 Chelsea Gootjes
Chelsea Gootjes Jaap Jan Zwaginga
Jaap Jan Zwaginga Bart O. Roep
Bart O. Roep Tatjana Nikolic
Tatjana Nikolic
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 04 May 2022
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.886736
This article is part of the Research Topic Genetic Basis of Tolerance Induction Defects Underlying the Development of Autoimmune Pathologies View all 11 articles
Type 1 diabetes (T1D) is an autoimmune disease that develops in the interplay between genetic and environmental factors. A majority of individuals who develop T1D have a HLA make up, that accounts for 50% of the genetic risk of disease. Besides these HLA haplotypes and the insulin region that importantly contribute to the heritable component, genome-wide association studies have identified many polymorphisms in over 60 non-HLA gene regions that also contribute to T1D susceptibility.
Combining the risk genes in a score (T1D-GRS), significantly improved the prediction of disease progression in autoantibody positive individuals. Many of these minor-risk SNPs are associated with immune genes but how they influence the gene and protein expression and whether they cause functional changes on a cellular level remains a subject of investigation. A positive correlation between the genetic risk and the intensity of the peripheral autoimmune response was demonstrated both for HLA and non-HLA genetic risk variants. We also observed epigenetic and genetic modulation of several of these T1D susceptibility genes in dendritic cells (DCs) treated with vitamin D3 and dexamethasone to acquire tolerogenic properties as compared to immune activating DCs (mDC) illustrating the interaction between genes and environment that collectively determines risk for T1D. A notion that targeting such genes for therapeutic modulation could be compatible with correction of the impaired immune response, inspired us to review the current knowledge on the immune-related minor risk genes, their expression and function in immune cells, and how they may contribute to activation of autoreactive T cells, Treg function or β-cell apoptosis, thus contributing to development of the autoimmune disease.
For several decades the incidence of Type 1 diabetes (T1D) has been increasing worldwide (1). This disease is characterized by the infiltration of immune cells in the islets of Langerhans (2, 3) ultimately leading to the loss of insulin producing β-cells with insulin replacement as the only available option to prevent fatal outcomes in all patients. Curative treatments are lacking for several reasons, one being that the events in humans leading to a selective β-cell dysfunction and loss is hard to detect. Although the analyses of fresh and cryopreserved tissues in the past decade, enabled by the nPOD initiative (www.JDRFnPOD.org), have significantly contributed to our understanding of the important local players in the process (2–5), many questions remain. So far, adaptive immune cells are indisputably involved in the β-cell destruction by their tissue specificity. The clinically approved therapies modulate immunity in general, the more preferable antigen-specific immune therapies show promising results but are not ready for general clinical application (6).
While autoimmune T1D is not completely inherited and environmental factors show a significant contribution to the pathogenesis (7), certain genetic polymorphisms do critically increase the predisposition for T1D (8). Polymorphisms in HLA and insulin (INS) regions were first described and contribute strongly to the disease risk (9, 10). Later, genome-wide association studies (GWAS) have identified many additional SNPs in so called non-HLA risk genes, which show a small but clear individual contribution to the increased risk for T1D (11). When included in a cumulative score (T1D-GRS), it significantly improved the capacity to discriminate T1D from T2D or healthy subjects, and to discriminate monogenic from autoimmune T1D (12–14). The exact functional contribution of many of these SNPs to the T1D-GRS remains to be characterized. We have observed a positive correlation between the non-HLA genetic risk, in addition to, but independently of HLA, and the intensity of the peripheral autoimmune response in T1D patients (15). Indeed, many of the associated T1D risk genes are controlled by lymphoid enhancers or involved in immune networks (11, 16). Our studies focusing on the differential transcriptome of tolerogenic (tolDC) versus inflammatory dendritic cells (mDCs) showed that a tolerogenic modulation of monocytes by 1,25(OH)2 vitamin D3 (VitD3) induced a stable change in the expression of sets of these non-HLA risk genes (17, 18), inspiring a hypothesis that quantitative and/or qualitative effects of the SNPs on the related gene products may reflect in a change of the immune regulatory vs. an immune activating balance. Here, we aim to review the knowledge of functional consequences of T1D risk SNPs on the regulation, expression and function of linked risk genes and further contemplate how this may impact the functionality of the effector vs. regulatory T cells, changing the balance between immune activation and suppression in the pancreas that is critical to attenuate chronic inflammation and an uncontrolled damage of insulin producing β-cells.
The exact order of immune events that cause human T1D has not been established. Hurdles such as that circulating blood cells poorly represent what is going on in the human pancreas, as well as the inability to directly analyze the target organ have significantly delayed our understanding of this autoimmune disease. Literature supports different scenarios describing the initiating events, involving an altered thymic selection of T cells that recognize β-cell antigens, viral infections that mark β-cells as the targets for destruction, enhanced expression of neo-antigens by β-cells due to cell stress, or an increased sensitivity of β-cells to inflammation (19–21). In all cases, β-cells seem critically involved in the process while the (auto)immune system is essential to execute the destructive insult resulting eventually in disease (22). The proposed initiating events are not mutually exclusive and likely cause the exposure of β-cell (neo-)antigens, which are taken-up by antigen presenting cells and presented to T cells in the context of high-risk human leukocyte antigen (HLA) molecules. The ‘first hit’ could occur when such presentation of β-cell antigens activates a destructive autoimmune response that may inflict some damage in the pancreatic islets but the disease is delayed as long as the immune regulation keeps the autoimmunity in check. The ‘second hit’ may occur when a regulatory checkpoint is bypassed such as upon an anti-cancer stimulating immunotherapy by check point inhibition or when the stressed (or infected) islets of Langerhans start releasing cytokines and chemokines, attracting immune cells where they target and eliminate functional β-cells to the point of no return.
T1D patients donating tissues for research helped to collect a significant evidence that T1D pathogenesis can follow different individual scenarios but also that mechanisms described in animal models are not all or not just as strongly present in the human immune system. For example, the infiltration of immune cells around the islet of Langerhans, designated as insulitis, in which activated CD4+ T cells control macrophages to induce killing of the β-cells by cytokines and reactive oxygen species, is clearly evident in mice (23) but not evident in human pancreas (24, 25). Cytotoxic CD8+ T cells are essential for the ultimate destruction of β-cells, while their antigen specificity varies between patients (4, 26). Hypothetically, techniques that discriminate relevant antigens and cells in the target tissue, allowing to separate primary immune aggressors from those only guilty by association will help solve this puzzle. Such bystanders may be the autoantibodies, which role in the immunopathology of human T1D is proved dispensable although they represent a good biomarker of an ongoing and in time often increasingly complex autoimmunity (27, 28). These antibodies can be found months to years before the clinical symptoms (29), help an early diagnosis of the disease and may prove valuable to identify individuals that will benefit from new curative treatments.
Time will tell whether the knowledge on the specificity of T1D autoantibodies to insulin (INS), 65 kDa glutamic acid decarboxylase (GAD65), insulinoma-associated protein2 (IA2) and zinc transporter 8(Znt8) (30) has helped or derailed the investigation of the β-cell specific targets of T cell autoimmunity (26, 31). More recent findings point to alternative transcripts and (neo-)antigens created by stressed or damaged β-cells, which are normally invisible to the immune system, as more likely to drive the T-cell mediated pathogenic destruction (21, 32–37). The contribution of the originally described antigens could be different, namely to secure immune regulation through a negative selection of high-affinity autoreactive T cells (38), or to establish peripheral tolerance through low-affinity self-peptide recognizing regulatory T cells (Tregs) (39, 40). The existence of autoantibodies may hence be a sign of a regulation ‘gone wrong’ as a consequence of a genetically imprinted or environmentally caused impaired T cell selection, effector activation or reduced Treg function, such as demonstrated in T1D patients (41, 42).
Certain genetic polymorphisms associate with a higher risk to develop an autoimmune disease, which is most often expressed as an odds ratio (OR) that measures the strength of association between carrying a gene variant X (exposure) and development of T1D (outcome) (43). Specific HLA haplotypes and SNPs in the insulin gene (INS) strongly increase the odds to develop T1D and are hence designated as major susceptibility genes (44, 45). The HLA region was first associated with the risk of developing T1D, which is in line with a critical role of HLA in shaping the adaptive immunity (46). In the population of Caucasian origin, more than 90% of patients that develop T1D before puberty are carriers of one or both HLA haplotypes, namely HLA-DR3/DQ2 (DRB1*0301-DQA1*0501-DQB1*0201) or HLA-DR4/DQ8 (DRB1*0401-DQA1*0301- DQB1*03020 (47). In fact, heterozygotes carriers of both DRB1*03 and DRB1*04 carry up to 40 times higher risk to develop T1D than individuals with other HLA genotypes (48, 49). This synergic effect is likely caused by the formation of highly susceptible trans-encoded HLA-DQ (α1, β1) heterodimer molecules (48, 50), which efficiently bind and present β-cell derived peptides, increasing the number of different peptides that could trigger a pathogenic CD4+ T cell responses (51). Furthermore, the risk variant specific epigenetic modulation of the HLA expression could contribute to the disease pathogenesis (52).
A stable HLA molecule on the cell surface, however, does not exist without a peptide. Hence the contribution of HLA should be considered in combination with antigens/peptides that they present. The so far well-established β-cell antigens that are targeted by both B and T cell responses are INS, GAD65, IA2 and Znt8 but the list of target antigens is increasing (34, 35, 53). Of the β-cell proteins targeted as autoantigens, only SNPs in the INS gene are associated with an increased risk for T1D. The increased risk was first attributed to the polymorphism in variable number of tandem repeats (VNTR) in the insulin promotor (54, 55), determining the differential insulin expression between thymus and islets and leading to a faulty selection of the autoreactive T cells in thymus. While this may explain a part of the association, alternatives have been also explored, one being that other SNPs in the 3’ UTR of the INS gene (rs3842752 (56) and rs3842753 (57)) actually functionally contributes to the increased risk. Namely, these SNPs are expressed when an alternative translation start in the INS mRNA is used, creating a new protein sequence called INS-DRIP. Interestingly, a few T1D patients carrying the protective allele (C-H) demonstrated no autoreactivity to INS-DRIP unlike the carriers of the susceptible (R-P) version (36). Which insulin-related SNP is causal and whether the increased risk is a consequence of the expression of 3’SNPs in INS-DRIP or it reflects the 5’ INS promotor polymorphism remains unresolved, given the strong linkage disequilibrium between the 5’and 3’ regions of INS, and the exact underlying mechanism is currently under investigation.
Despite the critical role of CD8+ T cells, the contribution of HLA class I molecules to the disease propensity is less obvious and affected by the high linkage disequilibrium between HLA class I and II genes. For instance, 50-70% of T1D patients carry HLA-A2 (0201), which turns this HLA class I allele as the most frequent amongst patients; yet, this variant is also present in 30-40% of the general population, affecting the statistical significance. HLA-B*39 has been identified as single HLA class I allele standing out in its association with T1D, but this variant is relatively rare (58). In our view, this indicates a more important role of HLA class II and antigen presentation in establishing and control of the immune regulation than in the actual β-cell destruction.
For many risk genes variants, there is still insufficient understanding of whether and how they functionally impact the initiation and progression of the autoimmune process causing T1D. The functional outcomes of the coding T1D risk variants have been reviewed recently (59), and a fine mapping of the 10 known susceptibility regions combined with functional analyses provided further insight in potentially causal missense and non-coding SNP variants (60). Many of these risk genes were differentially expressed in dendritic cells upon tolerogenic modulation (17, 18). Hence, we here consider the functional roles in immune regulation of the minor T1D risk genes as such or when influenced by the SNP. We mainly focus on the genes for which functional data on human cells are available to allow a discussion on the consequences of the causal SNPs for the autoreactive T cell activation, Treg function or β-cell apoptosis that may support the autoimmune disease (Table 1 and Figure 1).
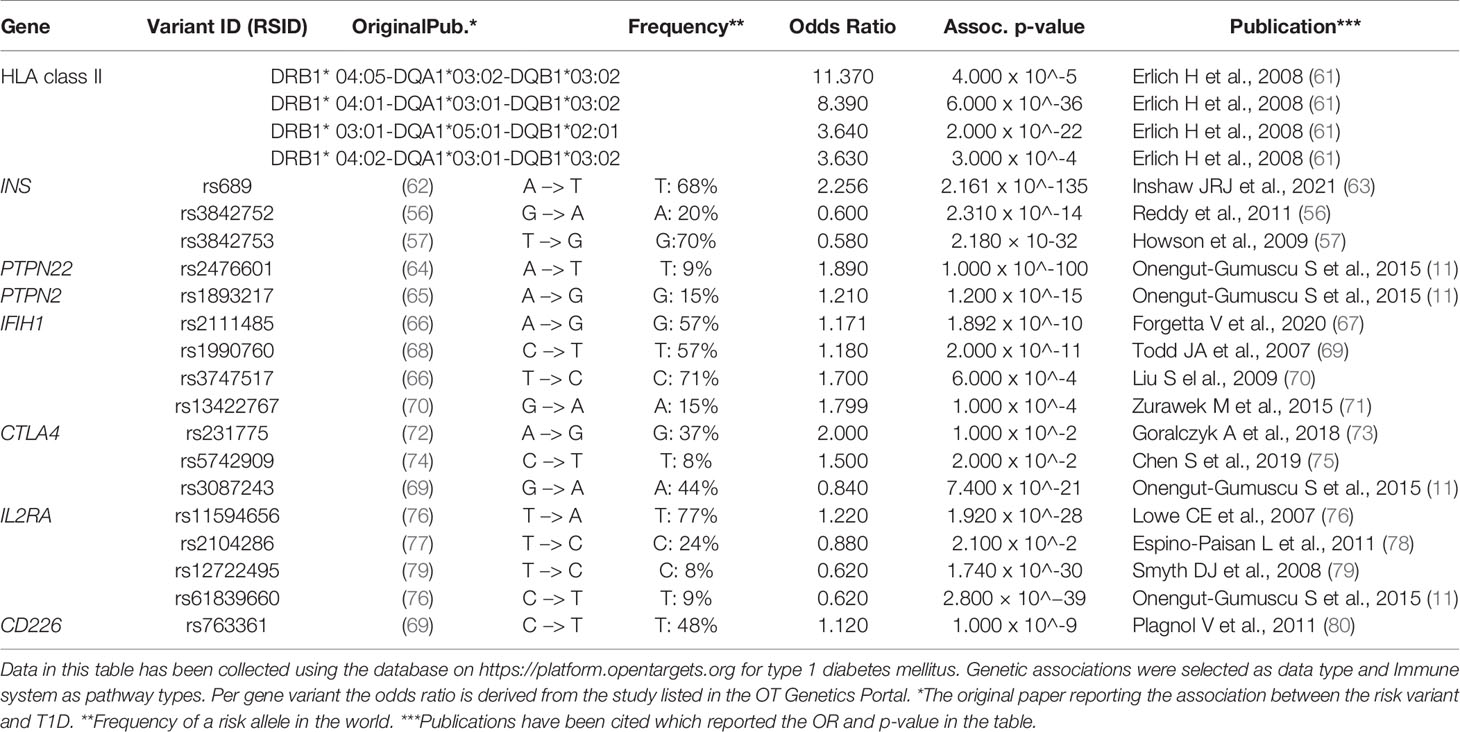
Table 1 Risk gene variants associated with T1D (discussed in this review). For each gene variant the variant ID, risk allele frequency and odd ratio are presented.
