 Can Gan
Can Gan Qiuyu Cai
Qiuyu Cai Chengwei Tang
Chengwei Tang Jinhang Gao
Jinhang Gao- 1Lab of Gastroenterology and Hepatology, West China Hospital, Sichuan University, Chengdu, China
- 2Department of Gastroenterology, West China Hospital, Sichuan University, Chengdu, China
Inflammasomes are multiprotein complexes that can sense danger signals and activate caspase-1 to mediate pro-inflammatory cytokines release and pyroptotic cell death. There are two main canonical and non-canonical signaling pathways that trigger inflammasome activation. Inflammasomes are expressed and assembled in parenchymal and nonparenchymal cells in response to liver injury in the liver. Additionally, the hepatocytes, biliary epithelial cells (cholangiocytes), hepatic stellate cells (HSCs), hepatic macrophages, and liver sinusoidal endothelial cells (LSECs) contribute to liver fibrosis via different mechanisms. However, the underlying mechanism of the inflammasome and pyroptosis in these liver cells in liver fibrosis remains elusive. This review summarizes the activation and function of inflammasome complexes and then discusses the association between inflammasomes, pyroptosis, and liver fibrosis. Unlike other similar reviewers, we will focus on the effect of inflammasome activation and pyroptosis in the various liver cells during the development of liver fibrosis. We will also highlight the latest progress of pharmacological intervention in inflammasome-mediated liver fibrosis.
1 Introduction
Inflammasomes are signaling platforms in response to infectious diseases or chronic sterile inflammation. These multimeric complexes respond to molecular patterns from pathogens and cellular damage by releasing pro-inflammatory cytokines and inducing pyroptotic cell death (1). In general, inflammasome complexes are composed of a pattern-recognition receptor, an effector caspase-1, and an adaptor connecting these two components. There are two groups of inflammasomes in terms of receptors: the NLR family containing NLRP1, 2, 3, 6, NLRC4 and NLRP12; pyrin and HIN domain-containing (PYHIN) family including absent in melanoma 2 (AIM2) and pyrin (2). The inflammasomes could either aggravate inflammation via interleukin (IL)-1β and IL-18 or induce pyroptosis via gasdermin D (GSDMD). Although the inflammasome and its related pyroptosis have been well studied in infectious diseases, the role and mechanism of inflammasome and pyroptosis in liver fibrosis remain unclear.
Liver fibrosis is characterized by excessive extracellular matrix (ECM) deposition in response to chronic liver injury, including virus infection, non-alcoholic steatohepatitis (NASH), alcoholic liver disease (ALD), and autoimmune diseases (3, 4). Generally, activated HSCs are primary myofibroblasts that produce and secrete ECM. In addition, hepatocytes, Kupffer cells (KCs), LSECs, cholangiocytes, and recruited cell types (e.g., bone marrow-derived macrophages) also contribute to liver fibrosis (5, 6). The mechanisms of liver fibrosis are complicated and involve different cells, signaling pathways, and cross-talk between individual cells. Recent studies have shown that inflammasomes and inflammasome-related pyroptosis are involved in liver fibrogenesis from various pathologies (7). The detailed mechanism of the inflammasome and pyroptosis in liver fibrosis is not well-defined. Collaboration of multiple liver cells maintains hepatic homeostasis in health and contributes to disturbed hepatic homeostasis in liver fibrosis (8). This review summarizes the progress of inflammasome and inflammasome-related pyroptosis in liver fibrosis. The roles and mechanisms of inflammasomes and pyroptosis in various liver cells and their cross-talks will also be described. Eventually, the potential therapeutic targets and future directions will also be implied based on the current progress.
2 Inflammasome Biology and Activation
There are five main kinds of inflammasome sensor complexes in terms of receptors, NLRP1, NLRP3, NLRC4, AIM2, and pyrin (Figure 1). The inflammasome receptor oligomerizes and recruits the adaptor ASC and effector caspase-1 to assemble inflammasome complexes after sensing pathogens or host-derived injuries. After activation, the cleaved caspase-1 mediates the maturation and secretion of IL-1β and IL-18 and pyroptotic cell death (9). In this section, we will compare the structures of different inflammasomes and discuss the mechanism of inflammasome activation.
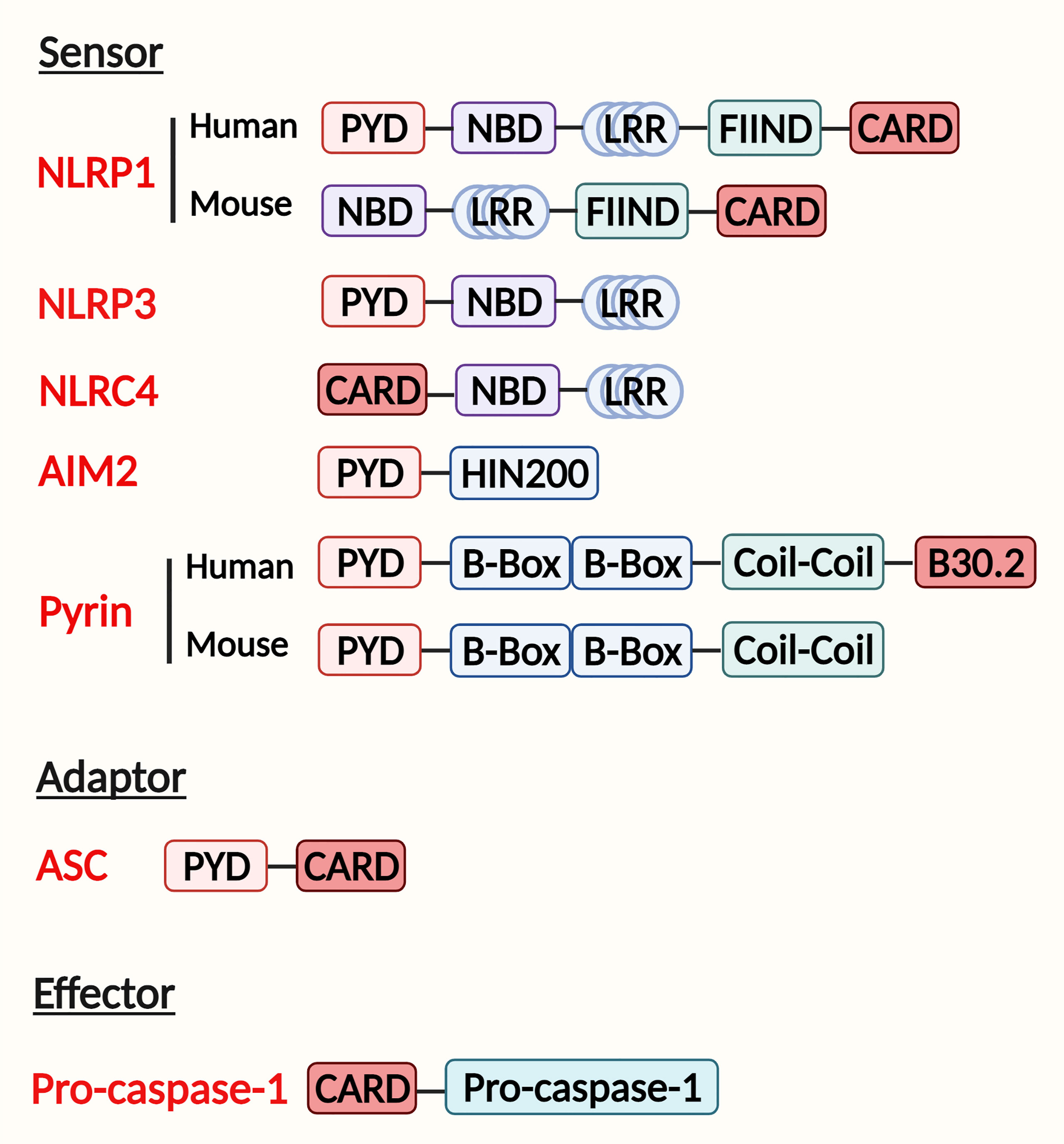
Figure 1 Domain structure and inflammasome assembly. The inflammasome complex contains a sensor, adapter, and effector protein. There are two kinds of sensors: the NOD-like receptor (NLR) family and the PYHIN (pyrin and HIN200 (hematopoietic interferon-inducible nuclear antigens with 200 amino-acid repeats) domain-containing protein) family. The NLR family includes NLRP1, NLRP3, NLRC4 sensors. They contain a nucleotide-binding domain (NBD), carboxy-terminal leucine-rich repeat (LRR), either a pyrin domain (PYD) or a caspase activation and recruitment domain (CARD). Additionally, NLRP1 has a function-to-find domain (FIIND) at the C terminal. The PYHIN family AIM2 receptor has a PYD and HIN200 domain, whereas pyrin contains a PYD, two B boxes, and a coil-coil domain. Human pyrin has an additional B30.2 domain. Adaptor has a PYD to combine with the sensor and a CARD with the effector. The effector is composed of a CARD and pro-caspase-1, which will be cleaved when inflammasome assembly.
2.1 Inflammasome Assembly and Structure
2.1.1 NLRP1 Inflammasome
NLRP1 was the first inflammasome to be identified; however, little research has focused on NLRP1 inflammasome due to the complexity and differences in structures between humans and mice. Human NLRP1 inflammasome has two domains in NOD-like receptor (NLR): an N−terminal pyrin domain (PYD) and a C−terminal caspase activation and recruitment domain (10). In contrast, the mouse genome encodes Nlrp1a, Nlrp1b, and Nlrp1c, which activate caspase-1 without the help of an ASC adaptor due to the lack of a PYD domain in the receptor (11). But a function-to-find domain (FIIND) at the C−terminal is required in both humans and mice to participate in the activation of the NLRP1 inflammasome (Figure 1) (12).
Regarding inflammasome activation, Bacillus anthracis lethal toxin (LT) was involved in the NLRP1 inflammasome activation and subsequent caspase-1−dependent cytokine release and pyroptosis (13). Moreover, extracellular ATP was also demonstrated to trigger the NLRP1 inflammasome by activating the P2X7 receptor and potassium efflux (14, 15). As the NLRP1 inflammasome is less well-studied, the role of the NLRP1 inflammasome in chronic liver disease and liver fibrosis remains unclear.
2.1.2 NLRP3 Inflammasome
The NLRP3 inflammasome contains the NOD-like receptor NLRP3, the adaptor ASC, and the effector pro-caspase-1 (Figure 1). The NLRP3 inflammasome is the most widely studied and the best-characterized inflammasome in infectious and chronic inflammatory diseases (16).
The stimuli involved in NLRP3 activation include pathogen-associated molecular patterns (PAMPs) ligands (17), such as bacterial, fungal, and viral components, pore-forming toxins, nucleic acids; and damage-associated molecular patterns (DAMPs) such as extracellular ATP (18), uric acid crystals (19), and amyloid (20). Activation by these PAMPs and DAMPs indicates that the NLRP3 inflammasome is a common sensor of cellular stress or injury. Activation of the NLRP3 inflammasome requires two steps. First, extracellular stimuli prime cells, e.g., LPS binds to TLR4 to activate NF-κB and subsequent transcription and translation of IL−1β. Then secondary stimuli such as ATP induce inflammasome complex assembly and IL-1β cleavage and secretion. An increasing number of studies have confirmed the critical roles of NLRP3 inflammasome activation in pro-inflammatory cytokines release and pyroptosis initiation. The NLRP3 inflammasome-mediated cellular communication among different liver cell types in acute and chronic liver diseases from diverse liver injuries was also well-defined (21, 22), indicating the pivotal role of NLRP3 inflammasome in the development of liver diseases.
2.1.3 NLRC4 Inflammasome
The NLRC4 inflammasome is activated in response to Flagellin infection (23), Salmonella infection (24), and bacterial type 3 secretion systems (T3SSs) (25). These stimulators bind to NLRC4 indirectly. During infection, neuronal apoptosis inhibitory proteins (NAIPs) interact with the ligand and NLRC4 receptor, activating the assembly of the NLRC4 inflammasome (Figure 1) (26). A few studies showed that bacterial flagellin induced NLRC4 inflammasome activation in hepatocytes and KCs (27, 28). Moreover, the NLRC4-driven IL-1 release was also involved in liver fibrosis (27, 28). Though these studies show an important role of NLRC4 inflammasome in bacterial infection in liver cells, more studies are needed to explore the potential mechanisms of NLRC4 inflammasome activation and its role in liver fibrosis.
