 Bingran Wang1,2
Bingran Wang1,2 Jun Shen1,2*
Jun Shen1,2*- 1Division of Gastroenterology and Hepatology, Key Laboratory of Gastroenterology and Hepatology, Ministry of Health, Inflammatory Bowel Disease Research Center, Renji Hospital, School of Medicine, Shanghai Institute of Digestive Disease, Shanghai Jiao Tong University, Shanghai, China
- 2Ottawa-Shanghai Joint School of Medicine, Shanghai Jiao Tong University School of Medicine, Shanghai, China
Intestinal immunity and homeostasis are maintained through the regulation of cytokine trafficking, microbiota, necrosis and apoptosis. Intestinal immunity and homeostasis participate in host defenses and inflammatory responses locally or systemically through the gut-organ axis. NF-κB functions as a crucial transcription factor mediating the expression of proteins related to the immune responses. The activation of NF-κB involves two major pathways: canonical and non-canonical. The canonical pathway has been extensively studied and reviewed. Here, we present the current knowledge of NIK, a pivotal mediator of the non-canonical NF-κB pathway and its role in intestinal immunity and homeostasis. This review also discusses the novel role of NIK signaling in the pathogenesis and treatment of inflammatory bowel disease.
1 Introduction
The intestine is the largest component of the human immune system (1). Intestinal immunity and homeostasis are complex and sophisticatedly regulated by abundant innate and adaptive immune cells, mucous associated lymphoid tissue, and trillions of commensal microorganisms (2). Because of the complexity of intestinal immunity and its connection to the immune system, dysregulation of the intestinal immunity leads to local or systemic inflammatory responses, causing impaired absorptive function or even translocation of microbiota, and is involved in the progression of several inflammatory diseases. Disturbances in intestinal immunity and homeostasis emerge as the pathogenesis of inflammatory bowel disease (IBD) and systemic immune activation, or lead to the progression of chronic metabolic diseases, such as diabetes mellitus (3).
Nuclear factor-κB (NF-κB) is a family of transcription factors that serves to regulate inflammatory and immunological responses by controlling the expression of a large number of targeted genes in response to changes in the environment (4, 5). The activation of NF-κB is mediated by two major pathways, namely canonical and non-canonical, which mediate the signaling downstream of different receptors, have different signaling cascade component and implement different biological functions via activating different subtypes of NF-κB. NF-κB inducing kinase (NIK) is a crucial non-canonical mediator of the NF-κB signaling cascade, and mainly responds to signals transduced by the tumor necrosis factor receptor (TNFR) superfamily. In the past two decades, the role of the non-canonical NF-κB signaling pathway in mucosal immunity and homeostasis has been highlighted. Numerous research illustrate NIK is involved in the regulation of intestinal immunity and homeostasis through activating the development of effector T cells and IgA secretion (6–9), stimulation of the secretion of cytokines induced by microbiota (10), maintenance of microfold cell function (11), and sustaining the function of regulatory T cells (Tregs) and suppressor dendritic cells (DCs) (12, 13). Moreover, the close relationship between aberrant NIK signaling and pathogenesis of IBD has been extensively studied (14, 15). Consequently, this article reviews the signaling pathways mediated by NIK and its relevance to the pathogenesis and treatment of IBD, including ulcerative colitis (UC) and Crohn’s disease (CD).
2 NF-κB Is Activated by Canonical and Non-Canonical Pathways
NF-κB is a family of transcription factor, including NF-κB1 (p50/p105), NF-κB2 (p52/p100), RelA (p65), RelB, and c-Rel in mammals, and regulates the transcription of kappa chain in B cells via binding to κB enhancer as homodimers or heterodimers (16). NF-κB shares two common motifs: transcription activation domain (TAD) and N-terminal Rel homology domain (RHD). The former is responsible for positive regulation of gene expression by recruiting coactivators and the latter is responsible for NF-κB dimerization and DNA binding (17). The phosphorylation of TAD facilitates the recruitment of CREB binding protein (CBP)/p300 coactivator (18), which decreases the levels of histone deacetylases 3 (HADC3), resulting in the augmentation of histone acetylation and subsequent enhancement of the transcription of target genes (19).
NF-κB, exerts immunoregulatory function by enhancing the transcription of target genes under specific stimulations. In the physiological state, NF-κB is sequestered in the cytoplasm and is deprived of the nuclear translocation by inhibitors of NF-κB (IκB), among which the most representative IκB is IκB. p105 and p100, as precursors of p50 and p52, respectively, also exhibit an IκB-like structure in their C-terminal portion, and perform similar functions as IκB (20). NF-κB activation is mediated by both canonical and non-canonical pathways, depending on the types of stimulus (Figure 1) (21).
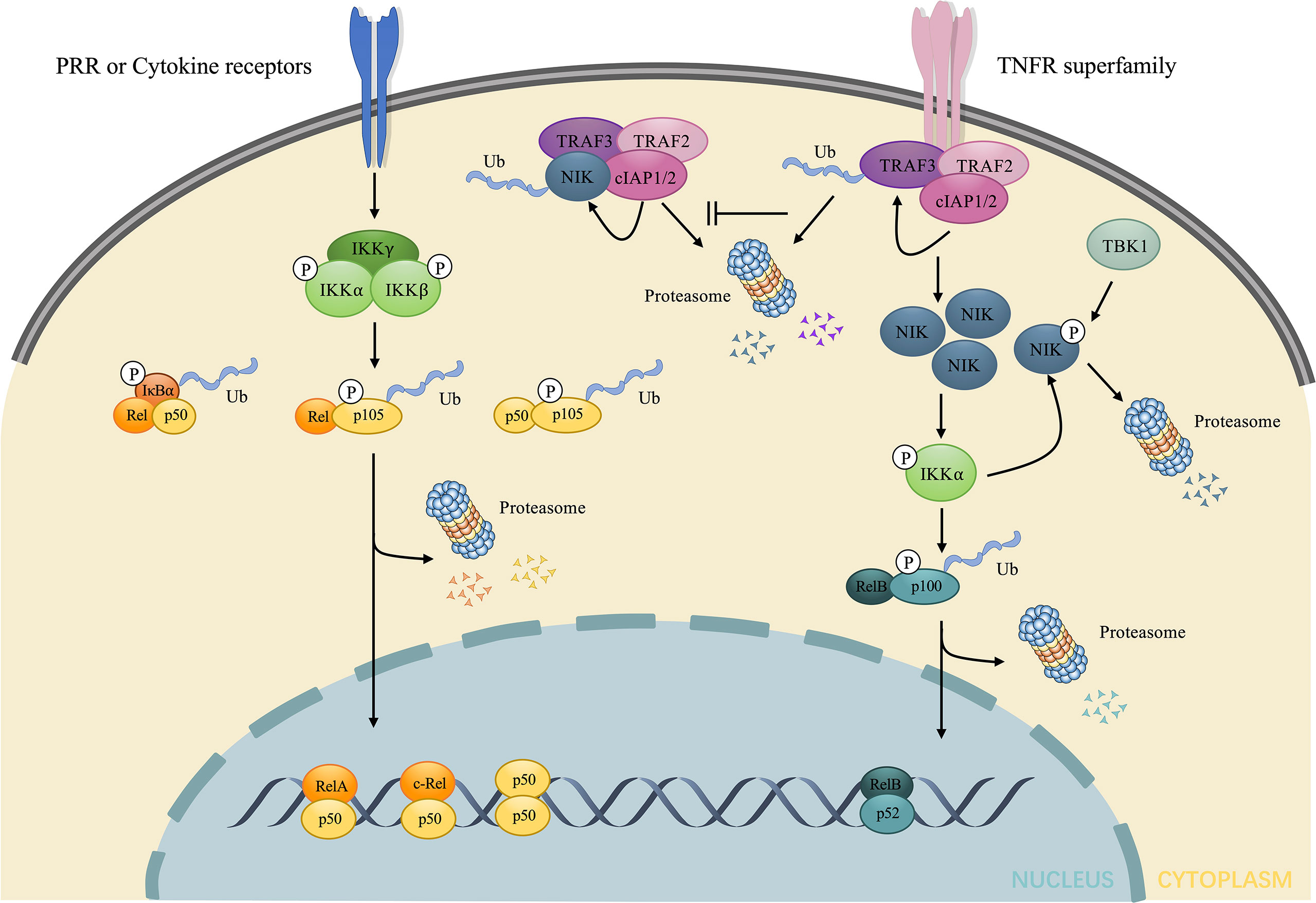
Figure 1 Brief illustration of the activation and regulation of the two major NF-κB signaling pathway. The canonical pathway is induced downstream of the stimulation of LPS and cytokines. Such stimuli mediate the activation of IKK, consisting of three subunits: IKKα, IKKβ, and IKKγ, which in turn phosphorylates and elicits the ubiquitin dependent processing of IκB and p105, leading to the nuclear translocation of RelA/p50, c-Rel/p50, p50/p50 dimers and the regulation of target gene expression. The non-canonical pathway is induced downstream of TNFR superfamily. In the absence of stimuli, NIK undergoes ubiquitin-dependent degradation mediated by NIK ubiquitin ligase, composed of TRAF3, TRAF2 and cIAP1/2. Under stimuli, TRAF3 undergoes ubiquitin-dependent degradation, which allows the accumulation of NIK. NIK directly phosphorylates and activates IKKα, contributing to the phosphorylation and ubiquitin dependent processing of p100 to p52. The nuclear accumulation of p52/RelB dimers changes transcriptional activity of target genes. The excessive NIK can also be evacuated via negative feedback mechanisms. IKKα can phosphorylate NIK, resulting in the direct proteasome-mediated degradation without the involvement of NIK ubiquitin ligase. TBK1 downstream of CD40 and BAFF also phosphorylates NIK and induces degradation. LPS, lipopolysaccharides; IKK, IκB kinase; TNFR, tumor necrosis factor receptor; NIK, NF-κB inducing kinase; TBK1, TANK-binding kinase 1; BAFF, B-cell activating factor belonging to the TNF family.
2.1 Canonical Pathway
Under numerous stimuli, such as ligands for cytokine receptors, NF-κB is activated via the canonical pathway. IκB kinase (IKK), consists of the catalytic subunits: IKKα, IKKβ, and a regulatory subunit: IKKγ, also known as NF-κB essential modulator (NEMO) (22, 23), which plays a key role in the canonical pathway by phosphorylating IκB, inducing its ubiquitination and proteasomal degradation. This process promotes the nuclear translocation of the NF-κB dimer, predominantly p50/RelA and p50/c-Rel, which enhances the transcription of proinflammatory cytokines.
2.2 Non-Canonical Pathway
Under the stimulation of certain receptors, such as the lymphotoxin-β receptor (LTβR) (8), TNFR superfamily, CD40 (24), and B-cell activating factor (BAFF) receptor (BAFFR) (25), the activation of NF-κB is triggered by a non-canonical pathway. NIK, as a prototypical activator of the non-canonical pathway (26) maintains a low level without stimuli as a result of ubiquitin-dependent degradation mediated by TNF receptor-associated factor-3 (TRAF3), which provides ubiquitination substrate binding sites and complexes with TRAF2 and cellular inhibitor of apoptosis1/2 (cIAP1/2) to form NIK ubiquitin ligase (27). However, the stimulation of certain receptors leads to the degradation of TRAF3 after ubiquitylated by cIAP1/2, thus contributing to the accumulation of NIK (28). Since NIK is degraded via NIK ubiquitin ligase upon synthesized, the accumulation of NIK takes time for de novo synthesis, which explains why the non-canonical pathway is slow and dependent on protein synthesis compared to the canonical pathway (29). Subsequently, NIK activates IKKα via phosphorylation and complexation with IKKα and p100 (30) to enhance the phosphorylation of p100 at Ser866 and Ser870 by IKKα without the help of IKKβ and IKKγ subunit (31). The phosphorylation of p100 creates sites that are bound by TrCP and induces the ubiquitination and proteasome limited degradation, not only producing mature p52 (NF-κB2) but also initiating nuclear translocation of p52 (32). The mature p52 prefers interacting with RelB. Consequently, the predominant NF-κB dimer in the non-canonical signaling is p52/RelB.
