beyond. Acta Biochim Biophys Sin (Shanghai) (2020) 52:5467–74. doi: 10.1093/abbs/gmaa016
148. Delmaghani S, Defourny J, Aghaie A, Beurg M, Dulon D, Thelen N, et al. Hypervulnerability to Sound Exposure Through Impaired Adaptive Proliferation of Peroxisomes. Cell (2015) 163:4894–906. doi: 10.1016/j.cell.2015.10.023
149. Kazmierczak M, Kazmierczak P, Peng AW, Harris SL, Shah P, Puel JL, et al. Pejvakin, a Candidate Stereociliary Rootlet Protein, Regulates Hair Cell Function in a Cell-Autonomous Manner. J Neurosci (2017) 37:133447–64. doi: 10.1523/JNEUROSCI.2711-16.2017
150. Vande Walle L, Lamkanfi M. Pyroptosis. Curr Biol (2016) 26:13R568–R72. doi: 10.1016/j.cub.2016.02.019
151. Jorgensen I, Miao EA. Pyroptotic Cell Death Defends Against Intracellular Pathogens. Immunol Rev (2015) 265:1130–42. doi: 10.1111/imr.12287
152. Monack DM, Raupach B, Hromockyj AE, Falkow S. Salmonella Typhimurium Invasion Induces Apoptosis in Infected Macrophages. Proc Natl Acad Sci U S A (1996) 93:189833–8. doi: 10.1073/pnas.93.18.9833
153. Chen Y, Smith MR, Thirumalai K, Zychlinsky A. A Bacterial Invasin Induces Macrophage Apoptosis by Binding Directly to ICE. EMBO J (1996) 15:153853–60. doi: 10.1002/j.1460-2075.1996.tb00759.x
154. Hersh D, Monack DM, Smith MR, Ghori N, Falkow S, Zychlinsky A. The Salmonella Invasin SipB Induces Macrophage Apoptosis by Binding to Caspase-1. Proc Natl Acad Sci USA (1999) 96:52396–401. doi: 10.1073/pnas.96.5.2396
155. Jorgensen I, Zhang Y, Krantz BA, Miao EA. Pyroptosis Triggers Pore-Induced Intracellular Traps (PITs) That Capture Bacteria and Lead to Their Clearance by Efferocytosis. J Exp Med (2016) 213:102113–28. doi: 10.1084/jem.20151613
156. Chen X, He WT, Hu L, Li J, Fang Y, Wang X, et al. Pyroptosis Is Driven by Non-Selective Gasdermin-D Pore and its Morphology is Different From MLKL Channel-Mediated Necroptosis. Cell Res (2016) 26:91007–20. doi: 10.1038/cr.2016.100
157. Kovacs SB, Miao EA. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol (2017) 27:9673–84. doi: 10.1016/j.tcb.2017.05.005
158. Davis MA, Fairgrieve MR, Den Hartigh A, Yakovenko O, Duvvuri B, Lood C, et al. Calpain Drives Pyroptotic Vimentin Cleavage, Intermediate Filament Loss, and Cell Rupture That Mediates Immunostimulation. Proc Natl Acad Sci U S A (2019) 116:115061–70. doi: 10.1073/pnas.1818598116
159. Andersson U, Tracey KJ. HMGB1 Is a Therapeutic Target for Sterile Inflammation and Infection. Annu Rev Immunol (2011) 29:139–62. doi: 10.1146/annurev-immunol-030409-101323
160. Broderick S, Wang X, Simms N, Page-McCaw A. Drosophila Ninjurin A Induces Nonapoptotic Cell Death. PLoS One (2012) 7:9:e44567. doi: 10.1371/journal.pone.0044567
161. Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, et al. FDA-Approved Disulfiram Inhibits Pyroptosis by Blocking Gasdermin D Pore Formation. Nat Immunol (2020) 21:7736–45. doi: 10.1038/s41590-020-0669-6
162. Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, et al. Succination Inactivates Gasdermin D and Blocks Pyroptosis. Science (2020) 369:65111633–7. doi: 10.1126/science.abb9818
163. Van Opdenbosch N, Lamkanfi M. Caspases in Cell Death, Inflammation, and Disease. Immunity (2019) 50:61352–64. doi: 10.1016/j.immuni.2019.05.020
164. Tsuchiya K. Inflammasome-Associated Cell Death: Pyroptosis, Apoptosis, and Physiological Implications. Microbiol Immunol (2020) 64:4252–69. doi: 10.1111/1348-0421.12771
165. Fink SL, Cookson BT. Caspase-1-Dependent Pore Formation During Pyroptosis Leads to Osmotic Lysis of Infected Host Macrophages. Cell Microbiol (2006) 8:111812–25. doi: 10.1111/j.1462-5822.2006.00751.x
166. Ralston KS, Petri WA. The Ways of a Killer: How Does Entamoeba histolytica Elicit Host Cell Death? Essays Biochem (2011) 51:193–210. doi: 10.1042/bse0510193
167. Ravdin JI, Sperelakis N, Guerrant RL. Effect of Ion Channel Inhibitors on the Cytopathogenicity of Entamoeba histolytica. J Infect Dis (1982) 146:3335–40. doi: 10.1093/infdis/146.3.335
