 Mingkai Yu1,2†
Mingkai Yu1,2† Yuejie Zhu3†Yujiao Li4Zhiqiang Chen1,2Zhiwei Li5Jing Wang6Zheng Li6Fengbo Zhang7,8*
Yuejie Zhu3†Yujiao Li4Zhiqiang Chen1,2Zhiwei Li5Jing Wang6Zheng Li6Fengbo Zhang7,8* Jianbing Ding1,2*
Jianbing Ding1,2*- 1Department of Immunology, School of Basic Medical Sciences, Xinjiang Medical University, Urumqi, China
- 2Xinjiang Key Molecular Biology Laboratory of Endemic Disease, Xinjiang Medical University, Urumqi, China
- 3Reproductive Medicine Center, The First Affiliated Hospital of Xinjiang Medical University, Urumqi, China
- 4Department of Blood Transfusion, The First Affiliated Hospital of Xinjiang Medical University, Urumqi, China
- 5Clinical Laboratory Center, Xinjiang Uygur Autonomous Region People’s Hospital, Urumqi, China
- 6Xinjiang Laboratory of Respiratory Disease Research, Traditional Chinese Medicine Hospital Affiliated to Xinjiang Medical University, Urumqi, China
- 7Department of Clinical Laboratory, The First Affiliated Hospital of Xinjiang Medical University, Urumqi, China
- 8State Key Laboratory of Pathogenesis, Prevention, Treatment of Central Asian High Incidence Diseases, The First Affiliated Hospital of Xinjiang Medical University, Urumqi, China
The development of an effective multivalent vaccine against SARS-CoV-2 variants is an important means to improve the global public health situation caused by COVID-19. In this study, we identified the antigen epitopes of the main global epidemic SARS-CoV-2 and mutated virus strains using immunoinformatics approach, and screened out 8 cytotoxic T lymphocyte epitopes (CTLEs), 17 helper T lymphocyte epitopes (HTLEs), 9 linear B-cell epitopes (LBEs) and 4 conformational B-cell epitopes (CBEs). The global population coverage of CTLEs and HTLEs was 93.16% and 99.9% respectively. These epitopes were spliced together by corresponding linkers and recombined into multivalent vaccine. In silico tests, the vaccine protein was a non-allergen and the docking with TLR-3 molecule showed a strong interaction. The results of immune simulation showed that the vaccine may be helpful to initiate both cellular and humoral immunity against all VOC. The optimistic immunogenicity of the vaccine was confirmed in vivo and in vitro finally. Therefore, our vaccine may have potential protection against SARS-CoV-2 and its variants.
1 Introduction
In December 2019, the world witnessed the first coronavirus pandemic in human history. The coronavirus is a novel virus which was named as the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) by the International Committee on Taxonomy of Viruses (ICTV), and caused corona virus disease 2019 (COVID-19) which was named by the World Health Organization (WHO) (1). The SARS-CoV-2 can invade lung and other multiple organs, causing severe pathological damage (2). Most of the young adults have no serious clinical symptoms, but the elderly and patients with underlying diseases are more serious after infected by SARS-CoV-2 (3, 4). The human individual is generally vulnerable to the SARS-CoV-2. The Worldometer database showed that more than 240 million novel coronavirus pneumonia cases had been confirmed and the cumulative death toll was more than four million all over the world as of October 2021. The COVID-19 had triggered serious public health problem and additional financial burdens around the world (5, 6).
The SARS-CoV-2 is a single and positive stranded RNA virus with an enveloped structure (7). The length of the virus genome is 26-32 kb and diameter of virus particles is between 70-120 nm. The SARS-CoV-2 encodes replicase, spike protein (S protein), envelope protein (E protein), membrane protein (M protein) and nucleocapsid protein (N protein) successively in sequence from genome 5’ to 3’ end (8). The S protein, especially, plays a key role in SARS-CoV-2 infection for it can bind human angiotensin converting enzyme 2 (ACE2) to infect target cells. As RNA virus, SARS-CoV-2 mutation probability is much higher than DNA virus (9). Up to now, some mutants of SARS-CoV-2 have attracted great attention. They are uniformly called variants of human concern (VOC), because the transmission, virulence and sensitivity of VOC have changed significantly compared with the original strain (10).
The variant named B.1.1.7 first appeared in the United Kingdom and had 10 characteristic mutations in S protein, including D614G and N501Y mutations in ACE2 receptor binding domain (RBD) and two characteristic mutations in Protein N (11, 12). It was reported that the transmission rate of B.1.1.7 variant increased by 35-45% and the mortality of patients infected with B.1.1.7 variant can be increased by 30% (13, 14). The variant B.1.351 had three mutations combination of N501Y, E484K and K417N on RBD in South Africa and then spread around the world (15). It was later confirmed that the binding strength of B.1.351 variant to ACE2 was three times stronger than that of the wild type (16). The variant P.1 first reported in Brazil and Japan that derived from Lineage B.1.1.28, and it had 12 S protein missense mutations, including three RBD mutations of E484K, K417T and N501Y (17). Paiva et al. (18) recently found that P.1 variant existed in cases of reinfection with SARS-CoV-2, which suggested that P.1 can escape from the antibody produced by the infection of the original strain. The variant B.1.429 in California contained four missense mutations, of which the L452R mutation is located in RBD (11). Recently, researchers at the University of California found that the variant B.1.429 can affect the neutralization efficiency of protective antibodies, so the B.1.429 variant is more contagious in people. Los Alamos National Laboratory in New Mexico also found that B.1.1.7 and B.1.429 variant can recombine and fuse with each other, which warns us that people may have entered a new phase of the SARS-CoV-2 pandemic. The variant B.1.617.2 with 13 amino acid mutations was first discovered in India in October 2020, including E484Q, L452R and P681R in its spike protein (19). Therefore, the mutations weakened the binding between SARS-CoV-2 S protein and existing antibodies, and potentially reduced the protection of the vaccine (13, 20).
At present, there are many kinds of vaccines on the market globally, such as BioNTech vaccine (Pfizer Corp), mRNA vaccine (Moderna Corp), AZD1222 adenovirus-based vaccine (Astrazeneca Corp), JNJ-78436735 adenovirus-based vaccine (Johnson & Johnson Corp), NVX-CoV2373 nanoparticle-based vaccine (Novavax Corp), etc. However, with the emergence of SARS-CoV-2 variants, the epitopes of original virus have also changed, threatening the effectiveness of existing vaccines (21). In view of the variation characteristics of SARS-CoV-2, it is a particularly urge task to further develop efficient COVID-19 vaccine for preventing this serious infectious disease (22). Currently, it has been proved that the SARS-CoV-2 variants can escape the humoral immunity induced by the vaccine, so the development of specific vaccines against virus mutants is particularly important for the prevention and control of COVID-19 epidemic. The research and development of global vaccine involve a variety of technical schemes, among which the recombinant protein vaccine made in vitro is the most widely used (23). Recombinant protein vaccine can contain different specific pathogen proteins and stimulate human body to produce corresponding antibodies. Because the spike protein coupled with ACE2 receptor mediates the viral entry, it plays a crucial function in the SARS-CoV-2 infection and has been used the top candidate antigen for the development of vaccine. With the immunoinformatics, the activation of immunogenic epitopes on the S protein may be a more useful approach. We have researched scientifically rigorous strategy of multi-epitope peptides based on different proteins against parasitic and bacteria diseases, such as hydatid and brucellosis (24–28). In this study, we have used immunoinformatics to predict and design a multivalent and multi-epitope vaccine that derived from the S proteins of prevalent SARS-CoV-2 and its variants for conferring optimistic protection.
2 Materials and Methods
2.1 The Acquisition of Vaccine Candidate Antigens
2.1.1 Screening SARS-CoV-2 Variants
China National Center for Bio-information (CNCB) (https://bigd.big.ac.cn/ncov/variation/annotation) had classified the SARS-CoV-2 variants. This study was graded according to the assessment of CNCB website, and the Class I variants (the population incidence was greater than 0.05) were selected to screen antigens for vaccine design. Finally, we selected 5 SARS-CoV-2 variants of different lineages with the best quality evaluation.
2.1.2 Obtaining S Protein Sequences
Because of the important role of S protein in the SARS-CoV-2 infecting host process, the Protein S of SARS-CoV-2 and its variants were used to identify antigen epitopes and to design vaccine. The S protein sequences of Wuhan-Hu-1 and B.1.1.7, B.1.351, P.1, B.1.429, B.1.617.2 etc. variants were obtained from National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/). In addition, antigenicity of all S proteins was predicted using online software VaxiJen 2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html). The VaxiJen was the first server that did not rely on protein sequence alignment and only classified proteins according to their physicochemical properties, so as to predict protective antigens (29).
2.1.3 The Sequence Alignment of Protein S
The sequence alignment of all S proteins of Wuhan-Hu-1 and B.1.1.7, B.1.351, P.1, B.1.429 B.1.617.2 variants were performed using software SnapGene, which could screen out the mutated amino acid sites.
2.2 The Prediction of Signal Peptide
To determine whether there are signal peptide regions in the candidate antigen proteins, the signal peptides of S proteins in different SARS-CoV-2 lineages were predicted using SignalP-5.0 Server (http://www.cbs.dtu.dk/services/SignalP/) (30).
2.3 The Identification of Antigen Epitopes
2.3.1 The Identification of CTL Epitopes
The antigenic epitopes were restrictive in binding to human leukocyte antigen (HLA) molecules. In order to make the vaccine effect cover the global population more widely, we expanded the selection of HLA allele. According to the HLA allele reference set from the IEDB website (http://tools.iedb.org/mhci/) database (31), there are 27 high frequency HLA-I alleles (HLA-A*01:01, HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-A*03:01, HLA-A*11:01, HLA-A*23:01, HLA-A*24:02, HLA-A*26:01, HLA-A*30:01, HLA-A*30:02, HLA-A*31:01, HLA-A*32:01, HLA-A*33:01, HLA-A*68:01, HLA-A*68:02, HLA-B*07:02, HLA-B*08:01, HLA-B*15:01, HLA-B*35:01, HLA-B*40:01, HLA-B*44:02, HLA-B*44:03, HLA-B*51:01, HLA-B*53:01, HLA-B*57:01, HLA-B*58:01) were used to predict the SARS-CoV-2 cytotoxic T lymphocyte (CTL) antigenic epitopes. In this study, the online software NetCTLpan was used to predict the CTL epitopes (CTLEs). The online Server NetCTLpan 1.1 (http://www.cbs.dtu.dk/services/NetCTLpan/) used artificial neural networks (ANNs), and it can simulate the binding between antigen peptide and major histocompatibility complex (MHC) for the prediction of MHC class I antigenic epitopes (32). The signal peptide sequences were removed when predicting epitopes in S proteins. In each virus strain, the CTLEs that appeared in four HLA-I alleles at the same time were selected as the dominant epitopes. And the dominant epitopes of six virus strains were compared and integrated for vaccine design. The antigenicity of epitopes screened out was further predicted using VaxiJen 2.0, and the antigen epitopes that its antigenicity value greater than threshold were used for the construction of the final vaccine.
2.3.2 The Identification of HTL Epitopes
According to the HLA allele reference set from IEDB (http://tools.iedb.org/mhcii/) (32), there were also 27 high frequency HLA-II alleles (HLA-DRB1*01:01, HLA-DRB1*03:01, HLA-DRB1*04:01, HLA-DRB1*04:05, HLA-DRB1*07:01, HLA-DRB1*08:02, HLA-DRB1*09:01, HLA-DRB1*11:01, HLA-DRB1*12:01, HLA-DRB1*13:02, HLA-DRB1*15:01, HLA-DRB3*01:01, HLA-DRB3*02:02, HLA-DRB4*01:01, HLA-DRB5*01:01, HLA-DQA1*05:01/DQB1*02:01, HLA-DQA1*05:01/DQB1*03:01, HLA-DQA1*03:01/DQB1*03:02, HLA-DQA1*04:01/DQB1*04:02, HLA-DQA1*01:01/DQB1*05:01, HLA-DQA1*01:02/DQB1*06:02, HLA-DPA1*02:01/DPB1*01:01, HLA-DPA1*01:03/DPB1*02:01, HLA-DPA1*01:03/DPB1*04:01,HLA-DPA1*03:01/DPB1*04:02, HLA-DPA1*02:01/DPB1*05:01, HLA-DPA1*02:01/DPB1*14:01) were used to predict the SARS-CoV-2 helper T lymphocyte (HTL) antigen epitopes. The online Server NetMHCIIpan 4.0 (https://services.healthtech.dtu.dk/service.php?NetMHCIIpan-4.0) (33) was used to predict the HTL epitopes (HTLEs). In each virus strain,the HTLEs that appeared in six HLA-II alleles at the same time were selected as the dominant epitopes and the prediction method of the final epitope was the same as that of CTLEs.
2.3.3 The Identification of Linear B-Cell Epitopes
The online Server BepiPred-2.0 (https://services.healthtech.dtu.dk/service.php?BepiPred-2.0) (34) was used to predict the B-cell epitopes (BEs). The BEs were divided into linear epitopes and conformational epitopes. And the BepiPred-2.0 predicted that was linear B-cell epitopes (LBEs). The BEs of six virus strains were compared and synthesized, and the finally screened epitopes were used for vaccine construction. The signal peptide sequences were removed when predicting epitopes in S proteins.
2.3.4 The Identification of Conformational B-Cell Epitopes
The online software IEDB (http://tools.iedb.org/ellipro/) was used to predict the conformational B-cell epitopes (CBEs). The predicted conformational epitopes of six virus strains were compared and synthesized. The predicted conformational epitopes of six virus strains were compared and synthesized, and finally used in the construction of vaccine. And the signal peptide sequences were removed when predicting epitopes in S proteins.
2.4 Design and Construction of Vaccine
All the filtered CTLEs, HLTEs, LBEs and CBEs were placed and used in the multivalent vaccine construct. In light of the research of Dong (35), our study selected reasonable Linker-sequence to connect the SARS-CoV-2 antigen epitopes respectively. First, the AAY linkers were inserted between CTLEs, the GPGPG linkers were inserted between HTLEs and the KK linkers were inserted between BEs. Then the TAT-sequence (TGALLAAGAAA) was attached to the C-terminal of merged epitopes, which can make the vaccine deliver intracellularly (36). The recombinant SARS-CoV-2 multivalent epitope vaccine was named as rSMEV in the study.
2.5 Secondary and Tertiary Structure Prediction of rSMEV
When the antigen epitopes were connected sequentially by Linkers, the secondary structure of rSMEV was predicted by online software SOMPA (npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sopma.html). The SOMPA adopted the self-optimized prediction method (SOPM) to improve the accuracy of prediction by multiple alignment with protein sequences of the same family (37). And then the tertiary structure was predicted by software Rosettafold. The Rosettafold had ultra-high tertiary structure prediction accuracy by successively converting and integrating the three-track network information of 1D sequence, 2D distance and 3D coordinates (38).
2.6 Structure Optimization and Quality Validation
After primary 3D modeling, the initial vaccine 3D model was further optimized by GalaxyRefine Server (http://galaxy.seoklab.org/). The GalaxyRefine adopted ab initio modeling, and refined the loop or terminus regions in the primary protein 3D model (39). At the same time, the rationality of the optimized protein tertiary structure needs to be further verified. The ProSA-web (https://prosa.services.came.sbg.ac.at/prosa.php) and the Structure Assessment service of SWISS-MODEL (https://swissmodel.expasy.org/assess) were used to verify the tertiary structure quality of vaccine construct. The ProSA-web could evaluate the overall model quality by Z-Score as well as the local model quality. In addition, the SWISS-MODEL had drawn Ramachandran plot that displayed favorable areas of backbone dihedral angles against amino acid residues in tertiary structure (40).