2.1.4 AIM2 Inflammasome
The AIM2 inflammasome is a cytosolic receptor that senses double-stranded DNA (dsDNA). It contains an N-terminal PYD domain and a C-terminal hematopoietic interferon-inducible nuclear protein with a 200-amino acid repeat (HIN200) domain (Figure 1) (29). When dsDNA binds to the AIM2 receptor, ASC and pro-caspase-1 are recruited for inflammasome complex formation and activation (30, 31). Previous studies have found that the AIM2 inflammasome induction in hepatocytes and macrophages participates in chronic liver diseases via the pyroptosis pathway (32–35). However, the mechanism of AIM2 inflammasome regulation is not well-studied.
2.1.5 Pyrin Inflammasome
Pyrin, encoded by the MEFV gene, contains a PYD, two B−boxes, a coiled-coil domain, and a SPRY/PRY domain (Figure 1). Pyrin was recently recognized as an inflammasome-forming and pyroptosis-initiating protein. Pyrin inflammasome was reported to stimulate pyroptosis, but there is no evidence showing its direct effect on the progression of liver fibrosis (36, 37).
2.2 Functions of Effector Components in Inflammasomes
Inflammasome assembly activates caspase-1, which cleaves the pro-IL-1β and pro-IL-18 into active IL-1β and IL-18. Meanwhile, GSDMD is cleaved by caspase-1 to N-terminal fragments (GSDMD-NT), which are inserted into the cell lipid membrane to assemble arc‐ and slit‐like oligomers and grow into large and stable ring‐like oligomers to form transmembrane pores. This process leads to cell membrane rupture and resultant pyroptosis (38). We next explored the functions of these effector components in inflammasomes.
2.2.1 IL Maturation and Secretion
Activation of inflammasome complexes cleaves pro-caspase-1 and triggers IL-1β and IL-18 maturation and secretion. IL−1β and IL−18 are crucial cytokines involved in immune responses and trigger the inflammatory cascade (39). However, there have been many debates about how IL-1 is secreted out of cells. Recent studies have shown that Gsdmd knockout macrophages mature IL−1β normally but fail to secrete it due to lack of pyroptosis, suggesting that IL-1β is secreted through cell membrane rupture and lysis (40). However, whether IL-1 is released via pyroptosis in other cell types remains unknown; further studies are needed.
2.2.2 Pyroptosis
Pyroptosis is a distinct type of programmed cell death characterized by the formation of cell membrane pores, the release of intracellular contents, nuclear condensation, and cell lysis (41). Compared with pyroptosis, apoptosis and necroptosis are caspase-3/7 and receptor-interacting serine-threonine kinase-1/3 (RIPK-1/3) mediated programmed cell death, respectively (42). Pyroptosis depends on inflammatory caspase-1 to cleave gasdermins to form membrane pores. Of all gasdermins, GSDMD has been demonstrated to play a central role in pyroptotic cell death. Active caspases cleave GSDMD to GSDMD-NT, which causes membrane pores and induces pyroptosis via both canonical and non-canonical signaling pathways (41, 43). However, the mechanism by which GSDMD executes cell death is poorly explored.
2.3 Canonical and Non-Canonical Signaling Pathways of Inflammasome Activation
As previously reviewed, inflammasomes are initiated and assembled via a canonical or non-canonical signaling pathway (2). There are two steps to process inflammasome activation in the canonical signaling pathway (Figure 2) (44). In the first step, PAMPs, such as LPS, bind to TLRs and elicit the downstream MyD88–NF-κB signaling pathway to produce pro-IL-1β and pro-IL-18. In the second step, endogenous danger signals activate inflammasome assembly and caspase-1. A combination of PRR, ASC, and pro-caspase-1 helps activate caspase-1, which cleaves GSDMD into GSDMD-NT as well as cleaves pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18. There are three main mechanisms involved in the second step. Firstly, extracellular ATP binds to the P2X7 receptor and opens a pore on the cell membrane through pannexin 1 protein, causing potassium efflux and NLRP3 activation (18). Secondly, uric acid crystals and amyloid endocytosed by phagosome lysosomes contribute to lysosome rupture and release of cathepsin B, which helps inflammasome assembly (19). Thirdly, thioredoxin-interacting protein (TXNIP) detached from thioredoxin and mitochondrial DNA (mtDNA) accumulation in a ROS-dependent manner activates inflammasomes (45).
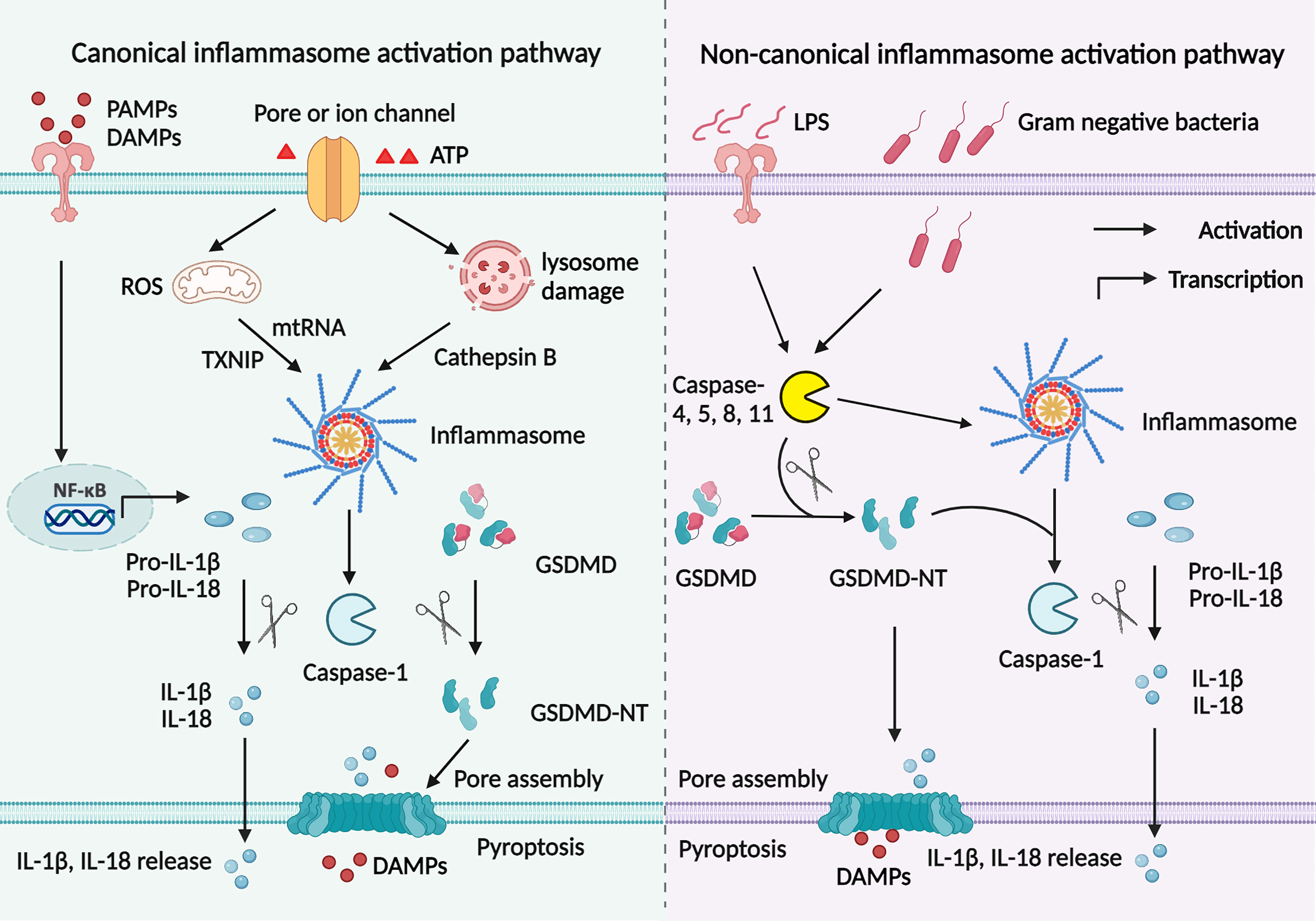
Figure 2 Canonical and non-canonical signaling pathways to activate inflammasomes. Canonical signaling pathway to activate inflammasomes is mediated by two signaling steps. The priming step involves PAMPs or DAMPs binding to TLRs to initiate NF- κB signaling pathway, which promotes pro-IL-1β and pro-IL-18 production. Then endogenous danger signals activate inflammasome assembly and caspase-1 in the second step. Active caspase-1 cleaves GSDMD into GSDMD-NT; cut pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18. The GSDMD breaks a pore on the cell membrane to help IL-1β and IL-18 secretion. There are three main mechanisms involved in inflammasome activation. Extracellular ATP binds to the P2X7 receptor and causes potassium efflux and NLRP3 activation. Uric acid and amyloid are endocytosed in a lysosome-dependent manner, promoting lysosome rupture and cathepsin B release and resultant inflammasome assembly. TXNIP and mtDNA accumulation in a ROS-dependent manner activates inflammasomes. However, in the non-canonical signaling pathway, intracellular LPS or toxin initiates caspase-11, promoting inflammasomes assembly and caspase-1 activation. Then both active caspase-11 and caspase-1 cleave GSDMD into GSDMD-NT, leading to cascaded reactions similar to the canonical signaling pathway.
Apart from the canonical signaling pathway, inflammasomes are also activated by the non-canonical signaling pathway (Figure 2). In this process, intracellular LPS or toxin initiates caspase-11, promoting inflammasome assembly and caspase-1 activation. Then both active caspase-11 and caspase-1 cleave GSDMD into GSDMD-NT, leading to cascaded reactions similar to the canonical signaling pathway (46). Inflammasome activation is a complex process that involves multiple proteins. It remains unclear whether the cross-talk of the canonical signaling pathway and the non-canonical signaling pathway also revokes inflammasome activation.
3 Inflammasome and Pyroptosis in Liver Fibrosis
Liver fibrosis is a dynamic process characterized by an imbalance in ECM deposition and degradation due to chronic liver injury of various etiologies (5). During this process, quiescent HSCs transdifferentiate into fibrogenic activated phenotype, representing the dominant collagen-producing myofibroblasts in different chronic injury models (5, 7, 47). Stimulators of chronic liver injuries, such as fatty acid, alcohol, toxin, and virus infection, affect hepatocytes, hepatic macrophages, LSECs, and cholangiocytes. Damaged liver cells interact with HSCs by releasing pro-inflammatory and pro-fibrotic factors, promoting HSC activation and resultant liver fibrosis (48–50). It is worth noting that inflammasome activation and pyroptosis play a central role in the inflammatory cascade (51). Previous studies have demonstrated that inflammasome components are widely expressed in these liver cell types in response to liver injury (52–54). Moreover, inflammasomes could induce liver fibrosis both directly and indirectly. Inflammasome activation in HSCs directly leads to HSCs activation, which is responsible for ECM formation and liver fibrosis. Indirectly, pro-inflammatory cytokine release or pyroptotic cell death in hepatocytes and other nonparenchymal cells induces HSC activation and eventually leads to liver fibrosis (Figure 3) (55).
