 Fiorella Rossi
Fiorella Rossi Uriel Y. Moreno-Nieves
Uriel Y. Moreno-Nieves
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 02 June 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.886429
This article is part of the Research Topic Innate Immune Cell Therapy of Cancer View all 11 articles
In recent years, immunotherapy for cancer has become mainstream with several products now authorized for therapeutic use in the clinic and are becoming the standard of care for some malignancies. Chimeric antigen receptor (CAR)-T cell therapies have demonstrated substantial efficacy for the treatment of hematological malignancies; however, they are complex and currently expensive to manufacture, and they can generate life-threatening adverse events such as cytokine release syndrome (CRS). The limitations of current CAR-T cells therapies have spurred an interest in alternative immunotherapy approaches with safer risk profiles and with less restrictive manufacturing constraints. Natural killer (NK) cells are a population of immune effector cells with potent anti-viral and anti-tumor activity; they have the capacity to swiftly recognize and kill cancer cells without the need of prior stimulation. Although NK cells are naturally equipped with cytotoxic potential, a growing body of evidence shows the added benefit of engineering them to better target tumor cells, persist longer in the host, and be fitter to resist the hostile tumor microenvironment (TME). NK-cell-based immunotherapies allow for the development of allogeneic off-the-shelf products, which have the potential to be less expensive and readily available for patients in need. In this review, we will focus on the advances in the development of engineering of NK cells for cancer immunotherapy. We will discuss the sourcing of NK cells, the technologies available to engineer NK cells, current clinical trials utilizing engineered NK cells, advances on the engineering of receptors adapted for NK cells, and stealth approaches to avoid recipient immune responses. We will conclude with comments regarding the next generation of NK cell products, i.e., armored NK cells with enhanced functionality, fitness, tumor-infiltration potential, and with the ability to overcome tumor heterogeneity and immune evasion.
Cancer is a major public health problem worldwide and is the second leading cause of death in the United States, after cardiovascular disease (1, 2). Although a reduction in smoking and improvements in early detection and treatment have lowered the death rate of certain malignancies, such as non-small cell lung cancer (NSCLC), the overall survival of patients with late-stage malignancies remains poor—substantiating the need for novel therapeutic options. Treatment regimens for cancer have long consisted of surgery, radiation, and chemotherapy; but recently, a new approach has been on the rise: cancer immunotherapy.
Inspired by the natural capacity of our immune system to recognize and prevent tumor progression, via the concerted action of an array of immune cells and humoral factors, multiple immunotherapies have been developed (3). Early clinical studies of the therapeutic potential of stimulating T cells in cancer patients evidenced that administration of interleukin-2 (IL-2) can lead to durable tumor regressions in advanced cancers (4). Approval of IL-2 treatment for metastatic melanoma and renal cancer by the US Federal and Drug Administration (FDA) kickstarted a new era of cancer therapy and brought the immunotherapy in the spotlight (4, 5). The immuno-therapeutic arsenal for cancer continued to expand with the approval of immune checkpoint inhibitors (ICIs), which are directed against inhibitory molecules expressed on the surface of T cells or antigen-presenting cells (APCs) such as PD-1 and PD-L1, respectively, in several indications (6). More recently, chimeric antigen receptor (CAR)-T cells have shown therapeutic efficacy in treating refractory hematological malignancies, and several CAR-T cell therapy products have been approved by the FDA (7).
Despite their efficacy in treating cancer, current CAR-T cells therapies have limitations including life-threatening adverse events such as cytokine release syndrome (CRS) and neurotoxicity; additionally, CAR-T cell therapies are potentially susceptible to antigen escape of tumor cells and can cause detrimental on-target off-tumor effects (8). Additional limitations of current CAR-T cell therapy include complex and personalized manufacturing with time-sensitive and complex vein-to-vein logistics and the need of specialized centers for its application to patients.
Natural killer (NK) cells are a population of immune effector cells with potent anti-viral and anti-tumor activity and have recently emerged as a candidate for cancer immunotherapy. The biology of NK cells could potentially overcome some of the limitations of existing CAR-T cell products. For example, NK cells can recognize and kill tumor cells without the need of prior stimulation (9, 10). In addition, the response of NK cells is not major histocompatibility complex (MHC) restricted but relies on the ligation of multiple germline-encoded receptors, which enables CAR-NK cells to recognize and eliminate tumor cells even in the event of antigen loss or downregulation. Furthermore, multiple clinical trials have shown that NK cells are well tolerated and may have a safer profile than T-cell-based therapies (11).
In this review, we discuss advances on the development of engineered NK cells for cancer immunotherapy, including the sourcing of NK cells and the move towards off-the-shelf modalities, technologies to engineer NK cells, current clinical trials using engineered NK cells, advances on the engineering of receptors, and stealth approaches to avoid host immune response. In addition, we will comment on the next generation of NK cell products, i.e., armored NK cells with enhanced functionality, fitness, tumor-infiltration potential, and with the ability to overcome tumor heterogeneity and immune evasion.
NK cells belong to group 1 innate lymphoid cells (ILCs) (12–15) and are characterized by their innate capacity to swiftly detect and kill aberrant cells, such as virus-infected cells and cancer cells (9, 10, 16). NK cells exhibit potent anti-tumor responses through multiple mechanisms and are functionally similar to cytotoxic CD8+ T cells.
Several populations of NK cells have been described. Most of our knowledge about human NK cells comes from peripheral blood NK (PBNK) cells, given that this population can be easily sampled. PBNK cells can be divided into two distinct subsets based on their expression of CD56 (also known as neural cell adhesion molecule 1): CD56dim NK cells represent a mature population characterized by their innate cytotoxic potential, and CD56bright NK cells represent a less mature population characterized by their immunomodulatory potential owing to their ability to secrete large amounts of cytokines upon stimulation (9, 16, 17). Additional populations of NK cells have been identified in multiple tissues where they play specialized immune functions. Tissue-resident NK cells have been observed in tissues such as the liver, gut, lungs, uterus, and kidney [reviewed elsewhere (18–21)].
Although NK cells belong to the innate immune system, certain populations of NK cells can display features attributed to the adaptive immune system such as antigen specificity and memory-like responses. Indeed, NK-cell populations mediating robust memory-like responses have been reported in the context of viral infections, contact hypersensitivity reactions, and after pro-inflammatory cytokine stimulation (22–25).
Unlike T cells, NK cells lack an antigen-specific T-cell receptor (TCR); instead, their activity is controlled by an array of germline-encoded receptors, both activating and inhibitory, that enable NK cells to sense their environment (26). The response of NK cells is modulated by the integration of signals delivered via activating and inhibitory receptors, and the balance of these signals determines the outcome of the interaction between NK cells and their environment, i.e., killing of target cells, secretion of cytokines, or no response.
Several activating receptors have been identified in NK cells; they recognize ligands expressed upon cell stress, viral infection, or tumor transformation. The major activating receptors involved in target cell recognition include NKG2D and natural cytotoxicity receptors (NCRs; namely, NKp46, NKp30, and NKp44). In addition, NK cells express the low-affinity Fc receptor CD16, which enables them to detect antibody-coated target cells and to exert antibody-dependent cell cytotoxicity (ADCC) (10, 26). NK cells are negatively regulated by inhibitory receptors, most of which ligate MHC class I molecules and gauge the level of expression of self-molecules on adjacent cells (10, 26–28). Human leukocyte antigen class I (HLA-I)–specific inhibitory receptors include the killer cell immunoglobulin-like receptors (KIRs) and the lectin-like CD94-NKG2A heterodimers. The interaction between self-MHC molecules and their cognate KIRs during NK cell development provides essential inhibitory signals for NK cell maturation and contributes to their education, i.e., their acquisition of functional competency (27, 28).
NK cells can kill target cells in a perforin-dependent manner, where following the formation of a lytic immunological synapse, preformed lytic granules containing perforin and granzymes converge toward the synapse and are released into the synaptic cleft (29). Perforin molecules form pores in the postsynaptic membrane of target cells allowing granzymes to enter the target cell and activate caspases, resulting in the apoptosis of the target cell (29–31). NK cells are protected from perforin-mediated autolysis by densely packed and highly ordered presynaptic lipid membranes (32). NK cells can also kill target cells in a perforin-independent manner, via the expression of FAS ligand (FasL) and tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) (9). Additionally, NK cells have immunomodulatory potential owing to their ability to secrete cytokines, chemokines, and growth factors, including interferon gamma (IFN-γ), TNF-α, CCL5, XCL1, and granulocyte-macrophage colony-stimulating factor (GM-CSF). As such, NK cells can positively or negatively influence the anti-tumor responses by modulating innate and adaptive immune cells (30, 33, 34).
NK cells are key players of tumor immunosurveillance—they have the innate ability to differentiate healthy from malignant cells and mount an immune response following recognition of transformed cells. The clinical relevance of NK cells in cancer has been investigated, both in hematological and solid malignancies.
The abundance of NK cells has been correlated with prognosis in some hematological conditions, such as chronic lymphocytic leukemia (CLL), diffuse large B-cell lymphoma (DLBCL), T-cell lymphoma, and multiple myeloma (MM) (35). However, in acute lymphoid leukemia (ALL), the presence of NK cells in the bone marrow of children at the time of diagnosis was associated with favorable response to treatment and survival (36). A retrospective study of Hodgkin’s lymphoma (HL) patients showed that low numbers of infiltrating NK cells were associated with unfavorable clinical outcome (37). A prospective study of the relationship between the phenotype of NK cells at the time of ALL diagnosis and the minimal residual disease (MRD) at the end of induction chemotherapy showed that the presence of NK cells with a strong effector phenotype was associated with better leukemia control (38).
Additionally, evidence of the clinical relevance of NK cells in hematological malignancies comes from studies that linked mutations in genes essential for NK cell anti-tumor function with occurrence of cancer. As such, mutations of PRF1 (encoding for perforin) are frequently found in patients with anaplastic large cell lymphoma (ALCL) and ALL, and mutation of FASLG (encoding for FasL) was observed in lymphoma patients (39–41). The secretion of IFN-γ by NK cells was found to be a positive prognostic marker in chronic myeloid leukemia (CML) (35), while reduced NK cell activity was observed in multiple malignancies, including advanced MM (42), and was associated with high-risk myelodysplastic syndrome (MDS) (43).
Concerning solid tumors, multiple studies in the early 2000s showed that increased infiltration of NK cells, based on CD57 expression, served as a positive prognosis factor in patients suffering from various malignancies. It was shown that increased infiltration of NK cells in squamous cell lung carcinoma, colorectal carcinoma, gastric carcinoma, and pulmonary adenocarcinoma correlated with better prognosis and survival of patients (44–47). In addition, in an 11-year follow-up prospective study of a cohort of Japanese general population, it was found that higher NK cell cytotoxicity was associated with reduced cancer risk, whereas low cytotoxicity was associated with increased cancer risk—in this cohort, the most frequent cancers identified were stomach, lung, and intestine (48).
More recently, a meta-analysis highlighted the important prognostic value of NK cell infiltration into a variety of solid tumors. High levels of NK cell markers in solid tumor tissues predicted favorable prognosis for solid tumor patients (49). Indeed, increased NK cell infiltration correlated with decreased risk of death; and in terms of localization, intraepithelial infiltration was more predictive of survival than NK cell infiltration into the tumor-adjacent stroma (50).
Cursons et al. showed that patients with metastatic melanoma have an improved survival rate if their tumor has a gene signature predicting NK cell infiltration, and high expression of IL-15 was associated with higher survival (51). IL-15 is an important cytokine for NK cell homeostasis and activation, and the presence of IL-15 within the TME was associated with NK cells with high anti-tumor function in head and neck squamous cell carcinoma (HNSCC) (52).
Additionally, NK cells were identified as a robust prognostic and predicative factor for chemotherapy outcome in gastric cancer (53). In metastatic melanoma patients undergoing ICI therapy, NK cell infiltration into tumor was correlated with favorable response to anti-PD-1 therapy, even in the event of tumor MHC-I downregulation (54).
Considering the association of NK cells with prognosis in several tumor malignancies and that NK cells can target tumor cells that have downregulated HLA-I molecules—an immune evasion mechanism often correlated with worse prognosis of cancer patients (55–57)—there is a window of opportunity to evaluate NK cell therapies, particularly in patients who have failed conventional immunotherapy due to tumors displaying reduced or non-existent HLA class I expression (58, 59).
In the early 1980s, seminal studies by Rosenberg and colleagues showed that exposure of patient-derived peripheral blood mononuclear cells (PBMCs) to IL-2 alone generated cells that were able to recognize and kill tumor cells in vitro (4, 60, 61). These cells, which were named lymphokine-activated killer (LAK) cells, represented a heterogeneous mixture of T cells and NK cells. Phillips and Lanier later showed that a substantial part of the LAK anti-tumor activity was attributed to NK cells (62). Preclinical studies showed that adoptive transfer of LAK cells into tumor-bearing mice resulted in anti-tumor activity, and the concomitant administration of IL-2 with LAK cells increased in vivo anti-tumor activity of LAK cells (4, 63–65). These findings spurred an interest in this novel immunotherapy approach, resulting in clinical trials for advanced cancers using autologous LAK cells in combination with IL-2 infusions. However, in 1993, results from a randomized trial of 181 patients with advanced melanoma or renal cancer comparing treatment with IL-2 alone or in conjunction with LAK cells showed that the observed anti-tumor effects tended to be due to IL-2 alone (66), thereby hampering clinical studies of LAK cells.
Similarly, early clinical studies testing IL-2-activated autologous NK cells for treatment of patients with metastatic melanoma, renal cell carcinoma, relapsed lymphoma, and metastatic breast cancer were ineffective (67, 68), suggesting that in autologous settings, inhibitory signals from self-MHC molecules in tumor cells are likely to suppress NK cell function in the absence of activating signals. These investigators also noted a lack of persistence of the infused NK cells as a potential limitation.
In patients with acute myeloid leukemia (AML) undergoing hematopoietic stem cell transplant (HSCT), Ruggeri et al. showed a correlation between KIR profile and the outcome following HSCT (69), therefore suggesting that stratification of patients by their KIR ligand mismatch can select for patients with alloreactive NK cells that protect from AML relapse. Similar observations were made in additional studies, reporting decreased relapse and increased survival when patients received either HLA- or KIR-mismatched transplants (70–73).
In a landmark study in patients with poor-prognosis AML, Miller et al. found that the adoptive transfer of allogeneic IL-2-activated NK cells combined with lymphodepleting therapy resulted in a marked increase in endogenous IL-15, expansion of donor-derived NK cells, and induction of complete remissions in 26% of the patients (74). This study also highlighted the importance of lymphodepleting therapy in the creation of a cytokine milieu supportive of NK cell expansion. Using similar approaches, clinical responses were also observed when allogeneic NK cells were adoptively transferred into patients with refractory lymphoma and advanced MM (75, 76).
While the initial promising results from allogeneic NK cell adoptive transfer to cancer patients demonstrated the potential of NK cells, recent success of CAR-T cell therapies proved the value of targeting tumors with engineered receptors. These observations fused with the rapid progression of technologies available to engineer NK cells (Table 1) to set the stage for clinical trials of CAR-engineered NK cells in cancer (109).