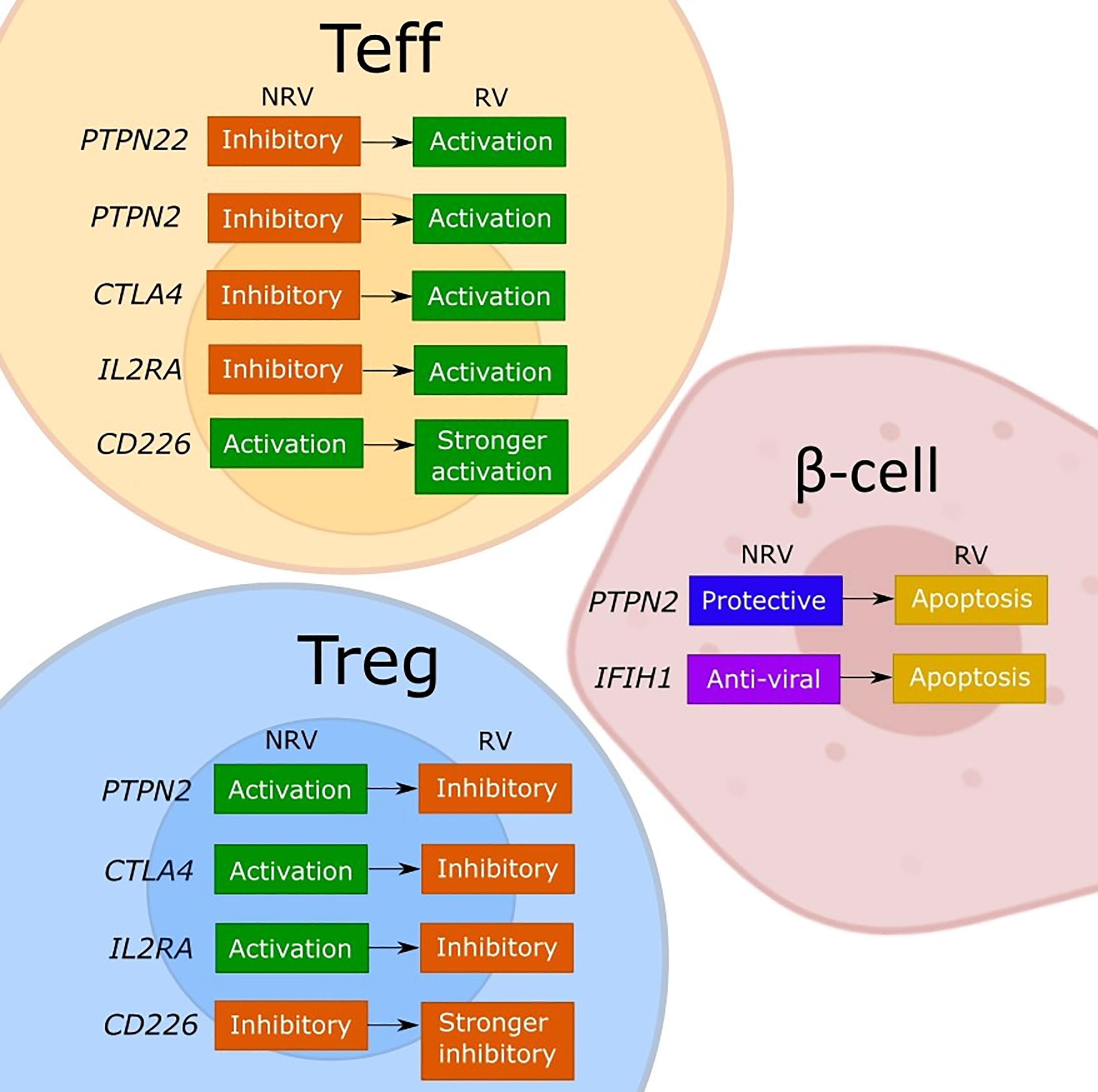
Figure 1 Model of the discussed effects of T1D risk variants on cellular functions. The figure depicts our interpretation of the consequences for effector T cell (Teff), regulatory T cell (Tregs), and β-cells of the described or assumed change in the gene function caused by a T1D risk variant (RV) as compared to the non-risk variant (NRV) SNP as discussed in the manuscript. While the LYP protein normally controls the effector T cells by a downstream signaling inhibition, the risk variant (rs2476601) induces a change in PTPN22 that promotes Teff responses. The functional effects of PTPN22 remain unclear. The PTPN2 protein plays an anti-apoptotic role in β-cells and controls T cells via IL-2, which may favor Tregs due to a strong sensitivity to IL-2. Indirectly, a good activity of Tregs keeps the effector T cells under control. The PTPN2 risk variant (rs1893217) causes a decrease in PTPN2 expression and contributes to the sensitivity of β-cells to immune- or virus-mediated apoptosis. The risk variant also reduces IL-2 receptor signaling, which decreases FOXP3+ Tregs in T1D patients, and thus dysregulating Treg function. The PTPN2 deficiency (mimicking the rs1893217 variant) results in increased Teff proliferation. The MDA5 (encoded by IFIH1) normally functions to activate stress- and anti-viral response, and by increasing the activity of MDA5, the risk variant (rs1990760) increases the basal IFN-I production leading to β-cell apoptosis. CTLA-4 functions normally to promote Treg function and inhibit Teff activation. The risk variant for CTLA4 (rs231775) results in decreased expression of CTLA-4 on T cells, releasing the control of a Teff cell activation and reducing the suppressive Treg potency. The IL2RA risk variants impair the expression of CD25 and thus the IL-2 response and with the associated lower FOXP3 expression impacts primarily Tregs and their suppressive function. The resulting reduced Treg potency will indirectly release the control on Teff promoting the activation. The CD226 is an activating T cell molecule that promotes the inflammatory activity of Teff and reduces the suppression of Tregs. The CD226 risk variant (rs763361) results in an isoform of CD226 with increased activity, which further increases Teff and CD226+ Tregs, thereby further reducing the overall suppressive capacity of Tregs.
Protein tyrosine phosphatase, non-receptor type 22 (PTPN22) encodes protein Lymphoid-tyrosine phosphatase (LYP) (81). The PTPN22 allele C1858T has a single amino acid substitution R620W (arginine to tryptophan; rs2476601, OR= 1.890), and has been associated with T1D, Hashimoto’s thyroiditis, rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), vitiligo and Graves disease (82). The linkage with several autoimmune diseases and the observation that individuals with this variant are protected from pulmonary tuberculosis or cancer (83, 84), suggests a role in promoting effector responses at cost of immune regulation (85–88). LYP protein inhibits T cell and B cell activation by dephosphorylation of tyrosine residues in Src family kinases. The interaction between C-terminal Src kinase (CSK) and the P1 motifs on LYP are important to regulate the inhibitory activity of LYP. Next to lymphocytes, LYP plays a role in the control of activation and migration of innate myeloid cells (monocytes, macrophages, DCs and neutrophils) (89–91).
The molecular consequences of the PTPN22 mutation and the impact on T1D risk have been discussed extensively before (92, 93). The debate regarding the impact of the T1D risk variant on T cells remains as the results support both gain-of-function and loss-of-function as a mechanism (94). Following the gain-of-function hypothesis, the R620W mutation blunts the TCR signaling allowing autoreactive thymocytes to escape selection (95). The same group reported later that R620W is located in the P1 motif and prevents the binding of LYP to CSK (96), directing towards a loss-of-function hypothesis that may affect TCR signaling and certainly applies for the regulation of LFA-1 signaling. A deletion of Ptpn22 in mice, mimicking the loss-of-function, demonstrated increased Treg levels (97) which is in line with a study that shows a direct correlation between having the PTPN22 R620W variant and elevated circulating Treg frequency in humans (98). Recently, Perry et al. showed a higher expression of PTPN22 in Tregs than in conventional T cells (Tconv) at rest irrespective of the genotype, but a lower impact of the risk variant on the suppression of distal TCR signaling in both subsets and permitting a stronger proliferation of Tconvs. The consequences for Tregs in this study were less evident implying a differential contribution of PTPN22 risk variant to Tconv and Treg (99).
In myeloid cells, PTPN22 is involved in the downstream signaling of TLR4, TLR7/8, NOD2 and cytokine receptors (reviewed in (92)). In this case, LYP does not work as a phosphatase but promotes TRAF3 ubiquitination and TLR-induced upregulation of type I interferons (IFNs). The PTPN22 R620W variant failed to support this type I IFN response (100). Additionally, antigen presenting cells with the PTPN22 R620W variant are more sensitive to NLRP3 and secrete more IL-1b in response to TLR-stimulation (101). Combined with the dampened type I IFN signal, this could affect the response of myeloid cells to infections (102) and the subsequent activation of T cells.
Protein Tyrosine Phosphatase, Non-Receptor Type 2 (PTPN2) is ubiquitously expressed, including β-cells and hematopoietic cells. PTPN2 takes part in a broad range of signaling pathways regulating the response to hormones, cytokines and inflammation (93, 103–105). The β-cells upregulate PTPN2 in response to cytokines or polyI:C (mimicking viral infection) (106, 107). Judging by the effects of knockdown in primary rats and human β-cells, which exacerbated cytokine induced pro-apoptotic signaling via STAT1, JNK1, and BIM and enhanced apoptosis, PTPN2 plays a protective and anti-apoptotic role in β-cells (106–108). The risk SNP rs1893217 (OR=1.210) is an intronic non-coding variant which may contribute to the sensitivity of β-cells to immune- or virus-mediated apoptosis (107).
The risk variant of PTPN2 is associated with decreased PTPN2 expression in CD4+ memory T cells and reduced IL-2 receptor signaling via STAT5 phosphorylation, which correlated with reduced FOXP3 expression in Tregs (109) suggesting that PTPN2 indirectly modulates IL-2 responsiveness in T cells and thus can work independent of the susceptible IL2RA gene variant. This dysregulation of Tregs can contribute to the faulty maintenance of autoreactive T cells and B cells and thus sustain the vicious circle of uncontrolled autoimmune response (93). Indeed, antigen-specific effector T cells (Th1 and Tfh) in Ptpn2 deficient mice show increased proliferation (110). Cell cultures of human myeloid cells showed that a loss of PTPN2 enhances IFN-g, IL-6 and MCP-1 secretion (103), implicating PTPN2 in the regulation of inflammation through antigen presenting cells as well.
Interferon Induced with Helicase C Domain 1 (IFIH1) encodes for melanoma differentiation-associated gene 5 (MDA5). MDA5 is a cytoplasmic receptor for double stranded RNA (dsRNA) and detects viral RNA (106, 111, 112). Detection of dsRNA will activate a cascade of antiviral responses in the innate immune system by the production of IFN (113, 114). There are four SNPs in the IFIH1 gene (rs1990760, OR=1.180; rs3747517, OR=1.700; rs2111485, OR=1.171; and rs13422767, OR=1.799) which are associated with T1D (70, 71). Variants rs2111485 and rs13422767 are located in an intergenic region of the 2q24 locus (13–23 kb 3′ of IFIH1), but it is not known whether the DNA sequences in this region act as a transcriptional silencer or enhancer. Winkler et al. showed that children at risk and islet-autoantibody positive with the rs2111485 variant genotype progressed faster to T1D (115). The contribution of other SNPs in the disease progression was not validated in this study. Variants rs1990760 and rs3747517 are located within the binding site of transcription factors and could therefore influence the expression of IFIH1 (116).
Human PBMCs and cell lines with the IFIH1 rs1990760 variant (coding an amino acid substitution A946T) have heightened basal and ligand-triggered IFN-I production (117). This SNP was thus characterized as a gain-of-function variant with a capacity to protect the carriers against specific viral challenges while promoting the risk for autoimmune diseases. This confirmed a hypothesis based on the results from previous studies in healthy individuals carrying the rs1990760 variant and animal models (118, 119), that this variant enlarges the risk for autoimmune disease by increasing the basal activity of IFN-stimulated genes through the recognition of self-dsRNAs without the need for a concomitant viral challenge.
MDA5 activation in DCs mediates cell maturation, increasing antigen processing and presentation through the expression of MHC class I chemokine receptors and co-stimulatory molecules (120), thus promoting the activation and expansion of inflammatory T cells (119). Hence, a heightened MDA5 activation can support the induction of autoimmunity via agitated DCs presenting the islet antigens to T cells in a pro-inflammatory rather than an anti-inflammatory context.
Next to the viral or cytoplasmic dsRNA, mitochondrial dsRNA released after β-cell stress could trigger the production of proinflammatory cytokines in individuals carrying the IFIH1 risk variants (70, 121). Namely, the normal processing of the transcribed mitochondrial genome increases under stress causing a leakage of the mitochondrial dsRNA remnants into the cytosol (121, 122), where MDA5 recognizes these as damage-associated molecular patterns (DAMP). Hence, metabolic stress in β-cells that causes mitochondrial dysfunction might also contribute to the heightened IFN response and apoptosis of β-cells (123).
