 Di Zheng
Di Zheng Tejasvini Bhuvan
Tejasvini Bhuvan Natalie L. Payne
Natalie L. Payne Tracy S. P. Heng
Tracy S. P. Heng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 16 June 2022
Sec. Alloimmunity and Transplantation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.892443
This article is part of the Research Topic Next Generation MSC Therapy Manufacturing, Potency and Mechanism of Action Analysis View all 15 articles
Mesenchymal stromal cells (MSCs) have demonstrated therapeutic potential in inflammatory models of human disease. However, clinical translation has fallen short of expectations, with many trials failing to meet primary endpoints. Failure to fully understand their mechanisms of action is a key factor contributing to the lack of successful commercialisation. Indeed, it remains unclear how the long-ranging immunomodulatory effects of MSCs can be attributed to their secretome, when MSCs undergo apoptosis in the lung shortly after intravenous infusion. Their apoptotic fate suggests that efficacy is not based solely on their viable properties, but also on the immune response to dying MSCs. The secondary lymphoid organs (SLOs) orchestrate immune responses and play a key role in immune regulation. In this review, we will discuss how apoptotic cells can modify immune responses and highlight the importance of MSC-immune cell interactions in SLOs for therapeutic outcomes.
Mesenchymal stem cells, more accurately known as mesenchymal stromal cells (MSCs), are one of the most widely investigated therapeutic cell types, owing to their ease of accessibility and expansion from tissues free of ethical concerns, as well as their immunomodulatory capacity in various preclinical disease models (1–3) . MSCs can be sourced from the stroma of almost all tissues, but most commonly from bone marrow (BM), adipose tissue (AD) and umbilical cord (UC) (4, 5). MSCs are contained within a heterogeneous population expressing CD105, CD73 and CD90 while lacking CD45, CD11b, CD19 and HLA-DR. They are characterized by their adherence to plastic and ability to differentiate into osteoblasts, adipocytes and chondrocytes in vitro (6).
With increasing clinical translation, however, this minimal set of criteria is now recognized as inadequate for defining MSCs. It does not reflect MSC potency, which is largely based on broad immunomodulatory properties (7), rather than their self-renewal or multipotential capacity (8, 9). It does not address changes in marker expression levels or biological properties due to culture expansion and cell manufacturing processes (10), including cryopreservation and freeze-thawing (11). Additionally, it does not identify the risk profile of the MSC product, particularly with regard to hemocompatibility of intravenously (IV) delivered cells and their potential to trigger adverse thromboembolic events (12). Thus, proposed amendments to the criteria have included functional potency assays based on the immunomodulatory activity of MSCs (13), and profiling of the hemocompatibility of the diverse array of MSC products now in clinical use (14).
The preclinical efficacy of MSCs in various unrelated conditions such as graft-versus-host disease (GvHD), Crohn’s disease, kidney transplantation, myocardial infarction, stroke, diabetes, acute respiratory distress syndrome (ARDS), multiple sclerosis, and brain and spinal cord injury, mainly relates to immune regulation (15–20). In vitro studies have shown that MSCs can modulate adaptive and innate immune responses. MSCs suppress T cell proliferation, cytokine response (e.g. IFN-γ production) and cytotoxic activity in response to antigen-specific stimuli (21–23) whist promoting regulatory T cells (Tregs), via their production of soluble factors (Figure 1) such as nitric oxide (NO), indolamine 2,3 dioxygenase (IDO) and transforming growth factor-beta (TGF-β) (24, 30–32). MSCs can also downregulate the cytokine responses of innate immune cells, including dendritic cells (DCs) and monocytes, via the expression of prostaglandin E2 (PGE2) (33). How these in vitro findings relate to their mode of action remains to be clarified, given the complexity of immune responses in vivo and the short persistence of MSCs following infusion.
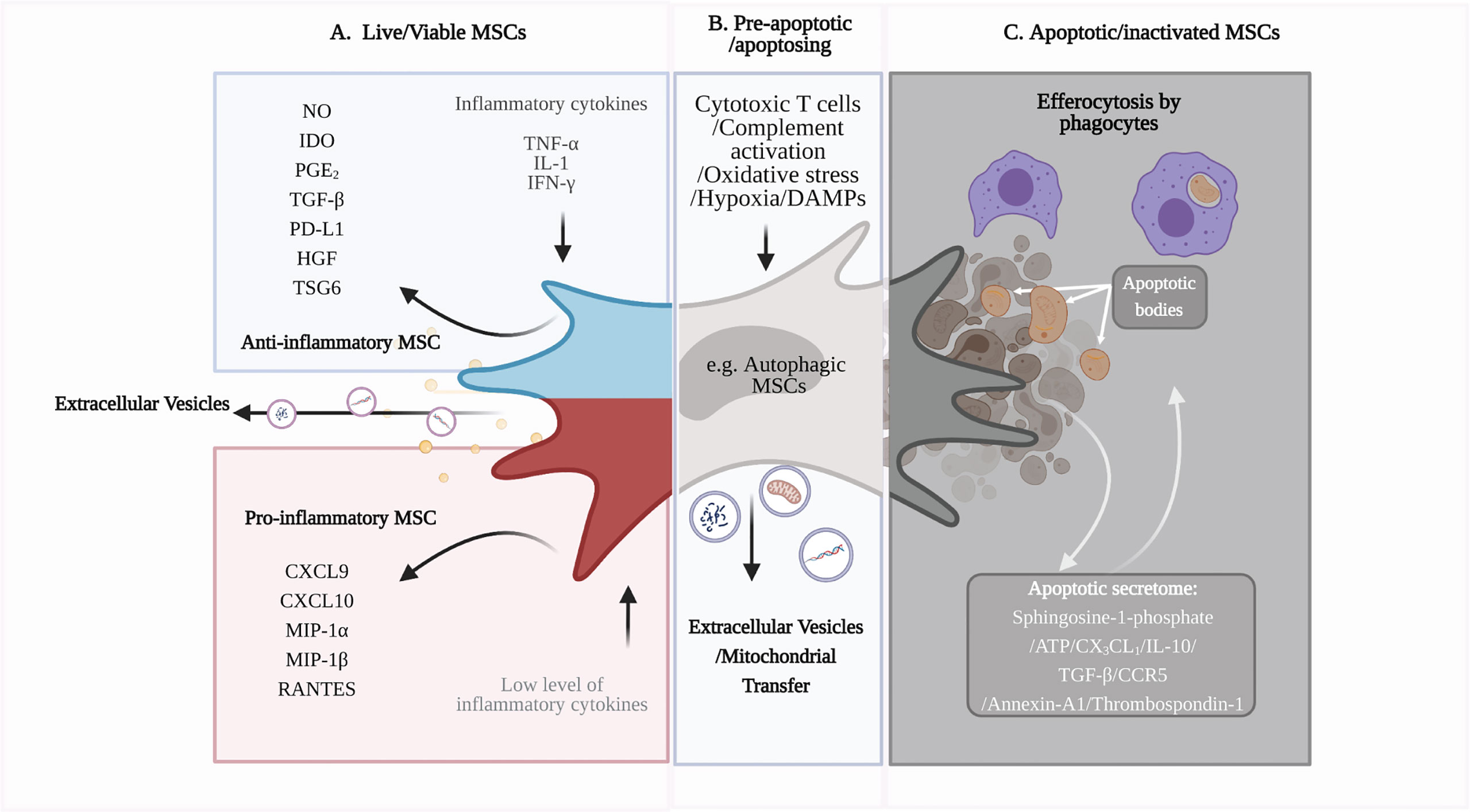
Figure 1 Immunomodulatory capacity of viable and apoptotic MSCs. (A) Live or viable MSCs can sense the microenvironment and respond to cytokine signals by polarizing into ‘pro-inflammatory’ or ‘anti-inflammatory’ phenotypes (24). ‘Anti-inflammatory’ MSCs produce anti-inflammatory soluble factors, including IDO and TGF-β, to modulate immune cell function and dampen inflammation. MSCs also produce extracellular vesicles (EV), such as exosomes, that can be immunomodulatory, depending on their cargo (19, 25). (B) Excessive inflammatory stress or the presence of cytotoxic immune cells will induce apoptosis of MSCs (26). MSCs can undergo a pre-apoptotic stage, known as autophagy. Autophagic MSCs, as well as the EVs produced during this stage, may have roles in immunosuppression (27). (C) As MSCs undergo apoptosis, their apoptotic secretome can promote an anti-inflammatory microenvironment and attract phagocytes for efferocytosis (28). Phagocytes that have engulfed the apoptotic MSCs become immunomodulatory and have downstream regulatory effects on adaptive immune cells, such as T cells (29). Figure was created with BioRender.com.
To date, studies have investigated different routes of MSC administration in order to maximise therapeutic efficacy. The different administration routes result in variations in MSC survival. IV infusion has been the most commonly used and studied method for MSC delivery because it is convenient, minimally invasive and reproducible (4). However, MSCs administered via the IV route are entrapped in the lung and rapidly cleared, with few traces detected in other tissues (34, 35). Studies using various MSC detection techniques, including MSCs constitutively expressing fluorescent proteins or luciferase, or labelled with fluorescent dyes or radioactive tracers, showed that viable MSCs are detected in the lung within 24 hours of IV administration but not at 72 hours (35–37). The decrease in detectable viable cells is directly proportional to the increase in dead cells in the lungs, indicating that IV-infused MSCs undergo cell death (37). Activation of caspase 3, a hallmark of apoptosis, in MSCs further indicates that MSCs undergo programmed cell death following their entrapment in the lung (38, 39).
The survival of IV-delivered MSCs can also be compromised if cells trigger the instant blood-mediated inflammatory reaction (IBMIR) due to incompatibility with blood (40). Expression of secreted and cell surface immunogenic factors (e.g. tissue factor (TF)/CD142) vary across MSC tissue sources and cell manufacturing conditions, including freeze-thawing and culture passaging (12, 14). These factors can activate the innate immune system and trigger the coagulation and complement cascades, which limit MSC engraftment and efficacy, but also increase the potential for adverse thromboembolic events (41).
Other injection routes, including intraperitoneal (IP), subcutaneous (SC), and intramuscular (IM), which bypass the lung, have also been examined. Prolonged detection of MSCs has been observed following IP and SC injection (42, 43). Following IM injection, a dwell time of up to 5-month was observed (44). Other studies have administered MSCs directly into the diseased tissue, for example, intratracheal administration in models of lung inflammation or intrathecal administration in models of spinal cord injury or neuroinflammatory disease (45–48). In these studies, MSCs are directly exposed to an inflammatory environment, which can influence MSCs survival and therapeutic efficacy (26, 49). Pre-conditioning of MSCs via hypoxia or serum deprivation for instance, can promote cell survival when subsequently exposed to an ischemic environment (50–52). Although the quantity, duration and the type of stress insult matter, overall, excessive stress will likely predispose MSCs to cell death (26).
In some disease settings, dead or dying MSCs and their associated ‘by-products’ can contribute to therapeutic efficacy (38, 39, 53–55) raising additional questions about their mode of action. How are MSCs killed in different microenvironments? How does the dying process contribute to therapeutic efficacy in different disease settings? Importantly, how do IV administered MSCs dying in the lung exert anti-inflammatory effects in distal organs in disease settings that seemingly do not involve the lung?
The molecular pathway that induces the death of MSCs in vivo is unclear and complicated by pre-existing disease, inflammatory cell infiltrate and the presence of different pathogens (26). The stress signals from the inflammatory microenvironment can trigger the apoptosis of MSCs. In a mouse model of GvHD, the presence of elevated numbers of cytotoxic CD8+ T cells and CD56+ natural killer (NK) cells in the lung caused MSC apoptosis (38). Settings that do not involve cytotoxic cell infiltrate likely involve other mechanisms. We recently showed that disabling the intrinsic (mitochondrial) pathway of apoptosis in MSCs prevented caspase 3 activation in the lung shortly after IV injection, indicating that MSCs were predominantly killed via the intrinsic pathway in non-GvHD settings (39). Activation of coagulation and complement by infused MSCs has also been shown to damage and reduce viability of MSCs (40, 56). Stronger activation of these proteolytic cascades is demonstrated by freeze-thawed (without culture recovery) and high-passage MSCs compared with fresh culture-derived, minimally expanded cells. This is due to variations in expression of immunogenic triggers, which may subsequently impact their in vivo therapeutic function (11).
In general, apoptosis is an ordered event that creates a transient immunosuppressive microenvironment via the release of anti-inflammatory mediators, including IL-10, TGF-β, CCR5, annexin-A1 and thrombospondin-1 (Figure 1) (28). Besides these secreted factors, cells can undergo a pre-apoptotic stage known as autophagy when they sense danger or stress signals from the microenvironment (57). By culturing MSCs in a stressed environment, autophagic MSCs can be pre-engineered to secrete immunomodulatory factors, such as TGF-β, to regulate T cell proliferation (58). Interestingly, MSCs have been shown to communicate with damaged cells via bidirectional transfer of mitochondria to increase mitochondrial biogenesis and rescue the cellular function of damaged cells (27). In preclinical models of myocardial infarction and respiratory disorders, mitochondrial transfer has been shown to contribute to the therapeutic effects of MSCs (Figure 1) (27, 59–61). MSCs can transfer mitochondria to macrophages via tunnelling nanotubules, cell fusion or extracellular vesicles (EV), which influence the macrophage function to modulate inflammation (59, 62). Whilst mitochondrial transfer has yet to be demonstrated in apoptotic MSCs, apoptotic bodies, which are a distinct type of extracellular vesicles formed when apoptotic cells disassemble into fragments (63), may also contribute to therapeutic efficacy upon engulfment by macrophages (64). Thus, MSCs are susceptible to cell death post-administration, which contributes to the anti-inflammatory effects of MSC therapy (10, 39).
