 Jack T. Stapleton
Jack T. Stapleton- 1Medicine Service, Iowa City Veterans Administration Healthcare, Iowa City, IA, United States
- 2Departments of Internal Medicine, Microbiology & Immunology, University of Iowa, Iowa City, IA, United States
Two groups identified a novel human flavivirus in the mid-1990s. One group named the virus hepatitis G virus (HGV) and the other named it GB Virus type C (GBV-C). Sequence analyses found these two isolates to be the same virus, and subsequent studies found that the virus does not cause hepatitis despite sharing genome organization with hepatitis C virus. Although HGV/GBV-C infection is common and may cause persistent infection in humans, the virus does not appear to directly cause any other known disease state. Thus, the virus was renamed “human pegivirus 1” (HPgV-1) for “persistent G” virus. HPgV-1 is found primarily in lymphocytes and not hepatocytes, and several studies found HPgV-1 infection associated with prolonged survival in people living with HIV. Co-infection of human lymphocytes with HPgV-1 and HIV inhibits HIV replication. Although three viral proteins directly inhibit HIV replication in vitro, the major effects of HPgV-1 leading to reduced HIV-related mortality appear to result from a global reduction in immune activation. HPgV-1 specifically interferes with T cell receptor signaling (TCR) by reducing proximal activation of the lymphocyte specific Src kinase LCK. Although TCR signaling is reduced, T cell activation is not abolished and with sufficient stimulus, T cell functions are enabled. Consequently, HPgV-1 is not associated with immune suppression. The HPgV-1 immunomodulatory effects are associated with beneficial outcomes in other diseases including Ebola virus infection and possibly graft-versus-host-disease following stem cell transplantation. Better understanding of HPgV-1 immune escape and mechanisms of inflammation may identify novel therapies for immune-based diseases.
Introduction and Background
Following the discovery of hepatitis C Virus (HCV), it became clear that there are cases of post-transfusion hepatitis that are not caused by hepatitis B virus (HBV) or HCV (1). Since none of the hepatitis viruses are readily cultivated in laboratory cell culture systems, progress in identifying additional etiologies of post-transfusion hepatitis proved difficult. In the mid-1990s, two companies independently used recently developed molecular methodologies to identify a human RNA virus in samples obtained from individuals with non-A, non-B, non-C post-transfusion hepatitis (2–4). The viral genome sequence and organization shared many similarities with HCV and other members of the Flaviviridae (2, 4–6), and one of the groups discovering the virus named it Hepatitis G Virus (HGV) (4), while the other was called GB virus type C (GBV-C) (2). “G.B.” were the initials of a surgeon who developed fulminant hepatitis in the 1960s, and whose serum caused transmissible hepatitis in marmosets [reviewed in (1, 7]. This “GB” serum was passaged in marmosets, and two viruses were identified in these materials that were called GB virus A and B (GBV-A, GBV-B) (3). Using these viral sequences as a guide, PCR methods were developed that identified a human virus and this virus was called GBV-C (2).
The “GB” serum reagents were extensively studied in the 1960s and 70s under the premise that they may contain the elusive cause of post-transfusion hepatitis, which ultimately proved to be HCV and not the GB agent (8–10). Following identification of HGV/GBV-C, virologic and serologic studies found that this virus is common in humans and that infection correlates with exposure to blood-born and sexually transmitted infections (4, 11–23). The related GBV-A and GBV-B proved to be non-human primate viruses, and there is no evidence that the surgeon “GB” was infected with GBV-C (reviewed in (7, 24). Further, numerous studies demonstrated that HGV is not specifically associated with acute or chronic hepatitis (14, 21, 25–28). In the majority of infections viremia persists for more than one year and may persist for decades in a subset of individuals (17, 18, 29). The underlying host factor(s) responsible for viral clearance remain unknown.
Since the virus does not appear to cause hepatitis, and there is no evidence that the surgeon G.B. was infected with this virus, the two names describing this virus were not logical. A 2011 proposal to assign the “GB viruses” into a novel genus named “Pegivirus” this was accepted by the International Committee on the Taxonomy of Viruses (ICTV) in 2013 (7, 30). Pegiviruses were comprised of viruses that demonstrated distant sequence relatedness to other members of the family Flaviviridae based on phylogenetic alignments of the RNA dependent, RNA polymerase. In addition to having a distinct phylogenetic position most closely related with the hepaciviruses within the Flaviviridae, pegiviruses differ from other hepaciviruses in genome organization features including an apparent lack of a nucleocapsid protein, a different internal ribosomal entry site (IRES) structure in the 5’ non-coding genome region, and less predicted glycosylation of the envelope glycoprotein E2 (reviewed in (7). The name “pegivirus” was derived from “Pe” for “persistent” and “G” representing the historical naming of the virus as HGV and GBV-C (7).
Although initially found only in primates and bats, the host range of pegiviruses has greatly expanded following the advent of next-generation sequencing (31–34). The genus currently has at least 11 host species (Pegivirus A to K) and additional host species are continuing to be identified. Pegivirus A members infect New World primate monkeys (35–37), and bats (38), while members of Pegivirus C infect humans and chimpanzees (2, 4, 39, 40) and other Old World monkeys (41, 42). Human pegivirus is regularly called HPgV-1 although this has not been formally assigned by the ICTV. Excluding HPgV-1 (pegivirus C), species B through J infect a wide range of primates and non-primate hosts including horses, domestic cats, lemurs, bats, dolphins, pigs, and rodent species (31, 38, 43–50). Figure 1 outlines the chronology of Pegivirus discovery.

Figure 1 History of Pegivirus Discovery.
Limited information suggests that pegiviruses do not transmit readily between host species; however, HPgV-1 can infect chimpanzees (51), and a baboon isolate of Pegivirus C can infect rhesus macaques (42, 52). Remarkably, novel viruses sharing pegivirus and hepacivirus features are found in non-mammalian hosts including sharks, lizards (Australian gecko), and geese (53–56). The goose pegivirus is reported to replicate in goslings, embryonated goose eggs, and primary goose embryo fibroblasts, and thus is the first pegivirus efficiently cultured in vitro (55). Until the goose pegivirus was recently described, no pegivirus has been described that replicate efficiently in cell culture systems and this has greatly hampered understanding of the biology of this enigmatic virus. The ability of all of the pegiviruses to cause persistent infection has not been confirmed.
A second human pegivirus closely associated with HCV infection was discovered in 2015 (57, 58). This virus is referred to as HPgV-2 or HHPgV-1, while the original HGV/GBV-C is referred to as HPgV-1. HPgV-2/HHPgV-1 is closely associated with HCV and is a different virus (not a different genotype) than HPgV-1. HPgV-2 is much less prevalent. Although there are few reports elucidating the pathogenic outcomes of this infection, in contrast with HPgV-1, the virus shares some virologic features with HCV including a predicted type IV IRES and highly glycosylated E2 protein (58). Nevertheless, a recent study shows that, like HPgV-1, HPgV-2 appears to be lymphotropic. More study of this virus is needed (59). Since current knowledge related to HPgV-2 is limited, the virus is not in the same phylogenetic lineage as HPgV-1 and is not known to influence other diseases, HPgV-2 will not be discussed further in this review.
HPgV-1 infection occurs globally, and phylogenetic studies identify 7 genotypes that are geographically distributed (60–64). The virus is thought to be ancient based on several lines of evidence. First, genotypes are distributed in patterns that mimic human migration from Africa (7, 61) and the genetic distance between genotypes is similar to that of mitochondrial DNA between peoples of different races (61). South American populations comprised of mixed European and indigenous ancestry have a mixture of genotypes 2 (European/N. American) and 3 (Asian), while indigenous South Americans have genotype 3. Further, the prevalence of the virus in indigenous populations is similar to that of other regions of the world (61, 65–67). Finally, HPgV-1 is closely related to both old world and new world pegiviruses, suggesting that the virus was in primates prior to the split of the continents (61). Taken together, it has been suggested that HPgV-1 was present in primordial humans. Of note, recombination between genotypes has been described, which may complicate phylogenetic analyses and interpretation of genotypic distributions (68).
HPgV-1 shares many features with HCV. The viral genome is ~ 9.4 kB long, has a positive polarity and serves as the messenger RNA being translated off of its 5’ non-coding IRES (2, 4, 6, 35, 69, 70). The genome includes a long open reading frame that is translated into a C-terminal polyprotein with structural proteins post-translationally processed by cellular signal peptidase, and nonstructural proteins processed by two viral proteases (NS2 and NS3) and the NS3 co-factor (NS4A) (71, 72). A schematic of the viral genome, translation product polyprotein, and final protein composition is shown in Figure 2.
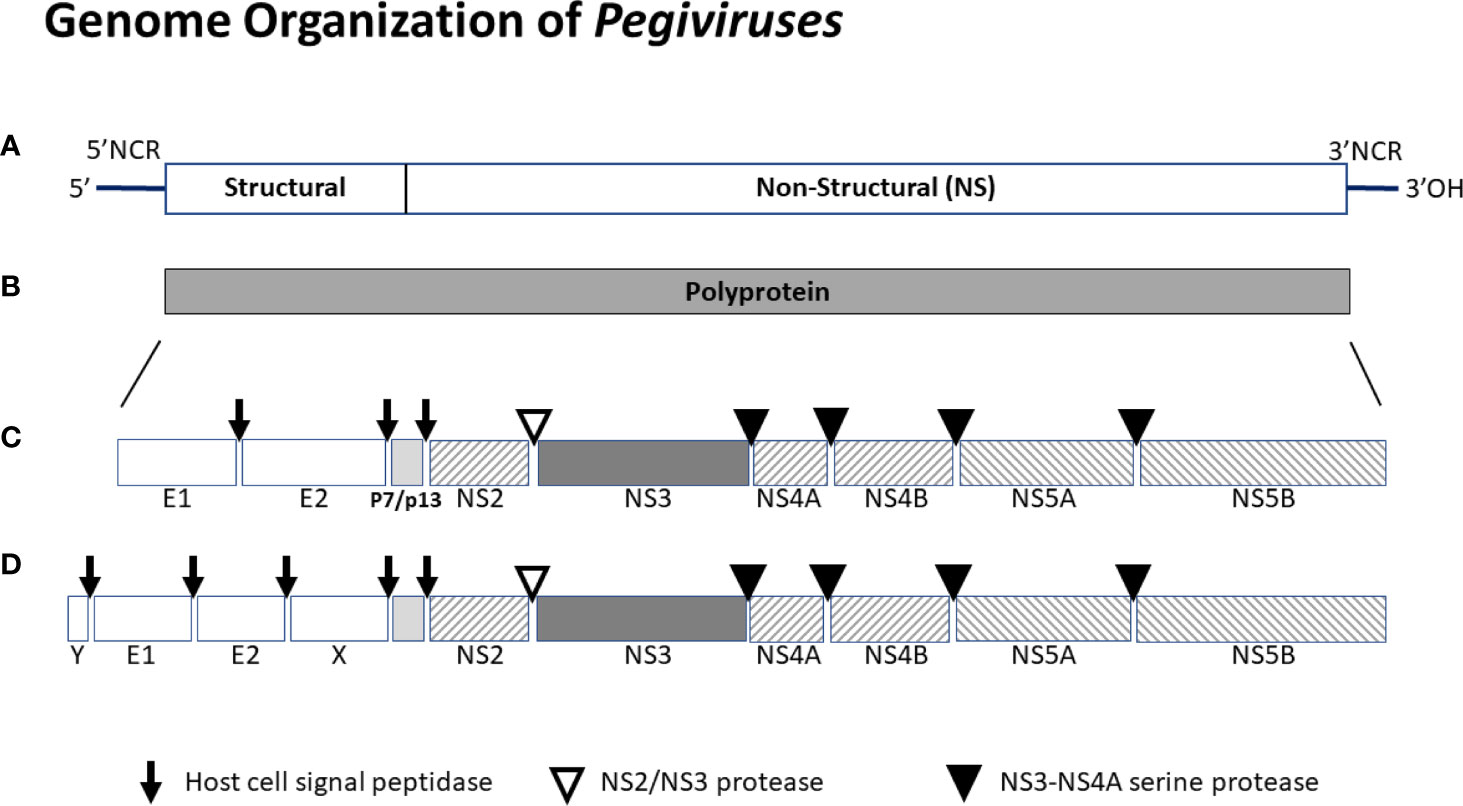
Figure 2 Schematic of the genome organization of pegiviruses. Pegivirus genomes range from 8.9 to 11.3 kb, and those with longer genomes (e.g. HPgV-2) produce two additional structural proteins (X and Y). The genome (A) has a highly structured 5’ noncoding region (NCR) that contains an internal ribosomal entry site directing translation, and a 3’ NCR. The positive sense viral genome is translated and encodes a single polyprotein (B) that is co- and post-translationally cleaved into the final viral proteins (C or D). Structural proteins are cleaved by host cell signal peptidases. Nonstructural proteins NS2-NS3 are cleaved by the NS2-NS3 autoprotease while NS3 to NS5 proteins are cleaved by the NS3-NS4A protease complex. All pegiviruses have two structural proteins (envelope glycoproteins E1 and E2), and those with longer genomes have two additional predicted structural proteins - a basic protein of unknown function upstream of E1 (Y) and some pegiviruses contain a predicted glycoprotein downstream of E2 (X). The RNA dependent, RNA polymerase resides in NS5B. The human pegivirus (HPgV-1) genome is approximately 9.4 kb and the final protein organization is shown in the middle panel (B).
Transmission and Epidemiology
HPgV-1 is transmitted via blood exposure, sexual exposure, and from mother to child (11, 12, 17, 21, 73–79). Based on transfusion studies, HPgV-1 infection typically lasts for > 6 months and is cleared in more than 50% of infections within 2 years (12, 18, 80). In a subset of individuals, viremia persists for more than two years, and for longer in some individuals (29, 81). In those who clear viremia, antibodies to the major viral envelope glycoprotein (E2) are usually detected and these antibodies are somewhat, though not completely protective (82–84). Interestingly, more than 90% of viremic individuals do not have detectable E2 antibodies, and no reproducible antibody pattern against any viral protein is elicited, suggesting a mechanism of immune evasion not described for other virus infections (reviewed in (7, 24, 28).
Based on E2 antibody and viremia prevalence studies, HPgV-1 is extremely common (12, 17, 21, 76, 84, 85). Viremia prevalence among healthy blood donors in developed countries ranges from 1.8% to 6% and E2 antibody prevalence ranges from 10 to 15% (7, 29, 67, 84, 86–89). The prevalence of infection in developed countries is estimated to be as high as 20% based on viremia and antibody determination, and infection is two to five times more frequent in developing countries and in individuals with other sexually transmitted or bloodborne infections (7, 29, 67, 84, 86–89). Since antibodies are lost in some individuals over time, prevalence estimates based on viremia and antibody detection likely represent an underestimate of true infection rates. A recent analysis of 79 studies including a meta-analysis of 63 of these found the global prevalence in healthy volunteer blood donors to be 3.1%, with geographic differences ranging from 1.7% in North America to 9.1% in South America (88). Complicating determinations of prevalence, reinfection of people who have lost antibodies is described (29, 90), and a small proportion of viremic individuals have detectable E2 antibodies (29, 91). The mechanisms underlying viral clearance and the combination of viremia with E2 antibodies are unclear.
Of note, no country screens blood donors for HPgV, despite the fact that viremia is common (2, 4). The argument against screening includes the fact that roughly one in 30 donations would need to be discarded in the U.S., greatly reducing blood availability for an infection, HPgV-1, that does not appear to cause significant disease. A recent study found that there are more than 17.9 million units of blood products transfused in the U.S. in 2017 (92), suggesting that approximately 1,000 blood product units containing HPgV-1 are transfused every day in the United States (92).
As noted above, HPgV-1 prevalence is higher among individuals who are highly exposed to bloodborne or sexually transmitted infections compared to healthy blood donors (21, 67, 76, 93, 94). In people living with HIV (PLWH) for example, 39% were viremic and 45% had detectable E2 antibody in the Multicenter AIDS Cohort study group, suggesting near universal infection (29). The proportion of PLWH who cleared viremia over six years of observation also appears to be lower than that of healthy blood donors (29, 81). Viremia rates in PLWH vary between studies, ranging from 15% to 41% [reviewed in (84, 95–97]. Further evidence of reduced clearance in PLWH is drawn from the finding that the ratio of E2 antibody to viremia is higher among people living with HCV infection (98). Given the rate of HPgV-1 viremia in healthy blood donors and the increase prevalence among people with other transmission risks, it appears that perhaps one third of humans experience HPgV-1 infection during their lifetime (7, 84).
