 Clara Franco-Jarava1,2,3
Clara Franco-Jarava1,2,3 Irene Valenzuela4,5
Irene Valenzuela4,5 Jacques G. Riviere3,6,7Marina Garcia-Prat3,6,7
Jacques G. Riviere3,6,7Marina Garcia-Prat3,6,7 Mónica Martínez-Gallo1,2,3Romina Dieli-Crimi1,2,3Neus Castells4,5Laura Batlle-Masó3,6
Mónica Martínez-Gallo1,2,3Romina Dieli-Crimi1,2,3Neus Castells4,5Laura Batlle-Masó3,6 Pere Soler-Palacin3,6,7*
Pere Soler-Palacin3,6,7* Roger Colobran1,2,3,4*
Roger Colobran1,2,3,4*- 1Immunology Division, Vall d’Hebron University Hospital, Universitat Autònoma de Barcelona (UAB), Barcelona, Spain
- 2Translational Immunology Research Group, Vall d’Hebron Research Institute (VHIR), Vall d’Hebron University Hospital, Barcelona, Spain
- 3Jeffrey Modell Diagnostic and Research Center for Primary Immunodeficiencies, Barcelona, Spain
- 4Department of Clinical and Molecular Genetics, Vall d’Hebron University Hospital, Barcelona, Spain
- 5Medicine Genetics Research Group, Vall d’Hebron Research Institute (VHIR), Vall d’Hebron University Hospital, Barcelona, Spain
- 6Infection in Immunocompromised Pediatric Patients Research Group, Vall d’Hebron Research Institute (VHIR), Vall d’Hebron University Hospital, Barcelona, Spain
- 7Pediatric Infectious Diseases and Immunodeficiencies Unit, Vall d’Hebron University Hospital, Barcelona, Spain
Syndromic immunodeficiencies are a heterogeneous group of inborn errors of immunity that can affect the development of non-immune organs and systems. The genetic basis of these immunodeficiencies is highly diverse, ranging from monogenic defects to large chromosomal aberrations. Antibody deficiency is the most prevalent immunological abnormality in patients with syndromic immunodeficiencies caused by chromosomal rearrangements, and usually manifests as a common variable immunodeficiency (CVID)-like phenotype. Here we describe a patient with a complex phenotype, including neurodevelopmental delay, dysmorphic features, malformations, and CVID (hypogammaglobulinemia, reduced pre-switch and switch memory B cells, and impaired vaccine response). Microarray-based comparative genomic hybridization (aCGH) revealed a 13-Mb deletion on chromosome 4q22.2-q24 involving 53 genes, some of which were related to the developmental manifestations in our patient. Although initially none of the affected genes could be linked to his CVID phenotype, subsequent reanalysis identified NFKB1 haploinsufficiency as the cause. This study underscores the value of periodic reanalysis of unsolved genetic studies performed with high-throughput technologies (eg, next-generation sequencing and aCGH). This is important because of the ongoing incorporation of new data establishing the relationship between genes and diseases. In the present case, NFKB1 had not been associated with human disease at the time aCGH was performed. Eight years later, reanalysis of the genes included in the chromosome 4 deletion enabled us to identify NFKB1 haploinsufficiency as the genetic cause of our patient’s CVID. In the future, other genes included in the deletion may be linked to human disease, allowing us to better define the molecular basis of our patient’s complex clinical phenotype.
Introduction
Inborn errors of immunity (IEI) are a large heterogeneous group of disorders affecting various components of the immune system. To date, more than 450 genes causing inborn errors of immunity have been identified, involving several molecular disease mechanisms (gain of function, loss of function, dominant negative, haploinsufficiency) and modes of inheritance (X-linked, autosomal dominant, and recessive) (1, 2). The clinical phenotype of primary immunodeficiencies can include immune-related conditions (greatly varying, but related to susceptibility to infection, autoimmunity, inflammation, allergy, immune dysregulation, or malignancy), or clinical conditions that are not directly related to immune system dysfunction and affect other organ systems. These latter conditions are collectively termed syndromic immunodeficiencies. The genetic bases of syndromic immunodeficiencies vary considerably, but they can be classified into two major categories. First, monogenic defects affecting single genes that are usually pleiotropic, meaning they control or have an effect on two or more seemingly unrelated phenotypic traits. The International Union of Immunological Societies (IUIS) Expert Committee of Inborn Errors of Immunity lists the conditions caused by these genes under the category: combined immunodeficiencies with syndromic features. In the last IUIS update, the category included 63 genes causing 58 different disorders (1). And second, chromosomal rearrangements involving different genes. Here, the spectrum of genetic defects is huge, encompassing aneuploidies such as Down syndrome (trisomy 21) and Turner syndrome (monosomy X), both known to be associated with immunodeficiency (3, 4), as well as more restricted defects such as 22q11 microdeletions (5). In addition to these well-known conditions, other private and sporadic (ie, affecting only a single patient) chromosomal rearrangements involving immunodeficiency have been described. In 2016 we participated in a European Society for Immunodeficiencies (ESID) working group that published a retrospective, observational survey study describing 46 patients with chromosomal aberrations associated with immunodeficiency (largest reported cohort to date) (6). In some cases the specific gene/s causing the immune phenotype can be identified (7), but in many others, they remain unknown (6). According to the ESID survey published by Schatorjé et al, antibody deficiency is the most prevalent immunological abnormality in syndromic immunodeficiency caused by chromosomal rearrangements, usually manifesting as a common variable immunodeficiency (CVID)-like phenotype (6).
