
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 10 June 2022
Sec. Immunological Tolerance and Regulation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.899975
 Siawosh K. Eskandari1,2*†
Siawosh K. Eskandari1,2*† Hazim Allos1†
Hazim Allos1† Basmah S. Al Dulaijan1†Gandolina Melhem1Ina Sulkaj1,3Juliano B. Alhaddad1
Basmah S. Al Dulaijan1†Gandolina Melhem1Ina Sulkaj1,3Juliano B. Alhaddad1 Anis J. Saad1
Anis J. Saad1 Christa Deban1Philip Chu1
Christa Deban1Philip Chu1 John Y. Choi1
John Y. Choi1 Branislav Kollar4,5Bohdan Pomahac4,6
Branislav Kollar4,5Bohdan Pomahac4,6 Leonardo V. Riella1,7
Leonardo V. Riella1,7 Stefan P. Berger2
Stefan P. Berger2 Jan S. F. Sanders2Judy Lieberman8,9Li Li10
Jan S. F. Sanders2Judy Lieberman8,9Li Li10 Jamil R. Azzi1,10*
Jamil R. Azzi1,10*Regulatory T cells (Tregs) have shown great promise as a means of cellular therapy in a multitude of allo- and auto-immune diseases—due in part to their immunosuppressive potency. Nevertheless, the clinical efficacy of human Tregs in patients has been limited by their poor in vivo homeostasis. To avert apoptosis, Tregs require stable antigenic (CD3ζ/T-cell-receptor-mediated), co-stimulatory (CD28-driven), and cytokine (IL-2-dependent) signaling. Notably, this sequence of signals supports an activated Treg phenotype that includes a high expression of granzymes, particularly granzyme B (GrB). Previously, we have shown that aside from the functional effects of GrB in lysing target cells to modulate allo-immunity, GrB can leak out of the intracellular lysosomal granules of host Tregs, initiating pro-apoptotic pathways. Here, we assessed the role of inhibiting mechanistic target of rapamycin complex 1 (mTORC1), a recently favored drug target in the transplant field, in regulating human Treg apoptosis via GrB. Using ex vivo models of human Treg culture and a humanized mouse model of human skin allotransplantation, we found that by inhibiting mTORC1 using rapamycin, intracytoplasmic expression and functionality of GrB diminished in host Tregs; lowering human Treg apoptosis by in part decreasing the phosphorylation of S6K and c-Jun. These findings support the already clinically validated effects of mTORC1 inhibition in patients, most notably their stabilization of Treg bioactivity and in vivo homeostasis.
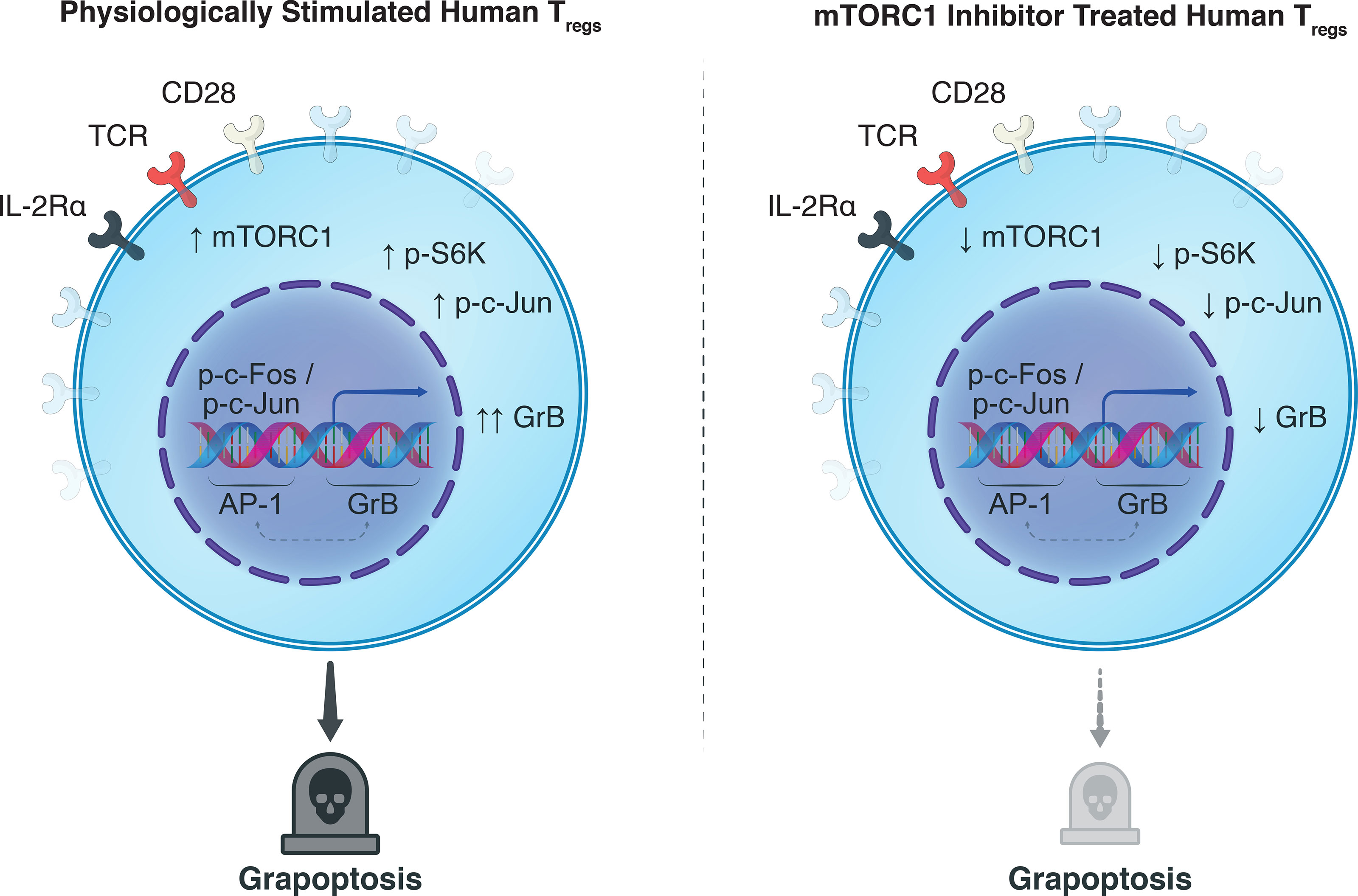
Graphical Abstract
The first description of cells residing in the lymphatics with an immunoregulatory phenotype dates back to studies by Gershon et al. in 1972 (1). The formal phenotypic characterization of what we now know as CD4+ regulatory T cells (Tregs), however, was not established until 1995 and 2003, when interleukin 2 receptor alpha (IL-2Rα; CD25) and FoxP3 were respectively identified as essential Treg markers in mice (2–4). In humans, additional studies later found that IL-7Rα (CD127) expression on CD4+ T cells inversely correlated with the human Treg phenotype, such that CD4+ CD25highCD127low T cells marked the purest human Treg population (5, 6). IL-7 is notably a memory cytokine that promotes the survival of human CD4+ T cells (7), where it is hypothesized that we humans acquire specialized memory CD4+ CD25hiCD127hi non-Tregs over the course of our lives in response to a variety of serendipitous pathogen exposures. Since the discovery of human CD4+ CD25hiCD127lo FoxP3+ Tregs, a myriad of clinical applications have been explored for auto-immune diseases and organ and tissue transplantation with varying degrees of success (8).
In general, therapeutic strategies involving Tregs begin with the isolation of Tregs from the peripheral blood of a patient, followed by an ex vivo expansion protocol, and ultimately adoptive re-infusion back into the patient (9, 10). Nonetheless, despite evolving techniques in isolating, handling, and expanding Tregs, Treg immunotherapy continues to be thwarted by invariably short Treg lifespan in patients and the unstable expression of the transcriptional immunoregulator FoxP3 (11). Additionally, considering the plasticity of the Treg phenotype, a worrisome complication of Treg therapy is the conversion of Tregs into pathologic T-cells secreting pro-inflammatory cytokines such as IL-17, which notoriously mediate auto-immune processes (12).
