 Yuming Peng
Yuming Peng Qiang Yin1,2*
Qiang Yin1,2*
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 28 July 2022
Sec. Molecular Innate Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.891868
This article is part of the Research Topic The Role of Immune Cells in Hepatic Ischemia Reperfusion View all 13 articles
Liver ischemia-reperfusion injury (IRI) is a major complication of liver trauma, resection, and transplantation. IRI may lead to liver dysfunction and failure, but effective approach to address it is still lacking. To better understand the cellular and molecular mechanisms of liver IRI, functional roles of numerous cell types, including hepatocytes, Kupffer cells, neutrophils, and sinusoidal endothelial cells, have been intensively studied. In contrast, hepatic stellate cells (HSCs), which are well recognized by their essential functions in facilitating liver protection and repair, have gained less attention in their role in IRI. This review provides a comprehensive summary of the effects of HSCs on the injury stage of liver IRI and their associated molecular mechanisms. In addition, we discuss the regulation of liver repair and regeneration after IRI by HSCs. Finally, we highlight unanswered questions and future avenues of research regarding contributions of HSCs to IRI in the liver.
Liver diseases have become one of the leading causes of death worldwide in the past few decades. It was estimated that over two million deaths were attributed to major liver diseases, such as cirrhosis and liver cancer (1), highlighting the demand for liver transplantation, which is currently the optimal treatment for end-stage liver diseases. Upon restoration of blood supply after interruption, the liver subjects to further injury that aggravates the initial injury caused by ischemia. This pathophysiological process is called liver ischemia-reperfusion injury (IRI) (2). Liver IRI can be classified into warm and cold IRI, which share similar mechanisms with differences mainly in the clinical settings (3). Warm IRI, initiated by hepatocellular damage, develops in situ during liver trauma and transplantation where hepatic blood flow falls transiently. Cold IRI, initiated by liver sinusoidal endothelial cells (LSECs) damage and microcirculatory disruption, occurs ex vivo during cold storage of the liver before transplantation surgery (4). Liver IRI is a critical complication in several clinical settings including liver trauma, resection, and transplantation (5–7). The degree of liver IRI depends on the period, methods of ischemia, and baseline liver condition (8). Continuous occlusion as short as 5 min can still lead to liver damage 1 d postoperatively in rat model of liver IRI, and IRI is much exacerbated in steatotic liver (9). IRI is an important cause of liver dysfunction (10), yet no reliable treatment option has been discovered. A better understanding of the cellular and molecular mechanisms of liver IRI may lead to improvements to clinical outcomes of liver disease patients, particularly those undergoing liver transplantation.
Substantial knowledge has been accumulated in regard to mechanisms underlying hepatic ischemic injury due to success of animal models. It is proposed that liver IRI consists of initial and late phases with distinct pathophysiological characteristics. Initial phase of liver IRI occurs 1-3 h after reperfusion (11–13), and manifests as rapid Kupffer cell activation after reperfusion (11, 14). Reactive oxygen species (ROS) is released by Kupffer cells, leading to oxidative stress and liver injury. Subsequently, the early liver injury triggers the release of a series of pro-inflammatory cytokines, such as TNF-α and IL-1β, inducing immune cell recruitment and more severe liver injury (15). The late phase of liver IRI, which occurs at 6-24 h after reperfusion (11, 12), is characterized by the recruitment of neutrophils to the liver and subsequent damage to hepatocytes (14).
Multiple cell types, including hepatocytes, liver sinusoidal endothelial cells (LSECs), Kupffer cells, hepatic stellate cells (HSCs) extrahepatic macrophages, neutrophils, and platelets, are involved in the progression of liver IRI (14, 16). Hepatocytes and LSECs are the main cell types subject to IRI induced cell death (17). Extensive studies indicate that Kupffer cells play a critical role in regulating IRI by promoting inflammatory injury mediated by cytokines and chemokines (2, 17). Neutrophils act as the main actor of cell injury during liver IRI following their recruitment to the liver regulated by Kupffer cells releasing of chemokines. Upon migration and infiltration to the liver, neutrophils respond to signals released by injured hepatocytes, inducing release of ROS and degranulation to cause further injury (17, 18). HSCs, which reside in the perisinusoidal space of liver and are known for their essential function of regulating hepatic fibrosis (19), has not been long investigated in liver IRI. As more recent studies shed light in the role of HSCs in liver IRI, we aimed to summarize the effects of HSCs on regulation of liver IRI in both injury and repair/regeneration stages, their intercellular communications with other cell types during IRI, and the associated molecular mechanisms.
HSCs are localized in the subendothelial space of Disse between hepatocytes and LSECs and comprise approximately 15% of total cells in human liver (20). Due to anatomic features, intercellular crosstalk can occur between adjacent cell types including hepatocytes, Kupffer cells, bone marrow-derived macrophages, LSECs, infiltrating leukocytes, and nerve cells (20, 21). HSCs are identified by expression of both mesenchymal and neuronal markers including desmin, vimentin, nestin, and glial fibrillary acidic protein (GFAP) (22). Under physiological circumstance, HSCs sustain a non-proliferative and quiescent phenotype with angular, rounded cell bodies, extended cytoplasmic processes, and unique vitamin A storage in lipid droplets (23). In normal liver, HSCs contribute to liver regeneration, regulation of sinusoidal circulation, and vitamin A storage and release (24–26). HSCs are important sources of myofibroblasts during liver damage (27). When liver injury occurs, however, HSCs become activated and transdifferentiate into proliferative, contractile, and inflammatory myofibroblasts, which are characterized by secretion of extracellular matrix (ECM) molecules (28, 29). In this condition, HSCs are marked by expression of α-smooth muscle actin (α-SMA) (30). Activated HSCs secrete endothelin-1 (ET-1), which is a molecule with potent vesoconstricting effect, promoting proliferation and fibrogenesis, and thus is supposed to contribute to portal hypertension (31, 32). HSCs have been identified as a critical driver of fibrosis in liver injury (19, 33). It is postulated that during liver IRI, HSCs are activated by TNF-α, IL-6, and nitric oxide (NO), followed by transdifferentiation into myofibroblast phenotype (17). Activation of HSCs results in secretion of matrix metalloproteinases (MMPs), cytokines, and chemokines, leading to ECM destruction, further activation of HSC, and infiltration of neutrophils and platelets (17). These effects imply HSCs can play an important role in regulating hepatic inflammation during IRI.
