 May Y. Choi1,2,3*
May Y. Choi1,2,3* Karen H. Costenbader1
Karen H. Costenbader1- 1Brigham and Women’s Hospital and Harvard Medical School, Boston, MA, United States
- 2Department of Medicine, University of Calgary, Calgary, AB, Canada
- 3Cumming School of Medicine, McCaig Institute for Bone and Joint Health, Calgary, AB, Canada
There is growing evidence that preceding the diagnosis or classification of systemic lupus erythematosus (SLE), patients undergo a preclinical phase of disease where markers of inflammation and autoimmunity are already present. Not surprisingly then, even though SLE management has improved over the years, many patients will already have irreversible disease-related organ damage by time they have been diagnosed with SLE. By gaining a greater understanding of the pathogenesis of preclinical SLE, we can potentially identify patients earlier in the disease course who are at-risk of transitioning to full-blown SLE and implement preventative strategies. In this review, we discuss the current state of knowledge of SLE preclinical pathogenesis and propose a screening and preventative strategy that involves the use of promising biomarkers of early disease, modification of lifestyle and environmental risk factors, and initiation of preventative therapies, as examined in other autoimmune diseases such as rheumatoid arthritis and type 1 diabetes.
1 Introduction: Prediction and Possibly Prevention of SLE in the Near Future
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterized by immune dysregulation and systemic inflammation, leading to progressive and irreversible multi-organ damage. Although SLE is relatively uncommon [SLE affects ~25 to 50 per 100,000 persons in the United States (1, 2)], it disproportionately affects young women during their prime reproductive years, particularly those of non-White ancestry (3, 4). SLE remains among the leading causes of mortality in young females, underscoring its impact as an important public health issue (5, 6). With the discovery of more risk factors for SLE including genetics and environmental/lifestyle risk factors our ability to estimate SLE risk is improving, and thus so is the identification of patients who are at high versus low risk of this complex autoimmune disease.
A better understanding of SLE pathogenesis may enable earlier and more accurate identification of at-risk patients, as well as the discovery of therapeutic targets, and the design of prevention trials. However, since the breakthrough and serendipitous discovery of the Lupus Erythematous (LE) cell and its role in SLE pathogenesis in 1948 (7), are we any closer to achieving this goal? The LE cell provided evidence that autoantibodies are a key player in SLE pathogenesis, which are generated by a dysregulated immune system leading to immune complex formation and deposition, and subsequent inflammation and organ damage. In the 75 years that followed the LE cell identification, there was an explosion of serologic tests and technologies developed to detect autoantibodies, most centrally the antinuclear antibody (ANA) test, to aid in the diagnosis or classification of SLE [reviewed in (8)].
SLE is notoriously difficult to diagnose and classify because of the heterogeneity and non-specificity of clinical signs and symptoms in early disease. The diagnosis of SLE is thus frequently delayed such that by the time a formal diagnosis is confirmed, irreversible organ damage has already occurred. There are reports that the diagnosis of SLE is delayed by a median of 47 months, with patients submitting to an average of 10 consultations and evaluation by three different physicians before a diagnosis is finally made (9). A delay in SLE diagnosis has been associated with worse outcomes including higher disease activity, organ damage, lower quality of life, and remarkably increased healthcare costs (9). Organ damage occurring early in the disease course also has a negative impact on SLE patients, as it is associated with further damage, development of comorbidities and early mortality (10, 11). The classification criteria for SLE have been through several iterations to improve sensitivity and specificity, with the most recent criteria being the American College of Rheumatology (ACR)/EULAR (European League Against Rheumatism) 2019 criteria (12, 13). Unlike the others, one of the major differences with the new criteria is that it uses the “ANA at a titer of ≥1:80 on HEp-2 cells or an equivalent positive test at least once” as an entry criterion.
Despite advances in therapy, such as the recent approval of several new drugs (anifrolumab, voclosporin, and a new indication for belimumab) (14–16), without timely and accurate diagnosis to allow the initiation of evidence-based therapy, patients with SLE will continue to be at increased risk for morbidity, disability, and premature death secondary to cardiovascular events (e.g., strokes and myocardial infarction), malignancy, and infection, driven by uncontrolled inflammation (6, 17). Furthermore, antimalarials continue to be the mainstay therapy in SLE. Hydroxychloroquine (HCQ) has been shown to reduce SLE flares (lupus nephritis in particular), organ damage, pregnancy complications, cardiovascular events and survival (18–23). There is also evidence to suggest it can delay the onset of SLE, prompting a clinical trial that is currently underway to answer whether it can be used as a preventative therapy (18).
Emerging research suggests that our increasing knowledge about risk factors and biomarkers for SLE could lead to the identification of those at highest risk, and potentially then to early interventions prior to the onset of symptoms, to intercept and prevent this often-devastating disease. We review how current understanding of the development of SLE is contributing to progress in the identification of those who are developing disease, and how genetic and population risk factor studies are leading to the potential for disease prevention through early identification, environmental or lifestyle changes, and therapeutic interventions.
2 The pathogenesis of pre-clinical SLE and important biomarkers and risk factors
Understanding of the etiopathogenesis of SLE is evolving [reviewed in (24)]. The currently accepted model for multiple complex autoimmune diseases is that development takes place over time prior to diagnosis and in several stages (Figure 1). This next section will review the three phases that precede the diagnosis of SLE: 1) genetic risk, 2) asymptomatic autoimmunity and inflammation, and 3) early symptoms of lupus. As we discuss each phase, we will describe potential avenues of disease prevention including biomarkers for early disease detection and modifiable risk factors.
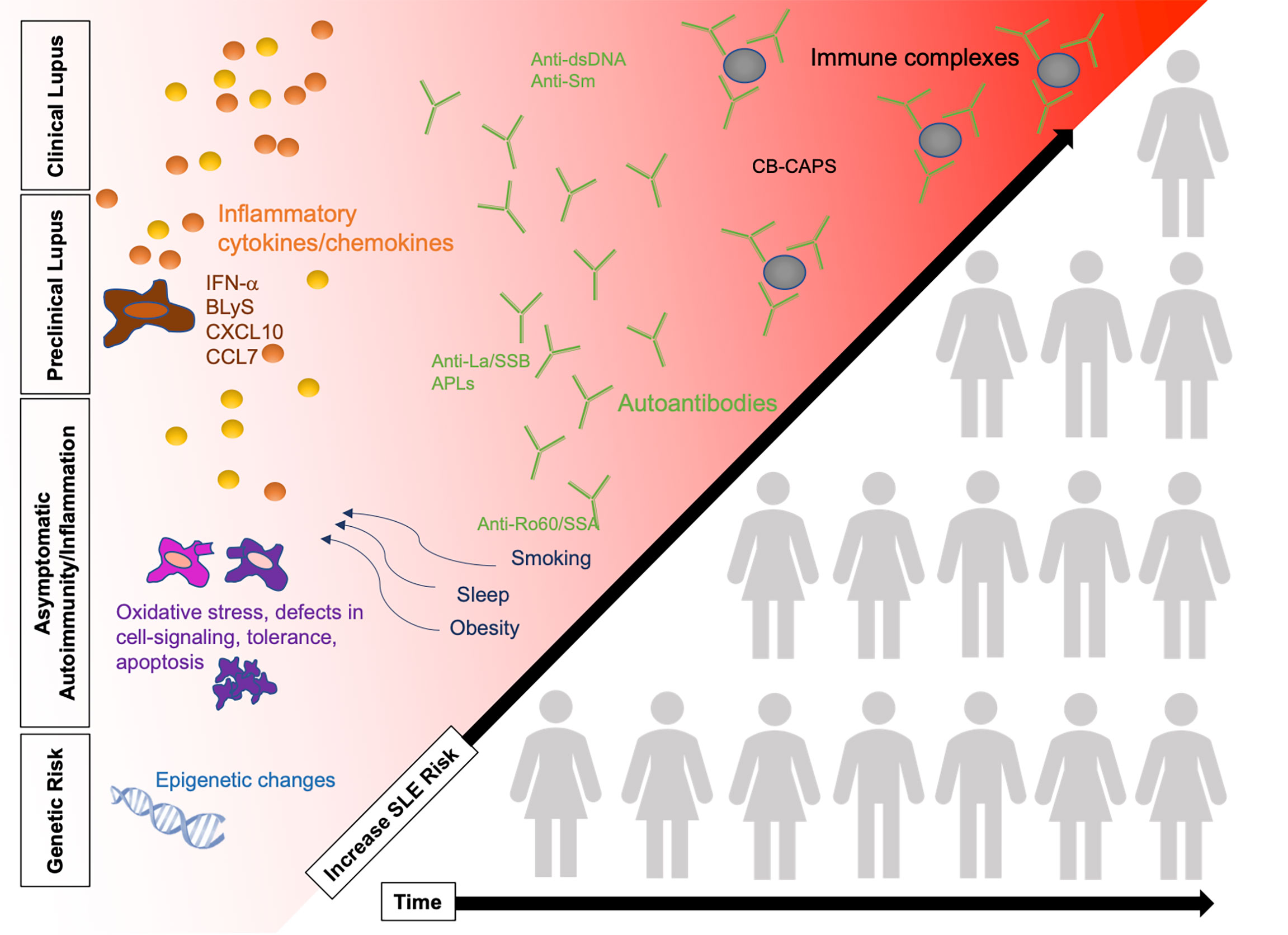
Figure 1 SLE pathogenesis in four phases, increasing in SLE risk over time as patients accumulate risk factors. Changes in the immune system are detected prior to the diagnosis of clinical SLE including presence of autoantibodies, cytokines, and immune complex deposition. Some patients (illustration not representative of actual pre-clinical/clinical SLE population) will progress over time to clinical SLE while others remain in the earlier stages of preclinical SLE. Refer to Figure 2 for potential points for early risk assessment and intervention opportunities. BLyS, B-cell lymphocyte stimulator; IFN, interferon; SLE, systemic lupus erythematosus.
2.1 Genetic Risk
SLE likely begins and is accelerated by a complex interplay between genetic risk, lifestyle and environmental risk factors and immune dysregulation. When individuals who possess SLE genetic risk alleles are exposed to environmental risk factors throughout their lives, synergistic interactions may take place, accelerating the onset of autoimmunity and inflammation. About 5-12% of subjects with a first-degree relative with SLE will develop the disease in their lifetime, whereas in persons with a congenital deficiency of the complement component C4, this risk can increase to 90% (25). Children who develop SLE appear to have a larger contribution of known SLE genetic risk, in particular non-HLA genes, than do adults with SLE, and thus the contribution of environmental exposures to SLE susceptibility may be increasingly important with advancing age (26, 27).
A series of landmark genome-wide association studies (GWAS) over the past decade in SLE have greatly expanded our understanding of the genetic basis of SLE [reviewed in (28, 29)]. To date, over 100 SLE susceptibility loci have been identified, predominantly in European and Asian populations, explaining up to 30% of SLE heritability (30–44). These include alleles in the Major Histocompatibility Complex (MHC) region (multiple genes), some of the Fcγ receptors, ATG5, BLK, BANK1, IRF5 (interferon regulatory factor 5), ITGAM, PDCD1, PTPN22, PXK, SPP1, STAT4, TNFSF4, TNFAIP3, XKR6, and deficiencies in complement components (29). Many of these genes belong to important pathways involved in immune complex clearance, host immune signal transduction, and pathways involving interferon (IFN), a key driving cytokine in many cases of SLE.
SLE-associated genes involved in the innate immune system have been gaining interest because of the “IFN signature”. Patients with SLE and high levels of IFN-α tend to have more severe disease manifestations (45). Normally, type I IFNs are produced during early response to viral infections and promote dendritic maturation and proinflammatory cytokines. This has several important effects on the immune system including the stimulation of the Th1 pathways, promotion of B-cell activation for autoantibody production, and regulation of apoptosis. One of these genes is IRF5, which regulates type I IFN-responsive genes. Outside of the MHC, it is one of the most strongly and consistently SLE-associated with a modest contribution to SLE risk (odds ratio 1.5) (46). The rs7574865 SNP risk variant of STAT4 has also shown to confer increased sensitivity to IFN-α signaling in peripheral blood mononuclear cells of SLE patients, and is associated with more severe disease, early disease onset and production of antibodies to double-stranded DNA (dsDNA) (47). Together, IRF5 and STAT4 have an additive effect for increased risk of SLE development (48). Additional genes that influence the IFN pathway and innate immune signaling include IRAK1, which is found on the X chromosome and therefore is thought to contribute increased SLE risk among females (49), and osteopontin, which is also associated with early disease onset (50), as well as IRF7, IFIH1, and TYK2.
Other risk factors for SLE development are genes linked to the MHC, primarily HLA-DRB1 in the MHC class II region (51). HLA molecules play a key role in autoantibody production as demonstrated by one Japanese study that identified both SLE risk signature and autoantibodies to ribonucleoprotein (RNP), SSA/Ro60, SSB/La, cardiolipin were localized to the peptide binding groove of HLA-DRB1 and anti-Sm to HLA-DPB1 (52). Multiple genes involved in the adaptive immune response and autoantibody production have also been linked to SLE risk such as PTPN22 (53) and BANK1 with three functional variants that lead to an altered B cell activation threshold to increase SLE risk (41).
Given that SLE is multifactorial and multigenic, an individual’s risk for SLE development cannot be well estimated using only known genetic risk factors. Several similar weighted genetic risk scores (GRS) have been developed to try to estimate an individual’s cumulative genetic susceptibility to SLE risk (54). A high GRS has been associated with earlier onset SLE and more severe disease phenotypes (55). Overall, men with SLE also appear to have slightly higher GRS than do women with SLE, suggesting that there is a stronger genetic component of disease among families with male SLE patients and perhaps that environmental or hormonal factors contribute to lowering the threshold for the development of SLE more among females than males (or, conversely, environmental, or hormonal factors may raise this threshold in males) (56). Other studies have also demonstrated greater SLE risk when genetic and environmental interactions are combined such as vitamin D status in those with CYP24A1 alleles (57), current/recent smoking and GRS (54).
Future genetic studies will likely reveal increased numbers of genetic biomarkers, further refining our understanding of SLE risk and pathogenesis. Large genetic studies in more diverse racial and ethnic groups are still necessary, as most SLE GWAS to date have studied subjects of European or Asian ancestry. Research and development of models that incorporate environmental risk factors will hopefully hone our ability to identify those who are at high risk of developing SLE, and lead to new therapeutic targets.
2.2 Asymptomatic Autoimmunity and Inflammation
Some individuals genetically susceptible to SLE will transition into a period of asymptomatic autoimmunity and inflammation prior to the development of overt clinical manifestations. Which individuals will progress and why? These are key questions that we are still trying to answer. Thus far, studies have pointed to environmental risk factors, some known and others yet to be discovered, as potential triggers for this transition. These events likely act by both separate and overlapping biological pathways, including but not limited to increasing oxidative stress, loss of immune tolerance, autoantibody formation, complement activation and immune complex deposition, epigenetic modifications, and upregulation in cytokine expression (58). In this pre-symptomatic phase where there is already evidence of early immune changes, can we use this our advantage to identify these at-risk patients earlier? And if we could identify the earliest changes of SLE, could we “turn it off” or move a person “backwards” on their trajectory towards SLE? In this next section, we will highlight important biomarkers and potential interventions as we review the different pathways of autoimmunity and inflammation in SLE pathogenesis.