One avenue of optimizing Treg therapy involves studying the physiological working mechanisms of Tregs to allow for engineered Tregs that can withstand unfavorable in vivo milieus. Notably, Tregs employ a host of cell-contact dependent and independent methods to induce immunosuppression, where each immunoregulatory method is reliant on circumstantial factors such as the target cells, the context of immune response, and the anatomical site of suppression. Among these mechanisms of Treg action, granzyme B (GrB-)mediated cytolysis (also, granolysis) of target cells is one of the vital pathways with which Tregs block effector T-cell proliferation (13, 14). While GrB is essential in targeting effector cells, however, our group has previously shown that this serine protease can also play a role in destabilizing host Tregs, thus, predisposing them to granzyme-B-induced apoptosis (grapoptosis) (15, 16). Particularly, we found that GrB, which is physiologically stored in intracytoplasmic lysosomal granules prior to being exocytosed for targeted lysis, can leak from these granules upon T-cell-receptor-dependent activation and precipitate both caspase dependent and independent cell death (16). The exact mechanisms underlying granzyme leakage from the lysosomal granules remain to be fully understood.
Separate studies looking at novel drug-based immunotherapies have recently led to the understanding that there are Treg-favorable and -unfavorable immunosuppressants depending on the drugs’ impact on the Treg compartment (17, 18). With regards to the Treg-favorable drugs, a growing body of evidence attributes Treg-sparing qualities to inhibitors of mechanistic target of rapamycin complex 1 (mTORC1) such as rapamycin (also known as sirolimus)—although the mechanisms behind this effect are incompletely understood (19).
Here, we studied the interplay between grapoptosis and mTORC1 signaling in human Tregs. Notably, using the mTORC1 inhibitor rapamycin, we observed an intrinsic link between the mTORC1 pathway and GrB activity in which mTORC1 inhibition reduced intracellular GrB reserves and spared Treg viability.
Rapamycin solution (#S-015) and cyclosporin A solution (#C-093) were both purchased from Millipore Sigma (Burlington, MA, USA). Recombinant human IL-2 (#212-12) was purchased from PeproTech (Rocky Hill, NJ, USA). Soluble human anti-CD3 (OKT3, #16-0037-85) and soluble human anti-CD28 (28.2, # 16-0289-85) were purchased from Invitrogen (Carlsbad, CA, United States).
Leukapheresed blood from healthy individuals was obtained from the Brigham and Women’s Hospital Blood Bank. Blood samples were processed for peripheral blood mononuclear cell (PBMCs) isolation within 24 hours by SepMate tubes (#85450, STEMCELL Technologies).
Healthy skin was obtained from consenting patients undergoing cosmetic procedures at the Brigham and Women’s Hospital Plastic Surgery department. The study protocol was approved by an Institutional Review Board at the Brigham and Women’s Hospital and was performed in accordance with the principles of the Declaration of Helsinki.
Cells were cultured in complete Dulbecco’s Modified Eagle’s medium (#10-013-CV, Corning) supplemented with 9% pooled GemCell U.S. Origin Human Serum AB (#100-512, GeminiBio), 2 mM L-glutamine (#25030081, Gibco), and 1% penicillin-streptomycin (#15140122, Gibco).
Functional grade purified anti-human CD3 (OKT3) and functional grade purified anti-human CD28 (CD28.2; eBioscience) were added in suspension at a concentration of 4 μg/mL together with 200 ng/mL recombinant human IL-2 (PeproTech).
Differences between two normally distributed groups were analyzed with independent samples two-tailed Student t-tests, and non-parametric Mann-Whitney U-tests when the assumption of homoscedasticity could not be met. Statistical analyses of multiple groups were performed with one-way analyses of variance followed by Holm-Šídák multiple comparison tests, or mixed-effects model analyses with the Geisser-Greenhouse correction followed by Holm-Šídák multiple comparison tests for experimental groups with matched data points across multiple time points or concentrations. P < 0.05 was considered significant for all analyses. Data analysis and graphing were performed with Prism 9.3.1 (GraphPad Software). Graphs show boxplots with median, interquartile range, minimum, maximum, and all individual data points of the denoted experimental groups.
Additional experimental details are described in the Supplementary Methods.
While an expanding fund of knowledge has linked granzyme (GrB) production, mTORC1 signaling, and CD8+ T cell activation (20–22)—cytotoxic CD8+ T cells being known for producing vast quantities of GrB (23)—it was unknown if the same intrinsic link existed in Tregs. First, to recapitulate earlier findings that human Tregs upregulate GrB and markers of apoptosis (16), we isolated human CD4+ CD25hiCD127lo Tregs from the peripheral blood mononuclear cells (PBMCs) of healthy donors (Figures S1A–C). Next, we expanded them ex vivo for three days in the presence of α-CD3, α-CD28, and IL-2—thus, providing antigenic, co-stimulatory, and cytokine signals. Importantly, throughout our experiments we opted for a minimum viable cell culture medium (see Methods) as opposed to an optimized Treg medium (24) to obviate potential compensatory and confounding glycolytic and metabolic effects exacted by exogenous cell culture reagents such as non-essential amino acids. This was most relevant for the experiments including mTORC1 inhibition, due to mTORC1’s role in both the immune system and cellular metabolism (23). Using flow cytometry to identify the activated CD4+ CD25hiCD127lo FoxP3+ Treg subset (Figures S2A, B), we observed 36.9% GrB+ and 39.0% Annexin V+ expressing Tregs on average (Figures S2C, D). Importantly, staining the cell surface with Annexin V is a sensitive method for measuring the rate of apoptosis in many cell types including T cells (25). Further bisecting the Treg population into GrB- and GrB+ subsets, we confirmed that the GrB+ Tregs were characterized by an increased rate of Annexin V+ apoptosis compared to their GrB- counterparts (16.6% vs. 82.9%, GrB- vs. GrB+ Tregs, P < 0.0001; Figures S2E, F).
To assess the effect of mTORC1 inhibition on Tregs, GrB production, and the rate of Treg apoptosis, we repeated the experiments in the presence of the specific mTORC1 inhibitor rapamycin, where rapamycin is known as a potent immunosuppressant with Treg-sparing effects (26–28). Using flow cytometry Figure S3A), we observed a reduction in the CD25hiCD127lo population of CD4+ T cells in the context of rapamycin treatment (63.4% vs. 53.7%, control (CT) vs. rapamycin (Rapa), P < 0.0001; Figure 1A). Conversely, Rapa treatment yielded an increase in the FoxP3+ subset (76.5% vs. 83.5%, CT vs. Rapa, P = 0.0346; Figure 1B). Within the FoxP3+ Treg population, we found that the increase in GrB expression in CT Tregs was abated with Rapa treatment (54.0% vs. 4.14%, CT vs. Rapa, P = 0.0022; Figure 1C) as well as the rate of apoptosis (34.5% vs. 6.55%, CT vs. Rapa, P < 0.0001; Figure 1D). Besides reduced percentages of GrB and Annexin V, the mean fluorescence intensities (MFIs) of GrB and Annexin V were also dimmed in the Rapa-treated Tregs compared to the CT Tregs Figures S3B, C). Synthesizing the decreased CD25hiCD127lo expression, increased Foxp3 percentage, and reduced Annexin V positivity, we found that Rapa treatment yielded 10% more viable CD4+ CD25hiCD127lo FoxP3+ Tregs in our ex vivo model compared to the regularly treated CT Tregs (31.8% vs. 41.9%, CT vs. Rapa, P < 0.0001; Figure S3D)—these combined effects potentially explaining rapamycin’s “Treg-sparing” moniker.
