 Srijon K. Banerjee
Srijon K. Banerjee Ayan Chatterjee
Ayan Chatterjee Shamba Gupta3
Shamba Gupta3 Abhinit Nagar
Abhinit Nagar
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 12 May 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.896353
This article is part of the Research Topic Molecular and Cellular Basis for NLRP3 Sensing of Infectious and Sterile Stimuli View all 7 articles
Nod-Like Receptor (NLR) is the largest family of Pathogen Recognition Receptors (PRRs) that patrols the cytosolic environment. NLR engagement drives caspase-1 activation that cleaves pro-IL-1B which then gets secreted. Released IL-1B recruits immune cells to the site of infection/injury. Caspase-1 also cleaves Gasdermin-D (GSDM-D) that forms pores within the plasma membrane driving inflammatory cell death called pyroptosis. NLRP3 is the most extensively studied NLR. The NLRP3 gene is encoded by 9 exons, where exon 1 codes for pyrin domain, exon 3 codes for NACHT domain, and Leucine Rich Repeat (LRR) domain is coded by exon 4-9. Exon 2 codes for a highly disorganized loop that connects the rest of the protein to the pyrin domain and may be involved in NLRP3 regulation. The NLRP3 inflammasome is activated by many structurally divergent agonists of microbial, environmental, and host origin. Activated NLRP3 interacts with an adaptor protein, ASC, that bridges it to pro-Caspase-1 forming a multi-protein complex called inflammasome. Dysregulation of NLRP3 inflammasome activity is a hallmark of pathogenesis in several human diseases, indicating its highly significant clinical relevance. In this review, we summarize the existing knowledge about the mechanism of activation of NLRP3 and its regulation during activation by infectious and sterile triggers.
Nod-Like Receptor (NLR) is the largest family of Pathogen Recognition Receptors (PRRs) that patrols the cytosolic environment. NLR engagement drives caspase-1 activation that cleaves pro-IL-1β which then gets secreted (1). Released IL-1β recruits immune cells to the site of infection/injury (2–8). Caspase-1 also cleaves Gasdermin-D (GSDM-D) that forms pores within the plasma membrane driving inflammatory cell death called pyroptosis (9–12). NLRP3 is the most extensively studied NLR (10–17). The NLRP3 gene is encoded by 9 exons, where exon 1 codes for pyrin domain, exon 3 codes for NACHT domain, and Leucine Rich Repeat (LRR) domain is coded by exon 4-9. Exon 2 codes for a highly disorganized loop that connects the rest of the protein to the pyrin domain and may be involved in NLRP3 regulation (18). The NLRP3 inflammasome is activated by many structurally divergent agonists of microbial, environmental, and host origin (11, 19). Activated NLRP3 interacts with an adaptor protein, ASC, that bridges it to pro-Caspase-1 forming a multi-protein complex called inflammasome (2–8). Dysregulation of NLRP3 inflammasome activity is a hallmark of pathogenesis in several human diseases (20–24), indicating its highly significant clinical relevance. In this review, we summarize the existing knowledge about the mechanism of activation of NLRP3 and its regulation during activation by infectious and sterile triggers (Figure 1).
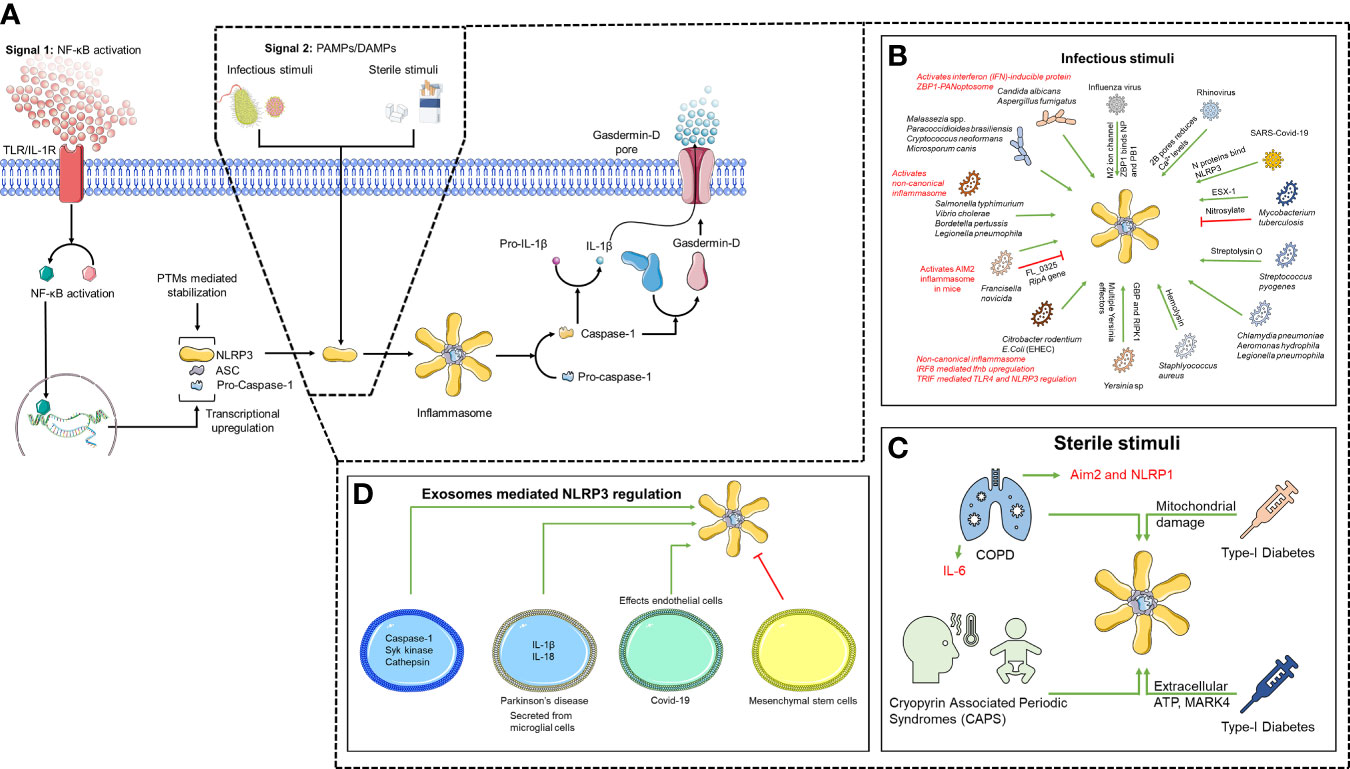
Figure 1 Two-step mechanism of NLRP3 activation. (A) TLR/IL1-R engagement leads to NF-κB activation driving transcriptional upregulation of inflammasome components (Signal 1). NLRP3 is then activated upon sensing unknown direct agonists (Signal 2), which can be (B) infectious, (C) sterile and can also be mediated through (D) exosomes.
NLRP3 inflammasome formation follows a two-signal process (25). The engagement of TLR or cytokine receptors resulting in activation of NF-κB is called the priming (signal 1). NF-κB activation induces the transcription of NLRP3, ASC, pro-caspase-1 and pro-IL-1β regulating their cellular levels allowing NLRP3 activation specifically during pathogen invasion and endogenous threats. Apart from transcriptional regulation, NLRP3 is also stabilized by priming-induced post-translation modifications (PTM) (25, 26).