The Cytotoxic T-Lymphocyte Associated Protein 4 (CTLA4) genes encodes a transmembrane co-receptor expressed on the surface of T cells. CTLA-4 functions as a negative regulator of T cell activation which can mediate T cell regulation or apoptosis by interacting with B7, a co-stimulatory molecule present on antigen presenting cells (124–127).
Genetic studies on CTLA4 in T1D have been focusing on three gene variants: the A49G SNP (rs231775, OR=2.000) in exon 1, the SNP rs3087243 (OR=0.840) which is in high linkage disequilibrium with the dinucleotide (ATn) repeat in the 3’- untranslated regions (UTR) and the coding C318T SNP (rs5742909, OR=1.500) in the CTLA4 promotor (128).
The first SNP rs231775 is in exon 1 at position 49 from A to G (A49G) of the CTLA4 gene (129, 130). Meta-analysis of 76 studies showed that the rs231775 variant is more prevalent in T1D patients with Caucasian and South Asian origin and is associated with Type 2 Diabetes (T2D) in East Asians and South Asians (75). The A49G SNP causes the amino acid replacement of threonine to alanine and influences the posttranslational modification of CTLA-4. These modifications result in an inefficient CTLA-4 glycosylation and decreased expression of CTLA-4 on T cells, leading to uncontrolled T cell activation, including the autoreactive T cells (75, 131). The rs231775 variant was also associated with reduced production of soluble CTLA-4 (sCTLA-4), which can inhibit T cell proliferation by binding/blocking B7 (132). This has been confirmed in Ctla4 KO NOD mice (133), in which the posttranscriptional silencing of sCTLA-4 reduced Treg potency and accelerated T1D onset. Interestingly, sCTLA4 suppressed proliferation of committed islet autoreactive T cell clones isolated from the blood of T1D patients in a dose-dependent manner, but it was unable to suppress naïve alloreactive T cells in an MLR (134), indicating a differential role for sCTLA4 in the control of memory versus primary immune responses.
The second SNP rs3087243 affects the size of dinucleotide (AT)n repeats in the 3’-UTR and the CTLA4 mRNA stability through a post-transcriptional control (135), influencing the rate of translation (38, 136, 137). De Jong et al. showed that autoreactive T cells with long variants of the (AT)n repeat in the 3’-UTR region have reduced CTLA4 mRNA levels (138), thus variations in the length of (AT)n repeats influence CTLA4 expression contributing to the development of T1D. Also a rare genetic variation (rs13384548) within the 3’-UTR of the CTLA4 mRNA disrupted the miR-302a* binding site reducing the capacity to control CTLA4 mRNA (139).
The SNP rs5742909 in the CTLA4 promotor region cause a C to T mutation at position 318. Individuals carrying the minor 318T allele have a higher promotor activity than the 318C allele, resulting in an increased expression of CTLA-4 by T cells (140). While this suggests that the C to T transition increases a regulatory function, the consequences for the T cell response and the effect of this gene variant on the development of T1D is not clear yet.
The protein IL-2Rα (CD25) is a high-affinity subunit of the IL-2 receptor that forms a complex with IL-2Rβ- and γ-chain to activate intracellular signaling upon interaction with IL-2 (141). IL-2RA is constitutively expressed on Tregs and can be induced upon activation in other (effector) T cells (142). Polymorphisms in the genes encoding for the IL-2 receptor, IL2RA (rs2104286, rs61839660, rs10795791, and rs41295121) and IL2RB (rs743777), are associated with T1D (69, 76, 143, 144). DNA methylation at CpGs (−373 and −456) within the promotor of the IL2RA gene was slightly higher in T1D patients than in controls (142), indicating that epigenetic changes in the IL2RA promotor might participate in the IL2RA risk allele for T1D. Indeed, methylation at CpG-373 was correlated with 16 SNPs in the IL2RA gene, both with the protective alleles (rs12722495, rs2104286, rs61839660) and the susceptible allele at rs11594656 (Table 1) (11, 69, 145, 146).
Regarding the functional consequences for T cells, Dendrou et al. showed that individuals with the SNP rs12722495 (OR=0.620) had a higher CD25 expression on CD4+ memory T cells, while the carriers of the SNP rs2104286 (OR=0.880) showed a lower CD25 expression on naïve CD4+ T cells, compared to the non-carriers (147). They further demonstrated that individuals with the protective variant (rs12722495) consistently had higher proportion of activated IL-2 producing CD69+ CD4+ memory T cells compared to individuals with a susceptible allele, supporting the hypothesis that cells with a higher surface CD25 are more responsive to IL-2R mediated activation (147). This is consistent with the earlier observed defects in IL-2 production in T1D patients (148, 149). Cerosaletti et al. challenged the view that expression levels of CD25 functionally contribute to the susceptibility and showed a reduced signaling from IL-2R (measured by a phosphorylation of STAT5) in CD4+ CD25hi T cells of T1D patients and healthy individuals carrying the rs2104286 risk haplotype (150). The unexpected higher expression of CD25 on naive Tregs in T1D patients and healthy controls with the rs2104286 risk haplotype compared to the carriers of the protective variant, was not explained in this study. The rs2104286 haplotype also correlated with increased soluble IL-2RA levels, suggesting that shedding of the IL-2RA may account for the reduced IL-2R signaling in these individuals. Alternative hypothesis explaining the protective effect of the SNP rs12722495 and the contribution of polymorphisms in IL-2R-pathway in general was through the effects on nTregs (151). Given their constitutive expression of CD25 and a strong sensitivity to IL-2, lower IL-2 signaling measured by the STAT5 phosphorylation reduces the IL-2 response, impacting the FOXP3 expression and thereby affecting the inhibitory function of Tregs (151, 152).
The SNP rs61839660 (OR=0.620) is located within the IL2RA gene and it is a non-coding causal SNP variant for T1D (11, 60). This SNP is co-inherited with a so-called group-A protective T1D haplotype that also includes the rs12722495 (153). Interestingly, a rare variation in the group-A haplotype causing the loss of the protective allele only at SNP rs61839660 was sufficient to counteract the high IL2RA mRNA and surface CD25 expression (153). The mechanistic studies revealed that the minor SNP variant reduces the IL2RA enhancer activity (154, 155), which is stimulation-responsive causing a delay in CD25 expression upon T cell activation, and that a deletion of this enhancer diverted the effective Treg polarization in mice (155).
Monocytes-derived and myeloid DCs express CD25 both as a surface-bound and soluble molecule when stimulated with prostaglandin E2 (PGE2) (156). Also, tumor-associated DCs co-express CD25 and the inhibitory molecule IDO (156). In our hands, tolDC express lower IL2RA mRNA and lack the surface-bound CD25 compared to mDCs (17). We did not measure whether tolDC also release less soluble CD25. Taken together, the surface-bound CD25 may enable mDCs to catch IL-2 and use it to stimulate T cells, while the soluble CD25 molecule could work to block IL-2 and help the regulation of T cell responses (157). The contribution of IL2RA risk variants to the DC function has not been investigated. As the effects described so far in T cells predominantly impact the downstream IL-2R signaling and DCs do not express other two proteins of the IL-2R complex, the functional contribution of genetic polymorphisms in IL2RA is more likely to show through the surface expression or production of soluble IL-2RA than to impact DC differentiation.
CD226 or DNAX-accessory molecule-1 (DNAM-1) is a transmembrane receptor expressed on T cells, NK cells, NKT cells, platelets and a subset of B cells (69, 158), and aids their activation and differentiation through co-stimulation (159). The inhibitory counterpart of CD226 is T cell Immunoreceptor with Ig and ITIM domains (TIGIT), which is a negative regulator molecule expressed in Tregs and NK cells (160). TIGIT binds CD155 on DCs, driving them towards a tolerogenic phenotype. Disturbance of the TIGIT/CD226 axis could therefore contribute to the development of autoimmunity (161).
The SNP rs763361 (Gly307Ser, OR=1.120) in the CD226 gene is associated with multiple autoimmune diseases, such as T1D, multiple sclerosis (MS), autoimmune thyroid disease, RA, SLE and systemic sclerosis (162). This SNP results in a missense mutation at position 307 (glycine to serine) and is located in two intracellular phosphorylation sites of the protein (residue 322 and 329) (159, 163). The SNP rs763361 may alter RNA splicing by disrupting splice site enhancers or silencers, resulting in an isoform of CD226 with altered function (69, 159, 163) and increased CD226 activity in T cells (164).
Indeed, Gaud et al. showed that in vitro anti-CD226 and anti-CD3 co-activation of human primary CD4+ T cells of individuals carrying the rs763361 risk variant induces enhanced p-ERK (164). The ERK pathway regulates T cell activation and differentiation. The rs763361 variant is associated with skewing to Th17 and Th17.1 cells after stimulation in vitro (164). Indeed, T1D patients carrying the rs763361 risk variant had greater frequency of GAD antibody and low C-peptide levels, reflecting a more aggressive disease pattern in a Brazilian population (165). Wallace et al. observed that the rs763361 risk variant correlated with reduced CD226 mRNA levels in monocytes and which could reduce cell activation and thus alter the interactions between monocytes and lymphocytes (166). When Cd226 was deleted in NOD mice, this decreased disease incidence and insulitis as compared to WT mice (167), but the deletion also increased the number of CD8+ thymocytes and splenocytes. The CD226 deficient CD8+ T cells showed decreased reactivity to the β-cell specific antigen IGRP, from which Shapiro et al. concluded that CD226 plays a role in the development of T1D by modulating thymic selection and affecting activation of CD8+ T cells (167). The effect of the rs763361 risk variant has not been studied in human CD8+ T cells or Tregs. The majority of human Tregs highly express TIGIT, but a Treg subset co-expresses CD226 (168). These CD226+ Tregs were associated with reduced suppressive capacity. Hypothetically, the rs763361 variant, which increases CD226 activity in T cells, will increase the proportion of CD226+ Tregs and thereby reduce the overall suppressive capacity of Tregs. Studying further the expression and function of CD226 in humans is needed for a better understanding of whether the rs763361 risk variant contributes through T cell activation only or also by affecting the interaction between monocytes and lymphocytes.
Gene expression can be changed by genetic engineering or using bioactive small molecules, for which aim the specific targeting of the scarce autoreactive T cells seems difficult. The targeting through DCs seems more viable and allows also antigen-specific immune modulation (169). The active form of VitD3 functions as a transcription factor upon binding to the vitamin D receptor (VDR) (170), creating a complex that binds with retinoid-X receptor (RXR) to enable the attachment to vitamin D response elements (VDRE) (171, 172). The VDR complex has a large effect on more than 3000 target genes, which includes forty-seven transcription factors and thus leaving hardly any immune pathway unaffected by VitD3 (173). This natural immunomodulator influences the development and function of T cells, B cells and monocytes (172, 174, 175), and controls the ability of the immune system to dampen inflammation. In two independent studies we found that about a third of the transcripts encoded by non-HLA T1D risk genes were differentially expressed between inflammatory mDCs and VitD3-derived tolDCs (17, 18). Interestingly, only five of these genes were also reported as direct targets of VDR (170), leaving others to an indirect control by VDR-targeted transcription factors. Of the direct VitD3-targets, the expression of ORMDL3, SH2B3, IKZF1, PTPN2 and IFIH1 genes was lower in tolDC while RAC2 and PTPN22 were higher in tolDC than in the inflammatory mDCs (17, 18).
The protein encoded by ORMDL3 is an enzyme involved in sphingolipid synthesis and lipid metabolism without a clear function in the immune response but interestingly the T1D patients who were the carriers of the linked polymorphism (rs12150079) showed a lower intensity of autoreactive T cell responses in T1D (11, 60, 176). The SH2B3 encodes LNK (lymphocyte adaptor protein) that takes parts in several signaling pathways controlling the hematopoiesis, cytokine and integrin signaling and cell migration (177). The functional consequences of the T1D risk variant (rs3184504) that causes a missense mutation remain speculative (11, 59, 146), one study using human cells that reports an augmented lymphocyte proliferation that correlates with the predisposing gene variant (178) Interestingly though, a recent study shows that the T1D risk-gene variant associates with a reduced mortality from sepsis in individuals with a European decent and suggest based on a mouse model that augmented phagocytosis and myelopoiesis may be underlying mechanisms (179). The gene IKZF1 codes for the transcription factor Ikaros (180), and the associated SNPs (rs10277986, rs62447205) are protective for T1D (11). How these SNPs affect the expression or function of Ikaros has not been described. Ikaros is a regulator of dendritic cell differentiation and immune homeostasis, and IKZF1 deficiency causes less inflammatory cytokines secrection by human monocytes (181), which is in line with the observed lower expression in our tolDCs. Finally, RAC2 encodes a protein from a Rho family of GTPases involved in cytoskeletal reorganization (e.g. needed for phagocytosis) but the effect of the described SNP variant (rs229533) increasing the risk for T1D is still unknown (11).
In our view, the polymorphisms in immune genes as are discussed in this review can influence both immune activation and regulation through a change in gene expression or in function (Figure 1). The consequences may differ between the cell types depending on the expression level or an implicated cellular function of a given gene. Indeed, by changing the expression of a target protein, some risk variants cause different functional effects in conventional T cells, Tregs or β-cells, depending on the implicated cellular function (Figure 1). For most of the evaluated genes, both the non-risk variant and the risk variant show opposing functional consequences in conventional T cells compared to Tregs. Namely, the risk genes for which a non-risk variant supports immune regulation (PTPN2, CTLA4 and IL2RA) are indeed activating for Tregs and work to suppress the effector T cells. The risk-variant SNPs of these genes change the function in the same manner irrespective of the cell type but the end result differs so the lower expression and signaling through PTPN2, CTLA-4 or IL-2RA will simultaneously impair the function of Tregs and release the tight control of the effector T cell. Similarly, the activation-promoting function of CD226 in effector T cells, enhanced by the risk-SNP, suppressed the inhibitory function of Tregs. Further, based on the regular function of LYP (PTPN22) to control the post-TCR signaling events, the activating contribution of the risk mutation in Teff is evident but the consequences for the human Tregs remain to be confirmed. The risk-SNP induced modulation of PTPN2 and IFIH1 will increase β-cell apoptosis, which increases the antigen release, and thus contribute to the development of T1D. In DCs the PTPN22 risk variant fails to promote upregulation of type I IFN which might result in diminished human host-protecting responses when dealing with viral infections. The PTPN2 risk variant may dysregulates the production of inflammatory cytokines and thus the maintenance of immune tolerance by DCs. The IFIH1 risk variants causes an increased IFN response, stimulating antigen presentation, while the IL2RA risk variant may inhibit the capacity of DCs to suppress T cell proliferation and cytokine production. In summary, the functional consequences of the causal T1D-risk variants have been extensively investigated and seem to paint a clear picture regarding the individual contributions but it is difficult from this information to consider how the polymorphisms may cumulatively modify the cell interactions that promote the impaired β-cell specific immune responses in individuals with high T1D risk-scores.
