 Marco Pio La Manna1,2†
Marco Pio La Manna1,2† Mojtaba Shekarkar Azgomi1,2†
Mojtaba Shekarkar Azgomi1,2† Bartolo Tamburini1,2
Bartolo Tamburini1,2 Giusto Davide Badami1,2Leila Mohammadnezhad1,2
Giusto Davide Badami1,2Leila Mohammadnezhad1,2 Francesco Dieli1,2‡
Francesco Dieli1,2‡ Nadia Caccamo1,2*‡
Nadia Caccamo1,2*‡- 1Central Laboratory of Advanced Diagnosis and Biomedical Research (CLADIBIOR) Azienda Ospedaliera Universitaria Policlinico (A.O.U.P.) Paolo Giaccone, University of Palermo, Palermo, Italy
- 2Department of Biomedicine, Neurosciences and Advanced Diagnostic (Bi.N.D.), University of Palermo, Palermo, Italy
The immune system, smartly and surprisingly, saves the exposure of a particular pathogen in its memory and reacts to the pathogen very rapidly, preventing serious diseases.
Immunologists have long been fascinated by understanding the ability to recall and respond faster and more vigorously to a pathogen, known as “memory”.
T-cell populations can be better described by using more sophisticated techniques to define phenotype, transcriptional and epigenetic signatures and metabolic pathways (single-cell resolution), which uncovered the heterogeneity of the memory T-compartment. Phenotype, effector functions, maintenance, and metabolic pathways help identify these different subsets. Here, we examine recent developments in the characterization of the heterogeneity of the memory T cell compartment. In particular, we focus on the emerging role of CD8+ TRM and TSCM cells, providing evidence on how their immunometabolism or modulation can play a vital role in their generation and maintenance in chronic conditions such as infections or autoimmune diseases.
Introduction
T lymphocytes are an essential component of adaptive immunity known for their role in protecting against infectious diseases caused by invasive pathogens and cancer cells but also for their crucial role as important target cells in immunotherapy (1–5).
Memory T cells differ in their naïve counterparts for several functional characteristics identified in booster reactions due to a second encounter with the pathogen. These T memory populations are organized in a developmental hierarchy, where stem cell memory T-cells (TSCM) renew themselves and generate long-lived central memory T-cells (TCM) and short-lived memory T-cells (TEM) (6–10). However, the mechanisms underlying the increased multipotency of TSCM cells relative to TCM cells were not clearly defined in the molecular terms (11).
The principal characteristics of the memory T-cell responses include several aspects such as an increased pool of specific memory T cells thanks to the recognition of pathogen-derived-antigens by TCR, a faster and more potent response to the infection; a pre-programming to generate a “tailor-made” set of effector cell types optimized to combat pathogens, which includes the role of T follicular helper cell (TFH) responses involved in helping humoral immunity; the presence of memory T cells in barrier tissues as sentinels in detecting and fighting infection. This last characteristic is dominated by resident memory T cells (TRM) that are maintained in non-lymphoid tissues and do not exchange with populations in the circulation. Since this population is not represented in the blood, it is difficult to evaluate its generation and persistence.
Once T cells are not properly primed or generated, they gradually lose their ability to resolve the chronic infection. Such T-cell memory populations are likely to disappear over time or become permanently dysfunctional, favoring the persistence of the pathogen. As a result, T lymphocyte responses can become dangerous, resulting in excessive tissue damage, collectively referred to as immunopathology, which may determine the immune system’s persistent systemic alteration, affecting the development of memory cells and their response to the re-encounter with the same pathogen.
The aim of this review is to focus on the role of TSCM and TRM cell subsets in the course of infectious and autoimmune diseases and how the metabolic program can impact the features of the immune response.
The Basic Concept of CD8+ T Cell Memory Subsets
The major T cell subsets are classified into CD4+ T and CD8+ T cells based on cell functions and cell surface markers. CD4+ T cells, also known as T helper (Th) cells, play an essential role in the adaptive immune response to pathogens due to their multiple roles in immune response orchestration (12), such as helping B cells to produce antibodies, recruiting granulocytes to infected sites, and producing cytokines and chemokines. Many distinct CD4+ T cell subsets have been identified since the identification of Th1 by Mosmann et al. (12–15). CD8+ T cells are essential for immune defense against intracellular pathogens (16, 17) and tumoral cells.
For several years, memory T cells were divided into two main principal subsets: central memory T cells (TCM) and effector memory T cells (TEM) (18). This initial classification was based on the expression of the lymphoid-homing molecules CCR7 and L-selectin (CD62L), while TEM were identified on the bases of the reduced expression of these receptors relative to T naive (TN) and TCM and for their presence as recirculating cells between the blood and peripheral tissues. After this first characterization of the different subsets, additional surface markers have been added, such as the mutually exclusive expression of the two isoforms of the CD45R molecule, CD45RA or CD45RO. In different species, such as mice, TEM has been shown to retain lower expression of molecules involved in long-term persistence (19), human primates (20, 21) and humans (22, 23), while upregulating transcription factors (TFs) mediating terminal differentiation with decreased self-renewal and multipotent capacity compared with TCM (7).
Furthermore, TEM cells have limited expansion potential and are unable to enter the lymph nodes from the blood, but are marked by the expression of genes associated with cytotoxicity and can perform rapid effector functions when renewing TCR signalling (24). For a long time, TEM cells were thought to be superior for penetrating and examining peripheral tissues; however, recent research indicates that TEM cells and TEM-like cells are largely excluded from human and mouse non-lymphoid tissues (NLTs) (23, 25–27).
TCM cells can be distinguished by the high-level expression of CD62L and CCR7 lymphoid localization markers and their capacity to recirculate between blood and lymphoid tissues. They are considered to be multipotent, and at least one subset of this cell pool shows increased expansion potential when the antigen is encountered (8). Two novel memory subsets have emerged during the past decade, further highlighting the inherent diversity among these subtypes. In particular, a noncirculating lineage of T cells, the TRM, express tissue-residency markers, CD69 and CD103, and reside in peripheral tissues (28–30). This subset is located at the entry point of invading pathogens to provide the first rapid defense line (28–30). While, TSCM, defined in humans by surface expression of CD122, CD95, and CXCR3 within the CD45RA+CD45RO–CCR7+CD62L+ naive-like compartment, are characterized by increased homeostatic capacity for automatic renewal, increased proliferation capacity, and multipotent development potential compared to TCM and TEM; moreover, this subset is capable of generating differentiated in vitro in cells, including TCM and TEM or as adoptive transfer in xenogeneic mouse models, as well as in humans (8, 9, 20, 31–33). Although this subset was initially identified in mice as part of an allogeneic bone marrow transplantation (34) or induced by T naive (TN) activated by the inhibition of glycogen synthase kinase 3b (GSK-3b) (35).
Although TSCM and TCM have the ability to retain the expression of associated naive genes while simultaneously displaying the rapid production of cytokine upon stimulating T-cell receptor (TCR), the T memory subsets as a whole are often represented as having mutually exclusive properties, primarily a potential similar stem-like potential (TSCM and TCM) versus immediacy of effector functions/cytotoxicity (TEM) (7, 8, 22, 36, 37).
An important aspect of CD8+ T cells is their ability to adapt their functions to the basic type of pathogens encountered. Firstly, the recognition of the antigen, T cells, through the clonal expansion process, become biased to recognize pathogens to which they have already been exposed (38). Moreover, remodelling of the epigenetic landscape enables the memory cells that form in this process to perform effective functions more rapidly (39). The distribution of the memory compartment of the CD8+ T cells at different body sites maximizes the chances of early recognition of pathogens during a renewed infection (40). In accordance with the concept that the CD8+ storage T-cell pool can provide fast effector functions and has the ability to renew clonal expansion, this cell pool is very diverse at epigenetic, transcriptional and protein expression levels.
It is believed that these memory populations are organized in a developmental hierarchy, according to which TSCM renew themselves and generate a long-lasting TCM, TEM cells, and TRM cells (6–10) (Figure 1). However, the mechanisms underlying the improved multi-potency of TSCM cells compared to TCM cells have not been clearly defined in molecular terms (11).
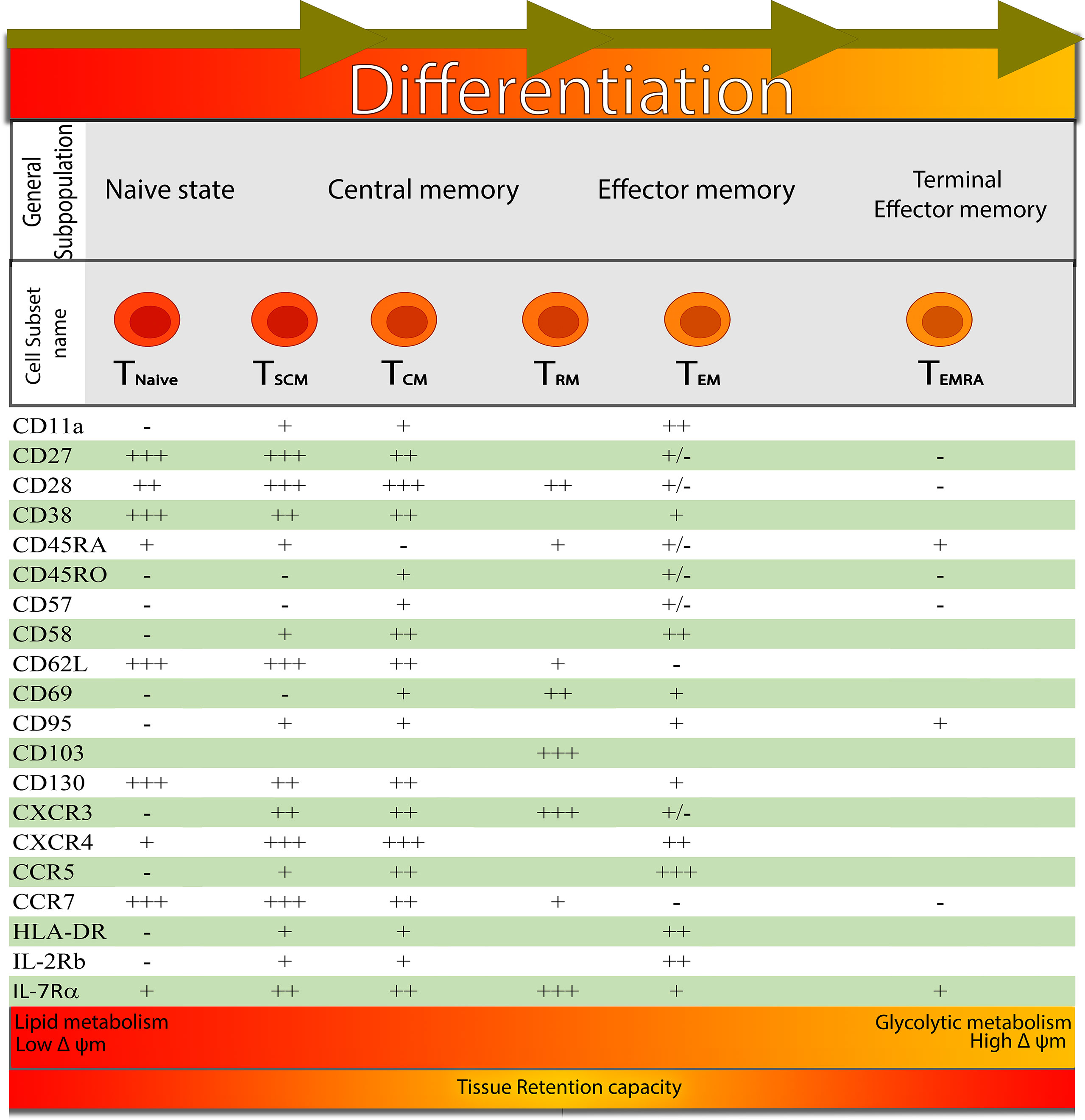
Figure 1 Signatures for distinguishing the different developmental T cell stages: from left to right, it shows the different development stages of CD8+ T cell-based on the expression of surface markers. At the bottom of the figure, the main metabolic pathway and mitochondrial membrane potential that are favoured at the functional and phenotypic stage are represented by different coloured gradients (from red to orange), indicating that TRM cells have a larger tissue retention capacity.
In addition to the CD8+ systemic memory T-cell pool, one can distinguish a TRM cell pool from CD8+ in tissues that permanently reside in non-lymphoid tissues (NLTs).
By a continuous migration and monitoring process that is limited to distinct anatomical compartments, such as stroma or organ parenchyma, TRM cells patrol tissues to scan for foreign invaders (41, 42). After encountering the antigen, TRM cells quickly induce a local alarm state, resulting in the recruitment of other immune cells and local production of antibacterial and antiviral proteins by epithelial cells (43, 44).
In keeping with this “pathogen alert” function, TRM cells produce not only cytotoxic molecules, such as granzyme B and perforin but also cytokines such as IFN-γ and TNF, which may affect the behaviour of neighbouring cells (45–50). In addition, the existence of TRM cells that express minimal levels of cytolytic molecules and therefore rely primarily on this pathogen, warning function, has been reported in several human tissues (51–54).
It has been proven that the differentiation of T cells from memory can be corrupted under conditions of persistent antigenic stimulation, this finding was primarily observed in chronic viral infections and progressive malignant tumours, which promote a state of T-cell exhaustion which is characterized by an organized loss of effective function, altered proliferation and increased regulation of inhibitor receptors (55). This dynamic process takes place over a period of weeks after initiation (56, 57) and involves a genome-wide accumulation of epigenetic changes (58, 59).
Recent studies have shown that exhausted T-cell (TEX) populations are developmental and functionally heterogeneous, incorporating stem-like progenitors that express T-cell factor 1 (TCF1), which produce highly differentiated TEX cells that are basically dysfunctional and do not have TCF1 (60–63). It is important to note that the therapeutic benefits of blocking immune checkpoints in the context of chronic viral infections and various cancers are believed to function via these TCF1+ progenitors, which appear to be susceptible to interventions that specifically target the inhibitory receptor programmed death-1 (PD-1) (60, 62, 64–67).Current data suggest that exhausted and functional memory T cells originate from distinct populations of stem-like progenitors engaged in distinct destinies. However, the precise nature of these stem-like progenitors, which shape the adaptive immune response and influence the outcome of many globally relevant diseases, has remained unclear. Very recently, Galletti et al. (68), with the aim of a complete and impartial approach, have mapped the origins of dysfunctional and functional CD8+ human memory T-cells. Their data allow the identification of two distinct subsets of CCR7+ progenitors in healthy individuals that are distinguished by the expression of PD-1 and TIGIT. They identified a progenitor committed the cells to generate a dysfunctional subset with an exhausted progeny expressing these two inhibitor receptors. In contrast, they identified another progenitor cell committed to generating a more functional progeny where the absence of these inhibitory receptors was observed. Therefore, the transcriptional assessment of the distinct subset PD-1+TIGIT+ also explained most of the differences between the TSCM and TCM cells, providing a clearer view of how human CD8+ memory T-cells differentiate.