2.2.1 Increased Oxidative Stress
Oxidative stress, which is defined by an imbalance between the production and neutralization of reactive oxygen intermediates (ROI), is normally utilized by phagocytic cells to eliminate pathogenic organisms. However, in SLE, this is increased leading to abnormal activation and processing of cell-death signals and autoantibody production [reviewed in (59)]. Endogenous sources of oxidative stress include increased ROI production in mitochondria, NADPH oxidase enzymes in phagocytes, endothelial cells, T cells, and B cells (60, 61). Ultra-violet (UV) radiation, viral and bacterial infections, and chemical exposure have been implicated to be environmental sources of oxidative stress. Oxidative stress not only induces T-cell dysfunction and propagation of oxidative modification of self-antigens leading to systemic inflammation, but it also damages various organ systems resulting in renal, cardiovascular, and cutaneous disease/comorbidities in SLE (62–64).
Currently, there are no biomarkers of oxidative stress in routine clinical use. Potential biomarkers that have been correlated with disease activity in established SLE patients include increased modification of serum albumin (65), urinary levels of F2 isoprostane (66), and serum nitric oxide levels (67). Future studies are still needed to determine if these biomarkers and others can help diagnose pre-symptomatic disease. Potential antioxidant therapies for SLE include N-acetylcysteine and rapamycin, but their role in preclinical disease is unclear (68, 69). On the other hand, dietary intake of antioxidant vitamins (vitamins A, C, and E and α-carotene, β-carotene, cryptoxanthin, lycopene, lutein, or zeaxanthin) has not been found to decrease SLE risk in epidemiologic studies (70, 71).
2.2.2 Break in Immunological Tolerance
Loss of self-tolerance occurs in SLE when autoantibodies target nuclear self-antigens that are released into the extracellular space and exposed to the immune system [reviewed in (72)]. Abnormalities in apoptosis, NETosis, and histone modifications are thought to be involved in this process. Apoptosis is an important source of autoantigens in SLE and it has been shown that many of the nuclear autoantigens (e.g., DNA, Ro, La, and small nuclear RNP) that are targeted in SLE are clustered in blebs at the surface of apoptotic cells where oxidative modification can occur (63, 73). NETosis is a specialized form of neutrophil cell death that has also been implicated as another potential source of autoantigens (74). During NETosis, structures termed neutrophil extracellular traps (NETs) are extruded by neutrophils to entrap and dismantle bacteria, viruses, fungi, and parasites. These NETs include fibrillary networks of DNA, citrullinated histones, and granule peptides such as cathepsin G, neutrophil elastase, and myeloperoxidase. In SLE, apoptosis and NETosis are increased, resulting in an excess load of nuclear autoantibodies (72, 74).
However, these on their own are unlikely to break immunological tolerance as several studies were not able to induce immune activation by immunizing mice with apoptotic cells/blebs or NETs (75, 76). A deficiency in clearance of apoptotic cells and/or NETs due to intrinsic phagocyte defects and absent/deficient serum factors are thought to lead to an enduring exposure of modified proteins such as histones in the immune system (77). These modified proteins are regarded as neoantigens that are no longer perceived as endogenous and subsequently elicit an autoimmune response. It can also stimulate an inflammatory response through the activation of nucleic acid recognition receptors (e.g., members of the Toll-like receptor (TLR) family), which are important in viral and bacterial defense and associated with type I IFN production (discussed in 2.2.4 Cytokines/Chemokines). Improving the clearance of apoptotic cells and/or NETs may therefore be potential therapeutic targets for SLE or SLE prevention.
2.2.3 Autoantibodies
In addition to apoptotic cells and NETs, other important sources of autoantigens include neoantigens generated from necrotic cells under the influence of processes like oxidation and cleavage and infectious agents (e.g., single-stranded RNA, double-stranded RNA, and DNA). Autoantibodies and cytokines are produced by B lymphocytes that process and present these antigens. Autoantibodies can form immune complexes with their antigen, which can lead to organ damage through immune complex deposition and local and systemic inflammation. In a positive feedback loop, autoantibodies can then induce NETosis, and immune complexes can stimulate plasmacytoid dendritic cells to produce pro-inflammatory cytokines including IFN-α which can incite further NETosis. In SLE, intrinsic abnormalities of B-cell and T-cell interaction also contributes to the production of autoantibodies [reviewed in (78)]. In SLE, these cells are hyperresponsive to stimuli resulting in the production of higher quantities of autoantibodies and cytokines. Furthermore, defects in immune tolerance permit the survival of dangerous autoreactive B cells that lead to further production and diversification of harmful autoantibodies in a process called epitope spreading (79, 80). Early in the disease course, an antibody response might begin with a particular epitope, and this is then later followed by a spread of the response to other epitopes in the same polypeptide (intramolecular) and in other distinct but structural similar molecules (intermolecular) (81). In Table 1, we summarize common SLE autoantibodies, their clinical associations, and onset prior to the diagnosis of SLE (82–85).

Table 1 SLE autoantibodies, clinical significance, and time to SLE onset.
SLE is thus a paradigmatic autoimmune disease, with formation and detection of a wide range of autoantibodies, some of which are more SLE-specific and more pathologic than others. Autoantibody detection has long been a valuable and effective approach to the diagnosis, classification and prognostication with a wide range of established systemic autoimmune rheumatic diseases (SARD), including SLE (87). However, the exact contribution of autoantibody testing to the identification of subclinical and very early SLE is still to be determined. In a seminal study by Arbuckle et al. (83), a serum biobank and database established by the American military was queried and SLE-related autoantibodies were found in stored blood up to 9.4 years (mean 3.3 years) before the onset of SLE symptoms and eventual diagnosis. Other studies have confirmed similar findings (84, 88–92). Anti-SSA/Ro60 antibodies typically appeared first (83, 91, 92). Anti-SSB/La and anti-phospholipid antibodies have been reported to appear next (83). IgG and/or IgM anti-cardiolipin antibodies were detected in 18.5% of patients with mean onset of 3.0 years prior to the diagnosis of SLE and up to 7.6 years before SLE diagnosis (93). Anti-dsDNA anti-Sm, and anti-RNP antibodies (mean 3.4 vs. 1.2 years; p=0.005) appear later (83, 91, 92). Other studies have also demonstrated that anti-dsDNA and anti-Sm antibodies in non-SLE or early undifferentiated connective tissue disease patients are predictive of SLE evolution (88, 94, 95). A positive ANA test, a test used to screen for the presence of autoantibodies, has been reported to appear up to 9.2 years (mean 2.25 years) prior to SLE diagnosis or classification. As SLE progressed before and after diagnosis or classification, new autoantibodies steadily accumulated, consistent with other literature supporting increased epitope spread over time (85, 92, 96, 97).
The absence of specific autoantibodies in SLE or the presence of others may also help to identify those who are at lower risk of progression to SLE. ANAs are non-specific and found in up to 20% of healthy subjects, and are more common in females, with increasing age, and in the setting of infection, lung, and autoimmune thyroid disease (98–100). Anti-dense fine speckled 70 (DFS70) antibodies may be a useful biomarker to rule out the diagnosis of SLE as they are rarely found in SLE patients. In an international study of 1137 patients with SLE followed from inception in the Systemic Lupus International Collaborating Clinics (SLICC) cohort, only 1.1% had monospecific (no other detectable autoantibodies) anti-DFS70 antibodies (101). Thus, the presence of anti-DFS70 antibodies may help to discriminate between those who are ANA-positive healthy subjects versus those with SLE. Anti-C1q autoantibodies, which are associated with lupus nephritis (102), were infrequently found in patients with incomplete SLE in a small cross-sectional study of 70 patients (86). The authors suggest that although it remains undetermined whether this autoantibody could be a predictor of SLE risk, the presence of an elevated anti-C1q antibody in a patient with incomplete SLE might raise concerns for SLE or more specifically, lupus nephritis (86).
One of the challenges of identifying novel predictive autoantibodies for SLE development is that although over 200 different autoantibodies have been described in SLE, only 10% have been made widely available as diagnostic assays approved by regulatory authorities; most are still for research purposes only (10). Furthermore, most studies of these novel autoantibodies in SLE have been small and cross-sectional in design, without consideration of hallmarks of early disease or variable longitudinal disease course and outcomes, even though autoantibody test results may vary over time. The parameters associated with this longitudinal variation, such as the impact of medical therapies on antibody responses, also have not been well studied.
There has been a call for future exploration of novel autoantibody biomarkers given the non-specificity of ANA for SLE (11, 12). Investigators at the University of Toronto examined approximately 200 ANA-positive patients without established SARD, using a custom antigen microarray of 144 established and novel autoantibodies (85). They found that the majority of patients who tested negative for most current commercially available autoantibodies were positive for autoantibodies on their custom microarray. Anti-Ro52/Tripartite motif containing-21 (TRIM21) autoantibodies were predictive of SARD progression over the next two years (defined by the 1997 ACR criteria for SLE (103), 2013 ACR-EULAR criteria for systemic sclerosis (104) or 2016 ACR–EULAR criteria for Sjögren’s syndrome (105)), with positive predictive value of 46% and negative predictive value of 89%. To close the ‘seronegative gap’, more studies of novel disease-specific autoantibody biomarkers are needed and will help to identify valid predictors of disease evolution, potentially enabling identification and treatment of patients with SLE in these early stages (10).
2.2.4 Cytokines/Chemokines
Increased IFN-α activity is an important contributor to SLE pathogenesis because of its involvement in the induction of B-lymphocyte stimulator (BLyS) and DNA- and RNA- protein binding autoantibody specificities. BLyS plays a key role in regulating B cell survival and differentiation, which is central to autoantibody production and class switching. Drugs blocking BLyS activity (belimumab), and more recently, the type I IFN receptor subunit 1 (anifrolumab), have reduced disease activity in patients with SLE in large clinical trials and are now approved therapies for SLE treatment (14, 16).
In a case-control study by Munroe et al. of SLE patients and matched healthy controls, serum collected prior to and at/after SLE classification were analyzed (92). Prior to SLE classification (average timespan of 4.3 years), upregulation of IFN-associated mediators, as observed with autoantibodies, accumulated over a period of years, and then plateaued close to the time of disease classification (p<0.001). The most important predictor of increased IFN-α activity was the number of positive autoantibodies (p<0.001). Increased circulating IFN-α activity and BLyS levels were also detected shortly before subjects met SLE classification criteria (p≤0.005), suggesting that this may be a turning point in SLE pathogenesis where immune dysregulation is amplified by positive feed-forward mechanisms. Other studies have also showed that early SLE patients have exacerbated type I IFN signatures, their autoantibodies specificities have already class-switched to IgG isotypes (106), and autoantibody containing immune complexes drive type I IFN activation (107–110).
Although IFN-α activity may be an important contributor to SLE progression, not all SLE patients (only ~25%) have increased IFN-α activity preceding SLE diagnosis or classification (92). Hence, other forms of immune dysregulation likely accompany IFN-α activity, such as type II IFN (IFN-γ). IFN-γ is important in mediating the crosstalk between innate cells and lymphocytes, breaking self-tolerance and enabling the activation and persistence of autoreactive B cells (111). It modulates TLR regulation to facilitate autoantibody production, antigen presentation, and recruitment of lymphocytes to germinal centers (111). It can also drive the production of IFN-α and BLyS levels, leading to inflammation, B cell activation and autoantibody production. Munroe et al. further found increased levels of circulating IFN-γ in pre-clinical SLE patients prior to detectable upregulation of IFN-α and autoantibody positivity, as well as dysregulation of the chemokines IP-10 (CXCL10) and MCP-3 (CCL7) (92). Other mediators that have been implicated in SLE pathogenesis and are elevated years before SLE classification include IL (interleukin)-12p70, MIG, IL-4, IL-5, and IL-6 (91). These chemokines, which aid in the recruitment of cells to sites of inflammation, may also be important biomarkers in early pathogenesis of SLE.
2.2.5 Complement Activation
Complement activation is responsible for much of the systemic inflammation and tissue damage in SLE [reviewed (112)]. All three pathways of complement activation are involved in SLE, with the classical pathway, activated by antigen-antibody complexes, being the most important in SLE pathogenesis. Low complement C3, C4 and CH50, levels are diagnostic and disease activity biomarkers in SLE (113). However, they are not always reliable as they are influenced by the acute phase response, individual differences in complement gene copy number and expression, and variability in protein catabolism and synthesis (114).
To overcome the limitations of measuring C3 and C4, assays to measure cell-bound activation (split) products (CB-CAPS), such as erythrocyte-bound C4d (EC4d) and B lymphocyte-bound C4d (BC4d), have recently been developed. These are formed upon activation of the complement cascade and reflect complement activation rather than the levels of the individual protein. These are measured using EDTA anti-coagulated blood by flow cytometry which can be labor intensive, but on the other hand, sample processing is usually minimal, no centrifugation is needed, and it does not require low temperature for storage and transportation.
CB-CAPS are promising SLE biomarkers, shown to be more sensitive than C3, C4, and anti-dsDNA for the SLE diagnosis (115, 116), and more prevalent in patients with probable SLE. When used in combination with a proprietary panel of other autoantibodies, one study reported these biomarkers were able to identify patients with a greater than three-fold increased risk of developing SLE and were slightly better than complements or anti-dsDNA alone at predicting transition to SLE among patients with undifferentiated connective tissue disease [reviewed in (117, 118)]. These results suggest that complement activation may also occur early in the evolution of SLE and be an important feature in patients with suspected SLE.
2.2.6 Lifestyle and Environmental Risk Factors Related to SLE Risk (With a Focus on Those That Are Potentially Modifiable)
The number of factors beyond age, race, sex, family history, and genetics that are strongly associated with risk of developing SLE has been growing in recent years. Multiple large cohort studies have contributed to our understanding of how lifestyle, behavioral, psychosocial, and environmental risk factors may converge and synergize with underlying genetic risk. This likely leads to an acceleration of underlying and brewing autoimmunity, allowing it to manifest in SLE. These factors include current cigarette smoking, obesity (in particular, at younger ages), childhood and adult trauma, stress, post-traumatic stress disorder, low or no alcohol intake, environmental air pollution, environmental silica, and hormonal exposures and reproductive factors among women [reviewed in (58, 119)]. While is it not known whether these environmental risk factors work via similar or disparate biologic pathways, nor whether they are perhaps also inextricably linked to other societal risk factors that are more difficult to measure, the picture of how and the extent to which they contribute to SLE susceptibility is coming into focus. Gene-environment interactions likely contribute to SLE risk, and only a handful of these specific interactions have been discovered to date (54, 57).