At the moment, no therapies have been developed that target PTPN22, PTPN2, IFIH1 and CD226. Therapies targeting CTLA4 (CTLA-4-Ig, abatacept) has been proven as safe and efficient to inhibit naïve T cell activation and therefore this approach is more selective namely inhibiting only T cell responses as compared to general/broad immunosuppression. The safety of abatacept as a subcutaneous (SC) and intravenous (IV) therapy is evaluated in RA (182, 183) and T1D patients, reporting no therapy-specific adverse effects (184, 185). Abatacept treatment showed good efficacy in prevention complete loss of β-cell function in T1D patients as is shown by preservation of C-peptide levels and insulin sensitivity improvement (184, 186, 187).
To compensate for the impaired IL-2 signaling due to lower expression or dysfunction of the IL2R gene, specifically in Tregs, ultra-low-dose IL-2 therapy has been tested, thus avoiding potential toxic effects of systemic IL-2. A phase II study has already been completed to determine the optimal IL-2 dose to use as a treatment in T1D patients (188). Participants did not exhibit severe adverse effects, a minimal NK cell expansion was observed after IL-2 treatment and no detrimental changes in glucose metabolism were observed, guaranteeing the safe use of IL-2 (188). Also, there was a dose-dependent increase in Tregs in all patients, and the low dose of IL-2 upregulated CD25 and FOXP3 expression on Tregs while CD4+ T effector memory cells were unchanged (189). Even though the latter trial showed some interesting effects of IL-2 therapy, the insulin secretion was not measured so the clinical efficacy of this therapy (i.e. on β-cell preservations) could not be determined. A newer alternative to avoid the influence on effector T cells uses the IL-2 mutant proteins (190), which has advanced to the clinical testing in GvHD (ClinicalTrials.gov Identifier: NCT03422627), though it remains a systemic antigen-independent approach.
Even though these therapies targeting CTLA4 and IL2RA seem promising, it remains to be seen whether they are beneficial for all patients or only those carrying the affected variant, and to what extent such therapies may influence the immune system and health in general given the generic and pleiotropic effects of IL-2. It is tempting to investigate whether therapies targeting specific pathways in which a risk gene of interest is involved or epigenetic targeting of a single of multiple risk gene variants will be more beneficial.
A treatment that simultaneously tackles multiple changes in the risk genes allowing a correction towards the non-risk variant function could potentially aid as curative intervention. The natural immunomodulator VitD3 seems a good candidate since it reduced the expression of IKZF1, PTPN2, IL2RA, CD226 and IFIH1 while increased RAC2, and PTPN22 in tolDC as compared to mDC. Considering the effects of the discussed risk-SNPs in these genes, the modulating action of VitD3 could counteract the immune-activating effects of risk-mutations in IL2RA, CD226, IFIH1 and PTPN22 while supporting the protective effects of IKZF1.
The potential clinical benefit of the treatment with Vitamin D, has been recognized earlier. The initial trial with VitD3-modulated tolDC in T1D patients confirmed safety and the clinical benefit of the treatment remains to be tested (191). VitD3 modulates T cells (172, 175, 192, 193) and a trial testing the combined treatment of T1D patients with VitD3 and GAD antigen did not show significant change overall but a particular β-cell preservation in individuals with the HLA DR3-DQ2 haplotype (194). Trials testing vitamin D supplementation (195–200), showed some clinical benefit such as improving diabetes control (HbA1c or insulin dose), reducing complications (195, 196), some indications of β-cell protection or immune regulation (197, 198), none of the study monitored the clinical and immunological effect simultaneously. Finally, VitD3 can contribute to T1D prevention since early postnatal VitD3 administration seems to protect from T1D (199), even though reduced circulating VitD3 levels do not increase T1D risk (200).
Understanding the role of genetic risk-variants in the T1D pathogenesis can have important implications for better understanding disease pathogenesis and heterogeneity, as well as the development of specific/selective disease intervention strategies. Models have been generated suggesting that different T1D risk-loci contribute to successive pathogenic checkpoints, which detection could allow timely and appropriate modulation of the autoimmunity and increase the chance for curative interventions. The mechanisms involved in the immunotherapy of cancer (201, 202), teach us about genetic variants that increase the risk for the development of autoimmune disease but positively impact the survival after cancer treatment (203). Hence, polygenic risk scores (204) not only help to predict disease but also to predict when a specific patient is more or less likely to respond to immunotherapy directed at the involved pathways.
The genetic risk score could allow early identification of individuals who will develop T1D allowing earlier curative interventions. Butty et al. studied the frequency of non-HLA risk alleles among individuals at risk of developing T1D (DTP-1 trial), of which about one-third progressed to the clinical disease (205). They concluded that immune risk gene variants more likely condition the initial development of autoimmunity, resulting in a detectable auto-Ab response, but less critically contribute to the events leading to disease onset (205). Hence immune modulation therapy makes more sense prior to the onset of autoimmunity, which will be possible when a prediction of T1D is improved. The recently reported improved cumulative risk score (T1D-GRS2) that includes 67 SNPs (all HLA-DQ haplotypes, non-DR-DQ loci within the HLA region and non-HLA loci) indeed enabled a sensitive discrimination of T1D from T2D and controls (13), but also improved the prediction of future T1D in infants. Still, around 10% of all infants would have to be monitored to capture 77% of future T1D cases. In this study it remains unclear whether successive application of the HLA-score, followed by a non-HLA score would have further improved the prediction sensitivity. Importantly, this GRS failed to predict T1D in patients with different ethnicities, underscoring the need to study all-inclusive cohorts (206).
Alternatively, fine mapping genetic studies of previously known autoimmune loci will also help to find relevant genetic variants with strong effect on the development of T1D (11, 60, 207). The availability of large human whole-genome sequencing data sets, also allows detecting rare SNPs with large effect size on complex traits (208, 209). Forgetta et al. recently discovered three novel risk gene variants in large human whole-genome sequencing data sets of T1D patients (67). Hence, studying the human whole-genome sequencing data might lead to the discovery of gene variants, which will give a better understanding of the genetics behind the development of T1D and possibly predict therapy responses.
Taken together, current literature only partially explains the functional implications of the risk-SNPs to the development of autoimmunity in T1D. The efficient in-depth analyses of the immune response that can detect and monitor low-frequent autoantigen-specific cells and a better understanding of immune tolerance are needed to investigate and understand the functional contributions of genetic polymorphisms in different cells of the immune system. The same polymorphism can have opposing functional consequences depending on the cell in which the linked gene is expressed. Gaining insight into how the human genetics impacts functional immunity is therefore important to allow discrimination of relevant and treatable targets and for selecting proper immunotherapy strategies with the most benefit for patients or individuals at risk of developing T1D.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
CG is supported by the Stichting DON and Dutch Diabetes Research Foundation (grant number 2020.10.011). TN is supported by Innovative Medicine Initiative 2 Joint Undertaking under grant agreement No 115797 (INNODIA), which receives support from the European Union’s Horizon 2020 research and innovation programme and EFPIA, JDRF and The Leona M. and Harry B Helmsley Charitable Trust. JZ is supported by the Dutch Arthritis Foundation (grant number LLP-16) and BR is supported by the Wanek Family Project for Type 1 Diabetes.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. VanBuecken D, Lord S, Greenbaum CJ, Feingold KR, Anawelt B, Boyce A, et al. Changing the Course of Disease in Type 1 Diabetes. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, editors. South Dartmouth (MA): MDText.com, Inc. (2000).
2. Richardson SJ, Willcox A, Bone AJ, Morgan NG, Foulis AK. Immunopathology of the Human Pancreas in Type-I Diabetes. Semin Immunopathol (2011) 33(1):9–21. doi: 10.1007/s00281-010-0205-0
3. Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TW, Atkinson MA, et al. Demonstration of Islet-Autoreactive CD8 T Cells in Insulitic Lesions From Recent Onset and Long-Term Type 1 Diabetes Patients. J Exp Med (2012) 209(1):51–60. doi: 10.1084/jem.20111187
4. Anderson AM, Landry LG, Alkanani AA, Pyle L, Powers AC, Atkinson MA, et al. Human Islet T Cells are Highly Reactive to Preproinsulin in Type 1 Diabetes. Proc Natl Acad Sci USA (2021) 118(41):e2107208118. doi: 10.1073/pnas.2107208118
5. Babon JA, DeNicola ME, Blodgett DM, Crevecoeur I, Buttrick TS, Maehr R, et al. Analysis of Self-Antigen Specificity of Islet-Infiltrating T Cells From Human Donors With Type 1 Diabetes. Nat Med (2016) 22(12):1482–7. doi: 10.1038/nm.4203
6. Roep BO, Wheeler DCS, Peakman M. Antigen-Based Immune Modulation Therapy for Type 1 Diabetes: The Era of Precision Medicine. Lancet Diabetes Endocrinol (2019) 7(1):65–74. doi: 10.1016/S2213-8587(18)30109-8
7. Knip M, Veijola R, Virtanen SM, Hyoty H, Vaarala O, Akerblom HK. Environmental Triggers and Determinants of Type 1 Diabetes. Diabetes (2005) 54 Suppl 2:S125–36. doi: 10.2337/diabetes.54.suppl_2.S125
8. Gregersen PK, Olsson LM. Recent Advances in the Genetics of Autoimmune Disease. Annu Rev Immunol (2009) 27:363–91. doi: 10.1146/annurev.immunol.021908.132653
9. Cudworth AG, Woodrow JC. HL-a System and Diabetes Mellitus. Diabetes (1975) 24(4):345–9. doi: 10.2337/diab.24.4.345
10. Nerup J, Platz P, Andersen OO, Christy M, Lyngsoe J, Poulsen JE, et al. HL-a Antigens and Diabetes Mellitus. Lancet (1974) 2(7885):864–6. doi: 10.1016/S0140-6736(74)91201-X
11. Onengut-Gumuscu S, Chen WM, Burren O, Cooper NJ, Quinlan AR, Mychaleckyj JC, et al. Fine Mapping of Type 1 Diabetes Susceptibility Loci and Evidence for Colocalization of Causal Variants With Lymphoid Gene Enhancers. Nat Genet (2015) 47(4):381–6. doi: 10.1038/ng.3245
12. Harrison JW, Tallapragada DSP, Baptist A, Sharp SA, Bhaskar S, Jog KS, et al. Type 1 Diabetes Genetic Risk Score is Discriminative of Diabetes in non-Europeans: Evidence From a Study in India. Sci Rep (2020) 10(1):9450. doi: 10.1038/s41598-020-65317-1
13. Sharp SA, Rich SS, Wood AR, Jones SE, Beaumont RN, Harrison JW, et al. Development and Standardization of an Improved Type 1 Diabetes Genetic Risk Score for Use in Newborn Screening and Incident Diagnosis. Diabetes Care (2019) 42(2):200–7. doi: 10.2337/dc18-1785
14. Patel KA, Oram RA, Flanagan SE, De Franco E, Colclough K, Shepherd M, et al. Type 1 Diabetes Genetic Risk Score: A Novel Tool to Discriminate Monogenic and Type 1 Diabetes. Diabetes (2016) 65(7):2094–9. doi: 10.2337/db15-1690
15. Claessens LA, Wesselius J, van Lummel M, Laban S, Mulder F, Mul D, et al. Clinical and Genetic Correlates of Islet-Autoimmune Signatures in Juvenile-Onset Type 1 Diabetes. Diabetologia (2020) 63(2):351–61. doi: 10.1007/s00125-019-05032-3