2.7 Analysis and Evaluation of rSMEV
2.7.1 Analysis of rSMEV Physicochemical Properties
The online tool ProtParam (https://web.expasy.org/protparam/) (41) was used to predict the physical and chemical properties of rSMEV. The important physicochemical properties that ProtParam had performed included molecular weight, theoretical isoelectric point (PI), charged polar, atomic composition, half-life, stability, hydrophobicity and so on. According to the physicochemical properties of vaccine protein, it can guide the preparation of vaccine solution and the vaccination strategy in vivo experiment.
2.7.2 Analysis of rSMEV Allergenicity and Antigenicity
In order to ensure the safety of the vaccine, we predicted its allergenicity. The online software AllerTOP 2.0 (http://www.ddg-pharmfac.net/AllerTOP/) was used to predict the allergenicity of rSMEV. This prediction tool is based on auto cross covariance (ACC) transformation of protein sequences into uniform equal-length vectors, and the target protein was compared with allergens and non-allergens data sets for predicting allergen properties (29). Meanwhile, the VaxiJen 2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) (34) was used to predict the antigenicity of rSMEV.
2.7.3 Evaluation of rSMEV Population Coverage
The alleles and genotype frequencies were different among different populations in the world, and the polymorphism of HLA alleles affects the binding ability of antigen peptides to HLA-I or HLA-II molecules (42). The rSMEV had contained both HLA-I and HLA-II antigen epitopes. In addition, a wide range of HLA-I and HLA-II alleles were selected when predicting antigen epitopes. In order to verify the coverage of rSMEV to the world population, the IEDB (http://tools.iedb.org/population/) (32) was used to analyze the rSMEV population coverage.
2.8 Molecular Docking of rSMEV With TLR-3
The toll like receptor (TLR) was a class of important protein molecules involved in innate immunity, and it was also a bridge between nonspecific immunity and specific immunity (43). The TLR-3 molecule was found in a wide range of antigen presenting cells (APCs), such as tissue dendritic cells and monocytes, and TLR-3 could activate the specific recognition response of body cells to RNA virus infection (44). In the study, the binding affinity that rSMEV with TLR-3 was confirmed by the molecular docking approach. The tertiary structure of TLR-3 molecular was obtained from the PDB database (http://www.wwpdb.org/). The molecular docking works were performed by ClusPro Server (https://cluspro.bu.edu/home.php) (45). And the interaction interface residues analysis was performed by software PyMol and Ligplot.
2.9 Molecular Dynamics Simulation
After molecular docking, the best-docked system was solvated in a rectangular box of TIP3P (46) waters extending up to minimum cutoff of 15 Å from the protein boundary. Cl- or Na+ ions were added into the protein surface to neutralize the total charges of the systems. The Amber ff14SB force field (47) was employed for the protein in all of the molecular dynamics (MD) simulations. The initial structures were fully minimized using combined steepest descent and conjugate gradient method. Then, the systems were gently annealed from 10 to 300 K under canonical ensemble for 0.2 ns with a weak restraint of 15 kcal/mol/Å. The 500 ps of density equilibration was performed under isothermal-isobaric ensemble at target temperature of 300K and the target pressure of 1.0 atm using Langevin-thermostat (48) and Berendsen barostat (49) with collision frequency of 0.002 ns and pressure-relaxation time of 0.001 ns. After proper minimizations and equilibrations, a productive MD run of 100 ns was performed for all the complex systems. The MD simulations were performed with GROMACS 2021.3 (50), and calculated MMGBSA through MMPBSA.py by Amber tools (51).
2.10 Immune Simulation
To detect the immune response of rSMEV to the host, the C-ImmSim Server (https://kraken.iac.rm.cnr.it/C-IMMSIM/) was used to perform the immune simulation (52). The C-ImmSim could simulate the cellular and humoral immunity in the immune response of vaccine to mammals. It provided a fast Position Specific Scoring Matrix (PSSM) to predict the epitopes that bound to HLA molecules (53). In this simulation work, the HLA heterozygous combination of host was HLA-A (HLA-A*01:01, HLA-A*03:01), HLA-B (HLA-B*15:01, HLA-B*35:01) and HLA-DR (HLA-DRB1*0401, HLA-DRB1*1302). The selection of host HLA was determined according to the highest frequency HLA molecules of all epitopes in rSMEV. The injection procedure was once every four weeks, a total of three injections. The time steps were 1, 84 and 168 (one time step corresponds to 8 h) (54). In addition, the Random Seed (12345), the Simulation Volume (10), the Adjuvant dose (100) and Number of antigens injected (1000) were system default. The simulation step was set as 1050 (53).
2.11 Codon Optimization
In order to better express the vaccine protein for subsequent animal in vivo experiment, we analyzed the adaptability and preference of the codon of rSMEV. The online tool JCat (JAVA Codon Adaptation Tool) (http://www.jcat.de/Start.jsp) was used to optimize the codon (55). The Escherichia coli (Strain K12) was chosen to express the rSMEV protein. Meanwhile, avoided rho-independent transcription terminators option, avoid prokaryotic ribosome binding sites option and avoided cleavage sites of restriction enzymes et al. options were selected for generating optimized DNA sequence corresponding to rSMEV.
2.12 In Silico Cloning of rSMEV
The pET-28a (+) was selected as the vector that expressed the rSMEV protein, and it was introduced into Escherichia coli. Before inserting the DNA sequence of rSMEV into the vector, it had been confirmed that there were no specific restriction enzyme recognition sites in the target gene sequence. Finally, the corresponding restriction endonuclease sequences were inserted at the N- and C-terminal of the DNA sequence of rSMEV. The in silico cloning work was performed by SnapGene. Finally, the rSMEV vaccine protein was synthesized and purified by SynPeptide Co Ltd (Shanghai, China).
2.13 In Vivo and In Vitro Experiments
2.13.1 Animal
The eight-week-old SPF BALB/c mice provided by the Animal Experiment Center of Xinjiang Medical University were randomly divided into rSMEV and Healthy control (HC) two groups. The mice were dripped intranasally with vaccine protein in rSMEV group (n=8), and the mice was dripped intranasally with normal saline in HC group (n=8). The concentration of vaccine protein solution was 500 μg/mL, each mouse was given 60 μL each time (30 μL each nostril), once every 2 weeks, 3 times in total and the vaccine protein was detected as endotoxin-negative by limulus amebocyte lysate (LAL) before using. Two weeks after the third immunization, the mouse splenocytes were collected aseptically in rSMEV and HC groups for experimental detection. The study was approved by the animal ethics committee of Xinjiang Medical University, and the whole animal experiment process was carried out in strict accordance with the experimental requirements and operation guidelines of the animal ethics committee of Xinjiang Medical University.
2.13.2 ELISPOT
The Enzyme-Linked Immunospot (ELISPOT) experiment was performed for evaluating the specific B-cell response in mice. First, the rSMEV vaccine protein solution (10 μg/mL) was coated on the ELISPOT plate and incubated overnight at 4 °C. Then the plate was washed for 5 times by Microplate washer (Thermo Fisher, FI-01620 Vantaa, Finland) and blocked with 200 μl RPMI-1640 medium (Sigma Aldrich, St. Louis, UA) containing 10% fetal calf serum (FCS) (Sigma) for 30 minutes at 37 °C. After the plate was washed by PBS containing 0.05% Tween 20, the vaccine protein and mice splenocytes suspension prepared (2×105) were added into the ELISPOT plate3. Three repeat wells were set for each sample and the plate was incubated for 20 hours in a 37 °C humidified incubator with 5% CO2. After the plate was washed for 5 times, the biotinylated detecting antibody (anti-mouse IgG) was added and the plate was incubated for 2 hours at room temperature. After the plate was washed, the Streptavidin-ALP was added and incubated at room temperature for 1 hour. The BCIP/NBT solution (MlBio) was added and incubated for chromogenic reaction. The plate was rinsed and dried, the antibody secreting cell (ASC) spot were counted using EliSpot Reader (AID, D 72479, Germany). In this experiment work, the Mouse IgG ELISpot BASIC kit (ALP) (3825-2A, Mabtech, Stockholm, Sweden) was adopted for the ELISPOT experiment.
2.13.3 ELISA
The enzyme-linked immunosorbent assay (ELISA) experiment was performed for evaluating the CD4+ T-cell response in mice. First, the splenocytes in HC and rSMEV groups were co-cultured with the rSMEV vaccine protein (5 μg/mL). The splenocytes were incubated with RPMI-1640 medium containing 10% FCS for 48 hours in a 37 °C under 5% CO2 humidified incubator. Then the culture supernatant was collected, and the samples and standards were added into the IFN-γ and IL-4 ELISA plates after plates were washed. The ELISA plates were incubated at 37 °C for 1.5 hours and washed for 5 times. The biotinylated detecting antibody was added to the plates and incubated at 37 °C for 1 hour. After the plates were washed for 5 times, the Streptavidin-ALP was added and incubated at 37 °C for 30 minutes. The BCIP/NBT solution were added for chromogenic reaction at 37 °C away from light for 15 minutes. Finally, the termination solution was added, and the OD450nm value was measured immediately by Spectrophotometer (Thermo Fisher, FI-01620 Vantaa, Finland) for detecting the corresponding level of IFN-γ and IL-4. The Mouse IFN-γ ELISA Kit (SEKM-0031, Solarbio Science & Technology Co., Ltd, Beijing, China) and Mouse IL-4 ELISA Kit (SEKM-0005, Solarbio Science & Technology Co., Ltd, Beijing, China) were adopted for ELISA experiment.
3 Results
The vaccine construction strategy of this study was shown in Figure 1.
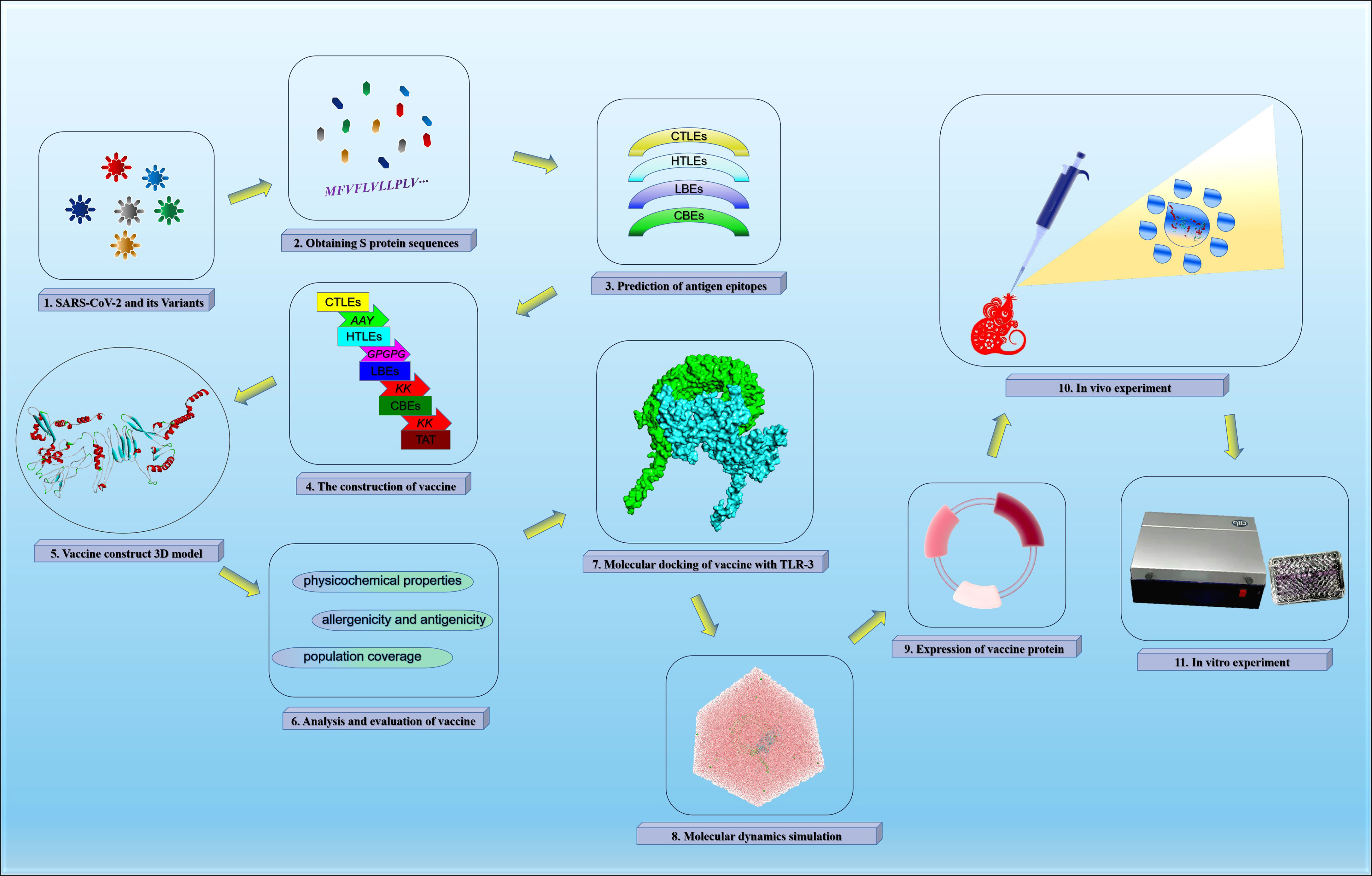
Figure 1 The vaccine construction strategy.
3.1 The Acquisition of Vaccine Candidate Antigens
3.1.1 Screening SARS-CoV-2 Variants
The GenBank search number of the six SARS-CoV-2 virus strains was NC_045512 (Wuhan-Hu-1), MW642026 (Lineage B.1.1.7), MW621453 (Lineage B.1.351), MW642250 (Lineage P.1), MW631888 (Lineage B.1.429) and MZ491545 (Lineage B.1.617.2) separately.
3.1.2 Obtaining S Protein Sequences
The GenBank accession number of S protein derived from six SARS-CoV-2 virus strains was YP_009724390 (Wuhan-Hu-1), QRX39358 (Lineage B.1.1.7), QRV12312 (Lineage B.1.351), QRX39425 (Lineage P.1), QRW35522 (Lineage B.1.429) and QXF88313 (Lineage B.1.617.2). The result was shown in Table 1 and the amino acid sequence of these proteins was shown in Supplementary Data.
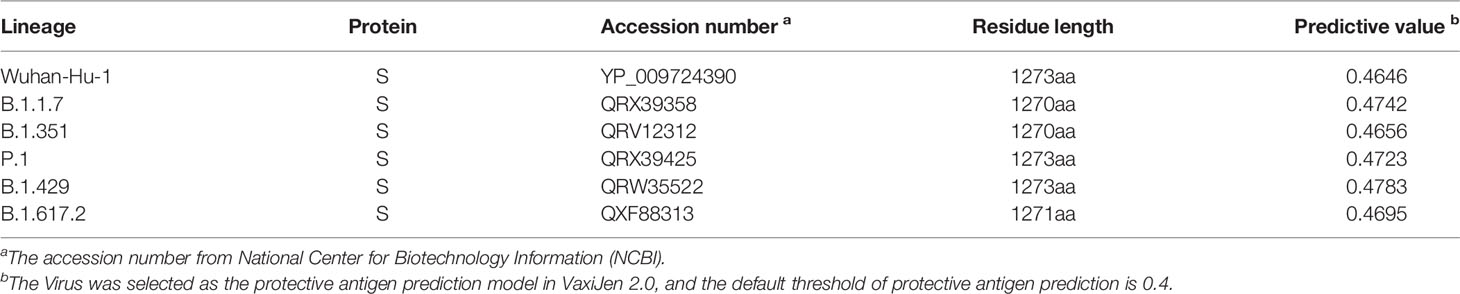
Table 1 The antigenic value of SARS-CoV-2 S proteins.