Disease Associations
HPgV-1 infection has been studied in a wide variety of people with and without chronic diseases. Taken together, studies do not show that the virus is clearly responsible for any acute or chronic disease (1, 28, 95, 98–105). Specifically, HPgV-1 is not associated with more extensive HCV-related disease, post-transplant liver disease or non-A to non-E hepatitis. This appears to be related to viral tropism, as evidence strongly suggests that the virus is lymphotropic. Supporting this hypothesis, removal of the donor liver in HCV/HPgV-1 co-infected individuals undergoing liver transplant results in a transient, but profound reduction in HCV viral load with no significant change in quantitative HPgV-1 viremia (106). HPgV-1 RNA is found in T bone marrow and spleen tissues (107–109), and in circulating T and B lymphocytes, monocytes, and NK cells (94, 110–116). Epidemiological studies also suggest that HPgV-1 is lymphotropic, and viral transmission mimics that of HIV (21, 76, 91, 94, 97, 117, 118).
Incubation of HPgV-1 with healthy donor peripheral blood mononuclear cells (PBMCs) or culture of PBMCs from HPgV-1 viremic individuals results in low level release of viral RNA into culture supernatant fluids, and T cell and NK cell functions are altered in vitro and in vivo (see below) (111–115, 119–121). The virus cannot be passaged for more than a few cycles in cell culture, and thus circulating PBMCs do not appear highly permissive for infection (94, 113). The primary cell type infected by HPgV-1 has not been identified, and there is no animal model of HPgV-1 infection to address this question directly. A pegivirus animal model was developed in rhesus macaques using a baboon pegivirus as the inoculum (42, 52). The distribution of baboon pegivirus RNA was consistent with studies of humans showing that bone marrow is the major location of both positive and negative (replicative) forms of HPgV-1 RNA (42, 109). Of note, this model may not be predictive of human pegivirus 1 infection, as there are sequence differences between simian and human viruses in critical regions of the genome.
Nevertheless, the specific cell type that is primarily infected and produces HPgV-1 in vivo remains a key unanswered question in HPgV-1 biology. Given the bone marrow location and distribution of virus RNA in human PBMCs, it has been speculated that a hematopoietic stem cell is the primary site of infection, and that a key replication factor (receptor, transcription factor, etc.) necessary for efficient and productive infection is lost during cell differentiation. Nevertheless, high total body levels of virus are present in infected individuals, with average plasma viral loads ranging from 1 x 105 genome equivalents/mL (GE/mL) to as high as 1 x 107 GE/mL (67, 122). Of note, viral load is distributed in a bi-phasic pattern. Approximately two thirds of healthy blood donors have an average viral load of 5 x 106 GE/mL while the remaining one third of individuals have a viral load of approximately 8 x 103 GE/mL (67). The reason for this biphasic distribution in viral load is unclear but may relate to host specific immunity, or perhaps to prior infection with some existing adaptive immune responses. It would be interesting to follow these subjects longitudinally to see if clearance rates over time differ based on viral load.
A variety of diseases have been studied to determine if there is an association with HPgV-1, and there is no clear association with fulminant hepatitis (98, 123, 124), aplastic anemia (15, 125, 126), hepatocellular carcinoma (103, 127, 128), multiple sclerosis (MS) (129), porphyria cutanea tarda (130), oral lichen planus or oral carcinoma (131), and others reviewed in (26–28, 95). However, there are two clinical situations in which HPgV-1 is associated with disease, non-Hodgkin’s lymphoma (NHL) and sporadic encephalitis.
In several studies including a meta-analysis, HPgV-1 infection is associated with approximately a 2.8-fold increased risk of developing NHL (132–135). HPgV-1 has also been associated with sporadic encephalitis, and viral RNA and antigens were detected in post-mortem brain tissues from two people with this condition (136, 137). A variant of HPgV-1 containing a deletion in the NS2 coding region was identified in the brain tissue of one subject, and this NS2 deletion virus appeared to replicate better in astrocytes than the wild type HPgV-1. HPgV-1 replication has recently been demonstrated in human astrocytes and microglia in vitro, though replication was not identified in neurons or an oligodendrocyte-derived cell line (137). HPgV-1 infection of human glia cells led to the suppression of peroxisome-associated gene expression which was accompanied by reduced expression of interferon-beta, interferon regulatory transcription factors 1 and 3, and mitochondrial antiviral signaling protein. These changes were consistent with findings in the brain tissue obtained from the patients with HPgV-1 and sporadic encephalitis (137). Given the high prevalence of HPgV, the high serum and PBMC viral load, and low rates of sporadic encephalitis, it remains unclear if HPgV-1 commonly infects neural tissue or has any direct neurological outcomes in those without encephalitis. Of note, in the rhesus model of pegivirus infection neural tissues did not demonstrate increased levels of viral RNA (42). It will be important to clarify the host feature(s) associated with putative HPgV-1-related encephalitis.
Among pegivirus infections in non-human species, no definitive association has been identified with a specific disease entity. Early reports suggested that a divergent member of the pegivirus genus found in horses (called Theiler’s disease-associated virus) was responsible for Theiler’s disease (44, 138). However, this conclusion was later shown to be spurious (139, 140). More recently, a novel pegivirus was identified in a research colony of common marmosets (Callithrix jacchus) in animals that died of lymphocytic enterocolitis (34). The investigators named this new virus the “southwest bike trail virus” (SOBV). Although identified in ill marmosets, a prospective analysis of a colony of 85 common marmosets failed to identify an association between SOBV infection and lymphocytic enterocolitis or any other disease. The prevalence of SOBV in common marmoset was high (34%) illustrating the difficulty in assigning disease causality for a prevalent virus (34). To date, SOBV has not been identified in common marmosets found in the wild and it is not clear if this species is the natural host for SOBV (34, 141).
Clinical Evidence of Viral Interference
In the first 5 years following the discovery of HPgV, more than 600 papers were published examining the potential clinical outcomes of infection, with most of these studies conducted by research groups whose focus was on viral hepatitis. Once there was clear evidence that HPgV-1 does not cause acute or chronic liver disease (26, 27), research interest and publication rates fell rapidly. However, small studies from Japan, Germany, the U.S. and France described the role of HPgV-1 in liver disease in HIV coinfection (76, 83, 142, 143). These reports confirmed that HPgV-1 was not associated with liver disease; however, people with HIV and HPgV-1 coinfection appeared to have prolonged survival compared to those with HIV alone. These data were subsequently confirmed in two large, cross-sectional studies of people with HIV infection. The relative risk of mortality was 3.7-fold (95% C.I. 2.5 – 5.4) higher among those without HPgV-1 infection in a U.S. study (94) and also significantly higher in a German study (122), although relative hazard rates and confidence intervals were not described for the latter. Interestingly, HPgV-1 viral loads were inversely related to HIV RNA levels, and HPgV-1 infection did not prevent CD4 depletion (67, 94, 122, 144). Nevertheless, CD4 counts were higher overall in those with HPgV-1 compared to those uninfected in several studies (67, 76, 94, 122, 145, 146).
A legitimate criticism of these early studies related to the fact that the duration of HIV infection was unknown at the time of HPgV-1 sampling. If HPgV-1 were acquired late in infection this would bias prevalence towards those living longer with HIV (118). An alternative explanation for the beneficial observation suggested that HPgV, increasingly recognized as a lymphotropic virus, was simply a marker of conserved CD4 cells. If so, CD4 depletion accompanying HIV infection would lead to a loss of HPgV-1 viremia (Van der Bij et al., 2005). This latter explanation was not consistent with the finding that survival was reduced in PLWH who presented with very low CD4 counts (< 50/mm3) (94, 147).
To address these limitations, two studies were conducted in HIV cohorts with known or an estimated time of HIV infection. In the Multicenter AIDS Cohort Study group, HPgV-1 viremia and E2 antibody were characterized in 271 individuals with documented HIV seroconversion within 12 months of the first positive HIV antibody test. In this “early” group, no survival benefit was observed; however, follow up testing 5-6 years following seroconversion found that those without HPgV-1 infection were 2.83-fold more likely to die in subsequent observation compared to those who were persistently HPgV-1 viremic (29). In addition, almost 20% of HPgV-1 viremic individuals cleared HPgV-1 in the first 5 to 6 years after seroconversion. Individuals who cleared viremia had significantly higher mortality than those who were persistently viremic (3.85-fold) suggesting either that HPgV-1 clearance results in the loss of a beneficial factor, or that clearance of viremia is related to a host variable that overcomes HPgV-1 immune escape mechanisms and which hastens HIV-related disease. Two additional studies confirmed that loss of HPgV-1 viremia was associated with significantly increased mortality (118, 148).
The Amsterdam HIV Study Group also examined the role of HPgV-1 on survival in people living with HIV. Although their data were highly consistent with the MACS study, the authors applied several statistical models to the data and came to different conclusions (118). Similar to the MACS study, the primary, unadjusted analysis found that mortality was significantly lower (RH 0.52, 95% CI 0.32-0.85) in those with persistent HPgV-1 infection. Three additional statistical models were applied to these data. In the first model, which was controlled for age at seroconversion, antiretroviral therapy, CCR5 genotype, baseline CD4, and HIV RNA 1 year after seroconversion, persistent HPgV-1 viremia was significantly associated with reduced mortality (RH 0.57, 95% CI 0.35-0.94). In the second model, the same variables plus time-varying HIV viral load were controlled and there remained a survival benefit (relative hazard of mortality 0.47, 95% CI 0.35 – 0.93) in those with persistent HPgV-1 infection. In the third model, time-varying CD4 count was added to the other cotrolled variables, and there remained a survival benefit (RH mortality 0.74, 95% CI 0.45 – 1.25); however, statistical significance was lost. The authors concluded from these data that HPgV-1 viremia is a marker of CD4 T cell number and that HPgV-1 is not a causative factor in the improved survival observed in the other models (118).
Limitations of the Amsterdam study include that the date of seroconversion was calculated and not measured directly. Since the calculated method of HIV infection has a relatively wide time window for seroconversion (149), the duration of HIV infection in this group was far less accurate than in the MACS study. Secondly, several studies demonstrate that CD4 decline is slowed in HPgV-1 infected PLWH (29, 76, 94, 122, 145). Since death in HIV-1 infection is caused by immune deficiency directly correlated to CD4 depletion, CD4 count is in the causal pathway for death. Controlling for time-varying CD4 removes the differences in CD4 decline in those with and without HPgV-1 infection, and from a statistical standpoint, controlling avariable in the causal pathway should remove any potential benefit of HPgV-1 (147). Nevertheless, there remained a 26% reduction in mortality in those with persistent HPgV in this model. This suggests that pegivirus viremia influences mortality by more than expected from its altered rate of CD4 depletion (147, 150). A meta-analysis of published studies meeting rigorous criteria confirmed a reduction in mortality in HIV infection for those with HPgV-1 co-infection (RH 0.41) (96).
Despite a large number of studies showing survival advantage, lower HIV VL, and higher CD4 in those with HIV and HPgV-1 coinfection, the interpretation of the Amsterdam data continues to reduce acceptance of the apparent beneficial effect of HPgV-1 on HIV survival. Since there is not a good animal model for HPgV, and human challenge experiments proposed to the FDA were not allowed to proceed (using licensed blood products), Koch’s postulates have not been confirmed. A close proxy to Koch’s postulates was assessed by Vahidnia et al., who tested plasma samples obtained from people living with HIV who had received blood transfusions in the 1980s and 1990s. Pre- and post-transfusion samples and samples from the transfused blood product were available for HPgV-1 viremia testing, and individuals who acquired HPgV-1 from transfusion were found to have a reduced risk of death (RH 0.22) compared to those who did not acquire HPgV-1 (151), confirming a direct role for HPgV-1 infection and prolonged survival in HIV infection.
Finally, two studies found that HPgV-1 infection in women is associated with reduced HIV transmission from mother to child in the pre-highly active antiretroviral therapy era. A U.S. study found an association between maternal HPgV-1 viremia in the third trimester and reduced HIV infection in the child (152). A large study in Thailand found that, among infants who acquired HPgV-1 during delivery, HIV transmission was reduced by 87%, further supporting an HPgV-related reductions in HIV disease outcomes (78).
Taken together, the data demonstrating a survival benefit among PLWH who have HPgV-1 coinfection is thus supported by numerous clinical studies. Two papers recommend studying HPgV-1 infection as a “biotherapy” for HIV infection (153, 154). To date, regulatory groups have not approved this approach. The following section will summarize data investigating mechanisms by which HPgV-1 might influence HIV disease outcomes.
Potential Mechanisms of HPgV-1 Interference With HIV
Evidence of a Direct Antiviral Effect
Infection of healthy human PBMCs with HPgV-1 for 24 hours prior to adding HIV results in more than a 90% reduction in HIV production (94, 119, 155). The extent of inhibition in HIV replication was reduced if HPgV-1 was added simultaneously or 24 hours after HIV infection, although HIV replication was still significantly reduced (94). Of note, PBMC infection with HPgV-1 is inefficient, and replication kinetics are extremely virus- and host-cell specific (113). Maintaining fresh PBMCs obtained from infected individuals in cell culture leads to low levels of HPgV-1 production over long periods of time, up to 10 weeks in many instances (114, 115). In this approach, the majority of PBMCs die in the first week but a small population of minimally metabolically active lymphocytes persist and release low levels of HPgV-1 RNA into the culture supernatants. Early studies found that expression of the HIV co-receptor CCR5 was reduced on CD4+ and CD8+ T cells in individuals with HPgV-1 and HIV co-infection compared to HIV mono-infected (156, 157). This result was recapitulated and extended in vitro (155), and incubation of PBMCs with HPgV-1 upregulated chemokine ligands for the two HIV co-receptors, CCR5 and CXCR4. Further, co-receptor expression was reduced under these conditions (155, 158).
Studies were designed to examine the ability of specific viral components to inhibit HIV with the hope of identifying novel antiviral agents that may have low risk of mutational escape. Expression of the NS5A protein in a CD4+ T cell line resulted in highly (>90%) suppressed HIV inhibition (159, 160). By making expressing a series of NS5A deletions, the inhibitory region was found to reside in a small (16 amino acid) E2 peptide region. HIV inhibition was observed with viruses representing different genotypes, and NS5A reduced CD4 and CXCR4 expression and increased production of the CXCR4 ligand SDF-1 (159, 161, 162). HIV inhibition required expression of the NS5A peptide region, as cells expressing NS5A coding RNA with a frameshift inserted to abolish protein synthesis did not inhibit HIV. (159) (Figure 3).