Functional roles of HSCs in injury stage of hepatic IRI has received much less attention than Kupffer cells, partly because functional inhibitor of Kupffer cells, gadolinium chloride, has enabled direct manipulation of these cells in experimental models (34). HSCs as a whole promote liver damage in the early phase of IRI, but they may mediate protective effect upon some pharmacological interventions or external stimuli as well. Thus HSCs may be regulated by specific signaling to combat IRI in the liver. Figure 1 summarizes the molecular mechanisms of liver IRI mediated by HSCs, which are discussed below.
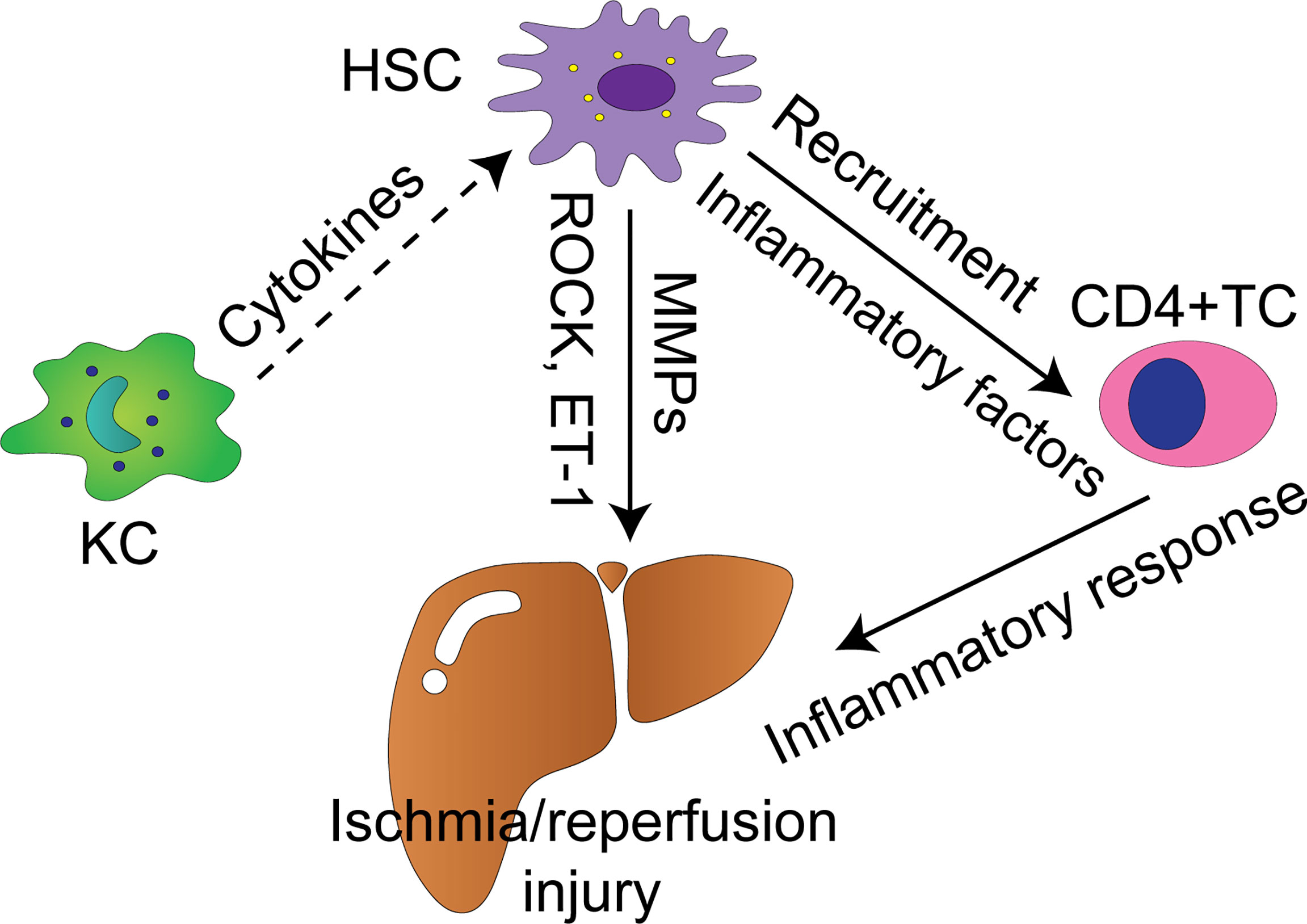
Figure 1 Cellular and molecular mechanisms by which HSCs modulate liver IRI. Solid arrows indicate positively regulatory effect with supporting experiment data, whereas dash arrows indicate putative positively regulatory effect. KC, Kupffer cell; TC, T cell; HSC, hepatic stellate cell; MMPs, matrix metalloproteinases; ROCK, Rho-associated coiled-coil forming protein serine/threonine kinase; ET-1, endothelin-1.
The involvement of HSCs in liver IRI was proposed based on preliminary observational studies. For instance, Takeda et al. found that heparin diminished serum levels of ET-1, aspartate transaminase (AST) and recovered hepatic IRI induced disturbance of oxidized and deoxidized hemoglobin after 1 h of IRI in rabbit model. Interestingly, electron microscopy revealed that IRI induced structural alteration of HSCs, which were target cells for ET-1, was normalized upon administration of heparin (35). These results suggested that HSCs mediated liver protective effect of heparin via microcirculation regulatory molecule ET-1. Further studies supported HSC’s role in hepatic IRI was partly mediated by regulation of microcirculation. HSCs play a key role in regulating hepatic microcirculation. In response to some stimuli, HSCs can contract or relax themselves, thus enlarging or shrinking the diameter of the sinusoidal lumen (36). Rho family of small GTPases regulates cell structure and motility mainly via rearrangement of actin cytoskeletons (37, 38). Rho-associated coiled-coil forming protein serine/threonine kinase (ROCK) was identified as one of the critical regulators of HSCs motility (39). In contrast to the scenario where HSCs appear to protect hepatocytes against IRI, HSCs drive liver injury mediated partly by ROCK. Administration of a specific inhibitor of ROCK named Y-27632 in rat attenuated IRI induced liver dysfunction manifested as increased deoxidized hemoglobin, decreased cytochrome oxidase, and elevated transaminase 1 h after reperfusion (40, 41). Consistently, Y-27632 resulted in relaxation of HSCs even in the presence of ET-1 in vitro (41). Liver protective effect of ROCK inhibition on IRI was also confirmed by another study in rat with steatotic liver. Kuroda et al. demonstrated that suppressing ROCK with specific inhibitor fasudil ameliorated IRI induced increase in portal perfusion pressure and liver damage at early stage of IRI in steatotic liver (42). In particular, Rho/ROCK signaling in HSCs from steatotic livers was activated and the activation was related to increased contractility and ET-1, making it more vulnerable to IRI (42). Specificity of the effect of ROCK regulator on HSCs was further supported by the findings that HSCs targeting inhibition of ROCK by vitamin A-coupled liposomes suppressed HSC activation, hepatic blood supply, portal perfusion pressure during early hepatic IRI, and improved survival rate after the damage in rat steatotic liver (43).