Recent advances in single-cell technologies at the proteomic and transcriptomic levels, along with genome-scale epigenetic profiling studies, provided new information that is refining our current understanding of gene expression programs and the developmental origin of memory T cells. In particular, single-cell transcriptomic has identified subsets of human and murine T cells that simultaneously co-express stem and effector genes. In addition, the epigenetic profiling of stem T cells has made it possible to identify several locus effectors in these cells which remain epigenetic, while having limited transcriptional activity, explaining their ability to quickly recall an effective response (69, 70).
T cell memory has a unique set of functional properties and potentials that set it apart from many of its sisters. This is not to say that every T-cell in memory is its own subset; rather, it is to say that one phenotypic or functional feature does not imply another.
Metabolic Regulation of Memory T-Cell Differentiation and Function
Very recently, the role of metabolism in the immune system has become more evident. Inactivated naive T cells migrate continuously under immune surveillance via secondary lymphoid organs at the expense of ATP and thus require basal replacement biosynthesis. Resting T cells are more dependent on energy-efficient processes such as the β-oxidation of fatty acids and the oxidation of pyruvate and glutamine via the tricarboxylic acid cycle (TCA cycle). To maintain this energy-providing basal metabolism, resting T cells need specific signals via TCR or cytokine receptors such as IL-7R (71). Furthermore, resting T cells do not have a static and well-determined metabolism but undergo dynamic regulation. For example, environmental signals can control the utilization of nutrients in resting T cells, causing them to become trophic and influencing the ability to initiate cell proliferation and resistance to apoptosis. IL-7R regulates glucose absorption mainly thanks to the PI3k-Akt-mTOR pathway, which promotes the trafficking of Glut1 on the cell surface (72, 73). As a result, IL-7R regulation of glycolysis is critical for T lymphocyte basal metabolism in vivo, as its deletion results in cell atrophy and the inability to maintain glycolysis in vivo (74).
Unlike naive T lymphocytes, T memory (TM) lymphocytes usually divide every 2-3 weeks undergoing intermittent cell division. These lymphocytes necessarily require the combination of IL-15 and IL-7 for their proliferation and survival to which a high level of CD127 and CD122 molecules is associated, respectively regulated by FOX01 and by, T-bet and eomesodermin (17, 75).
Furthermore, TM cells and their responses are dependent on lipid oxidation, and although the homeostasis of the various subgroups is very similar, there are important differences. In fact, CD4+ TM cells appear to be less dependent on IL-15 signal than CD8+ TM cells, and this is probably partly determined by the low expression of CD 122 on CD4+ TM cells (76).
Homeostasis of memory T cells is determined by IL-15 and/or IL-7, which keep them in a precarious state of quiescence (G1 phase of the cell cycle). Aerobic glycolysis and mitochondrial metabolism have important and well-defined roles in the differentiation of TM cells, but mitochondrial metabolism supports their quiescent, survival, and functions (77, 78). Increased mitochondrial function and acetate uptake mediate rapid recall responses by memory T cells, both in TCM and TRM cells. Thus, metabolism influences entry and exit from quiescence to regulate their generation, survival, and function (79, 80).
Although it is clear why the transition from quiescent and naive T cells to proliferating T cells results in a series of altered metabolic demands, it is less clear why differentiated active T cell pool memory cells necessitate a carefully orchestrated transition to invert the cell focus on vigorous expansion and equip emerging memory cells for sustained maintenance and the enhanced metabolic capacity required to mount a rapid recall response. Differentiating effectors involve a spectacular reprogramming of metabolic functions, where the requirements for increased biosynthesis and energy use are met in the most obvious way by induction aerobic glycolysis (oxidative phosphorylation, characteristic of the catabolic state of naive cells, is also maintained but plays a proportionately lesser role).
Cells need to reprogram their metabolism to leave rapidly expanding and reenter the memory pool (81). For the first time, it has been shown by pioneering studies that mTOR inhibition and enhanced fatty acid oxidation (FAO) induce an increase in CD8+ differentiated memory T cells (82–84).
Above all, memory cells’ metabolic status differs from that of naive T cells; changes in mitochondrial mass and FAO capacity (including alterations) result in memory cells that are much better able to undergo metabolic reprogramming. CD8+ memory T cells, for example, have increased mitochondrial mass and alternate breathing capacity (80, 85), which can be linked to the ability of cells to respond quickly to metabolic increases. Moreover, memory cells selectively express the carrier of Aquaporin-9 glycerol, which is involved in fatty acid storage (86).
The inability to achieve this metabolic change is related with decreased central memory retention or responsiveness (85, 86). Demonstrating that these “fit” memory cells appear to be favoured by mitochondrial remodelling (including networks of fused mitochondria) (78), and subsequent discoveries reveal that CD28 costimulation drives that process (87).
There is much more to be learned about how metabolic pathways regulate survival, differentiation and function in T lymphocytes; for example, the need for FAO in CD8+ TCM is satisfied in a way that is difficult to understand: TCM involves importing glucose and glycerol to synthesize and then metabolize triacylglycerides into what appears to be a futile metabolic cycle (i.e. those who consume as much energy as they produce) (88). Synthesis/storage (at times of abundance) and lipolysis (at times of need) ensure the continued survival of TCM, but why this is preferred to direct uptake of free fatty acids (FFA) is unclear.
CD4+ T lymphocytes also require glucose absorption for sustained maintenance in a pathway controlled by signalling through Notch (89). However, it is unclear how similar metabolic needs are for maintaining CD4+ and CD8+ TCM. The transcription activation of HIF-1α by hypoxia (or other indications) is associated with the promotion of the effector at the expense of the differentiation of memory (90, 91), but may also support epigenetic regulation (92);. Similarly, the regulation of aerobic glycolysis can influence epigenetic transcription and control of gene expression (93, 94).
Interestingly, recent data suggest that there are alternative metabolic strategies for memory T lymphocyte populations. Studies in which CD8+ T cells were prevented from switching between aerobic glycolysis and oxidative phosphorylation during an immune response (through sustained HIF activation) showed a surprisingly robust generation of memory cells in lymphoid sites - particularly TEM phenotype cells, suggesting that glycolysis dependency is sufficient to meet the metabolic requirements of at least some CD8+ recirculated memory T cells (95).
TRM appears to use another mechanism to treat their metabolic process. As stated above, TCM makes its own fatty acids for stimulating the FAO: the skin, whilst TRM, on the other hand, clearly use the more direct route of absorption of free fatty acids (involving the fatty acid-binding proteins Fabp4 and Fabp5) to meet their FAO requirements, as noted by the finding that Fabp4/Fabp5 deficiency selectively affects cutaneous TRM but not the generation of recirculated memory CD8+ T cells (45). It will be essential to determine whether this metabolic route is also used in other tissues. These findings illustrate the fact that there are obviously multiple ways to resolve metabolic needs for sustained maintenance of memory cells. It is unclear whether these alternatives will easily correspond to current subset designations.
General Background of TSCM Cell
While T cell development on the individual cell level is well known, we still lack critical information about how the T cell compartment is formed. Broadly, the T cell compartment is highly heterogeneous and different subsets of T cells based on phenotypical and functional distinct can be identified in the peripheral blood, which is classified, as previously described, as either naïve or memory cells based on their expression (or lack thereof) of various cell-surface markers and receptors. The frequency of memory T cells is highly age-dependent, and it dynamically changes throughout an individual’s lifetime. During the first two decades of life, all T cells in the peripheral blood are naïve. Memory T cells develop over time in response to exposure to diverse antigens, and eventually, circulating memory T cell frequencies reach a plateau and remain stable throughout adulthood (96, 97). Ultimately the proportion and the functionality of memory T cells become altered during immune senescence, starting at 65–70 years of age (97–99).
Recently a subset of memory T cells with stem cell-like properties has been identified with a superior survival potential and stemness function (100). TSCM in humans derived from naive T cells with a multipotent function that can further differentiate into other T cell subsets with a high proliferative capacity and self-renewal flair. Additionally, TSCM, defined in humans by surface expression of CD122 (also known as IL−2Rβ), CD95 (also known as FAS), and CXCR3 within the CD45RA+CD45RO–CCR7+CD62L+ naïve-like compartment, and they also express high levels of the co−stimulatory receptors CD27 and CD28, IL−7 receptor α−chain (IL−7Rα). These properties define their ‘stem cell-like’ capacities (100, 101).
TSCM cells have been described in peripheral blood and can be either CD4+ or CD8+. They are a small fraction of circulating T lymphocytes (≈2–3%). Notably, the frequency of circulating TSCM cells can vary because of the different factors that will be explained in this review, but there is evidence to be heritable and associated with single-nucleotide polymorphisms (SNPs) at a genetic locus containing CD95 (102) that showed a potential role of FAS-signalling pathway in the regulation of TSCM cell homeostasis. After many years of tracking the origin of different memory cells to address the stemness function, the studies recently showed that TSCM cells differentiate directly from naive precursors that can reconstitute the entire heterogeneity of memory T cell subsets (31, 32).
The Role of Metabolic Pathway of TSCM Cells in Infectious Diseases
T cells can be formidable soldiers in the fight against human diseases such as cancer and infection; there is a minority illustrious population due to the capacity to reconstitute a wide-ranging diversity of T cell compartment, robust proliferative potential, and extreme longevity such as TSCM, which make them an ideal cell population to use in adoptive immunotherapy or the development of new vaccines against infectious diseases, this might ultimately allow for the widespread application of adoptive immunotherapy based on multipotent, highly proliferative, tumour-specific TSCM. Finally, generating long-term T cell memory might be important for T cell-based vaccines designed to target intracellular pathogens, as well as immunotherapy. But we are still in the early stage of this intention because there are many outstanding questions about this very small population, such as their metabolic requirements, epigenetic and transcriptional programs also anatomical niches. So far, the clinical exploitation of TSCM has been limited by their low numbers in the circulation (100, 103) and the lack of robust, clinical-grade manufacturing protocols capable of generating and maintaining this cell type in vitro.
Here we will review all the available protocols in detail. In summary, all of these protocols are based on the fate decision that the naïve cell must make to go on a path for proliferation and effector production or memory development.
As explained before, the fate decision that naïve T cells have to make is highly dependent on cellular metabolism or transcriptional modification, but the key question in this critical decision time is how does an activated T cell retain the stem-like capacity to self-renew while simultaneously developing into more differentiated progeny?
Over the last decade, the existed protocol for isolation and in vitro expansion of TSCM have been accomplished using modified human T cell expansion protocols by targeting different cellular pathways, such as replacing IL-2 with IL-7 and IL-15, or IL-21 (104, 105), promoting Notch signalling (106) or limiting reactive oxygen species (ROS) metabolism upon antioxidant treatment (106), pharmacological inhibition of GSK-3b (107), AKT (108) or BET proteins (109) also takes to the point as additional strategies, which targeting the suppression of genes associated with terminal effector differentiation, such as Tbet, BATF, and EOMES, and in the maintenance of stem-like genes, such as those encoding TCF1 and lymphoid enhancer binding factor 1 (LEF1) to address the naïve fate decision and force it to generate TSCM (Figure 2).
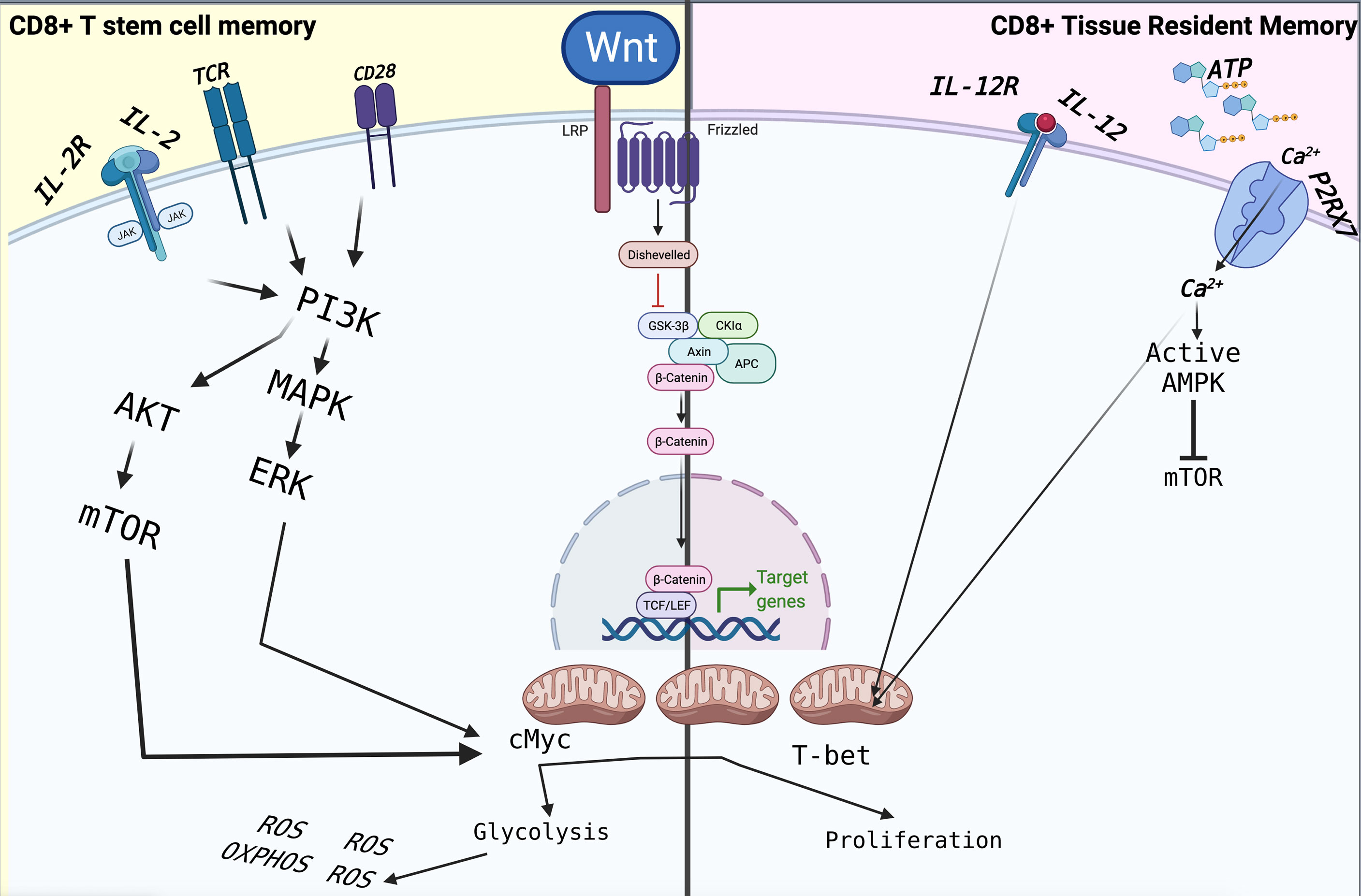
Figure 2 The immunometabolism of TSCM and TRM is driven by extracellular factors, which are mainly based on mitochondrial respiration through FAO for their homeostasis. In particular, the metabolism of TRMs is based on the detection of extracellular ATP mediated by P2XR7, which controls their function and their survival. Following stimulation through TCR and CD28 in the presence of IL-2 and IL-12, CD8+ T cells activate reprogramming, differentiating into effector cells that will lead to the elimination of the pathogen or tumour cell. The signalling pathways are those of MAPK and mTOR, which lead to cell proliferation assisted by intense metabolic activity such as glycolysis and OXPHOS. The latter leads to the increased production of ROS, which will induce terminal differentiation and ultimately apoptosis, limiting the overall antitumor effect of CD8+ T cells.