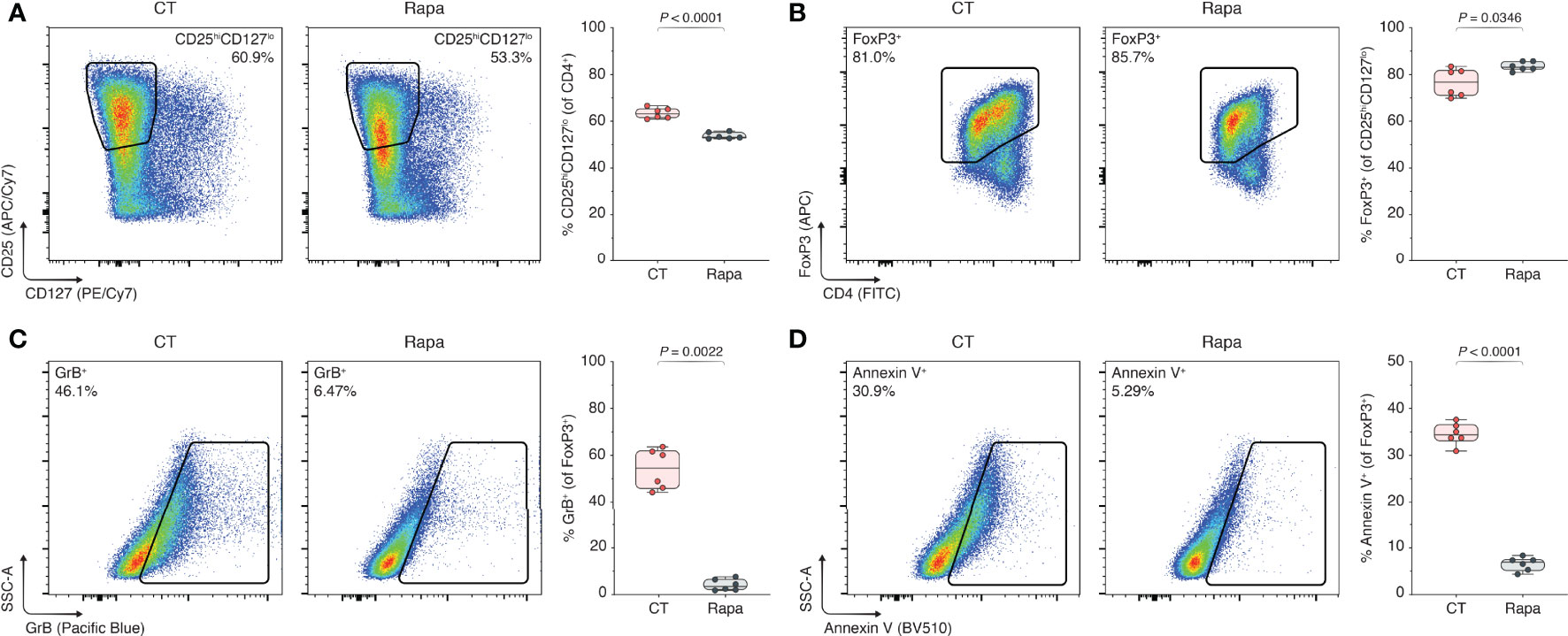
Figure 1 mTORC1 inhibition prevents GrB upregulation and rescues Tregs from apoptosis. Human Tregs were isolated and expanded for three days with α-CD3, α-CD28, and IL-2, without additional inclusions (CT), or with CT stimulants plus 100 nM rapamycin (Rapa). (A–D) Flow cytometric analyses of CD25hiCD127lo subset among CD4+ T cells (A), FoxP3+ subset among CD25hiCD127lo cells (B), and GrB+ (C) and Annexin V+ subsets (D) among FoxP3+ Tregs, including the respective box plots (n = 3 technical replicates/condition; 2 representative experiments of 5). Data represent boxplots with median, interquartile range, minimum, maximum, and all individual data points of the denoted experimental groups. P values were calculated with independent samples two-tailed Student’s t-tests, and non-parametric Mann-Whitney U-tests were performed when the assumption of homoscedasticity could not be met. CT, control; GrB, granzyme B; Rapa, rapamycin.
To understand the potential dose-dependency of rapamycin’s effect on the Treg compartment, we de-escalated our standard ex vivo rapamycin dose of 100 nM in 10Log steps, yielding a treatment of range of 0.1–10 nM of Rapa. Additionally, to ascertain whether the effects of rapamycin on GrB production and Annexin V apoptosis were due to the decreased activation of Tregs per se rather than a unique mTORC1-specific mechanism of action, we treated a group of human Tregs with cyclosporin A (CsA). Importantly, CsA belongs to a drug class known as calcineurin inhibitors (CNIs), which are archetypal immunosuppressants that continue to be used for solid organ transplantation to this day (28, 29). Using flow cytometry as described earlier Figure S3A), we found that the reduction in CD25hiCD127lo and increase in FoxP3 T cells indeed depended on the Rapa dose, however, not attaining statistical significance in increasing the FoxP3+ percentage in the range of 0.1–10 nM Rapa as with the 100 nM dosage Figures S4A, B). Treatment of Tregs with CsA decreased CD25hiCD127lo and increased FoxP3 as with Rapa treatment, but also without attaining statistical significance Figures S4A, B). Looking at the GrB expression in the FoxP3+ Tregs, intriguingly, we found that Rapa de-escalation significantly increased the GrB percentages and MFIs, returning them to the level of the CT Tregs at the lowest Rapa dose Figure S4D). On the contrary, CsA treatment did not diminish GrB expression, in fact, even increasing it (43.9% vs. 55.8%, CT vs. CsA, P = 0.0126; Figure S4D). Regarding the rate of apoptosis, Rapa de-escalation again resulted in a return to the CT Treg levels, while CsA treatment marginally decreased the Annexin V expression compared to CT Tregs (28.6% vs. 20.3%, CT vs. CsA, P = 0.0041; Figure S4E).
Next, we sought to determine if mTORC1 inhibition through rapamycin decreased GrB expression in activated Tregs per se or if it also affected the functionally active GrB escaping from the lysosomes into the cytoplasm. To measure intracytoplasmic GrB activity we used a specialized GranToxiLux assay (16, 30), which permits measurement of active GrB through surrogate green fluorescence emission. Importantly, green fluorescence is only emitted when the GrB-sensitive substrate in the GranToxiLux assay is cleaved (Figure 2A). To validate the GranToxiLux assay, we first isolated human CD4+ CD25hiCD127lo Tregs from the PBMCs of healthy donors and expanded them ex vivo for three days in the presence of α-CD3, α-CD28, and IL-2. Using flow cytometry Figure S5A), we identified the blastic (FSChiSSChi) lymphocyte subset, observing a significant increase in both the percentages of intracytoplasmically active GrB in the activated Treg condition compared to the freshly isolated, naïve Tregs (4.85% vs. 45.2%, naïve vs. activated Tregs, P < 0.0001; Figures S5B, C) as well as the MFIs (180 vs. 313, naïve vs. activated Tregs, P < 0.0001; Figure S5D).
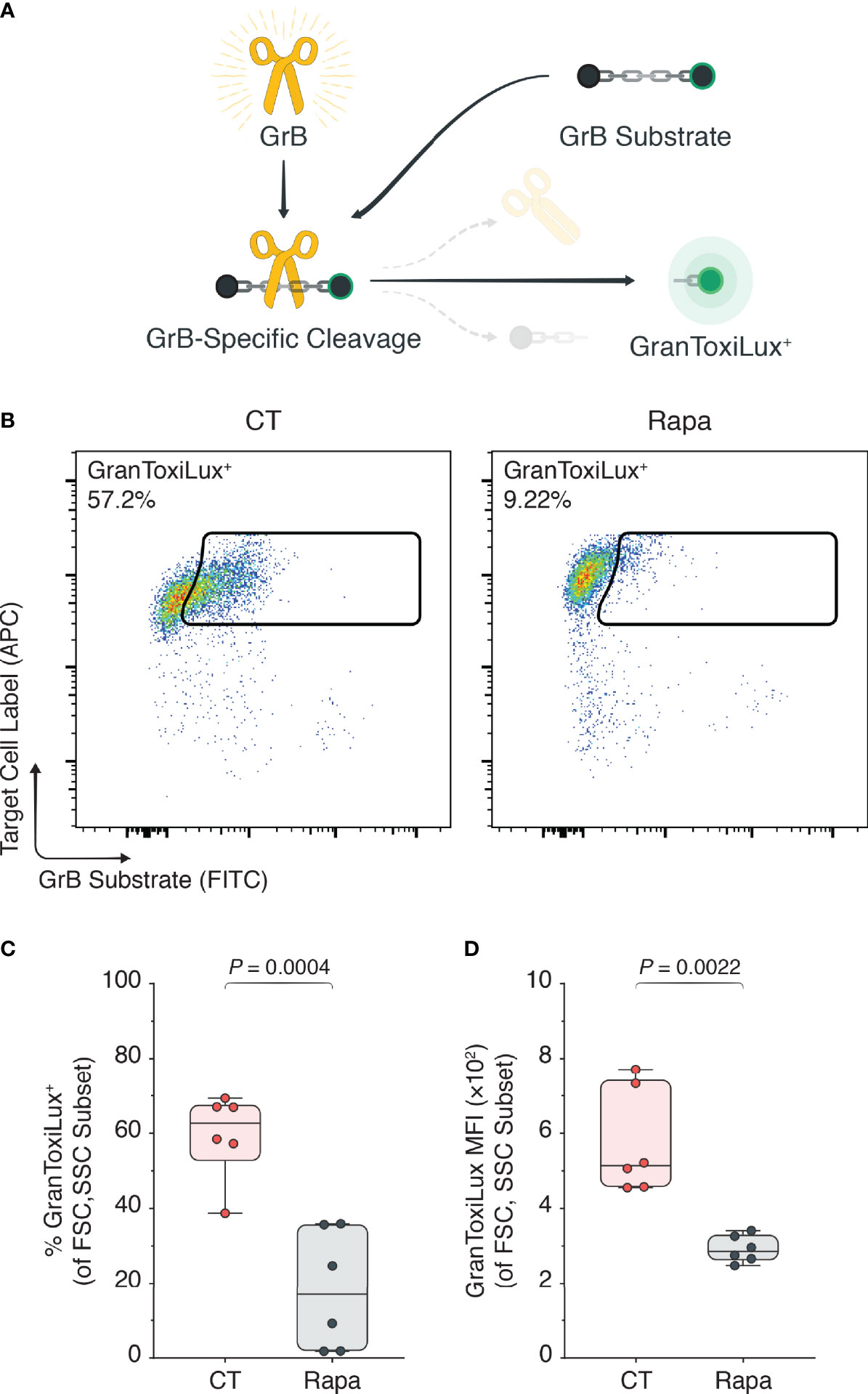
Figure 2 mTORC1 inhibition attenuates intracytoplasmically active GrB. Human Tregs were isolated and expanded for three days with α-CD3, α-CD28, and IL-2 (CT), or with CT stimulants plus 100 nM rapamycin (Rapa). (A) The GranToxiLux assay was used to determine functionally active GrB in the cytoplasmic Treg compartment, where active GrB cleaves the GrB-sensitive GranToxiLux substrate to yield a fluorescent GranToxiLux reagent detectable in the FITC channel. (B) Flow cytometric plots of active GrB in the Treg cytoplasm. (C, D) Box plots of GranToxiLux+ percentages (C) and mean fluorescence intensities (D) among the Treg population (n = 3 technical replicates/condition; 2 representative experiments of 4). Data represent boxplots with median, interquartile range, minimum, maximum, and all individual data points of the denoted experimental groups. P values were calculated with independent samples two-tailed Student’s t-tests, and non-parametric Mann-Whitney U-tests were performed when the assumption of homoscedasticity could not be met. CT, control; GrB, granzyme B; MFI, mean fluorescence intensity; Rapa, rapamycin; Tregs, regulatory T cells.