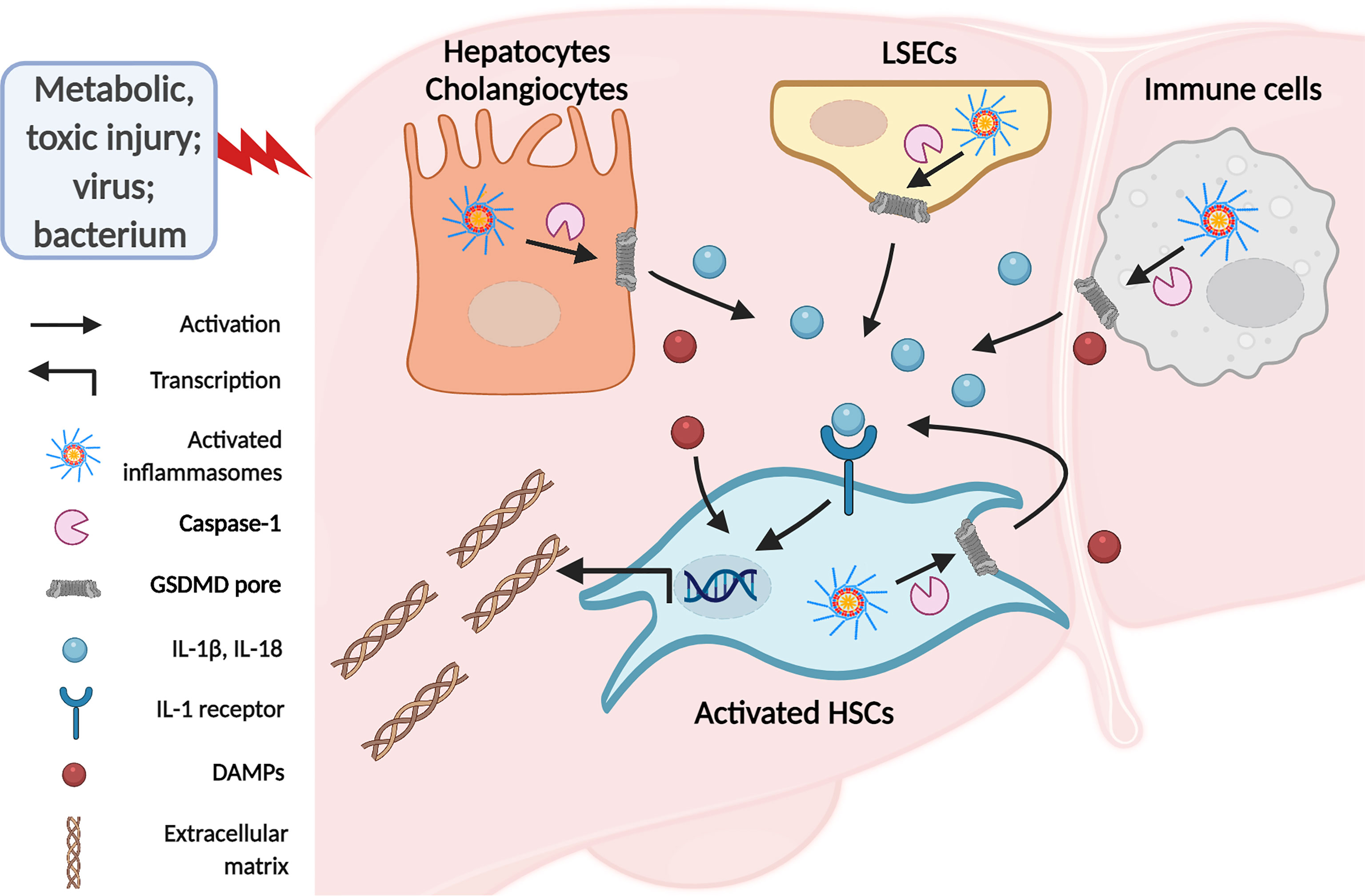
Figure 3 The inflammasomes in liver fibrosis. Activated HSCs are the primary source of extracellular matrix deposition in response to chronic liver injury. There are direct and indirect ways involved in inflammasome to stimulate HSCs. Activation of inflammasomes in hepatocytes and macrophages due to chronic liver injury leads to IL-1β and IL-18 release and pyroptotic cell death. Secreted IL-1β and IL-18 bind to IL-1 receptors on HSCs and induce HSCs activation and resultant ECM formation. In addition, DAMPs from pyroptotic cells promote HSCs activation and liver fibrosis. Meanwhile, PAMPs or DAMPs may directly target HSCs to induce inflammasome-dependent IL-1 release and pyroptosis, eliciting HSCs activation in an autocrine or paracrine manner.
3.1 Inflammasomes and Pyroptosis in Hepatocytes
Although inflammasomes are predominately expressed in immune cells, hepatocytes, which are the most abundant cell type in the liver and predisposed to liver injury, also express inflammasomes (52). Recently, increasing studies have demonstrated important roles of hepatocellular inflammasomes activation, the subsequent pyroptotic death, and inflammasome-implicated cross-talk with HSCs in chronic liver injury from various liver pathologies.
3.1.1 Hepatocellular NLRP3 Inflammasome in NAFLD and NASH-Related Liver Fibrosis
Hepatocellular NLRP3 inflammasome activation and caspase-1-mediated pyroptosis play a crucial role in the progression of non-alcoholic fatty liver disease (NAFLD). Lipid exposure resulted in hepatocellular NLRP3 inflammasome activation and cleavage of mature IL-1β and IL-18 by caspase-1 in high fructose-induced NAFLD mouse model. The inflammatory response aggravated hepatocellular lipid accumulation as well as impaired insulin sensitivity in JAK/STAT3-dependent (56) and PI3K/AKT-dependent manner (57), respectively. The evidence indicates that NLRP3 inflammasome mediates lipid and glucose metabolism in hepatocytes. Mechanically, NLRP3 inflammasome and cytokine production are induced by mitochondrial reactive oxygen species (ROS) in fatty hepatocytes (58, 59). Furthermore, the binding of IL-1β and IL-18 to HSC-derived IL-1 receptor, as well as DAMPs released from pyroptotic hepatocytes transdifferentiated HSCs into activated phenotype for fibrotic scar formation (57). Liver inflammation and ECM deposition are relieved in hepatocyte-specific caspase-11-deficient mice with high sucrose and high fat diet, where the production of IL-1β and GSDMD was blocked, further proving the role of inflammasomes in liver fibrosis (60). In summary, these studies have demonstrated that hepatocellular NLRP3 inflammasome and pyroptosis contribute to the development of NAFLD and liver fibrosis via the cross-talk between hepatocytes and HSCs.
3.1.2 Hepatocellular NLRP3 Inflammasome in Liver Fibrosis
Hepatocyte pyroptosis and the release of inflammasome particles caused by DAMPs and PAMPs were internalized by HSCs and further induced HSC activation and liver fibrosis (55, 61). It seems that the NLRP3 inflammasome in hepatocytes may play a pivotal role in developing liver fibrosis. Ethanol was shown to trigger hepatocyte NLRP3 inflammasome-dependent IL-1β production and pyroptosis by TXNIP overexpression (62) and the caspase-4/11–GSDMD non-canonical signaling pathway in ALD (63). Elevated inflammation accompanied apoptotic and necroptotic cell death in the Nlrp3-knockout acute BDL model. In contrast, decreased liver injury and bridge fibrosis in the Nlrp3-knockout chronic BDL model showed diverse roles of NLRP3 in acute and chronic cholestatic liver injury (64). Mechanically, endoplasmic reticulum (ER) stress is the central process involved in NLRP3 inflammasome activation in hepatocytes. The IRE1α–sXBP1–ER stress signaling pathway mediated hepatocellular NLRP3 inflammasome activation (65). In comparison, this NLRP3 inflammasome activation was relieved in XBP1 knockout mice (65). In the LPS- and CCl4-induced chronic liver injury model, NLRP3 inflammasome-mediated hepatocyte pyroptotic cell death was relieved after ER stress abrogation via either CHOP knockdown (66) or FXR regulation (67). The above studies demonstrate that ER stress-dependent inflammasome activation mediates chronic liver injury and fibrosis. However, further studies that use hepatocyte-specific NLRP3 deletion mice are urgently needed. Although many studies have revealed that NLPR3 is upregulated in hepatocytes, few studies have focused on the transcriptional regulation of NLRP3.
3.1.3 Other Hepatocellular Inflammasomes in Liver Fibrosis
In addition to NLRP3, other inflammasomes in hepatocytes are also involved in liver injury and liver fibrosis. AIM2 inflammasome activation by mtDNA resulted in increased expression of IL-1β and GSDMD in hepatocytes. The release of IL-1β promoted liver steatosis, inflammation, and activated IL-1 receptor on HSCs, contributing to the fibrogenic phenotype of HSCs (32); whereas HMGB1 redox status inhibited activation of caspase-1 through AIM2 inflammasome in chronic liver inflammation (33). In addition, NLRC4 inflammasome activation and pro-inflammatory cytokines induced by bacterial flagellin were rescued by hepatocyte Toll-Like Receptor 5 (27). The progression of NASH was attenuated by the deletion of NLRC4 inflammasome and the subsequent decrease in hepatocyte pyroptosis (68). The above studies show hepatocytes express and activate other inflammasomes, which participate in cellular communication with HSCs to induce liver fibrosis. Compared to the NLRP3 inflammasome, the effect of other inflammasomes remains unclear, and further studies are needed.
In summary, various chronic injuries lead to inflammasome assembly in hepatocytes via canonical and non-canonical signaling pathways. Then inflammasomes and pyroptosis in hepatocytes initiate liver fibrosis mainly via HSC activation.
3.2 Inflammasomes and Pyroptosis in Hepatic Macrophages
There are two phenotypes of macrophages in the liver: liver resident KCs and bone marrow-derived macrophages (BMDMs). KCs are the predominant macrophages in healthy conditions, whereas BMDMs infiltrate into the liver in response to danger signals (49, 69). Macrophages are recognized as the primary source of inflammasomes and pro-inflammatory cytokines (70). Generally, inflammasome activation and the subsequent pyroptosis in macrophages promote chronic liver inflammation by IL-1 and DAMPs release, which bind to receptors on HSCs and initiate HSCs-mediated fibrosis from various etiologies.
3.2.1 Hepatic Macrophage NLRP3 Inflammasome in NAFLD and NASH-Related Liver Fibrosis
In NASH models, the NLRP3 inflammasome activation in KCs or BMDMs caused inflammatory cytokine production, contributing to lipid synthesis in hepatocytes and fibrotic collagen production in HSCs (71–73). Furthermore, macrophage-specific Nrf2 knockout aggravated NASH progression by initiating ROS and IL-1β in a yes-associated protein (YAP)–NLRP3-dependent manner (74). This finding demonstrates the role of ROS in activating the NLRP3 inflammasome in NASH. Similarly, the activation of the kinase receptor-interacting protein 1 (RIP1) in BMDMs, which induced inflammasome assembly and pyroptotic cell death, also contributed to the pathogenesis of NASH (75). Mechanically, NLRP3-mediated M1 macrophage polarization was blocked by the TGR5 signaling pathway, which further restored liver steatosis, inflammatory infiltration, and liver fibrosis (76). TLR2 and palmitic acid cooperatively stimulated the NLRP3 inflammasome in KCs and BMDMs. Then the activated NLRP3 inflammasome enhanced macrophage autophagy via the release of IL-1β to induce HSC activation (77, 78). Accordingly, inhibiting the NLRP3 inflammasome by TIM-4 via LKB1–AMPKα-mediated autophagy in macrophages suppressed the progression of NAFLD (79).
Moreover, the caspase-1–IL-1β signaling pathway in macrophages leads to lipid accumulation, inflammatory infiltration, and liver fibrosis in NASH mouse models by mediating macrophage–hepatocyte and macrophage–HSC interactions (80). However, this progression was blocked by ezetimibe administration in an autophagy-dependent manner (80). Consistently, by exposing hepatocytes and KCs to cholesterol crystals, NLRP3 inflammasome activation was demonstrated to be involved in the development of NASH (81). The above investigations confirmed the vital role of autophagy in regulating macrophage-derived inflammasome assembly in NASH. Nevertheless, the role of macrophage-derived autophagy in inducing disrupted cross-talk in sinusoids and liver fibrosis in NASH remains unclear. Further studies that focus on cellular cross-talk in the sinusoids are urgently needed.
3.2.2 Hepatic Macrophage NLRP3 Inflammasome in Liver Fibrosis
NLRP3 inflammasome activation in KCs and BMDMs drove the caspase-1 signaling pathway, leading to IL-1 secretion and pyroptotic death in response to LPS-induced injury in vivo and in vitro (82, 83). However, NLRP3 inflammasome-derived IL-1β secretion and pyroptosis in macrophages and collagens deposition were blocked by pharmaceutical inhibition or genetic knockdown of COX-2 (84). Consistently, COX-2 promotes the development of liver cirrhosis (85) by inducing ROS (86, 87) and ER stress (88), indicating that COX-2 might contribute to liver cirrhosis via macrophage-derived inflammasomes. However, further studies are needed to determine how COX-2 regulates the activation of macrophage-derived inflammasomes and pyroptosis of macrophages in the context of liver fibrosis.