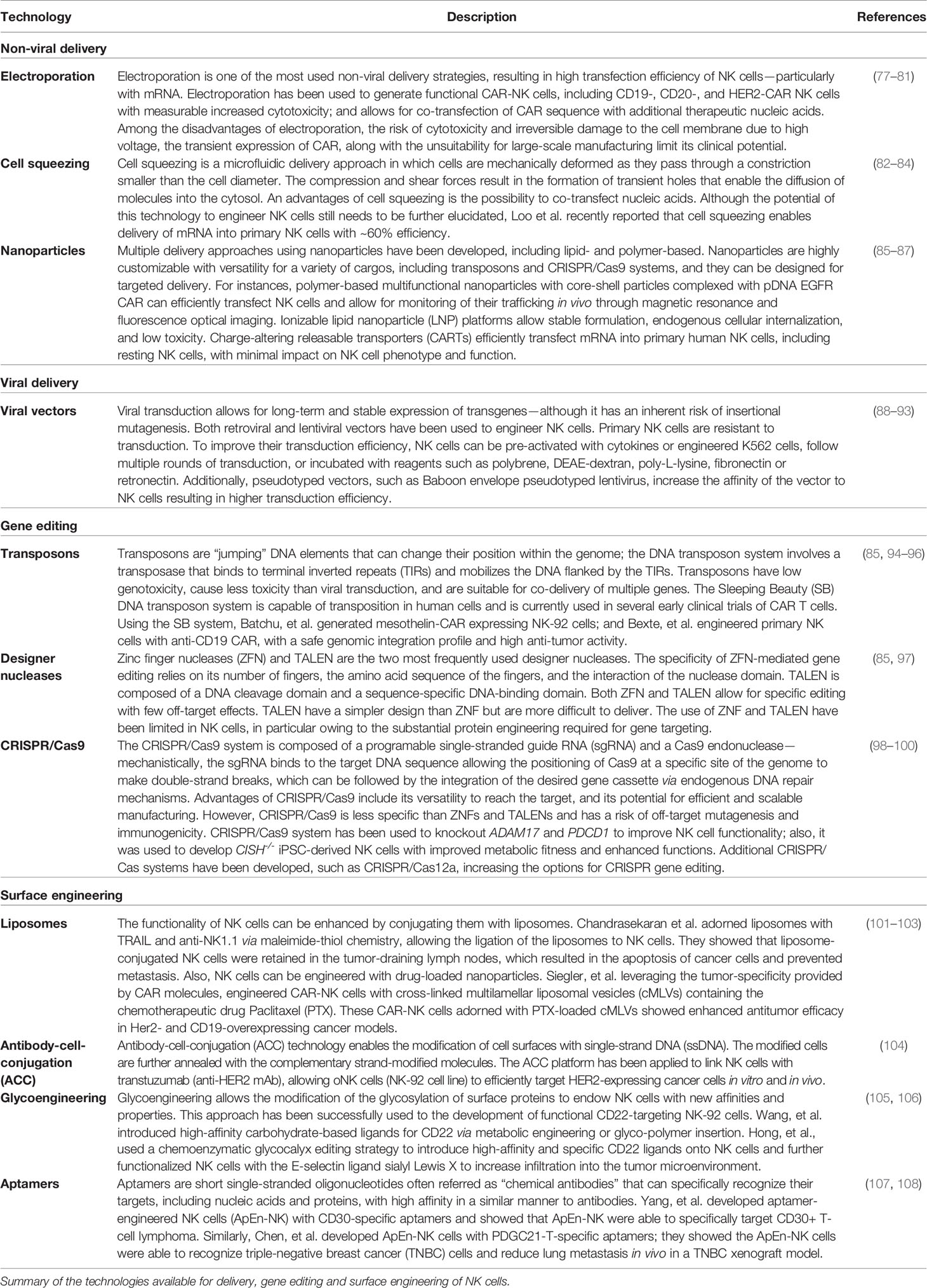
Table 1 Technologies for the engineering of NK cells.
To induce tumor-specific responses, CAR-based therapies rely on the targeting of tumor antigens (TAs) by immune effector cells. Multiple CAR approaches have been tested in preclinical studies of CAR-NK cells that target antigens including CD19, EGFR, HER2, EpCAM, GD2, Mesothelin, and HSP70 [reviewed in (77)].
In 2020, Liu et al. reported results from a phase 1/2 trial using HLA-mismatched, cord-blood derived, anti-CD19 CAR-NK cells for the treatment of relapsed or refractory B-cell malignancies and showed that 73% of patients responded, with seven of eight patients in complete remissions following therapy (110). They also reported that administration of CAR-NK cells was not associated with CRS, neurotoxicity, nor graft-versus-host disease (GvHD), therefore highlighting a safety profile of CAR-NK cell therapy.
The number of clinical trials exploring the potential of engineered NK cells for cancer therapy has steadily increased over time. In the current landscape, there are over 20 ongoing clinical trials using CAR-NK cells for hematological and solid indications (Table 2). One attractive CAR-NK cell strategy is the use of multiplexed induced pluripotent stem cells (iPSC)-derived NK (iNK) cells engineered to express CARs alongside additional edits, including IL-15 receptor fusion and high-affinity CD16 (114). Multiple clinical trials are evaluating iNK cells in hematological malignancies, such as MM, AML, CCL, and B-cell lymphoma, in combination with antibody therapy (daratumumab, elotuzumab, rituximab, and obinutuzumab, respectively), and iNK cells are being trialed in solid tumors, including ovarian cancer (Table 2).
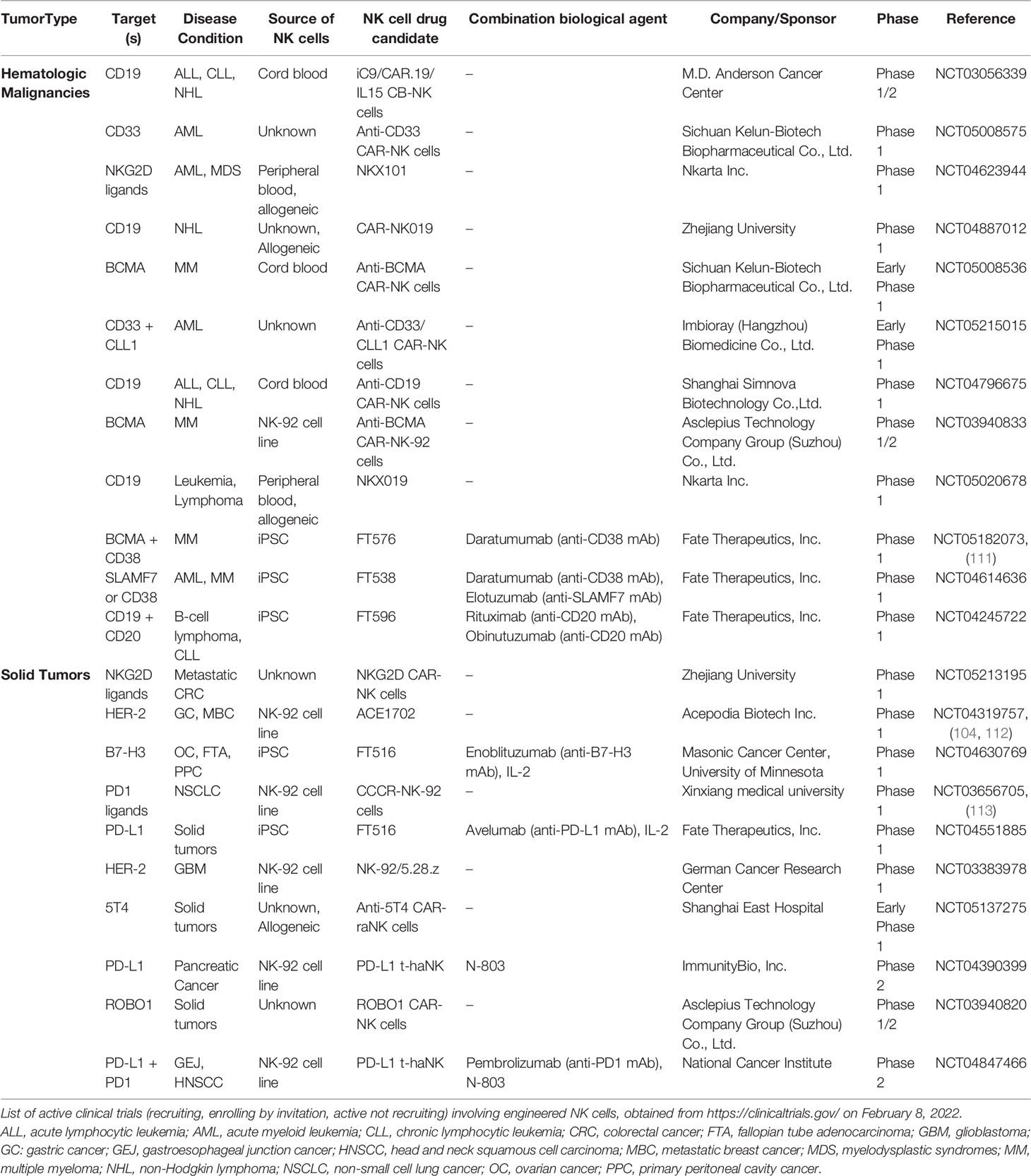
Table 2 List of ongoing clinical trials utilizing engineered NK cells.
Overall, clinical trials have demonstrated that NK cells possess potent anti-tumor effects without eliciting serious adverse effects associated with T-cell therapy, such as GvHD (115, 116), neurotoxicity (117), or cytokine release syndrome (118).
CAR-expressing NK cells are derived from a variety of sources, including peripheral blood (PB), umbilical cord blood (CB), iPSCs, and NK cell lines (Figure 1). Recent reviews have described the sources of NK cells in detail, with their advantages and caveats (77, 88).
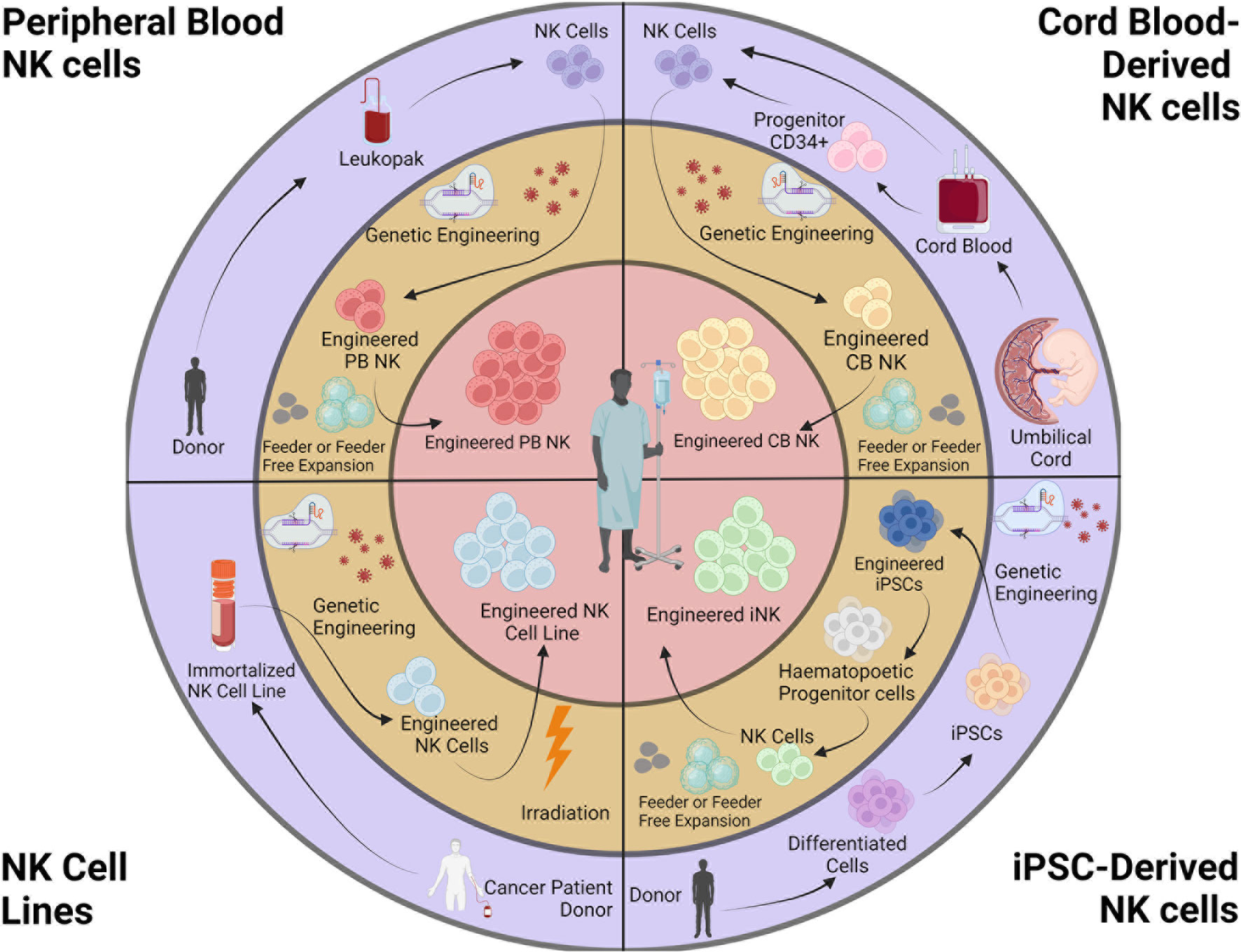
Figure 1 Sources and engineering of NK cell products. NK cells for cancer immunotherapy can be obtained from peripheral blood (autologous or allogeneic), cord blood, iPSCs, and NK cell lines. Isolated NK cells are genetically engineered and expanded. NK cell lines are irradiated before infusion. This figure was created using BioRender.
Whereas obtaining a suitable source of autologous PBNKs is a challenge because patients typically have received prior therapies, CBNKs typically exhibit a naive phenotype including lower expression of adhesion molecules (e.g., CD2, CD11a, CD18, and CD62L), CD16, KIRs, perforin, and granzyme B, resulting in decreased cytotoxicity (119, 120). NK cells can also be derived from cord-blood hematopoietic progenitor CD34+ cells (121, 122). However, these sources have the limitation of poor product standardization due to the heterogeneous nature of PB and CBNKs. Genetically modifying these primary NK cells remains highly variable using currently available technologies, resulting in difficulties developing consistent and reproducible engineered NK cells (123). Although the cancer-derived NK cell line NK-92 can overcome the challenges above, the obvious safety requirement for being mitotically inactivated by irradiation creates significant limitations to potential clinical use with patients (124). Without the ability for proliferation upon infusion, their anti-tumor activity is rapidly reduced overtime when compared to alternative NK cell therapies (74, 125). Indeed, a phase I study with NK-92 cells transduced with a CAR targeting CD33 were tested in patients with relapsed or refractory AML but showed no durable responses partly due to lower persistence and efficacy due to irradiation prior to treatment (126).
On the contrary, human iPSCs are a source for cell therapy that can be genetically engineered via established methods, expanded and produced indefinitely in a homogenous and limitless manner (127, 128), and can be differentiated into iNK cells (129). iPSC-derived NK cells have inherent safety considerations; indeed, given that iPSC can proliferate indefinitely, careful analysis of the final iNK product needs to be undertaken to ensure that it is free from residual iPSCs (130). These benefits can be translated into a highly versatile and standardized off-the-shelf iNK cell therapy to treat various malignancies (110, 131), with the possibility to make multiple precision edits at the single-cell level to produce banks of homogeneous NK-cell products with improved persistence, tumor targeting, homing, and functionality (132) (Figure 2).