A more clear landscape of functions of HSC in liver IRI is delineated by specific manipulating approaches for HSCs in vivo. Functional experiments in rodent model suggest that HSCs exacerbate injury during hepatic IRI mediated by TNF-α and ET-1. Exploiting genetically engineered mice expressing HSV-thymidine kinase under the GFAP promoter coupled with ganciclovir and CCL4 to eliminate actively proliferating HSCs, a seminal research by Stewart et al. showed that hepatic injury in both IRI and endotoxemia scenarios was attenuated in HSCs depleted mice (approximately 70%) (44, 45). The decreased injury was accompanied by significantly reduced hepatocyte pro-inflammatory cytokine TNF-α, neutrophil expression of chemoattractant CXCL1 and endothelin-A receptor (45). Of note, liver IRI and endotixin-induced acute injury might share similar cellular pathogenesis via HSC regulated inflammation. However, the time to evaluate the liver damage, mRNA and protein expression was not mentioned in this study, making it not feasible to infer whether HSCs regulate early or late liver IRI. Pharmacological approach alone has also been found to deplete HSCs in vivo. Gliotoxin induces apoptosis in both human and rat HSCs in vitro, and rat HSCs in vivo leading to resolution of fibrosis (46–48). Liver IRI in early phase was significantly reduced and sinusoidal perfusion was recovered by pretreatment with gliotoxin in HSCs decreased rat, suggesting HSCs exerted exacerbating effect on the magnitude of early liver ischemic injury (49).
Matrix metalloproteinases (MMPs) are a family of zinc-dependent proteases which are essential in the degradation of ECM to enable cellular movement and tissue reorganization (50). MMP activation and release are involved in liver IRI due to their profound effects on tissue integrity (51). It has been asserted that prolonged or over expression of MMP exerts harmful effects on the liver (52). Kupffer cells and HSCs are main sources of MMPs in the liver, although LSECs and leukocytes can also secrete MMPs (50, 53, 54). The involvement of MMPs in liver IRI is demonstrated by their concomitant expression and functional effects. HSCs contribute to MMP-9 production in the liver (55, 56). MMP-9 is increased 6 h after hepatic IRI in the steatotic rat orthotopic liver transplantation (OLT) model, and serum MMP-9 is elevated significantly 7 d post IRI in human OLT (57, 58). Furthermore, several MMPs, including MMP-9, are induced during early phase of liver IRI, and blocking MMPs with specific inhibitors reduces liver IRI and the release of proinflammatory cytokines (12, 59). MMP-9 deficiency and anti-MMP-9 neutralizing monoclonal antibodies also result in protection against damage during early phase of liver IRI in mice (60).
Despite the in-vivo data indicating HSCs exacerbate damage during IRI collectively, they may aid in or mediate liver protection via distinct signals. ROS have been proposed as key initiators of IRI response in the liver by causing direct cellular damage and inducing inflammatory response via high mobility group box‐1 (HMGB1) and NF-κB (61–63). An in-vitro study suggested that HSCs protected hepatocytes against ROS injury (64). In addition, pretreating mice with HSCs ameliorated liver IRI at 12 h after reperfusion in a regulatory T cells (Tregs)-dependent manner (65). It should be noted that, however, the HSCs administered were primary cell lines not subject to activation following hypoxia/reoxygenation (H/R) stress, which could largely explain the differences with HSC depletion results in-vivo. Post-conditioning with the volatile anesthetic drug sevoflurane protected the liver from IRI 1 d post reperfusion in a randomized controlled trial, and in-vitro study suggested that HSC might be the effector of the protection by reducing apoptosis of hepatocytes (66, 67). More specifically, supernatants of HSCs previously exposed to H/R induced apoptosis of hepatocytes, but this effect was attenuated with sevoflurane postconditioning (67). Fibroblast growth factor 10 (FGF10) belongs to the fibroblast growth factor (FGF) family, whose members play crucial roles in organ development, homeostasis, and repair (68). FGF10 binds to fibroblast growth factor receptor 2b (FGFR2b) and this signaling controls hepatoblast survival and liver size (69, 70). Li et al. demonstrated that HSCs secreted fibroblast growth factor 10 (FGF10) in vitro, which ameliorated inflammation and necrosis, and protected hepatocytes from apoptosis during early phase of liver IRI in vivo (71). These results elucidated the protective effects of FGF10 in early liver IRI, and strongly implied these effects were modulated by HSCs.
The liver has a large regenerative capacity following physical or functional loss, with the potential of hepatocyte proliferation to sustain liver function. Necrotic tissue in the postischemic liver is cleared and remodeled by phagocytes, HSCs, and other cells, followed by hepatocyte regeneration and reconstruction of functional liver architecture (14). Far less is elucidated about the mechanisms of these processes compared with the mechanisms of hepatic IRI. Particularly, the role of HSCs in the process of liver repair after IRI is not clear (72).
MMPs derived from HSCs may promote liver repair after IRI in the liver, although they have been shown to promote damage by destruction of ECM and recruitment of leukocytes (73). Specifically, reduction in liver damage at 24 h after reperfusion and significant delay of liver repair after 72 h of reperfusion were observed in MMP-9 knockout mice, compared with wild type mice (74). MMP-9 was found to increase TGF-β activation after IRI. In-vitro study showed that MMP-9 activated TGF-β secreted by HSCs, indicating involvement of HSC in liver repair (74). A recent study exquisitely examined pathology of liver fibrosis during the repair process after IRI and highlighted involvement of HSC and MMPs. Konishi et al. found that the number of activated HSCs increased along the damaged areas 1 wk after IRI (72). Liver fibrosis took place at the interface between necrotic site and regenerating liver associated with HSCs during the reparative process after IRI, and noticeably, the number of HSCs decreased shortly after resolution of injury and restoration of disrupted liver structure. They also investigated the expression of several MMPs related to degradation of extracellular matrix components and reported that the expression of MMP-2 and MMP-9 increased at 1 wk after liver IRI and diminished thereafter. In contrast, MMP-13 expression remained at low level 1 wk after IRI but significantly elevated after 2 wk and the trend was stable up to 8 wk post IRI. The trends of MMP-2 and MMP-9 expression were associated with resolution of liver fibrosis and concomitant increase and decrease thereafter in the number of activated HSCs (72). Akin to MMP-9, MMP-13 is expressed in and produced by HSCs (75, 76). In relation to the injury stage, MMP-9 plays both deleterious and protective roles in hepatic IRI, which is dependent on the timing (74). It can be inferred from the pathological findings and source of MMPs that HSCs participate in the reparative process after liver IRI.