CVID, the most common primary immunodeficiency, occurring in approximately 1:25,000 individuals, is an exclusion diagnosis based on clinical and immunological criteria. It is a clinically and genetically heterogeneous disorder characterized by susceptibility to infection, a poor vaccine response, hypogammaglobulinemia, and immune dysregulation (8, 9). Despite the increasing number of studies using next-generation sequencing tools, only 10% to 20% of CVID cases have a known underlying genetic cause (10, 11). In Europeans, pathogenic variants of the nuclear factor κB subunit 1 (NFKB1) gene are the most common monogenic cause of CVID, accounting for 4% of cases in a cohort of 390 unrelated CVID cases (12). More than 100 different NFKB1 variants have been described in CVID patients, mainly single nucleotide variants (SNVs) or small deletions/insertions (indels) causing a variety of deleterious effects on the protein (13).
Here, we report the clinical and molecular findings of a patient with a 13-Mb deletion in chromosome 4, comprising 53 genes. The patient has a complex phenotype, including neurodevelopmental delay, dysmorphic features, malformations, and immunodeficiency. Although initially none of the affected genes could be linked to his CVID phenotype, subsequent reanalysis allowed us to identify NFKB1 haploinsufficiency as the cause of these manifestations.
Case Description
The proband is a 25-year-old male, the only child of non-consanguineous Caucasian parents. Both parents were healthy and had no family history of congenital abnormalities, intellectual disability, or immune disorders. Fetal ultrasound in the third trimester of gestation detected a unilateral dysplastic kidney and oligohydramnios. A prenatal diagnosis was not performed. Delivery was induced at 38 weeks of gestation: birth weight was 2410g (-1.95SD), length 45cm (-2.75SD), and occipital-facial circumference 32cm (-1.38SD). At birth, he was noted to have facial dysmorphism with hypertelorism, a broad nasal tip and low-set ears, and a unilateral preauricular skin tag. Bilateral single palmar crease, 2-3 toe syndactyly, cryptorchidism, and hypospadias were also noted. Axial hypotonia was evident since birth. Abdominal ultrasound confirmed the unilateral dysplastic kidney. Echocardiography and transfontanellar ultrasound findings were normal. The electroencephalogram and ophthalmological examination were normal.
The patient was first admitted to our hospital within one month of life due to failure to thrive. Hypogammaglobulinemia and hypogonadism were detected at that time. He was seen in our Clinical Genetics Department at 3 months of age and was noted to have a narrow forehead, horizontal eyebrows, synophrys, deep-set eyes, epicanthus, wide nasal ridge, anteverted nares with underdeveloped alae nasi, and a thin upper lip vermilion with smooth philtrum (Figure 1). Karyotype testing and plasma 7-dehydrocholesterol (7DHC) analysis, based on the initial suspicion of Smith-Lemli-Opitz syndrome, were normal. Progressive developmental assessment showed a notable delay, as he walked independently at the age of 3, had poor fine motor control, and poor expressive language. At the age of 25 years, he remains unable to read or write.
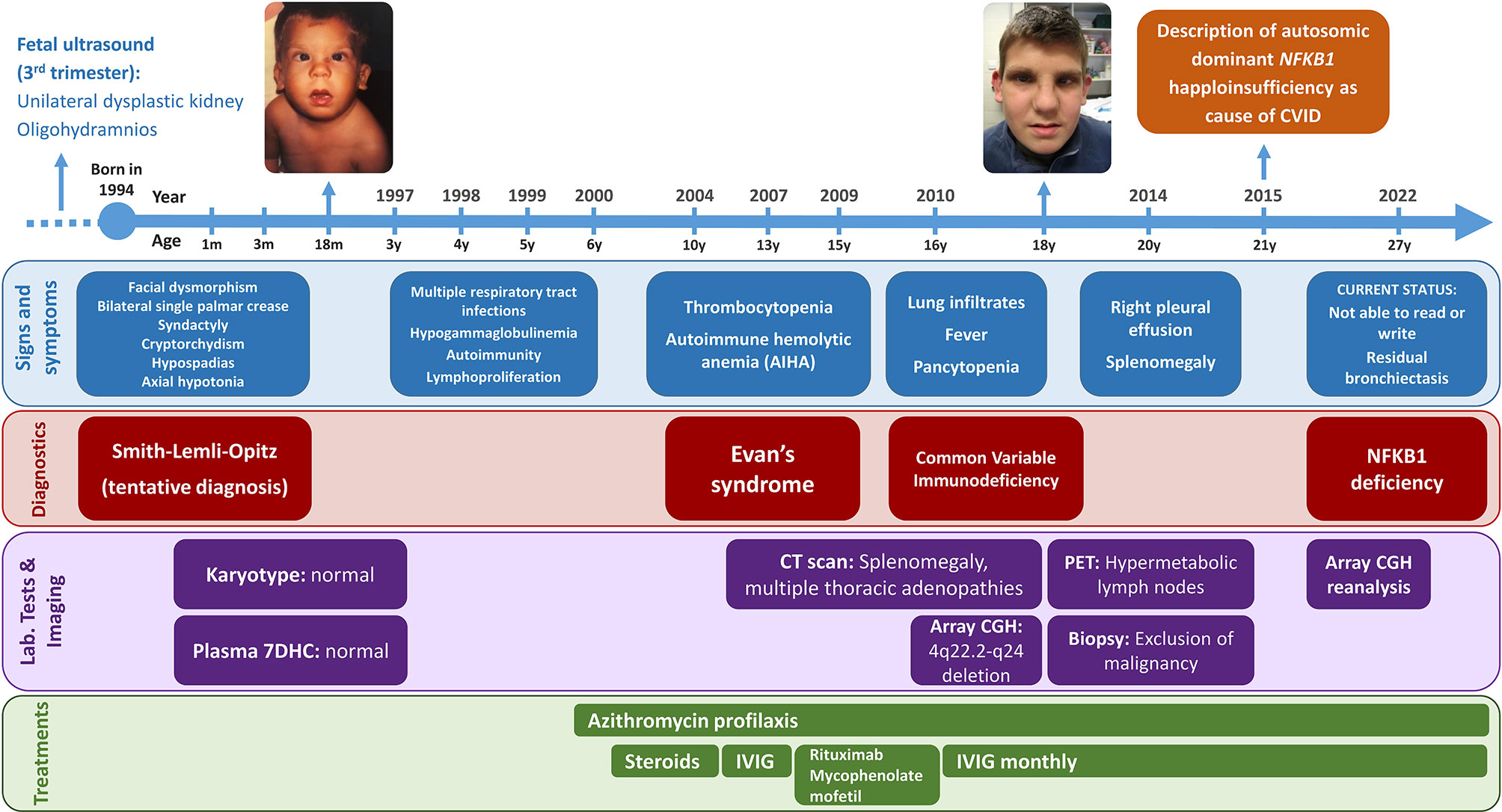
Figure 1 Timeline of the patient’s clinical history. Items are classified into signs and symptoms (blue), diagnoses (red), laboratory tests and imaging (purple), and treatments (green). The patient’s images are included with the explicit consent of the patient and his parents.