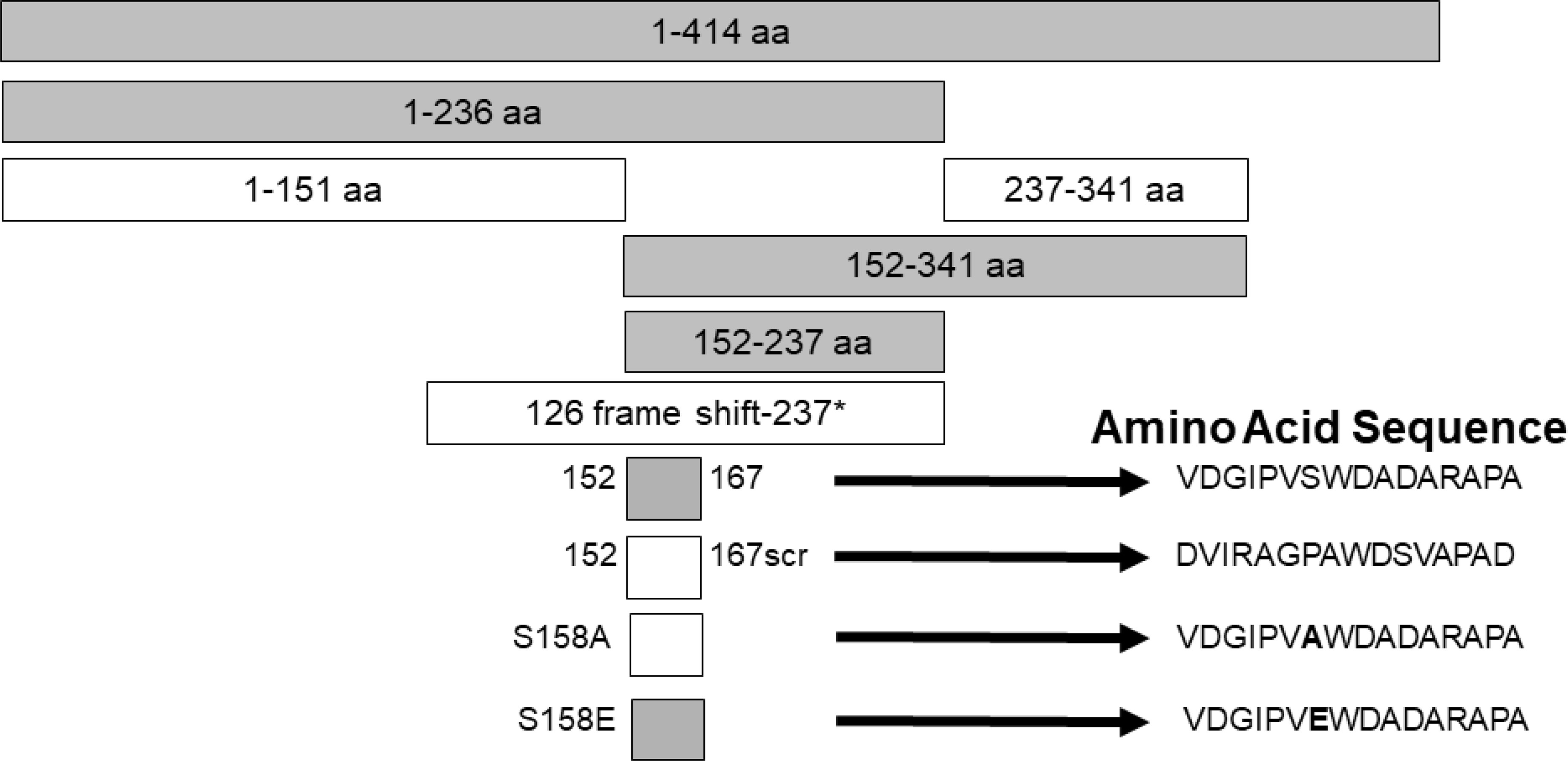
Figure 3 Expression of HPgV NS5A peptides inhibits HIV replication, adapted from References (159, 162). Schematic representation of the region of NS5A protein stably expressed in CD4+ T cells. The regions that inhibited HIV replication are shaded. Full-length NS5A protein (414 amino acids) is shown on top, and deletion proteins below. The 16 aa peptide from 152 – 167 was sufficient to inhibit HIV replication. Scrambling the order of the 16 amino acids or mutating the putative phosphorylation site (serine) at amino acid 158 abolished HIV inhibition (scr), while mutating this to a glutamic acid (phosphomimetic) restored HIV inhibition functions. FS represents the full-length RNA for NS5A in which a frame shift insertion was created to abolish NS5A protein expression.
In addition to NS5A, recombinant HPgV-1 envelope (E2) protein inhibited HIV replication in vitro (158, 163, 164). Similar to the approach used with NS5A, expression of HPgV-1 E2 in CD4+ T cell lines also potently inhibited HIV replication. Deletion mutagenesis identified a 17 amino acid E2 peptide previously shown to be involved in virus cellular binding was sufficient to inhibit HIV replication (McLinden et al., 2006;164, 165). As with NS5A, point mutations in this E2 peptide reversed the inhibition, and synthetic peptides containing the HIV TAT transduction domain peptide to provide entry into cells resulted in HIV inhibition compared to control peptides (164). Additional peptide regions of HPgV-1 E2 or E1 interfere with HIV replication (164, 166–174). Of note, E2 peptide regions shown to inhibit HIV in vitro are not consistent between different experimental systems; however, HPgV-1 E2- and E1-mediated HIV inhibition correlated with reduced HIV entry in most reports (163, 164, 166, 169–173).
In addition to reducing HIV entry, HPgV-1 E2 protein appears to have additional effects on HIV replication by inhibiting HIV GAG assembly and release (175). Specifically, expression of HPgV-1 E2 inhibited Gag processing with no change in the GAG precursor Pr55, but with reduced levels of the capsid regions CA24 and MAp17, both of which are phosphorylated motifs (175, 176). Glycosylated HPgV-1 E2 disrupted GAG trafficking to the plasma membrane, and reduced HIV particle production (175). Taken together HPgV-1 E2 protein and possibly an HPgV-1 fusion motif on E1 inhibits HIV replication in vitro.
The cellular receptor for HPgV-1 remains uncharacterized. An early study suggested that HPgV-1 E2 binds to CD81 (177), which serves as the major HCV E2 binding receptor on cells (178, 179), However, subsequent studies using CD81 deificient cell lines and competition experiments showed that CD81 is not required for E2 cell binding (180). Although these finding suggest that potential, peptide-based or small molecule HIV inhibitors based on HPgV-1 peptides might be developed, the widespread use of highly active, combination antiretroviral therapies essentially removed the need to pursue this novel therapeutic approach.
A third HPgV-1 protein has antiviral activity in vitro. The HPgV-1 NS3 serine protease inhibits HIV replication in vitro, and this inhibition requires protease activity (72). Further, HPgV-1 NS3 protease inhibits the induction of host Type 1 interferon responses independent of its antiviral activity (181). Type 1 interferons are important contributors to a complex, cross-regulatory immune network, thus HPgV-1 interference with this network may enhance viral replication by reducing innate immune responses, or be antiviral by reducing host immune activation, a key element in HIV-1 pathogenesis (182).While HPgV-1 viremia is associated with the greatest reduction in mortality in studies of people living with HIV (PLWH), two studies identified an association between the presence of HPgV-1 E2 antibody and improved survival in those without viremia or E2 antibody (29, 91). Characterization of HPgV-1 E2 antigenicity is limited, although anti-peptide antibodies were detected directed against inconsistent regions of the E2 protein (183, 184). These studies do not contain controls with and without HPgV-1 viremia, or pre- or post-HPgV-1 infection, and it is not clear that these tests can differentiate active or cleared viremia, thus their utility in diagnostic assays is untested.
Limited numbers of well characterized E2 monoclonal antibodies (MAbs) have been generated (185), and one of these antibodies reacts with the same 17 amino acid peptide region responsible for HIV inhibition in cell culture systems (164, 186). Seven of the eight human MAbs generated by DNA immunization recognize conformational epitopes on E2, and based on competition studies, bind to clustered and overlapping epitopes identifying a putative single, immunodominant antigenic site on the virus particle (185, 186). The one antibody that bound a linear epitope reacted with two overlapping peptides. Surprisingly the antibody did not bind to the 9 amino acid overlap domain and additional amino acids from either the N-terminus or C-terminus of this overlap was required for antibody binding, suggesting a conformational component to the antibody binding site (186).
Since two clinical studies found an association between HPgV-1 E2 antibodies and improved survival, HPgV-1 E2 MAbs were incubated with HIV and surprisingly these neutralized HIV infection (165). Rabbit and murine sera post-E2 immunization also neutralized HIV infectivity and precipitated HIV particles from diverse HIV lineages while pre-immunization sera did not (165). Thus, HPgV-1 E2 antibodies raised against recombinant protein or peptides have neutralizing activity against HIV and appear to react with non-gp120 regions of the HIV envelope (165). HPgV-1 E2 protein was included in a multivalent HIV vaccine candidate that shows promise against infection; however, the relative importance of this heterologous antigen remains to be elucidated (187).
Clinical and Laboratory Evidence for HPgV-1 Immune Modulation
Several lines of clinical evidence emerged suggesting that HPgV-1 modulates host immune responses, and that these immunomodulatory consequences of infection may influence HIV pathogenesis. Early studies found reduced levels of CCR5 levels on CD4+ and CD8+ T cells in HPgV-HIV co-infected individuals compared to those without HPgV-1 (156, 157, 188, 189). This was initially considered an antiviral effect reducing HIV entry; however, CCR5 is upregulated by immune activation, a key requirement for HIV replication and pathogenesis (190–193). As noted above, in vitro studies resulted in reduced CCR5 and or CXCR4 levels and upregulation of inhibitory chemokine ligands (155, 158, 159). The effect of HPgV-1 on HIV reactivation was evaluated using PBMCs obtained from PLWH who were virally suppressed for more than 6 months. HPgV-1 reduced IL-2 and PHA stimulated latent HIV reactivation in vitro, suggesting that HPgV-1 may hinder activation-induced therapeutic “cure” strategies for HIV, especially since up to 41% of PLWH HPgV-1 coinfection (84, 114, 194).
Several clinical studies suggest that HPgV-1 reduces immune activation. For example, longitudinal measurement of circulating cytokine levels in HIV infected individuals found that HPgV-1 coinfection is associated with maintenance of a Th1 profile while those with mono-infection become polarized towards a Th2 profile (145). This is supported in vitro as NS5A expression in a CD4+ T cell line downregulated CD4 and key Th2 cytokines (e.g. IL13) (195). Some data suggest that HPgV-1 genotypic differences may alter cytokine levels (196).
Recombinant IL-2 was tested as an adjunctive therapy for HIV in the 1990s (197). Although IL-2 therapy resulted in highly significant increases in CD4 counts, clinical endpoints were not improved and therapy had many side effects and was never pursued for clinical use (197). To assess the potential relationship between HPgV-1 infection and IL-2-mediated CD4 expansion, HPgV-1 RNA was characterized in samples obtained from HIV-positive subjects prospectively enrolled in a randomized trial of IL-2. Surprisingly, CD4 counts did not increase in those with HPgV-1 co-infection whereas those without HPgV-1 had a rise of more than 1,000 CD4 cells/mm3, suggesting that HPgV-1 interferes with CD4 expansion and IL-2 signaling (198). Examining this in vitro, recombinant HPgV-1 E2 protein reduced IL-2 receptor mediated STAT5 activation following IL-2 receptor stimulation (199). Further, HPgV-1 E2 protein reduced T cell proliferation and cell surface markers of T cell activation following stimulation through the T cell receptor (TCR) (121, 199).
Maidana-Giret et al. found that HPgV-1 was associated with reduced CD4+ and CD8+ T cell activation markers (CD38, HLA DR) during acute HIV infection (188). This was confirmed in individuals with chronic HIV infection, with HPgV-1 co-infected people having significantly reduced activation marker expression regardless of viral suppression, and also with reduced frequency of circulating double negative T cells (189, 200). Immune activation markers were not only reduced in T cells, as HPgV-1 coinfection was associated with reduced activation markers on B cells, monocytes, and NK cells (201). Since HIV induces chronic immune activation, and increased activation markers are highly predictive of HIV disease progression, these studies suggest that HPgV-1 mediates its associated survival benefit by dampening immune activation (202).
A series of CD4+ T cell lines expressing E2 protein or E2 protein deletions identified a 13 amino acid peptide region that was sufficient to reduce TCR-mediated activation (121). HPgV-1 reduced activation of the proximal tyrosine Src kinase (LCK) in the TCR-induced signaling cascade, and the region involved was independent of the peptide region that inhibited HIV replication (121, 164) (Figure 4). The E2 amino acid sequence required for TCR signaling inhibition is predicted to be a substrate for LCK, and mutation of the tyrosine in this peptide restored TCR function. Further, HPgV-1 E2 was phosphorylated by LCK in vitro, suggesting that E2 competitively inhibits TCR-mediated T cell activation and IL-2 release (121, 199). In addition, IL-12 receptor-mediated NK cell activation is reduced by HPgV-1 and recombinant E2 (203), consistent with clinical observations that NK cell activation is reduced in HPgV-HIV coinfected individuals compared to those with HIV monoinfection (201).
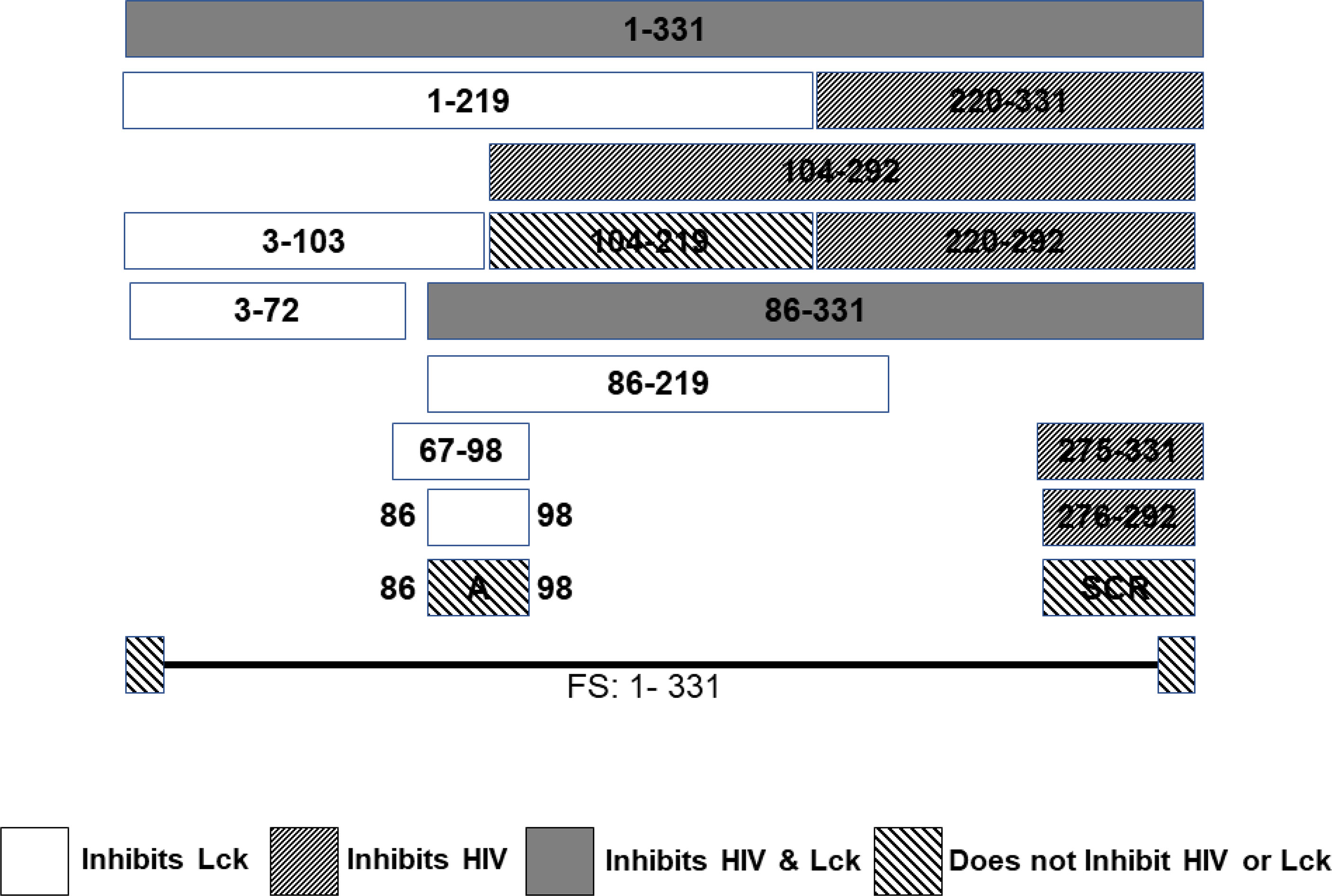
Figure 4 HPgV E2 protein has two discrete regions that interfere with either HIV replication or T cell activation adapted from References (121, 164). Schematic representation of the regions of E2 protein expressed by stably transfected CD4+ T cells that either inhibit HIV replication or blunts T cell receptor (TCR) signaling. HPgV E2 regions that both inhibited HIV and TCR signaling are shaded. Those that only interfere with TCR are white. E2 regions inhibiting HIV replication have narrow, forward slash shading, while regions that did not interfere with either HIV replication or TCR signaling are wide, backslash shaded. Full-length HPgV E2 protein with the C terminal transmembrane domain deleted (331 amino acids) is shown on top, and deletion proteins below. Scrambling the order of the 17 amino acids interfering with HIV replication abolished HIV inhibition. Mutation of a tyrosine in the TCR inhibitory peptide to an alanine abolished TCR inhibition. FS represents the full-length RNA for E2 in which a frame shift insertion was created to abolish E2 protein expression.
Of note, TCR signaling is not abolished by HPgV-1 or E2, and increasing the stimulus overcomes the inhibition in vitro. This is consistent with the finding that people who have HPgV_1 infection are not clinically immune compromised. Further, since HIV inhibition and TCR interference require different regions of the E2 protein, it suggests that both antiviral and reduced T cell activation influence HIV replication.