Rapid clearance of apoptotic debris by phagocytes is essential in maintaining body homeostasis. Known as efferocytosis, this physiological process ‘silently’ removes apoptotic cells, inducing peripheral tolerance and avoiding inflammation. Phagocytic cells, including macrophages and monocytes, play a critical role in MSC therapy, as demonstrated in disease models where depletion of macrophages with clodronate liposomes renders MSC therapy ineffective (33, 36, 38). Phagocytes that have engulfed apoptotic cells, including MSCs, display a regulatory phenotype characterized by upregulation of PD-L1, IDO, COX2 and CD206, increased production of IL-6, IL-10, TGF-β and PGE2, and decreased production of pro-inflammatory cytokines such as TNF-α and IL-12 (29, 37, 65). Phagocytes that have engulfed apoptotic MSCs can suppress T cell proliferation, downregulate CD4+ T cell activation and promote Foxp3+ Treg generation (29, 37). The inhibition of COX2 abrogated the immunosuppressive capacity of efferocytic monocytes that had engulfed apoptotic MSCs, highlighting the importance of the PGE2/COX2 axis in this immunosuppressive function (29).
Since MSC apoptosis and the subsequent response of immune cells to this process contributes to their immunomodulatory effects, an outstanding question is whether viable MSCs are still required for MSC therapy, or can pre-inactivated MSCs be used as an alternative?
Several studies have investigated the efficacy of in vitro induced apoptotic, dead or inactivated MSCs. Treatment outcomes can be influenced by the type and duration of stimulation used to inactivate the cells, and also the disease setting (26). Apoptotic MSCs (39, 53, 54), but not dead MSCs (36, 66) could inhibit lung inflammation. In certain disease settings, such as LPS-induced sepsis, inactivated or dead MSCs could replicate the immunomodulatory effects, as pre-inactivation enhanced the phagocytosis of MSCs (55, 67). In other settings, non-viable MSCs were ineffective or did not fully replicate the effects of viable MSCs (33, 42, 67). For instance, in GvHD, apoptotic MSCs had to be administered at a much higher dose than viable MSCs to achieve comparable therapeutic benefits (38). It is plausible that inflammatory diseases driven by innate immune or phagocytic cells are more likely to benefit from treatment with inactivated MSCs, while suppression of T cell responses require MSCs with an active secretome (67). Importantly, the type and stage of cell death at the time of MSC administration are key considerations, as apoptosis, but not necrosis or lytic cell death, induces anti-inflammatory responses (68).
Although MSC apoptosis and the subsequent host phagocytic response contribute to immunomodulation, there remains much to learn about the full mechanisms of MSC therapy. As MSC apoptosis and efferocytosis occurs in the lung post-infusion, how does this dampen inflammation in other organs or tissues? In vitro studies have shown that MSCs can regulate immune cell function via their paracrine activity, but such molecules must have a relatively long half-life and broad biodistribution for this to be a plausible mechanism in vivo. Immune responses are initiated and maintained in the SLOs, which play a key role in immune regulation during health and disease. Thus, our knowledge of how MSC therapy modulates immune responses would be improved by considering the known function of SLOs, their role in clearance of apoptotic cells and gaining a better understanding of the effects of MSCs in these organs.
Primary lymphoid organs, also known as central lymphoid organs, are the sites for the development and maturation of leukocytes. Primary lymphoid organs include the bone marrow and thymus (69–71). Lymphocytes (a class of leukocytes, e.g. T and B cells) generated in primary lymphoid organs then seed the SLOs where they initiate adaptive immune responses. SLOs include lymph nodes (LNs), spleen, tonsils, adenoids, Peyer’s patches, and mucosal-associated lymphoid tissues (MALT) (71). The localisation of lymphocytes in SLOs maximizes their interaction with foreign antigens that drain to the SLOs via blood or lymphatics. Antigen presenting cells, such as DCs, can also transport antigens to SLOs and activate lymphocytes there. Once activated, these lymphocytes undergo expansion and differentiate into effector or memory cells to provide antigen-specific responses. This review will focus on the spleen and LNs as the major SLOs.
The spleen is a network of branching arterial vessels that functions to filter blood, allowing for the capture of blood-borne pathogens and antigens, and is a key organ for iron metabolism and erythrocyte homeostasis (72, 73). Although early research suggested that excision of the spleen does not largely impact the human immune system, its functional significance has been demonstrated through various studies over the years. While the human and mouse spleen is similar in terms of gross structure and immune cell function, some key differences are known to exist and have been reported elsewhere (73–75). This section will focus on the mouse spleen.
The spleen is composed of the white pulp (WP) and red pulp (RP), with the marginal zone (MZ) situated in between (76, 77), (Figure 2). Arterial blood arrives at the MZ and runs through the cords in the RP, where the F4/80+ RP macrophages (RPMs) monitor and phagocytose incoming aged erythrocytes (85, 86). In addition, RPMs also extract any dead or opsonized cells from the circulation, while simultaneously surveying for pathogens and tissue damage (78). Other leukocyte populations located in the RP, including neutrophils, monocytes and γδ T cells, exert immune effector functions upon encountering an inflammatory insult (72, 76, 77). Blood is recollected in sinuses to form the venous sinusoidal system and ultimately enters the efferent vein for return to the systemic circulation (77, 79, 80).
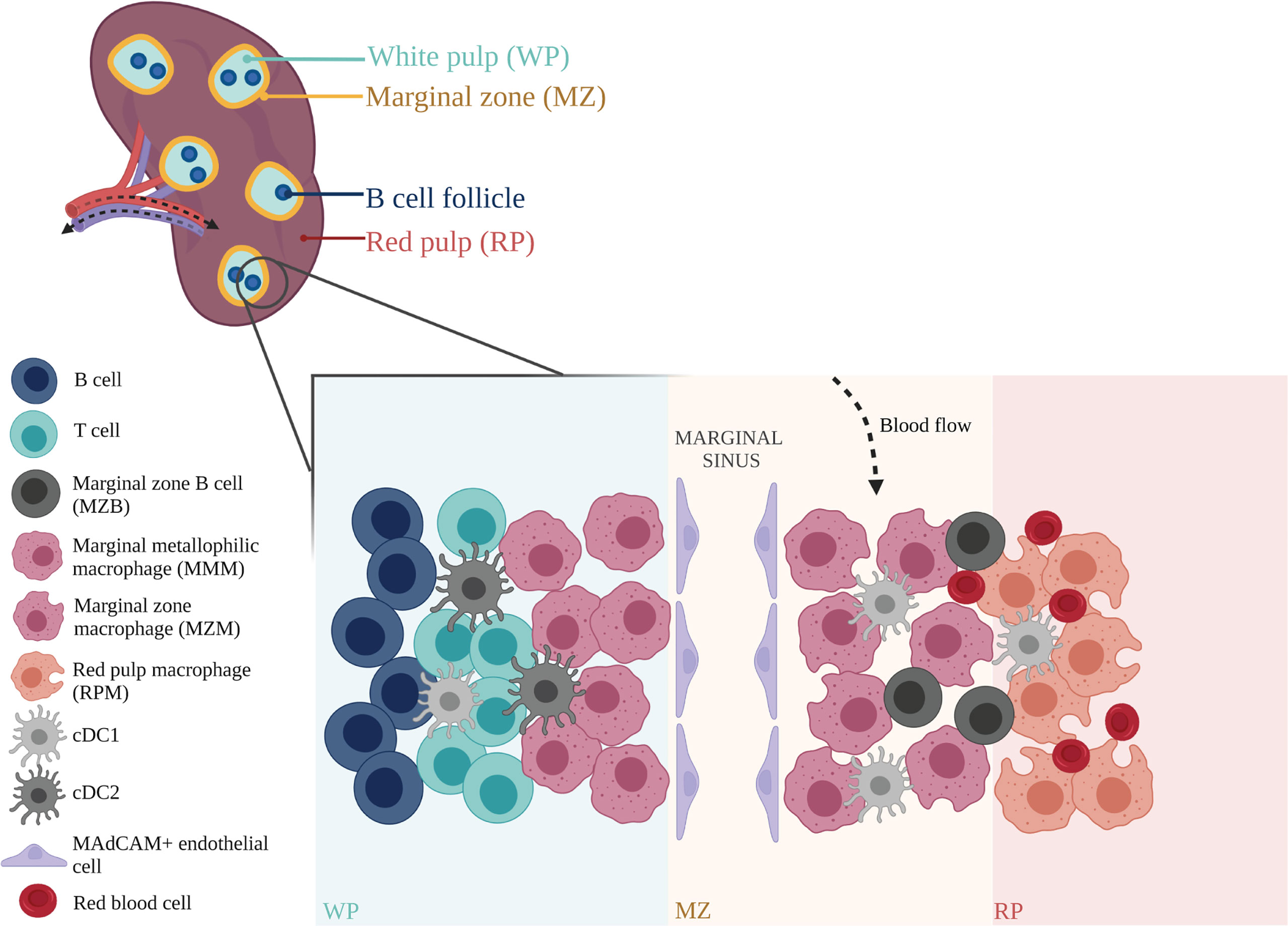
Figure 2 Anatomical structure and organization of mouse spleen. The splenic arterial network functions to filter blood and maintain erythrocyte homeostasis. Arterial blood, arriving at the marginal zone (MZ), passes through the red pulp (RP) cords, and is monitored by red pulp macrophages (RPMs) that survey for blood-borne antigens (78). Blood is recollected in sinuses before exiting the spleen through the efferent vein for return to the systemic circulation (79, 80). Within the spleen, adaptive immune responses to incoming systemic antigens are initiated in the white pulp (WP) which are largely driven by T and B cells (81, 82). Circulating lymphocytes arriving at the spleen may exit the bloodstream and enter the WP via the MZ (77). Marginal zone macrophages (MZMs) and marginal metallophilic macrophages (MMMs) are involved in the clearance of apoptotic cells, and maintenance of immune tolerance (83). Their functions are mediated by dendritic cell subsets, cDC1 and cDC2, which present antigens to T cells, and marginal zone B cells (MZBs) that help synchronize immune responses between the adaptive and innate arms (73, 84). This immune network is closely supported by stromal cells such as MAdCAM+ endothelial cells that line the marginal sinus, and help mediate tissue homeostasis (77). Figure was created with BioRender.com.
The white pulp is the site of lymphocyte differentiation and initiation of immune responses to blood borne antigens. Correct organization and maintenance of the WP is regulated by specific chemokines that attract T cells and B cells and establish their respective zones within the WP (81, 82). The continuous traffic of haematopoietic cells in and out of the spleen is an efficient way for these cells to survey the blood for pathogens and antigens.
The MZ is an important transit area for cells that are leaving the bloodstream and entering the WP. It contains a number of resident cells that have unique properties, including a subset of DCs and innate-like B cells called MZB cells (73, 84). Two specific subsets of macrophages are also found here: MZ macrophages (MZMs) and marginal metallophilic macrophages (MMMs) (76, 87). The MZMs line the outer ring of the WP and are characterized by expression of C-type lectin SIGNR1 and type I scavenger receptor MARCO (85, 88). The MMMs form the inner ring, located closer to the WP (89, 90). These macrophages are characterized by expression of the adhesion molecule SIGLEC1 (CD169) (82, 91–93).
While the spleen filters the blood and protects the host against blood-borne pathogens, LNs bring antigens draining from tissues together with lymphocytes circulating in the blood. There are more than 20 identified lymph nodes in mice and over 500 in humans, located at multiple sites throughout the lymphatic circulatory system (94, 95). Current understanding of LN structure and function is mainly gained from animal studies.
The parenchyma of the LN is compartmentalized by stromal cells and organized into the cortex at the outermost region containing B cell follicles, the paracortex in the inner region containing the T cell zone, and the medulla proximal to the efferent lymphatic vessels containing the medullary sinuses (95, 96), (Figure 3). Lymphocytes in the blood enter the LNs via high endothelial venules (HEVs) directly into the paracortex area. Lymph, containing soluble and cell-associated antigens, enters the LNs via afferent lymphatic vessels and is filtered through the lymphatic sinus (71, 95). Subcapsular sinus macrophages (SSMs) survey and capture lymph-borne antigens before they enter the cortex and paracortex, acting as gatekeepers that protect the host from aberrant immune responses and preventing the systemic spread of antigens (97–99, 102, 103). SSMs initiate innate immune responses by producing pro-inflammatory cytokines to recruit innate immune cells, and can also activate adaptive immunity by extending across the subcapsular sinus floor to present antigen to B cells in the follicles (99, 104).