3.1.3 The Sequence Alignment of Protein S
Compared with the original SARS-CoV-2 virus strain, the five virus variants had a total of 37 mutation sites. There were 25 mutation sites in the SARS-CoV-2 S1 region, among them, 20 mutation sites were in N-terminal domain (NTD) and five mutation sites in RBD. There were 17 mutation sites in the SARS-CoV-2 S2 region. Among them, two mutation sites were in heptad repeat region 1 (HR1) and one mutation site in HR2 (Figure 2).
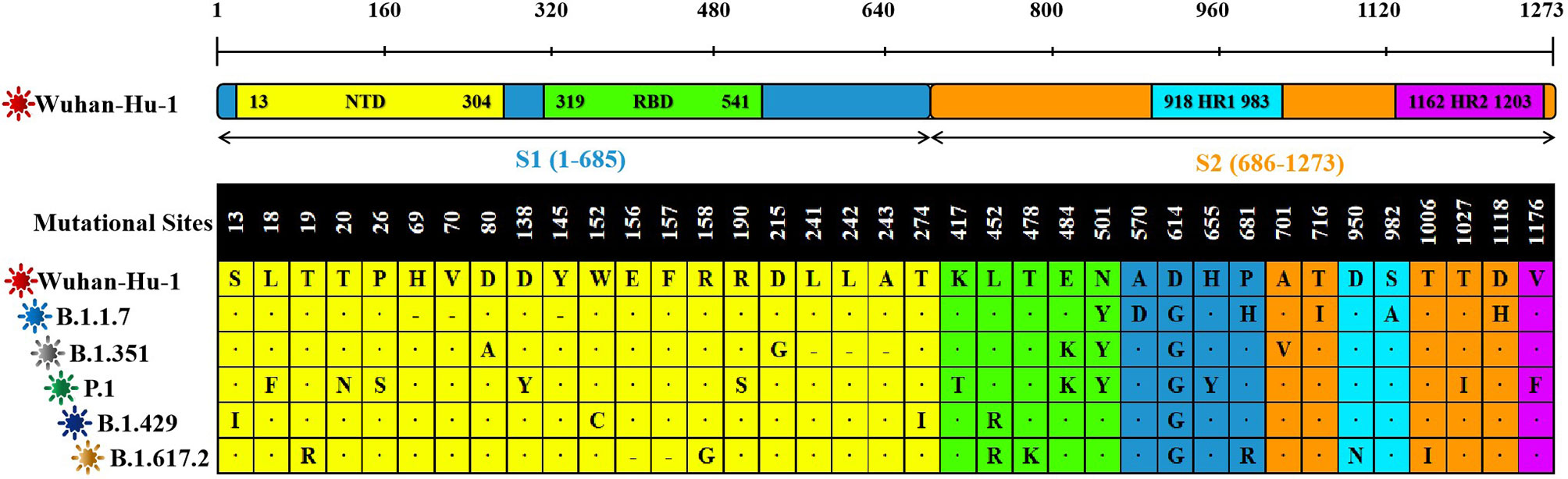
Figure 2 The sequence alignment of S proteins in different virus strains. The study had screened S proteins of six kinds of different SARS-CoV-2 virus strains. The Wuhan-Hu-1 strain was selected as the reference sequence in the sequence alignment. In this figure, we had used different colors to represent the different domains of the S protein. From the N-terminal to the C-terminal of the S protein, a total of 37 amino acid positions had been changed. Among these variations, “·” was omission, which represented the same amino acid as the reference sequence. “-” was loss, which represented the deletion of amino acid sequence. And the remaining mutation sites were amino acid missense mutation.
3.2 The Prediction of Signal Peptide
After the analysis, the S proteins of six different SARS-CoV-2 lineages all had signal peptide regions. The signal peptide region of Lineage Wuhan-Hu-1 was 1-15 (MFVFLVLLPLVSSQC), the signal peptide region of Lineage B.1.1.7 was 1-15 (mfvflvllplvssqc), the signal peptide region of Lineage B.1.351 (mfvflvllplvssqc), the signal peptide region of Lineage P.1 was (mfvflvllplvssqc), the signal peptide region of Lineage B.1.429 was 1-15 (mfvflvllplvsiqc), the signal peptide region of Lineage B.1.617.2 was (mfvflvllplvssqc) (Figure 3).
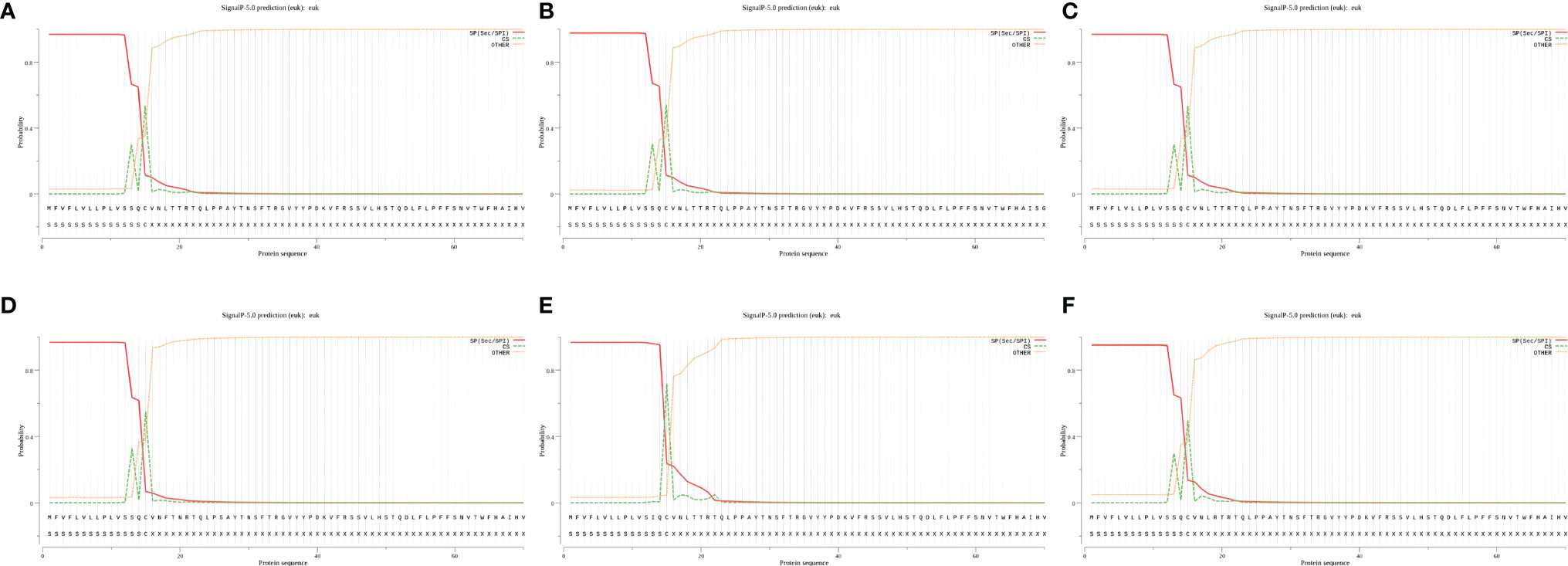
Figure 3 The signal peptides of S proteins. (A) The probability of signal peptide is 96.886%. (B) The probability of signal peptide is 97.653%. (C) The probability of signal peptide is 96.886%. (D) The probability of signal peptide is 96.685%. (E) The probability of signal peptide is 96.625%. (F) The probability of signal peptide is 95.053%. Because the virus strains Wuhan-Hu-1 and B.1.351 have high homology, and the server can only give the signal peptide probability of the first 60 amino acid residues, so (A) and (C) are very similar.
3.3 The Identification of Vaccine Antigen Epitopes
3.3.1 The Identification of CTL Epitopes
In this part, the CTLEs of six SARS-CoV-2 virus strains were analyzed by online Server NetCTLpan 1.1 with each virus strain having 27 kinds of CTLEs of HLA-I alleles. The Rank of CTLEs was less than 1% in the prediction using IEDB. All CTLEs were ranked from small to large according to the sequence start position (Figure 4A). For each virus strain, the CTLEs that appeared in four HLA-I alleles at the same time were selected as the dominant CTLEs (Figure 4B). And the dominant CTLEs of six virus strains were compared and integrated for vaccine design (Figure 4C). The antigenicity of epitopes screened out was further predicted, and the antigen epitopes that antigenicity value greater than the threshold were used for the construction of the final vaccine (Supplementary Table 1).
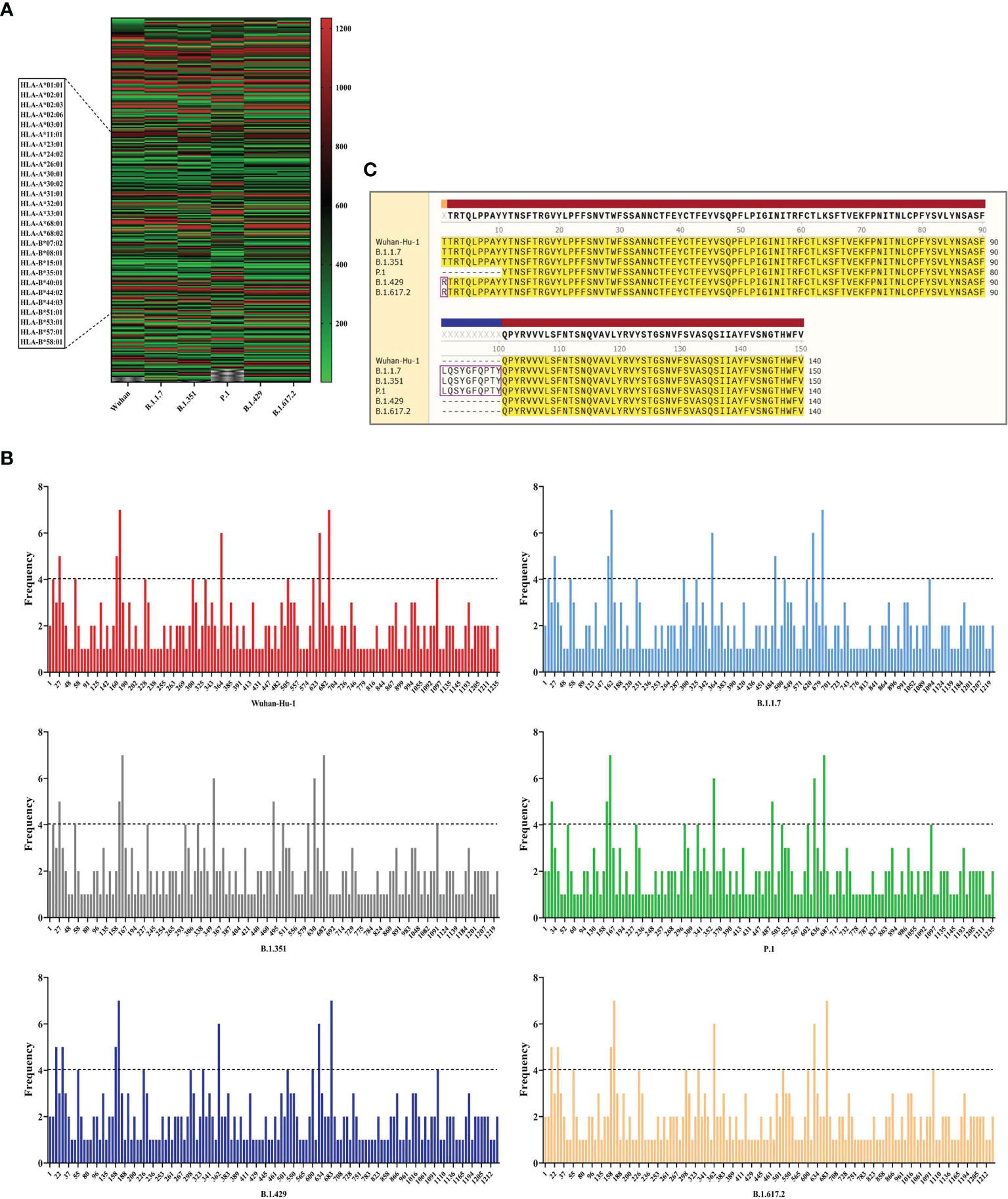
Figure 4 The identification of CTL epitopes. (A) After identifying the 27 kinds of HLA-I alleles, all the CTLEs of the S protein in the 6 SARS-CoV-2 strains were shown here. The abscissa represents the six virus strains and the ordinate represents the position of the beginning of the epitope peptide. (B) The abscissa represented the position of the beginning of the epitope, and the ordinate represented the frequency of the epitope in the 27 alleles. (C) All epitopes of each virus strain were connected in sequence according to the size of the starting position, and the epitopes that had undergone mutation were in the “pink” box. Through sequence alignment, all the common and variant epitopes of all strains were used for vaccine construction.
3.3.2 The Identification of HTL Epitopes
The HTLEs of six SARS-CoV-2 virus strains were analyzed by online Server NetMHCIIpan 4.0 and each virus strain also had 27 kinds of HTLEs of HLA-II alleles. The Rank of HTLEs was less than 2% in the prediction using IEDB. All HTLEs were ranked from small to large according to the sequence start position (Figure 5A). For each virus strain, the HTLEs that appeared in six HLA-II alleles at the same time were selected as the dominant HTLEs (Figure 5B). And the dominant HTLEs of six virus strains were compared and integrated for vaccine design (Figure 5C). The antigenicity of epitopes screened out was further predicted, and the antigen epitopes that antigenicity value greater than the threshold were used for the construction of the final vaccine (Supplementary Table 2).