Physiologically, the generation of effector T cells and T cells with stem cell-like properties are linked. In fact, inhibition of lymphocyte differentiation to maintain long-lived, self-renewing antigen-experienced T cells with stem cell-like properties are strictly correlated. A Recent protocol to generate CD8+ TSCM is by oxidative metabolism modulation using antioxidants such as N-acetylcysteine (NAC) that significantly increases the intracellular antioxidant molecule Glutathione and consequently decreases the ROS intracellular levels (106). NAC may regulate cellular behaviour in many ways, but the impressive observation is an increased level of Carnitine palmitoyl transferase 1 alfa (CPT1α) and TCF1 expression. From this study, it is now clear that the expression of TCF1 is marked as self-renewing human CD8+ T cells (110); furthermore, CPT1 is a transmembrane enzyme of the mitochondrial outer membrane, which converts long-chain fatty acyl-CoA to long-chain acylcarnitine, following which carnitine acylcarnitine translocase, transports the fatty acid across the inner mitochondrial membrane and then enter fatty acid β-oxidation. Therefore, when naïve T cell use via s fatty acid for energy via OxPhos and decreases dependence on glycolysis, it starts the long-live memory generation. A confirmation for this statement is the study of Seok et al. that demonstrated CPT1 can increase fatty acid oxidation and reduce mitochondrial membrane potential (ΔΨm) (111), which is clearly one of the indispensable characteristics of TSCM. The induction of TCF1 and its downstream target ID3 and LEF1 genes are thought to be responsible for the enhanced long-term persistence of stem-like memory T cells. Furthermore, NAC might scavenge ROS levels because it reacts slowly with free radicals such as O2-, H2O2 and NO3- also, NAC decreases mTOR activity, which it explained as a master regulator of multiple metabolic processes, including glycolysis in T cells. As a result, and according to this explanation, the modulation of mTOR signalling by the simple addition of an inexpensive antioxidant such as NAC in the range of 10–20 mM can triggers the formation of TSCM (112).
An additional strategy in the protocol development to induce in vitro cells with TSCM properties is the activation of the Wnt/β-catenin pathway in naïve T cells stimulated through T cell receptor by inhibition of glycogen synthase kinase-3b (Gsk-3b) (107). While the Wnt/β-catenin pathway has been shown to play an essential role in various developmental processes such as asymmetric cell division, stem cell pluripotency, and cell fate specification (113–115), in addition, the Wnt/β-catenin pathway is clearly marked as the essential role of development of T lymphocytes in thymus. Indeed, the role of Wnt proteins in regulating mature T cell function has remained a relatively understudied area. As different Wnt signalling pathways have been described, but our focus is on the canonical Wnt pathway. Actually, Wnt signalling and β-catenin dependent gene expression play a critical role during the formation of different body regions in the early embryo. One of the most important results of Wnt signalling and the high level of β-catenin in certain cell types is the maintenance of stem cell pluripotency (116). Broadly, β-catenin is considered the centre of the Wnt signalling pathway. In addition, the transcriptional activity of the T cell factor (TCF) is induced by direct association with β-catenin (115, 117). However, the expression of β-catenin levels can be carefully modulated by Gsk-3b. The significantly effective way to rapidly accumulate β-catenin to almost 1.7 fold is inhibition of Gsk-3b by a molecule known as TWS119, which initiates the Wnt signalling pathway and controls TCF and LEF transcription factors (115) which acts as the main downstream effectors of the Wnt signalling pathway. The idea behind this protocol is that Wnt signalling functions as a mechanism in T cells to establish a pool of undifferentiated progenitor or stem cells that are more developed than naïve T cells for further differentiation or proliferation. Hence, such applications also require careful study of the regulation of WNT signalling in T cell development and fate. Indeed, small-molecule inhibitors and activators of WNT–β-catenin such as TWS119 are being developed for TSCM generation as therapeutic applications. Primary studies clearly showed that inhibition of GSK3β by 3 μM of TWS119 can arrest CD8+ T cell’s development into effector cells, consequently maintaining TSCM phenotype, which can be used as a new vaccination design strategies and adoptive immunotherapies (107). As we explained before, maintaining the stemness function of T cells is dependent on cell metabolism and using more fatty acids as energy via OxPhos by decreasing mTOR. Taken together, an existence of cross-talk between mTOR and Wnt signalling comes to mind, which still remains unclear. Furthermore, Scholz et al. revealed that induction of TSCM is utterly independent from the Wnt signalling pathway (118), which has already been called into question as to the molecular mechanism of TWS119, but there is confirming evidence in mouse T cells that TWS119 inhibits mTORC1 (119). In any case, inhibition of mTORC1 can switch the metabolic program of activated naïve T cells toward an oxidative metabolism dependent on FAO, but in vitro experiment with TWS119 showed that all naïve T cells do not follow equally this issue (119). A point that needs to be addressed is how a cell’s intrinsic factors can stop differentiation upon mTORC1 inhibition. One of the important clues might be Krüppel-like-factor 2 (KLF2), which has been shown to maintain the expression of CCR7 and CD62L and has been suggested to be up-regulated upon mTORC1 inhibition (85, 120). Interestingly, KLF2 can positively regulate the expression of GSK3β, GATA6 and FOXP3 based on STRING gene interaction data. In addition, the low expression of KLF2 in naive T cells can make them a TSCM cell precursor potential (118).
These two investigations reveal that gene expression differs significantly between in vitro these two well-developed in vitro induced-TSCM protocols;in vitro TWS119-induced TSCM demonstrated an upregulation of LAMP3 (CD63), providing them with a robust co-stimulatory signal, as well as a remarkable elevation of interferon responsive gene expression such as IFIT2, IFIT3, IFI6, and IFI27 Interestingly, mTOR signalling modulated induced-TSCM, resulting in the upregulation of genes with cell metabolism control functions such as NQO1, TXNRD1, which can protect cells from oxidative stress and toxic quinones and may be linked to increased oxidative phosphorylation and fatty acid oxidation (118).
The Role of Long-Term Memory Cells During Chronic Infections
During past years, the role of pathogen-specific TSCM cells in human acute and chronic infections caused by viruses, bacteria, and parasites has been increasingly considered. However, the TSCM development and formation mechanisms during natural immune responses against foreign pathogens remain poorly understood. The critical point of the robust immune response against pathogens is to have effectors not exhausted T cells; this requires continuously replenishment by less differentiated T cell subsets such as TSCM (60, 62, 121, 122). TSCM has recently been linked to a variety of disorders, including autoimmune diseases such as systemic lupus erythematosus (SLE) (123) and infectious diseases (124) and human T cell leukaemia (125). Therefore those subtype of T cells is also taken into account for tumour immunotherapy (126, 127) and allogeneic hematopoietic stem cell transplantation (31, 32).
Longevity and stemness function of TSCM can act as a double-edged sword as protective or pathogenic, as the evidence shows disrupting TSCM cell reservoirs in retroviral infections such as CD4+ TSCM or in autoimmune diseases such as aplastic anaemia, rheumatoid arthritis, type I diabetes and SLE, might be a target in diseases controlling strategy. On the other hand, the protective role of TSCM in acute and chronic infections makes them optimal candidates for therapeutic exploitation in vaccination and adoptive T cell therapy against infectious diseases and cancer. One of the best examples of the double-edged role of TSCM in human infectious diseases is controlling HIV or developing AIDS.
However, there is increasing evidence demonstrating that TSCM cells play a crucial role in HIV as a reservoir regardless of successful antiretroviral therapy (ART) (128–131), but immunometabolism data and stem cell-specific pathways can play a very important role in virus resistance since hematopoietic stem cells seem to be largely resistant to HIV-1. Hence, this theory cannot be far from the idea that at least a part of TSCM could have defence mechanisms protecting this cell subset from retroviral infection. A small amount of TSCM showed low expression of CCR5, which can be protected from HIV infection, but physiologically CD4+ TSCM expresses CCR5 (R5)- and CXCR4 (X4), which make them usual reservoirs for HIV, because silent or latent infection of these long-lived, self-renewing cells could provide an extremely stable niche for the virus which is modulated by SAMHD1 expression (132). Accordingly, it would need to be targeted TSCM in efforts to eradicate the virus that is appealing for many years after the discovery of TSCM as the stem cells of cellular immune memory from controlling this subset by Wnt-β-catenin to CRISPR-edited stem cells technology (133, 134).
Up to now, the role of HIV-1-specific CD8+ TSCM in immune defence is uncertain even if there is evidence that HIV-1-specific TSCM may contribute to HIV-1 restriction and naturally control the infection. Therefore, the proportions of total CD8+ TSCM in untreated HIV patients are inversely correlated with plasma viremia levels (135). As mentioned earlier, cell immunometabolism is fundamental for CD8+ memory formation. Surprisingly, a recent study found that HIV patients who could naturally control HIV infection had HIV-specific CD8+ T imprinted in their memory program, which is associated with their ability to use diverse metabolic resources; in contrast, poor functionality of HIV-specific CD8+ was associated with reliance on glycolysis as a primary energy source (136). In addition, this dependency is in contrast whit memory formation with stem cell function and development. Mueller, et al. could convincingly establish that IL15 can enhance the survival and function of HIV specific CD8+ T cells by increasing mitochondrial biogenesis (137), which we considered as a fait decision factor for TSCM formation. Taken together, those evidence unveil the importance of metabolic activity regulation and HIV-specific CD8+ TSCM formation and consequently induce HIV remission and provide a target for therapeutic vaccine development.
Inducing a long-lasting stem cell-like memory CD8+ T cell is not only for protecting human from an infection but also could be a novel target to inducing long-term memory, as yellow fever vaccination evidence shows rapid detection of YF-specific CD8+ T cells, but frequency of effector cells decreasing with time while YF-specific CD8+ TSCM maintained for more than 25 years and was capable of self-renewal ex vivo. Additionally, 17 years after vaccination, TSCM can be prominently distinguished based on their marker maintenance, such as CD58, CD95, CXCR3, KLRG1, CD11a+ high, IL-18Rα, granzyme A and IL-2Rβ (138). Accordingly, TSCM thought to be a pivotal T cell subset that have different qualities in terms of effector function, proliferative capacity as well as immune reconstitution and memory formation and it is believed that a TSCM net production can enhance adoptive immunotherapy which even very low numbers of T cells from the memory stem cell pool such as TCMs or TSCMs can reconstitute robust and long term maintained immune responses (11, 139). Given the TSCM role, a major challenge in vaccine development is determining the relative importance of TSCM cells and defining if effective vaccines should induce antigen-specific TSCM for maximal protection.
TSCM and Autoimmune Diseases
TSCM cells provide long-term protective immunity for anti-tumour immunity, which is likely based on responsiveness to self-antigens; therefore, their involvement in autoimmunity is unavoidable (102, 140). The study of Hosokawa et al. showed that CD8+ TSCM cells played an essential role in the pathogenetic mechanism of aplastic anaemia (AA) and were expected to become a new therapeutic direction for this disease (141). An increased CD8+ TSCMs frequency at diagnosis is associated with responsiveness to immunosuppressive therapy, and an elevated CD8+ TSCMs population after immunosuppressive therapy correlates with treatment failure or relapse in AA patients.
A positive correlation between the frequencies of TSCM CD4+ and CD8+ cells is seen in AA, autoimmune uveitis and systemic lupus erythematosus (SLE) (141). SLE, a chronic, connective tissue multi-organ disorder that affects young women. Compared to healthy controls, there was a significant increase in the percentage of CD4+ and CD8+ TSCM cells in SLE patients. T Follicular helper cells (TFH) increase antibodies produced by B cells, and TSCM cells play a role in SLE pathogenesis by maintaining TFH cells (142).
Immune thrombocytopenia (ITP), an acquired autoimmune haemorrhagic disease, represents about 30% of all haemorrhagic diseases (143). Many researchers have conducted extensive studies on the pathogenic mechanisms of ITP. At present, there is growing evidence that abnormal cellular immunity plays a significant role in the development of IPT. Very recently, Cao et al., showed that the percentage of CD8+ TSCMs in peripheral blood before treatment in ITP patients was significantly higher than that in healthy controls, although some data overlapped in the two groups, whereas the percentages of the other T cell subsets did not exhibit significant differences (143).
Considering that TSCMs can generate all memory and effector T cells, the authors speculated that they could cause the progression of autoimmune diseases. Moreover, the variation in the percentage of TSCMs CD8+ is correlated with the effectiveness of the treatment. The percentage of CD8+ TSCM in peripheral blood of ITP patients was significantly reduced after glucocorticoid treatment, indicating that the imbalance of the ratio of CD8+ TSCM may be involved in the occurrence and development of ITP (143).
Type I diabetes (T1D) results from chronic autoimmune destruction of insulin-producing β-cells mediated by self-reactive T-cells. Many β-cell autoantigens are targets for autoimmunity. Among these, GAD65, (pro)insulin and the block-specific glucose-6-phosphatase subunit catalytic protein (IGRP) appear to be highly antigenic in man (144). The presence of a memory response to islet autoantigens may be related to the limited success of immunotherapies tested to date in T1D patients. Memory autoimmunity is highly resistant to modulation with immunosuppressant drugs and immunomodulatory molecules (145, 146), primarily designed to target activated and fast-proliferating T-effector cells. These raised the question of whether self-reactive and long-lasting TSCM precursors can serve as a reservoir of self-reactive clones in post-treatment autoimmune relapses (147). Identifying specific circulating TSCM for GAD65, insulin, and IGRP supports this assumption and provides a new target cell population to design novel immunotherapy approaches. Selective targeting of self-reactive TSCM clones may be used to suppress more differentiated T-cells permanently.