The molecular mechanism behind NLRP3 activation (signal 2) is believed to be the result of various cellular events involving K+ efflux, ROS generation, and release of cathepsin B due to phagosome rupture (10, 11, 19). It was believed that agonists are sensed by LRRs of NLRP3 inducing conformational changes leading to recruitment of inflammasome components (11),although, recent studies suggest that LRRs are dispensable for NLRP3 activation (18, 27). Given the variety of agonists, it is believed that NLRP3 actually senses an upstream unifying factor, e.g. cellular stress, commonly induced by all NLRP3 agonists (19, 25, 28). ROS was once believed to be a common upstream factor until recently where it was shown that ROS affects priming and not activation (29, 30). K+ efflux is one of the most accepted models of NLRP3 inflammasome activation (19, 25). NLRP3 agonists, nigericin and ATP, activates NLRP3 through K+ efflux. Nigericin is a K+/H+ ionophore while ATP-dependent NLRP3 activation engages P2X purinoceptor 7 receptor (P2X7R), a ligand-gated ion channel capable of K+ efflux (31–33). It was recently discovered that P2X7R couples with Two-pore domain Weak Inwardly rectifying K+ channel 2 (TWIK2) for K+ efflux, whereas P2X7R is responsible for influx of Na+ and Ca2+ (34, 35). P2X7R pores are believed to facilitate PAMPs/DAMPs entry to the cell (19, 36). However, some agonists of NLRP3 like monosodium urate crystals (MSU) and particulate asbestos are too large to be translocated through these pores (19). Moreover, several studies have shown K+ efflux-independent activation of NLRP3, demanding reexamination of the K+ efflux model (29, 37, 38). The lysosomal rupture model considers the size of agonists. Phagosome destabilization and rupture is caused by inefficient clearance of particulate agonists. The emanating release of Cathepsin B is believed to activate NLRP3 either directly or indirectly (11, 15, 16, 25). While several studies show cathepsin B inhibitor blocks lysosomal disruption and impair NLRP3 inflammasome (39), IL-1β processing in Cathepsin B deficient mice is comparable to that of wild-type mice (40, 41). Thus, cathepsin B inhibitors likely impair NLRP3 inflammasome activation by off-target effects. Lysosomal rupture also causes K+ efflux suggesting involvement of multiple pathways in activating NLRP3 (42–45). As most of NLRP3 agonists induces reactive oxygen species (ROS) and various ROS scavengers also impairs inflammasome activation (46, 47) suggesting that NLRP3 senses cellular stress (10, 11, 13, 15, 19, 25). NLRP3 activation requires interaction with Thioredoxin (TX) Interacting Protein (TXNIP) (48, 49), but, these finding could not be confirmed by others (50). Further, ROS activation also drives K+ efflux further suggesting that NLRP3 can be activated by multiple independent pathways. Moreover, the mitotic kinase, NEK7, facilitates NLRP3 activation and inflammasome assembly following activation by ATP and nigericin (30, 51, 52). NEK7 binds to LRR, hinge domain 2 (HD2) and the NACHT domain (53). Oridonin, the major active ingredient of the traditional Chinese medicinal herb Rabdosia rubescens, blocks NLRP3-NEK7 interaction by covalently modifying cysteine (C) 279 (54) on the interaction surface of NEK7 Moreover, NEK7 interacts with minimally active NLRP3 truncated mutant (NLRP3 1-686). NEK7 also interacts with NLRP3 1-665 which cannot be activated by various agonists (27). Thus, NEK7 interaction is not the primary requirement for NLRP3 activation, suggesting the possible involvement of other cellular factors. Lastly, various NLRP3 agonists cause mitochondrial dysfunction and release of mitochondrial ROS and mitochondrial DNA that activates NLRP3 (55–57). However, the direct or indirect nature of this interaction is yet to be investigated. Thus, it is likely that either a common upstream factor activates, or multiple pathways lead to inflammasome activation. It is also likely that a combination of factors leads to activation of NLRP3. Since, inflammasome formation follows prion-like polymerization leading to Supramolecular Organization Centers (SMOCs) it is also possible that some threshold effect is required to activate NLRP3 which may be achieved by involving multiple pathways. Such a mechanism would also account for rapid NLRP3 activation by multiple agonists.
Since NLRP3 activation mechanism remains elusive, it is plausible that a common upstream factor activates/regulates NLRP3. Most of the proteins interacting with NLRP3 binds to the NACHT-LRR domain suggesting the importance of these domains in NLRP3 regulation. Heat shock protein (Hsp) 90 chaperone complex binds to NLRP3-LRR domain (58), however, it is unclear whether the chaperone complex facilitates proper folding or unfolding of LRRs after stimulation. Further, redox sensitive TXNIP interacts with NACHT-LRR domain of NLRP3 and knocking down TXNIP also reduces NLRP3 inflammasome response (49). Interestingly, TXNIP-/- BMDMs showed no effect on inflammasome response (50). Mitotic kinase, NEK7, binding is shown critical for NLRP3 inflammasome response (30, 52, 53). Additionally, E3 ubiquitin ligase tripartite motif-containing protein 31 (TRIM31) promotes proteasomal degradation of NLRP3 thus attenuating NLRP3 inflammasome response (59). NLRP3 inflammasome response is impaired by binding of Pyrin only proteins (POPs) and CARD only proteins (COPs). POPs and COPs are reviewed in detail in Le H.T. et al., 2013 (60). Interestingly, a few interacting partners binds to PYD domain suggesting a possible involvement of PYD in NLRP3 inflammasome response. Mitochondrial anti-viral signaling protein (MAVS) interaction with NLRP3 PYD is critical for mitochondrial localization and NLRP3 inflammasome response (61). Further, microtubule-affinity regulating kinase 4 (MARK4) also interacts with the PYD domain of NLRP3 which is critical for positioning and translocation of NLRP3 to form the inflammasome (62). Although several binding partners of NLRP3 are discovered, the mechanism behind their action is not yet understood. One of the caveats with these studies are the use of special agonists and the importance of these protein-protein interactions is specific to the respective agonists. It is important to evaluate the role of these proteins under different conditions.
Various PTM regulate innate immune signaling through different cellular processes [Reviewed in (63)]. NLRP3 expression is regulated by both transcriptional and post-translational modifications (PTM) (26). NLRP3 is phosphorylated by Spleen tyrosine kinase (Syk) (64–67), Death-associated protein kinase (DAPK) (68), Transforming growth factor beta-activated kinase 1 (TAK1) (69) and Extracellular Signal-Regulated Kinase 1 (ERK1) (70) in infection models. It is unclear whether these kinases specifically phosphorylate NLRP3 or other inflammasome components. Dephosphorylation of NLRP3 at serine 5 by Protein phosphatase 2 (PP2A) (71), and tyrosine 859 by protein tyrosine phosphatase non-receptor type (PTPN22) primes NLRP3 for activation (72, 73). Interestingly, phosphorylation at serine 295 by Protein kinase D (PKD) activates NLRP3, whereas, by Protein kinase A (PKA) abrogates NLRP3 inflammasome activation (22, 72–74). It is unclear how the same PTM at the same site leads to two different outcomes. It is likely that a combination of PTMs rather than a single PTM regulate NLRP3 inflammasome. PYD, NACHT and LRRs are ubiquitinylated by TRIM31 (59, 75), Ariadne homolog 2 (ARIH2) (76), and membrane-associated RING finger protein 7 (MARCH7) (77), respectively promoting proteasomal degradation of NLRP3. Finally, deubiquitylation of LRRs by BRCA1/BRCA2-containing complex subunit 3 (BRCC3) is required for NLRP3 oligomerization and activation (78). In the resting state, mitochondrial E3 ubiquitin protein ligase 1 (MUL1) SUMOylates NLRP3 at multiple sites. Following activation, sentrin-specific protease 6 (SENP6) and 7 (SENP7) deSUMOylates NLRP3 promoting inflammasome function (79). However, further studies are required to confirm the role of SUMOylation in NLRP3 activation. Nitrosylation is associated with NLRP3 inhibition (80–82). Mao et al. demonstrated that treatment with nitric oxide donor, SNAP, inhibits inflammasome function in mouse peritoneal macrophages, THP1 cells and human peripheral blood mononuclear cells (PBMCs) (81). During mycobacterial infection of mice, NLRP3 is nitrosylated by IFN-γ induced nitric oxide synthase (iNOS) (82). NO-mediated inhibition is specific for NLRP3 as the AIM2 and NLRC4 inflammasomes are only moderately affected (80). NO-mediated inhibition operates through thiol modifications of cysteine residues. Further, a recent study has shown cysteine dependent NLRP3 inflammasome response to sterile agonists whereas response to Fransicella novicida U112 was cysteine independent (18). Cysteine thiol groups are strongly nucleophilic, and the availability of d-orbital electrons help attain multiple oxidation states (83–86). Such chemistry provides versatility of forming a molecular code that can efficiently respond to oxidative stress (87). Depending on the cell redox state, cysteines can undergo various reversible and irreversible modifications (83–86). While reversible modification of cysteine functions as a signaling intermediate, irreversible intermediates are rarely involved in signaling. NLRP3 has 43 cysteines and is regulated by nitrosylation (82), however, which cysteines are modified is still unknown. PTMs are very strong regulator of protein functions. Whether a single PTM regulate NLRP3 inflammasome or multiple PTMs works in tandem to regulate NLRP3 is yet to be determined. Further studies evaluating the PTMs on NLRP3 following stimulation by different agonists are required to fully understand the role of PTMs on NLRP3 activation.
Several studies have revealed a crucial role of the NLRP3 inflammasome in bacterial infections (1). The Pathogen associated molecular patterns of Gram-negative bacterial pathogens like Yersinia spp, Francisella spp and Salmonella spp are recognized by more than one PRR (2). However, NLRP3 activation is a common response to all 3 pathogens. Francisella novicida U112 activates human NLRP3, whereas in mice, AIM2 is the predominant inflammasome that responds to Francisella (Fn) infection (3), and a recent study shows this response is debilitating to the host (4). However, NLRP3 deficient mice display improved survival in Fn infection indicating the presence of a contrasting non-inflammasome role of NLRP3 (5). NLRP3 share structural similarity to CIITA which makes it likely to function as a transcriptional regulator as well. A non-inflammasome function of NLRP3 is interesting and requires further investigation. However, it is still perplexing why a single protein belonging to such a big super-family have multiple functions. Salmonella enterica serovar Typhimurium activates the NLRC4 and NLRP3 inflammasomes in macrophages and both are required for efficient IL1β release (6). Given the critical role that NLRP3 activation plays in mounting an effective immune response to bacterial infection, it is not surprising that bacterial pathogens have developed tools to subvert inflammasome activation (7). Majority of the evidence of inflammasome activation in Yersinia infections have been discovered while identifying virulence factors that disabled the host immune response. Yersinia effectors YopK, YopB, YopD, YopM and YopJ have all been implicated to regulate the NLRP3 inflammasome through different mechanisms (8–11). In case of Fn, the FL_0325 virulence factor and gene ripA were discovered to suppress NLRP3 and AIM2 activation and mutant strains lacking these genes generated a stronger innate immune response than wild type Fn (12–14). It is interesting that how effectors are targeted towards NLRP3 rather than blocking the end-product of inflammasome activation, i.e., caspase-1. This suggests that NLRP3 might have functions beyond caspase-1 activation. Among other Gram-negative pathogens known to activate the NLRP3 inflammasome are Aeromonas hydrophila, Bordetella pertussis, Vibrio cholerae and Legionella pneumophila (15–21). V. cholerae and EndotoxinB. pertussis activate more than one type of inflammasome including NLRP3 via canonical and non-canonical pathways (22, 23). Several host factors are also involved in the activation of the NLRP3 inflammasome during bacterial infections. For example, IRF8 promotes Ifnb transcription which in turn activates caspase-11 to trigger the NLRP3 inflammasome in murine macrophages infected with Citrobacter rodentium (24). In another study, TRIF was identified as an important bridge between TLR4 and NLRP3 in enterohemorrhagic Escherichia coli (EHEC) and C. rodentium infected cells (25). Noncanonical activation of NLRP3 was found to be caspase-4 dependent in macrophages infected by L. pneumophila, Y. pseudotuberculosis and S. Typhimurium (26). Further, in Yersinia infected cells, RIPK-1 and Guanylate binding proteins are critical regulators of pyroptosis or apoptosis (27, 28).
Examples of NLRP3 inflammasome response to virulence factors of Gram-positive bacterial pathogens include hemolysins secreted by Staphlyococcus aureus (Sa) which activates NLRP3 and induces IL-1β secretion (88, 89). However, another group showed that staphylococcal hemolysins are dispensable for NLRP3 activation (88, 89). Sa-associated PAMPs also cooperate with hemolysin to activate NLRP3 (90). Similarly, streptolysin O released from Streptococcus pyogenes is important for NLRP3 activation (91, 92). NLRP3-dependent IL-1β is also crucial for protection against Chlamydia pneumoniae infection (93). It is interesting to see how NLRP3 activation by gram-positive bacteria broadly use K+ efflux, whereas for Gram-negative bacteria the mechanism is more varied. Mycobacterium tuberculosis (Mtb) inhibits the inflammasome response activation by nitrosylating NLRP3 (82). ESX-I secretion system of both Mtb and Mycobacterium marinum activates NLRP3 activation (94–96). Some bacteria, like Listeria monocytogenes induce multiple inflammasomes, including NLRP3, Aim-2 and NLRC4, and trigger inflammasome formation by mixed NLRs (97–102). These observations highlight the importance of NLRP3 in detection of various bacteria and the importance of the inflammasome response during bacterial infections.
Many fungal species have been shown to activate the NLRP3 inflammasome that include C. albicans, A. fumigatus, Malassezia spp., Paracoccidioides brasiliensis, Cryptococcus neoformans and Microsporum canis (103). NLRP3 deficient mice are susceptible to C. albicans infection (104). Further, hypheal forms of fungi are more potent inducers of NLRP3 inflammasome than yeast which activates NLRP3 with candidalysin (105, 106). C. albicans and A. fumigatus also activates the interferon (IFN)-inducible protein ZBP1-PANoptosome, resulting in NLRP3 inflammasome activation and PANoptosis (107). In A. fumigatus, the activation of NLRP3 inflammasome is dependent on ROS production and K+ efflux (108). Syk kinase activity downstream of Card9 activation by the fungal sensor Dectin1, is required for NLRP3 activation (64, 109), however, the precise mechanism of NLRP3 activation by fungal PAMPs is unclear, contributing to the bottleneck in development of new drug targets to treat fungal infections. NLRP3 activation through fungal activation require further studies to elaborate the mechanism of activation.
NLRP3 is the only member of the NLR family that plays a role detecting viral RNA and proteins in viral infections. Both DNA and RNA viruses have been shown to activate the NLRP3 inflammasome with Sendai and Influenza virus being the first viruses discovered to do so (110). The interferon-inducible protein, thus activating the NLRP3 inflammasome (111). The cytosolic dsRNA sensor DHX33 which is a member of DExD/H-box helicase family, can interact with NLRP3, activating it (112, 113). Vivoporins are virus-encoded proteins with ion channel activity that cause changes in membrane stability which can be recognized by NLRP3 and activate the inflammasome (114). NLRP3 activation by influenza virus is proposed to be driven by the viral M2 ion channel that transports H+ ions out of trans-Golgi network but also leads to activation of other ion channels that drive K+ efflux (115, 116). In Rhinovirus, the 2B protein activates NLRP3 by creating pores and reducing the level of Ca2+ in ER and Golgi membranes (117). Inhibition of NLRP3 activation during Respiratory syncytial virus (RSV) infection decreases lung pathology in vivo (118). Recent study shows the SARS-CoV-2 N protein induces proinflammatory cytokines by promoting the assembly of NLRP3 inflammasome via direct interaction with NLRP3. Using a mouse model of infection, the authors have demonstrated that activation of NLRP3 by N protein has been associated with lung injury and a cytokine storm, the two hallmarks of Covid-19 infection (119, 120).
Cryopyrin Associated Periodic Syndromes (CAPS) are a spectrum of chronic inflammatory diseases caused by gain of function mutations in the NACHT and LRR domain of NLRP3 (20, 121–123). These diseases include Familial cold autoinflammatory syndrome (FCAS), Muckle-Wells Syndrome (MWS) and Neonatal onset multi-systemic inflammatory disease (NOMID)/Chronic infantile neurological cutaneous articular syndrome (CINCA). Macrophages and monocytes isolated from CAPS patients spontaneously secrete IL-1β in the absence of any inflammatory stimuli (124). While recurrent fever and joint-pains are clinical symptoms of FCAS and MWS (125), NOMID patients present severe neurological and developmental complications (125, 126).
COPD presents as a combination of emphysema, chronic bronchitis, and chronic airway obstruction (127, 128). While inflammation and IL1β release is central to progression of COPD, there are mixed reports of whether and which inflammasome activation causes severe damage to the lungs (129–131). Exposure to cigarette smoke and other particulate matter is a major cause of COPD and its role in activating the NLRP3 inflammasome is well documented (132, 133). Further, evidence of NLRP3 activation in stable and exacerbated COPD was found in sputum and plasma samples of patients, in an in vitro model of COPD and in patients with neutrophilic asthma (134–136). In contrast, one study found no induction of NLRP3 but an increase in inflammatory cytokines like IL-6 in the broncho alveolar lavage (BAL) fluid of patients with stable COPD (137). Another genetic study of polymorphisms in COPD patients, identified a single nucleotide polymorphism in NLRP1 which correlated with decreased lung function (138). Additionally, other studies discovered the activation of the AIM2 inflammasome consistent with IL1β release in BAL, lung tissue and peripheral blood mononuclear cells isolated from COPD patients (139, 140). Taken together, there is ample evidence of IL-1 cytokines and inflammasome activation in stable and exacerbated COPD but whether NLRP3 activation drives disease progression and the mechanism behind it is yet to be elucidated.
Type I (T1D) and Type II (T2D) diabetes differ in the mechanism by which insulin resistance develops (141), but the role of inflammasomes have been implicated in both. In a study from Brazil, 2 SNPs in the NLRP3 gene were found in pediatric patients with T1D (142). In a murine model of T1D, mitochondrial DNA from diabetic mice displayed the ability to induce IL1β which could be inhibited in NLRP3 -/- macrophages (143). Further, NLRP3 deficient mice were unable to develop T1D (144). Although the exact mechanism of how the NLRP3 inflammasome contributes to T1D is yet to be discovered, the existing data indicates a crucial role for the former. As for T2D, studies have been more illuminating. IL1β release and the activation of NLRP3 are central to T2D (21, 145, 146). Monocyte-derived macrophages from T2D patients displayed high NLRP3, IL-18 expression, caspase-1 cleavage and NLRP3 dependent IL1β secretion (147). High extracellular ATP in T2D results in the activation of the P2X7 receptor that in turn activates the NLRP3 inflammasome (148). Additionally, activation of NLRP3 can be controlled by the microtubule affinity regulating kinase (MARK4) which in turn is regulated by the E-74 like ETS transcription factor (ELF3). High glucose increased ELF3 expression in HUVECs and triggered the NLRP3 inflammasome (149). Diabetic markers such as saturated fatty acids and islet amyloid polypeptide also activate NLRP3 (50, 150). Recent clinical trials have shown potential in IL-1β blocking therapies further establishing an important role of IL-1β in progression of diabetes (151). However, treatment with Canakinumab, an IL1β inhibitor, did not reduce the risk of diabetes in patients with pre-diabetes (152).
Extracellular vesicles (EVs) are membrane enclosed nano-bodies (size 30-1000nm) that facilitate cargo transport and signal transduction regulating physiological and pathological processes (153). Numerous studies have established that exosomes can critically influence the progression of inflammatory diseases by modulating the NLRP3 inflammasome (154–159).
Inflammasome-associated EV facilitate inflammatory responses in neighboring and distantly located recipient cells (159, 160). Particulate NLRP3 activators, like calcium oxalate, monosodium urate and β-glucan can activate EV-mediated cargo release in human macrophages (154). EV cargo contains Caspase-1, Syk kinase and Cathepsin indicative of NLRP3 inflammasome associated EV release upon lysosomal damage (161). NLRP3 inflammasome derived exosomes promotes inflammation by transducing signal to neighboring cells (162). NLRP3 activation causes exosome release, carrying IL-1β and IL-18, from microglial cell membrane promoting neuro-inflammation in Parkinson’s disease (162). Latest reports show that exosomes isolated from a severe COVID-19 patient’s plasma can exert its effect on human endothelial cells and liver endothelial cells increasing the expression of NLRP3, IL-1β and caspase-1 mRNA (160). Interestingly, EVs can also have inhibitory effect on NLRP3 activation. Mesenchymal stem cells derived exosomes from umbilical cords can attenuate caspase-1 production, resulting in lower levels of IL-1β and IL-18 thus inhibiting NLRP3 activation (163). Stem cell-derived exosomes can repair ischemic muscle injury by inhibiting the Rb1-mediated NLRP3 inflammasome pathway highlighting anti-inflammatory potential of exosomes (164).
In summary, NLRP3 activation by sterile and infectious agents display significant differences in mechanism. Moreover, the activation mechanism between gram-positive and gram-negative bacteria also differs greatly. Further, bacteria induced NLRP3 activation mechanism is different from NLRP3 activation by fungi and viruses. Such a difference in activation mechanism account for versatility of NLRP3 to react to different threats. Such differences may also be important in channel downstream signaling for launching adaptive immune response to the threat. In the two-decades of inflammasome research, scientists have discovered various mechanism of NLRP3 activation. While these discoveries have significantly advanced the inflammasome biology field, they have opened several new questions that will require further attention.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Author AN was employed by Luminex Corporation.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Evavold CL, Kagan JC. Inflammasomes: Threat-Assessment Organelles of the Innate Immune System. Immunity (2019) 51(4):609–24. doi: 10.1016/j.immuni.2019.08.005
2. Dinarello CA. The IL-1 Family and Inflammatory Diseases. Clin Exp Rheumatol (2002) 20(5 Suppl 27):S1–13.
3. Dinarello CA. Infection, Fever, and Exogenous and Endogenous Pyrogens: Some Concepts Have Changed. J Endotoxin Res (2004) 10(4):201–22. doi: 10.1179/096805104225006129
4. Dinarello CA. Immunological and Inflammatory Functions of the Interleukin-1 Family. Annu Rev Immunol (2009) 27:519–50. doi: 10.1146/annurev.immunol.021908.132612
5. Dinarello CA. IL-1: Discoveries, Controversies and Future Directions. Eur J Immunol (2010) 40(3):599–606. doi: 10.1002/eji.201040319
6. Dinarello CA. Overview of the IL-1 Family in Innate Inflammation and Acquired Immunity. Immunol Rev (2018) 281(1):8–27. doi: 10.1111/imr.12621
7. Sims JE, Smith DE. The IL-1 Family: Regulators of Immunity. Nat Rev Immunol (2010) 10(2):89–102. doi: 10.1038/nri2691
8. Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and Chemokines: At the Crossroads of Cell Signalling and Inflammatory Disease. Biochim Biophys Acta (2014) 1843(11):2563–82. doi: 10.1016/j.bbamcr.2014.05.014
9. Horvath GL, Schrum JE, De Nardo CM, Latz E. Intracellular Sensing of Microbes and Danger Signals by the Inflammasomes. Immunol Rev (2011) 243(1):119–35. doi: 10.1111/j.1600-065X.2011.01050.x
10. Lamkanfi M, Dixit VM. Mechanisms and Functions of Inflammasomes. Cell (2014) 157(5):1013–22. doi: 10.1016/j.cell.2014.04.007
11. Latz E, Xiao TS, Stutz A. Activation and Regulation of the Inflammasomes. Nat Rev Immunol (2013) 13(6):397–411. doi: 10.1038/nri3452
12. Sharma D, Kanneganti TD. The Cell Biology of Inflammasomes: Mechanisms of Inflammasome Activation and Regulation. J Cell Biol (2016) 213(6):617–29. doi: 10.1083/jcb.201602089
13. Schroder K, Tschopp J. The Inflammasomes. Cell (2010) 140(6):821–32. doi: 10.1016/j.cell.2010.01.040
14. Martinon F, Mayor A, Tschopp J. The Inflammasomes: Guardians of the Body. Annu Rev Immunol (2009) 27:229–65. doi: 10.1146/annurev.immunol.021908.132715
15. Malik A, Kanneganti TD. Inflammasome Activation and Assembly at a Glance. J Cell Sci (2017) 130(23):3955–63. doi: 10.1242/jcs.207365
16. He Y, Hara H, Nunez G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci (2016) 41(12):1012–21. doi: 10.1016/j.tibs.2016.09.002
17. Atianand MK, Rathinam VA, Fitzgerald KA. SnapShot: Inflammasomes. Cell. (2013) 153(1):272–e1. doi: 10.1016/j.cell.2013.03.009
18. Rahman T, Nagar A, Duffy EB, Okuda K, Silverman N, Harton JA. NLRP3 Sensing of Diverse Inflammatory Stimuli Requires Distinct Structural Features. Front Immunol (2020) 11:1828. doi: 10.3389/fimmu.2020.01828
19. Tschopp J, Schroder K. NLRP3 Inflammasome Activation: The Convergence of Multiple Signalling Pathways on ROS Production? Nat Rev Immunol (2010) 10(3):210–5. doi: 10.1038/nri2725
20. Davis BK, Wen H, Ting JP. The Inflammasome NLRs in Immunity, Inflammation, and Associated Diseases. Annu Rev Immunol (2011) 29:707–35. doi: 10.1146/annurev-immunol-031210-101405
21. Menu P, Vince JE. The NLRP3 Inflammasome in Health and Disease: The Good, the Bad and the Ugly. Clin Exp Immunol (2011) 166(1):1–15. doi: 10.1111/j.1365-2249.2011.04440.x
22. Mortimer L, Moreau F, MacDonald JA, Chadee K. NLRP3 Inflammasome Inhibition is Disrupted in a Group of Auto-Inflammatory Disease CAPS Mutations. Nat Immunol (2016) 17(10):1176–86. doi: 10.1038/ni.3538
23. Ting JP, Kastner DL, Hoffman HM. CATERPILLERs, Pyrin and Hereditary Immunological Disorders. Nat Rev Immunol (2006) 6(3):183–95. doi: 10.1038/nri1788
24. Yang CA, Chiang BL. Inflammasomes and Human Autoimmunity: A Comprehensive Review. J Autoimmun (2015) 61:1–8. doi: 10.1016/j.jaut.2015.05.001
25. Swanson KV, Deng M, Ting JP. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
26. Yang J, Liu Z, Xiao TS. Post-Translational Regulation of Inflammasomes. Cell Mol Immunol (2017) 14(1):65–79. doi: 10.1038/cmi.2016.29
27. Hafner-Bratkovic I, Bencina M, Fitzgerald KA, Golenbock D, Jerala R. NLRP3 Inflammasome Activation in Macrophage Cell Lines by Prion Protein Fibrils as the Source of IL-1beta and Neuronal Toxicity. Cell Mol Life Sci (2012) 69(24):4215–28. doi: 10.1007/s00018-012-1140-0
28. Van Hauwermeiren F, Lamkanfi M. The NEK-Sus of the NLRP3 Inflammasome. Nat Immunol (2016) 17(3):223–4. doi: 10.1038/ni.3391
29. Gross CJ, Mishra R, Schneider KS, Medard G, Wettmarshausen J, Dittlein DC, et al. K(+) Efflux-Independent NLRP3 Inflammasome Activation by Small Molecules Targeting Mitochondria. Immunity (2016) 45(4):761–73. doi: 10.1016/j.immuni.2016.08.010
30. He Y, Zeng MY, Yang D, Motro B, Nunez G. NEK7 is an Essential Mediator of NLRP3 Activation Downstream of Potassium Efflux. Nature (2016) 530(7590):354–7. doi: 10.1038/nature16959
31. Perregaux D, Gabel CA. Interleukin-1 Beta Maturation and Release in Response to ATP and Nigericin. Evidence That Potassium Depletion Mediated by These Agents is a Necessary and Common Feature of Their Activity. J Biol Chem (1994) 269(21):15195–203. doi: 10.1016/S0021-9258(17)36591-2
32. Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. The Cytolytic P2Z Receptor for Extracellular ATP Identified as a P2X Receptor (P2x7). Science. (1996) 272(5262):735–8. doi: 10.1126/science.272.5262.735
33. Walev I, Reske K, Palmer M, Valeva A, Bhakdi S. Potassium-Inhibited Processing of IL-1 Beta in Human Monocytes. EMBO J (1995) 14(8):1607–14. doi: 10.1002/j.1460-2075.1995.tb07149.x
34. Di A, Xiong S, Ye Z, Malireddi RKS, Kometani S, Zhong M, et al. The TWIK2 Potassium Efflux Channel in Macrophages Mediates NLRP3 Inflammasome-Induced Inflammation. Immunity (2018) 49(1):56–65.e4. doi: 10.1016/j.immuni.2018.04.032
35. Samways DS, Li Z, Egan TM. Principles and Properties of Ion Flow in P2X Receptors. Front Cell Neurosci (2014) 8:6. doi: 10.3389/fncel.2014.00006
36. Kanneganti TD, Lamkanfi M, Nunez G. Intracellular NOD-Like Receptors in Host Defense and Disease. Immunity (2007) 27(4):549–59. doi: 10.1016/j.immuni.2007.10.002
37. Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity. (2016) 44(4):833–46. doi: 10.1016/j.immuni.2016.01.012
38. Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, Wheeler ML, et al. Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell. (2016) 166(3):624–36. doi: 10.1016/j.cell.2016.05.076
39. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica Crystals and Aluminum Salts Activate the NALP3 Inflammasome Through Phagosomal Destabilization. Nat Immunol (2008) 9(8):847–56. doi: 10.1038/ni.1631
40. Dostert C, Guarda G, Romero JF, Menu P, Gross O, Tardivel A, et al. Malarial Hemozoin is a Nalp3 Inflammasome Activating Danger Signal. PloS One (2009) 4(8):e6510. doi: 10.1371/journal.pone.0006510
41. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 Inflammasome is Involved in the Innate Immune Response to Amyloid-Beta. Nat Immunol (2008) 9(8):857–65. doi: 10.1038/ni.1636
42. Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, et al. Critical Role for Calcium Mobilization in Activation of the NLRP3 Inflammasome. Proc Natl Acad Sci USA (2012) 109(28):11282–7. doi: 10.1073/pnas.1117765109
43. Katsnelson MA, Lozada-Soto KM, Russo HM, Miller BA, Dubyak GR. NLRP3 Inflammasome Signaling is Activated by Low-Level Lysosome Disruption But Inhibited by Extensive Lysosome Disruption: Roles for K+ Efflux and Ca2+ Influx. Am J Physiol Cell Physiol (2016) 311(1):C83–C100. doi: 10.1152/ajpcell.00298.2015
44. Katsnelson MA, Rucker LG, Russo HM, Dubyak GR. K+ Efflux Agonists Induce NLRP3 Inflammasome Activation Independently of Ca2+ Signaling. J Immunol (2015) 194(8):3937–52. doi: 10.4049/jimmunol.1402658
45. Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) Efflux is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity (2013) 38(6):1142–53. doi: 10.1016/j.immuni.2013.05.016
46. Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 Inflammasome is Triggered by Low Intracellular Potassium Concentration. Cell Death Differ (2007) 14(9):1583–9. doi: 10.1038/sj.cdd.4402195
47. Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. ATP Activates a Reactive Oxygen Species-Dependent Oxidative Stress Response and Secretion of Proinflammatory Cytokines in Macrophages. J Biol Chem (2007) 282(5):2871–9. doi: 10.1074/jbc.M608083200
48. Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate Immune Activation Through Nalp3 Inflammasome Sensing of Asbestos and Silica. Science (2008) 320(5876):674–7. doi: 10.1126/science.1156995
49. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-Interacting Protein Links Oxidative Stress to Inflammasome Activation. Nat Immunol (2010) 11(2):136–40. doi: 10.1038/ni.1831
50. Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, et al. Activation of the NLRP3 Inflammasome by Islet Amyloid Polypeptide Provides a Mechanism for Enhanced IL-1beta in Type 2 Diabetes. Nat Immunol (2010) 11(10):897–904. doi: 10.1038/ni.1935
51. Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, et al. A Genome-wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) Screen Identifies NEK7 as an Essential Component of NLRP3 Inflammasome Activation. J Biol Chem (2016) 291(1):103–9. doi: 10.1074/jbc.C115.700492
52. Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, et al. NLRP3 Activation and Mitosis are Mutually Exclusive Events Coordinated by NEK7, A New Inflammasome Component. Nat Immunol (2016) 17(3):250–8. doi: 10.1038/ni.3333
53. Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, et al. Structural Mechanism for NEK7-Licensed Activation of NLRP3 Inflammasome. Nature (2019) 570(7761):338–43. doi: 10.1038/s41586-019-1295-z
54. He H, Jiang H, Chen Y, Ye J, Wang A, Wang C, et al. Oridonin is a Covalent NLRP3 Inhibitor With Strong Anti-Inflammasome Activity. Nat Commun (2018) 9(1):2550. doi: 10.1038/s41467-018-04947-6
55. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy Proteins Regulate Innate Immune Responses by Inhibiting the Release of Mitochondrial DNA Mediated by the NALP3 Inflammasome. Nat Immunol (2011) 12(3):222–30. doi: 10.1038/ni.1980
56. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome During Apoptosis. Immunity (2012) 36(3):401–14. doi: 10.1016/j.immuni.2012.01.009
57. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New Mitochondrial DNA Synthesis Enables NLRP3 Inflammasome Activation. Nature. (2018) 560(7717):198–203. doi: 10.1038/s41586-018-0372-z
58. Mayor A, Martinon F, De Smedt T, Petrilli V, Tschopp J. A Crucial Function of SGT1 and HSP90 in Inflammasome Activity Links Mammalian and Plant Innate Immune Responses. Nat Immunol (2007) 8(5):497–503. doi: 10.1038/ni1459
59. Song H, Liu B, Huai W, Yu Z, Wang W, Zhao J, et al. The E3 Ubiquitin Ligase TRIM31 Attenuates NLRP3 Inflammasome Activation by Promoting Proteasomal Degradation of NLRP3. Nat Commun (2016) 7:13727. doi: 10.1038/ncomms13727
60. Le HT, Harton JA. Pyrin- and CARD-Only Proteins as Regulators of NLR Functions. Front Immunol (2013) 4:275. doi: 10.3389/fimmu.2013.00275
61. Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The Adaptor MAVS Promotes NLRP3 Mitochondrial Localization and Inflammasome Activation. Cell. (2013) 153(2):348–61. doi: 10.1016/j.cell.2013.02.054
62. Li X, Thome S, Ma X, Amrute-Nayak M, Finigan A, Kitt L, et al. MARK4 Regulates NLRP3 Positioning and Inflammasome Activation Through a Microtubule-Dependent Mechanism. Nat Commun (2017) 8:15986. doi: 10.1038/ncomms15986
63. Liu J, Qian C, Cao X. Post-Translational Modification Control of Innate Immunity. Immunity. (2016) 45(1):15–30. doi: 10.1016/j.immuni.2016.06.020
64. Gross O, Poeck H, Bscheider M, Dostert C, Hannesschläger N, Endres S, et al. Syk Kinase Signalling Couples to the Nlrp3 Inflammasome for Anti-Fungal Host Defence. Nature (2009) 459(7245):433–6. doi: 10.1038/nature07965
65. Said-Sadier N, Padilla E, Langsley G, Ojcius DM. Aspergillus Fumigatus Stimulates the NLRP3 Inflammasome Through a Pathway Requiring ROS Production and the Syk Tyrosine Kinase. PloS One (2010) 5(4):e10008. doi: 10.1371/journal.pone.0010008
66. Shio MT, Eisenbarth SC, Savaria M, Vinet AF, Bellemare MJ, Harder KW, et al. Malarial Hemozoin Activates the NLRP3 Inflammasome Through Lyn and Syk Kinases. PloS Pathog (2009) 5(8):e1000559. doi: 10.1371/journal.ppat.1000559
67. Wong KW, Jacobs WR Jr. Critical Role for NLRP3 in Necrotic Death Triggered by Mycobacterium Tuberculosis. Cell Microbiol (2011) 13(9):1371–84. doi: 10.1111/j.1462-5822.2011.01625.x
68. Chuang YT, Lin YC, Lin KH, Chou TF, Kuo WC, Yang KT, et al. Tumor Suppressor Death-Associated Protein Kinase is Required for Full IL-1beta Production. Blood. (2011) 117(3):960–70. doi: 10.1182/blood-2010-08-303115
69. Gong YN, Wang X, Wang J, Yang Z, Li S, Yang J, et al. Chemical Probing Reveals Insights Into the Signaling Mechanism of Inflammasome Activation. Cell Res (2010) 20(12):1289–305. doi: 10.1038/cr.2010.135
70. Ghonime MG, Shamaa OR, Das S, Eldomany RA, Fernandes-Alnemri T, Alnemri ES, et al. Inflammasome Priming by Lipopolysaccharide is Dependent Upon ERK Signaling and Proteasome Function. J Immunol (2014) 192(8):3881–8. doi: 10.4049/jimmunol.1301974
71. Stutz A, Kolbe CC, Stahl R, Horvath GL, Franklin BS, van Ray O, et al. NLRP3 Inflammasome Assembly is Regulated by Phosphorylation of the Pyrin Domain. J Exp Med (2017) 214(6):1725–36. doi: 10.1084/jem.20160933
72. Spalinger MR, Kasper S, Gottier C, Lang S, Atrott K, Vavricka SR, et al. NLRP3 Tyrosine Phosphorylation is Controlled by Protein Tyrosine Phosphatase Ptpn22. J Clin Invest (2016) 126(11):4388. doi: 10.1172/JCI90897
73. Spalinger MR, Lang S, Gottier C, Dai X, Rawlings DJ, Chan AC, et al. PTPN22 Regulates NLRP3-Mediated IL1B Secretion in an Autophagy-Dependent Manner. Autophagy (2017) 13(9):1590–601. doi: 10.1080/15548627.2017.1341453
74. Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang L, et al. Bile Acids Control Inflammation and Metabolic Disorder Through Inhibition of NLRP3 Inflammasome. Immunity. (2016) 45(4):944. doi: 10.1016/j.immuni.2016.10.009
75. Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non-Transcriptional Priming and Deubiquitination Regulate NLRP3 Inflammasome Activation. J Biol Chem (2012) 287(43):36617–22. doi: 10.1074/jbc.M112.407130
76. Kawashima A, Karasawa T, Tago K, Kimura H, Kamata R, Usui-Kawanishi F, et al. ARIH2 Ubiquitinates NLRP3 and Negatively Regulates NLRP3 Inflammasome Activation in Macrophages. J Immunol (2017) 199(10):3614–22. doi: 10.4049/jimmunol.1700184
77. Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, et al. Dopamine Controls Systemic Inflammation Through Inhibition of NLRP3 Inflammasome. Cell (2015) 160(1-2):62–73. doi: 10.1016/j.cell.2014.11.047
78. Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 Critically Regulates Inflammasome Activity. Mol Cell (2013) 49(2):331–8. doi: 10.1016/j.molcel.2012.11.009
79. Barry R, John SW, Liccardi G, Tenev T, Jaco I, Chen CH, et al. SUMO-Mediated Regulation of NLRP3 Modulates Inflammasome Activity. Nat Commun (2018) 9(1):3001. doi: 10.1038/s41467-018-05321-2
80. Hernandez-Cuellar E, Tsuchiya K, Hara H, Fang R, Sakai S, Kawamura I, et al. Cutting Edge: Nitric Oxide Inhibits the NLRP3 Inflammasome. J Immunol (2012) 189(11):5113–7. doi: 10.4049/jimmunol.1202479
81. Mao K, Chen S, Chen M, Ma Y, Wang Y, Huang B, et al. Nitric Oxide Suppresses NLRP3 Inflammasome Activation and Protects Against LPS-Induced Septic Shock. Cell Res (2013) 23(2):201–12. doi: 10.1038/cr.2013.6
82. Mishra BB, Rathinam VA, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, et al. Nitric Oxide Controls the Immunopathology of Tuberculosis by Inhibiting NLRP3 Inflammasome-Dependent Processing of IL-1beta. Nat Immunol (2013) 14(1):52–60. doi: 10.1038/ni.2474
83. Chalker JM, Bernardes GJ, Lin YA, Davis BG. Chemical Modification of Proteins at Cysteine: Opportunities in Chemistry and Biology. Chem Asian J (2009) 4(5):630–40. doi: 10.1002/asia.200800427
84. Chung HS, Wang SB, Venkatraman V, Murray CI, Van Eyk JE. Cysteine Oxidative Posttranslational Modifications: Emerging Regulation in the Cardiovascular System. Circ Res (2013) 112(2):382–92. doi: 10.1161/CIRCRESAHA.112.268680
85. Gaston B. Nitric Oxide and Thiol Groups. Biochim Biophys Acta (1999) 1411(2-3):323–33. doi: 10.1016/S0005-2728(99)00023-7
86. Yang HY, Kwon J, Choi HI, Park SH, Yang U, Park HR, et al. In-Depth Analysis of Cysteine Oxidation by the RBC Proteome: Advantage of Peroxiredoxin II Knockout Mice. Proteomics. (2012) 12(1):101–12. doi: 10.1002/pmic.201100275
87. Sitia R, Molteni SN. Stress, Protein (Mis)Folding, and Signaling: The Redox Connection. Sci STKE (2004) 2004(239):pe27. doi: 10.1126/stke.2392004pe27
88. Craven RR, Gao X, Allen IC, Gris D, Bubeck Wardenburg J, McElvania-Tekippe E, et al. Staphylococcus Aureus Alpha-Hemolysin Activates the NLRP3-Inflammasome in Human and Mouse Monocytic Cells. PloS One (2009) 4(10):e7446. doi: 10.1371/journal.pone.0007446
89. Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, et al. Cryopyrin Activates the Inflammasome in Response to Toxins and ATP. Nature (2006) 440(7081):228–32. doi: 10.1038/nature04515
90. Anand PK, Malireddi RK, Kanneganti TD. Role of the Nlrp3 Inflammasome in Microbial Infection. Front Microbiol (2011) 2:12. doi: 10.3389/fmicb.2011.00012
91. Toma C, Higa N, Koizumi Y, Nakasone N, Ogura Y, McCoy AJ, et al. Pathogenic Vibrio Activate NLRP3 Inflammasome via Cytotoxins and TLR/Nucleotide-Binding Oligomerization Domain-Mediated NF-Kappa B Signaling. J Immunol (2010) 184(9):5287–97. doi: 10.4049/jimmunol.0903536
92. Harder J, Franchi L, Munoz-Planillo R, Park JH, Reimer T, Nunez G. Activation of the Nlrp3 Inflammasome by Streptococcus Pyogenes Requires Streptolysin O and NF-Kappa B Activation But Proceeds Independently of TLR Signaling and P2X7 Receptor. J Immunol (2009) 183(9):5823–9. doi: 10.4049/jimmunol.0900444
93. He X, Mekasha S, Mavrogiorgos N, Fitzgerald KA, Lien E, Ingalls RR. Inflammation and Fibrosis During Chlamydia Pneumoniae Infection is Regulated by IL-1 and the NLRP3/ASC Inflammasome. J Immunol (2010) 184(10):5743–54. doi: 10.4049/jimmunol.0903937
94. Carlsson F, Kim J, Dumitru C, Barck KH, Carano RA, Sun M, et al. Host-Detrimental Role of Esx-1-Mediated Inflammasome Activation in Mycobacterial Infection. PloS Pathog (2010) 6(5):e1000895. doi: 10.1371/journal.ppat.1000895
95. McElvania Tekippe E, Allen IC, Hulseberg PD, Sullivan JT, McCann JR, Sandor M, et al. Granuloma Formation and Host Defense in Chronic Mycobacterium Tuberculosis Infection Requires PYCARD/ASC But Not NLRP3 or Caspase-1. PloS One (2010) 5(8):e12320. doi: 10.1371/journal.pone.0012320
96. Mishra BB, Moura-Alves P, Sonawane A, Hacohen N, Griffiths G, Moita LF, et al. Mycobacterium Tuberculosis Protein ESAT-6 is a Potent Activator of the NLRP3/ASC Inflammasome. Cell Microbiol (2010) 12(8):1046–63. doi: 10.1111/j.1462-5822.2010.01450.x
97. Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM. Redundant Roles for Inflammasome Receptors NLRP3 and NLRC4 in Host Defense Against Salmonella. J Exp Med (2010) 207(8):1745–55. doi: 10.1084/jem.20100257
98. Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, et al. Cytosolic Flagellin Requires Ipaf for Activation of Caspase-1 and Interleukin 1beta in Salmonella-Infected Macrophages. Nat Immunol (2006) 7(6):576–82. doi: 10.1038/ni1346
99. Man SM, Hopkins LJ, Nugent E, Cox S, Gluck IM, Tourlomousis P, et al. Inflammasome Activation Causes Dual Recruitment of NLRC4 and NLRP3 to the Same Macromolecular Complex. Proc Natl Acad Sci USA (2014) 111(20):7403–8. doi: 10.1073/pnas.1402911111
100. Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, et al. Cytoplasmic Flagellin Activates Caspase-1 and Secretion of Interleukin 1beta via Ipaf. Nat Immunol (2006) 7(6):569–75. doi: 10.1038/ni1344
101. Warren SE, Mao DP, Rodriguez AE, Miao EA, Aderem A. Multiple Nod-Like Receptors Activate Caspase 1 During Listeria Monocytogenes Infection. J Immunol (2008) 180(11):7558–64. doi: 10.4049/jimmunol.180.11.7558
102. Wu J, Fernandes-Alnemri T, Alnemri ES. Involvement of the AIM2, NLRC4, and NLRP3 Inflammasomes in Caspase-1 Activation by Listeria Monocytogenes. J Clin Immunol (2010) 30(5):693–702. doi: 10.1007/s10875-010-9425-2
103. Briard B, Malireddi RKS, Kanneganti T-D. Role of Inflammasomes/Pyroptosis and PANoptosis During Fungal Infection. PloS pathogens (2021) 17(3):e1009358. doi: 10.1371/journal.ppat.1009358
104. Hise AG, Tomalka J, Ganesan S, Patel K, Hall BA, Brown GD, et al. An Essential Role for the NLRP3 Inflammasome in Host Defense Against the Human Fungal Pathogen Candida Albicans. Cell Host Microbe (2009) 5(5):487–97. doi: 10.1016/j.chom.2009.05.002
105. Rogiers O, Frising UC, Kucharíková S, Jabra-Rizk MA, van Loo G, Van Dijck P, et al. Candidalysin Crucially Contributes to Nlrp3 Inflammasome Activation by Candida Albicans Hyphae. mBio (2019) 10(1):e02221–18. doi: 10.1128/mBio.02221-18
106. Joly S, Ma N, Sadler JJ, Soll DR, Cassel SL, Sutterwala FS. Cutting Edge: Candida Albicans Hyphae Formation Triggers Activation of the Nlrp3 Inflammasome. J Immunol (Baltimore Md 1950) (2009) 183(6):3578–81. doi: 10.4049/jimmunol.0901323
107. Banoth B, Tuladhar S, Karki R, Sharma BR, Briard B, Kesavardhana S, et al. ZBP1 Promotes Fungi-Induced Inflammasome Activation and Pyroptosis, Apoptosis, and Necroptosis (PANoptosis). J Biol Chem (2020) 295(52):18276–83. doi: 10.1074/jbc.RA120.015924
108. Karki R, Man SM, Malireddi RKS, Gurung P, Vogel P, Lamkanfi M, et al. Concerted Activation of the AIM2 and NLRP3 Inflammasomes Orchestrates Host Protection Against Aspergillus Infection. Cell Host Microbe (2015) 17(3):357–68. doi: 10.1016/j.chom.2015.01.006
109. Kistowska M, Fenini G, Jankovic D, Feldmeyer L, Kerl K, Bosshard P, et al. Malassezia Yeasts Activate the NLRP3 Inflammasome in Antigen-Presenting Cells via Syk-Kinase Signalling. Exp Dermatol (2014) 23(12):884–9. doi: 10.1111/exd.12552
110. Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, et al. The NLRP3 Inflammasome Mediates In Vivo Innate Immunity to Influenza A Virus Through Recognition of Viral RNA. Immunity (2009) 30(4):556–65. doi: 10.1016/j.immuni.2009.02.005
111. Kuriakose T, Man SM, Malireddi RKS, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an Innate Sensor of Influenza Virus Triggering the NLRP3 Inflammasome and Programmed Cell Death Pathways. Sci Immunol (2016) 1(2):aag2045. doi: 10.1126/sciimmunol.aag2045
112. Mitoma H, Hanabuchi S, Kim T, Bao M, Zhang Z, Sugimoto N, et al. The DHX33 RNA Helicase Senses Cytosolic RNA and Activates the NLRP3 Inflammasome. Immunity (2013) 39(1):123–35. doi: 10.1016/j.immuni.2013.07.001
113. Chakrabarti A, Banerjee S, Franchi L, Loo Y-M, Gale M Jr., Núñez G, et al. RNase L Activates the NLRP3 Inflammasome During Viral Infections. Cell Host Microbe (2015) 17(4):466–77. doi: 10.1016/j.chom.2015.02.010
114. Farag NS, Breitinger U, Breitinger HG, El Azizi MA. Viroporins and Inflammasomes: A Key to Understand Virus-Induced Inflammation. Int J Biochem Cell Biol (2020) 122:105738–. doi: 10.1016/j.biocel.2020.105738
115. Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome Recognition of Influenza Virus is Essential for Adaptive Immune Responses. J Exp Med (2009) 206(1):79–87. doi: 10.1084/jem.20081667
116. Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, et al. Critical Role for Cryopyrin/Nalp3 in Activation of Caspase-1 in Response to Viral Infection and Double-Stranded RNA. J Biol Chem (2006) 281(48):36560–8. doi: 10.1074/jbc.M607594200
117. Triantafilou K, Kar S, van Kuppeveld FJ, Triantafilou M. Rhinovirus-Induced Calcium Flux Triggers NLRP3 and NLRC5 Activation in Bronchial Cells. Am J Respir Cell Mol Biol (2013) 49(6):923–34. doi: 10.1165/rcmb.2013-0032OC
118. Malinczak C-A, Schuler CF, Duran AJ, Rasky AJ, Mire MM, Núñez G, et al. NLRP3-Inflammasome Inhibition During Respiratory Virus Infection Abrogates Lung Immunopathology and Long-Term Airway Disease Development. Viruses (2021) 13(4):692. doi: 10.3390/v13040692
119. Pan P, Shen M, Yu Z, Ge W, Chen K, Tian M, et al. SARS-CoV-2 N Protein Promotes NLRP3 Inflammasome Activation to Induce Hyperinflammation. Nat Commun (2021) 12(1):4664. doi: 10.1038/s41467-021-25015-6
120. Yalcinkaya M, Liu W, Islam MN, Kotini AG, Gusarova GA, Fidler TP, et al. Modulation of the NLRP3 Inflammasome by Sars-CoV-2 Envelope Protein. Sci Rep (2021) 11(1):24432. doi: 10.1038/s41598-021-04133-7
121. Church LD, Cook GP, McDermott MF. Primer: Inflammasomes and Interleukin 1beta in Inflammatory Disorders. Nat Clin Pract Rheumatol (2008) 4(1):34–42. doi: 10.1038/ncprheum0681
122. Dodé C, Le Dû N, Cuisset L, Letourneur F, Berthelot J-M, Vaudour G, et al. New Mutations of CIAS1 That Are Responsible for Muckle-Wells Syndrome and Familial Cold Urticaria: A Novel Mutation Underlies Both Syndromes. Am J Hum Genet (2002) 70(6):1498–506. doi: 10.1086/340786
123. Miyamae T. Cryopyrin-Associated Periodic Syndromes: Diagnosis and Management. Paediatr Drugs (2012) 14(2):109–17. doi: 10.2165/11595040-000000000-00000
124. Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 Forms an IL-1beta-Processing Inflammasome With Increased Activity in Muckle-Wells Autoinflammatory Disorder. Immunity (2004) 20(3):319–25. doi: 10.1016/S1074-7613(04)00046-9
125. Hoffman HM. Therapy of Autoinflammatory Syndromes. J Allergy Clin Immunol (2009) 124(6):1129–38. doi: 10.1016/j.jaci.2009.11.001
126. Neven B, Prieur AM, Quartier dit Maire P. Cryopyrinopathies: Update on Pathogenesis and Treatment. Nat Clin Pract Rheumatol (2008) 4(9):481–9. doi: 10.1038/ncprheum0874
127. Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA, Calverley P, et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease: GOLD Executive Summary. Am J Respir Crit Care Med (2007) 176(6):532–55. doi: 10.1164/rccm.200703-456SO
128. Hosseinian N, Cho Y, Lockey RF, Kolliputi N. The Role of the NLRP3 Inflammasome in Pulmonary Diseases. Ther Adv Respir Disease (2015) 9(4):188–97. doi: 10.1177/1753465815586335
129. Bafadhel M, McKenna S, Terry S, Mistry V, Reid C, Haldar P, et al. Acute Exacerbations of Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med (2011) 184(6):662–71. doi: 10.1164/rccm.201104-0597OC
130. Beckett EL, Stevens RL, Jarnicki AG, Kim RY, Hanish I, Hansbro NG, et al. A New Short-Term Mouse Model of Chronic Obstructive Pulmonary Disease Identifies a Role for Mast Cell Tryptase in Pathogenesis. J Allergy Clin Immunol (2013) 131(3):752–62.e7. doi: 10.1016/j.jaci.2012.11.053
131. Gessner C, Scheibe R, Wötzel M, Hammerschmidt S, Kuhn H, Engelmann L, et al. Exhaled Breath Condensate Cytokine Patterns in Chronic Obstructive Pulmonary Disease. Respir Med (2005) 99(10):1229–40. doi: 10.1016/j.rmed.2005.02.041
132. Churg A, Zhou S, Wang X, Wang R, Wright JL. The Role of Interleukin-1β in Murine Cigarette Smoke–Induced Emphysema and Small Airway Remodeling. Am J Respir Cell Mol Biol (2009) 40(4):482–90. doi: 10.1165/rcmb.2008-0038OC
133. Hirota JA, Hirota SA, Warner SM, Stefanowicz D, Shaheen F, Beck PL, et al. The Airway Epithelium Nucleotide-Binding Domain and Leucine-Rich Repeat Protein 3 Inflammasome is Activated by Urban Particulate Matter. J Allergy Clin Immunol (2012) 129(4):1116–25.e6. doi: 10.1016/j.jaci.2011.11.033
134. Faner R, Sobradillo P, Noguera A, Gomez C, Cruz T, López-Giraldo A, et al. The Inflammasome Pathway in Stable COPD and Acute Exacerbations. ERJ Open Res (2016) 2:00002-2016. doi: 10.1183/23120541.00002-2016
135. Nachmias N, Langier S, Brzezinski RY, Siterman M, Stark M, Etkin S, et al. NLRP3 Inflammasome Activity Is Upregulated in an In-Vitro Model of COPD Exacerbation. PloS One (2019) 14(5):e0214622. doi: 10.1371/journal.pone.0214622
136. Simpson JL, Phipps S, Baines KJ, Oreo KM, Gunawardhana L, Gibson PG. Elevated Expression of the NLRP3 Inflammasome in Neutrophilic Asthma. Eur Respir J (2014) 43:1067–76. doi: 10.1183/09031936.00105013
137. Di Stefano A, Caramori G, Barczyk A, Vicari C, Brun P, Zanini A, et al. Innate Immunity But Not NLRP3 Inflammasome Activation Correlates With Severity of Stable COPD. Thorax (2014) 69:516–24. doi: 10.1136/thoraxjnl-2012-203062
138. Ozretić P, da Silva Filho MI, Catalano C, Sokolović I, Vukić-Dugac A, Šutić M, et al. Association of NLRP1 Coding Polymorphism With Lung Function and Serum IL-1β Concentration in Patients Diagnosed With Chronic Obstructive Pulmonary Disease (COPD). Genes (2019) 10(10):783. doi: 10.3390/genes10100783
139. Colarusso C, Terlizzi M, Molino A, Imitazione P, Somma P, Rega R, et al. AIM2 Inflammasome Activation Leads to IL-1α and TGF-β Release From Exacerbated Chronic Obstructive Pulmonary Disease-Derived Peripheral Blood Mononuclear Cells. Front Pharmacol (2019) 10. doi: 10.3389/fphar.2019.00257
140. Tran HB, Hamon R, Jersmann H, Ween MP, Asare P, Haberberger R, et al. AIM2 Nuclear Exit and Inflammasome Activation in Chronic Obstructive Pulmonary Disease and Response to Cigarette Smoke. J Inflamm (2021) 18(1):19. doi: 10.1186/s12950-021-00286-4
141. Khawandanah J. Double or Hybrid Diabetes: A Systematic Review on Disease Prevalence, Characteristics and Risk Factors. Nutr Diabetes. (2019) 9(1):33. doi: 10.1038/s41387-019-0101-1
142. Pontillo A, Brandao L, Guimaraes R, Segat L, Araujo J, Crovella S. Two SNPs in NLRP3 Gene are Involved in the Predisposition to Type-1 Diabetes and Celiac Disease in a Pediatric Population From Northeast Brazil. Autoimmunity (2010) 43(8):583–9. doi: 10.3109/08916930903540432
143. Carlos D, Costa FRC, Pereira CA, Rocha FA, Yaochite JNU, Oliveira GG, et al. Mitochondrial DNA Activates the NLRP3 Inflammasome and Predisposes to Type 1 Diabetes in Murine Model. Front Immunol (2017) 8. doi: 10.3389/fimmu.2017.00164
144. Hu C, Ding H, Li Y, Pearson JA, Zhang X, Flavell RA, et al. NLRP3 Deficiency Protects From Type 1 Diabetes Through the Regulation of Chemotaxis Into the Pancreatic Islets. Proc Natl Acad Sci (2015) 112:11318–23. doi: 10.1073/pnas.1513509112
145. Ding S, Xu S, Ma Y, Liu G, Jang H, Fang J. Modulatory Mechanisms of the NLRP3 Inflammasomes in Diabetes. Biomolecules. (2019) 9(12):850. doi: 10.3390/biom9120850
146. Grant R, Dixit V. Mechanisms of Disease: Inflammasome Activation and the Development of Type 2 Diabetes. Front Immunol (2013) 4. doi: 10.3389/fimmu.2013.00050
147. Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 Inflammasome Activation in Patients With Type 2 Diabetes. Diabetes. (2013) 62(1):194–204. doi: 10.2337/db12-0420
148. Wang D, Wang H, Gao H, Zhang H, Zhang H, Wang Q, et al. P2X7 Receptor Mediates NLRP3 Inflammasome Activation in Depression and Diabetes. Cell Biosci (2020) 10(1):28. doi: 10.1186/s13578-020-00388-1
149. Wang J, Shen X, Liu J, Chen W, Wu F, Wu W, et al. High Glucose Mediates NLRP3 Inflammasome Activation via Upregulation of ELF3 Expression. Cell Death Disease. (2020) 11(5):383. doi: 10.1038/s41419-020-2598-6
150. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty Acid-Induced NLRP3-ASC Inflammasome Activation Interferes With Insulin Signaling. Nat Immunol (2011) 12(5):408–15. doi: 10.1038/ni.2022
151. Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, et al. Interleukin-1-Receptor Antagonist in Type 2 Diabetes Mellitus. N Engl J Med (2007) 356(15):1517–26. doi: 10.1056/NEJMoa065213
152. Everett BM, Donath MY, Pradhan AD, Thuren T, Pais P, Nicolau JC, et al. Anti-Inflammatory Therapy With Canakinumab for the Prevention and Management of Diabetes. J Am Coll Cardiol (2018) 71(21):2392–401. doi: 10.1016/j.jacc.2018.03.002
153. Deb A, Gupta S, Mazumder PB. Exosomes: A New Horizon in Modern Medicine. Life Sci (2021) 264:118623. doi: 10.1016/j.lfs.2020.118623
154. Cypryk W, Nyman TA, Matikainen S. From Inflammasome to Exosome-Does Extracellular Vesicle Secretion Constitute an Inflammasome-Dependent Immune Response? Front Immunol (2018) 9:2188. doi: 10.3389/fimmu.2018.02188
155. Li C, Lin H, He H, Ma M, Jiang W, Zhou R. Inhibition of the NLRP3 Inflammasome Activation by Manoalide Ameliorates Experimental Autoimmune Encephalomyelitis Pathogenesis. Front Cell Dev Biol (2022) 10:822236. doi: 10.3389/fcell.2022.822236
156. Lorey MB, Rossi K, Eklund KK, Nyman TA, Matikainen S. Global Characterization of Protein Secretion From Human Macrophages Following Non-Canonical Caspase-4/5 Inflammasome Activation. Mol Cell Proteomics (2017) 16(4 suppl 1):S187–99. doi: 10.1074/mcp.M116.064840
157. Nyman TA, Lorey MB, Cypryk W, Matikainen S. Mass Spectrometry-Based Proteomic Exploration of the Human Immune System: Focus on the Inflammasome, Global Protein Secretion, and T Cells. Expert Rev Proteomics (2017) 14(5):395–407. doi: 10.1080/14789450.2017.1319768
158. Singhto N, Kanlaya R, Nilnumkhum A, Thongboonkerd V. Roles of Macrophage Exosomes in Immune Response to Calcium Oxalate Monohydrate Crystals. Front Immunol (2018) 9:316. doi: 10.3389/fimmu.2018.00316
159. Zhang Y, Liu F, Yuan Y, Jin C, Chang C, Zhu Y, et al. Inflammasome-Derived Exosomes Activate NF-kappaB Signaling in Macrophages. J Proteome Res (2017) 16(1):170–8. doi: 10.1021/acs.jproteome.6b00599
160. Sur S, Steele R, Isbell TS, Ray R, Ray RB. Circulatory Exosomes From COVID-19 Patients Trigger NLRP3 Inflammasome in Endothelial Cells. bioRxiv. (2022) 2022:02.03.479081. doi: 10.1101/2022.02.03.479081
161. Hornung V, Latz E. Critical Functions of Priming and Lysosomal Damage for NLRP3 Activation. Eur J Immunol (2010) 40(3):620–3. doi: 10.1002/eji.200940185
162. Troncoso-Escudero P, Parra A, Nassif M, Vidal RL. Outside in: Unraveling the Role of Neuroinflammation in the Progression of Parkinson's Disease. Front Neurol (2018) 9:860. doi: 10.3389/fneur.2018.00860
163. Cai X, Zhang ZY, Yuan JT, Ocansey DKW, Tu Q, Zhang X, et al. hucMSC-Derived Exosomes Attenuate Colitis by Regulating Macrophage Pyroptosis via the miR-378a-5p/NLRP3 Axis. Stem Cell Res Ther (2021) 12(1):416. doi: 10.1186/s13287-021-02492-6
Keywords: NLRP3, Inflammasome, Microbes, Sterile Inflammation, extracellular vesicles, post-translational modification
Citation: Banerjee SK, Chatterjee A, Gupta S and Nagar A (2022) Activation and Regulation of NLRP3 by Sterile and Infectious Insults. Front. Immunol. 13:896353. doi: 10.3389/fimmu.2022.896353
Received: 15 March 2022; Accepted: 19 April 2022;
Published: 12 May 2022.
Edited by:
Jianguo Wu, Wuhan University, ChinaReviewed by:
Sarang Tartey, IGM Biosciences, United StatesCopyright © 2022 Banerjee, Chatterjee, Gupta and Nagar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abhinit Nagar, YW5hZ2FyQGx1bWluZXhjb3JwLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.