16. Braenne I, Onengut-Gumuscu S, Chen R, Manichaikul AW, Rich SS, Chen WM, et al. Dynamic Changes in Immune Gene Co-Expression Networks Predict Development of Type 1 Diabetes. Sci Rep (2021) 11(1):22651. doi: 10.1038/s41598-021-01840-z
17. Nikolic T, Woittiez NJC, van der Slik A, Laban S, Joosten A, Gysemans C, et al. Differential Transcriptome of Tolerogenic Versus Inflammatory Dendritic Cells Points to Modulated T1D Genetic Risk and Enriched Immune Regulation. Genes Immun (2017) 18(3):176–83. doi: 10.1038/gene.2017.18
18. van Megen KM, Chen Z, Joosten AM, Laban S, Zwaginga JJ, Natarajan R, et al. 1,25-Dihydroxyvitamin D3 Induces Stable and Reproducible Therapeutic Tolerogenic Dendritic Cells With Specific Epigenetic Modifications. Cytotherapy (2021) 23(3):242–55. doi: 10.1016/j.jcyt.2020.12.003
19. Burrack AL, Martinov T, Fife BT. T Cell-Mediated Beta Cell Destruction: Autoimmunity and Alloimmunity in the Context of Type 1 Diabetes. Front Endocrinol (Lausanne) (2017) 8:343. doi: 10.3389/fendo.2017.00343
20. Op de Beeck A, Eizirik DL. Viral Infections in Type 1 Diabetes Mellitus–Why the Beta Cells? Nat Rev Endocrinol (2016) 12(5):263–73. doi: 10.1038/nrendo.2016.30
21. Piganelli JD, Mamula MJ, James EA. The Role of Beta Cell Stress and Neo-Epitopes in the Immunopathology of Type 1 Diabetes. Front Endocrinol (Lausanne) (2020) 11:624590. doi: 10.3389/fendo.2020.624590
22. Roep BO, Thomaidou S, van Tienhoven R, Zaldumbide A. Type 1 Diabetes Mellitus as a Disease of the Beta-Cell (do Not Blame the Immune System?). Nat Rev Endocrinol (2021) 17(3):150–61. doi: 10.1038/s41574-020-00443-4
23. Calderon B, Suri A, Unanue ER. In CD4+ T-Cell-Induced Diabetes, Macrophages are the Final Effector Cells That Mediate Islet Beta-Cell Killing: Studies From an Acute Model. Am J Pathol (2006) 169(6):2137–47. doi: 10.2353/ajpath.2006.060539
24. Richardson SJ, Pugliese A. 100 Years of Insulin: Pancreas Pathology in Type 1 Diabetes: An Evolving Story. J Endocrinol (2021) 252(2):R41–57. doi: 10.1530/JOE-21-0358
25. Rodriguez-Calvo T, Richardson SJ, Pugliese A. Pancreas Pathology During the Natural History of Type 1 Diabetes. Curr Diabetes Rep (2018) 18(11):124. doi: 10.1007/s11892-018-1084-3
26. Laban S, Suwandi JS, van Unen V, Pool J, Wesselius J, Hollt T, et al. Heterogeneity of Circulating CD8 T-Cells Specific to Islet, Neo-Antigen and Virus in Patients With Type 1 Diabetes Mellitus. PloS One (2018) 13(8):e0200818. doi: 10.1371/journal.pone.0200818
27. Bloem SJ, Roep BO. The Elusive Role of B Lymphocytes and Islet Autoantibodies in (Human) Type 1 Diabetes. Diabetologia (2017) 60(7):1185–9. doi: 10.1007/s00125-017-4284-5
28. Martin S, Wolf-Eichbaum D, Duinkerken G, Scherbaum WA, Kolb H, Noordzij JG, et al. Development of Type 1 Diabetes Despite Severe Hereditary B-cell Deficiency. N Engl J Med (2001) 345(14):1036–40. doi: 10.1056/NEJMoa010465
29. Katsarou A, Gudbjornsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, et al. Type 1 Diabetes Mellitus. Nat Rev Dis Primers (2017) 3:17016. doi: 10.1038/nrdp.2017.16
30. Lampasona V, Liberati D. Islet Autoantibodies. Curr Diabetes Rep (2016) 16(6):53. doi: 10.1007/s11892-016-0738-2
31. Rodriguez-Calvo T, Krogvold L, Amirian N, Dahl-Jorgensen K, von Herrath M. One in Ten Cd8(+) Cells in the Pancreas of Living Individuals With Recent-Onset Type 1 Diabetes Recognizes the Preproinsulin Epitope Ppi15-24. Diabetes (2021) 70(3):752–8. doi: 10.2337/db20-0908
32. Azoury ME, Samassa F, Buitinga M, Nigi L, Brusco N, Callebaut A, et al. Cd8(+) T Cells Variably Recognize Native Versus Citrullinated Grp78 Epitopes in Type 1 Diabetes. Diabetes (2021) 70(12):2879–91. doi: 10.2337/db21-0259
33. McLaughlin RJ, de Haan A, Zaldumbide A, de Koning EJ, de Ru AH, van Veelen PA, et al. Human Islets and Dendritic Cells Generate Post-Translationally Modified Islet Autoantigens. Clin Exp Immunol (2016) 185(2):133–40. doi: 10.1111/cei.12775
34. Rodriguez-Calvo T, Johnson JD, Overbergh L, Dunne JL. Neoepitopes in Type 1 Diabetes: Etiological Insights, Biomarkers and Therapeutic Targets. Front Immunol (2021) 12:667989. doi: 10.3389/fimmu.2021.667989
35. Roep BO, Kracht MJ, van Lummel M, Zaldumbide A. A Roadmap of the Generation of Neoantigens as Targets of the Immune System in Type 1 Diabetes. Curr Opin Immunol (2016) 43:67–73. doi: 10.1016/j.coi.2016.09.007
36. Kracht MJ, van Lummel M, Nikolic T, Joosten AM, Laban S, van der Slik AR, et al. Autoimmunity Against a Defective Ribosomal Insulin Gene Product in Type 1 Diabetes. Nat Med (2017) 23(4):501–7. doi: 10.1038/nm.4289
37. van Lummel M, Duinkerken G, van Veelen PA, de Ru A, Cordfunke R, Zaldumbide A, et al. Posttranslational Modification of HLA-DQ Binding Islet Autoantigens in Type 1 Diabetes. Diabetes (2014) 63(1):237–47. doi: 10.2337/db12-1214
38. Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, et al. Projection of an Immunological Self Shadow Within the Thymus by the Aire Protein. Sci (2002) 298(5597):1395–401. doi: 10.1126/science.1075958
39. Kyewski B, Klein L. A Central Role for Central Tolerance. Annu Rev Immunol (2006) 24:571–606. doi: 10.1146/annurev.immunol.23.021704.115601
40. Beringer DX, Kleijwegt FS, Wiede F, van der Slik AR, Loh KL, Petersen J, et al. T Cell Receptor Reversed Polarity Recognition of a Self-Antigen Major Histocompatibility Complex. Nat Immunol (2015) 16(11):1153–61. doi: 10.1038/ni.3271
41. Visperas A, Vignali DA. Are Regulatory T Cells Defective in Type 1 Diabetes and Can We Fix Them? J Immunol (2016) 197(10):3762–70. doi: 10.4049/jimmunol.1601118
42. Lindley S, Dayan CM, Bishop A, Roep BO, Peakman M, Tree TI. Defective Suppressor Function in CD4(+)CD25(+) T-Cells From Patients With Type 1 Diabetes. Diabetes (2005) 54(1):92–9. doi: 10.2337/diabetes.54.1.92
44. Redondo MJ, Steck AK, Pugliese A. Genetics of Type 1 Diabetes. Pediatr Diabetes (2018) 19(3):346–53. doi: 10.1111/pedi.12597
45. Bakay M, Pandey R, Hakonarson H. Genes Involved in Type 1 Diabetes: An Update. Genes (Basel) (2013) 4(3):499–521. doi: 10.3390/genes4030499
46. Rønningen KSR C, Diaz AO. HLA-Class II Associations in Insulin-Dependent Diabetes Mellitus Among Blacks, Caucasoids and Japanese. Oxford: University Press (1991).
47. Noble JA, Valdes AM, Thomson G, Erlich HA. The HLA Class II Locus DPB1 can Influence Susceptibility to Type 1 Diabetes. Diabetes (2000) 49(1):121–5. doi: 10.2337/diabetes.49.1.121
48. Koeleman BP, Lie BA, Undlien DE, Dudbridge F, Thorsby E, de Vries RR, et al. Genotype Effects and Epistasis in Type 1 Diabetes and HLA-DQ Trans Dimer Associations With Disease. Genes Immun (2004) 5(5):381–8. doi: 10.1038/sj.gene.6364106
49. Nepom GT. Cap. 15. HLA and Type 1 Diabetes In:Lechler R, Warrens Aeditors HLA in Health and Disease 2nd ed. London: Academic Press (2000). p. 231–8.
50. Nepom BS, Schwarz D, Palmer JP, Nepom GT. Transcomplementation of HLA Genes in IDDMHla-DQ Alpha- and Beta-Chains Produce Hybrid Molecules in DR3/4 Heterozygotes. Diabetes (1987) 36(1):114–7. doi: 10.2337/diab.36.1.114
51. van Lummel M, van Veelen PA, Zaldumbide A, de Ru A, Janssen GM, Moustakas AK, et al. Type 1 Diabetes-Associated HLA-DQ8 Transdimer Accommodates a Unique Peptide Repertoire. J Biol Chem (2012) 287(12):9514–24. doi: 10.1074/jbc.M111.313940
52. Miao F, Chen Z, Zhang L, Liu Z, Wu X, Yuan YC, et al. Profiles of Epigenetic Histone Post-Translational Modifications at Type 1 Diabetes Susceptible Genes. J Biol Chem (2012) 287(20):16335–45. doi: 10.1074/jbc.M111.330373
53. Roep BO, Peakman M. Antigen Targets of Type 1 Diabetes Autoimmunity. Cold Spring Harb Perspect Med (2012) 2(4):a007781. doi: 10.1101/cshperspect.a007781
54. Kennedy GC, German MS, Rutter WJ. The Minisatellite in the Diabetes Susceptibility Locus IDDM2 Regulates Insulin Transcription. Nat Genet (1995) 9(3):293–8. doi: 10.1038/ng0395-293
55. Lucassen AM, Screaton GR, Julier C, Elliott TJ, Lathrop M, Bell JI. Regulation of Insulin Gene Expression by the IDDM Associated, Insulin Locus Haplotype. Hum Mol Genet (1995) 4(4):501–6. doi: 10.1093/hmg/4.4.501
56. Reddy MV, Wang H, Liu S, Bode B, Reed JC, Steed RD, et al. Association Between Type 1 Diabetes and GWAS Snps in the Southeast US Caucasian Population. Genes Immun (2011) 12(3):208–12. doi: 10.1038/gene.2010.70
57. Howson JM, Walker NM, Smyth DJ, Todd JA, Type IDGC. Analysis of 19 Genes for Association With Type I Diabetes in the Type I Diabetes Genetics Consortium Families. Genes Immun (2009) 10 Suppl 1:S74–84. doi: 10.1038/gene.2009.96
58. Nejentsev S, Howson JM, Walker NM, Szeszko J, Field SF, Stevens HE, et al. Localization of Type 1 Diabetes Susceptibility to the MHC Class I Genes HLA-B and HLA-a. Nature (2007) 450(7171):887–92. doi: 10.1038/nature06406
59. Shapiro MR, Thirawatananond P, Peters L, Sharp RC, Ogundare S, Posgai AL, et al. De-Coding Genetic Risk Variants in Type 1 Diabetes. Immunol Cell Biol (2021) 99(5):496–508. doi: 10.1111/imcb.12438
60. Westra HJ, Martinez-Bonet M, Onengut-Gumuscu S, Lee A, Luo Y, Teslovich N, et al. Fine-Mapping and Functional Studies Highlight Potential Causal Variants for Rheumatoid Arthritis and Type 1 Diabetes. Nat Genet (2018) 50(10):1366–74. doi: 10.1038/s41588-018-0216-7
61. Erlich H, Valdes AM, Noble J, Carlson JA, Varney M, Concannon P, et al. Hla DR-DQ Haplotypes and Genotypes and Type 1 Diabetes Risk: Analysis of the Type 1 Diabetes Genetics Consortium Families. Diabetes (2008) 57(4):1084–92. doi: 10.2337/db07-1331
62. Cervin C, Lyssenko V, Bakhtadze E, Lindholm E, Nilsson P, Tuomi T, et al. Genetic Similarities Between Latent Autoimmune Diabetes in Adults, Type 1 Diabetes, and Type 2 Diabetes. Diabetes (2008) 57(5):1433–7. doi: 10.2337/db07-0299
63. Inshaw JRJ, Sidore C, Cucca F, Stefana MI, Crouch DJM, McCarthy MI, et al. Analysis of Overlapping Genetic Association in Type 1 and Type 2 Diabetes. Diabetologia (2021) 64(6):1342–7. doi: 10.1007/s00125-021-05428-0
64. Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, et al. A Functional Variant of Lymphoid Tyrosine Phosphatase is Associated With Type I Diabetes. Nat Genet (2004) 36(4):337–8. doi: 10.1038/ng1323
65. Carr EJ, Niederer HA, Williams J, Harper L, Watts RA, Lyons PA, et al. Confirmation of the Genetic Association of CTLA4 and PTPN22 With ANCA-associated Vasculitis. BMC Med Genet (2009) 10:121. doi: 10.1186/1471-2350-10-121
66. Qu HQ, Marchand L, Grabs R, Polychronakos C. The Association Between the IFIH1 Locus and Type 1 Diabetes. Diabetologia (2008) 51(3):473–5. doi: 10.1007/s00125-007-0895-6
67. Forgetta V, Manousaki D, Istomine R, Ross S, Tessier MC, Marchand L, et al. Rare Genetic Variants of Large Effect Influence Risk of Type 1 Diabetes. Diabetes (2020) 69(4):784–95. doi: 10.2337/db19-0831
68. Smyth DJ, Cooper JD, Bailey R, Field S, Burren O, Smink LJ, et al. A Genome-Wide Association Study of Nonsynonymous SNPs Identifies a Type 1 Diabetes Locus in the Interferon-Induced Helicase (IFIH1) Region. Nat Genet (2006) 38(6):617–9. doi: 10.1038/ng1800
69. Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, et al. Robust Associations of Four New Chromosome Regions From Genome-Wide Analyses of Type 1 Diabetes. Nat Genet (2007) 39(7):857–64. doi: 10.1038/ng2068
70. Liu S, Wang H, Jin Y, Podolsky R, Reddy MV, Pedersen J, et al. IFIH1 Polymorphisms are Significantly Associated With Type 1 Diabetes and IFIH1 Gene Expression in Peripheral Blood Mononuclear Cells. Hum Mol Genet (2009) 18(2):358–65. doi: 10.1093/hmg/ddn342
71. Zurawek M, Fichna M, Fichna P, Skowronska B, Dzikiewicz-Krawczyk A, Januszkiewicz D, et al. Cumulative Effect of IFIH1 Variants and Increased Gene Expression Associated With Type 1 Diabetes. Diabetes Res Clin Pract (2015) 107(2):259–66. doi: 10.1016/j.diabres.2014.11.008
72. Kavvoura FK, Ioannidis JP. CTLA-4 Gene Polymorphisms and Susceptibility to Type 1 Diabetes Mellitus: A HuGE Review and Meta-Analysis. Am J Epidemiol (2005) 162(1):3–16. doi: 10.1093/aje/kwi165
73. Goralczyk A. Genetic Susceptibility to Type 1 Diabetes in Children: Analysis of Polymorphisms rs1990760 – IFIH1, rs20541 – IL13, rs231775 – CTLA 4. ESPE Abstracts (2018) 89:P–P1-052.