To appreciate the impact of rapamycin on the GranToxiLux substrate conversion, we repeated the experiments in the presence of rapamycin with Tregs isolated from new PBMC donors. We observed a significant attenuation of the percentages of active GrB in the cytoplasm of Rapa-treated Tregs compared with CT Tregs (59.58% vs. 18.21%, naïve vs. activated Tregs, P = 0.0004; Figures 2B, C) as well as the GranToxiLux substrate MFIs (574 vs. 292, naïve vs. activated Tregs, P = 0.0022; Figure 2D).
To explore the potential mechanistic link between mTORC1 and GrB, we studied the expression of phosphorylated S6 kinase 1 (p-S6K) and c-Jun (p-c-Jun) in human CD4+ CD25hiCD127lo Tregs isolated from the PBMCs of healthy donors. These particular targets were chosen on the basis that mTORC1 physiologically endorses its pleiotropic effects through phosphorylation of S6K—among other kinases— (31, 32) and GrB is known to be upregulated by a variety of transcription factors including activating protein (AP-)1, a heterodimer consisting of proteins including c-Jun (33). We cultured the isolated Tregs ex vivo in a 24-hour window without stimulants (NC), or with α-CD3, α-CD28, and IL-2 in the presence or absence of rapamycin (Rapa and CT respectively). Using flow cytometry Figure S6), we found that providing 24 hours of mitogenic stimuli to Tregs increased their expression of p-S6K relative to the non-stimulated Tregs (1.86×104 vs. 4.85×104, NC vs. CT Tregs, P = 0.0001; Figure 3A) as well as their expression of p-c-Jun (435 vs. 623, NC vs. CT Tregs, P < 0.0001; Figure 3B). Addition of rapamycin to the human Treg culture, however, impeded the upregulation of p-S6K relative to the CT Tregs as early as 6 hours post treatment (3.76×104 vs. 2.72×104, CT vs. Rapa Tregs, P = 0.0025; Figure 3A) and within 24 hours of treatment for the upregulation of p-c-Jun (623 vs. 550, CT vs. Rapa Tregs, P = 0.0051; Figure 3B).
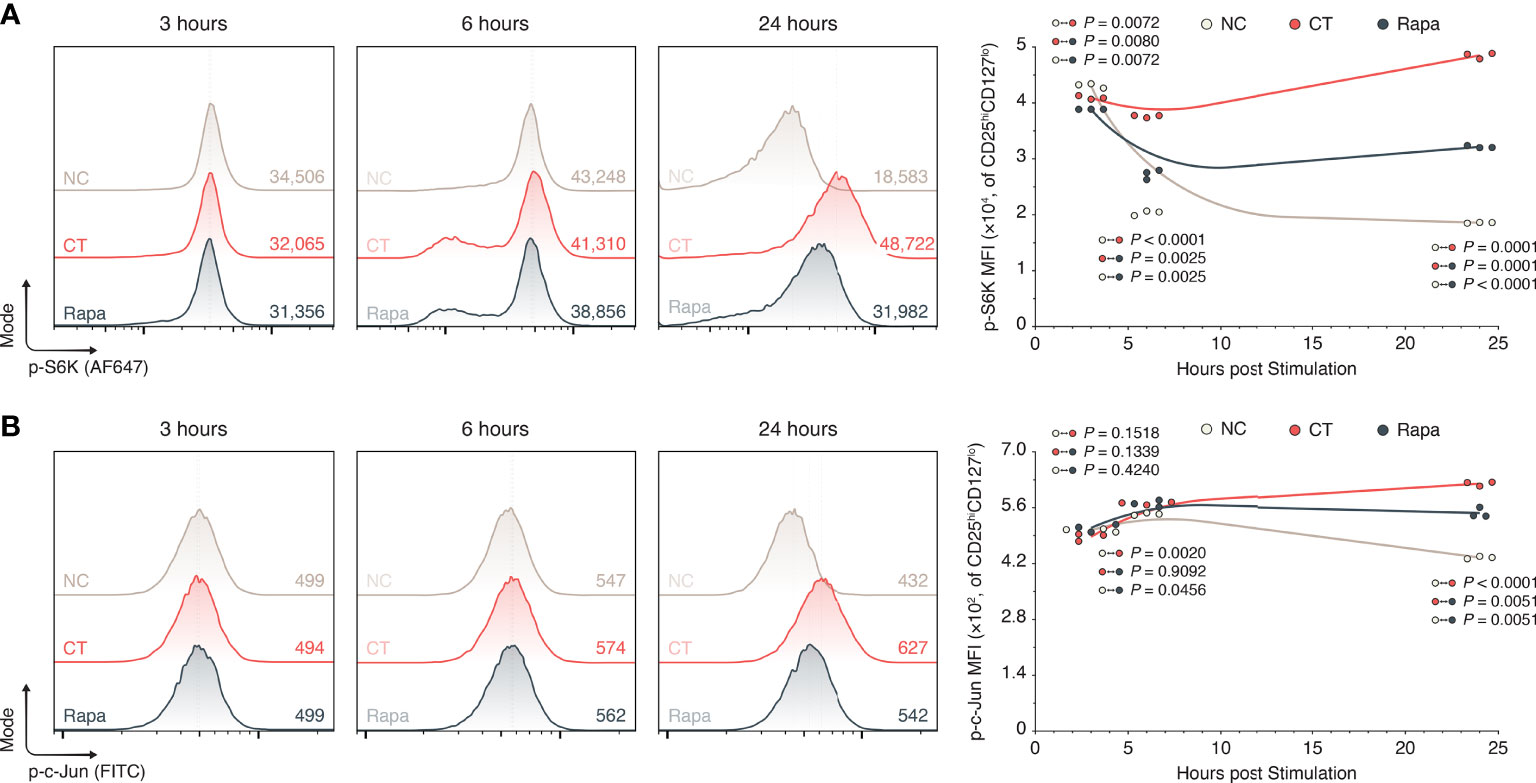
Figure 3 mTORC1 inhibition attenuates S6K and c-Jun phosphorylation. Human Tregs were isolated and expanded in a 24-hour cell culture window without inclusions (NC), with α-CD3, α-CD28, and IL-2 (CT), or with CT stimulants and 100 nM rapamycin (Rapa). (A) Flow histograms of p-S6K expression among CD25hiCD127lo Tregs, including inset mean fluorescence intensities (MFIs) and the 24-hour period XY plot (n = 3 technical replicates/condition/timepoint; 1 representative experiment of 3). (B) Flow histograms of p-c-Jun expression among CD25hiCD127lo Tregs, including inset mean fluorescence intensities (MFIs) and the 24-hour period XY plot (n = 3 technical replicates/condition/timepoint; 1 representative experiment of 3). All individual datapoints are displayed. P values were calculated using mixed-effects model analyses with the Geisser-Greenhouse correction followed by Holm-Šídák multiple comparison tests. AF647, Alexa Fluor 647; CT, control; MFI, mean fluorescence intensity; NC, negative control; Rapa, rapamycin.