Recent innovative works involving Yes-associated protein (YAP) also underline the critical role of HSC in liver repair and regeneration after IRI. YAP and transcriptional coactivator with PDZ-binding motif (TAZ) are downstream effectors of the Hippo signalling pathway, which have been identified as essential regulators controlling cellular proliferation and organ size (77). Marked activation and proliferation of HSCs was observed at both injury and repair/regeneration phases after liver IRI in mice, with concurrent selective activation of YAP and TAZ and expression of their target genes. Inhibiting YAP and TAZ after injury phase significantly diminished HSC and hepatocyte proliferation, suggesting the dependence of liver repair and regeneration after IRI on HSC (78). Liu et al. demonstrated that YAP inhibition prior to ischemia and reperfusion operation delayed liver repair and increased hepatic fibrosis at 7 days after IRI. These changes were associated with enhanced HSC stimulation manifested as fibrogenic and contractile characteristics in-vitro (79). This study indicated HSC might serve as a regulator of liver repair and fibrogenesis in an YAP dependent manner.
HSCs contribute to liver fibrosis during reparative process after IRI. ECM accumulation generated by HSCs potentiates at the boundary between necrotic and hepatocyte regenerating region. Resolution of liver fibrosis is associated with decreased activation of HSC (72). In fibrotic liver, HSCs may also promote liver repair after IRI. Fibrotic liver shows more severe injury but more rapid repair and regeneration compared with nonfibrotic liver in mice, which are accompanied by prominent accumulation of HSCs in fibrotic liver in early reparative phase (80).
The signaling cascades leading to hepatic damage are various and complex, involving interactions between hepatocytes, Kupffer cells, HSCs, LSECs, recruited neutrophils, macrophages, and platelets (81). HSCs are highly versatile cells with complex crosstalk with residential hepatic cells and circulating immune cells, including hepatocytes, Kupffer cells, LSECs, natural killer cells (NK cells), T cells, and B cells (26, 27, 82, 83). This notion is demonstrated with enormous evidence mainly in the context of chronic hepatic injury leading to hepatic fibrosis, such as viral infection and alcoholic liver disease, but only a few works elucidate the crosstalk involving HSCs in liver IRI. As mentioned above, Kupffer cells are fundamental drivers of the early hepatic IRI. The crosstalk between Kupffer cells and HSCs were validated by in-vitro model (84, 85). This crosstalk was mediated by H2O2 and IL-6 (84). Furthermore, Kupffer cells can activate HSCs in vitro and in vivo mediated by IL-1 and TNF during fibrogenesis (86). It is believed that TNF-α and IL-6 released by Kupffer cells activate HSCs in the early phase of liver IRI (17). As liver fibrosis is a component of liver repair after IRI (72), Kupffer cells may induce activation and proliferation of HSCs in the recovery of IRI. In the scenario of liver fibrosis following chronic liver injury, activation and proliferation of HSCs are induced by TNF-α, IL-6, TGF-β, platelet-derived growth factor (PDGF), and ROS secreted by Kupffer cells (87, 88). CD4+ T cells are essential in promoting pro-inflammatory immune response in the liver and play an important role in hepatic IRI (89–91). Reifart et al. reported that CD4+ T cells interacted with HSCs along their migration to the liver in vivo. Depletion of HSCs diminished CD4+ T cell recruitment to the postischemic tissue and protected the liver from IRI (92). LSECs form the vascular wall of the liver sinusoid and play crucial protective roles in vascular homeostasis, and inflammation. LSECs are prominently vulverable to IRI, making them one of a key factors leading to hepatic IRI (81). LSECs suffering from ischemic challenge decrease production of NO, and together with elevated ET-1 production, contribute to contraction of HSCs. These events lead to narrowing of the sinusoidal lumen and microcirculatory dysfunction (81, 93). Cellular and molecular mechanisms by which HSCs regulate hepatic IRI are shown in Figure 1.
Despite decades of research into the development of liver IRI and its intervention, liver IRI is still a major cause of mortality and morbidity after hepatic surgery and transplantation. Much less attention has been focused on the roles of HSCs in liver IRI compared to other cell types involved. HSCs become activated and proliferate in response to IRI, likely through signals from Kupffer cells. HSCs promote early phase hepatic IRI by constraining hepatic microcirculation mediated by ROCK, effects of ET-1 signalling, and pro-inflammatory cascades initiated by TNF-α. MMPs derived from HSCs may also increase damage by destruction of ECM and recruitment of leukocytes. HSCs can mediate hepatic protective effect via external stimuli such as sevoflurane, and FGF10. During the repair and regeneration stage, HSCs play an fundamental role in potentiating liver recovery. Molecular mechanisms involve activation of TGF-β signalling pathway by MMP-9, activation of YAP and TAZ. During the reparative stage of liver IRI, HSCs also regulate fibrogenesis, the extent of which may be critical to functional recovery of the liver.
Future research regarding involvement of HSCs in liver IRI can be aimed at three directions to aid in better understanding of the pathophysiology of IRI and development of novel therapeutic interventions. Intercellular communications between HSCs and other cell types should be studied using in vivo visualization techniques and cell-type specific genetic animal models. Furthermore, it is clinically useful to identify HSCs derived biomarkers predictive of transplantation outcomes with less expensive modern multi-omics technologies. It is of paramount importance to screen and identify novel agents to ameliorate hepatic IRI, given that clinical trials of many drugs targeting HSCs for anti-fibrosis are completed or under way (26). An update of the clinical trials and drugs is shown in Table 1 (94–99). Because HSCs contribute to damage and repair of liver IRI, it is likely that anti-fibrotic drug has an effect on combating IRI.