74. Qu HQ, Bradfield JP, Grant SF, Hakonarson H, Polychronakos C, Type IDGC. Remapping the Type I Diabetes Association of the CTLA4 Locus. Genes Immun (2009) 10 Suppl 1:S27–32. doi: 10.1038/gene.2009.88
75. Chen M, Li S. Associations Between Cytotoxic T-lymphocyte-associated Antigen 4 Gene Polymorphisms and Diabetes Mellitus: A Meta-Analysis of 76 Case-Control Studies. Biosci Rep (2019) 39(5):BSR20190309. doi: 10.1042/BSR20190309
76. Lowe CE, Cooper JD, Brusko T, Walker NM, Smyth DJ, Bailey R, et al. Large-Scale Genetic Fine Mapping and Genotype-Phenotype Associations Implicate Polymorphism in the IL2RA Region in Type 1 Diabetes. Nat Genet (2007) 39(9):1074–82. doi: 10.1038/ng2102
77. Maier LM, Lowe CE, Cooper J, Downes K, Anderson DE, Severson C, et al. IL2RA Genetic Heterogeneity in Multiple Sclerosis and Type 1 Diabetes Susceptibility and Soluble Interleukin-2 Receptor Production. PloS Genet (2009) 5(1):e1000322. doi: 10.1371/journal.pgen.1000322
78. Espino-Paisan L, de la Calle H, Fernandez-Arquero M, Figueredo MA, de la Concha EG, Urcelay E, et al. Study of Polymorphisms in 4q27, 10p15, and 22q13 Regions in Autoantibodies Stratified Type 1 Diabetes Patients. AutoImmun (2011) 44(8):624–30. doi: 10.3109/08916934.2011.592515
79. Smyth DJ, Plagnol V, Walker NM, Cooper JD, Downes K, Yang JH, et al. Shared and Distinct Genetic Variants in Type 1 Diabetes and Celiac Disease. N Engl J Med (2008) 359(26):2767–77. doi: 10.1056/NEJMoa0807917
80. Plagnol V, Howson JM, Smyth DJ, Walker N, Hafler JP, Wallace C, et al. Genome-Wide Association Analysis of Autoantibody Positivity in Type 1 Diabetes Cases. PloS Genet (2011) 7(8):e1002216. doi: 10.1371/journal.pgen.1002216
81. Cohen S, Dadi H, Shaoul E, Sharfe N, Roifman CM. Cloning and Characterization of a Lymphoid-Specific, Inducible Human Protein Tyrosine Phosphatase, Lyp. Blood (1999) 93(6):2013–24. doi: 10.1182/blood.V93.6.2013.406k25_2013_2024
82. Lei ZX, Chen WJ, Liang JQ, Wang YJ, Jin L, Xu C, et al. The Association Between rs2476601 Polymorphism in PTPN22 Gene and Risk of Alopecia Areata: A Meta-Analysis of Case-Control Studies. Med (Baltimore) (2019) 98(20):e15448. doi: 10.1097/MD.0000000000015448
83. Cubas R, Khan Z, Gong Q, Moskalenko M, Xiong H, Ou Q, et al. Autoimmunity Linked Protein Phosphatase PTPN22 as a Target for Cancer Immunotherapy. J Immunother Cancer (2020) 8(2):e001439. doi: 10.1136/jitc-2020-001439
84. Maine CJ, Teijaro JR, Marquardt K, Sherman LA. PTPN22 Contributes to Exhaustion of T Lymphocytes During Chronic Viral Infection. Proc Natl Acad Sci U S A (2016) 113(46):E7231–E9. doi: 10.1073/pnas.1603738113
85. Burn GL, Svensson L, Sanchez-Blanco C, Saini M, Cope AP. Why is PTPN22 a Good Candidate Susceptibility Gene for Autoimmune Disease? FEBS Lett (2011) 585(23):3689–98. doi: 10.1016/j.febslet.2011.04.032
86. Gomez LM, Anaya JM, Martin J. Genetic Influence of PTPN22 R620W Polymorphism in Tuberculosis. Hum Immunol (2005) 66(12):1242–7. doi: 10.1016/j.humimm.2005.11.008
87. Boechat AL, Ogusku MM, Sadahiro A, dos Santos MC. Association Between the PTPN22 1858C/T Gene Polymorphism and Tuberculosis Resistance. Infect Genet Evol (2013) 16:310–3. doi: 10.1016/j.meegid.2013.02.019
88. Lamsyah H, Rueda B, Baassi L, Elaouad R, Bottini N, Sadki K, et al. Association of PTPN22 Gene Functional Variants With Development of Pulmonary Tuberculosis in Moroccan Population. Tissue Antigens (2009) 74(3):228–32. doi: 10.1111/j.1399-0039.2009.01304.x
89. Stanford SM, Rapini N, Bottini N. Regulation of TCR Signalling by Tyrosine Phosphatases: From Immune Homeostasis to Autoimmunity. Immunol (2012) 137(1):1–19. doi: 10.1111/j.1365-2567.2012.03591.x
90. Pao LI, Lam KP, Henderson JM, Kutok JL, Alimzhanov M, Nitschke L, et al. B Cell-Specific Deletion of Protein-Tyrosine Phosphatase Shp1 Promotes B-1a Cell Development and Causes Systemic Autoimmunity. Immun (2007) 27(1):35–48. doi: 10.1016/j.immuni.2007.04.016
91. Clarke F, Jordan CK, Gutierrez-Martinez E, Bibby JA, Sanchez-Blanco C, Cornish GH, et al. Protein Tyrosine Phosphatase PTPN22 is Dispensable for Dendritic Cell Antigen Processing and Promotion of T-cell Activation by Dendritic Cells. PloS One (2017) 12(10):e0186625. doi: 10.1371/journal.pone.0186625
92. Armitage LH, Wallet MA, Mathews CE. Influence of PTPN22 Allotypes on Innate and Adaptive Immune Function in Health and Disease. Front Immunol (2021) 12:636618. doi: 10.3389/fimmu.2021.636618
93. Cerosaletti K, Buckner JH. Protein Tyrosine Phosphatases and Type 1 Diabetes: Genetic and Functional Implications of PTPN2 and PTPN22. Rev Diabetes Stud (2012) 9(4):188–200. doi: 10.1900/RDS.2012.9.188
94. Bottini N, Peterson EJ. Tyrosine Phosphatase PTPN22: Multifunctional Regulator of Immune Signaling, Development, and Disease. Annu Rev Immunol (2014) 32:83–119. doi: 10.1146/annurev-immunol-032713-120249
95. Vang T, Congia M, Macis MD, Musumeci L, Orru V, Zavattari P, et al. Autoimmune-Associated Lymphoid Tyrosine Phosphatase is a Gain-of-Function Variant. Nat Genet (2005) 37(12):1317–9. doi: 10.1038/ng1673
96. Vang T, Liu WH, Delacroix L, Wu S, Vasile S, Dahl R, et al. LYP Inhibits T-cell Activation When Dissociated From CSK. Nat Chem Biol (2012) 8(5):437–46. doi: 10.1038/nchembio.916
97. Maine CJ, Hamilton-Williams EE, Cheung J, Stanford SM, Bottini N, Wicker LS, et al. PTPN22 Alters the Development of Regulatory T Cells in the Thymus. J Immunol (2012) 188(11):5267–75. doi: 10.4049/jimmunol.1200150
98. Valta M, Gazali AM, Viisanen T, Ihantola EL, Ekman I, Toppari J, et al. Type 1 Diabetes Linked PTPN22 Gene Polymorphism is Associated With the Frequency of Circulating Regulatory T Cells. Eur J Immunol (2020) 50(4):581–8. doi: 10.1002/eji.201948378
99. Perry DJ, Peters LD, Lakshmi PS, Zhang L, Han Z, Wasserfall CH, et al. Overexpression of the PTPN22 Autoimmune Risk Variant LYP-620W Fails to Restrain Human Cd4(+) T Cell Activation. J Immunol (2021) 207(3):849–59. doi: 10.4049/jimmunol.2000708
100. Zhang J, Zahir N, Jiang Q, Miliotis H, Heyraud S, Meng X, et al. The Autoimmune Disease-Associated PTPN22 Variant Promotes Calpain-Mediated Lyp/Pep Degradation Associated With Lymphocyte and Dendritic Cell Hyperresponsiveness. Nat Genet (2011) 43(9):902–7. doi: 10.1038/ng.904
101. Spalinger MR, Kasper S, Gottier C, Lang S, Atrott K, Vavricka SR, et al. NLRP3 Tyrosine Phosphorylation is Controlled by Protein Tyrosine Phosphatase PTPN22. J Clin Invest (2016) 126(5):1783–800. doi: 10.1172/JCI83669
102. Boettler T, von Herrath M. Protection Against or Triggering of Type 1 Diabetes? Different Roles for Viral Infections. Expert Rev Clin Immunol (2011) 7(1):45–53. doi: 10.1586/eci.10.91
103. Scharl M, Hruz P, McCole DF. Protein Tyrosine Phosphatase non-Receptor Type 2 Regulates IFN-gamma-induced Cytokine Signaling in THP-1 Monocytes. Inflamm Bowel Dis (2010) 16(12):2055–64. doi: 10.1002/ibd.21325
104. Spalinger MR, Kasper S, Chassard C, Raselli T, Frey-Wagner I, Gottier C, et al. PTPN2 Controls Differentiation of CD4(+) T Cells and Limits Intestinal Inflammation and Intestinal Dysbiosis. Mucosal Immunol (2015) 8(4):918–29. doi: 10.1038/mi.2014.122
105. Spalinger MR, Manzini R, Hering L, Riggs JB, Gottier C, Lang S, et al. Ptpn2 Regulates Inflammasome Activation and Controls Onset of Intestinal Inflammation and Colon Cancer. Cell Rep (2018) 22(7):1835–48. doi: 10.1016/j.celrep.2018.01.052
106. Colli ML, Moore F, Gurzov EN, Ortis F, Eizirik DL. MDA5 and PTPN2, Two Candidate Genes for Type 1 Diabetes, Modify Pancreatic Beta-Cell Responses to the Viral by-Product Double-Stranded RNA. Hum Mol Genet (2010) 19(1):135–46. doi: 10.1093/hmg/ddp474
107. Moore F, Colli ML, Cnop M, Esteve MI, Cardozo AK, Cunha DA, et al. PTPN2, a Candidate Gene for Type 1 Diabetes, Modulates Interferon-Gamma-Induced Pancreatic Beta-Cell Apoptosis. Diabetes (2009) 58(6):1283–91. doi: 10.2337/db08-1510
108. Santin I, Moore F, Colli ML, Gurzov EN, Marselli L, Marchetti P, et al. PTPN2, a Candidate Gene for Type 1 Diabetes, Modulates Pancreatic Beta-Cell Apoptosis Via Regulation of the BH3-only Protein Bim. Diabetes (2011) 60(12):3279–88. doi: 10.2337/db11-0758
109. Long SA, Cerosaletti K, Wan JY, Ho JC, Tatum M, Wei S, et al. An Autoimmune-Associated Variant in PTPN2 Reveals an Impairment of IL-2R Signaling in CD4(+) T Cells. Genes Immun (2011) 12(2):116–25. doi: 10.1038/gene.2010.54
110. Wiede F, Brodnicki TC, Goh PK, Leong YA, Jones GW, Yu D, et al. T-Cell-Specific PTPN2 Deficiency in NOD Mice Accelerates the Development of Type 1 Diabetes and Autoimmune Comorbidities. Diabetes (2019) 68(6):1251–66. doi: 10.2337/db18-1362
111. Kumar H, Kawai T, Akira S. Pathogen Recognition in the Innate Immune Response. Biochem J (2009) 420(1):1–16. doi: 10.1042/BJ20090272
112. Skog O, Korsgren O, Frisk G. Modulation of Innate Immunity in Human Pancreatic Islets Infected With Enterovirus In Vitro. J Med Virol (2011) 83(4):658–64. doi: 10.1002/jmv.21924
113. Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare Variants of IFIH1, a Gene Implicated in Antiviral Responses, Protect Against Type 1 Diabetes. Sci (2009) 324(5925):387–9. doi: 10.1126/science.1167728
114. Downes K, Pekalski M, Angus KL, Hardy M, Nutland S, Smyth DJ, et al. Reduced Expression of IFIH1 is Protective for Type 1 Diabetes. PloS One (2010) 5(9):e12646. doi: 10.1371/journal.pone.0012646
115. Winkler C, Lauber C, Adler K, Grallert H, Illig T, Ziegler AG, et al. An Interferon-Induced Helicase (IFIH1) Gene Polymorphism Associates With Different Rates of Progression From Autoimmunity to Type 1 Diabetes. Diabetes (2011) 60(2):685–90. doi: 10.2337/db10-1269
116. Looney BM, Xia CQ, Concannon P, Ostrov DA, Clare-Salzler MJ. Effects of Type 1 Diabetes-Associated IFIH1 Polymorphisms on MDA5 Function and Expression. Curr Diabetes Rep (2015) 15(11):96. doi: 10.1007/s11892-015-0656-8
117. Gorman JA, Hundhausen C, Errett JS, Stone AE, Allenspach EJ, Ge Y, et al. The A946T Variant of the RNA Sensor IFIH1 Mediates an Interferon Program That Limits Viral Infection But Increases the Risk for Autoimmunity. Nat Immunol (2017) 18(7):744–52. doi: 10.1038/ni.3766
118. Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. Isoforms of RNA-Editing Enzyme ADAR1 Independently Control Nucleic Acid Sensor MDA5-Driven Autoimmunity and Multi-organ Development. Immun (2015) 43(5):933–44. doi: 10.1016/j.immuni.2015.11.001
119. Lincez PJ, Shanina I, Horwitz MS. Reduced Expression of the MDA5 Gene IFIH1 Prevents Autoimmune Diabetes. Diabetes (2015) 64(6):2184–93. doi: 10.2337/db14-1223
120. Baccala R, Hoebe K, Kono DH, Beutler B, Theofilopoulos AN. TLR-Dependent and TLR-independent Pathways of Type I Interferon Induction in Systemic Autoimmunity. Nat Med (2007) 13(5):543–51. doi: 10.1038/nm1590
121. Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, Rotig A, et al. Mitochondrial Double-Stranded RNA Triggers Antiviral Signalling in Humans. Nat (2018) 560(7717):238–42. doi: 10.1038/s41586-018-0363-0
122. Imagawa A, Hanafusa T, Miyagawa J, Matsuzawa Y, Osaka IDDM Study Group. A Novel Subtype of Type 1 Diabetes Mellitus Characterized by a Rapid Onset and an Absence of Diabetes-Related Antibodies. N Engl J Med (2000) 342(5):301–7. doi: 10.1056/NEJM200002033420501
123. Lee JW, Kim WH, Yeo J, Jung MH. ER Stress is Implicated in Mitochondrial Dysfunction-Induced Apoptosis of Pancreatic Beta Cells. Mol Cells (2010) 30(6):545–9. doi: 10.1007/s10059-010-0161-5
124. Robey E, Allison JP. T-Cell Activation: Integration of Signals From the Antigen Receptor and Costimulatory Molecules. Immunol Today (1995) 16(7):306–10. doi: 10.1016/0167-5699(95)80140-5
125. Gribben JG, Freeman GJ, Boussiotis VA, Rennert P, Jellis CL, Greenfield E, et al. CTLA4 Mediates Antigen-Specific Apoptosis of Human T Cells. Proc Natl Acad Sci USA (1995) 92(3):811–5. doi: 10.1073/pnas.92.3.811
126. Green JM, Noel PJ, Sperling AI, Walunas TL, Gray GS, Bluestone JA, et al. Absence of B7-dependent Responses in CD28-deficient Mice. Immun (1994) 1(6):501–8. doi: 10.1016/1074-7613(94)90092-2
127. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative Disorders With Early Lethality in Mice Deficient in Ctla-4. Sci (1995) 270(5238):985–8. doi: 10.1126/science.270.5238.985
128. Nisticò L, Cascino I, Buzzetti R. CTLA-4 in Type 1 Diabetes Mellitus. In: Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience (2013). Available from: https://www.ncbi.nlm.nih.gov/books/NBK5997/.