Although ex vivo experiments provide great insights into the working mechanisms of Treg-affecting drugs, we also sought to investigate if mTORC1 inhibition could affect GrB and Annexin V expression of human Tregs in an in vivo humanized mouse model (Figure S7A). In this model, we transplanted healthy human skin obtained from consenting patients undergoing cosmetic surgery onto the trunks of NOD-scid IL-2 receptor-γnull (NSG) mice. Seven days post-transplant, 5.0×106 human PBMCs and 1.0×106 human Tregs were adoptively transferred into the NSG mice to respectively initiate and suppress the allo-immune responses (Figure S7B). To assess the effects of mTORC1 inhibition on the Tregs we compared daily intraperitoneal injections of the phosphate-buffered saline vehicle (PBS) to 1 mg/kg rapamycin injections. Twenty-one days post transplantation, we euthanized the mice and studied the splenocytes by flow cytometry (Figure S7B). Notably, this model has several limitations. First, the read-out sites are restricted to the splenic compartment as Tregs from the skin grafts cannot be extracted from the dermal-epidermal interface without disrupting the Treg surface phenotype—due to the required enzymatic digestion—and there are no secondary lymphoid tissues (SLT) apart from the spleen due to SLT atrophy in the NSG mice. Secondly, there are methodological restrictions to attaining large enough human Treg quantities for survival studies as well as being able to include a multitude of conditions. Nevertheless, this model does allow us to study the effect of rapamycin on Tregs in four dimensions, namely in three-dimensional space and over time, permitting the assessment of GrB expression and Annexin V positivity on in vivo derived human Tregs.
Using flow cytometry to identify the human Tregs residing within in the spleen (Figures S7C, D), we found that rapamycin treatment marginally increased the percentage of CD4+ CD25+FoxP3+ Tregs relative to PBS-treated Tregs, but not significantly so (34.6% vs. 47.3%, PBS vs. Rapa, P = 0.0959; Figure S7D). Additionally, looking at the CD4+ CD25++FoxP3+ Treg subset we found that rapamycin treatment did not affect this subset relative to the PBS treatment group (2.1% vs. 2.3%, PBS vs. Rapa, P = 0.8678; Figure S7D). At the same time, however, the GrB expression among the splenic Tregs of the Rapa-treated mice was significantly lowered compared to the splenic PBS Tregs (53.4% vs. 34.1%, PBS vs. Rapa, P = 0.0043; Figures S7E, F). Additionally, the Annexin V expression in the Rapa condition was significantly attenuated compared to the PBS condition (56.0% vs. 33.8%, PBS vs. Rapa, P = 0.0042; Figures S7G, H). Thus, although the percentages of CD4+ CD25+FoxP3+ Tregs were not significantly different between the treatment groups, looking at the viable CD4+ CD25+FoxP3+ Tregs, we found significantly more live Tregs in the Rapa condition compared to the PBS group (16.7% vs. 32.5%, PBS vs. Rapa, P = 0.0123; Figure S7I). Finally, to assess if the observed effects in the splenic compartment of the Rapa-treated group were due to mTORC1 inhibition, we stained the splenocytes for phosphorylated 4E-BP. Notably, the phosphorylation of 4E-BP is physiologically repressed by mTORC1 (32), thus, expecting p-4E-BP to be increased in mTORC1-inhibited cells. Indeed, we found that the splenic Tregs of the Rapa-treated mice expressed higher percentages of p-4E-BP compared with the Tregs of PBS-treated mice (28.8% vs. 50.1%, PBS vs. Rapa, P = 0.0269; Figure S7J), indicating a veritable effect of the Rapa treatment on these Tregs.
A growing body of evidence supports the influence of granzyme (GrB) secretion by activated Tregs on the suppression of effector lymphocytes and regulation of overall allograft tolerance (13–15, 34). While GrB is vital for the effector functionality of Tregs, however, we recently showed that activated Tregs also have the propensity to leak GrB in the intracytoplasmic compartment of host Tregs, leading to caspase-3 dependent and independent cell death (16). This finding also proved clinically relevant, as we found that Tregs in the peripheral blood of renal allografts recipients with T-cell-mediated rejection expressed higher levels of GrB—thus, making them more prone to granzyme-B-induced apoptosis (grapoptosis) (16). Since the poor in vivo homeostasis of Tregs handicaps the clinical application of these cells as an immunosuppressive cytotherapy, insights on the factors that moderate Treg survival are much needed to improve the efficacy of adoptively transferred Tregs in patients (35). Here, we explored the potential role of GrB in regulating Treg homeostasis in association with mTORC1 signaling, considering recent lines of inquiry have attributed Treg-sparing qualities to mTORC1 inhibitors (36, 37). mTORC1 inhibitors notably suppress pro-inflammatory T cells while sparing or in certain cases even inducing proliferation of Tregs, making them an attractive target to study in the context of grapoptosis.
While several studies in the literature have previously hinted at mTORC1 inhibitors reducing GrB expression in activated Tregs (15, 16, 34, 38), the relationship between decreased GrB expression and increased viability was not yet explored. In our experiments, we found that mTORC1 inhibition could indeed spare Treg homeostasis by decreasing both intracellular GrB expression and activity, and protect Tregs from pro-apoptotic pathways marked by the expression of Annexin-V-specific phospholipids on the Treg surface. Accordingly, we speculate that the treatment of patients with mTORC1 inhibitors could be lowering the apoptotic rate of Tregs by, in part, decreasing intracytoplasmic GrB activity.
Intriguingly, while the treatment of human Tregs with rapamycin in our ex vivo models lead to reduced GrB levels and increased viability, we did not observe the same effect upon treating human Tregs with the calcineurin inhibitor (CNI) cyclosporin A. CNIs are notably a mainstay of the immunosuppressive regime used after solid organ transplantation (28), realizing immunosuppression in a T cell agnostic fashion by eliminating both Tregs and Tcons, and possibly even inhibiting Tregs to a greater degree (15, 34, 38). mTORC1 inhibitors, in contrast, selectively downregulate the serine/threonine kinase mTOR—which physiologically promotes the survival, proliferation, and accentuation of T cell function downstream of the T-cell-receptor (TCR), CD28, and IL-2 signaling pathways (27, 39, 40). Mounting evidence now suggests that mTORC1 inhibitors preferentially suppress Tcon proliferation over that of Tregs, although rapamycin is not agnostic to the Treg population and can similarly inhibit Tregs at higher concentrations (41). Notably, however, Tregs can alternatively rely on different kinases for cell growth and activation, thus maintaining their activity and proliferative capacity despite mTORC1 inhibition (26–28). We hypothesize that Tregs may possess these redundancies to mTORC1 signaling to be able to survive in a wide range of milieus, including competitive niches such as inflammatory sites and the tumor microenvironment. Notably, Tregs can metabolically adapt to these harsh environments by switching to aerobic glycolysis, upregulating FoxP3 in response to hypoxia, and outcompeting other T cells subsets under acidic, low glucose, and high lactate conditions (42–44). In fact, it seems Tregs have not only evolved to survive reducing environments but are now even known to employ extracellular redox metabolites to suppress effectors T cells (45). Considering mTORC1 sits at the interface of immune responses and metabolic activities—a burgeoning domain known as immunometabolism—mTORC1 inhibition is perhaps only likely to affect Treg homeostasis. Indeed, in clinical trials with kidney and transplant recipients, mTORC1 inhibitors benefitted Treg counts in the peripheral circulation when compared to CNIs (46, 47), while in another trial, liver transplant recipients who were converted from tacrolimus to sirolimus—a CNI to an mTORC1 inhibitor—demonstrated elevated numbers of Tregs in their blood and liver grafts (48).
Unsurprisingly, mTORC1 inhibitors such as sirolimus and everolimus have earned the reputation of being Treg-friendly compared to alternative immunosuppressive strategies (49). The effect of mTORC1 inhibition on Tregs, however, is not entirely unambiguous. Experiments by Procaccini and colleagues have previously indicated that mTORC1 is locked in a complex, oscillatory dance with Tregs—mTORC1 being a context-dependent regulatory checkpoint for CD4+ CD25hiCD127lo Treg activity, proliferation and fate (36). Human Tregs can even express elevated levels of phosphorylated translational regulators of mTORC1 including p70S6K and S6 compared to the effector CD4+ CD25lo T cell population (18, 36). This finding was further elaborated on with mice deficient in Raptor, an mTORC1 adaptor protein, which displayed disrupted Treg responses—indicating a contextually protective role for mTORC1 in certain autoimmune diseases (18). Additionally, work by Sun and colleagues showed that mTORC1 is highly expressed in the effector cluster of murine Tregs compared to the “resting reservoir” of central Tregs, positing that mTORC1 activity is required for the conversion of central Tregs to effector Tregs and can be inhibited with rapamycin (50). Considering the potential disparate roles of mTORC1 in the murine Treg compartment, further research towards the relevance of the murine findings in human Tregs and the role of mTORC1 in the different Treg subsets is warranted.