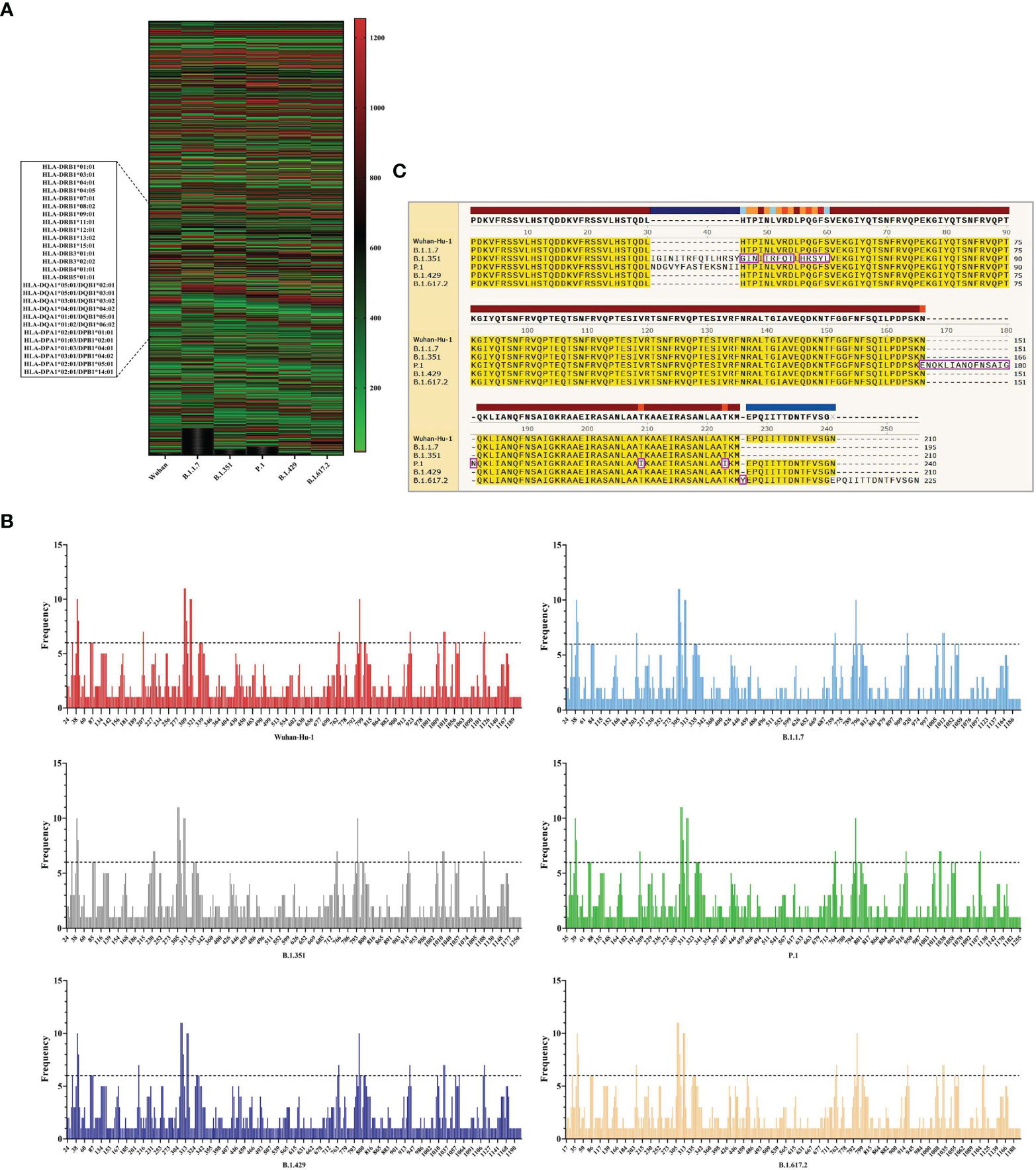
Figure 5 The identification of HTL epitopes. (A) After identifying the 27 kinds of HLA-II alleles, all the HTLEs of the S protein in the 6 SARS-CoV-2 strains were shown here. The abscissa represents the six virus strains and the ordinate represents the position of the beginning of the epitope peptide. (B) The abscissa represented the position of the beginning of the epitope, and the ordinate represented the frequency of the epitope in the 27 alleles. (C) All epitopes of each virus strain were connected in sequence according to the size of the starting position, and the epitopes that had undergone mutation were in the “pink” box. Through sequence alignment, all the common and variant epitopes of all strains were used for vaccine construction.
3.3.3 The Identification of Linear B-Cell Epitopes
The BepiPred-2.0 predicted the LBEs. In the result outputted from BepiPred-2.0, when the threshold of the peptide was greater than 0.8, the peptide sequence was recognized as an epitope (Figure 6A). The BEs of six virus strains were compared and synthesized, and the finally screened epitopes were identified as the dominant LBEs (Figure 6B). After all dominant LBEs were predicted for their antigenicity, the antigen epitopes that antigenicity value greater than the threshold were used for the construction of the final vaccine (Supplementary Table 3).
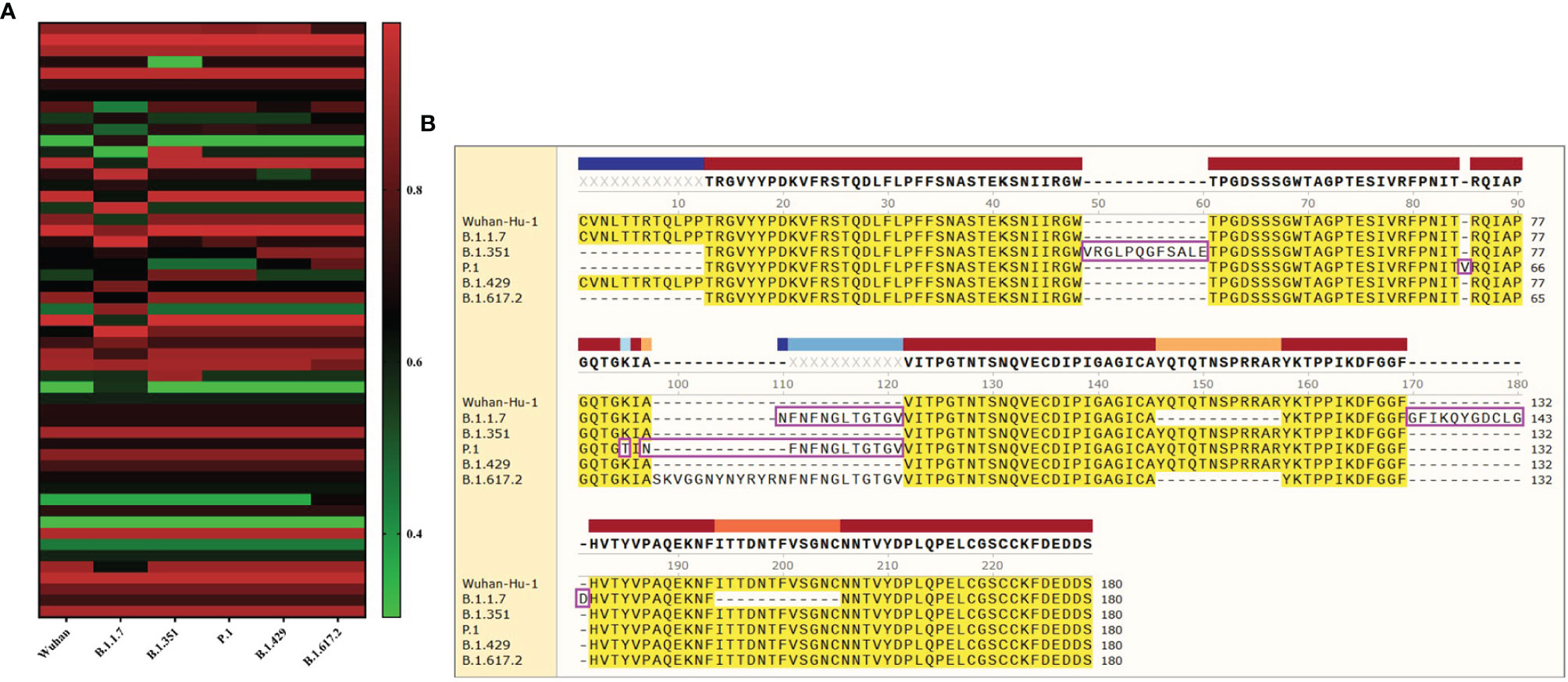
Figure 6 The identification of B-cell epitopes. (A) All the LBEs of the S protein in the 6 SARS-CoV-2 strains were shown here. The abscissa represents the six virus strains and the ordinate represents the Rank of the epitope peptide. (B) All epitopes of each virus strain were connected in sequence according to the size of the starting position, and the epitopes that had undergone mutation were in the “pink” box. Through sequence alignment, all the common and variant epitopes of all strains were used for vaccine construction.
3.3.4 The Identification of Conformational B-Cell Epitopes
The software IEDB had predicted the CBEs of S proteins from different SARS-CoV-2 lineages. We selected the best ranked CBEs among the six virus strains (Figure 7). After comparison, the sequences of CBEs were used to the construction of final vaccine (Supplementary Table 4).

Figure 7 The conformational B-cell epitopes of six SARS-CoV-2 lineages. (A) The “yellow” region is the predicted best conformational B-cell epitope of S protein of virus strain Wuhan-Hu-1. (B) The “yellow” region is the predicted best conformational B-cell epitope of S protein of virus strain B.1.1.7. (C) The “yellow” region is the predicted best conformational B-cell epitope of S protein of virus strain B.1.351. (D) The “yellow” region is the predicted best conformational B-cell epitope of S protein of virus strain P.1. (E) The “yellow” region is the predicted best conformational B-cell epitope of S protein of virus strain B.1.429. (F) The “yellow” region is the predicted best conformational B-cell epitope of S protein of virus strain B.1.617.2.
3.4 Design and Construction of Vaccine
The rSMEV construct contained the best candidate antigen epitopes. There were 8 CTLEs, 17 HTLEs, 9 LBEs and 4 CBEs in the rSMEV. The Linker-AAY had connected all CTLEs, which can enhance the presentation of epitopes and help to produce suitable sites for antigen epitopes to bind to TAP transporters (35). The Linker-GPGPG had connected all HTLEs, the Linker-KK had connected all LBEs and CBEs (Figure 8). All the antigen epitopes for construction of rSMEV were shown in the Table 2. The number of vaccine residues of final designed multivalent vaccine was 614 aa.
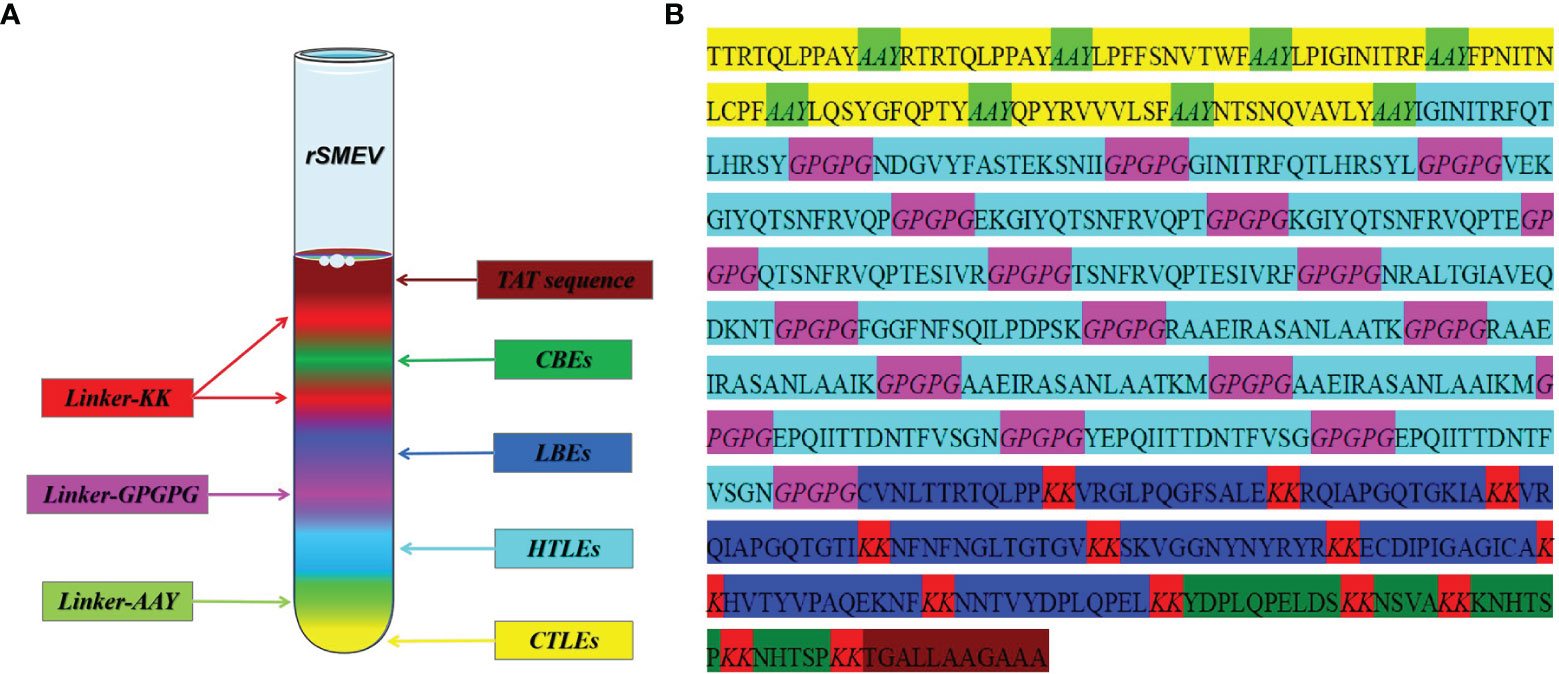
Figure 8 The design and construction of rSMEV. (A) Schematic diagram of all the components needed in vaccine construction, and the different colors represented different epitopes and linkers. (B) The specific amino acid sequence of all the epitopes and linkers required for vaccine construction, and the color of each part corresponded to the color in the (A).
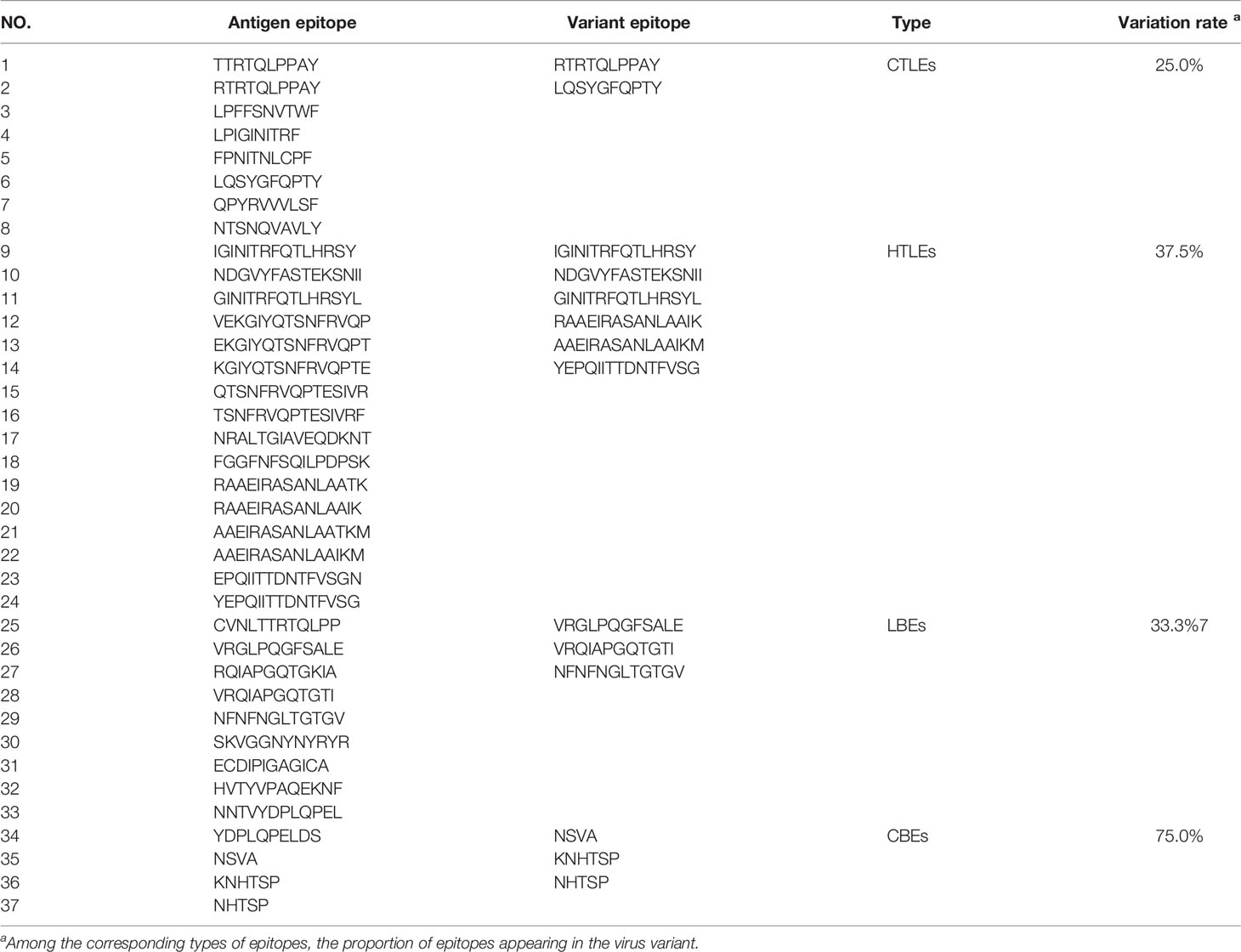
Table 2 The antigen epitopes for vaccine construction.