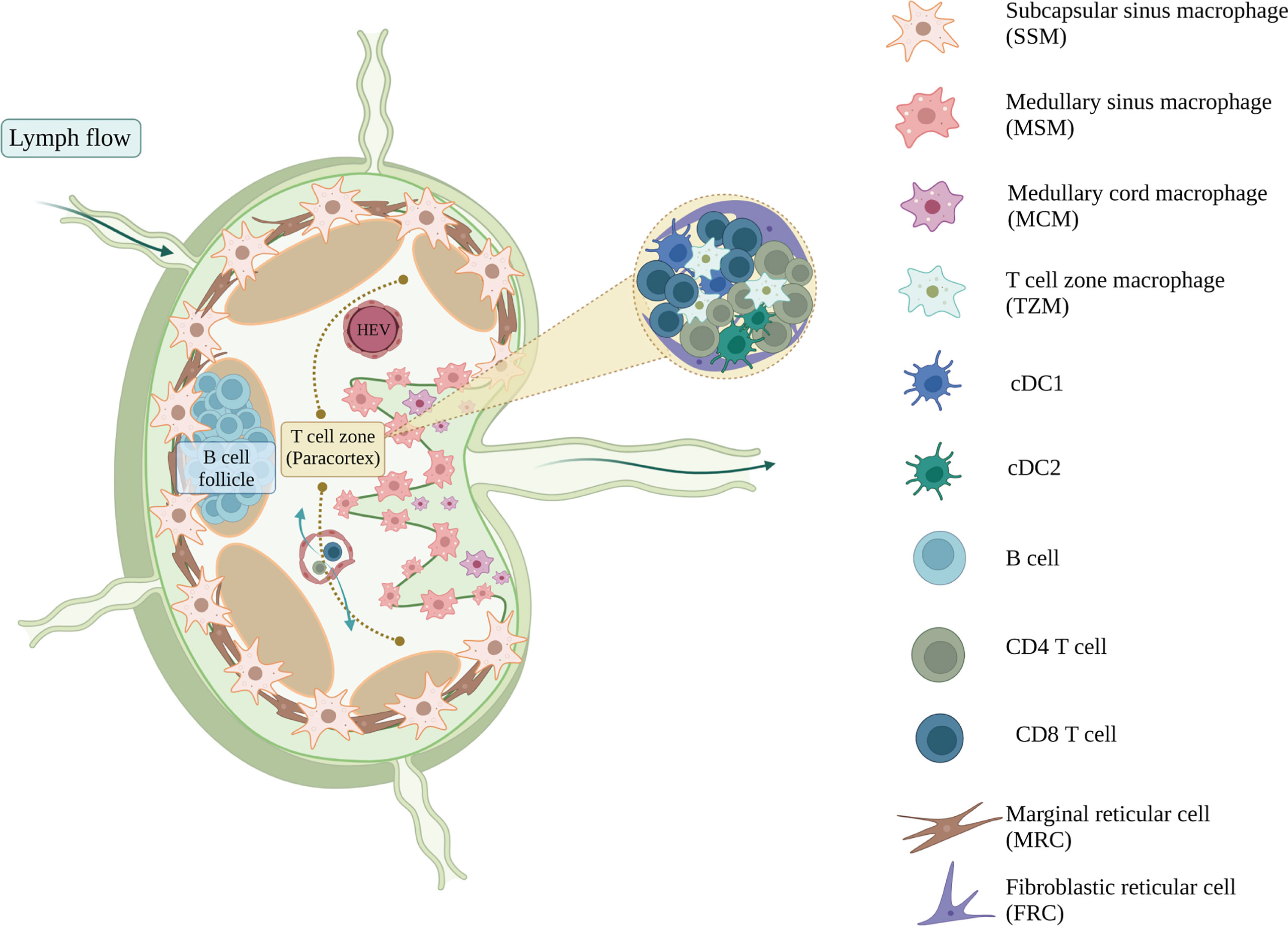
Figure 3 The immune and stromal cell composition of mouse lymph node. Lymph nodes filter the lymph and respond to the lymph-borne antigens, from which T and B cells will get activated, proliferate, and provide adaptive immune responses. Myeloid populations of lymph nodes support and regulate this adaptive response and maintain the homeostasis of lymph nodes (97). Subcapsular sinus macrophages (SSM) survey the infiltrating lymph before they transduce the activation signals towards the B cells sitting in the B cell follicles (98, 99), whereas cDC1 and cDC2 present the antigens that activate the CD4+ and CD8+ T cells inside the T cell zone, respectively (95). Subsets of lymph node macrophages, including medullary cord macrophages (MCM), T cell zone macrophages (TZM) and tingible body macrophages (TBM), are known for their efferocytotic ability (97, 100). They clear the apoptotic cell debris and maintain the homeostasis of lymph nodes. Lymph node stromal cells also help in regulating tissue homeostasis. Marginal reticular cells (MRC) organize and regulate the B cell follicles, whereas fibroblastic reticular cells (FRC) maintain the T cell homeostasis within the paracortex (101). Figure was created with BioRender.com.
Lymph passing by the subcapsular sinus will encounter another type of sinus macrophage, known as medullary sinus macrophages (MSMs). MSMs are located along the sinus of the medulla region, interacting with the lymph and also lymphocytes that are egressing the LN (97). Functionally, MSMs can actively phagocytose antigens, as well as apoptotic immune cells in the lymph (105–107). As the lymph passes through the medullary sinus, it then exits the LNs via the efferent lymphatic vessel and eventually return to the blood circulation.
The LN contains other macrophages that are also important in maintaining tissue homeostasis and clearing apoptotic debris. Medullary cord macrophages (MCMs) support plasma cell survival and efferocytose apoptotic debris, while tingible body macrophages (TBMs) are involved in the clearance of apoptotic B cells in the germinal centre (97). A recent study has identified a resident macrophage population in the T cell zone, known as T cell zone macrophages (TZMs), that efferocytose apoptotic DCs draining from the periphery (100). As efferocytosis induces an immunomodulatory phenotype in phagocytes, these LN resident macrophages play an important role in immune regulation.
Essentially all tissues undergo programmed cell death, known as apoptosis, evident through the constant turnover of cells. Under homeostatic conditions, apoptotic cells rarely accumulate in tissues due to the efficient efferocytosis by tissue phagocytes. The clearance of apoptotic cells is linked to an anti-inflammatory response that also induces immunological tolerance (108, 109). The precise apoptotic cell machinery and phagocytic components involved in apoptotic cell-induced immunosuppression are reviewed extensively elsewhere (109–112). However, it is clear that the efficient clearance of apoptotic cells can be attributed to the redundancy in the mechanisms of apoptotic cell recognition.
Splenic and LN macrophages are known to participate in the efferocytosis of apoptotic cells and maintenance of immune tolerance. TZMs and TBMs in the LN are known to engulf apoptotic debris silently without stimulating T cells, in order to maintain local immune homeostasis (100). The splenic MZ is also involved in the clearance of apoptotic cells from the circulation. IV-injected apoptotic cells drain to the splenic MZ, where they are rapidly engulfed by macrophages in the marginal zone (83). MZMs are known to be critical for particulate trapping in the spleen (72). Depletion studies (in which both MZMs and MMMs are depleted) have also indicated a crucial role for MZ macrophages in apoptotic cell-driven immunomodulation. The initial engulfment of apoptotic cells by macrophages in the MZ is vital in the generation of tolerance to self, whereby delayed clearance of apoptotic cells results in reduced immune tolerance to apoptotic cell-associated antigens (113). For example, depletion of MZ macrophages led to development of systemic tolerance breakdown in mouse models of systemic lupus erythematosus (SLE) and induced inflammatory responses towards apoptotic cell antigens (113).
The infusion of apoptotic cells has been reported to induce immunosuppression in experimental inflammatory diseases, autoimmunity and transplantation. In animal models of autoimmune arthritis, when apoptotic cells were infused not at the joint, but via the IV (114–116) or IP route (114, 117), the resolution of arthritic inflammation was conserved at the joint. In mouse models of transplantation, IV infusion of donor apoptotic splenocytes was shown to promote donor-specific immunosuppression, prolonging the survival of heart allografts (118, 119). Clinical studies have also shown that infusion of leukocytes rendered non-viable, either via a chemical cross-linker, 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (ECDI) or extracorporeal photochemotherapy, is safe and potentially beneficial in multiple sclerosis (120) and cutaneous T cell lymphoma (121). In apoptotic cell-based therapies, the spleen plays a pivotal role as splenic macrophages and DCs are involved in the phagocytosis of apoptotic leukocytes administrated via the IV route (122, 123). Together, these findings point to a critical role for the SLOs in the clearance of apoptotic cells and establishment of a tolerogenic state.
SLOs have a crucial role in host immune defense. In SLOs, antigen priming and immune cell activation occurs, followed by clonal expansion of antigen-specific effector lymphocytes, and the formation of immunological memory and tolerance. The importance of the spleen in host immunity has been demonstrated in various disease settings. In a long-term follow-up study, patients whose spleen had been surgically removed had an increased risk of developing bacterial infections (124). In malaria, the spleen is important in controlling the blood stage infection, clearance of parasitized red blood cells, induction of memory lymphocytes and replenishment of healthy red blood cells (125, 126). Splenectomized patients infected with malaria experienced enhanced parasitic burden, severe disease symptoms, and higher mortality rate (126).
Although SLOs are protective during infection, they may also promote the establishment and progression of some inflammatory diseases. LNs are the key sites for the initiation of GvHD and acute colitis (127, 128). In GvHD, donor cells migrate to the recipient LNs via their expression of lymphoid homing molecules, CD62L (L-selectin) and CCR7 (C-C chemokine receptor 7) (129). Upon recognition of alloantigen on the donor cells, T cells in the recipient LNs are activated and migrate to tissues where they cause damage (commonly in skin, gut and liver) (127). In acute colitis, intestinal migratory DCs drain to the mesenteric LNs where they present antigen and induce Th1 or Th17 responses (128, 130, 131). Pathogenic Th17 cells can also migrate from the mesenteric LNs to the gut and cause inflammatory bowel disease (131). Some studies have shown that lymphadenectomy (surgical removal of a group of lymph nodes and surrounding lymphatic tissues) protected rats from GvHD (132, 133). Lymphadenectomy is rarely investigated in clinical studies for the purpose of reducing inflammation, although it is performed on cancer patients to stop the spread of tumor metastases via the lymphatic system, or to remove the tumor cells in the lymphatic tissues (134, 135).
The spleen can also promote inflammatory responses in some disease settings. For example, splenectomy showed protective effects (e.g. reduced infarct volume) in rat models of brain, liver, kidney and intestine ischemic injury (136–140). In animal models of ischemic brain injury and also in patients with stroke, spleen atrophy (reduction in splenic weight and size) is commonly observed and thought to be a result of splenic leukocytes egressing via the blood circulation and into the injured brain (141). These circulating leukocytes are composed of monocytes, macrophages, neutrophils and lymphocytes, and their migration to the brain exacerbates inflammation and neurodegeneration (142, 143). Splenectomy performed on a rat stroke model (middle cerebral artery occlusion, MCAO), prior to injury, showed neuroprotective effects, with a reduction in brain infarct volume, peripheral immune cells and activated microglia in the brain infarct tissue (140, 142). Apart from the infiltration of splenic immune cells into the brain injury sites, the spleen also contributes to brain inflammation by producing pro-inflammatory signals, such as IFN-γ (140).
The involvement of SLOs and peripheral immune cells in the progression of central nervous system (CNS) inflammation has also been highlighted in other disease models, including spinal cord injury and experimental autoimmune encephalomyelitis (EAE, mouse model of multiple sclerosis) (144–146). In these disease settings, peripheral T cells and myeloid cells are recruited to the CNS and promote disease symptoms by enhancing inflammation. The crucial contribution of splenic T cells in promoting neuroinflammation is further demonstrated in neuropathic pain studies, in which the adoptive transfer of splenic T cells elevated neuropathic pain symptoms in the recipient (144).
Understanding how SLOs regulate the inflammatory response in such disease settings is important in defining the role of these SLOs in mediating the immunomodulatory effects of cell therapy.
Given the importance of SLOs in disease progression and tolerance induction, there is emerging evidence for their involvement in cell therapy in experimental disease models. A notable example is stroke, where IV administration of different therapeutic cell types, such as umbilical cord blood cells, haematopoietic stem cells, amnion epithelial cells, bone marrow stromal cells and multipotent adult progenitor cells, were shown to induce neuroprotective effects (147–150). IV-injected therapeutic cells were detected in the spleen and induced an anti-inflammatory environment that promoted repair in the CNS (e.g. infarct size, peripheral immune cell infiltration) (149–151). Of note, a study using neural stem cells to treat stroke showed that the direct interaction between IV-administered stem cells with splenocytes, particularly CD11b+ cells, was necessary for the treatment to be neuroprotective, with splenectomy abolishing the beneficial effect (152). In line with this, the improvement in functional recovery from stroke in MAPC-treated animals was also not evident in the absence of a spleen, supporting a critical role for the spleen (153).
Despite the central role of the spleen and lymph nodes in the blood and lymph network respectively, MSCs are rarely detected in the SLOs. When reported, the presence of MSCs in the SLOs is usually minimal and requires sensitive detection techniques (Figure 4). Small numbers of MSCs were detected in spleen and lymph nodes 24 hours post-IV infusion in naïve mice (154), and for up to 5 weeks in a mouse model of sclerodermatous GvHD (155). Similarly, localization within the spleen and LNs may also occur in the days after IP, but not SC or IM, infusion (44), although MSC biodistribution was variable following this delivery route and attachment of MSC aggregates to the peritoneal walls has been reported (157).