In a recent, large, prospective evaluation of healthy lifestyle behaviors and SLE risk using the Nurses’ Health Study (NHS) and NHSII, adherence to multiple healthy behaviors (healthy diet (highest 40th percentile of the Alternative Healthy Eating Index), regular exercise (performing at least 19 metabolic equivalent hours of exercise per week), never smoker or past smoker, moderate alcohol consumption [drinking ≥5 gm/day alcohol), and maintaining a healthy body weight (body mass index <25 kg/m2)] was associated with a lower risk of SLE development overall (120). There was a 19% reduction for each additional healthy behavior and an even greater reduction (22%) was observed for the risk of dsDNA positive SLE. Strikingly, the risk of SLE was half as high among those with the best adherence to healthy lifestyle behaviors compared to among those with the poorest adherence. Overall, the population attributable risk, or the proportion of the risk in this population that could be attributed to these five modifiable lifestyle risk factors was 47.7% [95% confidence interval (CI) 23.1-66.6%]. These results suggest that lifestyle behaviors likely work synergistically to influence the risk of SLE and potentially produce stronger effects together than individually via common biological pathways including production of autoantibodies and dysregulation of pro-inflammatory cytokines. Moreover, although much work remains to be done in disentangling the specific pathways by which these environmental risk factors may be related to SLE pathogenesis, this also suggest that much of SLE may be preventable with lifestyle change, a somewhat revolutionary concept.
Many potential biologic mechanisms and synergies are possible. For example, exposure to obesity and toxic components of cigarette smoke both cause oxidative stress (121). This, in turn, increases intracellular levels of reactive oxygen species to damage DNA forming immunogenic DNA adducts, thereby promoting dsDNA antibody production (section 2.2.3) (122–124). In the NHS and NHSII cohorts, cigarette smoking was associated with a higher risk of anti-dsDNA positive SLE than never smokers [hazard ratio 1.86 (95%CI 1.14-13.04)] (125), a finding confirmed in other studies (126, 127). In addition to causing oxidative stress (section 2.2.1), the by-products of smoking could also augment autoreactive B cells in the native repertoire (126) and induce pulmonary ANA in the lungs of exposed mice (128). Alcohol consumption, on the other hand, contains several compounds such as ethanol and antioxidants, that can potentially counteract the changes induced by smoking and obesity including inhibiting key enzymes in DNA synthesis (129, 130). Moderate alcohol intake (≥5 gm/day or >0.5 drinks/day) was associated with a decreased risk of incident SLE in NHS and other studies [hazard ratio 0.61 (95%CI 0.41-0.89)] (131).
Although the association between SLE risk and various diets is less clear in humans (132–134), murine models have demonstrated that low dietary fiber intake and Western-type diet (i.e., high in sugar, fat, refined grains, and red meat) were associated with increased autoantibody production in SLE-prone mice (135, 136). A murine study also demonstrated that in mice genetically susceptible to SLE, sleep deprivation was associated with an earlier onset of disease and accelerated production of autoantibodies (137). Among women followed in the Black Women’s Health study, a diet high in carbohydrates was associated with increased risk of developing SLE (132). The association between lack of sleep (less than the recommended 7 hours a night) and SLE risk in humans has been reported in several studies (138, 139). In a prospective study of 436 non-SLE relatives of SLE patients, relatives were more likely to transition to SLE if they reported sleeping less than seven hours a day [odds ratio 2.8 (95%CI 1.6-5.1)] (138).
Many lifestyle factors associated with SLE development increase levels of pro-inflammatory cytokines (section 2.2.4). Smoking increases BLyS expression (128), Tumor necrosis factor alpha (TNF-α), and IL-6 (140, 141). Among positive ANA women, elevated BLyS and lower IL-10 (an anti-inflammatory cytokine) levels could be found among current smokers (142). Both TNF-α and IL-6 also play important roles in the modulation of insulin resistance (121). Adipose tissue, in particular visceral fat, secretes pro-inflammatory adipocyte-derived cytokines and exhibit higher levels of C-reactive protein (CRP), TNF-α receptor 2, and IL-6 than non-obese individuals (143). Alcohol, on the other hand, suppresses TNF-α, IL-6, IL-8, and IFN-γ to counteract systemic inflammation (129, 130). In sleep-deprived individuals, increased levels of IL-6, TNF-α have been observed in addition to its role in impairing the function of T cells and CD4 regulatory T cells, which are important in self-tolerance (section 2.2.2) (144–148). Sleep disturbances in individuals who have had childhood or adult trauma, post-traumatic stress disorder or occupational stress from working nightshifts or rotating shifts, may also explain why these factors have also been linked to SLE onset (149–155). Systemic inflammation with elevated TNF, IL-6 and CRP levels is also found in these conditions (150, 156–164).
Other environmental and occupational related risk factors, including chemical and physical exposures, have also been linked to SLE onset and mechanisms involving stimulation of cellular necrosis and relate to intracellular antigens with resulting inflammation and IFN upregulation. These exposures include crystalline silica dust (165–168), air pollution and other respiratory particulates (169, 170), heavy metals such as mercury (149), and agricultural pesticides (149, 171, 172). UV radiation is also thought to trigger SLE onset, and it has been shown in SLE patients and lupus-prone mice, that there is a rise in type I IFN signaling and expansion and prolonged activation of T cells following UVB exposure (173–175). The association of UV radiation and SLE risk however is likely complicated by its role in vitamin D3 synthesis in the skin, which has been hypothesized to reduce SLE risk (176). A more detailed discussion about vitamin D and its role in preventing SLE is found in section 3.
Use of exogenous hormones, oral contraceptive pills, and hormone replacement therapy have been associated with risk of SLE (177–179). Among recent oral contraceptive pill users, a dose response between oral contraceptive pill dose of ethinyl estradiol and SLE risk has been demonstrated (178). Estrogen is thought to induce autoreactivity by upregulating several genes involved in B cell activation and survival (cd22, shp-1, bcl-2, and vcam-1) and preventing B cell receptor-mediated apoptosis (180).
The association between infection and SLE is the Epstein-Barr virus (EBV) has been of interest for many years. The data on whether prior EBV infection is a risk factor for SLE development are still unclear [reviewed in (181)]. The release of EBV-encoded small RNA from infected cells is thought to induce type 1 interferon and proinflammatory cytokines via activating TLR-3 signaling (182). Another potential mechanism is through molecular mimicry between EBV and SLE antigens and epitope spreading. In a systematic review and meta-analysis of 25 case-control studies, a higher seroprevalence of anti-viral capsid antigen IgG [odds ratio 2.08 (95%CI 1.15-3.76)] and anti-early antigen antibody, a marker of viral replication, was observed in patients with existing SLE compared to health or nonhealthy controls [odds ratio 4.5 (95%CI 3.00-11.06)] (183). However, the results should be interpreted with caution given there was publication bias regarding recruitment, matching and reporting of blinded laboratory analysis and these studies do not address whether EBV is causally related to SLE. On the other hand, in a Danish population-based study, it was the EBV-serologic negative individuals that had an increased risk for SLE, particularly one to four years after serologic testing [standardized incidence rate 6.6 (95%CI 3.3–13.2)] (184). This may reflect surveillance bias as those patients who go on to develop SLE may have had EBV testing as part of their workup for early SLE symptoms. More recently, there are data to suggest that EBV reactivation is associated with SLE disease onset. In a prospective study of unaffected relatives of SLE patients (n=436), SLE relatives who transitioned to classifiable SLE had increased levels of EBV IgG antibodies prior to SLE transition compared to relatives who did not transition (185). Furthermore, increasing levels of EBV antibodies were associated with SLE disease transitioning, particularly among those with variants in genes that are associated with SLE and implicated in EBV infection.
The association between vaccinations and SLE risk remains to be elucidated, but thus far, epidemiological studies in SLE suggest that there is no association (186). It is thought that vaccines could potentially trigger autoimmunity through molecular mimicry, autoantibodies, and response to adjuvants in the vaccine. There have been emerging reports of new-onset autoimmune diseases including rheumatoid arthritis (187), immune thrombotic thrombocytopenia (188), autoimmune liver disease (189), IgA nephropathy (190), and Guillain-Barré Syndrome [reviewed in (191)] after vaccination. However, the evidence is from mainly case reports or cross-sectional studies demonstrating a temporal association. There have also been a few case reports of SLE and lupus nephritis 1-2 weeks following COVID-19 vaccination (192–194). Without more substantive evidence, however, individuals should be encouraged to get vaccinated as it remains one of the most effective interventions to prevent COVID-19 infection and related morbidity and mortality.
2.3 Early or Preclinical SLE
During the next phase of SLE pathogenesis, still pre-diagnosis, individuals may start to develop early non-specific symptoms of SLE, but not yet enough to be diagnosed or classified with the disease (12, 103). These patients are sometimes referred to as incomplete lupus or undifferentiated connective tissue disease (195). Eventually, some people with early and non-specific breakdown of immune tolerance and signs and symptoms of systemic inflammation and autoimmunity will develop more disease features and organ damage and diagnosed or classified as SLE. The duration of this early phase is highly variable from individual to individual. Some may have smoldering disease onset over years, while others experience a rapidly explosive onset of SLE with multiple simultaneous and severe clinical manifestations and autoantibodies. The rapidity of SLE onset likely relates to the specific combination of genetic and environmental SLE risk factors and their interactions, and has been shown to vary by racial ancestry (196). Depending on the cohort and setting, it has been reported that up to half of undifferentiated SARD patients with very early connective tissue disease evolve to fulfill diagnostic and classification criteria of a SARD, including SLE (197). Identifying those at high risk of developing SLE, or in early phases of its development, would enable a “window of opportunity” whereby interventions could be targeted at intercepting disease and halting or slowing the progression to SLE (87).
3 Discussion: Proposal of a Clinical Care Pathway to Screen and Prevent SLE
Even before patients are diagnosed with SLE, some may suffer irreversible organ damage, including pulmonary arterial hypertension, cardiovascular disease, renal, and neurological damage (198). Studies have also demonstrated that prior to being diagnosed by an astute clinician or meeting formal classification criteria for SLE, patients are already at higher risk of hospitalizations and lupus-related complications (199, 200). If these patients who are developing SLE could be identified at an early stage, decision‐making regarding preventative strategies and therapeutic interventions could be improved.
An appropriate screening and prevention program for SLE has great potential to improve public health outcomes. When organized effectively, it would be targeted to identifying those at risk for SLE to prevent disease development, reduce disability, and cut mortality through early detection and treatment. This will be challenging however, given that SLE is a rare disease in the general population. Here we proposed a clinical care pathway for the screening and prevention of SLE (Figure 2) involving four different levels that start with targeting patients who are at genetic risk, the asymptomatic autoimmunity stage, pre-clinical, and finally clinical disease states as discussed in the section above.
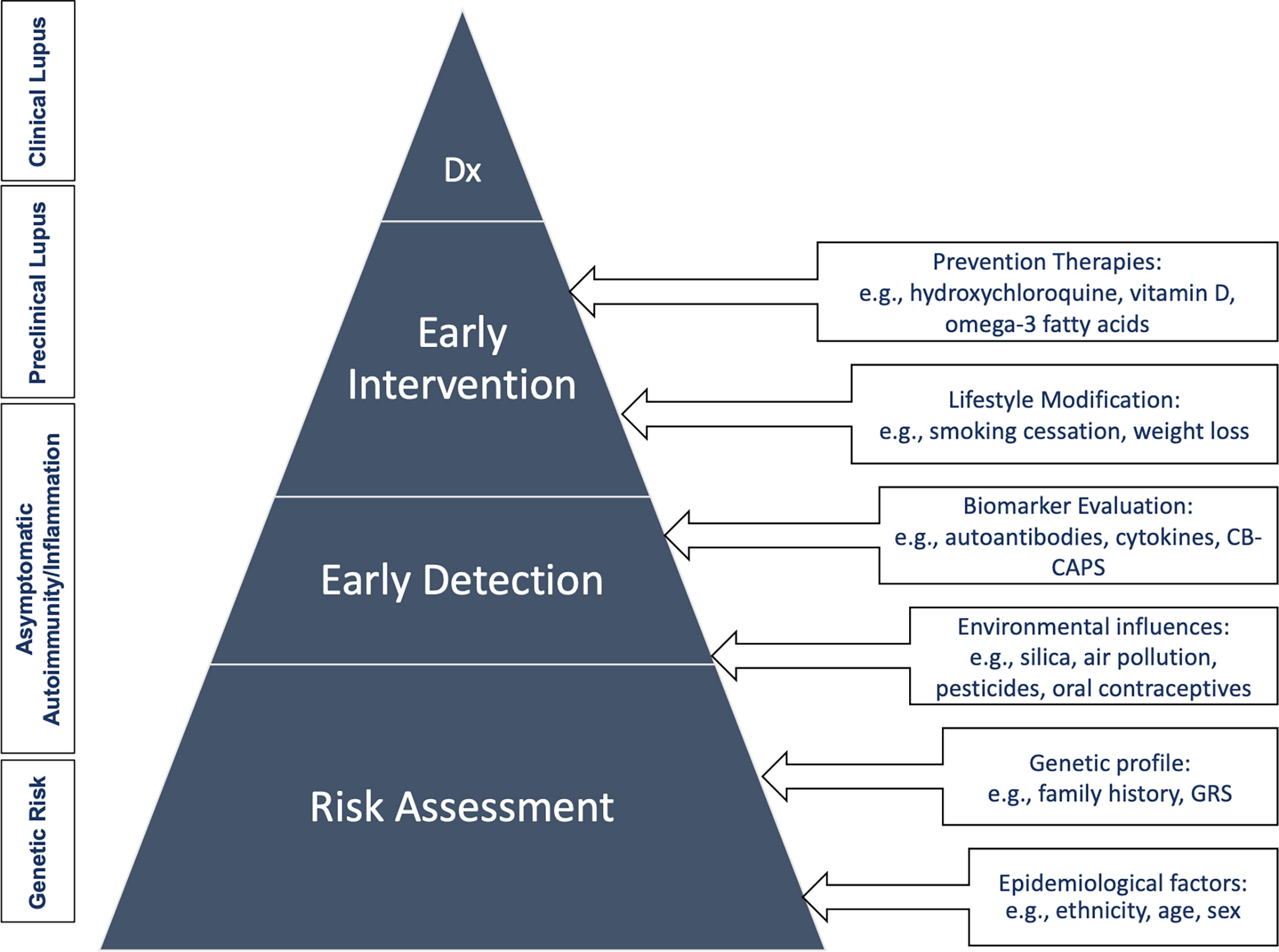
Figure 2 Clinical care pathway for the screening and prevention of SLE. Dx, diagnosis; GRS, genetic risk score.
3.1 Risk Assessment and Early Detection
Currently, there is no consensus concerning how to identify individuals at high risk for SLE or at what preclinical phase of disease should a patient be referred to see a rheumatologist. Given that SLE is a relatively rare disease with an incidence of about 1/2000 in the general population, most hypothetical screening programs would have to rely on inexpensive, readily available, and accurate tests (201). Population studies have used a 30-item questionnaire that can be completed within 30 minutes called the Connective Tissue Disease Screening Questionnaire (CSQ) to screen populations for SLE and other connective tissue diseases (202). It has high sensitivity for SLE (96%, 95%CI 90-99%) but moderate specificity (86%, 95%CI 81-91%) and has been validated among African American women (203). It is best employed in a two-stage screening method followed by medical record review or in-person assessment and should not be used as a test on its own due to the high false-positive rate. The ANA is the biomarker that we utilize today to “screen” for autoimmune connective tissue diseases, including SLE (204–206). However, as the ANA test is most usually performed when patients already have symptoms, it is not really a population-based screening test. As some patients may already have organ damage, ideally, those patients should be caught earlier in the asymptomatic autoimmunity and early preclinical phases, prior to clinical signs and symptoms. In this review, we highlighted numerous biomarkers that have shown promise in the identification of at-risk patients that could be detected in these earlier phases (section 2). These included autoantibodies such as ANA and anti-SSA/Ro60, genetic susceptibility loci, and upregulated cytokines/chemokines that coincide with timing of the initial appearance of autoantibodies, as well as markers of complement activation.