168. Huston CD, Houpt ER, Mann BJ, Hahn CS, Petri WA Jr. Caspase 3-Dependent Killing of Host Cells by the Parasite Entamoeba histolytica. Cell Microbiol (2000) 2:6617–25. doi: 10.1046/j.1462-5822.2000.00085.x
169. Becker SM, Cho KN, Guo X, Fendig K, Oosman MN, Whitehead R, et al. Epithelial Cell Apoptosis Facilitates Entamoeba histolytica Infection in the Gut. Am J Pathol (2010) 176:31316–22. doi: 10.2353/ajpath.2010.090740
170. Mascarenhas DPA, Cerqueira DM, Pereira MSF, Castanheira FVS, Fernandes TD, Manin GZ, et al. Inhibition of Caspase-1 or Gasdermin-D Enable Caspase-8 Activation in the Naip5/NLRC4/ASC Inflammasome. PLoS Pathog (2017) 13:8:e1006502. doi: 10.1371/journal.ppat.1006502
171. Van Opdenbosch N, Van Gorp H, Verdonckt M, Saavedra PHV, de Vasconcelos NM, Goncalves A, et al. Caspase-1 Engagement and TLR-Induced C-FLIP Expression Suppress ASC/caspase-8-Dependent Apoptosis by Inflammasome Sensors NLRP1b and NLRC4. Cell Rep (2017) 21:123427–44. doi: 10.1016/j.celrep.2017.11.088
172. Akhter A, Gavrilin MA, Frantz L, Washington S, Ditty C, Limoli D, et al. Caspase-7 Activation by the Nlrc4/Ipaf Inflammasome Restricts Legionella Pneumophila Infection. PLoS Pathog (2009) 5:4:e1000361. doi: 10.1371/journal.ppat.1000361
173. Erener S, Petrilli V, Kassner I, Minotti R, Castillo R, Santoro R, et al. Inflammasome-Activated Caspase 7 Cleaves PARP1 to Enhance the Expression of a Subset of NF-kappaB Target Genes. Mol Cell (2012) 46:2200–11. doi: 10.1016/j.molcel.2012.02.016
174. Kang SJ, Wang S, Kuida K, Yuan J. Distinct Downstream Pathways of Caspase-11 in Regulating Apoptosis and Cytokine Maturation During Septic Shock Response. Cell Death Differ (2002) 9:101115–25. doi: 10.1038/sj.cdd.4401087
175. Sagulenko V, Vitak N, Vajjhala PR, Vince JE, Stacey KJ. Caspase-1 is an Apical Caspase Leading to Caspase-3 Cleavage in the AIM2 Inflammasome Response, Independent of Caspase-8. J Mol Biol (2018) 430:2238–47. doi: 10.1016/j.jmb.2017.10.028
176. Akino K, Toyota M, Suzuki H, Imai T, Maruyama R, Kusano M, et al. Identification of DFNA5 as a Target of Epigenetic Inactivation in Gastric Cancer. Cancer Sci (2007) 98:188–95. doi: 10.1111/j.1349-7006.2006.00351.x
177. Kim MS, Lebron C, Nagpal JK, Chae YK, Chang X, Huang Y, et al. Methylation of the DFNA5 Increases Risk of Lymph Node Metastasis in Human Breast Cancer. Biochem Biophys Res Commun (2008) 370:138–43. doi: 10.1016/j.bbrc.2008.03.026
178. Kim MS, Chang X, Yamashita K, Nagpal JK, Baek JH, Wu G, et al. Aberrant Promoter Methylation and Tumor Suppressive Activity of the DFNA5 Gene in Colorectal Carcinoma. Oncogene (2008) 27:253624–34. doi: 10.1038/sj.onc.1211021
179. Blader IJ, Coleman BI, Chen CT, Gubbels MJ. Lytic Cycle of Toxoplasma gondii: 15 Years Later. Annu Rev Microbiol (2015) 69:463–85. doi: 10.1146/annurev-micro-091014-104100
180. Hu RS, He JJ, Elsheikha HM, Zou Y, Ehsan M, Ma QN, et al. Transcriptomic Profiling of Mouse Brain During Acute and Chronic Infections by Toxoplasma gondii Oocysts. Front Microbiol (2020) 11:570903. doi: 10.3389/fmicb.2020.570903
181. Channon JY, Miselis KA, Minns LA, Dutta C, Kasper LH. Toxoplasma gondii Induces Granulocyte Colony-Stimulating Factor and Granulocyte-Macrophage Colony-Stimulating Factor Secretion by Human Fibroblasts: Implications for Neutrophil Apoptosis. Infect Immun (2002) 70:116048–57. doi: 10.1128/IAI.70.11.6048-6057.2002
182. Aga E, Katschinski DM, van Zandbergen G, Laufs H, Hansen B, Muller K, et al. Inhibition of the Spontaneous Apoptosis of Neutrophil Granulocytes by the Intracellular Parasite Leishmania Major. J Immunol (2002) 169:2898–905. doi: 10.4049/jimmunol.169.2.898
183. van Zandbergen G, Klinger M, Mueller A, Dannenberg S, Gebert A, Solbach W, et al. Cutting Edge: Neutrophil Granulocyte Serves as a Vector for Leishmania Entry Into Macrophages. J Immunol (2004) 173:116521–5. doi: 10.4049/jimmunol.173.11.6521
184. Conos SA, Lawlor KE, Vaux DL, Vince JE, Lindqvist LM. Cell Death is Not Essential for Caspase-1-Mediated Interleukin-1beta Activation and Secretion. Cell Death Differ (2016) 23:111827–38. doi: 10.1038/cdd.2016.69