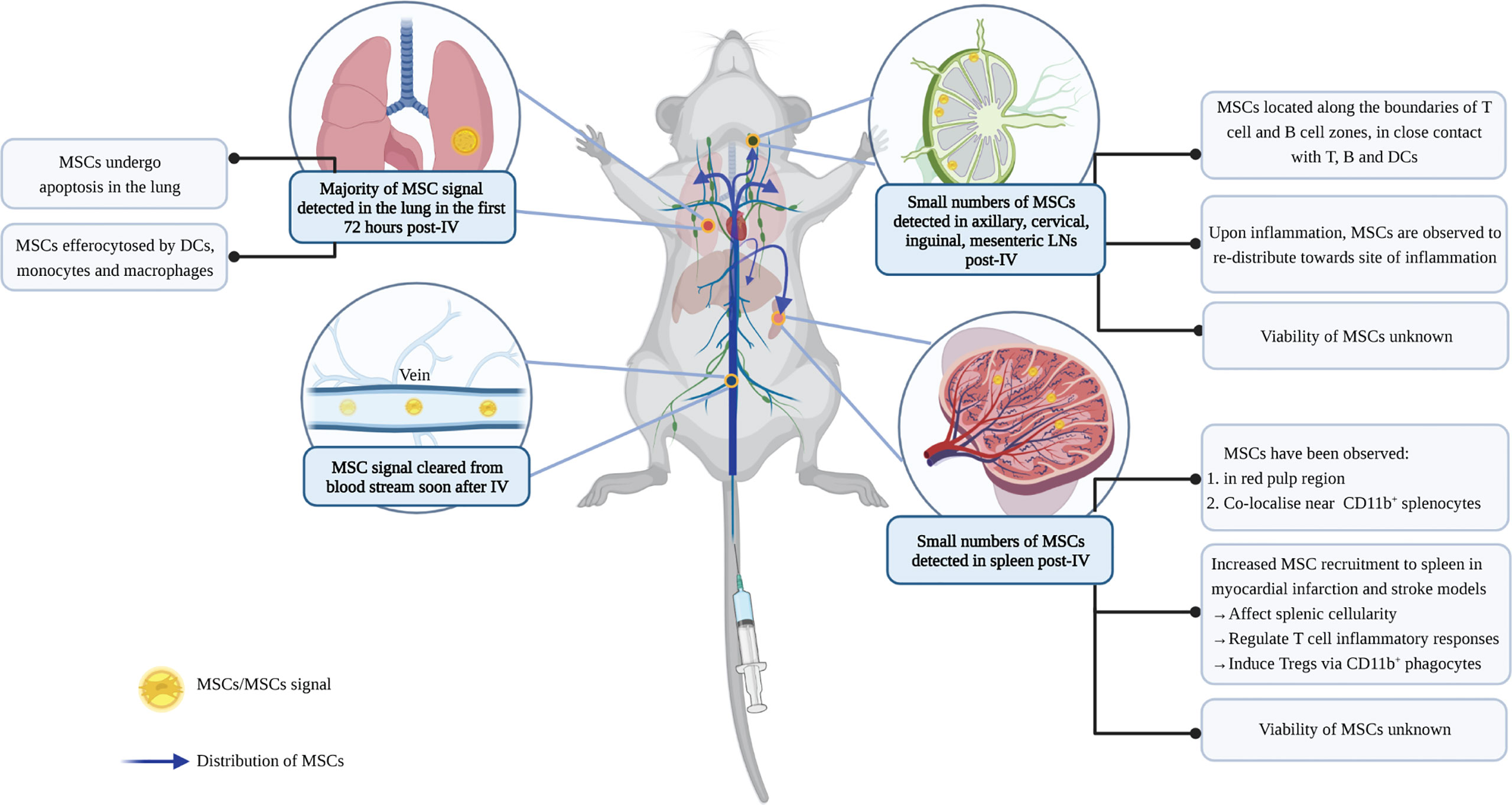
Figure 4 Biodistribution of MSCs post-IV infusion. IV administration is the most commonly used method for MSC delivery. To study the biodistribution of MSCs following IV infusion, studies have used different labelling approaches to track cell signals. Depending on the sensitivity of the techniques, MSCs (or signals of labels) have been found in lung, liver, and SLOs following IV infusion (37, 154). MSCs are cleared rapidly from the blood, and the majority of them are then detected in the lung where they undergo cell death (35). Some signals can also be found in the liver, although these are mostly dead MSCs (37, 154). Dead MSCs in the lung are cleared by lung phagocytes to avoid inflammation (37). Recent studies identified small numbers of MSCs in the SLOs, however, it is unclear whether they remain viable inside the SLOs (154–156). In LNs, MSCs are observed to localize along the boundaries of the germinal center and paracortical area; while in the spleen, MSC signals are detected in the red pulp region, co-localizing with CD11b+cells (155, 156). MSCs in the SLOs can regulate T cell response and induce Tregs, and their recruitment is influenced by inflammation. However, the extent by which these immunomodulatory effects in the SLOs are induced by efferocytosis of dying MSCs, or direct contact with viable MSCs or their secretome, remains unknown. Figure was created with BioRender.com.
It is thought that the presence of MSCs in SLOs is a result of their active migration, since they express higher levels of homing molecules, including CCR7, CD62L and intercellular adhesion molecule-1 (ICAM-1), compared to other fibroblastic cells (154, 158, 159). MSCs engineered to overexpress CCR7 or ICAM-1 showed improved MSC migration to the spleen and LNs post-IV injection in a GvHD model (158, 159). The increased distribution of MSCs within SLOs correlated with an improvement in their immunomodulatory effects and clinical outcomes in these disease models (158, 159).
The presence of inflammation can influence the biodistribution of MSCs in SLOs. In a myocardial infarction (MI) model, the presence of inflammation increased the recruitment of IV-infused MSCs to the spleen compared to controls without MI (160). In contrast, inflammation can reduce the presence of MSCs in the LNs. In experimental models of colitis and delayed-type hypersensitivity (DTH) (154, 161), accumulation of MSCs was reduced in the LNs and redistributed towards the inflamed tissues.
However, it is important to note the caveats of the labelling approaches used in such studies. For example, membrane dyes are retained when cells lose membrane integrity upon cell death, making it hard to discriminate signals from viable cells, cellular debris, or redistribution by phagocytes that had engulfed apoptotic cells (162). Moreover, studies have repeatedly shown that the small numbers of detectable MSCs eventually get cleared (35, 39, 163), yet their interaction with the host immune system within minutes to hours post-IV infusion is likely critical to therapeutic outcomes. For instance, complement activation by MSCs can adversely trigger IBMIR, but has also been found to upregulate the expression of CD11b on blood myeloid cells, which mediate the immunosuppressive effects of MSCs (164). Clarification of whether the detection of MSC signals in SLOs indicates the presence of viable MSCs or apoptotic cell clearance therefore has important implications for our understanding of the mechanisms of MSC therapy.
In several disease settings, the cellular composition of SLOs and function of their immune cells are changed upon cell therapy. The presence of MSCs is also found to associate with a change in the total cellularity of the SLO (154, 158, 160, 165), (Figure 4). In a model of myocardial infarction, MSC recruitment to the spleen was shown to decrease splenic natural killer cells and neutrophils (160). In a stroke model, the infusion of human umbilical cord blood cells altered the T cell and monocyte/macrophage composition in the spleen (165). In GvHD and DTH models, MSC recruitment to the LNs was also observed to regulate the survival and activation of lymphocytes, in particular, T cells (154, 158).
The splenic T cell profile is further modulated following cell therapy. Splenic T cells from animals that had received cell therapy were composed of a less pro-inflammatory population (with reduced IFN-γ+ and IL-17+ CD4+ T cells), which exhibited a reduced pro-inflammatory response when restimulated in vitro (165, 166). In an autoimmune uveitis model (166), T cells from MSC-treated groups showed reduced proliferative response and produced less pro-inflammatory Th1 and Th17 cytokines, but more anti-inflammatory IL-10, upon antigen restimulation.
Apart from modulating the T cell profile and inflammatory responses, MSC treatment can also induce Tregs in SLOs. In studies of autoimmune disease and allograft transplantation, MSCs inhibited inflammation by inducing an increase in FoxP3+ Tregs in the draining LNs and the spleen (156, 166–169). Similarly, an increase in splenic Tregs after MSC treatment was observed in ischemic kidney injury models. The importance of Treg induction in this model was highlighted when the therapeutic effects of MSCs were abrogated following the depletion of Tregs or the complete excision of the spleen (170).
Studies have established that co-culturing different myeloid cell populations with MSCs induces regulatory phenotypes, which can modulate immune responses. For example, DCs co-cultured with MSCs downregulated their expression of co-stimulatory molecules and were found to be less stimulatory in activating T cell responses in vitro (171–173). Monocytes and macrophages exposed to MSCs were less pro-inflammatory (produced less TNF and IL-12, and more IL-10 and IL-6) (174), more phagocytic and had increased bacterial killing capacity (33, 174–176). Macrophages with an immunoregulatory profile could further maintain an anti-inflammatory microenvironment and influence Treg generation (174, 177). Induction of amphiregulin in MSC-primed macrophages was recently identified as one pathway leading to induction of Tregs and decreased Th1 responses (178). The phenotype and morphology of macrophages ‘re-educated’ by MSCs share some similarities with myeloid-derived suppressor cells (MDSCs), which are differentiated from immature myeloid cells via PGE2 and IL-10-dependent mechanisms, and have immunosuppressive function (177). However, these in vitro findings need to be contextualized within the complex structural organization and cellular composition of the SLOs.
Studies examining immune cell changes in SLOs following MSC therapy support a role for myeloid cells in mediating the immunomodulatory effects of MSCs. When infused in a GvHD model, MSCs overexpressing the homing molecule, ICAM-1, were found in greater numbers in the SLOs (compared to control MSCs) and were found to inhibit splenic DC activation and maturation, suppress CD4+ T cell differentiation, and increase the splenic Treg/effector T cell ratio (159). Whilst it remains to be established that the change in regulatory/effector T cell balance is a direct consequence of DC function altered by MSCs in the spleen, studies have linked the induction of Tregs to CD11b+ phagocytic cells. Co-culture of splenocytes with MSCs induced Tregs, but not in the absence of CD11b+ cells (156). In a model of enterocolitis, the increase in Tregs in the SLOs underlies MSC therapeutic efficacy, which is abrogated upon the depletion of CD11b+ cells by clodronate-filled liposomes (156). Importantly, adoptive transfer of CD11b+ cells that had been co-cultured with MSCs was sufficient to increase Tregs in the SLOs and protect against disease (156). In another study using an osteosarcoma xenograft model in mice lacking T cells, clodronate depletion of CD11b+ splenic macrophages increased the amount of bioluminescence signal of luciferase-expressing MSCs found in the spleen after IV injection, and subsequently facilitated MSC delivery to the tumor (179). The data suggest that MSCs are phagocytosed by macrophages in the spleen, and overcoming this barrier promotes tissue-targeted delivery of MSCs. Thus, while the tolerogenic outcome of efferocytosis can contribute to the anti-inflammatory effects of MSC therapy, settings that require efficient delivery of MSCs to tissues may need to employ strategies that avoid splenic macrophage clearance.
MSCs exhibit broad spectrum immunomodulatory effects in various inflammatory diseases, but do not persist for significant periods in any part of the body following IV infusion. Instead, their lung entrapment and rapid clearance has shifted the focus to lung phagocytic cells as mediators of their therapeutic effects. Although traces of MSCs have been found in SLOs, along with changes in immune cell composition and function, limitations in labelling and imaging techniques make it difficult to establish with certainty that the small numbers detected are viable MSCs, cell debris or label redistribution following phagocytic uptake. There are several cell types with phagocytic capacity in the SLOs, including migratory cells from the circulation. Cells found in different compartments of the spleen and LNs perform different function, which depends crucially upon their spatial interactions with other cells and chemical cues. Furthermore, SLOs comprise not just immune cells, but also a variety of stromal populations that have functional roles in health and disease (180, 181), including the capacity to efferocytose apoptotic cells (182). Whether these stromal populations have a role in MSC therapy is an open area to explore.
DZ, TB, NP, and TH contributed conception and design of the manuscript. DZ wrote the first draft of the manuscript and made the figures. TB, NP, and TH wrote sections of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
DZ and TB are recipients of the Australian Government Research Training Program (RTP) Scholarship. TH is supported by funding from the National Health and Medical Research Council of Australia (GNT1107188, GNT1162499, GNT2012290) and the Australian Research Council (IC190100026). The Australian Regenerative Medicine Institute is supported by grants from the State Government of Victoria and the Australian Government.
TSPH received funding from Regeneus Ltd outside of this work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank Ivan Poon and Scott Mueller for helpful discussion.
