 Geoffrey Parriott
Geoffrey Parriott Barbara L. Kee
Barbara L. Kee
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 20 April 2022
Sec. T Cell Biology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.885144
This article is part of the Research Topic Molecular Switches of the Immune System: The E Protein/Id Axis in Hematopoietic Development and Function View all 11 articles
T Lymphocyte Acute Lymphoblastic Leukemia (ALL) is an aggressive disease arising from transformation of T lymphocytes during their development. The mutation spectrum of T-ALL has revealed critical regulators of the growth and differentiation of normal and leukemic T lymphocytes. Approximately, 60% of T-ALLs show aberrant expression of the hematopoietic stem cell-associated helix-loop-helix transcription factors TAL1 and LYL1. TAL1 and LYL1 function in multiprotein complexes that regulate gene expression in T-ALL but they also antagonize the function of the E protein homodimers that are critical regulators of T cell development. Mice lacking E2A, or ectopically expressing TAL1, LYL1, or other inhibitors of E protein function in T cell progenitors, also succumb to an aggressive T-ALL-like disease highlighting that E proteins promote T cell development and suppress leukemogenesis. In this review, we discuss the role of E2A in T cell development and how alterations in E protein function underlie leukemogenesis. We focus on the role of TAL1 and LYL1 and the genes that are dysregulated in E2a-/- T cell progenitors that contribute to human T-ALL. These studies reveal novel mechanisms of transformation and provide insights into potential therapeutic targets for intervention in this disease.
T cell acute lymphoblastic leukemia (T-ALL) is an aggressive disease that accounts for 15% of pediatric and 25% of adult leukemia cases (1). These leukemias can be grouped into subtypes based on their unique gene expression profiles and response to therapy with subtypes mirroring known stages of T cell development (2–4). Consistent with these heterogeneous phenotypes, mutations in, or altered expression of, genes encoding multiple transcription factors (cMYC, IKZF1, GATA3, TCF7, LEF1), epigenetic regulators (EZH2 and PHF6), cytokine receptors (IL7R and FLT3), cell cycle regulators (CDKN2A and CDKN2B), and signaling proteins (PTEN) have been identified in subsets of human T-ALL (5–7). Despite this heterogeneity, there are oncogenic pathways that are dysregulated broadly across multiple T-ALL subtypes. In particular, mutations that impact the Notch signaling pathway are present in approximately 80% of T-ALL (8). Approximately 60% of T-ALL cases also have mutations that augment expression of the transcription factors T acute lymphoblastic leukemia antigen 1 (TAL1), also called Stem Cell Leukemia (SCL), and lymphocytic leukemia antigen 1 (LYL1) (9). The high frequency of NOTCH1, TAL1 and LYL1 dysregulation indicate that these pathways impact key processes underlying T cell homeostasis at multiple stage of development and make them attractive targets for therapeutic intervention. Indeed, NOTCH signaling inhibitors have been designed and tested in clinical trials and show some utility for treatment of T-ALL (10). Unfortunately, the broad expression of NOTCH1 has limited the utility of NOTCH1 inhibition as a single agent for treatment of T-ALL, although recently described inhibitors have had some success (10). Nonetheless, ongoing studies taking advantage of partial Notch signaling inhibition combined with targeting other essential pathways is a promising approach for future therapeutics (11). A better understanding of the mechanisms leading to leukemogenesis and the sensitivity of leukemias to inhibition of oncogenic pathways will help to develop novel therapeutics to intervene in this disease.
TAL1 and LYL1 are basic helix-loop-helix (bHLH) proteins that bind DNA in association with the E protein transcription factors (12). The E proteins form homodimers that are critical for B and T lymphocyte development and dimerization with TAL1 or LYL1 alters E protein DNA binding specificity and promotes interactions with unique transcriptional complexes (13–15). Therefore, while TAL1:E protein or LYL1:E protein dimers in T-ALL regulate leukemia-associated genes, expression of TAL1 and LYL1 can also interrupt the function of E protein homodimers. There is substantial evidence in mice to suggest that inhibition of E protein function is sufficient to promote T cell progenitor transformation (16–19). These mouse models recapitulate features of their human T-ALL counterparts such as recurrent mutations in the Notch1 gene and a requirement for Notch signaling for their survival (20–22). In this review we will discuss the mechanisms driving the genesis and maintenance of T-ALL with a focus on the insights gained through studies in E2a-deficieint mice.
The Tcf3 (E2a) gene encodes 2 bHLH proteins (E12 and E47) through alternative splicing of exons encoding the bHLH domain (13). The HLH domain is involved in dimerization with other HLH proteins and the basic region is largely responsible for DNA binding, although some DNA contacts are made with the HLH domain (12). The bHLH domains of E12 and E47 share approximately 80% identity and they bind the same DNA motif, although with differing affinity, and interact with the same proteins (23, 24). There are two additional genes encoding for E proteins in humans and mice, TCF12 (HEB) and TCF4 (E2-2), that each code for two E box binding proteins through alternative transcription start sites, resulting in proteins with differing activation domains but identical bHLH domains (25). Other proteins that dimerize with E proteins include the Class IV HLH proteins (ID1-4), which lack a DNA binding domain and therefore prevent E proteins from stably binding DNA, and Class II bHLH proteins, which are largely cell type specific (i.e. MYOD in muscle cells and TAL1 in hematopoietic stem cells) (12). E proteins are broadly and constitutively expressed and are generally found in complexes with tissue-restricted Class II proteins. However, E proteins function as homodimers in lymphocytes; E2A homodimers predominating in B lymphocytes and dimers of E2A and HEB being prevalent in T lymphocytes (13, 25). Consistent with this, E2a-/- mice have severe defects in lymphopoiesis with a complete lack of B lymphocytes and a 3-5X decrease in thymocyte numbers prior to the onset of leukemogenesis (16, 17, 26, 27). Tcf12-deficiency or Tcf4-deficiency also impacts T cell development but to date, neither of these deficiencies is sufficient to promote T-ALL like disease (25, 28–30).
An advantage of studying T-ALL in mice is that we can track progenitors prior to the onset of disease and discern the impact of these mutations on T cell development as well as on leukemogenesis (Figure 1). TAL1, and by analogy its E protein partners, plays a critical role in specification of hematopoietic stem cells (HSCs) from hemangiogenic endothelium but TAL1 is not essential for HSC survival after HSC development (31, 32). However, post HSC specification, TAL1 plays important roles in megakaryocyte differentiation and erythropoiesis (33, 34). LYL1 is not essential for HSC specification, although it becomes essential when TAL1 is limiting, indicating that these proteins have some redundant functions (35). ChIP-seq experiments with TAL1 and LYL1 in HSC-like cell lines revealed extensive overlap in their binding sites indicating that these proteins regulate an overlapping set of genes (36). In E2a-/- mice, HSC specification is intact but lymphopoiesis is impacted at the stage when HSCs become specified to the lymphoid fate with fewer lympho-myeloid primed progenitors (LMPPs) and a failure to initiate expression of multiple lymphoid genes (37, 38). In LMPPs E2A likely functions in cooperation with LYL1 since Lyl1-/- mice have a strikingly similar phenotype to E2a-/- mice at this stage (39). In contrast, TAL1 antagonizes T lymphocyte specification within the HSC and LMPP populations (40). Therefore, despite the similarity in TAL1 and LYL1 structure and their overlapping function in HSC specification, these proteins function in an opposing manner to regulate lymphoid specification. Given the roles of E2A, TAL1 and LYL1 in T-ALL, we anticipate that understanding how these proteins control lymphocyte development will provide insights into the mechanisms that drive lymphopoietic alterations and transformation.
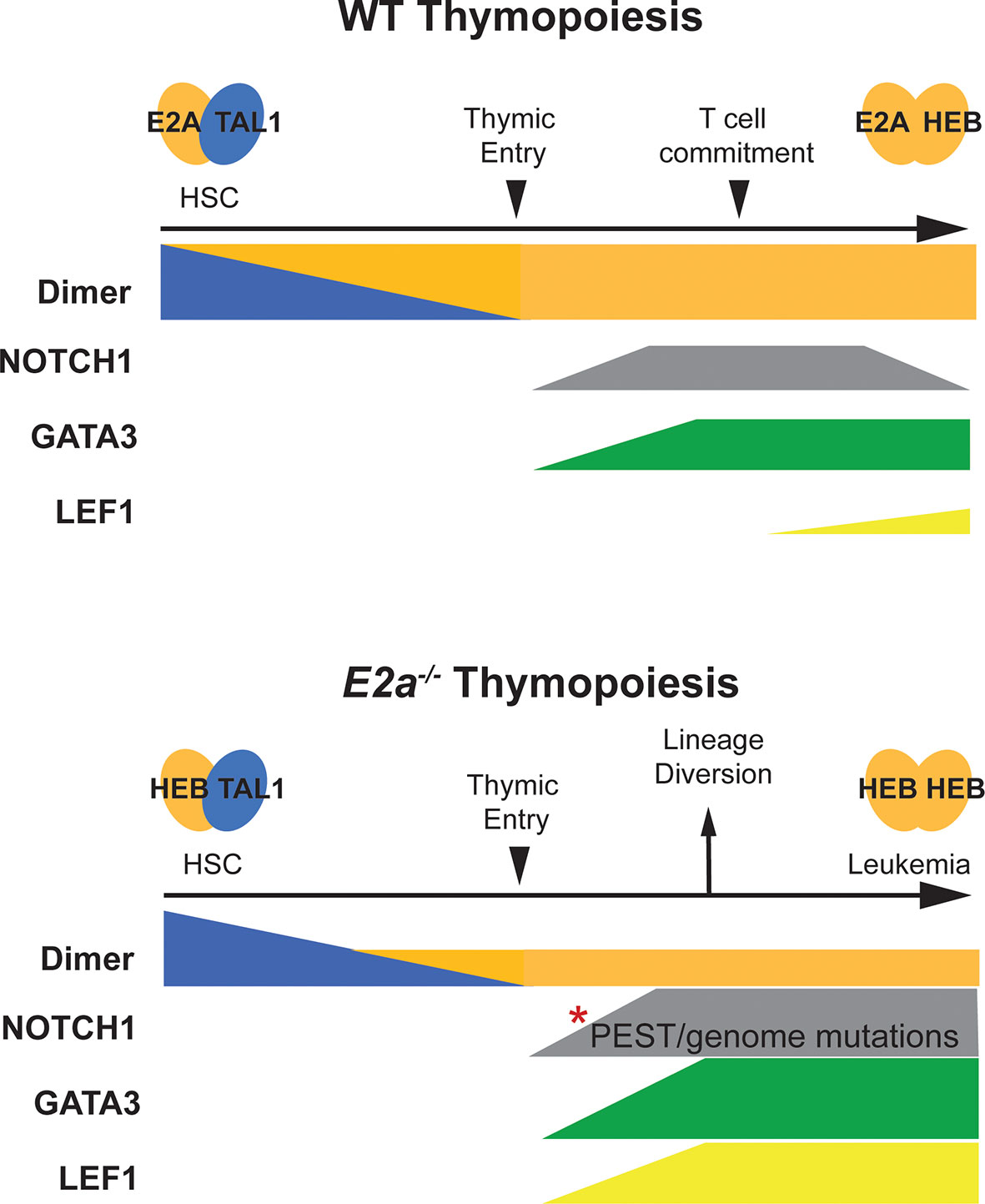
Figure 1 Schematic representation of T cell development in WT and E2a-/- mice. T cell development in WT mice is depicted in the top half of the figure. Tal1:E2A (or E protein) dimers are prevalent in HSCs and decline as progenitors differentiate toward the T cell lineage. At the time of thymic commitment E2a:HEB dimers dominate. NOTCH1 and GATA3 start to be expressed upon thymic entry, along with TCF1 (not depicted) under the influence of NOTCH1 ligands in the thymus. LEF1 expression initiates with commitment to the T lymphocyte lineage but remains low throughout T cell development. T cell development in E2a-/- mice is depicted on the lower half of the figure. Here, Tal1:HEB or E2-2 dimers control early hematopoiesis resulting in a reduced number of thymic seeding progenitors that can only express HEB : HEB or E2-2 dimers after extinction of TAL1 expression. GATA3 expression is high and diverts progenitors toward the ILC/NK fate. Mutations in the Notch1 gene accumulate and LEF1 is expressed at very high levels. Combined, these alterations in NOTCH1, GATA3 and LEF1 contribute to T cell transformation.