Studies confirmed that NLRP3 activation was not only required for hepatic inflammation and fibrosis but also as an essential mediator to amplify and perpetuate programmed inflammatory pyroptotic cell death (55). LPS from the damaged gut barrier and endogenous danger signals from hepatocytes damaged by ethanol (extracellular ATP and uric acid) are responsible for macrophage recruitment and subsequent inflammatory cytokine processing and pyroptotic cell death (89–92). The process is termed classically activated (M1) macrophage polarization (93). Increased NLRP3 and subsequent IL-1β maturation and secretion in macrophages exacerbated liver inflammation and fibrosis in ALD via induction of IL-1 receptor expression on HSCs and facilitated ECM secretion and formation (91). However, macrophage-specific Atg5 knockout promoted caspase-1 activation, and pro-inflammatory M1 macrophage polarization, resulting in liver fibrosis during chronic ethanol exposure. Autophagy, a conserved cellular process to remove the damaged or unnecessary component, plays an important role in negatively regulating NLRP3 inflammasome in macrophages by degrading mitochondrial-derived DAMPs and inflammasome complexes. Research has demonstrated that autophagy in macrophages is protective against alcohol-induced liver injury in an NLRP3 inflammasome-dependent manner (94). Taken together, NLRP3 inflammasome induction in macrophages polarizes M1 phenotype, which is under the negative regulation of the autophagy signaling pathway.
Patients with chronic HCV infection and cirrhosis have elevated serum Il-1β and IL-18 levels (95, 96). Increased serum IL-1β and IL-18 are the components of NLRP3 inflammasome activation in response to HCV uptake by liver macrophages (95, 96). The current results confirm that NLRP3 inflammasome assembly and activation are central to mitigating HCV-related inflammation and fibrosis (95–97). Besides, NLRP3 inflammasomes expressed in KCs cause inflammatory cytokine production and fibrotic collagen formation in schistosomiasis-induced liver fibrosis (SSLF) (98) and idiosyncratic liver injury-induced fibrosis (99, 100). The above observation confirms that the NLRP3 inflammasomes in hepatic macrophages are crucial in initiating and propagating liver fibrosis from different etiologies.
3.2.3 Other Hepatic Macrophage Inflammasomes in Liver Fibrosis
Not only NLRP3 inflammasome but also other inflammasome complexes are involved in liver injury and liver fibrosis. Assembly of the AIM2 inflammasome in KCs led to the processing of IL-1β and IL-18 in response to hepatitis B virus infection, aggregating the development of liver cirrhosis (34, 101). Moreover, perfluorooctane sulfonate (PFOS), a chemical that causes chronic systematic inflammation, activated the AIM2 inflammasome via the Ca(2+)–PKC–NF-κB/JNK–BAX/BAK axis (35). In contrast, deletion of the AIM2 suppressed PFOS-induced inflammation and fibrosis in the liver and other organs (35). This study shows the critical role of the AIM2 inflammasome in toxin-induced chronic inflammation (35).
The NLRP6 inflammasome promotes the onset of hepatic granuloma and collagen deposition, indicating that NLRP6 is a crucial trigger for SSLF (102). Activation of the NLRC4 inflammasome and the subsequent pyroptosis and IL-18 and IL-1β secretion in macrophages promotes cross-talk with HSCs, exacerbating inflammation and fibrosis development in NAFLD (28). Furthermore, the NLRP12 inflammasome negatively modulated inflammatory responses by blocking the NF-κB and MAPK signaling pathways as well as IL-1β release from BMDMs in mouse liver and spleen against the infection of Brucella abortus, which is a kind of Gram-negative bacterium infection causing innate immunity and subsequent chronic inflammation in the host (103).
In summary, inflammasome activation mediates the polarization of liver macrophages and subsequent interaction with HSCs, contributing to the progression of liver fibrosis.
3.3 Inflammasomes and Pyroptosis in Cholangiocytes
Cholangiopathies, e.g., primary sclerosing cholangitis (PSC) and primary biliary cholangitis (PBC), represent an autoimmune inflammatory liver disease characterized by chronic inflammation and subsequent sclerosis and destruction of intrahepatic small bile ducts (104). Increased NLRP3 expression in reactive cholangiocytes resulted in pro-inflammatory IL-18 production and influenced the epithelial integrity of cholangiocytes in both murine PSC model and patients with PSC (105). Meanwhile, inflammasome activation in cholangiocytes interacts with nuclear translocation of pSer675β-catenin and transcriptional activation, which recruits M1 macrophages in a CXC-chemokine ligand-1/10/12 (CXCL-1/10/12) dependent manner and activates HSCs in a transforming growth factor-β (TGF-β) dependent manner. These liver cell phenotype changes initiate biliary inflammation and fibrosis (106, 107). Furthermore, galectin-3, a pleiotropic lectin that mediates cell-cell adhesion, is secreted by these inflammatory macrophages and exacerbates cholangiocyte injury. While inflammasome activation and PBC-induced fibrosis can be improved by deleting macrophage-derived galectin-3 expression in the mouse model (108).
3.4 Inflammasomes and Pyroptosis in HSCs
In addition to cross-talk through inflammasomes between other liver cell types and HSCs, HSC-derived inflammasomes can be remarkably induced in pathological conditions. Inflammasome activation in HSCs initiates a range of functional changes such as transdifferentiating into a fibrogenic activated phenotype, as well as inhibition of chemotaxis (109).
3.4.1 HSC NLRP3 Inflammasome in Liver Fibrosis
Palmitic acid upregulated the NLRP3–IL-1β axis in HSCs via the TLR4–MyD88–NF-κB signaling pathway (110) and hedgehog signaling pathway (111). Then the activated NLRP3 facilitates IL-1 receptor expression on HSCs and the consequent fibrotic induction with the development of NASH (111). Alternatively, the HSC-derived NLRP3 inflammasome could also be activated via the PDGFβR–NLRP3–caspase-1 signaling pathway (112), and result in increased expression of fibrotic markers alpha-smooth muscle actin (α-SMA), connective tissue growth factor (CTGF), and tissue inhibitors of matrix metalloproteinase 1 (TIMP1) in the CCl4 mouse model. Mechanically, many molecules are involved in the activation of the NLRP3 inflammasome. ROS plays a vital role in activating the NLRP3–IL-1β signaling pathway in HSCs (113). Angiotensin II-mediated NLRP3 inflammasome assembly contributed to cholestatic liver fibrosis (114), whereas Nlrp3 knockout or inhibition of Ang- (1–7) reduced ECM synthesis and deposition (115). The vitamin D receptor (VDR) agonist calcipotriol alleviated cholestatic fibrosis via YAP1 mediated inactivation of the NLRP3 inflammasome and caspase-1 (116). The P2X7 receptor, which binds with endogenous danger extracellular ATP, is involved in the immune response and inflammation by activating the NLRP3 inflammasome and increasing IL-1β production and pyroptosis in HSCs during the development of chronic alcoholic liver fibrosis (117). Meanwhile, the release of inflammasome components from pyroptotic HSCs, in turn, activates quiescent HSCs (117). Cysteine–cysteine chemokine ligand 5 (CCL5) secreted from macrophages, activated HSC-derived NLRP3, IL-1β, and IL-6 and upregulated liver fibrosis markers α-SMA and TGF-β1 (118). In contrast, blocking antibodies against CCL5 inhibited HSCs activation and HCV-related liver fibrosis (118).
3.4.2 HSC Inflammasomes in Infection-Related and Hormone-Related Liver Fibrosis
Brucella abortus infection not only affected macrophages but also triggered NLRP3 and AIM2 inflammasome assembly in HSCs, which eventually led to collagens deposition in mouse liver (119). Schistosoma Japonicum is a parasite that causes granulomatous inflammation and tissue damage. Infection with Schistosoma Japonicum stimulated the NLRP3 inflammasome via Dectin-1, and JNK signaling pathways, contributing to SSLF (120). Aldosterone, the main mineralocorticoid steroid hormone secreted by the adrenal gland, was shown to play a role in regulating myofibroblast contraction and proliferation by assembly of NLRP3 inflammasome in HSCs. NLRP3 depletion in primary mouse HSCs attenuated liver fibrosis in the presence of aldosterone, indicating the crucial role of NLRP3 inflammasome in aldosterone-mediated liver fibrosis (121).
The above research indicates that the NLRP3 inflammasome is the most well-studied inflammasome in HSCs. Whether other inflammasomes in HSCs are also involved in the development of liver fibrosis remains unclear.
3.5 Inflammasomes and Pyroptosis in Other Liver Cells
Apart from the abovementioned cells, inflammasome activation has also existed in other liver cells. DAMPs from fatty hepatocytes initiate NLRP3 inflammasome complex assembly via the P2X7 receptor on sinusoidal lining cells such as LSECs (122), resulting in NASH-associated fibrosis (123). Inflammasome activation in natural killer (NK) cells promotes HSC apoptosis and alleviates the progression of liver fibrosis in a TRAIL-dependent degranulation manner (124). In contrast, natural killer T (NKT) cells had dual roles in regulating liver fibrosis via activating the NLRP3 inflammasome (125). Besides, the NLRC4 inflammasome in neutrophils initiated auto-inflammatory disease, and these effects were attenuated in Asc knockout mice or after IL-1 receptor inhibitor administration (126).
The impact of the different inflammasomes in individual cells varies in liver diseases and animal models. Research that uses high-throughput technologies might help us establish the landscape of inflammasomes in liver cells cross-talk. Additionally, more attention should be paid to the mechanism of inflammasome-induced pyroptosis of individual liver cells during different pathological stimulations.
4 Inflammasome-Targeting Therapies in Liver Fibrosis
An increasing number of studies have confirmed the involvement of inflammasomes in the development of chronic inflammation-induced liver fibrosis, indicating the possibility of therapies targeting inflammasome complex activation as well as signaling pathways involved in IL-1β and IL-18. Here, we summarized the pharmacological therapies targeting inflammasome- and pyroptosis-related signaling pathways on chronic liver injury-induced fibrosis from clinical trials and pre-clinical experimental studies (Table 1).
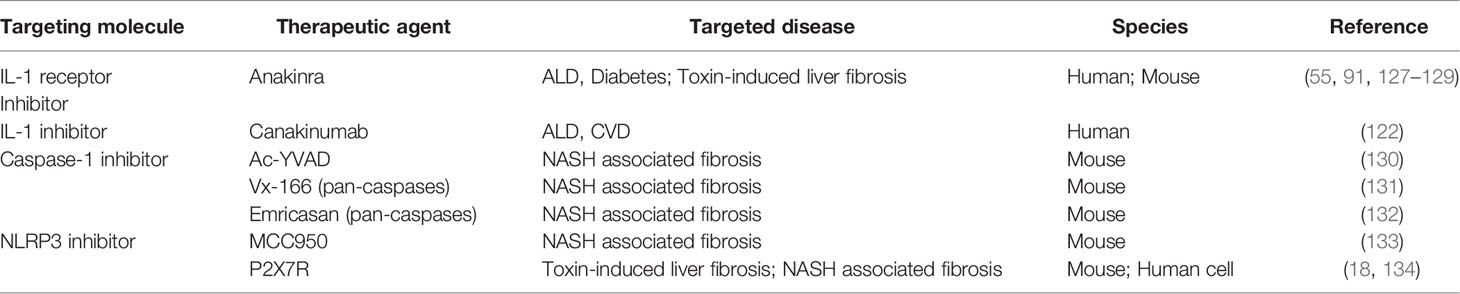
Table 1 Potential Therapeutic Agents for liver fibrosis.