1. Rendra E, Scaccia E, Bieback K. Recent Advances in Understanding Mesenchymal Stromal Cells. F1000Res (2020) 9:156. doi: 10.12688/f1000research.21862.1
2. Jossen V, van den Bos C, Eibl R, Eibl D. Manufacturing Human Mesenchymal Stem Cells at Clinical Scale: Process and Regulatory Challenges. Appl Microbiol Biotechnol (2018) 102:3981–94. doi: 10.1007/s00253-018-8912-x
3. Martin I, Galipeau J, Kessler C, Le Blanc K, Dazzi F. Challenges for Mesenchymal Stromal Cell Therapies. Sci Transl Med (2019) 11(480):eaat2189. doi: 10.1126/scitranslmed.aat2189
4. Kabat M, Bobkov I, Kumar S, Grumet M. Trends in Mesenchymal Stem Cell Clinical Trials 2004-2018: Is Efficacy Optimal in a Narrow Dose Range? Stem Cells Transl Med (2020) 9:17–27. doi: 10.1002/sctm.19-0202
5. Mendicino M, Bailey AM, Wonnacott K, Puri RK, Bauer SR. MSC-Based Product Characterization for Clinical Trials: An FDA Perspective. Cell Stem Cell (2014) 14:141–5. doi: 10.1016/j.stem.2014.01.013
6. Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal Criteria for Defining Multipotent Mesenchymal Stromal Cells. The International Society for Cellular Therapy Position Statement. Cytotherapy (2006) 8:315–7. doi: 10.1080/14653240600855905
7. Krampera M, Galipeau J, Shi Y, Tarte K, Sensebe L. Therapy MSCCotISfC. Immunological Characterization of Multipotent Mesenchymal Stromal Cells–The International Society for Cellular Therapy (ISCT) Working Proposal. Cytotherapy (2013) 15:1054–61. doi: 10.1016/j.jcyt.2013.02.010
8. Bianco P, Cao X, Frenette PS, Mao JJ, Robey PG, Simmons PJ, et al. The Meaning, the Sense and the Significance: Translating the Science of Mesenchymal Stem Cells Into Medicine. Nat Med (2013) 19:35–42. doi: 10.1038/nm.3028
9. Robey P. “Mesenchymal Stem Cells”: Fact or Fiction, and Implications in Their Therapeutic Use. F1000Res (2017) 6:524. doi: 10.12688/f1000research.10955.1
10. Cherian DS, Bhuvan T, Meagher L, Heng TSP. Biological Considerations in Scaling Up Therapeutic Cell Manufacturing. Front Pharmacol (2020) 11:654. doi: 10.3389/fphar.2020.00654
11. Cottle C, Porter AP, Lipat A, Turner-Lyles C, Nguyen J, Moll G, et al. Impact of Cryopreservation and Freeze-Thawing on Therapeutic Properties of Mesenchymal Stromal/Stem Cells and Other Common Cellular Therapeutics. Curr Stem Cell Rep (2022) 8:72–92. doi: 10.1007/s40778-022-00212-1
12. Moll G, Ankrum JA, Olson SD, Nolta JA. Improved MSC Minimal Criteria to Maximize Patient Safety: A Call to Embrace Tissue Factor and Hemocompatibility Assessment of MSC Products. Stem Cells Trans Med (2022) 11:2–13. doi: 10.1093/stcltm/szab005
13. Galipeau J, Krampera M, Barrett J, Dazzi F, Deans RJ, DeBruijn J, et al. International Society for Cellular Therapy Perspective on Immune Functional Assays for Mesenchymal Stromal Cells as Potency Release Criterion for Advanced Phase Clinical Trials. Cytotherapy (2016) 18:151–9. doi: 10.1016/j.jcyt.2015.11.008
14. Moll G, Ankrum JA, Kamhieh-Milz J, Bieback K, Ringdén O, Volk HD, et al. Intravascular Mesenchymal Stromal/Stem Cell Therapy Product Diversification: Time for New Clinical Guidelines. Trends Mol Med (2019) 25:149–63. doi: 10.1016/j.molmed.2018.12.006
15. Hoogduijn MJ, Roemeling-van Rhijn M, Engela AU, Korevaar SS, Mensah FK, Franquesa M, et al. Mesenchymal Stem Cells Induce an Inflammatory Response After Intravenous Infusion. Stem Cells Dev (2013) 22:2825–35. doi: 10.1089/scd.2013.0193
16. Moll G, Geissler S, Catar R, Ignatowicz L, Hoogduijn MJ, Strunk D, et al. Cryopreserved or Fresh Mesenchymal Stromal Cells: Only a Matter of Taste or Key to Unleash the Full Clinical Potential of MSC Therapy? Adv Exp Med Biol (2016) 951:77–98. doi: 10.1007/978-3-319-45457-3_7
17. Ankrum JA, Ong JF, Karp JM. Mesenchymal Stem Cells: Immune Evasive, Not Immune Privileged. Nat Biotechnol (2014) 32:252–60. doi: 10.1038/nbt.2816
18. Thomi G, Surbek D, Haesler V, Joerger-Messerli M, Schoeberlein A. Exosomes Derived From Umbilical Cord Mesenchymal Stem Cells Reduce Microglia-Mediated Neuroinflammation in Perinatal Brain Injury. Stem Cell Res Ther (2019) 10:105. doi: 10.1186/s13287-019-1207-z
19. Lu Y, Zhou Y, Zhang R, Wen L, Wu K, Li Y, et al. Bone Mesenchymal Stem Cell-Derived Extracellular Vesicles Promote Recovery Following Spinal Cord Injury via Improvement of the Integrity of the Blood-Spinal Cord Barrier. Front Neurosci (2019) 13:209. doi: 10.3389/fnins.2019.00209
20. Gama KB, Santos DS, Evangelista AF, Silva DN, de Alcantara AC, Dos Santos RR, et al. Conditioned Medium of Bone Marrow-Derived Mesenchymal Stromal Cells as a Therapeutic Approach to Neuropathic Pain: A Preclinical Evaluation. Stem Cells Int (2018) 2018:8179013–8179013. doi: 10.1155/2018/8179013
21. Di Nicola M, Carlo-Stella C, Magni M, Milanesi M, Longoni PD, Matteucci P, et al. Human Bone Marrow Stromal Cells Suppress T-Lymphocyte Proliferation Induced by Cellular or Nonspecific Mitogenic Stimuli. Blood (2002) 99:3838–43. doi: 10.1182/blood.V99.10.3838
22. Bartholomew A, Sturgeon C, Siatskas M, Ferrer K, McIntosh K, Patil S, et al. Mesenchymal Stem Cells Suppress Lymphocyte Proliferation In Vitro and Prolong Skin Graft Survival In Vivo. Exp Hematol (2002) 30:42–8. doi: 10.1016/S0301-472X(01)00769-X
23. Krampera M. Mesenchymal Stromal Cell 'Licensing': A Multistep Process. Leukemia (2011) 25:1408–14. doi: 10.1038/leu.2011.108
24. Bernardo ME, Fibbe WE. Mesenchymal Stromal Cells: Sensors and Switchers of Inflammation. Cell Stem Cell (2013) 13:392–402. doi: 10.1016/j.stem.2013.09.006
25. Del Fattore A, Luciano R, Pascucci L, Goffredo BM, Giorda E, Scapaticci M, et al. Immunoregulatory Effects of Mesenchymal Stem Cell-Derived Extracellular Vesicles on T Lymphocytes. Cell Transplant (2015) 24:2615–27. doi: 10.3727/096368915X687543
26. Weiss DJ, English K, Krasnodembskaya A, Isaza-Correa JM, Hawthorne IJ, Mahon BP. The Necrobiology of Mesenchymal Stromal Cells Affects Therapeutic Efficacy. Front Immunol (2019) 10:1228. doi: 10.3389/fimmu.2019.01228
27. Mahrouf-Yorgov M, Augeul L, Da Silva CC, Jourdan M, Rigolet M, Manin S, et al. Mesenchymal Stem Cells Sense Mitochondria Released From Damaged Cells as Danger Signals to Activate Their Rescue Properties. Cell Death Differ (2017) 24:1224–38. doi: 10.1038/cdd.2017.51
28. Saas P, Daguindau E, Perruche S. Concise Review: Apoptotic Cell-Based Therapies-Rationale, Preclinical Results and Future Clinical Developments. Stem Cells (2016) 34:1464–73. doi: 10.1002/stem.2361
29. Cheung TS, Galleu A, von Bonin M, Bornhauser M, Dazzi F. Apoptotic Mesenchymal Stromal Cells Induce Prostaglandin E2 in Monocytes: Implications for the Monitoring of Mesenchymal Stromal Cell Activity. Haematologica (2019) 104:e438–41. doi: 10.3324/haematol.2018.214767
30. Uccelli A, Moretta L, Pistoia V. Mesenchymal Stem Cells in Health and Disease. Nat Rev Immunol (2008) 8:726–36. doi: 10.1038/nri2395
31. Sato K, Ozaki K, Oh I, Meguro A, Hatanaka K, Nagai T, et al. Nitric Oxide Plays a Critical Role in Suppression of T-Cell Proliferation by Mesenchymal Stem Cells. Blood (2007) 109:228–34. doi: 10.1182/blood-2006-02-002246
32. Ren G, Zhang L, Zhao X, Xu G, Zhang Y, Roberts AI, et al. Mesenchymal Stem Cell-Mediated Immunosuppression Occurs via Concerted Action of Chemokines and Nitric Oxide. Cell Stem Cell (2008) 2:141–50. doi: 10.1016/j.stem.2007.11.014
33. Nemeth K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, et al. Bone Marrow Stromal Cells Attenuate Sepsis via Prostaglandin E(2)-Dependent Reprogramming of Host Macrophages to Increase Their Interleukin-10 Production. Nat Med (2009) 15:42–9. doi: 10.1038/nm.1905
34. Lee RH, Pulin AA, Seo MJ, Kota DJ, Ylostalo J, Larson BL, et al. Intravenous hMSCs Improve Myocardial Infarction in Mice Because Cells Embolized in Lung are Activated to Secrete the Anti-Inflammatory Protein TSG-6. Cell Stem Cell (2009) 5:54–63. doi: 10.1016/j.stem.2009.05.003
35. Eggenhofer E, Benseler V, Kroemer A, Popp FC, Geissler EK, Schlitt HJ, et al. Mesenchymal Stem Cells are Short-Lived and do Not Migrate Beyond the Lungs After Intravenous Infusion. Front Immunol (2012) 3:297. doi: 10.3389/fimmu.2012.00297
36. Mathias LJ, Khong SM, Spyroglou L, Payne NL, Siatskas C, Thorburn AN, et al. Alveolar Macrophages are Critical for the Inhibition of Allergic Asthma by Mesenchymal Stromal Cells. J Immunol (2013) 191:5914–24. doi: 10.4049/jimmunol.1300667
37. de Witte SFH, Luk F, Sierra Parraga JM, Gargesha M, Merino A, Korevaar SS, et al. Immunomodulation By Therapeutic Mesenchymal Stromal Cells (MSC) Is Triggered Through Phagocytosis of MSC By Monocytic Cells. Stem Cells (2018) 36:602–15. doi: 10.1002/stem.2779
38. Galleu A, Riffo-Vasquez Y, Trento C, Lomas C, Dolcetti L, Cheung TS, et al. Apoptosis in Mesenchymal Stromal Cells Induces In Vivo Recipient-Mediated Immunomodulation. Sci Transl Med (2017) 9(416):eaam7828. doi: 10.1126/scitranslmed.aam7828
39. Pang SHM, D'Rozario J, Mendonca S, Bhuvan T, Payne NL, Zheng D, et al. Mesenchymal Stromal Cell Apoptosis is Required for Their Therapeutic Function. Nat Commun (2021) 12:6495. doi: 10.1038/s41467-021-26834-3
40. Moll G, Rasmusson-Duprez I, von Bahr L, Connolly-Andersen AM, Elgue G, Funke L, et al. Are Therapeutic Human Mesenchymal Stromal Cells Compatible With Human Blood? Stem Cells (2012) 30:1565–74. doi: 10.1002/stem.1111
41. Caplan H, Olson SD, Kumar A, George M, Prabhakara KS, Wenzel P, et al. Mesenchymal Stromal Cell Therapeutic Delivery: Translational Challenges to Clinical Application. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.01645
42. Giri J, Galipeau J. Mesenchymal Stromal Cell Therapeutic Potency is Dependent Upon Viability, Route of Delivery, and Immune Match. Blood Adv (2020) 4:1987–97. doi: 10.1182/bloodadvances.2020001711
43. Kallmeyer K, Andre-Levigne D, Baquie M, Krause KH, Pepper MS, Pittet-Cuenod B, et al. Fate of Systemically and Locally Administered Adipose-Derived Mesenchymal Stromal Cells and Their Effect on Wound Healing. Stem Cells Transl Med (2020) 9:131–44. doi: 10.1002/sctm.19-0091
44. Braid LR, Wood CA, Wiese DM, Ford BN. Intramuscular Administration Potentiates Extended Dwell Time of Mesenchymal Stromal Cells Compared to Other Routes. Cytotherapy (2018) 20:232–44. doi: 10.1016/j.jcyt.2017.09.013
45. Chen G, Park CK, Xie RG, Ji RR. Intrathecal Bone Marrow Stromal Cells Inhibit Neuropathic Pain via TGF-Beta Secretion. J Clin Invest (2015) 125:3226–40. doi: 10.1172/JCI80883
46. Schafer S, Berger JV, Deumens R, Goursaud S, Hanisch UK, Hermans E. Influence of Intrathecal Delivery of Bone Marrow-Derived Mesenchymal Stem Cells on Spinal Inflammation and Pain Hypersensitivity in a Rat Model of Peripheral Nerve Injury. J Neuroinflamm (2014) 11:157. doi: 10.1186/s12974-014-0157-8
47. Sun J, Han ZB, Liao W, Yang SG, Yang Z, Yu J, et al. Intrapulmonary Delivery of Human Umbilical Cord Mesenchymal Stem Cells Attenuates Acute Lung Injury by Expanding CD4+CD25+ Forkhead Boxp3 (FOXP3)+ Regulatory T Cells and Balancing Anti- and Pro-Inflammatory Factors. Cell Physiol Biochem (2011) 27:587–96. doi: 10.1159/000329980
48. Gupta N, Su X, Popov B, Lee JW, Serikov V, Matthay MA. Intrapulmonary Delivery of Bone Marrow-Derived Mesenchymal Stem Cells Improves Survival and Attenuates Endotoxin-Induced Acute Lung Injury in Mice. J Immunol (2007) 179:1855–63. doi: 10.4049/jimmunol.179.3.1855
49. Abreu SC, Hampton TH, Hoffman E, Dearborn J, Ashare A, Singh Sidhu K, et al. Differential Effects of the Cystic Fibrosis Lung Inflammatory Environment on Mesenchymal Stromal Cells. Am J Physiol Lung Cell Mol Physiol (2020) 319:L908–25. doi: 10.1152/ajplung.00218.2020
50. Moya A, Larochette N, Paquet J, Deschepper M, Bensidhoum M, Izzo V, et al. Quiescence Preconditioned Human Multipotent Stromal Cells Adopt a Metabolic Profile Favorable for Enhanced Survival Under Ischemia. Stem Cells (2017) 35:181–96. doi: 10.1002/stem.2493
51. Liu J, Hao H, Huang H, Tong C, Ti D, Dong L, et al. Hypoxia Regulates the Therapeutic Potential of Mesenchymal Stem Cells Through Enhanced Autophagy. Int J Low Extrem Wounds (2015) 14:63–72. doi: 10.1177/1534734615573660
52. Lv B, Hua T, Li F, Han J, Fang J, Xu L, et al. Hypoxia-Inducible Factor 1 Alpha Protects Mesenchymal Stem Cells Against Oxygen-Glucose Deprivation-Induced Injury via Autophagy Induction and PI3K/AKT/mTOR Signaling Pathway. Am J Transl Res (2017) 9:2492–9.