E2A proteins are required for proper expression of Notch1 at the inception of T cell development (37, 41). Consistent with this, when E2A-/- multipotent progenitors are cultured under T cell differentiation conditions in vitro they fail to generate T cells unless they are transduced with a NOTCH1 producing retroviral vector (42). E2a-/- DN2 thymocytes struggle to enter the T cell lineage and fail to control the expression of GATA3, which is substantially elevated in E2a-/- DN2 and DN3 thymocytes (43). This elevated expression of GATA3 contributes to diversion of these cells toward the innate lymphoid lineages, which is particularly evident when E2a and Heb are both deleted or when ID1 is over expressed in T cell progenitors (43–46). Indeed, heterozygous deletion of Gata3 restores differentiation of E2a-/- DN2 cells into the T cell lineage (43). Ectopic expression of GATA3 under the control of the CD2 promoter is able to promote T cell transformation suggesting that this failure to repress Gata3 could be a key event in the generation of E2a-/- leukemias (47). In established E2a-/- leukemia lines re-expression of E2A proteins alters the transcription of numerous genes including Gata3, which is indirectly regulated by E2A-mediated induction of GFI1B (48). Whether GFI1B, alone or in combination with the related transcription factor GFI1, functions to dampen Gata3 expression at the inception of T cell development remains to be fully explored but it is notable that both Gfi1b-/- and Gfi1-/- mice have defects in T cell development that overlap with those of E2a-/- mice (49, 50). During B cell development Gata3 is repressed by EBF1 suggesting that GFI1/GFI1B and EBF1 might play similar roles in progenitors prior to their entry into the T and B cell developmental pathway with EBF1 leading to more severe, or sustained, repression of Gata3 (51).
The few T cell progenitors that develop from E2a-/- DN2 thymocytes highly express LEF1, an effector of the Wnt signaling pathway, and LEF1 is required for the survival of E2a-/- leukemias (52, 53). LEF1 is not essential for T cell development owing to the high expression of the related transcription factor TCF1 in T cell progenitors (54). Tcf7 (encoding TCF1) is regulated by NOTCH1 and plays a major role in T cell lineage specification (55, 56). TCF1 is expressed in E2a-/- thymocytes despite the increased expression of LEF1; nonetheless, LEF1 impacts E2A-/- T cell development. Indeed, deletion of Lef1 from E2a-/- T cell progenitors results in a profound loss of DN3 thymocytes while, surprisingly, not affecting overall T cell numbers (53). These findings suggest that LEF1 plays a role in controlling the maturation of E2a-/- T cells. Lef1 mRNA is elevated in multiple mouse models that develop T-ALL, and as described later in this review, LEF1 can play both oncogenic and tumor suppressor roles in these models depending on the timing of its expression (53, 57–60). In the following sections we will discuss the known contributions of the E2A interacting proteins TAL1 and LYL1 and the genes dysregulated in E2a-/- thymocytes to human and murine T-ALL.
TAL1 was identified as a gene involved in the t(1:14) and t(1;7) chromosomal translocations in T-ALL, which place TAL1 under the control of the TCRA/TCRD or TCRB locus, respectively (61–63). LYL1 was also identified through a chromosomal translocation in T-ALL in which LYL1 on chromosome 19 is juxtaposed to the TCRB constant regions on Chromosome 7 (64). These translocations are found in approximately 3-7% of TAL1/LYL1+ T-ALL cases, however, there are frequent alterations at the TAL1 locus in T-ALL including deletions such as TAL1d, which arises from a site-specific DNA recombination event causing a 90kb deletion upstream of TAL1 (62). These deletions place the coding region of the TAL1 gene downstream of regulatory elements in the SCL interrupting locus (STIL). The STIL regulatory elements are constitutively active in thymocytes, resulting in ectopic TAL1 expression. These TAL1 upstream deletions are specific for T-ALL cells and are most likely caused by erroneous V(D)J recombinase activity (65, 66). Alterations in the TAL1 gene that result in ectopic T lymphocyte expression of TAL1 are now recognized to be present in as many as 60% of T-ALL (1). While these genomic alterations account for a majority of T-ALL associated TAL1 expression, a subset of patients have ectopic TAL1 expression without these alterations. Studies into the mechanisms of TAL1 deregulation in these patients revealed small insertions (<20bp) in a region 8kb upstream of TAL1 that create a de novo MYB binding site that results in strong enhancer activity in these leukemias (67). Chromatin immunoprecipitation followed by high throughput sequencing (ChIP-seq) experiments showed that MYB binds to this novel site along with chromatin remodelers and other components of the DNA transcriptional machinery. Deletion of the novel MYB binding site abrogated MYB binding and significantly reduced TAL1 expression. MYB is highly expressed in thymocytes and MYB is often dysregulated in cancer and thus this mutation can lead to robust TAL1 transcription in leukemic cells (68). Taken together, these findings outline multiple mechanisms leading to the errant expression of TAL1 in T-ALL (Figure 2).
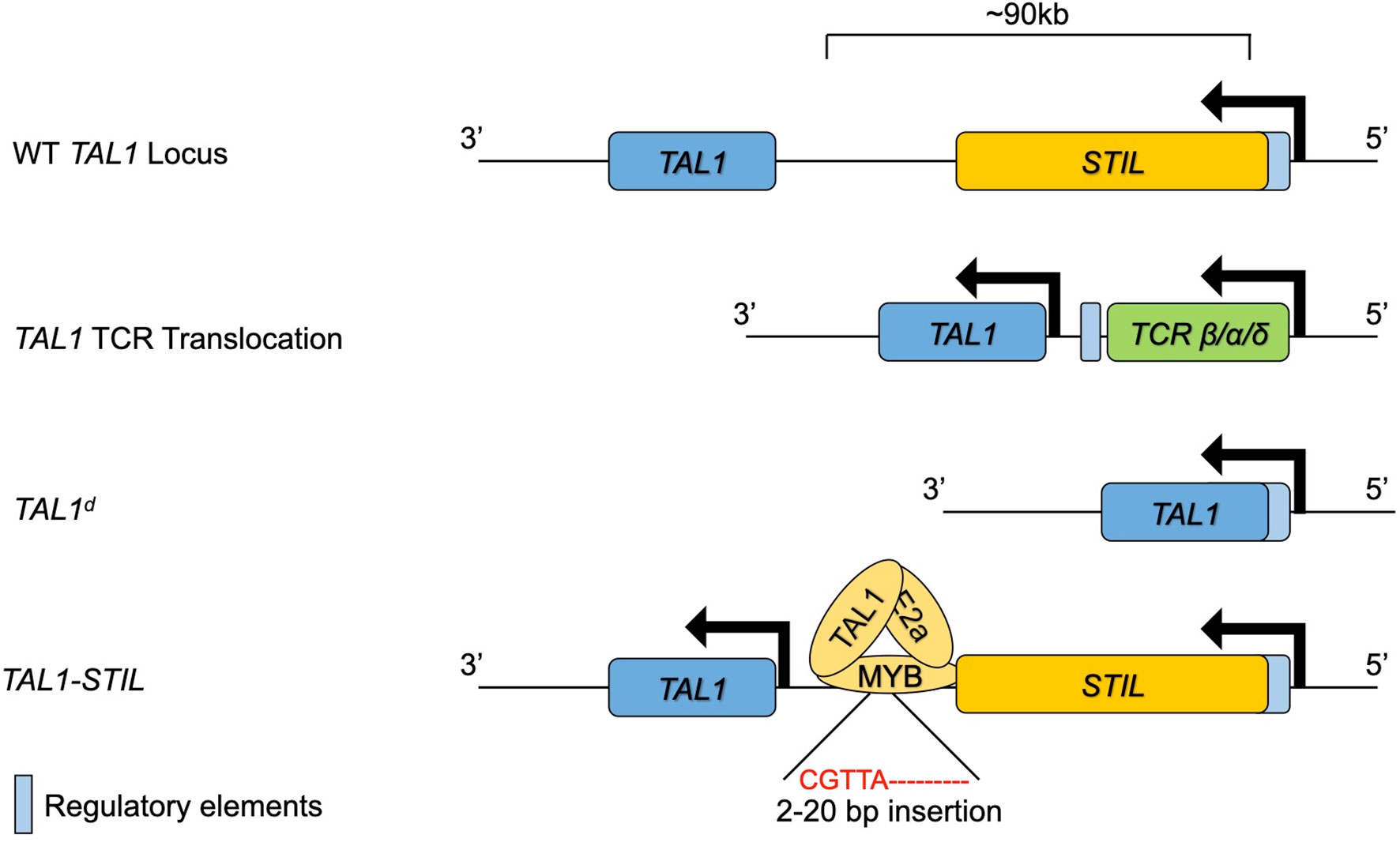
Figure 2 The TAL1 genomic locus and mutations leading to ectopic expression in T lymphocytes and T-ALL. The WT TAL1 locus is depicted downstream of the STIL gene. TAL1 transcription is increased in T lymphocytes through genomic translocation into the TCRB/A/D locus, through genomic deletions bringing the STIL enhancer close to TAL1 or through insertions that create a novel MYB binding enhancer upstream of TAL1.
