 Mathias Busch
Mathias Busch Haribaskar Ramachandran
Haribaskar Ramachandran Tina Wahle
Tina Wahle Andrea Rossi
Andrea Rossi Roel P. F. Schins
Roel P. F. Schins- Institut für Umweltmedizinische Forschung (IUF) – Leibniz-Research Institute for Environmental Medicine, Duesseldorf, Germany
The NLRP3 inflammasome plays an important role in intestinal homeostasis as well as inflammation. However, in vivo studies investigating the role of the NLRP3 inflammasome in inflammatory bowel disease (IBD) report contrasting results, leaving it unclear if the NLRP3 inflammasome augments or attenuates intestinal inflammation. To investigate the role of the NLRP3/caspase-1 pathway in a model of acute intestinal inflammation, we modified a previously established in vitro triple culture model of the healthy and inflamed intestine (Caco-2/HT29-MTX-E12/THP-1). Using THP-1 knockout cell lines, we analyzed how the NLRP3 inflammasome and its downstream enzyme caspase-1 (CASP1) affect inflammatory parameters including barrier integrity and cytotoxicity, as well as gene expression and secretion of pro-inflammatory cytokines and mucus. Furthermore, we investigated differences in inflammation-mediated cytotoxicity towards enterocyte-like (Caco-2) or goblet-like (HT29-MTX-E12) epithelial cells. As a complementary approach, inflammation-related cytotoxicity and gene expression of cytokines was analyzed in intestinal tissue explants from wildtype (WT) and Nlrp3-/- mice. Induction of intestinal inflammation impaired the barrier, caused cytotoxicity, and altered gene expression of pro-inflammatory cytokines and mucins in vitro, while the knockout of NLRP3 and CASP1 in THP 1 cells led to attenuation of these inflammatory parameters. The knockout of CASP1 tended to show a slightly stronger attenuating effect compared to the NLRP3 knockout model. We also found that the inflammation-mediated death of goblet-like cells is NLRP3/caspase-1 dependent. Furthermore, inflammation-related cytotoxicity and upregulation of pro-inflammatory cytokines was present in ileal tissue explants from WT, but not Nlrp3-/- mice. The here presented observations indicate a pro-inflammatory and adverse role of the NLRP3 inflammasome in macrophages during acute intestinal inflammation.
Introduction
Inflammatory bowel disease (IBD), namely Crohn’s disease (CD) or ulcerative colitis (UC) are characterized by chronic, relapsing inflammation of the gastrointestinal tract (1). IBD is thought to originate from an exaggerated immune response towards constituents of the mucosal microbiome, but the precise etiology is still not known (2, 3). As a central activator of the innate immune system, the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome is a crucial regulator of intestinal homeostasis and has been implicated in the pathogenesis of IBD (4, 5). After activation via pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs), the NLRP3 inflammasome oligomerizes and recruits procaspase-1, which leads to the formation of the active caspase-1 (CASP1) (6). CASP1 cleaves the inactive pro-IL-1β, which results in the secretion of mature IL-1β (7). As an early pro-inflammatory cytokine, IL-1β induces subsequent inflammatory cascades (8), including the activation of the transcription factor NF-κB (9). In turn, NF-κB regulates secondary inflammatory cytokines like IL-6 and IL-8 (10, 11).
However, the exact role of the NLRP3 inflammasome in IBD is not clear (12), as different studies report conflicting results: genetic polymorphisms decreasing NLRP3 expression in patients were linked to higher susceptibility towards CD (13), while Nlrp3 knockout mice developed less severe DSS-induced colitis compared to the wild type (WT) (14). The latter effect could be reversed by cohousing Nlrp3-/- mice with WT mice (15), which implies a crucial role of the microbiome in IBD susceptibility. Other studies reported increased colitis symptoms in Nlrp3-/- mice when compared to WT mice (16, 17).
The gastrointestinal epithelium is covered in mucus, which plays an important protective role in intestinal homeostasis. Mucus primarily consists of different mucins, highly glycosylated proteins mainly produced and secreted by goblet cells, but also in parts by enterocytes. Mucins can be divided into two classes: secreted mucins and membrane-associated mucins. Secreted mucins, such as MUC2 and MUC5AC, and also transmembrane mucins, such as MUC1, MUC13, and MUC20, have been shown to be altered or aberrantly expressed in patients suffering from IBD (18–23). This indicates a major role of mucus not only during homeostasis, but also during intestinal inflammation.
We have previously developed an in vitro triple culture model of the healthy and inflamed intestine, comprised of the human cell lines Caco-2, HT29-MTX-E12, and THP-1. This system is capable of replicating several hallmarks of intestinal inflammation, including impairment of barrier integrity, cytotoxicity, an increase of pro-inflammatory cytokine secretion, reduction of acidic mucus secretion, and DNA damage in epithelial cells after prolonged inflammation (24, 25). In these previous studies, this model was applied in toxicological research investigating particle effects in the context of inflammation.
The aim of the present study was to investigate the role of the NLRP3 inflammasome and its downstream enzyme CASP1 in macrophages during initial acute intestinal inflammation by using the in vitro triple culture model with different THP-1 cell lines (WT, CASP1-/- and NLRP3-/-). As a complementary approach, we studied short-term inflammation-related cytotoxicity and gene expression of pro-inflammatory cytokines in intestinal tissue explants from WT and Nlrp3-/- mice.
Materials and Methods
Materials
The cell culture media (DMEM, MEM, RPMI 1640), 2-mercaptoethanol (ME), horse serum, sodium pyruvate, and phosphate-buffered saline (PBS) were purchased from Thermo Fisher Scientific. Fetal calf serum (FCS) for Caco-2 and HT29-MTX-E12, non-essential amino acids (NEAA), L-glutamine, D-glucose, penicillin/streptomycin (P/S), trypsin, phorbol 12-myristate 13-acetate (PMA), interferon-gamma (IFN-γ), lipopolysaccharides (LPS), Accutase, β-nicotinamide adenine dinucleotide sodium salt (NAD), lithium L-lactate, phenazine methosulfate (PMS), iodonitrotetrazolium chloride (INT), Triton X-100, bovine serum albumin (BSA), and 50 nm amine-modified polystyrene nanobeads (PS-NH2) were purchased from Sigma-Aldrich/Merck. Paraformaldehyde (PFA) was ordered from Carl Roth (Germany). The DQ12 quartz sample used in this study was kindly provided by the Institute of Occupational Medicine (IOM, Edinburgh, UK).
Cell Culture
Caco-2 (DSMZ, ACC169) cells were cultured in MEM-based cell culture medium substituted with 20 % FCS, 1 % P/S and 1 % L-glutamine. HT29-MTX-E12 (ECACC, 12040401) cells were cultured in DMEM-based cell culture medium substituted with 10 % FCS, 1 % P/S, and 1 % NEAA. Caco-2 and HT29-MTX-E12 cells were regularly split at roughly 80 % confluence and used at passages 5 – 25 for experiments. THP-1 (ATCC, TIB-202) cells were cultured in RPMI 1640-based cell culture medium (containing L-glutamine and 25 mM HEPES) substituted with 10 % FCS, 1 % P/S, 1 nM sodium pyruvate, 0.7 % D-glucose and 50 nM ME and maintained between 2*105 and 8*105 cells/ml. For experiments, THP-1 cells were used at passages 5 – 15.
Generation of CASP1-Deficient and NLRP3-Deficient THP-1 Cell Lines
CASP1 and NLRP3 THP-1 mutant cells were generated as previously described (26). Briefly, gRNAs were designed using the CRISPR design tool CHOPCHOP (http://chopchop.cbu.uib.no/) and cloned into a modified PX458 plasmid (Addgene #48138). The resulting bicistronic vector encoded the respective gRNA, Cas9 nuclease, and a GFP selection marker. gRNAs efficiency was assessed using High-Resolution Melt Analysis (HRMA). A gRNA targeting CASP1 exon 5 (5’-TAATGAGAGCAAGACGTGTG-3’) and NLRP3 exon 2 (5’-GCTAATGATCGACTTCAATG-3’) were chosen for further experiments (HRMA primers, CASP1: Fwd 5’-CACCGTAATGAGAGCAAGACGTGTG-3’ Rev 5’-AAACCACACGTCTTGCTCTCATTAC-3’; NLRP3: Fwd 5’-CAGACCATGTGGATCTAGCC-3 Rev 5’-TGTTGATCGCAGCGAAGAT-3’). THP-1 cells were electroporated using a Neon transfection system (Thermofisher) according to the manufacturer’s instructions. After 48 h, cells were FACS sorted and plated as single cells into 96-well plates. Cells were duplicated into maintenance and lysis plates after a week. Clones were then lysed with proteinase K and genotyped by PCR followed by deep sequencing using a miSeq Illumina sequencer and a V2 Nano cassette.
Investigation of the Cytokine Profile of THP-1 ko Cell Lines
THP-1 cells (WT, CASP1-/- and NLRP3-/-, at the same passage number) were seeded into T25 flasks at 6*105 cells/ml, differentiated with 100 nM PMA for 24 h, detached with accutase, and seeded in 24-well plates at 120.000 cells/well. Cells were allowed to re-attach for 1.5 h and were treated with different NLRP3 inflammasome activators in RPMI-based medium containing 1% FCS: 10 µg/cm² amino-modified polystyrene nanospheres (PS-NH2), 200 µg/cm² crystalline silica (DQ12), 10 ng/ml LPS or a combination of 10 ng/ml LPS and 10 ng/mL IFN-γ. After 24 h, supernatants were collected and stored at -20°C until analysis via ELISA.
Stable and Inflamed Triple Cultures
The set-up of the stable and inflamed triple cultures was established as described previously (24). In this context, the terms “stable” and “inflamed” relate to the physiological states of the intestine that the model aims to represent. Briefly, Caco-2 and HT29-MTX-E12 cells were seeded in transwell inserts (12-well format) and grown for 21 days to allow differentiation of Caco-2 and mucus production by HT29-MTX-E12 cells. For the stable triple culture, the inserts were transferred onto PMA-differentiated THP-1 cells seeded in 12-well plates (WT, CASP1-/- and NLRP3-/-, at the same passage number). For the inflamed triple culture, epithelial cells were primed with 10 ng/ml IFN-γ for 24 h and then transferred onto PMA-differentiated THP-1 cells, which were pre-activated with 10 ng/ml LPS/IFN-γ for 4 h. Stable and inflamed triple cultures were started in parallel and maintained for 48 h.