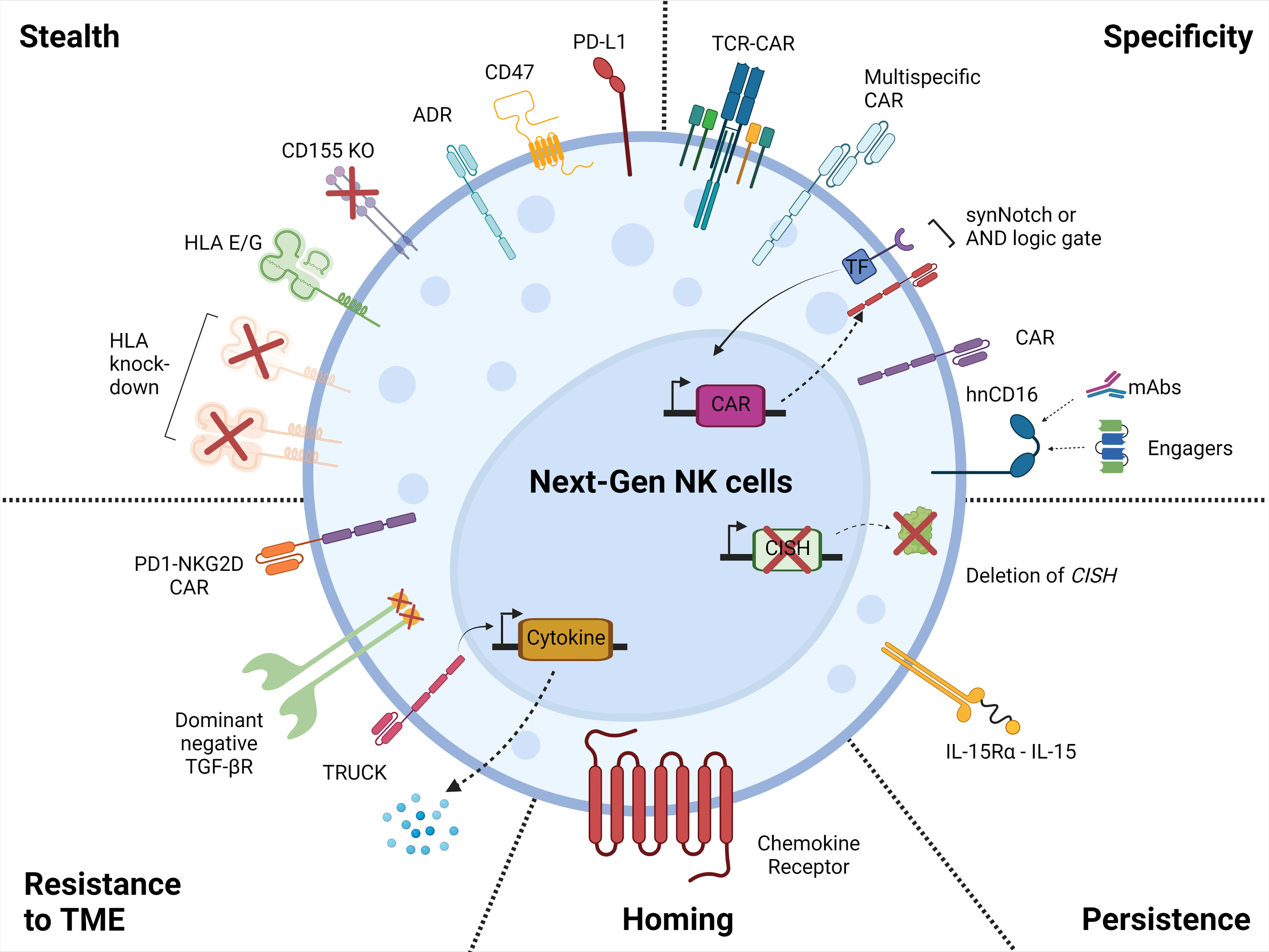
Figure 2 Next generation NK cell products. Illustration of the attributes of the next-generation NK cell products with increased tumor-targeting specificity, persistence, homing, resistance to the tumor microenvironment, and with stealth capabilities. This figure was created using BioRender.
Engineering CARs onto an immune cell redirects their specificity onto a particular antigen. The first proof of concept for antigen specificity stemmed from combining the signaling of a T cell with the antigen specificity of an antibody via fusing the variable regions of an antibody with the constant region of TCR (133, 134). This CAR technology was developed further by utilizing the commonly utilized antigen-binding domain known as single-chain variable fragment (scFv; containing the variable heavy and light chains from an antibody) leading to “first-generation” CAR constructs. These have proven to be capable of eliciting tumor-specific cytotoxicity, as demonstrated by human epidermal growth factor receptor 2 (HER2)-specific CARs using scFv recognizing HER2 fused to a CD3ζ signaling domain for activation (signal 1) (135, 136). However, these first-generation CARs failed to elicit significant anti-tumor responses due to limited persistence (137) and expansion in vivo (138).
Endogenous TCR cellular signaling requires both the TCR together with costimulatory (signal 2) or accessory molecules to elicit a robust response, which can be adapted into a single CAR molecule via molecular engineering. By taking advantage of the modular nature of CAR receptor technology, this allowed for the continual refinement and modification of these engineered proteins to optimize functionality, leading to improved second- and third-generation CARs containing one or two costimulatory domains, respectively. Despite promising results for various hematological malignancies (110, 139–143), the therapeutic efficacy of CAR-T approaches was limited in both hematological (143, 144) and solid tumors (145, 146). These limitations have been related to diminished cytotoxicity, inefficient trafficking, and infiltration of tumors in an immunosuppressive environment where heterogeneity in tumor target expression can often occur. Initiatives to overcome these shortcomings have led to various strategies including engineering fourth-generation CARs to produce and release a transgenic product, such as pro-inflammatory cytokine IL-12, which is constitutively or inducibly expressed upon CAR activation (147, 148). T cells that are transduced with these fourth-generation CARs are generally referred to as T cells redirected for universal cytokine-mediated killing (TRUCKs) and will be further discussed in later sections.
The development of next-generation NK cells for cancer therapy has been facilitated by the emergence of sophisticated technologies enabling the engineering of NK cells (Table 1) in terms of delivery (viral vectors, electroporation, cell squeezing, and nanoparticles), gene editing (transposons, designer nucleases, and CRISPR/Cas systems), and surface engineering (liposomes, antibody–cell conjugation, glycoengineering, and aptamers).
Engineering genetic modifications to augment persistence and functionality can be easily performed using a single-cell iPSC engineering platform. This was demonstrated in iNK cells with the addition of a modified high-affinity version of CD16a (149). NK cells naturally express CD16a, which binds the Fc portion of immunoglobulin G (IgG) attached to target cells and induces ADCC. Engagement of CD16a alone is sufficient for inducing NK cell activation, leading to secretion of various inflammatory cytokines and chemokines for the recruitment and activation of other immune cells (150–152). However, CD16a undergoes rapid proteolytic cleavage upon stimulation mediated by ADAM17 (153–155). Substitution of serine at position 197 located in the middle of the cleavage region with proline (S197P) effectively prevents cleavage of the intact and functional receptor (156). In addition, the binding affinity of CD16a varies between allelic variants (157). This led to the discovery of a high-affinity CD16a variant with valine at position 158 (158V) (158); patients with this variant had greater objective responses and progression-free survival when treated with mAb therapy such as rituximab (159), cetuximab (160), or trastuzumab (161). Engineered iNK cells with a high-affinity non-cleavable version of CD16a (hnCD16) displayed enhanced ADCC effector function when combined with anti-tumor mAbs (131).
NK cells do not naturally produce signal 3 cytokines such as IL-2, IL-7, or IL-15, which is in contrast to polyclonal CAR-T cell therapies that, upon CAR engagement, produce significant amounts of IL-2 that supports their expansion (162). Because NK cells rely on signal 3 for expansion, survival, and cytotoxicity, investigators have focused on ways to provide this signal to NK cell therapies, either exogenously or by design in the NK cell product. Optimizing CAR signaling with the addition of signal 3, which includes the transgenic expression of cytokines or a chimeric cytokine receptor, further enhances cytotoxicity and persistence of NK cells. The TME is often deficient in signal 3, thus hampering the potential degree of anti-tumor response of cell therapies. As previously mentioned, fourth-generation TRUCK CAR-T cells have been engineered to minimize systemic toxicity and to induce targeted accumulation of cytokines at the tumor site by providing signal 3. To enhance both NK durability and host immune responses, initial clinical trials focused on the administration of cytokines such as IL-2 and IL-15 for safety and efficacy. However, IL-2 administration resulted in severe systemic toxicity (163), which prompted the focus of investigations onto IL-15. In contrast to IL-2, treatment with IL-15 did not promote expansion of inhibitory Tregs (164) and activation-induced cell death (AICD) of T cells (165). Clinical trials combining NK cell therapy and IL-15 cytokine support resulted in limited anti-tumor responses in patients due to a short half-life after delivery (166, 167). To enhance and maintain the therapeutic effectiveness of IL-15, a membrane-bound IL-15/IL-15 receptor fusion (IL-15RF) was generated, resulting in increased persistence, proliferation, and enhanced cytotoxicity in iNK cells (131, 168). Deletion of cytokine-inducible SH2-containing protein (CIS, encoded by CISH), a negative regulator of IL-15 signaling, further improved cytotoxic effector function, metabolic fitness, and in vivo persistence of iNK cells (98, 169).
Heterogeneous antigen expression in tumor cells led to the development of CAR-T cells targeting simultaneously two or more tumor-associated antigens either by a tandem (170–172) or split CAR construct configuration (173). Indeed, both anti-CD19 and anti-CD22 CAR-T cell therapies have shown impressive efficacy; however, patients have shown reduced antigen density at relapse, suggesting tumor-antigen-specific downregulation as a mechanism for immune escape (140, 174). Currently, the optimal strategy for multi-targeting is being investigated between tandem or separate CAR constructs in T cells regarding safety and efficacy of these constructs (170, 172, 175). However, having a heterogeneous mixed product resulting from transducing T cells with either a tandem or bicistronic CAR has proven to be a complex manufacturing procedure and makes mechanistic investigations difficult. This could potentially be overcome by utilizing a renewable source for consistent and selective gene editing (131). In another example, targeting B-cell maturation antigen (BCMA) has shown extremely promising and potentially durable objective response rates in MM (139). However, gamma-secretase (GS)-mediated cleavage of BCMA releases soluble BCMA fragments that have been shown to be capable of inhibiting BCMA-CAR function, leading to the testing of combinatorial therapy with GS inhibitors to prevent antigen escape (176). Targeting pan-cancer antigens such as CD276 (B7-H3) has also shown tremendous success in treating various solid malignancies; however, their success is largely dependent on the level of surface antigen density (177). Although multi-targeting or combinatorial cleavage inhibition may address tumor immune evasion through antigen loss, it does not address other resistance mechanisms such as inhibition by engagement of PD-L1 on tumor cells (178) or navigating through the immunosuppressive TME.
While these multi-specific CAR strategies are developed to target tumor-associated antigens—which may be also expressed in healthy tissues—they could result in severe on-target off-tumor toxicities similar to those seen in specific cases with some CAR-T cell therapy trials (179–181). Strategies to circumvent this issue have led to engineering approaches to insert logic gates into the cells, resulting in activation in the presence of combinations of target antigens. Utilizing a split CAR model by separating signal 1 and signal 2 into different CARs, each with a binder for a different antigen, has now led to the development of an AND logic gate (182). Although intracellular CD3ζ domain for signal 1 by itself is sufficient for signaling, Kloss et al. modified the CD3ζ domain to make it insufficient to produce an activating signal without the co-stimulation by a secondary CAR with signal 2 (183). Another strategy of utilizing AND logic gating is by inducing expression of the primary tumor-targeting CAR only in response to a secondary antigen such as the SynNotch system (184). To overcome the potential off-target recognition of healthy cells, inhibitory NOT logic gates can be engineered to ignore cells that express a particular antigen that is expressed in healthy tissue but not in tumors. The NOT logic CAR was initially conceived by fusing the intracellular domain of PD1 or CTLA4 with a targeting domain recognizing antigens expressed on healthy tissues, thus allowing the primary tumor-targeting CAR to selectively kill tumor cells (185). Naturally, for any of these approaches to work requires identification of combinatorial expression patterns that are truly unique to tumors. In addition, a distinct separation between healthy and tumor cells must be present; otherwise, unintended activation or inactivation may still potentially occur. The recent improvements in engineering multiple edits into cell therapy products demonstrate the benefits of logic circuitry if the appropriate antigen combinations can be identified.
Beyond the use of exquisite, but synthetic, logic circuitry for efficient tumor targeting comes augmenting cells with CARs based on natural cytotoxicity ligands native to NK cells to target heterogeneous tumors. Several groups have demonstrated the use of NKG2D-based CAR therapy with unique intracellular signaling domains, such as a second-generation CD3ζ/CD28 (186) or NK-inspired DAP12 (187), to target various tumor and immunosuppressive cells. Alternatively, NKp46 (188), NKp44 (189), and NKp30 (190) have each been fused to generate second-generation CAR-T cells that further demonstrates the benefit of tumor recognition capability of natural NK ligands for anti-tumor efficacy. This concept was taken further by utilizing an scFv targeting B7H6, a ligand for NKp30, to create a CAR that targets multiple tumor cells while demonstrating an impressive safety profile, since B7H6 is not constitutively expressed on healthy tissues (191). Indeed, utilizing NK ligands for the recognition of heterogeneous tumor targets provides a selective but effective way to overcome evasion by tumor cells to T-cell therapies. Alternatively, NK-insensitive cancer cell lines that are non-targetable by NKG2D-mediated cytotoxicity have resulted in the generation of NK cells expressing a TCR-CAR (192). This allows the TCR-CAR to redirect cytotoxic effector cells such as NK-92 cells by endowing them the ability to recognize tumor cells typically elusive to NK cell detection. Indeed, expressing the TCR in NK cells could greatly enhance the range of targetable antigens by enabling recognition of tumorigenic neoantigens within the intracellular proteome. However, this strategy can potentially give rise to TCR-mediated GvHD. There is also a limitation due to the diversity of MHC alleles and the likelihood that a particular TCR will only recognize a neoantigen when it is presented by a specific MHC allele. Thus, TCRs would have to be selected to recognize a neoantigen presented in a wide range of MHC alleles to minimize GvHD. One potential clone, MC.7.G5, appeared to recognize a currently unknown cancer-specific molecule presented by the non-polymorphic protein MHC-related 1 (MR1) (193). Since MR1 is a member of a family of non-classical MHC proteins, this TCR should enable recognition of cancer cells in a wide range of patients while minimizing off-target effects. Indeed, further investigation will be required for TCR-CARs to assess whether their off-target potential is worth their clinical impact.
Successful treatment of solid tumors has been elusive in part due to the immunosuppressive nature of the TME. Tumor cells can evade immune surveillance by secreting or promoting the secretion of transforming growth factor beta (TGF-β) (194). Because of its suppressive role in the TME, TGF-β has been targeted to boost cell therapy anti-tumor response. TGF-β mediates downregulation of NKG2D, NKp30, TRAIL, and DNAM1 receptors on activated NK cells (195, 196). To shield adoptive NK cell therapies from the suppressive effects of TGF-β, introduction of a dominant negative form of TGF-β type II receptor (TGF-βRII) efficiently blocked TGF-β signaling and maintained cell surface expression of receptors and cytotoxicity in NK and T cells (197–199). Elegant strategies embracing the inhibitory cytokine and converting it into a potent stimulatory signaling have been created by rewiring the recognition domain into a second-generation CAR-T cell to orchestrate upregulation of cytokine production and proliferation (200). Similarly, expressing a CAR with a TGF-βRII extracellular and transmembrane domains combined with the intracellular domain of NKG2D on NK-92 cells converted the immunosuppressive signal into increased cytotoxicity while preventing downregulation of NKG2D surface expression (201). This strategy has also been applied to other inhibitory receptors such as PD-1, generating a PD-1 CAR with NK-tailored endodomains such as NKG2D or DAP10/NKG2D to mediate cytotoxicity by NK cells against solid malignancies in the TME (202, 203).