TAL1 positive leukemias frequently express CD4 and CD8 and have a cortical phenotype similar to what is observed in E2a-/- mice (2, 16, 17, 69). In mice, expression of TAL1 under the control of the Lck promoter, which drives gene expression early in T cell development, is sufficient to predispose mice in T-ALL-like disease (70). That TAL1 functions through sequestration of E protein is implied by studies that showed that TAL1-driven leukemia is not dependent on the DNA binding ability of TAL1 (18). Moreover, transgenic expression of ID1 or ID2, which prevent E proteins from binding to DNA, also predisposes mice to develop a T-ALL- like disease (19, 71). It is also notable that Lck-TAL1 promotes leukemia in a dose-dependent manner that is augmented by deletion of the E proteins encoded by Heb/Tcf12 indicating that E protein dose is a major determinant of leukemogenesis in this model (18). Analogous to what is observed in Lck-TAL1 transgenic mice, ectopic expression of LYL1 in T cell progenitors blocks the formation of E-protein homodimers, suppresses the expression of E2A-dependent genes, and leads to T-ALL like disease (69, 72). These findings suggest that at least a part of the mechanism through which TAL1 and LYL1 promote leukemogenesis is through inhibition of E protein homodimer function. However, LYL1+ and TAL1+ leukemias have unique gene expression profiles and LYL1+ leukemias tend to be related to immature CD4-CD8- T cell progenitors (2). These observations suggest that LYL1 and TAL1 have unique functions or that they are expressed in different cellular contexts, either distinct stages of development or stages of transformation. Interestingly, nearly 30% of pediatric TAL1+ T-ALL patients have heterozygous loss-of-function mutations in USP7, a deubiquitinating enzyme that interacts with E proteins, and other leukemia-associated proteins, and is associated with decreased E protein target gene expression and increased cell growth (73). Therefore, there appears to be multiple mechanisms contributing to reduced E protein function in T-ALL.
While inhibition of E protein function is sufficient to predispose T cell progenitors to transformation, TAL1 and LYL1 may contribute to transformation through their participation in transcriptional complexes that activate or inhibit gene expression. In hematopoietic progenitors and T-ALL, both TAL1 and LYL1 bind DNA in large complexes that include the LMO (LIM only), LIM domain binding (LDB1), and GATA protein families (74–78). Indeed, LMO proteins are critical members of the TAL1 complex, acting as bridging factors that connect TAL1 to other DNA binding proteins like GATA1/2/3 (79). This is of particular interest because LMO proteins are overexpressed in approximately 10% of T-ALLs and these leukemias frequently have TAL1 overexpression (1). In a subset of pediatric (3.7%) and adult (5.5%) T-ALL the LMO2 gene contains intronic indels that result in de novo binding sites for the leukemia-associated transcription factors MYB, ETS1 or RUNX1 and thus dysregulated LMO2 expression (80). Interestingly, both TAL1 and LMO1 or LMO2 are required to induce reporter activity in T-ALL cell lines (81). Experiments in human T-ALL cell lines have been vital to elucidating the core components of the TAL1 complex in leukemia. ChIP-seq in these lines has identified E2A, GATA3, LMO1/LMO2, RUNX1, and MYB as co-bound to TAL1 bound regions suggesting multi-transcription factor complex formation (82). Using siRNA to knock down TAL1 or other members of the TAL1 complex, Sanda et al. identified genes regulated by this complex in leukemias (83). Interestingly, expression of the genes that make up the TAL1 complex is decrease upon TAL1 siRNA knockdown. This finding suggests a positive feed-forward mechanism where the oncogenic TAL1 complex promotes expression of itself, in addition to promoting expression of other known oncogenes. TAL1-dependent genes include MYB, which positively regulates cell cycle and anti-apoptotic genes, TRIB2, which supports the survival of T-ALL cell lines, and ARID5, a gene associated with a variety of leukemias (83–85). Of note, the oncogenic microRNA miR-223 is also decreased after reducing TAL1 and ChIP-seq revealed that the TAL1 complex binds to a putative enhancer near this gene (86).
There is evidence indicating that LYL1 forms oncogenic complexes similar to TAL1. Indeed, Jurkat cells forced to express LYL1, LMO2 and LDB1 induced robust target gene expression that was dependent on LMO2 and LDB1 (77). Further, LMO2 is frequently overexpressed in TAL1 expressing leukemias but in LMO2 transgenic mice TAL1 is dispensable for leukemogenesis (87). In contrast, deletion of Lyl1 significantly increases leukemia latency in LMO2 transgenic mice suggesting that LYL1 supports transformation. Microarray analysis revealed that LMO2 expressing thymocytes have higher Lyl1 expression compared to wild type thymocytes indicating a potential feed-forward mechanism reminiscent of the mechanism seen in TAL1 expressing leukemias. Consistent with this idea, the LYL1 promoter contains ETS and GATA binding sites, which promote the expression of LYL1 in HSCs and both ETS1 and GATA3 are implicated in T-ALL (88–90). Taken together, these data suggest that LYL1 may function in a manner analogous to TAL1 during T cell progenitor transformation.
NOTCH1 is constitutively activated in a majority of T-ALL, including those that overexpress TAL1, and in leukemias from E2a-/- mice (20, 91). NOTCH1 functions as both a surface receptor and transcription factor that is essential for T cell development (8, 92). The ligands for NOTCH1 are members of the Jagged and Delta-like family, with DELTA-LIKE 4 (DLL4) being the most important in the thymus (93). NOTCH1 is translated as a single protein that is cleaved in the Golgi to create extracellular and intracellular components that are held together in the membrane by heterodimerization domains (HD) (8). Upon ligand binding the extracellular portion undergoes a conformational change that allows cleavage by a disintegrin and metalloprotease, which exposes a cleavage site for γ-secretase, which then cleaves and liberates the intracellular domain of NOTCH1 (called ICN). The ICN translocates to the nucleus, where it complexes with the DNA bound transcriptional repressor CBF1/RBP-Jκ and recruits Mastermind (MAML) proteins to initiate the transcription of multiple genes that promote T cell specification (8). Activation of NOTCH1 is transient owing to the presence of a PEST sequence at the 3’ end that is recognized by the FBW7 ubiquitin ligase and targets ICN for proteasomal degradation (94). Targets of the ICN/CBF1/MAML complex in murine T cells include Hes1 and Tcf7, both of all of which play critical roles in T cell development (56, 95, 96). Interestingly, Notch signaling also shuts down transcription of Ccr9, which encodes a chemokine receptor involved in thymic homing of LMPPs (97, 98). Repression of Ccr9 may trap lymphoid progenitors in the cortex of the thymus as they undergo commitment to the T cell lineage. Repression of Ccr9 may also explain why ectopic expression of ICN in murine HSCs promotes T cell transformation without accumulation of these leukemic cells in the thymus (99).
NOTCH1 was identified as an oncogene in T-ALL by its involvement in a t(7;9)(q34;q34.3) translocation that placed the 3’ end of the NOTCH1 gene under control of the TCRB locus, resulting in constitutive activation of NOTCH1 in T cell progenitors (100). This translocation is present in approximately 2% of leukemias, however, it is now appreciated that > 60% of all human T-ALLs have mutations in NOTCH1 (8). These mutations cluster in the heterodimerization domain (HD) and in the PEST domain. Mutations affecting the HD domains prevent the association of the extracellular and intracellular portions of NOTCH1 thus allowing for spontaneous γ-secretase mediated cleavage to produce active ICN. The PEST domain mutations promote stabilization of ICN by removing the phosphorylation sites that lead to docking of FBW7. These mutations are not mutually exclusive, with ~20% of human leukemias having mutations in both domains (101). Additional mutations have been identified that inactivate FBW7 resulting in the constitutive stabilization of ICN (8, 102, 103). The TAL1 complex also represses FBWX7 through miRNA-223 suggesting that there may be numerous mechanisms contributing to stabilization of NOTCH1 in T-ALL (86).
Leukemias arising in E2a-/- mice have mutations in the PEST domain of NOTCH1 but no mutations have been identified in the HD domain (20). How then is NOTCH1 activated in these leukemias? Insight into this question came when it was revealed that alternative transcription initiation sites are used at the Notch1 gene in Ikzf1-/- and E2a-/- leukemias (104, 105). Multiple transcripts were identified that initiate from a cryptic promoter upstream of exon 26 leading to a protein that lacks the extracellular domain of NOTCH1. This cyptic promoter can be activated by deletion of the promoter upstream of exon 1, which occurs through a RAG1-dependent mechanism (106). Surprisingly, in Ikzf1-/- mice, deletion of the first exon of Notch1 did not impact T cell development, unlike what is seen in Ikzf1+/+ mice, due to use of this alternative mechanism for transcribing Notch1 in the absence of IKAROS. Ikzf1-/- thymocytes have increased histone acetylation near IKAROS binding sites located near the alternative Notch1 promoter raising the possibility that IKAROS represses the use of this alternative mechanism through epigenetic modification (104, 105). Potential E2A binding sites are also present within the alternate promoter and these alternative NOTCH1 isoforms are expressed in E2a-/- leukemias indicating that E proteins may cooperate with IKAROS to repress alternative promoter use (104). While E2A may repress the alternative promoter, E2A promotes Notch1 expression in thymic seeding progenitors (42). This deficiency in NOTCH1 could provide a strong selective pressure for NOTCH1 mutation or altered transcription initiation site used to support T cell development. Genomic deletions have also been found in mouse leukemic cells that result in splicing of Exon 1 to downstream exons and again result in proteins that lack the extracellular domain and are constitutively active but dependent on γ-secretase (8, 106).
The mechanisms by which NOTCH1 promotes leukemogenesis have been studied intensively. Interestingly, Notch signaling can impact expression of E2A, at least in mice, where it has been shown that mitogen activated protein kinase phosphorylation of E2A leads to NOTCH1-dependent ubiquitination and proteasomal degradation of E2A (107, 108). Whether this mechanism contributes to ICN induced leukemogenesis in mice or humans requires further investigation. In human T-ALL, a major target of Notch signaling is c-MYC, which itself is oncogenic in T lymphocyte progenitors (8, 109). ICN binds to an enhancer 140 Mb downstream of c-MYC, whose activity correlates with responsiveness to NOTCH1 inhibitors (110). Moreover, mutation of this enhancer prevents leukemogenesis by ectopic expression of ICN demonstrating that it is an essential target. This enhancer is also regulated by NOTCH3 in NOTCH3-dependent leukemias (111). It is likely that there are many essential targets of NOTCH1 in T-ALL. Indeed, in E2a-/- leukemias c-MYC expression is stably amplified through trisomy at chromosome 15 and therefore does not require ICN for expression yet these leukemias are still dependent on Notch1 signaling (16, 52).
ChIP-seq analysis for ICN has revealed multiple novel targets of NOTCH1 in leukemia (112). ICN bound regions are in close proximity to RUNX, ETS, and ZNF143 binding motifs and these regions have extensive histone acetylation and H3K4me3 chromatin modifications, indicative of open chromatin and active gene transcription (60, 112). Thus, it is possible that NOTCH1 promotes accessibility to target gene regulatory regions, which allows other T cell specific transcription factors or DNA transcriptional machinery to bind and promote gene expression. Consistent with this idea, NOTCH1 was required for recruitment of RUNX1 and MYB to enhancers located within the TCRG and TCRB locus (113).
Many ICN target genes, including DTX1, IGF1R, IL7R, and GIMAP, have been identified by evaluating changes in gene expression after treatment of leukemias with γ-secretase inhibitors (60, 109). Importantly, many of these genes are co-regulated by T cell specific factors like RUNX and ETS1. Deletion of RUNX1 in DN2/3 thymocytes impairs IL7R expression (114), and expression of dominant-negative RUNX1 and NOTCH1 inhibitors (RUNT and DN-MAML, respectively) suppressed IL7R mRNA expression (60). Further, ETS1 binds to multiple NOTCH1 occupied sites in T-ALL (89). Indeed, mice overexpressing NOTCH1 fail to develop leukemia when lacking functional ETS1 suggesting that both of these factors are required for leukemia initiation. ETS1 is frequently over expressed in human T-ALL samples and cell lines indicating that ETS1 may act in concert with NOTCH1 in the human disease as well. Indeed, shRNA–mediated knockdown of ETS1 in human T-ALL lines promoted cell death and significantly down-regulated expression of the oncogenes c-MYC and IGFR1, as well as other NOTCH1 target genes like HES1 and DELTEX1 (89). Understanding of the spectrum of genes induced by NOTCH1 and identifying co-regulators may reveal mechanisms that could be targeted for treatment of T-ALL.
GATA3 is essential for T cell specification and in its absence multipotent progenitors fail to generate committed T lymphocytes (115–117). Ectopic expression of GATA3 can also derail T cell development and force T cell progenitors down alternative lineages, such as the mast cell lineage (118). However, transgenic expression of GATA3, under the control of the CD2 promoter, which drives expression in all lymphocytes, predisposes mice to develop T-ALL-like disease with trisomy of chromosome 15 and activation of NOTCH1, similar to what is seen in E2a-/- leukemias, although with longer latency (47, 119). GATA3 is elevated in E2a-/- T cell progenitors and has a negative impact on the ability of DN2 cells to generate T lineage-restricted cells (43). These findings support a role for GATA3 in T cell leukemogenesis and implicate it as a potential contributing factor to T cell transformation in E2a-/- mice even though this has not been formally demonstrated. Indeed, in non-ETP-ALL GATA3 expression is elevated compared to T cells from healthy donors and defines a stem-like progenitor (120). The mechanism by which GATA3 promotes thymocyte transformation and leukemia survival is not well understood. One potential mechanism involves GATA3’s association with TAL1 as a member of the oncogenic TAL1 complex. Indeed, siRNA knockdown of GATA3 in T-ALL cells represses transcription of TAL1 target genes suggesting that GATA3 is required for proper TAL1 complex function (83). GATA3 and other members of the TAL1 oncogenic complex also bind to the NOTCH1-regulated enhancer downstream of c-Myc (90, 110). Mutating the GATA3 binding sites in this enhancer impacted nucleosome eviction and chromatin accessibility, resulting in decreased c-MYC expression and abrogated leukemia development in mice (90). These observations indicate that GATA3 cooperates with TAL1 and NOTCH1 to promote transformation through regulation of c-MYC.
In contrast to these cases of increased GATA3 expression, 5% of T-ALL patients have silencing mutations in the GATA3 gene (1). Consistent with this, another study found that 33% of patients in their cohort with the ETP-ALL subtype had reduced GATA3 expression associated with increased methylation throughout the GATA3 gene (120). Thus, GATA3 may play multiple distinct roles in T-ALL development, suppressing ETP-ALL or promoting T-ALL at later stages. Decreased GATA3 expression in ETP-ALL is consistent with GATA3’s function in promoting T cell lineage differentiation as GATA3 silencing could contribute to a developmental block at the ETP stage that supports transformation. It also seems likely that GATA3 is not a driver mutation and its function may be dependent on the spectrum of additional mutations that occur during transformation.
The NOTCH1 target gene Tcf7, encoding the protein TCF1, is also implicated as a suppressor of T cell transformation (57, 121). TCF1 is a member of the HMG box family of proteins along with the closely related protein LEF1. Both TCF1 and LEF1 can promote transcription in response to canonical WNT signaling activation or repress transcription through recruitment of the Groucho related co-repressors such as TLE3 (122, 123). In the absence of TCF1, thymocytes have a developmental block at the ETP, DN2, and ISP stages whereas mice lacking LEF1 have no obvious defects in DN thymocytes (54–56, 124). Combined deletion of Tcf7 and Lef1 exacerbates the phenotype seen in Tcf7-deficient mice, leading to a nearly complete block in T cell development (54). This observation indicates that TCF1 and LEF1 have overlapping functions and that LEF1 partially compensates for the loss of TCF1. In addition to the defects seen in T lymphopoiesis, approximately 50% of Tcf7-/- mice develop T-ALL (57, 121). Tcf7-/- leukemias are heterogeneous; phenotypically resembling DN3, DN4, and DP thymocytes. Despite this cell surface phenotype, RNA profiling revealed that the transcriptome of Tcf7-/- T-ALLs is related to that of human ETP-ALLs, which is consistent with the early requirement for TCF1 in T cell development (57). Tcf7-/- leukemias have activated NOTCH signaling and inhibiting this pathway with GSI at least partially impacts their viability (121). Further, Tcf7-/- leukemias highly express ID2 and LEF1, particularly in a subset of T cell progenitors with a gene signature predictive of high leukemic potential, suggesting that that suppression of E protein activity may be a feature of transformation in this model (57, 58). Indeed, Tcf7-/-Id2-/- mice showed an increased latency of leukemogenesis consistent with this hypothesis.
Like Tcf7-/- leukemias, E2a-/- leukemias have high expression of Lef1 and LEF1 is required for the survival and proliferation of these leukemias (52). LEF1 is an oncogene in acute myeloid leukemia and in multiple forms of B lymphocyte leukemia and it is suppressed by TCF1 (125–128). Ectopic expression of LEF1 in HSCs induced acute myeloid leukemia-like or B cell ALL-like disease in mice, demonstrating LEF1’s oncogenic potential (127). In an adult cohort of T-ALL patients, high LEF1 expression was associated with increased expression of the oncogenes encoding c-MYC and CYCLIN D1 suggesting that LEF1 is positively associated with T cell leukemia (129). Moreover, 4 unique mutations that augment LEF1 function were found in these patients. In contrast, approximately 11% of pediatric T-ALL patients were found to have inactivating mutations in the LEF1 gene (7, 130). These mutations consist of deletions or truncation mutations, both resulting in lower LEF1 function. These conflicting findings suggest that LEF1 can play multiple roles in T cell leukemia. Indeed, while E2a-/- leukemias are dependent on LEF1, inactivation of Lef1 in E2a-/- mice prior to transformation did not prevent transformation; rather, it reduced leukemia latency and resulted in leukemias with a unique gene expression program compared to E2a-/- leukemias (53). Taken together, these experiments reveal that the timing of genetic alterations in the evolution of T-ALL can determine latency, phenotype and genetic susceptibilities within these cells.
T-ALL is a heterogeneous disease that is associated with mutations in numerous T cell specific transcription factors, epigenetic regulators, and signaling pathways. Despite this heterogeneity, many of the mutations seen in patients impact the function of E proteins and their target genes. Recent studies looking at the clonal evolution of T-ALL have further indicated that TAL1 upregulation is a founding event in the human disease, preceding mutations in NOTCH1 (131). Thus, understanding how reduced E protein function impacts T cell development and leukemogenesis is highly relevant to the human disease. The E2a-/- mouse model has revealed connections between T cell developmental alterations and the transformation process. For example, Notch1, Gata3, and Lef1/Tcf7 expression are altered early in T cell development in E2a-/- mice and contribute to transformation. The pathways regulated by these proteins are promising candidates for therapeutic intervention in T-ALL. Indeed, inhibitors of NOTCH1 activation or function have been in clinical trials, however many of these inhibitors have adverse side effects due to the many cell types that rely on Notch signaling (11, 132). Novel NOTCH1 inhibitors that prevent the formation of the ICN transcriptional activation complex show decreased off target effects in vivo compared to inhibitors that target NOTCH1 activation, but still induce mild intestinal toxicity (133). Further investigation of oncogenic and tumor suppressive pathways in murine T-ALL models, including GATA3 and TCF1/LEF1, and their application to human leukemia may identify novel targets that alone, or in combination with other targets, will have fewer side effects without sacrificing anti-leukemia efficacy.
GP and BK wrote the manuscript and approved the final version. All authors contributed to the article and approved the submitted version.
Our studies on E2a-/- T cell leukemia were supported by the NIH/NIAID (R01 AI106352, R21 AI119894, R21 AI096350, and R03 AI137605) and the Janet Rowley Fund (University of Chicago Comprehensive Cancer Center).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank past and present members of the Kee lab, Fotini Gounari, Warren Pear and Adolfo Ferrando for helpful discussions.
1. Belver L, Ferrando A. The Genetics and Mechanisms of T Cell Acute Lymphoblastic Leukaemia. Nat Rev Cancer (2016) 16(8):494–507. doi: 10.1038/nrc.2016.63
2. Ferrando AA, Look AT. Gene Expression Profiling in T-Cell Acute Lymphoblastic Leukemia. Semin Hematol (2003) 40(4):274–80. doi: 10.1016/s0037-1963(03)00195-1
3. Noronha EP, Marques LVC, Andrade FG, Thuler LCS, Terra-Granado E, Pombo-de-Oliveira MS, et al. The Profile of Immunophenotype and Genotype Aberrations in Subsets of Pediatric T-Cell Acute Lymphoblastic Leukemia. Front Oncol (2019) 9:316. doi: 10.3389/fonc.2019.00316
4. Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D, et al. Early T-Cell Precursor Leukaemia: A Subtype of Very High-Risk Acute Lymphoblastic Leukaemia. Lancet Oncol (2009) 10(2):147–56. doi: 10.1016/S1470-2045(08)70314-0
5. Van Vlierberghe P, Ferrando A. The Molecular Basis of T Cell Acute Lymphoblastic Leukemia. J Clin Invest (2012) 122(10):3398–406. doi: 10.1172/JCI61269
6. Iacobucci I, Kimura S, Mullighan CG. Biologic and Therapeutic Implications of Genomic Alterations in Acute Lymphoblastic Leukemia. J Clin Med (2021) 10(17):3792–816. doi: 10.3390/jcm10173792
7. Girardi T, Vicente C, Cools J, De Keersmaecker K. The Genetics and Molecular Biology of T-All. Blood (2017) 129(9):1113–23. doi: 10.1182/blood-2016-10-706465
8. Aster JC, Blacklow SC, Pear WS. Notch Signalling in T-Cell Lymphoblastic Leukaemia/Lymphoma and Other Haematological Malignancies. J Pathol (2011) 223(2):262–73. doi: 10.1002/path.2789
9. Sanda T, Leong WZ. Tal1 as a Master Oncogenic Transcription Factor in T-Cell Acute Lymphoblastic Leukemia. Exp Hematol (2017) 53:7–15. doi: 10.1016/j.exphem.2017.06.001
10. Ferreira A, Aster JC. Notch Signaling in Cancer: Complexity and Challenges on the Path to Clinical Translation. Semin Cancer Biol (2021) S1044-579X(21)00109-7. doi: 10.1016/j.semcancer.2021.04.008
11. Zheng R, Li M, Wang S, Liu Y. Advances of Target Therapy on Notch1 Signaling Pathway in T-Cell Acute Lymphoblastic Leukemia. Exp Hematol Oncol (2020) 9(1):31. doi: 10.1186/s40164-020-00187-x
12. Massari ME, Murre C. Helix-Loop-Helix Proteins: Regulators of Transcription in Eucaryotic Organisms. Mol Cell Biol (2000) 20(2):429–40. doi: 10.1128/MCB.20.2.429-440.2000
14. Hsu HL, Cheng JT, Chen Q, Baer R. Enhancer-Binding Activity of the Tal-1 Oncoprotein in Association With the E47/E12 Helix-Loop-Helix Proteins. Mol Cell Biol (1991) 11(6):3037–42. doi: 10.1128/mcb.11.6.3037-3042.1991
15. Miyamoto A, Cui X, Naumovski L, Cleary ML. Helix-Loop-Helix Proteins Lyl1 and E2a Form Heterodimeric Complexes With Distinctive DNA-Binding Properties in Hematolymphoid Cells. Mol Cell Biol (1996) 16(5):2394–401. doi: 10.1128/MCB.16.5.2394
16. Bain G, Engel I, Robanus Maandag EC, te Riele HP, Voland JR, Sharp LL, et al. E2a Deficiency Leads to Abnormalities in Alphabeta T-Cell Development and to Rapid Development of T-Cell Lymphomas. Mol Cell Biol (1997) 17(8):4782–91. doi: 10.1128/mcb.17.8.4782
17. Yan W, Young AZ, Soares VC, Kelley R, Benezra R, Zhuang Y. High Incidence of T-Cell Tumors in E2a-Null Mice and E2a/Id1 Double-Knockout Mice. Mol Cell Biol (1997) 17(12):7317–27. doi: 10.1128/mcb.17.12.7317
18. O'Neil J, Shank J, Cusson N, Murre C, Kelliher M. Tal1/Scl Induces Leukemia by Inhibiting the Transcriptional Activity of E47/Heb. Cancer Cell (2004) 5(6):587–96. doi: 10.1016/j.ccr.2004.05.023
19. Kim D, Peng XC, Sun XH. Massive Apoptosis of Thymocytes in T-Cell-Deficient Id1 Transgenic Mice. Mol Cell Biol (1999) 19(12):8240–53. doi: 10.1128/mcb.19.12.8240
20. Reschly EJ, Spaulding C, Vilimas T, Graham WV, Brumbaugh RL, Aifantis I, et al. Notch1 Promotes Survival of E2a-Deficient T Cell Lymphomas Through Pre-T Cell Receptor-Dependent and -Independent Mechanisms. Blood (2006) 107(10):4115–21. doi: 10.1182/blood-2005-09-3551
21. Tatarek J, Cullion K, Ashworth T, Gerstein R, Aster JC, Kelliher MA. Notch1 Inhibition Targets the Leukemia-Initiating Cells in a Tal1/Lmo2 Mouse Model of T-All. Blood (2011) 118(6):1579–90. doi: 10.1182/blood-2010-08-300343
22. Sharma VM, Draheim KM, Kelliher MA. The Notch1/C-Myc Pathway in T Cell Leukemia. Cell Cycle (2007) 6(8):927–30. doi: 10.4161/cc.6.8.4134
23. Murre C, McCaw PS, Baltimore D. A New DNA Binding and Dimerization Motif in Immunoglobulin Enhancer Binding, Daughterless, Myod, and Myc Proteins. Cell (1989) 56(5):777–83. doi: 10.1016/0092-8674(89)90682-x
24. Murre C, McCaw PS, Vaessin H, Caudy M, Jan LY, Jan YN, et al. Interactions Between Heterologous Helix-Loop-Helix Proteins Generate Complexes That Bind Specifically to a Common DNA Sequence. Cell (1989) 58(3):537–44. doi: 10.1016/0092-8674(89)90434-0
25. Braunstein M, Anderson MK. Heb in the Spotlight: Transcriptional Regulation of T-Cell Specification, Commitment, and Developmental Plasticity. Clin Dev Immunol (2012) 2012:678705. doi: 10.1155/2012/678705
26. Zhuang Y, Soriano P, Weintraub H. The Helix-Loop-Helix Gene E2a Is Required for B Cell Formation. Cell (1994) 79(5):875–84. doi: 10.1016/0092-8674(94)90076-0
27. Bain G, Maandag EC, Izon DJ, Amsen D, Kruisbeek AM, Weintraub BC, et al. E2a Proteins Are Required for Proper B Cell Development and Initiation of Immunoglobulin Gene Rearrangements. Cell (1994) 79(5):885–92. doi: 10.1016/0092-8674(94)90077-9
28. Barndt RJ, Dai M, Zhuang Y. Functions of E2a-Heb Heterodimers in T-Cell Development Revealed by a Dominant Negative Mutation of Heb. Mol Cell Biol (2000) 20(18):6677–85. doi: 10.1128/MCB.20.18.6677-6685.2000
29. Wikstrom I, Forssell J, Penha-Goncalves MN, Bergqvist I, Holmberg D. A Role for E2-2 at the Dn3 Stage of Early Thymopoiesis. Mol Immunol (2008) 45(11):3302–11. doi: 10.1016/j.molimm.2008.02.012
30. Bergqvist I, Eriksson M, Saarikettu J, Eriksson B, Corneliussen B, Grundstrom T, et al. The Basic Helix-Loop-Helix Transcription Factor E2-2 Is Involved in T Lymphocyte Development. Eur J Immunol (2000) 30(10):2857–63. doi: 10.1002/1521-4141(200010)30:10<2857::AID-IMMU2857>3.0.CO;2-G
31. Schlaeger TM, Mikkola HK, Gekas C, Helgadottir HB, Orkin SH. Tie2cre-Mediated Gene Ablation Defines the Stem-Cell Leukemia Gene (Scl/Tal1)-Dependent Window During Hematopoietic Stem-Cell Development. Blood (2005) 105(10):3871–4. doi: 10.1182/blood-2004-11-4467
32. Mikkola HK, Klintman J, Yang H, Hock H, Schlaeger TM, Fujiwara Y, et al. Haematopoietic Stem Cells Retain Long-Term Repopulating Activity and Multipotency in the Absence of Stem-Cell Leukaemia Scl/Tal-1 Gene. Nature (2003) 421(6922):547–51. doi: 10.1038/nature01345
33. Gekas C, Rhodes KE, Gereige LM, Helgadottir H, Ferrari R, Kurdistani SK, et al. Mef2c Is a Lineage-Restricted Target of Scl/Tal1 and Regulates Megakaryopoiesis and B-Cell Homeostasis. Blood (2009) 113(15):3461–71. doi: 10.1182/blood-2008-07-167577
34. Chagraoui H, Kassouf M, Banerjee S, Goardon N, Clark K, Atzberger A, et al. Scl-Mediated Regulation of the Cell-Cycle Regulator P21 Is Critical for Murine Megakaryopoiesis. Blood (2011) 118(3):723–35. doi: 10.1182/blood-2011-01-328765
35. Souroullas GP, Salmon JM, Sablitzky F, Curtis DJ, Goodell MA. Adult Hematopoietic Stem and Progenitor Cells Require Either Lyl1 or Scl for Survival. Cell Stem Cell (2009) 4(2):180–6. doi: 10.1016/j.stem.2009.01.001
36. Tijssen MR, Cvejic A, Joshi A, Hannah RL, Ferreira R, Forrai A, et al. Genome-Wide Analysis of Simultaneous Gata1/2, Runx1, Fli1, and Scl Binding in Megakaryocytes Identifies Hematopoietic Regulators. Dev Cell (2011) 20(5):597–609. doi: 10.1016/j.devcel.2011.04.008
37. Dias S, Mansson R, Gurbuxani S, Sigvardsson M, Kee BL. E2a Proteins Promote Development of Lymphoid-Primed Multipotent Progenitors. Immunity (2008) 29(2):217–27. doi: 10.1016/j.immuni.2008.05.015
38. Yang Q, Kardava L, St Leger A, Martincic K, Varnum-Finney B, Bernstein ID, et al. E47 Controls the Developmental Integrity and Cell Cycle Quiescence of Multipotential Hematopoietic Progenitors. J Immunol (2008) 181(9):5885–94. doi: 10.4049/jimmunol.181.9.5885
39. Zohren F, Souroullas GP, Luo M, Gerdemann U, Imperato MR, Wilson NK, et al. The Transcription Factor Lyl-1 Regulates Lymphoid Specification and the Maintenance of Early T Lineage Progenitors. Nat Immunol (2012) 13(8):761–9. doi: 10.1038/ni.2365
40. de Pooter RF, Dias S, Chowdhury M, Bartom ET, Okoreeh MK, Sigvardsson M, et al. Cutting Edge: Lymphomyeloid-Primed Progenitor Cell Fates Are Controlled by the Transcription Factor Tal1. J Immunol (2019) 202(10):2837–42. doi: 10.4049/jimmunol.1801220
41. Pereira de Sousa A, Berthault C, Granato A, Dias S, Ramond C, Kee BL, et al. Inhibitors of DNA Binding Proteins Restrict T Cell Potential by Repressing Notch1 Expression in Flt3-Negative Common Lymphoid Progenitors. J Immunol (2012) 189(8):3822–30. doi: 10.4049/jimmunol.1103723
42. Ikawa T, Kawamoto H, Goldrath AW, Murre C. E Proteins and Notch Signaling Cooperate to Promote T Cell Lineage Specification and Commitment. J Exp Med (2006) 203(5):1329–42. doi: 10.1084/jem.20060268
43. Xu W, Carr T, Ramirez K, McGregor S, Sigvardsson M, Kee BL. E2a Transcription Factors Limit Expression of Gata3 to Facilitate T Lymphocyte Lineage Commitment. Blood (2013) 121(9):1534–42. doi: 10.1182/blood-2012-08-449447
44. Miyazaki M, Miyazaki K, Chen K, Jin Y, Turner J, Moore AJ, et al. The E-Id Protein Axis Specifies Adaptive Lymphoid Cell Identity and Suppresses Thymic Innate Lymphoid Cell Development. Immunity (2017) 46(5):818–34 e4. doi: 10.1016/j.immuni.2017.04.022
45. Wang HC, Qian L, Zhao Y, Mengarelli J, Adrianto I, Montgomery CG, et al. Downregulation of E Protein Activity Augments an Ilc2 Differentiation Program in the Thymus. J Immunol (2017) 198(8):3149–56. doi: 10.4049/jimmunol.1602009
46. Qian L, Bajana S, Georgescu C, Peng V, Wang HC, Adrianto I, et al. Suppression of Ilc2 Differentiation From Committed T Cell Precursors by E Protein Transcription Factors. J Exp Med (2019) 216(4):884–99. doi: 10.1084/jem.20182100
47. Nawijn MC, Ferreira R, Dingjan GM, Kahre O, Drabek D, Karis A, et al. Enforced Expression of Gata-3 During T Cell Development Inhibits Maturation of Cd8 Single-Positive Cells and Induces Thymic Lymphoma in Transgenic Mice. J Immunol (2001) 167(2):715–23. doi: 10.4049/jimmunol.167.2.715
48. Xu W, Kee BL. Growth Factor Independent 1b (Gfi1b) Is an E2a Target Gene That Modulates Gata3 in T-Cell Lymphomas. Blood (2007) 109(10):4406–14. doi: 10.1182/blood-2006-08-043331
49. Yucel R, Karsunky H, Klein-Hitpass L, Moroy T. The Transcriptional Repressor Gfi1 Affects Development of Early, Uncommitted C-Kit+ T Cell Progenitors and Cd4/Cd8 Lineage Decision in the Thymus. J Exp Med (2003) 197(7):831–44. doi: 10.1084/jem.20021417
50. Doan LL, Porter SD, Duan Z, Flubacher MM, Montoya D, Tsichlis PN, et al. Targeted Transcriptional Repression of Gfi1 by Gfi1 and Gfi1b in Lymphoid Cells. Nucleic Acids Res (2004) 32(8):2508–19. doi: 10.1093/nar/gkh570
51. Banerjee A, Northrup D, Boukarabila H, Jacobsen SE, Allman D. Transcriptional Repression of Gata3 Is Essential for Early B Cell Commitment. Immunity (2013) 38(5):930–42. doi: 10.1016/j.immuni.2013.01.014
52. Spaulding C, Reschly EJ, Zagort DE, Yashiro-Ohtani Y, Beverly LJ, Capobianco A, et al. Notch1 Co-Opts Lymphoid Enhancer Factor 1 for Survival of Murine T-Cell Lymphomas. Blood (2007) 110(7):2650–8. doi: 10.1182/blood-2007-04-084202
53. Carr T, McGregor S, Dias S, Verykokakis M, LeBeau MM, Xue HH, et al. Oncogenic and Tumor Suppressor Functions for Lymphoid Enhancer Factor I in a Murine Model of T Acute Lymphoblastic Leukemia. Front Immunol (2022) 13:845488. doi: 10.3389/fimmu.2022.845488
54. Okamura RM, Sigvardsson M, Galceran J, Verbeek S, Clevers H, Grosschedl R. Redundant Regulation of T Cell Differentiation and Tcralpha Gene Expression by the Transcription Factors Lef-1 and Tcf-1. Immunity (1998) 8(1):11–20. doi: 10.1016/s1074-7613(00)80454-9
55. Weber BN, Chi AW, Chavez A, Yashiro-Ohtani Y, Yang Q, Shestova O, et al. A Critical Role for Tcf-1 in T-Lineage Specification and Differentiation. Nature (2011) 476(7358):63–8. doi: 10.1038/nature10279
56. Germar K, Dose M, Konstantinou T, Zhang J, Wang H, Lobry C, et al. T-Cell Factor 1 Is a Gatekeeper for T-Cell Specification in Response to Notch Signaling. Proc Natl Acad Sci USA (2011) 108(50):20060–5. doi: 10.1073/pnas.1110230108
57. Yu S, Zhou X, Steinke FC, Liu C, Chen SC, Zagorodna O, et al. The Tcf-1 and Lef-1 Transcription Factors Have Cooperative and Opposing Roles in T Cell Development and Malignancy. Immunity (2012) 37(5):813–26. doi: 10.1016/j.immuni.2012.08.009
58. Wang F, Qi Z, Yao Y, Yu G, Feng T, Zhao T, et al. Exploring the Stage-Specific Roles of Tcf-1 in T Cell Development and Malignancy at Single-Cell Resolution. Cell Mol Immunol (2021) 18(3):644–59. doi: 10.1038/s41423-020-00527-1
59. Geimer Le Lay AS, Oravecz A, Mastio J, Jung C, Marchal P, Ebel C, et al. The Tumor Suppressor Ikaros Shapes the Repertoire of Notch Target Genes in T Cells. Sci Signal (2014) 7(317):ra28. doi: 10.1126/scisignal.2004545
60. Wang H, Zang C, Taing L, Arnett KL, Wong YJ, Pear WS, et al. Notch1-Rbpj Complexes Drive Target Gene Expression Through Dynamic Interactions With Superenhancers. Proc Natl Acad Sci USA (2014) 111(2):705–10. doi: 10.1073/pnas.1315023111
61. Fitzgerald TJ, Neale GA, Raimondi SC, Goorha RM. C-Tal, a Helix-Loop-Helix Protein, Is Juxtaposed to the T-Cell Receptor-Beta Chain Gene by a Reciprocal Chromosomal Translocation: T(1;7)(P32;Q35). Blood (1991) 78(10):2686–95. doi: 10.1182/blood.V78.10.2686.2686
62. Brown L, Cheng JT, Chen Q, Siciliano MJ, Crist W, Buchanan G, et al. Site-Specific Recombination of the Tal-1 Gene Is a Common Occurrence in Human T Cell Leukemia. EMBO J (1990) 9(10):3343–51. doi: 10.1002/j.1460-2075.1990.tb07535.x
63. Begley CG, Aplan PD, Davey MP, Nakahara K, Tchorz K, Kurtzberg J, et al. Chromosomal Translocation in a Human Leukemic Stem-Cell Line Disrupts the T-Cell Antigen Receptor Delta-Chain Diversity Region and Results in a Previously Unreported Fusion Transcript. Proc Natl Acad Sci USA (1989) 86(6):2031–5. doi: 10.1073/pnas.86.6.2031
64. Mellentin JD, Smith SD, Cleary ML. Lyl-1, a Novel Gene Altered by Chromosomal Translocation in T Cell Leukemia, Codes for a Protein With a Helix-Loop-Helix DNA Binding Motif. Cell (1989) 58(1):77–83. doi: 10.1016/0092-8674(89)90404-2
65. Breit TM, Mol EJ, Wolvers-Tettero IL, Ludwig WD, van Wering ER, van Dongen JJ. Site-Specific Deletions Involving the Tal-1 and Sil Genes Are Restricted to Cells of the T Cell Receptor Alpha/Beta Lineage: T Cell Receptor Delta Gene Deletion Mechanism Affects Multiple Genes. J Exp Med (1993) 177(4):965–77. doi: 10.1084/jem.177.4.965
66. Aplan PD, Lombardi DP, Ginsberg AM, Cossman J, Bertness VL, Kirsch IR. Disruption of the Human Scl Locus by "Illegitimate" V-(D)-J Recombinase Activity. Science (1990) 250(4986):1426–9. doi: 10.1126/science.2255914
67. Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, et al. Oncogene Regulation. An Oncogenic Super-Enhancer Formed Through Somatic Mutation of a Noncoding Intergenic Element. Science (2014) 346(6215):1373–7. doi: 10.1126/science.1259037
68. Liu X, Xu Y, Han L, Yi Y. Reassessing the Potential of Myb-Targeted Anti-Cancer Therapy. J Cancer (2018) 9(7):1259–66. doi: 10.7150/jca.23992
69. Zhong Y, Jiang L, Hiai H, Toyokuni S, Yamada Y. Overexpression of a Transcription Factor Lyl1 Induces T- and B-Cell Lymphoma in Mice. Oncogene (2007) 26(48):6937–47. doi: 10.1038/sj.onc.1210494
70. Kelliher MA, Seldin DC, Leder P. Tal-1 Induces T Cell Acute Lymphoblastic Leukemia Accelerated by Casein Kinase Iialpha. EMBO J (1996) 15(19):5160–6. doi: 10.1002/j.1460-2075.1996.tb00900.x
71. Morrow MA, Mayer EW, Perez CA, Adlam M, Siu G. Overexpression of the Helix-Loop-Helix Protein Id2 Blocks T Cell Development at Multiple Stages. Mol Immunol (1999) 36(8):491–503. doi: 10.1016/s0161-5890(99)00071-1
72. Lukov GL, Rossi L, Souroullas GP, Mao R, Goodell MA. The Expansion of T-Cells and Hematopoietic Progenitors as a Result of Overexpression of the Lymphoblastic Leukemia Gene, Lyl1 Can Support Leukemia Formation. Leuk Res (2011) 35(3):405–12. doi: 10.1016/j.leukres.2010.07.023
73. Shaw TI, Dong L, Tian L, Qian C, Liu Y, Ju B, et al. Integrative Network Analysis Reveals Usp7 Haploinsufficiency Inhibits E-Protein Activity in Pediatric T-Lineage Acute Lymphoblastic Leukemia (T-All). Sci Rep (2021) 11(1):5154. doi: 10.1038/s41598-021-84647-2
74. Wadman IA, Osada H, Grutz GG, Agulnick AD, Westphal H, Forster A, et al. The Lim-Only Protein Lmo2 Is a Bridging Molecule Assembling an Erythroid, DNA-Binding Complex Which Includes the Tal1, E47, Gata-1 and Ldb1/Nli Proteins. EMBO J (1997) 16(11):3145–57. doi: 10.1093/emboj/16.11.3145
75. Schlaeger TM, Schuh A, Flitter S, Fisher A, Mikkola H, Orkin SH, et al. Decoding Hematopoietic Specificity in the Helix-Loop-Helix Domain of the Transcription Factor Scl/Tal-1. Mol Cell Biol (2004) 24(17):7491–502. doi: 10.1128/MCB.24.17.7491-7502.2004
76. Wilson NK, Miranda-Saavedra D, Kinston S, Bonadies N, Foster SD, Calero-Nieto F, et al. The Transcriptional Program Controlled by the Stem Cell Leukemia Gene Scl/Tal1 During Early Embryonic Hematopoietic Development. Blood (2009) 113(22):5456–65. doi: 10.1182/blood-2009-01-200048
77. Layer JH, Christy M, Placek L, Unutmaz D, Guo Y, Dave UP. Ldb1 Enforces Stability on Direct and Indirect Oncoprotein Partners in Leukemia. Mol Cell Biol (2020) 40(12):e00652–19. doi: 10.1128/MCB.00652-19
78. Layer JH, Alford CE, McDonald WH, Dave UP. Lmo2 Oncoprotein Stability in T-Cell Leukemia Requires Direct Ldb1 Binding. Mol Cell Biol (2016) 36(3):488–506. doi: 10.1128/MCB.00901-15
79. Hoang T, Lambert JA, Martin R. Scl/Tal1 in Hematopoiesis and Cellular Reprogramming. Curr Top Dev Biol (2016) 118:163–204. doi: 10.1016/bs.ctdb.2016.01.004
80. Rahman S, Magnussen M, Leon TE, Farah N, Li Z, Abraham BJ, et al. Activation of the Lmo2 Oncogene Through a Somatically Acquired Neomorphic Promoter in T-Cell Acute Lymphoblastic Leukemia. Blood (2017) 129(24):3221–6. doi: 10.1182/blood-2016-09-742148
81. Ono Y, Fukuhara N, Yoshie O. Tal1 and Lim-Only Proteins Synergistically Induce Retinaldehyde Dehydrogenase 2 Expression in T-Cell Acute Lymphoblastic Leukemia by Acting as Cofactors for Gata3. Mol Cell Biol (1998) 18(12):6939–50. doi: 10.1128/MCB.18.12.6939
82. Tan TK, Zhang C, Sanda T. Oncogenic Transcriptional Program Driven by Tal1 in T-Cell Acute Lymphoblastic Leukemia. Int J Hematol (2019) 109(1):5–17. doi: 10.1007/s12185-018-2518-z
83. Sanda T, Lawton LN, Barrasa MI, Fan ZP, Kohlhammer H, Gutierrez A, et al. Core Transcriptional Regulatory Circuit Controlled by the Tal1 Complex in Human T Cell Acute Lymphoblastic Leukemia. Cancer Cell (2012) 22(2):209–21. doi: 10.1016/j.ccr.2012.06.007
84. Ciciro Y, Sala A. Myb Oncoproteins: Emerging Players and Potential Therapeutic Targets in Human Cancer. Oncogenesis (2021) 10(2):19. doi: 10.1038/s41389-021-00309-y
85. Tan SH, Yam AW, Lawton LN, Wong RW, Young RA, Look AT, et al. Trib2 Reinforces the Oncogenic Transcriptional Program Controlled by the Tal1 Complex in T-Cell Acute Lymphoblastic Leukemia. Leukemia (2016) 30(4):959–62. doi: 10.1038/leu.2015.195
86. Mansour MR, Sanda T, Lawton LN, Li X, Kreslavsky T, Novina CD, et al. The Tal1 Complex Targets the Fbxw7 Tumor Suppressor by Activating Mir-223 in Human T Cell Acute Lymphoblastic Leukemia. J Exp Med (2013) 210(8):1545–57. doi: 10.1084/jem.20122516
87. McCormack MP, Shields BJ, Jackson JT, Nasa C, Shi W, Slater NJ, et al. Requirement for Lyl1 in a Model of Lmo2-Driven Early T-Cell Precursor All. Blood (2013) 122(12):2093–103. doi: 10.1182/blood-2012-09-458570
88. Chan WY, Follows GA, Lacaud G, Pimanda JE, Landry JR, Kinston S, et al. The Paralogous Hematopoietic Regulators Lyl1 and Scl Are Coregulated by Ets and Gata Factors, But Lyl1 Cannot Rescue the Early Scl-/- Phenotype. Blood (2007) 109(5):1908–16. doi: 10.1182/blood-2006-05-023226
89. McCarter AC, Gatta GD, Melnick A, Kim E, Sha C, Wang Q, et al. Combinatorial Ets1-Dependent Control of Oncogenic Notch1 Enhancers in T-Cell Leukemia. Blood Cancer Discov (2020) 1(2):178–97. doi: 10.1158/2643-3230.bcd-20-0026
90. Belver L, Yang AY, Albero R, Herranz D, Brundu FG, Quinn SA, et al. Gata3-Controlled Nucleosome Eviction Drives Myc Enhancer Activity in T-Cell Development and Leukemia. Cancer Discov (2019) 9(12):1774–91. doi: 10.1158/2159-8290.CD-19-0471
91. O'Neil J, Calvo J, McKenna K, Krishnamoorthy V, Aster JC, Bassing CH, et al. Activating Notch1 Mutations in Mouse Models of T-All. Blood (2006) 107(2):781–5. doi: 10.1182/blood-2005-06-2553
92. Radtke F, Wilson A, Stark G, Bauer M, van Meerwijk J, MacDonald HR, et al. Deficient T Cell Fate Specification in Mice With an Induced Inactivation of Notch1. Immunity (1999) 10(5):547–58. doi: 10.1016/s1074-7613(00)80054-0
93. Koch U, Fiorini E, Benedito R, Besseyrias V, Schuster-Gossler K, Pierres M, et al. Delta-Like 4 Is the Essential, Nonredundant Ligand for Notch1 During Thymic T Cell Lineage Commitment. J Exp Med (2008) 205(11):2515–23. doi: 10.1084/jem.20080829
94. Crusio KM, King B, Reavie LB, Aifantis I. The Ubiquitous Nature of Cancer: The Role of the Scf(Fbw7) Complex in Development and Transformation. Oncogene (2010) 29(35):4865–73. doi: 10.1038/onc.2010.222
95. De Obaldia ME, Bell JJ, Wang X, Harly C, Yashiro-Ohtani Y, DeLong JH, et al. T Cell Development Requires Constraint of the Myeloid Regulator C/Ebp-Alpha by the Notch Target and Transcriptional Repressor Hes1. Nat Immunol (2013) 14(12):1277–84. doi: 10.1038/ni.2760
96. Harly C, Kenney D, Ren G, Lai B, Raabe T, Yang Q, et al. The Transcription Factor Tcf-1 Enforces Commitment to the Innate Lymphoid Cell Lineage. Nat Immunol (2019) 20(9):1150–60. doi: 10.1038/s41590-019-0445-7
97. Krishnamoorthy V, Carr T, de Pooter RF, Emanuelle AO, Gounari F, Kee BL. Repression of Ccr9 Transcription in Mouse T Lymphocyte Progenitors by the Notch Signaling Pathway. J Immunol (2015) 194(7):3191–200. doi: 10.4049/jimmunol.1402443
98. Zlotoff DA, Sambandam A, Logan TD, Bell JJ, Schwarz BA, Bhandoola A. Ccr7 and Ccr9 Together Recruit Hematopoietic Progenitors to the Adult Thymus. Blood (2010) 115(10):1897–905. doi: 10.1182/blood-2009-08-237784
99. Allman D, Karnell FG, Punt JA, Bakkour S, Xu L, Myung P, et al. Separation of Notch1 Promoted Lineage Commitment and Expansion/Transformation in Developing T Cells. J Exp Med (2001) 194(1):99–106. doi: 10.1084/jem.194.1.99
100. Ellisen LW, Bird J, West DC, Soreng AL, Reynolds TC, Smith SD, et al. Tan-1, the Human Homolog of the Drosophila Notch Gene, Is Broken by Chromosomal Translocations in T Lymphoblastic Neoplasms. Cell (1991) 66(4):649–61. doi: 10.1016/0092-8674(91)90111-b
101. Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, et al. Activating Mutations of Notch1 in Human T Cell Acute Lymphoblastic Leukemia. Science (2004) 306(5694):269–71. doi: 10.1126/science.1102160
102. Thompson BJ, Buonamici S, Sulis ML, Palomero T, Vilimas T, Basso G, et al. The Scffbw7 Ubiquitin Ligase Complex as a Tumor Suppressor in T Cell Leukemia. J Exp Med (2007) 204(8):1825–35. doi: 10.1084/jem.20070872
103. O'Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, et al. Fbw7 Mutations in Leukemic Cells Mediate Notch Pathway Activation and Resistance to Gamma-Secretase Inhibitors. J Exp Med (2007) 204(8):1813–24. doi: 10.1084/jem.20070876
104. Gomez-del Arco P, Kashiwagi M, Jackson AF, Naito T, Zhang J, Liu F, et al. Alternative Promoter Usage at the Notch1 Locus Supports Ligand-Independent Signaling in T Cell Development and Leukemogenesis. Immunity (2010) 33(5):685–98. doi: 10.1016/j.immuni.2010.11.008
105. Jeannet R, Mastio J, Macias-Garcia A, Oravecz A, Ashworth T, Geimer Le Lay AS, et al. Oncogenic Activation of the Notch1 Gene by Deletion of Its Promoter in Ikaros-Deficient T-All. Blood (2010) 116(25):5443–54. doi: 10.1182/blood-2010-05-286658
106. Ashworth TD, Pear WS, Chiang MY, Blacklow SC, Mastio J, Xu L, et al. Deletion-Based Mechanisms of Notch1 Activation in T-All: Key Roles for Rag Recombinase and a Conserved Internal Translational Start Site in Notch1. Blood (2010) 116(25):5455–64. doi: 10.1182/blood-2010-05-286328
107. Nie L, Perry SS, Zhao Y, Huang J, Kincade PW, Farrar MA, et al. Regulation of Lymphocyte Development by Cell-Type-Specific Interpretation of Notch Signals. Mol Cell Biol (2008) 28(6):2078–90. doi: 10.1128/MCB.00844-07
108. Nie L, Xu M, Vladimirova A, Sun XH. Notch-Induced E2a Ubiquitination and Degradation Are Controlled by Map Kinase Activities. EMBO J (2003) 22(21):5780–92. doi: 10.1093/emboj/cdg567
109. Chiang MY, Wang Q, Gormley AC, Stein SJ, Xu L, Shestova O, et al. High Selective Pressure for Notch1 Mutations That Induce Myc in T-Cell Acute Lymphoblastic Leukemia. Blood (2016) 128(18):2229–40. doi: 10.1182/blood-2016-01-692855
110. Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, et al. A Notch1-Driven Myc Enhancer Promotes T Cell Development, Transformation and Acute Lymphoblastic Leukemia. Nat Med (2014) 20(10):1130–7. doi: 10.1038/nm.3665
111. Choi SH, Severson E, Pear WS, Liu XS, Aster JC, Blacklow SC. The Common Oncogenomic Program of Notch1 and Notch3 Signaling in T-Cell Acute Lymphoblastic Leukemia. PloS One (2017) 12(10):e0185762. doi: 10.1371/journal.pone.0185762
112. Wang H, Zou J, Zhao B, Johannsen E, Ashworth T, Wong H, et al. Genome-Wide Analysis Reveals Conserved and Divergent Features of Notch1/Rbpj Binding in Human and Murine T-Lymphoblastic Leukemia Cells. Proc Natl Acad Sci USA (2011) 108(36):14908–13. doi: 10.1073/pnas.1109023108
113. Rodriguez-Caparros A, Garcia V, Casal A, Lopez-Ros J, Garcia-Mariscal A, Tani-Ichi S, et al. Notch Signaling Controls Transcription Via the Recruitment of Runx1 and Myb to Enhancers During T Cell Development. J Immunol (2019) 202(8):2460–72. doi: 10.4049/jimmunol.1801650
114. Egawa T, Tillman RE, Naoe Y, Taniuchi I, Littman DR. The Role of the Runx Transcription Factors in Thymocyte Differentiation and in Homeostasis of Naive T Cells. J Exp Med (2007) 204(8):1945–57. doi: 10.1084/jem.20070133
115. Garcia-Ojeda ME, Klein Wolterink RG, Lemaitre F, Richard-Le Goff O, Hasan M, Hendriks RW, et al. Gata-3 Promotes T-Cell Specification by Repressing B-Cell Potential in Pro-T Cells in Mice. Blood (2013) 121(10):1749–59. doi: 10.1182/blood-2012-06-440065
116. Van de Walle I, Dolens AC, Durinck K, De Mulder K, Van Loocke W, Damle S, et al. Gata3 Induces Human T-Cell Commitment by Restraining Notch Activity and Repressing Nk-Cell Fate. Nat Commun (2016) 7:11171. doi: 10.1038/ncomms11171
117. Scripture-Adams DD, Damle SS, Li L, Elihu KJ, Qin S, Arias AM, et al. Gata-3 Dose-Dependent Checkpoints in Early T Cell Commitment. J Immunol (2014) 193(7):3470–91. doi: 10.4049/jimmunol.1301663
118. Taghon T, Yui MA, Rothenberg EV. Mast Cell Lineage Diversion of T Lineage Precursors by the Essential T Cell Transcription Factor Gata-3. Nat Immunol (2007) 8(8):845–55. doi: 10.1038/ni1486
119. van Hamburg JP, de Bruijn MJ, Dingjan GM, Beverloo HB, Diepstraten H, Ling KW, et al. Cooperation of Gata3, C-Myc and Notch in Malignant Transformation of Double Positive Thymocytes. Mol Immunol (2008) 45(11):3085–95. doi: 10.1016/j.molimm.2008.03.018
120. Fransecky L, Neumann M, Heesch S, Schlee C, Ortiz-Tanchez J, Heller S, et al. Silencing of Gata3 Defines a Novel Stem Cell-Like Subgroup of Etp-All. J Hematol Oncol (2016) 9(1):95. doi: 10.1186/s13045-016-0324-8
121. Tiemessen MM, Baert MR, Schonewille T, Brugman MH, Famili F, Salvatori DC, et al. The Nuclear Effector of Wnt-Signaling, Tcf1, Functions as a T-Cell-Specific Tumor Suppressor for Development of Lymphomas. PloS Biol (2012) 10(11):e1001430. doi: 10.1371/journal.pbio.1001430
122. Staal FJ, Clevers HC. Wnt Signaling in the Thymus. Curr Opin Immunol (2003) 15(2):204–8. doi: 10.1016/s0952-7915(03)00003-7
123. Brantjes H, Roose J, van De Wetering M, Clevers H. All Tcf Hmg Box Transcription Factors Interact With Groucho-Related Co-Repressors. Nucleic Acids Res (2001) 29(7):1410–9. doi: 10.1093/nar/29.7.1410
124. Verbeek S, Izon D, Hofhuis F, Robanus-Maandag E, te Riele H, van de Wetering M, et al. An Hmg-Box-Containing T-Cell Factor Required for Thymocyte Differentiation. Nature (1995) 374(6517):70–4. doi: 10.1038/374070a0
125. Staal FJ, Clevers H. Tales of the Unexpected: Tcf1 Functions as a Tumor Suppressor for Leukemias. Immunity (2012) 37(5):761–3. doi: 10.1016/j.immuni.2012.10.008
126. Feder K, Edmaier-Schroger K, Rawat VPS, Kirsten N, Metzeler K, Kraus JM, et al. Differences in Expression and Function of Lef1 Isoforms in Normal Versus Leukemic Hematopoiesis. Leukemia (2020) 34(4):1027–37. doi: 10.1038/s41375-019-0635-1
127. Petropoulos K, Arseni N, Schessl C, Stadler CR, Rawat VP, Deshpande AJ, et al. A Novel Role for Lef-1, a Central Transcription Mediator of Wnt Signaling, in Leukemogenesis. J Exp Med (2008) 205(3):515–22. doi: 10.1084/jem.20071875
128. Santiago L, Daniels G, Wang D, Deng FM, Lee P. Wnt Signaling Pathway Protein Lef1 in Cancer, as a Biomarker for Prognosis and a Target for Treatment. Am J Cancer Res (2017) 7(6):1389–406.
129. Guo X, Zhang R, Liu J, Li M, Song C, Dovat S, et al. Characterization of Lef1 High Expression and Novel Mutations in Adult Acute Lymphoblastic Leukemia. PloS One (2015) 10(5):e0125429. doi: 10.1371/journal.pone.0125429
130. Gutierrez A, Sanda T, Ma W, Zhang J, Grebliunaite R, Dahlberg S, et al. Inactivation of Lef1 in T-Cell Acute Lymphoblastic Leukemia. Blood (2010) 115(14):2845–51. doi: 10.1182/blood-2009-07-234377
131. Furness CL, Mansur MB, Weston VJ, Ermini L, van Delft FW, Jenkinson S, et al. The Subclonal Complexity of Stil-Tal1+ T-Cell Acute Lymphoblastic Leukaemia. Leukemia (2018) 32(9):1984–93. doi: 10.1038/s41375-018-0046-8
132. Moore G, Annett S, McClements L, Robson T. Top Notch Targeting Strategies in Cancer: A Detailed Overview of Recent Insights and Current Perspectives. Cells (2020) 9(6):1503–649. doi: 10.3390/cells9061503
Keywords: Leukemia, E protein, TAL1, LYL1, murine, T lymphocyte
Citation: Parriott G and Kee BL (2022) E Protein Transcription Factors as Suppressors of T Lymphocyte Acute Lymphoblastic Leukemia. Front. Immunol. 13:885144. doi: 10.3389/fimmu.2022.885144
Received: 27 February 2022; Accepted: 23 March 2022;
Published: 20 April 2022.
Edited by:
David L. Wiest, Fox Chase Cancer Center, United StatesReviewed by:
Pieter Van Vlierberghe, Ghent University, BelgiumCopyright © 2022 Parriott and Kee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Barbara L. Kee, YmtlZUBic2QudWNoaWNhZ28uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.