53. Liu FB, Lin Q, Liu ZW. A Study on the Role of Apoptotic Human Umbilical Cord Mesenchymal Stem Cells in Bleomycin-Induced Acute Lung Injury in Rat Models. Eur Rev Med Pharmacol Sci (2016) 20:969–82.
54. Sung PH, Chang CL, Tsai TH, Chang LT, Leu S, Chen YL, et al. Apoptotic Adipose-Derived Mesenchymal Stem Cell Therapy Protects Against Lung and Kidney Injury in Sepsis Syndrome Caused by Cecal Ligation Puncture in Rats. Stem Cell Res Ther (2013) 4:155. doi: 10.1186/scrt385
55. Luk F, de Witte SF, Korevaar SS, Roemeling-van Rhijn M, Franquesa M, Strini T, et al. Inactivated Mesenchymal Stem Cells Maintain Immunomodulatory Capacity. Stem Cells Dev (2016) 25:1342–54. doi: 10.1089/scd.2016.0068
56. Li Y, Lin F. Mesenchymal Stem Cells are Injured by Complement After Their Contact With Serum. Blood (2012) 120:3436–43. doi: 10.1182/blood-2012-03-420612
57. Ghanta S, Tsoyi K, Liu X, Nakahira K, Ith B, Coronata AA, et al. Mesenchymal Stromal Cells Deficient in Autophagy Proteins Are Susceptible to Oxidative Injury and Mitochondrial Dysfunction. Am J Respir Cell Mol Biol (2017) 56:300–9. doi: 10.1165/rcmb.2016-0061OC
58. Gao L, Cen S, Wang P, Xie Z, Liu Z, Deng W, et al. Autophagy Improves the Immunosuppression of CD4+ T Cells by Mesenchymal Stem Cells Through Transforming Growth Factor-Beta1. Stem Cells Transl Med (2016) 5:1496–505. doi: 10.5966/sctm.2015-0420
59. Jackson MV, Morrison TJ, Doherty DF, McAuley DF, Matthay MA, Kissenpfennig A, et al. Mitochondrial Transfer via Tunneling Nanotubes is an Important Mechanism by Which Mesenchymal Stem Cells Enhance Macrophage Phagocytosis in the In Vitro and In Vivo Models of ARDS. Stem Cells (2016) 34:2210–23. doi: 10.1002/stem.2372
60. Tseng N, Lambie SC, Huynh CQ, Sanford B, Patel M, Herson PS, et al. Mitochondrial Transfer From Mesenchymal Stem Cells Improves Neuronal Metabolism After Oxidant Injury In Vitro: The Role of Miro1. J Cereb Blood Flow Metab (2021) 41:761–70. doi: 10.1177/0271678X20928147
61. Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, et al. Mitochondrial Transfer From Bone-Marrow-Derived Stromal Cells to Pulmonary Alveoli Protects Against Acute Lung Injury. Nat Med (2012) 18:759–65. doi: 10.1038/nm.2736
62. Phinney DG, Di Giuseppe M, Njah J, Sala E, Shiva S, St Croix CM, et al. Mesenchymal Stem Cells Use Extracellular Vesicles to Outsource Mitophagy and Shuttle microRNAs. Nat Commun (2015) 6:8472. doi: 10.1038/ncomms9472
63. Phan TK, Ozkocak DC, Poon IKH. Unleashing the Therapeutic Potential of Apoptotic Bodies. Biochem Soc Trans (2020) 48:2079–88. doi: 10.1042/BST20200225
64. Liu J, Qiu X, Lv Y, Zheng C, Dong Y, Dou G, et al. Apoptotic Bodies Derived From Mesenchymal Stem Cells Promote Cutaneous Wound Healing via Regulating the Functions of Macrophages. Stem Cell Res Ther (2020) 11:507. doi: 10.1186/s13287-020-02014-w
65. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages That Have Ingested Apoptotic Cells In Vitro Inhibit Proinflammatory Cytokine Production Through Autocrine/Paracrine Mechanisms Involving TGF-Beta, PGE2, and PAF. J Clin Invest (1998) 101:890–8. doi: 10.1172/JCI1112
66. Kavanagh H, Mahon BP. Allogeneic Mesenchymal Stem Cells Prevent Allergic Airway Inflammation by Inducing Murine Regulatory T Cells. Allergy (2011) 66:523–31. doi: 10.1111/j.1398-9995.2010.02509.x
67. Weiss ARR, Lee O, Eggenhofer E, Geissler E, Korevaar SS, Soeder Y, et al. Differential Effects of Heat-Inactivated, Secretome-Deficient MSC and Metabolically Active MSC in Sepsis and Allogenic Heart Transplantation. Stem Cells (2020) 38:797–807. doi: 10.1002/stem.3165
68. Kim EH, Wong SW, Martinez J. Programmed Necrosis and Disease:We Interrupt Your Regular Programming to Bring You Necroinflammation. Cell Death Differ (2019) 26:25–40. doi: 10.1038/s41418-018-0179-3
69. Burrell BE, Ding Y, Nakayama Y, Park KS, Xu J, Yin N, et al. Tolerance and Lymphoid Organ Structure and Function. Front Immunol (2011) 2:64. doi: 10.3389/fimmu.2011.00064
70. Boehm T, Hess I, Swann JB. Evolution of Lymphoid Tissues. Trends Immunol (2012) 33:315–21. doi: 10.1016/j.it.2012.02.005
71. Ruddle NH, Akirav EM. Secondary Lymphoid Organs: Responding to Genetic and Environmental Cues in Ontogeny and the Immune Response. J Immunol (2009) 183:2205–12. doi: 10.4049/jimmunol.0804324
72. Borges da Silva H, Fonseca R, Pereira RM, Cassado Ados A, Alvarez JM, D'Imperio Lima MR. Splenic Macrophage Subsets and Their Function During Blood-Borne Infections. Front Immunol (2015) 6:480. doi: 10.3389/fimmu.2015.00480
73. Lewis SM, Williams A, Eisenbarth SC. Structure and Function of the Immune System in the Spleen. Sci Immunol (2019) 4:eaau6085. doi: 10.1126/sciimmunol.aau6085
74. Steiniger BS. Human Spleen Microanatomy: Why Mice do Not Suffice. Immunology (2015) 145:334–46. doi: 10.1111/imm.12469
75. Garraud O, Borhis G, Badr G, Degrelle S, Pozzetto B, Cognasse F, et al. Revisiting the B-Cell Compartment in Mouse and Humans: More Than One B-Cell Subset Exists in the Marginal Zone and Beyond. BMC Immunol (2012) 13:63. doi: 10.1186/1471-2172-13-63
76. Bronte V, Pittet MJ. The Spleen in Local and Systemic Regulation of Immunity. Immunity (2013) 39:806–18. doi: 10.1016/j.immuni.2013.10.010
77. Mebius RE, Kraal G. Structure and Function of the Spleen. Nat Rev Immunol (2005) 5:606–16. doi: 10.1038/nri1669
78. Kurotaki D, Uede T, Tamura T. Functions and Development of Red Pulp Macrophages. Microbiol Immunol (2015) 59:55–62. doi: 10.1111/1348-0421.12228
79. Satodate R, Tanaka H, Sasou S, Sakuma T, Kaizuka H. Scanning Electron Microscopical Studies of the Arterial Terminals in the Red Pulp of the Rat Spleen. Anat Rec (1986) 215:214–6. doi: 10.1002/ar.1092150304
80. Cesta MF. Normal Structure, Function, and Histology of the Spleen. Toxicol Pathol (2006) 34:455–65. doi: 10.1080/01926230600867743
81. Nolte MA, Belien JA, Schadee-Eestermans I, Jansen W, Unger WW, van Rooijen N, et al. A Conduit System Distributes Chemokines and Small Blood-Borne Molecules Through the Splenic White Pulp. J Exp Med (2003) 198:505–12. doi: 10.1084/jem.20021801
82. Nolte MA, Arens R, Kraus M, van Oers MH, Kraal G, van Lier RA, et al. B Cells are Crucial for Both Development and Maintenance of the Splenic Marginal Zone. J Immunol (2004) 172:3620–7. doi: 10.4049/jimmunol.172.6.3620
83. McGaha TL, Chen Y, Ravishankar B, van Rooijen N, Karlsson MC. Marginal Zone Macrophages Suppress Innate and Adaptive Immunity to Apoptotic Cells in the Spleen. Blood (2011) 117:5403–12. doi: 10.1182/blood-2010-11-320028
84. You Y, Myers RC, Freeberg L, Foote J, Kearney JF, Justement LB, et al. Marginal Zone B Cells Regulate Antigen Capture by Marginal Zone Macrophages. J Immunol (2011) 186:2172–81. doi: 10.4049/jimmunol.1002106
85. den Haan JM, Mebius RE, Kraal G. Stromal Cells of the Mouse Spleen. Front Immunol (2012) 3:201. doi: 10.3389/fimmu.2012.00201
86. Schmidt EE, MacDonald IC, Groom AC. Microcirculation in Mouse Spleen (Nonsinusal) Studied by Means of Corrosion Casts. J Morphol (1985) 186:17–29. doi: 10.1002/jmor.1051860103
87. van Vliet E, Melis M, van Ewijk W. Marginal Zone Macrophages in the Mouse Spleen Identified by a Monoclonal Antibody. Anatomical Correlation With a B Cell Subpopulation. J Histochem Cytochem (1985) 33:40–4. doi: 10.1177/33.1.3880783
88. Geijtenbeek TB, Groot PC, Nolte MA, van Vliet SJ, Gangaram-Panday ST, van Duijnhoven GC, et al. Marginal Zone Macrophages Express a Murine Homologue of DC-SIGN That Captures Blood-Borne Antigens. vivo. Blood (2002) 100:2908–16. doi: 10.1182/blood-2002-04-1044
89. Crocker PR, Gordon S. Mouse Macrophage Hemagglutinin (Sheep Erythrocyte Receptor) With Specificity for Sialylated Glycoconjugates Characterized by a Monoclonal Antibody. J Exp Med (1989) 169:1333–46. doi: 10.1084/jem.169.4.1333
90. Kraal G, Janse M. Marginal Metallophilic Cells of the Mouse Spleen Identified by a Monoclonal Antibody. Immunology (1986) 58:665–9.