In addition to improving effector function within a hostile immunosuppressive environment, NK cell trafficking and retention within tumor sites is essential for optimal anti-tumor efficacy. Inducing expression of CCR7 on NK cells enhanced migration and homing to the lymph node-associated chemokine CCL19 for CD16 and rituximab-mediated ADCC against hematological malignancies (78, 204). Augmenting CAR-NK cells targeting the glioma antigen epidermal growth factor variant III (EGFRvIII) with CXCR4 expression conferred enhanced chemotaxis to U87-MG glioblastoma cells that secrete CXCL12/SDF-1α, a CXC chemokine that binds to receptors CXCR4 and CXCR7 (205). Furthermore, inducing expression of CXCR1 in NK cells with a NKG2D CAR were shown to significantly increase anti-tumor responses in subcutaneous and intraperitoneal xenograft models along with an intravenous injection model against established peritoneal ovarian cancer xenografts (206).
NK cells have an advantage over T cells for allogeneic cell therapies because they bypass the risk of GvHD driven by αβ T-cell receptors. However, host-versus-graft (HvG) immune rejection by recipient immune cells and antibodies could limit the expansion and/or persistence of allogeneic NK cells, thus impairing their efficacy. Allogeneic HvG is the rejection of non-self donor cells due to genetic polymorphisms between the donor and recipient. In humans, this response is primarily directed against polymorphic HLA genes (major mismatch), polymorphisms in non-HLA proteins leading to “non-self” peptides presented on shared HLA alleles (minor mismatch), and red blood cell antigens (such as ABO and Rh antigens). Since NK cells do not express red blood cell antigens (207), strategies to avoid HvG for NK cell therapies are focused on avoiding rejection due to major and minor mismatch mechanisms, which are driven by CD8+ T cells, CD4+ T cells, antibodies, and to a lesser extent NK cells (208, 209).
Decades of experience in allogeneic transplantation have shown that immune rejection can occur rapidly after cell or organ transplantation, sometimes within hours if patients have pre-existing antibodies against donor HLA types (210). Allogeneic, haplo-identical (50% HLA-matched) NK cells administered to patients pre-treated with lymphodepletion usually do not persist over a month, sometimes even being eliminated within several days in lower intensity lymphodepletion regimens (74, 75, 211–213). The loss of transferred NK cells tends to coincide with the return of patient lymphocytes to baseline levels, and one study has confirmed de novo generation of an anti-donor T-cell response 9–14 days after NK cell transfer (75). Occasionally allogeneic donor NK cells can engraft and persist long-term in patients, which may be related to the enhanced levels of IL-15 after intense lymphodepletion (74), or the transgenic inclusion of IL-15 in the donor NK cells (110). However, another study showed that systemic IL-15, intended to support donor NK expansion, actually accelerated their loss by stimulating patient CD8+ T cells that may have eliminated the transferred NK cells (212). One approach to mitigate HvG rejection is to further deplete the immune system of the patient, for instance by adding anti-CD52 antibodies to the lymphodepletion regimen. Because CD52 is expressed in all lymphocytes (including NK cells), an engineering strategy to remove CD52 in the transferred cell therapy is required. This method has been used for allogeneic CAR-T cells and could conceivably be extended to allogeneic NK cells, but the deep and sustained depletion of the patient’s immune system can lead to severe toxicities (214). Other researchers have proposed suppressing the HvG response by engineering the cell therapy product with a 4-1BB-targeting CAR that can eliminate activated recipient lymphocytes (215). This alloimmune defense receptor (ADR) has the potential benefits of not requiring additional lymphodepleting agents and enhancing co-stimulation of the transferred cells; however, the safety and feasibility of targeting 4-1BB+ endogenous cells in patients is still unknown.
Many investigators have instead focused on engineering NK cells to be immunologically silent and evade the HvG response, sometimes termed “stealth.” HLA class I contains the polymorphic HLA-A, HLA-B, and HLA-C surface proteins. These molecules are heterodimers that consist of two polypeptide chains: the polymorphic HLA-encoded α chain and β2-microglobulin (β2M). HLA class I molecules are expressed in all nucleate cells and are the anchors to present intracellular peptides to CD8+ T cells (216). If the NK cell therapy is from a donor that does not share all the HLA-A, B, and C alleles of the patient, the patient’s CD8+ T cells will recognize the donor’s HLA molecules as foreign and mount a rejection response (major mismatch). Even if the donor and patient are perfectly HLA matched, patient CD8+ T cells can respond to non-self-peptides presented on the shared HLA class I molecules (minor mismatch). Thus, one mechanism to avoid both major and minor CD8+ T cell HvG is to delete or silence β2M because it is required for the surface expression of all HLA class I molecules. It has been shown that β2M-knockout (KO) NK cells do not induce an allogeneic CD8+ T cell response (217), a finding which has been shown with other cell types, including iPSCs and their derivatives (218, 219).
However, targeting β2M expression creates a problem because HLA class I molecules have an important function as inhibitory ligands for NK cells (220). HLA-C and certain HLA-A and HLA-B alleles are ligands for KIRs. HLA-E, a non-polymorphic HLA class I molecule expressed on all healthy cells, is a ligand for the NKG2A/CD94 receptor. HLA class I interactions with KIRs and NKG2A/CD94 play a major role in self-tolerance of NK cells, such that when these interactions are lost, the balance between activating and inhibitory signals on NK cells is shifted towards activation, resulting in “missing-self” lysis of target cells (26). In addition to potentially making a cell therapy product susceptible to patient NK cells, HLA class I reduction in an NK cell therapy could induce fratricide that limits the expansion and/or survival of the modified NK cells.
One strategy to solve this problem is to force expression of molecules that can inhibit NK cells, such as a transgenic HLA-E-β2M fusion protein (217). Not all NK cells express NKG2A, so other investigators have combined the HLA-E-β2M evasion strategy with genetic deletion of CD155, a ligand for the activating receptor DNAM-1, which further reduces the number of patient NK cells that are triggered by β2M-KO cell therapies (219). This finding was described with iPSC-derived T cells, so whether CD155-KO would also be relevant for NK cell therapies is an open question. Overexpression of HLA-G has also been proposed as a strategy to limit patient NK cell activation, which may also help suppress effector CD8+ T cell responses (221). Another strategy proposed to prevent NK missing-self responses of HLA class I deficient cells is to overexpress CD47 (222). CD47 is a transmembrane protein with a well-described role as a “don’t eat me” signal due to its binding to signal regulatory protein α (SIRPα) on myeloid cells (223) and high CD47 expression on tumor cells is thought to protect tumor cells from immune responses (224). Recently it was found that IL-2 stimulated NK cells upregulate SIRPα and can be inhibited through high levels of CD47 expression on β2M-KO target cells (225). Altogether, the concept to express ligands for inhibitory receptors is a promising strategy to evade patient NK cells but requires careful evaluation to ensure that trans-inhibition does not limit the function of the NK cell therapy product.
An alternative approach to avoid CD8+ T cell HvG while minimizing the induction of NK “missing-self” is to specifically delete the HLA-A, HLA-B, and HLA-C genes while leaving β2M and HLA-E intact to engage NKG2A on patient NK cells (218). This strategy still results in a loss of inhibitory KIR signaling, so it could be enhanced with additional immunosuppressive molecules like PD-L1 and CD47. In another modification of this approach, only HLA-A and HLA-B are deleted, thus providing HLA-C-driven KIR signaling in addition to HLA-E-driven NKG2A signaling (226). Although this approach opens the door to allogeneic CD8+ T cell responses to HLA-C, 12 separate banks could be made that each retain a common HLA-C allele to allow matching with >90% of patients.
Activated NK cells express HLA class II, which, like HLA class I, is also highly polymorphic. During an allogeneic encounter, CD4+ T cells become activated through major or minor mismatch with HLA class II, leading to both enhancement of allo-reactive CD8+ T cells and direct cytotoxicity by CD4+ T cells. Additional genome editing of NK cell therapies could include the disruption of the MHC II trans-activator (CIITA) gene, a required component for HLA class II gene transcription. Several research groups have described using CRISPR technology to generate CIITA-KO hypoimmunogenic iPSC lines, either alone or in combination with β2M-KO (218, 219, 225, 227). Importantly, a cell therapy deficient for both HLA class I and II (e.g., β2M KO plus CIITA KO) will completely avoid host CD8+ and CD4+ T cell responses, in addition to evading anti-donor HLA antibodies that may exist or be generated in the patient.
Myeloid cells are crucial members of the innate immune system, where they are the first responders against infection. However, much is still unknown about the full contribution of these cells during an allogeneic response in humans; for instance, do they specifically respond to allogeneic non-self, or do they simply facilitate host T and B cell responses to allogeneic cells? A study done by Dai et al. showed that naive myeloid cells, specifically monocytes and macrophages, retain innate immune memory after the first encounter with allogeneic cells (228). This specific memory was acquired by the binding of the paired Ig-like receptor-A (PIR-A) on murine monocytes and macrophages with the MHC class I molecule on allogeneic cells. The human ortholog of PIR-A is the leukocyte IgG-like receptor (LILR) family, which contains 11 functional genes that encode five activating (LILRA1, 2, 4–6) receptors, five inhibitory (LILRB1-5) receptors, and one soluble protein (LILRA3) (229, 230). Barkal et al. showed that binding of MHC class I with the inhibitory receptor LILRB1 suppresses macrophage phagocytic activity (231). Given that some LILR genes are highly polymorphic (232), an intriguing possibility is that human myeloid cells may respond to allogeneic cells more vigorously than autologous cells and potentially acquire memory capabilities. Whether this mechanism could limit the persistence of allogeneic NK cell therapies is unknown, but one method proposed to prevent myeloid cell phagocytosis is the overexpression of CD47 (225).
Overall, there are many proposed strategies to avoid the HvG response, and their efficacy will need to be determined in clinical trials. Importantly, better characterization of patient immune responses against administered allogeneic NK cell therapies will facilitate improved stealth approaches in the future.
NK cells have numerous features that make them a promising cell therapy strategy for the treatment of cancer. Many trials using adoptively transferred allogeneic NK cells have demonstrated their favorable safety profile, so the main challenge for NK cell therapies is to enhance their efficacy to the level expected from CAR-T cells. A seminal study has demonstrated that allogeneic NK cells engineered to express a CAR and IL-15 have encouraging anti-tumor activity in lymphoid malignancies (110), but obstacles still remain for scalability, activity in solid tumors, and reliable persistence. Engineered iNK cells offer a highly scalable, off-the-shelf cell therapy. Modifying iNK cells with cytokine signaling and immune-evasion modules will boost their expansion and persistence, further improving clinical benefit from these therapies. Because NK cells integrate multiple receptor–ligand interactions to recognize and destroy target cells, they are naturally suited to limit the chance of tumor antigen escape, synergize with antibody therapeutics, and incorporate logic gates that can fine-tune the specificity of their response. Rapid advances in single-cell sequencing and CRISPR screening promise to deepen knowledge of NK cell signaling networks, enabling future improvements in NK cell therapies that build upon the advantageous biology of NK cells.
MA and UM-N contributed equally to this work. FR, MA, and UM-N wrote the manuscript. NF created the illustrations. All authors contributed to the article and approved the submitted version.