Regarding the specific interplay between mTORC1 and GrB it is worthy to note that these axes have previously been linked in cytotoxic CD8 T cells (CTLs) (20–22). Notably, GrB is highly expressed by CTLs, being the tenth most produced protein by CTLs overall (23), while the overexpression (22) and downregulation (22, 23) of mTORC1 in CD8 T cells respectively resulted in the upregulation and attenuation of GrB. Taking an excursion to the disparate domain of fruit flies, a recent study by Jouandin and colleagues in Science interestingly tied the lysosomal compartment and TORC1 responses together (51). While this study did not explore the link between GrB stores in the lysosomes and TORC1, it did intrinsically link the autolysosome to the latter, reiterating the possible connection between GrB and mTORC1.
Synthesizing our findings with the knowledge garnered in the literature: Antigenic signatures are relayed to Tregs from the extracellular milieu through the T-cell receptor (CD3) with co-stimulatory (CD28) and cytokine (IL-2) signaling, activating host Tregs and mTORC1 (Figure 4A). We believe this permits a cascade of proliferative stimuli including S6K and c-Jun phosphorylation, resulting in the escalation of Treg activation. Nuclear translocation of AP-1 heterodimers, including p-c-Jun, can then drive GrB transcription (Figure 4B), which can ultimately destabilize the autolysosome harboring synthesized stores of GrB and lead to their escape before being exocytosed (Figure 4C). In the same manner that GrB causes granolysis of target cells, nuclear translocation of GrB in host Tregs can cause granzyme-B-induced apoptosis—in part, explaining the poor homeostasis of adoptively transferred human Tregs through grapoptosis. Notably, we found that treatment of human Tregs with an mTORC1 inhibitor decreased S6K and c-Jun phosphorylation and attenuated both intracellular GrB expression and Treg apoptosis (Figure 4D). Although further research efforts are warranted to deepen our understanding of the complex interplay between mTORC1 inhibition and grapoptosis, our findings provide preliminary insights towards another piece of the Treg homeostasis puzzle.
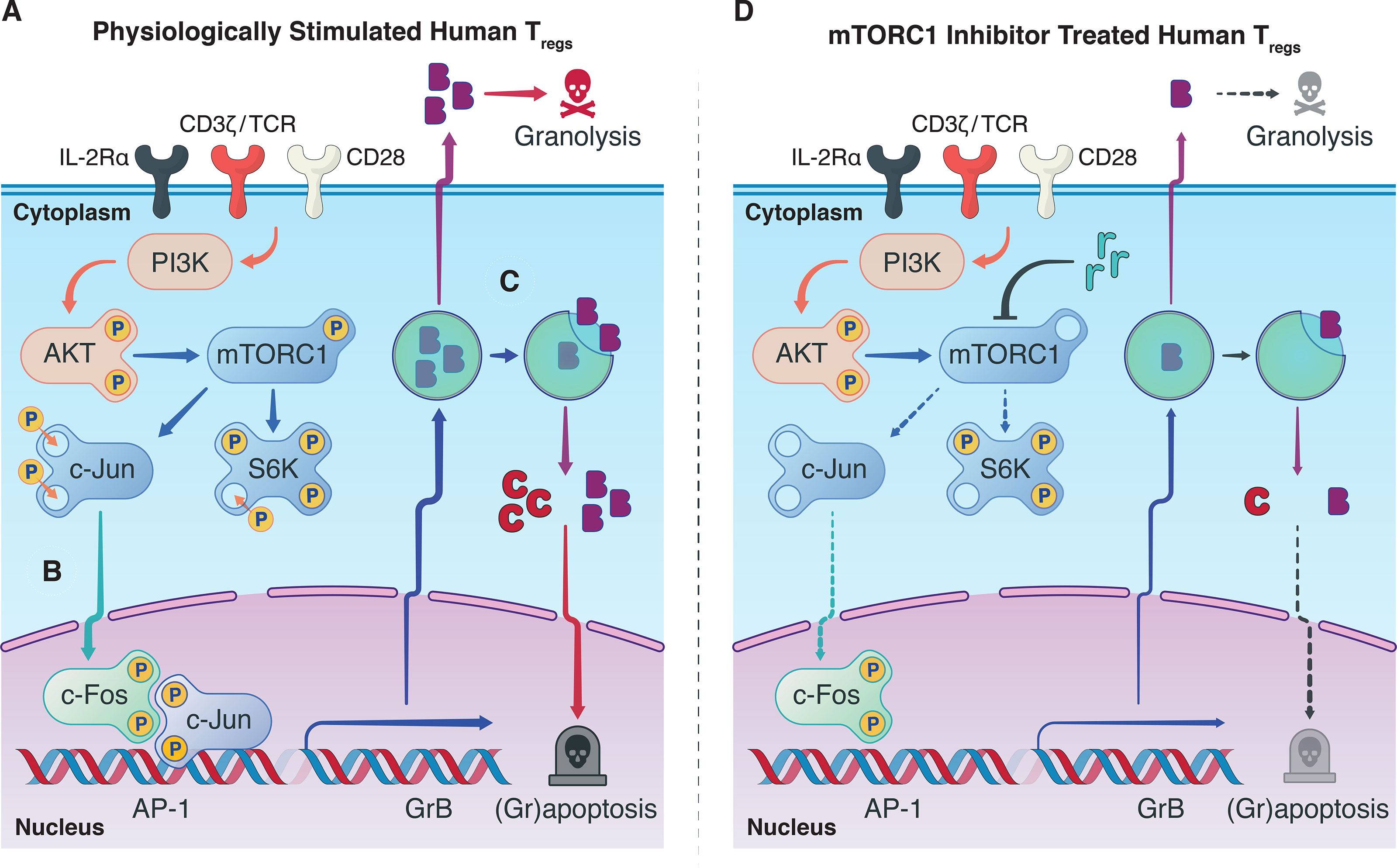
Figure 4 Observed interplay between mTORC1 and GrB pathways. (A) In a physiological setting, antigenic Treg stimulation via the T-cell receptor, co-stimulatory CD28-based activation, and IL-2Rα (CD25)-dependent cytokine signaling, sets in motion a phosphorylation cascade involving phosphoinositide 3-kinase (PI3K), Akt, mTORC1, S6 kinase 1 (S6K), and c-Jun. Phosphorylated c-Jun is known to permit its nuclear translocation where it can undergo dimerization with p-c-Fos and generate transcription factor Activator Protein-1 (AP-1). (B) Our hypothesis is that AP-1 can unwind heterochromatic DNA in the promotor region of GrB in Tregs to boost the transcription of GrB mRNA. Conventionally, upon mRNA translation, the mature GrB proteins are stored in designated cytotoxic granules to save their potent cytolytic activity for target effector T cells. (C) In the context of protracted antigenic stimulation, however, GrB can inadvertently leak from its harboring lysosomes to precipitate caspase-dependent and independent cell death. (D) Treatment of Tregs with mTORC1 inhibitors such as rapamycin provide a multipronged effect, notably, decreasing phosphorylation of p-S6K and p-c-Jun, attenuating GrB expression and activity, and finally diminishing Treg apoptosis. The exact biochemical constituents that drive this phenotypic and functional shift in the Treg population upon mTORC1 inhibition remain to be elucidated and warrant further research efforts.
As with all human Treg studies, attaining large numbers of Tregs for ex vivo and in vivo experiments is restricted by methodological constraints, limiting the number of testable conditions. Development of alternative cell culture models and use of fluorescence-activated cell sorting methods would likely permit greater exploration of the exact mechanisms of action underpinning mTORC1-centric regulation of the human Treg phenotype. Alternatively, murine models could be employed to decipher the specifics behind mTORC1 signaling and grapoptosis—even though human Tregs are a heterogeneous population of cells that are metabolically and phenotypically distinct cells from their murine counterparts (52–54).
Additionally, although the Tregs in this study were harvested from diverse donors, and each donor pool of human T cells responds differently to mitogenic stimuli (due to genetic and health factors pertaining to the donor), GrB was upregulated in all activated Tregs, and this increase could consistently be neutralized with mTORC1 inhibition. Nonetheless, the clinical application of these findings, as with all human studies, remains to be validated in settings with clinically-relevant patient sample sizes.