91. Fujiyama S, Nakahashi-Oda C, Abe F, Wang Y, Sato K, Shibuya A. Identification and Isolation of Splenic Tissue-Resident Macrophage Sub-Populations by Flow Cytometry. Int Immunol (2019) 31:51–6. doi: 10.1093/intimm/dxy064
92. Kleinclauss F, Perruche S, Masson E, de CarvalhoBittencourt M, Biichle S, Remy-Martin JP, et al. Intravenous Apoptotic Spleen Cell Infusion Induces a TGF-Beta-Dependent Regulatory T-Cell Expansion. Cell Death Differ (2006) 13:41–52. doi: 10.1038/sj.cdd.4401699
93. Khanna KM, Lefrancois L. Geography and Plumbing Control the T Cell Response to Infection. Immunol Cell Biol (2008) 86:416–22. doi: 10.1038/icb.2008.22
94. Willard-Mack CL. Normal Structure, Function, and Histology of Lymph Nodes. Toxicol Pathol (2006) 34:409–24. doi: 10.1080/01926230600867727
95. Grant SM, Lou M, Yao L, Germain RN, Radtke AJ. The Lymph Node at a Glance - How Spatial Organization Optimizes the Immune Response. J Cell Sci (2020) 133:jcs241828. doi: 10.1242/jcs.241828
96. Camara A, Cordeiro OG, Alloush F, Sponsel J, Chypre M, Onder L, et al. Lymph Node Mesenchymal and Endothelial Stromal Cells Cooperate via the RANK-RANKL Cytokine Axis to Shape the Sinusoidal Macrophage Niche. Immunity (2019) 50:1467–1481e6. doi: 10.1016/j.immuni.2019.05.008
97. Gray EE, Cyster JG. Lymph Node Macrophages. J Innate Immun (2012) 4:424–36. doi: 10.1159/000337007
98. Asano K, Kikuchi K, Tanaka M. CD169 Macrophages Regulate Immune Responses Toward Particulate Materials in the Circulating Fluid. J Biochem (2018) 164:77–85. doi: 10.1093/jb/mvy050
99. Louie DAP, Liao S. Lymph Node Subcapsular Sinus Macrophages as the Frontline of Lymphatic Immune Defense. Front Immunol (2019) 10:347. doi: 10.3389/fimmu.2019.00347
100. Baratin M, Simon L, Jorquera A, Ghigo C, Dembele D, Nowak J, et al. T Cell Zone Resident Macrophages Silently Dispose of Apoptotic Cells in the Lymph Node. Immunity (2017) 47:349–362 e345. doi: 10.1016/j.immuni.2017.07.019
101. Alexandre YO, Mueller SN. Stromal Cell Networks Coordinate Immune Response Generation and Maintenance. Immunol Rev (2018) 283:77–85. doi: 10.1111/imr.12641
102. Grabowska J, Lopez-Venegas MA, Affandi AJ, den Haan JMM. CD169(+) Macrophages Capture and Dendritic Cells Instruct: The Interplay of the Gatekeeper and the General of the Immune System. Front Immunol (2018) 9:2472. doi: 10.3389/fimmu.2018.02472
103. Kuka M, Iannacone M. The Role of Lymph Node Sinus Macrophages in Host Defense. Ann N Y Acad Sci (2014) 1319:38–46. doi: 10.1111/nyas.12387
104. Junt T, Moseman EA, Iannacone M, Massberg S, Lang PA, Boes M, et al. Subcapsular Sinus Macrophages in Lymph Nodes Clear Lymph-Borne Viruses and Present Them to Antiviral B Cells. Nature (2007) 450:110–4. doi: 10.1038/nature06287
105. Steer HW, Foot RA. Changes in the Medulla of the Parathymic Lymph Nodes of the Rat During Acute Gastro-Intestinal Inflammation. J Anat (1987) 152:23–36.
106. Fossum S. The Architecture of Rat Lymph Nodes. IV. Distribution of Ferritin and Colloidal Carbon in the Draining Lymph Nodes After Foot-Pad Injection. Scand J Immunol (1980) 12:433–41. doi: 10.1111/j.1365-3083.1980.tb00087.x
107. Nossal GJ, Abbot A, Mitchell J. Antigens in Immunity. XIV. Electron Microscopic Radioautographic Studies of Antigen Capture in the Lymph Node Medulla. J Exp Med (1968) 127:263–76. doi: 10.1084/jem.127.2.263
108. Kuang R, Perruche S, Chen W. Apoptotic Cell-Linked Immunoregulation: Implications for Promoting Immune Tolerance in Transplantation. Cell Biosci (2015) 5:27. doi: 10.1186/s13578-015-0019-9
109. Morelli AE, Larregina AT. Concise Review: Mechanisms Behind Apoptotic Cell-Based Therapies Against Transplant Rejection and Graft Versus Host Disease. Stem Cells (2016) 34:1142–50. doi: 10.1002/stem.2326
110. Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic Cell Clearance: Basic Biology and Therapeutic Potential. Nat Rev Immunol (2014) 14:166–80. doi: 10.1038/nri3607
111. Elliott MR, Ravichandran KS. The Dynamics of Apoptotic Cell Clearance. Dev Cell (2016) 38:147–60. doi: 10.1016/j.devcel.2016.06.029
112. Green DR, Oguin TH, Martinez J. The Clearance of Dying Cells: Table for Two. Cell Death Differ (2016) 23:915–26. doi: 10.1038/cdd.2015.172
113. Miyake Y, Asano K, Kaise H, Uemura M, Nakayama M, Tanaka M. Critical Role of Macrophages in the Marginal Zone in the Suppression of Immune Responses to Apoptotic Cell-Associated Antigens. J Clin Invest (2007) 117:2268–78. doi: 10.1172/JCI31990
114. Gray M, Miles K, Salter D, Gray D, Savill J. Apoptotic Cells Protect Mice From Autoimmune Inflammation by the Induction of Regulatory B Cells. Proc Natl Acad Sci U.S.A. (2007) 104:14080–5. doi: 10.1073/pnas.0700326104
115. Notley CA, Brown MA, Wright GP, Ehrenstein MR. Natural IgM is Required for Suppression of Inflammatory Arthritis by Apoptotic Cells. J Immunol (2011) 186:4967–72. doi: 10.4049/jimmunol.1003021
116. Bonnefoy F, Daoui A, Valmary-Degano S, Toussirot E, Saas P, Perruche S. Apoptotic Cell Infusion Treats Ongoing Collagen-Induced Arthritis, Even in the Presence of Methotrexate, and is Synergic With Anti-TNF Therapy. Arthritis Res Ther (2016) 18:184. doi: 10.1186/s13075-016-1084-0
117. Perruche S, Saas P, Chen W. Apoptotic Cell-Mediated Suppression of Streptococcal Cell Wall-Induced Arthritis Is Associated With Alteration of Macrophage Function and Local Regulatory T-Cell Increase: A Potential Cell-Based Therapy? Arthritis Res Ther (2009) 11:R104. doi: 10.1186/ar2750
118. Sun E, Gao Y, Chen J, Roberts AI, Wang X, Chen Z, et al. Allograft Tolerance Induced by Donor Apoptotic Lymphocytes Requires Phagocytosis in the Recipient. Cell Death Differ (2004) 11:1258–64. doi: 10.1038/sj.cdd.4401500
119. Wang Z, Larregina AT, Shufesky WJ, Perone MJ, Montecalvo A, Zahorchak AF, et al. Use of the Inhibitory Effect of Apoptotic Cells on Dendritic Cells for Graft Survival via T-Cell Deletion and Regulatory T Cells. Am J Transplant (2006) 6:1297–311. doi: 10.1111/j.1600-6143.2006.01308.x
120. Lutterotti A, Yousef S, Sputtek A, Sturner KH, Stellmann JP, Breiden P, et al. Antigen-Specific Tolerance by Autologous Myelin Peptide-Coupled Cells: A Phase 1 Trial in Multiple Sclerosis. Sci Transl Med (2013) 5:188ra175. doi: 10.1126/scitranslmed.3006168
121. Edelson R, Berger C, Gasparro F, Jegasothy B, Heald P, Wintroub B, et al. Treatment of Cutaneous T-Cell Lymphoma by Extracorporeal Photochemotherapy. Preliminary Results. N Engl J Med (1987) 316:297–303. doi: 10.1056/NEJM198702053160603
122. Morelli AE, Larregina AT, Shufesky WJ, Zahorchak AF, Logar AJ, Papworth GD, et al. Internalization of Circulating Apoptotic Cells by Splenic Marginal Zone Dendritic Cells: Dependence on Complement Receptors and Effect on Cytokine Production. Blood (2003) 101:611–20. doi: 10.1182/blood-2002-06-1769
123. Wang Z, Shufesky WJ, Montecalvo A, Divito SJ, Larregina AT, Morelli AE. In Situ-Targeting of Dendritic Cells With Donor-Derived Apoptotic Cells Restrains Indirect Allorecognition and Ameliorates Allograft Vasculopathy. PloS One (2009) 4:e4940. doi: 10.1371/journal.pone.0004940
124. Robinette CD, Fraumeni JF Jr. Splenectomy and Subsequent Mortality in Veterans of the 1939-45 War. Lancet (1977) 2:127–9. doi: 10.1016/S0140-6736(77)90132-5
125. Chotivanich K, Udomsangpetch R, McGready R, Proux S, Newton P, Pukrittayakamee S, et al. Central Role of the Spleen in Malaria Parasite Clearance. J Infect Dis (2002) 185:1538–41. doi: 10.1086/340213
126. Ghosh D, Stumhofer JS. The Spleen: “Epicenter” in Malaria Infection and Immunity. J Leukoc Biol (2021) 110:753–69. doi: 10.1002/JLB.4RI1020-713R
127. Portero-Sainz I, Gomez-Garcia deSoria V, Cuesta-Mateos C, Fernandez-Arandojo C, Vega-Piris L, Royg M, et al. A High Migratory Capacity of Donor T-Cells in Response to the Lymph Node Homing Receptor CCR7 Increases the Incidence and Severity of GvHD. Bone Marrow Transplant (2017) 52:745–52. doi: 10.1038/bmt.2016.342
128. Acedo SC, Gotardo EM, Lacerda JM, de Oliveira CC, de Oliveira Carvalho P, Gambero A. Perinodal Adipose Tissue and Mesenteric Lymph Node Activation During Reactivated TNBS-Colitis in Rats. Dig Dis Sci (2011) 56:2545–52. doi: 10.1007/s10620-011-1644-8
129. Yakoub-Agha I, Saule P, Depil S, Micol JB, Grutzmacher C, Boulanger-Villard F, et al. A High Proportion of Donor CD4+ T Cells Expressing the Lymph Node-Homing Chemokine Receptor CCR7 Increases Incidence and Severity of Acute Graft-Versus-Host Disease in Patients Undergoing Allogeneic Stem Cell Transplantation for Hematological Malignancy. Leukemia (2006) 20:1557–65. doi: 10.1038/sj.leu.2404308
130. Tamoutounour S, Henri S, Lelouard H, de Bovis B, de Haar C, van derWoude CJ, et al. CD64 Distinguishes Macrophages From Dendritic Cells in the Gut and Reveals the Th1-Inducing Role of Mesenteric Lymph Node Macrophages During Colitis. Eur J Immunol (2012) 42:3150–66. doi: 10.1002/eji.201242847
131. Bsat M, Chapuy L, Rubio M, Wassef R, Richard C, Schwenter F, et al. Differential Pathogenic Th17 Profile in Mesenteric Lymph Nodes of Crohn's Disease and Ulcerative Colitis Patients. Front Immunol (2019) 10:1177. doi: 10.3389/fimmu.2019.01177
132. Clark CL, Price BA, Malcolm P, Lear PA, Wood RF. Graft Versus Host Disease in Small Bowel Transplantation. Br J Surg (1991) 78:1077–9. doi: 10.1002/bjs.1800780915
133. Brouha PC, Perez-Abadia G, Francois CG, Laurentin-Perez LA, Gorantla V, Vossen M, et al. Lymphadenectomy Prior to Rat Hind Limb Allotransplantation Prevents Graft-Versus-Host Disease in Chimeric Hosts. Transpl Int (2004) 17:341–50. doi: 10.1111/j.1432-2277.2004.tb00453.x
134. Neubauer NL, Lurain JR. The Role of Lymphadenectomy in Surgical Staging of Endometrial Cancer. Int J Surg Oncol (2011) 2011:814649. doi: 10.1155/2011/814649
135. Giuliano AE, Jones RC, Brennan M, Statman R. Sentinel Lymphadenectomy in Breast Cancer. J Clin Oncol (1997) 15:2345–50. doi: 10.1200/JCO.1997.15.6.2345
136. Li M, Li F, Luo C, Shan Y, Zhang L, Qian Z, et al. Immediate Splenectomy Decreases Mortality and Improves Cognitive Function of Rats After Severe Traumatic Brain Injury. J Trauma (2011) 71:141–7. doi: 10.1097/TA.0b013e3181f30fc9
137. Das M, Leonardo CC, Rangooni S, Pennypacker KR, Mohapatra S, Mohapatra SS. Lateral Fluid Percussion Injury of the Brain Induces CCL20 Inflammatory Chemokine Expression in Rats. J Neuroinflamm (2011) 8:148. doi: 10.1186/1742-2094-8-148
138. Savas MC, Ozguner M, Ozguner IF, Delibas N. Splenectomy Attenuates Intestinal Ischemia-Reperfusion-Induced Acute Lung Injury. J Pediatr Surg (2003) 38:1465–70. doi: 10.1016/S0022-3468(03)00497-4
139. Okuaki Y, Miyazaki H, Zeniya M, Ishikawa T, Ohkawa Y, Tsuno S, et al. Splenectomy-Reduced Hepatic Injury Induced by Ischemia/Reperfusion in the Rat. Liver (1996) 16:188–94. doi: 10.1111/j.1600-0676.1996.tb00726.x
140. Seifert HA, Leonardo CC, Hall AA, Rowe DD, Collier LA, Benkovic SA, et al. The Spleen Contributes to Stroke Induced Neurodegeneration Through Interferon Gamma Signaling. Metab Brain Dis (2012) 27:131–41. doi: 10.1007/s11011-012-9283-0