The innate antiviral immune response influences HIV disease progression, so potentially HPgV-1 might activate innate immunity in PLWH and thus slow disease progression. Lalle et al. found enhanced activation of the interferon system and circulating dendritic cells including promotion of interferon-gamma and activation and maturation of circulating dendritic cells in HPgV-1 co-infected individuals compared to HIV-mono infection (204). In contrast, Hoseini et al. did not find any differences in interferon producing cells among HPgV-1 – HIV coinfected individuals (205), and serum cytokines revealed a reduction in proinflammatory cytokines one year after HPgV-1 infection, although in the acute phase of HPgV-1 (first three months after infection), pro-apoptotic cytokines were upregulated (206). Chronic HPgV-1 coinfection in HIV was associated with significantly lower numbers of Fas-expressing CD4+ T cells when compared to HIV mono-infected (207). Since Fas expression correlates with Fas-mediated apoptosis, HPgV-1 may potentially slow CD4+ depletion by reducing apoptosis and T cell activation (115, 116, 156, 157, 188, 189, 199, 200, 203, 207).
Like HCV, HPgV-1 viremia can be cleared in individuals treated with alpha-interferon (208–212), though factors predicting interferon susceptibility have not been identified. Like HCV, sequence polymorphisms in the HPgV-1 NS5A protein correlate with interferon sensitivity, (213–215). Two HPgV-1 proteins have direct anti-interferon activities. NS5A blocks dimerization of PKR following interferon stimulation (215), and as noted above, the NS3 serine protease blocks the induction of Type 1 interferons and this requires active HPgV-1 protease activity (181).
Taken together, clinical studies illustrate that HPgV-1 infection is associated with reduced immune activation despite the presence of high viral loads, and laboratory studies identify several mechanisms by which HPgV-1 interferes with immune cell functions. Mechanisms supported by in vitro or in vivo evidence are summarized in Figure 5. Since HPgV-1 is not associated with immune suppression, the effects of HPgV-1 appear to reduce but not block immunologic hyper-activation. Since HIV disease progression is largely driven by chronic immune activation, it is likely that these immunologic effects of HPgV-1 contribute to the beneficial outcomes observed in numerous clinical studies. Since immune activation can be stimulated with sufficient stimulus, HPgV-1 appears to provide a beneficial dampening of immune activation without resultant immune deficiency.
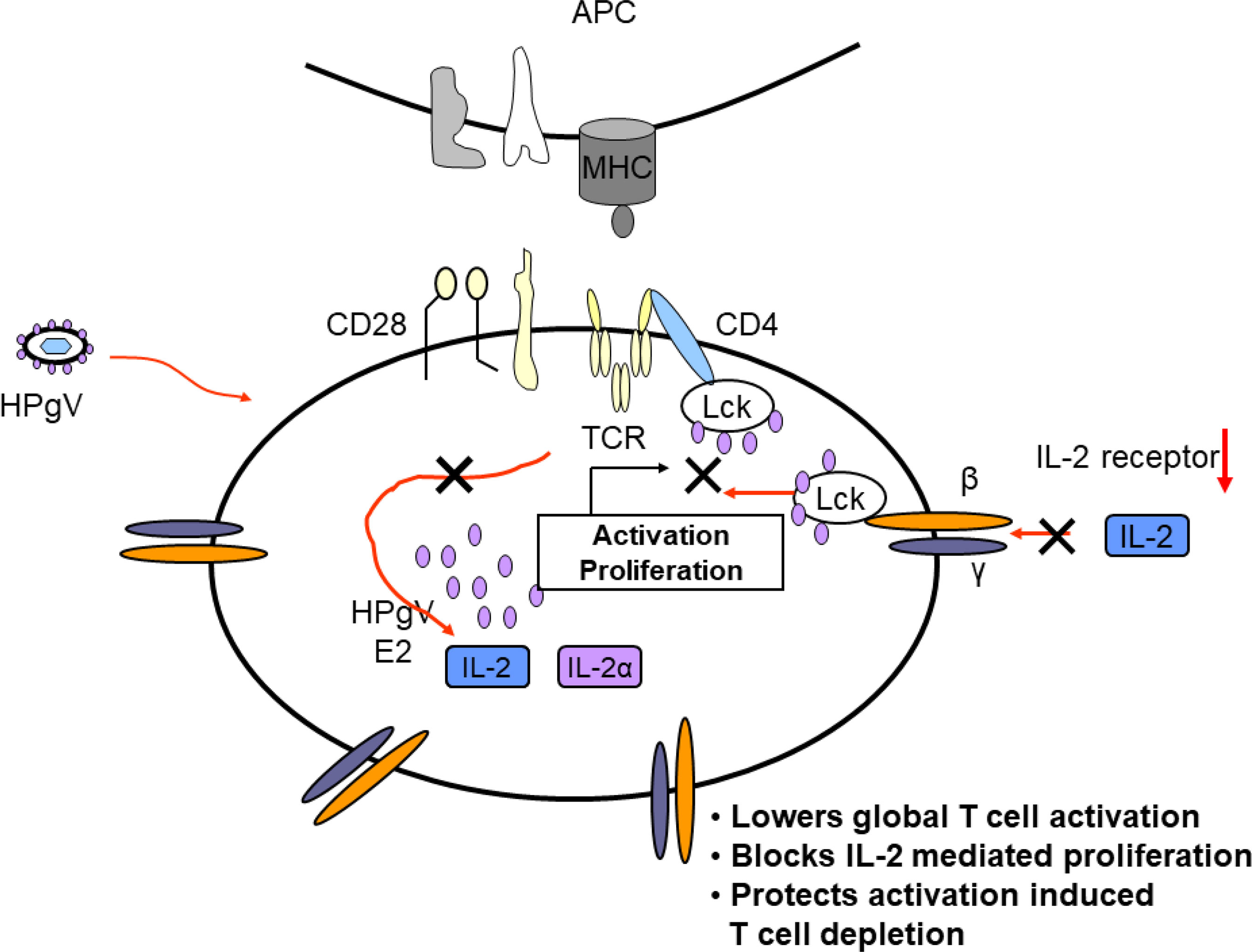
Figure 5 Proposed model of mechanisms by which HPgV interferes with T cell activation, proliferation, and depletion. Virus particles containing HPgV E2 protein interact with T cell and other immune cells and genomic RNA is translated in the cytoplasm, producing additional E2 protein. The effects of HPgV particles and/or intracellular E2 proteins interfere with T cell receptor-mediated activation of Lck, reducing cellular IL-2 release. HPgV also interferes with IL-2 receptor function further reducing T cell activation and proliferation. These effects can be overcome with sufficient TCR stimulus.
HPgV-1 Influences Other Immune-Mediated Disease States
Viral-mediated interference with T cell activation may be a common feature of viral immune modulation (Stapleton JT, 2014), and in addition to HIV-1, there are at least three additional diseases in which HPgV-1 infection is association with beneficial clinical outcomes. Although studies of HCV and HPgV-1 did not identify synergistic pathology, a beneficial role for HPgV-1 infection was found in individuals with triple infection with HIV, HCV and HPgV-1 compared to those with HIV and HCV coinfection (216). Specifically, HPgV-1 infection was associated with slower progression of HCV-related liver disease, and also with reduced LCK activation in intrahepatic T cells (216, 217). Since HCV pathogenesis is driven by immunologic destruction of infected hepatocytes, reducing T cell activation and proliferation likely contributes to the improved outcomes in HCV-induced liver disease (24, 218).
Similarly, Ebola virus infection is associated with cytokine storm and high mortality rates (219). Studies in West Africans with Ebola found that mortality in those with HPgV-1 coinfection was 47% compared to 78% in those without HPgV-1 coinfection, presumably due to the effects of HPgV-1 on immune activation (219). Although there have not been studies reported to date to address this, it would be of interest to see if primary COVID-19 outcomes are ameliorated by HPgV-1 infection since severe SARS-CoV-2 infection is associated with cytokine storm (220).
One additional immune-mediated clinical disease may be influenced by HPgV-1 infection. Vu et al. found that, following stem cell transplantation, both the rate of severe graft-versus-host-disease and overall mortality were reduced in individuals with HPgV-1 infection (221). This finding requires confirmation, but given the effects of HPgV-1 on immune activation, there is biologic plausibility supporting a beneficial interaction (222).
Conclusions and Key Questions Regarding HPgV
HPgV-1 is associated with an increased risk of lymphoma and possibly with sporadic encephalitis. However, given the high prevalence of infection and lack of commonly recognized disease states, in concert with the finding that persistent infection is beneficial in HIV coinfected individuals (and possibly other disease states), HPgV appears to represent a largely symbiotic infection within the human virome that has little or no pathogenicity. Several direct anti-retroviral actions of HPgV-1 have been identified, and these likely contribute to the improved survival observed in people living with HIV. Examination of the structural components of anti-HIV peptides may facilitate the development of cellular-based, small molecule HIV inhibitors, and identifying the cross-reactive HIV antigen(s) recognized by HPgV-1 E2 antibody may facilitate development of HIV vaccines (159, 164, 165, 168, 169, 171, 173).
In addition to developing HIV therapeutics, HPgV-1 is highly associated with immune modulation of human T cells, B cells, NK cells and monocytes, suggesting that this aspect of the virus provides beneficial, anti-inflammatory effects without inducing clinically evident immune compromise (188, 189, 201). In persistently viremic individuals, high rates of viral replication are observed concurrently with reduced markers of immune activation and in most, no detectable anti-HPgV-1 antibodies detected (7).
As a relatively non-pathogenic, lymphotropic virus that infects and preferentially replicates in bone marrow and spleen, HPgV-1 potentially could be used as either a novel gene therapy delivery system or as a bio-vaccine for HIV (153, 154). Further study of the role of HPgV infection on disease outcomes in immune-mediated diseases could provide insights into the natural history of many immune-mediated diseases, including most recently COVID-19. The effect, if any of HPgV-1 on SARS-CoV-2 infection has not been examined at the time of writing.
Currently, the biggest problem hindering HPgV-1 development for therapeutic use relates to the difficulties in growing the virus in vitro. While virus replicates in lymphocytes, NK cells, and neural cells in vitro (110–113, 115, 116, 120, 137, 203), infection cannot be maintained in cell culture for more than handful of passages, suggesting either abortive infection or slow infection with dilutional loss of infected cells. Specifically, most of the cells in PBMC cultures obtained from HPgV-1 infected individuals die rapidly, leaving a small subset of primary mononuclear cells that survive for weeks to months producing low levels of virus (115). In CD4+ Jurkat T cell lines, intracellular viral RNA and antigens are detected following infection (113, 119); however, the amount virus released slowly decays, and after several passes of supernatant fluids to new, uninfected cells, the infection is lost. Since HPgV-1 slows cellular proliferation induced by IL-2 or TCR-stimulation (121, 199), this may reflect a virus-driven interference with Src kinase-mediated cellular proliferation, resulting in gradual loss of infected cells that produce such low levels of virus that infection of new cells is gradually lost.
Many approaches to identify specific cell types that support HPgV-1 replication have been attempted including use of CD34+ human stem cells, interferon-resistant cells, and other innate knockout cell lines without success (116). The recent finding that goose pegivirus replicates well in embryonated goose eggs and primary goose embryo fibroblasts raises hope that novel culture systems can be developed for HPgV-1 (55).
Another important limitation in HPgV-1 research is the validated association of HPgV-1 infection with the development of lymphoma and the recent suggestion of an association with encephalitis. Given the effects of HPgV-1 on immune activation, the most logical explanation for increased risk of lymphoma relates to a reduction in immune surveillance, though a direct causal effect on lymphoma needs to be excluded (133–135). The two to three-fold increase in risk of lymphoma in HPgV-1 viremic individuals is significant, but does not explain the vast majority of lymphoma cases. Further, the age of HPgV-1 infection (sexually active adolescents to late-mid life) is the opposite that of maximal lymphoma risk (223).
In summary, HPgV-1 is a highly prevalent, low- or non-virulent human infection that reaches high viral loads in plasma and is associate with reductions in human immune activation markers. Pegiviruses are increasingly being identified in diverse hosts, and their apparent longevity in humans and non-human primates suggests that they convey an overarching beneficial effect on health. Future studies to determine the breadth and magnitude of these effects are warranted.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This study was supported in part by a Veterans Administration Merit Review grant (BX000207).
Conflict of Interest
The author JS has patents related to the use of GB virus C in the potential treatment of HIV-1 infection.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The author thanks Drs. Jinhua Xiang, James H. McLinden, Nirjal Bhattari, Ernest Chivero, Emma Mohr, Sabina Wuenschmann, Robert (Bo) Rydze, and Ms. Qing Chang for their creative and enthusiastic laboratory work, Dr. Gibran Horemheb Rubio Quintinares for helpful discussions, and Dr. Jeff Meier and the entire University of Iowa HIV Program for their support, cooperation, assistance and encouragement related to studies of HPgV-1 infection in people living with HIV/AIDS.
References
1. Simons JN, Desai SM, Mushahwar IK. The GB Viruses. Curr Top Microbiol Immunol (2000) 242:341–75. doi: 10.1007/978-3-642-59605-6_16
2. Simons JN, Leary TP, Dawson GJ, Pilot-Matias TJ, Muerhoff AS, Schlauder GG, et al. Isolation of Novel Virus-Like Sequences Associated With Human Hepatitis. Nat Med (1995) 1:564–9. doi: 10.1038/nm0695-564
3. Simons JN, Pilot-Matias TJ, Leary TP, Dawson GJ, Desai GJ, Desai SM, et al. Identification of Two Flavivirus-Like Genomes in the GB Hepatitis Agent. Proc Natl Acad Sci USA (1995) 92:3401–5. doi: 10.1073/pnas.92.8.3401
4. Linnen J, Wages J, Zhang-Keck ZY, Fry KE, Krawczynski KZ, Alter H, et al. Molecular Cloning and Disease Association of Hepatitis G Virus: A Transfusion-Transmissible Agent. Science (1996) 271:505–8. doi: 10.1126/science.271.5248.505
5. Leary TP, Muerhoff AS, Simons JN, Pilot-Matias TJ, Erker JC, Chalmers ML, et al. Sequence and Genomic Organization of GBV-C: A Novel Member of the Flaviviridae Associated With Human non-A-E Hepatitis. J.Med.Virol (1996) 48:60–7. doi: 10.1002/(SICI)1096-9071(199601)48:1<60::AID-JMV10>3.0.CO;2-A
6. Okamoto H, Nakao H, Inoue T, Fukuda M, Kishimoto J, Iizuka H, et al. The Entire Nucleotide Sequence of Two GB Virus C/hepatitis G Virus Isolates of Distinct Genotypes From Japan. J.Gen.Virol (1997) 78:737–45. doi: 10.1099/0022-1317-78-4-737
7. Stapleton JT, Foung S, Muerhoff AS, Bukh J, Simmonds P. The GB Viruses: A Review and Proposed Classification of GBV-A, GBV-C(HGV), and GBV-D in Genus Pegivirus Within the Family Flaviviridae. J.Gen.Virol (2011) 92:233–46. doi: 10.1099/vir.0.027490-0
8. Deinhardt F, Holmes AW, Capps RB, Popper H. Studies on the Transmission of Human Viral Hepatitis to Marmoset Monkeys. I. Transmission of Disease, Serial Passages, and Description of Liver Lesions. J.Exp.Med (1967) 125:673–88. doi: 10.1084/jem.125.4.673
9. Zuckerman AJ. The History of Viral Hepatitis From Antiquity to the Present. In: Deinhardt F, Deinhardt J, editors. Viral Hepatitis: Laboratory and Clinical Science. New York: Marcel Dekker (1983). p. 3–32.
10. Deinhardt F, Deinhardt JB. Animal Models. In: Gerety RJ, editor. Hepatitis a. London: Academic Press (1984). p. 185–204.
