 Ana Cheong
Ana Cheong Zachary D. Nagel
Zachary D. Nagel- Department of Environmental Health, Harvard T.H. Chan School of Public Health, Boston, MA, United States
DNA damage constantly threatens genome integrity, and DNA repair deficiency is associated with increased cancer risk. An intuitive and widely accepted explanation for this relationship is that unrepaired DNA damage leads to carcinogenesis due to the accumulation of mutations in somatic cells. But DNA repair also plays key roles in the function of immune cells, and immunodeficiency is an important risk factor for many cancers. Thus, it is possible that emerging links between inter-individual variation in DNA repair capacity and cancer risk are driven, at least in part, by variation in immune function, but this idea is underexplored. In this review we present an overview of the current understanding of the links between cancer risk and both inter-individual variation in DNA repair capacity and inter-individual variation in immune function. We discuss factors that play a role in both types of variability, including age, lifestyle, and environmental exposures. In conclusion, we propose a research paradigm that incorporates functional studies of both genome integrity and the immune system to predict cancer risk and lay the groundwork for personalized prevention.
1 Introduction
Why some individuals are more susceptible to cancer than others remains a fundamental unanswered question in cancer biology. Both immunodeficiency and DNA repair deficiency are associated with elevated cancer risk. The canonical hypothesis regarding DNA repair deficiency is that unrepaired DNA damage leads to increased somatic mutations and malignant transformation of somatic cells. An alternative, underexplored hypothesis is that DNA repair deficiency increases cancer risk, at least in part, by leading to impaired immune cell function. Immunodeficiency is associated with profound defects in some DNA repair pathways, but for some, like nucleotide excision repair, how they contribute to immune function is not yet understood. Furthermore, it remains unclear how inter-individual variation in immune function and DNA repair capacity (DRC) among the general population collectively contribute to cancer risk. We propose that integrating blood-based genome integrity assays and immunophenotyping could afford improved predictions of cancer risk and ultimately open new opportunities for precision prevention and treatment of cancer.
Here we provide an overview of the current understanding of the origins of inter-individual variation in both DNA repair and immune function, and the extent to which they have been shown to contribute to cancer risk. We have structured two sections on inter-individual variation in DNA repair (Section 2) and immune function (Section 3) similarly to underscore the many parallels between two fields that have largely developed independently. We discuss the role of each process in cancer risk, as well as genetic and non-genetic mechanisms contributing to inter-individual variation. After discussing the potential for integrating immunophenotyping and genome integrity assays into cancer risk prediction (Section 4), we highlight emerging technologies that are increasingly making such analyses feasible (Section 5), and close with a list of open questions recommendations for future studies (Section 6) and a brief synopsis (Section 7).
2 Variation in DNA Repair and Its Relationship to Cancer Risk and Carcinogenesis
2.1 DNA Repair Protects Against Cancer
Genome integrity is constantly threatened by endogenous and environmental DNA damaging agents. These agents include reactive oxygen species (ROS) generated by normal cellular metabolism, errors in DNA replication, ultraviolet (UV) light, ionizing radiation, and mutagenic chemicals (1). While unrepaired DNA damage can lead to disease by promoting cell death, transcriptional stress, senescence, and mutations (2), DNA repair limits these processes by maintaining genome integrity. Depending on the agent, DNA can be damaged in numerous ways. The types of DNA damage include base damage, single strand breaks, inter- and intra-strand crosslinks, bulky adducts, methylated DNA adducts, mismatches, and double-strand DNA breaks (DSBs). Complexes of DNA repair proteins form DNA repair machines that specialize in the removal of particular types of DNA damage, and defects in one or more of the DNA repair pathways increase the frequency of specific types of mutations in the genome (3) (Figure 1). As DNA damage and repair have been extensively reviewed elsewhere (1, 4, 5), we will not cover the detailed mechanisms here.
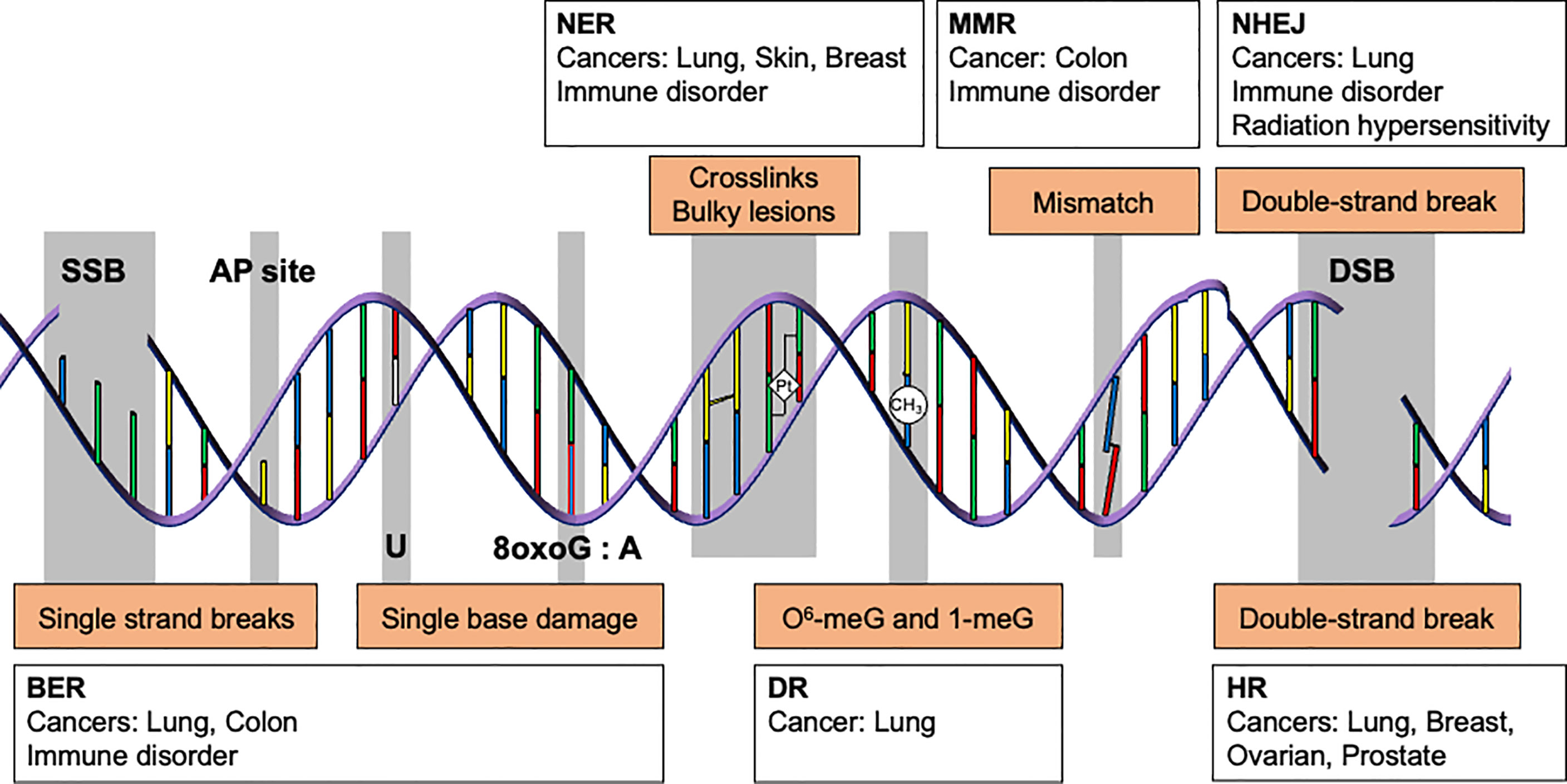
Figure 1 DNA repair pathways and their association with cancer and immune disorders. Genome integrity is maintained by multiple DNA repair pathways. Depending on the type of DNA damage, specific subsets of DNA repair proteins recognize and repair the damage. For instance, single strand breaks, abasic sites, and single base lesions are primarily repaired by base excision repair (BER). Some types of alkylation damage, such as O6-methylguanine and 1-methylguanine, are repaired by direct reversal (DR). Intra-strand crosslinks and bulky lesions are repaired by nucleotide excision repair (NER). Mismatched bases are repaired by mismatch repair (MMR), whereas double strand breaks are resolved by homologous recombination (HR) or non-homologous end joining (NHEJ). Unrepaired DNA lesions may give rise to somatic mutations and cancer. Deficiency in BER, NER, MMR, and NHEJ is also associated with immunodeficiency, which increases cancer risk(s).
2.2 Defects in DNA Repair Are Linked to Cancer-Prone Genetic Disorders
Genome instability is a hallmark of cancer, and nearly all cancers are caused by one or more somatic mutations induced by DNA replication in the presence of DNA damage (6, 7). As our understanding of the etiology of cancer mutation signatures advances rapidly, it is becoming evident that genomic alterations in individual cancers can often be attributed to specific DNA damaging agents and DNA repair defects (3, 8–11). Historically, much has been learned from constitutional DNA repair deficiency syndromes that are associated with elevated cancer risk in humans. Below we highlight examples for several DNA repair pathways. In subsequent sections, we discuss variability in DNA repair in the general population, which is emerging as a potential predictor for cancer risk (12, 13) (14) (15).
Nucleotide excision repair defects in xeroderma pigmentosum (XP) patients are associated with an extremely high risk of skin cancers due to the inability to repair UV-induced DNA damage (16). Early studies revealed seven complementation groups that correspond to the DNA repair genes XPA, XPB, XPC, XPD, XPE, XPF, and XPG. Deficiency in the translesion DNA polymerase eta (aka POLH or XPV) also causes XP in humans (17) (18). Cells from these individuals exhibit normal NER, but are deficient for accurate replicative bypass of unrepaired UV-induced DNA damage, resulting in an increased rate of UV-induced mutagenesis. In the case of combined XP and Cockayne syndrome (XP-CS), mutations in XPB, XPF, XPD, or XPG have been detected among patients. These patients display a mild XP phenotype. Yet, despite the universality of DNA repair deficiency, skin cancers are rare except in those with mutations in XPB or XPD (19). XP-CS patients with mutations in XPG are photosensitive and have skin freckling, but skin cancers are rare. This may be in part due to very early mortality, but the severe photosensitivity phenotype that commonly accompanies XPG-CS also leads to early diagnosis and better sun protection for these patients.
Numerous diseases are associated with defects in double strand break repair. Fanconi Anemia is caused by mutations in a group of genes involved in both DSB repair and the repair of DNA inter-strand cross-links (20). Patients commonly experience immunodeficiency due to bone marrow failure and are at increased risk of acute myeloid leukemia (21). Mutation in Werner syndrome protein (WRN) predisposes to cancer. WRN is a RecQ family DNA helicase with well-established roles in both non-homologous end joining (NHEJ) and homologous recombination (HR), as well as emerging roles in base excision repair (BER) and nucleotide excision repair (NER) (22). Patients with Werner syndrome display premature aging, and have higher risks of cancer and cardiovascular disease (23–26). WRN patients develop thyroid epithelial neoplasms, melanoma, and soft tissue sarcomas, as well as leukemia and primary bone neoplasms (27). RECQL4 is involved in NHEJ (28, 29), HR (30), NER, and BER (31), and its mutation is known to induce trisomy, aneuploidy, and chromosomal rearrangements. RECQL4 deficiency is associated with several diseases, including Rothmund-Thomson syndrome (RTS), RAPADILINO syndrome, and Baller-Gerold syndrome (BGS) (32). Patients with RTS or RAPADILINO have higher risk for osteosarcoma and lymphoma (33, 34). LIG4 syndrome is caused by deficiency in Ligase IV, which is essential for NHEJ (35). Patients with LIG4 syndrome exhibit severe combined immunodeficiency due to the role of NHEJ in V(D)J recombination, a key process for antibody diversification (36). Ataxia telangiectasia (A-T) is a DNA damage response disorder caused by mutations in the Ataxia telangiectasia mutated (ATM) gene. Among other symptoms, patients with A-T experience immunodeficiency and are at increased risk for cancer, particularly lymphoid malignancies (37).
Several diseases are associated with defects in base excision repair and single strand break repair (38) (39). MutY DNA glycosylase (MUTYH) -associated polyposis (MAP) arises from germline mutation of MUTYH. Characterized mainly by the biallelic germline mutations of Y165C or G382D in MUTYH, MAP is associated with colorectal adenomas and carcinomas (40, 41). As a BER protein, MUTYH functions to remove adenine opposite 8-oxo-7,8-dihydroguanine (OG), which is left unrepaired by 8-oxoguanine DNA glycosylase (OGG1), and thereby prevent G:C to T:A transversion mutations (42). Some MUTYH variants are associated with diminished OG:A repair (43), leading to higher colorectal cancer (CRC) risk (44) (45). Defects in uracil DNA glycosylase (UNG) result in an extreme immunodeficiency known as Hyper-IgM syndrome due to the central role of this enzyme in antibody diversification (46). While UNG deficiency is too rare to allow reliable estimates of its consequences for cancer risk, in general Hyper-IgM patients suffer from higher rates of malignancy (47). Similarly, deficiency in the Nth like DNA glycosylase 1 (NTHL1) is associated with a tumor syndrome that is dominated by colorectal cancer but includes several other malignancies (48–50).
Constitutional mismatch repair deficiency is an extremely rare disease that is associated with increased risk of a wide range of malignancies (51). Lynch syndrome is another DNA repair deficiency syndrome associated with cancer. It arises from the presence of one or more mismatch repair (MMR) gene mutations (52). While the normal tissues in Lynch syndrome patients often do not exhibit detectable MMR defects, Lynch syndrome is associated with MMR-deficient cancers with high microsatellite instability (MSI) (53).
2.3 Factors That Contribute to Variation in DRC
While much has been learned from diseases associated with DNA repair deficiency, they are relatively rare and represent the extremes of inefficient DNA repair in human populations. In the general population, DNA repair gene polymorphisms, age, environmental exposures, and lifestyle are several major factors thought to give rise to inter-individual variation (12). Variation in DRC is a consequence of the collective influence of these factors.
2.3.1 Genetics
A large number of polymorphisms have been identified in DNA repair genes, and their associations with cancer imply functional consequences. While relatively few studies have investigated functional significance directly, accumulating research supports genetic variation as an important driver of inter-individual variation in DRC (Table 1). For example, variant alleles of X-Ray repair cross complementing 3 (XRCC3) are associated with higher levels of bulky DNA adducts (59). XRCC1 variants may be associated with either higher or lower BER repair activities (60, 62–64). XPD polymorphisms decrease XPD expression, with the most pronounced effect seen in older individuals (55). Some OGG1 variants are associated with higher percentage tail DNA measured using comet assays (% tail DNA). Variant genotypes of BER and NER genes have also been associated with a wide variety of markers of genome instability. These include micronuclei and baseline %TD (58, 61, 66), chromosome breaks (62), sister chromatid exchanges (56, 60, 61), deletions and dicentric chromosomes (56), DNA adduct levels (59), overall BER repair activities (65), repair of radiation-induced damage (54) (58) (56), and repair of oxidative damage (54) (43), with cumulative effects for individuals with variant alleles in multiple DNA repair genes (57). While genetic determinants of DRC might be presumed exert similar effects on all tissues, this may not be true in light of evidence from animal models indicating tissue-dependent allele specific expression (67).
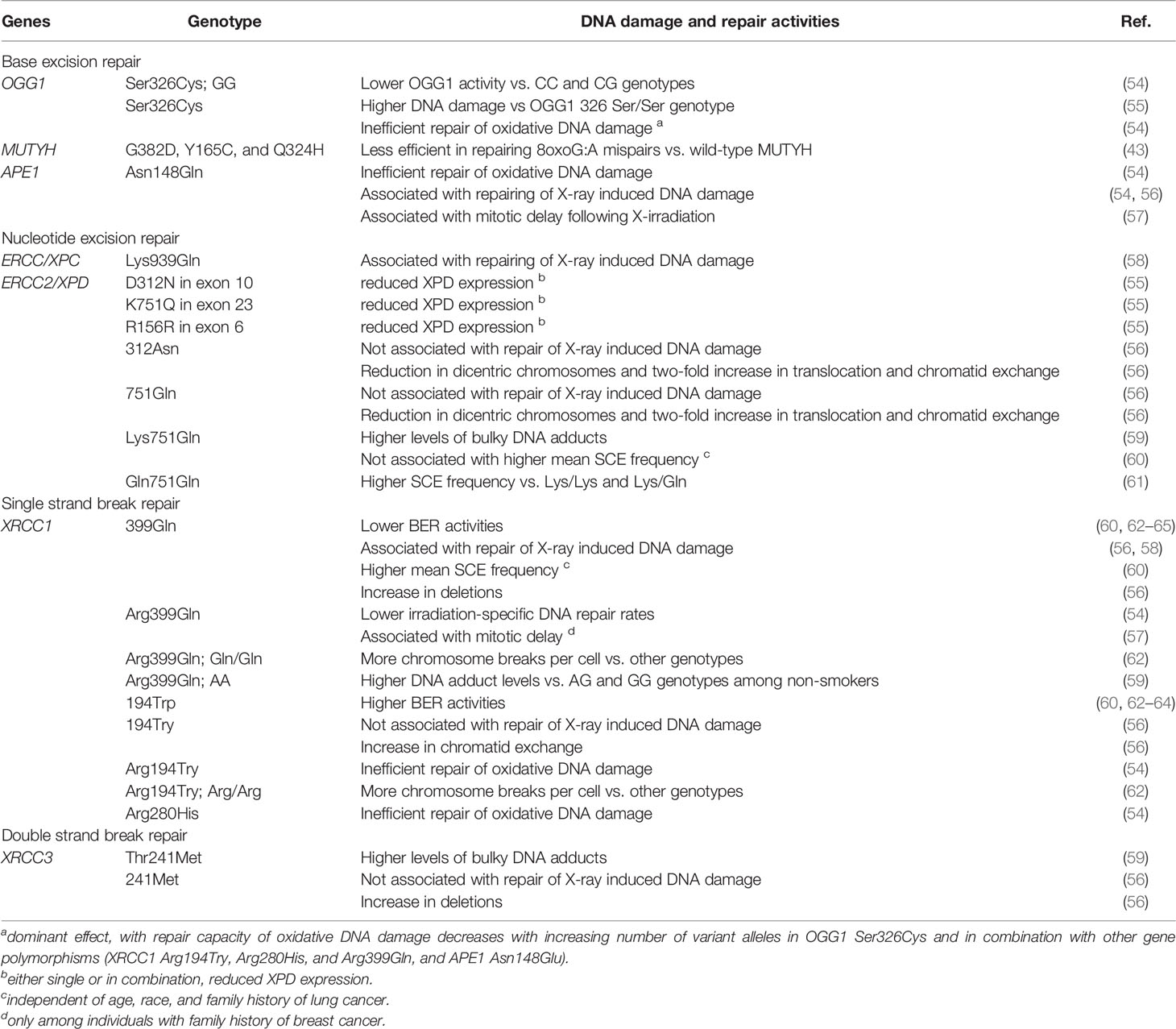
Table 1 Polymorphism in DNA repair genes and their association with genome integrity.
2.3.2 Aging
An age-dependent decline in DRC and DNA damage accumulation has been proposed as a key mechanism underlying aging (68), and ongoing studies are beginning to uncover interventions that may mitigate the effects of compromised genome integrity in older individuals (69). The presence of age-dependent changes and the potential for interventions that may reverse them underline the likely role for age in inter-individual variation in DRC. Here we highlight studies testing this idea directly in human populations.
Assays that measure the accumulation of DNA damage provide indirect evidence for age-dependent changes in DRC. Peripheral blood mononuclear cells (PBMCs) isolated from older individuals have a higher frequency of dicentric and ring chromosomes (70) and a higher degree of negative supercoiling (71). Levels of single-strand breaks (SSBs) and oxidized bases (detected as formamidopyrimidine DNA glycosylase (FPG)-sensitive sites) in PBMCs are lower in younger individuals (age <65 years) when compared with older individuals (age >65 years) (72), although basal levels of SSBs and alkali sensitive sites in lymphocytes were age-independent in a separate study (73).
Direct measurements of DNA repair provide further insights into age-dependent changes in genome integrity. A study that used neutral comet assays to measure double strand break (DSB) repair and fluorometric analysis of DNA unwinding (FADU) to measure SSB repair in X-irradiated lymphocytes found diminished DSB repair in older individuals (74). Another study found that while overall rates of repair were similar, a subpopulation of repair deficient lymphocytes was significantly more abundant in older individuals (73, 74). Higher levels of DNA damage might intuitively be interpreted to reflect inefficient DNA repair, but the situation may be more complex. For example, one study found that the level of SSBs correlated positively with OGG1 activity (72), which was higher in older individuals. The higher levels of SSBs may thus reflect the accumulation of unresolved repair intermediates downstream of BER initiation, and phenomenon that has been termed BER imbalance (75–79). Elevated OGG1 activity in lymphocytes from older individuals has been observed in additional studies (72, 80). Nevertheless, in another study OGG1 repair activity in lymphocytes was reported to decrease with age (81). The decrease in OGG1 activity was more pronounced among individuals with Cys/Cys, Ser/Cys, than with Ser/Ser genotypes at position 326, suggesting that study design and the genetic makeup of cohorts may at least partially explain the differences among studies. By contrast with OGG1, AP site incision capacity is not associated with age (82).
Evidence for age-dependent changes in DRC have also come from studies wherein cells have been challenged with DNA damaging agents. Repair replication declines in lymphocytes irradiated with ultra-violet light (UV), with the rate of UV-induced decrease in DRC estimated to be about 30% from 20 to 90 years (83). By contrast, repair replication in UV-irradiated keratinocytes is comparable between infant and older adults, suggesting that age effects may be heterogeneous across human tissues (84). Similar to the decline in repair of UV-induced damage, rejoining of chromosomes following X-irradiation decreases with age in human leukocytes (85). Consistent with higher rates of BER initiation following oxidative DNA damage, a study that compared individuals in three groups based on age of 35-39 years (Group 1), 50-54 years (Group 2), and 65-69 years (Group 3) using an ELISA assay in PBMCs following challenge with hydrogen peroxide revealed significantly higher levels of single stranded DNA in Group 3, but not Group 2, when compared to Group 1 (86). This finding is consistent with a second study that made use of comet assays (80),, as well as those in the previous section finding elevated OGG1 activity in older individuals. A rare in vivo study in which the epidermis of subjects was subjected to UV-irradiation followed by skin biopsies found that the efficiency in removing irradiation-induced cyclobutane pyrimidine dimers (CPD) is lower in older subjects, consistent with ex vivo studies in cultured primary cells (87).