4.1 IL-1 Inhibitors
Macrophages, hepatocytes, and HSCs are the major cells that produce inflammasome-driven IL-1β due to ethanol and LPS stimulation. Recombinant IL-1R antagonist administration in mice inhibited the IL-1 signaling pathway and reversed alcohol-induced liver steatosis, inflammation, and fibrosis (91, 129). Anakinra, a recombinant form of human IL-1R antagonist, is FDA approved medicine to treat rheumatoid arthritis and neonatal-onset multisystem inflammatory disease (135). It was shown that Anakinra could relieve macrophage infiltration, lipid accumulation, and liver fibrosis in a mouse model of ALD (129). Moreover, Anakinra treatment alleviated liver injury and inflammation without affecting fibrosis in a mouse model of CCl4-induced liver fibrosis, as shown by a decrease in caspase-1 and IL-1β with unchanged CTGF and TIMP expression (55). Since good efficiency in treating experimental liver diseases, Anakinra combined with zinc and pentoxifylline is used to treat patients with severe alcoholic hepatitis and type 2 diabetes. Unfortunately, Anakinra did not affect 30-day mortality in research focused on 3-month and 6-month survival rates (ClinicalTrials.gov, AH/NCT01809132) (127). In another clinical trial, Anakinra was administered to patients with type 2 diabetes. They found a decrease in insulin resistance and systematic inflammation, which also participate in the progression of NASH (128). Further clinical trials on Anakinra in patients with NASH are needed. Canakinumab, a monoclonal antibody against IL-1β, was beneficial in patients with cardiovascular diseases (122), and a clinical trial about its effect on patients with severe alcoholic hepatitis is ongoing (ClinicalTrials.gov, AH/NCT03775109). Although many basic studies have shown beneficial effects by inhibiting the IL-1 singling pathway, few have been translated into clinical treatment of chronic liver diseases and liver fibrosis. More clinical trials are needed to explore the role of IL-1 in liver fibrosis.
4.2 Caspase-1 Inhibitors
The pan-caspase inhibitors, Vx-166 and Emricasan, showed a beneficial role in liver inflammation and fibrosis in NASH mice by decreasing the expression of IL-1β and IL-18 and inactivating HSCs (131, 132). A caspase-1 specific inhibitor, Ac-YVAD, was demonstrated to block liver steatosis and fibrosis in mice fed with HFD (130). In view of the excellent effect on animal models, clinical trials of caspase-1 inhibitors on chronic liver diseases are urgently needed in the future.
4.3 Inflammasomes and Their Upstream Inhibitors
The NLRP3 inflammasome inhibitor MCC950 was reported to relieve liver inflammation via polarizing macrophages into M2 phenotype in CCl4-induced liver injury (136) and fibrosis via decreasing plasma and hepatic IL-1β and IL-6 in a mouse model of NASH (133), suggesting the pivotal role of MCC950 in attenuating liver inflammation from various etiologies. As the upstream molecule of the inflammasome, the P2X7 receptor induces inflammasome activation (18, 117). P2X7 receptor pharmacological inhibitor SGM-1019 was shown to block IL-1β secretion in KCs, HSC activation, and collagen deposition in human cells from NASH and in the primate model from CCl4-induced liver fibrosis (134). Although inflammasome inhibitors showed promising effects in experimental models, whether these inhibitors could improve liver inflammation and fibrosis in patients with liver cirrhosis remains obscure.
5 Future Perspectives
Inflammasome, a double-edged sword in liver injury, could protect the liver from pathogen infection, metabolism syndrome, and oxidative stress by eliminating the initial cause of cell injury and promoting wound healing. However, excessive and chronic inflammasome activation contributes to the pathogenesis of various liver diseases, which is the primary topic of our review. In summary, the pathological role of inflammasomes in liver fibrosis has gained substantial recognition from diverse chronic liver injury models. Exogenous and endogenous danger signals activate inflammasomes via canonical or non-canonical signaling pathways, leading to increased IL-1β, IL-18, and pyroptotic cell death. In vivo and in vitro studies over the recent years have evidenced central roles of IL-1β, IL-18, and pyroptotic cell death in NLRP3 inflammasome-induced biological responses, but the functions of the non-canonical inflammasome activation signaling pathway and other NLR genes in liver fibrosis remains poorly understood. Further studies are needed to explore the cellular source of other inflammasomes and their roles in liver fibrosis. In addition, the mechanism of GSDMD assembly leading to cell lysis is largely unknown. Apart from IL-1, inflammasomes also regulate the release of other immune factors, such as leukotrienes and prostaglandins (137), which could regulate liver fibrosis progression (85). Therefore, further studies are required to explore the potential mechanisms.
Quiescent HSCs change phenotypes and have different cell fates after stimuli, including activation, senescence, and inactivation. These phenotypes can be transdifferentiated from each other (5). Most studies illustrate that HSCs are activated by paracrine profibrogenic cytokines and danger signals, while other molecules from pyroptotic cells that regulate HSC fate have scarcely been investigated. Specifically, it remains unclear whether inhibition of inflammasome-mediated cell interactions with HSCs could regress liver fibrosis. Although IL-1 from inflammatory hepatic macrophages could activate HSCs, the release of matrix metalloproteinases (MMPs) from these inflammatory macrophages degrades ECM and resolves fibrosis (138). Inflammasome-activated NK cells and NKT cells secreted inflammatory cytokine IL-1 to promote liver fibrogenesis, whereas IFN-γ secreted from the two kinds of cells have an antifibrotic role and trigger HSC apoptosis, promoting the regression of liver fibrosis (139, 140). Therefore, the cell-specific mechanisms of intercellular cross-talk in HSC activation need in-depth investigation.
In recent years, the pharmacological treatment of inflammasomes in chronic liver injury mainly focuses on IL-1β and IL-1 receptors. Research on blocking other molecules in the inflammasome pathway, such as caspase-1 and GSDMD, is missing. Though a recent study has shown that IL-18 inhibitor attenuates renal fibrosis in the ischemia-reperfusion murine model (141), there is no research on IL-18 inhibitors treating liver fibrosis in either animal models or human. Further studies are urgent to explore the potential of blocking IL-18, as well as caspase-1 and GSDMD in treating liver fibrosis. Although inflammasomes have been confirmed to have critical effects in many experimental animal models and in vitro cell lines, no direct evidence demonstrates that inflammasomes mediate liver fibrosis in humans. Organoids are self-organized 3D tissue cultures from stem cells that could recapitulate the function of the represented organ (142). Hence, organoids may be a novel research method to explore the mechanism of the inflammasome in regulating liver fibrosis. It is shown that bone marrow-derived mesenchymal stem cells (BM-MSCs) injection could decrease inflammasomes in the ischemic stroke rat model (143). Therefore, cell therapy by in vivo delivery of BM-MSCs may be helpful to mitigate inflammasomes during the development of liver fibrosis. Further studies are needed to target the novel direction. In summary, targeting inflammasome signaling pathways to treat liver fibrosis is promising, and more basic research and clinical trials are demanded in the future.
Author Contributions
CT and JG designed and coordinated the study. CG, QC, CT, and JG analyzed articles and finalized the figures. CG, QC, and JG wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Fund of China (82170623, 82170625, U1702281, 81873584, 82000613, and 82000574), National Key R&D Program of China (2017YFA0205404), Sichuan Science and Technology Program (2020YJ0084 and 2021YFS0147), and the 135 projects for disciplines of excellence of West China Hospital, Sichuan University (ZYGD18004).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Broz P, Dixit VM. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat Rev Immunol (2016) 16(7):407–20. doi: 10.1038/nri.2016.58
2. Christgen S, Place DE, Kanneganti TD. Toward Targeting Inflammasomes: Insights Into Their Regulation and Activation. Cell Res (2020) 30(4):315–27. doi: 10.1038/s41422-020-0295-8
3. Zeng XH, Yuan XL, Cai QY, Tang CW, Gao JH. Circular Rna as an Epigenetic Regulator in Chronic Liver Diseases. Cells (2021) 10(8):1945–59. doi: 10.3390/cells10081945
4. Cai Q, Gan C, Tang C, Wu H, Gao J. Mechanism and Therapeutic Opportunities of Histone Modifications in Chronic Liver Disease. Front Pharmacol (2021) 12:784591. doi: 10.3389/fphar.2021.784591
5. Kisseleva T, Brenner D. Molecular and Cellular Mechanisms of Liver Fibrosis and Its Regression. Nat Rev Gastroenterol Hepatol (2021) 18(3):151–66. doi: 10.1038/s41575-020-00372-7
6. Koyama Y, Brenner DA. Liver Inflammation and Fibrosis. J Clin Invest (2017) 127(1):55–64. doi: 10.1172/jci88881
7. Tsuchida T, Friedman SL. Mechanisms of Hepatic Stellate Cell Activation. Nat Rev Gastroenterol Hepatol (2017) 14(7):397–411. doi: 10.1038/nrgastro.2017.38
8. Cao S, Liu M, Sehrawat TS, Shah VH. Regulation and Functional Roles of Chemokines in Liver Diseases. Nat Rev Gastroenterol Hepatol (2021) 18(9):630–47. doi: 10.1038/s41575-021-00444-2
9. Vanaja SK, Rathinam VA, Fitzgerald KA. Mechanisms of Inflammasome Activation: Recent Advances and Novel Insights. Trends Cell Biol (2015) 25(5):308–15. doi: 10.1016/j.tcb.2014.12.009
10. Martínez-Cardona C, Lozano-Ruiz B, Bachiller V, Peiró G, Algaba-Chueca F, Gómez-Hurtado I, et al. Aim2 Deficiency Reduces the Development of Hepatocellular Carcinoma in Mice. Int J Cancer (2018) 143(11):2997–3007. doi: 10.1002/ijc.31827
11. Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, et al. Reconstituted Nalp1 Inflammasome Reveals Two-Step Mechanism of Caspase-1 Activation. Mol Cell (2007) 25(5):713–24. doi: 10.1016/j.molcel.2007.01.032