11. Dawson GJ, Schlauder GG, Pilot-Matias TJ, Thiele D, Leary TP, Murphy P, et al. Prevalence Studies of GB Virus-C Using Reverse-Transcriptase-Polymerase Chain Reaction. J.Med.Virol (1996) 50:97–103. doi: 10.1002/(SICI)1096-9071(199609)50:1<97::AID-JMV16>3.0.CO;2-V
12. Pilot-Matias TJ, Carrick RJ, Coleman PF, Leary TP, Surowy TK, Simons JN, et al. Expression of the GB Virus C E2 Glycoprotein Using the Semliki Forest Virus Vector System and its Utility as a Serologic Marker. Virology (1996) 225:282–92. doi: 10.1006/viro.1996.0602
13. Alter HJ, Conry-Cantilena C, Melpolder J, Tan D, Vanraden M, Herion D, et al. Hepatitis C in Asymptomatic Blood Donors. Hepatology (1997) 26:29S–33S. doi: 10.1002/hep.510260705
14. Alter HJ, Nakatsuji Y, Melpolder J, Wages J, Wesley R, Shih JWK, et al. The Incidence of Transfusion-Associated Hepatitis G Virus Infection and its Relation to Liver Disease. N Engl J Med (1997) 336:747–54. doi: 10.1056/NEJM199703133361102
15. Alter MJ, Gallagher M, Morris TT, Moyer LA, Meeks EL, Krawczynski K, et al. Acute non-A-E Hepatitis in the United States and the Role of Hepatitis G Virus Infection. N Engl J Med (1997) 336:741–6. doi: 10.1056/NEJM199703133361101
16. Bhardwaij B, Qian K, Detmer J, Mizokami M, Kolberg JA, Urdea MS, et al. Detection of GB Virus-C/hepatitis G Virus RNA in Serum by Reverse Transcription Polymerase Chain Reaction. J Med Virol (1997) 52:92–6. doi: 10.1002/(SICI)1096-9071(199705)52:1<92::AID-JMV15>3.0.CO;2-2
17. Gutierrez RA, Dawson GJ, Mushahwar IK. ELISA for Detection of Antibody to the E2 Protein of GB Virus C. J Virol Methods (1997) 69:1–6. doi: 10.1016/S0166-0934(97)00140-7
18. Tacke M, Schmolke S, Schlueter V, Sauleda S, Esteban JI, Tanaka E, et al. Humoral Immune Response to the E2 Protein of Hepatitis G Virus is Associated With Long-Term Recovery From Infection and Reveals a High Frequency of Hepatitis G Virus Exposure Among Healthy Blood Donors. Hepatol (1997) 26:1626–33. doi: 10.1002/hep.510260635
19. Thomas DL, Nakatsuji Y, Shih JW, Alter HJ, Nelson KE, Astemborski JA, et al. Persistence and Clinical Significance of Hepatitis G Virus Infections in Injecting Drug Users. J.Infect.Dis (1997) 176:586–92. doi: 10.1086/514078
20. Bjorkman P, Sundstrom G, Widell A. Hepatitis C Virus and GB Virus C/hepatitis G Virus Viremia in Swedish Blood Donors With Different Alanine Aminotransferase Levels. Transfusion (1998) 38:378–84. doi: 10.1046/j.1537-2995.1998.38498257377.x
21. Heuft HG, Berg T, Schreier E, Kunkel U, Tacke M, Schwella N, et al. Epidemiological and Clinical Aspects of Hepatitis G Virus Infection in Blood Donors and Immunocompromised Recipients of HGV-Contaminated Blood. Vox Sanguinis (1998) 74:161–7. doi: 10.1046/j.1423-0410.1998.7430161.x
22. Tanaka E, Tacke M, Kobayashi M, Nakatsuji Y, Kiyosawa K, Schmolke S, et al. Past and Present Hepatitis G Virus Infections in Areas Where Hepatitis C is Highly Endemic and Those Where it is Not Endemic. J.Clin.Microbiol (1998) 36:110–4. doi: 10.1128/JCM.36.1.110-114.1998
23. Kupfer B, Ruf T, Matz B, Nattermann J, Spengler U, Rockstroh JK, et al. Comparison of GB Virus C, HIV, and HCV Infection Markers in Hemophiliacs Exposed to non-Inactivated or Inactivated Factor Concentrates. J Clin Virol (2005) 34:42–7. doi: 10.1016/j.jcv.2005.01.008
24. Bhattarai N, Stapleton JT. GB Virus C: The Good Boy Virus? Trends Microbiol (2012) 20:124–30. doi: 10.1016/j.tim.2012.01.004
25. Alter HJ. Clinical, Virological and Epidemiological Basis for the Treatment of Chronic non-A, non-B Hepatitis. J.Hepatol (1990) 11 Suppl. 1:S19–25. doi: 10.1016/0168-8278(90)90158-n
26. Alter HJ. G-Pers Creepers, Where'd You Get Those Papers? A Reassessment of the Literature on the Hepatitis G Virus. Transfusion (1997) 37:569–72. doi: 10.1046/j.1537-2995.1997.37697335149.x
27. Theodore D, Lemon SM. GB Virus C, Hepatitis G Virus, or Human Orphan Flavivirus? Hepatol (1997) 25:1285–6. doi: 10.1002/hep.510250541
28. Stapleton JT. GB Virus Type C/Hepatitis G Virus. Semin Liver Dis (2003) 23:137–48. doi: 10.1055/s-2003-39943
29. Williams CF, Klinzman D, Yamashita TE, Xiang J, Polgreen PM, Rinaldo C, et al. Persistent GB Virus C Infection and Survival in HIV-Infected Men. N Engl J Med (2004) 350:981–90. doi: 10.1056/NEJMoa030107
30. Adams MJ, King AM, Carstens EB. Ratification Vote on Taxonomic Proposals to the International Committee on Taxonomy of Viruses, (2013). Arch Virol (2013) 158:2023–30. doi: 10.1007/s00705-013-1688-5
31. Hartlage AS, Cullen JM, Kapoor A. The Strange, Expanding World of Animal Hepaciviruses. Annu Rev Virol (2016) 3:53–75. doi: 10.1146/annurev-virology-100114-055104
32. Smith DB, Becher P, Bukh J, Gould EA, Meyers G, Monath T, et al. Proposed Update to the Taxonomy of the Genera Hepacivirus and Pegivirus Within the Flaviviridae Family. J Gen Virol (2016) 97:2894–907. doi: 10.1099/jgv.0.000612
33. Simmonds P, Becher P, Bukh J, Gould EA, Meyers G, Monath T, et al. ICTV Virus Taxonomy Profile: Flaviviridae. J Gen Virol (2017) 98:2–3. doi: 10.1099/jgv.0.000672
34. Heffron AS, Lauck M, Somsen ED, Townsend EC, Bailey AL, Sosa M, et al. Discovery of a Novel Simian Pegivirus in Common Marmosets (Callithrix Jacchus) With Lymphocytic Enterocolitis. Microorganisms (2020) 8:1509. doi: 10.3390/microorganisms8101509
35. Muerhoff AS, Leary TP, Simons JN, Pilot-Matias TJ, Dawson GJ, Erker JC, et al. Genomic Organization of GB Viruses A and B: Two New Members of the Flaviviridae Associated With GB Agent Hepatitis. J.Virol (1995) 69:5621–30. doi: 10.1128/jvi.69.9.5621-5630.1995
36. Bukh J, Apgar CL. Five New or Recently Discovered (GBV-A) Virus Species are Indigenous to New World Monkeys and may Constitute a Seperate Genus of the Flaviviridae. Virology (1997) 229:429–36. doi: 10.1006/viro.1997.8461
37. Leary TP, Desai SM, Yamaguchi J, Chalmers MJ, Schlauder GG, Dawson GJ, et al. Species-Specific Variants of GB Virus A in Captive Monkeys. J.Virol (1997) 70:9028–30. doi: 10.1128/jvi.70.12.9028-9030.1996
38. Quan PL, Firth C, Conte JM, Williams SH, Zambrana-Torrelio CM, Anthony SJ, et al. Bats are a Major Natural Reservoir for Hepaciviruses and Pegiviruses. Proc Natl Acad Sci U.S.A. (2013) 110:8194–9. doi: 10.1073/pnas.1303037110
39. Adams NJ, Prescott LE, Jarvis LM, Lewis JCM, Mcclure MO, Smith DB, et al. Detection in Chimpanzees of a Novel Flavivirus Related to GB Virus-C/hepatitis G Virus. J.Gen.Virol (1998) 79:1871–7. doi: 10.1099/0022-1317-79-8-1871
40. Birkenmeyer LG, Desai SM, Muerhoff AS, Leary TP, Simons JN, Montes CC, et al. Isolation of a GB Virus-Related Genome From a Chimpanzee. J.Med.Virol (1998) 56:44–51. doi: 10.1002/(SICI)1096-9071(199809)56:1<44::AID-JMV8>3.0.CO;2-N
41. Sibley SD, Lauck M, Bailey AL, Hyeroba D, Tumukunde A, Weny G, et al. Discovery and Characterization of Distinct Simian Pegiviruses in Three Wild African Old World Monkey Species. PloS One (2014) 9:e98569. doi: 10.1371/journal.pone.0098569
42. Bailey AL, Lauck M, Mohns M, Peterson EJ, Beheler K, Brunner KG, et al. Durable Sequence Stability and Bone Marrow Tropism in a Macaque Model of Human Pegivirus Infection. Sci Transl Med (2015) 7:305ra144. doi: 10.1126/scitranslmed.aab3467
43. Epstein JH, Quan PL, Briese T, Street C, Jabado O, Conlan S, et al. Identification of GBV-D, a Novel GB-Like Flavivirus From Old World Frugivorous Bats (Pteropus Giganteus) in Bangladesh. PloS Pathog (2010) 5:e1000972–1000978. doi: 10.1371/journal.ppat.1000972
44. Chandriani S, Skewes-Cox P, Zhong W, Ganem DE, Divers TJ, Van Blaricum AJ, et al. Identification of a Previously Undescribed Divergent Virus From the Flaviviridae Family in an Outbreak of Equine Serum Hepatitis. Proc Natl Acad Sci U.S.A. (2013) 110:E1407–1415. doi: 10.1073/pnas.1219217110
45. Kapoor A, Simmonds P, Cullen JM, Scheel TK, Medina JL, Giannitti F, et al. Identification of a Pegivirus (GB Virus-Like Virus) That Infects Horses. J Virol (2013) 87:7185–90. doi: 10.1128/JVI.00324-13
46. Kapoor A, Simmonds P, Scheel TK, Hjelle B, Cullen JM, Burbelo PD, et al. Identification of Rodent Homologs of Hepatitis C Virus and Pegiviruses. mBio (2013) 4:e00216–00213. doi: 10.1128/mBio.00216-13
47. Firth C, Bhat M, Firth MA, Williams SH, Frye MJ, Simmonds P, et al. Detection of Zoonotic Pathogens and Characterization of Novel Viruses Carried by Commensal Rattus Norvegicus in New York City. mBio (2014) 5:e01933–01914. doi: 10.1128/mBio.01933-14
48. Canuti M, Williams CV, Sagan SM, Oude Munnink BB, Gadi S, Verhoeven JTP, et al. Virus Discovery Reveals Frequent Infection by Diverse Novel Members of the Flaviviridae in Wild Lemurs. Arch Virol (2019) 164:509–22. doi: 10.1007/s00705-018-4099-9
49. Rodrigues TCS, Subramaniam K, Mcculloch SD, Goldstein JD, Schaefer AM, Fair PA, et al. Genomic Characterization of a Novel Pegivirus Species From Free-Ranging Bottlenose Dolphins (Tursiops Truncatus) in the Indian River Lagoon, Florida. Virus Res (2019) 263:98–101. doi: 10.1016/j.virusres.2019.01.002
50. Nishizawa T, Usui R, Narabu Y, Takahashi M, Murata K, Okamoto H. Identification of a Novel Pegivirus in Pet Cats (Felis Silvestris Catus) in Japan. Virus Res (2021) 301:198452. doi: 10.1016/j.virusres.2021.198452
51. Bukh J, Kim JP, Govindarajan S, Apgar CL, Foung SK, Wages J Jr., et al. Experimental Infection of Chimpanzees With Hepatitis G Virus and Genetic Analysis of the Virus. J.Inf.Dis (1998) 177:855–62. doi: 10.1086/515255
52. Bailey AL, Buechler CR, Matson DR, Peterson EJ, Brunner KG, Mohns MS, et al. Pegivirus Avoids Immune Recognition But Does Not Attenuate Acute-Phase Disease in a Macaque Model of HIV Infection. PloS Pathog (2017) 13:e1006692. doi: 10.1371/journal.ppat.1006692
53. Shi M, Lin XD, Vasilakis N, Tian JH, Li CX, Chen LJ, et al. Divergent Viruses Discovered in Arthropods and Vertebrates Revise the Evolutionary History of the Flaviviridae and Related Viruses. J Virol (2016) 90:659–69. doi: 10.1128/JVI.02036-15
54. Porter AF, Pettersson JH, Chang WS, Harvey E, Rose K, Shi M, et al. Novel Hepaci- and Pegi-Like Viruses in Native Australian Wildlife and non-Human Primates. Virus Evol (2020) 6:veaa064. doi: 10.1093/ve/veaa064
55. Wu Z, Wu Y, Zhang W, Merits A, Simmonds P, Wang M, et al. The First Nonmammalian Pegivirus Demonstrates Efficient In Vitro Replication and High Lymphotropism. J Virol (2020) 94:e01150–20. doi: 10.1128/JVI.01150-20
56. Zhen W, Wu Y, Zhang W, Wang M, Jia R, Zhu D, et al. Emergence of a Novel Pegivirus Species in Southwest China Showing a High Rate of Coinfection With Parvovirus and Circovirus in Geese. Poult Sci (2021) 100:101251. doi: 10.1016/j.psj.2021.101251
57. Berg MG, Lee D, Coller K, Frankel M, Aronsohn A, Cheng K, et al. Discovery of a Novel Human Pegivirus in Blood Associated With Hepatitis C Virus Co-Infection. PloS Pathog (2015) 11:e1005325. doi: 10.1371/journal.ppat.1005325
58. Kapoor A, Kumar A, Simmonds P, Bhuva N, Singh Chauhan L, Lee B, et al. Virome Analysis of Transfusion Recipients Reveals a Novel Human Virus That Shares Genomic Features With Hepaciviruses and Pegiviruses. mBio (2015) 6:e01466–01415. doi: 10.1128/mBio.01466-15
59. Wan Z, Liu J, Hu F, Shui J, Li L, Wang H, et al. Evidence That the Second Human Pegivirus (HPgV-2) is Primarily a Lymphotropic Virus and can Replicate Independent of HCV Replication. Emerg Microbes Infect (2020) 9:485–95. doi: 10.1080/22221751.2020.1730247
60. Muerhoff AS, Smith DB, Leary TP, Erker JC, Desai SM, Mushawar IK. Indentification of GB Virus C Variants by Phylogentic Analysis of 5'-Untranslated and Coding Region Sequences. J Virol (1998) 71:6501–8. doi: 10.1128/jvi.71.9.6501-6508.1997
61. Smith DB, Simmonds P, Frost S, Haydon D, Cuceanu N, Prescott L, et al. Phylogenetic Analysis of GBV-C/hepatitis G Virus. J.Gen.Virol (2000) 81:769–80. doi: 10.1099/0022-1317-81-3-769
62. Muerhoff AS, Dawson GJ, Desai SM. A Previously Unrecognized Sixth Genotype of GB Virus C Revealed by Analysis of 5'-Untranslated Region Sequences. J.Med.Virol (2006) 78:105–11. doi: 10.1002/jmv.20510
63. Feng Y, Zhao W, Feng Y, Dai J, Li Z, Zhang X, et al. A Novel Genotype of GB Virus C: Its Identification and Predominance Among Injecting Drug Users in Yunnan, China. PloS One (2011) 6:e21151. doi: 10.1371/journal.pone.0021151
64. Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, et al. Expanded Classification of Hepatitis C Virus Into 7 Genotypes and 66 Subtypes, Updated Criteria and Assignment Web Resource. Hepatology (2013) 59:318–27. doi: 10.1002/hep.26744
65. Gonzalez-Perez MA, Norder H, Bergstrom A, Lopez E, Visona KA, Manius LL. High Prevalence of GB Virus C Strains Genetically Related to Strains With Asian Origin in Nicaraguan Hemophiliacs. J.Med.Virol (1997) 52:149–55. doi: 10.1002/(SICI)1096-9071(199706)52:2<149::AID-JMV5>3.0.CO;2-3
66. Pujol FH, Khudyakov YE, Devesa M, Cong ME, Loureiro CL, Blitz L, et al. Hepatitis G Virus Infection in Amerindians and Other Venezuelan High-Risk Groups. J Clin Microbiol (1998) 36:470–4. doi: 10.1128/JCM.36.2.470-474.1998
67. Horemheb-Rubio G, Ramos-Cervantes P, Arroyo-Figueroa H, Avila-Rios S, Garcia-Morales C, Reyes-Teran G, et al. High HPgV Replication is Associated With Improved Surrogate Markers of HIV Progression. PloS One (2017) 12:e0184494. doi: 10.1371/journal.pone.0184494
68. Blackard JT, Ma G, Polen C, Dubois JC, Gast J, Radens CM, et al. Recombination Among GB Virus C (GBV-C) Isolates in the United States. J Gen Virol (2016) 97:1537–44. doi: 10.1099/jgv.0.000477
69. Simons JN, Desai SM, Schultz DE, Lemon SM, Mushahwar IK. Translation Initiation in GB Viruses A and C: Evidence for Internal Ribosome Entry and Implications for Genomic Organization. J.Virol (1996) 70:6126–35. doi: 10.1128/jvi.70.9.6126-6135.1996
70. Kim JP, Fry KE. Molecular Characterization of the Hepatitis G Virus. J Viral Hepat (1997) 4:77–9. doi: 10.1111/j.1365-2893.1997.tb00208.x