Cytokine Quantification by Enzyme-Linked Immuno-Sorbent Assay
The release of IL-1β, IL-8, and TNF-α into the supernatant of THP-1 monocultures (t=24 h) or the basolateral supernatant of triple cultures (t=48 h) was analyzed using R&D systems DuoSet ELISA kits for these proinflammatory cytokines. Antibodies were diluted according to the manufacturer’s protocol and high-protein-binding 96-well plates were incubated with capture antibody in coating buffer (0.1 M NaHCO3, pH 8.2) overnight. After blocking with 3% BSA/PBS, 100 µl of supernatants, diluted if necessary (IL-1β: 1:5 and IL-8: 1:10), were incubated for 2 h. Detection antibody, horseradish peroxidase (1:40 in 1% BSA/PBS), and BioRad TMB Peroxidase EIA Substrate was consecutively incubated for 2 h, 0.5 h, and 5-20 min, respectively, before stopping the reaction with 50 µl 1 M H2SO4. Absorbance was measured at 450 nm and 540 nm. The standard curve was plotted as a four-parameter log fit.
Measurement of Transepithelial Electric Resistance
Barrier integrity during triple cultures was measured as TEER using a voltohmmeter (EVOM, World Precision Instruments) with a chopstick electrode (STX2). Before measurements, the electrode was sterilized in 70 % ethanol and washed with PBS and MEM. TEER measurements were performed at t = 0,4,20,24,44,48 after the start of the triple cultures and were corrected for blank value and transwell filter area (0.9 cm²).
LDH Assay
To investigate cytotoxicity in the epithelial cells after 48 h of triple culture, 50 µl of apical supernatant was transferred onto 96-well plates and 150 µl of reaction mix containing 50 µl Tris buffer (200 mM, pH 8), 50 µl Li-Lactate solution (50 mM), 46 µl NAD+ solution (5 mM), 2 µl INT solution (65 mM) and 2 µl PMS solution (29 mM) was added per well. After 5 min incubation, the reaction was stopped with 50 µl 1 M H2SO4, and absorbance was measured at 490 nm and 680 nm.
Analysis of Cell-Specific Cytotoxicity During Inflammation
To analyze differences in cytotoxicity between Caco-2 and HT29-MTX-E12 cells during inflammation, 104 Caco-2 or HT29-MTX-E12 cells per well were seeded in 96-well plates. After 48 h of growth, cells were treated with basolateral supernatants [t=48h; relative concentrations of 0.25, 0.5 and 1; diluted with RPMI-based medium (e.g. concentration of 0.25: 25 % supernatant in medium; 1: undiluted supernatant)]. Treatment with 0.5% Triton-X 100 for 15 min served as positive control, supernatant at the relative concentrations without cells served as blanks. After 48 h incubation, the LDH assay was performed as described above. Cytotoxicity was calculated as follows: .
Immunofluorescent MUC5AC Staining
To further investigate the cell-specificity of inflammation-derived cytotoxicity, the epithelial cell layer was stained for MUC5AC, a mucin produced by the goblet-like HT29-MTX-E12 cell line, but not Caco-2 (27). After 48 h of triple culture, the filters were fixed in 4 % paraformaldehyde, permeabilized in 0.1 % Triton-X 100/PBS, and blocked with 3 % BSA/PBS. Filters were incubated with MUC5AC primary antibody (mouse; 2 µg/ml) in 1 % BSA/PBS for 1 h at RT and with Alexa 488 goat-anti-mouse antibody (1:300) and Hoechst 33342 (0.5 µg/ml) for 30 min at 37°C. Filters were placed on microscopy slides, mounted with Prolong Gold Antifade, and sealed with a cover slip. Microscopic evaluation was performed using a Zeiss Axio Imager.M2 at 100x magnification. For each condition, 20 images were taken and MUC5AC coverage was quantified as fluorescent area [%], using a macro in ImageJ version 1.53c (see Supplementary Material).
Gene Expression Analysis
Following 48 h of triple culture, gene expression was investigated in the epithelial cell layer and THP-1 cells separately. RNA was isolated using the Roche High Pure RNA Isolation kit. Filters containing epithelial cells were cut out of the inserts using a scalpel and placed in 200 µl ice-cold PBS. THP-1 cells were collected from the well bottom using a cell scraper in 200 µl PBS. 400 µl lysis buffer was added to each sample and vortexed for 15 s. Subsequently, the RNA isolation kit was used according to the manufacturer’s instructions. RNA quantification, DNase I digestion with the Amplification Grade DNase I Kit (Sigma-Aldrich), reverse transcription with the iScript cDNA Synthesis Kit (Bio-Rad), and qRT-PCR were performed as described previously (25). The expression of IL-1β, IL-8, and TNF-α was investigated in both epithelial and THP-1 cells and the expression of MUC1, MUC2, MUC5AC, MUC13, and MUC20 was analyzed in epithelial cells only. β-actin was used as reference gene. Primers are listed in Supplementary Table 1. Changes in gene expression dependent on inflammation status, or dependent on THP-1 cell line were calculated using the ΔΔCT method (28).
Cultivation and Treatment of Ileal Tissue Explants
Ileal tissue was collected from healthy mice (C57BL6/J) under the study approval by the Landesamt für Natur, Umwelt und Verbraucherschutz (LANUV, Northrhine Westphalia, Germany, reference number 81-02.05.50.17.018). WT and Nlrp3-/- mice, pairs of female siblings from heterozygous parents, were sacrificed by cervical dislocation, transferred to a semi-sterile lab bench, and dissected. The ileum was isolated, rinsed with HBSS+/+ supplemented with 1% P/S, cut open length-wise and cut into 12 equally sized pieces (~2 mm²). Tissues were separately placed in the wells of a 24-well plate containing 1 ml HBSS+/+ supplemented with 1% P/S and the 24-well plate was transferred to a sterile lab bench. Using sterile forceps, the explants were transferred to a new 24-well plate containing 1 ml explant medium (DMEM, high glucose, supplemented with 10% horse serum, 1% P/S, 1 µM hydrocortisone, 14.3 nM glucagon, 1 nM 3,3’,5-triiodo-L-thyronine, 200 µM ascorbate-2-phosphate, 20 µM linoleic acid and 10 nM β-estradiol). This procedure and medium composition were adapted from Bareiss et al. (29). 6 of the 12 explants were immediately treated with 1 µg/ml LPS and murine IFN-γ and incubated at 37 °C and 5% CO2 for 6 h. Pilot studies indicated that the viability of these tissue explants decreased significantly when cultivated for longer than 6 h. After 1, 3, and 6 h, 100 µl of supernatant was collected, stored at -20°C, and replaced with 100 µl of fresh medium. The LDH assay was performed with 50 µl of supernatants as described above. 50 µl of medium without tissue served as negative control. After 6 h, tissue explants were snap-frozen in liquid nitrogen and stored at -80°C until further analysis. RNA was isolated from the intestinal tissue explants using the Roche High Pure RNA tissue kit according to the manufacturer’s instructions. Reverse transcription and qRT-PCR was performed as described above. The expression of Il-1β, Il-6, Kc, Mip-2 and Tnf-α was analyzed using β-actin as reference gene and compared to the untreated control of the respective genotype. Primers are listed in Supplementary Table 2.
Statistical Analysis
Statistical analysis and illustration of results were performed using GraphPad Prism 8.3. Unless stated otherwise, experiments were performed in three independent runs with two biological replicates. Statistical analysis of triple culture experiments in stable and inflamed state was performed with two-way ANOVA and Tukey’s test. A p-value of <0.05 was considered statistically significant.
Results
First, we confirmed the successful knockout of CASP1 and NLRP3 in THP-1 cells by quantifying the release of pro-inflammatory cytokines after treatment with different NLRP3 activators. Next, the role of CASP1 and the NLRP3 inflammasome during intestinal inflammation was investigated in an advanced in vitro triple culture model by analyzing inflammation parameters including barrier integrity, cytotoxicity, gene expression, and secretion of cytokines and mucins. Furthermore, we investigated differences in susceptibility of enterocyte- and goblet-like cell types towards inflammation-derived cytotoxicity and investigated inflammation-related cytotoxicity and gene expression in intestinal tissue explants from WT and Nlrp3-/- mice.
Reaction of THP-1 Knockout Cell Lines to Inflammatory Stimuli
To investigate the reaction of the CASP1-/- and NLRP3-/- cells towards pro-inflammatory stimuli, monocultures were treated with the different NLRP3 activators PS-NH2 (30), DQ12 quartz (31), LPS (32) and the combination of LPS and IFN-γ, the activators used to induce the inflamed state in the triple cultures (33). The pro-inflammatory cytokines IL-1β, IL-8, and TNF-α were analyzed after 24 h via ELISA (Figure 1).