12. Finger JN, Lich JD, Dare LC, Cook MN, Brown KK, Duraiswami C, et al. Autolytic Proteolysis Within the Function to Find Domain (Fiind) Is Required for Nlrp1 Inflammasome Activity. J Biol Chem (2012) 287(30):25030–7. doi: 10.1074/jbc.M112.378323
13. Moayeri M, Sastalla I, Leppla SH. Anthrax and the Inflammasome. Microbes Infect (2012) 14(5):392–400. doi: 10.1016/j.micinf.2011.12.005
14. Pelegrin P, Surprenant A. Pannexin-1 Mediates Large Pore Formation and Interleukin-1beta Release by the Atp-Gated P2x7 Receptor. EMBO J (2006) 25(21):5071–82. doi: 10.1038/sj.emboj.7601378
15. Wickliffe KE, Leppla SH, Moayeri M. Anthrax Lethal Toxin-Induced Inflammasome Formation and Caspase-1 Activation Are Late Events Dependent on Ion Fluxes and the Proteasome. Cell Microbiol (2008) 10(2):332–43. doi: 10.1111/j.1462-5822.2007.01044.x
16. Latz E, Xiao TS, Stutz A. Activation and Regulation of the Inflammasomes. Nat Rev Immunol (2013) 13(6):397–411. doi: 10.1038/nri3452
17. Duncan JA, Gao X, Huang MT, O'Connor BP, Thomas CE, Willingham SB, et al. Neisseria Gonorrhoeae Activates the Proteinase Cathepsin B to Mediate the Signaling Activities of the Nlrp3 and Asc-Containing Inflammasome. J Immunol (2009) 182(10):6460–9. doi: 10.4049/jimmunol.0802696
18. Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, et al. Cryopyrin Activates the Inflammasome in Response to Toxins and Atp. Nature (2006) 440(7081):228–32. doi: 10.1038/nature04515
19. Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-Associated Uric Acid Crystals Activate the Nalp3 Inflammasome. Nature (2006) 440(7081):237–41. doi: 10.1038/nature04516
20. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The Nalp3 Inflammasome Is Involved in the Innate Immune Response to Amyloid-Beta. Nat Immunol (2008) 9(8):857–65. doi: 10.1038/ni.1636
21. Woolbright BL, Jaeschke H. Role of the Inflammasome in Acetaminophen-Induced Liver Injury and Acute Liver Failure. J Hepatol (2017) 66(4):836–48. doi: 10.1016/j.jhep.2016.11.017
22. Szabo G, Csak T. Inflammasomes in Liver Diseases. J Hepatol (2012) 57(3):642–54. doi: 10.1016/j.jhep.2012.03.035
23. Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, et al. Cytoplasmic Flagellin Activates Caspase-1 and Secretion of Interleukin 1beta Via Ipaf. Nat Immunol (2006) 7(6):569–75. doi: 10.1038/ni1344
24. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential Activation of the Inflammasome by Caspase-1 Adaptors Asc and Ipaf. Nature (2004) 430(6996):213–8. doi: 10.1038/nature02664
25. Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, et al. Innate Immune Detection of the Type Iii Secretion Apparatus Through the Nlrc4 Inflammasome. Proc Natl Acad Sci U S A (2010) 107(7):3076–80. doi: 10.1073/pnas.0913087107
26. Tenthorey JL, Kofoed EM, Daugherty MD, Malik HS, Vance RE. Molecular Basis for Specific Recognition of Bacterial Ligands by Naip/Nlrc4 Inflammasomes. Mol Cell (2014) 54(1):17–29. doi: 10.1016/j.molcel.2014.02.018
27. Etienne-Mesmin L, Vijay-Kumar M, Gewirtz AT, Chassaing B. Hepatocyte Toll-Like Receptor 5 Promotes Bacterial Clearance and Protects Mice Against High-Fat Diet-Induced Liver Disease. Cell Mol Gastroenterol Hepatol (2016) 2(5):584–604. doi: 10.1016/j.jcmgh.2016.04.007
28. Chen Y, Ma K. Nlrc4 Inflammasome Activation Regulated by Tnf-α Promotes Inflammatory Responses in Nonalcoholic Fatty Liver Disease. Biochem Biophys Res Commun (2019) 511(3):524–30. doi: 10.1016/j.bbrc.2019.02.099
29. Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, et al. The Aim2 Inflammasome Is Essential for Host Defense Against Cytosolic Bacteria and DNA Viruses. Nat Immunol (2010) 11(5):395–402. doi: 10.1038/ni.1864
30. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. Aim2 Recognizes Cytosolic Dsdna and Forms a Caspase-1-Activating Inflammasome With Asc. Nature (2009) 458(7237):514–8. doi: 10.1038/nature07725
31. Mambwe B, Neo K, Javanmard Khameneh H, Leong KWK, Colantuoni M, Vacca M, et al. Tyrosine Dephosphorylation of Asc Modulates the Activation of the Nlrp3 and Aim2 Inflammasomes. Front Immunol (2019) 10:1556. doi: 10.3389/fimmu.2019.01556
32. Xu L, Zhou J, Che J, Wang H, Yang W, Zhou W, et al. Mitochondrial DNA Enables Aim2 Inflammasome Activation and Hepatocyte Pyroptosis in Nonalcoholic Fatty Liver Disease. Am J Physiol Gastrointest Liver Physiol (2021) 320(6):G1034–g44. doi: 10.1152/ajpgi.00431.2020
33. Sun Q, Loughran P, Shapiro R, Shrivastava IH, Antoine DJ, Li T, et al. Redox-Dependent Regulation of Hepatocyte Absent in Melanoma 2 Inflammasome Activation in Sterile Liver Injury in Mice. Hepatology (2017) 65(1):253–68. doi: 10.1002/hep.28893
34. Lozano-Ruiz B, Bachiller V, García-Martínez I, Zapater P, Gómez-Hurtado I, Moratalla A, et al. Absent in Melanoma 2 Triggers a Heightened Inflammasome Response in Ascitic Fluid Macrophages of Patients With Cirrhosis. J Hepatol (2015) 62(1):64–71. doi: 10.1016/j.jhep.2014.08.027
35. Wang LQ, Liu T, Yang S, Sun L, Zhao ZY, Li LY, et al. Perfluoroalkyl Substance Pollutants Activate the Innate Immune System Through the Aim2 Inflammasome. Nat Commun (2021) 12(1):2915. doi: 10.1038/s41467-021-23201-0
36. Schnappauf O, Chae JJ, Kastner DL, Aksentijevich I. The Pyrin Inflammasome in Health and Disease. Front Immunol (2019) 10:1745. doi: 10.3389/fimmu.2019.01745
37. Heilig R, Broz P. Function and Mechanism of the Pyrin Inflammasome. Eur J Immunol (2018) 48(2):230–8. doi: 10.1002/eji.201746947
38. Mulvihill E, Sborgi L, Mari SA, Pfreundschuh M, Hiller S, Müller DJ. Mechanism of Membrane Pore Formation by Human Gasdermin-D. EMBO J (2018) 37(14):e98321–31. doi: 10.15252/embj.201798321
39. Rathinam VA, Fitzgerald KA. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell (2016) 165(4):792–800. doi: 10.1016/j.cell.2016.03.046
40. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D Is an Executor of Pyroptosis and Required for Interleukin-1β Secretion. Cell Res (2015) 25(12):1285–98. doi: 10.1038/cr.2015.139
41. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of Gsdmd by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature (2015) 526(7575):660–5. doi: 10.1038/nature15514
42. Kist M, Vucic D. Cell Death Pathways: Intricate Connections and Disease Implications. EMBO J (2021) 40(5):e106700. doi: 10.15252/embj.2020106700
43. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 Cleaves Gasdermin D for Non-Canonical Inflammasome Signalling. Nature (2015) 526(7575):666–71. doi: 10.1038/nature15541
44. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting Edge: Nf-Kappab Activating Pattern Recognition and Cytokine Receptors License Nlrp3 Inflammasome Activation by Regulating Nlrp3 Expression. J Immunol (2009) 183(2):787–91. doi: 10.4049/jimmunol.0901363
45. Zhou R, Yazdi AS, Menu P, Tschopp J. A Role for Mitochondria in Nlrp3 Inflammasome Activation. Nature (2011) 469(7329):221–5. doi: 10.1038/nature09663
46. Rühl S, Broz P. Caspase-11 Activates a Canonical Nlrp3 Inflammasome by Promoting K(+) Efflux. Eur J Immunol (2015) 45(10):2927–36. doi: 10.1002/eji.201545772
47. Parola M, Pinzani M. Liver Fibrosis: Pathophysiology, Pathogenetic Targets and Clinical Issues. Mol Aspects Med (2019) 65:37–55. doi: 10.1016/j.mam.2018.09.002
48. Marrone G, Shah VH, Gracia-Sancho J. Sinusoidal Communication in Liver Fibrosis and Regeneration. J Hepatol (2016) 65(3):608–17. doi: 10.1016/j.jhep.2016.04.018
49. Gao J, Wei B, Liu M, Hirsova P, Sehrawat TS, Cao S, et al. Endothelial P300 Promotes Portal Hypertension and Hepatic Fibrosis Through C-C Motif Chemokine Ligand 2-Mediated Angiocrine Signaling. Hepatology (2021) 73(6):2468–83. doi: 10.1002/hep.31617
50. Gao J, Wei B, de Assuncao TM, Liu Z, Hu X, Ibrahim S, et al. Hepatic Stellate Cell Autophagy Inhibits Extracellular Vesicle Release to Attenuate Liver Fibrosis. J Hepatol (2020) 73(5):1144–54. doi: 10.1016/j.jhep.2020.04.044
51. Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in Health and Disease. Nature (2012) 481(7381):278–86. doi: 10.1038/nature10759
52. Szabo G, Petrasek J. Inflammasome Activation and Function in Liver Disease. Nat Rev Gastroenterol Hepatol (2015) 12(7):387–400. doi: 10.1038/nrgastro.2015.94
53. Schuster-Gaul S, Geisler LJ, McGeough MD, Johnson CD, Zagorska A, Li L, et al. Ask1 Inhibition Reduces Cell Death and Hepatic Fibrosis in an Nlrp3 Mutant Liver Injury Model. JCI Insight (2020) 5(2):e123294–308. doi: 10.1172/jci.insight.123294
54. Luan J, Ju D. Inflammasome: A Double-Edged Sword in Liver Diseases. Front Immunol (2018) 9:2201. doi: 10.3389/fimmu.2018.02201
55. Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. Nlrp3 Inflammasome Activation Results in Hepatocyte Pyroptosis, Liver Inflammation, and Fibrosis in Mice. Hepatology (2014) 59(3):898–910. doi: 10.1002/hep.26592
56. Zhang X, Zhang JH, Chen XY, Hu QH, Wang MX, Jin R, et al. Reactive Oxygen Species-Induced Txnip Drives Fructose-Mediated Hepatic Inflammation and Lipid Accumulation Through Nlrp3 Inflammasome Activation. Antioxid Redox Signal (2015) 22(10):848–70. doi: 10.1089/ars.2014.5868
57. Wan X, Xu C, Lin Y, Lu C, Li D, Sang J, et al. Uric Acid Regulates Hepatic Steatosis and Insulin Resistance Through the Nlrp3 Inflammasome-Dependent Mechanism. J Hepatol (2016) 64(4):925–32. doi: 10.1016/j.jhep.2015.11.022
58. Zhang NP, Liu XJ, Xie L, Shen XZ, Wu J. Impaired Mitophagy Triggers Nlrp3 Inflammasome Activation During the Progression From Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis. Lab Invest (2019) 99(6):749–63. doi: 10.1038/s41374-018-0177-6
59. Yu X, Hao M, Liu Y, Ma X, Lin W, Xu Q, et al. Liraglutide Ameliorates Non-Alcoholic Steatohepatitis by Inhibiting Nlrp3 Inflammasome and Pyroptosis Activation Via Mitophagy. Eur J Pharmacol (2019) 864:172715. doi: 10.1016/j.ejphar.2019.172715
60. Zhu Y, Zhao H, Lu J, Lin K, Ni J, Wu G, et al. Caspase-11-Mediated Hepatocytic Pyroptosis Promotes the Progression of Nonalcoholic Steatohepatitis. Cell Mol Gastroenterol Hepatol (2021) 12(2):653–64. doi: 10.1016/j.jcmgh.2021.04.009