71. Belyaev AS, Chong S, Novikov A, Kongpachith A, Masiarz FR, Lim M, et al. Hepatitis G Virus Encodes Protease Activities Which can Effect Processing of the Virus Putative Nonstructural Proteins. J.Virol (1998) 72:868–72. doi: 10.1128/JVI.72.1.868-872.1998
72. George SL, Varmaz D, Tavis JE, Chowdhury A. The GB Virus C (GBV-C) NS3 Serine Protease Inhibits HIV-1 Replication in a CD4+ T Lymphocyte Cell Line Without Decreasing HIV Receptor Expression. PloS One (2012) 7:e30653. doi: 10.1371/journal.pone.0030653
73. Lin HH, Kao JH, Yeh KY, Liu DP, Chang MH, Chen PJ, et al. Mother-To-Infant Transmission of GB Virus C/hepatitis G Virus: The Role of High-Titered Maternal Viremia and Mode of Delivery. J Infect Dis (1998) 177:1202–6. doi: 10.1086/515264
74. Scallan MF, Clutterbuck D, Jarvis LM, Scott GR, Simmonds P. Sexual Transmission of GB Virus C/Hepatitis G Virus. J.Med.Virol (1998) 55:203–8. doi: 10.1002/(SICI)1096-9071(199807)55:3<203::AID-JMV4>3.0.CO;2-5
75. Zanetti AR, Tanzi E, Romano L, Principi N, Zuin G, Minola E, et al. Multicenter Trial on Mother-To-Infant Transmission of GBV-C Virus. J Med Virol (1998) 54:107–12. doi: 10.1002/(SICI)1096-9071(199802)54:2<107::AID-JMV7>3.0.CO;2-A
76. Lefrere JJ, Ferec C, Roudot-Thoraval F, Loiseau P, Cantaloube JF, Biagini P, et al. GBV-C/hepatitis G Virus (HGV) RNA Load in Immunodeficient Individuals and in Immunocompetent Individuals. J.Med.Virol (1999) 59:32–7. doi: 10.1002/(SICI)1096-9071(199909)59:1<32::AID-JMV6>3.0.CO;2-O
77. Frey SE, Homan SM, Solok-Anderson M, Cayco MT, Cortorreal P, Musial CE, et al. Evidence for Probable Sexual Transmission of the Hepatitis G Virus. Clin Infect Dis (2002) 34:1033–8. doi: 10.1086/339206
78. Supapol WB, Remis RS, Raboud J, Millson M, Tappero J, Kaul R, et al. Reduced Mother-To-Child Transmission of HIV Associated With Infant But Not Maternal GB Virus C Infection. J.Infect.Dis (2008) 197:1369–77. doi: 10.1086/587488
79. Santos LM, Lobato RC, Barral MFM, Goncalves CV, Da Hora VP, Martinez AMB. Prevalence and Vertical Transmission of Human Pegivirus Among Pregnant Women Infected With HIV. Int J Gynaecol Obstet (2017) 138:113–8. doi: 10.1002/ijgo.12175
80. Tanaka E, Kiyosawa K, Shimoda K, Hino K, Tacke M, Schmolke S, et al. Evolution of Hepatitis G Virus Infection and Antibody Response to Envelope Protein in Patients With Transfusion-Associated non-A, non-B Hepatitis. J Viral Hepat (1998) 5:153–9. doi: 10.1046/j.1365-2893.1998.00095.x
81. Gutierrez RA, Dawson GJ, Knigge MF, Melvin SL, Heynen CA, Kyrk CR, et al. Seroprevalence of GB Virus C and Persistence of RNA and Antibody. J Med Virol (1997) 53:167–73. doi: 10.1002/(SICI)1096-9071(199710)53:2<167::AID-JMV10>3.0.CO;2-G
82. Hassoba HM, Pessoa MG, Terrault NA, Lewis NJ, Hayden M, Hunt JC, et al. Antienvelope Antibodies are Protective Against GBV-C Reinfection: Evidence From the Liver Transplant Model. J.Med.Virol (1998) 56:253–8. doi: 10.1002/(SICI)1096-9071(199811)56:3<253::AID-JMV13>3.0.CO;2-F
83. Tillmann HL, Heringlake S, Trauwein C, Meissner D, Nashan B, Schlitt HJ, et al. Antibodies Against the GB Virus C Envelope 2 Protein Before Liver Transplantation Protect Against GB Virus C De Novo Infection. Hepatology (1998) 28:379–84. doi: 10.1002/hep.510280213
84. Mohr EL, Stapleton JT. GB Virus Type C Interactions With HIV: The Role of Envelope Glycoproteins. J Viral Hepat (2009) 16:757–8. doi: 10.1111/j.1365-2893.2009.01194.x
85. Dille BJ, Surowy TK, Gutierrez RA, Coleman PF, Knigge MF, Carrick RJ, et al. An ELISA for Detection of Antibodies to the E2 Protein of GB Virus. J Infect Dis (1997) 175:458–61. doi: 10.1093/infdis/175.2.458
86. Marano G, Franchini M, Farina B, Piccinini V, Pupella S, Vaglio S, et al. The Human Pegivirus: A New Name for an "Ancient" Virus. Can Transfusion Medicine Come Up With Something New? Acta Virol (2017) 61:401–12. doi: 10.4149/av_2017_402
87. Da Mota LD, Finger-Jardim F, Silva CM, Germano FN, Nader MM, Goncalves CV, et al. High Prevalence and Autochtonous Transmission of Human Pegivirus (HPgV-1) in Blood Donors in the Extreme Southern of Brazil. J Med Virol (2019) 91:31–7. doi: 10.1002/jmv.25291
88. Yang N, Dai R, Zhang X. Global Prevalence of Human Pegivirus-1 in Healthy Volunteer Blood Donors: A Systematic Review and Meta-Analysis. Vox Sang (2020) 115:107–19. doi: 10.1111/vox.12876
89. Yu Y, Wan Z, Wang JH, Yang X, Zhang C. Review of Human Pegivirus: Prevalence, Transmission, Pathogenesis, and Clinical Implication. Virulence (2022) 13:324–41. doi: 10.1080/21505594.2022.2029328
90. Defereux H, Sabin CA, Kinson Z, Brown D, Griffioen A, Dusheiko GM, et al. Influence of HIV-1 Infection on GBV-C Infection in Multiply Infected Haemophilic Patients. J.Med.Virol (1998) 56:316–20. doi: 10.1002/(SICI)1096-9071(199812)56:4<316::AID-JMV5>3.0.CO;2-K
91. Tillmann HL, Heiken H, Knapik-Botor A, Heringlake S, Ockenga J, Wilber JC, et al. Infection With GB Virus C and Reduced Mortality Among HIV-Infected Patients. N Engl J Med (2001) 345:715–24. doi: 10.1056/NEJMoa010398
92. Jones JM, Sapiano MRP, Savinkina AA, Haass KA, Baker ML, Henry RA, et al. Slowing Decline in Blood Collection and Transfusion in the United States - 2017. Transfusion (2020) 60 Suppl 2:S1–9. doi: 10.1111/trf.15604
93. Heringlake S, Ockenga J, Tillmann HL, Trautwein C, Meissner D, Stoll M, et al. GB Virus C/Hepatitis G Virus Infection: A Favorable Prognostic Factor in Human Immunodeficiency Virus-Infected Patients? J.Infect.Dis (1998) 177:1723–6. doi: 10.1086/517431
94. Xiang J, Wunschmann S, Diekema DJ, Klinzman D, Patrick KD, George SL, et al. Effect of Coinfection With GB Virus C (Hepatitis G Virus) on Survival Among Patients With HIV Infection. N Engl J Med (2001) 345:707–14. doi: 10.1056/NEJMoa003364
95. Polgreen PM, Xiang J, Chang Q, Stapleton JT. GB Virus Type C/Hepatitis G Virus: A Nonpathogenic Flavivirus Associated With Prolonged Survival in HIV-Infected Individuals. Microbes Infect (2003) 5:1255–61. doi: 10.1016/j.micinf.2003.08.006
96. Zhang W, Chaloner K, Tillmann HL, Williams CF, Stapleton JT. Effect of Early and Late GBV-C Viremia on Survival of HIV Infected Individuals: A Meta-Analysis. HIV Med (2006) 7:173–80. doi: 10.1111/j.1468-1293.2006.00366.x
97. Zimmerman J, Blackard JT. Human Pegivirus Type 1 Infection in Asia-A Review of the Literature. Rev Med Virol (2022) 32:e2257. doi: 10.1002/rmv.2257
98. Rambusch EG, Wedemeyer H, Tillmann HL, Heringlake S, Manns MP. Significance of Coinfection With Hepatitis G Virus for Chronic Hepatitis C - a Review of the Literature. Z.Gastroenterol (1998) 36:41–53.
99. Yoshiba M, Okamoto H, Mishiro S. Detection of the GBV-C Hepatitis Virus Genome in Serum From Patients With Fulminant Hepatitis of Unknown Eaetiology. Lancet (1995) 346:1131–2. doi: 10.1016/S0140-6736(95)91802-7
100. Heringlake S, Osterkamp S, Trautwein C, Tillmann HL, Boker K, Muerhoff S, et al. Association Between Fulminant Hepatic Failure and a Strain of GBV Virus C. Lancet (1996) 348:1626–9. doi: 10.1016/S0140-6736(96)04413-3
101. Brown KE, Tisdale J, Barrett AJ, Dunbar CE, Young NS. Hepatitis-Associated Aplastic Anemia. N Engl J Med (1997) 336:1059–64. doi: 10.1056/NEJM199704103361504
102. Fried MW, Khudyakov YE, Smallwood GA, Cong ME, Nichols B, Diaz E, et al. Hepatitis G Virus Co-Infection in Liver Transplant Recipients With Chronic Hepatitis C and non-Viral Chronic Liver Disease. Hepatology (1997) 25:1271–5. doi: 10.1002/hep.510250536
103. Kubo S, Nishiguchi S, Kuroki T, Hirohashi K, Tanaka H, Tsukamoto S, et al. Poor Association of GBV-C Viremia With Hepatocellular Carcinoma. J.Hepatol (1997) 27:91–5. doi: 10.1016/S0168-8278(97)80285-1
104. Pessoa MG, Terrault NA, Ferrell LD, Kim JP, Kolberg J, Detmer J, et al. Hepatitis G Virus in Patients With Cryptogenic Liver Disease Undergoing Liver Transplantation. Hepatology (1997) 25:1266–70. doi: 10.1002/hep.510250535
105. Hollingsworth RC, Minton EJ, Fraser-Moodie C, Metivier E, Rizzi PM, Irving WL, et al. Hepatitis G Infection: Role in Cryptogenic Chronic Liver Disease and Primary Liver Cell Cancer in the UK. Trent Hepatitis C Virus Study Group. J Viral Hepat (1998) 4:165–9. doi: 10.1046/j.1365-2893.1998.00102.x
106. Berg T, Muller AR, Platz KP, Hohne M, Bechstein WO, Hopf U, et al. Dynamics of GB Virus C Viremia Early After Orthotopic Liver Transplantation Indicates Extrahepatic Tissues as the Predominant Site of GB Virus C Replication. Hepatology (1999) 29:245–9. doi: 10.1002/hep.510290121
107. Laskus T, Radkowski M, Wang LF, Vargas H, Rakela J. Lack of Evidence for Hepatitis G Virus Replication in the Livers of Patients Coinfected With Hepatitis C Virus and G Virus. J.Virol (1997) 71:7804–6. doi: 10.1128/jvi.71.10.7804-7806.1997
108. Radkowski M, Wang LF, Cianciara J, Rakela J, Laskus T. Analysis of Hepatitis G Virus/GB Virus C Quasispecies and Replication Sites in Human Subjects. Biochem.Biophys.Res.Commun (1999) 258:296–9. doi: 10.1006/bbrc.1999.0632
109. Radkowski M, Kubicka J, Kisiel E, Cianciara J, Nowicki M, Rakela J, et al. Detection of Active Hepatits C Virus and Hepatits G Virus/GB Virus C Replication in Bone Marrow in Human Subjects. Transfusion Med Rev (2000) 95:3986–9. doi: 10.1182/blood.V95.12.3986.012k39_3986_3989
110. Madejon A, Fogeda M, Bartolome J, Pardo M, Gonzalez C, Cotonat T, et al. GB Virus C RNA in Serum, Liver and Peripheral Blood Mononuclear Cells From Patients With Chronic Hepatitis B, C and D. Gastroenterology (1997) 113:573–8. doi: 10.1053/gast.1997.v113.pm9247478
111. Fogeda M, Navas S, Martin J, Casqueiro M, Rodriguez E, Arocena C, et al. In Vitro Infection of Human Peripheral Blood Mononuclear Cells by GB Virus C/Hepatitis G Virus. J.Virol (1999) 73:4052–61. doi: 10.1128/JVI.73.5.4052-4061.1999
112. Fogeda M, Lopez-Alcorocho JM, Bartolome J, Arocena C, Martin MA, Carreno V. Existence of Distinct GB Virus C/Hepatitis G Virus Variants With Different Tropism. J.Virol (2000) 74:7936–42. doi: 10.1128/JVI.74.17.7936-7942.2000
113. George SL, Xiang J, Stapleton JT. Clinical Isolates of GB Virus Type C Vary in Their Ability to Persist and Replicate in Peripheral Blood Mononuclear Cell Cultures. Virology (2003) 316:191–201. doi: 10.1016/S0042-6822(03)00585-3
114. Rydze RT, Bhattarai N, Stapleton JT. GB Virus C Infection is Associated With a Reduced Rate of Reactivation of Latent HIV and Protection Against Activation-Induced T-Cell Death. Antivir Ther (2012) 17:1271–9. doi: 10.3851/IMP2309
115. Chivero ET, Bhattarai N, Rydze RT, Winters MA, Holodniy M, Stapleton JT. Human Pegivirus RNA is Found in Multiple Blood Mononuclear Cells In Vivo and Serum-Derived Viral RNA-Containing Particles are Infectious In Vitro. J Gen Virol (2014) 95:1307–19. doi: 10.1099/vir.0.063016-0
116. Chivero ET, Stapleton JT. Tropism of Human Pegivirus (Formerly Known as GB Virus C/hepatitis G Virus) and Host Immunomodulation: Insights Into a Highly Successful Viral Infection. J Gen Virol (2015) 96:1521–32. doi: 10.1099/vir.0.000086
117. Tucker TJ, Smuts HEM, Eedes C, Gideon DK, Eckhaus P, Robson SC, et al. Evidence That the GBV-C/Hepatitits C Virus is Primarily a Lymphotropic Virus. J.Med.Virol (2000) 61:52–8. doi: 10.1002/(SICI)1096-9071(200005)61:1<52::AID-JMV8>3.0.CO;2-L
118. Van Der Bij AK, Kloosterboer N, Prins M, Boeser-Nunnink B, Geskus RB, Lange JMA, et al. GB Virus C Coinfection and HIV-1 Disease Progression: The Amsterdam Cohort Study. J.Infect.Dis (2005) 191:678–85. doi: 10.1086/427559
119. Xiang J, Wunschmann S, Schmidt WN, Shao J, Stapleton JT. Full-Length GB Virus C (Hepatitis G Virus) RNA Transcripts are Infectious in Primary CD4-Positive T Cells. J.Virol (2000) 74:9125–33. doi: 10.1128/JVI.74.19.9125-9133.2000
120. George SL, Varmaz D, Stapleton JT. GB Virus C Replicates in Primary T and B Lymphocytes. J.Infect.Dis (2006) 193:451–4. doi: 10.1086/499435
121. Bhattarai N, Mclinden JH, Xiang J, Landay A, Chivero ET, Stapleton JT. GB Virus C Particles Inhibit T Cell Activation via Envelope E2 Protein-Mediated Inhibition of T Cell Receptor Signaling. J.Immunol (2013) 190:6351–9. doi: 10.4049/jimmunol.1300589
122. Tillmann HL, Manns MP. GB Virus-C Infection in Patients Infected With the Human Immunodeficiency Virus. Antiviral Res (2001) 52:83–90. doi: 10.1016/S0166-3542(01)00172-3
123. Sallie R, Shaw J, Mutimer D. GBV-C Virus and Fulminant Hepatic Failure. Lancet (1996) 347:1552. doi: 10.1016/S0140-6736(96)90704-7
124. Kanda T, Yokosuka O, Ehata T, Maru Y, Imazeki F, Saisho H, et al. Detection of GBV-C RNA in Patients With non-A-E Fulminant Hepatitis by Reverse-Transcription Polymerase Chain Reaction. Hepatology (1997) 25:1261–5. doi: 10.1002/hep.510250534
125. Kanda T, Yokosuka O, Imazeki F, Tagawa M, Ehata T, Saisho H, et al. GB Virus-C RNA in Japanese Patients With Hepatocellular Carcinoma and Cirrhosis. J.Hepatol (1997) 27:464–9. doi: 10.1016/S0168-8278(97)80349-2
126. Sarrazin C, Rüster B, Lee J-H, Kronenberger B, Roth WK, Zuezem S, et al. Prospective Follow-Up of Patients With GBV-C/HGV Infection: Specific Mutational Patterns, Clinical Outcome, and Genetic Diversity. J.Med.Virol (2000) 62:191–8. doi: 10.1002/1096-9071(200010)62:2<191::AID-JMV10>3.0.CO;2-7
127. Tagger A, Donato F, Ribero ML, Chiesa R, Tomasoni V, Portera G, et al. A Case-Control Study on GB Virus C/hepatitis G Virus Infection and Hepatocellular Carcinoma. Brescia HCC Study. Hepatology (1997) 26:1653–7. doi: 10.1002/hep.510260639
128. Zignego AL, Giannini C, Gentilini P, Bellesi G, Hadziyannis S, Ferris C. Could HGV Infection be Implicated in Lymphomagenesis? Br.J.Haematol (1997) 98:778–9. doi: 10.1046/j.1365-2141.1997.d01-3531.x
129. Kriesel JD, Hobbs MR, Jones BB, Milash B, Nagra RM, Fischer KF. Deep Sequencing for the Detection of Virus-Like Sequences in the Brains of Patients With Multiple Sclerosis: Detection of GBV-C in Human Brain. PloS One (2012) 7:e31886. doi: 10.1371/journal.pone.0031886
130. Fargion S, Sampietro M, Fracanzani AL, Molteni V, Ticozz M, Mattioli A, et al. Hepatitis G Virus in Patients With Porphyria Cutanea Tarda (PCT). J.Hepatol (1997) 26(S):207.
131. Nagao Y, Sata M, Noguchi S, Mizokami M, Kameyama T, Tanikawa K. GB Virus Infection in Patients With Oral Cancer and Oral Lichen Planus. J Oral Pathol Med (1997) 26:138–41. doi: 10.1111/j.1600-0714.1997.tb00037.x
132. Krajden M, Yu A, Braybrook H, Lai AS, Mak A, Chow R, et al. GBV-C/hepatitis G Virus Infection and non-Hodgkin Lymphoma: A Case Control Study. Int J Cancer (2010) 126:2885–92. doi: 10.1002/ijc.25035
133. Chang CM, Stapleton JT, Klinzman D, Mclinden JH, Purdue MP, Katki HA, et al. GBV-C Infection and Risk of NHL Among U.S. Adults. Cancer Res (2014) 74:5553–60. doi: 10.1158/0008-5472.CAN-14-0209
134. Fama A, Xiang J, Link BK, Allmer C, Klinzman D, Feldman AL, et al. Human Pegivirus Infection and Lymphoma Risk and Prognosis: A North American Study. Br J Haematol (2018) 182:644–53. doi: 10.1111/bjh.15416
135. Fama A, Larson MC, Link BK, Habermann TM, Feldman AL, Call TG, et al. Human Pegivirus Infection and Lymphoma Risk: A Systematic Review and Meta-Analysis. Clin Infect Dis (2020) 71:1221–8. doi: 10.1093/cid/ciz940
136. Balcom EF, Doan MAL, Branton WG, Jovel J, Blevins G, Edguer B, et al. Human Pegivirus-1 Associated Leukoencephalitis: Clinical and Molecular Features. Ann Neurol (2018) 84:781–7. doi: 10.1002/ana.25343
137. Doan MAL, Roczkowsky A, Smith M, Blevins G, Van Landeghem FKH, Gelman BB, et al. Infection of Glia by Human Pegivirus Suppresses Peroxisomal and Antiviral Signaling Pathways. J Virol (2021) 95:e0107421. doi: 10.1128/JVI.01074-21
138. De Souza AJS, Malheiros AP, De Sousa ERP, Moreira ACN, Silva AL, Das Chagas AAC, et al. First Report of Equine Pegivirus in South America, Brazil. Acta Trop (2015) 152:56–9. doi: 10.1016/j.actatropica.2015.08.014
139. Tomlinson JE, Kapoor A, Kumar A, Tennant BC, Laverack MA, Beard L, et al. Viral Testing of 18 Consecutive Cases of Equine Serum Hepatitis: A Prospective Stud-2018). J Vet Intern Med (2019) 33:251–7. doi: 10.1111/jvim.15368
140. Tomlinson JE, Tennant BC, Struzyna A, Mrad D, Browne N, Whelchel D, et al. Viral Testing of 10 Cases of Theiler's Disease and 37 in-Contact Horses in the Absence of Equine Biologic Product Administration: A Prospective Stud-2018). J Vet Intern Med (2019) 33:258–65. doi: 10.1111/jvim.15362
141. Mandl JN, Ahmed R, Barreiro LB, Daszak P, Epstein JH, Virgin HW, et al. Reservoir Host Immune Responses to Emerging Zoonotic Viruses. Cell (2015) 160:20–35. doi: 10.1016/j.cell.2014.12.003
142. Toyoda H, Fukuda Y, Hayakawa T, Takamatsu J, Saito H. Effect of GB Virus C/hepatitis G Virus Coinfection on the Course of HIV Infection in Hemophilia Patients in Japan. J Acquir Immune Defic Syndr (1998) 17:209–13. doi: 10.1097/00042560-199803010-00004
143. Yeo AET, Matsumoto A, Hisada M, Shih JW, Alter HJ, Goedert JJ. Effect of Hepatitis G Virus Infection on Progression of HIV Infection in Patients With Hemophilia. Ann.Intern.Med (2000) 132:959–63. doi: 10.7326/0003-4819-132-12-200006200-00006
144. Bjorkman P, Flamholc L, Molnegren V, Marshall A, Guner N, Widell A. Enhanced and Resumed GB Virus C Replication in HIV-1-Infected Individuals Receiving HAART. AIDS (2007) 21:1641–3. doi: 10.1097/QAD.0b013e32823bc9b7
145. Nunnari G, Nigro L, Palermo F, Attanasio M, Berger A, Doerr HW, et al. Slower Progression of HIV-1-Infection in Persons With GB Virus C Co-Infection Correlates With an Intact T-Helper 1 Cytokine Profile. Ann.Int.Med (2003) 139:26–30. doi: 10.7326/0003-4819-139-1-200307010-00009
146. N'guessan KF, Anderson M, Phinius B, Moyo S, Malick A, Mbangiwa T, et al. The Impact of Human Pegivirus on CD4 Cell Count in HIV-Positive Persons in Botswana. Open Forum Infect Dis (2017) 4:ofx222. doi: 10.1093/ofid/ofx222
147. Stapleton JT, Chaloner K, Williams CF. GB Virus C and Survival in the Amsterdam Cohort Study. J.Infect.Dis (2005) 191:2157–8. doi: 10.1086/430505
148. Bjorkman P, Flamholc L, Naucler A, Molnegren V, Wallmark E, Widell A. GB Virus C During the Natural Course of HIV-1 Infection: Viremia at Diagnosis Does Not Predict Mortality. AIDS (2004) 18:877–86. doi: 10.1097/00002030-200404090-00005
149. Geskus RB. On the Inclusion of Prevalent Cases in HIV/AIDS Natural History Studies Through a Marker-Based Estimate of Time Since Seroconversion. Stat.Med (2000) 19:1753–69. doi: 10.1002/1097-0258(20000715)19:13<1753::AID-SIM487>3.0.CO;2-F
150. George SL. Persistent GB Virus C Infection is Associated With Decreased HIV-1 Disease Progression in the Amsterdam Cohort Study. J.Infect.Dis (2005) 191:2156–7. doi: 10.1086/430504
151. Vahidnia F, Petersen M, Stapleton T, Rutherford GW, Busch M, Custer B. Acquisition of GB Virus Type C (HGV) and Lower Mortality in Patients With Advanced HIV Disease. Clin.Infect.Dis (2012) 55:1012–9. doi: 10.1093/cid/cis589
152. Handelsman E, Cheng I, Thompson B, Hershow R, Mofenson LM, Hollinger FB, et al. Impact of GB Virus Type C Infection on Mother-To-Child HIV Transmission in the Women and Infants Transmission Study Cohort. HIV Med (2007) 8:561–7. doi: 10.1111/j.1468-1293.2007.00510.x
153. Gretch D. Editorial Commentary: Advocating the Concept of GB Virus C Biotherapy Against AIDS. Clin Infect Dis (2012) 55:1020–1. doi: 10.1093/cid/cis591
154. Greenhalgh S, Schmidt R, Day T. Fighting the Public Health Burden of AIDS With the Human Pegivirus. Am J Epidemiol (2019) 188:1586–94. doi: 10.1093/aje/kwz139
155. Xiang J, George SL, Wunschmann S, Chang Q, Klinzman D, Stapleton JT. Inhibition of HIV-1 Replication by GB Virus C Infection Through Increases in RANTES, MIP-1α, MIP-1α, and SDF-1. Lancet (2004) 363:2040–6. doi: 10.1016/S0140-6736(04)16453-2
156. Nattermann J, Nischalke HD, Kupfer B, Rockstroh J, Hess L, Sauerbruch T, et al. Regulation of CC Chemokine Receptor 5 in Hepatitis G Virus Infection. AIDS (2003) 17:1457–62. doi: 10.1097/00002030-200307040-00006
157. Schwarze-Zander C, Neibecker M, Othman S, Tural C, Clotet B, Blackard JT, et al. GB Virus C Coinfection in Advanced HIV Type-1 Disease is Associated With Low CCR5 and CXCR4 Surface Expression on CD4(+) T-Cells. Antivir.Ther (2010) 15:745–52. doi: 10.3851/IMP1602
158. Jung S, Knauer O, Donhauser N, Eichenmuller M, Helm M, Fleckenstein B, et al. Inhibition of HIV Strains by GB Virus C in Cell Culture can be Mediated by CD4 and CD8 T-Lymphocyte Derived Soluble Factors. AIDS (2005) 19:12567–1272. doi: 10.1097/01.aids.0000180097.50393.df
159. Xiang J, Mclinden JH, Chang Q, Kaufman TM, Stapleton JT. An 85 Amino Acid Segment of the GB Virus Type C NS5A Phosphoprotein Inhibits HIV-1 Replication in CD4+ Jurkat T-Cells. Proc.Natl.Acad.Sci.U.S.A (2006) 103:15570–5. doi: 10.1073/pnas.0604728103
160. Xiang J, Mclinden JH, Chang Q, Jordan EL, Stapleton JT. Characterization of a Peptide Domain Within the GB Virus C NS5A Phosphoprotein That Inhibits HIV Replication. PloS One (2008) 3:e2580,2581-2510. doi: 10.1371/journal.pone.0002580
161. Xiang J, Sathar MA, Mclinden JH, Klinzman D, Chang Q, Stapleton JT. South African GB Virus C Isolates: Interactions Between Genotypes 1 and 5 GBV-C Isolates and the Human Immunodeficiency Virus. J.Infect.Dis (2005) 192:2147–51. doi: 10.1086/498170
162. Chang Q, Mclinden JH, Stapleton JT, Sathar MA, Xiang J. GB Virus C NS5A Protein From Genotypes 1, 2, 3, and 5 and a 30 Amino Acid NS5A Fragment Expression Inhibit HIV-1 Replication in a CD4+ T-Lymphocytes. J.Gen.Virol (2007) 88:3341–6. doi: 10.1099/vir.0.83198-0
163. Jung S, Eichenmueller M, Donhauser N, Neipel F, Engel AM, Hess G, et al. HIV Entry Inhibition by the Envelope 2 Glycoprotein of GB Virus C. AIDS (2007) 21:645–7. doi: 10.1097/QAD.0b013e32803277c7
164. Xiang J, Mclinden JH, Kaufman TM, Mohr EL, Bhattarai N, Chang Q, et al. Characterization of a Peptide Domain Within the GB Virus C Envelope Glycoprotein (E2) That Inhibits HIV Replication. Virol (2012) 430:53–62. doi: 10.1016/j.virol.2012.04.019
165. Mohr EL, Xiang J, Mclinden JH, Kaufman TM, Montefiori DC, Klinzman D, et al. GB Virus C Envelope Protein E2 Elicits Antibodies That React With a Cellular Antigen on HIV-1 Particles and Neutralize Diverse HIV-1 Isolates. J.Immunol (2010) 185:4496–505. doi: 10.4049/jimmunol.1001980
166. Herrera E, Gomara MJ, Mazzini S, Ragg E, Haro I. Synthetic Peptides of Hepatitis G Virus (GBV-C/HGV) in the Selection of Putative Peptide Inhibitors of the HIV-1 Fusion Peptide. J Phys Chem B (2009) 113:7383–91. doi: 10.1021/jp900707t
167. Herrera E, Tenckhoff S, Gomara MJ, Galatola R, Bleda MJ, Gil C, et al. Effect of Synthetic Peptides Belonging to E2 Envelope Protein of GB Virus C on Human Immunodeficiency Virus Type 1 Infection. J.Med.Chem (2010) 53:6054–63. doi: 10.1021/jm100452c
168. Haro I, Gomara MJ, Galatola R, Domenech O, Prat J, Girona V, et al. Study of the Inhibition Capacity of an 18-Mer Peptide Domain of GBV-C Virus on Gp41-FP HIV-1 Activity. Biochim Biophys Acta (2011) 1808:1567–73. doi: 10.1016/j.bbamem.2011.02.019
169. Koedel Y, Eissmann K, Wend H, Fleckenstein B, Reil H. Peptides Derived From a Distinct Region of GB Virus C Glycoprotein E2 Mediate Strain-Specific HIV-1 Entry Inhibition. J.Virol (2011) 85:7037–47. doi: 10.1128/JVI.02366-10
170. Sanchez-Martin MJ, Busquets MA, Girona V, Haro I, Alsina MA, Pujol M. Effect of E1(64-81) Hepatitis G Peptide on the In Vitro Interaction of HIV-1 Fusion Peptide With Membrane Models. Biochim Biophys Acta (2011) 1808:2178–88. doi: 10.1016/j.bbamem.2011.05.020
171. Sanchez-Martin MJ, Hristova K, Pujol M, Gomara MJ, Haro I, Alsina MA, et al. Analysis of HIV-1 Fusion Peptide Inhibition by Synthetic Peptides From E1 Protein of GB Virus C. J Colloid Interface Sci (2011) 360:124–31. doi: 10.1016/j.jcis.2011.04.053
172. Sanchez-Martin MJ, Urban P, Pujol M, Haro I, Alsina MA, Busquets MA. Biophysical Investigations of GBV-C E1 Peptides as Potential Inhibitors of HIV-1 Fusion Peptide. Chemphyschem (2011) 12:2816–22. doi: 10.1002/cphc.201100407
173. Gomara MJ, Sanchez-Merino V, Paus A, Merino-Mansilla A, Gatell JM, Yuste E, et al. Definition of an 18-Mer Synthetic Peptide Derived From the GB Virus C E1 Protein as a New HIV-1 Entry Inhibitor. Biochim Biophys Acta (2016) 1860:1139–48. doi: 10.1016/j.bbagen.2016.02.008
174. Hoffmann R, Ruegamer T, Schaubacher J, Rohrhofer A, Kirmess P, Fiebig KM, et al. Exploring Viral Interference Using Peptides: Molecular Determinants of HIV-1 Inhibition by a Peptide Derived From Human Pegivirus-1 Envelope Protein E2. ChemMedChem (2021) 16:1290–6. doi: 10.1002/cmdc.202000892
175. Timmons CL, Shao Q, Wang C, Liu L, Liu H, Dong X, et al. GB Virus Type C E2 Protein Inhibits Human Immunodeficiency Virus Type 1 Assembly Through Interference With HIV-1 Gag Plasma Membrane Targeting. J.Infect.Dis (2013) 207:1171–80. doi: 10.1093/infdis/jit001
176. Cartier C, Sivard P, Tranchat C, Decimo D, Desgranges C, Boyer V. Identification of Three Major Phosphorylation Sites Within HIV-1 Capsid. Role of Phosphorylation During the Early Steps of Infection. J Biol Chem (1999) 274:19434–40. doi: 10.1074/jbc.274.27.19434
177. Nattermann J, Nischalke HD, Feldmann G, Ahlenstiel G, Sauebruch T, Spengler U. Binding of HCV E2 to CD81 Induces RANTES Secretion and Internalization of CC Chemokine Receptor 5. J Viral Hepat (2006) 11:519–26. doi: 10.1111/j.1365-2893.2004.00545.x
178. Flint M, Maidens C, Loomis-Price LD, Shotton C, Dubuisson J, Monk P, et al. Characterization of Hepatitis C Virus E2 Glycoprotein Interaction With a Putative Cellular Receptor, Cd81. J Virol (1999) 73:6235–44. doi: 10.1128/JVI.73.8.6235-6244.1999