185. Chen KW, Gross CJ, Sotomayor FV, Stacey KJ, Tschopp J, Sweet MJ, et al. The Neutrophil NLRC4 Inflammasome Selectively Promotes IL-1beta Maturation Without Pyroptosis During Acute Salmonella Challenge. Cell Rep (2014) 8:2570–82. doi: 10.1016/j.celrep.2014.06.028
186. Brough D, Rothwell NJ. Caspase-1-Dependent Processing of Pro-Interleukin-1beta is Cytosolic and Precedes Cell Death. J Cell Sci (2007) 120:Pt 5772–81. doi: 10.1242/jcs.03377
187. Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity (2016) 44:4833–46. doi: 10.1016/j.immuni.2016.01.012
188. Martin-Sanchez F, Diamond C, Zeitler M, Gomez AI, Baroja-Mazo A, Bagnall J, et al. Inflammasome-Dependent IL-1beta Release Depends Upon Membrane Permeabilisation. Cell Death Differ (2016) 23:71219–31. doi: 10.1038/cdd.2015.176
189. Russo HM, Rathkey J, Boyd-Tressler A, Katsnelson MA, Abbott DW, Dubyak GR. Active Caspase-1 Induces Plasma Membrane Pores That Precede Pyroptotic Lysis and are Blocked by Lanthanides. J Immunol (2016) 197:41353–67. doi: 10.4049/jimmunol.1600699
190. Monteleone M, Stanley AC, Chen KW, Brown DL, Bezbradica JS, von Pein JB, et al. Interleukin-1beta Maturation Triggers its Relocation to the Plasma Membrane for Gasdermin-D-Dependent and -Independent Secretion. Cell Rep (2018) 24:61425–33. doi: 10.1016/j.celrep.2018.07.027
191. Cerretti DP, Kozlosky CJ, Mosley B, Nelson N, Van Ness K, Greenstreet TA, et al. Molecular Cloning of the Interleukin-1 Beta Converting Enzyme. Science (1992) 256:505397–100. doi: 10.1126/science.1373520
192. Garlanda C, Dinarello CA, Mantovani A. The Interleukin-1 Family: Back to the Future. Immunity (2013) 39:61003–18. doi: 10.1016/j.immuni.2013.11.010
193. Snyder JS, Soumier A, Brewer M, Pickel J, Cameron HA. Adult Hippocampal Neurogenesis Buffers Stress Responses and Depressive Behaviour. Nature (2011) 476:7361458–61. doi: 10.1038/nature10287
194. Nickel W, Rabouille C. Mechanisms of Regulated Unconventional Protein Secretion. Nat Rev Mol Cell Biol (2009) 10:2148–55. doi: 10.1038/nrm2617
195. Rabouille C, Malhotra V, Nickel W. Diversity in Unconventional Protein Secretion. J Cell Sci (2012) 125:Pt 225251–5. doi: 10.1242/jcs.103630
196. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion From Living Macrophages. Immunity (2018) 48:135–44.e6. doi: 10.1016/j.immuni.2017.11.013
197. Zanoni I, Tan Y, Di Gioia M, Springstead JR, Kagan JC. By Capturing Inflammatory Lipids Released From Dying Cells, the Receptor CD14 Induces Inflammasome-Dependent Phagocyte Hyperactivation. Immunity (2017) 47:4697–709.e3. doi: 10.1016/j.immuni.2017.09.010
198. Ruhl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. ESCRT-Dependent Membrane Repair Negatively Regulates Pyroptosis Downstream of GSDMD Activation. Science (2018) 362:6417956–60. doi: 10.1126/science.aar7607
199. Andrews NW, Almeida PE, Corrotte M. Damage Control: Cellular Mechanisms of Plasma Membrane Repair. Trends Cell Biol (2014) 24:12734–42. doi: 10.1016/j.tcb.2014.07.008
200. Bilodeau PS, Urbanowski JL, Winistorfer SC, Piper RC. The Vps27p Hse1p Complex Binds Ubiquitin and Mediates Endosomal Protein Sorting. Nat Cell Biol (2002) 4:7534–9. doi: 10.1038/ncb815
201. Keefe D, Shi L, Feske S, Massol R, Navarro F, Kirchhausen T, et al. Perforin Triggers a Plasma Membrane-Repair Response That Facilitates CTL Induction of Apoptosis. Immunity (2005) 23:3249–62. doi: 10.1016/j.immuni.2005.08.001
202. Jimenez AJ, Maiuri P, Lafaurie-Janvore J, Divoux S, Piel M, Perez F. ESCRT Machinery is Required for Plasma Membrane Repair. Science (2014) 343:61741247136. doi: 10.1126/science.1247136
203. Scheffer LL, Sreetama SC, Sharma N, Medikayala S, Brown KJ, Defour A, et al. Mechanism of Ca(2)(+)-Triggered ESCRT Assembly and Regulation of Cell Membrane Repair. Nat Commun (2014) 5:5646. doi: 10.1038/ncomms6646
204. McNeil PL, Kirchhausen T. An Emergency Response Team for Membrane Repair. Nat Rev Mol Cell Biol (2005) 6:6499–505. doi: 10.1038/nrm1665