Host cell reactivation assays have provided important insights into age-dependent changes in DRC. For instance, in one study skin fibroblasts from younger donors had higher efficiency in repairing UV-irradiated plasmids than those from the older donors (88). However, the same study found no relationship between age and the removal of genomic UV-induced adducts, and a second study found the repair UV-induced induced plasmid lesions was similar in skin fibroblasts from donors of age 21-96 years (89). The differences between the HCR studies in fibroblasts might reflect the relatively small samples sizes (N=8-10), which limit statistical power; a somewhat larger study (N=20) using host cell reactivation assays in fibroblasts did find an age-dependent decrease in DRC (90). In lymphocytes, repair of UV irradiated plasmids decreases with age, with an estimated average 0.61% decrease per year between 20 and 60 years of age (91). These results were consistent with a second study using HCR in lymphocytes that found an age-dependent decline in repair of UV-induced damage, which was notably absent among basal cell carcinoma cases, for whom DRC was lower than in controls at younger age (92). Another study that stands out for its analysis of pathways other than NER using host cell reactivation assays in primary skin fibroblasts indicated that both NHEJ and HR decline strikingly with age (93). Taken together, the findings suggest that age-dependent changes in DRC may depend in a complex manner on the cell type, DNA repair pathway, and the health status of the study participants.
Age-associated changes in DRC may be explained in part by the differential expression of DNA repair genes. The expression levels of excision repair cross-complementing group 1 (ERCC1) (94, 95), XPA (94), XPF (95), XRCC4, ligase 4 (LIG4), LIG3 (93), DNA polymerase delta 1 (POLD1) (88, 96), POLE, replication factor C (RFC) (88), and replication protein A (RPA) (94) decrease with age. On the contrary, the expression levels of CSA and XPG seemed to increase with age, but the change could not be confirmed by qPCR (88). There is no difference in the expression levels of proliferating cell nuclear antigen (PCNA) (88), NHEJ factors DNA PKcs, artemis, XRCC4-like factor (XLF) (93),, Ku70 and Ku80 (93, 97) and HR factors breast cancer associated gene 2 (BRCA2), meiotic recombination 11 (MRE11), RAD51, Nijmegen breakage syndrome 1 (NBS1), and RAD51 (93) among different age groups. While these studies were performed in either human PBMCs and primary fibroblasts, whether these changes in the expression of DNA repair factors resemble those in other tissues from the same individual have not been studied.
Though a detailed review of animal models is beyond the scope of this article, we note that they recapitulate several aspects of human aging biology with respect to genome integrity, including age-dependent increases in DNA damage levels (98) (99), accumulation of mutations (100, 101), and dysregulation of DNA repair (102) (103) (104) (105) (106).
2.3.3 Environmental Factors
Mounting evidence indicates that environmental exposures can alter DRC. Here we focus on two well-established examples, namely arsenic and smoking. Like tobacco smoke, arsenic is an environmental agent classified as carcinogenic to humans by the International Agency for Research on Cancer (107), and causes cancer at least in part by directly inducing DNA damage (108, 109). Exposure to arsenic is associated with chromosome aberrations in human PBMCs (110) and DNA damage (111, 112). Children with in utero exposure to arsenic have higher salivary 8-hydroxydeoxyguanosine (8-OHdG), a biomarker of DNA damage caused by oxidative stress, than their unexposed counterparts (113) (114). Consistent with a key role for DNA damage in the etiology of arsenic associated malignancies, arsenic exposure is associated with a distinct mutational signature (115). Furthermore, individuals with lower DRC and those with select polymorphisms in DNA repair genes are more susceptible to arsenic induced skin lesions (109) (108) (116, 117) (110) (118).
Population studies provide extensive evidence in support of the concept that arsenic exposure leads to alterations in DNA repair. Arsenic exposure is associated with decreased expression of MutS homolog 2 (MSH2) and mutL homolog 1 (MLH1), though not PMS1 homolog 2 (PMS2) (119). Urinary arsenic concentrations are positively associated with MLH1 promoter methylation, which is consistent with an epigenetic mechanism of arsenic-induced dysregulation of MMR (120). Arsenic exposure also leads overexpression of excision repair cross complementation group 2 (ERCC2/XPD) and less efficient NER (121). ERCC1 expression may be influenced by arsenic exposure (122) (111), but there appear to be complex dependencies on dose, duration, and speciation of arsenic exposure (122), as well as the age of the exposed population (94, 95). Diminished expression of XPF and XPB, but increased in XPG expression have been associated with arsenic exposure (123). At the functional level, repair of DNA damage induced by hydrogen peroxide, ionizing radiation and 2-acetylaminofluorene (2-AAF) is impaired in arsenic-exposed individuals relative to unexposed controls (112) (117) (111). These population studies are broadly consistent with in vitro studies indicating that arsenic exposure synergizes with the DNA damaging effect of UV (124, 125) and inhibits repair of DNA damage induced by a variety of agents (124) (126) (127). Collectively, these findings indicate that, in addition to the direct induction of DNA damage, arsenic exposure likely sensitizes cells to the DNA damaging effects of other mutagenic agents.
Exposure to environmental tobacco smoke (ETS), also known as passive smoking, compromises genomic stability. Passive smokers have higher levels of several types of DNA damage than unexposed individuals (128). They also excrete higher levels of 5-hydroxymethyluracil (129), which is not directly induced by tobacco smoke but may result from ETS-induced oxidative stress. Though passive smoking has not been correlated with levels of 8-OHdG in serum or leukocyte DNA (128, 130), lymphocytes from passive smokers have a longer comet tail length, more Fpg-sensitive sites, and are slower in repairing H2O2-induced DNA damage (131). Furthermore, buccal epithelial cells of passive smokers have higher micronuclei frequency when compared to non-smokers (132). Interestingly, allele variants and expression levels of several DNA repair genes have been associated with lung cancer risk and genome instability among never-smokers, including XRCC1 (132), OGG1, XPD (133), and AGT (134). A study using nasal epithelial cells further revealed that NER was among the top 6 pathways with altered gene expression in association with third hand smoking (135). While these data underscore the potential role of environmental exposures in modulating DRC, the mechanism by which passive smoking affects the activity of specific DNA repair pathways is incompletely understood.
2.3.4 Circadian Rhythm, Lifestyle, and Dietary Factors
Lifestyle factors have been shown to influence DRC. One of the most studied factors is circadian rhythm, which has been reviewed extensively (136–138) (139). It has recently been shown directly that individuals subjected to a night shift schedule exhibit diminished DRC (140). Diet can also affect the efficiency of DNA repair (141). Mounting evidence indicates that calorie restriction is associated with changes in DNA damage and repair (142). While these phenomena await more detailed study in human populations, animal models provide substantial support for the influence of diet on DNA repair. In mice, calorie restriction enhances NHEJ (143), and increases the fidelity of DNA polymerase and DNA excision repair in the liver (144). It also reverses the age-related decline in BER in brain, liver, spleen, and testis, and lowers their mutation frequency (145). In rat hepatocytes, caloric restriction altered the induction and repair of DNA damage in a manner that depended on age (146). Findings from an Ercc1Δ/- mouse model of premature aging further show that dietary restriction from 10% to 30% could preserve genome integrity, mitigate premature-aging associated decline in gene transcription, and prolong their lifespan (147). This supports the hypothesis that dietary restriction may attenuate the aging process. Similarly, chronic supplementation of melatonin reduces DNA damage by upregulating APE and OGG1 (148). The underlying mechanism and whether additional DNA repair pathways are affected require further investigation. Overall, the findings in humans and animal models support a role for lifestyle and circadian rhythm in DNA repair, adding a layer of complexity to the origins of inter-individual differences.
3 Variation in Immune Function and Its Relationship to Cancer Risk and Carcinogenesis
The immune system defends against both infection and malignancy. Based on the response time, mode of initiation, and the cell types involved, there are two immune subsystems. The innate immune system is activated rapidly upon recognition of pathogenic antigens and stress signals. It is, in part, comprised of dendritic cells (DC), monocytes, macrophages, granulocytes, and natural killer cells (NK). These cells phagocytose pathogens and activate inflammation signaling pathways and the complement cascade. The adaptive immune system, on the contrary, is more flexible in recognizing antigens. Its cellular components, including T lymphocytes (T cells) and B lymphocytes (B cells) can undergo mutagenesis to create novel and specific antigen receptors. T cells can be further subdivided into naïve T cells that recirculate between blood and lymph nodes to scout for specific antigens and memory T cells that are long lived and can mount a response to previously encountered immunogenic stimuli. Cytotoxic T cells (or CD8+ T cells) secrete granzymes to induce apoptosis in target cells and pore-forming perforin to punch holes in the target cell membrane for granzymatic actions. T helper cells (or CD4+ T helper cells) secrete cytokines to activate macrophages and further activate cytotoxic T cells. B cells express membrane-bound and secretory antigen-specific immunoglobulins (or antibodies) to defend against pathogens. Like NK cells, they are also involved in the activation of CD4+ T cells (149). Thus, immune response to foreign antigens depends on the specific functions of and interplay between the two immune subsystems that are comprised of a wide variety of immune cells.
Due to the presence of neoantigens that arise from genome instability and can be presented on the cell surface, cancer cells can be immunogenic. They can accordingly be recognized and eliminated by immune cells in the process of immune-surveillance (2). However, cancer cells are capable of escaping surveillance by altering antigen expression and hijacking the immune system to favor tumor growth. Through cytokine secretion, they can induce the differentiation of myeloid suppressor cells, which are inflammatory monocytes capable of inhibiting the activities of cytotoxic T and NK cells, as well as DC maturation (150). Moreover, as innate immune cells, including macrophages and neutrophils, infiltrate into tumors through chemotaxis, they can be polarized towards a pro-tumor phenotype and increase the secretion of proinflammatory cytokines to support, rather than suppress, tumor growth (151).
Current cancer immunotherapies that leverage the cytotoxicity of immune cells have proven efficacy in suppressing tumor growth. For example, NK and NKT cell populations expanded and activated in vitro have demonstrated potent cytotoxicity against liver cancer (152). T cells engineered with chimeric antigen receptors (CAR-T) are highly effective in targeting CD19-expressing tumors (153). DC vaccines that capitalize on the cytotoxicity of monocyte-derived DCs induce a tumor-specific immune response, although the effects differ by vaccination route and do not correlate with overall survival in phase I/II clinical trials (154). To date, immune checkpoint blockade therapies that target the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed death 1 (PD-1) and its ligand PD-L1 have demonstrated improved responses and better overall survival for multiple cancers (155). Pembroluzuimab, which is an anti-PD-1 antibody, has been approved by the Food and Drug Administration to treat patients with metastatic melanoma. Another anti-PD-1 antibody, Nivolumab, has also been approved to treat patients with metastatic melanoma and patients who are previously treated for advanced or metastatic non-small cell lung cancer. These emerging therapeutic strategies form the basis for numerous ongoing clinical trials (156). For the purpose of this review, we highlight them as evidence in support of immune control of cancers.
3.1 Defects in Immune Function Are Linked to Cancer
Impaired immune function has been linked to increased cancer risk. By analogy to genetic diseases of DNA repair deficiency, patients with impaired immune function have provided insights on the role of the immune system in cancer. Numerous primary immunodeficiency disorders are associated with increased risk of malignancy (157) (158) (159) (160). Notably, since the DNA repair machinery plays integral roles in multiple aspects of immune function, some immunodeficiency disorders are caused by genetic defects in DNA repair as outlined in Section 2.2. In the general population, individuals with low cytotoxic activity of peripheral blood lymphocytes have higher risk of cancer (161). Immunosuppression due to organ transplantation and some viral infections are likewise factors that impair the immune response. Patients receiving immunosuppressants to prevent organ rejection have higher risk for non-melanoma skin cancer (162). This may explain why transplant recipients are generally more likely to develop cancer than those without organ transplantation (163–166). Cancers in transplant patients are also more aggressive and are associated with poor overall survival (167–169). Viral infection can suppress the immune system and increase cancer risk. Human immunodeficiency virus (HIV) -infected patients develop more aggressive cancer (164) and have higher risk for Kaposi’s sarcoma, B-cell non-Hodgkin’s lymphoma, and multiple myeloma (170).
Despite the strong evidence in support of a role for the immune system in controlling cancers, there are notable exceptions. Individuals with severe combined immunodeficiency due to loss of LIG4 function and those with dendritic cell deficiency tend to be susceptible to hematologic malignancies, but are not notably predisposed to solid malignancies (160, 171). Similarly, immunodeficient mice do not necessarily develop cancer. NOD scid gamma (NSG) mice have a relatively low risk of developing tumors over a life-span of about 89 weeks (172), and nude mice do not frequently develop spontaneous tumors (173), despite being highly susceptible to infection (174, 175).
3.2 Factors That Contribute to Variation in Immune Function
Inter-individual variation in immune function has been postulated as a driver of variations in cancer susceptibility. While age appears to be the most prominent intrinsic driving factor for variation in immune function, environmental exposures can also have a significant impact. Genetic variation associated with autoimmunity, inflammatory disease, and susceptibility to infection, is estimated to explain to 20-40% of the immune variation (176), leaving the remainder to be explained by other mechanisms. In this section, we review how immune function can be affected by heritable factors, and describe how environmental exposures may further explain inter-individual variation in the immune response across populations.
3.2.1 Genetics
Reminiscent of the situation for DRC, significant inter-individual differences in immune function have been reported. In a recent detailed repeated measures study, inter-individual variation in immune cell composition and plasma cytokine levels revealed that differences between individuals are generally larger than longitudinal variability within person (177). Plasma levels of the chemokine CC chemokine ligand 20 (CCL20) are negatively associated with the proportion of central memory and effector memory cells in CD4+ and CD8+ T cell lineages, and individuals with extremely high counts of these immune subsets are found to have low levels of plasma CCL20 and CCL22. Plasma levels of IL-16 are also negatively associated with the proportion of central memory T cells in CD4+ and CD8+ lineages, and CD56dim NK cells. Overall, plasma levels of 21 proteins accounted for nearly 80% of the variation in the abundance of central memory T cells. In a separate study, the abundance of CD8+CD45RO+ memory T cells and CD3+CD56+ NKT cells was found to vary significantly between individuals in repeated measures taken from 25 individuals over at least a three-week interval, but levels were largely stable within-person (178) Of note, differences in immunophenotype have been associated with age, sex, body mass index, and race (177, 179–183). Environmental exposure, vaccination (184, 185), and infection (186–189) can furthermore lead to within-person variation. Nevertheless, the observation that the immune cell composition and cytokine levels of an individual are relatively stable throughout a year (177) suggests that some variability may be determined by genetics or processes that occur during development.
Several lines of evidence support a role for genetics in human variation with respect to immune function. Although studies in monozygotic and dizygotic twins indicate that immune responses are dominated by non-heritable factors, numerous parameters including serum proteins and immune cell population composition are heritable (190). Single nucleotide polymorphisms (SNPs) in the IL-12B gene, which codes for IL-12p40, are associated with immune-related diseases such as psoriasis (191) and asthma (192). Eight SNPs have also been identified to be associated with IL-10 levels (181). Furthermore, studies in twins indicate that ex vivo lipopolysaccharide (LPS)-induced IL-1β production, as a measure of innate immunity, is heritable (180). This suggests that varying levels of LPS-induced secretion of tumor necrosis factor alpha (TNFα), which ranges widely from 0.187 to 2.714 ng/ml in healthy blood donors may be explained at least in part by genetics (193). In further support of genetic variation as a driver of differences in immune responses, a functional study using toll-like receptor (TLR) ligand-stimulated cord blood mononuclear cells has detailed the association between cytokine production and SNPs in innate immune genes (182). Taken together, the available data support a role for genetics in inter-individual variation in immune function in the general population.
3.2.2 Aging
It has long been appreciated that the immune system undergoes age-related changes, which are collectively referred to as immunosenescence and notably include the accumulation of DNA damage in immune cells (194). Although age-dependent changes in immune cell function have been reported in bone marrow (187), bronchoalveolar lavage (179), and thymus (195), this review will focus on PBMCs because they are most immediately amenable to population studies. Several studies have found age-dependent changes in total leukocyte counts (196) or the composition of leukocyte subtypes (179, 197, 198) (199) (183, 198) (Table 2).
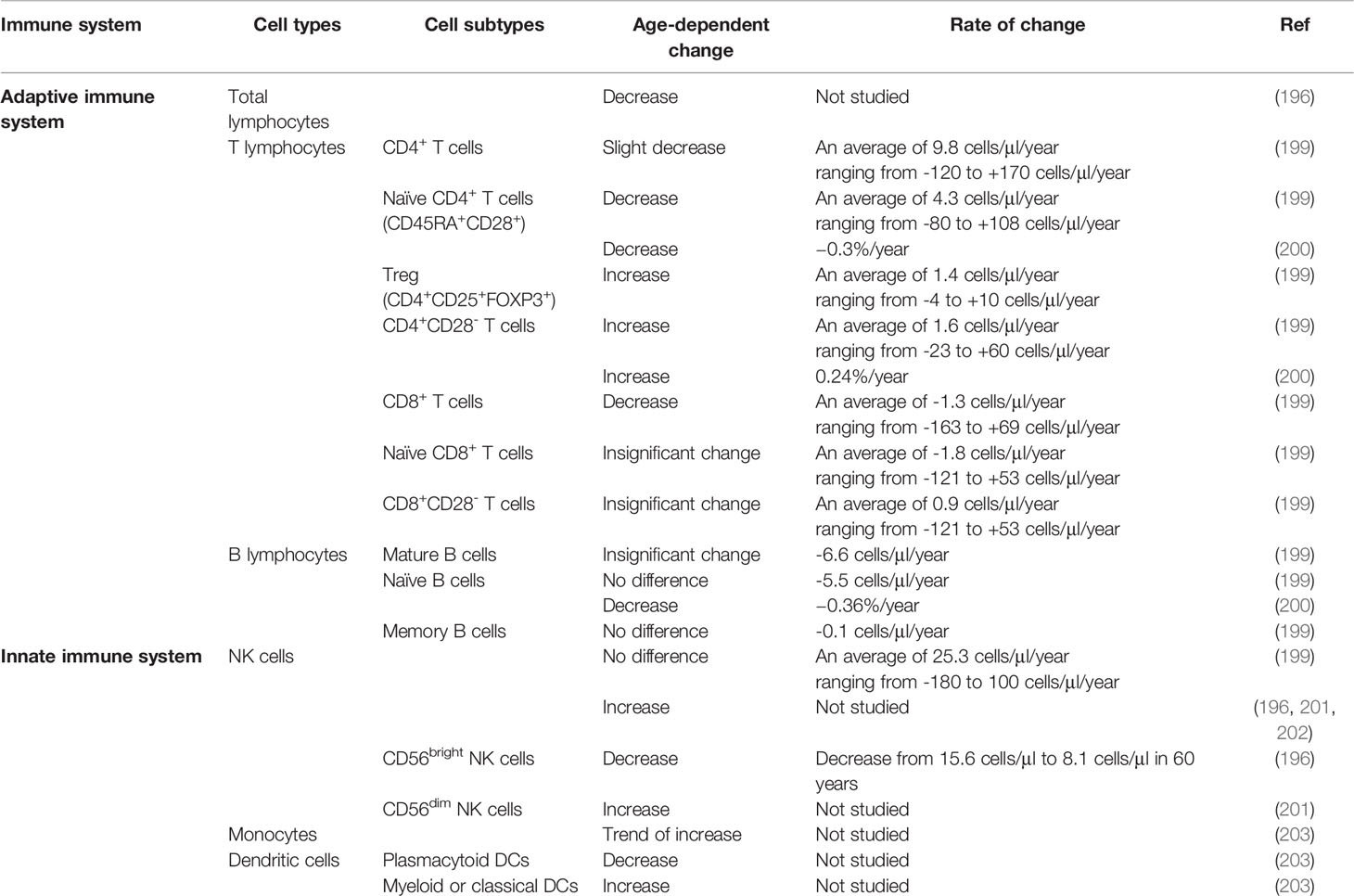
Table 2 Age-dependent changes in the population of immune cell subtypes.
Age-related changes in adaptive immune cells have been noted. A major study involving 177 individuals, who were sampled every six months for three years, has identified an age-dependent decrease in CD4+ and CD8+ recent thymic emigrant T cells and transitional B cells (183). This decrease coincides with the reduction in thymus and bone marrow activity and an increase in the inflammatory population and CD8+ T cells. In particular, the proportion of CD4+ T cells decreases with age (198, 199) whereas that of CD4+ NKT cells increases with age [(198); Table 2]. Based on the expression of CD27 and CD28, T cells can be further subdivided into naïve and early-differentiated cells (CD27+CD28+) and fully differentiated (CD27-CD28-) CD4+ and CD8+ T cells (204, 205). A younger cohort had a significantly larger CD27+CD28+ subpopulation when compared to an older cohort (206). Similarly, based on the expression of a leukocyte common antigen isoform, CD45RA, and chemokine receptor CCR7, T cells can be subdivided into naïve (CD45RA+CCR7+), central memory (CD45RA-CCR7+), effector memory (CD45RA-CCR7-), and terminally differentiated effector memory (CD45RA+CCR7-) cells (207). Within the CD8+ subset, older individuals had fewer CD45RA+ naïve T cells and more central memory and terminally differentiated effector cells when compared to younger individuals (208). In contrast, within the CD4+ subset, a decrease in the CD27+CD28+ cells was the only difference observed in older individuals. These findings imply that the naïve T cells shift towards a more terminally differentiated subpopulation upon aging. This may limit the plasticity of the naïve T cells to differentiate and respond to novel antigens. The concomitant loss of the central memory and terminally differentiated CD8+ T subsets suggests that regardless of the activation by CD4+ T cells, the cytotoxic T cell response is compromised in the elderly.
Age-dependent, subset-specific changes in innate immune cell counts have been documented. The proportion of NK cells increases with age (196, 199, 201, 202) (Table 2) Based on the expression level of the pathogen recognition receptor CD56 (209), NK cells can be further divided into CD56bright, which resides in the lymph node (210) and are immunoregulators due to their cytokine production capacity (211), and CD56dim NK cells, which have cytotoxic potential (212). The CD56bright subset is less abundant in cord blood when compared to adult blood (202), and decreases further with age (196). By contrast, the CD56dim subset increases with age (201, 203). Similarly, the proportion of monocytes increases with age (203). This monocyte population includes the classical, transitional, and CD14+CD16+ non-classical subsets. Among the non-monocytes, the proportion of myeloid-derived DCs increases with age, whereas that of plasmacytoid DCs decreases with age. In view of the age-dependent changes in immune cell composition, associating the cell count and their functions will help map the landscape of immunophenotype throughout life. Integrating this information may help identify phenotypic and functional biomarkers for immunosenescence, treatment response, or higher susceptibility to diseases including infections and cancers.
Molecular markers of immune function, including cytokine production and response to antigenic stimuli also change with age. In particular, the production of cytokines interferon gamma (IFN-γ), interleukin (IL) -4 (IL-4) and IL-6 has been shown to increase with age whereas that of IL-2, IL-10, and TNF-α decreases with age (97, 180, 183, 213). Since the cytokine production capacity of CD4+ T cells is invariant with age, changes in T cell subtype composition are proposed to explain age-related changes in function (206). Interestingly, expression levels of IL-7 are lower in nonagenarians than middle aged individuals (214). Genes in the IL7R gene network are also differentially expressed between the age groups. The fact that higher IL-7R expression level is associated with better prospective survival suggests a role for cytokines and immune response in longevity.
The ability of T cells to respond to mitogenic stimuli is also affected by age. Aging attenuates the proliferation of PBMCs induced by stimuli including phytohemagglutinin (PHA), concanavalin A, pokeweed mitogen, and anti-CD3 (aCD3) or anti-CD28 (aCD28) monoclonal antibodies either alone or in combination (97, 197, 213). In particular, CD4+ T cells from elderly individuals have a lower proliferative response to staphylococcal enterotoxin B (206). Activated T cells also have lower induction of nuclear factor kappa B (NFkB) in response to anti-CD3, phorbol myristate acetate (PMA), and TNFα (215). Notably, treatment with phorbol dibutyrate and calcium ionophore A23187 induces higher nuclear translocation of NFkB in neonatal than adult T cells, though the composition of NFkB is similar between the two groups (216). Collectively, these results imply that T cells from older individuals are less sensitive to stimuli.
Similar to T cells, NK cells isolated from elderly individuals have diminished proliferation activity and CD69 induction following treatment with IL-2 when compared to the younger group (201). The response of CD8+ CD45RO+ memory T cells and CD3+CD56+ NKT cells to IL-23 also decreases with increasing age. T cell receptor repertoire diversity decreases and clonality increases with age (217). Taken together, the findings support the notion that age-dependent decrease in immune cell function, based on proliferation and cytokine production induced by antigenic stimuli, and cytotoxicity, has an impact on cancer risk. How age-dependent changes in immune function modify cancer risk warrants further investigation.
3.2.3 The Environment
Exposure to environmental agents can have major effects on the immune system (218). Given that the exposure effects have been well documented, to underscore parallels with environmental agents that affect DNA repair, we will focus on how the immune system is impacted by the same cancer-causing agents (arsenic and smoking) that were discussed in Section 2.3.3. We will review how environmental exposure may contribute to inter-individual variation in immune function. As with DNA damaging agents, extensive experimentation has been carried out in vitro and with animal models to understand the biological mechanisms underlying the immune effects of environmental exposures, but they are beyond the scope of this review.
Arsenic-induced changes in the immune system are implicated by epidemiological studies. Subjects exposed to higher levels of arsenic have higher serum levels of immunoglobulin (Ig) A (219). Urinary arsenic levels are also positively associated with the number T helper (Th) 17 cells (220), whereas nail arsenic levels are associated with lower counts of CD56+ NK cells (221), after adjusting for confounding factors. Consistent with impairment of the immune system, lymphocytes isolated from arsenic-exposed individuals have a longer average doubling time in vitro (222). They also secrete lower levels of cytokines, including IL-2, IL-4, IL-6, IL-10, tumor necrosis factor alpha (TNFα), and IFNγ under basal conditions (223) and following stimulation with concanavalin A (Con A) (224). Monocyte-derived macrophages isolated from the exposed individuals display abnormal morphology, diminished adhesion, and have reduced phagocytic capacity (225). These findings indicate arsenic exposure disrupts both innate and adaptive immune responses. Notably, arsenic exposure often leads to skin lesions (219) but not necessarily cancer. Whether the immunomodulation induced by arsenic contributes to excess cancer risk in exposed populations awaits further investigation.
Early life exposure to arsenic may also impact the immune system. Children with prenatal exposure to arsenic have higher risk of respiratory illness (226), and diminished cell-mediated immune function (227). Prenatal arsenic exposure alters cord blood immune cell composition, increases the proliferation of effector T and T cells, and reduces the suppression by T regulatory (Treg) cells in a dose-dependent manner (228) (229). Prenatal arsenic exposure is also inversely associated with the percentage of naive and activated T helper memory cells in cord blood, with notable sex-dependent differences in the strength of the association (230). Moreover, lymphocytes isolated from children with prenatal arsenic exposure secrete lower levels of CX3CL and tumor necrosis factor alpha following PHA stimulation (231). Proteomic analyses of cord blood further revealed aberrant levels of chemokine (C-X-C motif) ligands, macrophage migration inhibitor factor (232), and interleukins (233). This implies that the prenatal period may be a critical window of susceptibility for disruption of immune responses by environmental arsenic exposure. Nevertheless, further studies are needed to determine whether these arsenic effects can be causally linked to higher cancer risk later in life.
Smoking suppresses the immune system (234), but the impact of passive smoking is less studied. One study involving non-smoking adult volunteers has shown that serum levels of the nicotine metabolite cotinine correlate with an increase in the naïve CD3+ and CD4+ T cell subsets and a decrease in the memory CD3+ and CD4+ T cell subsets in peripheral blood (235). Other studies have focused on immune cells in the saliva and nasal lavage, which are primary target tissues due to their proximity to the exposure route of ETS. For instance, ETS is associated with a higher percentage of phagocytic cells in the saliva (236). ETS is also correlated with the level of immunoglobulin E and immunoglobulin A in nasal lavage following exposure to ragweed (237). By contrast, ETS has no effect on the levels of cytokines IL-2, IL-5, IL-13, and IFNγ in the nasal lavage. These findings indicate that ETS has differential effects on the subsets of peripheral T cells, and may induce inflammatory responses. Interestingly, parental smoking dose-dependently decreases IFNγ production in mitogen stimulated PBMC and is associated with active wheezing in children (238). In view of the above findings, exposure to ETS in children is suggested to be associated with asthma and cancer (239). In summary, the findings reported in this section underscore the significant impacts of two exemplar environmental exposures that can modify immune function, and which are associated with increased cancer risk.
2.3.4 Circadian Rhythm, Lifestyle, and Dietary Factors
As is the case with DNA repair, accumulating evidence indicates that immune function can vary substantially within an individual over the course of the day. Circulating immune cell populations undergo cyclic diurnal changes (240) (241). Among the immune cell subpopulations investigated, rhythmic changes are strongest among naïve CD4+ and CD8+ T cells (242, 243), and weakest, albeit still significant, among B cells (240). These observations have been made in both humans and mouse models (244), which have provided insights into how circadian rhythm regulates the trafficking of immune cells (245) (246). Notably, mice immunized with T cell dependent antigen trinitrophenyl-ovalbumin (OVA) in the evening have higher serum levels of antibodies when compared to those immunized in the morning (247). Consistent with these findings in animals, individuals receiving bacillus Calmette-Guerin vaccination in the morning exhibit stronger trained immunity and adaptive response when compared to those vaccinated in the evening (248). It is thus postulated that the timing of immunotherapy or cancer vaccine administration may affect the tumor suppressing effect. With these considerations in mind, chronotherapy is emerging as a novel research field that may improve the efficacy of cancer treatment (138).
Rhythmic changes in the immune cells are associated with levels of hormones and regulated by changes in cytokine levels and the expression of molecular clock genes (249–252). Levels of the stress hormone cortisol level peak near the time of awakening and then decline throughout the day (240). Its serum level negatively correlates with the abundance of circulating T cell subsets, including total CD4+ and CD8+ T cells (243). In vitro treatment with cortisol further shows that the suppression is most pronounced in native T cells, when compared to central memory and effector memory T cells. By contrast, the effector CD4+ and CD8+ T cells remain unaffected. Melatonin is the pineal hormone responsible for circadian synchronization (253) and its level peaks at night (240). Treatment in vitro with melatonin does not affect T cell proliferation upon simulation with Con A (254). However, higher salivary melatonin levels measured in the morning are associated with a higher percentage of HLA-DR+ monocytes and CD16+ lymphocytes, a higher CD4/CD8 ratio, lower lactate dehydrogenase activity in lymphocytes, and fewer CD3+ and CD8+ cells when compared to low salivary melatonin levels (255). High salivary melatonin levels in the evening are associated with a different constellation of immune system characteristics including lower phagocytic activity of granulocytes, lower CD4/CD8 ratios, and lower circulating levels of HLA-DR monocytes and CD16+ lymphocytes. Moreover, melatonin inhibits the secretion of T-cell independent antibodies (IgM, IgG1, IgG2b, and IgG3) in mice (247). These findings indicate that hormonal disruption of circadian rhythm can impact the immune response in complex ways.
Similar to the situation with DNA repair (139), animal models reveal that immune cells are subject to regulation by a circadian clock at a molecular level. For instance, rhythmic changes in the expression of clock genes including brain and muscle ARNT-like 1 (Bmal1), nuclear receptor superfamily 1, group D, member 1 (Rev-erbα) Period circadian regulator 1 (Per1), Per2, and Clock have been identified in mouse bone marrow derived macrophages (256), peritoneal macrophages (257), splenic macrophages, DCs, and B cells (256). In human CD4+ T cells, rhythmic changes in the expression of clock genes are synchronized with the production of IFNγ, IL-2, IL-4, and CD40L (258). In wild type mice, serum levels of LPS-induced cytokines display rhythmic changes (259), which are lost in Bmal1 deficient mice. Similarly, rhythmic change in serum levels of IL-6 is lost in Rev-erbα−/− mice (259). These findings reveal that the rhythmic control of immune function is tightly regulated by an intrinsic circadian clock, and the available data currently support a stronger role of the circadian clock in the innate immune response.
How nutrition modifies the immune system is a continually evolving field of research. Early studies focused primarily on the effects of vitamins and trace elements on the immune function have been reviewed (260). For instance, deficiency in vitamin B6 impairs lymphocyte maturation, proliferation, antibody production, and activity of T cells. It also attenuates NK cell activities. Deficiency in folate attenuates proliferation of CD8+ T cells and NK cell activities. Deficiency in vitamin B12 reduces total lymphocyte counts and the number of CD8+ cells. Vitamin C has also been shown to stimulate neutrophil chemotaxis, but its anti-inflammatory effects remain incompletely understood. Deficiency in vitamin A impairs phagocytosis and increases production of IL-12 and TNFα, which promotes inflammation. Deficiency in vitamin D impairs the innate immune response. Deficiency in trace elements including selenium, zinc, copper, and iron, can also disrupt the immune system. Comparisons between high and low fat diets have revealed impacts on cytokine levels that may impact the homeostatic balance between Treg and Th17 cells (261). Children following a Mediterranean diet for a year have higher salivary levels of an anti-inflammatory cytokine IL-10 and lower levels of IL-17 (262). A variety of dietary components, including red grape polyphenols, prebiotics, probiotics and symbiotics have been suggested to boost immune function in older individuals (263). Taken together, these findings establish an important role for diet-dependent immune-modulation, which may affect cancer susceptibility, as has been recently reviewed (263–265).
Several lines of evidence support a role for exercise in modulating immune function. Regular exercise and physical fitness can delay the onset of immunosenescence and tumorigenesis (265). Exercise improves the circulation and function of innate immune cells (266–268). Although the increase in immune cells is transient, it leads to a 40-50% decrease in the number of days with upper respiratory tract infection among adults during winter season (269). By contrast, exercise routines that induce muscle and tissue injury are pro-inflammatory and suppresses immune response transiently (265). Thus, the effects of exercise on the immune system appear complex and require further investigation.
Collectively, the data presented in this section outline numerous potential non-heritable sources of inter-individual variation in immune function. Taken together with the effects of aging, genetics, and the environment, these findings are consistent with a highly dynamic model of immune function. As with DNA repair, assessments of immune function at the individual level may provide important insights into disease susceptibility, but must be carried out in a manner that takes the many sources of variability into account. In the next section, we discuss a possible strategy for surveying both immunophenotype and genome integrity in human populations.
4 Potential for Simultaneous Profiling of Immunophenotype and Genome Integrity for More Accurate Assessments of Cancer Risk
Although DNA repair and immune function are distinct biological processes, they are subject to many of the same influences, and they both play important roles in cancer susceptibility. It has long been appreciated that several DNA repair pathways play integral roles in the immune system (270) (271). Furthermore, one of the most acute consequences of exposure to DNA damaging agents is suppression of the immune system (272) (273) (273). As outlined in this review, environmental exposures such as arsenic and passive smoking, circadian disruption, and lifestyle factors can modulate both DNA repair and immune function. It is noteworthy that defects in DNA repair and immune function are two of the most prominent hallmarks of cancer (2). Accordingly, efforts are underway to perform functional profiling in human populations, with the goal of identifying biomarkers that could be used for personalized prevention and treatment of cancer. While the idea of functional profiling has been framed independently in the context of genome integrity (13), and immune function (274), we propose that considering both simultaneously would increase the accuracy and robustness of cancer susceptibility predictions (Figure 2).
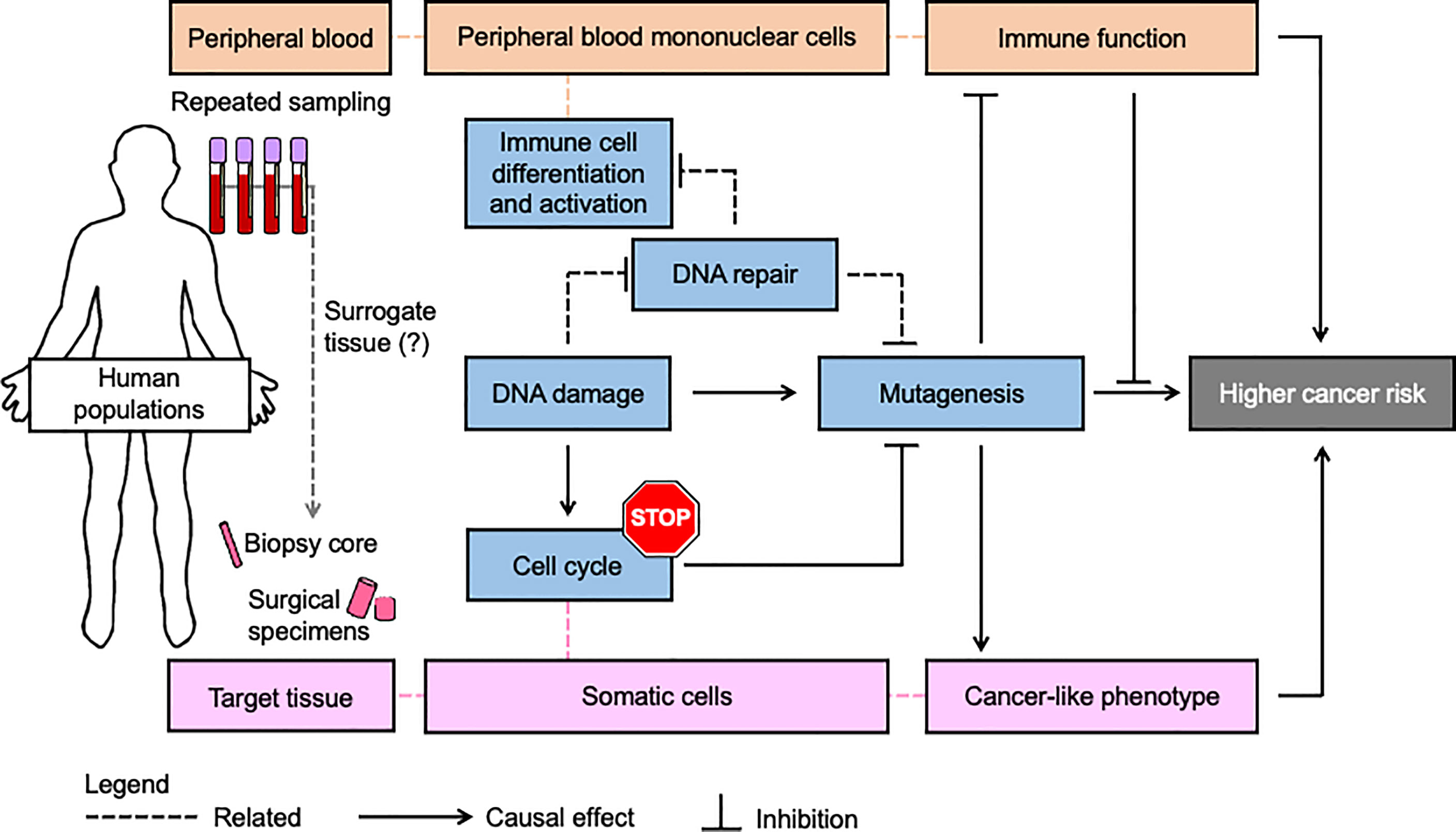
Figure 2 Simultaneous assessment of genome integrity and immune function may be a more robust strategy for personalized prevention and treatment of cancer. Most population studies use blood samples to assess genome integrity and immune function because blood draws are less invasive than the procedures for collecting other tissues from human subjects. A key assumption is that fundamental processes in cancer etiology (blue boxes) as measured in blood (red boxes) are sufficiently related to be considered a surrogate for the corresponding target tissue (pink boxes). Since blood and its components are heavily involved in immune processes, this tissue can provide extensive insights into immunophenotype. Likewise, lymphocytes provide extensive insights into inter-individual variation in genome integrity mechanisms, including those underlying risk of numerous solid malignancies as reviewed herein. In addition to its role in preventing mutagenesis and immunosuppression that can be induced by DNA damage, DNA repair is extensively involved in the differentiation and activation of immune cells. Nevertheless, variation in immune function and genome integrity pathways is independent and challenging to predict from genetics and indirect genomic markers. Therefore simultaneous functional assessment of DNA repair activities and immune function in studies using blood may improve the accuracy and precision of cancer risk estimates beyond what is possible when considering either process alone.
Patients with defects in nucleotide excision repair provide an excellent example of elevated cancer risk in individuals who are deficient in DNA repair and, perhaps, immune function (275–277). XP patients have a massively higher risk of developing UV- induced skin cancers (278), but also have an increased risk of developing internal tumors including glioblastoma, leukemia, lymphoma, and lung cancer (279, 280). The prevailing hypothesis regarding cancer susceptibility, both in XP patients and among those with lower NER capacity in the general population (14), has been that increased genome instability leads to higher rates of mutation and thus greater cancer susceptibility. However, it was noted in early case reports and small studies that XP patients also suffer from some forms of immune dysfunction (281) (275). Lymphocytes from XP patients have a larger clone size in response to allogeneic leukocytes (282), suggesting that lymphocytes of XP patients are more diverse, possibly due to a higher somatic mutational burden. Earlier studies have shown that lymphocytes from XP patients are less responsive to stimulation with mitogens (275, 277). Notably, serum from XP patients can attenuate the response of normal lymphocytes to PHA (275). A case study has also shown that DCs isolated from a trichothiodystrophy (TTD) patient with an XPD mutation have lower expression of CD86 co-stimulatory molecules and HLA glycoproteins, and are defective in stimulating native T lymphocytes (277). Notably, TTD patients commonly suffer from infections and there are several documented cases of immunodeficiency (283). Since some TTD patients do not appear to exhibit defects in DNA repair, these findings raise the possibility that NER proteins could have a role in immune function that is distinct from their role in DNA repair. NK cells from XP patients of multiple complementation groups display impaired lytic activity and lower IFNγ production in response to poly I:C stimulation, though the total NK cell count is normal (276). Moreover, XP patients have higher tolerance to the grafting of skin from a normal HLA-incompatible donor (275). Taken together, these findings suggest that innate and adaptive immune cell function may be defective in patients with nucleotide excision repair defects, but the underlying mechanism and the extent to which these findings may extend generally to patients with XP and other NER deficiency disorders remain unknown. Additional, comprehensive studies in larger cohorts of patients with NER deficiency are needed to assess whether their cancer-prone phenotypes can be explained in part by an accompanying immune defect. Such studies would also illuminate whether inter-individual variation in NER can be expected contribute to variation in immune function in the general population.
In the case of xeroderma pigmentosum variant (XP-V), patients express a truncated POLH, which reduces the expression and activity of DNA polymerase η (Pol η) (284). POLH is involved in translesion synthesis (TLS), which promotes tolerance of CPDs, thymidine dimers, and 8-oxoguanine lesions (285, 286). Loss of POLH leads to error prone-repair of CPDs by mutagenic polymerases zeta, kappa, and iota (287). Interestingly, UVA irradiation induces a mutational signature that suggests a role for basal mutagenesis induced by oxidative damage in the elevated risk for internal cancers in XPV patients (288). XP-V patients also have lower frequency of A/T mutation and higher frequency of deletion in Ig genes in activated B cells, which likely reflects the role of POLH in somatic hypermutation in B cells (289). POLH deficiency may thus drive the higher cancer incidence among XP-V patients via multiple mechanisms. In contrast with the severe combined immunodeficiency often associated with LIG4 syndrome due to disrupted V(D)J recombination (35), XPV patients do not present with pronounced immunodeficiency, possibly due to compensatory activities of other polymerases in somatic hypermutation. A small group of patients deficient for a subunit of another polymerase (POLE) does exhibit immunodeficiency and points to the possibility for additional rare polymerase deficiency disorders yet to be discovered (290).
Population studies offer numerous opportunities for simultaneous investigation of immune function and genome integrity. In identifying cancer risks and associating genome instability with cancer outcomes, these studies almost exclusively rely on blood samples due to its safe and relatively less invasive sampling method when compared with other types of biopsies. Furthermore, the multitude of cellular and molecular markers of immune function in blood represent a rich source of information that can be paired with analyses of genome integrity in lymphocytes. Some studies have already taken advantage of the opportunity to measure both genome integrity and immune function in a single population. For example, it has been observed that immunosuppressive drugs suppress DNA repair in human PBMCs (291, 292). As discussed in the following section, emerging technologies have greatly increased the feasibility of simultaneous profiling of DRC and immunophenotype in human populations.
5 Technological Advancements That Will Help Shape the Future of Precision Medicine
Significant technological advances have recently yielded functional tools for the interrogation of genome integrity and immune function. Here we review a sampling of emerging technologies that hold promise for enabling combined phenotyping with respect to DNA repair and the immune system in human populations.
As has been reviewed recently, several technologies are now available for analyses of genome integrity in human populations (13). Fluorescence-based multiplex flow-cytometric host cell reactivation (FM-HCR) assay measures the ability of live cells to repair site-specific DNA lesions (293). The assay is designed to have each fluorescent plasmid engineered to incorporate a specific type of DNA damage, including mismatches, abasic sites, oxidized bases, or DSB. The use of multiple fluorescent proteins enables multiplexing analyses for DNA repair activities. FM-HCR has thus been applied in a variety of settings, including in primary human lymphocytes (294–299).