From the immunological perspective, the patient showed marked immune system dysregulation early in life, with infections, autoimmunity and lymphoproliferation. Between 3 months and 6 years of age, he was repeatedly hospitalized for recurrent upper and lower respiratory tract infections caused by respiratory viruses and encapsulated bacteria (including various episodes of pneumonia), which led to residual bronchiectasis. At that point, prophylactic treatment with azithromycin was started. Later, the patient presented with Evans syndrome. Initially, at 10 years of age, he experienced only thrombocytopenia, which had a good response to steroids. At the age of 13 years, he showed autoimmune hemolytic anemia together with thrombocytopenia and received intravenous immunoglobulin therapy. Two years later, because of recurrent thrombocytopenia episodes, the patient received rituximab and mycophenolate mofetil, with a good clinical response. At the age of 16 years, he was hospitalized because of lung infiltrates, fever, and pancytopenia. Computed tomography study revealed splenomegaly and multiple thoracic lymph nodes. After ruling out ALPS-related disorders, a diagnosis of CVID was established based on the patient’s autoimmune cytopenia, sustained hypogammaglobulinemia, and absent isohemagglutinins; immunoglobulin replacement therapy was started. He also showed an impaired response to Haemophilus influenzae type b (Hib) conjugate vaccine and tetanus toxoid vaccine, and a normal response to pneumococcal polysaccharide vaccine. At the age of 20 years, the pulmonary condition progressed to right pleural effusion, and positron emission tomography detected hypermetabolic lymph nodes on both sides of the diaphragm, including increased spleen uptake. Malignancy and clonal rearrangements were excluded by lymph node biopsy (Figure 1).
Extended Immunological Characterization of the Patient
At the age of 16 years when the patient was diagnosed with CVID, he showed hypogammaglobulinemia with low IgG and IgM, and absent IgA. Absolute levels of T and B cell lymphocyte subpopulations were low, despite normal percentages (Table 1). T cell immunophenotyping revealed that the decreased total numbers of CD4 T cells are mostly due to the decrease in naive CD4 T cells. Absolute numbers of CD8 T cells were also reduced. Distribution of CD4 T helper subpopulations showed a Th1-skewed profile. B cell immunophenotyping revealed a maturation stop at naïve subpopulations. Most B cells were naïve, and there was a marked reduction in pre-switch and switch memory B cells (Table 1).
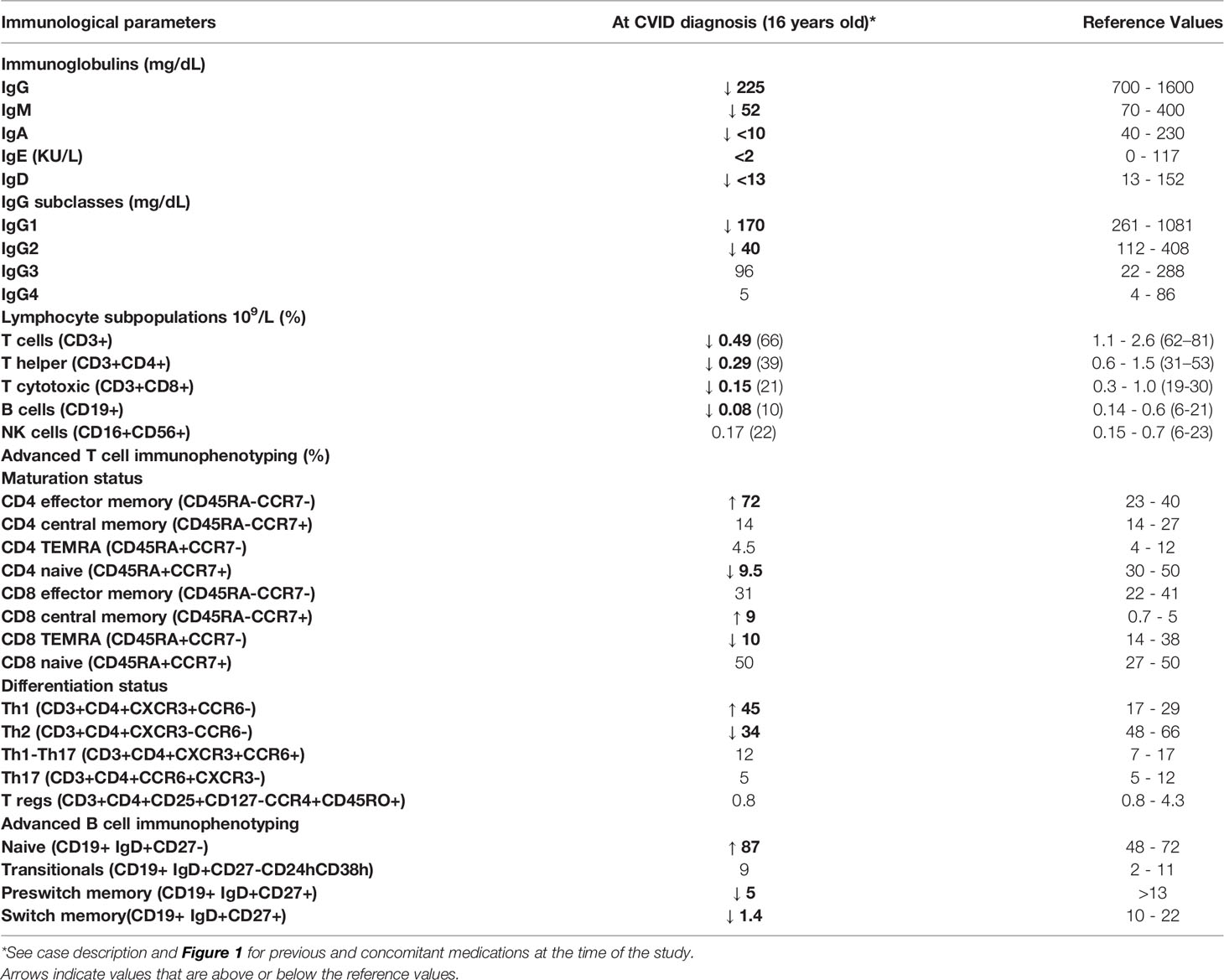
Table 1 Immunological findings in a patient with a 13-Mb deletion (including the NFKB1 gene) in chromosome 4q22.2-q24.