Further efforts are needed to clarify changes in health and disease stages among the different TSCM cell states.
General Background of TRM Cells
TRM cells are an essential part of the immune survey system, sensing alteration in homeostasis throughout the body and leading immune surveillance of most non-lymphoid tissues (148–151). TRM cells are involved in protection from infection and cancer, but also likely promoting autoimmunity, allergy, and inflammatory diseases and impede successful transplantation. The residence signature that marks TRM cells in several tissues is characterized by both increased expression of proteins that promote tissue retention and reduced expression of proteins involved in the recirculation in the blood and lymphatic vessels.
For instance, TRM cells exhibit reduced expression of the surface molecules such as S1PR1 and CCR7 cells, which promote T cells to leave the NLT, an observation that is explained by a decreased expression of the transcription factor KLF2, which leads to transcription of S1PR1 and CCR7 (152). On the other hand, TRM cells express CD69 and, in the case of TRM cells located in epithelial tissues, the E-cadherin αE integrin binding (CD103, encoded by ITGAE gene), which both promote tissue retention (29).
TRM cells as all the memory T cells express CD44 and lack CD62L, while TRM cells are exclusively CD44hi and CD62Llow they can display several tissue-specific markers, so that no single marker can unique identify a TRM population in all tissues.
Thus, the expression of CD69 and CD103 should be considered an imperfect marker for inferring tissue residence because the absence of their expression does not exclude the potential to leave NLTs (151, 153–156).
CD103, CD49a (VLA-1) and CD69 are the most commonly utilized markers to distinguish TRM cells from circulating memory cells (157). CD103 is a ligand for E-cadherin, widely expressed in the junctions between epithelial cells at barrier sites (158). CD49a is an integrin ligand for collagen IV which is located in the lamina dense of barriers tissues (159). Moreover, CD49a has an anti-apoptotic function for the collagen bounding T lymphocytes (160). CD69 is an S1P1 antagonist, countering signals for lymphocyte egression and circulation (161, 162). CD69 is expressed by the majority of TRM, CD103 is mainly expressed primarily by CD8+ TRM cells (163). Many other cell surface markers are associated with TRM cells, including CXCR6, CD101, PD-1, and CX3CR1.
Nevertheless, a large part of our current understanding of TRM cells relies on analyses of CD69+CD103+ TRM cells in epithelial tissues.
Role of TRM in Chronic Infectious Diseases
Consistent with their role as local sentinels, it has been demonstrated that TRM cells both prevent and exacerbate pathologies. For example, TRM cells protect local recurrent pathogens (148, 164) but these cells can also provide protection against the development of skin malignancies (165–167).
TRM cells can lead to immunotherapy-induced colitis (168), autoimmune disorders of the skin vitiligo (169) and psoriasis (170, 171) as well as other autoimmune and allergic diseases, and can play a central role in allograft rejection (172).
TRM constantly perform surveillance of the cells surrounding them and intimately interact with the components of their environment including epithelial cells and the extracellular matrix. For this reason, TRM are among the first line of defence and are early activated by the encountering of antigens. The involvement of TRM cells in a series of human diseases makes the design of therapeutic strategies that can modulate both their production and their activity an attractive objective, and to realize this goal, understanding how this cell pool is formed is essential (173).
While studies on the role of TRMs in cancer are very extensive, the role of TRMs in the context of chronic infections is less explored. In general, there is strong evidence that chronic tissue-specific infection is associated with a non-functional alteration of the TRM population evidenced by an exhausted phenotype. However, even in this context some studies have highlighted the possibility of using this subset for vaccination or therapeutic purposes against infectious diseases such as HIV and tuberculosis.
For instance, a recent study showed that in HIV patients, CD8+ T cells HIV specific with a TRM phenotype are abundant in HIV infected lymph nodes compared to peripheral blood. These CD8+ TRM cells show different specificity toward different HIV antigens, and this finding results in a different clonotype observed in different HIV infected lymph nodes (155).
The presence of HIV specific TRM cells in lymph nodes of HIV infected subjects makes these cells suitable targets of a vaccine against HIV. A recent study on murine model based on subcutaneous microneedles delivery array (MA) vaccination with recombinant E1, E3 deleted human adenovirus type 5 (AdHu5) vector encoding HIV-1 CN54gag (AdHu5-CN54gag), induced in the skin the a subset of specific memory T CD8+ cells with a TRM phenotype (CXCR3+, CD103+, CD49a+, CD69+) and a polyfunctional profile (174).
In a recent study on the tuberculosis pleural effusion (TPE), the authors found a heterogeneous population of T cells in which CD103+CD8+ T cells were the predominant subset of CD103+ lymphocytes. Moreover, the frequency of CD103+ CD69+ memory CD8+ T cells in TPE was significantly higher than the frequency of these cells in peripheral blood. Altogether, CD103 and CD69 expressions define in TPE distinct CD8+ TRM -like subsets with different phenotypes and functions (175).
The consistent presence of TRM in lungs focused the attention of many scientists on these cells as a target for vaccine or host direct therapy against Mycobacterium tuberculosis infection (Mtb).
Moreover, different studies on possible mucosal TB vaccines against TB have been carried out on mouse models, revealing the marked presence of both CD8+ and CD4+ TRM in the lungs. In a study on murine models, the intranasal boosting with a Sendai virus vectored tuberculosis vaccine, SeV85AB, induced antigen-specific CD103+CD8+ T-cell protective response in the lung, associated with a reduction of mycobacterial burden both in lung and spleen compared to subcutaneous BCG alone vaccination (176). Nanoparticles (NPs) are an emerging vaccine technology, with recent oncology and infectious diseases successes. Hart et al., have associated Mtb control by NPs due to the enhanced cellular immunity compared to BCG, including superior CD4+ and CD8+ T cell proliferation, TRM seeding in the lungs, and cytokine polyfunctionality (177). In another study, Bacillus subtilis spores coated with a fusion protein 1 (“FP1”) consisting of Mtb antigens Ag85B, ACR, and HBHA have been tested in a murine low-dose Mtb aerosol challenge model (178). This vaccination model demonstrated an enhanced pulmonary control of Mtb, as evidenced by reduced bacterial burdens in the lungs. Spore-FP1 immunization generated superior antigen-specific memory T-cell proliferation in both CD4+ and CD8+ compartments. Moreover, CD69+CD103+ TRM cells were found within the lung parenchyma after mucosal immunization, confirming the advantages of mucosal delivery (178). Finally, liposome-based subunit vaccine formulation of phosphatidylserine encapsulating, Mtb antigens Ag85B, and ESAT-6, given mucosally, could generate TRM cells, that have been found in the lung parenchyma that are likely to be antigen specific, even if no formal evidence of their antigen specificity was demonstrated (179).
The administration of BCG by intranasal rote elicited the generation of both CD8+ CD69+CD103+ CXCR3+ TRMs, able to improve the protection against Mtb challenge when compared to the simple administration of subcutaneous BCG (180).
Adoptive transfer of these cells to naive, TCRα−/− transgenic mice, prior to Mtb infection, demonstrated parenchymal T-cells, which expressed the TRM CXCR3+KLRG1− phenotype, had far greater activity than circulating T-cells (18-fold vs 4-fold reduction in lung bacterial burden) (181).
An interesting finding regarding TRM cells in the tuberculosis murine model is the expression of PD-1, which is a key molecule to modulate the IFN-γ production and thus, prevent lethal lung disease. This finding was assessed by a recent study showing the generation of protective KLRG1−PD1+ TRM cells in lung after mucosal vaccination with BCG, but not after intradermal BCG administration (182).
TRM Generation and Homeostasis in the Barrier and Mucosal Tissues
TRM cells are a dedicated population of T cells that continuously recirculate through tissues or whether they can permanently reside and show decisive functions, organizing local protective immune responses. This migration, a feature of classical memory T cell subsets, is the basis of the differential expression of CCR7 (22). Once the pathogen is encountered, it expands in situ, acquiring marked cytotoxic activities, promoting the formation of a tissue antiviral environment and releasing numerous cytokines (183, 184). Indeed, TRM cells’ maturation foresees the encounter with the antigen at the tissue level resulting in the downregulation of CCR7 in the activated cells, and this process is a prerequisite for the formation of TRM (185–187). Furthermore, a study on a mouse model, based on the intranasal or intramuscular administration of CpG and nucleoprotein peptides (NP) from influenza A/HKx31 (H3N2) virus, showed the formation of NP-specific TRM in the respiratory tract tissues only in mice that had had intranasal administration of the NP (188).
Those functions generally require fundamental immunometabolism changes, metabolic homoeostasis, and remodelling in T cells during life and ageing. All of this remains unexplored, but it is now clear that during humans ageing, progressive deterioration leads to impaired function and continual decline that results in an increased risk of disease and death (189); as an example, a recent study clearly shows how over the years there is a variation in the passage of immune cells in the lungs and how these cells respond specifically against classic influenza and SARS-CoV-2 virus (190). In fact, most immune cells in human lungs remain stable with age, unlike memory CD8+ T cells that decrease, particularly the TRM cells (191, 192). As seen in Nguyen et al. studies, a subgroup of CD103+ and CD69+ TRM cells in the lungs of donors under the age of 50 accounts for about 48% of the total memory CD8+ T cells, but in subjects over 50 years of age, it represents only 25% of CD8+ memory T cells (190).
These observations can be realized through ageing and T cell immunometabolism; for example, ageing is associated with reduced TCR diversity (193) and shifting from naïve to memory subset, and these phenotypic changes result in an overall compromised response to new antigens. An example to prove this fact is that the overall frequency of influenza-specific CD8+ pulmonary memory T lymphocytes does not show variations with the donor’s age, while the subgroup of influenza-specific TRM lymphocytes with phenotype CD103+, CD69+ decreases with age. This decrease coincides with the reciprocal increase in the CD103- and CD69- population of CD8+ TRM T lymphocytes, influencing the quality of the early inflammatory response following exposure to the virus (190).
Homoeostasis and remodelling in T cells during life and ageing leads to a marked susceptibility of the elderly to the most serious forms of virus infection. For example, in influenza, specific CD8+ TRM T cells in the lung rapidly acquire proinflammatory effector functions directly in situ, thus being decisive in fighting a viral infection (194, 195).
The Role of Metabolic Pathway of TRM in Infectious Diseases
Barrier tissues and their microenvironments have different trophic and pH conditions that TRM cells should face. Moreover, the cell populations, the extracellular matrix, and the collagen type are different. For this reason, TRM cells can exploit diverse sources of energy by different metabolic pathways.
Metabolic reprogramming and metabolism changes are associated with adapting specific T cell subunits for survival and cell function, supporting cell function through energy supply and biosynthetic precursors. For example, cellular and transcriptional changes during an infection are responsible for developing different memory CD8+ T cells and antigen-specific CD8+ T cells. In addition, many changes can control CD8+ T cell metabolism in response to infection, driven by extracellular metabolites, such as transcriptional, translational, and epigenetic changes.
During acute infection, one of the most abundant CD8+ TRM cells become embedded in lymphoid and non-lymphoid peripheral tissues (151, 196). However, after pathogen clearance, only a few T cells remain as long-lived memory CD8+ T cells such as TRM or TSCM, which are critically dependent on cellular metabolism to regulate CD8+ T cell differentiation and memory formation (197–200). For example, an essential requirement for the differentiation of TRM cells and TCM CD8+ T cells is purinergic receptor P2RX7 (201), which polymorphism on this gene plays a role in susceptibility to TB in the Asian population (202). In fact, non-lethal P2XR7 maintains mitochondrial homeostasis and TGF-β dependent programs of tissue residency by increasing AMPK activation compared with other tissue CD8+ T cells. CD8+ TRM appears to possess a distinct functional and metabolic profile that can depend on their environment and adapt to it. CD8+ TRM maintains long-term survival by using FAs which closely depend on extracellular factors such as extracellular ATP, which could influence the TRM metabolism and expression of CD69 and CD103 (201).
As an example, P2XR7 can affect the mTOR pathway to switching from glycolysis to OXPHOS (203). Unlike circulating memory CD8+ T that mainly depends on glycolysis to respond to pathogenic antigen (80), TRM depends on the uptake of exogenous FAs for subsequent FAO engagement and subsequently mitochondrial membrane potential and fatty acid metabolism for their homeostasis. Many microenvironment factors such as FA-rich sites, extracellular ATP (eATP) and many receptors can affect gene upregulation which eventually changes effector function and lipid metabolism on TRM. For example, the low-affinity P2RX7 receptor to eATP promotes the NLRP3 inflammasome activation (204). In contrast, P2RX7 can promote CD8+ TRM cell subsets through mitochondrial health (205) (Figure 2). Moreover, due to increased infected cell death or gene expression upregulation to change effector function in infection sites, the extracellular NAD+ concentration increases, promoting CD8+ TRM cell death by P2RX7 (206).
Another pathway used by TRMs exploits free fatty acid (FFA). TRM cells showed a high expression of the fatty acid-binding receptors FABP4, FABP5, low-density lipid receptor, ApoE, and the CD36 scavenger receptor. These molecules are useful to enhance lipolysis for rapid exploiting the direct pathway of FFA uptake (45). The FFA can be used in oxidative phosphorylation and in FAO (45, 207). Thus, it is likely that TRM can switch their metabolic pathway from oxidative phosphorylation and glycolysis to FAO after antigen encounter and subsequent activation.
The master transcriptional regulators of TRM cells differentiation in different tissues are Hobit, Blimp1 (163), and Runx3 (208) at the transcriptomic level. The first two transcriptional factors are negative regulators of tissue egresses (209) and are involved in the TRM cells cytotoxic capability (210). Runx3 enhances the expression of CD103 and CD69 tissue retention molecules (208).
In conclusion, the fate decision by metabolism change in TRM cells remains largely unknown. Those cells could be a fascinating target not only for vaccination efficiency or controlling infection but also in tumour and autoimmune tissue disorders due to the double-edged sword character. Given this content, understanding in detail metabolic reprogramming to possible manipulation to enhance or decrease TRM longevity and function is crucial.
TRM Cells and Autoimmune Disease
TRM cells have been shown to play a role in autoimmune diseases, especially autoimmune skin diseases. In vitiligo, it showed the presence of TRM cells in the epidermis and dermis of lesioned areas. These cells expressed CXCR3, whose binding with its ligands CXCL9 and CXCL10 drive the production of IFN-γ, which represents an effector cytokine of this pathology. Moreover, these TRM cells produced high quantities of IFN-γ and GRZB after in vitro stimulation (211). The phenotype of vitiligo associated with TRM cells also displays the α chain of IL- 15 receptor and CD122, which play an essential role in the pathological activity of TRM; in fact, the blocking of CD122 by monoclonal antibody leads to the repigmentation of vitiligo areas in an experimental mouse model (169).