However, upon phosphorylated by NIK, IKKα will also phosphorylate NIK at Ser809, Ser812, and Ser815 (33), which disturbs the stability of NIK, resulting in direct proteasome mediated degradation independent of cIAP1/2 and ubiquitylation (34). TANK-binding kinase 1 (TBK1), induced by anti-CD40 and BAFF, also phosphorylates NIK at Ser862, which is located in the degradation-determination region, and thus triggers the degradation of excessive NIK without the involvement of NIK ubiquitin ligase (35). Unlike TRAF-cIAP ubiquitin E3 complex mediated physiological degradation of NIK in the unstimulated state, both pathways mentioned above show a negative feedback of the NIK axis after stimulation, aimed at inhibiting excessive stimulation and thus preventing immune disorders or oncogenesis. Impaired negative feedback of the non-canonical NF-κB signaling is associated with autoimmune diseases, such as systemic lupus erythematosus (SLE), and IBD (14, 36, 37) and the sustained NIK signaling is considered as an oncogenic event in diffuse large B-cell lymphoma (38). In conclusion, NIK is the central core of the non-canonical NF-κB signaling (Figure 1).
3 NIK in Intestinal Immunity and Homeostasis
3.1 NIK Modulates Adaptive Immunity
NIK is activated in several immune cells under the stimuli of microbial invasion, functioning as an indispensable component of adaptive immunity. It has been reported that NIK participates in the development of T cells (39), regelation of IgA secretion (7), microfold cells (M cells) maintenance (11) and B cells migration (40, 41). The role of the NIK in intestinal adaptive immunity includes different parts (Figure 2).
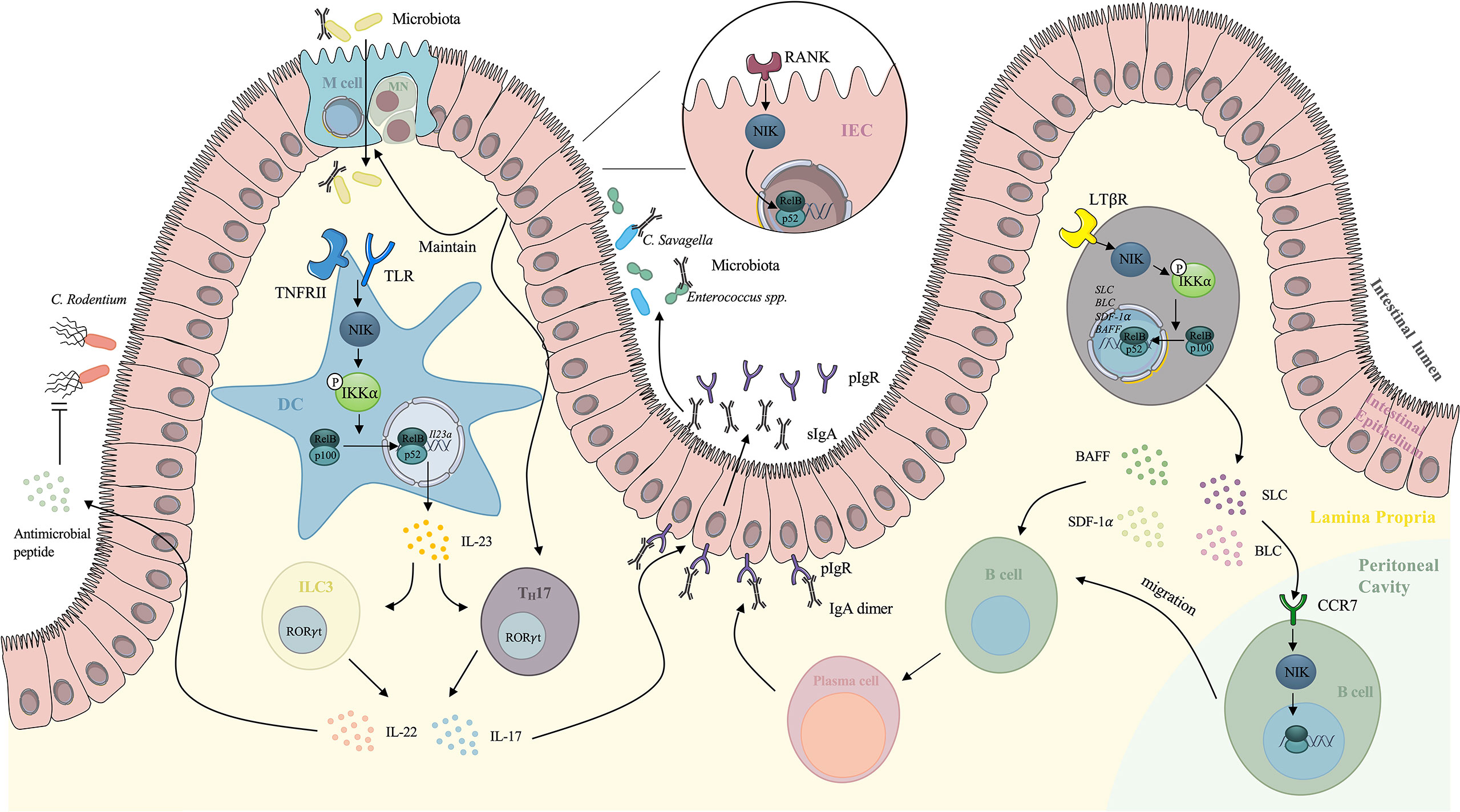
Figure 2 Mechanism of NIK signaling-mediated regulation of intestinal adaptive immunity. TLR, together with TNFRII induces NIK signaling in DCs. The nuclear translocation of RelB/p52 enhances the expression of IL-23, which maintains the TH17 cells and ILC3s. IL-17 secreted by TH17 cells and ILC3s stimulates the expression of pIgR on the basal surface of IECs, which increases the secretion of IgA to intestinal lumen. IL-22 secreted by TH17 cells and ILC3s also stimulates antimicrobial peptide against Citrobacter rodentium. NIK signaling downstream of RANK on IECs mediates the maintenance and differentiation of M cells, which facilitates the delivery of antigens to immune cells. NIK in IECs also induces IL-17 expression and IgA secretion, which enhances the intestinal immunotolerance. NIK signaling also modulates the migration of peritoneal cavity B cells to GALTs. Under the stimulation of SLC and BLC, NIK is activated in peritoneal cavity B cells and it mediates the migration. NIK participates in the downstream of LTβR in the intestinal stromal cells as well via upregulation of SDF-1 and BAFF, providing the microenvironment for B cells maturation. NIK also upregulates SLC and BLC, inducing B cells migration. NIK, NF-κB inducing kinase; DCs, dendritic cells; ILC3, type 3 innate lymphoid cells; IECs, intestinal epithelial cells; M cells, microfold cells; GALT, gut associated lymphoid tissue; SLC, secondary lymphoid tissue chemokine; BLC, B lymphocyte chemoattractant; BAFF, B-cell activating factor belonging to the TNF family.
3.1.1 NIK Facilitates T Cells Development in Peyer’s Patches and Intestinal Secretory IgA Production
Peyer’s Patches (PPs), as secondary lymphoid tissues (SLTs) of the intestine, play an essential role in maintaining the microenvironment and maturation of lymphocytes. NIK is believed to be indispensable for cell mediated immunity and SLTs formation (42). The NIK signaling downstream of LTβR plays an essential role in formation of PPs. Mice with disrupted genes encoding LTβR lack PPs (43). Yilmaz et al. indicated that relB-/- and nfkb2-/- mice showed rudimentary PPs, and they further validated that the NIK/IKKα/p52-RelB axis mediates the development of PPs downstream of LTβR in intestinal stromal cells (44). Recent studies have elucidated the mechanisms by which the NIK stimulates the maturation of T lymphocytes in PPs (7). Instead of directly functioning in T lymphocytes, the NIK induced non-canonical NF-κB signaling mediates IL-23 induction in DCs (6) when Toll-like receptors (TLRs) are activated. This in turn maintains TH17 cells and type 3 innate lymphoid cells (ILC3s), both of which have the ability to express the transcription factor RORt and secrete IL-17 and IL-22 and finally enhance the expression of polymeric immunoglobulin receptor (pIgR) in the intestinal epithelium, leading to an increase in IgA secretion into the intestinal lumen independent of microbiota (7). Cytokines involved in the trafficking play a significant role in regulating intestinal immunity and homeostasis. IL-22 and ILC3s induced by IL-23 are thought to participate in the early phase of host defense against Citrobacter rodentium (C. rodentium). However, the overstimulation of NIK in DCs contributes to the exacerbation of colitis, as a consequence of IL-17 overexpression. The contribution of the IL-23/IL-17 axis has been recognized in the pathogenesis of IBD, as IL-17 induced by IL-23 elicits the release of proinflammatory cytokines, such as IL-6 and IL-8, releasing in myofibroblasts and epithelial cells which causes the recruitment of neutrophils and epithelial cell injury (45).
3.1.2 NIK Maintains M Cell Function
Another player associated with the NIK signaling in the Peyer’s Patches is the M cell. M cells are specialized intestinal epithelial cell that mediates the internalization of dietary antigens and microbiota into the mucosa, initiating antigen-specific immune responses (46). Glycoprotein 2(GP2) expression on the apical surface of M cells helps the recognition of type 1 pilus-containing bacteria and the transcytosis of bacteria, which facilitates antigen recognition and processing by immune cells (47). IgA receptors on the apical surface of M cells also functions as receptors for IgA coated bacteria and mediate the engulfment (48). 1 integrin is also expressed in M cells, targeting Yersinia enterocolitica and inducing antigen internalization (49). Consequently, M cells play an essential role in regulating intestinal immunity and homeostasis and are related to inflammatory responses (50). Recent research has shown that the epithelial NIK signaling is involved in the M-cell maintenance, as Nik-/- mice exhibit loss of M cells in PPs (51). Ramakrishnan et al. demonstrated that epithelial RANKL mediates M-cell differentiation in duodenal and colon enteroids via the NIK signaling (11). Epithelial NIK signaling also elicits IL-17 and IgA which not only protect the intestine from colitis but also facilitate antigen uptake and processing by M-cells via IgA coating of commensal bacteria (11). All of these factors enhance the immunotolerance of the intestine and reduce inflammatory conditions, contributing to normal intestinal immunity and homeostasis. Moreover, increased levels of circulating IL-17 and IgA also protect against sepsis. However, prolonged stimulation of the NIK leads to chronic elevated IL-17 and IgA levels, resulting in intestinal injury as reviewed above. In addition, overexpression of NIK results in the ectopic expression of M cells, which worsens intestinal inflammation (52). Clinical data shows overstimulated NIK among patients with IBD, validating that uncontrolled stimulation of the NIK signaling mediates the intestinal inflammatory response (53). Consequently, intestinal homeostasis is maintained via the balanced activation of the NIK signaling.
3.1.3 NIK Augments Peritoneal Cavity Cell Migration
One of the classical in vivo models to investigate the NIK signaling is the alymphoplasia (aly) mice, carrying a point mutation in the gene encoding NIK. As a result, impaired lymphocyte function and stromal compartment can be observed in aly mice (54). A special feature is that aly mice have a higher B1/B2 cell ratio in the peritoneal cavity (PEC) than normal mice, suggesting that antigen-specific B cells in the lamina propria (LP) are derived from PEC cells (55). Frequent migration between PEC cells and gut-associated lymphoid tissue (GALT) exists, as half of the IgA plasma cells of intestinal LP were proven to be derived from PEC cells (55). Evidence also showed that under the stimulation of secondary lymphoid tissue chemokine (SLC), NIK signaling mediated the cell migration from PEC to GALT in LP. Fagarasan et al. confirmed that aly PEC cells have a lower migration rate and reduced chemotactic activity of SLC and B lymphocyte chemoattractant (BLC) either at rest or under the stimulation of lipopolysaccharides (LPS) (41). Furthermore, NIK also mediates the migration of PEC cells downstream of the SLC receptor. The stromal cells in Peyer’s Patches are involved in the migration of PEC cells. The NIK signaling also modulates the secretion of BLC and SLC to provide microenvironment for B cells migration and class switch (56). Dejardin and colleagues reported that the activated LTβR in stromal cells can elicit the NIK/IKKα axis, resulting in the processing of p100, a precursor of p52, which leads to the nuclear translocation of p52. After dimerized with RelB, p52 upregulates SLC, BLC, stromal cell derived factor-1 (SDF1-α), and BAFF (40). SLC and BLC mediate the migration of PECs to GALT. SDF1-α and BAFF provide the microenvironment for B cell maturation (57, 58), thus facilitating the migration of B cells. Kunisawa et al. examined cytokines secretion related to peritoneal B cell trafficking in aly stromal cells, and confirmed that the NIK signaling in stromal cells facilitated B cell emigration from the peritoneal cavity by enhancing the expression levels of VCAM-1 and ICAM-1 on stromal cells and regulating the balance of CXCL13 expression (59).
3.1.4 NIK Mediates Naïve B Cell Migration to Intestinal Lamina Propria
PPs function as secondary lymphoid tissues in the gut, where stromal cells provide microenvironment for B cells differentiation and homing. It has been proven that the majority of IgA plasma cells in LP are derived from PPs, and IgA+ B cells generated in PPs prefer to migrate to the intestinal LP (60). Suzuki et al. applied NIK deficient aly mice and showed that the NIK signaling downstream of LTβR in intestinal stromal cells mediates the naive B cells migration to intestinal LP. However, NIK signaling is not involved in the migration of B cells in PPs to LP. (61)
3.2 NIK Contributes to Innate Immunity
The intestinal epithelium contributes to the defense against pathogen invasion, and the microbiota lying on the surface of the intestinal lumen interacts with the epithelium, both of which contribute to intestinal innate immunity. Here, we demonstrate concrete mechanisms of intestinal innate immunity driven by the NIK (Figure 3).
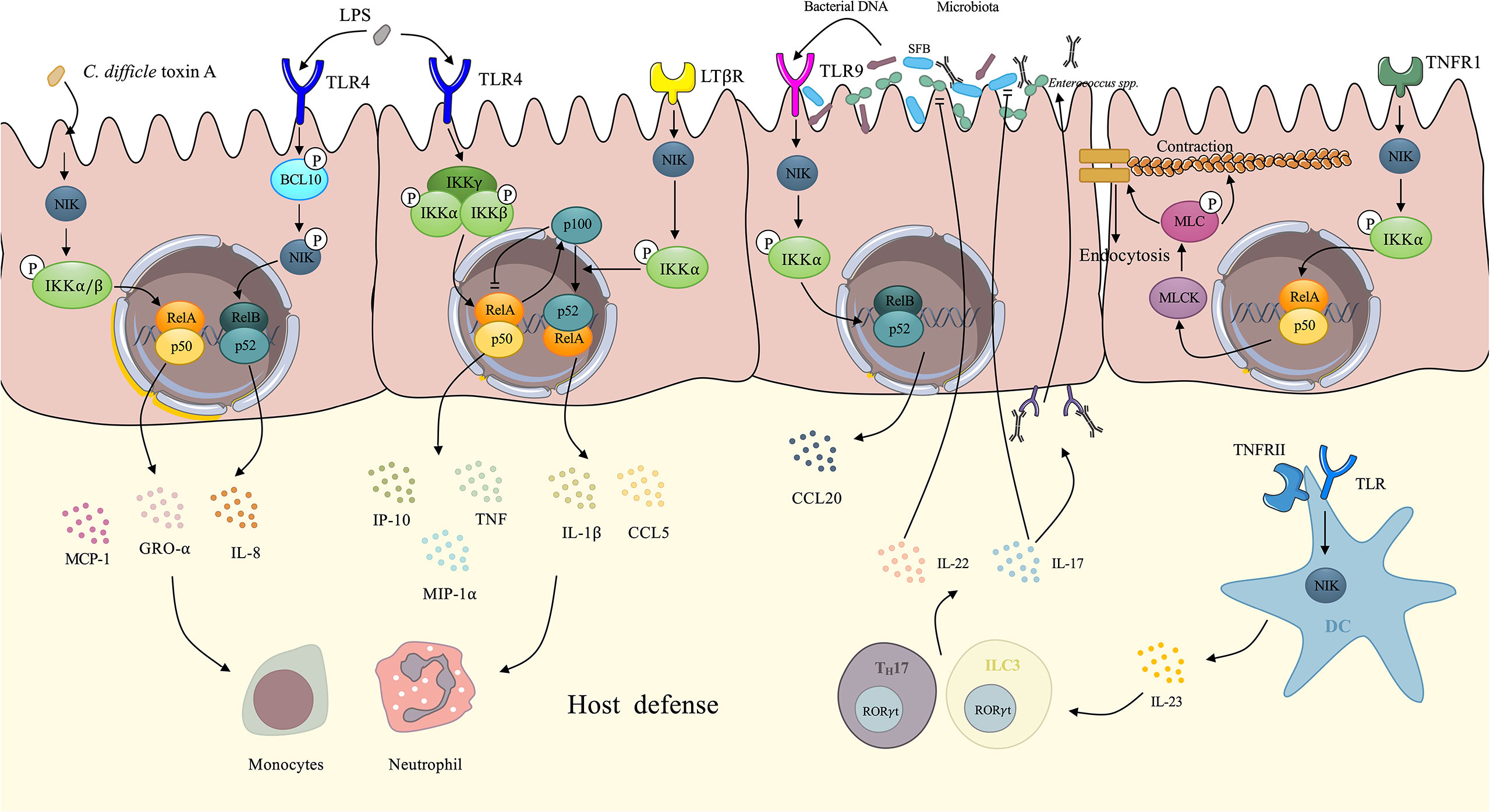
Figure 3 Demonstration of NIK regulates intestinal innate immunity. IECs upregulate several proinflammatory cytokines via NIK signaling. Upon the stimulation of Clostridium difficile toxin A, IECs activates the NIK/IKKα axis, resulting in the nuclear translocation of RelA/p50, which facilitates the expression of IL-8, GRO-α, and MCP-1. Under the stimulation of LPS, TLR4 on the IECs activates NIK by phosphorylation of BCL-10, which facilitates the nuclear translocation of RelB/p52 and enhances the expression of IL-8. The canonical IKK complex also mediates the signaling transduction downstream of TLR4, eliciting transcription of TNF, GRO-α, and MCP-1. In the meantime, RelA induces the expression of p100 as negative feedback to cease canonical NF-κB signaling. However, p100 can be utilized by NIK signaling downstream of LTβR, not only evacuating the inhibitory effect of p100, but also promoting the processing of p100 to p52. After dimerized with RelB, p52 upregulates IL-1 and CCL5. These proinflammatory cytokines initiates host defense via recruitment of neutrophils and monocytes. TNF has the ability to increase intestinal tight junction permeability through the activation of NIK/IKKα axis, which induces the nuclear translocation of RelA/p50, and activates MLCK/MLC axis, contributing to the loss of the tight junction proteins: occludin and claudin-1. Finally, the contraction of actomyosin filament leads to the opening of tight junction. Increased secretion of IgA induced by NIK signaling in DCs changes the microbiota. With IgA coating, evident downregulation of Enterococci and SFB can be observed. IL-22 and IL-17 induced by NIK signaling in DCs also has an inhibitory effect on Enterococci and SFB dysbiosis respectively. The microbiota DNA also stimulates TLR9 on IECs induces CCL20 secretion via NIK signaling. NIK, NF-κB inducing kinase; IECs, intestinal epithelial cells; DCs, dendritic cells; TNF, tumor necrosis factor; SFB, segmented filamentous bacteria.
3.2.1 NIK Mediates Cytokine Release in the Intestinal Epithelium
The intestinal epithelium is a single layer of cells lining on the intestinal lumen, functioning as a barrier against pathogens and a coordinating hub for immune defense events such as cytokine trafficking (62). IECs are reported to upregulate several cytokines under the stimulation of bacterial toxin. Research has also related the NIK to the bacterial toxin-induced expression of cytokines by IECs.
Kim et al. elucidated that the NIK signaling mediates Clostridium difficile (C. difficile) toxin A-induced cytokines expression by IECs (63). Upon the stimulation of C. difficile toxin A, phosphorylated NIK activates IKKα/β. IKKα and IKKβ then directly phosphorylate serine residues on IκB, resulting in the degradation of IκB and nuclear translocation of the NF-κB dimer, p65/p50 and p65/p65. Proinflammatory cytokines such as IL-8, growth regulated protein-α(GRO-α), and monocyte chemoattractant protein-1(MCP-1) are upregulated by transcription factor p65/p50 and p65/p65 dimers, contributing to the innate immunity of the intestinal tract (64). The engagement of non-canonical NF-κB in mediating LPS-induced immune response is well recognized (65). It has been reported that the NIK mediates intestinal innate immunity by transducing signals downstream TLR4 induced by LPS in IECs. After activation by LPS, TLR4 transduces signals by phosphorylating B cell lymphoma/leukemia (BCL)-10 at Ser138, which phosphorylates NIK without changing the level of NIK. This further leads to the activation of IKKα and nuclear translocation of the RelB-p52 dimer and subsequent induction of IL-8 secretion and inflammatory responses (66). Banoth et al. revealed that IEC LTβR induced NIK/IKKα signaling provided a co-stimulatory signaling to sustain canonical RelA/NF-κB signaling by TLR4 in response to C. rodentium (67). Researchers has shown that activation of the NIK/IKKα signaling downstream of LTβR not only led to a rapid augmentation of canonical NF-κB targeted pro-inflammatory genes, including TNF, interferon inducible protein-10 (IP-10), and macrophage inflammatory protein-1α (MIP-1α), but also sustained the prolonged accumulation of IL-1 and C-C chemokine ligand 5 (CCL5) downstream of TLR4 after activation by LPS (67). The canonical NF-κB component, RelA, elicits the expression of p100 as negative feedback by utilizing its inhibitory domain to terminate sustained activation (68) downstream of TLR4. However, p100 also dimerizes with RelB as a precursor of non-canonical NF-κB (69), which is utilized by the NIK signaling downstream of LTβR, preventing the inhibitory effect of p100 as well as enhancing the non-canonical NF-κB signaling and finally forming a positive loop to maintain proinflammatory responses to counter bacteria (67). In agreement with previous findings, a recent publication reported that non-canonical NF-κB signaling enhanced canonical RelA mediated inflammatory responses in IECs and exacerbated colitis (70). They also revealed an upregulated non-canonical NF-κB signaling in IBD patients, indicating that uncontrolled IEC-specific NIK signaling involves in the pathogenesis and progression of IBD (70). In physiological state, intestinal inflammatory response is self-limiting rather than fulminant due to the negative regulatory function of NIK signaling. It has been reported that IKKα induced by NIK participates in the negative regulation of inflammation via its kinase property (71, 72). Lawrence et al. elucidated that IKKα accelerated canonical c-Rel and RelA turnover and dissociation from promoters of pro-inflammatory genes in macrophages through direct phosphorylation (72). IKKα also negatively regulates apoptosis-associated specklike protein containing a CARD (ASC) through phosphorylating at Ser193 and Ser16 and interferes with the assembly of nod-like receptor P3 (NLRP3) inflammasome (71). These negative regulation mechanisms in NIK plays an essential role in the reduction of uncontrolled inflammation and the maintenance of intestinal homeostasis. These findings not only elucidated the correlations between the NIK signaling and IEC-mediated innate immunity, but also revealed the crosstalk between non-canonical and canonical NF-κB. (Figure 3). These findings seem contradictory that NIK signaling not only induces intestinal inflammation but also negatively regulates inflammatory responses. Actually, NIK plays its role in regulating intestinal homeostasis in a cell-specific way. Under the stress of pathogen invasion, NIK signaling in IECs promotes adaptive immunity against pathogens while macrophages inhibits excessive inflammation through NIK signaling to protect intestinal tissue from damage (72).
3.2.2 NIK Modulates Intestinal Tight Junction
The intestinal tight junction (TJ) plays a significant role in gut immunity and homeostasis (73). Evidence has shown that paracellular permeation of intestinal antigens mediated by defective TJs can induce or propagate inflammatory responses. Intestinal TJ permeability which is considered an etiological factor in CD, is significantly increased in patients with CD (74). Studies have shown that proinflammatory cytokines [e.g., TNF-α, IL-1 (75), IFN-γ (76)] are related to increased intestinal TJ permeability. TNF- α and IL-1 modulates intestinal TJ by enhancing the expression of myosin light chain kinase (MLCK) (77, 78). TNF-α also increases epithelial cell apoptosis (79), whereas IFN-γ manipulates TJ permeability via micropinocytosis of tight junction proteins, such as occludin, and claudin-1 instead of inducing apoptosis (80). Clinical research has also shown that an increase in intestinal TJ permeability induced by TNF-α can be observed in patients with IBD (81), and that IL-1 is also elevated in the intestinal tissues of patients with CD (82). Recently, Al-Sadi et al. defined the NIK signaling as being involved in TNF- α modulation of intestinal TJ permeability, both in vitro and in vivo (83). Research has shown that TNF causes an increase in phosphorylated NIK, and induces the phosphorylation of IKKα at Ser176 without the involvement of IKKα. Surprisingly, NIK signaling is mediated by the canonical pathway NF-κB, p50-RelA dimer, which finally results in the activation of MLCK gene and thus increasing the TJ permeability (83). MLCK was shown to catalyze the phosphorylation of MLC, contributing to the activation of Mg2+–Myosin ATPase, which finally results in the contraction of the peri-junctional actomyosin filament, thus leading to the tension-induced opening of the TJ barrier and the increased permeability (78). However, IL-1-induced increase in intestinal TJ permeability is mediated by MEKK-1 pathway, rather than the NIK signaling. Recent research confirms that IL-1 induced MEKK-1/IKKα canonical NF-κB pathway, and the activation of MLCK gene, without the involvement of NIK (84). In conclusion, TNF-α induced the modulation of MCLK and thus the increased intestinal TJ permeability was mediated by NIK/IKKα axis activated NF-κB p50/RelA dimer, while IL-1 achieves the same property via the NIK-independent canonical pathway. These findings indicate that the selectivity of canonical or non-canonical pathways targeting NF-κB is mediated by different cytokines and ultimately achieves the same physiological process: increase in intestinal TJ permeability (Figure 3).
3.2.3 NIK Rescues Intestinal Epithelium Injury
NIK signaling is activated upon IECs detachment to reduce apoptosis and maintain intestinal homeostasis. It has been reported that IL-1, IL-1R type II and IL-6 is upregulated upon IEC detachment (85, 86). Yan et al. revealed that IECs detachment activated NIK and increased IKKα phosphorylation, resulting in the activation and nuclear translocation of p65. The p65 activation induced by NIK signaling resulted in induction of IL-1, IL-1R type II and IL-6 and reduced caspase activation and apoptosis (87). Moreover, NIK signaling is activated to alleviate intestinal injuries by facilitating regeneration (88). Tumor progression locus-2 (Tpl2), a mitogen-activated protein kinase kinase kinase 8 (MAP3K8) activates NIK signaling through phosphorylation (89). Roulis et al. demonstrated an intestinal myofibroblast (IMF)-specific pathway mediated by Tpl2 maintained intestinal homeostasis via promoting epithelium regeneration under injuries (88). IMF sensed the penetrated bacteria via TLR4. Tpl2 interacted with p105 and mediated the phosphorylation of ERK downstream of TLR4, which enhanced the expression of prostaglandin E2 (PGE2) by inducing enzymatic activity of cyclooxygenase-2 (88). PGE2 plays an essential role in intestinal proliferation by sustaining Wnt signaling of stem cells, which rescued the injured intestinal epithelium (90). As a result, reduced level of Tpl2 is associated with disturbance of intestinal homeostasis and IBD pathogenesis (91).
3.3 NIK Maintains Intestinal Regulatory Microenvironment
Intestinal homeostasis is the result of balance of pro-inflammatory and regulatory anti-inflammatory immune cells. Consequently, the imbalance of TH17 cells and Tregs has been considered as the pathogenic mechanism of CD (92). The regulatory microenvironment of intestine is maintained by IECs (93), CD103+ suppressor migration DCs (94) and different populations of Tregs and supported by tissue-resident macrophages (95). Multiple studies have shown that NIK signaling sustains intestinal regulatory microenvironment. Previous studies confirmed that the homeostasis of peripheral Tregs is mediated by NIK signaling since overexpression of NIK increases Tregs function (12). Researchers also found that conditional depletion of nfkb2 in Tregs led to increasing number of peripheral Tregs with impaired suppressive capacity. As the result, Tregs-specific nfkb2-/- mice displayed localized immune infiltrations in the colons with increased CD4+ and CD8+ T cells expressing IFN-γ, while additional depletion of RelB reversed the phenotype (96). Further mechanism study showed that downstream of T cell receptor (TCR), increased p100 synthesis had an inhibitory effect of RelB. However, non-canonical NF-κB signaling downstream of OX40 or GITR contributed to the processing of p100 to p52 which enhanced nuclear translocation of RelB, resulting in the impaired suppressive function of Tregs (96). nfkb2-/- mice showed massive inflammation in colons, with other tissues intact, for example lungs, kidney and liver. Depletion of nfkb2 in T cells selectively affected the peripheral Tregs without evident effects on Tregs generated in thymus, indicating that NIK signaling has specific effect on peripheral Tregs (96). In addition to Tregs-intrinsic role, NIK is involved in the maintenance of Tregs through modulating the secretion of IL-10. Serebrennikova et al. revealed that deletion of Tpl2, a MAP3K8 which regulated NIK and IKKα by phosphorylation led to impaired IL-10 expression of macrophages and DCs via inhibition of mTOR/Stat3 signaling downstream of TLRs. Consequently, peripheral Foxp3+ inducible Tregs failed to achieve suppressive function without sufficient IL-10, and shaped a pro-inflammatory microenvironment of intestinal mucosal and accelerated intestinal inflammation and oncogenesis (97).
Several researches showed that NIK signaling mediates the suppressive function of DCs through inducing expression of a key enzyme, indoleamine 2,3-dioxygenase (IDO) (13). Non-canonical NF-κB signaling mediated by NIK and IKKα promotes the expression of IDO in DCs downstream of CD40, which promotes the differentiation of T cells with regulatory properties (98). Under sustained exposure to LPS, increased level of IDO can be found in DCs, which leads to overexpression of immunoregulatory molecules, including programmed death ligand 1 (PD-L1), PD-L2, and IL-10 and finally induces DCs with tolerogenic phenotype to alleviate tissue damage or allergic reaction caused by prolonged inflammatory response. Since the interaction between RelB and kynurenine, a metabolite of IDO, DCs favors NIK signaling rather than canonical NF-κB signaling downstream of TLR4 (99). Consequently, the involvement of NIK signaling in endotoxin tolerance is mediated by interaction with IDO signaling. Yu et al. elucidated that RelB/p52 directly bound to IDO gene promoter in myeloid-derived suppressor cells of breast cancer microenvironment mediated by NIK signaling downstream of STAT3 activation (100). That is, NIK signaling is involved in the activation of IDO, which plays a central role in the suppression of immune responses. Far deeper mechanisms of interactions between NIK and IDO in shaping the intestinal regulatory immune microenvironment should be deciphered.
NIK signaling is a crucial mediator of intestinal immune tolerance. Nik-/-, relb-/-, and nfkb2-/- mice spontaneously develop autoimmunity (101). NIK essentially modulates immune regulation in a tissue-specific way. Andreas et al. confirmed that RelB deletion in DCs contributed to accumulation of tissue Tregs and showed a protective role in autoimmune encephalomyelitis (102). However, DC-specific deletion of RelB reduced intestinal RORt+ Tregs leading to defective tolerance of microbiome and oral antigens (102). In addition, DC-specific deletion of TRAF6, an upstream regulator of canonical NF-κB impairs intestinal Tregs differentiation, resulting in defective tolerance and aberrant type 2 allergic response after oral antigen uptake (103).
3.4 NIK and Microbiota
Gut microbiota are a large group of commensal bacteria that occur in the gastrointestinal tract, do not exhibit pathogenicity, and modulate intestinal immunity and homeostasis (10). The number of commensal bacteria is ten-fold more than the cells in the human body. Microbiota interacts with innate immune system, not only by creating a protective barrier on the intestinal lumen, thus preventing adhesion of pathogenic bacteria, but also by stimulating the secretion of certain chemokines or cytokines to maintain intestinal immunity (104). Currently, NIK signaling is associated with the cytokine secretion induced by commensal bacteria. Dutta et al. reported that both commensal and pathogenic bacterial DNA-induced CCL20 secretion by TLR9 in colonic epithelial cells is mediated by both NIK/IKKα/p100 (NF-κB2) phosphorylation and the MEK/ERK pathway (105). However, there was a temporal difference between these two pathways. The MEK/ERK signaling axis induces the transcription factor AP1 (cjun/cfos), which predominantly participates in the early stage of CCL20 gene transcription, while the NIK/IKKα signaling axis induces non-canonical NF-κB2 (p52/RelB), which is predominantly involved in the late phase of CCL20 expression downstream of TLR9 stimulated by bacterial DNA. CCL20 is a chemokine with antimicrobial effects against Staphylococcus aureus (S. aureus) and Escherichia (E. coli) (106) and modulates the trafficking of several immune cells such as DCs, T cells, and B cells by stimulating C-C chemokine receptor (CCR) 6 (107). Jie et al. demonstrated altered intestinal microbiota, with significantly elevated Enterococci and segmented filamentous bacteria (SFB), for example, C. savagella in mice with DC specific depletion of NIK (7). The overexpression of Enterococci leads to opportunistic infection (108), and SFB stimulates immune cell activation and differentiation, which may exacerbate autoimmune responses (109). It was further illustrated that the IL-23/IL-17, IL-22/IgA axis induced by the NIK signaling pathway in DCs contributes to the decreased abundance of Enterococci and SFB, in agreement with recent research showing that IL-22 derived from ILCs inhibits Enterococci expansion (110). IL-17 regulates the abundance of SFB, which maintains the intestinal homeostasis and protects the intestine from inflammatory responses (Figure 3) (111). NIK participates in the induction of secretory IgA in both DCs and intestinal epithelial cells downstream of TLR and RANK, respectively (7, 11). Secretary IgA provides a coating for microbiota and prevents the accumulation of opportunistic pathogenic microbes under certain conditions, thus exhibiting a protective role in infection and inflammation (112). Consequently, the loss of NIK is related to increased susceptibility to colitis (11). However, sustained activation of NIK leads to high IgA coating bacteria which causes a shift to colitogenic dysbiosis and exacerbate inflammatory responses in patients with UC and CD (112). Increased level of IgA levels lead to IgA-coated microbiota enrichment, which promote the TH17 dependent local or systemic inflammation, worsening peripheral spondylarthritis, an extraintestinal manifestation of CD (113).
3.5 NF-κB Independent Roles for NIK in Intestinal Homeostasis
Several mechanisms has also been reported that NIK signaling exerts its function in maintaining intestinal homeostasis without the involvement of NF-κB. As reviewed above, IKKα, a kinase induced by NIK interferes with NLRP3 inflammasome assembly by phosphorylating ASC rather than activating RelB and p52, thereby influencing intestinal homeostasis and immunity (71). It is reported that NIK phosphorylated receptor-interacting protein kinase 1 (RIP1) and induced tissue destruction downstream of TNFR1 independent of NF-κB (114). Jung et al. revealed that overstimulated NIK induced the fission of mitochondria and cell invasion by regulation of DRP1 phosphorylation in the absence of IKKα and NF-κB, which emerges as a novel mechanism of oncogenesis (115). Upregulated NIK signaling is also observed in IBD patients (70). Combined together, these studies provide us with a possible mechanism of inflammation-cancer transition in IBD patients.
4 Role of NIK Signaling in the Pathogenesis of IBD
We have reviewed the role of NIK signaling in intestinal immunity and homeostasis. In this part, we will summarize mechanisms of aberrant NIK signaling in the pathogenesis of IBD through breaking intestinal homeostasis. On the one hand, upregulated non-canonical NF-κB signaling is observed in IBD patients (70). Overstimulated NIK signaling in DCs and M cells leads to increased IL-17 secretion which plays a central role in intestinal injury and exacerbate colitis (7, 11). Sustained activation of NIK also shapes microbiome containing high IgA coating bacteria which causes a shift to colitogenic dysbiosis and exacerbate inflammatory responses in patients with UC and CD (112). Non-canonical NF-κB signaling is reported to enhance canonical RelA mediated inflammatory responses in IECs and exacerbated colitis via crosstalk mechanisms (70). On the other hand, NIK signaling participates in the maintenance of regulatory microenvironment and intestinal tissue repair. As a result, lack of NIK signaling also contributes to the pathogenesis of IBD as well. Consequently, it has been shown that Tpl2, as an activator of NIK has significantly reduced level in IBD patients (91).
5 Perspectives
Currently, NIK, as the pivotal component of the non-canonical NF-κB pathway, integrates significant physiological event such as cytokine trafficking, survival signaling, and apoptosis, and has been confirmed to maintain intestinal innate and adaptive immune response of the intestinal tract. Depletion of NIK leads to impaired PP and inadequate T cell development (44). However, overstimulation of the NIK signaling pathway also contributes to the disturbance of homeostasis, leading to prolonged inflammation, which is associated with IBD pathogenesis (11). In addition, the pathogenesis and progression of several autoimmune disease are also related to NIK overexpression (116, 117). NIK inhibitors have also been designed as novel therapy for SLE (118). It is the balanced activation of NIK that maintains intestinal immunity and homeostasis. Consequently, optimizing NIK to within the normal range is a promising area for pharmacological discovery.
There are also questions that remain to be resolved regarding the NIK signaling pathway. The upregulation of the non-canonical NF-κB pathway has been reported in patients with IBD treated with anti-TNF antibody, which is associated with regression of drug efficacy (53). Patients resistant to anti-TNF treatment showed a significant upregulation of NIK and downregulation of NLRP12, a negative regulator of both canonical and non-canonical NF-κB pathways (53). Consequently, whether recent targeted therapies for IBD such as TNF-α antibody, have an effect on the NIK signaling, how the NIK signaling pathway mediates the resistance in the novel medication of IBD, and what could be the salvage of resistance to targeted therapy of IBD become major problems to be resolved.
The canonical NF-κB pathway is overstressed in mediating the response to anti-TNF therapies such as infliximab and adalimumab (119). However, an aberrant non-canonical pathway was demonstrated in patients resistant to anti-TNF treatment (53), combined with what we reviewed above, suggesting the involvement of NIK in drug resistance. Consequently, resistance to drug therapy may be mediated by such crosstalk mechanisms. Savinova et al. reported a crosstalk mechanism between the canonical and non-canonical pathways mediated by NIK (68). Sustained canonical NF-κB signaling activation leads to negative feedback that RelA induces the production of p100, which contains an inhibitory domain of RelA and functions as an IκB protein (120). However, p100 also functions as the precursor of non-canonical NF-κB, and is further processed to p52 mediated by NIK. Consequently, cooling of the canonical NF-κB pathway is possible to reverse downregulation through the NIK signaling via crosstalk mechanisms. The crosstalk mechanism also reminds investigators to use NIK inhibitors to reverse anti-TNF therapy resistance. Unfortunately, the crosstalk mechanism between the two pathways has not yet been fully understood and requires further investigation.
6 Conclusion
NIK is the central core of non-canonical NF-κB signaling and participates in the intestinal immunity and homeostasis. Both excessive and impaired NIK signaling cause the disturbance of intestinal immunity and homeostasis. NIK signaling regulates intestinal homeostasis in a spatiotemporal heterogeneous way. NIK signaling in different tissues and cells plays different roles even contradictory roles. Different activation pattern of NIK signaling can also be seen at early and late phase response to pathogens. Advanced knowledge of the NIK in the regulation of intestinal immunity and homeostasis can provide new perspectives on inflammatory diseases. NIK is closely related to the pathogenesis and drug resistance of IBD, and is considered as a novel target for treatment of IBD and carcinoma in gastrointestinal tract. Consequently, more sophisticated molecular mechanisms of NIK signaling in IBD are urgently needed.
Author Contributions
BW drafted the article and JS revised it critically for intellectual content. All authors read and approved the final manuscript.
Funding
Supported by grants from Shanghai Science and Technology Innovation Initiative (21SQBS02302), Cultivated Funding for Clinical Research Innovation, Renji Hospital, School of Medicine, Shanghai Jiaotong University (RJPY-LX-004) and National Natural Science Foundation of China (No. 81770545).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
IBD, Inflammatory bowel disease; UC, Ulcerative colitis; CD, Crohn’s disease; NF-κB, Nuclear factor-κB; NIK, NF-κB inducing kinase; IκB, Inhibitors of NF-κB; IKK, IκB kinase; NEMO, NF-κB essential modulator; LTβR, Lymphotoxin-β receptor; TNF, Tumor necrosis factor; TNFR, Tumor necrosis factor receptor; TAD, Transcription activation domain; RHD, Rel homology domain; CBP, CREB binding protein; HADC3, Histone deacetylases 3; TRAF, TNF receptor-associated factor; cIAP1/2, Cellular inhibitor of apoptosis1/2; TBK1, TANK-binding kinase 1; M cells, Microfold cells; DCs, Dendritic cells; TLR, Toll-like receptor; ILC3, Type 3 innate lymphoid cells; IECs, Intestinal epithelial cells; pIgR, Polymeric immunoglobulin receptor; GP2, Glycoprotein 2; aly, Alymphoplasia; PEC, Peritoneal cavity; LP, Lamina propria; GALT, Gut associated lymphoid tissue; SLC, Secondary lymphoid tissue chemokine; BLC, B lymphocyte chemoattractant; BAFF, B-cell activating factor; LPS, Lipopolysaccharide; SDF1-α Stromal cell derived factor-1α; GRO-α, Growth regulated protein-α; MCP-1, Monocyte chemoattractant protein-1; BCL, B cell lymphoma/leukemia; MIP-1α, Macrophage inflammatory protein-1α; CCL, C-C chemokine ligand; ASC, Apoptosis-associated specklike protein containing a CARD; NLRP3, Nod-like receptor P3; TJ, Tight junction; Tpl2, Tumor progression locus-2; MAP3K8, Mitogen-activated protein kinase kinase kinase 8; IMF, Intestinal myofibroblast; PGE2, Prostaglandin E2; Tregs, Regulatory T cells; TCR, T cell receptor; IDO, Indoleamine 2,3-dioxygenase; PD-L1, Programmed death ligand 1; MLCK, Myosin light chain kinase; CCR, C-C chemokine receptor; RIP1, Receptor-interacting protein kinase 1.
References
1. Mowat AM, Agace WW. Regional Specialization Within the Intestinal Immune System. Nat Rev Immunol (2014) 14(10):667–85. doi: 10.1038/nri3738
2. Gomaa EZ. Human Gut Microbiota/Microbiome in Health and Diseases: A Review. Antonie Van Leeuwenhoek (2020) 113(12):2019–40. doi: 10.1007/s10482-020-01474-7
3. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic Endotoxemia Initiates Obesity and Insulin Resistance. Diabetes (2007) 56(7):1761–72. doi: 10.2337/db06-1491
4. Sun SC. The Noncanonical NF-kappaB Pathway. Immunol Rev (2012) 246(1):125–40. doi: 10.1111/j.1600-065X.2011.01088.x
5. Sun SC. The non-Canonical NF-kappaB Pathway in Immunity and Inflammation. Nat Rev Immunol (2017) 17(9):545–58. doi: 10.1038/nri.2017.52
6. Hofmann J, Mair F, Greter M, Schmidt-Supprian M, Becher B. NIK Signaling in Dendritic Cells But Not in T Cells is Required for the Development of Effector T Cells and Cell-Mediated Immune Responses. J Exp Med (2011) 208(9):1917–29. doi: 10.1084/jem.20110128
7. Jie Z, Yang JY, Gu M, Wang H, Xie X, Li Y, et al. NIK Signaling Axis Regulates Dendritic Cell Function in Intestinal Immunity and Homeostasis. Nat Immunol (2018) 19(11):1224–35. doi: 10.1038/s41590-018-0206-z
8. Lan J, Heneghan AF, Sano Y, Jonker MA, Omata J, Xu W, et al. Parenteral Nutrition Impairs Lymphotoxin Beta Receptor Signaling via NF-Kappab. Ann Surg (2011) 253(5):996–1003. doi: 10.1097/SLA.0b013e31821224eb
9. Russo MP, Schwabe RF, Sartor RB, Jobin C. NF-kappaB-Inducing Kinase Restores Defective IkappaB Kinase Activity and NF-kappaB Signaling in Intestinal Epithelial Cells. Cell Signal (2004) 16(6):741–50. doi: 10.1016/j.cellsig.2003.11.007
10. Yousefi B, Eslami M, Ghasemian A, Kokhaei P, Salek Farrokhi A, Darabi N. Probiotics Importance and Their Immunomodulatory Properties. J Cell Physiol (2019) 234(6):8008–18. doi: 10.1002/jcp.27559
11. Ramakrishnan SK, Zhang H, Ma X, Jung I, Schwartz AJ, Triner D, et al. Intestinal non-Canonical NFkappaB Signaling Shapes the Local and Systemic Immune Response. Nat Commun (2019) 10(1):660. doi: 10.1038/s41467-019-08581-8
12. Chen X, Willette-Brown J, Wu X, Hu Y, Howard OM, Hu Y, et al. Ikkα is Required for the Homeostasis of Regulatory T Cells and for the Expansion of Both Regulatory and Effector CD4 T Cells. FASEB J (2015) 29(2):443–54. doi: 10.1096/fj.14-259564
13. Mellor AL, Munn DH. IDO Expression by Dendritic Cells: Tolerance and Tryptophan Catabolism. Nat Rev Immunol (2004) 4(10):762–74. doi: 10.1038/nri1457
14. Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, et al. NLRP12 Suppresses Colon Inflammation and Tumorigenesis Through the Negative Regulation of Noncanonical NF-κb Signaling. Immunity (2012) 36(5):742–54. doi: 10.1016/j.immuni.2012.03.012
15. Zhang J, Webster JD, Dugger DL, Goncharov T, Roose-Girma M, Hung J, et al. Ubiquitin Ligases Ciap1 and Ciap2 Limit Cell Death to Prevent Inflammation. Cell Rep (2019) 27(9):2679–2689 e2673. doi: 10.1016/j.celrep.2019.04.111
16. Shen HM, Tergaonkar V. NFkappaB Signaling in Carcinogenesis and as a Potential Molecular Target for Cancer Therapy. Apoptosis (2009) 14(4):348–63. doi: 10.1007/s10495-009-0315-0
17. Bartuzi P, Hofker MH, van de Sluis B. Tuning NF-κb Activity: A Touch of COMMD Proteins. Biochim Biophys Acta (2013) 1832(12):2315–21. doi: 10.1016/j.bbadis.2013.09.014
18. Chen LF, Williams SA, Mu Y, Nakano H, Duerr JM, Buckbinder L, et al. NF-kappaB RelA Phosphorylation Regulates RelA Acetylation. Mol Cell Biol (2005) 25(18):7966–75. doi: 10.1128/MCB.25.18.7966-7975.2005
19. Hoberg JE, Popko AE, Ramsey CS, Mayo MW. IkappaB Kinase Alpha-Mediated Derepression of SMRT Potentiates Acetylation of RelA/p65 by P300. Mol Cell Biol (2006) 26(2):457–71. doi: 10.1128/MCB.26.2.457-471.2006
20. Beinke S, Ley SC. Functions of NF-Kappab1 and NF-Kappab2 in Immune Cell Biology. Biochem J (2004) 382(Pt 2):393–409. doi: 10.1042/BJ20040544
21. Oh H, Ghosh S. NF-Kappab: Roles and Regulation in Different CD4(+) T-Cell Subsets. Immunol Rev (2013) 252(1):41–51. doi: 10.1111/imr.12033
22. Häcker H, Karin M. Regulation and Function of IKK and IKK-Related Kinases. Sci STKE (2006) 357):re13. doi: 10.1126/stke.3572006re13
23. Sethi G, Sung B, Aggarwal BB. Nuclear factor-kappaB Activation: From Bench to Bedside. Exp Biol Med (Maywood) (2008) 233(1):21–31. doi: 10.3181/0707-MR-196
24. Zhang S, Breidenbach JD, Russell BH, George J, Haller ST. CD40/CD40L Signaling as a Promising Therapeutic Target for the Treatment of Renal Disease. J Clin Med (2020) 9(11):3653. doi: 10.3390/jcm9113653
25. Morrison MD, Reiley W, Zhang M, Sun SC. An Atypical Tumor Necrosis Factor (TNF) Receptor-Associated Factor-Binding Motif of B Cell-Activating Factor Belonging to the TNF Family (BAFF) Receptor Mediates Induction of the Noncanonical NF-kappaB Signaling Pathway. J Biol Chem (2005) 280(11):10018–24. doi: 10.1074/jbc.M413634200
26. Sun SC. Controlling the Fate of NIK: A Central Stage in Noncanonical NF-kappaB Signaling. Sci Signal (2010) 3(123):pe18. doi: 10.1126/scisignal.3123pe18
27. Liao G, Zhang M, Harhaj EW, Sun SC. Regulation of the NF-kappaB-Inducing Kinase by Tumor Necrosis Factor Receptor-Associated Factor 3-Induced Degradation. J Biol Chem (2004) 279(25):26243–50. doi: 10.1074/jbc.M403286200
28. Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, et al. Noncanonical NF-kappaB Activation Requires Coordinated Assembly of a Regulatory Complex of the Adaptors Ciap1, Ciap2, TRAF2 and TRAF3 and the Kinase NIK. Nat Immunol (2008) 9(12):1371–8. doi: 10.1038/ni.1676
29. Qing G, Qu Z, Xiao G. Stabilization of Basally Translated NF-kappaB-Inducing Kinase (NIK) Protein Functions as a Molecular Switch of Processing of NF-Kappab2 P100. J Biol Chem (2005) 280(49):40578–82. doi: 10.1074/jbc.M508776200
30. Xiao G, Fong A, Sun SC. Induction of P100 Processing by NF-kappaB-Inducing Kinase Involves Docking IkappaB Kinase Alpha (IKKalpha) to P100 and IKKalpha-Mediated Phosphorylation. J Biol Chem (2004) 279(29):30099–105. doi: 10.1074/jbc.M401428200
31. Liang C, Zhang M, Sun SC. Beta-TrCP Binding and Processing of NF-Kappab2/P100 Involve its Phosphorylation at Serines 866 and 870. Cell Signal (2006) 18(8):1309–17. doi: 10.1016/j.cellsig.2005.10.011
32. Fong A, Sun SC. Genetic Evidence for the Essential Role of Beta-Transducin Repeat-Containing Protein in the Inducible Processing of NF-Kappa B2/P100. J Biol Chem (2002) 277(25):22111–4. doi: 10.1074/jbc.C200151200
33. Razani B, Zarnegar B, Ytterberg AJ, Shiba T, Dempsey PW, Ware CF, et al. Negative Feedback in Noncanonical NF-kappaB Signaling Modulates NIK Stability Through IKKalpha-Mediated Phosphorylation. Sci Signal (2010) 3(123):ra41. doi: 10.1126/scisignal.2000778
34. Gray CM, McCorkell KA, Chunduru SK, McKinlay MA, May MJ. Negative Feedback Regulation of NF-κb-Inducing Kinase is Proteasome-Dependent But Does Not Require Cellular Inhibitors of Apoptosis. Biochem Biophys Res Commun (2014) 450(1):341–6. doi: 10.1016/j.bbrc.2014.05.122
35. Jin J, Xiao Y, Chang JH, Yu J, Hu H, Starr R, et al. The Kinase TBK1 Controls IgA Class Switching by Negatively Regulating Noncanonical NF-kappaB Signaling. Nat Immunol (2012) 13(11):1101–9. doi: 10.1038/ni.2423
36. Zhang J, Roschke V, Baker KP, Wang Z, Alarcón GS, Fessler BJ, et al. Cutting Edge: A Role for B Lymphocyte Stimulator in Systemic Lupus Erythematosus. J Immunol (2001) 166(1):6–10. doi: 10.4049/jimmunol.166.1.6
37. Li Y, Yang JY, Xie X, Jie Z, Zhang L, Shi J, et al. Preventing Abnormal NF-κb Activation and Autoimmunity by Otub1-Mediated P100 Stabilization. Cell Res (2019) 29(6):474–85. doi: 10.1038/s41422-019-0174-3
38. Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, et al. Mutations of Multiple Genes Cause Deregulation of NF-kappaB in Diffuse Large B-Cell Lymphoma. Nature (2009) 459(7247):717–21. doi: 10.1038/nature07968
39. Li Y, Wang H, Zhou X, Xie X, Chen X, Jie Z, et al. Cell Intrinsic Role of NF-κb-Inducing Kinase in Regulating T Cell-Mediated Immune and Autoimmune Responses. Sci Rep (2016) 6:22115. doi: 10.1038/srep22115
40. Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, et al. The Lymphotoxin-Beta Receptor Induces Different Patterns of Gene Expression via Two NF-kappaB Pathways. Immunity (2002) 17(4):525–35. doi: 10.1016/S1074-7613(02)00423-5
41. Fagarasan S, Shinkura R, Kamata T, Nogaki F, Ikuta K, Tashiro K, et al. Alymphoplasia (Aly)-Type Nuclear Factor kappaB-Inducing Kinase (NIK) Causes Defects in Secondary Lymphoid Tissue Chemokine Receptor Signaling and Homing of Peritoneal Cells to the Gut-Associated Lymphatic Tissue System. J Exp Med (2000) 191(9):1477–86. doi: 10.1084/jem.191.9.1477
42. Hofmann J, Greter M, Du Pasquier L, Becher B. B-Cells Need a Proper House, Whereas T-Cells are Happy in a Cave: The Dependence of Lymphocytes on Secondary Lymphoid Tissues During Evolution. Trends Immunol (2010) 31(4):144–53. doi: 10.1016/j.it.2010.01.003
43. Matsumoto M. Role of TNF Ligand and Receptor Family in the Lymphoid Organogenesis Defined by Gene Targeting. J Med Invest (1999) 46(3-4):141–50.
44. Yilmaz ZB, Weih DS, Sivakumar V, Weih F. RelB is Required for Peyer's Patch Development: Differential Regulation of P52-RelB by Lymphotoxin and TNF. EMBO J (2003) 22(1):121–30. doi: 10.1093/emboj/cdg004
45. Fitzpatrick LR. Novel Pharmacological Approaches for Inflammatory Bowel Disease: Targeting Key Intracellular Pathways and the IL-23/IL-17 Axis. Int J Inflam (2012) 2012:389404. doi: 10.1155/2012/389404
46. Kucharzik T, Lügering N, Rautenberg K, Lügering A, Schmidt MA, Stoll R, et al. Role of M Cells in Intestinal Barrier Function. Ann N Y Acad Sci (2000) 915:171–83. doi: 10.1111/j.1749-6632.2000.tb05240.x
47. Ohno H, Hase K. Glycoprotein 2 (GP2): Grabbing the FimH Bacteria Into M Cells for Mucosal Immunity. Gut Microbes (2010) 1(6):407–10. doi: 10.4161/gmic.1.6.14078
48. Rey J, Garin N, Spertini F, Corthésy B. Targeting of Secretory IgA to Peyer's Patch Dendritic and T Cells After Transport by Intestinal M Cells. J Immunol (2004) 172(5):3026–33. doi: 10.4049/jimmunol.172.5.3026
49. Clark MA, Hirst BH, Jepson MA. M-Cell Surface Beta1 Integrin Expression and Invasin-Mediated Targeting of Yersinia Pseudotuberculosis to Mouse Peyer's Patch M Cells. Infect Immun (1998) 66(3):1237–43. doi: 10.1128/IAI.66.3.1237-1243.1998
50. Nakamura Y, Mimuro H, Kunisawa J, Furusawa Y, Takahashi D, Fujimura Y, et al. Microfold Cell-Dependent Antigen Transport Alleviates Infectious Colitis by Inducing Antigen-Specific Cellular Immunity. Mucosal Immunol (2020) 13(4):679–90. doi: 10.1038/s41385-020-0263-0
51. Hahn M, Macht A, Waisman A, Hövelmeyer N. NF-κb-Inducing Kinase is Essential for B-Cell Maintenance in Mice. Eur J Immunol (2016) 46(3):732–41. doi: 10.1002/eji.201546081
52. Bennett KM, Parnell EA, Sanscartier C, Parks S, Chen G, Nair MG, et al. Induction of Colonic M Cells During Intestinal Inflammation. Am J Pathol (2016) 186(5):1166–79. doi: 10.1016/j.ajpath.2015.12.015
53. Nguyen VQ, Eden K, Morrison HA, Sammons MB, Knight KK, Sorrentino S, et al. Noncanonical NF-kappaB Signaling Upregulation in Inflammatory Bowel Disease Patients is Associated With Loss of Response to Anti-TNF Agents. Front Pharmacol (2021) 12:655887. doi: 10.3389/fphar.2021.655887
54. Yamada T, Mitani T, Yorita K, Uchida D, Matsushima A, Iwamasa K, et al. Abnormal Immune Function of Hemopoietic Cells From Alymphoplasia (Aly) Mice, a Natural Strain With Mutant NF-Kappa B-Inducing Kinase. J Immunol (2000) 165(2):804–12. doi: 10.4049/jimmunol.165.2.804
55. Bos NA, Bun JC, Popma SH, Cebra ER, Deenen GJ, van der Cammen MJ, et al. Monoclonal Immunoglobulin A Derived From Peritoneal B Cells is Encoded by Both Germ Line and Somatically Mutated VH Genes and is Reactive With Commensal Bacteria. Infect Immun (1996) 64(2):616–23. doi: 10.1128/iai.64.2.616-623.1996
56. Müller G, Lipp M. Concerted Action of the Chemokine and Lymphotoxin System in Secondary Lymphoid-Organ Development. Curr Opin Immunol (2003) 15(2):217–24. doi: 10.1016/S0952-7915(03)00014-1
57. Schiemann B, Gommerman JL, Vora K, Cachero TG, Shulga-Morskaya S, Dobles M, et al. An Essential Role for BAFF in the Normal Development of B Cells Through a BCMA-Independent Pathway. Science (2001) 293(5537):2111–4. doi: 10.1126/science.1061964
58. Egawa T, Kawabata K, Kawamoto H, Amada K, Okamoto R, Fujii N, et al. The Earliest Stages of B Cell Development Require a Chemokine Stromal Cell-Derived Factor/Pre-B Cell Growth-Stimulating Factor. Immunity (2001) 15(2):323–34. doi: 10.1016/S1074-7613(01)00185-6
59. Kunisawa J, Gohda M, Kurashima Y, Ishikawa I, Higuchi M, Kiyono H. Sphingosine 1-Phosphate-Dependent Trafficking of Peritoneal B Cells Requires Functional NFkappaB-Inducing Kinase in Stromal Cells. Blood (2008) 111(9):4646–52. doi: 10.1182/blood-2007-10-120071
60. Kang HS, Chin RK, Wang Y, Yu P, Wang J, Newell KA, et al. Signaling via LTbetaR on the Lamina Propria Stromal Cells of the Gut is Required for IgA Production. Nat Immunol (2002) 3(6):576–82. doi: 10.1038/ni795
61. Suzuki K, Meek B, Doi Y, Honjo T, Fagarasan S. Two Distinctive Pathways for Recruitment of Naive and Primed IgM+ B Cells to the Gut Lamina Propria. Proc Natl Acad Sci USA (2005) 102(7):2482–6. doi: 10.1073/pnas.0409539102
62. Allaire JM, Crowley SM, Law HT, Chang SY, Ko HJ, Vallance BA. The Intestinal Epithelium: Central Coordinator of Mucosal Immunity. Trends Immunol (2018) 39(9):677–96. doi: 10.1016/j.it.2018.04.002
63. Kim JM, Lee JY, Yoon YM, Oh YK, Youn J, Kim YJ. NF-Kappa B Activation Pathway is Essential for the Chemokine Expression in Intestinal Epithelial Cells Stimulated With Clostridium Difficile Toxin a. Scand J Immunol (2006) 63(6):453–60. doi: 10.1111/j.1365-3083.2006.001756.x
64. Kim JM, Cho SJ, Oh YK, Jung HY, Kim YJ, Kim N. Nuclear Factor-Kappa B Activation Pathway in Intestinal Epithelial Cells is a Major Regulator of Chemokine Gene Expression and Neutrophil Migration Induced by Bacteroides Fragilis Enterotoxin. Clin Exp Immunol (2002) 130(1):59–66. doi: 10.1046/j.1365-2249.2002.01921.x
65. Hofer S, Rescigno M, Granucci F, Citterio S, Francolini M, Ricciardi-Castagnoli P. Differential Activation of NF-Kappa B Subunits in Dendritic Cells in Response to Gram-Negative Bacteria and to Lipopolysaccharide. Microbes Infect (2001) 3(4):259–65. doi: 10.1016/S1286-4579(01)01378-8
66. Bhattacharyya S, Borthakur A, Dudeja PK, Tobacman JK. Lipopolysaccharide-Induced Activation of NF-kappaB non-Canonical Pathway Requires BCL10 Serine 138 and NIK Phosphorylations. Exp Cell Res (2010) 316(19):3317–27. doi: 10.1016/j.yexcr.2010.05.004
67. Banoth B, Chatterjee B, Vijayaragavan B, Prasad MV, Roy P, Basak S. Stimulus-Selective Crosstalk via the NF-kappaB Signaling System Reinforces Innate Immune Response to Alleviate Gut Infection. Elife (2015) 4:e05648. doi: 10.7554/eLife.05648
68. Savinova OV, Hoffmann A, Ghosh G. The Nfkb1 and Nfkb2 Proteins P105 and P100 Function as the Core of High-Molecular-Weight Heterogeneous Complexes. Mol Cell (2009) 34(5):591–602. doi: 10.1016/j.molcel.2009.04.033
69. Sun SC. The Noncanonical NF-κb Pathway. Immunol Rev (2012) 246(1):125–40. doi: 10.1111/j.1600-065X.2011.01088.x
70. Chawla M, Mukherjee T, Deka A, Chatterjee B, Sarkar UA, Singh AK, et al. An Epithelial Nfkb2 Pathway Exacerbates Intestinal Inflammation by Supplementing Latent RelA Dimers to the Canonical NF-κb Module. Proc Natl Acad Sci USA (2021) 118(25):2024828118. doi: 10.1073/pnas.2024828118
71. Martin BN, Wang C, Willette-Brown J, Herjan T, Gulen MF, Zhou H, et al. Ikkα Negatively Regulates ASC-Dependent Inflammasome Activation. Nat Commun (2014) 5:4977. doi: 10.1038/ncomms5977
72. Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. IKKalpha Limits Macrophage NF-kappaB Activation and Contributes to the Resolution of Inflammation. Nature (2005) 434(7037):1138–43. doi: 10.1038/nature03491
73. Suzuki T. Regulation of the Intestinal Barrier by Nutrients: The Role of Tight Junctions. Anim Sci J (2020) 91(1):e13357. doi: 10.1111/asj.13357
74. May GR, Sutherland LR, Meddings JB. Is Small Intestinal Permeability Really Increased in Relatives of Patients With Crohn's Disease? Gastroenterology (1993) 104(6):1627–32. doi: 10.1016/0016-5085(93)90638-S
75. Al-Sadi RM, Ma TY. IL-1beta Causes an Increase in Intestinal Epithelial Tight Junction Permeability. J Immunol (2007) 178(7):4641–9. doi: 10.4049/jimmunol.178.7.4641
76. Boivin MA, Roy PK, Bradley A, Kennedy JC, Rihani T, Ma TY. Mechanism of Interferon-Gamma-Induced Increase in T84 Intestinal Epithelial Tight Junction. J Interferon Cytokine Res (2009) 29(1):45–54. doi: 10.1089/jir.2008.0128
77. Al-Sadi R, Ye D, Dokladny K, Ma TY. Mechanism of IL-1beta-Induced Increase in Intestinal Epithelial Tight Junction Permeability. J Immunol (2008) 180(8):5653–61. doi: 10.4049/jimmunol.180.8.5653
78. Ma TY, Boivin MA, Ye D, Pedram A, Said HM. Mechanism of TNF-{Alpha} Modulation of Caco-2 Intestinal Epithelial Tight Junction Barrier: Role of Myosin Light-Chain Kinase Protein Expression. Am J Physiol Gastrointest Liver Physiol (2005) 288(3):G422–430. doi: 10.1152/ajpgi.00412.2004
79. Florian P, Schöneberg T, Schulzke JD, Fromm M, Gitter AH. Single-Cell Epithelial Defects Close Rapidly by an Actinomyosin Purse String Mechanism With Functional Tight Junctions. J Physiol (2002) 545(2):485–99. doi: 10.1113/jphysiol.2002.031161
80. Bruewer M, Utech M, Ivanov AI, Hopkins AM, Parkos CA, Nusrat A. Interferon-Gamma Induces Internalization of Epithelial Tight Junction Proteins via a Macropinocytosis-Like Process. FASEB J (2005) 19(8):923–33. doi: 10.1096/fj.04-3260com
81. Chang J, Leong RW, Wasinger VC, Ip M, Yang M, Phan TG. Impaired Intestinal Permeability Contributes to Ongoing Bowel Symptoms in Patients With Inflammatory Bowel Disease and Mucosal Healing. Gastroenterology (2017) 153(3):723–731 e721. doi: 10.1053/j.gastro.2017.05.056
82. Mahida YR, Wu K, Jewell DP. Enhanced Production of Interleukin 1-Beta by Mononuclear Cells Isolated From Mucosa With Active Ulcerative Colitis of Crohn's Disease. Gut (1989) 30(6):835–8. doi: 10.1136/gut.30.6.835
83. Al-Sadi R, Guo S, Ye D, Rawat M, Ma TY. TNF-Alpha Modulation of Intestinal Tight Junction Permeability Is Mediated by NIK/IKK-Alpha Axis Activation of the Canonical NF-kappaB Pathway. Am J Pathol (2016) 186(5):1151–65. doi: 10.1016/j.ajpath.2015.12.016
84. Al-Sadi R, Ye D, Said HM, Ma TY. IL-1beta-Induced Increase in Intestinal Epithelial Tight Junction Permeability is Mediated by MEKK-1 Activation of Canonical NF-kappaB Pathway. Am J Pathol (2010) 177(5):2310–22. doi: 10.2353/ajpath.2010.100371
85. Waterhouse CC, Stadnyk AW. Rapid Expression of IL-1beta by Intestinal Epithelial Cells. vitro Cell Immunol (1999) 193(1):1–8. doi: 10.1006/cimm.1999.1468
86. Miller TL, McGee DW. Epithelial Cells Respond to Proteolytic and non-Proteolytic Detachment by Enhancing Interleukin-6 Responses. Immunology (2002) 105(1):101–10. doi: 10.1046/j.0019-2805.2001.01352.x
87. Yan SR, Joseph RR, Rosen K, Reginato MJ, Jackson A, Allaire N, et al. Activation of NF-kappaB Following Detachment Delays Apoptosis in Intestinal Epithelial Cells. Oncogene (2005) 24(43):6482–91. doi: 10.1038/sj.onc.1208810
88. Roulis M, Nikolaou C, Kotsaki E, Kaffe E, Karagianni N, Koliaraki V, et al. Intestinal Myofibroblast-Specific Tpl2-Cox-2-PGE2 Pathway Links Innate Sensing to Epithelial Homeostasis. Proc Natl Acad Sci U S A (2014) 111(43):E4658–67. doi: 10.1073/pnas.1415762111
89. Tsatsanis C, Patriotis C, Tsichlis PN. Tpl-2 Induces IL-2 Expression in T-Cell Lines by Triggering Multiple Signaling Pathways That Activate NFAT and NF-Kappab. Oncogene (1998) 17(20):2609–18. doi: 10.1038/sj.onc.1202460
90. Goessling W, North TE, Loewer S, Lord AM, Lee S, Stoick-Cooper CL, et al. Genetic Interaction of PGE2 and Wnt Signaling Regulates Developmental Specification of Stem Cells and Regeneration. Cell (2009) 136(6):1136–47. doi: 10.1016/j.cell.2009.01.015
91. Gantke T, Sriskantharajah S, Sadowski M, Ley SC. Iκb Kinase Regulation of the TPL-2/ERK MAPK Pathway. Immunol Rev (2012) 246(1):168–82. doi: 10.1111/j.1600-065X.2012.01104.x
92. Guan Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J Immunol Res (2019) 2019:7247238. doi: 10.1155/2019/7247238
93. Wells JM, Rossi O, Meijerink M, van Baarlen P. Epithelial Crosstalk at the Microbiota-Mucosal Interface. Proc Natl Acad Sci U S A (2011) 108 Suppl 1(Suppl 1):4607–14. doi: 10.1073/pnas.1000092107
94. Scott CL, Aumeunier AM, Mowat AM. Intestinal CD103+ Dendritic Cells: Master Regulators of Tolerance? Trends Immunol (2011) 32(9):412–9. doi: 10.1016/j.it.2011.06.003
95. Chen W, Liu D, Ren C, Su X, Wong CK. Yang R. A Special Network Comprised of Macrophages, Epithelial Cells, and Gut Microbiota for Gut Homeostasis. Cells (2022) 11(2):307. doi: 10.3390/cells11020307
96. Grinberg-Bleyer Y, Caron R, Seeley JJ, De Silva NS, Schindler CW, Hayden MS, et al. The Alternative NF-κb Pathway in Regulatory T Cell Homeostasis and Suppressive Function. J Immunol (2018) 200(7):2362–71. doi: 10.4049/jimmunol.1800042
97. Serebrennikova OB, Tsatsanis C, Mao C, Gounaris E, Ren W, Siracusa LD, et al. Tpl2 Ablation Promotes Intestinal Inflammation and Tumorigenesis in Apcmin Mice by Inhibiting IL-10 Secretion and Regulatory T-Cell Generation. Proc Natl Acad Sci USA (2012) 109(18):E1082–91. doi: 10.1073/pnas.1115098109
98. Tas SW, Vervoordeldonk MJ, Hajji N, Schuitemaker JH, van der Sluijs KF, May MJ, et al. Noncanonical NF-kappaB Signaling in Dendritic Cells is Required for Indoleamine 2,3-Dioxygenase (IDO) Induction and Immune Regulation. Blood (2007) 110(5):1540–9. doi: 10.1182/blood-2006-11-056010
99. Salazar F, Awuah D, Negm OH, Shakib F, Ghaemmaghami AM. The Role of Indoleamine 2,3-Dioxygenase-Aryl Hydrocarbon Receptor Pathway in the TLR4-Induced Tolerogenic Phenotype in Human DCs. Sci Rep (2017) 7:43337. doi: 10.1038/srep43337
100. Yu J, Wang Y, Yan F, Zhang P, Li H, Zhao H, et al. Noncanonical NF-κb Activation Mediates STAT3-Stimulated IDO Upregulation in Myeloid-Derived Suppressor Cells in Breast Cancer. J Immunol (2014) 193(5):2574–86. doi: 10.4049/jimmunol.1400833
101. Brown KD, Claudio E, Siebenlist U. The Roles of the Classical and Alternative Nuclear factor-kappaB Pathways: Potential Implications for Autoimmunity and Rheumatoid Arthritis. Arthritis Res Ther (2008) 10(4):212. doi: 10.1186/ar2457
102. Andreas N, Potthast M, Geiselhöringer AL, Garg G, de Jong R, Riewaldt J, et al. RelB Deficiency in Dendritic Cells Protects From Autoimmune Inflammation Due to Spontaneous Accumulation of Tissue T Regulatory Cells. J Immunol (2019) 203(10):2602–13. doi: 10.4049/jimmunol.1801530
103. Han D, Walsh MC, Cejas PJ, Dang NN, Kim YF, Kim J, et al. Dendritic Cell Expression of the Signaling Molecule TRAF6 is Critical for Gut Microbiota-Dependent Immune Tolerance. Immunity (2013) 38(6):1211–22. doi: 10.1016/j.immuni.2013.05.012
104. Nishida A, Inoue R, Inatomi O, Bamba S, Naito Y, Andoh A. Gut Microbiota in the Pathogenesis of Inflammatory Bowel Disease. Clin J Gastroenterol (2018) 11(1):1–10. doi: 10.1007/s12328-017-0813-5
105. Dutta P, Ta A, Thakur BK, Dasgupta N, Das S. Biphasic Ccl20 Regulation by Toll-Like Receptor 9 Through the Activation of ERK-AP-1 and non-Canonical NF-kappaB Signaling Pathways. Biochim Biophys Acta Gen Subj (2017) 1861(1 Pt A):3365–77. doi: 10.1016/j.bbagen.2016.08.019
106. Hoover DM, Boulegue C, Yang D, Oppenheim JJ, Tucker K, Lu W, et al. The Structure of Human Macrophage Inflammatory Protein-3alpha /CCL20. Linking Antimicrobial and CC Chemokine Receptor-6-Binding Activities With Human Beta-Defensins. J Biol Chem (2002) 277(40):37647–54. doi: 10.1074/jbc.M203907200
107. Schutyser E, Struyf S, Van Damme J. The CC Chemokine CCL20 and its Receptor CCR6. Cytokine Growth Factor Rev (2003) 14(5):409–26. doi: 10.1016/S1359-6101(03)00049-2
108. Ben Braïek O, Smaoui S. Enterococci: Between Emerging Pathogens and Potential Probiotics. BioMed Res Int (2019) 2019:5938210. doi: 10.1155/2019/5938210
109. Flannigan KL, Denning TL. Segmented Filamentous Bacteria-Induced Immune Responses: A Balancing Act Between Host Protection and Autoimmunity. Immunology (2018) 154(4):537–46. doi: 10.1111/imm.12950
110. Pham TA, Clare S, Goulding D, Arasteh JM, Stares MD, Browne HP, et al. Epithelial IL-22RA1-Mediated Fucosylation Promotes Intestinal Colonization Resistance to an Opportunistic Pathogen. Cell Host Microbe (2014) 16(4):504–16. doi: 10.1016/j.chom.2014.08.017
111. Kumar P, Monin L, Castillo P, Elsegeiny W, Horne W, Eddens T, et al. Intestinal Interleukin-17 Receptor Signaling Mediates Reciprocal Control of the Gut Microbiota and Autoimmune Inflammation. Immunity (2016) 44(3):659–71. doi: 10.1016/j.immuni.2016.02.007
112. Bunker JJ, Bendelac A. IgA Responses to Microbiota. Immunity (2018) 49(2):211–24. doi: 10.1016/j.immuni.2018.08.011
113. Viladomiu M, Kivolowitz C, Abdulhamid A, Dogan B, Victorio D, Castellanos JG, et al. IgA-Coated E. Coli Enriched in Crohn's Disease Spondyloarthritis Promote T(H)17-Dependent Inflammation. Sci Transl Med (2017) 9(376):eaaf9655. doi: 10.1126/scitranslmed.aaf9655
114. Boutaffala L, Bertrand MJ, Remouchamps C, Seleznik G, Reisinger F, Janas M, et al. NIK Promotes Tissue Destruction Independently of the Alternative NF-κb Pathway Through TNFR1/RIP1-Induced Apoptosis. Cell Death Differ (2015) 22(12):2020–33. doi: 10.1038/cdd.2015.69
115. Jung JU, Ravi S, Lee DW, McFadden K, Kamradt ML, Toussaint LG, et al. NIK/MAP3K14 Regulates Mitochondrial Dynamics and Trafficking to Promote Cell Invasion. Curr Biol (2016) 26(24):3288–302. doi: 10.1016/j.cub.2016.10.009
116. Aya K, Alhawagri M, Hagen-Stapleton A, Kitaura H, Kanagawa O, Novack DV. NF-(Kappa)B-Inducing Kinase Controls Lymphocyte and Osteoclast Activities in Inflammatory Arthritis. J Clin Invest (2005) 115(7):1848–54. doi: 10.1172/JCI23763
117. Enzler T, Bonizzi G, Silverman GJ, Otero DC, Widhopf GF, Anzelon-Mills A, et al. Alternative and Classical NF-Kappa B Signaling Retain Autoreactive B Cells in the Splenic Marginal Zone and Result in Lupus-Like Disease. Immunity (2006) 25(3):403–15. doi: 10.1016/j.immuni.2006.07.010
118. Brightbill HD, Suto E, Blaquiere N, Ramamoorthi N, Sujatha-Bhaskar S, Gogol EB, et al. NF-κb Inducing Kinase is a Therapeutic Target for Systemic Lupus Erythematosus. Nat Commun (2018) 9(1):179. doi: 10.1038/s41467-017-02672-0
119. Billmeier U, Dieterich W, Neurath MF, Atreya R. Molecular Mechanism of Action of Anti-Tumor Necrosis Factor Antibodies in Inflammatory Bowel Diseases. World J Gastroenterol (2016) 22(42):9300–13. doi: 10.3748/wjg.v22.i42.9300
Keywords: NIK, non-canonical NF-κB, intestinal immunity, intestinal homeostasis, IBD – inflammatory bowel disease
Citation: Wang B and Shen J (2022) NF-κB Inducing Kinase Regulates Intestinal Immunity and Homeostasis. Front. Immunol. 13:895636. doi: 10.3389/fimmu.2022.895636
Received: 14 March 2022; Accepted: 31 May 2022;
Published: 27 June 2022.
Edited by:
Koen Venken, Ghent University, BelgiumReviewed by:
Yanchuan Li, University of Texas MD Anderson Cancer Center, United StatesSantasabuj Das, National Institute of Cholera and Enteric Diseases (ICMR), India
Copyright © 2022 Wang and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Shen, c2hlbmp1bl9tZWQyMDAwQDE2My5jb20=