Extended activation of Tregs with antigenic, co-stimulatory, and cytokine signals upregulates a pro-apoptotic protein signature that includes the intracytoplasmic expression of granzyme B. Inhibition of mTORC1 in Tregs can neutralize this signature by in part decreasing the expression and activity of intracellular GrB, thus improving Treg homeostasis. Whether mTORC1 inhibition solely decreases grapoptosis in host Tregs or also diminishes granolysis of target cells, warrants future research efforts.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by Institutional Review Board at the Brigham and Women’s Hospital. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Authors are listed alphabetically: JRA formulated the overarching research aims. HA, JRA, and SE designed the experimental models and analyzed and interpreted the data. BD, GM, HA, IS, SE. performed experiments and collected data. BK, BP, JC provided study materials, reagents, patient samples, instrumentation, or other analysis tools. HA, JRA, and SE wrote the initial draft of the manuscript. AS, CD, JBA, JS, JL, LL, LVR, PC, and SB critically reviewed the manuscript and provided commentary. JRA, and SE revised the manuscript and prepared it for publication. JRA acquired funding for the work presented in the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the American Heart Association (AHA Award 13FTF17000018 to JRA), American Diabetes Association (ADA Award 1-17-IBS-206 to JRA), Qatar Foundation Grant (NPRP8-1744-3-357X to JRA), and the National Institutes of Health (RO1 AI134842 to JRA).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank our colleagues at the Schuster Family Transplantation Research Center at the Brigham and Women’s Hospital for their assistance and collaboration, and we thank the patients who made this work possible in trusting us with their donated skin and blood samples.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.899975/full#supplementary-material
Supplementary Data Sheet 1 | Original Data.
2. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic Self-Tolerance Maintained by Activated T Cells Expressing IL-2 Receptor Alpha-Chains (CD25). Breakdown of a Single Mechanism of Self-Tolerance Causes Various Autoimmune Diseases. J Immunol (1995) 155:1151–64.
3. Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu T-T, et al. Regulatory T-Cell Suppressor Program Co-Opts Transcription Factor IRF4 to Control T(H)2 Responses. Nature (2009) 458:351–6. doi: 10.1038/nature07674
4. Yu F, Sharma S, Edwards J, Feigenbaum L, Zhu J. Dynamic Expression of Transcription Factors T-Bet and GATA-3 by Regulatory T Cells Maintains Immunotolerance. Nat Immunol (2015) 16:197–206. doi: 10.1038/ni.3053
5. Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 Expression Inversely Correlates With FoxP3 and Suppressive Function of Human CD4+ T Reg Cells. J Exp Med (2006) 203:1701–11. doi: 10.1084/jem.20060772
6. Yu N, Li X, Song W, Li D, Yu D, Zeng X, et al. CD4(+)CD25 (+)CD127 (Low/-) T Cells: A More Specific Treg Population in Human Peripheral Blood. Inflammation (2012) 35:1773–80. doi: 10.1007/s10753-012-9496-8
7. Chetoui N, Boisvert M, Gendron S, Aoudjit F. Interleukin-7 Promotes the Survival of Human CD4+ Effector/Memory T Cells by Up-Regulating Bcl-2 Proteins and Activating the JAK/STAT Signalling Pathway. Immunology (2010) 130:418–26. doi: 10.1111/j.1365-2567.2009.03244.x
8. Duggleby R, Danby RD, Madrigal JA, Saudemont A. Clinical Grade Regulatory CD4(+) T Cells (Tregs): Moving Toward Cellular- Based Immunomodulatory Therapies. Front Immunol (2018) 9:252. doi: 10.3389/fimmu.2018.00252
9. Riley JL, June CH, Blazar BR. Human T Regulatory Cell Therapy: Take a Billion or So and Call Me in the Morning. Immunity (2009) 30:656–65. doi: 10.1016/j.immuni.2009.04.006
10. McDonald-Hyman C, Turka LA, Blazar BR. Advances and Challenges in Immunotherapy for Solid Organ and Hematopoietic Stem Cell Transplantation. Sci Transl Med (2015) 7:280rv2. doi: 10.1126/scitranslmed.aaa6853
11. Hatzioannou A, Boumpas A, Papadopoulou M, Papafragkos I, Varveri A, Alissafi T, et al. Regulatory T Cells in Autoimmunity and Cancer: A Duplicitous Lifestyle. Front Immunol (2021) 12:731947. doi: 10.3389/fimmu.2021.731947
12. Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martinez-Llordella M, Ashby M, et al. Instability of the Transcription Factor Foxp3 Leads to the Generation of Pathogenic Memory T Cells In Vivo. Nat Immunol (2009) 10:1000–7. doi: 10.1038/ni.1774
13. Gondek DC, Lu L-F, Quezada SA, Sakaguchi S, Noelle RJ. Cutting Edge: Contact-Mediated Suppression by CD4+CD25+ Regulatory Cells Involves a Granzyme B-Dependent, Perforin-Independent Mechanism. J Immunol (2005) 174:1783–6. doi: 10.4049/jimmunol.174.4.1783
14. Vignali DAA, Collison LW, Workman CJ. How Regulatory T Cells Work. Nat Rev Immunol (2008) 8:523–32. doi: 10.1038/nri2343
15. Azzi J, Skartsis N, Mounayar M, Magee CN, Batal I, Ting C, et al. Serine Protease Inhibitor 6 Plays a Critical Role in Protecting Murine Granzyme B-Producing Regulatory T Cells. J Immunol (2013) 191:2319–27. doi: 10.4049/jimmunol.1300851
16. Sula Karreci E, Eskandari SK, Dotiwala F, Routray SK, Kurdi AT, Assaker JP, et al. Human Regulatory T Cells Undergo Self-Inflicted Damage via Granzyme Pathways Upon Activation. JCI Insight (2017) 2. doi: 10.1172/jci.insight.91599
17. Battaglia M, Stabilini A, Roncarolo M-G. Rapamycin Selectively Expands CD4+CD25+FoxP3+ Regulatory T Cells. Blood (2005) 105:4743–8. doi: 10.1182/blood-2004-10-3932
18. Chapman NM, Chi H. mTOR Signaling, Tregs and Immune Modulation. Immunotherapy (2014) 6:1295–311. doi: 10.2217/imt.14.84
19. Raimondi G, Sumpter TL, Matta BM, Pillai M, Corbitt N, Vodovotz Y, et al. Mammalian Target of Rapamycin Inhibition and Alloantigen-Specific Regulatory T Cells Synergize to Promote Long-Term Graft Survival in Immunocompetent Recipients. J Immunol (2010) 184:624–36. doi: 10.4049/jimmunol.0900936
20. Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR Kinase Determines Effector Versus Memory CD8+ T Cell Fate by Regulating the Expression of Transcription Factors T-Bet and Eomesodermin. Immunity (2010) 32:67–78. doi: 10.1016/j.immuni.2009.10.010
21. Li Q, Rao RR, Araki K, Pollizzi K, Odunsi K, Powell JD, et al. A Central Role for mTOR Kinase in Homeostatic Proliferation Induced CD8+ T Cell Memory and Tumor Immunity. Immunity (2011) 34:541–53. doi: 10.1016/j.immuni.2011.04.006
22. Pollizzi KN, Patel CH, Sun I-H, Oh M-H, Waickman AT, Wen J, et al. Mtorc1 and Mtorc2 Selectively Regulate CD8+ T Cell Differentiation. J Clin Invest (2015) 125:2090–108. doi: 10.1172/JCI77746
23. Hukelmann JL, Anderson KE, Sinclair LV, Grzes KM, Murillo AB, Hawkins PT, et al. The Cytotoxic T Cell Proteome and its Shaping by the Kinase mTOR. Nat Immunol (2016) 17:104–12. doi: 10.1038/ni.3314
24. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In Vitro-Expanded Antigen-Specific Regulatory T Cells Suppress Autoimmune Diabetes. J Exp Med (2004) 199:1455–65. doi: 10.1084/jem.20040139
25. Crowley LC, Marfell BJ, Scott AP, Waterhouse NJ. Quantitation of Apoptosis and Necrosis by Annexin V Binding, Propidium Iodide Uptake, and Flow Cytometry. Cold Spring Harb Protoc (2016) 2016:953–57. doi: 10.1101/pdb.prot087288
26. Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo M-G. Rapamycin Promotes Expansion of Functional CD4+CD25+FOXP3+ Regulatory T Cells of Both Healthy Subjects and Type 1 Diabetic Patients. J Immunol (2006) 177:8338–47. doi: 10.4049/jimmunol.177.12.8338
27. Singh K, Stempora L, Harvey RD, Kirk AD, Larsen CP, Blazar BR, et al. Superiority of Rapamycin Over Tacrolimus in Preserving Nonhuman Primate Treg Half-Life and Phenotype After Adoptive Transfer. Am J Transplant Off J Am Soc Transplant Am Soc Transpl Surg (2014) 14:2691–703. doi: 10.1111/ajt.12934
28. Matas AJ, Smith JM, Skeans MA, Thompson B, Gustafson SK, Stewart DE, et al. OPTN/SRTR 2013 Annual Data Report: Kidney. Am J Transplant (2015) 15 Suppl 2:1–34. doi: 10.1111/ajt.13195
29. Azzi JR, Sayegh MH, Mallat SG. Calcineurin Inhibitors: 40 Years Later, Can’t Live Without. J Immunol (2013) 191:5785–91. doi: 10.4049/jimmunol.1390055
30. Migueles SA, Osborne CM, Royce C, Compton AA, Joshi RP, Weeks KA, et al. Lytic Granule Loading of CD8+ T Cells is Required for HIV-Infected Cell Elimination Associated With Immune Control. Immunity (2008) 29:1009–21. doi: 10.1016/j.immuni.2008.10.010
31. Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ. Phosphorylation of Ribosomal Protein S6 Is Inhibitory for Autophagy in Isolated Rat Hepatocytes. J Biol Chem (1995) 270:2320–6. doi: 10.1074/jbc.270.5.2320
32. Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J. Amino Acid Sufficiency and mTOR Regulate P70 S6 Kinase and eIF-4e BP1 Through a Common Effector Mechanism. J Biol Chem (1998) 273:14484–94. doi: 10.1074/jbc.273.23.14484
33. Karin M, Liu ZG, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol (1997) 9:240–6. doi: 10.1016/s0955-0674(97)80068-3
34. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and Perforin are Important for Regulatory T Cell-Mediated Suppression of Tumor Clearance. Immunity (2007) 27:635–46. doi: 10.1016/j.immuni.2007.08.014
35. Romano M, Fanelli G, Albany CJ, Giganti G, Lombardi G. Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity. Front Immunol (2019) 10:43. doi: 10.3389/fimmu.2019.00043
36. Procaccini C, De Rosa V, Galgani M, Abanni L, Cali G, Porcellini A, et al. An Oscillatory Switch in mTOR Kinase Activity Sets Regulatory T Cell Responsiveness. Immunity (2010) 33:929–41. doi: 10.1016/j.immuni.2010.11.024
37. Gallon L, Traitanon O, Yu Y, Shi B, Leventhal JR, Miller J, et al. Differential Effects of Calcineurin and Mammalian Target of Rapamycin Inhibitors on Alloreactive Th1, Th17, and Regulatory T Cells. Transplantation (2015) 99:1774–84. doi: 10.1097/TP.0000000000000717
38. Efimova OV, Kelley TW. Induction of Granzyme B Expression in T-Cell Receptor/CD28-Stimulated Human Regulatory T Cells Is Suppressed by Inhibitors of the PI3K-mTOR Pathway. BMC Immunol (2009) 10:59. doi: 10.1186/1471-2172-10-59
39. Garber K. Targeting mTOR: Something Old, Something New. J Natl Cancer Inst (2009) 101:288–90. doi: 10.1093/jnci/djp034
40. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. Mtorc1 Couples Immune Signals and Metabolic Programming to Establish T(reg)-Cell Function. Nature (2013) 499:485–90. doi: 10.1038/nature12297
41. Zeiser R, Leveson-Gower DB, Zambricki EA, Kambham N, Beilhack A, Loh J, et al. Differential Impact of Mammalian Target of Rapamycin Inhibition on CD4+CD25+Foxp3+ Regulatory T Cells Compared With Conventional CD4+ T Cells. Blood (2008) 111:453–62. doi: 10.1182/blood-2007-06-094482
42. Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, et al. Hypoxia-Inducible Factor-1 Alpha-Dependent Induction of FoxP3 Drives Regulatory T-Cell Abundance and Function During Inflammatory Hypoxia of the Mucosa. Proc Natl Acad Sci USA (2012) 109:E2784–93. doi: 10.1073/pnas.1202366109
43. Blagih J, Zani F, Chakravarty P, Hennequart M, Pilley S, Hobor S, et al. Cancer-Specific Loss of P53 Leads to a Modulation of Myeloid and T Cell Responses. Cell Rep (2020) 30:481–96.e6. doi: 10.1016/j.celrep.2019.12.028
44. Blagih J, Hennequart M, Zani F. Tissue Nutrient Environments and Their Effect on Regulatory T Cell Biology. Front Immunol (2021) 12:637960. doi: 10.3389/fimmu.2021.637960
45. Yan Z, Garg SK, Kipnis J, Banerjee R. Extracellular Redox Modulation by Regulatory T Cells. Nat Chem Biol (2009) 5:721–3. doi: 10.1038/nchembio.212
46. Ruggenenti P, Perico N, Gotti E, Cravedi P, D’Agati V, Gagliardini E, et al. Sirolimus Versus Cyclosporine Therapy Increases Circulating Regulatory T Cells, But Does Not Protect Renal Transplant Patients Given Alemtuzumab Induction From Chronic Allograft Injury. Transplantation (2007) 84:956–64. doi: 10.1097/01.tp.0000284808.28353.2c
47. Akimova T, Kamath BM, Goebel JW, Meyers KEC, Rand EB, Hawkins A, et al. Differing Effects of Rapamycin or Calcineurin Inhibitor on T-Regulatory Cells in Pediatric Liver and Kidney Transplant Recipients. Am J Transplant (2012) 12:3449–61. doi: 10.1111/j.1600-6143.2012.04269.x
48. Levitsky J, Mathew JM, Abecassis M, Tambur A, Leventhal J, Chandrasekaran D, et al. Systemic Immunoregulatory and Proteogenomic Effects of Tacrolimus to Sirolimus Conversion in Liver Transplant Recipients. Hepatology (2013) 57:239–48. doi: 10.1002/hep.25579
49. Shan J, Feng L, Li Y, Sun G, Chen X, Chen P. The Effects of Rapamycin on Regulatory T Cells: Its Potential Time- Dependent Role in Inducing Transplant Tolerance. Immunol Lett (2014) 162:74–86. doi: 10.1016/j.imlet.2014.07.006
50. Sun I-H, Oh M-H, Zhao L, Patel CH, Arwood ML, Xu W, et al. mTOR Complex 1 Signaling Regulates the Generation and Function of Central and Effector Foxp3(+) Regulatory T Cells. J Immunol (2018) 201:481–92. doi: 10.4049/jimmunol.1701477
51. Jouandin P, Marelja Z, Shih Y-H, Parkhitko AA, Dambowsky M, Asara JM, et al. Lysosomal Cystine Mobilization Shapes the Response of TORC1 and Tissue Growth to Fasting. Science (2022) 375:eabc4203. doi: 10.1126/science.abc4203
52. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional Delineation and Differentiation Dynamics of Human CD4+ T Cells Expressing The FoxP3 Transcription Factor. Immunity (2009) 30:899–911. doi: 10.1016/j.immuni.2009.03.019
53. Newton R, Priyadharshini B, Turka LA. Immunometabolism of Regulatory T Cells. Nat Immunol (2016) 17:618–25. doi: 10.1038/ni.3466
Keywords: granzyme B, mTORC1, rapamycin, grapoptosis, human tregs, treg homeostasis
Citation: Eskandari SK, Allos H, Al Dulaijan BS, Melhem G, Sulkaj I, Alhaddad JB, Saad AJ, Deban C, Chu P, Choi JY, Kollar B, Pomahac B, Riella LV, Berger SP, Sanders JSF, Lieberman J, Li L and Azzi JR (2022) mTORC1 Inhibition Protects Human Regulatory T Cells From Granzyme-B-Induced Apoptosis. Front. Immunol. 13:899975. doi: 10.3389/fimmu.2022.899975
Received: 19 March 2022; Accepted: 05 May 2022;
Published: 10 June 2022.
Edited by:
Anne Maria Pesenacker, University College London, United KingdomReviewed by:
Moanaro Biswas, Indiana University, United StatesCopyright © 2022 Eskandari, Allos, Al Dulaijan, Melhem, Sulkaj, Alhaddad, Saad, Deban, Chu, Choi, Kollar, Pomahac, Riella, Berger, Sanders, Lieberman, Li and Azzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Siawosh K. Eskandari, cy5lc2thbmRhcmlAdW1jZy5ubA==; Jamil R. Azzi, amF6emlAYndoLmhhcnZhcmQuZWR1
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.