Identification of a Large Deletion in Chromosome 4 Affecting 53 Genes
Because of the patient’s complex phenotype involving immunological abnormalities, dysmorphic features and developmental changes, microarray-based comparative genomic hybridization (aCGH) was performed in 2010 (Supplementary Methods), which identified a 13.03-Mb heterozygous deletion on chromosome 4q22.2-q24 (ISCN formula to date: arr [GRCh37] 4q22.2 q24 (94264597_107296595)x1) (Figure 2A). This large deletion involved 53 genes and was confirmed to be a de novo event by FISH analysis of both parents using a locus-specific BAC probe. A few genes in the 4q22.2-q24 deletion are reported to cause genetic disorders by haploinsufficiency and these could relate to the patient’s developmental manifestations: SMARCAD1 (Huriez syndrome, OMIM #181600), EIF4E (susceptibility to autism, OMIM #615091), and PPP3CA (developmental and epileptic encephalopathy, OMIM #617711). No previously described CVID-related gene defect was detected at that time and additional genetic studies were not performed.
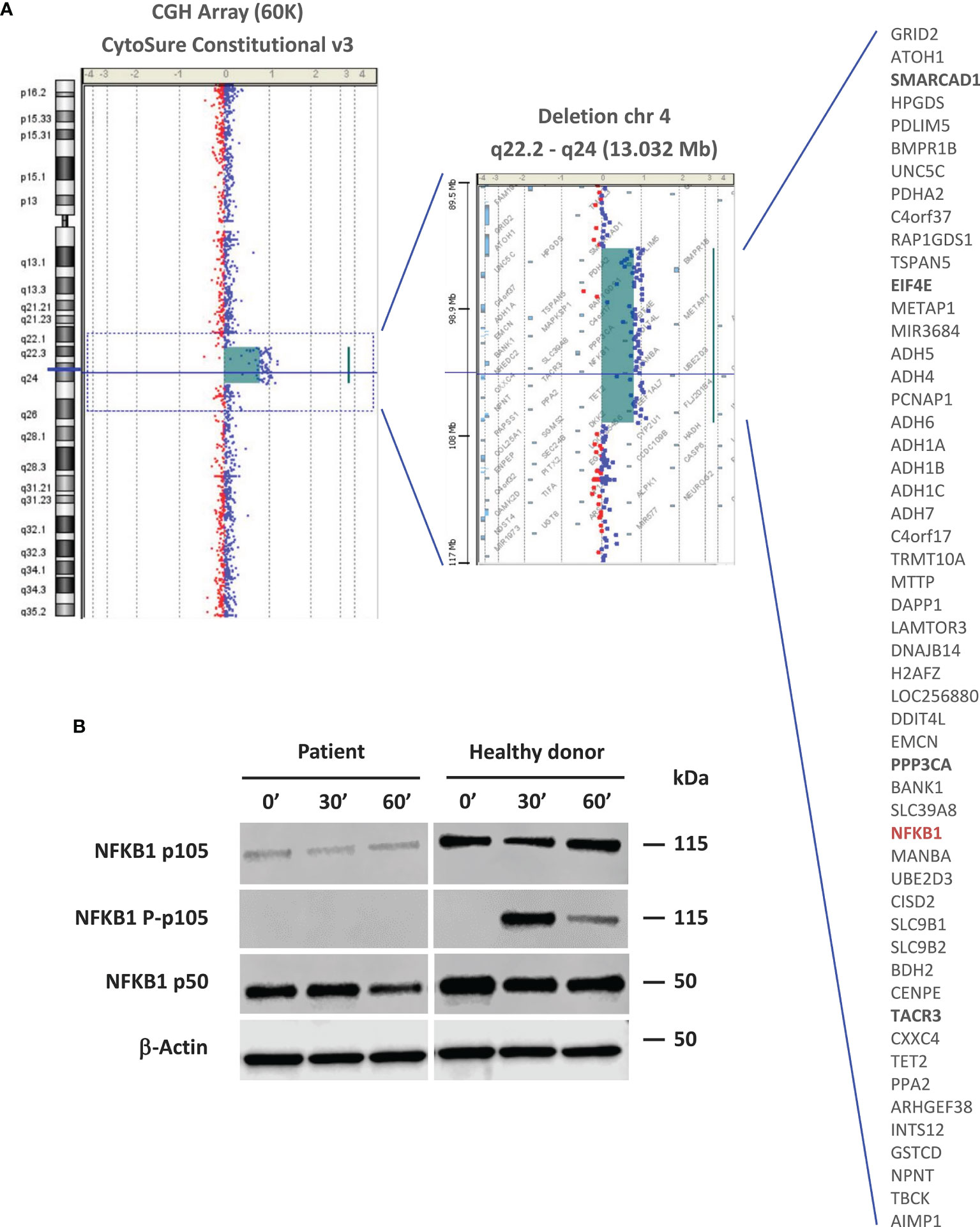
Figure 2 Molecular and functional studies. (A) Array CGH-based identification of the 13-Mb deletion in the 4q22.2-q24 chromosomal region. Patient DNA was labeled with cyanine 3 (Cy3, red dots) and control DNA with cyanine 5 (Cy5, blue dots). The image is reported as the ratio of Cy5 and Cy3 fluorescence intensity. The list of genes encompassed by this deletion is shown. Genes that are likely related to the patient’s non-immune clinical phenotype are in bold. The NFKB1 gene is highlighted in red. (B) NF-kB pathway evaluation by western blot. NFKB1 p105/p50 expression and activation were evaluated in PBMC-derived protein extracts from the patient and a healthy donor. For a better comparison between the healthy donor and patient, we quantified the p105 and p50 bands using the ImageJ software. The quantification reflects the relative amounts as a ratio of each protein band relative to the lane’s loading control (β-actin): p105 patient (0’: 0.24, 30’: 0.20, 60’: 0.22); p105 healthy donor (0’: 0.79, 30’: 0.76, 60’: 1.10). p50 patient (0’: 1.07, 30’: 1.12, 60’: 0.74); p50 healthy donor (0’: 1.39, 30’: 1.27, 60’: 1.41).