3.5 Secondary and Tertiary Structure Prediction of rSMEV
In the result of secondary prediction, there were 20.03% Alpha helix (Hh), 23.94% Extended strand (Ee), 6.51% Beta turn (Tt) and 49.51% Random coil (Cc) (Figure 9A). The tertiary structure was shown in secondary structure style (Figure 9B), and the proportion of Hh, Ee, Tt and Cc was consistent with the secondary structure. This indirectly showed that the prediction of secondary structure and tertiary structure was reasonable.
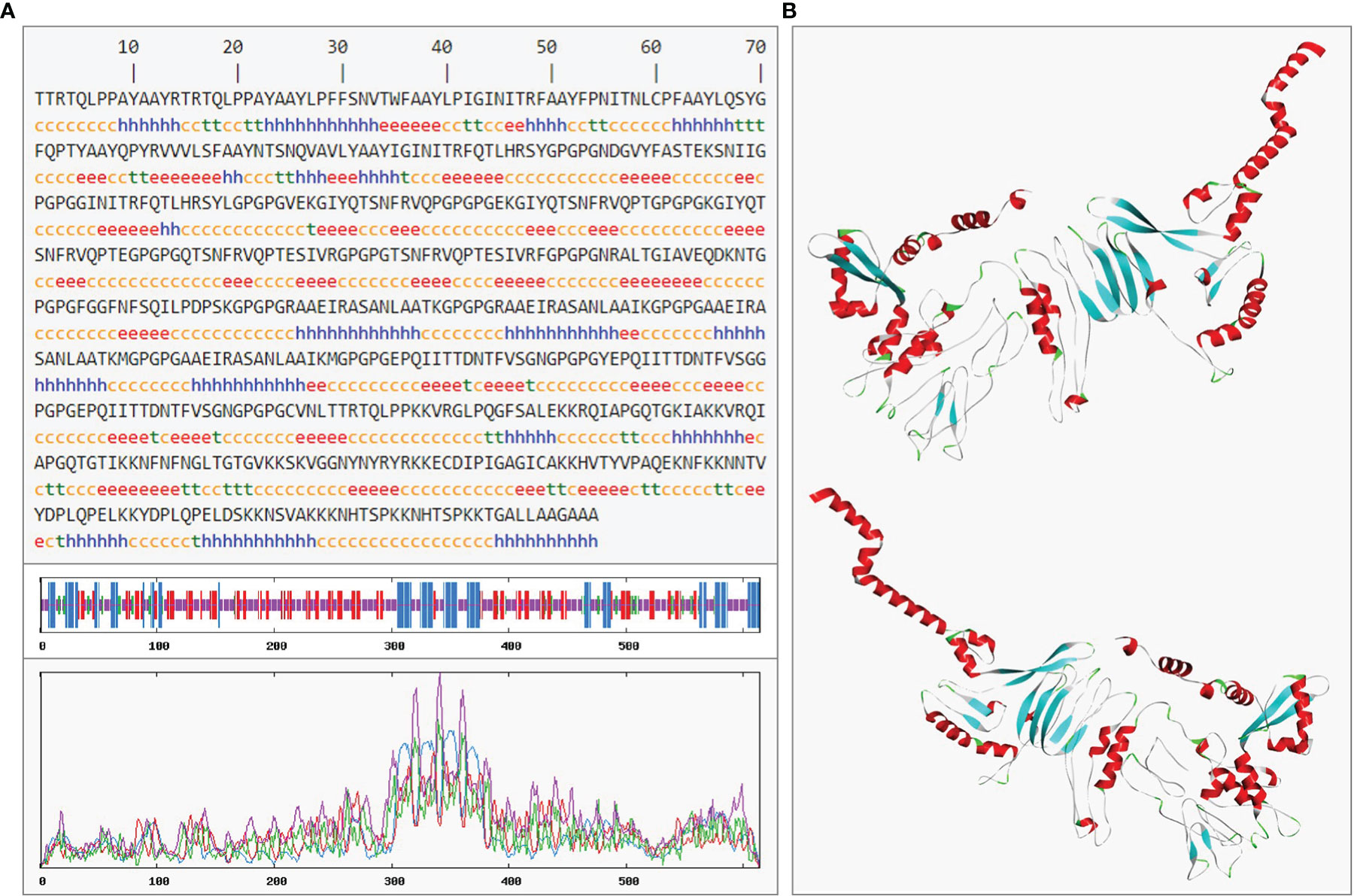
Figure 9 The prediction of rSMEV secondary and tertiary structure. (A) In the prediction of rSMEV secondary structure, the “h” represented the Alpha helix, the “e” represented the Extended strand, the “t” represented the Beta turn and the “c” represented the Random coil. (B) In this 3D model, the tertiary structure was displayed in front view and back view. The “red” part was Alpha helix, the “cyan” part was Extended strand, the “green” pare was Beta turn and the “gray” part was Random coil in 3D model.
3.6 Tertiary Structure Optimization and Quality Validation
After the initial tertiary structure was refined by GalaxyRefine Server, the server had outputted five optimized models. All models were further verified by ProSA-web, the website had calculated the Z-Score of models. The Z-Score of initial Model was −6.89, the optimized Model 1 was −6.88, the optimized Model 2 was −6.96, the optimized Model 3 was −6.93, the optimized Model 4 was −7.02 and the optimized Model 5 was also −6.87. Through observation, it was found that the optimized Model 4 was more in line with the Z-score range of similar protein size. So, the optimized Model 4 was selected as the final tertiary structure. The Ramachandran plot drawn by SWISS-MODEL showed that there were 91.03% Ramachandran favored region, 2.45% Ramachandran Outliers region and 0.00% Rotamer Outliers region in initial structure, and there were 92.65% Ramachandran favored region, 1.47% Ramachandran Outliers region and 0.21% Rotamer Outliers region in final structure (Figure 10).
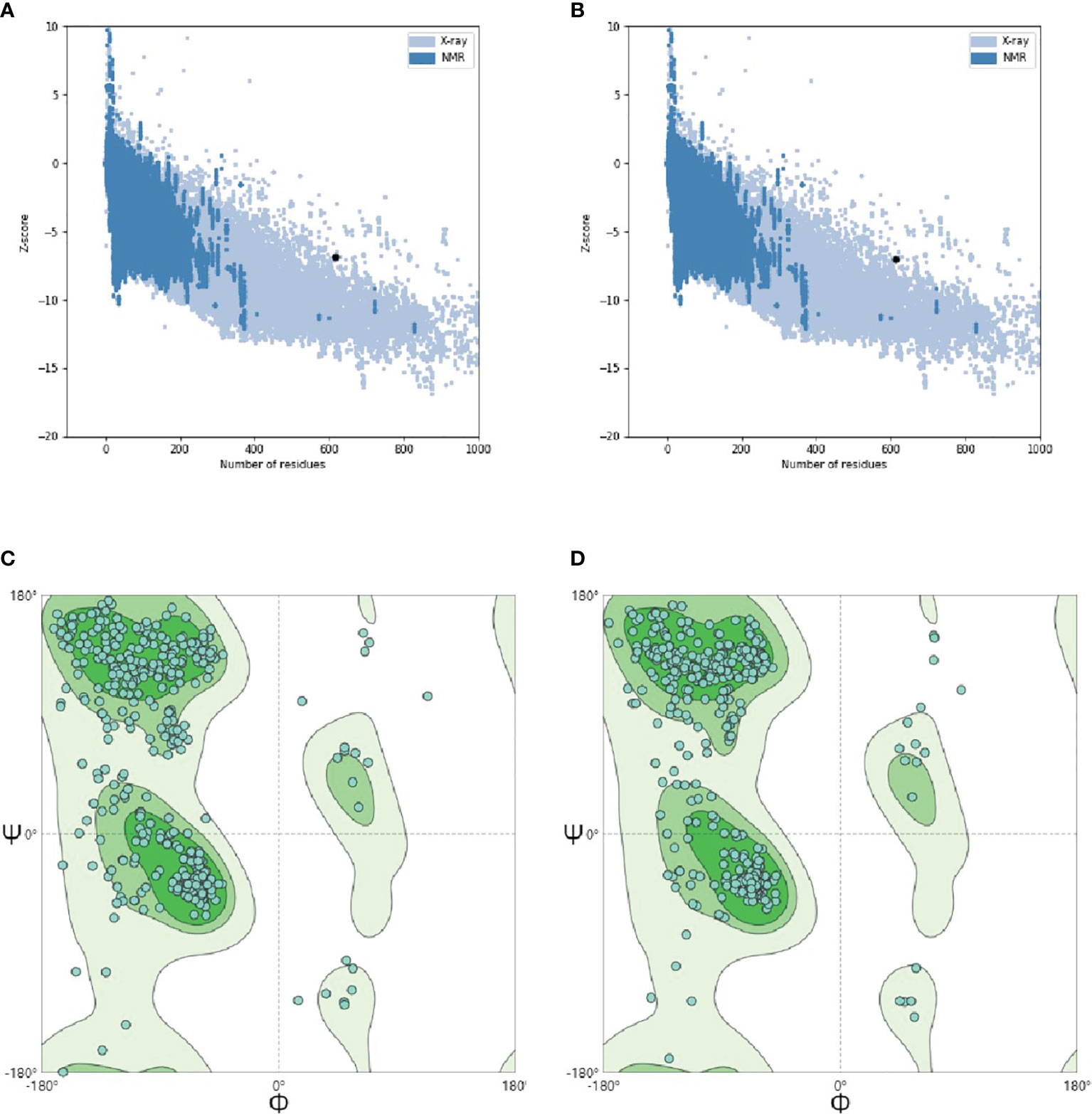
Figure 10 Tertiary structure optimization and quality validation. (A) and (B) are the Z-score evaluation results. The abscissa represents the number of amino acids of the protein and the ordinate represents the score. The region formed by blue and gray blots in the figure are the reasonable scoring areas of the protein, and the black dot represents the target protein. The good target protein with the corresponding number of amino acids should fall in reasonable area. (C) and (D) are Ramachandran plot. In the figure, dark green is favored regions, green is additional allowed regions, light green is generously allowed regions and white is disallowed regions. The rationality of amino acid residues in these regions decreased in turn. When evaluating the quality of 3D model, it is hoped that the proportion of amino acid residues in favored regions is larger.
3.7 Analysis and Evaluation of rSMEV
3.7.1 Analysis of rSMEV Physicochemical Properties
The vaccine protein had 614 amino acids, the total number of atoms was 9122 and the formula was C2904H4540N822O850S6. The molecular weight was 65 KD, and it was an acceptable vaccine since that the molecular weight of protein less than 110 KD could be easily purified (56). The theoretical pI was 10.04, and it included 29 acidic (negatively charged residues) amino acids (Asp + Glu) and 69 alkaline (positively charged residues) amino acids (Arg + Lys). The instability index (II) was computed to be 27.49 (when it<40, the protein is stable) (57), so the vaccine was classified as stable protein. The aliphatic index was 64.28 and Grand average of hydropathicity (GRAVY) was −0.481 (the range of GRAVY is −2∼2, negative value means that protein is hydrophilic) (58), so the vaccine belonged to hydrophilic protein.
3.7.2 Analysis of rSMEV Allergenicity and Antigenicity
The online software AllerTOP 2.0 defined the rSMEV as non-allergen, the UniProtKB accession number of the protein nearest to rSMEV was P46379 and P46379 was also probable non-allergen. The VaxiJen 2.0 showed that the overall prediction value for the protective antigen was 0.6858, the Virus was selected as the prediction model and the threshold for this model was 0.4. Therefore, in this allergenicity and antigenicity analysis, the rSMEV was identified as safe protective antigen.
3.7.3 Evaluation of rSMEV Population Coverage
In the analysis of population coverage, the Class I and Class II T-cell epitopes were calculated separately. The IEDB showed that the HLA-I T-cell epitope covered 93.16% and HLA-II T-cell epitope covered 99.9% world population in the rSMEV. The detailed results were showed in Supplementary Tables 5, 6 and Supplementary Figure 1.
3.8 Molecular Docking of rSMEV With TLR-3
The PDB ID of TLR-3 was 7c76 (59) in the PDB database and the TLR-3 molecular obtained was a complex that blended with UNC93B1. The TLR-3 was isolated from the complex using software Discovery Studio for molecular docking. In this molecular docking work, the TLR-3 molecules acted as the receptor, while the rSMEV acted as ligand. The ClusPro Server can output some clusters after docking work, and each cluster has a different number of members. In the rSMEV-TLR-3 complex formed by the combination of vaccine molecule rSMEV and antigen recognition molecule TLR-3, the lowest complex energy weighted score was −1175.3 cal/mol and all the docking results were shown in Supplementary Table 7. The interaction interface residues analysis from PyMol was 3D and Ligplot was 2D format. The results show that there were two ionic bonds and four hydrogen bonds at the docking interface, which participated in the interaction between the subunits of the complex (Figure 11).