Like vitiligo, psoriasis is an autoimmune skin disease, where IL-17 sustains its pathogenesis. CD8+ TRM cells from psoriatic lesions showed IL-17 production after in vitro restimulation. These cells display a specific phenotype (CD103+ CD49a- CCR6+) characterized by the expression of IL23R and the absence of CXCR3 (211, 212). Several studies reported the involvement of TRM cells in other autoimmune diseases. CD8+ TRM cells specific for astrocytes-expressed protein play a crucial role in multiple sclerosis (MS) pathogenesis in a mouse model (213, 214). Moreover, CD103+ cells were detected in the high inflammatory infiltrate in the lesioned central nervous system (CNS) zones of MS patients (215). But their role remains controversial as these cells, even if stimulated, do not produce cytokines (214). Another study reported the presence of IFN-γ, IL-18, and IL-22 producing CD8+ cells in lesioned Langerhans islet in TD1 patients (216).
Several factors should be considered about the mechanisms that trigger the pathological activation of CD8+ TRM cells in the tissues associated with autoimmune disease. For skin diseases, keratinocytes, dendritic cells, and fibroblasts, after stimulation, can determine the production of IL-7, IL-15, and IL-17 that can trigger CD8+ TRM cells’ activation and enhance their proliferation and the maintenance of a stable pool of activated cells (183). CD8+ TRM cells in response to IL-7 and IL-15 can rapidly be activated by the JACK/STAT5 pathway, which profoundly influences the formation of long-lived memory T cells to enhance the anti-apoptotic BCL-2 molecule (217–219). Also, the PI3K/AKT pathway activation is associated with long-lived memory T cells (217, 220, 221). An appropriate balance of the signals from these two pathways is critical either in activated CD8+ TRM cells or their survival (222).
It was demonstrated that cross-talk between Treg and TRM cells is linked to the ability of Treg to make TGF-β available for the TRM formation and corroborated by similar expression of CXCR3, which allows both cell types to migrate toward the site of infection and inflammation (223, 224). The role of Treg can prevent pathological activation of TRM cells. Nevertheless, it happens that impaired function of Treg in the skin leads to the formation of TRM cells with autoimmune activity. To confirm this hypothesis, it was observed that the checkpoint inhibitor therapy used in patients with melanoma is associated with an increase in vitiligo disease (169, 225).
About metabolic pathways used by TRM cells during their activation in autoimmune diseases, skin TRM cells can exploit FFA and mitochondrial oxidative metabolism. Together with the fact that skin autoimmune disorders are often concomitant with metabolic diseases, these findings suggest that the metabolic disorders determine a more likely uncontrolled activation of CD8+ TRM cells due to an enhanced FFA metabolism. Evident lipid accumulation in droplets (45) and high expression of fatty-acid binding proteins (FABPs) in TRM cells (45, 226) determine the higher expression of transcripts of enzymes involved in lipid metabolism, which supports this hypothesis. In conclusion, the presence of dysfunctional CD8+ TRM cells in lesions caused by autoimmune diseases makes the TRM cells subset an important player in the onset and chronic assessment of these diseases and, at the same time, a promising target for new therapeutic strategies in autoimmune diseases.
T Cell Memory Subsets in SARS-COV-2 Infection or Vaccination
Within the T cell memory compartment, TRM and TSCM CD8+ T cells play a different role in SARS-COV-2 infection or vaccination. The first subset has been detected in the respiratory tract following coronavirus infection, where they employ a protective function (227). Moreover, activated profiles of lung CD8+ and CD4+ TRM cells were reported in COVID-19 patients, and these cells were frequently detected even ten months after infection (228, 229). Recovered patients acquire TRM cells with Th1 phenotype against COVID-19 (229). It was shown that, although resident T cells are elicited by COVID-19 infection, they do not present sufficient protection against secondary viral infection (230). On the other side, CD8+ TRM cells can significantly contribute to lung damage/pathology development, determined by the prolonged activities after respiratory viral infection (231). In fact, lung CD103− CD69+ T cells of older patients with COVID-19 produce elevated inflammatory molecules associated with worse lung pathology and reduced lung function (232). It has been found that respiratory CD103- CD69+ T cells exacerbate lung pathology and reduce lung function (232). Therefore, several therapeutic strategies have been developed to target the TRM cells. A recent study has evaluated the role of Tofacitinib (the FDA approved oral JAK2/1/3 inhibitor) targeting the tissue memory CD8+ T cells, which may be an effective therapy against chronic lung diseases promoted by acute SARS-CoV-2 infection (233). SARS-CoV-2 specific memory TSCM cell responses are induced after recovery from SARS-CoV-2 infection or vaccination, according to increasing data. After ten months of infection, Jung et al. discovered a successful generation of TSCM cells in convalescent COVID-19 patients (234). Another study clearly showed that an antigen-specific population of TSCM is observed even after six months of vaccination with BNT162b2 vaccine, which provides a multipotent memory reservoir (235). It was recently revealed that stimulation of T cells shortly after BNT162b2 vaccination with spike epitopes leads to rapid production of TNF-α, which was independent of the neutralizing antibodies’ titer (236). The production of TNF-α, even within a few hours after TCR-stimulation, is most characteristic of naive-like CD8+ T cells, similar to TSCM (237). The high production of TNF-α is not always related to a good immunological response, due to its involvement in several inflammatory diseases or the severity of long COVID patients (238–240). In conclusion, TRM and TSCM cells represent a double-edge sward in SARS-Cov-2 infection, and further studies are needed in order to delineate their role better.
Concluded Remarks
The recent application of high-level single-cell technologies and epigenetic profiling approaches has revealed that the stem and the effector properties of CD8+ T cells are no longer reciprocal. Transcriptional programming that equips a T cell with a multipotent development capability can coexist with effector programs and establishes a population of long-lived memory T cells that can respond against a previously exposed pathogen. Recent studies investigating these properties in T cell responses to chronic antigen sources illustrate an essential role of these coexisting characteristics in establishing therapeutic modalities based on selected subsets of T lymphocytes. The great interest in studying the impact of the metabolic activity of pathways in immune cells has opened a new window in understanding the requirements of metabolic pathways for differentiation/maintenance of CD8+ T cells, which can lead to the discovery of molecular targets capable of developing new anti-infective and/or anti-inflammatory drugs applicable to different contexts (241–244).
The outstanding issues surrounding these fundamental properties of T cells are of great translational importance and represent a question of intense investigation. These processes will likely provide more information on the cellular and molecular mechanisms of memory T-cell differentiation and exhaustion in acute and chronic pathogenic challenges, allowing the development of new therapeutic approaches that use the exquisite specificity and longevity of T cells.
Author Contributions
Conceptualization and Supervision, FD, NC; Writing-Review & Editing, BT MPLM, MSA, LM, GDB; Writing-Original Draft Preparation, NC, FD, MPLM; BT, MSA, LM; Figure preparation: MPLM, BT, GDB, MSA, LM. All authors have read and agreed to the submitted version of the manuscript.
Funding
This work was supported by grants from the Horizon n2020 Programmes EMI-TB (contract no. 643558). The text represents the authors’ views and does not necessarily represents position of the European Commission, which will not be liable for the use made of such information.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, et al. Lineage Tracking Reveals Dynamic Relationships of T Cells in Colorectal Cancer. Nature (2018) 564(7735):268–72. doi: 10.1038/s41586-018-0694-x
2. Farh KK-H, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and Epigenetic Fine Mapping of Causal Autoimmune Disease Variants. Nature (2015) 518(7539):337–43. doi: 10.1038/nature13835
3. Farber DL, Yudanin NA, Restifo NP. Human Memory T Cells: Generation, Compartmentalization and Homeostasis. Nat Rev Immunol (2014) 14(1):24–35. doi: 10.1038/nri3567
4. Hu G, Cui K, Fang D, Hirose S, Wang X, Wangsa D, et al. Transformation of Accessible Chromatin and 3D Nucleome Underlies Lineage Commitment of Early T Cells. Immunity (2018) 48(2):227–42. doi: 10.1016/j.immuni.2018.01.013
5. Qin P, Pang Y, Hou W, Fu R, Zhang Y, Wang X, et al. Integrated Decoding Hematopoiesis and Leukemogenesis Using Single-Cell Sequencing and its Medical Implication. Cell Discov (2021) 7(1):1–17. doi: 10.1038/s41421-020-00223-4
6. Gattinoni L, Speiser DE, Lichterfeld M, Bonini C. T Memory Stem Cells in Health and Disease. Nat Med (2017) 23(1):18–27. doi: 10.1038/nm.4241
7. Lugli E, Galletti G, Boi SK, Youngblood BA. Stem, Effector, and Hybrid States of Memory CD8+ T Cells. Trends Immunol (2020) 41(1):17–28. doi: 10.1016/j.it.2019.11.004
8. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A Human Memory T Cell Subset With Stem Cell–Like Properties. Nat Med (2011) 17(10):1290–7. doi: 10.1038/nm.2446
9. Biasco L, Scala S, Basso Ricci L, Dionisio F, Baricordi C, Calabria A, et al. In Vivo Tracking of T Cells in Humans Unveils Decade-Long Survival and Activity of Genetically Modified T Memory Stem Cells. Sci Trans Med (2015) 7(273):273ra13–ra13. doi: 10.1126/scitranslmed.3010314
10. Mahnke YD, Brodie TM, Sallusto F, Roederer M, Lugli E. The Who’s Who of T-Cell Differentiation: Human Memory T-Cell Subsets. Eur J Immunol (2013) 43(11):2797–809. doi: 10.1002/eji.201343751
11. Graef P, Buchholz VR, Stemberger C, Flossdorf M, Henkel L, Schiemann M, et al. Serial Transfer of Single-Cell-Derived Immunocompetence Reveals Stemness of CD8(+) Central Memory T Cells. Immunity (2014) 41(1):116–26. doi: 10.1016/j.immuni.2014.05.018
12. Zhu J, Paul WE. CD4 T Cells: Fates, Functions, and Faults. Blood J Am Soc Hematol (2008) 112(5):1557–69. doi: 10.1182/blood-2008-05-078154
13. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two Types of Murine Helper T Cell Clone. I. Definition According to Profiles of Lymphokine Activities and Secreted Proteins. J Immunol (1986) 136(7):2348–57.
14. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic Self-Tolerance Maintained by Activated T Cells Expressing IL-2 Receptor Alpha-Chains (CD25). Breakdown of a Single Mechanism of Self-Tolerance Causes Various Autoimmune Diseases. J Immunol (1995) 155(3):1151–64.
15. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 Programs the Development and Function of CD4+ CD25+ Regulatory T Cells. Nat Immunol (2003) 4(4):330–6. doi: 10.1038/ni904
16. Suni MA, Ghanekar SA, Houck DW, Maecker HT, Wormsley SB, Picker LJ, et al. CD4+ CD8dim T Lymphocytes Exhibit Enhanced Cytokine Expression, Proliferation and Cytotoxic Activity in Response to HCMV and HIV-1 Antigens. Eur J Immunol (2001) 31(8):2512–20. doi: 10.1002/1521-4141(200108)31:8<2512::AID-IMMU2512>3.0.CO;2-M
17. Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, et al. Effector and Memory CD8+ T Cell Fate Coupled by T-Bet and Eomesodermin. Nat Immunol (2005) 6(12):1236–44. doi: 10.1038/ni1268
18. Sallusto F, Geginat J, Lanzavecchia A. Central Memory and Effector Memory T Cell Subsets: Function, Generation, and Maintenance. Annu Rev Immunol (2004) 22:745–63. doi: 10.1146/annurev.immunol.22.012703.104702
19. Masopust D, Vezys V, Marzo AL, Lefrançois L. Preferential Localization of Effector Memory Cells in Nonlymphoid Tissue. Science (2001) 291(5512):2413–7. doi: 10.1126/science.1058867
20. Lugli E, Dominguez MH, Gattinoni L, Chattopadhyay PK, Bolton DL, Song K, et al. Superior T Memory Stem Cell Persistence Supports Long-Lived T Cell Memory. J Clin Invest (2013) 123(2):594–9. doi: 10.1172/JCI66327
21. Pitcher CJ, Hagen SI, Walker JM, Lum R, Mitchell BL, Maino VC, et al. Development and Homeostasis of T Cell Memory in Rhesus Macaque. J Immunol (2002) 168(1):29–43. doi: 10.4049/jimmunol.168.1.29
22. Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two Subsets of Memory T Lymphocytes With Distinct Homing Potentials and Effector Functions. Nature (1999) 401(6754):708–12. doi: 10.1038/44385
23. Thome JJC, Yudanin N, Ohmura Y, Kubota M, Grinshpun B, Sathaliyawala T, et al. Spatial Map of Human T Cell Compartmentalization and Maintenance Over Decades of Life. Cell (2014) 159(4):814–28. doi: 10.1016/j.cell.2014.10.026
24. Olson JA, McDonald-Hyman C, Jameson SC, Hamilton SE. Effector-Like CD8+ T Cells in the Memory Population Mediate Potent Protective Immunity. Immunity (2013) 38(6):1250–60. doi: 10.1016/j.immuni.2013.05.009
25. Gerlach C, Moseman EA, Loughhead SM, Alvarez D, Zwijnenburg AJ, Waanders L, et al. The Chemokine Receptor CX3CR1 Defines Three Antigen-Experienced CD8 T Cell Subsets With Distinct Roles in Immune Surveillance and Homeostasis. Immunity (2016) 45(6):1270–84. doi: 10.1016/j.immuni.2016.10.018
26. Osborn JF, Mooster JL, Hobbs SJ, Munks MW, Barry C, Harty JT, et al. Enzymatic Synthesis of Core 2 O-Glycans Governs the Tissue-Trafficking Potential of Memory CD8+ T Cells. Sci Immunol (2017) 2(16):eaan6049. doi: 10.1126/sciimmunol.aan6049
27. Buggert M, Vella LA, Nguyen S, Wu VH, Chen Z, Sekine T, et al. The Identity of Human Tissue-Emigrant CD8+ T Cells. Cell (2020) 183(7):1946–61. doi: 10.1016/j.cell.2020.11.019
28. Schenkel JM, Masopust D. Tissue-Resident Memory T Cells. Immunity (2014) 41(6):886–97. doi: 10.1016/j.immuni.2014.12.007
29. Mueller SN, Mackay LK. Tissue-Resident Memory T Cells: Local Specialists in Immune Defence. Nat Rev Immunol (2016) 16(2):79–89. doi: 10.1038/nri.2015.3
30. Milner JJ, Goldrath AW. Transcriptional Programming of Tissue-Resident Memory CD8+ T Cells. Curr Opin Immunol (2018) 51:162–9. doi: 10.1016/j.coi.2018.03.017
31. Cieri N, Oliveira G, Greco R, Forcato M, Taccioli C, Cianciotti B, et al. Generation of Human Memory Stem T Cells After Haploidentical T-Replete Hematopoietic Stem Cell Transplantation. Blood (2015) 125(18):2865–74. doi: 10.1182/blood-2014-11-608539
32. Roberto A, Castagna L, Zanon V, Bramanti S, Crocchiolo R, McLaren JE, et al. Role of Naive-Derived T Memory Stem Cells in T-Cell Reconstitution Following Allogeneic Transplantation. Blood (2015) 125(18):2855–64. doi: 10.1182/blood-2014-11-608406
33. Oliveira G, Ruggiero E, Stanghellini MTL, Cieri N, D’Agostino M, Fronza R, et al. Tracking Genetically Engineered Lymphocytes Long-Term Reveals the Dynamics of T Cell Immunological Memory. Sci Trans Med (2015) 7(317):317ra198–317ra198. doi: 10.1126/scitranslmed.aac8265
34. Zhang Y, Joe G, Hexner E, Zhu J, Emerson SG. Host-Reactive CD8+ Memory Stem Cells in Graft-Versus-Host Disease. Nat Med (2005) 11(12):1299–305. doi: 10.1038/nm1326
35. Gattinoni L, Zhong X-S, Palmer DC, Ji Y, Hinrichs CS, Yu Z, et al. Wnt Signaling Arrests Effector T Cell Differentiation and Generates CD8+ Memory Stem Cells. Nat Med (2009) 15(7):808–13. doi: 10.1038/nm.1982
36. Hamann D, Baars PA, Rep MHG, Hooibrink B, Kerkhof-Garde SR, Klein MR, et al. Phenotypic and Functional Separation of Memory and Effector Human CD8+ T Cells. J Exp Med (1997) 186(9):1407–18. doi: 10.1084/jem.186.9.1407
37. Restifo NP, Dudley ME, Rosenberg SA. Adoptive Immunotherapy for Cancer: Harnessing the T Cell Response. Nat Rev Immunol (2012) 12(4):269–81. doi: 10.1038/nri3191
38. Obar JJ, Khanna KM, Lefrançois L. Endogenous Naive CD8+ T Cell Precursor Frequency Regulates Primary and Memory Responses to Infection. Immunity (2008) 28(6):859–69. doi: 10.1016/j.immuni.2008.04.010
39. Abdelsamed HA, Zebley CC, Youngblood B. Epigenetic Maintenance of Acquired Gene Expression Programs During Memory CD8 T Cell Homeostasis. Front Immunol (2018) 9:6. doi: 10.3389/fimmu.2018.00006
40. Jameson SC, Masopust D. Understanding Subset Diversity in T Cell Memory. Immunity (2018) 48(2):214–26. doi: 10.1016/j.immuni.2018.02.010
41. Dijkgraaf FE, Matos TR, Hoogenboezem M, Toebes M, Vredevoogd DW, Mertz M, et al. Tissue Patrol by Resident Memory CD8+ T Cells in Human Skin. Nat Immunol (2019) 20(6):756–64. doi: 10.1038/s41590-019-0404-3
42. Ariotti S, Beltman JB, Chodaczek G, Hoekstra ME, Van Beek AE, Gomez-Eerland R, et al. Tissue-Resident Memory CD8+ T Cells Continuously Patrol Skin Epithelia to Quickly Recognize Local Antigen. Proc Natl Acad Sci (2012) 109(48):19739–44. doi: 10.1073/pnas.1208927109
43. Ariotti S, Hogenbirk MA, Dijkgraaf FE, Visser LL, Hoekstra ME, Song J-Y, et al. Skin-Resident Memory CD8+ T Cells Trigger a State of Tissue-Wide Pathogen Alert. Science (2014) 346(6205):101–5. doi: 10.1126/science.1254803
44. Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D. Resident Memory CD8 T Cells Trigger Protective Innate and Adaptive Immune Responses. Science (2014) 346(6205):98–101. doi: 10.1126/science.1254536
45. Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, et al. Survival of Tissue-Resident Memory T Cells Requires Exogenous Lipid Uptake and Metabolism. Nature (2017) 543(7644):252–6. doi: 10.1038/nature21379
46. Masopust D, Vezys V, Wherry EJ, Barber DL, Ahmed R. Cutting Edge: Gut Microenvironment Promotes Differentiation of a Unique Memory CD8 T Cell Population. J Immunol (2006) 176(4):2079–83. doi: 10.4049/jimmunol.176.4.2079
47. Hoekstra ME, Vijver SV, Schumacher TN. Modulation of the Tumor Micro-Environment by CD8+ T Cell-Derived Cytokines. Curr Opin Immunol (2021) 69:65–71. doi: 10.1016/j.coi.2021.03.016
48. Hombrink P, Helbig C, Backer RA, Piet B, Oja AE, Stark R, et al. Programs for the Persistence, Vigilance and Control of Human CD8+ Lung-Resident Memory T Cells. Nat Immunol (2016) 17(12):1467–78. doi: 10.1038/ni.3589
49. Yang Q, Zhang M, Chen Q, Chen W, Wei C, Qiao K, et al. Cutting Edge: Characterization of Human Tissue-Resident Memory T Cells at Different Infection Sites in Patients With Tuberculosis. J Immunol (2020) 204(9):2331–6. doi: 10.4049/jimmunol.1901326
50. Steinbach K, Vincenti I, Kreutzfeldt M, Page N, Muschaweckh A, Wagner I, et al. Brain-Resident Memory T Cells Represent an Autonomous Cytotoxic Barrier to Viral Infection. J Exp Med (2016) 213(8):1571–87. doi: 10.1084/jem.20151916
51. Kiniry BE, Ganesh A, Critchfield JW, Hunt PW, Hecht FM, Somsouk M, et al. Predominance of Weakly Cytotoxic, T-Betloweomesneg CD8+ T-Cells in Human Gastrointestinal Mucosa: Implications for HIV Infection. Mucosal Immunol (2017) 10(4):1008–20. doi: 10.1038/mi.2016.100
52. Kiniry BE, Hunt PW, Hecht FM, Somsouk M, Deeks SG, Shacklett BL. Differential Expression of CD8+ T Cell Cytotoxic Effector Molecules in Blood and Gastrointestinal Mucosa in HIV-1 Infection. J Immunol (2018) 200(5):1876–88. doi: 10.4049/jimmunol.1701532
53. Seidel JA, Vukmanovic-Stejic M, Muller-Durovic B, Patel N, Fuentes-Duculan J, Henson SM, et al. Skin Resident Memory CD8+ T Cells are Phenotypically and Functionally Distinct From Circulating Populations and Lack Immediate Cytotoxic Function. Clin Exp Immunol (2018) 194(1):79–92. doi: 10.1111/cei.13189
54. Smolders J, Heutinck KM, Fransen NL, Remmerswaal E, Hombrink P, Ten Berge IJM, et al. Tissue-Resident Memory T Cells Populate the Human Brain. Nat Commun (2018) 9(1):1–14. doi: 10.1038/s41467-018-07053-9
56. Angelosanto JM, Blackburn SD, Crawford A, Wherry EJ. Progressive Loss of Memory T Cell Potential and Commitment to Exhaustion During Chronic Viral Infection. J Virol (2012) 86(15):8161–70. doi: 10.1128/JVI.00889-12
57. Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, et al. Tumor-Specific T Cell Dysfunction is a Dynamic Antigen-Driven Differentiation Program Initiated Early During Tumorigenesis. Immunity (2016) 45(2):389–401. doi: 10.1016/j.immuni.2016.07.011
58. Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, et al. Chromatin States Define Tumour-Specific T Cell Dysfunction and Reprogramming. Nature (2017) 545(7655):452–6. doi: 10.1038/nature22367
59. Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, et al. The Epigenetic Landscape of T Cell Exhaustion. Science (2016) 354(6316):1165–9. doi: 10.1126/science.aae0491
60. Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining CD8+ T Cells That Provide the Proliferative Burst After PD-1 Therapy. Nature (2016) 537(7620):417–21. doi: 10.1038/nature19330
61. Leong YA, Chen Y, Ong HS, Wu D, Man K, Deleage C, et al. CXCR5+ Follicular Cytotoxic T Cells Control Viral Infection in B Cell Follicles. Nat Immunol (2016) 17(10):1187–96. doi: 10.1038/ni.3543
62. Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, et al. T Cell Factor 1-Expressing Memory-Like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity (2016) 45(2):415–27. doi: 10.1016/j.immuni.2016.07.021
63. Brummelman J, Mazza EMC, Alvisi G, Colombo FS, Grilli A, Mikulak J, et al. High-Dimensional Single Cell Analysis Identifies Stem-Like Cytotoxic CD8+ T Cells Infiltrating Human Tumors. J Exp Med (2018) 215(10):2520–35. doi: 10.1084/jem.20180684
64. He R, Hou S, Liu C, Zhang A, Bai Q, Han M, et al. Follicular CXCR5-Expressing CD8+ T Cells Curtail Chronic Viral Infection. Nature (2016) 537(7620):412–6. doi: 10.1038/nature19317
65. Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T Cell States Associated With Response to Checkpoint Immunotherapy in Melanoma. Cell (2018) 175(4):998–1013. doi: 10.1016/j.cell.2018.10.038
66. Siddiqui I, Schaeuble K, Chennupati V, Marraco SAF, Calderon-Copete S, Ferreira DP, et al. Intratumoral Tcf1+ PD-1+ CD8+ T Cells With Stem-Like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity (2019) 50(1):195–211. doi: 10.1016/j.immuni.2018.12.021
67. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of Exhausted CD8+ T Cells Differentially Mediate Tumor Control and Respond to Checkpoint Blockade. Nat Immunol (2019) 20(3):326–36. doi: 10.1038/s41590-019-0312-6
68. Galletti G, De Simone G, Mazza EMC, Puccio S, Mezzanotte C, Bi TM, et al. Two Subsets of Stem-Like CD8+ Memory T Cell Progenitors With Distinct Fate Commitments in Humans. Nat Immunol (2020) 21(12):1552–62. doi: 10.1038/s41590-020-0791-5
69. Abdelsamed HA, Moustaki A, Fan Y, Dogra P, Ghoneim HE, Zebley CC, et al. Human Memory CD8 T Cell Effector Potential is Epigenetically Preserved During In Vivo Homeostasis. J Exp Med (2017) 214(6):1593–606. doi: 10.1084/jem.20161760
70. Akondy RS, Fitch M, Edupuganti S, Yang S, Kissick HT, Li KW, et al. Origin and Differentiation of Human Memory CD8 T Cells After Vaccination. Nature (2017) 552(7685):362–7. doi: 10.1038/nature24633
71. Tan JT, Dudl E, LeRoy E, Murray R, Sprent J, Weinberg KI, et al. IL-7 is Critical for Homeostatic Proliferation and Survival of Naive T Cells. Proc Natl Acad Sci (2001) 98(15):8732–7. doi: 10.1073/pnas.161126098
72. Rathmell JC, Farkash EA, Gao W, Thompson CB. IL-7 Enhances the Survival and Maintains the Size of Naive T Cells. J Immunol (2001) 167(12):6869–76. doi: 10.4049/jimmunol.167.12.6869
73. Barata JT, Silva A, Brandao JG, Nadler LM, Cardoso AA, Boussiotis VA. Activation of PI3K is Indispensable for Interleukin 7–Mediated Viability, Proliferation, Glucose Use, and Growth of T Cell Acute Lymphoblastic Leukemia Cells. J Exp Med (2004) 200(5):659–69. doi: 10.1084/jem.20040789
74. Jacobs SR, Michalek RD, Rathmell JC. IL-7 is Essential for Homeostatic Control of T Cell Metabolism In Vivo. J Immunol (2010) 184(7):3461–9. doi: 10.4049/jimmunol.0902593
75. Surh CD, Sprent J. Homeostasis of Naive and Memory T Cells. Immunity (2008) 29(6):848–62. doi: 10.1016/j.immuni.2008.11.002
76. Shin H, Blackburn SD, Blattman JN, Wherry EJ. Viral Antigen and Extensive Division Maintain Virus-Specific CD8 T Cells During Chronic Infection. J Exp Med (2007) 204(4):941–9. doi: 10.1084/jem.20061937
77. Sprent J, Surh CD. Normal T Cell Homeostasis: The Conversion of Naive Cells Into Memory-Phenotype Cells. Nat Immunol (2011) 12(6):478–84. doi: 10.1038/ni.2018
78. Buck MD, O’Sullivan D, Geltink RIK, Curtis JD, Chang C-H, Sanin DE, et al. Mitochondrial Dynamics Controls T Cell Fate Through Metabolic Programming. Cell (2016) 166(1):63–76. doi: 10.1016/j.cell.2016.05.035
79. Balmer ML, Ma EH, Bantug GR, Grählert J, Pfister S, Glatter T, et al. Memory CD8+ T Cells Require Increased Concentrations of Acetate Induced by Stress for Optimal Function. Immunity (2016) 44(6):1312–24. doi: 10.1016/j.immuni.2016.03.016
80. van der Windt GJW, O’Sullivan D, Everts B, Huang SC-C, Buck MD, Curtis JD, et al. CD8 Memory T Cells Have a Bioenergetic Advantage That Underlies Their Rapid Recall Ability. Proc Natl Acad Sci (2013) 110(35):14336–41. doi: 10.1073/pnas.1221740110
81. Pearce EL, Poffenberger MC, Chang C-H, Jones RG. Fueling Immunity: Insights Into Metabolism and Lymphocyte Function. Science (2013) 342(6155):1242454. doi: 10.1126/science.1242454
82. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR Regulates Memory CD8 T-Cell Differentiation. Nature (2009) 460(7251):108–12. doi: 10.1038/nature08155
83. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang L-S, et al. Enhancing CD8 T-Cell Memory by Modulating Fatty Acid Metabolism. Nature (2009) 460(7251):103–7. doi: 10.1038/nature08097
84. Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR Kinase Determines Effector Versus Memory CD8+ T Cell Fate by Regulating the Expression of Transcription Factors T-Bet and Eomesodermin. Immunity (2010) 32(1):67–78. doi: 10.1016/j.immuni.2009.10.010
85. van der Windt GJW, Everts B, Chang C-H, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial Respiratory Capacity is a Critical Regulator of CD8+ T Cell Memory Development. Immunity (2012) 36(1):68–78. doi: 1016/j.immuni.2011.12.007
86. Cui G, Staron MM, Gray SM, Ho P-C, Amezquita RA, Wu J, et al. IL-7-Induced Glycerol Transport and TAG Synthesis Promotes Memory CD8+ T Cell Longevity. Cell (2015) 161(4):750–61. doi: 10.1016/j.cell.2015.03.021
87. Geltink RIK, O’Sullivan D, Corrado M, Bremser A, Buck MD, Buescher JM, et al. Mitochondrial Priming by CD28. Cell (2017) 171(2):385–97. doi: 10.1016/j.cell.2017.08.018
88. O’Sullivan D, van der Windt GJW, Huang SC-C, Curtis JD, Chang C-H, Buck MD, et al. Memory CD8+ T Cells Use Cell-Intrinsic Lipolysis to Support the Metabolic Programming Necessary for Development. Immunity (2014) 41(1):75–88. doi: 10.1016/j.immuni.2014.06.005
89. Maekawa Y, Ishifune C, Tsukumo S-i, Hozumi K, Yagita H, Yasutomo K. Notch Controls the Survival of Memory CD4+ T Cells by Regulating Glucose Uptake. Nat Med (2015) 21(1):55–61. doi: 10.1038/nm.3758
90. Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, et al. Hypoxia-Inducible Factors Enhance the Effector Responses of CD8+ T Cells to Persistent Antigen. Nat Immunol (2013) 14(11):1173–82. doi: 10.1038/ni.2714
91. Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF Transcription Factors, Inflammation, and Immunity. Immunity (2014) 41(4):518–28. doi: 10.1016/j.immuni.2014.09.008
92. Tyrakis PA, Palazon A, Macias D, Lee K, Phan A, Veliça P, et al. S-2-Hydroxyglutarate Regulates CD8+ T-Lymphocyte Fate. Nature (2016) 540(7632):236–41. doi: 10.1038/nature20165
93. Chang C-H, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional Control of T Cell Effector Function by Aerobic Glycolysis. Cell (2013) 153(6):1239–51. doi: 10.1016/j.cell.2013.05.016
94. Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. Aerobic Glycolysis Promotes T Helper 1 Cell Differentiation Through an Epigenetic Mechanism. Science (2016) 354(6311):481–4. doi: 10.1126/science.aaf6284
95. Phan AT, Doedens AL, Palazon A, Tyrakis PA, Cheung KP, Johnson RS, et al. Constitutive Glycolytic Metabolism Supports CD8+ T Cell Effector Memory Differentiation During Viral Infection. Immunity (2016) 45(5):1024–37. doi: 10.1016/j.immuni.2016.10.017
96. Sathaliyawala T, Kubota M, Yudanin N, Turner D, Camp P, Thome JJ, et al. Distribution and Compartmentalization of Human Circulating and Tissue-Resident Memory T Cell Subsets. Immunity (2013) 38(1):187–97. doi: 10.1016/j.immuni.2012.09.020
97. Saule P, Trauet J, Dutriez V, Lekeux V, Dessaint JP, Labalette M. Accumulation of Memory T Cells From Childhood to Old Age: Central and Effector Memory Cells in CD4(+) Versus Effector Memory and Terminally Differentiated Memory Cells in CD8(+) Compartment. Mech Ageing Dev (2006) 127(3):274–81. doi: 10.1016/j.mad.2005.11.001
98. Cossarizza A, Ortolani C, Paganelli R, Barbieri D, Monti D, Sansoni P, et al. CD45 Isoforms Expression on CD4+ and CD8+ T Cells Throughout Life, From Newborns to Centenarians: Implications for T Cell Memory. Mech Ageing Dev (1996) 86(3):173–95. doi: 10.1016/0047-6374(95)01691-0
99. Goronzy JJ, Weyand CM. Understanding Immunosenescence to Improve Responses to Vaccines. Nat Immunol (2013) 14(5):428–36. doi: 10.1038/ni.2588
100. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A Human Memory T Cell Subset With Stem Cell–Like Properties. Nat Med (2011) 17(10):1290–7. doi: 10.1038/nm.2446
101. Gattinoni L, Klebanoff CA, Restifo NP. Paths to Stemness: Building the Ultimate Antitumour T Cell. Nat Rev Cancer (2012) 12(10):671–84. doi: 10.1038/nrc3322
102. Roederer M, Quaye L, Mangino M, Beddall MH, Mahnke Y, Chattopadhyay P, et al. The Genetic Architecture of the Human Immune System: A Bioresource for Autoimmunity and Disease Pathogenesis. Cell (2015) 161(2):387–403. doi: 10.1016/j.cell.2015.02.046
103. Lugli E, Gattinoni L, Roberto A, Mavilio D, Price DA, Restifo NP, et al. Identification, Isolation and In Vitro Expansion of Human and Nonhuman Primate T Stem Cell Memory Cells. Nat Protoc (2013) 8(1):33–42. doi: 10.1038/nprot.2012.143
104. Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, Provasi E, et al. IL-7 and IL-15 Instruct the Generation of Human Memory Stem T Cells From Naive Precursors. Blood (2013) 121(4):573–84. doi: 10.1182/blood-2012-05-431718
105. Zanon V, Pilipow K, Scamardella E, De Paoli F, De Simone G, Price DA, et al. Curtailed T-Cell Activation Curbs Effector Differentiation and Generates CD8+ T Cells With a Naturally-Occurring Memory Stem Cell Phenotype. Eur J Immunol (2017) 47(9):1468–76. doi: 10.1002/eji.201646732
106. Pilipow K, Scamardella E, Puccio S, Gautam S, De Paoli F, Mazza EMC, et al. Antioxidant Metabolism Regulates CD8+ T Memory Stem Cell Formation and Antitumor Immunity. JCI Insight (2018) 3(18). doi: 10.1172/jci.insight.122299
107. Gattinoni L, Zhong X-S, Palmer DC, Ji Y, Hinrichs CS, Yu Z, et al. Wnt Signaling Arrests Effector T Cell Differentiation and Generates CD8+ Memory Stem Cells. Nat Med (2009) 15(7):808–13. doi: 10.1038/nm.1982
108. Mousset CM, Hobo W, Ji Y, Fredrix H, De Giorgi V, Allison RD, et al. Ex Vivo AKT-Inhibition Facilitates Generation of Polyfunctional Stem Cell Memory-Like CD8+ T Cells for Adoptive Immunotherapy. OncoImmunology (2018) 7(10):e1488565. doi: 10.1080/2162402X.2018.1488565
109. Kagoya Y, Nakatsugawa M, Yamashita Y, Ochi T, Guo T, Anczurowski M, et al. BET Bromodomain Inhibition Enhances T Cell Persistence and Function in Adoptive Immunotherapy Models. J Clin Invest (2016) 126(9):3479–94. doi: 10.1172/JCI86437
110. Kratchmarov R, Magun AM, Reiner SL. TCF1 Expression Marks Self-Renewing Human CD8(+) T Cells. Blood Adv (2018) 2(14):1685–90. doi: 10.1182/bloodadvances.2018016279
111. Seok J, Jung HS, Park S, Lee JO, Kim CJ, Kim GJ. Alteration of Fatty Acid Oxidation by Increased CPT1A on Replicative Senescence of Placenta-Derived Mesenchymal Stem Cells. Stem Cell Res Ther (2020) 11(1):1. doi: 10.1186/s13287-019-1471-y
112. Pilipow K, Scamardella E, Lugli E. (2020). Generating Stem-Like Memory T Cells with Antioxidants for Adoptive Cell Transfer Immunotherapy of Cancer. Methods Enzymol 631:137–158. doi: 10.1016/bs.mie.2019.08.016
113. Kim W, Kim M, Jho E-h. Wnt/β-Catenin Signalling: From Plasma Membrane to Nucleus. Biochem J (2013) 450(1):9–21. doi: 10.1042/BJ20121284
114. Niehrs C. The Complex World of WNT Receptor Signalling. Nat Rev Mol Cell Biol (2012) 13(12):767–79. doi: 10.1038/nrm3470
115. Staal FJT, Luis TC, Tiemessen MM. WNT Signalling in the Immune System: WNT is Spreading its Wings. Nat Rev Immunol (2008) 8(8):581–93. doi: 10.1038/nri2360
116. Sokol SY. Maintaining Embryonic Stem Cell Pluripotency With Wnt Signaling. Development (2011) 138(20):4341. doi: 10.1242/dev.066209
117. Verbeek S, Izon D, Hofhuis F, Robanus-Maandag E, Te Riele H, van de Watering M, et al. An HMG-Box-Containing T-Cell Factor Required for Thymocyte Differentiation. Nature (1995) 374(6517):70–4. doi: 10.1038/374070a0
118. Scholz G, Jandus C, Zhang L, Grandclément C, Lopez-Mejia IC, Soneson C, et al. Modulation of mTOR Signalling Triggers the Formation of Stem Cell-Like Memory T Cells. EBioMedicine (2016) 4:50–61. doi: 10.1016/j.ebiom.2016.01.019
119. Xiao Z, Sun Z, Smyth K, Li L. Wnt Signaling Inhibits CTL Memory Programming. Mol Immunol (2013) 56(4):423–33. doi: 10.1016/j.molimm.2013.06.008
120. Chi H. Regulation and Function of mTOR Signalling in T Cell Fate Decisions. Nat Rev Immunol (2012) 12(5):325–38. doi: 10.1038/nri3198
121. Speiser DE, Utzschneider DT, Oberle SG, Münz C, Romero P, Zehn D. T Cell Differentiation in Chronic Infection and Cancer: Functional Adaptation or Exhaustion? Nat Rev Immunol (2014) 14(11):768–74. doi: 10.1038/nri3740
122. Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, et al. The TCF1-Bcl6 Axis Counteracts Type I Interferon to Repress Exhaustion and Maintain T Cell Stemness. Sci Immunol (2016) 1(6):eaai8593. doi: 10.1126/sciimmunol.aai8593
123. Wong CK, Ho CY, Li EK, Lam CWK. Elevation of Proinflammatory Cytokine (IL-18, IL-17, IL-12) and Th2 Cytokine (IL-4) Concentrations in Patients With Systemic Lupus Erythematosus. Lupus (2000) 9(8):589–93. doi: 10.1191/096120300678828703
124. Jaafoura S, de Goër de Herve MG, Hernandez-Vargas EA, Hendel-Chavez H, Abdoh M, Mateo MC, et al. Progressive Contraction of the Latent HIV Reservoir Around a Core of Less-Differentiated CD4+ Memory T Cells. Nat Commun (2014) 5:5407. doi: 10.1038/ncomms6407
125. Nagai Y, Kawahara M, Hishizawa M, Shimazu Y, Sugino N, Fujii S, et al. T Memory Stem Cells are the Hierarchical Apex of Adult T-Cell Leukemia. Blood (2015) 125(23):3527–35. doi: 10.1182/blood-2014-10-607465
126. Gattinoni L, Restifo NP. Moving T Memory Stem Cells to the Clinic. Blood (2013) 121(4):567–8. doi: 10.1182/blood-2012-11-468660
127. Flynn JK, Gorry PR. Stem Memory T Cells (TSCM)-Their Role in Cancer and HIV Immunotherapies. Clin Transl Immunol (2014) 3(7):e20–e. doi: 10.1038/cti.2014.16
128. Buzon MJ, Sun H, Li C, Shaw A, Seiss K, Ouyang Z, et al. HIV-1 Persistence in CD4+ T Cells With Stem Cell-Like Properties. Nat Med (2014) 20(2):139–42. doi: 10.1038/nm.3445
129. Chahroudi A, Silvestri G, Lichterfeld M. T Memory Stem Cells and HIV: A Long-Term Relationship. Curr HIV/AIDS Rep (2015) 12(1):33–40. doi: 10.1007/s11904-014-0246-4
130. Jaafoura S, de Goër de Herve MG, Hernandez-Vargas EA, Hendel-Chavez H, Abdoh M, Mateo MC, et al. Progressive Contraction of the Latent HIV Reservoir Around a Core of Less-Differentiated CD4+ Memory T Cells. Nat Commun (2014) 5(1):5407. doi: 10.1038/ncomms6407
131. Tabler CO, Lucera MB, Haqqani AA, McDonald DJ, Migueles SA, Connors M, et al. CD4+ Memory Stem Cells are Infected by HIV-1 in a Manner Regulated in Part by SAMHD1 Expression. J Virol (2014) 88(9):4976–86. doi: 10.1128/JVI.00324-14
132. Tabler CO, Lucera MB, Haqqani AA, McDonald DJ, Migueles SA, Connors M, et al. CD4<sup<+</sup< Memory Stem Cells Are Infected by HIV-1 in a Manner Regulated in Part by SAMHD1 Expression. J Virol (2014) 88(9):4976. doi: 10.1128/JVI.00324-14
133. McNamara LA, Collins KL. Hematopoietic Stem/Precursor Cells as HIV Reservoirs. Curr Opin HIV AIDS (2011) 6(1):43–8. doi: 10.1097/COH.0b013e32834086b3
134. Xu L, Wang J, Liu Y, Xie L, Su B, Mou D, et al. CRISPR-Edited Stem Cells in a Patient With HIV and Acute Lymphocytic Leukemia. New Engl J Med (2019) 381(13):1240–7. doi: 10.1056/NEJMoa1817426