aCGH Reanalysis Revealed NFKB1 Haploinsufficiency Underlying the CVID Phenotype
In light of the continuous accumulation of new data on the relationship between disease and genes/variants, we usually perform a periodic data reanalysis (every 2-5 years) in cases of unsolved genetic studies using high-throughput technologies (eg, next-generation sequencing or aCGH). In one of these reanalyses performed in 2018, we discovered that one of the genes included in the 13-Mb deletion was NFKB1 (Figure 2A). In 2015, the group led by Bodo Grimbacher reported for the first time monoallelic pathogenic mutations in NFKB1 as a cause of CVID by a mechanism of haploinsufficiency (14). As the patient’s clinical, laboratory and molecular findings concurred, we considered that NFKB1 haploinsufficiency was the cause of his CVID phenotype. To exclude pathogenic (P) or likely pathogenic (LP) variants in other genes associated with CVID we performed whole exome sequencing. We did not find any P or LP variants in IEI causing genes based on the IUIS classification (1, 2), including all reported CVID-related genes. Recently, autosomal recessive TET2 deficiency has been described causing an immune dysregulation syndrome including susceptibility to infection, lymphadenopathy, hepatosplenomegaly, developmental delay, autoimmunity, and lymphoma of B-cell origin (15). Since TET2 was one of the 53 genes included in the 13-Mb deletion, we analyzed WES data confirming that the other TET2 allele was present and showed the wild type sequence (without rare genetic variants).
NFKB1 encodes for a full-length precursor, p105 protein, which gives rise to the p50 active subunit implicated in the canonical NF-κB pathway (16). The effect of NFKB1 haploinsufficiency on p105/p50 expression and activation was evaluated by western blot (WB) of PBMC-derived protein extracts (Supplementary Methods), which showed detectable but markedly decreased p105 and p50 levels in the patient compared to those of a healthy donor. p105 phosphorylation was not detected following PMA and ionomycin stimulation (Figure 2B).
Discussion
Syndromic immunodeficiencies caused by private and sporadic chromosomal rearrangements are highly variable and the specific cause of the immunological defect sometimes remains unknown (6). Here, we describe a patient with a complex phenotype including neurodevelopmental delay and immunodeficiency. aCGH disclosed a large (13-Mb) deletion in chromosome 4q22.2-q24 involving 53 genes, which included NFKB1 as the cause of his CVID phenotype.
Interstitial deletions in the 4q21-q25 region have been reported in a small number of patients so far, and precise mapping of the breakpoints is described in only a few cases (17–21). Four of these patients had a deletion that considerably overlapped the one described in this study (molecular and clinical details summarized in Table 2). On analysis of the data from all these cases, we found that the most common clinical features were developmental delay/intellectual disability (100%), hypotonia (100%), congenital heart defects (60%), short stature (60%), and other non-specific dysmorphic features. Interestingly, the two patients with the most concordant deletions (Terada et al. and the current patient) both showed intrauterine growth retardation and genital malformation. Immune system abnormalities have not been described in any patients with interstitial deletions in the 4q21-q25 region. However, in only one case the deletion reported included the NFKB1 gene (patient 2 in Hilhorst-Hofstee et al.) and the patient’s latest examination took place at the age of 12 years. Two main features of NFKB1 deficiency are incomplete clinical penetrance (70%) and a highly variable age of onset. According to the largest published study, the mean age at which the first characteristic clinical manifestations occur (mainly infections, autoimmune manifestations, and inflammatory symptoms) is 17 years (ranging from birth to >60 years) (13). Therefore, the patient in the Hilhorst-Hofstee study was likely asymptomatic at the time the paper was written, but he may show symptoms later.