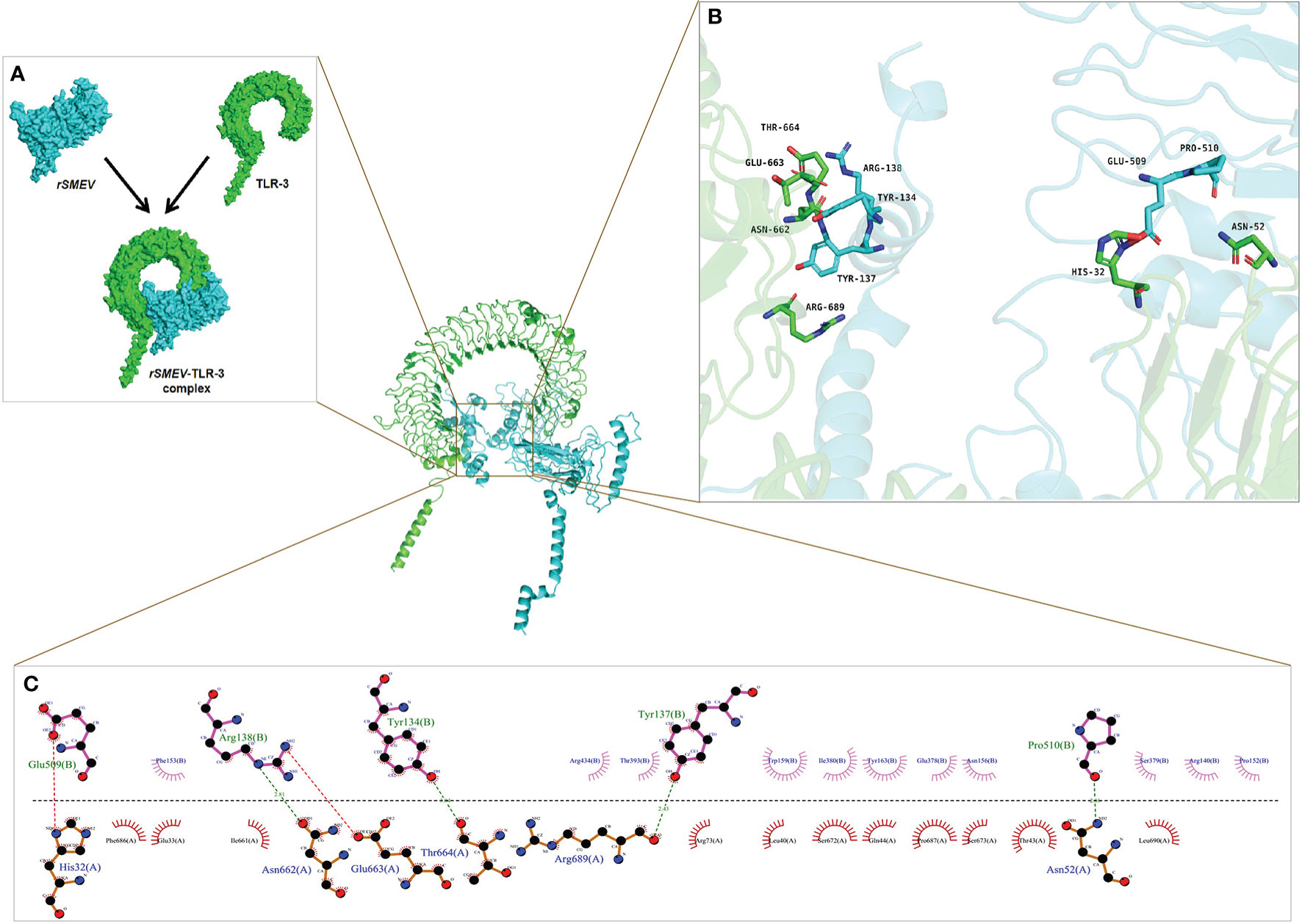
Figure 11 The molecular docking of rSMEV with TLR-3. (A) Schematic diagram of docking between vaccine molecule rSMEV and antigen recognition molecule TLR-3. The rSMEV is light blue and TLR-3 is green. After docking, the rSMEV-TLR-3 complex was formed. (B) The interaction interface predicted by the PyMol is three-dimensional. In this three-dimensional interaction interface, it is mainly ionic bond and hydrogen bond that participate in the interaction. (C) The interaction interface predicted by the Ligplot is two-dimensional. And in this two-dimensional interaction interface, the red dotted line represents ionic bond and the green dotted line represents hydrogen bond, so there are 2 ionic bonds and 4 hydrogen bonds involved in the interaction.
3.9 Molecular Dynamics Simulation
In order to evaluate the structural stability of rSMEV-TLR-3 complex, the best docking pose (Figure 12A) of rSMEV and TLR-3 was simulated by MD. The docking complex was solvated in a rectangular box of TIP3P waters (Figure 12B). After running the MD simulation of 100 ns, the results showed that the root mean square displacement (RMSD) value rose sharply to 1nm within 30 ns. After 30 ns, the RMSD value tended to be stable, and the floating range of the overall operation was about 1 nm (Figure 12C). The root mean square mobility (RMSF) value showed that the two chains of the complex had a lower value (<1 nm) between residues 0-650, while the residues after site 650 had a larger RMSF value (>1 nm) (Figure 12D). Next, the average binding free energy was −88.54 ± 6.91 kcal/mol of complex detected by MMPBSA tool of the GROMACS. (Supplementary Table 8).
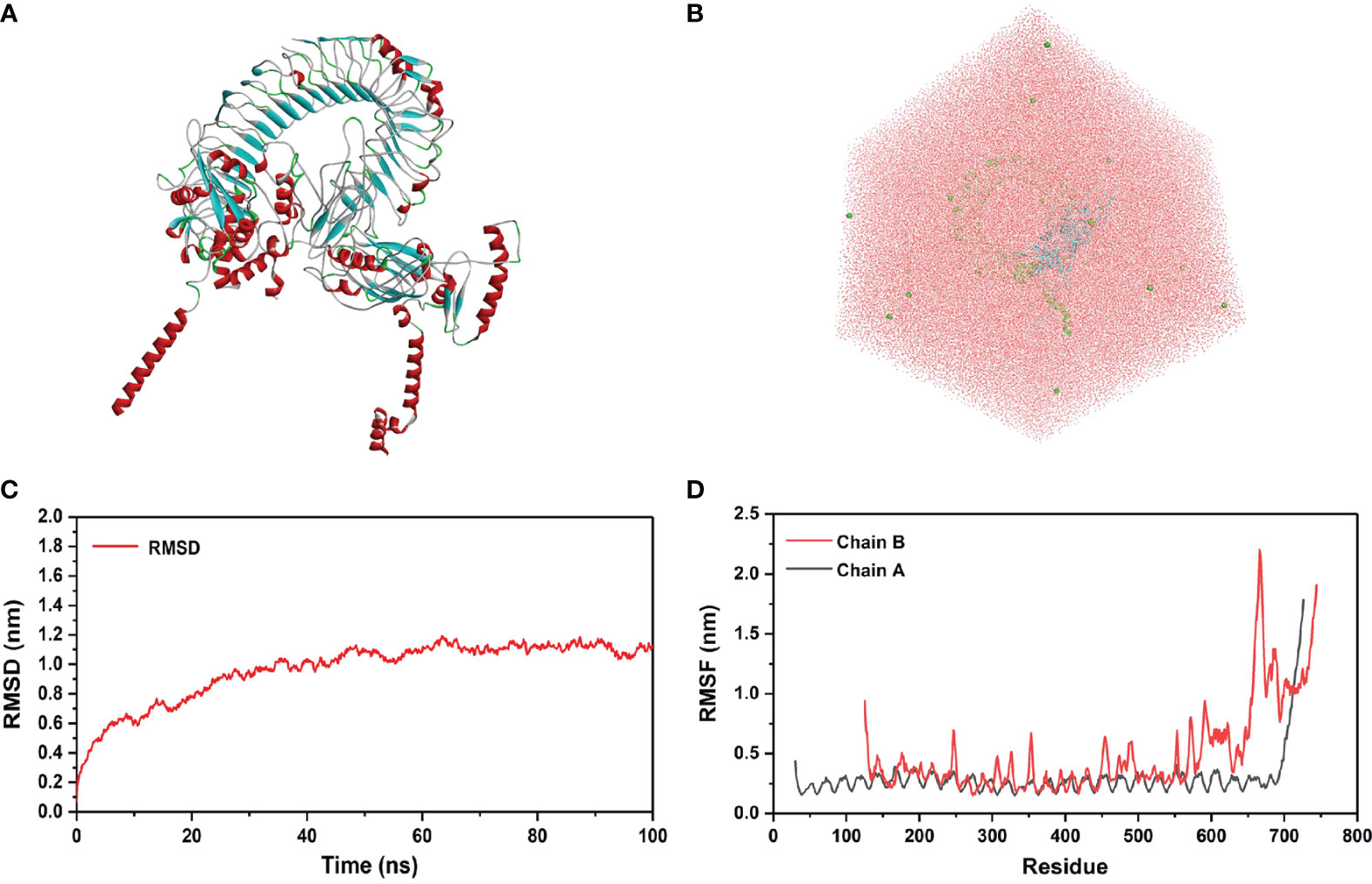
Figure 12 Molecular docking of rSMEV with TLR-3. (A) This is a three-dimensional model of rSMEV and TLR-3 complex, and its color is displayed in the form of secondary structure style. (B) This is the rectangular box of TIP3P waters, and it extends up to minimum cutoff of 15 Å from the protein boundary. After molecular docking, the best-docked system was solvated in it. (C) This is the RMSD running diagram of MD simulation. The abscissa represents the running time of MD simulation, and the ordinate is the value of RMSD. (D) This is the RMSF running diagram of MD simulation. The chain A is TLR-3 and the chain B is rSMEV. The abscissa represents each amino acid residue in the complex, and the ordinate represents the RMSF value.
3.10 Immune Simulation
The C-ImmSim Server conducted the immune stimulation of the rSMEV vaccine. The Immune simulation results showed that the antigen count decreased with the increase of antibody level in the immune response, which was mainly due to the production of total B-lymphocytes (Figure 13A) and T-lymphocytes. The IgM + IgG and IgM antibodies were found in the primary immunization. And the levels of IgM + IgG, IgM, IgG1 and IgG1 + IgG2 antibodies increased in the secondary and tertiary immune responses compared to primary immune response (Figure 13B). Besides B-cell, total and memory TH-cell (helper T cell) (Figure 13C) populations along with active TH-cell (Figure 13D) populations increased during the secondary and tertiary immune responses. The active TC-cell (cytotoxic T lymphocyte) count was sustained growth after each immunization (Figure 13E). And in the three stages after immunization, the level of IFN-γ had increased (Figure 13F).
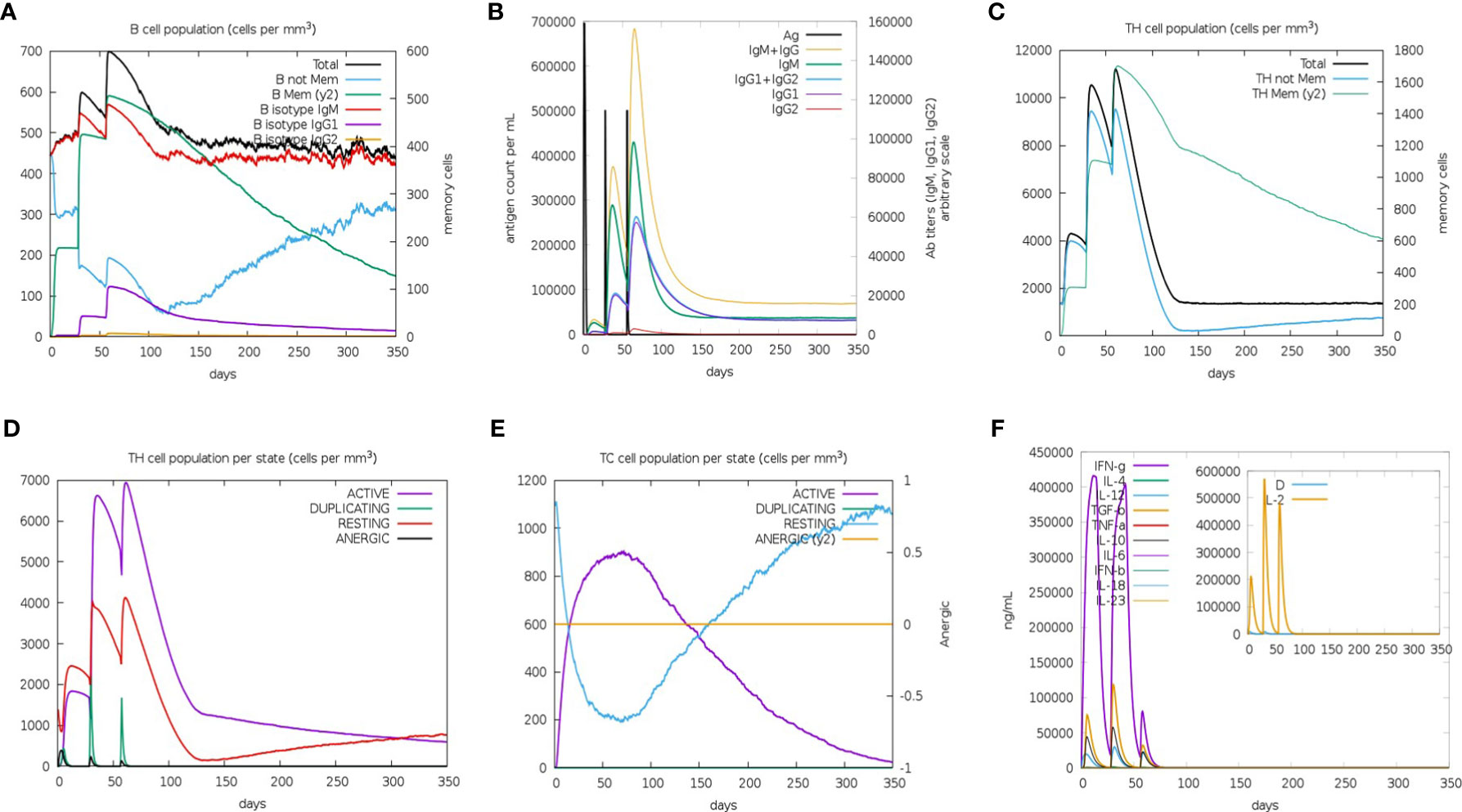
Figure 13 The immune simulated response spectrum. (A) The B-cell populations that could produce antibodies of various subtypes after vaccination. (B) The production of antibodies that represented the proliferation of the immune response after vaccination, and the various subtypes of immunoglobulins were shown in different colors. (C) The generation of memory and non-memory HTL populations after vaccination. (D) The HTL populations in various states. (E) The CTL populations in various states. (F) The cytokine profile showed the production of various cytokines after vaccination.
3.11 Codon Optimization
The rSMEV protein had 614 amino acids, and the DNA sequence of rSMEV had 1842 nucleotides after adaptability and preference analysis of codon. The codon adaptation index (CAI) of the improved sequence was 0.93. When the CAI-Value>0.8, the codon improved was considerate to have a high adaptability to the DNA sequence (Supplementary Figure 2). The GC-Content of the improved codon was 54.51% and the GC-Content of Escherichia coli strain K12 used in the study was 50.73%.
3.12 In Silico Cloning of rSMEV
The DNA sequence of rSMEV was inserted to the sites between XhoI (site 158) and BamHI (site 2006) in the pET-28a (+) vector. The DNA sequence of rSMEV was written in Supplementary Data. The XhoI and BamHI were common restriction endonucleases in molecular cloning, and the DNA sequence did not contain these two digestion sites (Supplementary Figure 3).
3.13 The Results of ELISPOT and ELISA Experiments
In the study, the ELISPOT experiment was performed to evaluate the rSMEV induced B-cell response. The splenocytes of rSMEV group mice could secrete specific antibodies induced by vaccine protein, while the splenocytes of HC group mice did not secrete specific antibodies (Figure 14A). After statistical test, the specific antibody secreting cells (ASCs) in rSMEV group was significantly higher than that in HC group, and the difference was statistically significant (P<0.001) (Figure 14B). This suggested that rSMEV protein could activate B-cell reaction. The ELISA experiment was performed to evaluate the rSMEV induced CD4+ T-cell response. The CD4+ T-cell was activated by vaccine protein, so the content of IFN-γ (Figure 14C) and IL-4 (Figure 14D) in rSMEV group was significantly higher than that in HC group, and the difference was statistically significant (P-value of IFN-γ was less than 0.0001 and P-value of IL-4 was less than 0.001). This suggested that rSMEV protein could activate CD4+ T-cell reaction.