While some of these tests are readily available and accessible, there are several questions related to their use for screening purposes that need to be clarified. To better understand what makes a screening program appropriate, there are ten principles laid out by the 1968 World Health Organization that prompt important discussion about the benefits, harm, costs and ethics of a screening and prevention programs (207). If a program for SLE were implemented today, it would likely satisfy many of the criteria such as 1) “the condition should be an important health problem”; 2) “there should be an accepted treatment for patients with recognized disease”; 3) “facilities for diagnosis and treatment should be available”. However, there is still uncertainty surrounding some of the other criteria. Specifically related to testing, for instance, it is unclear if a biomarker test or panel were administered to screen for SLE in the general population that, “the cost of case-finding (including a diagnosis and treatment of patients diagnosed) [would] be economically balanced in relation to possible expenditure on medical care as a whole.” We have yet to determine the population that should be targeted for screening. However, it may be reasonable to narrow the screening eligibility criteria, based on the evidence from epidemiological studies to individuals from high-risk populations.
Preliminary data using the NHS and NHSII cohorts demonstrate that a weighted GRS in combination with lifestyle and environmental risk factors predicted future SLE risk with a good area under the curve of 0.77 (208). Therefore, using a GRS in combination with other risk factors assessment may be a valuable tool that may feasibly be employed in at-risk populations for predicting disease (Table 2). Once these patients have been identified, they could then be referred and potentially enrolled in prevention trials (discussed in 3.2.2 Preventative Therapies). Other prevention efforts targeting individuals at high genetic risk for lifestyle modification type of prevention trials could also be envisioned.

Table 2 SLE risk stratification chart.
3.2 Early Intervention
3.2.1 Lifestyle Modification
We discussed several modifiable risk factors that health care providers should encourage their patients who may be at risk for SLE to address, including smoking cessation, moderate alcohol consumption, regular exercise, avoidance of certain occupational and environmental exposures, medications, and maintaining a healthy weight and good sleep hygiene. The cost-effectiveness of adopting a healthy lifestyle is clear in that it is not only the risk of SLE that would be reduced, but that of many other chronic and complex diseases. To test the effectiveness of lifestyle interventions in actually reducing SLE risk, a primary prevention clinical trial would be necessary, but would be very challenging.
It is important to recognize that while the evidence suggests providers should encourage patients to adhere to as many healthy behaviors as possible for the greatest reduction in SLE and other chronic disease risk, there are many structural and institutional factors that affect an individual’s ability to adhere or achieve a healthy lifestyle. These include poverty, pollution, toxins, stress, and institutional and structural racism, among others, which have disproportionately affected non-White groups in the United States, who are also the same groups with the highest incidence and severity of SLE. Future studies should examine how to improve adherence to lifestyle interventions and address barriers that prevent or limit ability to meet healthy goals, especially among sociodemographic groups that are medically vulnerable.
3.2.2 Preventative Therapies
The first prevention trial in SLE is the Study of Anti-Malarials in Incomplete Lupus Erythematosus (SMILE), a multi-center, randomized, double-blind, placebo-controlled trial of HCQ compared to placebo, a 24-month clinical study (209). The purpose of this trial is to evaluate the efficacy and safety of HCQ intervention to prevent future onset of clinically apparent SLE. The inclusion criteria are patients 15-49 years of age with a positive ANA and at least one (but not three or more) additional clinical or laboratory criterion from the 2012 SLICC classification criteria (210). This study is expected to be completed in 2023. This study was initiated after James et al. demonstrated in a retrospective study on 130 United States military personnel that individuals who were treated with HCQ prior to SLE diagnosis had delayed the onset of complete SLE compared to untreated patients (median: 1.08 years versus 0.29 years) (18). Furthermore, individuals who had received HCQ in that study had slower accumulation of new autoantibodies. Other small studies showed that patients with incomplete SLE or new-onset, mild SLE treated with HCQ had lower levels of IFN-inducible genes, serum BLyS levels (also known as B cell–activating factor or BAFF), anti-C1q antibodies, IL-9, and better self-reported health status scores (86, 211). These results support the hypothesis that HCQ could influence SLE disease progression. Therefore, the SMILE trial will not only inform clinicians as to whether HCQ can be used to prevent SLE, but it will be the first step towards testing feasibility of disease prevention studies in SLE.
Recently, the results of a large (25,871 participants) randomized, double-blind, placebo-controlled, two-by-two factorial design trial examined the impact of vitamin D (cholecalciferol; 2000 IU/day) and marine omega 3 fatty acids (1 g/day as a fish oil capsule containing 460 mg of eicosapentaenoic acid and 380 mg of docosahexaenoic acid) on the incidence of various autoimmune diseases (212). The investigators found a reduction in autoimmune disease by 22% with vitamin D supplementation for five years, with or without omega 3 fatty acids, reduction by 15% with omega-3 fatty acid supplementation with or without vitamin D (not statistically significant). While there were too few new cases of SLE to be examined in this older population (men age 50 and older and women age 55 and older), vitamin D deficiency is common in SLE (213) and is important for regulating numerous genes involved in inflammation and the immune system through IL-2 inhibition, antibody production, and proliferation of lymphocytes (214, 215). Additionally, prior small cohort studies in SLE on specialized pro-resolving mediators (SPMs), a family of omega-3 fatty acid-derived lipid mediators, suggest that specific SPMs, such as the resolvins and lipoxins, may counter-regulate the production of inflammatory mediators and promote resolution of inflammation (216, 217). Further studies to examine whether omega-3 fatty acid supplementation can affect SPM levels and thereby forestall the development of SLE in at-risk populations will be needed.
Another potential therapy to decrease SLE risk that has been proposed is melatonin. Disrupted melatonin production in nightshift workers has been proposed as an important mechanism of increasing risk for autoimmune diseases including SLE [reviewed in (218)]. In lupus-prone mice, abnormal circadian rhythm of melatonin levels in response to light/dark cycle has been observed (219). When melatonin was administered to lupus-prone mice, there was decreased levels of autoantibodies, inflammatory cytokines, reduce renal injury, and increased levels of anti-inflammatory cytokine IL-10 (220, 221), particularly for females. Further studies in humans are called for to investigate the mechanism by which melatonin may be related to SLE risk and whether it could be a potential therapeutic strategy.
It is important to recognize that there are significant barriers to conducting prevention trials in SLE. A major challenge faced by past SLE prevention trials is low patient recruitment and retention. A lack of enthusiasm among clinicians and patients due to risk aversiveness and misunderstanding or misinterpretation of the purpose of prevention trials have resulted in underenrollment and selective enrollment, poor adherence, and attrition in some studies (222–224). Whereas good health status, encouragement from one’s physicians, desire to learn and contribute to research are positive factors for participation in SLE prevention trials (225). Therefore, future prevention trials in SLE should employ strategies such as health education about the clinical problem and importance of the trial, and involving the patients personal physicians to improve recruitment of SLE patients into prevention trials (225).
4 Conclusion
Developing a deeper understanding of SLE pathogenesis, its preclinical stages, and risk factors, will ultimately enable effective screening and potentially prevention. This may appear to be a daunting task; however, tremendous progress has been made over the last few decades with greater insights into the etiopathogenesis of SLE, identification of novel biomarkers for early SLE detection, epidemiologic and genetic studies that have revealed important risk factors, and the first prevention trial in SLE is already underway. Well-designed prospective clinical studies to further elucidate the mechanisms of disease development and more clinical prevention trials are needed.
Author Contributions
All authors have participated drafting the work or revising it critically for important intellectual content, final approval of the version published, agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved,
Funding
This work was supported by the Lupus Foundation of America Gary S. Gilkeson Career Development Award and NIH K24 AR066109 and R01 AR057327, and McCaig Institute for Bone and Joint Health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Izmirly PM, Parton H, Wang L, McCune WJ, Lim SS, Drenkard C, et al. Prevalence of Systemic Lupus Erythematosus in the United States: Estimates From a Meta-Analysis of the Centers for Disease Control and Prevention National Lupus Registries. Arthritis Rheumatol (2021) 73(6):991–6. doi: 10.1002/art.41632
2. Cross M, Smith E, Hoy D, Carmona L, Wolfe F, Vos T, et al. The Global Burden of Rheumatoid Arthritis: Estimates From the Global Burden of Disease 2010 Study. Ann Rheum Dis (2014) 73(7):1316–22. doi: 10.1136/annrheumdis-2013-204627
3. Peschken CA, Esdaile JM. Rheumatic Diseases in North America's Indigenous Peoples. Semin Arthritis Rheumatol (1999) 28(6):368–91. doi: 10.1016/S0049-0172(99)80003-1
4. Pons-Estel GJ, Alarcon GS, Scofield L, Reinlib L, Cooper GS. Understanding the Epidemiology and Progression of Systemic Lupus Erythematosus. Semin Arthritis Rheumatol (2010) 39(4):257–68. doi: 10.1016/j.semarthrit.2008.10.007
5. Yen EY, Singh RR. Brief Report: Lupus-An Unrecognized Leading Cause of Death in Young Females: A Population-Based Study Using Nationwide Death Certificates, 2000-2015. Arthritis Rheumatol (2018) 70(8):1251–5. doi: 10.1002/art.40512
6. Walsh SJ, Rau LM. Autoimmune Diseases: A Leading Cause of Death Among Young and Middle-Aged Women in the United States. Am J Public Health (2000) 90(9):1463–6. doi: 10.2105/ajph.90.9.1463
7. Hargraves MM, Richmond H, Morton R. Presentation of Two Bone Marrow Elements; the Tart Cell and the L.E. Cell. Proc Staff Meet Mayo Clin (1948) 23(2):25–8.
8. Touma Z. Outcome Measures and Metrics in Systemic Lupus Erythematosus. Cham, Switzerland: Springer (2021).
9. Kernder A, Richter JG, Fischer-Betz R, Winkler-Rohlfing B, Brinks R, Aringer M, et al. Delayed Diagnosis Adversely Affects Outcome in Systemic Lupus Erythematosus: Cross Sectional Analysis of the LuLa Cohort. Lupus (2021) 30(3):431–8. doi: 10.1177/0961203320983445
10. Rahman P, Gladman DD, Urowitz MB, Hallett D, Tam LS. Early Damage as Measured by the SLICC/ACR Damage Index is a Predictor of Mortality in Systemic Lupus Erythematosus. Lupus (2001) 10(2):93–6. doi: 10.1191/096120301670679959
11. Urowitz MB, Gladman DD, Ibanez D, Sanchez-Guerrero J, Romero-Diaz J, Gordon C, et al. American College of Rheumatology Criteria at Inception, and Accrual Over 5 Years in the SLICC Inception Cohort. J Rheumatol (2014) 41(5):875–80. doi: 10.3899/jrheum.130704
12. Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R, et al. 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheumatol (2019) 71(9):1400–12. doi: 10.1002/art.40930
13. Aringer M, Brinks R, Dorner T, Daikh D, Mosca M, Ramsey-Goldman R, et al. European League Against Rheumatism (EULAR)/American College of Rheumatology (ACR) SLE Classification Criteria Item Performance. Ann Rheum Dis (2021). doi: 10.1136/annrheumdis-2021-221374
14. Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N Engl J Med (2020) 382(3):211–21. doi: 10.1056/NEJMoa1912196
15. Rovin BH, Teng YKO, Ginzler EM, Arriens C, Caster DJ, Romero-Diaz J, et al. Efficacy and Safety of Voclosporin Versus Placebo for Lupus Nephritis (AURORA 1): A Double-Blind, Randomised, Multicentre, Placebo-Controlled, Phase 3 Trial. Lancet (2021) 397(10289):2070–80. doi: 10.1016/S0140-6736(21)00578-X
16. Furie R, Rovin BH, Houssiau F, Malvar A, Teng YKO, Contreras G, et al. Two-Year, Randomized, Controlled Trial of Belimumab in Lupus Nephritis. N Engl J Med (2020) 383(12):1117–28. doi: 10.1056/NEJMoa2001180
17. Garen T, Lerang K, Hoffmann-Vold AM, Andersson H, Midtvedt O, Brunborg C, et al. Mortality and Causes of Death Across the Systemic Connective Tissue Diseases and the Primary Systemic Vasculitides. Rheumatology (Oxford) (2019) 58(2):313–20. doi: 10.1093/rheumatology/key285
18. James JA, Kim-Howard XR, Bruner BF, Jonsson MK, McClain MT, Arbuckle MR, et al. Hydroxychloroquine Sulfate Treatment is Associated With Later Onset of Systemic Lupus Erythematosus. Lupus (2007) 16(6):401–9. doi: 10.1177/0961203307078579
19. Kaiser R, Cleveland CM, Criswell LA. Risk and Protective Factors for Thrombosis in Systemic Lupus Erythematosus: Results From a Large, Multi-Ethnic Cohort. Ann Rheum Dis (2009) 68(2):238–41. doi: 10.1136/ard.2008.093013
20. Ruiz-Irastorza G, Egurbide MV, Pijoan JI, Garmendia M, Villar I, Martinez-Berriotxoa A, et al. Effect of Antimalarials on Thrombosis and Survival in Patients With Systemic Lupus Erythematosus. Lupus (2006) 15(9):577–83. doi: 10.1177/0961203306071872
21. Siso A, Ramos-Casals M, Bove A, Brito-Zeron P, Soria N, Munoz S, et al. Previous Antimalarial Therapy in Patients Diagnosed With Lupus Nephritis: Influence on Outcomes and Survival. Lupus (2008) 17(4):281–8. doi: 10.1177/0961203307086503
22. Tsakonas E, Joseph L, Esdaile JM, Choquette D, Senecal JL, Cividino A, et al. A Long-Term Study of Hydroxychloroquine Withdrawal on Exacerbations in Systemic Lupus Erythematosus. The Canadian Hydroxychloroquine Study Group. Lupus (1998) 7(2):80–5. doi: 10.1191/096120398678919778
23. Feldman CH, Zhang Z, Desai RJ, Lin TC, Collins JE, Subramanian SV, et al. Association Between Hydroxychloroquine Nonadherence and Adverse Outcomes Among Patients With Systemic Lupus Erythematosus. Arthritis Rheumatol (2017) 69(S10).