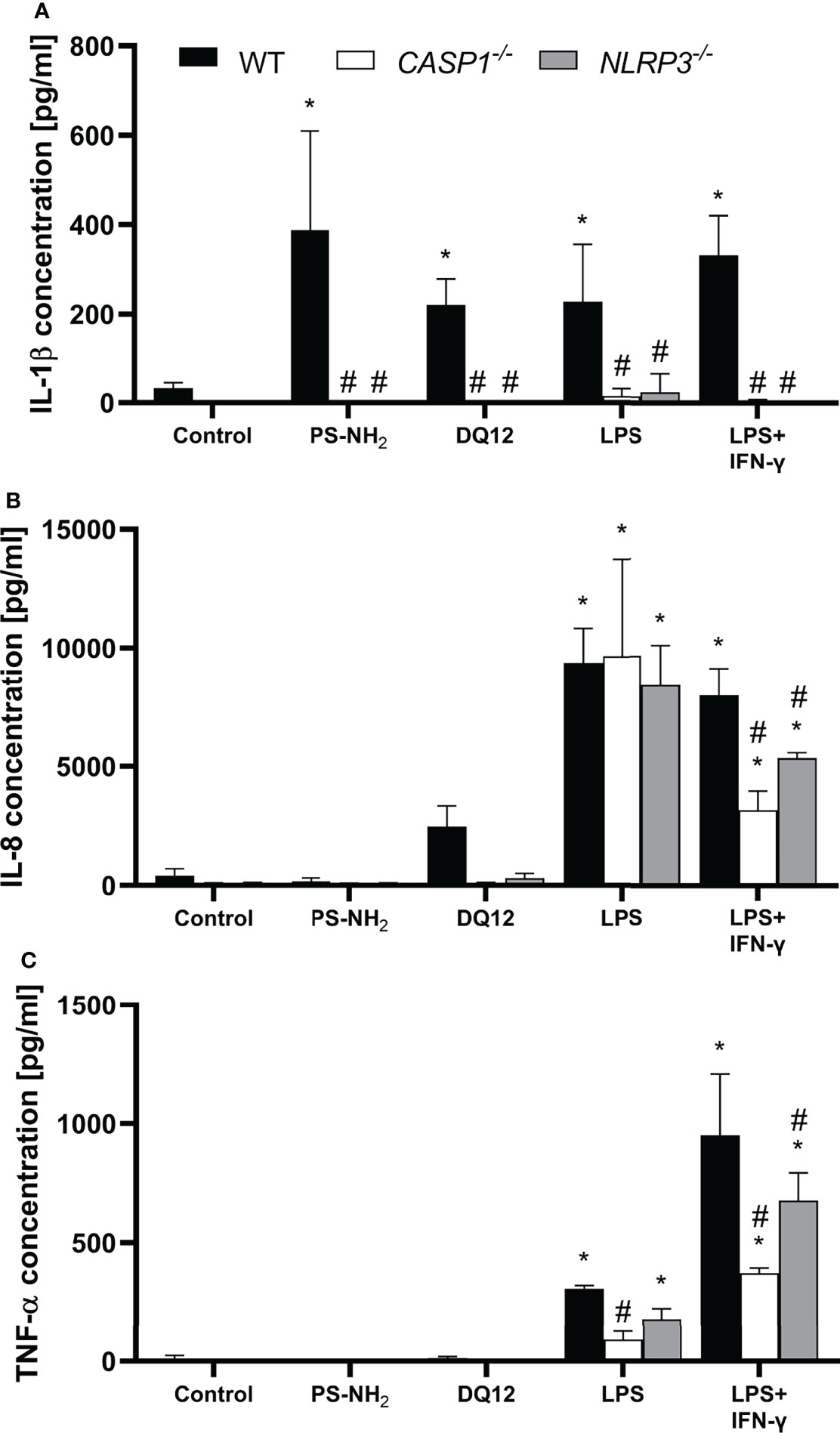
Figure 1 Secretion of (A) IL-1β, (B) IL-8 and (C) TNF-α into the supernatant by differentiated THP-1 cells (wild type, CASP1-/- or NLRP3-/-) after 24 h incubation with 10 µg/cm² amine-modified polystyrene nanobeads (PS-NH2), 200 µg/cm² crystalline silica (DQ12), LPS (10 ng/ml) or LSP/IFN-γ (both 10 ng/ml). Mean ± SD of N = 3; *p < 0.05, compared to the untreated control; #p < 0.05, compared to the WT of the same treatment group.
Treatment with all NLRP3 activators induced a significantly increased secretion of IL-1β by the WT cells, which was absent in both knockout cell lines (Figure 1A). Treatment with LPS alone induced a similarly strong increase in IL-8 secretion in all three cell lines, while increase in TNF-α secretion was statistically significant only in WT and NLRP3-/- THP-1 cells. Treatment with LPS in combination with IFN-γ resulted in a significantly increased secretion of IL-8 and TNF-α in all three cell lines compared to the respective control, but was significantly decreased in both knockout cell lines compared to the WT. The decrease was more prominent in the CASP1-/- THP-1 cells.
Transepithelial Electrical Resistance During Triple Culture
Next, intestinal triple cultures were established with either WT, CASP1-/- or NLRP3-/- THP-1 cells, in both stable or inflamed state. As a primary indicator of barrier integrity, TEER was measured after 24 and 48 h (Figure 2).
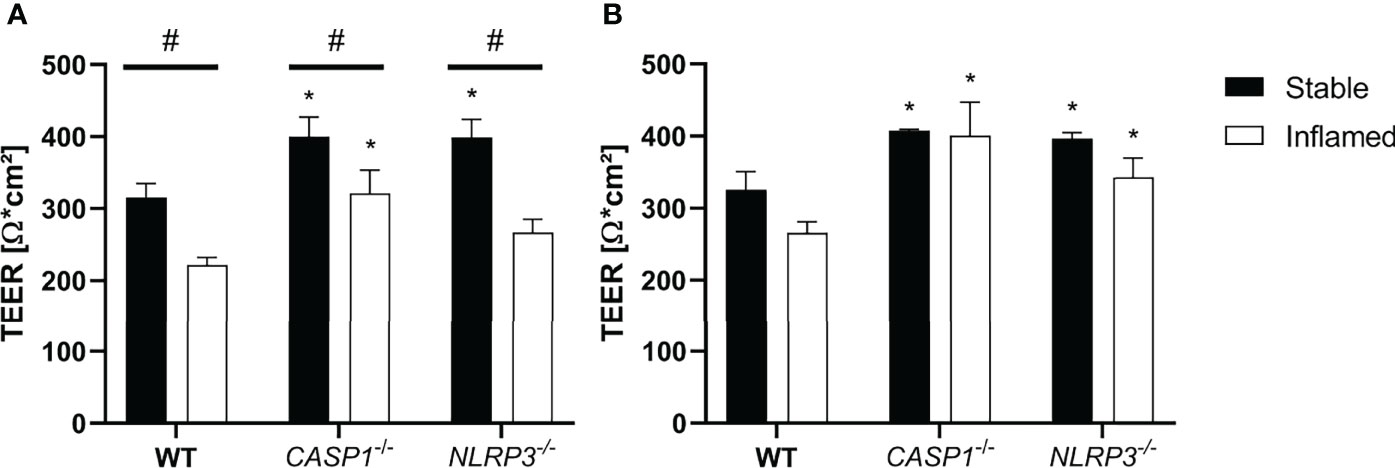
Figure 2 TEER at (A) t=24 h and (B) t=48 h of stable (full bars) and inflamed (open bars) triple-cultures with WT, CASP1-/- or NLRP3-/- THP-1 cells. Mean ± SD of N = 3; *p < 0.05, compared to the respective WT control; #p < 0.05 compared to the respective stable culture.
After 24 h, TEER in inflamed triple cultures was significantly decreased in all three models, compared to the respective stable cultures (Figure 2A). Compared to the WT cultures, TEER of CASP1-/- cultures was significantly increased in both states, while TEER of NLRP3-/- cultures was significantly increased only in stable state. After 48 h, the differences in TEER between stable and inflamed cultures of the same model were no longer statistically significant, but TEER of stable and inflamed CASP1-/- and NLRP3-/- cultures was significantly increased compared to the respective WT cultures (Figure 2B). A more detailed depiction of TEER development over 48 h is given in Supplementary Figure 1.
Cytotoxicity After 48 h of Triple Culture
To assess inflammation-derived cytotoxicity in the epithelial layer, the LDH assay was applied after 48 h of triple culture (Figure 3).
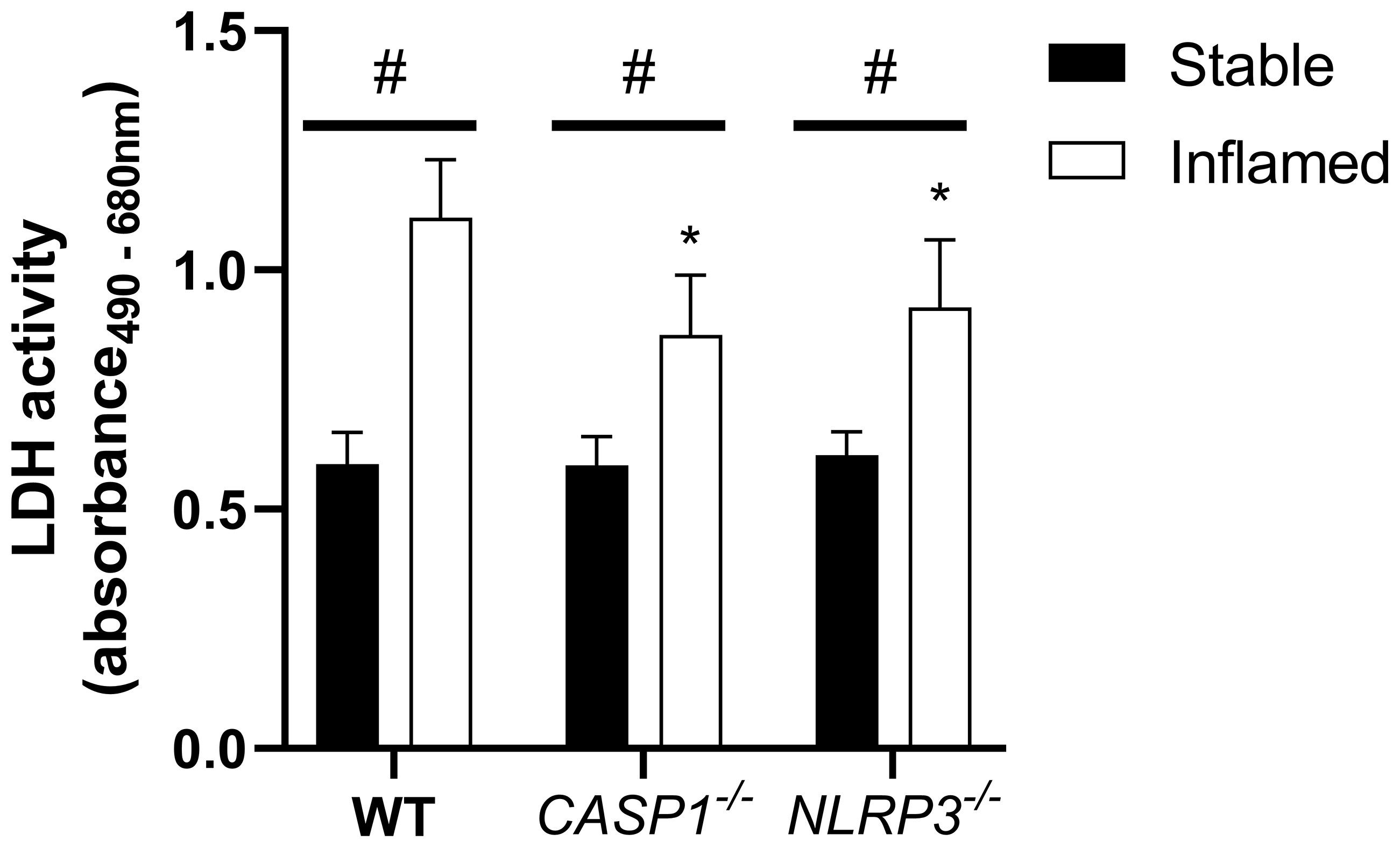
Figure 3 LDH activity in the apical compartment after 48 h of triple culture with WT, CASP1-/- or NLRP3-/- THP-1 cells. Mean + SD of N = 3. *p < 0.05 compared to the respective WT triple culture, #p < 0.05 compared to the respective stable culture.