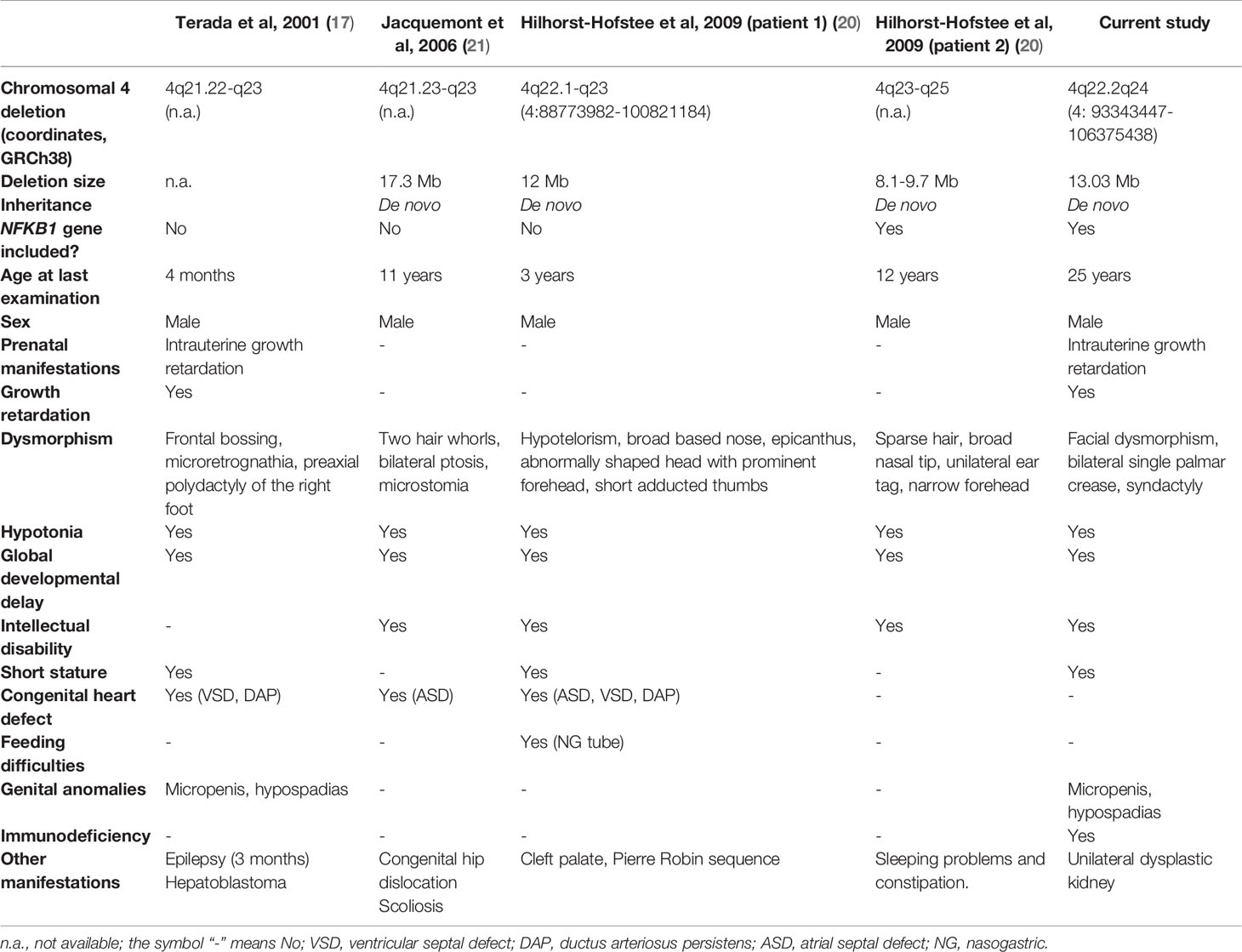
Table 2 Clinical and molecular features of reported patients with chromosomal deletions overlapping the 4q22.2-q24 region.
We extended our search of chromosomal deletions including NFKB1 to the Decipher database (https://www.deciphergenomics.org/) and found 11 patients with a range of deletions (from 900 Kb to 14.5 Mb) including NFKB1 (Supplementary Table 1). The age at their last clinical assessment ranged from 1 to 19 years. Interestingly, in one patient the available phenotypic information included decreased circulating antibody levels, concordant with NFKB1 deficiency.
To date, more than 100 heterozygous NFKB1variants have been reported in patients with a CVID-like phenotype (13, 22). Most pathogenic variants are predicted to cause loss of function, but only a few missense variants are truly pathogenic. In a large and comprehensive functional study, Li et al. found that only 57% of published NFKB1 variants were deleterious, whereas 43% were neutral (22). These authors also demonstrated that deleterious heterozygous NFKB1 variants cause disease by haploinsufficiency. Therefore, any new NFKB1 variants, especially missense variants, should be tested with a proper functional assay to determine the functional significance. Conversely, CVID patients carrying rare NFKB1 missense variants should not be diagnosed as having NFKB1 deficiency without a functional demonstration of the variant’s pathogenicity (22). Our patient is the first reported case of NFKB1 haploinsufficiency by complete absence of one NFKB1 allele, obviating the need for functional studies. However, we showed a marked reduction in p105 and p50 levels by WB. We could not detect p105 phosphorylation after PMA and ionomycin stimulation, probably due to a lack of sensitivity of our WB. A markedly reduced, but detectable, p105 phosphorylation is expected in individuals with NFKB1 haploinsufficiency.
Two additional patients harboring large NFKB1 deletions have been described (12). The deletion in the first patient extended from upstream the NFKB1 gene to intron 17 (del 103370996-103528207). The second patient’s deletion started at NFKB1 intron 1 and ran downstream of 3’UTR (del 103436974-103652655). Although neither of these deletions involve the whole NFKB1 gene, they are large enough to imply a de facto haploinsufficiency. In both reported patients and the patient described here the deletion was a de novo event and the first clinical signs of NFKB1 deficiency started during childhood.