FR, NF, AS, MA, and UM-N are current employees at Janssen Research and Development, LLC, Pharmaceutical Companies of Johnson & Johnson.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bray F, Laversanne M, Weiderpass E, Soerjomataram I. The Ever-Increasing Importance of Cancer as a Leading Cause of Premature Death Worldwide. Cancer (2021) 127:3029–30. doi: 10.1002/CNCR.33587
2. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA: A Cancer J Clin (2021) 71:7–33. doi: 10.3322/CAAC.21654
3. Esfahani K, Roudaia L, Buhlaiga N, del Rincon S V, Papneja N, Miller WH. A Review of Cancer Immunotherapy: From the Past, to the Present, to the Future. Curr Oncol (2020) 27:S87. doi: 10.3747/CO.27.5223
4. Rosenberg SA. IL-2: The First Effective Immunotherapy for Human Cancer. J Immunol (2014) 192:5451–8. doi: 10.4049/JIMMUNOL.1490019
5. Leko V, Rosenberg SA. Identifying and Targeting Human Tumor Antigens for T Cell-Based Immunotherapy of Solid Tumors. Cancer Cell (2020) 38:454–72. doi: 10.1016/j.ccell.2020.07.013
6. Wei SC, Duffy CR, Allison JP. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov (2018) 8:1069–86. doi: 10.1158/2159-8290.CD-18-0367
7. Majzner RG, Mackall CL. Clinical Lessons Learned From the First Leg of the CAR T Cell Journey. Nat Med (2019) 25:1341–55. doi: 10.1038/s41591-019-0564-6
8. Sterner RC, Sterner RM. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Sterner Sterner Blood Cancer J (2021) 11:69. doi: 10.1038/s41408-021-00459-7
9. Caligiuri MA. Human Natural Killer Cells. Blood (2008) 112:461–9. doi: 10.1182/blood-2007-09-077438
10. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of Natural Killer Cells. Nat Immunol (2008) 9:503–10. doi: 10.1038/ni1582
11. Daher M, Melo Garcia L, Li Y, Rezvani K. CAR-NK Cells: The Next Wave of Cellular Therapy for Cancer. Clin Trans Immunol (2021) 10. doi: 10.1002/CTI2.1274
12. Vivier E, Artis D, Colonna M, Diefenbach A, di Santo JP, Eberl G, et al. Innate Lymphoid Cells: 10 Years on. Cell (2018) 174:1054–66. doi: 10.1016/J.CELL.2018.07.017
13. Montaldo E, Vacca P, Vitale C, Moretta F, Locatelli F, Mingari MC, et al. Human Innate Lymphoid Cells. Immunol Lett (2016) 179:2–8. doi: 10.1016/j.imlet.2016.01.007
14. Eberl G, Colonna M, di Santo JP, McKenzie ANJ. Innate Lymphoid Cells: A New Paradigm in Immunology. Science (2015) 348:aaa6566. doi: 10.1126/science.aaa6566
15. Riggan L, Freud AG, O’Sullivan TE. True Detective: Unraveling Group 1 Innate Lymphocyte Heterogeneity. Trends Immunol (2019) 40:909–21. doi: 10.1016/J.IT.2019.08.005
16. Abel AM, Yang C, Thakar MS, Malarkannan S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front Immunol (2018) 9:1869. doi: 10.3389/fimmu.2018.01869
17. Michel T, Poli A, Cuapio A, Briquemont B, Iserentant G, Ollert M, et al. Human CD56bright NK Cells: An Update. J Immunol (2016) 196:2923–31. doi: 10.4049/jimmunol.1502570
18. Hashemi E, Malarkannan S. Tissue-Resident NK Cells: Development, Maturation, and Clinical Relevance. Cancers (2020) 12:1553. doi: 10.3390/CANCERS12061553
19. Peng H, Tian Z. Diversity of Tissue-Resident NK Cells. Semin Immunol (2017) 31:3–10. doi: 10.1016/j.smim.2017.07.006
20. Vitale M, Caligiuri MA, Sivori S. Editorial: Natural Killer Cells in Tissue Compartments. Front Immunol (2020) 11:258. doi: 10.3389/fimmu.2020.00258
21. Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The Broad Spectrum of Human Natural Killer Cell Diversity. Immunity (2017) 47:820–33. doi: 10.1016/j.immuni.2017.10.008
22. Pahl JHW, Cerwenka A, Ni J. Memory-Like NK Cells: Remembering a Previous Activation by Cytokines and NK Cell Receptors. Front Immunol (2018) 9:2796. doi: 10.3389/FIMMU.2018.02796
23. Sun JC, Beilke JN, Lanier LL. Adaptive Immune Features of Natural Killer Cells. Nature (2009) 457:557–61. doi: 10.1038/nature07665nature07665
24. Paust S, Gill HS, Wang BZ, Flynn MP, Moseman EA, Senman B, et al. Critical Role for the Chemokine Receptor CXCR6 in NK Cell-Mediated Antigen-Specific Memory of Haptens and Viruses. Nat Immunol (2010) 11:1127–35. doi: 10.1038/ni.1953
25. Cooper MA, Elliott JM, Keyel PA, Yang L, Carrero JA, Yokoyama WM. Cytokine-Induced Memory-Like Natural Killer Cells. Proc Natl Acad Sci USA (2009) 106:1915–9. doi: 10.1073/pnas.0813192106
26. Sivori S, Vacca P, del Zotto G, Munari E, Mingari MC, Moretta L. Human NK Cells: Surface Receptors, Inhibitory Checkpoints, and Translational Applications. Cell Mol Immunol (2019) 16:430–41. doi: 10.1038/s41423-019-0206-4
27. Shifrin N, Raulet DH, Ardolino M. NK Cell Self Tolerance, Responsiveness and Missing Self Recognition. Semin Immunol (2014) 26:138–44. doi: 10.1016/J.SMIM.2014.02.007
28. Boudreau JE, Hsu KC, Ca JB. Natural Killer Cell Education and the Response to Infection and Cancer Therapy: Stay Tuned. Trends Immunol (2018) 39: 222–39. doi: 10.1016/j.it.2017.12.001
29. Orange JS. Formation and Function of the Lytic NK-Cell Immunological Synapse. Nat Rev Immunol (2008) 8:713–25. doi: 10.1038/nri2381
30. Shimasaki N, Jain A, Campana D. NK Cells for Cancer Immunotherapy. Nat Rev Drug Discovery (2020) 19:200–18. doi: 10.1038/s41573-019-0052-1
31. Gwalani LA, Orange JS. Single Degranulations in NK Cells Can Mediate Target Cell Killing. J Immunol (2018) 200:3231–43. doi: 10.4049/jimmunol.1701500
32. Li Y, Orange JS. Degranulation Enhances Presynaptic Membrane Packing, Which Protects NK Cells From Perforin-Mediated Autolysis. PloS Biol (2021) 19:e3001328. doi: 10.1371/JOURNAL.PBIO.3001328
33. Guillerey C, Huntington ND, Smyth MJ. Targeting Natural Killer Cells in Cancer Immunotherapy. Nat Immunol 2016 17:9 (2016) 17:1025–36. doi: 10.1038/ni.3518
34. Sungur CM, Murphy WJ. Positive and Negative Regulation by NK Cells in Cancer. Crit Rev Oncog (2014) 19:57. doi: 10.1615/CritRevOncog.2014010805
35. Gonzalez-Rodriguez A, Villa-Álvarez M, Sordo-Bahamonde C, Lorenzo-Herrero S, Gonzalez S. NK Cells in the Treatment of Hematological Malignancies. J Clin Med (2019) 8:1557. doi: 10.3390/jcm8101557
36. Mizia-Malarz A, Sobol-Milejska G. NK Cells as Possible Prognostic Factor in Childhood Acute Lymphoblastic Leukemia. Dis Markers (2019) 2019. doi: 10.1155/2019/3596983
37. Álvaro-Naranjo T, Lejeune M, Salvadó-Usach MT, Bosch-Príncep R, Reverter-Branchat G, Jaén-Martínez J, et al. Tumor-Infiltrating Cells as a Prognostic Factor in Hodgkin’s Lymphoma: A Quantitative Tissue Microarray Study in a Large Retrospective Cohort of 267 Patients. Leuk Lymphoma (2005) 46:1581–91. doi: 10.1080/10428190500220654
38. Sullivan EM, Jeha S, Kang G, Cheng C, Rooney B, Holladay M, et al. NK Cell Genotype and Phenotype at Diagnosis of Acute Lymphoblastic Leukemia Correlate With Postinduction Residual Disease. Clin Cancer Res (2014) 20:5986–94. doi: 10.1158/1078-0432.CCR-14-0479
39. Yang L, Liu H, Zhao J, Da W, Zheng J, Wang L, et al. Mutations of Perforin Gene in Chinese Patients With Acute Lymphoblastic Leukemia. Leuk Res (2011) 35:196–9. doi: 10.1016/J.LEUKRES.2010.06.016
40. Cannella S, Santoro A, Bruno G, Pillon M, Mussolin L, Mangili G, et al. Germline Mutations of the Perforin Gene are a Frequent Occurrence in Childhood Anaplastic Large Cell Lymphoma. Cancer (2007) 109:2566–71. doi: 10.1002/CNCR.22718
41. Clementi R, Locatelli F, Dupré L, Garaventa A, Emmi L, Bregni M, et al. A Proportion of Patients With Lymphoma may Harbor Mutations of the Perforin Gene. Blood (2005) 105:4424–8. doi: 10.1182/BLOOD-2004-04-1477
42. Jurisic V, Srdic T, Konjevic G, Markovic O, Colovic M. Clinical Stage-Depending Decrease of NK Cell Activity in Multiple Myeloma Patients. Med Oncol (2007) 24:312–7. doi: 10.1007/S12032-007-0007-Y
43. Epling-Burnette PK, Bai F, Painter JS, Rollison DE, Salih HR, Krusch M, et al. Reduced Natural Killer (NK) Function Associated With High-Risk Myelodysplastic Syndrome (MDS) and Reduced Expression of Activating NK Receptors. Blood (2007) 109:4816–24. doi: 10.1182/BLOOD-2006-07-035519
44. Villegas FR, Coca S, Villarrubia VG, Jiménez R, Chillón MJ, Jareño J, et al. Prognostic Significance of Tumor Infiltrating Natural Killer Cells Subset CD57 in Patients With Squamous Cell Lung Cancer. Lung Cancer (2002) 35:23–8. doi: 10.1016/S0169-5002(01)00292-6
45. Coca S, Perez-Piqueras J, Martinez D, Colmenarejo A, Saez MA, Vallejo C, et al. The Prognostic Significance of Intratumoral Natural Killer Cells in Patients With Colorectal Carcinoma. Cancer (1997) 79:2320–8. doi: 10.1002/(sici)1097-0142(19970615)79:12<2320::aid-cncr5>3.0.co;2-p
46. Ishigami S, Natsugoe S, Tokuda K, Nakajo A, Che X, Iwashige H, et al. Prognostic Value of Intratumoral Natural Killer Cells in Gastric Carcinoma. Cancer (2000) 88:577–83. doi: 10.1002/(SICI)1097-0142(20000201)88:3<577::AID-CNCR13>3.0.CO;2-V
47. Takanami I, Takeuchi K, Giga M. The Prognostic Value of Natural Killer Cell Infiltration in Resected Pulmonary Adenocarcinoma. J Thorac Cardiovasc Surg (2001) 121:1058–63. doi: 10.1067/MTC.2001.113026
48. Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural Cytotoxic Activity of Peripheral-Blood Lymphocytes and Cancer Incidence: An 11-Year Follow-Up Study of a General Population. Lancet (2000) 356:1795–9. doi: 10.1016/S0140-6736(00)03231-1
49. Zhang S, Liu W, Hu B, Wang P, Lv X, Chen S, et al. Prognostic Significance of Tumor-Infiltrating Natural Killer Cells in Solid Tumors: A Systematic Review and Meta-Analysis. Front Immunol (2020) 11:1242. doi: 10.3389/fimmu.2020.01242
50. Nersesian S, Schwartz SL, Grantham SR, MacLean LK, Lee SN, Pugh-Toole M, et al. NK Cell Infiltration Is Associated With Improved Overall Survival in Solid Cancers: A Systematic Review and Meta-Analysis. Trans Oncol (2021) 14:100930. doi: 10.1016/J.TRANON.2020.100930
51. Cursons J, Souza-Fonseca-Guimaraes F, Foroutan M, Anderson A, Hollande F, Hediyeh-Zadeh S, et al. A Gene Signature Predicting Natural Killer Cell Infiltration and Improved Survival in Melanoma Patients. Cancer Immunol Res (2019) 7:1162–74. doi: 10.1158/2326-6066.CIR-18-0500
52. Moreno-Nieves UY, Tay JK, Saumyaa S, Horowitz NB, Shin JH, Mohammad IA, et al. Landscape of Innate Lymphoid Cells in Human Head and Neck Cancer Reveals Divergent NK Cell States in the Tumor Microenvironment. Proc Natl Acad Sci (2021) 118. doi: 10.1073/PNAS.2101169118
53. Li B, Jiang Y, Li G, Fisher GA, Li R. Natural Killer Cell and Stroma Abundance are Independently Prognostic and Predict Gastric Cancer Chemotherapy Benefit. JCI Insight (2020) 5. doi: 10.1172/JCI.INSIGHT.136570
54. Lee H, Quek C, Silva I, Tasker A, Batten M, Rizos H, et al. Integrated Molecular and Immunophenotypic Analysis of NK Cells in Anti-PD-1 Treated Metastatic Melanoma Patients. Oncoimmunology (2019) 8. doi: 10.1080/2162402X.2018.1537581
55. Roemer MGM, Advani RH, Redd RA, Pinkus GS, Natkunam Y, Ligon AH, et al. Classical Hodgkin Lymphoma With Reduced β2m/MHC Class I Expression Is Associated With Inferior Outcome Independent of 9p24.1 Status. Cancer Immunol Res (2016) 4:910–6. doi: 10.1158/2326-6066.CIR-16-0201
56. van Houdt IS, Sluijter BJR, Moesbergen LM, Vos WM, de Gruijl TD, Molenkamp BG, et al. Favorable Outcome in Clinically Stage II Melanoma Patients is Associated With the Presence of Activated Tumor Infiltrating T-Lymphocytes and Preserved MHC Class I Antigen Expression. Int J Cancer (2008) 123:609–15. doi: 10.1002/IJC.23543
57. Inoue M, Mimura K, Izawa S, Shiraishi K, Inoue A, Shiba S, et al. Expression of MHC Class I on Breast Cancer Cells Correlates Inversely With HER2 Expression. Oncoimmunology (2012) 1:1104. doi: 10.4161/ONCI.21056
58. Glas R, Sturmhöfel K, Hämmerling GJ, Kärre K, Ljunggren HG. Restoration of a Tumorigenic Phenotype by Beta 2-Microglobulin Transfection to EL-4 Mutant Cells. J Exp Med (1992) 175:843–6. doi: 10.1084/JEM.175.3.843
59. Malmberg KJ, Sohlberg E, Goodridge JP, Ljunggren HG. Immune Selection During Tumor Checkpoint Inhibition Therapy Paves Way for NK-Cell “Missing Self” Recognition. Immunogenetics (2017) 69:547–56. doi: 10.1007/S00251-017-1011-9/FIGURES/1
60. Grimm EA, Robb RJ, Roth JA, Neckers LM, Lachman LB, Wilson DJ, et al. Brief Definitive Report Lymphokine-Activated Killer Cell Phenomenon III. Evidence That IL-2 Is Sufficient for Direct Activation of Peripheral Blood Lymphocytes Into Lymphokine-Activated Killer Cells. J Exp Med (1983) 158:1356–61. doi: 10.1084/jem.158.4.1356
61. Grimm EA, Mazumder A, Zhang HZ, Rosenberg SA. Lymphokine-Activated Killer Cell Phenomenon. Lysis of Natural Killer-Resistant Fresh Solid Tumor Cells by Interleukin 2-Activated Autologous Human Peripheral Blood Lymphocytes. J Exp Med (1982) 155:1823–41. doi: 10.1084/JEM.155.6.1823
62. Phillips JH, Lanier LL. Dissection of the Lymphokine-Activated Killer Phenomenon Relative Contribution of Peripheral Blood Natural Killer Cells and T Lymphocytes to Cytolysis. J Exp Med (1986) 164:814–25. doi: 10.1084/jem.164.3.814
63. Mulé JJ, Shu S, Schwarz SL, Rosenberg SA. Adoptive Immunotherapy of Established Pulmonary Metastases With LAK Cells and Recombinant Interleukin-2. Science (1984) 225:1487–9. doi: 10.1126/SCIENCE.6332379
64. Ettinghausen SE, Rosenberg SA. Immunotherapy of Murine Sarcomas Using Lymphokine Activated Killer Cells: Optimization of the Schedule and Route of Administration of Recombinant Interleukin-2. Cancer Res (1986) 46:51–71. doi: 10.1007/BF00201905
65. Lafreniere R, Rosenberg SA. Successful Immunotherapy of Murine Experimental Hepatic Metastases With Lymphokine-Activated Killer Cells and Recombinant Interleukin 2. Cancer Res (1985) 45:3735–41.