In all three models, the LDH release into the apical supernatant in inflamed triple cultures was increased compared to the stable cultures. In both inflamed knockout triple cultures, LDH activity was significantly lower compared to the respective WT culture. No difference was observed between the stable cultures of the three models.
Cytokine Release
The cytokine concentrations in the basolateral supernatant of the three different triple cultures in stable or inflamed state were analyzed at t=48h (Figure 4).
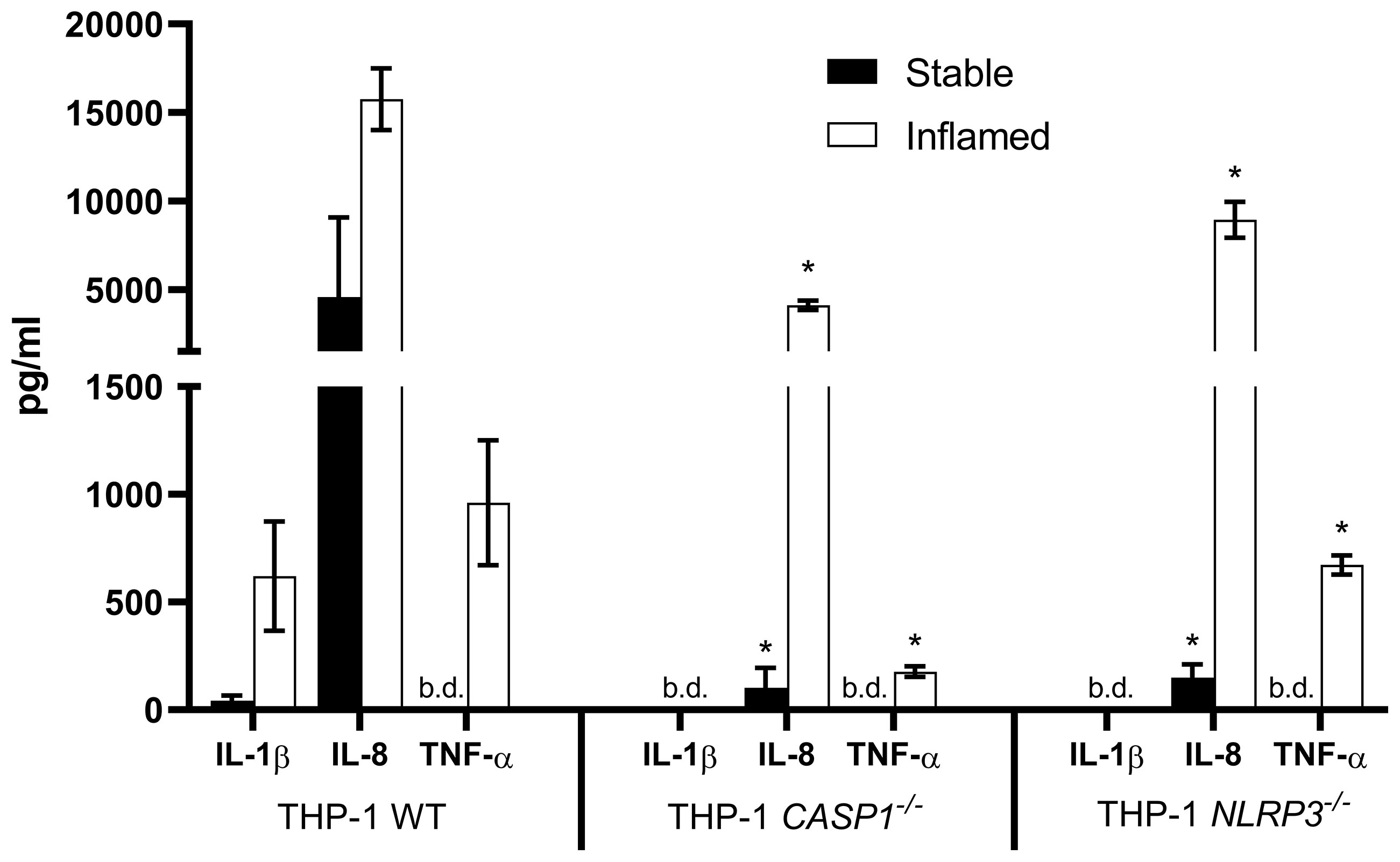
Figure 4 Cytokine release into the basolateral compartment after 48 h of stable (full bars) and inflamed (open bars) triple culture with WT, CASP1-/- or NLRP3-/- THP-1 cells, as assessed by ELISA. Mean ± SD of N = 3. *p < 0.05, compared to the respective WT triple culture. “b.d.” = below detection limit.
As expected, no IL-1β was detected in the CASP1-/- and NLRP3-/- triple cultures, while its release was present in the stable WT culture and strongly increased in the inflamed WT culture. In all three triple cultures, the pro-inflammatory cytokines IL-8 and TNF-α were either strongly increased or only present in the inflamed state. IL-8 concentrations in stable or inflamed state were significantly lower in both knockout cultures when compared to the respective WT culture. TNF-α concentrations were below the detection limit in all three stable cultures. In inflamed state, TNF-α levels were significantly lower in the CASP1-/- and NLRP3-/- triple cultures compared to the WT.
Gene Expression Analysis
After 48 h of triple culture, gene expression of pro-inflammatory cytokines and mucins was analyzed in the epithelium and THP-1 cells (Figure 5).
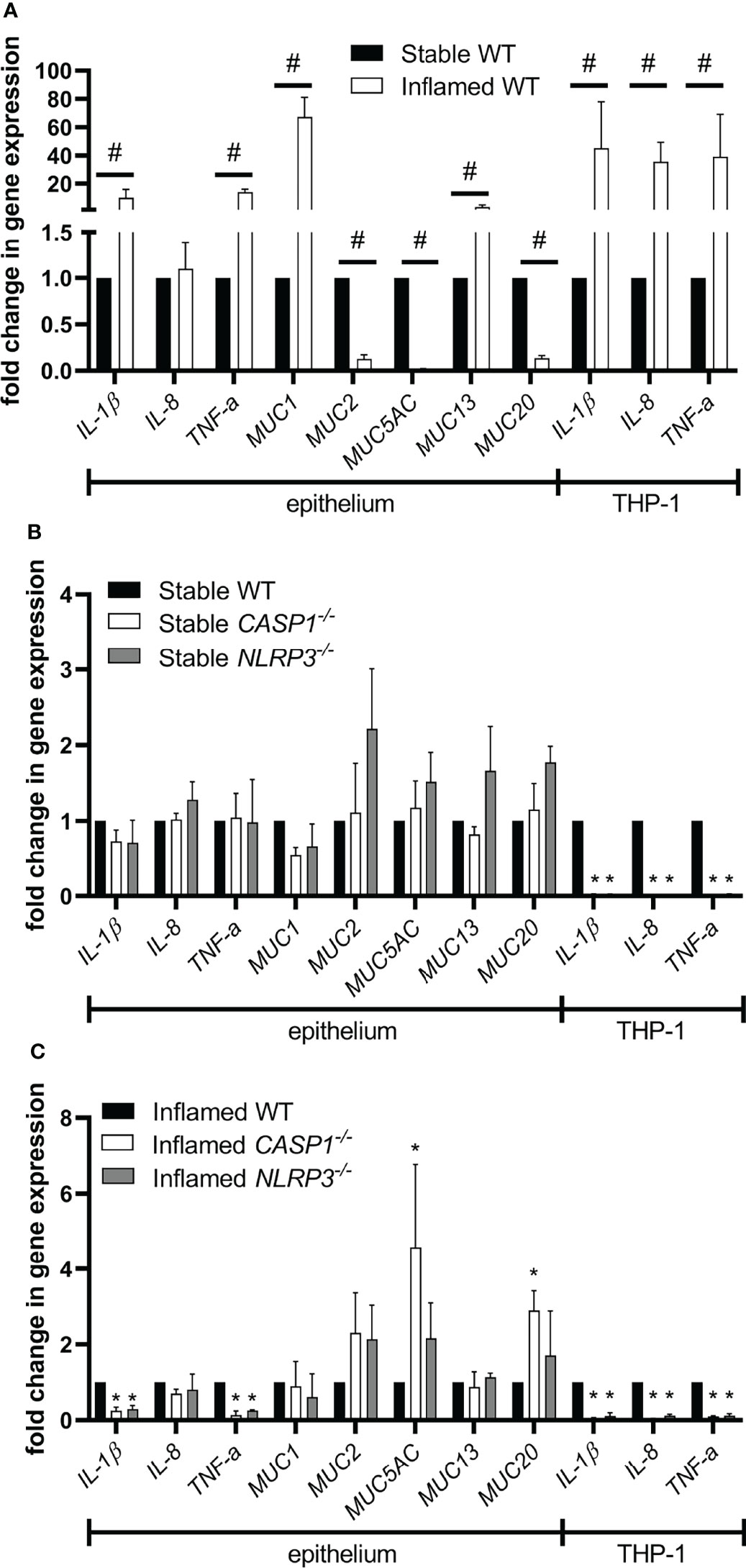
Figure 5 Gene expression in the epithelium or THP-1 cells after 48 h of triple culture. (A) Gene expression in the inflamed WT culture compared to the stable WT culture. (B) Gene expression in the stable knockout cultures compared to the stable WT culture. (C) Gene expression in the inflamed knockout cultures compared to the inflamed WT culture. Mean ± SD of N = 3. #p < 0.05, compared to the stable control; *p < 0.05, compared to the WT control.