NFKB1 deficient patients present considerable clinical and immunological heterogeneity but most (up to 90%) have hypogammaglobulinemia and this is the reason for the high incidence of bacterial infections, especially those affecting the upper and lower respiratory tract (13). Our patient presented with transient hypogammaglobulinemia at early infancy. Once the patient received rituximab, low B cells and hypogammaglobulinemia persisted thus making difficult to establish an appropriate diagnosis of CVID. It is well described that rituximab may cause long term immunological effects undistinguishable from CVID in susceptible individuals specially with low IgG and IgA levels before therapy (23, 24). Therefore, the role of NFKB1 as either causative or predisposing gene in this patient remains to be clarified. However, immunologically significant NFKB1 haploinsufficiency seems to reinforce the major role of this genetic defect in sight of the patient’s clinical and immunological features. Regarding T cell compartment, the Th1-skewed profile found in our patient has been previously described in NFKB1 deficient patients and associated with inflammatory symptoms (13, 25).
It is important to emphasize that we found the molecular defect associated with the patient’s CVID phenotype by reanalysis of aCGH data. There is now consensus that reanalysis of existing genomic data in unsolved cases increases the diagnostic yield (26, 27). Although systematic reanalysis is usually done with next-generation sequencing data (mainly whole exome or whole genome sequencing), data from other high-throughput technologies can also provide benefits. In the case reported here the large deletion found encompasses 53 genes, most of them still not associated with human disease. This was the case of NFKB1 at the time in which aCGH was performed in 2010. Eight years later, reanalysis of the genes included in the chromosome 4 deletion led us to define NFKB1 haploinsufficiency as the genetic defect causing or contributing to the patient’s CVID. In the future other genes included in the deletion may be linked to human disease, allowing us to better define the molecular basis of our patient’s complex clinical phenotype.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by Clinical Research Ethics Committee (CEIC), Vall d’Hebron Research Institute (VHIR). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
CF-J and MG-P designed, performed and analyzed the functional assays (western blot). IV, JR and PS-P provided patient care, and collected and provided clinical data. MM-G supervised and analyzed flow cytometry data. RC and LB-M supervised and analyzed sequencing data. NC performed and analyzed the comparative genome hybridization array. CF-J, IV and RC wrote the manuscript. PS-P and RC designed and supervised the project, provided resources and edited the manuscript. All co-authors reviewed, commented on, and approved the final version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by Instituto de Salud Carlos III, grants PI17/00660 and PI20/00761, cofinanced by the European Regional Development Fund (ERDF). This study was also funded by the Jeffrey Modell Foundation. This work is supported by the European Reference Network for Rare Immunodeficiency, Autoinflammatory and Autoimmune Diseases Network (ERN-RITA).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.897975/full#supplementary-material
References
1. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human Inborn Errors of Immunity: 2019 Update on the Classification From the International Union of Immunological Societies Expert Committee. J Clin Immunol (2020) 40(1):24–64. doi: 10.1007/s10875-019-00737-x
2. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. The Ever-Increasing Array of Novel Inborn Errors of Immunity: An Interim Update by the IUIS Committee. J Clin Immunol (2021) 41(3):666–79. doi: 10.1007/s10875-021-00980-1
3. Huggard D, Doherty DG, Molloy EJ. Immune Dysregulation in Children With Down Syndrome. Front Pediatr (2020) 8:73. doi: 10.3389/fped.2020.00073
4. Kanakatti Shankar R. Immunological Profile and Autoimmunity in Turner Syndrome. Horm Res Paediatr (2020) 93(7-8):415–22. doi: 10.1159/000512904
5. Morsheimer M, Brown Whitehorn TF, Heimall J, Sullivan KE. The Immune Deficiency of Chromosome 22q11. 2 deletion syndrome Am J Med Genet A (2017) 173(9):2366–72. doi: 10.1002/ajmg.a.38319
6. Schatorjé E, van der Flier M, Seppänen M, Browning M, Morsheimer M, Henriet S, et al. Primary Immunodeficiency Associated With Chromosomal Aberration - an ESID Survey. Orphanet J Rare Dis 2 (2016) 11(1):110. doi: 10.1186/s13023-016-0492-1
7. Franco-Jarava C, Wang H, Martin-Nalda A, Alvarez de la SD, García-Prat M, Bodet D, et al. TNFAIP3 Haploinsufficiency is the Cause of Autoinflammatory Manifestations in a Patient With a Deletion of 13Mb on Chromosome 6. Clin Immunol (2018) 191:44–51. doi: 10.1016/j.clim.2018.03.009
8. Cunningham-Rundles C, Bodian C. Common Variable Immunodeficiency: Clinical and Immunological Features of 248 Patients. Clin Immunol (1999) 92(1):34–48. doi: 10.1006/clim.1999.4725
9. Szczawinska-Poplonyk A, Schwartzmann E, Bukowska-Olech E, Biernat M, Gattner S, Korobacz T, et al. The Pediatric Common Variable Immunodeficiency - From Genetics to Therapy: A Review. Eur J Pediatr (2021) 23:1371–83. doi: 10.1007/s00431-021-04287-6