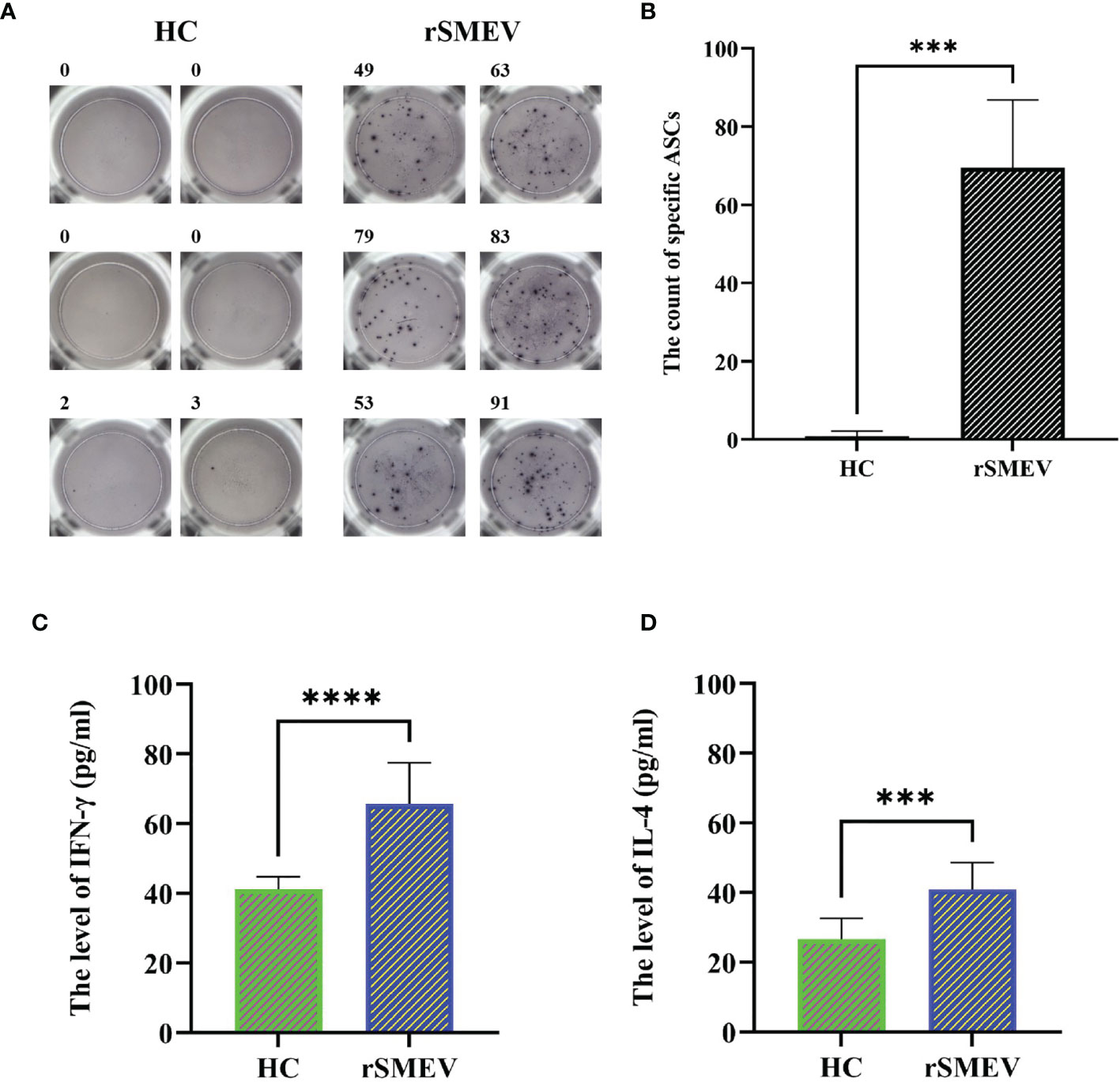
Figure 14 The results of ELISPOT and ELISA experiments. (A) The representative ELISPOT spot diagram in HC and rSMEV groups. (B) The count of ASCs in the HC and rSMEV groups were significantly different (***P<0.001) after using the two independent sample t-test, and the statistical test was two-tailed. The t-value was 9.671 and the degree of freedom was 5.059 (***P < 0.001). (C) The two independent sample t-test of the level of IFN-γ between HC group and rSMEV group, and the difference was statistically significant (****P<0.0001), the t-value was 6.247 and the degree of freedom was 18. (D) The two independent sample t-test of the level of IL-4 between HC group and rSMEV group, and the difference was statistically significant (***P<0.001), the t-value was 4.652 and the degree of freedom was 18.
4 Discussion
As you known, the decrease of vaccine protection ability elicited mainly by S protein mutation of SARS-CoV-2. There was no a multivalent epitope vaccine for S protein to resist the infection of SARS-CoV-2 and its variants at present. Therefore, we had contrived a multivalent epitope vaccine of S protein to cope with the problem of virus mutation more effectively. In this study, we took the VOC including Wuhan-Hu-1 and SARS-CoV-2 variants B.1.1.7, B.1.351, P.1, B.1.429, B.1.617.2, etc as the objects of study, and took S proteins of virus strains as the research target. Meanwhile, the immunogenicity of the vaccine was confirmed in vivo and in vitro study. Theoretically, the vaccine designed by this study has a broader protective effect for SARS-CoV-2 infection.
When recognizing the antigenic peptide presented by APCs or target cells, the T cell receptor (TCR) should recognize both the antigenic peptide and the polymorphic parts of its MHC molecules. Therefore, MHC molecular restriction was fully considered when screening the CD8+ and CD4+ T-cell epitopes for activating the cellular immunity. In order to expand population coverage of the vaccine, we predicted 27 kinds of HLA-I and 27 kinds of HLA-II T-cell epitopes respectively (60). By comparing the predicted epitope sequences of each virus strain, the common epitopes of different virus strains with the highest frequency were screened and selected. Ultimately, the global population coverage of CTLEs and HTLEs was 93.16% and 99.9% separately in our rSMEV. That means the epitopes-based vaccine might be employed by more population in different regions.
To ensure the SARS-CoV-2 vaccine more reliable and effective, we spliced together the chosen epitopes that included 8 CTLEs, 17 HTLEs, 9 LBEs and 4 CBEs by linker sequences. The appropriate linkers could avoid the formation of new epitopes (Linker-epitope) and enhance the presentation of antigen epitopes by ensuring the full exposure of the screened target epitopes (61, 62). Then, the TAT sequence (11aa) was attached to the N-terminal of vaccine construct simultaneously. The TAT sequence can carry macromolecular substances, making it easier to penetrate the cell membrane, so as to promote the phagocytosis of vaccine protein by APCs (36). After the design and construction of the vaccine, the secondary structure of rSMEV was analyzed. The secondary structure analysis showed that the vaccine protein had 6.51% Beta turn and 49.51% Random coil. Because the spatial structure of Beta turn and random coil is loose, it is easy to form epitopes. Therefore, the rSMEV that containing a large amount of this structure has a good vaccine structural basis.
In view of Rosettafold’s excellent performance in protein tertiary structure prediction (38), we used it to predict the high-quality tertiary structure of rSMEV. Meanwhile, the unreliable loops or termini of the predicted 3D structure were reconstructed and refined by the GalaxyRefine Server. The good tertiary structure of rSMEV would be used for later molecular docking and MD simulation. We further analyzed the physicochemical properties and allergenicity of rSMEV. The physicochemical properties results showed that molecular weight of rSMEV was 65 KD. Previous studies have reported that it is reasonable when the molecular weight of vaccine protein is less than 110 KD (56), which hinted that the rSMEV was suitable for vaccine. In addition, the analysis of theoretical pI, amino acid acidity, alkalinity, stability, hydrophilicity and allergenicity also displayed that the rSMEV was reasonable and safe.
When microorganisms break through the mucosal barrier, TLR can recognize them and initiating the adaptive immune response (63). Because TLR-3 molecule plays an important role in innate immunity against virus, the binding of antigen and TLR-3 can help APCs to present antigen and release local cytokines. For the sake of proving that the rSMEV had strong affinity with TLR-3 molecule, we conducted the molecular docking using ClusPro Server. The results showed that lowest energy value in the all outputted rSMEV-TLR-3 complexes was −1175.3 kcal/mol. The capacity of the system is lower, the structure of object is more stable (45). Therefore, the structure of rSMEV-TLR-3 complex was quite stable. Moreover, the interaction interfaces proved that the binding affinity between rSMEV and TLR-3 was ionic and hydrogen bonds mainly. Due to the interaction of ionic bond and hydrogen bond is strong, this also indirectly indicates that the binding between rSMEV and TLR-3 is very close (64).
We also used MD simulation to analyze the structural stability and agility of rSMEV-TLR-3 complex. The RMSD value represented the structural fluctuation of the overall structure of the vaccine and TLR-3 complex, which can evaluate the stability of the system during interaction. In this part, the floating range of 100 ns RMSD value was about 1 nm, suggesting that the structure of the complex is relatively stable. The RMSF value represented the flexibility of amino acid residues in the docking complex, which can evaluate the agility of each residues in the complex. The results suggested that the agility of receptor and ligand sites after 650 was higher than that of residues 0-650. The average binding free energy of the system calculated by the MMGBSA tool was −88.54 ± 6.91 kcal/mol. Therefore, MD simulation proved that rSMEV and TLR-3 docking complex in this study had reasonable structural stability and agility. The above results showed that rSMEV can closely bind to TLR-3, and the complex has high stability and agility.
That is more important, we had verified the immunogenicity of the rSMEV in vivo and in vitro. Firstly, the rSMEV can stimulate an excellent immune response confirmed by immune simulation. Then the mice were immunized with rSMEV through experiments in vivo and in vitro. The mice can produce specific ASCs and high levels of IFN-γ and IL-4, the result confirmed that the rSMEV can induce cellular and humoral immunity and has satisfied immunogenicity.
In many current studies that based on S proteins to design SARS-CoV-2 vaccine (46–51, 65), there are few studies on designing for multiple variants at the same time. Starting from considering the mutation of SARS-CoV-2, to the restriction of HLA alleles, and then to the binding between vaccine protein and TLR-3 molecule. Our research had made innovative and reasonable design in the above aspects for improving the immunogenicity and applicability of SARS-CoV-2 vaccine. At the same time, we also evaluated the immunogenicity of the vaccine by experiments in vivo and in vitro. Compared with other studies, our rSMEV is more significant in the current perspective. Obviously, the rSMEV designed by the study estimated that it may have a high potency against SARS-CoV-2 and its variants theoretically. However, this study did not include other antigens with important immunogenicity of SARS-CoV-2. At the same time, this study still needs to further carry out human clinical trials to ensure the effectiveness of the vaccine. All in all, we provided an effective immunological strategy and mean for the active prevention and control of COVID-19 worldwide.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics Statement
The animal study was reviewed and approved by The Animal Experiment Medical Ethics Committee of the First Affiliated Hospital of Xinjiang Medical University.
Author Contributions
MY, YZ, YL, ZC, ZWL, JW, and ZNL performed the experiments. MY and YZ wrote and edited the paper. YL, ZWL, JW, and ZNL collated and verified the experimental data. FZ and JD provided experimental design and ideas, and reviewed and modified the paper. All the authors agreed to publish the final version of the paper.
Funding
This study had been supported by National Natural Science Foundation of China [Grant number: 81960373, 81860352, 31560262] and The key R&D project of Xinjiang Autonomous Region: Research on key technologies of prevention and control for COVID-19 [Project number: 2021B03003-2].