Induction of the inflamed state in WT triple cultures resulted in a significant upregulation of IL-1β, TNF-α, MUC1, and MUC13 in the epithelium (Figure 5A). Furthermore, MUC2, MUC5AC, and MUC20 were significantly downregulated. In THP-1 cells, the gene expression of IL-1β, IL-8, and TNF-α was significantly increased in the inflamed state. When comparing the stable triple cultures, no differences in gene expression were observed in the epithelium (Figure 5B). At the same time, the expression of IL-1β, IL-8, and TNF-α was significantly lower in CASP1-/- and NLRP3-/- THP-1 cells in stable triple cultures. Comparing the inflamed triple cultures, the epithelial expression of IL-1β and TNF-α was significantly lower in both knockout cultures, while MUC5AC and MUC20 were upregulated only in inflamed CASP1-/- triple cultures (Figure 5C). In both inflamed knockout cultures, the expression of IL-1β, IL-8, and TNF-α was significantly downregulated in THP-1 cells.
MUC5AC Staining and Quantification
To specifically investigate the presence and distribution pattern of mucus after 48 h of triple culture, the epithelial cell layer was stained for MUC5AC, a mucin produced and secreted by HT29-MTX-E12 cells (Figure 6).
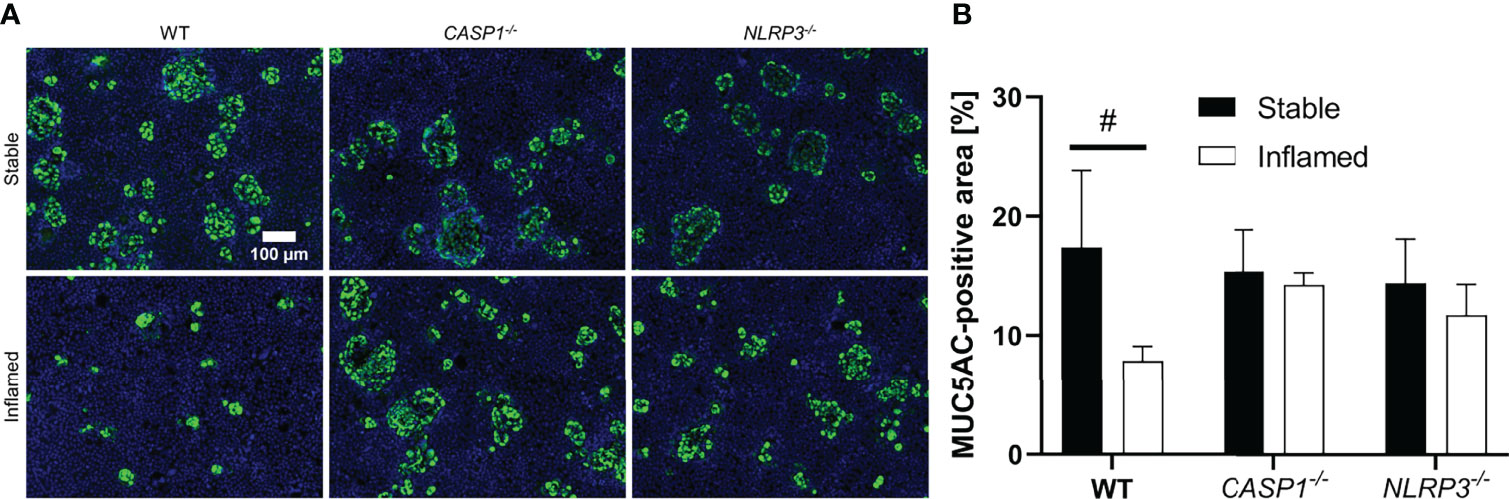
Figure 6 (A) Representative images of MUC5AC-stained epithelial layers of stable (top row) and inflamed (bottom row) triple cultures with WT, CASP1-/- or NLRP3-/- THP-1 cells. Blue: nuclei (Hoechst 33342), green: MUC5AC (Alexa Fluor 488). Images were acquired at 100x magnification. Scale bar (100 µm) applies to all images. (B) Quantitative analysis of MUC5AC-stained area in stable (full bars) or inflamed (open bars) triple cultures. Mean ± SD of N = 3, #p < 0.05 compared to the respective stable culture.
All stable cultures showed large areas of up to several dozen cells, positive for MUC5AC staining distributed across the epithelial layer (Figure 6A), which accounts for ~15% of the total area. No statistically significant difference was observed between stable WT, CASP1-/- and NLRP3-/- cultures (Figure 6B). In the inflamed WT cultures, however, these areas are drastically reduced in size, and the area covered in MUC5AC is significantly reduced (~8%) compared to the stable WT cultures. Such difference between the stable and inflamed state was not observed in the CASP1-/- or NLRP3-/- triple cultures.
Cytotoxic Effects of Triple Culture Supernatants on Epithelial Cell Monocultures
To investigate cell-specific, cytotoxic effects in triple cultures, monocultures of Caco-2 or HT29-MTX-E12 cells were treated with the basolateral supernatants of the triple cultures and the LDH assay was performed (Figure 7).
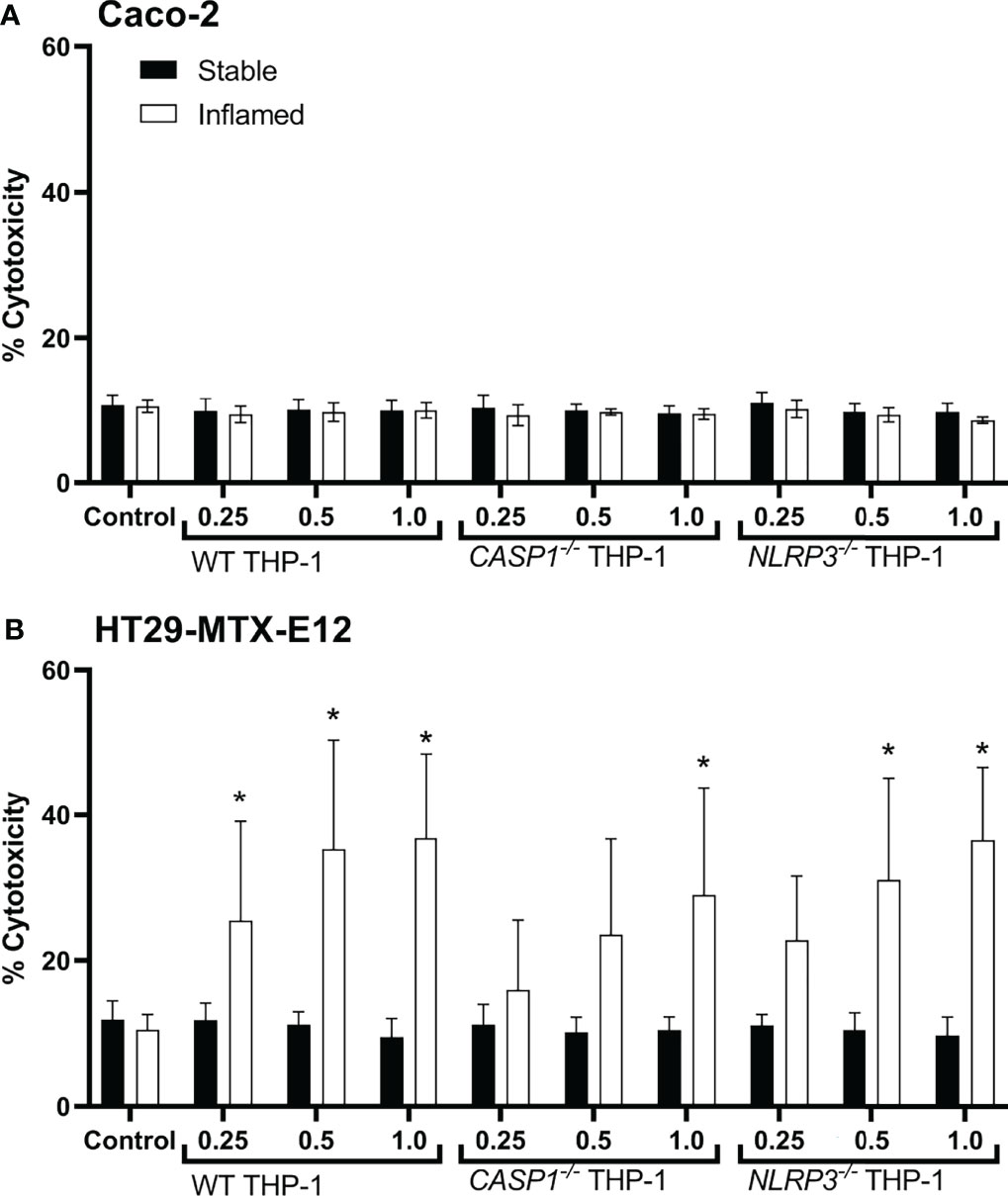
Figure 7 Cytotoxicity in Caco-2 (A) or HT29-MTX-E12 (B) monocultures after treatment with basolateral supernatants ( = 48h; relative concentration 0.25, 0.5 or 1) from stable (full bars) or inflamed (open bars) triple cultures (WT, CASP1-/- or NLRP3-/- THP-1 cells) for 48 (h) Cytotoxicity was assessed via LDH assay. Mean ± SD of N = 5, *p < 0.05 compared to the untreated control.
Treatment with triple culture supernatants did not cause any increased release of LDH in Caco-2 cells (Figure 7A). In HT29-MTX-E12 monocultures, however, the supernatants of all inflamed triple cultures resulted in cytotoxicity in a dose-dependent manner (Figure 7B). Treatment with WT supernatants caused a significant increase in cytotoxicity at a relative concentration as low as 0.25, treatment with NLRP3-/- supernatant at a concentration as low as 0.5, and treatment with CASP1-/- at a concentration of 1 (undiluted supernatant). No effects were observed in HT29-MTX-E12 monocultures when treated with the supernatants of stable triple cultures.
Inflammation-Related Cytotoxicity and Gene Expression of Pro-inflammatory Cytokines in Intestinal Tissue Explants From WT and Nlrp3-/- Mice
To further investigate the role of NLRP3 in intestinal inflammation and compare the in vitro results to ex vivo data, we analyzed the role of the NLRP3 inflammasome on inflammation-related cytotoxicity and gene expression of pro-inflammatory cytokines in murine intestinal tissue explants. Ileal tissue from WT and Nlrp3-/- mice was incubated with the same pro-inflammatory stimuli as the triple cultures (LPS and IFN-γ) and cytotoxicity was assessed after 1, 3, and 6 h, while gene expression was analyzed after 6 h (Figure 8).
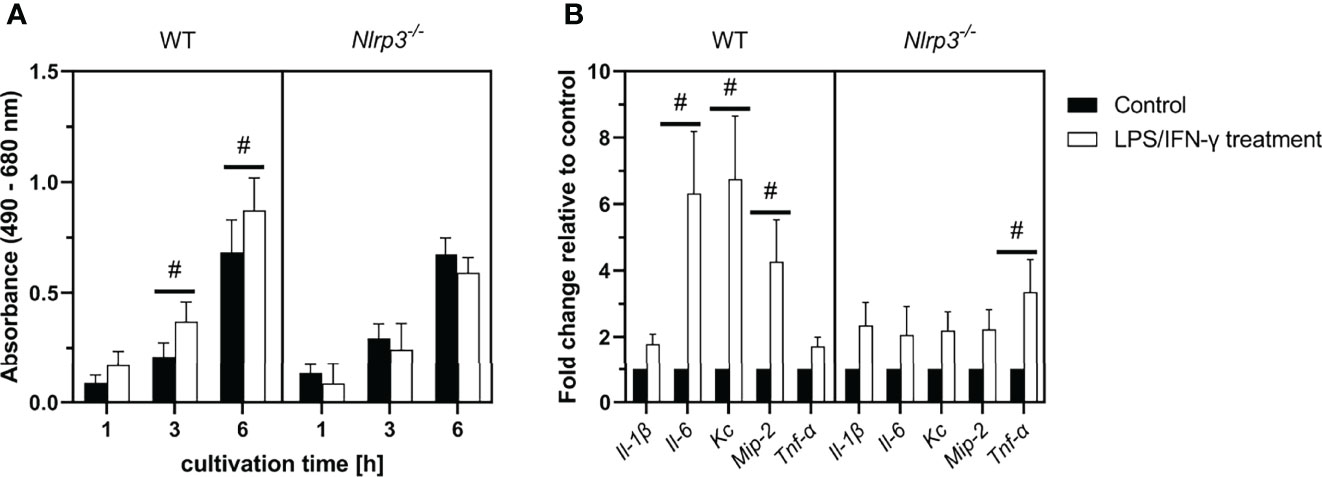
Figure 8 (A) LDH activity in the supernatants of LPS/IFN-γ-treated ileal tissue explants from WT or Nlrp3-/- mice after 1, 3, or 6 (h) (B) Gene expression in ileal tissue explants after 6 h of treatment. Mean ± SEM of N = 4, #p < 0.05 compared to the untreated control of the corresponding time point.
During cultivation, LDH activity in the supernatant of all explant cultures increased over time. No difference was observed between control tissue of WT or Nlrp3-/- mice. However, treatment with LPS and IFN-γ induced a significant increase of LDH activity after 3 and 6 h in the WT explants compared to the untreated control. This effect was not observed in the Nlrp3-/- explants.
In intestinal explants from WT mice, gene expression of the pro-inflammatory mediators Il-6, Kc and Mip-2 was significantly increased after treatment with LPS and IFN-γ. An upregulation of these genes was not observed in treated explants from Nlrp3-/- mice. However, the expression of Tnf-α was significantly increased in the Nlrp3-/-, but not the WT explants after treatment.
Discussion
The precise role of the NLRP3 inflammasome in intestinal inflammation is not understood yet. Although there is consensus about the crucial involvement of NLRP3 in intestinal homeostasis and inflammation, it is not clear if NLRP3 itself is a harmful or protective player (5). In vivo studies investigating DSS-induced colitis in Nlrp3-/- mice reported contrasting results on the severity of inflammation-related parameters compared to WT mice (12).
To investigate the role of NLRP3 in a different model of intestinal inflammation that does not involve animal experiments, we used an advanced in vitro human triple cell line culture of the healthy and inflamed intestine. Regarding inflammatory parameters, the inflamed version of the triple culture offers similar hallmarks as IBD, including an impaired barrier (34), inflammation-derived cytotoxicity (35), and increased concentrations of pro-inflammatory cytokines (36). IBD-associated loss of goblet cells has also been reported in rectal biopsies (37). We were able to replicate and detect all these effects when comparing the inflamed model with the stable, healthy model with WT THP-1 cells. Furthermore, we observed very specific changes in the mucin gene expression profile of the inflamed triple culture. MUC1 and MUC13, both transmembrane mucins, were strongly upregulated during inflammation in vitro. Both mucins have been shown to be upregulated in IBD patients (19, 21). The membrane-associated MUC20 was significantly downregulated during inflammation, which is in line with observations in IBD patients (23). MUC2 and MUC5AC, secreted mucins, were downregulated during inflammation in vitro. A reduced expression of muc2 has also been reported in colitis mice (38), while in contrast, an increased expression of muc5ac was observed in murine colitis (39). Apart from this discrepancy, the mucin expression profile of the inflamed triple culture model correlates very well with data from IBD patients and in vivo studies.
Investigating the effects of knocking out CASP1 or NLRP3 in the THP-1 cells, we found that both deficiencies significantly attenuated the aforementioned inflammatory parameters. Regarding barrier impairment, cytokine concentrations, and cytotoxicity towards goblet cells, the CASP1 knockout tended to show some slightly stronger attenuating effect than NLRP3. When comparing the mucin gene expression profiles of inflamed triple cultures, the knockout of CASP1 in THP-1 caused a significant upregulation of MUC5AC and MUC20. MUC5AC is predominantly secreted by HT29-MTX-E12 cells (27), which appear to be highly sensitive towards the contents of the basolateral supernatant, based on the cytotoxicity assessment in monocultures. This is in line with the MUC5AC staining of the epithelium, where MUC5AC-positive area is strongly reduced in the inflamed WT cultures. Taking these results together, it appears that HT29-MTX-E12 cells are proportionally more vulnerable during the inflamed state, compared to Caco-2 cells, which might explain the strong downregulation of MUC5AC. This downregulation is attenuated especially in CASP1-/- triple cultures, where the supernatant is less toxic towards HT29-MTX-E12 cells and more MUC5AC-positive cells remain on the filter after 48 h.
The expression and secretion of IL-1β, IL-8, and TNF-α, all crucial cytokines in inflammatory responses, is significantly reduced in LPS/IFN-γ treated CASP1-/- and NLRP3-/- THP-1 monocultures, as well as in inflamed CASP1-/- and NLRP3-/- triple cultures. This indicates that their release in THP-1 cells is directly dependent on (IL-1β) or influenced (IL-8 and TNF-α) by the NLRP3/CASP1 pathway. Furthermore, the gene expression of IL-1β and TNF-α is significantly lower in the epithelium of inflamed knockout cultures as well, meaning that the NLRP3/CASP1 pathway of the macrophage-like THP-1 cells affects the gene expression of the same cytokines in epithelial cells. While IL-1β is no longer detectable in knockout cultures, the concentrations of the cytokines IL-8 and TNF-α follow a gradient showing the highest concentrations in the WT cultures, followed by the NLRP3-/- and CASP1-/- cultures. Pro-apoptotic properties have been demonstrated for both IL-1β and TNF-α (40–42), while for IL-8, anti-apoptotic properties have been reported (43–45). Since IL-1β is completely absent in the knockout cultures, TNF-α might be the main cytokine responsible for apoptotic processes in the epithelium, which are still present, although to a lesser degree than in the WT cultures.
Our results show an enhanced susceptibility of goblet-like HT29-MTX-E12 cells towards inflammation-mediated cytotoxicity, compared to enterocyte-like Caco-2 cells. Strong synergistic cytotoxicity of TNF-α and IFN-γ towards HT29 cells has been described previously (46, 47). For our investigations, a high dose of IFN-γ is used to initially activate the THP-1 cells, so it is unlikely that the concentration of this cytokine differs significantly between the three inflamed cultures. However, analysis via ELISA revealed significant differences in TNF-α concentrations, which correlates to the cytotoxicity levels in inflamed triple cultures discussed above and HT29-MTX-E12 monocultures. Furthermore, TNF-α reportedly affects epithelial tight junctions and permeability (48). This might explain the lesser reduction of TEER in inflamed knockout cultures compared to the WT cultures. The relevance of TNF-α in intestinal inflammatory processes is well known, as anti-TNF-therapy is approved for the treatment of Crohn’s disease since 1998 (49). However, the interplay between the different cytokines, as well as other factors, is likely responsible for the differences in cytotoxicity and barrier impairment. Thus, further studies are necessary to investigate the exact role of TNF-α in this specific approach.
Summarizing the results of the triple culture experiments, the NLRP3/CASP1 pathway seems to play an adverse role in acute intestinal inflammation, as several inflammatory parameters are less severe in knockout cultures. The concentration of TNF-α, a secondary pro-inflammatory cytokine, is NLRP3-dependent and could be responsible for the alteration of specific cytotoxic processes during inflammation. The findings of the intestinal explant experiments support the harmful role of NLRP3: In contrast to tissues from WT mice, ileal tissues from Nlrp3-/- mice showed no signs of increased cytotoxicity after treatment with the same pro-inflammatory stimuli as the in vitro cultures. Furthermore, the gene expression of Il-6 and the murine IL-8 homologues Kc and Mip-2 was increased after treatment only in the WT tissue explants. Surprisingly, the expression of Tnf-α was significantly increased in treated Nlrp3-/- explants, but not WT explants. As Nlrp3-/- mice also show a constitutively increased expression of Tnf-α in ileal tissue when directly compared to WT mice (see Supplementary Figure 2), we hypothesize that the upregulation of Tnf-α acts as a compensation for the lack of Nlrp3 as a defence mechanism in the knockout mice.
Interestingly, different studies investigating the role of the NLRP3/CASP1 pathway in mice with DSS-induced colitis are either in line with our findings or report the complete opposite. Bauer et al. (14) and Elinav et al. (50) also describe a harmful role of NLRP3 based on data from Nlrp3-/- mice, while Allen et al. (16) and Hirota et al. (17) report a protective role. Siegmund et al. (51) observed complete protection against DSS-induced colitis in CASP1-/- mice, while Dupaul-Chicoine et al. (52) reported that CASP1-/- mice died rapidly after DSS administration. Factors responsible for these contradicting findings are currently discussed and could include differences in microbiome and genetic background of the mice, as well as different protocols to induce experimental colitis (5). The triple culture, however, exhibits several differences compared to mouse models, including a drastically reduced number of different cell types and the lack of a microbiome. The method of induction of the inflammation and the duration of it differ as well. One major difference is the knockout of NLRP3 only in macrophage-representing THP-1 cells, while in mouse models, all cell types are deficient of Nlrp3-/-. This means the mentioned in vivo studies investigated the role of NLRP3 during intestinal inflammation in general, while the presented triple culture experiments investigated the precise role of NLRP3 in macrophages. Zaki et al. (53) generated chimeric mice with Nlrp3-deficient hematopoietic cells (including macrophages) by transplanting bone marrow from Nlrp3-/- into WT mice, which represent a better in vivo analogue to our triple cultures with THP-1 knockouts. These mice were less susceptible to DSS-induced colitis than non-chimeric Nlrp3-/- mice, comparable to WT mice. The authors concluded that NLRP3 in non-hematopoietic cells (i.e. the epithelium) is more important for protection against intestinal inflammation than in hematopoietic cells. This is supported by the findings of Song-Zhao et al. (54); the authors showed that epithelial Nlrp3 offers early protection against bacterial-driven intestinal inflammation. Therefore, the NLRP3 inflammasome could have different, or even opposite roles during intestinal inflammation, depending on the cell type.
One limitation of the present study is the limited comparability to the in vivo studies. Inflammation itself is an in vivo phenomenon and only parts of it can be recreated in vitro. Furthermore, IBD is a chronic disease, while this in vitro model only allows investigations of initial, acute inflammatory responses. Nevertheless, our model exhibits several important hallmarks of the disease and compares well to data from intestinal tissue explants. Most importantly, it allows easy investigation and/or manipulation of separate cell types, which would be much more complex in an in vivo setup.
In conclusion, the use of THP-1 knockout cell lines in a triple culture model of the inflamed intestine presents a valid approach to investigate the role of the NLRP3/CASP1 pathway during acute intestinal inflammation. Our in vitro experiments suggest an adverse and pro-inflammatory role of NLRP3 in macrophages. The adverse role of NLRP3 was supported by data obtained from murine intestinal explants. The death of goblet cells and the subsequent changes in mucin expression and secretion appear to play an important role in acute intestinal inflammation, which seems to be indirectly linked to the activation of NLRP3.
Data Availability Statement
The data presented in the study are deposited in the Mendeley Data repository, accession number 10.17632/5hy2sr37mm.2.
Ethics Statement
The animal study was reviewed and approved by Landesamt für Natur, Umwelt und Verbraucherschutz (LANUV, Northrhine Westphalia, Germany, reference number 81-02.05.50.17.018).
Author Contributions
MB: Conceptualization, Investigation, Analysis, Visualization, Writing. HR: Methodology. TW: Methodology, Investigation. AR: Methodology. RS: Conceptualization, Supervision, Writing. All authors contributed to the article and approved the submitted version.
Funding
This project was funded by the Juergen Manchot Foundation, Duesseldorf, Germany through a Ph.D. scholarship for Mathias Busch.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors are thankful to Gerrit Bredeck of the IUF – Leibniz-Research Institute for Environmental Medicine, Duesseldorf, for critically reviewing the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.898039/full#supplementary-material
References
1. Ponder A, Long MD. A Clinical Review of Recent Findings in the Epidemiology of Inflammatory Bowel Disease. Clin Epidemiol (2013) 5:237–47. doi: 10.2147/CLEP.S33961
2. Bouma G, Strober W. The Immunological and Genetic Basis of Inflammatory Bowel Disease. Nat Rev Immunol (2003) 3:521–33. doi: 10.1038/nri1132
3. Cho JH, Brant SR. Recent Insights Into the Genetics of Inflammatory Bowel Disease. Gastroenterology (2011) 140:1704–12. doi: 10.1053/j.gastro.2011.02.046
4. Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, et al. Critical Role for NALP3/CIAS1/Cryopyrin in Innate and Adaptive Immunity Through its Regulation of Caspase-1. Immunity (2006) 24:317–27. doi: 10.1016/j.immuni.2006.02.004
5. Zhen Y, Zhang H. NLRP3 Inflammasome and Inflammatory Bowel Disease. Front Immunol (2019) 10:276. doi: 10.3389/fimmu.2019.00276
6. McKee CM, Coll RC. NLRP3 Inflammasome Priming: A Riddle Wrapped in a Mystery Inside an Enigma. J Leukoc Biol (2020) 108:937–52. doi: 10.1002/JLB.3MR0720-513R
7. Lopez-Castejon G, Brough D. Understanding the Mechanism of IL-1β Secretion. Cytokine Growth Factor Rev (2011) 22:189–95. doi: 10.1016/j.cytogfr.2011.10.001
8. Siegmund B. Interleukin-1β Converting Enzyme (Caspase-1) in Intestinal Inflammation. Biochem Pharmacol (2002) 64:1–8. doi: 10.1016/S0006-2952(02)01064-X
9. Parikh AA, Salzman AL, Kane CD, Fischer JE, Hasselgren P. IL-6 Production in Human Intestinal Epithelial Cells Following Stimulation With IL-1β Is Associated With Activation of the Transcription Factor NF-kB. J Surg Res (1997) 69:139–44. doi: 10.1006/jsre.1997.5061
10. Brasier AR. The Nuclear factor-kappaB-Interleukin-6 Signalling Pathway Mediating Vascular Inflammation. Cardiovasc Res (2010) 86:211–8. doi: 10.1093/cvr/cvq076
11. Kunsch C, Rosen CA. NF-Kappa B Subunit-Specific Regulation of the Interleukin-8 Promoter. Mol Cell Biol (1993) 13:6137–46. doi: 10.1128/mcb.13.10.6137-6146.1993
12. Perera AP, Sajnani K, Dickinson J, Eri R, Körner H. NLRP3 Inflammasome in Colitis and Colitis-Associated Colorectal Cancer. Mamm Genome (2018) 29:817–30. doi: 10.1007/s00335-018-9783-2
13. Villani A-C, Lemire M, Fortin G, Louis E, Silverberg MS, Collette C, et al. Common Variants in the NLRP3 Region Contribute to Crohn’s Disease Susceptibility. Nat Genet (2009) 41:71–6. doi: 10.1038/ng.285
14. Bauer C, Duewell P, Mayer C, Lehr HA, Fitzgerald KA, Dauer M, et al. Colitis Induced in Mice With Dextran Sulfate Sodium (DSS) is Mediated by the NLRP3 Inflammasome. Gut (2010) 59:1192–9. doi: 10.1136/gut.2009.197822
15. Bauer C, Duewell P, Lehr H-A, Endres S, Schnurr M. Protective and Aggravating Effects of Nlrp3 Inflammasome Activation in IBD Models: Influence of Genetic and Environmental Factors. Dig Dis (2012) 30(suppl 1):82–90. doi: 10.1159/000341681
16. Allen IC, TeKippe EM, Woodford R-MT, Uronis JM, Holl EK, Rogers AB, et al. The NLRP3 Inflammasome Functions as a Negative Regulator of Tumorigenesis During Colitis-Associated Cancer. J Exp Med (2010) 207:1045–56. doi: 10.1084/jem.20100050