10. Bogaert DJA, Dullaers M, Lambrecht BN, Vermaelen KY, De Baere E, Haerynck F. Genes Associated With Common Variable Immunodeficiency: One Diagnosis to Rule Them All? J Med Genet (2016) 53(9):575–90. doi: 10.1136/jmedgenet-2015-103690
11. de Valles-Ibáñez G, Esteve-Solé A, Piquer M, González-Navarro EA, Hernandez-Rodriguez J, Laayouni H, et al. Evaluating the Genetics of Common Variable Immunodeficiency: Monogenetic Model and Beyond. Front Immunol (2018) 9:636. doi: 10.3389/fimmu.2018.00636
12. Tuijnenburg P, Lango Allen H, Burns SO, Greene D, Jansen MH, Staples E, et al. Loss-Of-Function Nuclear Factor κb Subunit 1 (NFKB1) Variants are the Most Common Monogenic Cause of Common Variable Immunodeficiency in Europeans. J Allergy Clin Immunol (2018) 142(4):1285–96. doi: 10.1016/j.jaci.2018.01.039
13. Lorenzini T, Fliegauf M, Klammer N, Frede N, Proietti M, Bulashevska A, et al. Characterization of the Clinical and Immunologic Phenotype and Management of 157 Individuals With 56 Distinct Heterozygous NFKB1 Mutations. J Allergy Clin Immunol (2020) 146(4):901–11. doi: 10.1016/j.jaci.2019.11.051
14. Fliegauf M, Bryant VL, Frede N, Slade C, Woon ST, Lehnert K, et al. Haploinsufficiency of the NF-κb1 Subunit P50 in Common Variable Immunodeficiency. Am J Hum Genet (2015) 97(3):389–403. doi: 10.1016/j.ajhg.2015.07.008
15. Stremenova Spegarova J, Lawless D, Mohamad SMB, Engelhardt KR, Doody G, Shrimpton J, et al. Germline TET2 Loss of Function Causes Childhood Immunodeficiency and Lymphoma. Blood (2020) 136(9):1055–66. doi: 10.1182/blood.2020005844
16. Hayden MS, Ghosh S. NF-κb in Immunobiology. Cell Res (2011) 21(2):223–44. doi: 10.1038/cr.2011.13
17. Terada Y, Imoto I, Nagai H, Suwa K, Momoi M, Tajiri T, et al. An 8-cM Interstitial Deletion on 4q21-Q22 in DNA From an Infant With Hepatoblastoma Overlaps With a Commonly Deleted Region in Adult Liver Cancers. Am J Med Genet (2001) 103(2):176–80. doi: 10.1002/ajmg.1521
18. Harada N, Nagai T, Shimokawa O, Niikawa N, Matsumoto N. A 4q21-Q22 Deletion in a Girl With Severe Growth Retardation. Clin Genet (2002) 61(3):226–8. doi: 10.1034/j.1399-0004.2002.610311.x
19. Friedman JM, Baross A, Delaney AD, Ally A, Arbour L, Armstrong L, et al. Oligonucleotide Microarray Analysis of Genomic Imbalance in Children With Mental Retardation. Am J Hum Genet (2006) 79(3):500–13. doi: 10.1086/507471
20. Hilhorst-Hofstee Y, Tümer Z, Born P, Knijnenburg J, Hansson K, Yatawara V, et al. Molecular Characterization of Two Patients With De Novo Interstitial Deletions in 4q22-Q24. Am J Med Genet (2009) 149A(8):1830–3. doi: 10.1002/ajmg.a.32977
21. Jacquemont ML, Sanlaville D, Redon R, Raoul O, Cormier-Daire V, Lyonnet S, et al. Array-Based Comparative Genomic Hybridisation Identifies High Frequency of Cryptic Chromosomal Rearrangements in Patients With Syndromic Autism Spectrum Disorders. J Med Genet (2006) 43(11):843–9. doi: 10.1136/jmg.2006.043166
22. Li J, Lei WT, Zhang P, Rapaport F, Seeleuthner Y, Lyu B, et al. Biochemically Deleterious Human NFKB1 Variants Underlie an Autosomal Dominant Form of Common Variable Immunodeficiency. J Exp Med (2021) 218(11):e20210566. doi: 10.1084/jem.20210566
23. Labrosse R, Barmettler S, Derfalvi B, Blincoe A, Cros G, Lacombe-Barrios J, et al. Rituximab-Induced Hypogammaglobulinemia and Infection Risk in Pediatric Patients. J Allergy Clin Immunol (2021) 148(2):523–32.e8. doi: 10.1016/j.jaci.2021.03.041
24. Luterbacher F, Bernard F, Baleydier F, Ranza E, Jandus P, Blanchard-Rohner G. Case Report: Persistent Hypogammaglobulinemia More Than 10 Years After Rituximab Given Post-HSCT. Front Immunol (2021) 12:773853. doi: 10.3389/fimmu.2021.773853
25. Dieli-Crimi R, Martínez-Gallo M, Franco-Jarava C, Antolin M, Blasco L, Paramonov I, et al. Th1-Skewed Profile and Excessive Production of Proinflammatory Cytokines in a NFKB1-Deficient Patient With CVID and Severe Gastrointestinal Manifestations. Clin Immunol (2018) 195:49–58. doi: 10.1016/j.clim.2018.07.015
26. Tan NB, Stapleton R, Stark Z, Delatycki MB, Yeung A, Hunter MF, et al. Evaluating Systematic Reanalysis of Clinical Genomic Data in Rare Disease From Single Center Experience and Literature Review. Mol Genet Genomic Med (2020) 8(11):e1508. doi: 10.1002/mgg3.1508
Keywords: primary immunodeficiencies, syndromic immunodeficiencies, common variable immunodeficiency, nfkb1, chromosomal rearrangements
Citation: Franco-Jarava C, Valenzuela I, Riviere JG, Garcia-Prat M, Martínez-Gallo M, Dieli-Crimi R, Castells N, Batlle-Masó L, Soler-Palacin P and Colobran R (2022) Common Variable Immunodeficiency and Neurodevelopmental Delay Due to a 13Mb Deletion on Chromosome 4 Including the NFKB1 Gene: A Case Report. Front. Immunol. 13:897975. doi: 10.3389/fimmu.2022.897975
Received: 16 March 2022; Accepted: 25 May 2022;
Published: 17 June 2022.
Edited by:
Menno C. van Zelm, Monash University, AustraliaReviewed by:
Hanna IJspeert, Erasmus University Rotterdam, NetherlandsJavier Chinen, Baylor College of Medicine, United States
Copyright © 2022 Franco-Jarava, Valenzuela, Riviere, Garcia-Prat, Martínez-Gallo, Dieli-Crimi, Castells, Batlle-Masó, Soler-Palacin and Colobran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roger Colobran, cmNvbG9icmFuQHZoZWJyb24ubmV0;Pere Soler-Palacin, cHNvbGVyQHZoZWJyb24ubmV0