129. Chen Z, Fei M, Fu D, Zhang L, Ma Y, Wang Y, et al. Association Between Cytotoxic T Lymphocyte Antigen-4 Polymorphism and Type 1 Diabetes: A Meta-Analysis. Gene (2013) 516(2):263–70. doi: 10.1016/j.gene.2012.12.030
130. Wang K, Zhu Q, Lu Y, Lu H, Zhang F, Wang X, et al. Ctla-4 +49 G/A Polymorphism Confers Autoimmune Disease Risk: An Updated Meta-Analysis. Genet Test Mol Biomarkers (2017) 21(4):222–7. doi: 10.1089/gtmb.2016.0335
131. Anjos S, Nguyen A, Ounissi-Benkalha H, Tessier MC, Polychronakos C. A Common Autoimmunity Predisposing Signal Peptide Variant of the Cytotoxic T-lymphocyte Antigen 4 Results in Inefficient Glycosylation of the Susceptibility Allele. J Biol Chem (2002) 277(48):46478–86. doi: 10.1074/jbc.M206894200
132. Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, et al. Association of the T-cell Regulatory Gene CTLA4 With Susceptibility to Autoimmune Disease. Nature (2003) 423(6939):506–11. doi: 10.1038/nature01621
133. Gerold KD, Zheng P, Rainbow DB, Zernecke A, Wicker LS, Kissler S. The Soluble CTLA-4 Splice Variant Protects From Type 1 Diabetes and Potentiates Regulatory T-cell Function. Diabetes (2011) 60(7):1955–63. doi: 10.2337/db11-0130
134. Huurman VA, Unger WW, Koeleman BP, Oaks MK, Chandraker AK, Terpstra OT, et al. Differential Inhibition of Autoreactive Memory- and Alloreactive Naive T Cell Responses by Soluble Cytotoxic T Lymphocyte Antigen 4 (Sctla4), CTLA4Ig and LEA29Y. Clin Exp Immunol (2007) 150(3):487–93. doi: 10.1111/j.1365-2249.2007.03513.x
135. Yanagawa T, Hidaka Y, Guimaraes V, Soliman M, DeGroot LJ. CTLA-4 Gene Polymorphism Associated With Graves’ Disease in a Caucasian Population. J Clin Endocrinol Metab (1995) 80(1):41–5. doi: 10.1210/jcem.80.1.7829637
136. Hao S, Baltimore D. The Stability of mRNA Influences the Temporal Order of the Induction of Genes Encoding Inflammatory Molecules. Nat Immunol (2009) 10(3):281–8. doi: 10.1038/ni.1699
137. Anderson P. Post-Transcriptional Control of Cytokine Production. Nat Immunol (2008) 9(4):353–9. doi: 10.1038/ni1584
138. de Jong VM, Zaldumbide A, van der Slik AR, Laban S, Koeleman BP, Roep BO. Variation in the CTLA4 3’UTR has Phenotypic Consequences for Autoreactive T Cells and Associates With Genetic Risk for Type 1 Diabetes. Genes Immun (2016) 17(1):75–8. doi: 10.1038/gene.2015.51
139. de Jong VM, Zaldumbide A, van der Slik AR, Persengiev SP, Roep BO, Koeleman BP. Post-Transcriptional Control of Candidate Risk Genes for Type 1 Diabetes by Rare Genetic Variants. Genes Immun (2013) 14(1):58–61. doi: 10.1038/gene.2012.38
140. Ligers A, Teleshova N, Masterman T, Huang WX, Hillert J. CTLA-4 Gene Expression is Influenced by Promoter and Exon 1 Polymorphisms. Genes Immun (2001) 2(3):145–52. doi: 10.1038/sj.gene.6363752
141. Burchill MA, Yang J, Vang KB, Farrar MA. Interleukin-2 Receptor Signaling in Regulatory T Cell Development and Homeostasis. Immunol Lett (2007) 114(1):1–8. doi: 10.1016/j.imlet.2007.08.005
142. Belot MP, Fradin D, Mai N, Le Fur S, Zelenika D, Kerr-Conte J, et al. Cpg Methylation Changes Within the IL2RA Promoter in Type 1 Diabetes of Childhood Onset. PloS One (2013) 8(7):e68093. doi: 10.1371/journal.pone.0068093
143. Vella A, Cooper JD, Lowe CE, Walker N, Nutland S, Widmer B, et al. Localization of a Type 1 Diabetes Locus in the IL2RA/CD25 Region by Use of Tag Single-Nucleotide Polymorphisms. Am J Hum Genet (2005) 76(5):773–9. doi: 10.1086/429843
144. Qu HQ, Montpetit A, Hudson B, Polychronakos C. Toward Further Mapping of the Association Between the IL2RA Locus and Type 1 Diabetes. Diabetes (2007) 56:1174–6. doi: 10.2337/db06-1555
145. Bradfield JP, Qu HQ, Wang K, Zhang H, Sleiman PM, Kim CE, et al. A Genome-Wide Meta-Analysis of Six Type 1 Diabetes Cohorts Identifies Multiple Associated Loci. PloS Genet (2011) 7(9):e1002293. doi: 10.1371/journal.pgen.1002293
146. Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-Wide Association Study and Meta-Analysis Find That Over 40 Loci Affect Risk of Type 1 Diabetes. Nat Genet (2009) 41(6):703–7. doi: 10.1038/ng.381
147. Dendrou CA, Plagnol V, Fung E, Yang JH, Downes K, Cooper JD, et al. Cell-Specific Protein Phenotypes for the Autoimmune Locus IL2RA Using a Genotype-Selectable Human Bioresource. Nat Genet (2009) 41(9):1011–5. doi: 10.1038/ng.434
148. Zier KS, Leo MM, Spielman RS, Baker L. Decreased Synthesis of Interleukin-2 (IL-2) in Insulin-Dependent Diabetes Mellitus. Diabetes (1984) 33(6):552–5. doi: 10.2337/diab.33.6.552
149. Kaye WA, Adri MN, Soeldner JS, Rabinowe SL, Kaldany A, Kahn CR, et al. Acquired Defect in Interleukin-2 Production in Patients With Type I Diabetes Mellitus. N Engl J Med (1986) 315(15):920–4. doi: 10.1056/NEJM198610093151502
150. Cerosaletti K, Schneider A, Schwedhelm K, Frank I, Tatum M, Wei S, et al. Multiple Autoimmune-Associated Variants Confer Decreased IL-2R Signaling in CD4+ CD25(Hi) T Cells of Type 1 Diabetic and Multiple Sclerosis Patients. PloS One (2013) 8(12):e83811. doi: 10.1371/journal.pone.0083811
151. Garg G, Tyler JR, Yang JH, Cutler AJ, Downes K, Pekalski M, et al. Type 1 Diabetes-Associated IL2RA Variation Lowers IL-2 Signaling and Contributes to Diminished CD4+CD25+ Regulatory T Cell Function. J Immunol (2012) 188(9):4644–53. doi: 10.4049/jimmunol.1100272
152. Long SA, Cerosaletti K, Bollyky PL, Tatum M, Shilling H, Zhang S, et al. Defects in IL-2R Signaling Contribute to Diminished Maintenance of FOXP3 Expression in CD4(+)CD25(+) Regulatory T-cells of Type 1 Diabetic Subjects. Diabetes (2010) 59(2):407–15. doi: 10.2337/db09-0694
153. Rainbow DB, Pekalski M, Cutler AJ, Burren O, Walker N, Todd JA, et al. A Rare IL2RA Haplotype Identifies SNP rs61839660 as Causal for Autoimmunity. bioRxiv (2017) 1–11. doi: 10.1101/108126
154. Schwartz AM, Demin DE, Vorontsov IE, Kasyanov AS, Putlyaeva LV, Tatosyan KA, et al. Multiple Single Nucleotide Polymorphisms in the First Intron of the IL2RA Gene Affect Transcription Factor Binding and Enhancer Activity. Gene (2017) 602:50–6. doi: 10.1016/j.gene.2016.11.032
155. Simeonov DR, Gowen BG, Boontanrart M, Roth TL, Gagnon JD, Mumbach MR, et al. Discovery of Stimulation-Responsive Immune Enhancers With CRISPR Activation. Nature (2017) 549(7670):111–5. doi: 10.1038/nature23875
156. von Bergwelt-Baildon MS, Popov A, Saric T, Chemnitz J, Classen S, Stoffel MS, et al. CD25 and Indoleamine 2,3-Dioxygenase are Up-Regulated by Prostaglandin E2 and Expressed by Tumor-Associated Dendritic Cells In Vivo: Additional Mechanisms of T-cell Inhibition. Blood (2006) 108(1):228–37. doi: 10.1182/blood-2005-08-3507
157. Driesen J, Popov A, Schultze JL. CD25 as an Immune Regulatory Molecule Expressed on Myeloid Dendritic Cells. Immunobiology (2008) 213(9-10):849–58. doi: 10.1016/j.imbio.2008.07.026
158. Shibuya A, Campbell D, Hannum C, Yssel H, Franz-Bacon K, McClanahan T, et al. Dnam-1, a Novel Adhesion Molecule Involved in the Cytolytic Function of T Lymphocytes. Immun (1996) 4(6):573–81. doi: 10.1016/S1074-7613(00)70060-4
159. Shibuya K, Shirakawa J, Kameyama T, Honda S, Tahara-Hanaoka S, Miyamoto A, et al. Cd226 (Dnam-1) is Involved in Lymphocyte Function-Associated Antigen 1 Costimulatory Signal for Naive T Cell Differentiation and Proliferation. J Exp Med (2003) 198(12):1829–39. doi: 10.1084/jem.20030958
160. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The Surface Protein TIGIT Suppresses T Cell Activation by Promoting the Generation of Mature Immunoregulatory Dendritic Cells. Nat Immunol (2009) 10(1):48–57. doi: 10.1038/ni.1674
161. Lozano E, Dominguez-Villar M, Kuchroo V, Hafler DA. The TIGIT/CD226 Axis Regulates Human T Cell Function. J Immunol (2012) 188(8):3869–75. doi: 10.4049/jimmunol.1103627
162. Bai L, Jiang J, Li H, Zhang R. Role of CD226 Rs763361 Polymorphism in Susceptibility to Multiple Autoimmune Diseases. Immunol Invest (2020) 49(8):926–42. doi: 10.1080/08820139.2019.1703737
163. Shirakawa J, Shibuya K, Shibuya A. Requirement of the Serine at Residue 329 for Lipid Raft Recruitment of DNAM-1 (Cd226). Int Immunol (2005) 17(3):217–23. doi: 10.1093/intimm/dxh199
164. Gaud G, Roncagalli R, Chaoui K, Bernard I, Familiades J, Colacios C, et al. The Costimulatory Molecule CD226 Signals Through VAV1 to Amplify TCR Signals and Promote IL-17 Production by CD4(+) T Cells. Sci Signal (2018) 11(538):eaar3083. doi: 10.1126/scisignal.aar3083
165. Mattana TC, Santos AS, Fukui RT, Mainardi-Novo DT, Costa VS, Santos RF, et al. Cd226 rs763361 is Associated With the Susceptibility to Type 1 Diabetes and Greater Frequency of GAD65 Autoantibody in a Brazilian Cohort. Mediators Inflamm (2014) 2014:694948. doi: 10.1155/2014/694948
166. Wallace C, Rotival M, Cooper JD, Rice CM, Yang JH, McNeill M, et al. Statistical Colocalization of Monocyte Gene Expression and Genetic Risk Variants for Type 1 Diabetes. Hum Mol Genet (2012) 21(12):2815–24. doi: 10.1093/hmg/dds098
167. Shapiro MR, Yeh WI, Longfield JR, Gallagher J, Infante CM, Wellford S, et al. Cd226 Deletion Reduces Type 1 Diabetes in the NOD Mouse by Impairing Thymocyte Development and Peripheral T Cell Activation. Front Immunol (2020) 11:2180. doi: 10.3389/fimmu.2020.02180
168. Fuhrman CA, Yeh WI, Seay HR, Saikumar Lakshmi P, Chopra G, Zhang L, et al. Divergent Phenotypes of Human Regulatory T Cells Expressing the Receptors TIGIT and CD226. J Immunol (2015) 195(1):145–55. doi: 10.4049/jimmunol.1402381
169. Nikolic T, Roep BO. Regulatory Multitasking of Tolerogenic Dendritic Cells - Lessons Taken From Vitamin d3-treated Tolerogenic Dendritic Cells. Front Immunol (2013) 4:113. doi: 10.3389/fimmu.2013.00113
170. Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, et al. Gene Set Knowledge Discovery With Enrichr. Curr Protoc (2021) 1(3):e90. doi: 10.1002/cpz1.90