24. Tsokos GC, Lo MS, Costa Reis P, Sullivan KE. New Insights Into the Immunopathogenesis of Systemic Lupus Erythematosus. Nat Rev Rheumatol (2016) 12(12):716–30. doi: 10.1038/nrrheum.2016.186
25. Walport MJ. Complement and Systemic Lupus Erythematosus. Arthritis Res (2002) 4 Suppl 3(Suppl 3):S279–93. doi: 10.1186/ar586
26. Dominguez D, Kamphuis S, Beyene J, Wither J, Harley JB, Blanco I, et al. Relationship Between Genetic Risk and Age of Diagnosis in Systemic Lupus Erythematosus. J Rheumatol (2021) 48(6):852–8. doi: 10.3899/jrheum.200002
27. Webb R, Kelly JA, Somers EC, Hughes T, Kaufman KM, Sanchez E, et al. Early Disease Onset is Predicted by a Higher Genetic Risk for Lupus and is Associated With a More Severe Phenotype in Lupus Patients. Ann Rheum Dis (2011) 70(1):151–6. doi: 10.1136/ard.2010.141697
28. Kwon YC, Chun S, Kim K, Mak A. Update on the Genetics of Systemic Lupus Erythematosus: Genome-Wide Association Studies and Beyond. Cells (2019) 8(10):1180. doi: 10.3390/cells8101180
29. Moser KL, Kelly JA, Lessard CJ, Harley JB. Recent Insights Into the Genetic Basis of Systemic Lupus Erythematosus. Genes Immun (2009) 10(5):373–9. doi: 10.1038/gene.2009.39
30. Owen KA, Price A, Ainsworth H, Aidukaitis BN, Bachali P, Catalina MD, et al. Analysis of Trans-Ancestral SLE Risk Loci Identifies Unique Biologic Networks and Drug Targets in African and European Ancestries. Am J Hum Genet (2020) 107(5):864–81. doi: 10.1016/j.ajhg.2020.09.007
31. Langefeld CD, Ainsworth HC, Cunninghame Graham DS, Kelly JA, Comeau ME, Marion MC, et al. Transancestral Mapping and Genetic Load in Systemic Lupus Erythematosus. Nat Commun (2017) 8:16021. doi: 10.1038/ncomms16021
32. Chung SA, Brown EE, Williams AH, Ramos PS, Berthier CC, Bhangale T, et al. Lupus Nephritis Susceptibility Loci in Women With Systemic Lupus Erythematosus. J Am Soc Nephrol (2014) 25(12):2859–70. doi: 10.1681/ASN.2013050446
33. Sun C, Molineros JE, Looger LL, Zhou XJ, Kim K, Okada Y, et al. High-Density Genotyping of Immune-Related Loci Identifies New SLE Risk Variants in Individuals With Asian Ancestry. Nat Genet (2016) 48(3):323–30. doi: 10.1038/ng.3496
34. Morris DL, Sheng Y, Zhang Y, Wang YF, Zhu Z, Tombleson P, et al. Genome-Wide Association Meta-Analysis in Chinese and European Individuals Identifies Ten New Loci Associated With Systemic Lupus Erythematosus. Nat Genet (2016) 48(8):940–6. doi: 10.1038/ng.3603
35. Graham RR, Kyogoku C, Sigurdsson S, Vlasova IA, Davies LR, Baechler EC, et al. Three Functional Variants of IFN Regulatory Factor 5 (IRF5) Define Risk and Protective Haplotypes for Human Lupus. Proc Natl Acad Sci USA (2007) 104(16):6758–63. doi: 10.1073/pnas.0701266104
36. Fernando MM, Stevens CR, Sabeti PC, Walsh EC, McWhinnie AJ, Shah A, et al. Identification of Two Independent Risk Factors for Lupus Within the MHC in United Kingdom Families. PloS Genet (2007) 3(11):e192. doi: 10.1371/journal.pgen.0030192
37. Jacob CO, Reiff A, Armstrong DL, Myones BL, Silverman E, Klein-Gitelman M, et al. Identification of Novel Susceptibility Genes in Childhood-Onset Systemic Lupus Erythematosus Using a Uniquely Designed Candidate Gene Pathway Platform. Arthritis Rheumatol (2007) 56(12):4164–73. doi: 10.1002/art.23060
38. International Consortium for Systemic Lupus Erythematosus G, Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, et al. Genome-Wide Association Scan in Women With Systemic Lupus Erythematosus Identifies Susceptibility Variants in ITGAM, PXK, KIAA1542 and Other Loci. Nat Genet (2008) 40(2):204–10. doi: 10.1038/ng.81
39. Hom G, Graham RR, Modrek B, Taylor KE, Ortmann W, Garnier S, et al. Association of Systemic Lupus Erythematosus With C8orf13-BLK and ITGAM-ITGAX. N Engl J Med (2008) 358(9):900–9. doi: 10.1056/NEJMoa0707865
40. Nath SK, Han S, Kim-Howard X, Kelly JA, Viswanathan P, Gilkeson GS, et al. A Nonsynonymous Functional Variant in Integrin-Alpha(M) (Encoded by ITGAM) is Associated With Systemic Lupus Erythematosus. Nat Genet (2008) 40(2):152–4. doi: 10.1038/ng.71
41. Kozyrev SV, Abelson AK, Wojcik J, Zaghlool A, Linga Reddy MV, Sanchez E, et al. Functional Variants in the B-Cell Gene BANK1 are Associated With Systemic Lupus Erythematosus. Nat Genet (2008) 40(2):211–6. doi: 10.1038/ng.79
42. Sawalha AH, Webb R, Han S, Kelly JA, Kaufman KM, Kimberly RP, et al. Common Variants Within MECP2 Confer Risk of Systemic Lupus Erythematosus. PloS One (2008) 3(3):e1727. doi: 10.1371/journal.pone.0001727
43. Cunninghame Graham DS, Graham RR, Manku H, Wong AK, Whittaker JC, Gaffney PM, et al. Polymorphism at the TNF Superfamily Gene TNFSF4 Confers Susceptibility to Systemic Lupus Erythematosus. Nat Genet (2008) 40(1):83–9. doi: 10.1038/ng.2007.47
44. Graham RR, Cotsapas C, Davies L, Hackett R, Lessard CJ, Leon JM, et al. Genetic Variants Near TNFAIP3 on 6q23 are Associated With Systemic Lupus Erythematosus. Nat Genet (2008) 40(9):1059–61. doi: 10.1038/ng.200
45. Kirou KA, Lee C, George S, Louca K, Peterson MG, Crow MK. Activation of the Interferon-Alpha Pathway Identifies a Subgroup of Systemic Lupus Erythematosus Patients With Distinct Serologic Features and Active Disease. Arthritis Rheumatol (2005) 52(5):1491–503. doi: 10.1002/art.21031
46. Crow MK, Kirou KA. Interferon-Alpha in Systemic Lupus Erythematosus. Curr Opin Rheumatol (2004) 16(5):541–7. doi: 10.1097/01.bor.0000135453.70424.1b
47. Taylor KE, Remmers EF, Lee AT, Ortmann WA, Plenge RM, Tian C, et al. Specificity of the STAT4 Genetic Association for Severe Disease Manifestations of Systemic Lupus Erythematosus. PloS Genet (2008) 4(5):e1000084. doi: 10.1371/journal.pgen.1000084
48. Abelson AK, Delgado-Vega AM, Kozyrev SV, Sanchez E, Velazquez-Cruz R, Eriksson N, et al. STAT4 Associates With Systemic Lupus Erythematosus Through Two Independent Effects That Correlate With Gene Expression and Act Additively With IRF5 to Increase Risk. Ann Rheum Dis (2009) 68(11):1746–53. doi: 10.1136/ard.2008.097642
49. Jacob CO, Zhu J, Armstrong DL, Yan M, Han J, Zhou XJ, et al. Identification of IRAK1 as a Risk Gene With Critical Role in the Pathogenesis of Systemic Lupus Erythematosus. Proc Natl Acad Sci USA (2009) 106(15):6256–61. doi: 10.1073/pnas.0901181106
50. Kariuki SN, Moore JG, Kirou KA, Crow MK, Utset TO, Niewold TB. Age- and Gender-Specific Modulation of Serum Osteopontin and Interferon-Alpha by Osteopontin Genotype in Systemic Lupus Erythematosus. Genes Immun (2009) 10(5):487–94. doi: 10.1038/gene.2009.15
51. Relle M, Schwarting A. Role of MHC-Linked Susceptibility Genes in the Pathogenesis of Human and Murine Lupus. Clin Dev Immunol (2012) 2012:584374. doi: 10.1155/2012/584374
52. Molineros JE, Looger LL, Kim K, Okada Y, Terao C, Sun C, et al. Amino Acid Signatures of HLA Class-I and II Molecules are Strongly Associated With SLE Susceptibility and Autoantibody Production in Eastern Asians. PloS Genet (2019) 15(4):e1008092. doi: 10.1371/journal.pgen.1008092
53. Chung SA, Criswell LA. PTPN22: Its Role in SLE and Autoimmunity. Autoimmunity (2007) 40(8):582–90. doi: 10.1080/08916930701510848
54. Cui J, Raychaudhuri S, Karlson EW, Speyer C, Malspeis S, Guan H, et al. Interactions Between Genome-Wide Genetic Factors and Smoking Influencing Risk of Systemic Lupus Erythematosus. Arthritis Rheumatol (2020) 72(11):1863–71. doi: 10.1002/art.41414
55. Reid S, Alexsson A, Frodlund M, Morris D, Sandling JK, Bolin K, et al. High Genetic Risk Score is Associated With Early Disease Onset, Damage Accrual and Decreased Survival in Systemic Lupus Erythematosus. Ann Rheum Dis (2020) 79(3):363–9. doi: 10.1136/annrheumdis-2019-216227
56. Hughes T, Adler A, Merrill JT, Kelly JA, Kaufman KM, Williams A, et al. Analysis of Autosomal Genes Reveals Gene-Sex Interactions and Higher Total Genetic Risk in Men With Systemic Lupus Erythematosus. Ann Rheum Dis (2012) 71(5):694–9. doi: 10.1136/annrheumdis-2011-200385
57. Young KA, Munroe ME, Guthridge JM, Kamen DL, Niewold TB, Gilkeson GS, et al. Combined Role of Vitamin D Status and CYP24A1 in the Transition to Systemic Lupus Erythematosus. Ann Rheum Dis (2017) 76(1):153–8. doi: 10.1136/annrheumdis-2016-209157
58. Barbhaiya M, Costenbader KH. Environmental Exposures and the Development of Systemic Lupus Erythematosus. Curr Opin Rheumatol (2016) 28(5):497–505. doi: 10.1097/BOR.0000000000000318
59. Perl A. Oxidative Stress in the Pathology and Treatment of Systemic Lupus Erythematosus. Nat Rev Rheumatol (2013) 9(11):674–86. doi: 10.1038/nrrheum.2013.147
60. Gergely P Jr., Grossman C, Niland B, Puskas F, Neupane H, Allam F, et al. Mitochondrial Hyperpolarization and ATP Depletion in Patients With Systemic Lupus Erythematosus. Arthritis Rheumatol (2002) 46(1):175–90. doi: 10.1002/1529-0131(200201)46:1<175::AID-ART10015>3.0.CO;2-H
61. Gergely P Jr., Niland B, Gonchoroff N, Pullmann R Jr., Phillips PE, Perl A. Persistent Mitochondrial Hyperpolarization, Increased Reactive Oxygen Intermediate Production, and Cytoplasmic Alkalinization Characterize Altered IL-10 Signaling in Patients With Systemic Lupus Erythematosus. J Immunol (2002) 169(2):1092–101. doi: 10.4049/jimmunol.169.2.1092
62. Dhaun N, Kluth DC. Oxidative Stress Promotes Hypertension and Albuminuria During the Autoimmune Disease Systemic Lupus Erythematosus. Hypertension (2012) 59(5):e47. doi: 10.1161/HYPERTENSIONAHA.112.193276
63. Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens Targeted in Systemic Lupus Erythematosus are Clustered in Two Populations of Surface Structures on Apoptotic Keratinocytes. J Exp Med (1994) 179(4):1317–30. doi: 10.1084/jem.179.4.1317
64. Skaggs BJ, Hahn BH, McMahon M. Accelerated Atherosclerosis in Patients With SLE–mechanisms and Management. Nat Rev Rheumatol (2012) 8(4):214–23. doi: 10.1038/nrrheum.2012.14
65. Wang G, Pierangeli SS, Papalardo E, Ansari GA, Khan MF. Markers of Oxidative and Nitrosative Stress in Systemic Lupus Erythematosus: Correlation With Disease Activity. Arthritis Rheumatol (2010) 62(7):2064–72. doi: 10.1002/art.27442
66. Avalos I, Chung CP, Oeser A, Milne GL, Morrow JD, Gebretsadik T, et al. Oxidative Stress in Systemic Lupus Erythematosus: Relationship to Disease Activity and Symptoms. Lupus (2007) 16(3):195–200. doi: 10.1177/0961203306075802
67. Gilkeson G, Cannon C, Oates J, Reilly C, Goldman D, Petri M. Correlation of Serum Measures of Nitric Oxide Production With Lupus Disease Activity. J Rheumatol (1999) 26(2):318–24.
68. Lai ZW, Hanczko R, Bonilla E, Caza TN, Clair B, Bartos A, et al. N-Acetylcysteine Reduces Disease Activity by Blocking Mammalian Target of Rapamycin in T Cells From Systemic Lupus Erythematosus Patients: A Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheumatol (2012) 64(9):2937–46. doi: 10.1002/art.34502
69. Fernandez D, Bonilla E, Mirza N, Niland B, Perl A. Rapamycin Reduces Disease Activity and Normalizes T Cell Activation-Induced Calcium Fluxing in Patients With Systemic Lupus Erythematosus. Arthritis Rheumatol (2006) 54(9):2983–8. doi: 10.1002/art.22085
70. Costenbader KH, Kang JH, Karlson EW. Antioxidant Intake and Risks of Rheumatoid Arthritis and Systemic Lupus Erythematosus in Women. Am J Epidemiol (2010) 172(2):205–16. doi: 10.1093/aje/kwq089
71. Tam LS, Li EK, Leung VY, Griffith JF, Benzie IF, Lim PL, et al. Effects of Vitamins C and E on Oxidative Stress Markers and Endothelial Function in Patients With Systemic Lupus Erythematosus: A Double Blind, Placebo Controlled Pilot Study. J Rheumatol (2005) 32(2):275–82.
72. Pieterse E, van der Vlag J. Breaking Immunological Tolerance in Systemic Lupus Erythematosus. Front Immunol (2014) 5:164. doi: 10.3389/fimmu.2014.00164
73. Rosen A, Casciola-Rosen L, Ahearn J. Novel Packages of Viral and Self-Antigens are Generated During Apoptosis. J Exp Med (1995) 181(4):1557–61. doi: 10.1084/jem.181.4.1557
74. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil Extracellular Traps Kill Bacteria. Science (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
75. Fransen JH, Berden JH, Koeter CM, Adema GJ, van der Vlag J, Hilbrands LB. Effect of Administration of Apoptotic Blebs on Disease Development in Lupus Mice. Autoimmunity (2012) 45(4):290–7. doi: 10.3109/08916934.2012.664668
76. Liu CL, Tangsombatvisit S, Rosenberg JM, Mandelbaum G, Gillespie EC, Gozani OP, et al. Specific Post-Translational Histone Modifications of Neutrophil Extracellular Traps as Immunogens and Potential Targets of Lupus Autoantibodies. Arthritis Res Ther (2012) 14(1):R25. doi: 10.1186/ar3707
77. Dieker J, Muller S. Epigenetic Histone Code and Autoimmunity. Clin Rev Allergy Immunol (2010) 39(1):78–84. doi: 10.1007/s12016-009-8173-7
78. Daniel W, Bevra H. Dubois' Lupus Erythematosus and Related Syndromes. New York: Elseiver (2019).
79. Chan VS, Tsang HH, Tam RC, Lu L, Lau CS. B-Cell-Targeted Therapies in Systemic Lupus Erythematosus. Cell Mol Immunol (2013) 10(2):133–42. doi: 10.1038/cmi.2012.64
80. Monneaux F, Muller S. Epitope Spreading in Systemic Lupus Erythematosus: Identification of Triggering Peptide Sequences. Arthritis Rheumatol (2002) 46(6):1430–8. doi: 10.1002/art.10263
82. Choi MY, Fritzler MJ. Challenges and Advances in SLE Autoantibody Detection and Interpretation. In: Outcome Measures and Metrics in Systemic Lupus Erythematosus. Cham, Switzerland: Springer (2021). p. 67–91.
83. Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, et al. Development of Autoantibodies Before the Clinical Onset of Systemic Lupus Erythematosus. N Engl J Med (2003) 349(16):1526–33. doi: 10.1056/NEJMoa021933
84. Eriksson C, Kokkonen H, Johansson M, Hallmans G, Wadell G, Rantapaa-Dahlqvist S. Autoantibodies Predate the Onset of Systemic Lupus Erythematosus in Northern Sweden. Arthritis Res Ther (2011) 13(1):R30. doi: 10.1186/ar3258
85. Munoz-Grajales C, Prokopec SD, Johnson SR, Touma Z, Ahmad Z, Bonilla D, et al. Serological Abnormalities That Predict Progression to Systemic Autoimmune Rheumatic Diseases in Antinuclear Antibody Positive Individuals. Rheumatology (Oxford) (2022) 61(3):1092–105. doi: 10.1093/rheumatology/keab501
86. Olsen NJ, McAloose C, Carter J, Han BK, Raman I, Li QZ, et al. Clinical and Immunologic Profiles in Incomplete Lupus Erythematosus and Improvement With Hydroxychloroquine Treatment. Autoimmune Dis (2016) 2016:8791629. doi: 10.1155/2016/8791629
87. Choi MY, Fritzler MJ. Autoantibodies in SLE: Prediction and the P Value Matrix. Lupus (2019) 28(11):1285–93. doi: 10.1177/0961203319868531
88. Calvo-Alen J, Alarcon GS, Burgard SL, Burst N, Bartolucci AA, Williams HJ. Systemic Lupus Erythematosus: Predictors of its Occurrence Among a Cohort of Patients With Early Undifferentiated Connective Tissue Disease: Multivariate Analyses and Identification of Risk Factors. J Rheumatol (1996) 23(3):469–75.
89. Clegg DO, Williams HJ, Singer JZ, Steen VD, Schlegel S, Ziminski C, et al. Early Undifferentiated Connective Tissue Disease. II. The Frequency of Circulating Antinuclear Antibodies in Patients With Early Rheumatic Diseases. J Rheumatol (1991) 18(9):1340–3.
90. Mosca M, Tavoni A, Neri R, Bencivelli W, Bombardieri S. Undifferentiated Connective Tissue Diseases: The Clinical and Serological Profiles of 91 Patients Followed for at Least 1 Year. Lupus (1998) 7(2):95–100. doi: 10.1191/096120398678919787
91. Lu R, Munroe ME, Guthridge JM, Bean KM, Fife DA, Chen H, et al. Dysregulation of Innate and Adaptive Serum Mediators Precedes Systemic Lupus Erythematosus Classification and Improves Prognostic Accuracy of Autoantibodies. J Autoimmun (2016) 74:182–93. doi: 10.1016/j.jaut.2016.06.001
92. Munroe ME, Lu R, Zhao YD, Fife DA, Robertson JM, Guthridge JM, et al. Altered Type II Interferon Precedes Autoantibody Accrual and Elevated Type I Interferon Activity Prior to Systemic Lupus Erythematosus Classification. Ann Rheum Dis (2016) 75(11):2014–21. doi: 10.1136/annrheumdis-2015-208140
93. McClain MT, Arbuckle MR, Heinlen LD, Dennis GJ, Roebuck J, Rubertone MV, et al. The Prevalence, Onset, and Clinical Significance of Antiphospholipid Antibodies Prior to Diagnosis of Systemic Lupus Erythematosus. Arthritis Rheumatol (2004) 50(4):1226–32. doi: 10.1002/art.20120
94. Swaak T, Smeenk R. Detection of anti-dsDNA as a Diagnostic Tool: A Prospective Study in 441 non-Systemic Lupus Erythematosus Patients With anti-dsDNA Antibody (anti-dsDNA). Ann Rheum Dis (1985) 44(4):245–51. doi: 10.1136/ard.44.4.245
95. Danieli MG, Fraticelli P, Salvi A, Gabrielli A, Danieli G. Undifferentiated Connective Tissue Disease: Natural History and Evolution Into Definite CTD Assessed in 84 Patients Initially Diagnosed as Early UCTD. Clin Rheumatol (1998) 17(3):195–201. doi: 10.1007/BF01451046
96. Theander E, Jonsson R, Sjostrom B, Brokstad K, Olsson P, Henriksson G. Prediction of Sjogren's Syndrome Years Before Diagnosis and Identification of Patients With Early Onset and Severe Disease Course by Autoantibody Profiling. Arthritis Rheumatol (2015) 67(9):2427–36. doi: 10.1002/art.39214
97. Heinlen LD, McClain MT, Merrill J, Akbarali YW, Edgerton CC, Harley JB, et al. Clinical Criteria for Systemic Lupus Erythematosus Precede Diagnosis, and Associated Autoantibodies are Present Before Clinical Symptoms. Arthritis Rheumatol (2007) 56(7):2344–51. doi: 10.1002/art.22665
98. Satoh M, Chan EK, Ho LA, Rose KM, Parks CG, Cohn RD, et al. Prevalence and Sociodemographic Correlates of Antinuclear Antibodies in the United States. Arthritis Rheumatol (2012) 64(7):2319–27. doi: 10.1002/art.34380
99. Xavier RM, Yamauchi Y, Nakamura M, Tanigawa Y, Ishikura H, Tsunematsu T, et al. Antinuclear Antibodies in Healthy Aging People: A Prospective Study. Mech Ageing Dev (1995) 78(2):145–54. doi: 10.1016/0047-6374(94)01532-Q
100. Wandstrat AE, Carr-Johnson F, Branch V, Gray H, Fairhurst AM, Reimold A, et al. Autoantibody Profiling to Identify Individuals at Risk for Systemic Lupus Erythematosus. J Autoimmun (2006) 27(3):153–60. doi: 10.1016/j.jaut.2006.09.001
101. Choi MY, Clarke AE, St Pierre Y, Hanly JG, Urowitz MB, Romero-Diaz J, et al. The Prevalence and Determinants of Anti-DFS70 Autoantibodies in an International Inception Cohort of Systemic Lupus Erythematosus Patients. Lupus (2017) 26(10):1051–9. doi: 10.1177/0961203317692437
102. Orbai AM, Truedsson L, Sturfelt G, Nived O, Fang H, Alarcon GS, et al. Anti-C1q Antibodies in Systemic Lupus Erythematosus. Lupus (2015) 24(1):42–9. doi: 10.1177/0961203314547791
103. Hochberg MC. Updating the American College of Rheumatology Revised Criteria for the Classification of Systemic Lupus Erythematosus. Arthritis Rheumatol (1997) 40(9):1725. doi: 10.1002/art.1780400928
104. van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 Classification Criteria for Systemic Sclerosis: An American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. Arthritis Rheum (2013) 65(11):2737–47. doi: 10.1002/art.38098
105. Shiboski CH, Shiboski SC, Seror R, Criswell LA, Labetoulle M, Lietman TM, et al. 2016 American College of Rheumatology/European League Against Rheumatism Classification Criteria for Primary Sjogren's Syndrome: A Consensus and Data-Driven Methodology Involving Three International Patient Cohorts. Ann Rheum Dis (2017) 76(1):9–16. doi: 10.1136/annrheumdis-2016-210571
106. Li QZ, Zhou J, Lian Y, Zhang B, Branch VK, Carr-Johnson F, et al. Interferon Signature Gene Expression is Correlated With Autoantibody Profiles in Patients With Incomplete Lupus Syndromes. Clin Exp Immunol (2010) 159(3):281–91. doi: 10.1111/j.1365-2249.2009.04057.x
107. Eloranta ML, Barbasso Helmers S, Ulfgren AK, Ronnblom L, Alm GV, Lundberg IE. A Possible Mechanism for Endogenous Activation of the Type I Interferon System in Myositis Patients With Anti-Jo-1 or Anti-Ro 52/Anti-Ro 60 Autoantibodies. Arthritis Rheumatol (2007) 56(9):3112–24. doi: 10.1002/art.22860
108. Mathsson L, Ahlin E, Sjowall C, Skogh T, Ronnelid J. Cytokine Induction by Circulating Immune Complexes and Signs of in-Vivo Complement Activation in Systemic Lupus Erythematosus Are Associated With the Occurrence of Anti-Sjogren's Syndrome A Antibodies. Clin Exp Immunol (2007) 147(3):513–20. doi: 10.1111/j.1365-2249.2006.03313.x
109. Savarese E, Chae OW, Trowitzsch S, Weber G, Kastner B, Akira S, et al. U1 Small Nuclear Ribonucleoprotein Immune Complexes Induce Type I Interferon in Plasmacytoid Dendritic Cells Through Tlr7. Blood (2006) 107(8):3229–34. doi: 10.1182/blood-2005-07-2650
110. Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, et al. Toll-Like Receptor 9-Dependent Activation by DNA-Containing Immune Complexes is Mediated by HMGB1 and RAGE. Nat Immunol (2007) 8(5):487–96. doi: 10.1038/ni1457
111. Kil LP, Hendriks RW. Aberrant B Cell Selection and Activation in Systemic Lupus Erythematosus. Int Rev Immunol (2013) 32(4):445–70. doi: 10.3109/08830185.2013.786712
112. Weinstein A, Alexander RV, Zack DJ. A Review of Complement Activation in SLE. Curr Rheumatol Rep (2021) 23(3):16. doi: 10.1007/s11926-021-00984-1
113. Weinstein A, Bordwell B, Stone B, Tibbetts C, Rothfield NF. Antibodies to Native DNA and Serum Complement (C3) Levels. Application to Diagnosis and Classification of Systemic Lupus Erythematosus. Am J Med (1983) 74(2):206–16. doi: 10.1016/0002-9343(83)90613-7
114. Liu CC, Manzi S, Danchenko N, Ahearn JM. New Advances in Measurement of Complement Activation: Lessons of Systemic Lupus Erythematosus. Curr Rheumatol Rep (2004) 6(5):375–81. doi: 10.1007/s11926-004-0012-5
115. Kalunian KC, Chatham WW, Massarotti EM, Reyes-Thomas J, Harris C, Furie RA, et al. Measurement of Cell-Bound Complement Activation Products Enhances Diagnostic Performance in Systemic Lupus Erythematosus. Arthritis Rheumatol (2012) 64(12):4040–7. doi: 10.1002/art.34669
116. Putterman C, Furie R, Ramsey-Goldman R, Askanase A, Buyon J, Kalunian K, et al. Cell-Bound Complement Activation Products in Systemic Lupus Erythematosus: Comparison With Anti-Double-Stranded DNA and Standard Complement Measurements. Lupus Sci Med (2014) 1(1):e000056. doi: 10.1136/lupus-2014-000056
117. Ramsey-Goldman R, Li J, Dervieux T, Alexander RV. Cell-Bound Complement Activation Products in SLE. Lupus Sci Med (2017) 4(1):e000236. doi: 10.1136/lupus-2017-000236
118. Ramsey-Goldman R, Alexander RV, Massarotti EM, Wallace DJ, Narain S, Arriens C, et al. Complement Activation in Patients With Probable Systemic Lupus Erythematosus and Ability to Predict Progression to American College of Rheumatology-Classified Systemic Lupus Erythematosus. Arthritis Rheumatol (2020) 72(1):78–88. doi: 10.1002/art.41093
119. Woo JMP, Parks CG, Jacobsen S, Costenbader KH, Bernatsky S. The Role of Environmental Exposures and Gene-Environment Interactions in the Etiology of Systemic Lupus Erythematous. J Intern Med (2022) 291(6):755–78. doi: 10.1111/joim.13448
120. Choi MY, Hahn J, Malspeis S, Stevens EF, Karlson EW, Sparks JA, et al. Association of a Combination of Healthy Lifestyle Behaviors With Reduced Risk of Incident Systemic Lupus Erythematosus. Arthritis Rheumatol (2022) 74(2):274–83. doi: 10.1002/art.41935
121. Manna P, Jain SK. Obesity, Oxidative Stress, Adipose Tissue Dysfunction, and the Associated Health Risks: Causes and Therapeutic Strategies. Metab Syndr Relat Disord (2015) 13(10):423–44. doi: 10.1089/met.2015.0095
122. Włodarczyk M, Nowicka G. Obesity, DNA Damage, and Development of Obesity-Related Diseases. Int J Mol Sci (2019) 20(5):1146. doi: 10.3390/ijms20051146
123. Petruzzelli S, Celi A, Pulerà N, Baliva F, Viegi G, Carrozzi L, et al. Serum Antibodies to Benzo (a) Pyrene Diol Epoxide-DNA Adducts in the General Population: Effects of Air Pollution, Tobacco Smoking, and Family History of Lung Diseases. Cancer Res (1998) 58(18):4122–6.
124. Mooney LA, Perera FP, Van Bennekum AM, Blaner WS, Karkoszka J, Covey L, et al. Gender Differences in Autoantibodies to Oxidative DNA Base Damage in Cigarette Smokers. Cancer Epidemiol Prev Biomarkers (2001) 10(6):641–8.