Table 1 Clinical trials of drugs targeting HSC activation.
YP, MY, LC, XS, WX, JL, and QY contributed to the writing and editing of the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by Technical Innovation Project of Hunan Provincial Health Commission (No.: Memo [2018]187 of Xiangwei Medical Administration Office), International Talent Project of Hunan Children’s Hospital.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Byass P. The global burden of liver disease: A challenge for methods and for public health. BMC Med (2014), 12:159. doi: 10.1186/s12916-014-0159-5
2. Saidi RF, Kenari SKH. Liver ischemia/reperfusion injury: an overview. J Invest Surg (2014) 27(6):366–79. doi: 10.3109/08941939.2014.932473
3. Rampes S, Ma D. Hepatic ischemia-reperfusion injury in liver transplant setting: mechanisms and protective strategies. J BioMed Res (2019) 33(4):221–34. doi: 10.7555/JBR.32.20180087
4. Zhai Y, Petrowsky H, Hong JC, Busuttil RW, Kupiec-Weglinski JW. Ischaemia–reperfusion injury in liver transplantation–from bench to bedside. Nat Rev Gastroenterol Hepatol (2013) 10(2):79–89. doi: 10.1038/nrgastro.2012.225
5. Kim YI. Ischemia-reperfusion injury of the human liver during hepatic resection. J Hepatobil Pancreat Surg (2003) 10(3):195–9. doi: 10.1007/s00534-002-0730-x
6. Huguet C, Gavelli A, Bona S. Hepatic resection with ischemia of the liver exceeding one hour. J Am Coll Surg (1994) 178(5):454–8.
7. Lemasters JJ, Thurman RG. Reperfusion injury after liver preservation for transplantation. Annu Rev Pharmacol Toxicol (1997) 37:327–38. doi: 10.1146/annurev.pharmtox.37.1.327
8. Abu-Amara M, Yang SY, Tapuria N, Fuller B, Davidson B, Seifalian A. Liver ischemia/reperfusion injury: processes in inflammatory networks–a review. Liver Transpl (2010) 16(9):1016–32. doi: 10.1002/lt.22117
9. Kostakis ID, Sikalias N, Alexiou K, Mountzalia L, Papalois A, Karatzas T. How much ischemia can the severely steatotic rat liver tolerate? In Vivo (2018) 32(6):1381–6. doi: 10.21873/invivo.11390
10. Casillas-Ramírez A, Mosbah IB, Ramalho F, Roselló-Catafau J, Peralta C. Past and future approaches to ischemia-reperfusion lesion associated with liver transplantation. Life Sci (2006) 79(20):1881–94. doi: 10.1016/j.lfs.2006.06.024
11. Jaeschke H, Farhood A. Neutrophil and kupffer cell-induced oxidant stress and ischemia-reperfusion injury in rat liver. Am J Physiol (1991) 260(3 Pt 1):G355–362. doi: 10.1152/ajpgi.1991.260.3.G355
12. Cursio R, Mari B, Louis K, Rostagno P, Saint-Paul MC, Giudicelli J, et al. Rat liver injury after normothermic ischemia is prevented by a phosphinic matrix metalloproteinase inhibitor. FASEB J (2002) 16(1):93–5. doi: 10.1096/fj.01-0279fje
13. Crockett ET, Galligan JJ, Uhal BD, Harkema J, Roth R, Pandya K. Protection of early phase hepatic ischemia-reperfusion injury by cholinergic agonists. BMC Clin Pathol (2006) 6:3. doi: 10.1186/1472-6890-6-3
14. Konishi T, Lentsch AB. Hepatic Ischemia/Reperfusion: Mechanisms of tissue injury, repair, and regeneration. Gene Expr (2017) 17(4):277–87. doi: 10.3727/105221617X15042750874156
15. Granger DN, Kvietys PR. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol (2015) 6:524–51. doi: 10.1016/j.redox.2015.08.020
16. Hirao H, Nakamura K, Kupiec-Weglinski JW. Liver ischaemia-reperfusion injury: a new understanding of the role of innate immunity. Nat Rev Gastroenterol Hepatol (2022) 19(4):239–56. doi: 10.1038/s41575-021-00549-8
17. Dar WA, Sullivan E, Bynon JS, Eltzschig H, Ju C. Ischaemia reperfusion injury in liver transplantation: Cellular and molecular mechanisms. Liver Int (2019) 39(5):788–801. doi: 10.1111/liv.14091
18. Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology (2015) 62(2):600–14. doi: 10.1002/hep.27841
19. Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem (2000) 275(4):2247–50. doi: 10.1074/jbc.275.4.2247
20. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev (2008) 88(1):125–72. doi: 10.1152/physrev.00013.2007
21. Heymann F, Tacke F. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol (2016) 13(2):88–110. doi: 10.1038/nrgastro.2015.200
22. Asahina K. Hepatic stellate cell progenitor cells. J Gastroenterol Hepatol (2012) 27(s2):80–4. doi: 10.1111/j.1440-1746.2011.07001.x
23. Schachtrup C, Le Moan N, Passino MA, Akassoglou K. Hepatic stellate cells and astrocytes. Cell Cycle (2011) 10(11):1764–71. doi: 10.4161/cc.10.11.15828
24. Yin C, Evason KJ, Asahina K, Stainier DYR. Hepatic stellate cells in liver development, regeneration, and cancer. J Clin Invest (2013) 123(5):1902–10. doi: 10.1172/JCI66369
25. Zhang F, Wang F, He J, Lian N, Wang Z, Shao J, et al. Regulation of hepatic stellate cell contraction and cirrhotic portal hypertension by wnt/β-catenin signalling via interaction with Gli1. Br J Pharmacol (2021) 178(11):2246–65. doi: 10.1111/bph.15289
26. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol (2017) 14(7):397–411. doi: 10.1038/nrgastro.2017.38
27. Ezhilarasan D. Hepatic stellate cells in the injured liver: Perspectives beyond hepatic fibrosis. J Cell Physiol (2022) 237(1):436–49. doi: 10.1002/jcp.30582
28. Kisseleva T, Brenner DA. Hepatic stellate cells and the reversal of fibrosis. J Gastroenterol Hepatol (2006) 21 Suppl 3:S84–87. doi: 10.1111/j.1440-1746.2006.04584.x
29. Puche JE, Saiman Y, Friedman SL. Hepatic stellate cells and liver fibrosis. Compr Physiol (2013) 3(4):1473–92. doi: 10.1002/cphy.c120035
30. Cheng F, Li Y, Feng L, Li S. Hepatic stellate cell activation and hepatic fibrosis induced by ischemia/reperfusion injury. Transplant Proc (2008) 40(7):2167–70. doi: 10.1016/j.transproceed.2008.06.052
31. Li T, Shi Z, Rockey DC. Preproendothelin-1 expression is negatively regulated by IFNγ during hepatic stellate cell activation. Am J Physiol Gastrointest Liver Physiol (2012) 302(9):G948–957. doi: 10.1152/ajpgi.00359.2011
32. Rockey DC. Hepatic blood flow regulation by stellate cells in normal and injured liver. Semin Liver Dis (2001) 21(3):337–49. doi: 10.1055/s-2001-17551
33. Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Deliv Rev (2017) 121:27–42. doi: 10.1016/j.addr.2017.05.007
34. Suzuki S, Toledo-Pereyra LH, Rodriguez F, Lopez F. Role of kupffer cells in neutrophil activation and infiltration following total hepatic ischemia and reperfusion. Circ Shock (1994) 42(4):204–9.