61. Gaul S, Leszczynska A, Alegre F, Kaufmann B, Johnson CD, Adams LA, et al. Hepatocyte Pyroptosis and Release of Inflammasome Particles Induce Stellate Cell Activation and Liver Fibrosis. J Hepatol (2021) 74(1):156–67. doi: 10.1016/j.jhep.2020.07.041
62. Heo MJ, Kim TH, You JS, Blaya D, Sancho-Bru P, Kim SG. Alcohol Dysregulates Mir-148a in Hepatocytes Through Foxo1, Facilitating Pyroptosis Via Txnip Overexpression. Gut (2019) 68(4):708–20. doi: 10.1136/gutjnl-2017-315123
63. Khanova E, Wu R, Wang W, Yan R, Chen Y, French SW, et al. Pyroptosis by Caspase11/4-Gasdermin-D Pathway in Alcoholic Hepatitis in Mice and Patients. Hepatology (2018) 67(5):1737–53. doi: 10.1002/hep.29645
64. Frissen M, Liao L, Schneider KM, Djudjaj S, Haybaeck J, Wree A, et al. Bidirectional Role of Nlrp3 During Acute and Chronic Cholestatic Liver Injury. Hepatology (2021) 73(5):1836–54. doi: 10.1002/hep.31494
65. Lebeaupin C, Vallée D, Rousseau D, Patouraux S, Bonnafous S, Adam G, et al. Bax Inhibitor-1 Protects From Nonalcoholic Steatohepatitis by Limiting Inositol-Requiring Enzyme 1 Alpha Signaling in Mice. Hepatology (2018) 68(2):515–32. doi: 10.1002/hep.29847
66. Lebeaupin C, Proics E, de Bieville CH, Rousseau D, Bonnafous S, Patouraux S, et al. Er Stress Induces Nlrp3 Inflammasome Activation and Hepatocyte Death. Cell Death Dis (2015) 6(9):e1879. doi: 10.1038/cddis.2015.248
67. Han CY, Rho HS, Kim A, Kim TH, Jang K, Jun DW, et al. Fxr Inhibits Endoplasmic Reticulum Stress-Induced Nlrp3 Inflammasome in Hepatocytes and Ameliorates Liver Injury. Cell Rep (2018) 24(11):2985–99. doi: 10.1016/j.celrep.2018.07.068
68. Koh EH, Yoon JE, Ko MS, Leem J, Yun JY, Hong CH, et al. Sphingomyelin Synthase 1 Mediates Hepatocyte Pyroptosis to Trigger Non-Alcoholic Steatohepatitis. Gut (2021) 70(10):1954–64. doi: 10.1136/gutjnl-2020-322509
69. Kazankov K, Jørgensen SMD, Thomsen KL, Møller HJ, Vilstrup H, George J, et al. The Role of Macrophages in Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Nat Rev Gastroenterol Hepatol (2019) 16(3):145–59. doi: 10.1038/s41575-018-0082-x
70. Tacke F. Targeting Hepatic Macrophages to Treat Liver Diseases. J Hepatol (2017) 66(6):1300–12. doi: 10.1016/j.jhep.2017.02.026
71. Wang X, Gao Y, Song J, Tang C, Wang M, Que L, et al. The Tir/Bb-Loop Mimetic as-1 Prevents Non-Alcoholic Steatohepatitis and Hepatic Insulin Resistance by Inhibiting Nlrp3-Asc Inflammasome Activation. Br J Pharmacol (2017) 174(12):1841–56. doi: 10.1111/bph.13786
72. Wang F, Park JS, Ma Y, Ma H, Lee YJ, Lee GR, et al. Ginseng Saponin Enriched in Rh1 and Rg2 Ameliorates Nonalcoholic Fatty Liver Disease by Inhibiting Inflammasome Activation. Nutrients (2021) 13(3):856–72. doi: 10.3390/nu13030856
73. Mohammed S, Nicklas EH, Thadathil N, Selvarani R, Royce GH, Kinter M, et al. Role of Necroptosis in Chronic Hepatic Inflammation and Fibrosis in a Mouse Model of Increased Oxidative Stress. Free Radic Biol Med (2021) 164:315–28. doi: 10.1016/j.freeradbiomed.2020.12.449
74. Wang P, Ni M, Tian Y, Wang H, Qiu J, You W, et al. Myeloid Nrf2 Deficiency Aggravates Non-Alcoholic Steatohepatitis Progression by Regulating Yap-Mediated Nlrp3 Inflammasome Signaling. iScience (2021) 24(5):102427. doi: 10.1016/j.isci.2021.102427
75. Tao L, Yi Y, Chen Y, Zhang H, Orning P, Lien E, et al. Rip1 Kinase Activity Promotes Steatohepatitis Through Mediating Cell Death and Inflammation in Macrophages. Cell Death Differ (2021) 28(4):1418–33. doi: 10.1038/s41418-020-00668-w
76. Shi Y, Su W, Zhang L, Shi C, Zhou J, Wang P, et al. Tgr5 Regulates Macrophage Inflammation in Nonalcoholic Steatohepatitis by Modulating Nlrp3 Inflammasome Activation. Front Immunol (2020) 11:609060. doi: 10.3389/fimmu.2020.609060
77. Miura K, Yang L, van Rooijen N, Brenner DA, Ohnishi H, Seki E. Toll-Like Receptor 2 and Palmitic Acid Cooperatively Contribute to the Development of Nonalcoholic Steatohepatitis Through Inflammasome Activation in Mice. Hepatology (2013) 57(2):577–89. doi: 10.1002/hep.26081
78. Shen L, Yang Y, Ou T, Key CC, Tong SH, Sequeira RC, et al. Dietary Pufas Attenuate Nlrp3 Inflammasome Activation Via Enhancing Macrophage Autophagy. J Lipid Res (2017) 58(9):1808–21. doi: 10.1194/jlr.M075879
79. Liu W, Bai F, Wang H, Liang Y, Du X, Liu C, et al. Tim-4 Inhibits Nlrp3 Inflammasome Via the Lkb1/Ampkα Pathway in Macrophages. J Immunol (2019) 203(4):990–1000. doi: 10.4049/jimmunol.1900117
80. Kim SH, Kim G, Han DH, Lee M, Kim I, Kim B, et al. Ezetimibe Ameliorates Steatohepatitis Via Amp Activated Protein Kinase-Tfeb-Mediated Activation of Autophagy and Nlrp3 Inflammasome Inhibition. Autophagy (2017) 13(10):1767–81. doi: 10.1080/15548627.2017.1356977
81. Ioannou GN, Van Rooyen DM, Savard C, Haigh WG, Yeh MM, Teoh NC, et al. Cholesterol-Lowering Drugs Cause Dissolution of Cholesterol Crystals and Disperse Kupffer Cell Crown-Like Structures During Resolution of Nash. J Lipid Res (2015) 56(2):277–85. doi: 10.1194/jlr.M053785
82. Antonopoulos C, Russo HM, El Sanadi C, Martin BN, Li X, Kaiser WJ, et al. Caspase-8 as an Effector and Regulator of Nlrp3 Inflammasome Signaling. J Biol Chem (2015) 290(33):20167–84. doi: 10.1074/jbc.M115.652321
83. Zhang X, Wang G, Gurley EC, Zhou H. Flavonoid Apigenin Inhibits Lipopolysaccharide-Induced Inflammatory Response Through Multiple Mechanisms in Macrophages. PloS One (2014) 9(9):e107072. doi: 10.1371/journal.pone.0107072
84. Hua KF, Chou JC, Ka SM, Tasi YL, Chen A, Wu SH, et al. Cyclooxygenase-2 Regulates Nlrp3 Inflammasome-Derived Il-1β Production. J Cell Physiol (2015) 230(4):863–74. doi: 10.1002/jcp.24815
85. Gao JH, Wen SL, Feng S, Yang WJ, Lu YY, Tong H, et al. Celecoxib and Octreotide Synergistically Ameliorate Portal Hypertension Via Inhibition of Angiogenesis in Cirrhotic Rats. Angiogenesis (2016) 19(4):501–11. doi: 10.1007/s10456-016-9522-9
86. Tai Y, Zhao C, Zhang L, Tang S, Jia X, Tong H, et al. Celecoxib Reduces Hepatic Vascular Resistance in Portal Hypertension by Amelioration of Endothelial Oxidative Stress. J Cell Mol Med (2021) 25(22):10389–402. doi: 10.1111/jcmm.16968
87. Tang S, Huang Z, Jiang J, Gao J, Zhao C, Tai Y, et al. Celecoxib Ameliorates Liver Cirrhosis Via Reducing Inflammation and Oxidative Stress Along Spleen-Liver Axis in Rats. Life Sci (2021) 272:119203. doi: 10.1016/j.lfs.2021.119203
88. Su W, Tai Y, Tang SH, Ye YT, Zhao C, Gao JH, et al. Celecoxib Attenuates Hepatocyte Apoptosis by Inhibiting Endoplasmic Reticulum Stress in Thioacetamide-Induced Cirrhotic Rats. World J Gastroenterol (2020) 26(28):4094–107. doi: 10.3748/wjg.v26.i28.4094
89. Shang Y, Li XF, Jin MJ, Li Y, Wu YL, Jin Q, et al. Leucodin Attenuates Inflammatory Response in Macrophages and Lipid Accumulation in Steatotic Hepatocytes Via P2x7 Receptor Pathway: A Potential Role in Alcoholic Liver Disease. BioMed Pharmacother (2018) 107:374–81. doi: 10.1016/j.biopha.2018.08.009
90. Petrasek J, Iracheta-Vellve A, Saha B, Satishchandran A, Kodys K, Fitzgerald KA, et al. Metabolic Danger Signals, Uric Acid and Atp, Mediate Inflammatory Cross-Talk Between Hepatocytes and Immune Cells in Alcoholic Liver Disease. J Leukoc Biol (2015) 98(2):249–56. doi: 10.1189/jlb.3AB1214-590R
91. Mathews S, Gao B. Therapeutic Potential of Interleukin 1 Inhibitors in the Treatment of Alcoholic Liver Disease. Hepatology (2013) 57(5):2078–80. doi: 10.1002/hep.26336
92. Kim JW, Roh YS, Jeong H, Yi HK, Lee MH, Lim CW, et al. Spliceosome-Associated Protein 130 Exacerbates Alcohol-Induced Liver Injury by Inducing Nlrp3 Inflammasome-Mediated Il-1β in Mice. Am J Pathol (2018) 188(4):967–80. doi: 10.1016/j.ajpath.2017.12.010
93. Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage Polarization: Different Gene Signatures in M1(Lps+) Vs. Classically and M2(Lps-) Vs. Alternatively Activated Macrophages. Front Immunol (2019) 10:1084. doi: 10.3389/fimmu.2019.01084
94. Ilyas G, Cingolani F, Zhao E, Tanaka K, Czaja MJ. Decreased Macrophage Autophagy Promotes Liver Injury and Inflammation From Alcohol. Alcohol Clin Exp Res (2019) 43(7):1403–13. doi: 10.1111/acer.14041
95. Negash AA, Ramos HJ, Crochet N, Lau DT, Doehle B, Papic N, et al. Il-1β Production Through the Nlrp3 Inflammasome by Hepatic Macrophages Links Hepatitis C Virus Infection With Liver Inflammation and Disease. PloS Pathog (2013) 9(4):e1003330. doi: 10.1371/journal.ppat.1003330
96. Serti E, Werner JM, Chattergoon M, Cox AL, Lohmann V, Rehermann B. Monocytes Activate Natural Killer Cells Via Inflammasome-Induced Interleukin 18 in Response to Hepatitis C Virus Replication. Gastroenterology (2014) 147(1):209–20.e3. doi: 10.1053/j.gastro.2014.03.046
97. Negash AA, Olson RM, Griffin S, Gale M Jr. Modulation of Calcium Signaling Pathway by Hepatitis C Virus Core Protein Stimulates Nlrp3 Inflammasome Activation. PloS Pathog (2019) 15(2):e1007593. doi: 10.1371/journal.ppat.1007593
98. Zhang WJ, Fang ZM, Liu WQ. Nlrp3 Inflammasome Activation From Kupffer Cells Is Involved in Liver Fibrosis of Schistosoma Japonicum-Infected Mice Via Nf-κb. Parasit Vectors (2019) 12(1):29. doi: 10.1186/s13071-018-3223-8
99. Wang Y, Xu G, Wang Z, Li R, Zhan X, Liu H, et al. Psoralidin, a Major Component of Psoraleae Fructus, Induces Inflammasome Activation and Idiosyncratic Liver Injury. Int Immunopharmacol (2021) 92:107352. doi: 10.1016/j.intimp.2020.107352
100. Jia Y, Ma L, Wang Y, Wang W, Shen C, Wang X, et al. Nlrp3 Inflammasome and Related Cytokines Reflect the Immune Status of Patients With Hbv-Aclf. Mol Immunol (2020) 120:179–86. doi: 10.1016/j.molimm.2020.01.011
101. Zannetti C, Roblot G, Charrier E, Ainouze M, Tout I, Briat F, et al. Characterization of the Inflammasome in Human Kupffer Cells in Response to Synthetic Agonists and Pathogens. J Immunol (2016) 197(1):356–67. doi: 10.4049/jimmunol.1502301
102. Sanches RCO, Souza C, Marinho FV, Mambelli FS, Morais SB, Guimarães ES, et al. Nlrp6 Plays an Important Role in Early Hepatic Immunopathology Caused by Schistosoma Mansoni Infection. Front Immunol (2020) 11:795. doi: 10.3389/fimmu.2020.00795
103. Silveira TN, Gomes MT, Oliveira LS, Campos PC, Machado GG, Oliveira SC. Nlrp12 Negatively Regulates Proinflammatory Cytokine Production and Host Defense Against Brucella Abortus. Eur J Immunol (2017) 47(1):51–9. doi: 10.1002/eji.201646502
104. Lazaridis KN, LaRusso NF. The Cholangiopathies. Mayo Clin Proc (2015) 90(6):791–800. doi: 10.1016/j.mayocp.2015.03.017
105. Maroni L, Agostinelli L, Saccomanno S, Pinto C, Giordano DM, Rychlicki C, et al. Nlrp3 Activation Induces Il-18 Synthesis and Affects the Epithelial Barrier Function in Reactive Cholangiocytes. Am J Pathol (2017) 187(2):366–76. doi: 10.1016/j.ajpath.2016.10.010
106. Fabris L, Fiorotto R, Spirli C, Cadamuro M, Mariotti V, Perugorria MJ, et al. Pathobiology of Inherited Biliary Diseases: A Roadmap to Understand Acquired Liver Diseases. Nat Rev Gastroenterol Hepatol (2019) 16(8):497–511. doi: 10.1038/s41575-019-0156-4
107. Lan T, Qian S, Tang C, Gao J. Role of Immune Cells in Biliary Repair. Front Immunol (2022) 13:866040. doi: 10.3389/fimmu.2022.866040
108. Tian J, Yang G, Chen HY, Hsu DK, Tomilov A, Olson KA, et al. Galectin-3 Regulates Inflammasome Activation in Cholestatic Liver Injury. FASEB J (2016) 30(12):4202–13. doi: 10.1096/fj.201600392RR
109. Watanabe A, Sohail MA, Gomes DA, Hashmi A, Nagata J, Sutterwala FS, et al. Inflammasome-Mediated Regulation of Hepatic Stellate Cells. Am J Physiol Gastrointest Liver Physiol (2009) 296(6):G1248–57. doi: 10.1152/ajpgi.90223.2008
110. Dong Z, Zhuang Q, Ning M, Wu S, Lu L, Wan X. Palmitic Acid Stimulates Nlrp3 Inflammasome Activation Through Tlr4-Nf-κb Signal Pathway in Hepatic Stellate Cells. Ann Transl Med (2020) 8(5):168. doi: 10.21037/atm.2020.02.21
111. Duan NN, Liu XJ, Wu J. Palmitic Acid Elicits Hepatic Stellate Cell Activation Through Inflammasomes and Hedgehog Signaling. Life Sci (2017) 176:42–53. doi: 10.1016/j.lfs.2017.03.012
112. Wu X, Zhang F, Xiong X, Lu C, Lian N, Lu Y, et al. Tetramethylpyrazine Reduces Inflammation in Liver Fibrosis and Inhibits Inflammatory Cytokine Expression in Hepatic Stellate Cells by Modulating Nlrp3 Inflammasome Pathway. IUBMB Life (2015) 67(4):312–21. doi: 10.1002/iub.1348
113. Zhao Y, Wang Z, Feng D, Zhao H, Lin M, Hu Y, et al. P66shc Contributes to Liver Fibrosis Through the Regulation of Mitochondrial Reactive Oxygen Species. Theranostics (2019) 9(5):1510–22. doi: 10.7150/thno.29620
114. Ning ZW, Luo XY, Wang GZ, Li Y, Pan MX, Yang RQ, et al. Microrna-21 Mediates Angiotensin Ii-Induced Liver Fibrosis by Activating Nlrp3 Inflammasome/Il-1β Axis Via Targeting Smad7 and Spry1. Antioxid Redox Signal (2017) 27(1):1–20. doi: 10.1089/ars.2016.6669
115. Cai SM, Yang RQ, Li Y, Ning ZW, Zhang LL, Zhou GS, et al. Angiotensin-(1-7) Improves Liver Fibrosis by Regulating the Nlrp3 Inflammasome Via Redox Balance Modulation. Antioxid Redox Signal (2016) 24(14):795–812. doi: 10.1089/ars.2015.6498
116. Wang X, Wang G, Qu J, Yuan Z, Pan R, Li K. Calcipotriol Inhibits Nlrp3 Signal Through Yap1 Activation to Alleviate Cholestatic Liver Injury and Fibrosis. Front Pharmacol (2020) 11:200. doi: 10.3389/fphar.2020.00200
117. Shan L, Jiang T, Ci L, Liu Z, Lv X, Li J. Purine Signaling Regulating Hscs Inflammatory Cytokines Secretion, Activation, and Proliferation Plays a Critical Role in Alcoholic Liver Disease. Mol Cell Biochem (2020) 466(1-2):91–102. doi: 10.1007/s11010-020-03691-0
118. Sasaki R, Devhare PB, Steele R, Ray R, Ray RB. Hepatitis C Virus-Induced Ccl5 Secretion From Macrophages Activates Hepatic Stellate Cells. Hepatology (2017) 66(3):746–57. doi: 10.1002/hep.29170
119. Arriola Benitez PC, Pesce Viglietti AI, Gomes MTR, Oliveira SC, Quarleri JF, Giambartolomei GH, et al. Brucella Abortus Infection Elicited Hepatic Stellate Cell-Mediated Fibrosis Through Inflammasome-Dependent Il-1β Production. Front Immunol (2019) 10:3036. doi: 10.3389/fimmu.2019.03036
120. Lu YQ, Zhong S, Meng N, Fan YP, Tang WX. Nlrp3 Inflammasome Activation Results in Liver Inflammation and Fibrosis in Mice Infected With Schistosoma Japonicum in a Syk-Dependent Manner. Sci Rep (2017) 7(1):8120. doi: 10.1038/s41598-017-08689-1
121. Li Y, Zhang Y, Chen T, Huang Y, Zhang Y, Geng S, et al. Role of Aldosterone in the Activation of Primary Mice Hepatic Stellate Cell and Liver Fibrosis Via Nlrp3 Inflammasome. J Gastroenterol Hepatol (2020) 35(6):1069–77. doi: 10.1111/jgh.14961
122. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory Therapy With Canakinumab for Atherosclerotic Disease. N Engl J Med (2017) 377(12):1119–31. doi: 10.1056/NEJMoa1707914
123. Blasetti Fantauzzi C, Menini S, Iacobini C, Rossi C, Santini E, Solini A, et al. Deficiency of the Purinergic Receptor 2x(7) Attenuates Nonalcoholic Steatohepatitis Induced by High-Fat Diet: Possible Role of the Nlrp3 Inflammasome. Oxid Med Cell Longev (2017) 2017:8962458. doi: 10.1155/2017/8962458
124. Li T, Yang Y, Song H, Li H, Cui A, Liu Y, et al. Activated Nk Cells Kill Hepatic Stellate Cells Via P38/Pi3k Signaling in a Trail-Involved Degranulation Manner. J Leukoc Biol (2019) 105(4):695–704. doi: 10.1002/jlb.2a0118-031rr
125. Nilsson J, Hörnberg M, Schmidt-Christensen A, Linde K, Nilsson M, Carlus M, et al. Nkt Cells Promote Both Type 1 and Type 2 Inflammatory Responses in a Mouse Model of Liver Fibrosis. Sci Rep (2020) 10(1):21778. doi: 10.1038/s41598-020-78688-2
126. Nichols RD, von Moltke J, Vance RE. Naip/Nlrc4 Inflammasome Activation in Mrp8(+) Cells Is Sufficient to Cause Systemic Inflammatory Disease. Nat Commun (2017) 8(1):2209. doi: 10.1038/s41467-017-02266-w
127. Szabo G. Clinical Trial Design for Alcoholic Hepatitis. Semin Liver Dis (2017) 37(4):332–42. doi: 10.1055/s-0037-1608788
128. Larsen CM, Faulenbach M, Vaag A, Vølund A, Ehses JA, Seifert B, et al. Interleukin-1-Receptor Antagonist in Type 2 Diabetes Mellitus. N Engl J Med (2007) 356(15):1517–26. doi: 10.1056/NEJMoa065213
129. Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, et al. Il-1 Receptor Antagonist Ameliorates Inflammasome-Dependent Alcoholic Steatohepatitis in Mice. J Clin Invest (2012) 122(10):3476–89. doi: 10.1172/jci60777
130. Morrison MC, Mulder P, Salic K, Verheij J, Liang W, van Duyvenvoorde W, et al. Intervention With a Caspase-1 Inhibitor Reduces Obesity-Associated Hyperinsulinemia, Non-Alcoholic Steatohepatitis and Hepatic Fibrosis in Ldlr-/-. Leiden Mice Int J Obes (Lond) (2016) 40(9):1416–23. doi: 10.1038/ijo.2016.74
131. Witek RP, Stone WC, Karaca FG, Syn WK, Pereira TA, Agboola KM, et al. Pan-Caspase Inhibitor Vx-166 Reduces Fibrosis in an Animal Model of Nonalcoholic Steatohepatitis. Hepatology (2009) 50(5):1421–30. doi: 10.1002/hep.23167
132. Barreyro FJ, Holod S, Finocchietto PV, Camino AM, Aquino JB, Avagnina A, et al. The Pan-Caspase Inhibitor Emricasan (Idn-6556) Decreases Liver Injury and Fibrosis in a Murine Model of Non-Alcoholic Steatohepatitis. Liver Int (2015) 35(3):953–66. doi: 10.1111/liv.12570
133. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. Nlrp3 Inflammasome Blockade Reduces Liver Inflammation and Fibrosis in Experimental Nash in Mice. J Hepatol (2017) 66(5):1037–46. doi: 10.1016/j.jhep.2017.01.022
134. Baeza-Raja B, Goodyear A, Liu X, Lam K, Yamamoto L, Li Y, et al. Pharmacological Inhibition of P2rx7 Ameliorates Liver Injury by Reducing Inflammation and Fibrosis. PloS One (2020) 15(6):e0234038. doi: 10.1371/journal.pone.0234038
135. Gabay C, Lamacchia C, Palmer G. Il-1 Pathways in Inflammation and Human Diseases. Nat Rev Rheumatol (2010) 6(4):232–41. doi: 10.1038/nrrheum.2010.4
136. Yan W, Shen Y, Huang J, Lu L, Zhang Q. Mcc950 Ameliorates Acute Liver Injury Through Modulating Macrophage Polarization and Myeloid-Derived Suppressor Cells Function. Front Med (Lausanne) (2021) 8:752223. doi: 10.3389/fmed.2021.752223
137. von Moltke J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, van Rooijen N, et al. Rapid Induction of Inflammatory Lipid Mediators by the Inflammasome in Vivo. Nature (2012) 490(7418):107–11. doi: 10.1038/nature11351
138. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential Ly-6c Expression Identifies the Recruited Macrophage Phenotype, Which Orchestrates the Regression of Murine Liver Fibrosis. Proc Natl Acad Sci U S A (2012) 109(46):E3186–95. doi: 10.1073/pnas.1119964109
139. Weng H, Mertens PR, Gressner AM, Dooley S. Ifn-Gamma Abrogates Profibrogenic Tgf-Beta Signaling in Liver by Targeting Expression of Inhibitory and Receptor Smads. J Hepatol (2007) 46(2):295–303. doi: 10.1016/j.jhep.2006.09.014
140. Li X, Zhang M, Liu J, Huang Z, Zhao Q, Huang Y, et al. Intrahepatic Nk Cells Function Suppressed in Advanced Liver Fibrosis. Eur J Clin Invest (2016) 46(10):864–72. doi: 10.1111/eci.12669
141. Liang H, Xu F, Zhang T, Huang J, Guan Q, Wang H, et al. Inhibition of Il-18 Reduces Renal Fibrosis After Ischemia-Reperfusion. BioMed Pharmacother (2018) 106:879–89. doi: 10.1016/j.biopha.2018.07.031
142. Prior N, Inacio P, Huch M. Liver Organoids: From Basic Research to Therapeutic Applications. Gut (2019) 68(12):2228–37. doi: 10.1136/gutjnl-2019-319256
143. Sarmah D, Datta A, Kaur H, Kalia K, Borah A, Rodriguez AM, et al. Sirtuin-1 - Mediated Nf-κb Pathway Modulation to Mitigate Inflammasome Signaling and Cellular Apoptosis Is One of the Neuroprotective Effects of Intra-Arterial Mesenchymal Stem Cell Therapy Following Ischemic Stroke. Stem Cell Rev Rep (2022) 18(2):821–38. doi: 10.1007/s12015-021-10315-7
Keywords: inflammasome activation, pyroptosis, caspase-1, IL-1β, IL-18, liver cirrhosis
Citation: Gan C, Cai Q, Tang C and Gao J (2022) Inflammasomes and Pyroptosis of Liver Cells in Liver Fibrosis. Front. Immunol. 13:896473. doi: 10.3389/fimmu.2022.896473
Received: 15 March 2022; Accepted: 04 May 2022;
Published: 30 May 2022.
Edited by:
Alessandra Mortellaro, San Raffaele Telethon Institute for Gene Therapy (SR-Tiget), ItalyReviewed by:
Alessio Cantore, San Raffaele Scientific Institute (IRCCS), ItalyEdward N. Harris, University of Nebraska System, United States
Steven O’Reilly, STipe Therapeutics, Denmark
Copyright © 2022 Gan, Cai, Tang and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinhang Gao, R2FvLmppbmhhbmdAc2N1LmVkdS5jbg==; R2FvLmppbmhhbmdAcXEuY29t; Chengwei Tang, c2hjcWNkbWVkQDE2My5jb20=
†These authors contributed equally to this work