125. Barbhaiya M, Tedeschi SK, Lu B, Malspeis S, Kreps D, Sparks JA, et al. Cigarette Smoking and the Risk of Systemic Lupus Erythematosus, Overall and by Anti-Double Stranded DNA Antibody Subtype, in the Nurses' Health Study Cohorts. Ann Rheum Dis (2018) 77(2):196–202. doi: 10.1136/annrheumdis-2017-211675
126. Costenbader KH, Kim DJ, Peerzada J, Lockman S, Nobles-Knight D, Petri M, et al. Cigarette Smoking and the Risk of Systemic Lupus Erythematosus: A Meta-Analysis. Arthritis Rheumatol (2004) 50(3):849–57. doi: 10.1002/art.20049
127. Cozier YC, Barbhaiya M, Castro-Webb N, Conte C, Tedeschi SK, Leatherwood C, et al. Relationship of Cigarette Smoking and Alcohol Consumption to Incidence of Systemic Lupus Erythematosus in a Prospective Cohort Study of Black Women. Arthritis Care Res (Hoboken) (2019) 71(5):671–7. doi: 10.1002/acr.23703
128. Morissette MC, Gao Y, Shen P, Thayaparan D, Bérubé JC, Paré PD, et al. Role of BAFF in Pulmonary Autoantibody Responses Induced by Chronic Cigarette Smoke Exposure in Mice. Physiol Rep (2016) 4(24):e13057. doi: 10.14814/phy2.13057
129. Waldschmidt TJ, Cook RT, Kovacs EJ. Alcohol and Inflammation and Immune Responses: Summary of the 2006 Alcohol and Immunology Research Interest Group (AIRIG) Meeting. Alcohol (2008) 42(2):137–42. doi: 10.1016/j.alcohol.2007.11.003
130. Wirleitner B, Schroecksnadel K, Winkler C, Schennach H, Fuchs D. Resveratrol Suppresses Interferon-Gamma-Induced Biochemical Pathways in Human Peripheral Blood Mononuclear Cells In Vitro. Immunol Lett (2005) 100(2):159–63. doi: 10.1016/j.imlet.2005.03.008
131. Barbhaiya M, Lu B, Sparks JA, Malspeis S, Chang SC, Karlson EW, et al. Influence of Alcohol Consumption on the Risk of Systemic Lupus Erythematosus Among Women in the Nurses' Health Study Cohorts. Arthritis Care Res (Hoboken) (2017) 69(3):384–92. doi: 10.1002/acr.22945
132. Castro-Webb N, Cozier YC, Barbhaiya M, Ruiz-Narvaez EA, Li S, Costenbader KH, et al. Association of Macronutrients and Dietary Patterns With Risk of Systemic Lupus Erythematosus in the Black Women's Health Study. Am J Clin Nutr (2021) 114(4):1486–94. doi: 10.1093/ajcn/nqab224
133. Tedeschi SK, Barbhaiya M, Sparks JA, Karlson EW, Kubzansky LD, Roberts AL, et al. Dietary Patterns and Risk of Systemic Lupus Erythematosus in Women. Lupus (2020) 29(1):67–73. doi: 10.1177/0961203319888791
134. Barbhaiya M, Tedeschi S, Sparks JA, Leatherwood C, Karlson EW, Willett WC, et al. Association of Dietary Quality With Risk of Incident Systemic Lupus Erythematosus in the Nurses' Health Studies. Arthritis Care Res (Hoboken) (2021) 73(9):1250–8. doi: 10.1002/acr.24443
135. Schafer AL, Eichhorst A, Hentze C, Kraemer AN, Amend A, Sprenger DTL, et al. Low Dietary Fiber Intake Links Development of Obesity and Lupus Pathogenesis. Front Immunol (2021) 12:696810. doi: 10.3389/fimmu.2021.696810
136. Pan Y, Ke H, Yan Z, Geng Y, Asner N, Palani S, et al. The Western-Type Diet Induces Anti-HMGB1 Autoimmunity in Apoe(-/-) Mice. Atherosclerosis (2016) 251:31–8. doi: 10.1016/j.atherosclerosis.2016.05.027
137. Palma BD, Gabriel A Jr., Colugnati FA, Tufik S. Effects of Sleep Deprivation on the Development of Autoimmune Disease in an Experimental Model of Systemic Lupus Erythematosus. Am J Physiol Regul Integr Comp Physiol (2006) 291(5):R1527–32. doi: 10.1152/ajpregu.00186.2006
138. Young KA, Munroe ME, Harley JB, Guthridge JM, Kamen DL, Gilkensen GS, et al. Less Than 7 Hours of Sleep Per Night is Associated With Transitioning to Systemic Lupus Erythematosus. Lupus (2018) 27(9):1524–31. doi: 10.1177/0961203318778368
139. Hsiao YH, Chen YT, Tseng CM, Wu LA, Lin WC, Su VY, et al. Sleep Disorders and Increased Risk of Autoimmune Diseases in Individuals Without Sleep Apnea. Sleep (2015) 38(4):581–6. doi: 10.5665/sleep.4574
140. Bermudez EA, Rifai N, Buring JE, Manson JE, Ridker PM. Relation Between Markers of Systemic Vascular Inflammation and Smoking in Women. Am J Cardiol (2002) 89(9):1117–9. doi: 10.1016/S0002-9149(02)02284-1
141. Tracy RP, Psaty BM, Macy E, Bovill EG, Cushman M, Cornell ES, et al. Lifetime Smoking Exposure Affects the Association of C-Reactive Protein With Cardiovascular Disease Risk Factors and Subclinical Disease in Healthy Elderly Subjects. Arterioscler Thromb Vasc Biol (1997) 17(10):2167–76. doi: 10.1161/01.ATV.17.10.2167
142. Hahn J, Leatherwood C, Malspeis S, Liu X, Lu B, Roberts AL, et al. Associations Between Smoking and Systemic Lupus Erythematosus-Related Cytokines and Chemokines Among US Female Nurses. Arthritis Care Res (Hoboken) (2021) 73(11):1583–9. doi: 10.1002/acr.24370
143. Panagiotakos DB, Pitsavos C, Yannakoulia M, Chrysohoou C, Stefanadis C. The Implication of Obesity and Central Fat on Markers of Chronic Inflammation: The ATTICA Study. Atherosclerosis (2005) 183(2):308–15. doi: 10.1016/j.atherosclerosis.2005.03.010
144. Irwin MR, Wang M, Campomayor CO, Collado-Hidalgo A, Cole S. Sleep Deprivation and Activation of Morning Levels of Cellular and Genomic Markers of Inflammation. Arch Intern Med (2006) 166(16):1756–62. doi: 10.1001/archinte.166.16.1756
145. Vgontzas AN, Zoumakis E, Bixler EO, Lin HM, Follett H, Kales A, et al. Adverse Effects of Modest Sleep Restriction on Sleepiness, Performance, and Inflammatory Cytokines. J Clin Endocrinol Metab (2004) 89(5):2119–26. doi: 10.1210/jc.2003-031562
146. Bollinger T, Bollinger A, Skrum L, Dimitrov S, Lange T, Solbach W. Sleep-Dependent Activity of T Cells and Regulatory T Cells. Clin Exp Immunol (2009) 155(2):231–8. doi: 10.1111/j.1365-2249.2008.03822.x
147. Clinton JM, Davis CJ, Zielinski MR, Jewett KA, Krueger JM. Biochemical Regulation of Sleep and Sleep Biomarkers. J Clin Sleep Med (2011) 7(5 Suppl):S38–42. doi: 10.5664/JCSM.1360
148. Wilder-Smith A, Mustafa FB, Earnest A, Gen L, Macary PA. Impact of Partial Sleep Deprivation on Immune Markers. Sleep Med (2013) 14(10):1031–4. doi: 10.1016/j.sleep.2013.07.001
149. Cooper GS, Parks CG, Treadwell EL, St Clair EW, Gilkeson GS, Dooley MA. Occupational Risk Factors for the Development of Systemic Lupus Erythematosus. J Rheumatol (2004) 31(10):1928–33.
150. Calcagni E, Elenkov I. Stress System Activity, Innate and T Helper Cytokines, and Susceptibility to Immune-Related Diseases. Ann N Y Acad Sci (2006) 1069:62–76. doi: 10.1196/annals.1351.006
151. Roberts AL, Malspeis S, Kubzansky LD, Feldman CH, Chang SC, Koenen KC, et al. Association of Trauma and Posttraumatic Stress Disorder With Incident Systemic Lupus Erythematosus in a Longitudinal Cohort of Women. Arthritis Rheumatol (2017) 69(11):2162–9. doi: 10.1002/art.40222
152. Case SM, Feldman CH, Guan H, Stevens E, Kubzansky LD, Koenen KC, et al. Post-Traumatic Stress Disorder (PTSD) and Risk of Systemic Lupus Erythematosus (SLE) Among Medicaid Recipients. Arthritis Care Res (Hoboken) (2021). doi: 10.1002/acr.24758
153. Cozier YC, Barbhaiya M, Castro-Webb N, Conte C, Tedeschi S, Leatherwood C, et al. Association of Child Abuse and Systemic Lupus Erythematosus in Black Women During Adulthood. Arthritis Care Res (Hoboken) (2021) 73(6):833–40. doi: 10.1002/acr.24188
154. Feldman CH, Malspeis S, Leatherwood C, Kubzansky L, Costenbader KH, Roberts AL. Association of Childhood Abuse With Incident Systemic Lupus Erythematosus in Adulthood in a Longitudinal Cohort of Women. J Rheumatol (2019) 46(12):1589–96. doi: 10.3899/jrheum.190009
155. Roberts AL, Kubzansky LD, Malspeis S, Feldman CH, Costenbader KH. Association of Depression With Risk of Incident Systemic Lupus Erythematosus in Women Assessed Across 2 Decades. JAMA Psychiatry (2018) 75(12):1225–33. doi: 10.1001/jamapsychiatry.2018.2462
156. Sumner JA, Chen Q, Roberts AL, Winning A, Rimm EB, Gilsanz P, et al. Posttraumatic Stress Disorder Onset and Inflammatory and Endothelial Function Biomarkers in Women. Brain Behav Immun (2018) 69:203–9. doi: 10.1016/j.bbi.2017.11.013
157. Passos IC, Vasconcelos-Moreno MP, Costa LG, Kunz M, Brietzke E, Quevedo J, et al. Inflammatory Markers in Post-Traumatic Stress Disorder: A Systematic Review, Meta-Analysis, and Meta-Regression. Lancet Psychiatry (2015) 2(11):1002–12. doi: 10.1016/S2215-0366(15)00309-0
158. Gill J, Vythilingam M, Page GG. Low Cortisol, High DHEA, and High Levels of Stimulated TNF-Alpha, and IL-6 in Women With PTSD. J Trauma Stress (2008) 21(6):530–9. doi: 10.1002/jts.20372
159. Pace TW, Wingenfeld K, Schmidt I, Meinlschmidt G, Hellhammer DH, Heim CM. Increased Peripheral NF-kappaB Pathway Activity in Women With Childhood Abuse-Related Posttraumatic Stress Disorder. Brain Behav Immun (2012) 26(1):13–7. doi: 10.1016/j.bbi.2011.07.232
160. Lindqvist D, Wolkowitz OM, Mellon S, Yehuda R, Flory JD, Henn-Haase C, et al. Proinflammatory Milieu in Combat-Related PTSD is Independent of Depression and Early Life Stress. Brain Behav Immun (2014) 42:81–8. doi: 10.1016/j.bbi.2014.06.003
161. Slopen N, Kubzansky LD, McLaughlin KA, Koenen KC. Childhood Adversity and Inflammatory Processes in Youth: A Prospective Study. Psychoneuroendocrinology (2013) 38(2):188–200. doi: 10.1016/j.psyneuen.2012.05.013
162. Gola H, Engler H, Sommershof A, Adenauer H, Kolassa S, Schedlowski M, et al. Posttraumatic Stress Disorder is Associated With an Enhanced Spontaneous Production of Pro-Inflammatory Cytokines by Peripheral Blood Mononuclear Cells. BMC Psychiatry (2013) 13:40. doi: 10.1186/1471-244X-13-40
163. Hartwell KJ, Moran-Santa Maria MM, Twal WO, Shaftman S, DeSantis SM, McRae-Clark AL, et al. Association of Elevated Cytokines With Childhood Adversity in a Sample of Healthy Adults. J Psychiatr Res (2013) 47(5):604–10. doi: 10.1016/j.jpsychires.2013.01.008
164. Danese A, Moffitt TE, Pariante CM, Ambler A, Poulton R, Caspi A. Elevated Inflammation Levels in Depressed Adults With a History of Childhood Maltreatment. Arch Gen Psychiatry (2008) 65(4):409–15. doi: 10.1001/archpsyc.65.4.409
165. Boudigaard SH, Schlunssen V, Vestergaard JM, Sondergaard K, Toren K, Peters S, et al. Occupational Exposure to Respirable Crystalline Silica and Risk of Autoimmune Rheumatic Diseases: A Nationwide Cohort Study. Int J Epidemiol (2021) 50(4):1213–26. doi: 10.1093/ije/dyaa287
166. Parks CG, Cooper GS, Nylander-French LA, Sanderson WT, Dement JM, Cohen PL, et al. Occupational Exposure to Crystalline Silica and Risk of Systemic Lupus Erythematosus: A Population-Based, Case-Control Study in the Southeastern United States. Arthritis Rheumatol (2002) 46(7):1840–50. doi: 10.1002/art.10368
167. Finckh A, Cooper GS, Chibnik LB, Costenbader KH, Watts J, Pankey H, et al. Occupational Silica and Solvent Exposures and Risk of Systemic Lupus Erythematosus in Urban Women. Arthritis Rheumatol (2006) 54(11):3648–54. doi: 10.1002/art.22210
168. Cooper GS, Wither J, Bernatsky S, Claudio JO, Clarke A, Rioux JD, et al. Occupational and Environmental Exposures and Risk of Systemic Lupus Erythematosus: Silica, Sunlight, Solvents. Rheumatology (Oxford) (2010) 49(11):2172–80. doi: 10.1093/rheumatology/keq214
169. Bernatsky S, Smargiassi A, Barnabe C, Svenson LW, Brand A, Martin RV, et al. Fine Particulate Air Pollution and Systemic Autoimmune Rheumatic Disease in Two Canadian Provinces. Environ Res (2016) 146:85–91. doi: 10.1016/j.envres.2015.12.021
170. Bernatsky S, Smargiassi A, Johnson M, Kaplan GG, Barnabe C, Svenson L, et al. Fine Particulate Air Pollution, Nitrogen Dioxide, and Systemic Autoimmune Rheumatic Disease in Calgary, Alberta. Environ Res (2015) 140:474–8. doi: 10.1016/j.envres.2015.05.007
171. Parks CG, Walitt BT, Pettinger M, Chen JC, de Roos AJ, Hunt J, et al. Insecticide Use and Risk of Rheumatoid Arthritis and Systemic Lupus Erythematosus in the Women's Health Initiative Observational Study. Arthritis Care Res (Hoboken) (2011) 63(2):184–94. doi: 10.1002/acr.20335
172. Williams JN, Chang SC, Sinnette C, Malspeis S, Parks CG, Karlson EW, et al. Pesticide Exposure and Risk of Systemic Lupus Erythematosus in an Urban Population of Predominantly African-American Women. Lupus (2018) 27(13):2129–34. doi: 10.1177/0961203318805844
173. Sontheimer C, Liggitt D, Elkon KB. Ultraviolet B Irradiation Causes Stimulator of Interferon Genes-Dependent Production of Protective Type I Interferon in Mouse Skin by Recruited Inflammatory Monocytes. Arthritis Rheumatol (2017) 69(4):826–36. doi: 10.1002/art.39987