35. Matsumoto T, Yamaguchi M, Kikuchi H, Nakano H, Midorikawa T, Kumada K, et al. Heparin reduces serum levels of endothelin-1 and hepatic ischemia reperfusion injury in rabbits. Surg Today (2000) 30(6):523–5. doi: 10.1007/s005950070119
36. Zhang JX, Bauer M, Clemens MG. Vessel- and target cell-specific actions of endothelin-1 and endothelin-3 in rat liver. Am J Physiol (1995) 269(2 Pt 1):G269–277. doi: 10.1152/ajpgi.1995.269.2.G269
37. Hall A. Rho GTPases and the actin cytoskeleton. Science (1998) 279(5350):509–14. doi: 10.1126/science.279.5350.509
38. Schaks M, Giannone G, Rottner K. Actin dynamics in cell migration. Essays Biochem (2019) 63(5):483–95. doi: 10.1042/EBC20190015
39. Kawada N, Seki S, Kuroki T, Kaneda K. ROCK inhibitor y-27632 attenuates stellate cell contraction and portal pressure increase induced by endothelin-1. Biochem Biophys Res Commun (1999) 266(2):296–300. doi: 10.1006/bbrc.1999.1823
40. Ishizaki T, Uehata M, Tamechika I, Keel J, Nonomura K, Maekawa M, et al. Pharmacological properties of y-27632, a specific inhibitor of rho-associated kinases. Mol Pharmacol (2000) 57(5):976–83.
41. Mizunuma K, Ohdan H, Tashiro H, Fudaba Y, Ito H, Asahara T. Prevention of ischemia-reperfusion-induced hepatic microcirculatory disruption by inhibiting stellate cell contraction using rock inhibitor. Transplantation (2003) 75(5):579–86. doi: 10.1097/01.TP.0000052593.16876.AF
42. Kuroda S, Tashiro H, Igarashi Y, Tanimoto Y, Nambu J, Oshita A, et al. Rho inhibitor prevents ischemia-reperfusion injury in rat steatotic liver. J Hepatol (2012) 56(1):146–52. doi: 10.1016/j.jhep.2011.04.029
43. Kuroda S, Tashiro H, Kimura Y, Hirata K, Tsutada M, Mikuriya Y, et al. Rho-kinase inhibitor targeting the liver prevents ischemia/reperfusion injury in the steatotic liver without major systemic adversity in rats. Liver Transpl (2015) 21(1):123–31. doi: 10.1002/lt.24020
44. Geerts A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin Liver Dis (2001) 21(3):311–35. doi: 10.1055/s-2001-17550
45. Stewart RK, Dangi A, Huang C, Murase N, Kimura S, Stolz DB, et al. A novel mouse model of depletion of stellate cells clarifies their role in ischemia/reperfusion- and endotoxin-induced acute liver injury. J Hepatol (2014) 60(2):298–305. doi: 10.1016/j.jhep.2013.09.013
46. Wright MC, Issa R, Smart DE, Trim N, Murray GI, Primrose JN, et al. Gliotoxin stimulates the apoptosis of human and rat hepatic stellate cells and enhances the resolution of liver fibrosis in rats. Gastroenterology (2001) 121(3):685–98. doi: 10.1053/gast.2001.27188
47. Orr JG, Leel V, Cameron GA, Marek CJ, Haughton EL, Elrick LJ, et al. Mechanism of action of the antifibrogenic compound gliotoxin in rat liver cells. Hepatology (2004) 40(1):232–42. doi: 10.1002/hep.20254
48. Dekel R, Zvibel I, Brill S, Brazovsky E, Halpern Z, Oren R. Gliotoxin ameliorates development of fibrosis and cirrhosis in a thioacetamide rat model. Dig Dis Sci (2003) 48(8):1642–7. doi: 10.1023/A:1024792529601
49. Takahashi T, Yoshioka M, Uchinami H, Nakagawa Y, Otsuka N, Motoyama S, et al. Hepatic stellate cells play a functional role in exacerbating ischemia-reperfusion injury in rat liver. Eur Surg Res (2019) 60(1–2):74–85. doi: 10.1159/000499750
50. Wang X, Khalil RA. Matrix metalloproteinases, vascular remodeling, and vascular disease. Adv Pharmacol (2018) 81:241–330. doi: 10.1016/bs.apha.2017.08.002
51. Viappiani S, Sariahmetoglu M, Schulz R. The role of matrix metalloproteinase inhibitors in ischemia-reperfusion injury in the liver. Curr Pharm Des (2006) 12(23):2923–34. doi: 10.2174/138161206777947560
52. Palladini G, Ferrigno A, Richelmi P, Perlini S, Vairetti M. Role of matrix metalloproteinases in cholestasis and hepatic ischemia/reperfusion injury: A review. World J Gastroenterol (2015) 21(42):12114–24. doi: 10.3748/wjg.v21.i42.12114
53. Mormone E, George J, Nieto N. Molecular pathogenesis of hepatic fibrosis and current therapeutic approaches. Chem Biol Interact (2011) 193(3):225–31. doi: 10.1016/j.cbi.2011.07.001
54. Deleve LD, Wang X, Tsai J, Kanel G, Strasberg S, Tokes ZA. Sinusoidal obstruction syndrome (veno-occlusive disease) in the rat is prevented by matrix metalloproteinase inhibition. Gastroenterology (2003) 125(3):882–90. doi: 10.1016/S0016-5085(03)01056-4
55. Yp H. Matrix metalloproteinases, the pros and cons, in liver fibrosis. J Gastroenterol Hepatol (2006) 21 Suppl 3(Suppl 3):88–91. doi: 10.1111/j.1440-1746.2006.04586.x
56. Han YP, Yan C, Zhou L, Qin L, Tsukamoto H. A matrix metalloproteinase-9 activation cascade by hepatic stellate cells in trans-differentiation in the three-dimensional extracellular matrix. J Biol Chem (2007) 282(17):12928–39. doi: 10.1074/jbc.M700554200
57. Moore C, Shen XD, Gao F, Busuttil RW, Coito AJ. Fibronectin-alpha4beta1 integrin interactions regulate metalloproteinase-9 expression in steatotic liver ischemia and reperfusion injury. Am J Pathol (2007) 170(2):567–77. doi: 10.2353/ajpath.2007.060456