179. Wunschmann S, Klinzman D, Medh J, Klinzman D, Schmidt WN, Stapleton JT. Characterization of Hepatitis C Virus (HCV) and HCV E2 Interactions With CD81 and the Low Density Lipoprotein Receptor. J.Virol (2000) 74:10055–62. doi: 10.1128/JVI.74.21.10055-10062.2000
180. Kaufman TM, Mclinden JH, Xiang J, Engel AM, Stapleton JT. The GBV-C Envelope Glycoprotein E2 Does Not Interact Specifically With CD81. AIDS (2007) 21:1045–8. doi: 10.1097/QAD.0b013e3280f77412
181. Chowdhury AY, Tavis JE, George SL. Human Pegivirus (GB Virus C) NS3 Protease Activity Inhibits Induction of the Type I Interferon Response and is Not Inhibited by HCV NS3 Protease Inhibitors. Virology (2014) 456-457:300–9. doi: 10.1016/j.virol.2014.03.018
182. Hunt PW, Deeks SG. Immune-Based Therapy for HIV Infection: Are Acute and Chronic HIV Infection Different Diseases? J.Infect.Dis (2006) 194:1632–4. doi: 10.1086/509627
183. Pilot-Matias TJ, Muerhoff AS, Simons JN, Leary TP, Buijk SL, Chalmers ML, et al. Identification of Antigenic Regions in the GB Hepatitis Viruses GBV-A, GBV-B, and GBV-C. J.Med.Virol (1996) 48:329–38. doi: 10.1002/(SICI)1096-9071(199604)48:4<329::AID-JMV6>3.0.CO;2-9
184. Fernandez-Vidal M, Cubero MD, Ercilla G, Gomara MJ, Haro I. Application of a Chimeric Synthetic Peptide in the Development of a Serologic Method for the Diagnosis of Hepatitis G Virus Infection. Protein Pept Lett (2007) 14:865–70. doi: 10.2174/092986607782110239
185. Schmolke S, Tacke M, Schmitt U, Engel AM, Ofenloch-Haehnle B. Identification of Hepatitis G Virus Particles in Human Serum by E2-Specific Monoclonal Antibodies Generated by DNA Immunization. J.Virol (1998) 72:4541–5. doi: 10.1128/JVI.72.5.4541-4545.1998
186. Mclinden JH, Kaufman TM, Xiang J, Chang Q, Klinzman D, Engel AM, et al. Characterization of an Immunodominant Antigenic Site on GB Virus C Glycoprotein E2 That is Involved in Cell Binding. J.Virol (2006) 80:12131–40. doi: 10.1128/JVI.01206-06
187. Malherbe DC, Mendy J, Vang L, Barnette PT, Reed J, Lakhashe SK, et al. Combination Adenovirus and Protein Vaccines Prevent Infection or Reduce Viral Burden After Heterologous Clade C Simian-Human Immunodeficiency Virus Mucosal Challenge. J Virol (2018) 92:e01092–01117. doi: 10.1128/JVI.01092-17
188. Maidana Giret MT, Silva TM, Sauer MM, Tomiyama H, Levi JE, Bassichetto KC, et al. GBV-C Infection Modulates T Cell Activation in Recently HIV-Infected Subjects and is Independent of HIV-1 Viral Load. AIDS (2009) 23:2277–87. doi: 10.1097/QAD.0b013e32832d7a11
189. Stapleton JT, Chaloner K, Martenson JA, Zhang J, Klinzman D, Xiang J, et al. GB Virus C Infection is Associated With Altered Lymphocyte Subset Distribution and Reduced T Cell Activation and Proliferation in HIV-Infected Individuals. PloS One (2012) 7:e50563. doi: 10.1371/journal.pone.0050563
190. Mahalingam M, Peakman M, Davies ET, Pozniak A, Mcmanus TJ, Vergani D. T Cell Activation and Disease Severity in HIV Infection. Clin Exp Immunol (1993) 93:337–43. doi: 10.1111/j.1365-2249.1993.tb08182.x
191. Brenchley JM, Price DM, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial Translocation is a Cause of Systemic Immune Activation in Chronic HIV Infection. Nat.Med (2006) 12:1365–71. doi: 10.1038/nm1511
192. Hunt PW, Brenchley J, Sinclair E, Mccune JM, Roland M, Page-Shafer K, et al. Relationship Between T Cell Activation and CD4+ T Cell Count in HIV-Seropositive Individuals With Undetectable Plasma HIV RNA Levels in the Absence of Therapy. J.Infect.Dis (2008) 197:126–33. doi: 10.1086/524143
193. Richardson MW, Jadlowsky J, Didigu CA, Doms RW, Riley JL. Kruppel-Like Factor 2 Modulates CCR5 Expression and Susceptibility to HIV-1 Infection. J Immunol (2012) 189:3815–21. doi: 10.4049/jimmunol.1201431
194. Kim Y, Anderson JL, Lewin SR. Getting the "Kill" Into "Shock and Kill": Strategies to Eliminate Latent HIV. Cell Host Microbe (2018) 23:14–26. doi: 10.1016/j.chom.2017.12.004
195. Rydze RT, Xiang J, Mclinden JH, Stapleton JT. GB Virus C Infection Polarizes T Cell Cytokine Gene Expression Towards a Th1 Cytokine Profile via NS5A Protein Expression. J.Infect.Dis (2012) 206:69–72. doi: 10.1093/infdis/jis312
196. Blackard JT, Ma G, Welge JA, Taylor LE, Mayer KH, Klein RS, et al. Cytokine/chemokine Expression Associated With Human Pegivirus (HPgV) Infection in Women With HIV. J Med Virol (2017) 89:1904–11. doi: 10.1002/jmv.24836
197. Valdez H, Mitsuyasu R, Landay A, Sevin AD, Chan ES, Spritzler J, et al. Interleukin-2 Increases CD4+ Lymphocyte Numbers But Does Not Enhance Responses to Immunization: Results of A5046s. J Infect Dis (2003) 187:320–5. doi: 10.1086/346056
198. Stapleton JT, Chaloner K, Zhang J, Klinzman D, Souza IE, Xiang J, et al. GB Virus C Viremia is Associated With Reduced CD4 Expansion Following Interleukin 2 Therapy in HIV-Infected People Receiving HAART. AIDS (2009) 23:605–10. doi: 10.1097/QAD.0b013e32831f1b00
199. Bhattarai N, Mclinden JH, Xiang J, Kaufman TM, Stapleton JT. GB Virus C Envelope Protein E2 Inhibits TCR-Induced IL-2 Production and Alters IL-2-Signaling Pathways. J.Immunol (2012) 189:2211–6. doi: 10.4049/jimmunol.1201324
200. Bhattarai N, Rydze RT, Chivero ET, Stapleton JT. GBV-C Viremia is Associated With Higher Levels of Double Negative T Cells and Lower T Cell Activation in HIV-Infected Individuals on Antiretroviral Therapy. J.Infect.Dis (2012) 206:1469–72. doi: 10.1093/infdis/jis515
201. Stapleton JT, Martinson JA, Klinzman D, Xiang J, Desai SN, Landay A. GB Virus C Infection and B-Cell, Natural Killer Cell, and Moncyte Activation Markers in HIV-Infected Individuals. AIDS (2013) 27:1829–32. doi: 10.1097/QAD.0b013e328363089f
202. Hunt PW, Martin JN, Sinclair E, Bredt B, Hagos E, Lampiris H, et al. T Cell Activation is Associated With Lower CD4+ T Cell Gains in Human Immunodeficiency Virus-Infected Patients With Sustained Viral Suppression During Antiretroviral Therapy. J.Infect.Dis (2003) 187:1534–43. doi: 10.1086/374786
203. Chivero ET, Bhattarai N, Mclinden JH, Xiang J, Stapleton JT. Human Pegivirus (HPgV; Formerly Known as GBV-C) Inhibits IL-12 Dependent Natural Killer Cell Function. Virology (2015) 485:116–27. doi: 10.1016/j.virol.2015.07.008
204. Lalle E, Sacchi A, Abbate I, Vitale A, Martini F, D'offizi GD, et al. Activation of Interferon Response Genes and of Plasmacytoid Dendritic Cells in HIV-1 Positive Subjects With GB Virus C Co-Infection. Int.J.Immunopathol.Pharm (2008) 21:161–71. doi: 10.1177/039463200802100118
205. Haji Molla Hoseini M, Pourfathollah AA, Mohraz M, Soheili Z, Amini S, Aghaiepour M, et al. Evaluation of Circulating Natural Type 1 Interferon-Producing Cells in HIV/GBV-C and HIV/HCV Coinfected Patients: A Preliminary Study. Arch Med Res (2007) 38:868–75. doi: 10.1016/j.arcmed.2007.05.009
206. Lanteri MC, Vahidnia F, Tan S, Stapleton JT, Norris PJ, Heitman J, et al. Downregulation of Cytokines and Chemokines by GB Virus C After Transmission Via Blood Transfusion in HIV-Positive Blood Recipients. J Infect Dis (2015) 211:1585–96. doi: 10.1093/infdis/jiu660
207. Moenkemeyer M, Schmidt RE, Wedemeyer H, Tillmann HL, Heiken H. GBV-C Coinfection is Negatively Correlated to Fas Expression and Fas-Mediated Apoptosis in HIV-1 Infected Patients. J.Med.Virol (2008) 80:1933–40. doi: 10.1002/jmv.21305
208. Martinot M, Marcellin P, Boyer N, Detmer J, Pouteau M, Castelnau C, et al. Influence of Hepatitis G Virus Infection on the Severity of Liver Disease and Response to Interferon-À in Patients With Chronic Hepatitis C. Ann.Int.Med (1997) 126:874–81. doi: 10.7326/0003-4819-126-11-199706010-00004
209. Nagayama R, Miyake K, Okamoto H. Effect of Interferon on GB Virus C and Hepatitis C Virus in Hepatitis Patients With the Co-Infection. J.Med.Virol (1997) 52:156–60. doi: 10.1002/(SICI)1096-9071(199706)52:2<156::AID-JMV6>3.0.CO;2-2
210. Jarvis LM, Bell H, Simmonds P, Hawkins A, Hellum K, Harthug S, et al. The Effect of Treatment With Alpha-Interferon on Hepatitis G/GBV-C Viraemia. Scand J Gastroenterol (1998) 33:195–200. doi: 10.1080/00365529850166941
211. Lin R, Dutta U, Kaba S, Kench J. Effects of Hepatitis G Virus Coinfection on Severity of Hepatitis C: Relationship to Risk Factors and Response to Interferon Treatment. J.Gastro.Hepatol (1998) 13:773–80. doi: 10.1111/j.1440-1746.1998.tb00732.x
212. Lau DT, Miller KD, Detmer J, Kolberg J, Herpin B, Metcalf JA, et al. Hepatitis G Virus and Human Immunodeficiency Virus Coinfection: Response to Interferon-Alpha Therapy. J.Inf.Dis (1999) 180:1334–7. doi: 10.1086/315031
213. Kato T, Mizokami M, Orito E, Ohba K. Amino Acid Substitutions in NS5A Region of GB Virus C and Response to Interferon Therapy. J.Med.Virol (1999) 57:376–82. doi: 10.1002/(SICI)1096-9071(199904)57:4<376::AID-JMV9>3.0.CO;2-#
214. Fujisawa T, Horiike N, Michitaka K, Onji M. Influence of RNA Titre and Amino Acid Changes in the NS5A Region of GB Virus C/hepatitis G Virus on the Effectiveness of Interferon Therapy. J.Gastro.Hepatol (2000) 15:632–9. doi: 10.1046/j.1440-1746.2000.02196.x
215. Xiang J, Martinez-Smith C, Gale M Jr., Labrecque DR, Schmidt WN, Stapleton JT. GB Virus C NS5A Sequence Polymorphisms: Association With Interferon Susceptibility and Inhibition of PKR-Mediated eIF-2α Phosphorylation. J Interferon Cytokine Res (2005) 25:261–70. doi: 10.1089/jir.2005.25.261
216. Berzsenyi MD, Bowden DS, Kelly HA, Watson KM, Mijch AM, Hammond RA, et al. Reduction in Hepatitis C Related Liver Disease Associated With GB Virus C in Human Immunodeficiency Virus Co-Infection. Gastroenterology (2007) 133:1821–30. doi: 10.1053/j.gastro.2007.08.076
217. Berzsenyi MD, Woollard DJ, Mclean CA, Preiss S, Perreau VM, Beard MR, et al. Down-Regulation of Intra-Hepatic T-Cell Signaling Associated With GB Virus C in a HCV/HIV Co-Infected Group With Reduced Liver Disease. J Hepatol (2011) 55:536–44. doi: 10.1016/j.jhep.2010.12.021
218. Stapleton J. A New Variable Influencing HCV-Related Liver Disease in HIV-HCV Coinfected Individuals? Gastroenterology (2007) 133:2042–5. doi: 10.1053/j.gastro.2007.10.032
219. Lauck M, Bailey AL, Andersen KG, Goldberg Tl, Sabeti PC, O'Connor DH. GB Virus C Coinfections in West African Ebola Patients. J.Virol (2015) 89:2425–9. doi: 10.1128/JVI.02752-14
220. Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med (2020) 383:2255–73. doi: 10.1056/NEJMra2026131
221. Vu DL, Cordey S, Simonetta F, Brito F, Docquier M, Turin L, et al. Human Pegivirus Persistence in Human Blood Virome After Allogeneic Haematopoietic Stem-Cell Transplantation. Clin Microbiol Infect (2019) 25:225–32. doi: 10.1016/j.cmi.2018.05.004
222. Mrzljak A, Simunov B, Balen I, Jurekovic Z, Vilibic-Cavlek T. Human Pegivirus Infection After Transplant: Is There an Impact? World J Transplant (2022) 12:1–7. doi: 10.5500/wjt.v12.i1.1
Keywords: Pegivirus, GB virus, immune modulation, viral co-infection, Flavivirus
Citation: Stapleton JT (2022) Human Pegivirus Type 1: A Common Human Virus That Is Beneficial in Immune-Mediated Disease? Front. Immunol. 13:887760. doi: 10.3389/fimmu.2022.887760
Received: 02 March 2022; Accepted: 03 May 2022;
Published: 30 May 2022.
Edited by:
Imre Kovesdi, Unleash Immuno Oncolytics, United StatesReviewed by:
Marcelo A. Soares, National Cancer Institute (INCA), BrazilMahmoud Reza Pourkarim, KU Leuven, Belgium
Copyright © 2022 Stapleton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jack T. Stapleton, amFjay1zdGFwbGV0b25AdWlvd2EuZWR1