174. Yin Q, Xu X, Lin Y, Lv J, Zhao L, He R. Ultraviolet B Irradiation Induces Skin Accumulation of Plasmacytoid Dendritic Cells: A Possible Role for Chemerin. Autoimmunity (2014) 47(3):185–92. doi: 10.3109/08916934.2013.866105
175. Wolf SJ, Estadt SN, Theros J, Moore T, Ellis J, Liu J, et al. Ultraviolet Light Induces Increased T Cell Activation in Lupus-Prone Mice via Type I IFN-Dependent Inhibition of T Regulatory Cells. J Autoimmun (2019) 103:102291. doi: 10.1016/j.jaut.2019.06.002
176. Costenbader KH, Feskanich D, Holmes M, Karlson EW, Benito-Garcia E. Vitamin D Intake and Risks of Systemic Lupus Erythematosus and Rheumatoid Arthritis in Women. Ann Rheum Dis (2008) 67(4):530–5. doi: 10.1136/ard.2007.072736
177. Costenbader KH, Feskanich D, Stampfer MJ, Karlson EW. Reproductive and Menopausal Factors and Risk of Systemic Lupus Erythematosus in Women. Arthritis Rheumatol (2007) 56(4):1251–62. doi: 10.1002/art.22510
178. Bernier MO, Mikaeloff Y, Hudson M, Suissa S. Combined Oral Contraceptive Use and the Risk of Systemic Lupus Erythematosus. Arthritis Rheumatol (2009) 61(4):476–81. doi: 10.1002/art.24398
179. Lateef A, Petri M. Hormone Replacement and Contraceptive Therapy in Autoimmune Diseases. J Autoimmun (2012) 38(2-3):J170–6. doi: 10.1016/j.jaut.2011.11.002
180. Grimaldi CM, Cleary J, Dagtas AS, Moussai D, Diamond B. Estrogen Alters Thresholds for B Cell Apoptosis and Activation. J Clin Invest (2002) 109(12):1625–33. doi: 10.1172/JCI0214873
181. Jog NR, James JA. Epstein Barr Virus and Autoimmune Responses in Systemic Lupus Erythematosus. Front Immunol (2020) 11:623944. doi: 10.3389/fimmu.2020.623944
182. Iwakiri D, Zhou L, Samanta M, Matsumoto M, Ebihara T, Seya T, et al. Epstein-Barr Virus (EBV)-Encoded Small RNA is Released From EBV-Infected Cells and Activates Signaling From Toll-Like Receptor 3. J Exp Med (2009) 206(10):2091–9. doi: 10.1084/jem.20081761
183. Hanlon P, Avenell A, Aucott L, Vickers MA. Systematic Review and Meta-Analysis of the Sero-Epidemiological Association Between Epstein-Barr Virus and Systemic Lupus Erythematosus. Arthritis Res Ther (2014) 16(1):R3. doi: 10.1186/ar4429
184. Ulff-Moller CJ, Nielsen NM, Rostgaard K, Hjalgrim H, Frisch M. Epstein-Barr Virus-Associated Infectious Mononucleosis and Risk of Systemic Lupus Erythematosus. Rheumatology (Oxford) (2010) 49(9):1706–12. doi: 10.1093/rheumatology/keq148
185. Jog NR, Young KA, Munroe ME, Harmon MT, Guthridge JM, Kelly JA, et al. Association of Epstein-Barr Virus Serological Reactivation With Transitioning to Systemic Lupus Erythematosus in at-Risk Individuals. Ann Rheum Dis (2019) 78(9):1235–41. doi: 10.1136/annrheumdis-2019-215361
186. Grimaldi-Bensouda L, Le Guern V, Kone-Paut I, Aubrun E, Fain O, Ruel M, et al. The Risk of Systemic Lupus Erythematosus Associated With Vaccines: An International Case-Control Study. Arthritis Rheumatol (2014) 66(6):1559–67. doi: 10.1002/art.38429
187. Baimukhamedov C, Makhmudov S, Botabekova A. Seropositive Rheumatoid Arthritis After Vaccination Against SARS-CoV-2 Infection. Int J Rheum Dis (2021) 24(11):1440–1. doi: 10.1111/1756-185X.14220
188. Elrashdy F, Tambuwala MM, Hassan SS, Adadi P, Seyran M, Abd El-Aziz TM, et al. Autoimmunity Roots of the Thrombotic Events After COVID-19 Vaccination. Autoimmun Rev (2021) 20(11):102941. doi: 10.1016/j.autrev.2021.102941
189. Clayton-Chubb D, Schneider D, Freeman E, Kemp W, Roberts SK. Autoimmune Hepatitis Developing After the ChAdOx1 Ncov-19 (Oxford-AstraZeneca) Vaccine. J Hepatol (2021) 75(5):1249–50. doi: 10.1016/j.jhep.2021.06.014
190. Badier L, Toledano A, Porel T, Dumond S, Jouglen J, Sailler L, et al. IgA Vasculitis in Adult Patient Following Vaccination by ChadOx1 Ncov-19. Autoimmun Rev (2021) 20(11):102951. doi: 10.1016/j.autrev.2021.102951
191. Chen Y, Xu Z, Wang P, Li XM, Shuai ZW, Ye DQ, et al. New-Onset Autoimmune Phenomena Post-COVID-19 Vaccination. Immunology (2021) 165(4):386–401. doi: 10.1111/imm.13443
192. Patil S, Patil A. Systemic Lupus Erythematosus After COVID-19 Vaccination: A Case Report. J Cosmet Dermatol (2021) 20(10):3103–4. doi: 10.1111/jocd.14386
193. Zavala-Miranda MF, Gonzalez-Ibarra SG, Perez-Arias AA, Uribe-Uribe NO, Mejia-Vilet JM. New-Onset Systemic Lupus Erythematosus Beginning as Class V Lupus Nephritis After COVID-19 Vaccination. Kidney Int (2021) 100(6):1340–1. doi: 10.1016/j.kint.2021.09.009
194. Kreuter A, Burmann SN, Burkert B, Oellig F, Michalowitz AL. Transition of Cutaneous Into Systemic Lupus Erythematosus Following Adenoviral Vector-Based SARS-CoV-2 Vaccination. J Eur Acad Dermatol Venereol (2021) 35(11):e733–e5. doi: 10.1111/jdv.17514
195. Costenbader KH, Schur PH. We Need Better Classification and Terminology for "People at High Risk of or in the Process of Developing Lupus". Arthritis Care Res (Hoboken) (2015) 67(5):593–6. doi: 10.1002/acr.22484
196. Arbuckle MR, James JA, Dennis GJ, Rubertone MV, McClain MT, Kim XR, et al. Rapid Clinical Progression to Diagnosis Among African-American Men With Systemic Lupus Erythematosus. Lupus (2003) 12(2):99–106. doi: 10.1191/0961203303lu334oa
197. Bodolay E, Csiki Z, Szekanecz Z, Ben T, Kiss E, Zeher M, et al. Five-Year Follow-Up of 665 Hungarian Patients With Undifferentiated Connective Tissue Disease (UCTD). Clin Exp Rheumatol (2003) 21(3):313–20.
198. Bourn R, James JA. Preclinical Lupus. Curr Opin Rheumatol (2015) 27(5):433–9. doi: 10.1097/BOR.0000000000000199
199. Deane KD, El-Gabalawy H. Pathogenesis and Prevention of Rheumatic Disease: Focus on Preclinical RA and SLE. Nat Rev Rheumatol (2014) 10(4):212–28. doi: 10.1038/nrrheum.2014.6
200. Swaak AJ, van de Brink H, Smeenk RJ, Manger K, Kalden JR, Tosi S, et al. Incomplete Lupus Erythematosus: Results of a Multicentre Study Under the Supervision of the EULAR Standing Committee on International Clinical Studies Including Therapeutic Trials (ESCISIT). Rheumatology (Oxford) (2001) 40(1):89–94. doi: 10.1093/rheumatology/40.1.89
201. Organization WH. Screening Programmes: A Short Guide. Increase Effectiveness, Maximize Benefits and Minimize Harm Copenhagen, Denmark. (2020).
202. Karlson EW, Sanchez-Guerrero J, Wright EA, Lew RA, Daltroy LH, Katz JN, et al. A Connective Tissue Disease Screening Questionnaire for Population Studies. Ann Epidemiol (1995) 5(4):297–302. doi: 10.1016/1047-2797(94)00096-C
203. Karlson EW, Costenbader KH, McAlindon TE, Massarotti EM, Fitzgerald LM, Jajoo R, et al. High Sensitivity, Specificity and Predictive Value of the Connective Tissue Disease Screening Questionnaire Among Urban African-American Women. Lupus (2005) 14(10):832–6. doi: 10.1191/0961203305lu2227oa
204. Sciascia S, Roccatello D, Radin M, Parodis I, Yazdany J, Pons-Estel G, et al. Differentiating Between UCTD and Early-Stage SLE: From Definitions to Clinical Approach. Nat Rev Rheumatol (2022) 18(1):9–21. doi: 10.1038/s41584-021-00710-2
205. Choi MY, Barber MR, Barber CE, Clarke AE, Fritzler MJ. Preventing the Development of SLE: Identifying Risk Factors and Proposing Pathways for Clinical Care. Lupus (2016) 25(8):838–49. doi: 10.1177/0961203316640367
206. Gatto M, Saccon F, Zen M, Iaccarino L, Doria A. Preclinical and Early Systemic Lupus Erythematosus. Best Pract Res Clin Rheumatol (2019) 33(4):101422. doi: 10.1016/j.berh.2019.06.004
207. Wilson JMG, Jungner G, Organization WH. Principles and Practice of Screening for Disease Geneva, Switzerland. (1968).
208. Cui J, Malspeis S, Choi M, Lu B, Sparks JA, Yoshida K, et al. Risk Prediction Models for Incident Systemic Lupus Erythematosus Using Lifestyle/Environmental Risk Factors and a Genetic Risk Score. Arthritis Rheumatol (2021) 73(suppl 10).
209. Olsen NJ, James JA, Arriens C, Ishimori ML, Wallace DJ, Kamen DL, et al. Study of Anti-Malarials in Incomplete Lupus Erythematosus (SMILE): Study Protocol for a Randomized Controlled Trial. Trials (2018) 19(1):694. doi: 10.1186/s13063-018-3076-7
210. Petri M, Orbai AM, Alarcon GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and Validation of the Systemic Lupus International Collaborating Clinics Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheumatol (2012) 64(8):2677–86. doi: 10.1002/art.34473
211. Lambers WM, Westra J, Bootsma H, de Leeuw K. Hydroxychloroquine Suppresses Interferon-Inducible Genes and B Cell Activating Factor in Patients With Incomplete and New-Onset Systemic Lupus Erythematosus. J Rheumatol (2021) 48(6):847–51. doi: 10.3899/jrheum.200726
212. Hahn J, Cook NR, Alexander EK, Friedman S, Walter J, Bubes V, et al. Vitamin D and Marine Omega 3 Fatty Acid Supplementation and Incident Autoimmune Disease: VITAL Randomized Controlled Trial. BMJ (2022) 376:e066452. doi: 10.1136/bmj-2021-066452
213. Attar SM, Siddiqui AM. Vitamin D Deficiency in Patients With Systemic Lupus Erythematosus. Oman Med J (2013) 28(1):42–7. doi: 10.5001/omj.2013.10
214. Cutolo M, Otsa K, Paolino S, Yprus M, Veldi T, Seriolo B. Vitamin D Involvement in Rheumatoid Arthritis and Systemic Lupus Erythaematosus. Ann Rheum Dis (2009) 68(3):446–7. doi: 10.1136/ard.2008.093476
215. Iruretagoyena M, Hirigoyen D, Naves R, Burgos PI. Immune Response Modulation by Vitamin D: Role in Systemic Lupus Erythematosus. Front Immunol (2015) 6:513. doi: 10.3389/fimmu.2015.00513
216. Navarini L, Bisogno T, Margiotta DPE, Piccoli A, Angeletti S, Laudisio A, et al. Role of the Specialized Proresolving Mediator Resolvin D1 in Systemic Lupus Erythematosus: Preliminary Results. J Immunol Res (2018) 2018:5264195. doi: 10.1155/2018/5264195
217. Davis-Porada J, Serhan C, Norris P, Lipsky P, Salmon J. 3 Polyunsaturated Fatty Acids (PUFAs) and Specialized Pro-Resolving Mediators (SPMs) are Decreased in Plasma and Serum From SLE Patients Compared to Healthy Controls. Lupus Sci Med (2019) 6(Suppl 1):A4–A. doi: 10.1136/lupus-2019-lsm.3
218. Lin GJ, Huang SH, Chen SJ, Wang CH, Chang DM, Sytwu HK. Modulation by Melatonin of the Pathogenesis of Inflammatory Autoimmune Diseases. Int J Mol Sci (2013) 14(6):11742–66. doi: 10.3390/ijms140611742
219. Lechner O, Dietrich H, Oliveira dos Santos A, Wiegers GJ, Schwarz S, Harbutz M, et al. Altered Circadian Rhythms of the Stress Hormone and Melatonin Response in Lupus-Prone MRL/MP-Fas(Ipr) Mice. J Autoimmun (2000) 14(4):325–33. doi: 10.1006/jaut.2000.0375
220. Zhou LL, Wei W, Si JF, Yuan DP. Regulatory Effect of Melatonin on Cytokine Disturbances in the Pristane-Induced Lupus Mice. Mediators Inflamm (2010) 2010:951210. doi: 10.1155/2010/951210
221. Jimenez-Caliani AJ, Jimenez-Jorge S, Molinero P, Fernandez-Santos JM, Martin-Lacave I, Rubio A, et al. Sex-Dependent Effect of Melatonin on Systemic Erythematosus Lupus Developed in Mrl/Mpj-Faslpr Mice: It Ameliorates the Disease Course in Females, Whereas it Exacerbates it in Males. Endocrinology (2006) 147(4):1717–24. doi: 10.1210/en.2005-0648
222. Ferland D, Fortin PR. Recruitment Strategies in Superiority Trials in SLE: Lessons From the Study of Methotrexate in Lupus Erythematosus (SMILE). Lupus (1999) 8(8):606–11. doi: 10.1191/096120399680411371
223. Costenbader KH, Karlson EW, Gall V, de Pablo P, Finckh A, Lynch M, et al. Barriers to a Trial of Atherosclerosis Prevention in Systemic Lupus Erythematosus. Arthritis Rheumatol (2005) 53(5):718–23. doi: 10.1002/art.21441
224. Pope JE, Tingey DP, Arnold JM, Hong P, Ouimet JM, Krizova A. Are Subjects Satisfied With the Informed Consent Process? A Survey of Research Participants. J Rheumatol (2003) 30(4):815–24.
Keywords: systemic lupus erythematosus, prevention, biomarkers, risk factors, pathogenesis
Citation: Choi MY and Costenbader KH (2022) Understanding the Concept of Pre-Clinical Autoimmunity: Prediction and Prevention of Systemic Lupus Erythematosus: Identifying Risk Factors and Developing Strategies Against Disease Development. Front. Immunol. 13:890522. doi: 10.3389/fimmu.2022.890522
Received: 06 March 2022; Accepted: 04 May 2022;
Published: 03 June 2022.
Edited by:
David Karp, University of Texas Southwestern Medical Center, United StatesReviewed by:
Melissa E Munroe, Oklahoma Medical Research Foundation, United StatesRicardo Machado Xavier, Federal University of Rio Grande do Sul, Brazil
József Prechl, Diagnosticum Zrt., Hungary
Copyright © 2022 Choi and Costenbader. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: May Y. Choi, bWF5LmNob2lAdWNhbGdhcnkuY2E=