58. Kuyvenhoven JP, Ringers J, Verspaget HW, Lamers CBHW, van Hoek B. Serum matrix metalloproteinase MMP-2 and MMP-9 in the late phase of ischemia and reperfusion injury in human orthotopic liver transplantation. Transplant Proc (2003) 35(8):2967–9. doi: 10.1016/j.transproceed.2003.10.049
59. Shirahane K, Yamaguchi K, Koga K, Watanabe M, Kuroki S, Tanaka M. Hepatic ischemia/reperfusion injury is prevented by a novel matrix metalloproteinase inhibitor, ONO-4817. Surgery (2006) 139(5):653–64. doi: 10.1016/j.surg.2005.10.002
60. Hamada T, Fondevila C, Busuttil RW, Coito AJ. Metalloproteinase-9 deficiency protects against hepatic ischemia/reperfusion injury. Hepatology (2008) 47(1):186–98. doi: 10.1002/hep.21922
61. van Golen RF, van Gulik TM, Heger M. The sterile immune response during hepatic ischemia/reperfusion. Cytokine Growth Factor Rev (2012) 23(3):69–84. doi: 10.1016/j.cytogfr.2012.04.006
62. Reiniers MJ, van Golen RF, van Gulik TM, Heger M. Reactive oxygen and nitrogen species in steatotic hepatocytes: a molecular perspective on the pathophysiology of ischemia-reperfusion injury in the fatty liver. Antioxid Redox Signal (2014) 21(7):1119–42. doi: 10.1089/ars.2013.5486
63. Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology (2011) 54(1):133–44. doi: 10.1002/hep.24341
64. Jameel NM, Thirunavukkarasu C, Murase N, Cascio M, Prelich J, Yang S, et al. Constitutive release of powerful antioxidant-scavenging activity by hepatic stellate cells: protection of hepatocytes from ischemia/reperfusion injury. Liver Transpl (2010) 16(12):1400–9. doi: 10.1002/lt.22172
65. Feng M, Wang Q, Wang H, Wang M, Guan W, Lu L. Adoptive transfer of hepatic stellate cells ameliorates liver ischemia reperfusion injury through enriching regulatory T cells. Int Immunopharmacol (2014) 19(2):267–74. doi: 10.1016/j.intimp.2014.01.006
66. Beck-Schimmer B, Breitenstein S, Bonvini JM, Lesurtel M, Ganter M, Weber A, et al. Protection of pharmacological postconditioning in liver surgery: results of a prospective randomized controlled trial. Ann Surg (2012) 256(5):837–44; discission 844-845. doi: 10.1097/SLA.0b013e318272df7c
67. Beck-Schimmer B, Roth Z’graggen B, Booy C, Köppel S, Spahn DR, Schläpfer M, et al. Sevoflurane protects hepatocytes from ischemic injury by reducing reactive oxygen species signaling of hepatic stellate cells: translational findings based on a clinical trial. Anesth Analg (2018) 127(4):1058–65. doi: 10.1213/ANE.0000000000003692
68. Itoh N, Ornitz DM. Fibroblast growth factors: from molecular evolution to roles in development, metabolism and disease. J Biochem (2011) 149(2):121–30. doi: 10.1093/jb/mvq121
69. Watson J, Francavilla C. Regulation of FGF10 signaling in development and disease. Front Genet (2018) 9:500. doi: 10.3389/fgene.2018.00500
70. Berg T, Rountree CB, Lee L, Estrada J, Sala FG, Choe A, et al. Fibroblast growth factor 10 is critical for liver growth during embryogenesis and controls hepatoblast survival via β-catenin activation. Hepatology (2007) 46(4):1187–97. doi: 10.1002/hep.21814
71. Li S, Zhu Z, Xue M, Pan X, Tong G, Yi X, et al. The protective effects of fibroblast growth factor 10 against hepatic ischemia-reperfusion injury in mice. Redox Biol (2021) 40:101859. doi: 10.1016/j.redox.2021.101859
72. Konishi T, Schuster RM, Lentsch AB. Liver repair and regeneration after ischemia-reperfusion injury is associated with prolonged fibrosis. Am J Physiol Gastrointest Liver Physiol (2019) 316(3):G323–31. doi: 10.1152/ajpgi.00154.2018
73. Ji J. Dual role of matrix metalloprotease 9 in liver ischemia and reperfusion injury. J Surg Res (2013) 185(2):545–6. doi: 10.1016/j.jss.2012.12.028
74. Feng M, Wang H, Wang Q, Guan W. Matrix metalloprotease 9 promotes liver recovery from ischemia and reperfusion injury. J Surg Res (2013) 180(1):156–61. doi: 10.1016/j.jss.2012.09.042
75. Díaz-Sanjuán T, García-Ruiz I, Rodríguez-Juan C, Muñoz-Yagüe T, Solís-Muñoz P, Solís-Herruzo JA. Interferon alpha increases metalloproteinase-13 gene expression through a polyomavirus enhancer activator 3-dependent pathway in hepatic stellate cells. J Hepatol (2009) 50(1):128–39. doi: 10.1016/j.jhep.2008.07.034
76. George J, Tsutsumi M, Tsuchishima M. MMP-13 deletion decreases profibrogenic molecules and attenuates n-nitrosodimethylamine-induced liver injury and fibrosis in mice. J Cell Mol Med (2017) 21(12):3821–35. doi: 10.1111/jcmm.13304
77. Zhao B, Tumaneng K, Guan KL. The hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol (2011) 13(8):877–83. doi: 10.1038/ncb2303
78. Konishi T, Schuster RM, Lentsch AB. Proliferation of hepatic stellate cells, mediated by YAP and TAZ, contributes to liver repair and regeneration after liver ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol (2018) 314(4):G471–82. doi: 10.1152/ajpgi.00153.2017
79. Liu Y, Lu T, Zhang C, Xu J, Xue Z, Busuttil RW, et al. Activation of YAP attenuates hepatic damage and fibrosis in liver ischemia-reperfusion injury. J Hepatol (2019) 71(4):719–30. doi: 10.1016/j.jhep.2019.05.029