205. Walev I, Bhakdi SC, Hofmann F, Djonder N, Valeva A, Aktories K, et al. Delivery of Proteins Into Living Cells by Reversible Membrane Permeabilization With Streptolysin-O. Proc Natl Acad Sci U S A (2001) 98:63185–90. doi: 10.1073/pnas.051429498
206. Tam C, Idone V, Devlin C, Fernandes MC, Flannery A, He X, et al. Exocytosis of Acid Sphingomyelinase by Wounded Cells Promotes Endocytosis and Plasma Membrane Repair. J Cell Biol (2010) 189:61027–38. doi: 10.1083/jcb.201003053
207. Idone V, Tam C, Goss JW, Toomre D, Pypaert M, Andrews NW. Repair of Injured Plasma Membrane by Rapid Ca2+-Dependent Endocytosis. J Cell Biol (2008) 180:5905–14. doi: 10.1083/jcb.200708010
208. Holopainen JM, Medina OP, Metso AJ, Kinnunen PK. Sphingomyelinase Activity Associated With Human Plasma Low Density Lipoprotein. J Biol Chem (2000) 275:2216484–9. doi: 10.1074/jbc.275.22.16484
209. Gulbins E, Kolesnick R. Raft Ceramide in Molecular Medicine. Oncogene (2003) 22:457070–7. doi: 10.1038/sj.onc.1207146
210. Ramirez-Montiel F, Mendoza-Macias C, Andrade-Guillen S, Rangel-Serrano A, Paramo-Perez I, Rivera-Cuellar PE, et al. Plasma Membrane Damage Repair is Mediated by an Acid Sphingomyelinase in Entamoeba histolytica. PLoS Pathog (2019) 15:8:e1008016. doi: 10.1371/journal.ppat.1008016
211. Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-Induced Pyroptosis is an Innate Immune Effector Mechanism Against Intracellular Bacteria. Nat Immunol (2010) 11:121136–42. doi: 10.1038/ni.1960
212. Yu J, Nagasu H, Murakami T, Hoang H, Broderick L, Hoffman HM, et al. Inflammasome Activation Leads to Caspase-1-Dependent Mitochondrial Damage and Block of Mitophagy. Proc Natl Acad Sci U S A (2014) 111:4315514–9. doi: 10.1073/pnas.1414859111
213. Rivers-Auty J, Brough D. Potassium Efflux Fires the Canon: Potassium Efflux as a Common Trigger for Canonical and Noncanonical NLRP3 Pathways. Eur J Immunol (2015) 45:102758–61. doi: 10.1002/eji.201545958
214. Schmid-Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung V. Caspase-4 Mediates non-Canonical Activation of the NLRP3 Inflammasome in Human Myeloid Cells. Eur J Immunol (2015) 45:102911–7. doi: 10.1002/eji.201545523
215. Ruhl S, Broz P. Caspase-11 Activates a Canonical NLRP3 Inflammasome by Promoting K(+) Efflux. Eur J Immunol (2015) 45:102927–36. doi: 10.1002/eji.201545772
216. Platnich JM, Chung H, Lau A, Sandall CF, Bondzi-Simpson A, Chen HM, et al. Shiga Toxin/Lipopolysaccharide Activates Caspase-4 and Gasdermin D to Trigger Mitochondrial Reactive Oxygen Species Upstream of the NLRP3 Inflammasome. Cell Rep (2018) 25:61525–36.e7. doi: 10.1016/j.celrep.2018.09.071
217. Marie C, Verkerke HP, Theodorescu D, Petri WA. A Whole-Genome RNAi Screen Uncovers a Novel Role for Human Potassium Channels in Cell Killing by the Parasite Entamoeba histolytica. Sci Rep (2015) 5:13613. doi: 10.1038/srep13613
218. Roma EH, Macedo JP, Goes GR, Goncalves JL, Castro W, Cisalpino D, et al. Impact of Reactive Oxygen Species (ROS) on the Control of Parasite Loads and Inflammation in Leishmania Amazonensis Infection. Parasit Vectors (2016) 9:193. doi: 10.1186/s13071-016-1472-y
219. Cardoni RL, Antunez MI, Morales C, Nantes IR. Release of Reactive Oxygen Species by Phagocytic Cells in Response to Live Parasites in Mice Infected With Trypanosoma cruzi. Am J Trop Med Hyg (1997) 56:3329–34. doi: 10.4269/ajtmh.1997.56.329
220. Cardoni RL, Rottenberg ME, Segura EL. Increased Production of Reactive Oxygen Species by Cells From Mice Acutely Infected With Trypanosoma cruzi. Cell Immunol (1990) 128:111–21. doi: 10.1016/0008-8749(90)90002-9
221. Piacenza L, Alvarez MN, Peluffo G, Radi R. Fighting the Oxidative Assault: The Trypanosoma cruzi Journey to Infection. Curr Opin Microbiol (2009) 12:4415–21. doi: 10.1016/j.mib.2009.06.011
222. Pineyro MD, Arcari T, Robello C, Radi R, Trujillo M. Tryparedoxin Peroxidases From Trypanosoma cruzi: High Efficiency in the Catalytic Elimination of Hydrogen Peroxide and Peroxynitrite. Arch Biochem Biophys (2011) 507:2287–95. doi: 10.1016/j.abb.2010.12.014
223. Alvarez MN, Peluffo G, Piacenza L, Radi R. Intraphagosomal Peroxynitrite as a Macrophage-Derived Cytotoxin Against Internalized Trypanosoma cruzi: Consequences for Oxidative Killing and Role of Microbial Peroxiredoxins in Infectivity. J Biol Chem (2011) 286:86627–40. doi: 10.1074/jbc.M110.167247
224. Higuchi Mde L, Benvenuti LA, Martins Reis M, Metzger M. Pathophysiology of the Heart in Chagas' Disease: Current Status and New Developments. Cardiovasc Res (2003) 60:196–07. doi: 10.1016/s0008-6363(03)00361-4
225. Nagajyothi F, Machado FS, Burleigh BA, Jelicks LA, Scherer PE, Mukherjee S, et al. Mechanisms of Trypanosoma cruzi Persistence in Chagas Disease. Cell Microbiol (2012) 14:5634–43. doi: 10.1111/j.1462-5822.2012.01764.x
226. Tanowitz HB, Machado FS, Jelicks LA, Shirani J, de Carvalho AC, Spray DC, et al. Perspectives on Trypanosoma cruzi-Induced Heart Disease (Chagas Disease). Prog Cardiovasc Dis (2009) 51:6524–39. doi: 10.1016/j.pcad.2009.02.001
227. Tanowitz HB, Weiss LM, Montgomery SP. Chagas Disease has Now Gone Global. PloS Negl Trop Dis (2011) 5:4:e1136. doi: 10.1371/journal.pntd.0001136
228. Machado FS, Tyler KM, Brant F, Esper L, Teixeira MM, Tanowitz HB. Pathogenesis of Chagas Disease: Time to Move on. Front Biosci (Elite Ed) (2012) 4:1743–58. doi: 10.2741/495
229. Machado FS, Dutra WO, Esper L, Gollob KJ, Teixeira MM, Factor SM, et al. Current Understanding of Immunity to Trypanosoma cruzi Infection and Pathogenesis of Chagas Disease. Semin Immunopathol (2012) 34:6753–70. doi: 10.1007/s00281-012-0351-7
230. Silva GK, Costa RS, Silveira TN, Caetano BC, Horta CV, Gutierrez FR, et al. Apoptosis-Associated Speck-Like Protein Containing a Caspase Recruitment Domain Inflammasomes Mediate IL-1beta Response and Host Resistance to Trypanosoma cruzi Infection. J Immunol (2013) 191:63373–83. doi: 10.4049/jimmunol.1203293
231. Goncalves VM, Matteucci KC, Buzzo CL, Miollo BH, Ferrante D, Torrecilhas AC, et al. NLRP3 Controls Trypanosoma cruzi Infection Through a Caspase-1-Dependent IL-1R-Independent NO Production. PloS Negl Trop Dis (2013) 7:10:e2469. doi: 10.1371/journal.pntd.0002469
232. Deretic V, Saitoh T, Akira S. Autophagy in Infection, Inflammation and Immunity. Nat Rev Immunol (2013) 13:10722–37. doi: 10.1038/nri3532
233. Yu P, Wang HY, Tian M, Li AX, Chen XS, Wang XL, et al. Eukaryotic Elongation Factor-2 Kinase Regulates the Cross-Talk Between Autophagy and Pyroptosis in Doxorubicin-Treated Human Melanoma Cells In Vitro. Acta Pharmacol Sin (2019) 40:91237–44. doi: 10.1038/s41401-019-0222-z
234. Shio MT, Eisenbarth SC, Savaria M, Vinet AF, Bellemare MJ, Harder KW, et al. Malarial Hemozoin Activates the NLRP3 Inflammasome Through Lyn and Syk Kinases. PLoS Pathog (2009) 5:8:e1000559. doi: 10.1371/journal.ppat.1000559
235. Kalantari P, DeOliveira RB, Chan J, Corbett Y, Rathinam V, Stutz A, et al. Dual Engagement of the NLRP3 and AIM2 Inflammasomes by Plasmodium-Derived Hemozoin and DNA During Malaria. Cell Rep (2014) 6:1196–210. doi: 10.1016/j.celrep.2013.12.014
236. Pereira LMN, Assis PA, de Araujo NM, Durso DF, Junqueira C, Ataide MA, et al. Caspase-8 Mediates Inflammation and Disease in Rodent Malaria. Nat Commun (2020) 11:14596. doi: 10.1038/s41467-020-18295-x
237. Rauch I, Deets KA, Ji DX, von Moltke J, Tenthorey JL, Lee AY, et al. NAIP-NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion With Eicosanoid and IL-18 Release via Activation of Caspase-1 and -8. Immunity (2017) 46:4649–59. doi: 10.1016/j.immuni.2017.03.016
238. Correa G, Marques da Silva C, de Abreu Moreira-Souza AC, Vommaro RC, Coutinho-Silva R. Activation of the P2X(7) Receptor Triggers the Elimination of Toxoplasma gondii Tachyzoites From Infected Macrophages. Microbes Infect (2010) 12:6497–504. doi: 10.1016/j.micinf.2010.03.004
239. DeLaney AA, Berry CT, Christian DA, Hart A, Bjanes E, Wynosky-Dolfi MA, et al. Caspase-8 Promotes C-Rel-Dependent Inflammatory Cytokine Expression and Resistance Against Toxoplasma gondii. Proc Natl Acad Sci USA (2019) 116:2411926–35. doi: 10.1073/pnas.1820529116
240. Vince JE, Silke J. The Intersection of Cell Death and Inflammasome Activation. Cell Mol Life Sci (2016) 73:11–122349-67. doi: 10.1007/s00018-016-2205-2
241. Batista SJ, Still KM, Johanson D, Thompson JA, O'Brien CA, Lukens JR, et al. Gasdermin-D-Dependent IL-1alpha Release From Microglia Promotes Protective Immunity During Chronic Toxoplasma gondii Infection. Nat Commun (2020) 11:13687. doi: 10.1038/s41467-020-17491-z
242. Lima-Junior DS, Costa DL, Carregaro V, Cunha LD, Silva AL, Mineo TW, et al. Inflammasome-Derived IL-1beta Production Induces Nitric Oxide-Mediated Resistance to Leishmania. Nat Med (2013) 19:7909–15. doi: 10.1038/nm.3221
243. Lima-Junior DS, Mineo TWP, Calich VLG, Zamboni DS. Dectin-1 Activation During Leishmania Amazonensis Phagocytosis Prompts Syk-Dependent Reactive Oxygen Species Production to Trigger Inflammasome Assembly and Restriction of Parasite Replication. J Immunol (2017) 199:62055–68. doi: 10.4049/jimmunol.1700258
244. de Carvalho RVH, Andrade WA, Lima-Junior DS, Dilucca M, de Oliveira CV, Wang K, et al. Leishmania Lipophosphoglycan Triggers Caspase-11 and the non-Canonical Activation of the NLRP3 Inflammasome. Cell Rep (2019) 26:2429–37.e5. doi: 10.1016/j.celrep.2018.12.047
245. Farias Luz N, Balaji S, Okuda K, Barreto AS, Bertin J, Gough PJ, et al. RIPK1 and PGAM5 Control Leishmania Replication Through Distinct Mechanisms. J Immunol (2016) 196:125056–63. doi: 10.4049/jimmunol.1502492
246. Eskelinen EL, Kovacs AL. Double Membranes vs. Lipid Bilayers, and Their Significance for Correct Identification of Macroautophagic Structures. Autophagy (2011) 7:9931–2. doi: 10.4161/auto.7.9.16679
247. Panday A, Sahoo MK, Osorio D, Batra S. NADPH Oxidases: An Overview From Structure to Innate Immunity-Associated Pathologies. Cell Mol Immunol (2015) 12:15–23. doi: 10.1038/cmi.2014.89
248. Dhiman M, Garg NJ. P47phox-/- Mice are Compromised in Expansion and Activation of CD8+ T Cells and Susceptible to Trypanosoma cruzi Infection. PLoS Pathog (2014) 10:12:e1004516. doi: 10.1371/journal.ppat.1004516
249. Cunha-Neto E, Dzau VJ, Allen PD, Stamatiou D, Benvenutti L, Higuchi ML, et al. Cardiac Gene Expression Profiling Provides Evidence for Cytokinopathy as a Molecular Mechanism in Chagas' Disease Cardiomyopathy. Am J Pathol (2005) 167:2305–13. doi: 10.1016/S0002-9440(10)62976-8
250. Starkey J, Kobayashi N, Numaguchi Y, Moritani T. Cytotoxic Lesions of the Corpus Callosum That Show Restricted Diffusion: Mechanisms, Causes, and Manifestations. Radiographics (2017) 37:2562–76. doi: 10.1148/rg.2017160085
251. Heussler VT, Kuenzi P, Rottenberg S. Inhibition of Apoptosis by Intracellular Protozoan Parasites. Int J Parasitol (2001) 31:111166–76. doi: 10.1016/s0020-7519(01)00271-5
252. Matteucci KC, Pereira GJS, Weinlich R, Bortoluci KR. Frontline Science: Autophagy is a Cell Autonomous Effector Mechanism Mediated by NLRP3 to Control Trypanosoma cruzi Infection. J Leukoc Biol (2019) 106:3531–40. doi: 10.1002/JLB.HI1118-461R
253. Lamb CA, Yoshimori T, Tooze SA. The Autophagosome: Origins Unknown, Biogenesis Complex. Nat Rev Mol Cell Biol (2013) 14:12759–74. doi: 10.1038/nrm3696
254. Seveau S, Turner J, Gavrilin MA, Torrelles JB, Hall-Stoodley L, Yount JS, et al. Checks and Balances Between Autophagy and Inflammasomes During Infection. J Mol Biol (2018) 430:2174–92. doi: 10.1016/j.jmb.2017.11.006
255. Sun Q, Fan J, Billiar TR, Scott MJ. Inflammasome and Autophagy Regulation - a Two-Way Street. Mol Med (2017) 23:188–95. doi: 10.2119/molmed.2017.00077
256. Wang X, Li H, Li W, Xie J, Wang F, Peng X, et al. The Role of Caspase-1/GSDMD-Mediated Pyroptosis in Taxol-Induced Cell Death and a Taxol-Resistant Phenotype in Nasopharyngeal Carcinoma Regulated by Autophagy. Cell Biol Toxicol (2020) 36:5437–57. doi: 10.1007/s10565-020-09514-8
257. Rojas Marquez JD, Ana Y, Baigorri RE, Stempin CC, Cerban FM. Mammalian Target of Rapamycin Inhibition in Trypanosoma cruzi-Infected Macrophages Leads to an Intracellular Profile That is Detrimental for Infection. Front Immunol (2018) 9:313. doi: 10.3389/fimmu.2018.00313
258. Zhuo L, Chen X, Sun Y, Wang Y, Shi Y, Bu L, et al. Rapamycin Inhibited Pyroptosis and Reduced the Release of IL-1beta and IL-18 in the Septic Response. BioMed Res Int (2020) 2020:5960375. doi: 10.1155/2020/5960375
259. Sena-Dos-Santos C, Braga-da-Silva C, Marques D, Azevedo Dos Santos Pinheiro J, Ribeiro-Dos-Santos A, Cavalcante GC. Unraveling Cell Death Pathways During Malaria Infection: What do We Know So Far? Cells (2021) 10:2. doi: 10.3390/cells10020479
260. Kitchen AD, Chiodini PL. Malaria and Blood Transfusion. Vox Sang. (2006) 90:277–84. doi: 10.1111/j.1423-0410.2006.00733.x
261. Poespoprodjo JR, Fobia W, Kenangalem E, Hasanuddin A, Sugiarto P, Tjitra E, et al. Highly Effective Therapy for Maternal Malaria Associated With a Lower Risk of Vertical Transmission. J Infect Dis (2011) 204:101613–9. doi: 10.1093/infdis/jir558
262. Gazzinelli RT, Kalantari P, Fitzgerald KA, Golenbock DT. Innate Sensing of Malaria Parasites. Nat Rev Immunol (2014) 14:11744–57. doi: 10.1038/nri3742
263. Miller LH, Ackerman HC, Su XZ, Wellems TE. Malaria Biology and Disease Pathogenesis: Insights for New Treatments. Nat Med (2013) 19:2156–67. doi: 10.1038/nm.3073
264. de Carvalho RVH, Soares SMA, Gualberto ACM, Evangelista GCM, Duque JAM, Ferreira AP, et al. Plasmodium Berghei ANKA Infection Results in Exacerbated Immune Responses From C57BL/6 Mice Displaying Hypothalamic Obesity. Cytokine (2015) 76:2545–8. doi: 10.1016/j.cyto.2015.01.025
265. Coban C, Ishii KJ, Kawai T, Hemmi H, Sato S, Uematsu S, et al. Toll-Like Receptor 9 Mediates Innate Immune Activation by the Malaria Pigment Hemozoin. J Exp Med (2005) 201:119–25. doi: 10.1084/jem.20041836
266. Dey S, Bindu S, Goyal M, Pal C, Alam A, Iqbal MS, et al. Impact of Intravascular Hemolysis in Malaria on Liver Dysfunction: Involvement of Hepatic Free Heme Overload, NF-kappaB Activation, and Neutrophil Infiltration. J Biol Chem (2012) 287:3226630–46. doi: 10.1074/jbc.M112.341255
267. Pamplona A, Ferreira A, Balla J, Jeney V, Balla G, Epiphanio S, et al. Heme Oxygenase-1 and Carbon Monoxide Suppress the Pathogenesis of Experimental Cerebral Malaria. Nat Med (2007) 13:6703–10. doi: 10.1038/nm1586
268. Fernandez PL, Dutra FF, Alves L, Figueiredo RT, Mourao-Sa D, Fortes GB, et al. Heme Amplifies the Innate Immune Response to Microbial Molecules Through Spleen Tyrosine Kinase (Syk)-Dependent Reactive Oxygen Species Generation. J Biol Chem (2010) 285:4332844–51. doi: 10.1074/jbc.M110.146076
269. Fong KY, Wright DW. Hemozoin and Antimalarial Drug Discovery. Future Med Chem (2013) 5:121437–50. doi: 10.4155/fmc.13.113
270. Dostert C, Guarda G, Romero JF, Menu P, Gross O, Tardivel A, et al. Malarial Hemozoin Is a Nalp3 Inflammasome Activating Danger Signal. PLoS One (2009) 4:8e6510. doi: 10.1371/journal.pone.0006510
271. Dutra FF, Alves LS, Rodrigues D, Fernandez PL, de Oliveira RB, Golenbock DT, et al. Hemolysis-Induced Lethality Involves Inflammasome Activation by Heme. Proc Natl Acad Sci USA (2014) 111:39:E4110–8. doi: 10.1073/pnas.1405023111
272. Flegr J, Prandota J, Sovickova M, Israili ZH. Toxoplasmosis–a Global Threat. Correlation of Latent Toxoplasmosis With Specific Disease Burden in a Set of 88 Countries. PLoS One (2014) 9:3:e90203. doi: 10.1371/journal.pone.0090203
273. Tyebji S, Seizova S, Hannan AJ, Tonkin CJ. Toxoplasmosis: A Pathway to Neuropsychiatric Disorders. Neurosci Biobehav Rev (2019) 96:72–92. doi: 10.1016/j.neubiorev.2018.11.012
274. Frickel EM, Hunter CA. Lessons From Toxoplasma: Host Responses That Mediate Parasite Control and the Microbial Effectors That Subvert Them. J Exp Med (2021) 218:11. doi: 10.1084/jem.20201314
275. Rastogi S, Xue Y, Quake SR, Boothroyd JC. Differential Impacts on Host Transcription by ROP and GRA Effectors From the Intracellular Parasite Toxoplasma gondii. mBio (2020) 11:3. doi: 10.1128/mBio.00182-20
276. Andrade WA, Souza Mdo C, Ramos-Martinez E, Nagpal K, Dutra MS, Melo MB, et al. Combined Action of Nucleic Acid-Sensing Toll-Like Receptors and TLR11/TLR12 Heterodimers Imparts Resistance to Toxoplasma gondii in Mice. Cell Host Microbe (2013) 13:142–53. doi: 10.1016/j.chom.2012.12.003
277. Koblansky AA, Jankovic D, Oh H, Hieny S, Sungnak W, Mathur R, et al. Recognition of Profilin by Toll-Like Receptor 12 is Critical for Host Resistance to Toxoplasma gondii. Immunity (2013) 38:1119–30. doi: 10.1016/j.immuni.2012.09.016
278. Yarovinsky F. Innate Immunity to Toxoplasma gondii Infection. Nat Rev Immunol (2014) 14:2109–21. doi: 10.1038/nri3598
279. Fisch D, Bando H, Clough B, Hornung V, Yamamoto M, Shenoy AR, et al. Human GBP1 is a Microbe-Specific Gatekeeper of Macrophage Apoptosis and Pyroptosis. EMBO J (2019) 38:13:e100926. doi: 10.15252/embj.2018100926
280. Pandori WJ, Lima TS, Mallya S, Kao TH, Gov L, Lodoen MB. Toxoplasma gondii Activates a Syk-CARD9-NF-kappaB Signaling Axis and Gasdermin D-Independent Release of IL-1beta During Infection of Primary Human Monocytes. PLoS Pathog (2019) 15:8:e1007923. doi: 10.1371/journal.ppat.1007923
281. Kim B, Lee Y, Kim E, Kwak A, Ryoo S, Bae SH, et al. The Interleukin-1alpha Precursor is Biologically Active and is Likely a Key Alarmin in the IL-1 Family of Cytokines. Front Immunol (2013) 4:391. doi: 10.3389/fimmu.2013.00391
282. Dower SK, Kronheim SR, Hopp TP, Cantrell M, Deeley M, Gillis S, et al. The Cell Surface Receptors for Interleukin-1 Alpha and Interleukin-1 Beta Are Identical. Nature (1986) 324:6094266–8. doi: 10.1038/324266a0
283. Alvar J, Velez ID, Bern C, Herrero M, Desjeux P, Cano J, et al. Leishmaniasis Worldwide and Global Estimates of Its Incidence. PLoS One (2012) 7:5:e35671. doi: 10.1371/journal.pone.0035671
284. Chappuis F, Sundar S, Hailu A, Ghalib H, Rijal S, Peeling RW, et al. Visceral Leishmaniasis: What Are the Needs for Diagnosis, Treatment and Control? Nat Rev Microbiol (2007) 5:11873–82. doi: 10.1038/nrmicro1748
285. Lefevre L, Lugo-Villarino G, Meunier E, Valentin A, Olagnier D, Authier H, et al. The C-Type Lectin Receptors Dectin-1, MR, and SIGNR3 Contribute Both Positively and Negatively to the Macrophage Response to Leishmania infantum. Immunity (2013) 38:51038–49. doi: 10.1016/j.immuni.2013.04.010
286. Arango Duque G, Jardim A, Gagnon E, Fukuda M, Descoteaux A. The Host Cell Secretory Pathway Mediates the Export of Leishmania Virulence Factors Out of the Parasitophorous Vacuole. PLoS Pathog (2019) 15:7:e1007982. doi: 10.1371/journal.ppat.1007982
287. Shio MT, Christian JG, Jung JY, Chang KP, Olivier M. PKC/ROS-Mediated NLRP3 Inflammasome Activation is Attenuated by Leishmania Zinc-Metalloprotease During Infection. PLoS Negl Trop Dis (2015) 9:6:e0003868. doi: 10.1371/journal.pntd.0003868
288. Hergueta-Redondo M, Sarrio D, Molina-Crespo A, Megias D, Mota A, Rojo-Sebastian A, et al. Gasdermin-B Promotes Invasion and Metastasis in Breast Cancer Cells. PLoS One (2014) 9:3:e90099. doi: 10.1371/journal.pone.0090099
289. Xu B, Jiang M, Chu Y, Wang W, Chen D, Li X, et al. Gasdermin D Plays a Key Role as a Pyroptosis Executor of non-Alcoholic Steatohepatitis in Humans and Mice. J Hepatol (2018) 68:4773–82. doi: 10.1016/j.jhep.2017.11.040
290. Forstner G. Signal Transduction, Packaging and Secretion of Mucins. Annu Rev Physiol (1995) 57:585–605. doi: 10.1146/annurev.ph.57.030195.003101
Keywords: gasdermin, parasite, macrophage, Entamoba histolytica, innate immunity
Citation: Wang S, Moreau F and Chadee K (2022) Gasdermins in Innate Host Defense Against Entamoeba histolytica and Other Protozoan Parasites. Front. Immunol. 13:900553. doi: 10.3389/fimmu.2022.900553
Received: 20 March 2022; Accepted: 23 May 2022;
Published: 20 June 2022.
Edited by:
Lauren Webb, University of Washington, United StatesReviewed by:
Mingmin Lu, Nanjing Agricultural University, ChinaWilliam Petri, University of Virginia, United States
Copyright © 2022 Wang, Moreau and Chadee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kris Chadee, kchadee@ucalgary.ca