171. Darwish H, DeLuca HF. Vitamin D-regulated Gene Expression. Crit Rev Eukaryot Gene Expr (1993) 3(2):89–116.
172. Kongsbak M, Levring TB, Geisler C, von Essen MR. The Vitamin D Receptor and T Cell Function. Front Immunol (2013) 4:148. doi: 10.3389/fimmu.2013.00148
173. Warwick T, Schulz MH, Gunther S, Gilsbach R, Neme A, Carlberg C, et al. A Hierarchical Regulatory Network Analysis of the Vitamin D Induced Transcriptome Reveals Novel Regulators and Complete VDR Dependency in Monocytes. Sci Rep (2021) 11(1):6518. doi: 10.1038/s41598-021-86032-5
174. Vanherwegen AS, Eelen G, Ferreira GB, Ghesquiere B, Cook DP, Nikolic T, et al. Vitamin D Controls the Capacity of Human Dendritic Cells to Induce Functional Regulatory T Cells by Regulation of Glucose Metabolism. J Steroid Biochem Mol Biol (2019) 187:134–45. doi: 10.1016/j.jsbmb.2018.11.011
175. Baeke F, Korf H, Overbergh L, van Etten E, Verstuyf A, Gysemans C, et al. Human T Lymphocytes are Direct Targets of 1,25-Dihydroxyvitamin D3 in the Immune System. J Steroid Biochem Mol Biol (2010) 121(1-2):221–7. doi: 10.1016/j.jsbmb.2010.03.037
176. Holm LJ, Krogvold L, Hasselby JP, Kaur S, Claessens LA, Russell MA, et al. Abnormal Islet Sphingolipid Metabolism in Type 1 Diabetes. Diabetologia (2018) 61(7):1650–61. doi: 10.1007/s00125-018-4614-2
177. Devalliere J, Charreau B. The Adaptor Lnk (SH2B3): An Emerging Regulator in Vascular Cells and a Link Between Immune and Inflammatory Signaling. Biochem Pharmacol (2011) 82(10):1391–402. doi: 10.1016/j.bcp.2011.06.023
178. Lavrikova EY, Nikitin AG, Kuraeva TL, Peterkova VA, Tsitlidze NM, Chistiakov DA, et al. The Carriage of the Type 1 Diabetes-Associated R262W Variant of Human LNK Correlates With Increased Proliferation of Peripheral Blood Monocytes in Diabetic Patients. Pediatr Diabetes (2011) 12(2):127–32. doi: 10.1111/j.1399-5448.2010.00656.x
179. Allenspach EJ, Shubin NJ, Cerosaletti K, Mikacenic C, Gorman JA, MacQuivey MA, et al. The Autoimmune Risk R262W Variant of the Adaptor Sh2b3 Improves Survival in Sepsis. J Immunol (2021) 207(11):2710–9. doi: 10.4049/jimmunol.2100454
180. Swafford AD, Howson JM, Davison LJ, Wallace C, Smyth DJ, Schuilenburg H, et al. An Allele of IKZF1 (Ikaros) Conferring Susceptibility to Childhood Acute Lymphoblastic Leukemia Protects Against Type 1 Diabetes. Diabetes (2011) 60(3):1041–4. doi: 10.2337/db10-0446
181. Cytlak U, Resteu A, Bogaert D, Kuehn HS, Altmann T, Gennery A, et al. Ikaros Family Zinc Finger 1 Regulates Dendritic Cell Development and Function in Humans. Nat Commun (2018) 9(1):1239. doi: 10.1038/s41467-018-02977-8
182. Weinblatt ME, Moreland LW, Westhovens R, Cohen RB, Kelly SM, Khan N, et al. Safety of Abatacept Administered Intravenously in Treatment of Rheumatoid Arthritis: Integrated Analyses of Up to 8 Years of Treatment From the Abatacept Clinical Trial Program. J Rheumatol (2013) 40(6):787–97. doi: 10.3899/jrheum.120906
183. Mease PJ, Gottlieb AB, van der Heijde D, FitzGerald O, Johnsen A, Nys M, et al. Efficacy and Safety of Abatacept, a T-cell Modulator, in a Randomised, Double-Blind, Placebo-Controlled, Phase III Study in Psoriatic Arthritis. Ann Rheum Dis (2017) 76(9):1550–8. doi: 10.1136/annrheumdis-2016-210724
184. Orban T, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, et al. Co-Stimulation Modulation With Abatacept in Patients With Recent-Onset Type 1 Diabetes: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet (2011) 378(9789):412–9. doi: 10.1016/S0140-6736(11)60886-6
185. Roep BO. New Hope for Immune Intervention Therapy in Type 1 Diabetes. Lancet (2011) 378(9789):376–8. doi: 10.1016/S0140-6736(11)60977-X
186. Fang H, Consortium U-D, De Wolf H, Knezevic B, Burnham KL, Osgood J, et al. A Genetics-Led Approach Defines the Drug Target Landscape of 30 Immune-Related Traits. Nat Genet (2019) 51(7):1082–91. doi: 10.1038/s41588-019-0456-1
187. Orru V, Steri M, Sidore C, Marongiu M, Serra V, Olla S, et al. Complex Genetic Signatures in Immune Cells Underlie Autoimmunity and Inform Therapy. Nat Genet (2020) 52(10):1036–45. doi: 10.1038/s41588-020-0684-4
188. Hartemann A, Bensimon G, Payan CA, Jacqueminet S, Bourron O, Nicolas N, et al. Low-Dose Interleukin 2 in Patients With Type 1 Diabetes: A Phase 1/2 Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Diabetes Endocrinol (2013) 1(4):295–305. doi: 10.1016/S2213-8587(13)70113-X
189. Rosenzwajg M, Churlaud G, Mallone R, Six A, Derian N, Chaara W, et al. Low-Dose Interleukin-2 Fosters a Dose-Dependent Regulatory T Cell Tuned Milieu in T1D Patients. J Autoimmun (2015) 58:48–58. doi: 10.1016/j.jaut.2015.01.001
190. Khoryati L, Pham MN, Sherve M, Kumari S, Cook K, Pearson J, et al. An IL-2 Mutein Engineered to Promote Expansion of Regulatory T Cells Arrests Ongoing Autoimmunity in Mice. Sci Immunol (2020) 5(50):eaba5264. doi: 10.1126/sciimmunol.aba5264
191. Nikolic T, Zwaginga JJ, Uitbeijerse BS, Woittiez NJ, de Koning EJ, Aanstoot HJ, et al. Safety and Feasibility of Intradermal Injection With Tolerogenic Dendritic Cells Pulsed With Proinsulin Peptide-for Type 1 Diabetes. Lancet Diabetes Endocrinol (2020) 8(6):470–2. doi: 10.1016/S2213-8587(20)30104-2
192. Eerligh P, Koeleman BP, Dudbridge F, Jan Bruining G, Roep BO, Giphart MJ. Functional Genetic Polymorphisms in Cytokines and Metabolic Genes as Additional Genetic Markers for Susceptibility to Develop Type 1 Diabetes. Genes Immun (2004) 5(1):36–40. doi: 10.1038/sj.gene.6364036
193. van Etten E, Verlinden L, Giulietti A, Ramos-Lopez E, Branisteanu DD, Ferreira GB, et al. The Vitamin D Receptor Gene FokI Polymorphism: Functional Impact on the Immune System. Eur J Immunol (2007) 37(2):395–405. doi: 10.1002/eji.200636043
194. Ludvigsson J, Sumnik Z, Pelikanova T, Nattero Chavez L, Lundberg E, Rica I, et al. Intralymphatic Glutamic Acid Decarboxylase With Vitamin D Supplementation in Recent-Onset Type 1 Diabetes: A Double-Blind, Randomized, Placebo-Controlled Phase Iib Trial. Diabetes Care (2021) 44(7):1604–12. doi: 10.2337/dc21-0318
195. Nwosu BU, Parajuli S, Jasmin G, Fleshman J, Sharma RB, Alonso LC, et al. Ergocalciferol in New-onset Type 1 Diabetes: A Randomized Controlled Trial. J Endocr Soc (2022) 6(1):bvab179. doi: 10.1210/jendso/bvab179
196. Bogdanou D, Penna-Martinez M, Filmann N, Chung TL, Moran-Auth Y, Wehrle J, et al. T-Lymphocyte and Glycemic Status After Vitamin D Treatment in Type 1 Diabetes: A Randomized Controlled Trial With Sequential Crossover. Diabetes Metab Res Rev (2017) 33(3):e2865. doi: 10.1002/dmrr.2865
197. Savastio S, Cadario F, D’Alfonso S, Stracuzzi M, Pozzi E, Raviolo S, et al. Vitamin D Supplementation Modulates ICOS+ and ICOS- Regulatory T Cell in Siblings of Children With Type 1 Diabetes. J Clin Endocrinol Metab (2020) 105(12):dgaa588. doi: 10.1210/clinem/dgaa588
198. Treiber G, Prietl B, Frohlich-Reiterer E, Lechner E, Ribitsch A, Fritsch M, et al. Cholecalciferol Supplementation Improves Suppressive Capacity of Regulatory T-cells in Young Patients With New-Onset Type 1 Diabetes Mellitus - A Randomized Clinical Trial. Clin Immunol (2015) 161(2):217–24. doi: 10.1016/j.clim.2015.08.002
199. Miettinen ME, Niinisto S, Erlund I, Cuthbertson D, Nucci AM, Honkanen J, et al. Serum 25-Hydroxyvitamin D Concentration in Childhood and Risk of Islet Autoimmunity and Type 1 Diabetes: The TRIGR Nested Case-Control Ancillary Study. Diabetologia (2020) 63(4):780–7. doi: 10.1007/s00125-019-05077-4
200. Manousaki D, Harroud A, Mitchell RE, Ross S, Forgetta V, Timpson NJ, et al. Vitamin D Levels and Risk of Type 1 Diabetes: A Mendelian Randomization Study. PloS Med (2021) 18(2):e1003536. doi: 0.1371/journal.pmed.1003536
201. Lim YW, Chen-Harris H, Mayba O, Lianoglou S, Wuster A, Bhangale T, et al. Germline Genetic Polymorphisms Influence Tumor Gene Expression and Immune Cell Infiltration. Proc Natl Acad Sci USA (2018) 115(50):E11701–10. doi: 10.1073/pnas.1804506115
202. Jiao X, Wei X, Li S, Liu C, Chen H, Gong J, et al. A Genomic Mutation Signature Predicts the Clinical Outcomes of Immunotherapy and Characterizes Immunophenotypes in Gastrointestinal Cancer. NPJ Precis Oncol (2021) 5(1):36. doi: 10.1038/s41698-021-00172-5
203. Khan Z, Di Nucci F, Kwan A, Hammer C, Mariathasan S, Rouilly V, et al. Polygenic Risk for Skin Autoimmunity Impacts Immune Checkpoint Blockade in Bladder Cancer. Proc Natl Acad Sci USA (2020) 117(22):12288–94. doi: 10.1073/pnas.1922867117
204. Torkamani A, Wineinger NE, Topol EJ. The Personal and Clinical Utility of Polygenic Risk Scores. Nat Rev Genet (2018) 19(9):581–90. doi: 10.1038/s41576-018-0018-x
205. Butty V, Campbell C, Mathis D, Benoist C, Group DPTS. Impact of Diabetes Susceptibility Loci on Progression From Pre-Diabetes to Diabetes in at-Risk Individuals of the Diabetes Prevention Trial-Type 1 (DPT-1). Diabetes (2008) 57(9):2348–59. doi: 10.2337/db07-1736
206. Kaddis JS, Perry DJ, Vu AN, Rich SS, Atkinson MA, Schatz DA, et al. Improving the Prediction of Type 1 Diabetes Across Ancestries. Diabetes Care (2022) 45(3):e48–50. doi: 10.2337/dc21-1254
207. Robertson CC, Inshaw JRJ, Onengut-Gumuscu S, Chen WM, Santa Cruz DF, Yang H, et al. Fine-Mapping, Trans-Ancestral and Genomic Analyses Identify Causal Variants, Cells, Genes and Drug Targets for Type 1 Diabetes. Nat Genet (2021) 53(7):962–71. doi: 10.1038/s41588-021-00880-5
208. Manousaki D, Dudding T, Haworth S, Hsu YH, Liu CT, Medina-Gomez C, et al. Low-Frequency Synonymous Coding Variation in CYP2R1 has Large Effects on Vitamin D Levels and Risk of Multiple Sclerosis. Am J Hum Genet (2017) 101(2):227–38. doi: 10.1016/j.ajhg.2017.06.014
Keywords: type 1 diabetes, risk gene variants, immunoregulation, Tregs, tolerogenic dendritic cells, vitamin D
Citation: Gootjes C, Zwaginga JJ, Roep BO and Nikolic T (2022) Functional Impact of Risk Gene Variants on the Autoimmune Responses in Type 1 Diabetes. Front. Immunol. 13:886736. doi: 10.3389/fimmu.2022.886736
Received: 28 February 2022; Accepted: 08 April 2022;
Published: 04 May 2022.
Edited by:
Yi-Guang Chen, Medical College of Wisconsin, United StatesReviewed by:
Karen Cerosaletti, Benaroya Research Institute, United StatesCopyright © 2022 Gootjes, Zwaginga, Roep and Nikolic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chelsea Gootjes, Yy5nb290amVzQGx1bWMubmw=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.