The high throughput CometChip has been developed based on the established single gel electrophoresis assay (300, 301). Due to its 96-well format and automated image analysis, the CometChip is amenable to analyse large numbers of samples. It has recently been applied in a population study (302) and has been widely adopted for genotoxicity testing (303–305). CometChip technology has also been modified to interrogate DNA methylation status (306), levels of specific DNA adducts (307), and DNA damage in spheroids, which is also known as SpheroidChip (308).
A fluorescence-based unscheduled DNA synthesis (UDS) assay provides a substantially more convenient and user-friendly approach for measuring NER in populations. The original UDS assays used radio-labeled thymidine and autoradiography, making them laborious and inconvenient for routine clinical use (309). A new fluorescence-based method incorporates a thymidine analogue 5-ethynyl-2-deoxyuridine, which is conjugated to a fluorescent azide after UV irradiation and can be quantified by flow cytometry (310) (311). This technology is now being used to support the diagnosis of rare DNA repair deficiency disorders (312).
Single-cell whole-genome sequencing has opened up a new venue for studying somatic mutation and identifying mutational hotspots within the genome (313–316). This technology leverages single-cell multiple displacement amplification (SCMDA) procedure for detecting a full spectrum of base substitutions in a somatic cell. The technology has been used to reveal age-dependent changes in somatic mutations of B lymphocytes. The mutations in normal B lymphocytes not only resemble the COSMIC signatures in cancer (317), the data imply the age-dependent accumulation of somatic mutation is pivotal to the development B cell cancers (316). Thus, SCMDA, in combination with single-cell whole genome sequencing, is the tool for dissecting interindividual variation in mutation burdens influenced by genetics, age, environmental exposure, and lifestyle factors.
Single-cell RNA and DNA sequencing technology has advanced rapidly in recent years and found application in nearly every dimension of human biology (318). This technology analyzes the transcriptome of single cells within a heterogeneous population (319). It provides a powerful unbiased alternative to immunophenotyping approaches such as flow cytometry mass cytometry (CyTOF), which are less expensive but require labeling of surface markers and reveal little additional information at the single cell level (320) (321) (322). Single cell RNA sequencing enables the interrogation of cell-cell interactions, identification of changes during cell fate specification, and dissection of regulatory networks associated with cellular functions at single-cell level and based on cellular subtypes, which are not feasible in whole tissue analyses (323–326). Although single cell technologies remain expensive, continuous innovation raises the prospect of their eventual application in population studies. The emerging theory of clonal hematopoiesis of indeterminate potential (CHIP) describes the presence of somatic mutation in the cancer driver gene at a variant allele frequency of at least 2% in blood and bone marrow cells of a healthy individual (327–329). This process of clonal selection effectively amplifies mutations in a manner that makes them detectable by bulk sequencing. CHIP is induced by DNA damaging agents, and associated with increased risk of both leukemia and solid malignancies. It can thus be presumed to represent a molecular ruler that reflects both exposure to DNA damaging agents and the ability to repair DNA damage at the individual level.
Cellular indexing of transcriptomes and epitopes by sequencing, also known as CITE-seq, is a high throughput single-cell RNA sequencing analysis that is coupled with epitopes to interrogate expression of cell surface proteins (330). Since immune cell subtypes express specific surface markers, which can be captured by specific epitopes, CITE-seq has been widely used for determining the transcriptome profile of specific immune cells within a heterogeneous population (331, 332). Though CITE-seq and single-cell RNA sequencing serve similar purposes, CITE-seq has a shallower sequencing depth and relies heavily on the protein expression of specific cell surface marker. Its design better fits for studying immune cells.
Historically, it has not been feasible to perform functional screens of such nuanced phenotypes as those associated with modest defects in genome integrity or immune function. But these emerging technologies, particularly when used in combination, will enable such studies. Since blood samples are routinely collected for molecular epidemiological studies that focus on either genome integrity or immunophenotyping, the tissue could be maximally leveraged to understand how both processes may interact and contribute to cancer risk. Furthermore, studies combining immunophenotyping with genome integrity assays may shed light on whether mild DNA repair deficiencies in the general population lead to increased cancer risk, at least in part, by limiting the efficiency of immune responses.
6 Open Questions and Future Strategies for Population Studies
Here we briefly propose a framework for future studies aimed at understanding the joint influence of inter-individual variation in DRC and immune function on cancer risk. We pose several questions in the field that we view as important areas to investigate, followed by broad recommendations for pursuing population studies at the intersection of immune function and genome integrity.
6.1 Open Questions
1. Is blood a reliable surrogate for other tissues? Blood is an extremely rich source of data, including a variety of immune cells, cytokines, circulating DNA, and small molecules that can be analyzed to assess immune function, DNA damage and repair, and environmental exposures (Figure 3). Because it can be collected relatively easily and in a repeated manner, sampling blood is also among the most feasible approaches for population studies. Nevertheless, circulating immune cells may not reflect the biology of tissue resident immune cells and tissue-specific microenvironments. For these reasons, whenever possible, ideal studies would include sampling the tissue of interest and, in the case of cancer studies, the tumor as well.
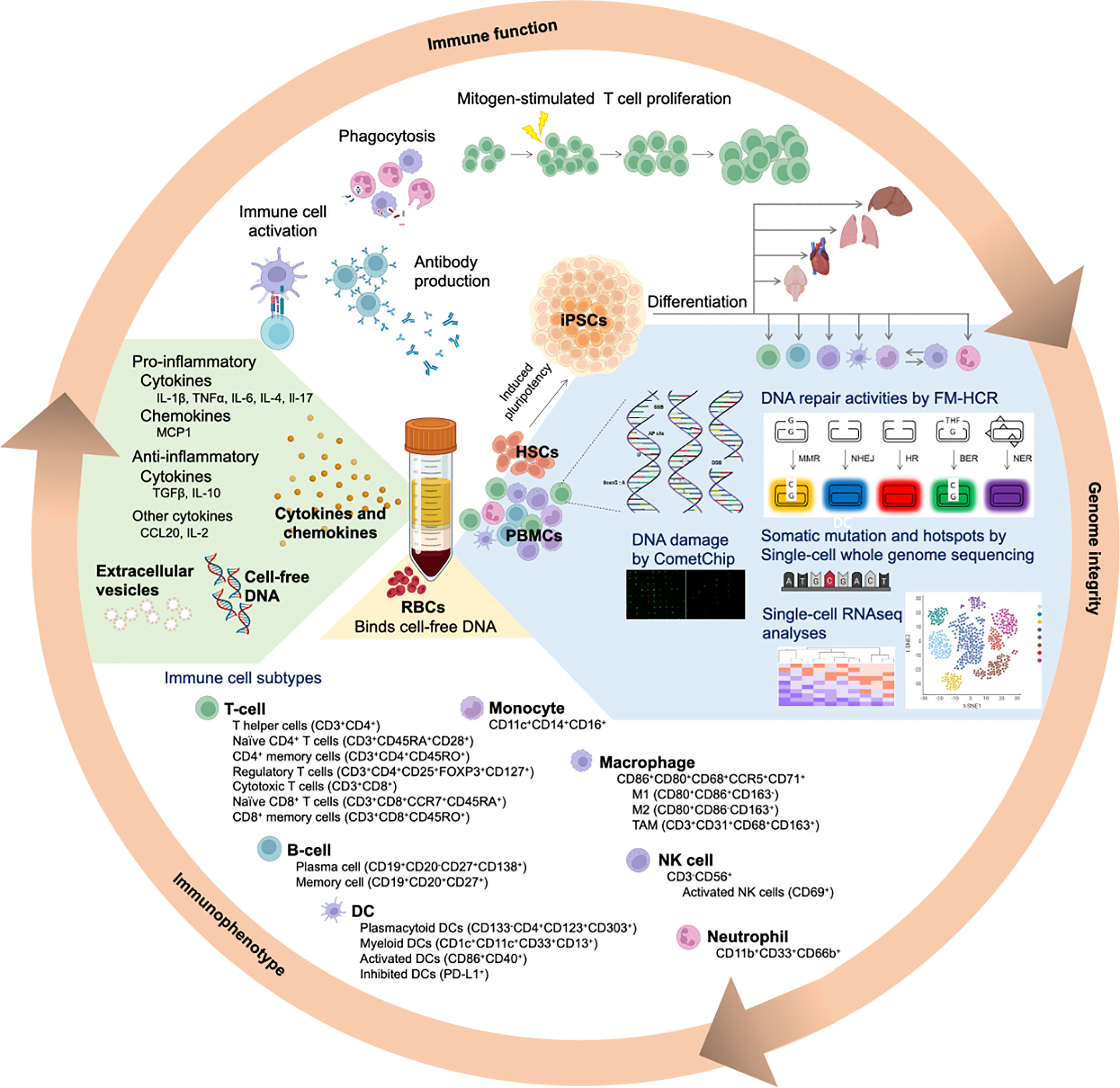
Figure 3 Simultaneous assessment of genome integrity and immune function using human blood samples. Following density gradient centrifugation of peripheral blood, peripheral blood mononuclear cells (PBMCs) are enriched in the buffy coat layer. Different immune cell subtypes within the PBMC population can be further identified based on their specific cell markers. Genomic integrity of the immune cell subtypes can be comprehensively evaluated by integrating various complementary approaches. Fluorescence-based multiplex host cell reactivation (FM-HCR) evaluates the ability of cells to repair specific DNA lesions. The CometChip assay reveals the magnitude of genomic DNA damage and repair kinetics in a high throughput manner. Single-cell whole genome sequencing identifies somatic mutations, whereas RNAseq (CITE-seq and single-cell RNAseq) measure the transcriptome. Moreover, hematopoietic stem cells isolated from the blood sample can potentially be used to generate induced pluripotent stem cells (iPSCs). Upon differentiating these iPSCs into a somatic cell type of interest, it becomes feasible to obtain large number of patient-derived, tissue-specific somatic cells, which may otherwise be scarce or not feasible to obtain. Red blood cells (RBCs), which are enriched in the bottom layer, bind cell-free DNA to minimize inflammatory responses. The plasma layer contains cytokines and chemokines secreted from the immune cells. These signaling molecules can be pro-inflammatory or anti-inflammatory, depending on the cellular status and presence of antigens. Notably, cell-free DNA and extracellular vesicles (EVs) are present in the plasma.
2. Which immune markers are the best predictors of cancer risk and outcomes? The emerging technologies described in the previous section provide an unprecedented opportunity for deep analysis of immunophenotypes, but because they have been developed so recently, they have only begun to be applied towards understanding the relationship between immune function and carcinogenesis. Studies surveying a broad array of immune markers are needed; these would include a census of circulating immune cells, measurements of cytokines, and tests for immune cell function.
3. Which combinations of functional assays are the best predictors of cancer risk and outcomes? Emerging functional assays described above and numerous established assays for immune cell activation and proliferation (333) integrate complex regulatory mechanisms and can complement ‘omics approaches (genotyping, transcriptional profiling, proteomics and DNA sequencing). Functional assays for DNA repair often outperform polygenetic cancer risk scores (334), and even stronger associations are seen in limited cases where multiple functional assays for different pathways have been applied to the same set of samples (335). But it is not possible to predict which functional biomarkers provide the most useful information to support mathematical models that would predict cancer risk or cancer outcomes. Thus, cancer case-control studies should be designed to integrate as many functional assays as is feasible for the same set of subjects. Given the practical constraints of funding and expertise, biological materials should be banked appropriately to enable future analyses.
4. How do markers of genome integrity and immune function change over the life course? As detailed in section 2 and section 3, the phenotypic markers we propose to survey with the goal of advancing personalized medicine are subject to time-dependent variation due to a variety of factors including lifestyle, environmental exposures, health status, and aging. To use these functional biomarkers as predictive tools, it will be necessary to carry out longitudinal studies wherein they are measured prospectively.
5. Does NER contribute to immune function? Numerous DNA repair pathways are already implicated in the mutagenic processes that occur during immune cell development and activation. In addition to those processes, emerging roles for DNA damage and DNA repair in gene regulation (336, 337), together with the growing recognition that many proteins “moonlight” in multiple roles within the cell (338), raise the possibility of as yet unrecognized mechanisms by which DNA repair pathway might contribute to immune function. By carrying out detailed immunophenotyping in individuals with profound defects in DNA repair, such as patients with XP, CS, and TTD, it can be determined whether NER deficiency, perhaps specifically which global genome (GG-NER) or transcription-coupled (TC-NER) NER subpathways, is associated with an immune disorder.
6. Can stem cell-derived cells recapitulate DRC of primary human tissues? A growing number of studies have found associations between DRC in blood cells and cancer risk, and the simplest interpretation is that the blood cells accurately represent genome maintenance in the tissue where the cancer develops. However, DNA repair varies with cell type and as a function of cell cycle and the tissue microenvironment. It is therefore possible that at least some of the associations between cancer risk and genome integrity as measured in immune cells is a reflection of immune cell function, rather than genome integrity in the target tissue. This question can in principle be unraveled by studies that measure DNA repair in multiple cell types from the same individual, but it likely will not be feasible to collect most tissues as part of a population study. By differentiating stem cells into cell types of interest, it may be possible to recapitulate physiological cell programming and make tissue-specific assessments of DRC on an individualized basis.
6.2 Recommendations
1. Focus on human studies: The framework we are proposing is at least in part discovery-based and centers human subjects, not biological model systems. This is a notable departure from the traditional approach more familiar to mechanistic biologists, wherein simple genetic models are used to test hypotheses before broaching the complexity of human systems. Instead, in this framework, one would first identify promising biomarkers in humans, and then follow up with confirmatory studies in model systems that best approximate the human biology. Taking the differences in telomere biology in mice and humans as an example (339), one can appreciate the value of prioritizing mechanistic characterization of biomarkers that have shown promise in human studies, and doing so in a model system that recapitulates the human biology. Though highly controlled genetic model systems such as CRISPR knockouts are not a feature of population studies, there are invaluable natural experiments and edge cases that can be leveraged for analogous purposes. For example, the phenotypes associated with rare genetic disorders that disrupt key aspects of genome maintenance and/or immune function such as those discussed in previous sections can be taken as upper or lower bounds for phenotypic variation in the general population. Likewise, biological samples from patients undergoing therapy with immunogens, immunosuppressants, or DNA damaging agents provide opportunities to understand physiological human responses to potentially carcinogenic real-world exposures. This is particularly so when the studies are conducted longitudinally, such that functional assays can be applied to samples collected before and after the exposure. Samples from individuals participating in studies that collect detailed personal environmental monitoring data present similar opportunities, and hold the advantage of avoiding the potential bias introduced by focusing on individuals with pre-existing health conditions, as is common in clinical studies.
2. Maximize the use and preservation of biological sample(s): The comprehensive functional characterization of human populations we are proposing is ambitious and may require some realignment of funding agency priorities and philosophies to reach its full potential. The prioritization of hypothesis-driven research commonly constrains the scope of projects and forces researchers to make decisions to severely limit the collection and analysis of biological samples. However, as illustrated in Figure 3, biological samples have extraordinary potential to provide insights into the many mechanisms driving human variation. To address this mismatch in the meantime, researchers should preserve biospecimens as comprehensively as possible. In the case of blood samples, this would entail banking each of the components and preserving them in a manner that is compatible with future downstream analyses, which may require live cells, for example.
3. Engage in team science: Population studies that make use of emerging technologies to characterize biological samples are inherently interdisciplinary. It is generally not within the capacity of a single investigator to have the expertise needed for establishing a human study cohort, developing and applying new technologies, interpreting biological data that span multiple fields, and, when applicable, treating and evaluating patients. In addition to a diverse group of scientific and medical experts who cover the technical expertise, the team should ideally include stakeholders who stand to benefit from the research. These stakeholders can also guide the focus of the study from its inception and ensure that vulnerable and underserved populations are included.
7 Closing Remarks
Many of the syndromes associated with defects in immune function or genome integrity have been discovered in recent years as genotyping technology has advanced. But these studies importantly relied upon functional characterization of variants of unknown significance, or the discovery of patients with a familiar disease of unknown etiology. The data suggest there are many more deficiency syndromes still to be discovered. Functional assays such as those outlined herein present powerful tools for identifying individuals with deficiencies in immunity or genome maintenance. By integrating these assays with modern genomics tools, it should be possible to accelerate the discovery and annotation of rare variants as well as functional associations with disease. Population studies are most easily carried out with blood, which contains the circulating cells and cytokines that can be used to define the immunophenotype. Therefore blood samples represent a largely untapped resource for analyzing both genome integrity and immune function simultaneously. Studies that compare these biological features between cancer patients and healthy counterparts will provide important clinical insights. Yet, simply surveying the complexity of the functional landscape across populations to define the range of variability is also a useful precursor to developing predictive models that incorporates the variability to explain disease susceptibility. Leveraging the advanced technologies and our current understanding of DRC, immune function, mutation, and cancer, it is timely to address these questions and improve the precision of strategies that assess and manage cancer risk for the welfare of population health.
Author Contributions
Both AC and ZN developed the concept and defined the scope of the review. AC performed a literature review, organized the results, drafted the review, and composed the figures and tables. ZN supervised the development of the review and contributed to the writing and editing of the manuscript and its components.
Funding
This work was supported by awards U01ES029520 and P30ES000002 (PI Weisskopf) to ZN. Both awards supported salary for ZN. An internal (Harvard T.H. Chan School of Public Health) award to ZN provided salary support for AC.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors gratefully acknowledge support from U01ES029520 and P30ES000002. Some figures generated are created with BioRender.com.
References
1. Tiwari V, Wilson D. DNA Damage and Associated DNA Repair Defects in Disease and Premature Aging. Am J Hum Genet (2019) 105(2):237–57. doi: 10.1016/j.ajhg.2019.06.005
2. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
3. Alexandrov LB, Stratton MR. Mutational Signatures: The Patterns of Somatic Mutations Hidden in Cancer Genomes. Curr Opin Genet Dev (2014) 24:52–60. doi: 10.1016/j.gde.2013.11.014
4. Chatterjee N, Walker GC. Mechanisms of DNA Damage, Repair, and Mutagenesis. Environ Mol Mutagen (2017) 58(5):235–63. doi: 10.1002/em.22087
5. Iyama T, Wilson DM 3rd. DNA Repair Mechanisms in Dividing and non-Dividing Cells. DNA Repair (Amst) (2013) 12(8):620–36. doi: 10.1016/j.dnarep.2013.04.015
6. Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discovery (2022) 12(1):31–46. doi: 10.1158/2159-8290.CD-21-1059
7. Stratton MR, Campbell PJ, Futreal PA. The Cancer Genome. Nature (2009) 458(7239):719–24. doi: 10.1038/nature07943
8. Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, et al. Mutational Signatures Associated With Tobacco Smoking in Human Cancer. Science (2016) 354(6312):618–22. doi: 10.1126/science.aag0299
9. Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Ng Tian AW, Wu Y, et al. The Repertoire of Mutational Signatures in Human Cancer. Nature (2020) 578(7793):94–101. doi: 10.1038/s41586-020-1943-3
10. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of Mutational Processes in Human Cancer. Nature (2013) 500(7463):415–21. doi: 10.1038/nature12477
11. Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) Database and Website. Br J Cancer (2004) 91(2):355–8. doi: 10.1038/sj.bjc.6601894
12. Nagel ZD, Chaim IA, Samson LD. Inter-Individual Variation in DNA Repair Capacity: A Need for Multi-Pathway Functional Assays to Promote Translational DNA Repair Research. DNA Repair (Amst) (2014) 19:199–213. doi: 10.1016/j.dnarep.2014.03.009
13. Nagel ZD, Engelward BP, Brenner DJ, Begley TJ, Sobol RW, Bielas JH, et al. Towards Precision Prevention: Technologies for Identifying Healthy Individuals With High Risk of Disease. Mutat Res (2017) 800-802:14–28. doi: 10.1016/j.mrfmmm.2017.03.007
14. Li C, Wang LE, Wei Q. DNA Repair Phenotype and Cancer Susceptibility–a Mini Review. Int J Cancer (2009) 124(5):999–1007. doi: 10.1002/ijc.24126
15. Decordier I, Loock KV, Kirsch-Volders M. Phenotyping for DNA Repair Capacity. Mutat Res (2010) 705(2):107–29. doi: 10.1016/j.mrrev.2010.05.002
16. Kraemer KH, DiGiovanna JJ. Forty Years of Research on Xeroderma Pigmentosum at the US National Institutes of Health. Photochem Photobiol (2015) 91(2):452–9. doi: 10.1111/php.12345
17. Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, et al. The XPV (Xeroderma Pigmentosum Variant) Gene Encodes Human DNA Polymerase Eta. Nature (1999) 399(6737):700–4. doi: 10.1038/21447
18. Cordonnier AM, Fuchs RP. Replication of Damaged DNA: Molecular Defect in Xeroderma Pigmentosum Variant Cells. Mutat Res (1999) 435(2):111–9. doi: 10.1016/S0921-8777(99)00047-6
19. Rapin I, Lindenbaum Y, Dickson DW, Kraemer KH, Robbins JH. Cockayne Syndrome and Xeroderma Pigmentosum. Neurology (2000) 55(10):1442–9. doi: 10.1212/WNL.55.10.1442
20. Ceccaldi R, Sarangi P, D'Andrea AD. The Fanconi Anaemia Pathway: New Players and New Functions. Nat Rev Mol Cell Biol (2016) 17(6):337–49. doi: 10.1038/nrm.2016.48
21. Nalepa G, Clapp DW. Fanconi Anaemia and Cancer: An Intricate Relationship. Nat Rev Cancer (2018) 18(3):168–85. doi: 10.1038/nrc.2017.116
22. Oshima J, Sidorova JM, Monnat RJ Jr. Werner Syndrome: Clinical Features, Pathogenesis and Potential Therapeutic Interventions. Ageing Res Rev (2017) 33:105–14. doi: 10.1016/j.arr.2016.03.002
23. Epstein CJ, Martin GM, Schultz AL, Motulsky AG. Werner's Syndrome a Review of its Symptomatology, Natural History, Pathologic Features, Genetics and Relationship to the Natural Aging Process. Med (Baltimore) (1966) 45(3):177–221. doi: 10.1097/00005792-196605000-00001
24. Goto M. Hierarchical Deterioration of Body Systems in Werner's Syndrome: Implications for Normal Ageing. Mech Ageing Dev (1997) 98(3):239–54. doi: 10.1016/S0047-6374(97)00111-5
25. Multani AS, Chang S. WRN at Telomeres: Implications for Aging and Cancer. J Cell Sci (2007) 120(Pt 5):713–21. doi: 10.1242/jcs.03397
26. Goto M, Ishikawa Y, Sugimoto M, Furuichi Y. Werner Syndrome: A Changing Pattern of Clinical Manifestations in Japan (1917~2008). Biosci Trends (2013) 7(1):13–22. doi: 10.5582/bst.2013.v7.1.13
27. Lauper JM, JM, Krause A, Vaughan TL, Monnat RJ Jr. Spectrum and Risk of Neoplasia in Werner Syndrome: A Systematic Review. PLoS One (2013) 8(4):e59709. doi: 10.1371/journal.pone.0059709
28. Fan W, Luo J. RecQ4 Facilitates UV Light-Induced DNA Damage Repair Through Interaction With Nucleotide Excision Repair Factor Xeroderma Pigmentosum Group A (XPA). J Biol Chem (2008) 283(43):29037–44. doi: 10.1074/jbc.M801928200
29. Shamanna RA, Singh DK, Lu H, Mirey G, Keijzers G, Salles B, et al. RECQ Helicase RECQL4 Participates in non-Homologous End Joining and Interacts With the Ku Complex. Carcinogenesis (2014) 35(11):2415–24. doi: 10.1093/carcin/bgu137
30. Arora A, Agarwal D, Abdel-Fatah TM, Lu H, Croteau DL, Moseley P, et al. RECQL4 Helicase has Oncogenic Potential in Sporadic Breast Cancers. J Pathol (2016) 238(4):495–501. doi: 10.1002/path.4681
31. Schurman SH, Hedayati M, Wang Z, Singh DK, Speina E, Zhang Y, et al. Direct and Indirect Roles of RECQL4 in Modulating Base Excision Repair Capacity. Hum Mol Genet (2009) 18(18):3470–83. doi: 10.1093/hmg/ddp291
32. Mann MB, Hodges CA, Barnes E, Vogel H, Hassold TJ, Luo G. Defective Sister-Chromatid Cohesion, Aneuploidy and Cancer Predisposition in a Mouse Model of Type II Rothmund-Thomson Syndrome. Hum Mol Genet (2005) 14(6):813–25. doi: 10.1093/hmg/ddi075
33. Lu L, Jin W, Liu H, Wang LL. RECQ DNA Helicases and Osteosarcoma. Adv Exp Med Biol (2014) 804:129–45. doi: 10.1007/978-3-319-04843-7_7
34. Mo D, Zhao Y, Balajee AS. Human RecQL4 Helicase Plays Multifaceted Roles in the Genomic Stability of Normal and Cancer Cells. Cancer Lett (2018) 413:1–10. doi: 10.1016/j.canlet.2017.10.021
35. Altmann T, Gennery AR. DNA Ligase IV Syndrome; a Review. Orphanet J Rare Dis (2016) 11(1):137. doi: 10.1186/s13023-016-0520-1
36. Schatz DG, Ji Y. Recombination Centres and the Orchestration of V(D)J Recombination. Nat Rev Immunol (2011) 11(4):251–63. doi: 10.1038/nri2941
37. Rothblum-Oviatt C, Wright J, Lefton-Greif MA, McGrath-Morrow SA, Crawford TO, Lederman HM. Ataxia Telangiectasia: A Review. Orphanet J Rare Dis (2016) 11(1):159. doi: 10.1186/s13023-016-0543-7
38. Stratigopoulou M, van Dam TP, Guikema JEJ. Base Excision Repair in the Immune System: Small DNA Lesions With Big Consequences. Front Immunol (2020) 11:1084. doi: 10.3389/fimmu.2020.01084
39. Wallace SS, Murphy DL, Sweasy JB. Base Excision Repair and Cancer. Cancer Lett (2012) 327(1-2):73–89. doi: 10.1016/j.canlet.2011.12.038
40. Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, et al. Inherited Variants of MYH Associated With Somatic G:C–>T:A Mutations in Colorectal Tumors. Nat Genet (2002) 30(2):227–32. doi: 10.1038/ng828
41. Cheadle JP, Sampson JR. MUTYH-Associated Polyposis–From Defect in Base Excision Repair to Clinical Genetic Testing. DNA Repair (Amst) (2007) 6(3):274–9. doi: 10.1016/j.dnarep.2006.11.001
42. David SS, O'Shea VL, Kundu S. Base-Excision Repair of Oxidative DNA Damage. Nature (2007) 447(7147):941–50. doi: 10.1038/nature05978
43. Raetz AG, Xie Y, Kundu S, Brinkmeyer MK, Chang C, David SS. Cancer-Associated Variants and a Common Polymorphism of MUTYH Exhibit Reduced Repair of Oxidative DNA Damage Using a GFP-Based Assay in Mammalian Cells. Carcinogenesis (2012) 33(11):2301–9. doi: 10.1093/carcin/bgs270
44. Theodoratou E, Campbell H, Tenesa A, Houlston R, Webb E, Lubbe S, et al. A Large-Scale Meta-Analysis to Refine Colorectal Cancer Risk Estimates Associated With MUTYH Variants. Br J Cancer (2010) 103(12):1875–84. doi: 10.1038/sj.bjc.6605966
45. Nieuwenhuis MH, Vogt S, Jones N, Nielsen M, Hes FJ, Sampson JR, et al. Evidence for Accelerated Colorectal Adenoma–Carcinoma Progression in MUTYH-Associated Polyposis? Gut (2012) 61(5):734–8. doi: 10.1136/gut.2010.229104
46. Imai K, Slupphaug G, Lee WI, Revy P, Nonoyama S, Catalan N, et al. Human Uracil-DNA Glycosylase Deficiency Associated With Profoundly Impaired Immunoglobulin Class-Switch Recombination. Nat Immunol (2003) 4(10):1023–8. doi: 10.1038/ni974
47. Etzioni A, Ochs HD. The Hyper IgM Syndrome–an Evolving Story. Pediatr Res (2004) 56(4):519–25. doi: 10.1203/01.PDR.0000139318.65842.4A
48. Weren RD, Ligtenberg MJ, Kets CM, Voer RM, Verwiel ET, Spruijt L, et al. A Germline Homozygous Mutation in the Base-Excision Repair Gene NTHL1 Causes Adenomatous Polyposis and Colorectal Cancer. Nat Genet (2015) 47(6):668–71. doi: 10.1038/ng.3287
49. Magrin L, Fanale D, Brando C, Fiorino A, Corsini LR, Sciacchitano R, et al. POLE, POLD1, and NTHL1: The Last But Not the Least Hereditary Cancer-Predisposing Genes. Oncogene (2021) 40(40):5893–901. doi: 10.1038/s41388-021-01984-2
50. Grolleman JE, Voer RM, Elsayed FA, Nielsen M, Weren RDA, Palles C, et al. Mutational Signature Analysis Reveals NTHL1 Deficiency to Cause a Multi-Tumor Phenotype. Cancer Cell (2019) 35(2):256–66.e5. doi: 10.1016/j.ccell.2018.12.011
51. Wimmer K, Kratz CP. Constitutional Mismatch Repair-Deficiency Syndrome. Haematologica (2010) 95(5):699–701. doi: 10.3324/haematol.2009.021626
52. Haraldsdottir S, Rafnar T, Frankel WL, Einarsdottir S, Sigurdsson A, Hampel H, et al. Comprehensive Population-Wide Analysis of Lynch Syndrome in Iceland Reveals Founder Mutations in MSH6 and PMS2. Nat Commun (2017) 8:14755. doi: 10.1038/ncomms14755
53. Peterson LM, Kipp BR, Halling KC, Kerr SE, Smith DI, Distad TJ, et al. Molecular Characterization of Endometrial Cancer: A Correlative Study Assessing Microsatellite Instability, MLH1 Hypermethylation, DNA Mismatch Repair Protein Expression, and PTEN, PIK3CA, KRAS, and BRAF Mutation Analysis. Int J Gynecol Pathol (2012) 31(3):195–205. doi: 10.1097/PGP.0b013e318231fc51
54. Vodicka P, Stetina R, Polakova V, Tulupova E, Naccarati A, Vodickova L, et al. Association of DNA Repair Polymorphisms With DNA Repair Functional Outcomes in Healthy Human Subjects. Carcinogenesis (2007) 28(3):657–64. doi: 10.1093/carcin/bgl187
55. Wolfe KJ, Wickliffe JK, Hill CE, Paolini M, Ammenheuser MM, Abdel-Rahman SZ. Single Nucleotide Polymorphisms of the DNA Repair Gene XPD/ERCC2 Alter mRNA Expression. Pharmacogenet Genomics (2007) 17(11):897–905. doi: 10.1097/FPC.0b013e3280115e63
56. Au WW, Salama SA, Sierra-Torres CH. Functional Characterization of Polymorphisms in DNA Repair Genes Using Cytogenetic Challenge Assays. Environ Health Perspect (2003) 111(15):1843–50. doi: 10.1289/ehp.6632
57. Hu JJ, Smith TR, Miller MS, Mohrenweiser HW, Golden A, Case LD. Amino Acid Substitution Variants of APE1 and XRCC1 Genes Associated With Ionizing Radiation Sensitivity. Carcinogenesis (2001) 22(6):917–22. doi: 10.1093/carcin/22.6.917
58. Cornetta T, Festa F, Testa A, Cozzi R. DNA Damage Repair and Genetic Polymorphisms: Assessment of Individual Sensitivity and Repair Capacity. Int J Radiat Oncol Biol Phys (2006) 66(2):537–45. doi: 10.1016/j.ijrobp.2006.06.037
59. Matullo G, Peluso M, Polidoro S, Guarrera S, Munnia A, Krogh V, et al. Combination of DNA Repair Gene Single Nucleotide Polymorphisms and Increased Levels of DNA Adducts in a Population-Based Study. Cancer Epidemiol Biomarkers Prev (2003) 12(7):674–7.
60. Duell EJ, Wiencke JK, Cheng TJ, Varkonyi A, Zuo ZF, Ashok TD, et al. Polymorphisms in the DNA Repair Genes XRCC1 and ERCC2 and Biomarkers of DNA Damage in Human Blood Mononuclear Cells. Carcinogenesis (2000) 21(5):965–71. doi: 10.1093/carcin/21.5.965
61. Zijno A, Verdina A, Galati R, Leopardi P, Marcon F, Andreoli C, et al. Influence of DNA Repair Polymorphisms on Biomarkers of Genotoxic Damage in Peripheral Lymphocytes of Healthy Subjects. Mutat Res (2006) 600(1-2):184–92. doi: 10.1016/j.mrfmmm.2006.04.004
62. Wang Y, Spitz MR, Zhu Y, Dong Q, Shete S, Wu X. From Genotype to Phenotype: Correlating XRCC1 Polymorphisms With Mutagen Sensitivity. DNA Repair (Amst) (2003) 2(8):901–8. doi: 10.1016/S1568-7864(03)00085-5
63. Lunn RM, Langlois RG, Hsieh LL, Thompson CL, Bell DA. XRCC1 Polymorphisms: Effects on Aflatoxin B1-DNA Adducts and Glycophorin A Variant Frequency. Cancer Res (1999) 59(11):2557–61.
64. Matullo G, Palli D, Peluso M, Guarrera S, Carturan S, Celentano E, et al. XRCC1, XRCC3, XPD Gene Polymorphisms, Smoking and (32)P-DNA Adducts in a Sample of Healthy Subjects. Carcinogenesis (2001) 22(9):1437–45. doi: 10.1093/carcin/22.9.1437
65. Patel AV, Calle EE, Pavluck AL, Feigelson HS, Thun MJ, Rodriguez C. A Prospective Study of XRCC1 (X-Ray Cross-Complementing Group 1) Polymorphisms and Breast Cancer Risk. Breast Cancer Res (2005) 7(6):R1168–73. doi: 10.1186/bcr1355
66. Erculj N, Zadel M, Dolzan V. Genetic Polymorphisms in Base Excision Repair in Healthy Slovenian Population and Their Influence on DNA Damage. Acta Chim Slov (2010) 57(1):182–8.
67. Chamberlain AJ, Jagt Vander CJ, Hayes BJ, Khansefid M, Marett LC, Millen CA, et al. Extensive Variation Between Tissues in Allele Specific Expression in an Outbred Mammal. BMC Genomics (2015) 16:993. doi: 10.1186/s12864-015-2174-0
68. Yousefzadeh M, Henpita C, Vyas R, Soto-Palma C, Robbins P, Niedernhofer L. DNA Damage-How and Why We Age? Elife (2021) 10:e62852. doi: 10.7554/eLife.62852
69. Li J, Bonkowski MS, Moniot S, Zhang D, Hubbard BP, Ling AJ, et al. A Conserved NAD(+) Binding Pocket That Regulates Protein-Protein Interactions During Aging. Science (2017) 355(6331):1312–7. doi: 10.1126/science.aad8242
70. Kishi K, Homma A, Kawa A, Kadowaki K. Age Related Change in the Frequency of Ara C-Induced Chromosome Aberrations in Human Peripheral Blood Lymphocytes. Mech Ageing Dev (1986) 37(3):211–9. doi: 10.1016/0047-6374(86)90038-2
71. Hartwig M, Korner IJ. Age-Related Changes of DNA Winding and Repair in Human Peripheral Lymphocytes. Mech Ageing Dev (1987) 38(1):73–8. doi: 10.1016/0047-6374(87)90111-4
72. Soares JP, Silva AM, Fonseca S, Oliveira MM, Peixoto F, Gaivao I, et al. How can Age and Lifestyle Variables Affect DNA Damage, Repair Capacity and Endogenous Biomarkers of Oxidative Stress? Exp Gerontol (2015) 62:45–52. doi: 10.1016/j.exger.2015.01.001
73. Singh NP, Danner DB, Tice RR, Brant L, Schneider EL. DNA Damage and Repair With Age in Individual Human Lymphocytes. Mutat Res (1990) 237(3-4):123–30. doi: 10.1016/0921-8734(90)90018-M
74. Garm C, Moreno-Villanueva M, Burkle A, Petersen I, Bohr VA, Christensen K, et al. Age and Gender Effects on DNA Strand Break Repair in Peripheral Blood Mononuclear Cells. Aging Cell (2013) 12(1):58–66. doi: 10.1111/acel.12019
75. Harrison JF, Hollensworth SB, Spitz DR, Copeland WC, Wilson GL, LeDoux SP. Oxidative Stress-Induced Apoptosis in Neurons Correlates With Mitochondrial DNA Base Excision Repair Pathway Imbalance. Nucleic Acids Res (2005) 33(14):4660–71. doi: 10.1093/nar/gki759
76. Leguisamo NM, Gloria HC, Kalil AN, Martins TV, Azambuja DB, Meira LB, et al. Base Excision Repair Imbalance in Colorectal Cancer has Prognostic Value and Modulates Response to Chemotherapy. Oncotarget (2017) 8(33):54199–214. doi: 10.18632/oncotarget.14909
77. Parrish MC, Chaim IA, Nagel ZD, Tannenbaum SR, Samson LD, Engelward BP. Nitric Oxide Induced S-Nitrosation Causes Base Excision Repair Imbalance. DNA Repair (Amst) (2018) 68:25–33. doi: 10.1016/j.dnarep.2018.04.008
78. Fu D, Calvo JA, Samson LD. Balancing Repair and Tolerance of DNA Damage Caused by Alkylating Agents. Nat Rev Cancer (2012) 12(2):104–20. doi: 10.1038/nrc3185
79. Cabelof DC, Raffoul JJ, Nakamura J, Kapoor D, Abdalla H, Heydari AR. Imbalanced Base Excision Repair in Response to Folate Deficiency is Accelerated by Polymerase Beta Haploinsufficiency. J Biol Chem (2004) 279(35):36504–13. doi: 10.1074/jbc.M405185200
80. Humphreys V, Martin RM, Ratcliffe B, Duthie S, Wood S, Gunnell D, et al. Age-Related Increases in DNA Repair and Antioxidant Protection: A Comparison of the Boyd Orr Cohort of Elderly Subjects With a Younger Population Sample. Age Ageing (2007) 36(5):521–6. doi: 10.1093/ageing/afm107
81. Chen SK, Hsieh WA, Tsai MH, Chen CC, Hong AI, Wei YH, et al. Age-Associated Decrease of Oxidative Repair Enzymes, Human 8-Oxoguanine DNA Glycosylases (Hogg1), in Human Aging. J Radiat Res (2003) 44(1):31–5. doi: 10.1269/jrr.44.31
82. Rossi O, Carrozzino F, Frosina G. Age-Independency of AP Site Incision Capacity in Women. Environ Mol Mutagen (1999) 34(4):256–9. doi: 10.1002/(SICI)1098-2280(1999)34:4<256::AID-EM5>3.0.CO;2-P
83. Lambert B, Ringborg U, Skoog L. Age-Related Decrease of Ultraviolet Light-Induced DNA Repair Synthesis in Human Peripheral Leukocytes. Cancer Res (1979) 39(7 Pt 1):2792–5.
84. Liu SC, Parsons CS, Hanawalt PC. DNA Repair Response in Human Epidermal Keratinocytes From Donors of Different Age. J Invest Dermatol (1982) 79(5):330–5. doi: 10.1111/1523-1747.ep12500087
85. Maillie HD, Baker JV, Simon W, Watts RJ, Quinn BR. Age Related Rejoining of Broken Chromosomes in Human Leukocytes Following X-Irradiation. Mech Ageing Dev (1992) 65(2-3):229–38. doi: 10.1016/0047-6374(92)90038-F
86. Barnett YA, King CM. An Investigation of Antioxidant Status, DNA Repair Capacity and Mutation as a Function of Age in Humans. Mutat Res (1995) 338(1-6):115–28. doi: 10.1016/0921-8734(95)00017-Z
87. Yamada M, Udono MU, Hori M, Hirose R, Sato S, Mori T, et al. Aged Human Skin Removes UVB-Induced Pyrimidine Dimers From the Epidermis More Slowly Than Younger Adult Skin In Vivo. Arch Dermatol Res (2006) 297(7):294–302. doi: 10.1007/s00403-005-0618-0
88. Takahashi Y, Moriwaki S, Sugiyama Y, Endo Y, Yamazaki K, Mori T, et al. Decreased Gene Expression Responsible for Post-Ultraviolet DNA Repair Synthesis in Aging: A Possible Mechanism of Age-Related Reduction in DNA Repair Capacity. J Invest Dermatol (2005) 124(2):435–42. doi: 10.1111/j.0022-202X.2004.23591.x
89. Merkle TJ, O'Brien K, Brooks PJ, Tarone RE, Robbins JH. DNA Repair in Human Fibroblasts, as Reflected by Host-Cell Reactivation of a Transfected UV-Irradiated Luciferase Gene, is Not Related to Donor Age. Mutat Res (2004) 554(1-2):9–17. doi: 10.1016/j.mrfmmm.2004.02.013
90. Moriwaki S, Ray S, Tarone RE, Kraemer KH, Grossman L. The Effect of Donor Age on the Processing of UV-Damaged DNA by Cultured Human Cells: Reduced DNA Repair Capacity and Increased DNA Mutability. Mutat Res (1996) 364(2):117–23. doi: 10.1016/0921-8777(96)00029-8
91. Wei Q, Matanoski GM, Farmer ER, Hedayati MA, Grossman L. DNA Repair and Aging in Basal Cell Carcinoma: A Molecular Epidemiology Study. Proc Natl Acad Sci U.S.A. (1993) 90(4):1614–8. doi: 10.1073/pnas.90.4.1614
92. Grossman L. Epidemiology of Ultraviolet-DNA Repair Capacity and Human Cancer. Environ Health Perspect (1997) 105 Suppl 4(Suppl 4):927–30. doi: 10.1289/ehp.97105s4927
93. Li Z, Zhang W, Chen Y, Guo W, Zhang J, Tang H, et al. Impaired DNA Double-Strand Break Repair Contributes to the Age-Associated Rise of Genomic Instability in Humans. Cell Death Differ (2016) 23(11):1765–77. doi: 10.1038/cdd.2016.65
94. Goukassian DA, Bagheri S, L, Eller MS, Gilchrest BA. DNA Oligonucleotide Treatment Corrects the Age-Associated Decline in DNA Repair Capacity. FASEB J (2002) 16(7):754–6. doi: 10.1096/fj.01-0829fje
95. Deng XD, Gao Q, Zhang W, Zhang B, Ma Y, Zhang LX, et al. The Age-Related Expression Decline of ERCC1 and XPF for Forensic Age Estimation: A Preliminary Study. J Forensic Leg Med (2017) 49:15–9. doi: 10.1016/j.jflm.2017.05.005
96. Wang JL, Guo HL, Wang PC, Liu CG. Age-Dependent Down-Regulation of DNA Polymerase Delta1 in Human Lymphocytes. Mol Cell Biochem (2012) 371(1-2):157–63. doi: 10.1007/s11010-012-1432-6
97. Frasca D, Barattini P, Tirindelli D, Guidi L, Bartoloni C, Errani A, et al. Effect of Age on DNA Binding of the Ku Protein in Irradiated Human Peripheral Blood Mononuclear Cells (PBMC). Exp Gerontol (1999) 34(5):645–58. doi: 10.1016/S0531-5565(99)00026-1
98. Heuser VD, Andrade VM, Peres A, Braga Macedo Gomes LM, Chies Bogo JA. Influence of Age and Sex on the Spontaneous DNA Damage Detected by Micronucleus Test and Comet Assay in Mice Peripheral Blood Cells. Cell Biol Int (2008) 32(10):1223–9. doi: 10.1016/j.cellbi.2008.07.005
99. Velegzhaninov I, Mezenceva V, Shostal O, Baranova A, Moskalev A. Age Dynamics of DNA Damage and CpG Methylation in the Peripheral Blood Leukocytes of Mice. Mutat Res (2015) 775:38–42. doi: 10.1016/j.mrfmmm.2015.03.006
100. Cabelof DC, Raffoul JJ, Yanamadala S, Ganir C, Guo Z, Heydari AR. Attenuation of DNA Polymerase Beta-Dependent Base Excision Repair and Increased DMS-Induced Mutagenicity in Aged Mice. Mutat Res (2002) 500(1-2):135–45. doi: 10.1016/S0027-5107(02)00003-9
101. Vaidya A, Mao Z, Tian X, Spencer B, Seluanov A, Gorbunova V. Knock-In Reporter Mice Demonstrate That DNA Repair by non-Homologous End Joining Declines With Age. PLoS Genet (2014) 10(7):e1004511. doi: 10.1371/journal.pgen.1004511
102. Hanaoka F, Sayato J, Arai H, Hasegawa N, Inui N, Mitsui Y, et al. Changes in DNA Polymerases Alpha, Beta and Gamma in Mouse Liver as a Function of Age. Mech Ageing Dev (1983) 23(3-4):315–27. doi: 10.1016/0047-6374(83)90032-5
103. Mikkelsen L, Bialkowski K, Risom L, Lohr M, Loft S, Moller P. Aging and Defense Against Generation of 8-Oxo-7,8-Dihydro-2'-Deoxyguanosine in DNA. Free Radic Biol Med (2009) 47(5):608–15. doi: 10.1016/j.freeradbiomed.2009.05.030
104. Intano GW, Cho EJ, McMahan CA, Walter CA. Age-Related Base Excision Repair Activity in Mouse Brain and Liver Nuclear Extracts. J Gerontol A Biol Sci Med Sci (2003) 58(3):205–11. doi: 10.1093/gerona/58.3.B205
105. Kovalchuk IP, Golubov A, Koturbash IV, Kutanzi K, Martin OA, Kovalchuk O. Age-Dependent Changes in DNA Repair in Radiation-Exposed Mice. Radiat Res (2014) 182(6):683–94. doi: 10.1667/RR13697.1
106. Szczesny B, Tann AW, Mitra S. Age- and Tissue-Specific Changes in Mitochondrial and Nuclear DNA Base Excision Repair Activity in Mice: Susceptibility of Skeletal Muscles to Oxidative Injury. Mech Ageing Dev (2010) 131(5):330–7. doi: 10.1016/j.mad.2010.03.009
107. Smith AH, Hopenhayn-Rich C, Bates MN, Goeden HM, Hertz-Picciotto I, Duggan HM, et al. Cancer Risks From Arsenic in Drinking Water. Environ Health Perspect (1992) 97:259–67. doi: 10.1289/ehp.9297259
108. Andrew AS, Mason RA, Kelsey KT, Schned AR, Marsit CJ, Nelson HH, et al. DNA Repair Genotype Interacts With Arsenic Exposure to Increase Bladder Cancer Risk. Toxicol Lett (2009) 187(1):10–4. doi: 10.1016/j.toxlet.2009.01.013
109. Applebaum KM, Karagas MR, Hunter DJ, Catalano PJ, Byler SH, Morris S, et al. Polymorphisms in Nucleotide Excision Repair Genes, Arsenic Exposure, and non-Melanoma Skin Cancer in New Hampshire. Environ Health Perspect (2007) 115(8):1231–6. doi: 10.1289/ehp.10096
110. Azizian-Farsani F, Rafiei G, Saadat M. Impact of Sodium Arsenite on Chromosomal Aberrations With Respect to Polymorphisms of Detoxification and DNA Repair Genes. Int J Toxicol (2014) 33(6):518–22. doi: 10.1177/1091581814557953
111. Andrew AS, Burgess JL, Meza MM, Demidenko E, Waugh MG, Hamilton JW, et al. Arsenic Exposure is Associated With Decreased DNA Repair In Vitro and in Individuals Exposed to Drinking Water Arsenic. Environ Health Perspect (2006) 114(8):1193–8. doi: 10.1289/ehp.9008
112. Jasso-Pineda Y, Diaz-Barriga F, Calderon J, Yanez L, Carrizales L, Perez-Maldonado IN. DNA Damage and Decreased DNA Repair in Peripheral Blood Mononuclear Cells in Individuals Exposed to Arsenic and Lead in a Mining Site. Biol Trace Elem Res (2012) 146(2):141–9. doi: 10.1007/s12011-011-9237-0
113. Ock CY, Kim EH, Choi DJ, Lee HJ, Hahm KB, Chung MH. 8-Hydroxydeoxyguanosine: Not Mere Biomarker for Oxidative Stress, But Remedy for Oxidative Stress-Implicated Gastrointestinal Diseases. World J Gastroenterol (2012) 18(4):302–8. doi: 10.3748/wjg.v18.i4.302
114. Hinhumpatch P, Navasumrit P, Chaisatra K, Promvijit J, Mahidol C, Ruchirawat M. Oxidative DNA Damage and Repair in Children Exposed to Low Levels of Arsenic In Utero and During Early Childhood: Application of Salivary and Urinary Biomarkers. Toxicol Appl Pharmacol (2013) 273(3):569–79. doi: 10.1016/j.taap.2013.10.002
115. Martinez VD, Thu KL, Vucic EA, Hubaux R, Adonis M, Gil L, et al. Whole-Genome Sequencing Analysis Identifies a Distinctive Mutational Spectrum in an Arsenic-Related Lung Tumor. J Thorac Oncol (2013) 8(11):1451–5. doi: 10.1097/JTO.0b013e3182a4dd8e
116. Banerjee M, Sarkar J, Das JK, Mukherjee A, Sarkar AK, Mondal L, et al. Polymorphism in the ERCC2 Codon 751 is Associated With Arsenic-Induced Premalignant Hyperkeratosis and Significant Chromosome Aberrations. Carcinogenesis (2007) 28(3):672–6. doi: 10.1093/carcin/bgl181
117. Banerjee M, Sarma N, Biswas R, Roy J, Mukherjee A, Giri AK, et al. DNA Repair Deficiency Leads to Susceptibility to Develop Arsenic-Induced Premalignant Skin Lesions. Int J Cancer (2008) 123(2):283–7. doi: 10.1002/ijc.23478
118. Breton CV, Kile ML, Catalano PJ, Hoffman E, Quamruzzaman Q, Rahman M, et al. GSTM1 and APE1 Genotypes Affect Arsenic-Induced Oxidative Stress: A Repeated Measures Study. Environ Health (2007) 6:39. doi: 10.1186/1476-069X-6-39
119. Bhattacharjee P, Sanyal T, Bhattacharjee S, Bhattacharjee P. Epigenetic Alteration of Mismatch Repair Genes in the Population Chronically Exposed to Arsenic in West Bengal, India. Environ Res (2018) 163:289–96. doi: 10.1016/j.envres.2018.01.002
120. Hossain MB, Vahter M, Concha G, Broberg K. Environmental Arsenic Exposure and DNA Methylation of the Tumor Suppressor Gene P16 and the DNA Repair Gene MLH1: Effect of Arsenic Metabolism and Genotype. Metallomics (2012) 4(11):1167–75. doi: 10.1039/c2mt20120h
121. Paul S, Banerjee N, Chatterjee A, Sau TJ, Das JK, Mishra PK, et al. Arsenic-Induced Promoter Hypomethylation and Over-Expression of ERCC2 Reduces DNA Repair Capacity in Humans by non-Disjunction of the ERCC2-Cdk7 Complex. Metallomics (2014) 6(4):864–73. doi: 10.1039/c3mt00328k
122. Mo J, Xia Y, Ning Z, Wade TJ, Mumford JL. Elevated ERCC1 Gene Expression in Blood Cells Associated With Exposure to Arsenic From Drinking Water in Inner Mongolia. Anticancer Res (2009) 29(8):3253–9.
123. Andrew AS, Karagas MR, Hamilton JW. Decreased DNA Repair Gene Expression Among Individuals Exposed to Arsenic in United States Drinking Water. Int J Cancer (2003) 104(3):263–8. doi: 10.1002/ijc.10968
124. Lee-Chen SF, Yu CT, Wu DR, Jan KY. Differential Effects of Luminol, Nickel, and Arsenite on the Rejoining of Ultraviolet Light and Alkylation-Induced DNA Breaks. Environ Mol Mutagen (1994) 23(2):116–20. doi: 10.1002/em.2850230207
125. Cooper KL, Yager JW, Hudson LG. Melanocytes and Keratinocytes Have Distinct and Shared Responses to Ultraviolet Radiation and Arsenic. Toxicol Lett (2014) 224(3):407–15. doi: 10.1016/j.toxlet.2013.11.010
126. Lynn S, Lai HT, Gurr JR, Jan KY. Arsenite Retards DNA Break Rejoining by Inhibiting DNA Ligation. Mutagenesis (1997) 12(5):353–8. doi: 10.1093/mutage/12.5.353
127. Yager JW, Wiencke JK. Enhancement of Chromosomal Damage by Arsenic: Implications for Mechanism. Environ Health Perspect (1993) 101 (Suppl 3):79–82. doi: 10.1289/ehp.93101s379
128. Collier AC, Dandge SD, Woodrow JE, Pritsos CA. Differences in DNA-Damage in non-Smoking Men and Women Exposed to Environmental Tobacco Smoke (ETS). Toxicol Lett (2005) 158(1):10–9. doi: 10.1016/j.toxlet.2005.02.005
129. Bianchini F, Donato F, Faure H, Ravanat JL, Hall J, Cadet J. Urinary Excretion of 5-(Hydroxymethyl) Uracil in Healthy Volunteers: Effect of Active and Passive Tobacco Smoke. Int J Cancer (1998) 77(1):40–6. doi: 10.1002/(SICI)1097-0215(19980703)77:1<40::AID-IJC8>3.0.CO;2-#
130. van Zeeland AA, AA, Groot AJ, Hall J, Donato F. 8-Hydroxydeoxyguanosine in DNA From Leukocytes of Healthy Adults: Relationship With Cigarette Smoking, Environmental Tobacco Smoke, Alcohol and Coffee Consumption. Mutat Res (1999) 439(2):249–57. doi: 10.1016/S1383-5718(98)00192-2
131. Fracasso ME, Doria D, Franceschetti P, Perbellini L, Romeo L. DNA Damage and Repair Capacity by Comet Assay in Lymphocytes of White-Collar Active Smokers and Passive Smokers (non- and Ex-Smokers) at Workplace. Toxicol Lett (2006) 167(2):131–41. doi: 10.1016/j.toxlet.2006.09.003
132. Chandirasekar R, Suresh K, Jayakumar R, Venkatesan R, Kumar Lakshman B, Sasikala K. XRCC1 Gene Variants and Possible Links With Chromosome Aberrations and Micronucleus in Active and Passive Smokers. Environ Toxicol Pharmacol (2011) 32(2):185–92. doi: 10.1016/j.etap.2011.05.002
133. Lodovici M, Luceri C, Filippo De C, Romualdi C, Bambi F, Dolara P. Smokers and Passive Smokers Gene Expression Profiles: Correlation With the DNA Oxidation Damage. Free Radic Biol Med (2007) 43(3):415–22. doi: 10.1016/j.freeradbiomed.2007.04.018
134. Cohet C, Borel S, Nyberg F, Mukeria A, Bruske-Hohlfeld I, Constantinescu V, et al. Exon 5 Polymorphisms in the O6-Alkylguanine DNA Alkyltransferase Gene and Lung Cancer Risk in non-Smokers Exposed to Second-Hand Smoke. Cancer Epidemiol Biomarkers Prev (2004) 13(2):320–3. doi: 10.1158/1055-9965.EPI-03-0120
135. Pozuelos GL, Kagda MS, Schick S, Girke T, Volz DC, Talbot P. Experimental Acute Exposure to Thirdhand Smoke and Changes in the Human Nasal Epithelial Transcriptome: A Randomized Clinical Trial. JAMA Netw Open (2019) 2(6):e196362. doi: 10.1001/jamanetworkopen.2019.6362
136. Sancar A, Lindsey-Boltz LA, Kang TH, Reardon JT, Lee JH, Ozturk N. Circadian Clock Control of the Cellular Response to DNA Damage. FEBS Lett (2010) 584(12):2618–25. doi: 10.1016/j.febslet.2010.03.017
137. Kettner NM, Katchy CA, Fu L. Circadian Gene Variants in Cancer. Ann Med (2014) 46(4):208–20. doi: 10.3109/07853890.2014.914808
138. Davis K, Roden LC, Leaner VD, van der Watt PJ. The Tumour Suppressing Role of the Circadian Clock. IUBMB Life (2019) 71(7):771–80. doi: 10.1002/iub.2005
139. Ashok Kumar PV, Dakup PP, Sarkar S, Modasia JB, Motzner MS, Gaddameedhi S. It's About Time: Advances in Understanding the Circadian Regulation of DNA Damage and Repair in Carcinogenesis and Cancer Treatment Outcomes. Yale J Biol Med (2019) 92(2):305–16.
140. Koritala BSC, Porter KI, Arshad OA, Gajula RP, Mitchell HD, Arman T, et al. Night Shift Schedule Causes Circadian Dysregulation of DNA Repair Genes and Elevated DNA Damage in Humans. J Pineal Res (2021) 70(3):e12726. doi: 10.1111/jpi.12726
141. Collins AR. Kiwifruit as a Modulator of DNA Damage and DNA Repair. Adv Food Nutr Res (2013) 68:283–99. doi: 10.1016/B978-0-12-394294-4.00016-X
142. Heydari AR, Unnikrishnan A, Lucente LV, Richardson A. Caloric Restriction and Genomic Stability. Nucleic Acids Res (2007) 35(22):7485–96. doi: 10.1093/nar/gkm860
143. Ke Z, Firsanov D, Spencer B, Seluanov A, Gorbunova V. Short-Term Calorie Restriction Enhances DNA Repair by non-Homologous End Joining in Mice. NPJ Aging Mech Dis (2020) 6:9. doi: 10.1038/s41514-020-00047-2
144. Srivastava VK, Busbee DL. Decreased Fidelity of DNA Polymerases and Decreased DNA Excision Repair in Aging Mice: Effects of Caloric Restriction. Biochem Biophys Res Commun (1992) 182(2):712–21. doi: 10.1016/0006-291X(92)91790-W
145. Cabelof DC, Yanamadala S, Raffoul JJ, Guo Z, Soofi A, Heydari AR. Caloric Restriction Promotes Genomic Stability by Induction of Base Excision Repair and Reversal of Its Age-Related Decline. DNA Repair (Amst) (2003) 2(3):295–307. doi: 10.1016/S1568-7864(02)00219-7
146. Shaddock JG, Feuers RJ, Chou MW, Pegram RA, Casciano DA. Effects of Aging and Caloric Restriction on the Genotoxicity of Four Carcinogens in the In Vitro Rat Hepatocyte/DNA Repair Assay. Mutat Res (1993) 295(1):19–30. doi: 10.1016/0921-8734(93)90008-Q
147. Vermeij WP, Dolle ME, Reiling E, Jaarsma D, Payan-Gomez C, Bombardieri CR, et al. Restricted Diet Delays Accelerated Ageing and Genomic Stress in DNA-Repair-Deficient Mice. Nature (2016) 537(7620):427–31. doi: 10.1038/nature19329
148. Damiani AP, Strapazzon G, Sardinha Oliveira TT, Rohr P, Gajski G, Pinho RA, et al. Melatonin Supplementation Over Different Time Periods Until Ageing Modulates Genotoxic Parameters in Mice. Mutagenesis (2020) 35(6):465–78. doi: 10.1093/mutage/geaa017
149. LeBien TW, Tedder TF. B Lymphocytes: How They Develop and Function. Blood (2008) 112(5):1570–80. doi: 10.1182/blood-2008-02-078071
150. Gallina G, Dolcetti L, Serafini P, Santo De C, Marigo I, Colombo MP, et al. Tumors Induce a Subset of Inflammatory Monocytes With Immunosuppressive Activity on CD8+ T Cells. J Clin Invest (2006) 116(10):2777–90. doi: 10.1172/JCI28828
151. Prestwich RJ, Errington F, Hatfield P, Merrick AE, Ilett EJ, Selby PJ, et al. The Immune System–is it Relevant to Cancer Development, Progression and Treatment? Clin Oncol (R Coll Radiol) (2008) 20(2):101–12. doi: 10.1016/j.clon.2007.10.011
152. Choi JW, Lee ES, Kim SY, Park SI, Oh S, Kang JH, et al. Cytotoxic Effects of Ex Vivo-Expanded Natural Killer Cell-Enriched Lymphocytes (MYJ1633) Against Liver Cancer. BMC Cancer (2019) 19(1):817. doi: 10.1186/s12885-019-6034-1
153. Labanieh L, Majzner RG, Mackall CL. Programming CAR-T Cells to Kill Cancer. Nat BioMed Eng (2018) 2(6):377–91. doi: 10.1038/s41551-018-0235-9
154. Huber A, Dammeijer F, Aerts J, Vroman H. Current State of Dendritic Cell-Based Immunotherapy: Opportunities for In Vitro Antigen Loading of Different DC Subsets? Front Immunol (2018) 9:2804. doi: 10.3389/fimmu.2018.02804
155. Sharma P, Allison JP. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies With Curative Potential. Cell (2015) 161(2):205–14. doi: 10.1016/j.cell.2015.03.030
156. Yi C, He Y, Xia H, Zhang H, Zhang P. Review and Perspective on Adjuvant and Neoadjuvant Immunotherapies in NSCLC. Onco Targets Ther (2019) 12:7329–36. doi: 10.2147/OTT.S218321
157. Mortaz E, Tabarsi P, Mansouri D, Khosravi A, Garssen J, Velayati A, et al. Cancers Related to Immunodeficiencies: Update and Perspectives. Front Immunol (2016) 7:365. doi: 10.3389/fimmu.2016.00365
158. Salavoura K, Kolialexi A, Tsangaris G, Mavrou A. Development of Cancer in Patients With Primary Immunodeficiencies. Anticancer Res (2008) 28(2b):1263–9.
159. Shapiro RS. Malignancies in the Setting of Primary Immunodeficiency: Implications for Hematologists/Oncologists. Am J Hematol (2011) 86(1):48–55. doi: 10.1002/ajh.21903
160. Bigley V, Barge D, Collin M. Dendritic Cell Analysis in Primary Immunodeficiency. Curr Opin Allergy Clin Immunol (2016) 16(6):530–40. doi: 10.1097/ACI.0000000000000322
161. Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural Cytotoxic Activity of Peripheral-Blood Lymphocytes and Cancer Incidence: An 11-Year Follow-Up Study of a General Population. Lancet (2000) 356(9244):1795–9. doi: 10.1016/S0140-6736(00)03231-1
162. Jung JW, Overgaard NH, Burke MT, Isbel N, Frazer IH, Simpson F, et al. Does the Nature of Residual Immune Function Explain the Differential Risk of non-Melanoma Skin Cancer Development in Immunosuppressed Organ Transplant Recipients? Int J Cancer (2016) 138(2):281–92. doi: 10.1002/ijc.29450
163. Miao Y, Everly JJ, Gross TG, Tevar AD, First MR, Alloway RR, et al. De Novo Cancers Arising in Organ Transplant Recipients are Associated With Adverse Outcomes Compared With the General Population. Transplantation (2009) 87(9):1347–59. doi: 10.1097/TP.0b013e3181a238f6
164. Shiels MS, Copeland G, Goodman MT, Harrell J, Lynch CF, Pawlish K, et al. Cancer Stage at Diagnosis in Patients Infected With the Human Immunodeficiency Virus and Transplant Recipients. Cancer (2015) 121(12):2063–71. doi: 10.1002/cncr.29324
165. Gonzalez JP, Zabaleta A, Sangro P, Basualdo JE, Burgos L, Paiva B, et al. Immunophenotypic Pattern of De Novo Malignancy After Liver Transplantation. Transplant Proc (2019) 51(1):77–9. doi: 10.1016/j.transproceed.2018.04.090
166. Morton LM, Gibson TM, Clarke CA, Lynch CF, Anderson LA, Pfeiffer R, et al. Risk of Myeloid Neoplasms After Solid Organ Transplantation. Leukemia (2014) 28(12):2317–23. doi: 10.1038/leu.2014.132
167. Genders RE, Weijns ME, Dekkers OM, Plasmeijer EI. Metastasis of Cutaneous Squamous Cell Carcinoma in Organ Transplant Recipients and the Immunocompetent Population: Is There a Difference? A Systematic Review and Meta-Analysis. J Eur Acad Dermatol Venereol (2019) 33(5):828–41. doi: 10.1111/jdv.15396
168. Gioco R, Corona D, Agodi A, Privitera F, Barchitta M, Giaquinta A, et al. De Novo Cancer Incidence and Prognosis After Kidney Transplantation: A Single Center Analysis. Transplant Proc (2019) 51(9):2927–30. doi: 10.1016/j.transproceed.2019.04.096
169. Horie K, Tsuchiya T, Iinuma K, Maekawa Y, Nakane K, Kato T, et al. Risk Factors and Incidence of Malignant Neoplasms After Kidney Transplantation at a Single Institution in Japan. Clin Exp Nephrol (2019) 23(11):1323–30. doi: 10.1007/s10157-019-01769-8