141. Chiu NL, Kaiser B, Nguyen YV, Welbourne S, Lall C, Cramer SC. The Volume of the Spleen and Its Correlates After Acute Stroke. J Stroke Cerebrovasc Dis (2016) 25:2958–61. doi: 10.1016/j.jstrokecerebrovasdis.2016.08.012
142. Ajmo CT Jr., Vernon DO, Collier L, Hall AA, Garbuzova-Davis S, Willing A, et al. The Spleen Contributes to Stroke-Induced Neurodegeneration. J Neurosci Res (2008) 86:2227–34. doi: 10.1002/jnr.21661
143. Pennypacker KR, Offner H. The Role of the Spleen in Ischemic Stroke. J Cereb Blood Flow Metab (2015) 35:186–7. doi: 10.1038/jcbfm.2014.212
144. Grace PM, Hutchinson MR, Bishop A, Somogyi AA, Mayrhofer G, Rolan PE. Adoptive Transfer of Peripheral Immune Cells Potentiates Allodynia in a Graded Chronic Constriction Injury Model of Neuropathic Pain. Brain Behav Immun (2011) 25:503–13. doi: 10.1016/j.bbi.2010.11.018
145. Barthelmes J, Tafferner N, Kurz J, de Bruin N, Parnham MJ, Geisslinger G, et al. Induction of Experimental Autoimmune Encephalomyelitis in Mice and Evaluation of the Disease-Dependent Distribution of Immune Cells in Various Tissues. J Vis Exp (2016) 8(111):53933. doi: 10.3791/53933
146. Zheng P, Fu H, Wei G, Wei Z, Zhang J, Ma X, et al. Antigen-Oriented T Cell Migration Contributes to Myelin Peptide Induced-EAE and Immune Tolerance. Clin Immunol (2016) 169:36–46. doi: 10.1016/j.clim.2016.06.004
147. Vendrame M, Cassady J, Newcomb J, Butler T, Pennypacker KR, Zigova T, et al. Infusion of Human Umbilical Cord Blood Cells in a Rat Model of Stroke Dose-Dependently Rescues Behavioral Deficits and Reduces Infarct Volume. Stroke (2004) 35:2390–5. doi: 10.1161/01.STR.0000141681.06735.9b
148. Vendrame M, Gemma C, Pennypacker KR, Bickford PC, Davis Sanberg C, Sanberg PR, et al. Cord Blood Rescues Stroke-Induced Changes in Splenocyte Phenotype and Function. Exp Neurol (2006) 199:191–200. doi: 10.1016/j.expneurol.2006.03.017
149. Schwarting S, Litwak S, Hao W, Bahr M, Weise J, Neumann H. Hematopoietic Stem Cells Reduce Postischemic Inflammation and Ameliorate Ischemic Brain Injury. Stroke (2008) 39:2867–75. doi: 10.1161/STROKEAHA.108.513978
150. Evans MA, Lim R, Kim HA, Chu HX, Gardiner-Mann CV, Taylor KWE, et al. Acute or Delayed Systemic Administration of Human Amnion Epithelial Cells Improves Outcomes in Experimental Stroke. Stroke (2018) 49:700–9. doi: 10.1161/STROKEAHA.117.019136
151. Acosta SA, Tajiri N, Hoover J, Kaneko Y, Borlongan CV. Intravenous Bone Marrow Stem Cell Grafts Preferentially Migrate to Spleen and Abrogate Chronic Inflammation in Stroke. Stroke (2015) 46:2616–27. doi: 10.1161/STROKEAHA.115.009854
152. Lee ST, Hamilton JA, Valenzuela KS, Bogaerts A, Xi X, Aronowski J, et al. Anti-Inflammatory Mechanism of Intravascular Neural Stem Cell Transplantation in Haemorrhagic Stroke. Brain (2008) 131:616–29. doi: 10.1093/brain/awm306
153. Yang B, Hamilton JA, Valenzuela KS, Bogaerts A, Xi X, Aronowski J, et al. Multipotent Adult Progenitor Cells Enhance Recovery After Stroke by Modulating the Immune Response From the Spleen. Stem Cells (2017) 35:1290–302. doi: 10.1002/stem.2600
154. Lim JH, Kim JS, Yoon IH, Shin JS, Nam HY, Yang SH, et al. Immunomodulation of Delayed-Type Hypersensitivity Responses by Mesenchymal Stem Cells Is Associated With Bystander T Cell Apoptosis in the Draining Lymph Node. J Immunol (2010) 185:4022–9. doi: 10.4049/jimmunol.0902723
155. Lim JY, Ryu DB, Lee SE, Park G, Min CK. Mesenchymal Stem Cells (MSCs) Attenuate Cutaneous Sclerodermatous Graft-Versus-Host Disease (Scl-GVHD) Through Inhibition of Immune Cell Infiltration in a Mouse Model. J Invest Dermatol (2017) 137:1895–904. doi: 10.1016/j.jid.2017.02.986
156. Parekkadan B, Upadhyay R, Dunham J, Iwamoto Y, Mizoguchi E, Mizoguchi A, et al. Bone Marrow Stromal Cell Transplants Prevent Experimental Enterocolitis and Require Host CD11b+ Splenocytes. Gastroenterology (2011) 140:966–75. doi: 10.1053/j.gastro.2010.10.013
157. Bazhanov N, Ylostalo JH, Bartosh TJ, Tiblow A, Mohammadipoor A, Foskett A, et al. Intraperitoneally Infused Human Mesenchymal Stem Cells Form Aggregates With Mouse Immune Cells and Attach to Peritoneal Organs. Stem Cell Res Ther (2016) 7:27. doi: 10.1186/s13287-016-0284-5
158. Li H, Jiang Y, Jiang X, Guo X, Ning H, Li Y, et al. CCR7 Guides Migration of Mesenchymal Stem Cell to Secondary Lymphoid Organs: A Novel Approach to Separate GvHD From GvL Effect. Stem Cells (2014) 32:1890–903. doi: 10.1002/stem.1656
159. Tang B, Li X, Liu Y, Chen X, Li X, Chu Y, et al. The Therapeutic Effect of ICAM-1-Overexpressing Mesenchymal Stem Cells on Acute Graft-Versus-Host Disease. Cell Physiol Biochem (2018) 46:2624–35. doi: 10.1159/000489689
160. Luger D, Lipinski MJ, Westman PC, Glover DK, Dimastromatteo J, Frias JC, et al. Intravenously Delivered Mesenchymal Stem Cells: Systemic Anti-Inflammatory Effects Improve Left Ventricular Dysfunction in Acute Myocardial Infarction and Ischemic Cardiomyopathy. Circ Res (2017) 120:1598–613. doi: 10.1161/CIRCRESAHA.117.310599
161. Lopez-Santalla M, Mancheno-Corvo P, Escolano A, Menta R, DelaRosa O, Abad JL, et al. Biodistribution and Efficacy of Human Adipose-Derived Mesenchymal Stem Cells Following Intranodal Administration in Experimental Colitis. Front Immunol (2017) 8:638. doi: 10.3389/fimmu.2017.00638
162. Krueger TEG, Thorek DLJ, Denmeade SR, Isaacs JT, Brennen WN. Concise Review: Mesenchymal Stem Cell-Based Drug Delivery: The Good, the Bad, the Ugly, and the Promise. Stem Cells Transl Med (2018) 7:651–63. doi: 10.1002/sctm.18-0024
163. von Bahr L, Batsis I, Moll G, Hagg M, Szakos A, Sundberg B, et al. Analysis of Tissues Following Mesenchymal Stromal Cell Therapy in Humans Indicates Limited Long-Term Engraftment and No Ectopic Tissue Formation. Stem Cells (2012) 30:1575–8. doi: 10.1002/stem.1118
164. Moll G, Jitschin R, von Bahr L, Rasmusson-Duprez I, Sundberg B, Lönnies L, et al. Mesenchymal Stromal Cells Engage Complement and Complement Receptor Bearing Innate Effector Cells to Modulate Immune Responses. PloS One (2011) 6:e21703. doi: 10.1371/journal.pone.0021703
165. Golden JE, Shahaduzzaman M, Wabnitz A, Green S, Womble TA, Sanberg PR, et al. Human Umbilical Cord Blood Cells Alter Blood and Spleen Cell Populations After Stroke. Transl Stroke Res (2012) 3:491–9. doi: 10.1007/s12975-012-0208-3
166. Zhang L, Zheng H, Shao H, Nian H, Zhang Y, Bai L, et al. Long-Term Therapeutic Effects of Mesenchymal Stem Cells Compared to Dexamethasone on Recurrent Experimental Autoimmune Uveitis of Rats. Invest Ophthalmol Vis Sci (2014) 55:5561–71. doi: 10.1167/iovs.14-14788
167. Ko JH, Lee HJ, Jeong HJ, Kim MK, Wee WR, Yoon SO, et al. Mesenchymal Stem/Stromal Cells Precondition Lung Monocytes/Macrophages to Produce Tolerance Against Allo- and Autoimmunity in the Eye. Proc Natl Acad Sci U.S.A. (2016) 113:158–63. doi: 10.1073/pnas.1522905113
168. Mancheno-Corvo P, Lopez-Santalla M, Menta R, DelaRosa O, Mulero F, Del Rio B, et al. Intralymphatic Administration of Adipose Mesenchymal Stem Cells Reduces the Severity of Collagen-Induced Experimental Arthritis. Front Immunol (2017) 8:462. doi: 10.3389/fimmu.2017.00462
169. Murphy N, Treacy O, Lynch K, Morcos M, Lohan P, Howard L, et al. TNF-α/IL-1β-Licensed Mesenchymal Stromal Cells Promote Corneal Allograft Survival via Myeloid Cell-Mediated Induction of Foxp3(+) Regulatory T Cells in the Lung. FASEB J (2019) 33:9404–21. doi: 10.1096/fj.201900047R
170. Hu J, Zhang L, Wang N, Ding R, Cui S, Zhu F, et al. Mesenchymal Stem Cells Attenuate Ischemic Acute Kidney Injury by Inducing Regulatory T Cells Through Splenocyte Interactions. Kidney Int (2013) 84:521–31. doi: 10.1038/ki.2013.114
171. Zhang Y, Ge XH, Guo XJ, Guan SB, Li XM, Gu Wc , et al. Bone Marrow Mesenchymal Stem Cells Inhibit the Function of Dendritic Cells by Secreting Galectin-1. BioMed Res Int (2017) 2017:3248605. doi: 10.1155/2017/3248605
172. Jiang XX, Zhang Y, Liu B, Zhang SX, Wu Y, Yu XD, et al. Human Mesenchymal Stem Cells Inhibit Differentiation and Function of Monocyte-Derived Dendritic Cells. Blood (2005) 105:4120–6. doi: 10.1182/blood-2004-02-0586
173. Aldinucci A, Rizzetto L, Pieri L, Nosi D, Romagnoli P, Biagioli T, et al. Inhibition of Immune Synapse by Altered Dendritic Cell Actin Distribution: A New Pathway of Mesenchymal Stem Cell Immune Regulation. J Immunol (2010) 185:5102–10. doi: 10.4049/jimmunol.1001332
174. Kim J, Hematti P. Mesenchymal Stem Cell-Educated Macrophages: A Novel Type of Alternatively Activated Macrophages. Exp Hematol (2009) 37:1445–53. doi: 10.1016/j.exphem.2009.09.004
175. Rabani R, Volchuk A, Jerkic M, Ormesher L, Garces-Ramirez L, Canton J, et al. Mesenchymal Stem Cells Enhance NOX2-Dependent Reactive Oxygen Species Production and Bacterial Killing in Macrophages During Sepsis. Eur Respir J (2018) 51(4):1702021. doi: 10.1183/13993003.02021-2017
176. Maggini J, Mirkin G, Bognanni I, Holmberg J, Piazzon IM, Nepomnaschy I, et al. Mouse Bone Marrow-Derived Mesenchymal Stromal Cells Turn Activated Macrophages Into a Regulatory-Like Profile. PloS One (2010) 5:e9252. doi: 10.1371/journal.pone.0009252
177. Eggenhofer E, Hoogduijn MJ. Mesenchymal Stem Cell-Educated Macrophages. Transplant Res (2012) 1:12. doi: 10.1186/2047-1440-1-12
178. Ko JH, Kim HJ, Jeong HJ, Lee HJ, Oh JY. Mesenchymal Stem and Stromal Cells Harness Macrophage-Derived Amphiregulin to Maintain Tissue Homeostasis. Cell Rep (2020) 30(11):3806-20.e6. doi: 10.1016/j.celrep.2020.02.062
179. Hasgur S, Desbourdes L, Relation T, Overholt KM, Stanek JR, Guess AJ, et al. Splenic Macrophage Phagocytosis of Intravenously Infused Mesenchymal Stromal Cells Attenuates Tumor Localization. Cytotherapy (2021) 23:411–22. doi: 10.1016/j.jcyt.2020.04.102
180. Fletcher AL, Heng TS. Lymph Node Stroma Join the Cancer Support Network. Cell Death Differ (2016) 23:1899–901. doi: 10.1038/cdd.2016.103
181. Fletcher AL, Elman JS, Astarita J, Murray R, Saeidi N, D'Rozario J. Lymph Node Fibroblastic Reticular Cell Transplants Show Robust Therapeutic Efficacy in High-Mortality Murine Sepsis. Sci Transl Med (2014) 6:249ra109. doi: 10.1126/scitranslmed.3009377
Keywords: mesenchymal stromal cells, cell therapy, immune responses, apoptosis, secondary lymphoid organs, spleen, lymph nodes, efferocytosis
Citation: Zheng D, Bhuvan T, Payne NL and Heng TSP (2022) Secondary Lymphoid Organs in Mesenchymal Stromal Cell Therapy: More Than Just a Filter. Front. Immunol. 13:892443. doi: 10.3389/fimmu.2022.892443
Received: 09 March 2022; Accepted: 19 May 2022;
Published: 16 June 2022.
Edited by:
Guido Moll, Charité Universitätsmedizin Berlin, GermanyReviewed by:
Lorena Braid, Simon Fraser University, CanadaCopyright © 2022 Zheng, Bhuvan, Payne and Heng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tracy S. P. Heng, VHJhY3kuSGVuZ0Btb25hc2guZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.