80. Konishi T, Schuster RM, Goetzman HS, Caldwell CC, Lentsch AB. Fibrotic liver has prompt recovery after ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol (2020) 318(3):G390–400. doi: 10.1152/ajpgi.00137.2019
81. Peralta C, Jiménez-Castro MB, Gracia-Sancho J. Hepatic ischemia and reperfusion injury: effects on the liver sinusoidal milieu. J Hepatol (2013) 59(5):1094–106. doi: 10.1016/j.jhep.2013.06.017
82. Fasbender F, Widera A, Hengstler JG, Watzl C. Natural killer cells and liver fibrosis. Front Immunol (2016) 7:19. doi: 10.3389/fimmu.2016.00019
83. Koda Y, Teratani T, Chu PS, Hagihara Y, Mikami Y, Harada Y, et al. CD8+ tissue-resident memory T cells promote liver fibrosis resolution by inducing apoptosis of hepatic stellate cells. Nat Commun (2021) 12(1):4474. doi: 10.1038/s41467-021-24734-0
84. Nieto N. Oxidative-stress and IL-6 mediate the fibrogenic effects of [corrected] kupffer cells on stellate cells. Hepatology (2006) 44(6):1487–501. doi: 10.1002/hep.21427
85. Cubero FJ, Nieto N. Ethanol and arachidonic acid synergize to activate kupffer cells and modulate the fibrogenic response via tumor necrosis factor alpha, reduced glutathione, and transforming growth factor beta-dependent mechanisms. Hepatology (2008) 48(6):2027–39. doi: 10.1002/hep.22592
86. Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology (2013) 58(4):1461–73. doi: 10.1002/hep.26429
87. Prosser CC, Yen RD, Wu J. Molecular therapy for hepatic injury and fibrosis: where are we? World J Gastroenterol (2006) 12(4):509–15. doi: 10.3748/wjg.v12.i4.509
88. Marrone G, Shah VH, Gracia-Sancho J. Sinusoidal communication in liver fibrosis and regeneration. J Hepatol (2016) 65(3):608–17. doi: 10.1016/j.jhep.2016.04.018
89. Zhai Y, Busuttil RW, Kupiec-Weglinski JW. Liver ischemia and reperfusion injury: new insights into mechanisms of innate-adaptive immune-mediated tissue inflammation. Am J Transplant (2011) 11(8):1563–9. doi: 10.1111/j.1600-6143.2011.03579.x
90. Shen X, Wang Y, Gao F, Ren F, Busuttil RW, Kupiec-Weglinski JW, et al. CD4 T cells promote tissue inflammation via CD40 signaling without de novo activation in a murine model of liver ischemia/reperfusion injury. Hepatology (2009) 50(5):1537–46. doi: 10.1002/hep.23153
91. Zhang Y, Ji H, Shen X, Cai J, Gao F, Koenig KM, et al. Targeting TIM-1 on CD4 T cells depresses macrophage activation and overcomes ischemia-reperfusion injury in mouse orthotopic liver transplantation. Am J Transplant (2013) 13(1):56–66. doi: 10.1111/j.1600-6143.2012.04316.x
92. Reifart J, Rentsch M, Mende K, Coletti R, Sobocan M, Thasler WE, et al. Modulating CD4+ T cell migration in the postischemic liver: hepatic stellate cells as new therapeutic target? Transplantation (2015) 99(1):41–7. doi: 10.1097/TP.0000000000000461
93. Serracino-Inglott F, Habib NA, Mathie RT. Hepatic ischemia-reperfusion injury. Am J Surg (2001) 181(2):160–6. doi: 10.1016/S0002-9610(00)00573-0
94. Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Or placebo for nonalcoholic steatohepatitis. N Engl J Med (2010) 362(18):1675–85. doi: 10.1056/NEJMoa0907929
95. Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall HU, Kipnes M, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology (2013) 145(3):574–82. doi: 10.1053/j.gastro.2013.05.042
96. Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet (2019) 394(10215):2184–96. doi: 10.1016/S0140-6736(19)33041-7
97. Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and -δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology (2016) 150(5):1147–1159.e5. doi: 10.1053/j.gastro.2016.01.038
98. Ratziu V, Sanyal A, Harrison SA, Wong VWS, Francque S, Goodman Z, et al. Cenicriviroc treatment for adults with nonalcoholic steatohepatitis and fibrosis: final analysis of the phase 2b CENTAUR study. Hepatology (2020) 72(3):892–905. doi: 10.1002/hep.31108
99. Chalasani N, Abdelmalek MF, Garcia-Tsao G, Vuppalanchi R, Alkhouri N, Rinella M, et al. Effects of belapectin, an inhibitor of galectin-3, in patients with nonalcoholic steatohepatitis with cirrhosis and portal hypertension. Gastroenterology (2020) 158(5):1334–1345.e5. doi: 10.1053/j.gastro.2019.11.296
Keywords: ischemia-reperfusion injury, hepatic stellate cells, hepatocytes, Kupffer cells, neutrophils, sinusoidal endothelial cells, liver transplantation, liver
Citation: Peng Y, Yin Q, Yuan M, Chen L, Shen X, Xie W and Liu J (2022) Role of hepatic stellate cells in liver ischemia-reperfusion injury. Front. Immunol. 13:891868. doi: 10.3389/fimmu.2022.891868
Received: 08 March 2022; Accepted: 27 June 2022;
Published: 28 July 2022.
Edited by:
Tao Qiu, Renmin Hospital of Wuhan University, ChinaReviewed by:
Edward N Harris, University of Nebraska System, United StatesCopyright © 2022 Peng, Yin, Yuan, Chen, Shen, Xie and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuming Peng, NDEyMTYyNTBAcXEuY29t; Qiang Yin, cWlhbmd5aW5AaG90bWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.