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
This study was very grateful to Dr. Maierdanjiang•Mamtimin, College of language and culture, Xinjiang Medical University and Professor Zhang Chuntao, Department of Microbiology, School of basic medicine, Xinjiang Medical University for their suggestions and help on the grammar rules of our article language.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.884433/full#supplementary-material
References
1. Ullah M, Wahab A, Saeed S, Khan SU, Fahad S. Coronavirus and its Terrifying Inning Around the Globe: The Pharmaceutical Cares at the Main Frontline. Chemosphere (2021) 275:129968. doi: 10.1016/j.chemosphere.2021.129968
2. Yang X, Yu Y, Xu J, Shu H, Liu H, Wu Y, et al. Clinical Course and Outcomes of Critically Ill Patients With SARS-CoV-2 Pneumonia in Wuhan, China: A Single-Centered, Retrospective, Observational Study. Lancet Respir Med (2020) 8:475–81. doi: 10.1016/S2213-2600(20)30079-5
3. Wang Y, Wang Y, Chen Y, Qin Q. Unique Epidemiological and Clinical Features of the Emerging 2019 Novel Coronavirus Pneumonia (COVID-19) Implicate Special Control Measures. J Med Virol (2020) 92:568–76. doi: 10.1002/jmv.25748
4. Niu S, Tian S, Lou J, Kang X, Zhang L, Lian H, et al. Clinical Characteristics of Older Patients Infected With COVID-19: A Descriptive Study. Arch Gerontol Geriatrics (2020) 0289:104058. doi: 10.1016/j.archger.2020.104058
5. Munster VJ, Koopmans M, van Doremalen N, van Riel D, de Wit E. A Novel Coronavirus Emerging in China - Key Questions for Impact Assessment. N Engl J Med (2020) 382:692–4. doi: 10.1056/NEJMp2000929
6. ElBagoury M, Tolba MM, Nasser HA, Jabbar A, Elagouz AM, Aktham Y, et al. The Find of COVID-19 Vaccine: Challenges and Opportunities. J Infect Public Health (2021) 14:389–416. doi: 10.1016/j.jiph.2020.12.025
7. Bchetnia M, Girard C, Duchaine C, Laprise C. The Outbreak of the Novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): A Review of the Current Global Status. J Infect Public Health (2020) 13:1601–10. doi: 10.1016/j.jiph.2020.07.011
8. Liang S, Liu X, Zhang S, Li M, Zhang Q, Chen J, et al. Binding Mechanism of Inhibitors to SARS-CoV-2 Main Protease Deciphered by Multiple Replica Molecular Dynamics Simulations. Phys Chem Chem Physics (2022) 24:1743–59. doi: 10.1039/d1cp04361g
9. Duffy S. Why are RNA Virus Mutation Rates So Damn High? PloS Biol (2018) 16:e3000003. doi: 10.1371/journal.pbio.3000003
10. Garcia-Beltran WF, Lam EC, St Denis K, Nitido AD, Garcia ZH, Hauser BM, et al. Multiple SARS-CoV-2 Variants Escape Neutralization by Vaccine-Induced Humoral Immunity. Cell (2021) 184:2372–83.e9. doi: 10.1016/j.cell.2021.04.006
11. Bager P, Wohlfahrt J, Fonager J, Rasmussen M, Albertsen M, Michaelsen TY, et al. Risk of Hospitalisation Associated With Infection With SARS-CoV-2 Lineage B.1.1.7 in Denmark: An Observational Cohort Study. Lancet Infect Dis (2021) 21:1507–17. doi: 10.1016/S1473-3099(21)00290-5
12. Kuzmina A, Khalaila Y, Voloshin O, Keren-Naus A, Boehm-Cohen L, Raviv Y, et al. SARS-CoV-2 Spike Variants Exhibit Differential Infectivity and Neutralization Resistance to Convalescent or Post-Vaccination Sera. Cell Host Microbe (2021) 29:522–528.e2. doi: 10.1016/j.chom.2021.03.008
13. Simmons G, Zmora P, Gierer S, Heurich A, Pöhlmann S. Proteolytic Activation of the SARS-Coronavirus Spike Protein: Cutting Enzymes at the Cutting Edge of Antiviral Research. Antiviral Res (2013) 100:605–14. doi: 10.1016/j.antiviral.2013.09.028
14. Washington NL, Gangavarapu K, Zeller M, Bolze A, Cirulli ET, Schiabor Barrett KM, et al. Emergence and Rapid Transmission of SARS-CoV-2 B.1.1.7 in the United States. Cell (2021) 184:2587–94.e7. doi: 10.1016/j.cell.2021.03.052
15. Wilton T, Bujaki E, Klapsa D, Majumdar M, Zambon M, Fritzsche M, et al. Rapid Increase of SARS-CoV-2 Variant B.1.1.7 Detected in Sewage Samples From England Between October 2020 and January 2021. Msystems (2021) 6:e0035321. doi: 10.1128/mSystems.00353-21
16. Laffeber C, de Koning K, Kanaar R, Lebbink JHG. Experimental Evidence for Enhanced Receptor Binding by Rapidly Spreading SARS-CoV-2 Variants. J Mol Biol (2021) 433:167058. doi: 10.1016/j.jmb.2021.167058
17. Resende PC, Bezerra JF, Teixeira Vasconcelos RH, Arantes I, Appolinario L, Mendonça AC, et al. Severe Acute Respiratory Syndrome Coronavirus 2 P.2 Lineage Associated With Reinfection Case, Brazil, June-October 2020. Emerging Infect Dis (2021) 27:1789–94. doi: 10.3201/eid2707.210401
18. Paiva MHS, Guedes DRD, Docena C, Bezerra MF, Dezordi FZ, Machado LC, et al. Multiple Introductions Followed by Ongoing Community Spread of SARS-CoV-2 at One of the Largest Metropolitan Areas of Northeast Brazil. Viruses (2020) 12:1414. doi: 10.3390/v12121414
19. Pascarella S, Ciccozzi M, Zella D, Bianchi M, Benedetti F, Benvenuto D, et al. SARS-CoV-2 B.1.617 Indian Variants: Are Electrostatic Potential Changes Responsible for a Higher Transmission Rate? J Med Virol (2021) 93:6551–6. doi: 10.1002/jmv.27210
20. Letko M, Marzi A, Munster V. Functional Assessment of Cell Entry and Receptor Usage for SARS-CoV-2 and Other Lineage B Betacoronaviruses. Nat Microbiol (2020) 5:562–9. doi: 10.1038/s41564-020-0688-y
21. Iacobucci G. Covid-19: New UK Variant may be Linked to Increased Death Rate, Early Data Indicate. BMJ (Clinical Res ed.) (2021) 372:n230. doi: 10.1136/bmj.n230
22. Shariare MH, Parvez MAK, Karikas GA, Kazi M. The Growing Complexity of COVID-19 Drug and Vaccine Candidates: Challenges and Critical Transitions. J Infect Public Health (2021) 14:214–20. doi: 10.1016/j.jiph.2020.12.009
23. Francis MJ. Recent Advances in Vaccine Technologies. Veterinary Clinics North America Small Anim Practice (2018) 48:231–41. doi: 10.1016/j.cvsm.2017.10.002
24. Yu M, Zhu Y, Li Y, Chen Z, Sha T, Li Z, et al. Design of a Novel Multi-Epitope Vaccine Against Echinococcus Granulosus in Immunoinformatics. Front Immunol (2021) 12:668492. doi: 10.3389/fimmu.2021.668492
25. Li Y, Zhu Y, Sha T, Chen Z, Yu M, Zhang F, et al. A Multi-Epitope Chitosan Nanoparticles Vaccine of Canine Against Echinococcus Granulosus. J BioMed Nanotechnol (2021) 17:910–20. doi: 10.1166/jbn.2021.3065
26. Li Z, Zhang F, Zhang C, Wang C, Lu P, Zhao X, et al. Immunoinformatics Prediction of OMP2b and BCSP31 for Designing Multi-Epitope Vaccine Against Brucella. Mol Immunol (2019) 114:651–60. doi: 10.1016/j.molimm.2019.09.013
27. Chen Z, Zhu Y, Sha T, Li Z, Li Y, Zhang F, et al. Design of a New Multi-Epitope Vaccine Against Brucella Based on T and B Cell Epitopes Using Bioinformatics Methods. Epidemiol Infect (2021) 149:e136. doi: 10.1017/S0950268821001229
28. Zhang F, Li Z, Jia B, Zhu Y, Pang P, Zhang C, et al. The Immunogenicity of OMP31 Peptides and Its Protection Against Brucella Melitensis Infection in Mice. Sci Rep (2019) 9:3512. doi: 10.1038/s41598-019-40084-w
29. Wold S, Jonsson J, Sjörström M, Sandberg M, Rännar S. DNAAnd Peptide Sequences and Chemical Processes Multivariately Modelled by Principal Component Analysis and Partial Least-Squares Projections to Latent Structures. Analytica Chimica Acta (1993) 277:239–53. doi: 10.1016/0003-2670(93)80437-P
30. Almagro Armenteros JJ, Tsirigos KD, Sønderby CK, Petersen TN, Winther O, Brunak S, et al. SignalP 5.0 Improves Signal Peptide Predictions Using Deep Neural Networks. Nat Biotechnol (2019) 37:420–3. doi: 10.1038/s41587-019-0036-z
31. Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, et al. The Immune Epitope Database (IEDB): 2018 Update. Nucleic Acids Res (2019) 47:D339–43. doi: 10.1093/nar/gky1006
32. Stranzl T, Larsen MV, Lundegaard C, Nielsen M. NetCTLpan: Pan-Specific MHC Class I Pathway Epitope Predictions. Immunogenetics (2010) 62:357–68. doi: 10.1007/s00251-010-0441-4
33. Reynisson B, Barra C, Kaabinejadian S, Hildebrand WH, Peters B, Nielsen M. Improved Prediction of MHC II Antigen Presentation Through Integration and Motif Deconvolution of Mass Spectrometry MHC Eluted Ligand Data. J Proteome Res (2020) 19:2304–15. doi: 10.1021/acs.jproteome.9b00874
34. Jespersen MC, Peters B, Nielsen M, Marcatili P. BepiPred-2.0: Improving Sequence-Based B-Cell Epitope Prediction Using Conformational Epitopes. Nucleic Acids Res (2017) 45:W24–9. doi: 10.1093/nar/gkx346
35. Dong R, Chu Z, Yu F, Zha Y. Contriving Multi-Epitope Subunit of Vaccine for COVID-19: Immunoinformatics Approaches. Front Immunol (2020) 11:1784. doi: 10.3389/fimmu.2020.01784
36. Frankel AD, Pabo CO. Cellular Uptake of the Tat Protein From Human Immunodeficiency Virus. Cell (1988) 55:1189–93. doi: 10.1016/0092-8674(88)90263-2
37. Combet C, Blanchet C, Geourjon C, Deléage G. NPS@: Network Protein Sequence Analysis. Trends Biochem Sci (2000) 25:147–50. doi: 10.1016/s0968-0004(99)01540-6
38. Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, et al. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science (2021) 373:871–6. doi: 10.1126/science.abj8754
39. Ko J, Park H, Heo L, Seok C. GalaxyWEB Server for Protein Structure Prediction and Refinement. Nucleic Acids Res (2012) 40:W294–7. doi: 10.1093/nar/gks493
40. Benkert P, Biasini M, Schwede T. Toward the Estimation of the Absolute Quality of Individual Protein Structure Models. Bioinformatics (2011) 27:343–50. doi: 10.1093/bioinformatics/btq662
41. Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, et al. Protein Identification and Analysis Tools in the ExPASy Server. Methods Mol Biol (1999) 112:531–52. doi: 10.1385/1-59259-584-7:531
42. Bui HH, Sidney J, Dinh K, Southwood S, Newman MJ, Sette A. Predicting Population Coverage of T-Cell Epitope-Based Diagnostics and Vaccines. BMC Bioinf (2006) 7:153. doi: 10.1186/1471-2105-7-153
43. Takeda K, Akira S. TLR Signaling Pathways. Semin Immunol (2004) 16:3–9. doi: 10.1016/j.smim.2003.10.003
44. Matsumoto M, Funami K, Tanabe M, Oshiumi H, Shingai M, Seto Y, et al. Subcellular Localization of Toll-Like Receptor 3 in Human Dendritic Cells. J Immunol (2003) 171:3154–62. doi: 10.4049/jimmunol.171.6.3154
45. Vajda S, Yueh C, Beglov D, Bohnuud T, Mottarella SE, Xia B, et al. New Additions to the ClusPro Server Motivated by CAPRI. Proteins (2017) 85:435–44. doi: 10.1002/prot.25219
46. Grifoni A, Sidney J, Zhang Y, Scheuermann RH, Peters B, Sette A. A Sequence Homology and Bioinformatic Approach Can Predict Candidate Targets for Immune Responses to SARS-CoV-2. Cell Host Microbe (2020) 27:671–80.e2. doi: 10.1016/j.chom.2020.03.002
47. Chen HZ, Tang LL, Yu XL, Zhou J, Chang YF, Wu X. Bioinformatics Analysis of Epitope-Based Vaccine Design Against the Novel SARS-CoV-2. Infect Dis Poverty (2020) 9:88. doi: 10.1186/s40249-020-00713-3
48. Kar T, Narsaria U, Basak S, Deb D, Castiglione F, Mueller DM, et al. A Candidate Multi-Epitope Vaccine Against SARS-CoV-2. Sci Rep (2020) 10:10895. doi: 10.1038/s41598-020-67749-1
49. Li W, Li L, Sun T, He Y, Liu G, Xiao Z, et al. Spike Protein-Based Epitopes Predicted Against SARS-CoV-2 Through Literature Mining. Med Novel Technol Devices (2020) 8:100048. doi: 10.1016/j.medntd.2020.100048
50. He J, Huang F, Zhang J, Chen Q, Zheng Z, Zhou Q, et al. Vaccine Design Based on 16 Epitopes of SARS-CoV-2 Spike Protein. J Med Virol (2021) 93:2115–31. doi: 10.1002/jmv.26596
51. Ahmed SF, Quadeer AA, McKay MR. Preliminary Identification of Potential Vaccine Targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies. Viruses (2020) 12:254. doi: 10.3390/v12030254
52. Rapin N, Lund O, Bernaschi M, Castiglione F. Computational Immunology Meets Bioinformatics: The Use of Prediction Tools for Molecular Binding in the Simulation of the Immune System. PloS One (2010) 5:e9862. doi: 10.1371/journal.pone.0009862
53. Bhatnager R, Bhasin M, Arora J, Dang AS. Epitope Based Peptide Vaccine Against SARS-COV2: An Immune-Informatics Approach. J Biomolecular Structure Dynamics (2021) 39:5690–705. doi: 10.1080/07391102.2020.1787227
54. Nain Z, Abdulla F, Rahman MM, Karim MM, Khan MSA, Sayed SB, et al. Proteome-Wide Screening for Designing a Multi-Epitope Vaccine Against Emerging Pathogen Elizabethkingia Anophelis Using Immunoinformatic Approaches. J Biomolecular Structure Dynamics (2020) 38:4850–67. doi: 10.1080/07391102.2019.1692072
55. Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC, et al. JCat: A Novel Tool to Adapt Codon Usage of a Target Gene to its Potential Expression Host. Nucleic Acids Res (2005) 33:W526–31. doi: 10.1093/nar/gki376
56. Barh D, Barve N, Gupta K, Chandra S, Jain N, Tiwari S, et al. Exoproteome and Secretome Derived Broad Spectrum Novel Drug and Vaccine Candidates in Vibrio Cholerae Targeted by Piper Betel Derived Compounds. PloS One (2013) 8:e52773. doi: 10.1371/journal.pone.0052773
57. Zhao X, Zhang F, Li Z, Wang H, An M, Li Y, et al. Bioinformatics Analysis of EgA31 and EgG1Y162 Proteins for Designing a Multi-Epitope Vaccine Against Echinococcus Granulosus. Infect Genet Evolution (2019) 73:98–108. doi: 10.1016/j.meegid.2019.04.017
58. Sha T, Li Z, Zhang C, Zhao X, Chen Z, Zhang F, et al. Bioinformatics Analysis of Candidate Proteins Omp2b, P39 and BLS for Brucella Multivalent Epitope Vaccines. Microbial Pathogenesis (2020) 147:104318. doi: 10.1016/j.micpath.2020.104318
59. Xu Y, Tao X, Shen B, Horng T, Medzhitov R, Manley JL, et al. Structural Basis for Signal Transduction by the Toll/interleukin-1 Receptor Domains. Nature (2000) 408:111–5. doi: 10.1038/35040600
60. Ayyagari VS, Venkateswarulu V, Srirama K. Design of a Multi-Epitope-Based Vaccine Targeting M-Protein of SARS-CoV2: An Immunoinformatics Approach. J Biomol Struct Dyn (2022) 40:2963–77. doi: 10.1080/07391102.2020.1850357
61. Nezafat N, Karimi Z, Eslami M, Mohkam M, Zandian S, Ghasemi Y. Designing an Efficient Multi-Epitope Peptide Vaccine Against Vibrio Cholerae via Combined Immunoinformatics and Protein Interaction Based Approaches. Comput Biol Chem (2016) 62:82–95. doi: 10.1016/j.compbiolchem.2016.04.006
62. Meza B, Ascencio F, Sierra-Beltrán AP, Torres J, Angulo C. A Novel Design of a Multi-Antigenic, Multistage and Multi-Epitope Vaccine Against Helicobacter Pylori: An in Silico Approach. Infect Genet Evolution (2017) 49:309–17. doi: 10.1016/j.meegid.2017.02.007
63. Nicodemus CF, Berek JS. TLR3 Agonists as Immunotherapeutic Agents. Immunotherapy (2010) 2:137–40. doi: 10.2217/imt.10.8
64. Jia S, Li C, Pan H, Wang M, Wang X, Lin Q. Preparation and Pore-Forming Mechanism of Hydrogen Bond and Ionic Bond Double-Driven Chitosan-Based Mesoporous Carbon. Int J Biol Macromol (2021) 179:519–31. doi: 10.1016/j.ijbiomac.2021.03.024
Keywords: SARS-CoV-2, COVID-19, variant, vaccine, epitope, immunoinformatics
Citation: Yu M, Zhu Y, Li Y, Chen Z, Li Z, Wang J, Li Z, Zhang F and Ding J (2022) Design of a Recombinant Multivalent Epitope Vaccine Based on SARS-CoV-2 and Its Variants in Immunoinformatics Approaches. Front. Immunol. 13:884433. doi: 10.3389/fimmu.2022.884433
Received: 26 February 2022; Accepted: 12 April 2022;
Published: 06 May 2022.
Edited by:
Yongjun Sui, National Cancer Institute (NIH), United StatesReviewed by:
Yusha Araf, Shahjalal University of Science and Technology, BangladeshSaba Ismail, National University of Medical Sciences (NUMS), Pakistan
Sajjad Ahmad, Abasyn University, Pakistan
Copyright © 2022 Yu, Zhu, Li, Chen, Li, Wang, Li, Zhang and Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianbing Ding, MTYwMTM3OTkzN0BxcS5jb20=; Fengbo Zhang, emZiMTMxQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship