 Tomoko Matsuda1
Tomoko Matsuda1 Naotomo Kambe1,2*Riko Takimoto-Ito2Yoko Ueki1
Naotomo Kambe1,2*Riko Takimoto-Ito2Yoko Ueki1 Satoshi Nakamizo2
Satoshi Nakamizo2 Megumu K. Saito3Syuji Takei4
Megumu K. Saito3Syuji Takei4 Nobuo Kanazawa5
Nobuo Kanazawa5- 1Department of Dermatology, Kansai Medical University, Hirakata, Japan
- 2Department of Dermatology, Kyoto University Graduate School of Medicine, Kyoto, Japan
- 3Department of Clinical Application, Center for iPS Cell Research and Application, Kyoto University, Kyoto, Japan
- 4Department of Pediatrics, Kagoshima University, Kagoshima, Japan
- 5Department of Dermatology, Hyogo Medical University, Nishinomiya, Japan
Blau syndrome is a systemic autoinflammatory granulomatous disease caused by mutations in the nucleotide-binding oligomerization domain 2 (NOD2) gene. NOD2 is an intracellular pathogen recognition receptor. Upon binding to muramyl dipeptide (MDP), NOD2 activates the NF-κB pathway, leading to the upregulation of proinflammatory cytokines. Clinical manifestations of Blau syndrome appear in patients before the age of four. Skin manifestations resolve spontaneously in some cases; however, joint and eye manifestations are progressive, and lead to serious complications, such as joint contracture and blindness. Currently, there is no specific curative treatment for the disease. Administration of high-dose oral steroids can improve clinical manifestations; however, treatments is difficult to maintain due to the severity of the side effects, especially in children. While several new therapies have been reported, including JAK inhibitors, anti-IL-6 and anti-IL-1 therapies, anti-TNF therapy plays a central role in the treatment of Blau syndrome. We recently performed an ex vivo study, using peripheral blood and induced pluripotent stem cells from patients. This study demonstrated that abnormal cytokine expression in macrophages from untreated patients requires IFNγ stimulation, and that anti-TNF treatment corrects the abnormalities associated with Blau syndrome, even in the presence of IFNγ. Therefore, although the molecular mechanisms by which the genetic mutations in NOD2 lead to granuloma formation remain unclear, it is possible that prior exposure to TNFα combined with IFNγ stimulation may provide the impetus for the clinical manifestations of Blau syndrome.
Introduction
Blau syndrome (MIM #186580) is a rare, systemic granulomatous disease caused by mutations in the nucleotide-binding oligomerization domain 2 (NOD2) gene and is inherited in an autosomal dominant manner. It is classified as an autoinflammatory syndrome, a concept that has received increasing attention in recent years.
In 1985, Blau (1) reported a family that caused granulomas in the skin, eyes, and joints over four generations. In 1990, Pastores et al. (2) reported a mother and daughter with similar symptoms and considered them to be from the same category as the diseases reported by Blau; thus, they named the disease Blau syndrome. Subsequently, a linkage analyses of a large pedigree were presumed, and discovered that Blau syndrome and Crohn’s disease have mutations in the same gene (3). In 2001, when mutations in the NOD2 gene were identified in Crohn’s disease (4, 5), Miceli-Richard et al. (6) examined the NOD2 gene in four families with Blau syndrome, and identified three gene mutations.
Idiopathic sarcoidosis pediatric cases are rare, yet it is known that in a small subset of patients, peak onset occurs before the age of four years (less than 0.5% of all sarcoidosis cases) (7). The clinical features in these cases were the absence of bilateral hilar lymphadenopathy, and the presence of joint symptoms. Hence, these cases were occasionally referred to as early-onset sarcoidosis (EOS), and the difference from idiopathic sarcoidosis debated (8). Initially no gene abnormalities were found in two solitary cases of EOS (6). We collected ten EOS cases in Japan and identified the same NOD2 mutations in these patients as those reported for Blau syndrome (9), and now Blau syndrome and EOS are considered to be the same disease (10, 11).
Molecular Mechanisms
NOD2 consists of 1,040 amino acids and has a three domain structure. The N-terminus contains two caspase activation and recruitment domain s (CARD), which are important as in signal transduction; the centrally located NOD region is involved in polymerization; and the C-terminus contains a leucine-rich repeat (LRR). The LRR recognizes muramyl dipeptide (MDP), a component of bacterial cell walls. In Blau syndrome, most patients have mutations in exon 4, the NOD region. Most of these mutations are missense, with single amino acid substitutions. The 334th amino acid of NOD2 is a hot spot for mutations. Typically, arginine (R) is mutated to glutamine (Q) or tryptophan (W) in this region (p.R334Q and p.R334W). Among the 50 patients with NOD2 mutations currently identified in Japan, the p.R334W mutation was most common (fifteen cases), followed by the p.R587C (nine cases), and then the p.R334Q (five cases) (12).
It is still unclear how the mutations identified in Blau syndrome are involved in the formation of granulomas. When NOD2 was shown to recognize MDP, an experimental system to overexpress the NOD2 genes into HEK293 cells was used, in which the mutant with a frameshift in the LRR frequently identified in Crohn’s disease was shown to be hyporesponsive to MDP (13). In contrast, mutants identified in Blau syndrome spontaneously promoted NF-κB transcription in the same experimental system, even without the addition of MDP (14). This system of assessing NF-κB transcriptional capacity using luciferase as an indicator is still today a very useful system for confirming whether identified mutations are those associated with Blau syndrome, and indeed all 15 mutants we identified showed spontaneous NF-κB transcriptional enhancement in the absence of MDP (9, 15). This ligand-independent NF-κB activation induced by mutations has been observed in another group of hereditary autoinflammatory syndromes known as cryopyrin-associated periodic syndromes (CAPS). CAPS is caused by mutations in the NLRP3 gene, another NOD family receptor (16). Interestingly, the p.R334W mutation in the NOD2 gene frequently found in Blau syndrome, corresponds to a missense mutation in an analogous position of the NLRP3 gene, the p.R260W mutation in CAPS. These mutations suggest a common molecular mechanism, inducing the autoactivation of NOD family receptors, between these two inflammatory diseases (17).
Nevertheless, we observe an overproduction of IL-1β in peripheral blood monocytes extracted from CAPS patients and the macrophages established from CAPS patients-derived induced pluripotent stem (iPS) cells compared to healthy controls (18, 19), whereas a reduced response to MDP to produce inflammatory cytokines in the peripheral blood of patients with Blau syndrome were reported (20, 21). This lower responsiveness to MDP at the cellular level was also observed in studies using iPS cells differentiated into macrophages established from our patients (22), or knock-in mice with a corresponding Nod2 mutation identified in Blau syndrome (23). Since granulomas may arise from the inability to eliminate foreign substances and pathogens, it is possible that a reduced response to MDP at the cellular level may induce chronic inflammation, leading to granuloma formation in Blau syndrome.
Clinical Manifestations
The clinical phenotype of Blau syndrome is characterized by a distinct triad of skin, joint and eye disorders.
Skin Rash
Skin lesions often present as a first symptom (12). The most frequent skin manifestations are scaly erythematous plaques with multiple lichenoid papules and no apparent subjective symptoms. Occasionally, a BCG vaccination is the trigger for a skin rash that closely resembles lichen scrofulous, a tuberculous rash. Histological findings are characterized by the presence of epithelioid granulomas, with giant cells in the dermis. Cutaneous manifestations may disappear spontaneously and are often overlooked without a proper diagnosis, due to the lack of subjective symptoms. Cases of erythema nodosum have also been reported (15).
Joint Symptoms
The skin rash is followed by joint symptoms (12). Symmetrical polyarthritis occurs in small joints, such as the fingers and toes; and large joints, such as the hands, elbows, knees, feet; and rarely in the shoulders (24, 25). Painless, cystic swelling of the dorsal surfaces of the wrist and ankle, sausage-like swelling of the fingers and toes especially toward the base, and camptodactyly are characteristic findings.
The absence of arthralgia in the presence of joint swelling, the lack of an initial limitation of movement, and the presence of cystic swelling on the dorsum of the hands and feet, without pain, are of high diagnostic value and are important in differentiating the disease from juvenile idiopathic arthritis (JIA). In Blau syndrome, inflammation of the joint synovium is rare, and in the early stages of the disease, the tendon sheath synovium is damaged, leading to edema around the synovium and a limitation of movement. Joint sonography (26, 27) and MRI are useful to identify the main site of inflammation. However, in children, it is difficult to distinguish synovial thickening due to the lack of ossification in the cartilage on the surface of the bone, and a large area of low-density echoes.
Ocular Manifestations
Ocular manifestations appear later than skin and joint symptoms (12). The most common ocular manifestation is a bilateral uveitis (28–30). Other symptoms include posterior iris adhesion, conjunctivitis, retinitis, and optic nerve atrophy, all of which affect the entire eye. If the lesions persist for an extended time, secondary cataracts and glaucoma may develop, leading to blindness, which greatly affects the prognosis.
Fever
Although fever is not included in the triad of symptoms, fever is an important clinical feature of Blau syndrome. Analysis of the clinical manifestations from fifty patients in Japan showed that in about half of the cases (26 out of 50), fever occurred relatively early in the course of the disease (12). Among these patients, ten had intermittent fever, while seventeen had persistent, which includes two patients that exhibited both types. The frequency of the fever was reported to be once every few days to once a month (31, 32), and the duration varied from a few hours to ten days (31, 33). The range of the fever was mostly high (38-40°C) (31, 32, 34–36), with one case of a mild fever (37°C) reported (34).
Fever is important in Blau syndrome diagnosis. De Rose et al. (37) reported that it is essential to keep Blau syndrome in mind as a cause of unknown fever in infants up to four years old. Interestingly, Rosé et al. (38) demonstrated that all three Blau syndrome patients with the NOD2 p.R587C mutation presented with fever, consistent with our study (12), where eight of nine patients with the p.R587C mutation had fever, compared to four out of 14 patients with p.R334W mutation had fever. In CAPS, there is a clear genotype-phenotype relationship depending on the intensity of the NLRP3 mutation. However, for NOD2 mutations, differential enhancement of NF-κB transcription was dependent on the site of the NOD2 mutation when evaluated in HEK293 cells (15) and the differences did not necessarily correlate with the severity of clinical symptoms in Blau syndrome. Considering that the frequency of cases that present with fever are higher in the p.R587C mutation than in the other mutations, fever may be the clinical symptom that best reflects the ability of NOD2 variants.
Treatment
Blau syndrome is currently treated with various therapies, with multiple reports on their effectiveness (Table 1). However, there is no specific cure for the disease. Currently, the relationship between the variant of the genetic mutation and the response to treatment is not clear.

Table 1 Characteristics results of different treatment methods for Blau syndrome.
Steroids
High doses of steroids can improve clinical manifestations when there is a rapid worsening of joint or ocular symptoms (43, 65); however, prolonged treatment, especially in children, is difficult due to the side effects, including hypertension, growth retardation (48), iatrogenic Cushing syndrome, and elevated intraocular pressure (51). The most common initial diagnosis in our study (12) was JIA with 16 patients. Some of these cases were treated with oral steroids and because these cases were considered relatively mild for JIA, small doses of steroids were continued for an extended time, resulting in a delay in the appropriate therapeutic intervention, and the progression to more severe symptoms, such as joint contractures and blindness.
Topical steroids are often used for skin lesions but are not effective. One patient (12), initially diagnosed with atopic dermatitis, had been treated with topical steroids for an extended time, despite a poor therapeutic response. After a skin biopsy was performed, and granulomas were detected, the patient was subsequently diagnosed with Blau syndrome. Therefore, if a skin rash is not responsive to topical steroids, the possibility of granulomatous diseases, including Blau syndrome, needs to be considered. However, since rashes tends to disappear spontaneously, it can be difficult to evaluate the effectiveness of treatment. In contrast, topical steroid injections and steroid eye drops improved ocular symptoms (45).
Methotrexate and Other Oral Compounds
Methotrexate (MTX) is effective for joint symptoms (33), and useful for steroid sparing (39), although it often needs to be combined with other treatments (12, 43, 65). In our study (12), excluding the seven patients with no treatment information, out of 43 patients, 25 patients were treated with MTX (seven were in combination with biologics, seven in combination with prednisolone [PSL] and biologics, five in combination with PSL, four with MTX alone, and two in combination with PSL, tacrolimus, and biologics).
Other reports suggest that thalidomide may be effective in the treatment of Blau syndrome (36, 48), but the number of cases is limited, and further studies are needed to evaluate the long-term efficacy and side effects.
In 2018, there was an encouraging report of a significant response to the use of tofacitinib, a Janus kinase inhibitor (JAKi), in patients with cutaneous idiopathic sarcoidosis (66). The patient achieved clinical and histological remission, suggesting that JAK- signal transducer and activation of transcription (STAT) signaling plays a role in the pathogenesis of sarcoidosis and other granuloma diseases. In skin lesion samples taken before treatment, phosphorylated STAT1 (pSTAT1) was identified consistently in granulomas composed of CD68-positive cells, and pSTAT3 was found within inflammatory cells surrounding the lesion; however, these signals disappeared from the granulomas after ten months of treatment with tofacitinib. RNA sequencing results showed that not only interferon-γ (IFNγ), which is dependent on the JAK-STAT pathway, but also tumor necrosis factor-α (TNFα), which is not directly mediated by the JAK-STAT pathway, was elevated in skin lesions before treatment. Expression of these factors was also reduced upon treatment. Based on the successful treatment of sarcoidosis with a JAKi, this treatment has been applied in Blau syndrome and has been reported to be effective. Zhang et al. (55) reported that a single dose of tofacitinib suppressed TNFα and IL-6 production, along with the production of other inflammatory cytokines. Substantial improvements in clinical symptoms and laboratory parameters in the tofacitinib-treated patients were observed.
Biologics
Interleukin (IL)-1, a downstream product of NOD2 through NF-κB transcription, is anecdotally reported to be elevated in some patients with Blau syndrome, and anti-IL-1β therapy was shown to be effective (52, 56). In general, however, IL-1β cannot be detected in serum from patients with Blau syndrome or in cultures from MDP-stimulated MNC (20, 67). Martin et al. (20) showed that IL-1β is not overexpressed in patients of Blau syndrome and the patients were unresponsive to IL-1β therapy.
Similarly, IL-6, another downstream product of NOD2 activation, increases in some patients with Blau syndrome, and Lu et al. (32) mentioned good responses to tocilizumab, especially for patients with fever, lymphadenopathy, and hepatosplenomegaly. Conversely, Nagakura et al. (57) reported that tocilizumab treatment was discontinued because of recurrent arthritis and the development of anti-tocilizumab IgE antibodies due to the absence of co-therapy with MTX.
TNF-Targeting Therapy
When the disease is not controlled by steroids or MTX, biologic agents may be useful and anti-TNF agents are primarily used. Infliximab is effective in treating joint symptoms and may prevent the onset of ocular symptoms (68). Adalimumab is indicated for the treatment of non-infectious uveitis and effective for ocular symptoms, joint symptoms, and systemic symptoms (60, 62, 69, 70). However, etanercept-induced myelopathy has been reported in pediatric patients with Blau syndrome (64).
From the patient cohort in Japan (12), 26 out of 43 patients were treated with biologics, all of which were anti-TNF agents (18 patients were treated with adalimumab, five patients with infliximab, two patients with golimumab, and one patient with etanercept). Focusing on the prognosis of ocular symptoms, of the 26 patients that were treated with anti-TNF therapies, only one was blind. In this case, since adalimumab treatment began, the condition in the right eye did not deteriorate. Conversely, of the 14 patients that did not use biologics, five were blind. Of the remaining patients, three were under observation without treatment, seven had unknown treatment details and one was blind. Four patients who had been treated with biologics for JIA prior to their Blau syndrome diagnosis maintained their ocular status despite a relatively advanced age. Thus, we conclude that early treatment targeting TNF is necessary to avoid irreversible ocular symptoms.
Discussion
An investigation into the cellular phenotypes of Blau syndrome may be necessary to evaluate the efficacy of anti-TNF therapy since the pharmacological mechanism behind their effectiveness in Blau syndrome is unknown.
The IFNγ signal has been reported to be increased in localized skin lesions of idiopathic sarcoidosis and Blau syndrome (71) and IFNγ upregulates the expression of NOD2, acting as a priming signal, as previously reported (22). This was established through iPS cells from patients with Blau syndrome that were differentiated into macrophages and confirmed that IFNγ treatment enhanced the expression of NOD2. The enhanced expression of NOD2 through IFNγ priming was observed regardless of the presence or absence of the NOD2 mutations. However, the spontaneous transcriptional enhancement of NF-κB and the production of proinflammatory cytokines were observed only in cells that contained the NOD2 mutation associated with Blau syndrome. These effects were not found in cells where the NOD2 mutation was corrected to the wild-type amino acid using the CRISPR-Cas9 system. Of interest, skin lesions have appeared following BCG vaccination in some patients with Blau syndrome. Upon review of fifty patients in the cohort from Japan, the attending physician provided information indicating that BCG vaccination may have induced symptoms in nine patients (12). Since IFNγ is a cytokine that is highly associated with BCG-mediated immune responses, BCG vaccination may be involved in NOD2-mediated inflammatory responses through the induction of NOD2 expression.
While verifying the studies using Blau syndrome patient-derived iPS cells, additional, surprising results were obtained (72). Macrophages, differentiated from the peripheral blood of Blau syndrome who did not receive anti-TNF treatment, released inflammatory cytokines after being primed with IFNγ, as described above. In contrast, macrophages differentiated from patients treated with anti-TNF, did not produce proinflammatory cytokines, similar to macrophages differentiated from healthy subjects. This was despite anti-TNF patient macrophages being primed with IFNγ and being differentiated ex vivo for one week with unchanged culture conditions. A comprehensive gene expression analysis of all three groups indicated that the cells from the anti-TNF treatment patient group had a nearly identical gene expression pattern as the cells differentiated from healthy individuals, without the NOD2 mutation. Furthermore, when primed with IFNγ, cells from these two groups (healthy individuals and anti-TNF treated patients) behaved in the same manner and had gene expression patterns that differed from macrophages differentiated from patients not receiving an anti-TNF agent. This study suggests that prior exposure to a proinflammatory cytokine, such as TNFα, at the monocytic or earlier progenitor cell stage is an important determinant for the macrophage response to IFNγ. Thus, the use of anti-TNF antibodies for Blau syndrome treatment is appropriate in the sense that long-term administration of anti-TNF antibodies may correct the abnormalities that occur in the early progenitor stage by blocking the autoinflammatory loop and restoring the threshold for which IFNγ stimulation triggers an inflammatory response in macrophages (Figure 1).
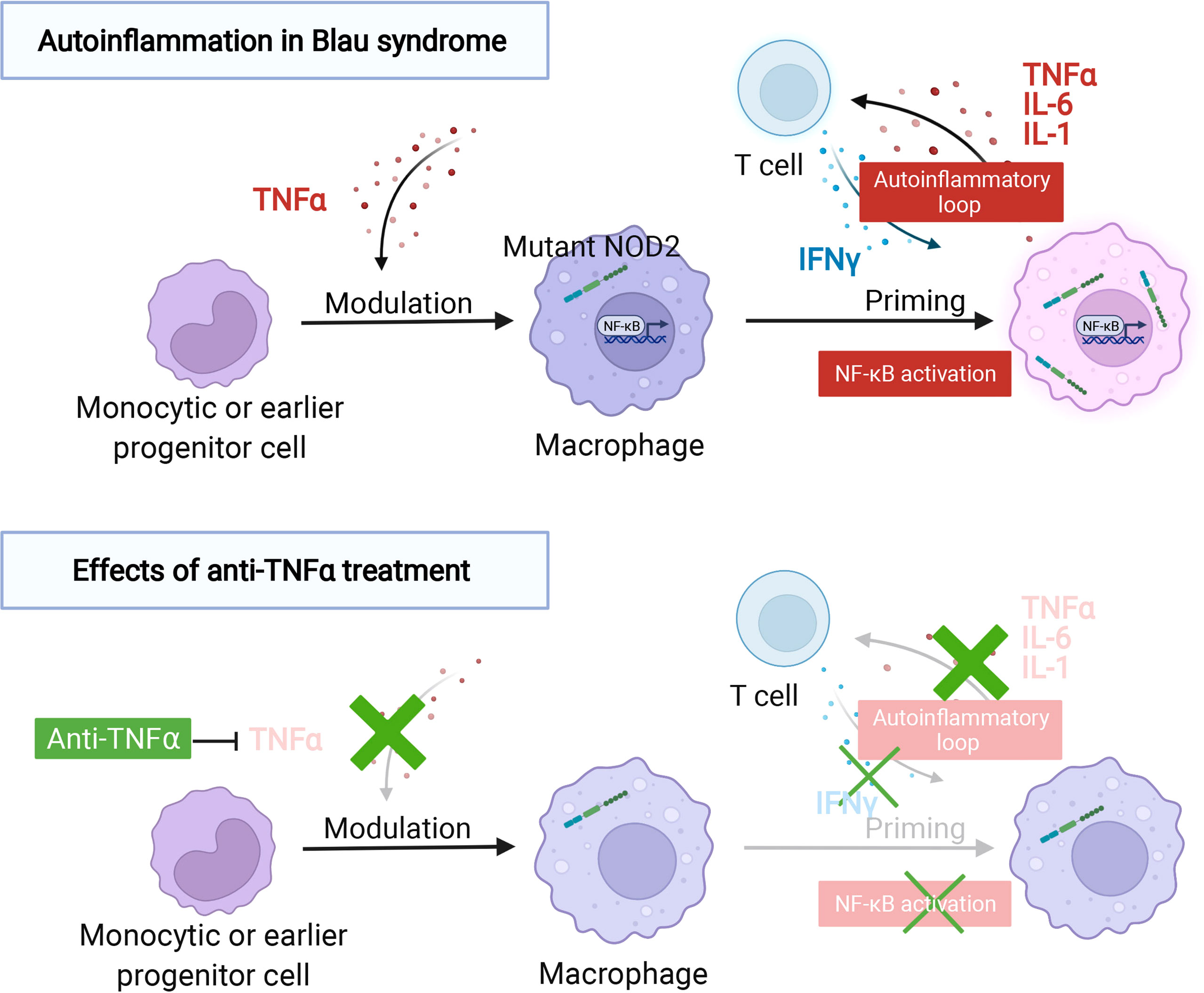
Figure 1 The autoinflammatory response in Blau syndrome and the effect of anti-TNFα treatment. Autoinflammation in Blau syndrome. The expression of NOD2 is induced by IFNγ with or without a NOD2 mutation. The production of proinflammatory cytokines, such as TNFα, IL-6, and IL-1, can only be confirmed in macrophages with NOD2 mutations associated with Blau syndrome, in the absence of muramyl dipeptide (MDP) that activate NOD2. These proinflammatory cytokines further activate T cells and induce the production of IFNγ, thereby establishing a self-amplifying autoinflammatory loop. The effect of anti-TNFα treatment. TNFα-targeted therapy inhibits T cell activation, indirectly suppressing IFNγ production in T cells, and as a result, the autoinflammatory loop is suppressed. Macrophages differentiated from blood collected from patients with Blau syndrome and treated with a TNFα-targeted therapy behaved similarly to wild-type NOD2-expressing cells, despite the presence of the Blau syndrome-associated NOD2 mutations. This suggests that TNFα may exert a modulatory effect on monocytic or early prongenitor cells before they are induced to differentiate into macrophages and adopt a more inflammation-specific form.
At present, no specific treatment for Blau syndrome exists, based on its etiology, and the disease is treated empirically. However, early diagnosis will allow for the prediction of future symptoms that are likely to appear, and prompt treatment will prevent or delay the onset of severe symptoms (joint contractures and blindness), which significantly impair the patient’s quality of life. Future accumulation of a large number of patients, with detailed information on disease progression, treatment, and prognosis, would be advantageous to analyze disease pathogenesis and establish a specific treatment based on disease etiology.
Author Contributions
TM, NaK, RT-I, YU, SN, MS, ST, and NoK contributed to conception and design of the study. TM and NaK wrote the first draft of the manuscript. NaK and MS created the first draft of the figure, while RT-I created the final figure. All authors have read, contributed to revisions of the manuscript, and approved the submitted version.
Funding
This work was supported in part by the Core Center for iPS Cell Research of Research Center Network for Realization of Regenerative Medicine (JP21bm0104001); the Acceleration Program for Intractable Diseases Research Utilizing Disease-Specific iPS Cells (17935423); the Practical Research Project for Rare/Intractable Diseases (17929899) from AMED (MS); a research grant from MHLW (NaK and NoK); a JSPS KAKENHI Grant Number 19K08784, 22K08380; and a Lexi’s Legacy Research Grant from Cure Blau syndrome Foundation (NaK).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgment
The figures were created with BioRender software, ©biorender.com.
References
1. Blau EB. Familial Granulomatous Arthritis, Iritis, and Rash. J Pediatr (1985) 107(5):689–93. doi: 10.1016/s0022-3476(85)80394-2
2. Pastores GM, Michels VV, Stickler GB, Su WP, Nelson AM, Bovenmyer DA. Autosomal Dominant Granulomatous Arthritis, Uveitis, Skin Rash, and Synovial Cysts. J Pediatr (1990) 117(3):403–8. doi: 10.1016/s0022-3476(05)81080-7
3. Tromp G, Kuivaniemi H, Raphael S, Ala-Kokko L, Christiano A, Considine E, et al. Genetic Linkage of Familial Granulomatous Inflammatory Arthritis, Skin Rash, and Uveitis to Chromosome 16. Am J Hum Genet (1996) 59(5):1097–107.
4. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, et al. Association of NOD2 Leucine-Rich Repeat Variants With Susceptibility to Crohn's Disease. Nature (2001) 411(6837):599–603. doi: 10.1038/35079107
5. Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A Frameshift Mutation in NOD2 Associated With Susceptibility to Crohn's Disease. Nature (2001) 411(6837):603–6. doi: 10.1038/35079114
6. Miceli-Richard C, Lesage S, Rybojad M, Prieur AM, Manouvrier-Hanu S, Hafner R, et al. CARD15 Mutations in Blau Syndrome. Nat Genet (2001) 29(1):19–20. doi: 10.1038/ng720
8. Kaufman KP, Becker ML. Distinguishing Blau Syndrome From Systemic Sarcoidosis. Curr Allergy Asthma Rep (2021) 21(2):10. doi: 10.1007/s11882-021-00991-3
9. Kanazawa N, Okafuji I, Kambe N, Nishikomori R, Nakata-Hizume M, Nagai S, et al. Early-Onset Sarcoidosis and CARD15 Mutations With Constitutive Nuclear Factor-kappaB Activation: Common Genetic Etiology With Blau Syndrome. Blood (2005) 105(3):1195–7. doi: 10.1182/blood-2004-07-2972
10. Becker ML, Rose CD. Blau Syndrome and Related Genetic Disorders Causing Childhood Arthritis. Curr Rheumatol Rep (2005) 7(6):427–33. doi: 10.1007/s11926-005-0046-3
11. Caso F, Costa L, Rigante D, Vitale A, Cimaz R, Lucherini OM, et al. Caveats and Truths in Genetic, Clinical, Autoimmune and Autoinflammatory Issues in Blau Syndrome and Early Onset Sarcoidosis. Autoimmun Rev (2014) 13(12):1220–9. doi: 10.1016/j.autrev.2014.08.010
12. Matsuda T, Kambe N, Ueki Y, Kanazawa N, Izawa K, Honda Y, et al. Clinical Characteristics and Treatment of 50 Cases of Blau Syndrome in Japan Confirmed by Genetic Analysis of the. Ann Rheum Dis (2020) 79(11):1492–9. doi: 10.1136/annrheumdis-2020-217320
13. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a General Sensor of Peptidoglycan Through Muramyl Dipeptide (MDP) Detection. J Biol Chem (2003) 278(11):8869–72. doi: 10.1074/jbc.C200651200
14. Chamaillard M, Philpott D, Girardin SE, Zouali H, Lesage S, Chareyre F, et al. Gene-Environment Interaction Modulated by Allelic Heterogeneity in Inflammatory Diseases. Proc Natl Acad Sci USA (2003) 100(6):3455–60. doi: 10.1073/pnas.0530276100
15. Okafuji I, Nishikomori R, Kanazawa N, Kambe N, Fujisawa A, Yamazaki S, et al. Role of the NOD2 Genotype in the Clinical Phenotype of Blau Syndrome and Early-Onset Sarcoidosis. Arthritis Rheum (2009) 60(1):242–50. doi: 10.1002/art.24134
16. Booshehri LM, Hoffman HM. CAPS and NLRP3. J Clin Immunol (2019) 39(3):277–86. doi: 10.1007/s10875-019-00638-z
17. McDonald C, Inohara N, Nunez G. Peptidoglycan Signaling in Innate Immunity and Inflammatory Disease. J Biol Chem (2005) 280(21):20177–80. doi: 10.1074/jbc.R500001200
18. Saito M, Fujisawa A, Nishikomori R, Kambe N, Nakata-Hizume M, Yoshimoto M, et al. Somatic Mosaicism of CIAS1 in a Patient With Chronic Infantile Neurologic, Cutaneous, Articular Syndrome. Arthritis Rheum (2005) 52(11):3579–85. doi: 10.1002/art.21404
19. Seki R, Ohta A, Niwa A, Sugimine Y, Naito H, Nakahata T, et al. Induced Pluripotent Stem Cell-Derived Monocytic Cell Lines From a NOMID Patient Serve as a Screening Platform for Modulating NLRP3 Inflammasome Activity. PloS One (2020) 15(8):e0237030. doi: 10.1371/journal.pone.0237030
20. Martin TM, Zhang Z, Kurz P, Rose CD, Chen H, Lu H, et al. The NOD2 Defect in Blau Syndrome Does Not Result in Excess Interleukin-1 Activity. Arthritis Rheum (2009) 60(2):611–8. doi: 10.1002/art.24222
21. Son S, Lee J, Woo CW, Kim I, Kye Y, Lee K, et al. Altered Cytokine Profiles of Mononuclear Cells After Stimulation in a Patient With Blau Syndrome. Rheumatol Int (2010) 30(8):1121–4. doi: 10.1007/s00296-009-1342-4
22. Takada S, Kambe N, Kawasaki Y, Niwa A, Honda-Ozaki F, Kobayashi K, et al. Pluripotent Stem Cell Models of Blau Syndrome Reveal an IFN-Gamma-Dependent Inflammatory Response in Macrophages. J Allergy Clin Immunol (2018) 141(1):339–49.e11. doi: 10.1016/j.jaci.2017.04.013
23. Dugan J, Griffiths E, Snow P, Rosenzweig H, Lee E, Brown B, et al. Blau Syndrome-Associated Nod2 Mutation Alters Expression of Full-Length NOD2 and Limits Responses to Muramyl Dipeptide in Knock-in Mice. J Immunol (2015) 194(1):349–57. doi: 10.4049/jimmunol.1402330
24. Rose CD, Wouters CH, Meiorin S, Doyle TM, Davey MP, Rosenbaum JT, et al. Pediatric Granulomatous Arthritis: An International Registry. Arthritis Rheum (2006) 54(10):3337–44. doi: 10.1002/art.22122
25. Rose CD, Pans S, Casteels I, Anton J, Bader-Meunier B, Brissaud P, et al. Blau Syndrome: Cross-Sectional Data From a Multicentre Study of Clinical, Radiological and Functional Outcomes. Rheumatol (Oxf) (2015) 54(6):1008–16. doi: 10.1093/rheumatology/keu437
26. Ikeda K, Seto Y, Narita A, Kawakami A, Kawahito Y, Ito H, et al. Ultrasound Assessment of Synovial Pathologic Features in Rheumatoid Arthritis Using Comprehensive Multiplane Images of the Second Metacarpophalangeal Joint: Identification of the Components That are Reliable and Influential on the Global Assessment of the Whole Joint. Arthritis Rheumatol (2014) 66(3):523–32. doi: 10.1002/art.38280
27. Ikeda K, Kambe N, Satoh T, Matsue H, Nakajima H. Preferentially Inflamed Tendon Sheaths in the Swollen But Not Tender Joints in a 5-Year-Old Boy With Blau Syndrome. J Pediatr (2013) 163(5):1525.e1. doi: 10.1016/j.jpeds.2013.05.059
28. Kurokawa T, Kikuchi T, Ohta K, Imai H, Yoshimura N. Ocular Manifestations in Blau Syndrome Associated With a CARD15/Nod2 Mutation. Ophthalmology (2003) 110(10):2040–4. doi: 10.1016/S0161-6420(03)00717-6
29. Pillai P, Sobrin L. Blau Syndrome-Associated Uveitis and the NOD2 Gene. Semin Ophthalmol (2013) 28(5-6):327–32. doi: 10.3109/08820538.2013.825285
30. Sarens IL, Casteels I, Anton J, Bader-Meunier B, Brissaud P, Chédeville G, et al. Blau Syndrome-Associated Uveitis: Preliminary Results From an International Prospective Interventional Case Series. Am J Ophthalmol (2018) 187:158–66. doi: 10.1016/j.ajo.2017.08.017
31. Takeuchi Y, Shigemura T, Kobayashi N, Kaneko N, Iwasaki T, Minami K, et al. Early Diagnosis of Early-Onset Sarcoidosis: A Case Report With Functional Analysis and Review of the Literature. Clin Rheumatol (2017) 36(5):1189–96. doi: 10.1007/s10067-017-3544-6
32. Lu L, Shen M, Jiang D, Li Y, Zheng X, Li Z, et al. Blau Syndrome With Good Reponses to Tocilizumab: A Case Report and Focused Literature Review. Semin Arthritis Rheum (2018) 47(5):727–31. doi: 10.1016/j.semarthrit.2017.09.010
33. PaÇ Kisaarslan A, SÖzerİ B, Şahİn N, Özdemİr ÇİÇekS, GÜndÜz Z, Demİrkaya E, et al. Blau Syndrome and Early-Onset Sarcoidosis: A Six Case Series and Review of the Literature. Arch Rheumatol (2020) 35(1):117–27. doi: 10.5606/ArchRheumatol.2020.7060
34. Matsuo T, Yashiro M, Yamasaki O, Tanaka T, Manki A. Bilateral Optic Disc Swelling as a Plausible Common Ocular Sign of Autoinflammatory Diseases: Report of Three Patients With Blau Syndrome or Cryopyrin-Associated Periodic Syndrome. Life (Basel) (2021) 11(12). doi: 10.3390/life11121433
35. Imayoshi M, Ogata Y, Yamamoto S. A Case of Sporadic Blau Syndrome With an Uncommon Clinical Course. Case Rep Rheumatol (2018) 2018:6292308. doi: 10.1155/2018/6292308
36. Yasui K, Yashiro M, Tsuge M, Manki A, Takemoto K, Yamamoto M, et al. Thalidomide Dramatically Improves the Symptoms of Early-Onset Sarcoidosis/Blau Syndrome: Its Possible Action and Mechanism. Arthritis Rheum (2010) 62(1):250–7. doi: 10.1002/art.25035
37. De Rose DU, Coppola M, Gallini F, Maggio L, Vento G, Rigante D. Overview of the Rarest Causes of Fever in Newborns: Handy Hints for the Neonatologist. J Perinatol (2021) 41(3):372–82. doi: 10.1038/s41372-020-0744-8
38. Rosé CD, Aróstegui JI, Martin TM, Espada G, Scalzi L, Yagüe J, et al. NOD2-Associated Pediatric Granulomatous Arthritis, an Expanding Phenotype: Study of an International Registry and a National Cohort in Spain. Arthritis Rheum (2009) 60(6):1797–803. doi: 10.1002/art.24533
39. Gedalia A, Khan TA, Shetty AK, Dimitriades VR, Espinoza LR. Childhood Sarcoidosis: Louisiana Experience. Clin Rheumatol (2016) 35(7):1879–84. doi: 10.1007/s10067-015-2870-9
40. Punzi L, Furlan A, Podswiadek M, Gava A, Valente M, De Marchi M, et al. Clinical and Genetic Aspects of Blau Syndrome: A 25-Year Follow-Up of One Family and a Literature Review. Autoimmun Rev (2009) 8(3):228–32. doi: 10.1016/j.autrev.2008.07.034
41. Milman N, Andersen CB, Hansen A, van Overeem Hansen T, Nielsen FC, Fledelius H, et al. Favourable Effect of TNF-Alpha Inhibitor (Infliximab) on Blau Syndrome in Monozygotic Twins With a De Novo CARD15 Mutation. APMIS (2006) 114(12):912–9. doi: 10.1111/j.1600-0463.2006.apm_522.x
42. Arostegui JI, Arnal C, Merino R, Modesto C, Antonia Carballo M, Moreno P, et al. NOD2 Gene-Associated Pediatric Granulomatous Arthritis: Clinical Diversity, Novel and Recurrent Mutations, and Evidence of Clinical Improvement With Interleukin-1 Blockade in a Spanish Cohort. Arthritis Rheum (2007) 56(11):3805–13. doi: 10.1002/art.22966
43. Arvesen KB, Herlin T, Larsen DA, Koppelhus U, Ramsing M, Skytte AB, et al. Diagnosis and Treatment of Blau Syndrome/Early-Onset Sarcoidosis, an Autoinflammatory Granulomatous Disease, in an Infant. Acta Derm Venereol (2017) 97(1):126–7. doi: 10.2340/00015555-2485
44. Latkany PA, Jabs DA, Smith JR, Rosenbaum JT, Tessler H, Schwab IR, et al. Multifocal Choroiditis in Patients With Familial Juvenile Systemic Granulomatosis. Am J Ophthalmol (2002) 134(6):897–904. doi: 10.1016/s0002-9394(02)01709-9
45. Nascimento H, Sousa JM, Fernández DG, Salomão GHA, Sato EH, Muccioli C, et al. Blau-Jabs Syndrome in a Tertiary Ophthalmologic Center. Ophthalmic Surg Lasers Imaging Retina (2018) 49(1):70–5. doi: 10.3928/23258160-20171215-12
46. Okazaki F, Wakiguchi H, Korenaga Y, Nakamura T, Yasudo H, Uchi S, et al. A Novel Mutation in Early-Onset Sarcoidosis/Blau Syndrome: An Association With Propionibacterium Acnes. Pediatr Rheumatol Online J (2021) 19(1):18. doi: 10.1186/s12969-021-00505-5
47. Marín-Noriega MA, Muñoz-Ortiz J, Mosquera C, de-la-Torre A. Ophthalmological Treatment of Early-Onset Sarcoidosis/Blau Syndrome in a Colombian Child: A Case Report. Am J Ophthalmol Case Rep (2020) 18:100714. doi: 10.1016/j.ajoc.2020.100714
48. Wang W, Zhong LQ, Li WD, Wu SJ, Song HM. Thalidomide may be an Effective Drug for Blau Syndrome: A Case Report. Ann Palliat Med (2021). doi: 10.21037/apm-21-2216
49. Iriqat S, Safieh MA, Fatouleh M, Alkaiyat A. Blau Syndrome: A Case Report From Palestine. Pediatr Rheumatol Online J (2021) 19(1):138. doi: 10.1186/s12969-021-00633-y
50. Naik AU, Annamalai R, Biswas J. Uveitis in Sporadic Blau Syndrome: Long-Term Follow-Up of a Refractory Case Treated Successfully With Adalimumab. Indian J Ophthalmol (2018) 66(10):1483–5. doi: 10.4103/ijo.IJO_629_18
51. Achille M, Ilaria P, Teresa G, Roberto C, Ilir A, Piergiorgio N, et al. Successful Treatment With Adalimumab for Severe Multifocal Choroiditis and Panuveitis in Presumed (Early-Onset) Ocular Sarcoidosis. Int Ophthalmol (2016) 36(1):129–35. doi: 10.1007/s10792-015-0135-x
52. Simonini G, Xu Z, Caputo R, De Libero C, Pagnini I, Pascual V, et al. Clinical and Transcriptional Response to the Long-Acting Interleukin-1 Blocker Canakinumab in Blau Syndrome-Related Uveitis. Arthritis Rheum (2013) 65(2):513–8. doi: 10.1002/art.37776
53. Fretzayas A, Moustaki M, Vougiouka O. The Puzzling Clinical Spectrum and Course of Juvenile Sarcoidosis. World J Pediatr (2011) 7(2):103–10. doi: 10.1007/s12519-011-0261-0
54. Babu K, Rao AP. Clinical Profile in Genetically Proven Blau Syndrome: A Case Series From South India. Ocul Immunol Inflamm (2021) 29(2):250–6. doi: 10.1080/09273948.2020.1746353
55. Zhang S, Cai Z, Mo X, Zeng H. Tofacitinib Effectiveness in Blau Syndrome: A Case Series of Chinese Paediatric Patients. Pediatr Rheumatol Online J (2021) 19(1):160. doi: 10.1186/s12969-021-00634-x
56. Papatesta EM, Kossiva L, Tsolia M, Maritsi D. Persistent Tenosynovitis, Steroid Dependency and a Hyperpigmented Scaly Macular Rash in a Child With Juvenile Idiopathic Arthritis. Cureus (2020) 12(10):e11208. doi: 10.7759/cureus.11208
57. Nagakura T, Wakiguchi H, Kubota T, Yamatou T, Yamasaki Y, Nonaka Y, et al. Tumor Necrosis Factor Inhibitors Provide Longterm Clinical Benefits in Pediatric and Young Adult Patients With Blau Syndrome. J Rheumatol (2017) 44(4):536–8. doi: 10.3899/jrheum.160672
58. Ikeda K, Kambe N, Takei S, Nakano T, Inoue Y, Tomiita M, et al. Ultrasonographic Assessment Reveals Detailed Distribution of Synovial Inflammation in Blau Syndrome. Arthritis Res Ther (2014) 16(2):R89. doi: 10.1186/ar4533
59. Otsubo Y, Okafuji I, Shimizu T, Nonaka F, Ikeda K, Eguchi K. A Long-Term Follow-Up of Japanese Mother and Her Daughter With Blau Syndrome: Effective Treatment of Anti-TNF Inhibitors and Useful Diagnostic Tool of Joint Ultrasound Examination. Mod Rheumatol (2017) 27(1):169–73. doi: 10.3109/14397595.2014.964388
60. DeSouza PJ, Shah R. Characterization of Blau Syndrome Panuveitis With Wide-Field Fluorescein Angiography. Am J Ophthalmol Case Rep (2019) 14:92–4. doi: 10.1016/j.ajoc.2019.03.006
61. Şahin N, Çiçek S, Kısaarslan AP, Gündüz Z, Poyrazoğlu MH, Düşünsel R. Unexpected Condition in a Rare Disease: Encephalopathy in Early-Onset Sarcoidosis. Turk J Pediatr (2021) 63(2):323–8. doi: 10.24953/turkjped.2021.02.018
62. Jindal AK, Pilania RK, Suri D, Gupta A, Gattorno M, Ceccherini I, et al. A Young Female With Early Onset Arthritis, Uveitis, Hepatic, and Renal Granulomas: A Clinical Tryst With Blau Syndrome Over 20 Years and Case-Based Review. Rheumatol Int (2021) 41(1):173–81. doi: 10.1007/s00296-019-04316-6
63. Cuesta IA, Moore EC, Rabah R, Bawle EV. Blau Syndrome (Familial Granulomatous Arthritis, Iritis, and Rash) in an African-American Family. J Clin Rheumatol (2000) 6(1):30–4. doi: 10.1097/00124743-200002000-00005
64. Caracseghi F, Izquierdo-Blasco J, Sanchez-Montanez A, Melendo-Perez S, Roig-Quilis M, Modesto C. Etanercept-Induced Myelopathy in a Pediatric Case of Blau Syndrome. Case Rep Rheumatol (2011) 2011:134106. doi: 10.1155/2011/134106
65. Takada S, Saito MK, Kambe N. Blau Syndrome: NOD2-Related Systemic Autoinflammatory Granulomatosis. G Ital Dermatol Venereol (2020) 155(5):537–41. doi: 10.23736/S0392-0488.19.06524-6
66. Damsky W, Thakral D, Emeagwali N, Galan A, King B. Tofacitinib Treatment and Molecular Analysis of Cutaneous Sarcoidosis. N Engl J Med (2018) 379(26):2540–6. doi: 10.1056/NEJMoa1805958
67. Galozzi P, Negm O, Greco E, Alkhattabi N, Gava A, Sfriso P, et al. Ex Vivo and In Vitro Production of Pro-Inflammatory Cytokines in Blau Syndrome. Reumatismo (2015) 66(4):277–84. doi: 10.4081/reumatismo.2014.772
68. Chen J, Luo Y, Zhao M, Wu D, Yang Y, Zhang W, et al. Effective Treatment of Tnfα Inhibitors in Chinese Patients With Blau Syndrome. Arthritis Res Ther (2019) 21(1):236. doi: 10.1186/s13075-019-2017-5
69. Chauhan K, Michet C. A Case of Blau Syndrome. Case Rep Rheumatol (2014) 2014:216056. doi: 10.1155/2014/216056
70. Bravo-Ljubetic L, Peralta-Calvo J, Noval S, Pastora-Salvador N, Abelairas-Gomez J, Merino R. Adalimumab Therapy for Refractory Childhood Uveitis. J AAPOS (2013) 17(5):456–9. doi: 10.1016/j.jaapos.2013.06.009
71. Janssen CE, Rose CD, De Hertogh G, Martin TM, Bader Meunier B, Cimaz R, et al. Morphologic and Immunohistochemical Characterization of Granulomas in the Nucleotide Oligomerization Domain 2-Related Disorders Blau Syndrome and Crohn Disease. J Allergy Clin Immunol (2012) 129(4):1076–84. doi: 10.1016/j.jaci.2012.02.004
Keywords: Blau syndrome, NOD2, granuloma, IFNγ, TNF
Citation: Matsuda T, Kambe N, Takimoto-Ito R, Ueki Y, Nakamizo S, Saito MK, Takei S and Kanazawa N (2022) Potential Benefits of TNF Targeting Therapy in Blau Syndrome, a NOD2-Associated Systemic Autoinflammatory Granulomatosis. Front. Immunol. 13:895765. doi: 10.3389/fimmu.2022.895765
Received: 14 March 2022; Accepted: 02 May 2022;
Published: 27 May 2022.
Edited by:
Tomoyuki Mukai, Kawasaki Medical School, JapanReviewed by:
Ruth J. Napier, Oregon Health and Science University, United StatesQingping Yao, Stony Brook University, United States
Copyright © 2022 Matsuda, Kambe, Takimoto-Ito, Ueki, Nakamizo, Saito, Takei and Kanazawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Naotomo Kambe, bmthbWJlQGt1aHAua3lvdG8tdS5hYy5qcA==