170. Goedert JJ. The Epidemiology of Acquired Immunodeficiency Syndrome Malignancies. Semin Oncol (2000) 27(4):390–401.
171. Chistiakov DA, Voronova NV, Chistiakov AP. Ligase IV Syndrome. Eur J Med Genet (2009) 52(6):373–8. doi: 10.1016/j.ejmg.2009.05.009
172. Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, et al. Human Lymphoid and Myeloid Cell Development in NOD/LtSz-Scid IL2R Gamma Null Mice Engrafted With Mobilized Human Hemopoietic Stem Cells. J Immunol (2005) 174(10):6477–89. doi: 10.4049/jimmunol.174.10.6477
173. Gershwin ME, Merchant B, Gelfand MC, Vickers J, Steinberg AD, Hansen CT. The Natural History and Immunopathology of Outbred Athymic (Nude) Mice. Clin Immunol Immunopathol (1975) 4(3):324–40. doi: 10.1016/0090-1229(75)90002-1
174. Tamura T, Taguchi F, Ueda K, Fujiwara K. Persistent Infection With Mouse Hepatitis Virus of Low Virulence in Nude Mice. Microbiol Immunol (1977) 21(12):683–91. doi: 10.1111/j.1348-0421.1977.tb00337.x
175. Watts CJ, Hahn BL, Sohnle PG. Resistance of Athymic Nude Mice to Experimental Cutaneous Bacillus Anthracis Infection. J Infect Dis (2009) 199(5):673–9. doi: 10.1086/596631
176. Liston A, Carr EJ, Linterman MA. Shaping Variation in the Human Immune System. Trends Immunol (2016) 37(10):637–46. doi: 10.1016/j.it.2016.08.002
177. Lakshmikanth T, Muhammad SA, Olin A, Chen Y, Mikes J, Fagerberg L, et al. Human Immune System Variation During 1 Year. Cell Rep (2020) 32(3):107923. doi: 10.1016/j.celrep.2020.107923
178. Shen H, Zhang W, Abraham C, Cho JH. Age and CD161 Expression Contribute to Inter-Individual Variation in Interleukin-23 Response in CD8+ Memory Human T Cells. PLoS One (2013) 8(3):e57746. doi: 10.1371/journal.pone.0057746
179. Mund E, Christensson B, Larsson K, Gronneberg R. Sex Dependent Differences in Physiological Ageing in the Immune System of Lower Airways in Healthy non-Smoking Volunteers: Study of Lymphocyte Subsets in Bronchoalveolar Lavage Fluid and Blood. Thorax (2001) 56(6):450–5. doi: 10.1136/thx.56.6.450
180. de Craen AJ, Posthuma D, Remarque EJ, Biggelaar den van AH, Westendorp RG, Boomsma DI. Heritability Estimates of Innate Immunity: An Extended Twin Study. Genes Immun (2005) 6(2):167–70. doi: 10.1038/sj.gene.6364162
181. Tekola Ayele F, Doumatey A, Huang H, Zhou J, Charles B, Erdos M, et al. Genome-Wide Associated Loci Influencing Interleukin (IL)-10, IL-1Ra, and IL-6 Levels in African Americans. Immunogenetics (2012) 64(5):351–9. doi: 10.1007/s00251-011-0596-7
182. Cho P, Gelinas L, Corbett NP, Tebbutt SJ, Turvey SE, Fortuno ES 3rd, et al. Association of Common Single-Nucleotide Polymorphisms in Innate Immune Genes With Differences in TLR-Induced Cytokine Production in Neonates. Genes Immun (2013) 14(4):199–211. doi: 10.1038/gene.2013.5
183. Carr EJ, Dooley J, Garcia-Perez JE, Lagou V, Lee JC, Wouters C, et al. The Cellular Composition of the Human Immune System Is Shaped by Age and Cohabitation. Nat Immunol (2016) 17(4):461–8. doi: 10.1038/ni.3371
184. Ovsyannikova IG, Haralambieva IH, Kennedy RB, Pankratz VS, Vierkant RA, Jacobson RM, et al. Impact of Cytokine and Cytokine Receptor Gene Polymorphisms on Cellular Immunity After Smallpox Vaccination. Gene (2012) 510(1):59–65. doi: 10.1016/j.gene.2012.08.021
185. Tsang JS, Schwartzberg PL, Kotliarov Y, Biancotto A, Xie Z, Germain RN, et al. Global Analyses of Human Immune Variation Reveal Baseline Predictors of Postvaccination Responses. Cell (2014) 157(2):499–513. doi: 10.1016/j.cell.2014.03.031
186. Weinberger B, Lazuardi L, Weiskirchner I, Keller M, Neuner C, Fischer KH, et al. Healthy Aging and Latent Infection With CMV Lead to Distinct Changes in CD8+ and CD4+ T-Cell Subsets in the Elderly. Hum Immunol (2007) 68(2):86–90. doi: 10.1016/j.humimm.2006.10.019
187. Herndler-Brandstetter D, Landgraf K, Tzankov A, Jenewein B, Brunauer R, Laschober GT, et al. The Impact of Aging on Memory T Cell Phenotype and Function in the Human Bone Marrow. J Leukoc Biol (2012) 91(2):197–205. doi: 10.1189/jlb.0611299
188. Arnold KB, Szeto GL, Alter G, Irvine DJ, Lauffenburger DA. CD4+ T Cell-Dependent and CD4+ T Cell-Independent Cytokine-Chemokine Network Changes in the Immune Responses of HIV-Infected Individuals. Sci Signal (2015) 8(399):ra104. doi: 10.1126/scisignal.aab0808
189. Lei Q, Li T, Kong L, Li L, Ding X, Wang X, et al. HBV-Pol is Crucial for HBV-Mediated Inhibition of Inflammasome Activation and IL-1beta Production. Liver Int (2019) 39(12):2273–84. doi: 10.1111/liv.14214
190. Brodin P, Jojic V, Gao T, Bhattacharya S, Angel CJ, Furman D, et al. Variation in the Human Immune System Is Largely Driven by non-Heritable Influences. Cell (2015) 160(1-2):37–47. doi: 10.1016/j.cell.2014.12.020
191. Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, et al. Genome-Wide Scan Reveals Association of Psoriasis With IL-23 and NF-kappaB Pathways. Nat Genet (2009) 41(2):199–204. doi: 10.1038/ng.311
192. Morahan G, Huang D, Wu M, Holt BJ, White GP, Kendall GE, et al. Association of IL12B Promoter Polymorphism With Severity of Atopic and non-Atopic Asthma in Children. Lancet (2002) 360(9331):455–9. doi: 10.1016/S0140-6736(02)09676-9
193. Ojeda Ojeda M, Silva CV, JARM, Fernandez-Ortega C. TNFalpha Production in Whole Blood Cultures From Healthy Individuals. Biochem Biophys Res Commun (2002) 292(2):538–41. doi: 10.1006/bbrc.2002.6688
194. Weyand CM, Goronzy JJ. Aging of the Immune System. Mechanisms and Therapeutic Targets. Ann Am Thorac Soc (2016) 13 Suppl 5(Suppl 5):S422–s428. doi: 10.1513/AnnalsATS.201602-095AW
195. Gui J, Mustachio LM, Su DM, Craig RW. Thymus Size and Age-Related Thymic Involution: Early Programming, Sexual Dimorphism, Progenitors and Stroma. Aging Dis (2012) 3(3):280–90.
196. Chidrawar SM, Khan N, Chan YL, Nayak L, Moss PA. Ageing Is Associated With a Decline in Peripheral Blood CD56bright NK Cells. Immun Ageing (2006) 3:10. doi: 10.1186/1742-4933-3-10
197. Lokhorst HM, van der Linden JA, Schuurman HJ, Gmelig Meyling FH, Bast EJ, de Gast GC. Immune Function During Ageing in Man: Relation Between Serological Abnormalities and Cellular Immune Status. Eur J Clin Invest (1983) 13(3):209–14. doi: 10.1111/j.1365-2362.1983.tb00089.x
198. Jing Y, Gravenstein S, Chaganty NR, Chen N, Lyerly KH, Joyce S, et al. Aging is Associated With a Rapid Decline in Frequency, Alterations in Subset Composition, and Enhanced Th2 Response in CD1d-Restricted NKT Cells From Human Peripheral Blood. Exp Gerontol (2007) 42(8):719–32. doi: 10.1016/j.exger.2007.01.009
199. Lin Y, Kim J, Metter EJ, Nguyen H, Truong T, Lustig A, et al. Changes in Blood Lymphocyte Numbers With Age In Vivo and Their Association With the Levels of Cytokines/Cytokine Receptors. Immun Ageing (2016) 13:24. doi: 10.1186/s12979-016-0079-7
200. Lin Y, Damjanovic A, Metter EJ, Nguyen H, Truong T, Najarro K, et al. Age-Associated Telomere Attrition of Lymphocytes In Vivo is Co-Ordinated With Changes in Telomerase Activity, Composition of Lymphocyte Subsets and Health Conditions. Clin Sci (Lond) (2015) 128(6):367–77. doi: 10.1042/CS20140481
201. Borrego F, Alonso MC, Galiani MD, Carracedo J, Ramirez R, Ostos B, et al. NK Phenotypic Markers and IL2 Response in NK Cells From Elderly People. Exp Gerontol (1999) 34(2):253–65. doi: 10.1016/S0531-5565(98)00076-X
202. Le Garff-Tavernier M, Beziat V, Decocq J, Siguret V, Gandjbakhch F, Pautas E, et al. Human NK Cells Display Major Phenotypic and Functional Changes Over the Life Span. Aging Cell (2010) 9(4):527–35. doi: 10.1111/j.1474-9726.2010.00584.x
203. Stervbo U, Meier S, Malzer JN, Baron U, Bozzetti C, Jurchott K, et al. Effects of Aging on Human Leukocytes (Part I): Immunophenotyping of Innate Immune Cells. Age (Dordr) (2015) 37(5):92. doi: 10.1007/s11357-015-9828-3
204. Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GM, Papagno L, et al. Memory CD8+ T Cells Vary in Differentiation Phenotype in Different Persistent Virus Infections. Nat Med (2002) 8(4):379–85. doi: 10.1038/nm0402-379
205. Yue FY, Kovacs CM, Dimayuga RC, Parks P, Ostrowski MA. HIV-1-Specific Memory CD4+ T Cells are Phenotypically Less Mature Than Cytomegalovirus-Specific Memory CD4+ T Cells. J Immunol (2004) 172(4):2476–86. doi: 10.4049/jimmunol.172.4.2476
206. Kovaiou RD, Weiskirchner I, Keller M, Pfister G, Cioca DP, Grubeck-Loebenstein B. Age-Related Differences in Phenotype and Function of CD4+ T Cells are Due to a Phenotypic Shift From Naive to Memory Effector CD4+ T Cells. Int Immunol (2005) 17(10):1359–66. doi: 10.1093/intimm/dxh314
207. Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two Subsets of Memory T Lymphocytes With Distinct Homing Potentials and Effector Functions. Nature (1999) 401(6754):708–12. doi: 10.1038/44385
208. Koch S, Larbi A, Derhovanessian E, Ozcelik D, Naumova E, Pawelec G. Multiparameter Flow Cytometric Analysis of CD4 and CD8 T Cell Subsets in Young and Old People. Immun Ageing (2008) 5:6. doi: 10.1186/1742-4933-5-6
209. Ziegler S, Weiss E, Schmitt AL, Schlegel J, Burgert A, Terpitz U, et al. CD56 Is a Pathogen Recognition Receptor on Human Natural Killer Cells. Sci Rep (2017) 7(1):6138. doi: 10.1038/s41598-017-06238-4
210. Cooper MA, Fehniger TA, Caligiuri MA. The Biology of Human Natural Killer-Cell Subsets. Trends Immunol (2001) 22(11):633–40. doi: 10.1016/S1471-4906(01)02060-9
211. Poli A, Michel T, Theresine M, Andres E, Hentges F, Zimmer J. CD56bright Natural Killer (NK) Cells: An Important NK Cell Subset. Immunology (2009) 126(4):458–65. doi: 10.1111/j.1365-2567.2008.03027.x
212. Van Acker HH, Capsomidis A, Smits EL, Van Tendeloo VF. CD56 in the Immune System: More Than a Marker for Cytotoxicity? Front Immunol (2017) 8:892. doi: 10.3389/fimmu.2017.00892
213. Frasca D, Barattini P, Goso C, Pucci S, Rizzo G, Bartoloni C, et al. Cell Proliferation and Ku Protein Expression in Ageing Humans. Mech Ageing Dev (1998) 100(2):197–208. doi: 10.1016/S0047-6374(97)00137-1
214. Passtoors WM, van den Akker EB, Deelen J, Maier AB, van der Breggen R, Jansen R, et al. IL7R Gene Expression Network Associates With Human Healthy Ageing. Immun Ageing (2015) 12:21. doi: 10.1186/s12979-015-0048-6
215. Trebilcock GU, Ponnappan U. Evidence for Lowered Induction of Nuclear Factor Kappa B in Activated Human T Lymphocytes During Aging. Gerontology (1996) 42(3):137–46. doi: 10.1159/000213785
216. Kilpinen S, Henttinen T, Lahdenpohja N, Hulkkonen J, Hurme M. Signals Leading to the Activation of NF-Kappa B Transcription Factor are Stronger in Neonatal Than Adult T Lymphocytes. Scand J Immunol (1996) 44(1):85–8. doi: 10.1046/j.1365-3083.1996.d01-277.x
217. Yoshida K, Cologne JB, Cordova K, Misumi M, Yamaoka M, Kyoizumi S, et al. Aging-Related Changes in Human T-Cell Repertoire Over 20years Delineated by Deep Sequencing of Peripheral T-Cell Receptors. Exp Gerontol (2017) 96:29–37. doi: 10.1016/j.exger.2017.05.015
218. Kreitinger JM, Beamer CA, Shepherd DM. Environmental Immunology: Lessons Learned From Exposure to a Select Panel of Immunotoxicants. J Immunol (2016) 196(8):3217–25. doi: 10.4049/jimmunol.1502149
219. Islam LN, Nabi AH, Rahman MM, Zahid MS. Association of Respiratory Complications and Elevated Serum Immunoglobulins With Drinking Water Arsenic Toxicity in Human. J Environ Sci Health A Tox Hazard Subst Environ Eng (2007) 42(12):1807–14. doi: 10.1080/10934520701566777
220. Lauer FT, Parvez F, Factor-Litvak P, Liu X, Santella RM, Islam T, et al. Changes in Human Peripheral Blood Mononuclear Cell (HPBMC) Populations and T-Cell Subsets Associated With Arsenic and Polycyclic Aromatic Hydrocarbon Exposures in a Bangladesh Cohort. PLoS One (2019) 14(7):e0220451. doi: 10.1371/journal.pone.0220451
221. Prasad P, Sarkar N, Sinha D. Effect of Low- and High-Level Groundwater Arsenic on Peripheral Blood and Lung Function of Exposed Rural Women. Regul Toxicol Pharmacol (2020) 115:104684. doi: 10.1016/j.yrtph.2020.104684
222. Ostrosky-Wegman P, Gonsebatt ME, Montero R, Vega L, Barba H, Espinosa J, et al. Lymphocyte Proliferation Kinetics and Genotoxic Findings in a Pilot Study on Individuals Chronically Exposed to Arsenic in Mexico. Mutat Res (1991) 250(1-2):477–82. doi: 10.1016/0027-5107(91)90204-2
223. Parvez F, Lauer FT, Factor-Litvak P, Liu X, Santella RM, Islam T, et al. Assessment of Arsenic and Polycyclic Aromatic Hydrocarbon (PAH) Exposures on Immune Function Among Males in Bangladesh. PLoS One (2019) 14(5):e0216662. doi: 10.1371/journal.pone.0216662
224. Biswas R, Ghosh P, Banerjee N, Das JK, Sau T, Banerjee A, et al. Analysis of T-Cell Proliferation and Cytokine Secretion in the Individuals Exposed to Arsenic. Hum Exp Toxicol (2008) 27(5):381–6. doi: 10.1177/0960327108094607
225. Banerjee N, Banerjee S, Sen R, Bandyopadhyay A, Sarma N, Majumder P, et al. Chronic Arsenic Exposure Impairs Macrophage Functions in the Exposed Individuals. J Clin Immunol (2009) 29(5):582–94. doi: 10.1007/s10875-009-9304-x
226. Farzan SF, Li Z, Korrick SA, Spiegelman D, Enelow R, Nadeau K, et al. Infant Infections and Respiratory Symptoms in Relation to in Utero Arsenic Exposure in a U.S. Cohort. Environ Health Perspect (2016) 124(6):840–7. doi: 10.1289/ehp.1409282
227. Ahmed S, Moore SE, Kippler M, Gardner R, Hawlader MD, Wagatsuma Y, et al. Arsenic Exposure and Cell-Mediated Immunity in Pre-School Children in Rural Bangladesh. Toxicol Sci (2014) 141(1):166–75. doi: 10.1093/toxsci/kfu113
228. Nadeau KC, Li Z, Farzan S, Koestler D, Robbins D, Fei DL, et al. In Utero Arsenic Exposure and Fetal Immune Repertoire in a US Pregnancy Cohort. Clin Immunol (2014) 155(2):188–97. doi: 10.1016/j.clim.2014.09.004
229. Kile ML, Houseman EA, Baccarelli AA, Quamruzzaman Q, Rahman M, Mostofa G, et al. Effect of Prenatal Arsenic Exposure on DNA Methylation and Leukocyte Subpopulations in Cord Blood. Epigenetics (2014) 9(5):774–82. doi: 10.4161/epi.28153
230. Nygaard UC, Li Z, Palys T, Jackson B, Subbiah M, Malipatlolla M, et al. Cord Blood T Cell Subpopulations and Associations With Maternal Cadmium and Arsenic Exposures. PLoS One (2017) 12(6):e0179606. doi: 10.1371/journal.pone.0179606
231. Parvez F, Akhtar E, Khan L, Haq MA, Islam T, Ahmed D, et al. Exposure to Low-Dose Arsenic in Early Life Alters Innate Immune Function in Children. J Immunotoxicol (2019) 16(1):201–9. doi: 10.1080/1547691X.2019.1657993
232. Bailey KA, Laine J, Rager JE, Sebastian E, Olshan A, Smeester L, et al. Prenatal Arsenic Exposure and Shifts in the Newborn Proteome: Interindividual Differences in Tumor Necrosis Factor (TNF)-Responsive Signaling. Toxicol Sci (2014) 139(2):328–37. doi: 10.1093/toxsci/kfu053
233. Smeester L, Bommarito PA, Martin EM, Recio-Vega R, Gonzalez-Cortes T, Olivas-Calderon E, et al. Chronic Early Childhood Exposure to Arsenic is Associated With a TNF-Mediated Proteomic Signaling Response. Environ Toxicol Pharmacol (2017) 52:183–7. doi: 10.1016/j.etap.2017.04.007
234. Qiu F, Liang CL, Liu H, Zeng YQ, Hou S, Huang S, et al. Impacts of Cigarette Smoking on Immune Responsiveness: Up and Down or Upside Down? Oncotarget (2017) 8(1):268–84. doi: 10.18632/oncotarget.13613
235. Vardavas CI, Plada M, Tzatzarakis M, Marcos A, Warnberg J, Gomez-Martinez S, et al. Passive Smoking Alters Circulating Naive/Memory Lymphocyte T-Cell Subpopulations in Children. Pediatr Allergy Immunol (2010) 21(8):1171–8. doi: 10.1111/j.1399-3038.2010.01039.x
236. Numabe Y, Ogawa T, Kamoi H, Kiyonobu K, Sato S, Kamoi K, et al. Phagocytic Function of Salivary PMN After Smoking or Secondary Smoking. Ann Periodontol (1998) 3(1):102–7. doi: 10.1902/annals.1998.3.1.102
237. Diaz-Sanchez D, Rumold R, Gong H Jr. Challenge With Environmental Tobacco Smoke Exacerbates Allergic Airway Disease in Human Beings. J Allergy Clin Immunol (2006) 118(2):441–6. doi: 10.1016/j.jaci.2006.04.047
238. Tebow G, Sherrill DL, Lohman IC, Stern DA, Wright AL, Martinez FD, et al. Effects of Parental Smoking on Interferon Gamma Production in Children. Pediatrics (2008) 121(6):e1563-9. doi: 10.1542/peds.2007-2795
239. Ng SP, Zelikoff JT. Smoking During Pregnancy: Subsequent Effects on Offspring Immune Competence and Disease Vulnerability in Later Life. Reprod Toxicol (2007) 23(3):428–37. doi: 10.1016/j.reprotox.2006.11.008
240. Mazzoccoli G, Sothern RB, Cata De A, Giuliani F, Fontana A, Copetti M, et al. A Timetable of 24-Hour Patterns for Human Lymphocyte Subpopulations. J Biol Regul Homeost Agents (2011) 25(3):387–95.
241. Aroca-Crevillen A, Adrover JM, Hidalgo A. Circadian Features of Neutrophil Biology. Front Immunol (2020) 11:576. doi: 10.3389/fimmu.2020.00576
242. Born J, Lange T, Hansen K, Molle M, Fehm HL. Effects of Sleep and Circadian Rhythm on Human Circulating Immune Cells. J Immunol (1997) 158(9):4454–64.
243. Dimitrov S, Benedict C, Heutling D, Westermann J, Born J, Lange T. Cortisol and Epinephrine Control Opposing Circadian Rhythms in T Cell Subsets. Blood (2009) 113(21):5134–43. doi: 10.1182/blood-2008-11-190769
244. Druzd D, Matveeva O, Ince L, Harrison U, He W, Schmal C, et al. Lymphocyte Circadian Clocks Control Lymph Node Trafficking and Adaptive Immune Responses. Immunity (2017) 46(1):120–32. doi: 10.1016/j.immuni.2016.12.011
245. Nguyen KD, Fentress SJ, Qiu Y, Yun K, Cox JS, Chawla A. Circadian Gene Bmal1 Regulates Diurnal Oscillations of Ly6C(hi) Inflammatory Monocytes. Science (2013) 341(6153):1483–8. doi: 10.1126/science.1240636
246. Hemmers S, Rudensky AY. The Cell-Intrinsic Circadian Clock Is Dispensable for Lymphocyte Differentiation and Function. Cell Rep (2015) 11(9):1339–49. doi: 10.1016/j.celrep.2015.04.058
247. Cernysiov V, Gerasimcik N, Mauricas M, Girkontaite I. Regulation of T-Cell-Independent and T-Cell-Dependent Antibody Production by Circadian Rhythm and Melatonin. Int Immunol (2010) 22(1):25–34. doi: 10.1093/intimm/dxp109
248. de Bree LCJ, Mourits VP, Koeken VA, Moorlag SJ, Janssen R, Folkman L, et al. Circadian Rhythm Influences Induction of Trained Immunity by BCG Vaccination. J Clin Invest (2020) 130(10):5603–17. doi: 10.1172/JCI133934
249. Arjona A, Silver AC, Walker WE, Fikrig E. Immunity's Fourth Dimension: Approaching the Circadian-Immune Connection. Trends Immunol (2012) 33(12):607–12. doi: 10.1016/j.it.2012.08.007
250. Labrecque N, Cermakian N. Circadian Clocks in the Immune System. J Biol Rhythms (2015) 30(4):277–90. doi: 10.1177/0748730415577723
251. Waggoner SN. Circadian Rhythms in Immunity. Curr Allergy Asthma Rep (2020) 20(1):2. doi: 10.1007/s11882-020-0896-9
252. Hergenhan S, Holtkamp S, Scheiermann C. Molecular Interactions Between Components of the Circadian Clock and the Immune System. J Mol Biol (2020) 432(12):3700–13. doi: 10.1016/j.jmb.2019.12.044
253. Brown GM. Light, Melatonin and the Sleep-Wake Cycle. J Psychiatry Neurosci (1994) 19(5):345–53.
254. Rogers N, van den Heuvel C, Dawson D. Effect of Melatonin and Corticosteroid on In Vitro Cellular Immune Function in Humans. J Pineal Res (1997) 22(2):75–80. doi: 10.1111/j.1600-079X.1997.tb00306.x
255. Litvinenko GI, Shurlygina AV, Malysheva OA, Kudaeva OT, Shirinskii VS, Kozlov VA, et al. Circadian Variations of Melatonin Concentration in Saliva and Blood Content of Immunocompetent Cells in Healthy Individuals. Bull Exp Biol Med (2002) 133(5):500–2. doi: 10.1023/A:1019882325663
256. Collins EJ, Cervantes-Silva MP, Timmons GA, JR, Curtis AM, Hurley JM. Post-Transcriptional Circadian Regulation in Macrophages Organizes Temporally Distinct Immunometabolic States. Genome Res (2021) 31(2):171–85. doi: 10.1101/2020.02.28.970715
257. Keller M, Mazuch J, Abraham U, Eom GD, Herzog ED, Volk HD, et al. A Circadian Clock in Macrophages Controls Inflammatory Immune Responses. Proc Natl Acad Sci U.S.A. (2009) 106(50):21407–12. doi: 10.1073/pnas.0906361106
258. Bollinger T, Leutz A, Leliavski A, Skrum L, Kovac J, Bonacina L, et al. Circadian Clocks in Mouse and Human CD4+ T Cells. PLoS One (2011) 6(12):e29801. doi: 10.1371/journal.pone.0029801
259. Gibbs JE, Blaikley J, Beesley S, Matthews L, Simpson KD, Boyce SH, et al. The Nuclear Receptor REV-ERBalpha Mediates Circadian Regulation of Innate Immunity Through Selective Regulation of Inflammatory Cytokines. Proc Natl Acad Sci U.S.A. (2012) 109(2):582–7. doi: 10.1073/pnas.1106750109
260. Wintergerst ES, Maggini S, Hornig DH. Contribution of Selected Vitamins and Trace Elements to Immune Function. Ann Nutr Metab (2007) 51(4):301–23. doi: 10.1159/000107673
261. Peluso I, Villano DV, Roberts SA, Cesqui E, Raguzzini A, Borges G, et al. Consumption of Mixed Fruit-Juice Drink and Vitamin C Reduces Postprandial Stress Induced by a High Fat Meal in Healthy Overweight Subjects. Curr Pharm Des (2014) 20(6):1020–4. doi: 10.2174/138161282006140220144802
262. Vitale E, Jirillo E, Magrone T. Determination of Body Mass Index and Physical Activity in Normal Weight Children and Evaluation of Salivary Levels of Interleukin 10 and Interleukin 17. Clin Immunol Endocrine Metab Drugs (2014) 1(2):81–8.
263. Soldati L, Di Renzo L, Jirillo E, Ascierto PA, Marincola FM, De Lorenzo A. The Influence of Diet on Anti-Cancer Immune Responsiveness. J Transl Med (2018) 16(1):75. doi: 10.1186/s12967-018-1448-0
264. Christ A, Lauterbach M, Latz E. Western Diet and the Immune System: An Inflammatory Connection. Immunity (2019) 51(5):794–811. doi: 10.1016/j.immuni.2019.09.020
265. Nieman DC, Lila MA, Gillitt ND. Immunometabolism: A Multi-Omics Approach to Interpreting the Influence of Exercise and Diet on the Immune System. Annu Rev Food Sci Technol (2019) 10:341–63. doi: 10.1146/annurev-food-032818-121316
266. Nieman DC, Henson DA, Austin MD, Brown VA. Immune Response to a 30-Minute Walk. Med Sci Sports Exerc (2005) 37(1):57–62. doi: 10.1249/01.MSS.0000149808.38194.21
267. Adams GR, Zaldivar FP, Nance DM, Kodesh E, Radom-Aizik S, Cooper DM. Exercise and Leukocyte Interchange Among Central Circulation, Lung, Spleen, and Muscle. Brain Behav Immun (2011) 25(4):658–66. doi: 10.1016/j.bbi.2011.01.002
268. Bigley AB, Rezvani K, Pistillo M, Reed J, Agha N, Kunz H, et al. Acute Exercise Preferentially Redeploys NK-Cells With a Highly-Differentiated Phenotype and Augments Cytotoxicity Against Lymphoma and Multiple Myeloma Target Cells. Part II: Impact of Latent Cytomegalovirus Infection and Catecholamine Sensitivity. Brain Behav Immun (2015) 49:59–65. doi: 10.1016/j.bbi.2014.12.027
269. Nieman DC, Henson DA, Austin MD, Sha W. Upper Respiratory Tract Infection is Reduced in Physically Fit and Active Adults. Br J Sports Med (2011) 45(12):987–92. doi: 10.1136/bjsm.2010.077875
270. Gennery AR, Cant AJ, Jeggo PA. Immunodeficiency Associated With DNA Repair Defects. Clin Exp Immunol (2000) 121(1):1–7. doi: 10.1046/j.1365-2249.2000.01257.x
271. Woodbine L, Gennery AR, Jeggo PA. The Clinical Impact of Deficiency in DNA non-Homologous End-Joining. DNA Repair (Amst) (2014) 16:84–96. doi: 10.1016/j.dnarep.2014.02.011
273. Krumbhaar EB, Krumbhaar HD. The Blood and Bone Marrow in Yelloe Cross Gas (Mustard Gas) Poisoning: Changes Produced in the Bone Marrow of Fatal Cases. J Med Res (1919) 40(3):497–508.3.
274. Brodin P, Davis MM. Human Immune System Variation. Nat Rev Immunol (2017) 17(1):21–9. doi: 10.1038/nri.2016.125
275. Dupuy JM, Lafforet D. A Defect of Cellular Immunity in Xeroderma Pigmentosum. Clin Immunol Immunopathol (1974) 3(1):52–8. doi: 10.1016/0090-1229(74)90022-1
276. Gaspari AA, Fleisher TA, Kraemer KH. Impaired Interferon Production and Natural Killer Cell Activation in Patients With the Skin Cancer-Prone Disorder, Xeroderma Pigmentosum. J Clin Invest (1993) 92(3):1135–42. doi: 10.1172/JCI116682
277. Racioppi L, Cancrini C, Romiti ML, Angelini F, Cesare Di S, Bertini E, et al. Defective Dendritic Cell Maturation in a Child With Nucleotide Excision Repair Deficiency and CD4 Lymphopenia. Clin Exp Immunol (2001) 126(3):511–8. doi: 10.1046/j.1365-2249.2001.01625.x
278. Cleaver JE. Defective Repair Replication of DNA in Xeroderma Pigmentosum. Nature (1968) 218(5142):652–6. doi: 10.1038/218652a0
280. Rizza ERH, DiGiovanna JJ, Khan SG, Tamura D, Jeskey JD, Kraemer KH. Xeroderma Pigmentosum: A Model for Human Premature Aging. J Invest Dermatol (2021) 141(4S):976–84. doi: 10.1016/j.jid.2020.11.012
281. Morison WL, Bucana C, Hashem N, Kripke ML, Cleaver JE, German JL. Impaired Immune Function in Patients With Xeroderma Pigmentosum. Cancer Res (1985) 45(8):3929–31.
282. Levis WR, Tanner DJ. Xeroderma Pigmentosum and Immune Diversity. N Engl J Med (1980) 302(9):523. doi: 10.1056/NEJM198002283020916
283. Faghri S, Tamura D, Kraemer KH, Digiovanna JJ. Trichothiodystrophy: A Systematic Review of 112 Published Cases Characterises a Wide Spectrum of Clinical Manifestations. J Med Genet (2008) 45(10):609–21. doi: 10.1136/jmg.2008.058743
284. Borroni RG, Diegoli M, Grasso M, Concardi M, Agozzino M, Vignini M, et al. Rare Exon 10 Deletion in POLH Gene in a Family With Xeroderma Pigmentosum Variant Correlating With Protein Expression by Immunohistochemistry. G Ital Dermatol Venereol (2020) 155(3):349–54. doi: 10.23736/S0392-0488.16.05158-0
285. Powers KT, Washington MT. Eukaryotic Translesion Synthesis: Choosing the Right Tool for the Job. DNA Repair (Amst) (2018) 71:127–34. doi: 10.1016/j.dnarep.2018.08.016
286. Quinet A, Lerner LK, Martins DJ, Menck CFM. Filling Gaps in Translesion DNA Synthesis in Human Cells. Mutat Res Genet Toxicol Environ Mutagen (2018) 836(Pt B):127–42. doi: 10.1016/j.mrgentox.2018.02.004
287. Waters LS, Minesinger BK, Wiltrout ME, D'Souza S, Woodruff RV, Walker GC. Eukaryotic Translesion Polymerases and Their Roles and Regulation in DNA Damage Tolerance. Microbiol Mol Biol Rev (2009) 73(1):134–54. doi: 10.1128/MMBR.00034-08
288. Moreno NC, de Souza TA, Garcia CCM, Ruiz NQ, Corradi C, Castro LP, et al. Whole-Exome Sequencing Reveals the Impact of UVA Light Mutagenesis in Xeroderma Pigmentosum Variant Human Cells. Nucleic Acids Res (2020) 48(4):1941–53. doi: 10.1093/nar/gkz1182
289. Lerner LK, Nguyen TV, Castro LP, Vilar JB, Munford V, Guillou Le M, et al. Large Deletions in Immunoglobulin Genes are Associated With a Sustained Absence of DNA Polymerase Eta. Sci Rep (2020) 10(1):1311. doi: 10.1038/s41598-020-58180-7
290. Logan CV, Murray JE, Parry DA, Robertson A, Bellelli R, Tarnauskaitė Ž, et al. DNA Polymerase Epsilon Deficiency Causes IMAGe Syndrome With Variable Immunodeficiency. Am J Hum Genet (2018) 103(6):1038–44. doi: 10.1016/j.ajhg.2018.10.024
291. Herman-Edelstein M, Rozen-Zvi B, Zingerman B, Lichtenberg S, Malachi T, Gafter U, et al. Effect of Immunosuppressive Drugs on DNA Repair in Human Peripheral Blood Mononuclear Cells. BioMed Pharmacother (2012) 66(2):111–5. doi: 10.1016/j.biopha.2011.11.008
292. Ori Y, Herman-Edelstein M, Zingerman B, Rozen-Zvi B, Gafter U, Malachi T, et al. Effect of Immunosuppressive Drugs on Spontaneous DNA Repair in Human Peripheral Blood Mononuclear Cells. BioMed Pharmacother (2012) 66(6):409–13. doi: 10.1016/j.biopha.2012.06.001
293. Nagel ZD, Margulies CM, Chaim IA, McRee SK, Mazzucato P, Ahmad A, et al. Multiplexed DNA Repair Assays for Multiple Lesions and Multiple Doses via Transcription Inhibition and Transcriptional Mutagenesis. Proc Natl Acad Sci U.S.A. (2014) 111(18):E1823–32. doi: 10.1073/pnas.1401182111
294. Nagel ZD, Kitange GJ, Gupta SK, Joughin BA, Chaim IA, Mazzucato P, et al. DNA Repair Capacity in Multiple Pathways Predicts Chemoresistance in Glioblastoma Multiforme. Cancer Res (2017) 77(1):198–206. doi: 10.1158/0008-5472.CAN-16-1151
295. Chaim IA, Nagel ZD, Jordan JJ, Mazzucato P, Ngo LP, Samson LD. In Vivo Measurements of Interindividual Differences in DNA Glycosylases and APE1 Activities. Proc Natl Acad Sci USA (2017) 114(48):E10379–88. doi: 10.1073/pnas.1712032114
296. Nagel ZD, Beharry AA, Mazzucato P, Kitange GJ, Sarkaria JN, Kool ET, et al. Fluorescent Reporter Assays Provide Direct, Accurate, Quantitative Measurements of MGMT Status in Human Cells. PLoS One (2019) 14(2):e0208341. doi: 10.1371/journal.pone.0208341
297. Lee KJ, Piett CG, Andrews JF, Mann E, Nagel ZD, Gassman NR. Defective Base Excision Repair in the Response to DNA Damaging Agents in Triple Negative Breast Cancer. PLoS One (2019) 14(10):e0223725. doi: 10.1371/journal.pone.0223725
298. Lee KJ, Mann E, Wright G, Piett CG, Nagel ZD, Gassman NR. Exploiting DNA Repair Defects in Triple Negative Breast Cancer to Improve Cell Killing. Ther Adv Med Oncol (2020) 12:1758835920958354. doi: 10.1177/1758835920958354
299. Toprani SM, Bitounis D, Huang Q, Oliveira N, Ng KW, Tay CY, et al. High-Throughput Screening Platform for Nanoparticle-Mediated Alterations of DNA Repair Capacity. ACS Nano (2021) 15(3):4728–46. doi: 10.1021/acsnano.0c09254
300. Ge J, Prasongtanakij S, Wood DK, Weingeist DM, Fessler J, Navasummrit P, et al. CometChip: A High-Throughput 96-Well Platform for Measuring DNA Damage in Microarrayed Human Cells. J Vis Exp (2014) 92):e50607. doi: 10.3791/50607
301. Ge J, Chow DN, Fessler JL, Weingeist DM, Wood DK, Engelward BP. Micropatterned Comet Assay Enables High Throughput and Sensitive DNA Damage Quantification. Mutagenesis (2015) 30(1):11–9. doi: 10.1093/mutage/geu063
302. Ngo LP, Kaushal S, Chaim IA, Mazzucato P, Ricciardi C, Samson LD, et al. CometChip Analysis of Human Primary Lymphocytes Enables Quantification of Inter-Individual Differences in the Kinetics of Repair of Oxidative DNA Damage. Free Radic Biol Med (2021) 174:89–99. doi: 10.1016/j.freeradbiomed.2021.07.033
303. Li N, Parrish M, Chan TK, Yin L, Rai P, Yoshiyuki Y, et al. Influenza Infection Induces Host DNA Damage and Dynamic DNA Damage Responses During Tissue Regeneration. Cell Mol Life Sci (2015) 72(15):2973–88. doi: 10.1007/s00018-015-1879-1
304. Chan TK, Loh XY, Peh HY, Tan WNF, Tan WSD, Li N, et al. House Dust Mite-Induced Asthma Causes Oxidative Damage and DNA Double-Strand Breaks in the Lungs. J Allergy Clin Immunol (2016) 138(1):84-96.e1. doi: 10.1016/j.jaci.2016.02.017
305. Sykora P, Witt KL, Revanna P, Smith-Roe SL, Dismukes J, Lloyd DG, et al. Next Generation High Throughput DNA Damage Detection Platform for Genotoxic Compound Screening. Sci Rep (2018) 8(1):2771. doi: 10.1038/s41598-018-20995-w
306. Townsend TA, Parrish MC, Engelward BP, Manjanatha MG. The Development and Validation of EpiComet-Chip, a Modified High-Throughput Comet Assay for the Assessment of DNA Methylation Status. Environ Mol Mutagen (2017) 58(7):508–21. doi: 10.1002/em.22101
307. Ngo LP, Owiti NA, Swartz C, Winters J, Su Y, Ge J, et al. Sensitive CometChip Assay for Screening Potentially Carcinogenic DNA Adducts by Trapping DNA Repair Intermediates. Nucleic Acids Res (2020) 48(3):e13. doi: 10.1093/nar/gkz1077
308. Chao C, Ngo Le P, Engelward BP. SpheroidChip: Patterned Agarose Microwell Compartments Harboring HepG2 Spheroids are Compatible With Genotoxicity Testing. ACS Biomater Sci Eng (2020) 6(4):2427–39. doi: 10.1021/acsbiomaterials.9b01951
309. Latimer JJ, Kelly CM. Unscheduled DNA Synthesis: The Clinical and Functional Assay for Global Genomic DNA Nucleotide Excision Repair. Methods Mol Biol (2014) 1105:511–32. doi: 10.1007/978-1-62703-739-6_36
310. Pimpley MR, Foley ML, Latimer JJ. New Perspectives on Unscheduled DNA Synthesis: Functional Assay for Global Genomic DNA Nucleotide Excision Repair. Methods Mol Biol (2020) 2102:483–507. doi: 10.1007/978-1-0716-0223-2_27
311. Wienholz F, Vermeulen W, Marteijn JA. Amplification of Unscheduled DNA Synthesis Signal Enables Fluorescence-Based Single Cell Quantification of Transcription-Coupled Nucleotide Excision Repair. Nucleic Acids Res (2017) 45(9):e68. doi: 10.1093/nar/gkw1360
312. Friedman J, Bird LM, Haas R, Robbins SL, Nahas SA, Dimmock DP, et al. Ending a Diagnostic Odyssey: Moving From Exome to Genome to Identify Cockayne Syndrome. Mol Genet Genomic Med (2021) 9(7):e1623. doi: 10.1002/mgg3.1623
313. Brazhnik K, Sun S, Alani O, Kinkhabwala M, Wolkoff AW, Maslov AY, et al. Single-Cell Analysis Reveals Different Age-Related Somatic Mutation Profiles Between Stem and Differentiated Cells in Human Liver. Sci Adv (2020) 6(5):eaax2659. doi: 10.1126/sciadv.aax2659
314. Li W, Calder RB, Mar JC, Vijg J. Single-Cell Transcriptogenomics Reveals Transcriptional Exclusion of ENU-Mutated Alleles. Mutat Res (2015) 772:55–62. doi: 10.1016/j.mrfmmm.2015.01.002
315. Sun S, Brazhnik K, Lee M, Maslov AY, Zhang Y, Huang Z, et al. Single-Cell Analysis of Somatic Mutation Burden in Mammary Epithelial Cells of Pathogenic BRCA1/2 Mutation Carriers. J Clin Invest (2022) 132(5):e148113. doi: 10.1172/JCI148113
316. Zhang L, Dong X, Lee M, Maslov AY, Wang T, Vijg J. Single-Cell Whole-Genome Sequencing Reveals the Functional Landscape of Somatic Mutations in B Lymphocytes Across the Human Lifespan. Proc Natl Acad Sci U.S.A. (2019) 116(18):9014–9. doi: 10.1073/pnas.1902510116
317. Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res (2019) 47(D1):D941–7. doi: 10.1093/nar/gky1015
318. Stuart T, Satija R. Integrative Single-Cell Analysis. Nat Rev Genet (2019) 20(5):257–72. doi: 10.1038/s41576-019-0093-7
319. Wang X, He Y, Zhang Q, Ren X, Zhang Z. Direct Comparative Analyses of 10X Genomics Chromium and Smart-Seq2. Genomics Proteomics Bioinf (2021). doi: 10.1016/j.gpb.2020.02.005
320. Maecker HT, McCoy JP, Nussenblatt R. Standardizing Immunophenotyping for the Human Immunology Project. Nat Rev Immunol (2012) 12(3):191–200. doi: 10.1038/nri3158
321. Mahnke YD, Roederer M. Optimizing a Multicolor Immunophenotyping Assay. Clin Lab Med (2007) 27(3):469–85. doi: 10.1016/j.cll.2007.05.002
322. Sahaf B, Rahman A, Maecker HT, Bendall SC. High-Parameter Immune Profiling With CyTOF. Methods Mol Biol (2020) 2055:351–68. doi: 10.1007/978-1-4939-9773-2_16
323. Buganim Y, Faddah DA, Cheng AW, Itskovich E, Markoulaki S, Ganz K, et al. Single-Cell Expression Analyses During Cellular Reprogramming Reveal an Early Stochastic and a Late Hierarchic Phase. Cell (2012) 150(6):1209–22. doi: 10.1016/j.cell.2012.08.023
324. Wills QF, Livak KJ, Tipping AJ, Enver T, Goldson AJ, Sexton DW, et al. Single-Cell Gene Expression Analysis Reveals Genetic Associations Masked in Whole-Tissue Experiments. Nat Biotechnol (2013) 31(8):748–52. doi: 10.1038/nbt.2642
325. Arsenio J, Kakaradov B, Metz PJ, Kim SH, Yeo GW, Chang JT. Early Specification of CD8+ T Lymphocyte Fates During Adaptive Immunity Revealed by Single-Cell Gene-Expression Analyses. Nat Immunol (2014) 15(4):365–72. doi: 10.1038/ni.2842
326. Moignard V, Woodhouse S, Haghverdi L, Lilly AJ, Tanaka Y, Wilkinson AC, et al. Decoding the Regulatory Network of Early Blood Development From Single-Cell Gene Expression Measurements. Nat Biotechnol (2015) 33(3):269–76. doi: 10.1038/nbt.3154
327. Gibson CJ, Steensma DP. New Insights From Studies of Clonal Hematopoiesis. Clin Cancer Res (2018) 24(19):4633–42. doi: 10.1158/1078-0432.CCR-17-3044
328. Steensma DP. Clinical Implications of Clonal Hematopoiesis. Mayo Clin Proc (2018) 93(8):1122–30. doi: 10.1016/j.mayocp.2018.04.002
329. Libby P, Ebert BL. CHIP (Clonal Hematopoiesis of Indeterminate Potential): Potent and Newly Recognized Contributor to Cardiovascular Risk. Circulation (2018) 138(7):666–8. doi: 10.1161/CIRCULATIONAHA.118.034392
330. Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, et al. Simultaneous Epitope and Transcriptome Measurement in Single Cells. Nat Methods (2017) 14(9):865–8. doi: 10.1038/nmeth.4380
331. Cadot S, Valle C, Tosolini M, Pont F, Largeaud L, Laurent C, et al. Longitudinal CITE-Seq Profiling of Chronic Lymphocytic Leukemia During Ibrutinib Treatment: Evolution of Leukemic and Immune Cells at Relapse. Biomark Res (2020) 8(1):72. doi: 10.1186/s40364-020-00253-w
332. Gayoso A, Steier Z, Lopez R, Regier J, Nazor KL, Streets A, et al. Joint Probabilistic Modeling of Single-Cell Multi-Omic Data With totalVI. Nat Methods (2021) 18(3):272–82. doi: 10.1038/s41592-020-01050-x
333. Albert-Vega C, Tawfik DM, Trouillet-Assant S, Vachot L, Mallet F, Textoris J. Immune Functional Assays, From Custom to Standardized Tests for Precision Medicine. Front Immunol (2018) 9:2367. doi: 10.3389/fimmu.2018.02367
334. Wu HC, Kehm R, Santella RM, Brenner DJ, Terry MB. DNA Repair Phenotype and Cancer Risk: A Systematic Review and Meta-Analysis of 55 Case-Control Studies. Sci Rep (2022) 12(1):3405. doi: 10.1038/s41598-022-07256-7
335. Machella N, Terry MB, Zipprich J, Gurvich I, Liao Y, Senie RT, et al. Double-Strand Breaks Repair in Lymphoblastoid Cell Lines From Sisters Discordant for Breast Cancer From the New York Site of the BCFR. Carcinogenesis (2008) 29(7):1367–72. doi: 10.1093/carcin/bgn140
336. Bordin DL, Lirussi L, Nilsen H. Cellular Response to Endogenous DNA Damage: DNA Base Modifications in Gene Expression Regulation. DNA Repair (Amst) (2021) 99:103051. doi: 10.1016/j.dnarep.2021.103051
337. Fleming AM, Burrows CJ. Oxidative Stress-Mediated Epigenetic Regulation by G-Quadruplexes. NAR Cancer (2021) 3(3):zcab038. doi: 10.1093/narcan/zcab038
338. Jeffery CJ. Protein Moonlighting: What is it, and Why is it Important? Philos Trans R Soc Lond B Biol Sci (2018) 373(1738):20160523. doi: 10.1098/rstb.2016.0523
Keywords: DNA repair, immunity, inter-individual variation, cancer risk, personalized medicine
Citation: Cheong A and Nagel ZD (2022) Human Variation in DNA Repair, Immune Function, and Cancer Risk. Front. Immunol. 13:899574. doi: 10.3389/fimmu.2022.899574
Received: 18 March 2022; Accepted: 20 June 2022;
Published: 22 July 2022.
Edited by:
Carlos F. M. Menck, University of São Paulo, BrazilReviewed by:
Clarissa Ribeiro Reily Rocha, Federal University of São Paulo, BrazilLigia Castro, University of São Paulo, Brazil
Copyright © 2022 Cheong and Nagel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zachary D. Nagel, em5hZ2VsQGhzcGguaGFydmFyZC5lZHU=