17. Hirota SA, Ng J, Lueng A, Khajah M, Parhar K, Li Y, et al. NLRP3 Inflammasome Plays a Key Role in the Regulation of Intestinal Homeostasis. Inflamm Bowel Dis (2011) 17:1359–72. doi: 10.1002/ibd.21478
18. Forgue-Lafitte M-E, Fabiani B, Levy PP, Maurin N, Fléjou J-F, Bara J. Abnormal Expression of M1/MUC5AC Mucin in Distal Colon of Patients With Diverticulitis, Ulcerative Colitis and Cancer. Int J Canc (2007) 121:1543–9. doi: 10.1002/ijc.22865
19. Furr AE, Ranganathan S, Finn OJ. Aberrant Expression of MUC1 Mucin in Pediatric Inflammatory Bowel Disease. Pediatr Dev Pathol (2010) 13:24–31. doi: 10.2350/08-06-0479.1
20. Larsson JMH, Karlsson H, Crespo JG, Johansson MEV, Eklund L, Sjövall H, et al. Altered O-Glycosylation Profile of MUC2 Mucin Occurs in Active Ulcerative Colitis and Is Associated With Increased Inflammation. Inflamm Bowel Dis (2011) 17:2299–307. doi: 10.1002/ibd.21625
21. Sheng YH, Lourie R, Lindén SK, Jeffery PL, Roche D, Tran TV, et al. The MUC13 Cell-Surface Mucin Protects Against Intestinal Inflammation by Inhibiting Epithelial Cell Apoptosis. Gut (2011) 60:1661–70. doi: 10.1136/gut.2011.239194
22. Sheng YH, Hasnain SZ, Florin THJ, McGuckin MA. Mucins in Inflammatory Bowel Diseases and Colorectal Cancer. JGH Open (2012) 27:28–38. doi: 10.1111/j.1440-1746.2011.06909.x
23. Yamamoto-Furusho JK, Ascaño-Gutiérrez I, Furuzawa-Carballeda J, Fonseca-Camarillo G. Differential Expression of MUC12, MUC16, and MUC20 in Patients With Active and Remission Ulcerative Colitis. Mediator Inflamm (2015) 2015:659018. doi: 10.1155/2015/659018
24. Busch M, Bredeck G, Kämpfer AA, Schins RP. Investigations of Acute Effects of Polystyrene and Polyvinyl Chloride Micro- and Nanoplastics in an Advanced In Vitro Triple Culture Model of the Healthy and Inflamed Intestine. Environ Res (2021) 193:110536. doi: 10.1016/j.envres.2020.110536
25. Kämpfer AAM, Busch M, Büttner V, Bredeck G, Stahlmecke B, Hellack B, et al. Model Complexity as Determining Factor for In Vitro Nanosafety Studies: Effects of Silver and Titanium Dioxide Nanomaterials in Intestinal Models. Small (2021) 17(15):2004223. doi: 10.1002/smll.202004223
26. Ramachandran H, Martins S, Kontarakis Z, Krutmann J, Rossi A. Fast But Not Furious: A Streamlined Selection Method for Genome-Edited Cells. Life Sci Allianc (2021) 4:e202101051. doi: 10.26508/lsa.202101051
27. Elzinga J, van der Lugt B, Belzer C, Steegenga WT. Characterization of Increased Mucus Production of HT29-MTX-E12 Cells Grown Under Semi-Wet Interface With Mechanical Stimulation. PloS One (2021) 16:e0261191. doi: 10.1371/journal.pone.0261191
28. Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the DDCT Method. Methods (2001) 25:402–8. doi: 10.1006/meth.2001.1262
29. Bareiss PM, Metzger M, Sohn K, Rupp S, Frick JS, Autenrieth IB, et al. Organotypical Tissue Cultures From Adult Murine Colon as an In Vitro Model of Intestinal Mucosa. Histochem Cell Biol (2008) 129:795–804. doi: 10.1007/s00418-008-0405-z
30. Lunov O, Syrovets T, Loos C, Nienhaus GU, Mailänder V, Landfester K, et al. Amino-Functionalized Polystyrene Nanoparticles Activate the NLRP3 Inflammasome in Human Macrophages. ACS Nano (2011) 5:9648–57. doi: 10.1021/nn203596e
31. Peeters PM, Eurlings IMJ, Perkins TN, Wouters EF, Schins RPF, Borm PJA, et al. Silica-Induced NLRP3 Inflammasome Activation In Vitro and in Rat Lungs. Part Fibre Toxicol (2014) 11:58. doi: 10.1186/s12989-014-0058-0
32. Schumann RR, Belka C, Reuter D, Lamping N, Kirschning CJ, Weber JR, et al. Lipopolysaccharide Activates Caspase-1 (Interleukin-1–Converting Enzyme) in Cultured Monocytic and Endothelial Cells. Blood (1998) 91:577–84. doi: 10.1182/blood.V91.2.577
33. Kämpfer AAM, Urbán P, Gioria S, Kanase N, Stone V, Kinsner-Ovaskainen A. Development of an In Vitro Co-Culture Model to Mimic the Human Intestine in Healthy and Diseased State. Toxicol In Vitro (2017) 45:31–43. doi: 10.1016/j.tiv.2017.08.011
34. Maloy KJ, Powrie F. Intestinal Homeostasis and Its Breakdown in Inflammatory Bowel Disease. Nature (2011) 474:298–306. doi: 10.1038/nature10208
35. Kappeler A, Mueller C. The Role of Activated Cytotoxic T Cells in Inflammatory Bowel Disease. Histol Histopathol (2000) 15:167–72. doi: 10.14670/HH-15.167
36. Neurath MF. Cytokines in Inflammatory Bowel Disease. Nat Rev Immunol (2014) 14:329–42. doi: 10.1038/nri3661
37. Strugala V, Dettmar PW, Pearson JP. Thickness and Continuity of the Adherent Colonic Mucus Barrier in Active and Quiescent Ulcerative Colitis and Crohn’s Disease. Int J Clin Pract (2008) 62:762–9. doi: 10.1111/j.1742-1241.2007.01665.x
38. Itani S, Watanabe T, Nadatani Y, Sugimura N, Shimada S, Takeda S, et al. NLRP3 Inflammasome has a Protective Effect Against Oxazolone-Induced Colitis: A Possible Role in Ulcerative Colitis. Sci Rep (2016) 6:39075. doi: 10.1038/srep39075
39. Olli KE, Rapp C, O’Connell L, Collins CB, McNamee EN, Jensen O, et al. Muc5ac Expression Protects the Colonic Barrier in Experimental Colitis. Inflamm Bowel Dis (2020) 26:1353–67. doi: 10.1093/ibd/izaa064
40. Friedlander RM, Galiardini V, Rotello RJ, Yuan J. Functional Role of Interleukin 1β (IL-1β) in IL-1β-Converting Enzyme-Mediated Apoptosis. J Exp Med (1996) 184:717–24. doi: 10.1084/jem.184.2.717
41. Shen J, Xu S, Zhou H, Liu H, Jiang W, Hao J, et al. IL-1β Induces Apoptosis and Autophagy via Mitochondria Pathway in Human Degenerative Nucleus Pulposus Cells. Sci Rep (2017) 7:41067. doi: 10.1038/srep41067
42. van Antwerp DJ, Martin SJ, Verma IM, Green DR. Inhibition of TNF-Induced Apoptosis by NF-κb. Trends Cell Biol (1998) 8:107–11. doi: 10.1016/S0962-8924(97)01215-4
43. Guo Y, Zang Y, Lv L, Cai F, Qian T, Zhang G, et al. IL−8 Promotes Proliferation and Inhibition of Apoptosis via STAT3/AKT/Nf−κb Pathway in Prostate Cancer. Mol Med Rep (2017) 16:9035–42. doi: 10.3892/mmr.2017.7747
44. Kettritz R, Gaido ML, Haller H, Luft FC, Jennette CJ, Falk RJ. Interleukin-8 Delays Spontaneous and Tumor Necrosis Factor-Alpha-Mediated Apoptosis of Human Neutrophils. Kidney Int (1998) 53:84–91. doi: 10.1046/j.1523-1755.1998.00741.x
45. Osawa Y, Nagaki M, Banno Y, Brenner DA, Asano T, Nozawa Y, et al. Tumor Necrosis Factor Alpha-Induced Interleukin-8 Production via NF-kappaB and Phosphatidylinositol 3-Kinase/Akt Pathways Inhibits Cell Apoptosis in Human Hepatocytes. Infect Immun (2002) 70:6294–301. doi: 10.1128/IAI.70.11.6294-6301.2002
46. Deem R, Shanahan F, Targan S. Triggered Human Mucosal T Cells Release Tumour Necrosis Factor-Alpha and Interferon-Gamma Which Kill Human Colonic Epithelial Cells. Clin Exp Immunol (1991) 83:79–84. doi: 10.1111/j.1365-2249.1991.tb05592.x
47. Woznicki JA, Saini N, Flood P, Rajaram S, Lee CM, Stamou P, et al. TNF-α Synergises With IFN-γ to Induce Caspase-8-JAK1/2-STAT1-Dependent Death of Intestinal Epithelial Cells. Cell Death Dis (2021) 12:864. doi: 10.1038/s41419-021-04151-3
48. Mullin JM, Snock KV. Effect of Tumor Necrosis Factor on Epithelial Tight Junctions and Transepithelial Permeability. Cancer Res (1990) 50:2172–6.
49. Adegbola SO, Sahnan K, Warusavitarne J, Hart A, Tozer P. Anti-TNF Therapy in Crohn’s Disease. Int J Mol Sci (2018) 19. doi: 10.3390/ijms19082244
50. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 Inflammasome Regulates Colonic Microbial Ecology and Risk for Colitis. Cell (2011) 145:745–57. doi: 10.1016/j.cell.2011.04.022
51. Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA. IL-1b-Converting Enzyme (Caspase-1) in Intestinal Inflammation. Proc Natl Acad Sci (2001) 98:13249–54. doi: 10.1073/pnas.23147399
52. Dupaul-Chicoine J, Yeretssian G, Doiron K, Bergstrom KSB, McIntire CR, LeBlanc PM, et al. Control of Intestinal Homeostasis, Colitis, and Colitis-Associated Colorectal Cancer by the Inflammatory Caspases. Immunity (2010) 32:367–78. doi: 10.1016/j.immuni.2010.02.012
53. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti T-D. The NLRP3 Inflammasome Protects Against Loss of Epithelial Integrity and Mortality During Experimental Colitis. Immunity (2010) 32:379–91. doi: 10.1016/j.immuni.2010.03.003
Keywords: caco-2, HT29-MTX-E12, gastrointestinal tract, IBD, barrier, inflammatory disease, mucus, macrophages
Citation: Busch M, Ramachandran H, Wahle T, Rossi A and Schins RPF (2022) Investigating the Role of the NLRP3 Inflammasome Pathway in Acute Intestinal Inflammation: Use of THP-1 Knockout Cell Lines in an Advanced Triple Culture Model. Front. Immunol. 13:898039. doi: 10.3389/fimmu.2022.898039
Received: 17 March 2022; Accepted: 16 June 2022;
Published: 13 July 2022.
Edited by:
Laura Stronati, Sapienza University of Rome, ItalyReviewed by:
Salah Amasheh, Freie Universität Berlin, GermanyRoberta Vitali, Energy and Sustainable Economic Development (ENEA), Italy
Copyright © 2022 Busch, Ramachandran, Wahle, Rossi and Schins. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roel P. F. Schins, Um9lbC5TY2hpbnNASVVGLUR1ZXNzZWxkb3JmLmRl