66. Rosenberg SA, Lotze MT, Yang JC, Topalian SL, Chang AE, Schwartzentruber DJ, et al. Prospective Randomized Trial of High-Dose Interleukin-2 Alone or in Conjunction With Lymphokine-Activated Killer Cells for the Treatment of Patients With Advanced Cancer. JNCI: J Natl Cancer Institute (1993) 85:622–32. doi: 10.1093/JNCI/85.8.622
67. Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive Transfer of Autologous Natural Killer Cells Leads to High Levels of Circulating Natural Killer Cells But Does Not Mediate Tumor Regression. Clin Cancer Res (2011) 17:6287–97. doi: 10.1158/1078-0432.CCR-11-1347
68. Burns LJ, Weisdorf DJ, DeFor TE, Vesole DH, Repka TL, Blazar BR, et al. IL-2-Based Immunotherapy After Autologous Transplantation for Lymphoma and Breast Cancer Induces Immune Activation and Cytokine Release: A Phase I/II Trial. Bone Marrow Transplant (2003) 32:177–86. doi: 10.1038/sj.bmt.1704086
69. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of Donor Natural Killer Cell Aloreactivity in Mismatched Hematopoietic Transplants. Science (2002) 295:2097–100. doi: 10.1126/science.1068440
70. Giebel S, Locatelli F, Lamparelli T, Velardi A, Davies S, Frumento G, et al. Survival Advantage With KIR Ligand Incompatibility in Hematopoietic Stem Cell Transplantation From Unrelated Donors. Blood (2003) 102:814–9. doi: 10.1182/blood-2003-01-0091
71. Cooley S, Weisdorf DJ, Guethlein LA, Klein JP, Wang T, Le CT, et al. Donor Selection for Natural Killer Cell Receptor Genes Leads to Superior Survival After Unrelated Transplantation for Acute Myelogenous Leukemia. Blood (2010) 116:2411–9. doi: 10.1182/BLOOD-2010-05-283051
72. Giebel S, Nowak I, Dziaczkowska J, Czerw T, Wojnar J, Krawczyk-Kulis M, et al. Activating Killer Immunoglobulin-Like Receptor Incompatibilities Enhance Graft-Versus-Host Disease and Affect Survival After Allogeneic Hematopoietic Stem Cell Transplantation. Eur J Haematol (2009) 83:343–56. doi: 10.1111/J.1600-0609.2009.01280.X
73. Symons HJ, Leffell MS, Rossiter ND, Zahurak M, Jones RJ, Fuchs EJ. Improved Survival With Inhibitory Killer Immunoglobulin Receptor (KIR) Gene Mismatches and KIR Haplotype B Donors After Nonmyeloablative, HLA-Haploidentical Bone Marrow Transplantation. Biol Blood Marrow Transplant (2010) 16:533–42. doi: 10.1016/J.BBMT.2009.11.022
74. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful Adoptive Transfer and In Vivo Expansion of Human Haploidentical NK Cells in Patients With Cancer. Blood (2005) 105:3051–7. doi: 10.1182/BLOOD-2004-07-2974
75. Shi J, Tricot G, Szmania S, Rosen N, Garg TK, Malaviarachchi PA, et al. Infusion of Haplo-Identical Killer Immunoglobulin-Like Receptor Ligand Mismatched NK Cells for Relapsed Myeloma in the Setting of Autologous Stem Cell Transplantation. Br J Haematol (2008) 143:641–53. doi: 10.1111/J.1365-2141.2008.07340.X
76. Bachanova V, Burns LJ, McKenna DH, Curtsinger J, Panoskaltsis-Mortari A, Lindgren BR, et al. Allogeneic Natural Killer Cells for Refractory Lymphoma. Cancer Immunol Immunother (2010) 59:1739–44. doi: 10.1007/S00262-010-0896-Z/FIGURES/1
77. Dezfouli AB, Yazdi M, Pockley AG, Khosravi M, Kobold S, Wagner E, et al. NK Cells Armed With Chimeric Antigen Receptors (CAR): Roadblocks to Successful Development. Cells (2021) 10:3390. doi: 10.3390/CELLS10123390
78. Carlsten M, Levy E, Karambelkar A, Li L, Reger R, Berg M, et al. Efficient mRNA-Based Genetic Engineering of Human NK Cells With High-Affinity CD16 and CCR7 Augments Rituximab-Induced ADCC Against Lymphoma and Targets NK Cell Migration Toward the Lymph Node-Associated Chemokine CCL19. Front Immunol (2016) 7:105. doi: 10.3389/FIMMU.2016.00105
79. Boissel L, Betancur M, Wels WS, Tuncer H, Klingemann H. Transfection With mRNA for CD19 Specific Chimeric Antigen Receptor Restores NK Cell Mediated Killing of CLL Cells. Leukemia Res (2009) 33:1255–9. doi: 10.1016/J.LEUKRES.2008.11.024
80. Liu H, Yang B, Sun T, Lin L, Hu Y, Deng M, et al. Specific Growth Inhibition of ErbB2-Expressing Human Breast Cancer Cells by Genetically Modified NK-92 Cells. Oncol Rep (2015) 33:95–102. doi: 10.3892/OR.2014.3548/HTML
81. Boissel L, Betancur M, Lu W, Wels WS, Marino T, van Etten RA, et al. Comparison of mRNA and Lentiviral Based Transfection of Natural Killer Cells With Chimeric Antigen Receptors Recognizing Lymphoid Antigens. Leukemia Lymphoma (2012) 53:958–65. doi: 10.3109/10428194.2011.634048
82. Sharei A, Zoldan J, Adamo A, Sim WY, Cho N, Jackson E, et al. A Vector-Free Microfluidic Platform for Intracellular Delivery. Proc Natl Acad Sci USA (2013) 110:2082–7. doi: 10.1073/PNAS.1218705110
83. Loo J, Sicher I, Goff A, Kim O, Clary N, Alexeev A, et al. Microfluidic Transfection of mRNA Into Human Primary Lymphocytes and Hematopoietic Stem and Progenitor Cells Using Ultra-Fast Physical Deformations. Sci Rep (2021) 11:1–11. doi: 10.1038/s41598-021-00893-4
84. DiTommaso T, Cole JM, Cassereau L, Buggé JA, Sikora Hanson JL, Bridgen DT, et al. Cell Engineering With Microfluidic Squeezing Preserves Functionality of Primary Immune Cells In Vivo. Proc Natl Acad Sci USA (2018) 115:E10907–14. doi: 10.1073/PNAS.1809671115
85. Atsavapranee ES, Billingsley MM, Mitchell MJ. Delivery Technologies for T Cell Gene Editing: Applications in Cancer Immunotherapy. EBioMedicine (2021) 67:103354. doi: 10.1016/J.EBIOM.2021.103354
86. Wilk AJ, Weidenbacher NLB, Vergara R, Haabeth OAW, Levy R, Waymouth RM, et al. Charge-Altering Releasable Transporters Enable Phenotypic Manipulation of Natural Killer Cells for Cancer Immunotherapy. Blood Adv (2020) 4:4244–55. doi: 10.1182/BLOODADVANCES.2020002355
87. Kim KS, Han JH, Park JH, Kim HK, Choi SH, Kim GR, et al. Multifunctional Nanoparticles for Genetic Engineering and Bioimaging of Natural Killer (NK) Cell Therapeutics. Biomaterials (2019) 221:119418. doi: 10.1016/J.BIOMATERIALS.2019.119418
88. El-Mayta R, Zhang Z, Hamilton AG, Mitchell MJ. Delivery Technologies to Engineer Natural Killer Cells for Cancer Immunotherapy. Cancer Gene Ther (2021) 28:1–13. doi: 10.1038/s41417-021-00336-2
89. Streltsova MA, Barsov E, Erokhina SA, Kovalenko EI. Retroviral Gene Transfer Into Primary Human NK Cells Activated by IL-2 and K562 Feeder Cells Expressing Membrane-Bound IL-21. J Immunol Methods (2017) 450:90–4. doi: 10.1016/J.JIM.2017.08.003
90. Guven H, Konstantinidis KV, Alici E, Aints A, Abedi-Valugerdi M, Christensson B, et al. Efficient Gene Transfer Into Primary Human Natural Killer Cells by Retroviral Transduction. Exp Hematol (2005) 33:1320–8. doi: 10.1016/J.EXPHEM.2005.07.006
91. Lowe E, Truscott LC, de Oliveira SN. In Vitro Generation of Human NK Cells Expressing Chimeric Antigen Receptor Through Differentiation of Gene-Modified Hematopoietic Stem Cells. Methods Mol Biol (2016) 1441:241–51. doi: 10.1007/978-1-4939-3684-7_20
92. Denning W, Das S, Guo S, Xu J, Kappes JC, Hel Z. Optimization of the Transductional Efficiency of Lentiviral Vectors: Effect of Sera and Polycations. Mol Biotechnol (2012) 53:308–14. doi: 10.1007/S12033-012-9528-5
93. Colamartino ABL, Lemieux W, Bifsha P, Nicoletti S, Chakravarti N, Sanz J, et al. Efficient and Robust NK-Cell Transduction With Baboon Envelope Pseudotyped Lentivector. Front Immunol (2019) 10:2873. doi: 10.3389/fimmu.2019.02873
94. Bexte T, Botezatu L, Miskey C, Campe J, Reindl LM, Gebel V, et al. Non-Viral Sleeping Beauty Transposon Engineered CD19-CAR-NK Cells Show a Safe Genomic Integration Profile and High Antileukemic Efficiency. Blood (2021) 138:2797–7. doi: 10.1182/BLOOD-2021-153999
95. Batchu RB, Gruzdyn OV, Tavva PS, Kolli BK, Dachepalli R, Weaver DW, et al. Engraftment of Mesothelin Chimeric Antigen Receptor Using a Hybrid Sleeping Beauty/minicircle Vector Into NK-92MI Cells for Treatment of Pancreatic Cancer. Surgery (2019) 166:503–8. doi: 10.1016/J.SURG.2019.05.047
96. Hudecek M, Ivics Z. Non-Viral Therapeutic Cell Engineering With the Sleeping Beauty Transposon System. Curr Opin Genet Dev (2018) 52:100–8. doi: 10.1016/J.GDE.2018.06.003
97. Kim MS, Kini AG. Engineering and Application of Zinc Finger Proteins and TALEs for Biomedical Research. Mol Cells (2017) 40:533. doi: 10.14348/MOLCELLS.2017.0139
98. Zhu H, Blum RH, Bernareggi D, Ask EH, Wu Z, Hoel HJ, et al. Metabolic Reprograming via Deletion of CISH in Human iPSC-Derived NK Cells Promotes In Vivo Persistence and Enhances Anti-Tumor Activity. Cell Stem Cell (2020) 27:224–37.e6. doi: 10.1016/j.stem.2020.05.008
99. Pomeroy EJ, Hunzeker JT, Kluesner MG, Lahr WS, Smeester BA, Crosby MR, et al. A Genetically Engineered Primary Human Natural Killer Cell Platform for Cancer Immunotherapy. Mol Ther (2019) 28:52–63. doi: 10.1016/J.YMTHE.2019.10.009
100. Sander JD, Joung JK. CRISPR-Cas Systems for Editing, Regulating and Targeting Genomes. Nat Biotechnol (2014) 32(4):347–55. doi: 10.1038/nbt.2842
101. Kim KS, Kim DH, Kim DH. Recent Advances to Augment NK Cell Cancer Immunotherapy Using Nanoparticles. Pharmaceutics (2021) 52513:525. doi: 10.3390/PHARMACEUTICS13040525
102. Chandrasekaran S, Chan MF, Li J, King MR. Super Natural Killer Cells That Target Metastases in the Tumor Draining Lymph Nodes. Biomaterials (2016) 77:66–76. doi: 10.1016/J.BIOMATERIALS.2015.11.001
103. Siegler EL, Kim YJ, Chen X, Siriwon N, Mac J, Rohrs JA, et al. Combination Cancer Therapy Using Chimeric Antigen Receptor-Engineered Natural Killer Cells as Drug Carriers. Mol Ther (2017) 25:2607–19. doi: 10.1016/J.YMTHE.2017.08.010
104. Li HK, Hsiao CW, Yang SH, Yang HP, Wu TS, Lee CY, et al. A Novel Off-the-Shelf Trastuzumab-Armed NK Cell Therapy (ACE1702) Using Antibody-Cell-Conjugation Technology. Cancers (2021) 13:2724. doi: 10.3390/CANCERS13112724
105. Hong S, Yu C, Wang P, Shi Y, Cao W, Cheng B, et al. Glycoengineering of NK Cells With Glycan Ligands of CD22 and Selectins for B-Cell Lymphoma Therapy. Angewandte Chemie - Int Edition (2021) 60:3603–10. doi: 10.1002/ANIE.202005934
106. Wang X, Lang S, Tian Y, Zhang J, Yan X, Fang Z, et al. Glycoengineering of Natural Killer Cells With CD22 Ligands for Enhanced Anticancer Immunotherapy. ACS Appl Mater Interf (2020) 2020:389. doi: 10.1021/ACSCENTSCI.9B00956
107. Chen Z, Zeng Z, Wan Q, Liu X, Qi J, Zu Y. Targeted Immunotherapy of Triple-Negative Breast Cancer by Aptamer-Engineered NK Cells. Biomaterials (2022) 280:121259. doi: 10.1016/J.BIOMATERIALS.2021.121259
108. Yang S, Wen J, Li H, Xu L, Liu Y, Zhao N, et al. Aptamer-Engineered Natural Killer Cells for Cell-Specific Adaptive Immunotherapy. Small (2019) 15:1900903. doi: 10.1002/SMLL.201900903
109. Wendel P, Reindl LM, Bexte T, Künnemeyer L, Särchen V, Albinger N, et al. Arming Immune Cells for Battle: A Brief Journey Through the Advancements of T and NK Cell Immunotherapy. Cancers (Basel) (2021) 13:1481. doi: 10.3390/cancers13061481
110. Liu E, Marin D, Banerjee P, MacApinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med (2020) 382:545–53. doi: 10.1056/NEJMoa1910607
111. Goodridge JP, Bjordahl R, Mahmood S, Reiser J, Gaidarova S, Blum R, et al. FT576: Multi-Specific Off-The-Shelf CAR-NK Cell Therapy Engineered for Enhanced Persistence, Avoidance of Self-Fratricide and Optimized Mab Combination Therapy to Prevent Antigenic Escape and Elicit a Deep and Durable Response in Multiple Myeloma. Blood (2020) 136:4–5. doi: 10.1182/BLOOD-2020-142750
112. Cheng ZF, Li HK, Yang HP, Lee CY, Tang SW, Lin YL, et al. A Novel Endogenous CD16-Expressing Natural Killer Cell for Cancer Immunotherapy. Biochem Biophysics Rep (2021) 26:100935. doi: 10.1016/J.BBREP.2021.100935
113. Lu C, Guo C, Chen H, Zhang H, Zhi L, Lv T, et al. A Novel Chimeric PD1-NKG2D-41BB Receptor Enhances Antitumor Activity of NK92 Cells Against Human Lung Cancer H1299 Cells by Triggering Pyroptosis. Mol Immunol (2020) 122:200–6. doi: 10.1016/J.MOLIMM.2020.04.016
114. Saetersmoen ML, Hammer Q, Valamehr B, Kaufman DS, Malmberg KJ. Off-The-Shelf Cell Therapy With Induced Pluripotent Stem Cell-Derived Natural Killer Cells. Semin Immunopathol (2019) 41:59–68. doi: 10.1007/s00281-018-0721-x
115. Rambaldi A, Biagi E, Bonini C, Biondi A, Introna M. Cell-Based Strategies to Manage Leukemia Relapse: Efficacy and Feasibility of Immunotherapy Approaches. Leukemia (2014) 29:1–10. doi: 10.1038/leu.2014.189
116. Castagna L, Sarina B, Bramanti S, Perseghin P, Mariotti J, Morabito L. Donor Lymphocyte Infusion After Allogeneic Stem Cell Transplantation. Transfusion Apheresis Sci (2016) 54:345–55. doi: 10.1016/J.TRANSCI.2016.05.011
117. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric Antigen Receptor T-Cell Therapy — Assessment and Management of Toxicities. Nat Rev Clin Oncol (2017) 15(1):47–62. doi: 10.1038/nrclinonc.2017.148
118. Maude SL, Pulsipher MA, Boyer MW, Grupp SA, Davies SM, Phillips CL, et al. Efficacy and Safety of CTL019 in the First US Phase II Multicenter Trial in Pediatric Relapsed/Refractory Acute Lymphoblastic Leukemia: Results of an Interim Analysis. Blood (2016) 128:2801–1. doi: 10.1182/BLOOD.V128.22.2801.2801
119. Passweg JR, Tichelli A, Meyer-Monard S, Heim D, Stern M, Kühne T, et al. Purified Donor NK-Lymphocyte Infusion to Consolidate Engraftment After Haploidentical Stem Cell Transplantation. Leukemia (2004) 18(11):1835–8. doi: 10.1038/sj.leu.2403524
120. Sarvaria A, Jawdat D, Madrigal JA, Saudemont A. Umbilical Cord Blood Natural Killer Cells, Their Characteristics, and Potential Clinical Applications. Front Immunol (2017) 8:329. doi: 10.3389/FIMMU.2017.00329
121. Spanholtz J, Preijers F, Tordoir M, Trilsbeek C, Paardekooper J, de Witte T, et al. Clinical-Grade Generation of Active NK Cells From Cord Blood Hematopoietic Progenitor Cells for Immunotherapy Using a Closed-System Culture Process. PloS One (2011) 6:e20740. doi: 10.1371/journal.pone.0020740
122. Carayol G, Robin C, Bourhis JH, Bennaceur-Griscelli A, Chouaib S, Coulombel L, et al. NK Cells Differentiated From Bone Marrow, Cord Blood and Peripheral Blood Stem Cells Exhibit Similar Phenotype and Functions. Eur J Immunol (1998) 28:1991–2002. doi: 10.1002/(SICI)1521-4141(199806)28:06<1991::AID-IMMU1991>3.0.CO;2-7
123. Carlsten M, Childs RW. Genetic Manipulation of NK Cells for Cancer Immunotherapy: Techniques and Clinical Implications. Front Immunol (2015) 6:266. doi: 10.3389/FIMMU.2015.00266
124. Klingemann HG, Wong E, Maki G. A Cytotoxic NK-Cell Line (NK-92) for Ex Vivo Purging of Leukemia From Blood. Biol Blood Marrow Transplant (1996) 2:68–75.
125. Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, McKenna DH, et al. Clearance of Acute Myeloid Leukemia by Haploidentical Natural Killer Cells Is Improved Using IL-2 Diphtheria Toxin Fusion Protein. Blood (2014) 123:3855–63. doi: 10.1182/BLOOD-2013-10-532531
126. Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, et al. First-In-Man Clinical Trial of CAR NK-92 Cells: Safety Test of CD33-CAR NK-92 Cells in Patients With Relapsed and Refractory Acute Myeloid Leukemia. Am J Cancer Res (2018) 8:1083.
127. Knorr DA, Ni Z, Hermanson D, Hexum MK, Bendzick L, Cooper LJN, et al. Clinical-Scale Derivation of Natural Killer Cells From Human Pluripotent Stem Cells for Cancer Therapy. Stem Cells Trans Med (2013) 2:274–83. doi: 10.5966/sctm.2012-0084
128. Valamehr B, Robinson M, Abujarour R, Rezner B, Vranceanu F, Le T, et al. Platform for Induction and Maintenance of Transgene-Free hiPSCs Resembling Ground State Pluripotent Stem Cells. Stem Cell Rep (2014) 2:366–81. doi: 10.1016/J.STEMCR.2014.01.014
129. Hermanson DL, Bendzick L, Pribyl L, McCullar V, Vogel RI, Miller JS, et al. Induced Pluripotent Stem Cell-Derived Natural Killer Cells for Treatment of Ovarian Cancer. Stem Cells (2016) 34:93–101. doi: 10.1002/stem.2230
130. Sato Y, Bando H, di Piazza M, Gowing G, Herberts C, Jackman S, et al. Tumorigenicity Assessment of Cell Therapy Products: The Need for Global Consensus and Points to Consider. Cytotherapy (2019) 21:1095–111. doi: 10.1016/J.JCYT.2019.10.001
131. Woan KV, Kim H, Bjordahl R, Davis ZB, Gaidarova S, Goulding J, et al. Harnessing Features of Adaptive NK Cells to Generate iPSC-Derived NK Cells for Enhanced Immunotherapy. Cell Stem Cell (2021) 28:2062–75.e5. doi: 10.1016/J.STEM.2021.08.013
132. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-Derived Natural Killer Cells Engineered With Chimeric Antigen Receptors Enhance Anti-Tumor Activity. Cell Stem Cell (2018) 23:181–92. doi: 10.1016/j.stem.2018.06.002
133. Kuwana Y, Asakura Y, Utsunomiya N, Nakanishi M, Arata Y, Itoh S, et al. Expression of Chimeric Receptor Composed of Immunoglobulin-Derived V Resions and T-Cell Receptor-Derived C Regions. Biochem Biophys Res Commun (1987) 149:960–8. doi: 10.1016/0006-291X(87)90502-X
134. Gross G, Waks T, Eshhar Z. Expression of Immunoglobulin-T-Cell Receptor Chimeric Molecules as Functional Receptors With Antibody-Type Specificity. Proc Natl Acad Sci (1989) 86:10024–8. doi: 10.1073/PNAS.86.24.10024
135. Stancovski I, Schindler DG, Waks T, Yarden Y, Sela M, Eshhar Z. Targeting of T Lymphocytes to Neu/HER2-Expressing Cells Using Chimeric Single Chain Fv Receptors. J Immunol (1993) 151:6577–82.
136. Eshhar Z, Waks T, Gross G, Schindler DG. Specific Activation and Targeting of Cytotoxic Lymphocytes Through Chimeric Single Chains Consisting of Antibody-Binding Domains and the Gamma or Zeta Subunits of the Immunoglobulin and T-Cell Receptors. Proc Natl Acad Sci (1993) 90:720–4. doi: 10.1073/PNAS.90.2.720
137. Brocker T, Karjalainen K. Signals Through T Cell Receptor-Zeta Chain Alone are Insufficient to Prime Resting T Lymphocytes. J Exp Med (1995) 181:1653. doi: 10.1084/JEM.181.5.1653
138. Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 Costimulation Improves Expansion and Persistence of Chimeric Antigen Receptor–Modified T Cells in Lymphoma Patients. J Clin Invest (2011) 121:1822–6. doi: 10.1172/JCI46110
139. Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-Cell Therapy Bb2121 in Relapsed or Refractory Multiple Myeloma. N Engl J Med (2019) 380:1726–37. doi: 10.1056/NEJMOA1817226
140. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults With B-Cell Lymphoblastic Leukemia. N Engl J Med (2018) 378:439–48. doi: 10.1056/NEJMOA1709866
141. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang ML, Arnason JE, et al. Pivotal Safety and Efficacy Results From Transcend NHL 001, a Multicenter Phase 1 Study of Lisocabtagene Maraleucel (Liso-Cel) in Relapsed/Refractory (R/R) Large B Cell Lymphomas. Blood (2019) 134:241–1. doi: 10.1182/BLOOD-2019-127508
142. Berdeja JG, Madduri D, Usmani SZ, Jakubowiak A, Agha M, Cohen AD, et al. Ciltacabtagene Autoleucel, a B-Cell Maturation Antigen-Directed Chimeric Antigen Receptor T-Cell Therapy in Patients With Relapsed or Refractory Multiple Myeloma (CARTITUDE-1): A Phase 1b/2 Open-Label Study. Lancet (2021) 398:314–24. doi: 10.1016/S0140-6736(21)00933-8
143. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med (2017) 377:2531–44. doi: 10.1056/NEJMOA1707447
144. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N Engl J Med (2013) 368:1509–18. doi: 10.1056/NEJMOA1215134
145. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor Activity and Long-Term Fate of Chimeric Antigen Receptor–Positive T Cells in Patients With Neuroblastoma. Blood (2011) 118:6050–6. doi: 10.1182/BLOOD-2011-05-354449
146. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of Glioblastoma After Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med (2016) 375:2561–9. doi: 10.1056/NEJMOA1610497
147. Chmielewski M, Hombach AA, Abken H. Of CARs and TRUCKs: Chimeric Antigen Receptor (CAR) T Cells Engineered With an Inducible Cytokine to Modulate the Tumor Stroma. Immunol Rev (2014) 257:83–90. doi: 10.1111/IMR.12125
148. Chmielewski M, Abken H. CAR T Cells Transform to Trucks: Chimeric Antigen Receptor-Redirected T Cells Engineered to Deliver Inducible IL-12 Modulate the Tumour Stroma to Combat Cancer. Cancer Immunol Immunother (2012) 61:1269–77. doi: 10.1007/S00262-012-1202-Z/FIGURES/2
149. Zhu H, Blum RH, Bjordahl R, Gaidarova S, Rogers P, Lee TT, et al. Pluripotent Stem Cell–Derived NK Cells With High-Affinity Noncleavable CD16a Mediate Improved Antitumor Activity. Blood (2020) 135:399–410. doi: 10.1182/BLOOD.2019000621
150. Bryceson YT, March ME, Ljunggren HG, Long EO. Synergy Among Receptors on Resting NK Cells for the Activation of Natural Cytotoxicity and Cytokine Secretion. Blood (2006) 107:159–66. doi: 10.1182/BLOOD-2005-04-1351
151. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or Adaptive Immunity? The Example of Natural Killer Cells. Science (2011) 331:44–9. doi: 10.1126/science.1198687
152. Fauriat C, Long EO, Ljunggren HG, Bryceson YT. Regulation of Human NK-Cell Cytokine and Chemokine Production by Target Cell Recognition. Blood (2010) 115:2167–76. doi: 10.1182/BLOOD-2009-08-238469
153. Harrison D, Phillips JH, Lanier LL. Involvement of a Metalloprotease in Spontaneous and Phorbol Ester-Induced Release of Natural Killer Cell-Associated Fc Gamma RIII (CD16-Ii). J Immunol (1991) 147:3459–65.
154. Tosi MF, Zakem H. Surface Expression of Fc Gamma Receptor III (CD16) on Chemoattractant-Stimulated Neutrophils is Determined by Both Surface Shedding and Translocation From Intracellular Storage Compartments. J Clin Invest (1992) 90:462. doi: 10.1172/JCI115882
155. Romee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankarlo D, et al. NK Cell CD16 Surface Expression and Function is Regulated by a Disintegrin and Metalloprotease-17 (ADAM17). Blood (2013) 121:3599–608. doi: 10.1182/blood-2012-04-425397
156. Jing Y, Ni Z, Wu J, Higgins LA, Markowski TW, Kaufman DS, et al. Identification of an ADAM17 Cleavage Region in Human CD16 (Fcγriii) and the Engineering of a Non-Cleavable Version of the Receptor in NK Cells. PloS One (2015) 10:e0121788. doi: 10.1371/JOURNAL.PONE.0121788
157. Koene HR, Kleijer M, Algra J, Roos D, von dem Borne AEGK, de Haas M. Fcγriiia-158V/F Polymorphism Influences the Binding of IgG by Natural Killer Cell Fcγriiia, Independently of the Fcγriiia-48L/R/H Phenotype. Blood (1997) 90:1109–14. doi: 10.1182/BLOOD.V90.3.1109
158. Wu J, Edberg JC, Redecha PB, Bansal V, Guyre PM, Coleman K, et al. A Novel Polymorphism of FcgammaRIIIa (CD16) Alters Receptor Function and Predisposes to Autoimmune Disease. J Clin Invest (1997) 100:1059–70. doi: 10.1172/JCI119616
159. Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, et al. Therapeutic Activity of Humanized Anti-CD20 Monoclonal Antibody and Polymorphism in IgG Fc Receptor Fcγriiia Gene. Blood (2002) 99:754–8. doi: 10.1182/BLOOD.V99.3.754
160. Bibeau F, Lopez-Crapez E, di FF, Thezenas S, Ychou M, Blanchard F, et al. Impact of Fcγriia-Fcγriiia Polymorphisms and KRAS Mutations on the Clinical Outcome of Patients With Metastatic Colorectal Cancer Treated With Cetuximab Plus Irinotecan. J Clin Oncol (2009) 27:1122–9. doi: 10.1200/JCO.2008.18.0463
161. Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, et al. Immunoglobulin G Fragment C Receptor Polymorphisms and Clinical Efficacy of Trastuzumab-Based Therapy in Patients With HER-2/Neu-Positive Metastatic Breast Cancer. J Clin Oncol (2008) 26:1789–96. doi: 10.1200/JCO.2007.14.8957
162. Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric Receptors Containing CD137 Signal Transduction Domains Mediate Enhanced Survival of T Cells and Increased Antileukemic Efficacy In Vivo. Mol Ther: J Am Soc Gene Ther (2009) 17:1453. doi: 10.1038/MT.2009.83
163. Rosenberg SA. Interleukin-2 and the Development of Immunotherapy for the Treatment of Patients With Cancer. Cancer J Sci Am (2000) 6(suppl 1):S2–7.
164. Ito S, Bollard CM, Carlsten M, Melenhorst JJ, Biancotto A, Wang E, et al. Ultra-Low Dose Interleukin-2 Promotes Immune-Modulating Function of Regulatory T Cells and Natural Killer Cells in Healthy Volunteers. Mol Ther (2014) 22:1388–95. doi: 10.1038/MT.2014.50
165. Marks-Konczalik J, Dubois S, Losi JM, Sabzevari H, Yamada N, Feigenbaum L, et al. IL-2-Induced Activation-Induced Cell Death is Inhibited in IL-15 Transgenic Mice. Proc Natl Acad Sci (2000) 97:11445–50. doi: 10.1073/PNAS.200363097
166. Kobayashi H, Carrasquillo JA, Paik CH, Waldmann TA, Tagaya Y. Differences of Biodistribution, Pharmacokinetics, and Tumor Targeting Between Interleukins 2 and 15. Cancer Res (2000) 60:3577–83.
167. Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, et al. Redistribution, Hyperproliferation, Activation of Natural Killer Cells and CD8 T Cells, and Cytokine Production During First-in-Human Clinical Trial of Recombinant Human Interleukin-15 in Patients With Cancer. J Clin Oncol (2015) 33:74–82. doi: 10.1200/JCO.2014.57.3329
168. Imamura M, Shook D, Kamiya T, Shimasaki N, Chai SMH, Coustan-Smith E, et al. Autonomous Growth and Increased Cytotoxicity of Natural Killer Cells Expressing Membrane-Bound Interleukin-15. Blood (2014) 124:1081–8. doi: 10.1182/blood-2014-02
169. Delconte RB, Kolesnik TB, Dagley LF, Rautela J, Shi W, Putz EM, et al. CIS is a Potent Checkpoint in NK Cell–Mediated Tumor Immunity. Nat Immunol (2016) 17:816–24. doi: 10.1038/ni.3470
170. Schultz LM, Davis KL, Baggott C, Chaudry C, Marcy AC, Mavroukakis S, et al. Phase 1 Study of CD19/CD22 Bispecific Chimeric Antigen Receptor (CAR) Therapy in Children and Young Adults With B Cell Acute Lymphoblastic Leukemia (ALL). Blood (2018) 132:898–8. doi: 10.1182/BLOOD-2018-99-117445
171. Hossain N, Sahaf B, Abramian M, Spiegel JY, Kong K, Kim S, et al. Phase I Experience With a Bi-Specific CAR Targeting CD19 and CD22 in Adults With B-Cell Malignancies. Blood (2018) 132:490. doi: 10.1182/BLOOD-2018-99-110142
172. Schneider D, Xiong Y, Wu D, Nölle V, Schmitz S, Haso W, et al. A Tandem CD19/CD20 CAR Lentiviral Vector Drives on-Target and Off-Target Antigen Modulation in Leukemia Cell Lines. J ImmunoTherapy Cancer (2017) 5:42. doi: 10.1186/S40425-017-0246-1
173. Amrolia PJ, Wynn R, Hough R, Vora A, Bonney D, Veys P, et al. Simultaneous Targeting of CD19 and CD22: Phase I Study of AUTO3, a Bicistronic Chimeric Antigen Receptor (CAR) T-Cell Therapy, in Pediatric Patients With Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia (R/R B-ALL): Amelia Study. Blood (2018) 132:279–9. doi: 10.1182/BLOOD-2018-99-118616
174. Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-Targeted CAR T Cells Induce Remission in B-ALL That is Naive or Resistant to CD19-Targeted CAR Immunotherapy. Nat Med (2017) 24:20–8. doi: 10.1038/nm.4441
175. Wang N, Hu X, Cao W, Li C, Xiao Y, Cao Y, et al. Efficacy and Safety of CAR19/22 T-Cell Cocktail Therapy in Patients With Refractory/Relapsed B-Cell Malignancies. Blood (2020) 135:17–27. doi: 10.1182/BLOOD.2019000017
176. Pont MJ, Hill T, Cole GO, Abbott JJ, Kelliher J, Salter AI, et al. γ-Secretase Inhibition Increases Efficacy of BCMA-Specific Chimeric Antigen Receptor T Cells in Multiple Myeloma. Blood (2019) 134:1585–97. doi: 10.1182/BLOOD.2019000050
177. Majzner RG, Theruvath JL, Nellan A, Heitzeneder S, Cui Y, Mount CW, et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin Cancer Res (2019) 25:2560–74. doi: 10.1158/1078-0432.CCR-18-0432
178. Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, et al. Targeted Delivery of a PD-1-Blocking scFv by CAR-T Cells Enhances Anti-Tumor Efficacy In Vivo. Nat Biotechnol (2018) 36:847–56. doi: 10.1038/nbt.4195
179. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DAN, Feldman SA, et al. T Cells Targeting Carcinoembryonic Antigen can Mediate Regression of Metastatic Colorectal Cancer But Induce Severe Transient Colitis. Mol Ther (2011) 19:620–6. doi: 10.1038/MT.2010.272
180. Lamers CHJ, Sleijfer S, Vulto AG, Kruit WHJ, Kliffen M, Debets R, et al. Treatment of Metastatic Renal Cell Carcinoma With Autologous T-Lymphocytes Genetically Retargeted Against Carbonic Anhydrase IX: First Clinical Experience. J Clin Oncol (2006) 24. doi: 10.1200/JCO.2006.05.9964
181. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol Ther (2010) 18:843–51. doi: 10.1038/MT.2010.24
182. Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH, et al. Chimeric Antigen Receptor T Cells With Dissociated Signaling Domains Exhibit Focused Antitumor Activity With Reduced Potential for Toxicity. In Vivo Cancer Immunol Res (2013) 1:43–53. doi: 10.1158/2326-6066.CIR-13-0008
183. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial Antigen Recognition With Balanced Signaling Promotes Selective Tumor Eradication by Engineered T Cells. Nat Biotechnol (2013) 31:71–5. doi: 10.1038/nbt.2459
184. Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, et al. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell (2016) 164:770–9. doi: 10.1016/J.CELL.2016.01.011
185. Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-Based Inhibitory Chimeric Antigen Receptors (iCARs) Divert Off-Target Immunotherapy Responses. Sci Trans Med (2013) 5. doi: 10.1126/SCITRANSLMED.3006597
186. Leivas A, Valeri A, Córdoba L, García-Ortiz A, Ortiz A, Sánchez-Vega L, et al. NKG2D-CAR-Transduced Natural Killer Cells Efficiently Target Multiple Myeloma. Blood Cancer J (2021) 11:1–11. doi: 10.1038/s41408-021-00537-w
187. Xiao L, Cen D, Gan H, Sun Y, Huang N, Xiong H, et al. Adoptive Transfer of NKG2D CAR mRNA-Engineered Natural Killer Cells in Colorectal Cancer Patients. Mol Ther (2019) 27:1114–25. doi: 10.1016/J.YMTHE.2019.03.011
188. Tal Y, Yaakobi S, Horovitz-Fried M, Safyon E, Rosental B, Porgador A, et al. An NCR1-Based Chimeric Receptor Endows T-Cells With Multiple Anti-Tumor Specificities. Oncotarget (2014) 5:10949. doi: 10.18632/ONCOTARGET.1919
189. Eisenberg V, Shamalov K, Meir S, Hoogi S, Sarkar R, Pinker S, et al. Targeting Multiple Tumors Using T-Cells Engineered to Express a Natural Cytotoxicity Receptor 2-Based Chimeric Receptor. Front Immunol (2017) 8:1212. doi: 10.3389/FIMMU.2017.01212
190. Zhang T, Wu M-R, Sentman CL. An NKp30-Based Chimeric Antigen Receptor Promotes T Cell Effector Functions and Antitumor Efficacy In Vivo. J Immunol (2012) 189:2290–9. doi: 10.4049/JIMMUNOL.1103495
191. Wu MR, Zhang T, DeMars LR, Sentman CL. B7H6-Specific Chimeric Antigen Receptors Lead to Tumor Elimination and Host Antitumor Immunity. Gene Ther (2015) 22:675–84. doi: 10.1038/gt.2015.29
192. Walseng E, Köksal H, Sektioglu IM, Fåne A, Skorstad G, Kvalheim G, et al. A TCR-Based Chimeric Antigen Receptor. Sci Rep (2017) 7:1–10. doi: 10.1038/s41598-017-11126-y
193. Crowther MD, Dolton G, Legut M, Caillaud ME, Lloyd A, Attaf M, et al. Genome-Wide CRISPR–Cas9 Screening Reveals Ubiquitous T Cell Cancer Targeting via the Monomorphic MHC Class I-Related Protein MR1. Nat Immunol (2020) 21:178–85. doi: 10.1038/s41590-019-0578-8
194. Pickup M, Novitskiy S, Moses HL. The Roles of Tgfβ in the Tumour Microenvironment. Nat Rev Cancer (2013) 13:788–99. doi: 10.1038/nrc3603
195. Tran HC, Wan Z, Sheard MA, Sun J, Jackson JR, Malvar J, et al. Tgfβr1 Blockade With Galunisertib (LY2157299) Enhances Anti-Neuroblastoma Activity of the Anti-GD2 Antibody Dinutuximab (Ch14.18) With Natural Killer Cells. Clin Cancer Res (2017) 23:804–13. doi: 10.1158/1078-0432.CCR-16-1743
196. Castriconi R, Cantoni C, della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming Growth Factor β1 Inhibits Expression of NKp30 and NKG2D Receptors: Consequences for the NK-Mediated Killing of Dendritic Cells. Proc Natl Acad Sci (2003) 100:4120–5. doi: 10.1073/PNAS.0730640100
197. Yvon ES, Burga R, Powell A, Cruz CR, Fernandes R, Barese C, et al. Cord Blood Natural Killer Cells Expressing a Dominant Negative TGF-β Receptor: Implications for Adoptive Immunotherapy for Glioblastoma. Cytotherapy (2017) 19:408–18. doi: 10.1016/J.JCYT.2016.12.005
198. Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol Ther (2018) 26:1855–66. doi: 10.1016/J.YMTHE.2018.05.003
199. Bollard CM, Tripic T, Cruz CR, Dotti G, Gottschalk S, Torrano V, et al. Tumor-Specific T-Cells Engineered to Overcome Tumor Immune Evasion Induce Clinical Responses in Patients With Relapsed Hodgkin Lymphoma. J Clin Oncol (2018) 36:1128–39. doi: 10.1200/JCO.2017.74.3179
200. Chang ZL, Lorenzini MH, Chen X, Tran U, Bangayan NJ, Chen YY. Rewiring T-Cell Responses to Soluble Factors With Chimeric Antigen Receptors. Nat Chem Biol (2018) 14:317–24. doi: 10.1038/nchembio.2565
201. Wang Z, Guo L, Song Y, Zhang Y, Lin D, Hu B, et al. Augmented Anti-Tumor Activity of NK-92 Cells Expressing Chimeric Receptors of TGF-βr II and NKG2D. Cancer Immunol Immunother (2017) 66:537–48. doi: 10.1007/S00262-017-1959-1/FIGURES/6
202. Guo C, Wang X, Zhang H, Zhi L, Lv T, Li M, et al. Structure-Based Rational Design of a Novel Chimeric PD1-NKG2D Receptor for Natural Killer Cells. Mol Immunol (2019) 114:108–13. doi: 10.1016/J.MOLIMM.2019.07.009
203. Li M, Zhi L, Yin M, Guo C, Zhang H, Lu C, et al. A Novel Bispecific Chimeric PD1-DAP10/NKG2D Receptor Augments NK92-Cell Therapy Efficacy for Human Gastric Cancer SGC-7901 Cell. Biochem Biophys Res Commun (2020) 523:745–52. doi: 10.1016/J.BBRC.2020.01.005
204. Somanchi SS, Somanchi A, Cooper LJN, Lee DA. Engineering Lymph Node Homing of Ex Vivo–Expanded Human Natural Killer Cells via Trogocytosis of the Chemokine Receptor CCR7. Blood (2012) 119:5164–72. doi: 10.1182/BLOOD-2011-11-389924
205. Müller N, Michen S, Tietze S, Töpfer K, Schulte A, Lamszus K, et al. Engineering NK Cells Modified With an EGFRvIII-Specific Chimeric Antigen Receptor to Overexpress CXCR4 Improves Immunotherapy of CXCL12/SDF-1α-Secreting Glioblastoma. J Immunother (2015) 38:197–210. doi: 10.1097/CJI.0000000000000082
206. Ng YY, Tay JCK, Wang S. CXCR1 Expression to Improve Anti-Cancer Efficacy of Intravenously Injected CAR-NK Cells in Mice With Peritoneal Xenografts. Mol Ther - Oncolytics (2020) 16:75–85. doi: 10.1016/J.OMTO.2019.12.006
207. Mollicone R, Caillard T, le Pendu J, Francois A, Sansonetti N, Villarroya H, et al. Expression of ABH and X (Lex) Antigens on Platelets and Lymphocytes. Blood (1988) 71:1113–9. doi: 10.1182/BLOOD.V71.4.1113.1113
208. de Wolf S, Sykes M. Alloimmune T Cells in Transplantation. J Clin Invest (2017) 127:2473–81. doi: 10.1172/JCI90595
209. Lanza R, Russell DW, Nagy A. Engineering Universal Cells That Evade Immune Detection. Nat Rev Immunol 2019 19:12 (2019) 19:723–33. doi: 10.1038/s41577-019-0200-1
210. Montgomery RA, Cozzi E, West LJ, Warren DS. Humoral Immunity and Antibody-Mediated Rejection in Solid Organ Transplantation. Semin Immunol (2011) 23:224–34. doi: 10.1016/J.SMIM.2011.08.021
211. Curti A, Ruggeri L, D’Addio A, Bontadini A, Dan E, Motta MR, et al. Successful Transfer of Alloreactive Haploidentical KIR Ligand-Mismatched Natural Killer Cells After Infusion in Elderly High Risk Acute Myeloid Leukemia Patients. Blood (2011) 118:3273–9. doi: 10.1182/BLOOD-2011-01-329508
212. Berrien-Elliott MM, Becker-Hapak M, Cashen AF, Jacobs MT, Wong P, Foster M, et al. Systemic IL-15 Promotes Allogeneic Cell Rejection in Patients Treated With Natural Killer Cell Adoptive Therapy. Blood (2021) 139:1177–83. doi: 10.1182/BLOOD.2021011532
213. Rubnitz JE, Inaba H, Ribeiro RC, Pounds S, Rooney B, Bell T, et al. NKAML: A Pilot Study to Determine the Safety and Feasibility of Haploidentical Natural Killer Cell Transplantation in Childhood Acute Myeloid Leukemia. J Clin Oncol (2010) 28:955–9. doi: 10.1200/JCO.2009.24.4590
214. Benjamin R, Graham C, Yallop D, Jozwik A, Mirci-Danicar OC, Lucchini G, et al. Genome-Edited, Donor-Derived Allogeneic Anti-CD19 Chimeric Antigen Receptor T Cells in Paediatric and Adult B-Cell Acute Lymphoblastic Leukaemia: Results of Two Phase 1 Studies. Lancet (2020) 396:1885–94. doi: 10.1016/S0140-6736(20)32334-5
215. Mo F, Watanabe N, McKenna MK, Hicks MJ, Srinivasan M, Gomes-Silva D, et al. Engineered Off-the-Shelf Therapeutic T Cells Resist Host Immune Rejection. Nat Biotechnol (2021) 39:56–63. doi: 10.1038/s41587-020-0601-5
216. Marino J, Paster J, Benichou G. Allorecognition by T Lymphocytes and Allograft Rejection. Front Immunol (2016) 7:582. doi: 10.3389/FIMMU.2016.00582
217. Hoerster K, Uhrberg M, Wiek C, Horn PA, Hanenberg H, Heinrichs S. HLA Class I Knockout Converts Allogeneic Primary NK Cells Into Suitable Effectors for “Off-The-Shelf” Immunotherapy. Front Immunol (2021) 11:586168. doi: 10.3389/FIMMU.2020.586168
218. Han X, Wang M, Duan S, Franco PJ, Kenty JHR, Hedrick P, et al. Generation of Hypoimmunogenic Human Pluripotent Stem Cells. Proc Natl Acad Sci USA (2019) 116:10441–6. doi: 10.1073/pnas.1902566116
219. Wang B, Iriguchi S, Waseda M, Ueda N, Ueda T, Xu H, et al. Generation of Hypoimmunogenic T Cells From Genetically Engineered Allogeneic Human Induced Pluripotent Stem Cells. Nat BioMed Eng (2021) 5:429–40. doi: 10.1038/S41551-021-00730-Z
220. Öberg L, Johansson S, Michaëlsson J, Tomasello E, Vivier E, Kärre K, et al. Loss or Mismatch of MHC Class I is Sufficient to Trigger NK Cell-Mediated Rejection of Resting Lymphocytes In Vivo – Role of KARAP/DAP12-Dependent and -Independent Pathways. Eur J Immunol (2004) 34:1646–53. doi: 10.1002/EJI.200424913
221. Zhao L, Teklemariam T, Hantash BM. Heterelogous Expression of Mutated HLA-G Decreases Immunogenicity of Human Embryonic Stem Cells and Their Epidermal Derivatives. Stem Cell Res (2014) 13:342–54. doi: 10.1016/J.SCR.2014.08.004
222. Deuse T, Hu X, Gravina A, Wang D, Tediashvili G, De C, et al. Hypoimmunogenic Derivatives of Induced Pluripotent Stem Cells Evade Immune Rejection in Fully Immunocompetent Allogeneic Recipients. Nat Biotechnol (2019) 37:252–8. doi: 10.1038/s41587-019-0016-3
223. Takizawa H, Manz MG. Macrophage Tolerance: CD47–SIRP-α–Mediated Signals Matter. Nat Immunol 2007 8:12 (2007) 8:1287–9. doi: 10.1038/ni1207-1287
224. Liu X, Kwon H, Li Z, Fu YX. Is CD47 an Innate Immune Checkpoint for Tumor Evasion? J Hematol Oncol (2017) 10:1–7. doi: 10.1186/S13045-016-0381-Z/TABLES/1
225. Deuse T, Hu X, Agbor-Enoh S, Jang MK, Alawi M, Saygi C, et al. The Sirpα–CD47 Immune Checkpoint in NK Cells. J Exp Med (2021) 218. doi: 10.1084/jem.20200839
226. Xu H, Wang B, Ono M, Kagita A, Fujii K, Sasakawa N, et al. Targeted Disruption of HLA Genes via CRISPR-Cas9 Generates iPSCs With Enhanced Immune Compatibility. Cell Stem Cell (2019) 24:566–78:e7. doi: 10.1016/J.STEM.2019.02.005
227. Romano E, Trionfini P, Giampietro R, Benigni A, Tomasoni S. Generation of a Homozygous CIITA Knockout iPS Cell Line Using the CRISPR-Cas9 System. Stem Cell Res (2021) 57:102580. doi: 10.1016/j.scr.2021.102580
228. Dai H, Lan P, Zhao D, Abou-Daya K, Liu W, Chen W, et al. PIRs Mediate Innate Myeloid Cell Memory to Nonself MHC Molecules. Science (2020) 368:1122–7. doi: 10.1126/science.aax4040
229. Al-Moussawy M, Abdelsamed HA, Lakkis FG. Immunoglobulin-Like Receptors and the Generation of Innate Immune Memory. Immunogenetics (2022) 74:179–95. doi: 10.1007/s00251-021-01240-7
230. Hirayasu K, Arase H. Functional and Genetic Diversity of Leukocyte Immunoglobulin-Like Receptor and Implication for Disease Associations. J Hum Genet (2015) 60:703–8. doi: 10.1038/JHG.2015.64
231. Barkal AA, Weiskopf K, Kao KS, Gordon SR, Rosental B, Yiu YY, et al. Engagement of MHC Class I by the Inhibitory Receptor LILRB1 Suppresses Macrophages and Is a Target of Cancer Immunotherapy Article. Nat Immunol (2018) 19:76–84. doi: 10.1038/s41590-017-0004-z
Keywords: NK cell, iPSC (induced pluripotent stem cell), CAR (chimeric antigen receptor), engineering, stealth
Citation: Rossi F, Fredericks N, Snowden A, Allegrezza MJ and Moreno-Nieves UY (2022) Next Generation Natural Killer Cells for Cancer Immunotherapy. Front. Immunol. 13:886429. doi: 10.3389/fimmu.2022.886429
Received: 28 February 2022; Accepted: 25 April 2022;
Published: 02 June 2022.
Edited by:
Rizwan Romee, Dana–Farber Cancer Institute, United StatesReviewed by:
Subramaniam Malarkannan, Medical College of Wisconsin, United StatesCopyright © 2022 Rossi, Fredericks, Snowden, Allegrezza and Moreno-Nieves. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Uriel Y. Moreno-Nieves, dW1vcmVub25AaXRzLmpuai5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.