135. Ribeiro SP, Milush JM, Cunha-Neto E, Kallas EG, Kalil J, Somsouk M, et al. The CD8+ Memory Stem T Cell (T(SCM)) Subset is Associated With Improved Prognosis in Chronic HIV-1 Infection. J Virol (2014) 88(23):13836–44. doi: 10.1128/JVI.01948-14
136. Angin M, Volant S, Passaes C, Lecuroux C, Monceaux V, Dillies M-A, et al. Metabolic Plasticity of HIV-Specific CD8+ T Cells is Associated With Enhanced Antiviral Potential and Natural Control of HIV-1 Infection. Nat Metab (2019) 1(7):704–16. doi: 10.1038/s42255-019-0081-4
137. Mueller YM, Bojczuk PM, Halstead ES, Kim AHJ, Witek J, Altman JD, et al. IL-15 Enhances Survival and Function of HIV-Specific CD8+ T Cells. Blood (2003) 101(3):1024–9. doi: 10.1182/blood-2002-07-1957
138. Fuertes Marraco SA, Soneson C, Cagnon L, Gannon PO, Allard M, Maillard SA, et al. Long-Lasting Stem Cell–Like Memory CD8<sup<+</sup< T Cells With a Naïve-Like Profile Upon Yellow Fever Vaccination. Sci Trans Med (2015) 7(282):282ra48. doi: 10.1126/scitranslmed.aaa3700
139. Flossdorf M, Rössler J, Buchholz VR, Busch DH, Höfer T. CD8(+) T Cell Diversification by Asymmetric Cell Division. Nat Immunol (2015) 16(9):891–3. doi: 10.1038/ni.3235
140. Gao S, Liang X, Wang H, Bao B, Zhang K, Zhu Y, et al. Stem Cell-Like Memory T Cells: A Perspective From the Dark Side. Cell Immunol (2021) 361:104273. doi: 10.1016/j.cellimm.2020.104273
141. Hosokawa K, Muranski P, Feng X, Townsley DM, Liu B, Knickelbein J, et al. Memory Stem T Cells in Autoimmune Disease: High Frequency of Circulating CD8+ Memory Stem Cells in Acquired Aplastic Anemia. J Immunol (2016) 196(4):1568–78. doi: 10.4049/jimmunol.1501739
142. Lee YJ, Park JA, Kwon H, Choi YS, Jung KC, Park SH, et al. Role of Stem Cell–Like Memory T Cells in Systemic Lupus Erythematosus. Arthritis Rheumatol (2018) 70(9):1459–69. doi: 10.1002/art.40524
143. Cao J, Zhang C, Han X, Cheng H, Chen W, Qi K, et al. Emerging Role of Stem Cell Memory-Like T Cell in Immune Thrombocytopenia. ScandInavian J Immunol (2019) 89(3):e12739. doi: 10.1111/sji.12739
144. Di Lorenzo TP, Peakman M, Roep BO. Translational Mini-Review Series on Type 1 Diabetes: Systematic Analysis of T Cell Epitopes in Autoimmune Diabetes. Clin Exp Immunol (2007) 148(1):1–16. doi: 10.1111/j.1365-2249.2006.03244.x
145. Sherry N, Hagopian W, Ludvigsson J, Jain SM, Wahlen J, Ferry RJ Jr., et al. Teplizumab for Treatment of Type 1 Diabetes (Protégé Study): 1-Year Results From a Randomised, Placebo-Controlled Trial. Lancet (2011) 378(9790):487–97. doi: 10.1016/S0140-6736(11)60931-8
146. Monti P, Brigatti C, Heninger AK, Scirpoli M, Bonifacio E. Disengaging the IL-2 Receptor With Daclizumab Enhances IL-7–Mediated Proliferation of CD4+ and CD8+ T Cells. Am J Transplantation (2009) 9(12):2727–35. doi: 10.1111/j.1600-6143.2009.02825.x
147. Monti P, Heninger A-K, Bonifacio E. Differentiation, Expansion, and Homeostasis of Autoreactive T Cells in Type 1 Diabetes Mellitus. Curr Diabetes Rep (2009) 9(2):113–8. doi: 10.1007/s11892-009-0020-y
148. Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS. Skin Infection Generates non-Migratory Memory CD8+ TRM Cells Providing Global Skin Immunity. Nature (2012) 483(7388):227–31. doi: 10.1038/nature10851
149. Wakim LM, Woodward-Davis A, Bevan MJ. Memory T Cells Persisting Within the Brain After Local Infection Show Functional Adaptations to Their Tissue of Residence. Proc Natl Acad Sci (2010) 107(42):17872–9. doi: 10.1073/pnas.1010201107
150. Fernandez-Ruiz D, Ng WY, Holz LE, Ma JZ, Zaid A, Wong YC, et al. Liver-Resident Memory CD8+ T Cells Form a Front-Line Defense Against Malaria Liver-Stage Infection. Immunity (2016) 45(4):889–902. doi: 10.1016/j.immuni.2016.08.011
151. Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyártó BZ, et al. Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell (2015) 161(4):737–49. doi: 10.1016/j.cell.2015.03.031
152. Skon CN, Lee J-Y, Anderson KG, Masopust D, Hogquist KA, Jameson SC. Transcriptional Downregulation of S1pr1 is Required for the Establishment of Resident Memory CD8+ T Cells. Nat Immunol (2013) 14(12):1285–93. doi: 10.1038/ni.2745
153. Walsh DA, Da Silva HB, Beura LK, Peng C, Hamilton SE, Masopust D, et al. The Functional Requirement for CD69 in Establishment of Resident Memory CD8+ T Cells Varies With Tissue Location. J Immunol (2019) 203(4):946–55. doi: 10.4049/jimmunol.1900052
154. Beura LK, Wijeyesinghe S, Thompson EA, Macchietto MG, Rosato PC, Pierson MJ, et al. T Cells in Nonlymphoid Tissues Give Rise to Lymph-Node-Resident Memory T Cells. Immunity (2018) 48(2):327–38. doi: 10.1016/j.immuni.2018.01.015
155. Buggert M, Nguyen S, Salgado-Montes de Oca G, Bengsch B, Darko S, Ransier A, et al. Identification and Characterization of HIV-Specific Resident Memory CD8+ T Cells in Human Lymphoid Tissue. Sci Immunol (2018) 3(24):eaar4526. doi: 10.1126/sciimmunol.aar4526
156. Fonseca R, Beura LK, Quarnstrom CF, Ghoneim HE, Fan Y, Zebley CC, et al. Developmental Plasticity Allows Outside-in Immune Responses by Resident Memory T Cells. Nat Immunol (2020) 21(4):412–21. doi: 10.1038/s41590-020-0607-7
157. Molodtsov A, Turk MJ. Tissue Resident CD8 Memory T Cell Responses in Cancer and Autoimmunity. Front Immunol (2018) 2810. doi: 10.3389/fimmu.2018.02810
158. Pauls K, Schön M, Kubitza RC, Homey B, Wiesenborn A, Lehmann P, et al. Role of Integrin αe (CD103) β7 for Tissue-Specific Epidermal Localization of CD8+ T Lymphocytes. J Invest Dermatol (2001) 117(3):569–75. doi: 10.1046/j.0022-202x.2001.01481.x
159. Bank I, Book M, Ware R. Functional Role of VLA-1 (CD49A) in Adhesion, Cation-Dependent Spreading, and Activation of Cultured Human T Lymphocytes. Cell Immunol (1994) 156(2):424–37. doi: 10.1006/cimm.1994.1187
160. Richter MV, Topham DJ. The α1β1 Integrin and TNF Receptor II Protect Airway CD8+ Effector T Cells From Apoptosis During Influenza Infection. J Immunol (2007) 179(8):5054–63. doi: 10.4049/jimmunol.179.8.5054
161. Lee Y-T, Suarez-Ramirez JE, Redman JM, Aguila CC, Hadley GA, Cauley LS. CD69 and CD103 Cooperatively Regulate CD8 T Cell Responses in the Lungs After Viral Infection (39.25). J Immunol (2009) 182(1 Supplement):39.25.
162. Lee Y-T, Suarez-Ramirez JE, Wu T, Redman JM, Bouchard K, Hadley GA, et al. Environmental and Antigen Receptor-Derived Signals Support Sustained Surveillance of the Lungs by Pathogen-Specific Cytotoxic T Lymphocytes. J Virol (2011) 85(9):4085–94. doi: 10.1128/JVI.02493-10
163. Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, et al. Human Tissue-Resident Memory T Cells are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep (2017) 20(12):2921–34. doi: 10.1016/j.celrep.2017.08.078
164. Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T Cells in Nonlymphoid Tissue That Provide Enhanced Local Immunity During Infection With Herpes Simplex Virus. Nat Immunol (2009) 10(5):524–30. doi: 10.1038/ni.1718
165. Enamorado M, Iborra S, Priego E, Cueto FJ, Quintana JA, Martínez-Cano S, et al. Enhanced Anti-Tumour Immunity Requires the Interplay Between Resident and Circulating Memory CD8+ T Cells. Nat Commun (2017) 8(1):1–11. doi: 10.1038/ncomms16073
166. Park SL, Buzzai A, Rautela J, Hor JL, Hochheiser K, Effern M, et al. Tissue-Resident Memory CD8+ T Cells Promote Melanoma–Immune Equilibrium in Skin. Nature (2019) 565(7739):366–71. doi: 10.1038/s41586-018-0812-9
167. Strickley JD, Messerschmidt JL, Awad ME, Li T, Hasegawa T, Ha DT, et al. Immunity to Commensal Papillomaviruses Protects Against Skin Cancer. Nature (2019) 575(7783):519–22. doi: 10.1038/s41586-019-1719-9
168. Luoma AM, Suo S, Williams HL, Sharova T, Sullivan K, Manos M, et al. Molecular Pathways of Colon Inflammation Induced by Cancer Immunotherapy. Cell (2020) 182(3):655–71. doi: 10.1016/j.cell.2020.06.001
169. Richmond JM, Strassner JP, Zapata J, Garg M, Riding RL, Refat MA, et al. Antibody Blockade of IL-15 Signaling has the Potential to Durably Reverse Vitiligo. Sci Trans Med (2018) 10(450):eaam7710. doi: 10.1126/scitranslmed.aam7710
170. Kurihara K, Fujiyama T, Phadungsaksawasdi P, Ito T, Tokura Y. Significance of IL-17A-Producing CD8+ CD103+ Skin Resident Memory T Cells in Psoriasis Lesion and Their Possible Relationship to Clinical Course. J Dermatol Sci (2019) 95(1):21–7. doi: 10.1016/j.jdermsci.2019.06.002
171. Sérézal IG, Hoffer E, Ignatov B, Martini E, Zitti B, Ehrström M, et al. A Skewed Pool of Resident T Cells Triggers Psoriasis-Associated Tissue Responses in Never-Lesional Skin From Patients With Psoriasis. J Allergy Clin Immunol (2019) 143(4):1444–54. doi: 10.1016/j.jaci.2018.08.048
172. de Leur K, Dieterich M, Hesselink DA, Corneth OBJ, Dor FJMF, de Graav GN, et al. Characterization of Donor and Recipient CD8+ Tissue-Resident Memory T Cells in Transplant Nephrectomies. Sci Rep (2019) 9(1):1–12. doi: 10.1038/s41598-019-42401-9
173. Dijkgraaf FE, Kok L, Schumacher TNM. Formation of Tissue-Resident CD8+ T-Cell Memory. Cold Spring Harbor Perspect Biol (2021) 13(8):a038117. doi: 10.1101/cshperspect.a038117
174. Zaric M, Becker PD, Hervouet C, Kalcheva P, Yus BI, Cocita C, et al. Long-Lived Tissue Resident HIV-1 Specific Memory CD8+ T Cells are Generated by Skin Immunization With Live Virus Vectored Microneedle Arrays. J Controlled Release (2017) 268:166–75. doi: 10.1016/j.jconrel.2017.10.026
175. Yu S, Lao S, Yang B, Wu C. Tissue-Resident Memory-Like CD8+ T Cells Exhibit Heterogeneous Characteristics in Tuberculous Pleural Effusion. J Immunol Res (2021) 2021:6643808. doi: 10.1155/2021/6643808
176. Hu Z, Wong K-W, Zhao H-M, Wen H-L, Ji P, Ma H, et al. Sendai Virus Mucosal Vaccination Establishes Lung-Resident Memory CD8 T Cell Immunity and Boosts BCG-Primed Protection Against TB in Mice. Mol Ther (2017) 25(5):1222–33. doi: 10.1016/j.ymthe.2017.02.018
177. Hart P, Copland A, Diogo GR, Harris S, Spallek R, Oehlmann W, et al. Nanoparticle-Fusion Protein Complexes Protect Against Mycobacterium Tuberculosis Infection. Mol Ther (2018) 26(3):822–33. doi: 10.1016/j.ymthe.2017.12.016
178. Copland A, Diogo GR, Hart P, Harris S, Tran AC, Paul MJ, et al. Mucosal Delivery of Fusion Proteins With Bacillus Subtilis Spores Enhances Protection Against Tuberculosis by Bacillus Calmette-Guérin. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.00346
179. Diogo GR, Hart P, Copland A, Kim M-Y, Tran AC, Poerio N, et al. Immunization With Mycobacterium Tuberculosis Antigens Encapsulated in Phosphatidylserine Liposomes Improves Protection Afforded by BCG. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.01349
180. Perdomo C, Zedler U, Kühl AA, Lozza L, Saikali P, Sander LE, et al. Mucosal BCG Vaccination Induces Protective Lung-Resident Memory T Cell Populations Against Tuberculosis. MBio (2016) 7(6):e01686-16. doi: 10.1128/mBio.01686-16
181. Sakai S, Kauffman KD, Schenkel JM, McBerry CC, Mayer-Barber KD, Masopust D, et al. Cutting Edge: Control of Mycobacterium Tuberculosis Infection by a Subset of Lung Parenchyma–Homing CD4 T Cells. J Immunol (2014) 192(7):2965–9. doi: 10.4049/jimmunol.1400019
182. Bull NC, Stylianou E, Kaveh DA, Pinpathomrat N, Pasricha J, Harrington-Kandt R, et al. Enhanced Protection Conferred by Mucosal BCG Vaccination Associates With Presence of Antigen-Specific Lung Tissue-Resident PD-1+ KLRG1– CD4+ T Cells. Mucosal Immunol (2019) 12(2):555–64. doi: 10.1038/s41385-018-0109-1
183. Park SL, Zaid A, Hor JL, Christo SN, Prier JE, Davies B, et al. Local Proliferation Maintains a Stable Pool of Tissue-Resident Memory T Cells After Antiviral Recall Responses. Nat Immunol (2018) 19(2):183–91. doi: 10.1038/s41590-017-0027-5
184. Beura LK, Mitchell JS, Thompson EA, Schenkel JM, Mohammed J, Wijeyesinghe S, et al. Intravital Mucosal Imaging of CD8(+) Resident Memory T Cells Shows Tissue-Autonomous Recall Responses That Amplify Secondary Memory. Nat Immunol (2018) 19(2):173–82. doi: 10.1038/s41590-017-0029-3
185. Bromley SK, Thomas SY, Luster AD. Chemokine Receptor CCR7 Guides T Cell Exit From Peripheral Tissues and Entry Into Afferent Lymphatics. Nat Immunol (2005) 6(9):895–901. doi: 10.1038/ni1240
186. Debes GF, Arnold CN, Young AJ, Krautwald S, Lipp M, Hay JB, et al. Chemokine Receptor CCR7 Required for T Lymphocyte Exit From Peripheral Tissues. Nat Immunol (2005) 6(9):889–94. doi: 10.1038/ni1238
187. Jennrich S, Lee MH, Lynn RC, Dewberry K, Debes GF. Tissue Exit: A Novel Control Point in the Accumulation of Antigen-Specific CD8 T Cells in the Influenza a Virus-Infected Lung. J Virol (2012) 86(7):3436–45. doi: 10.1128/JVI.07025-11
188. McMaster SR, Wein AN, Dunbar PR, Hayward SL, Cartwright EK, Denning TL, et al. Pulmonary Antigen Encounter Regulates the Establishment of Tissue-Resident CD8 Memory T Cells in the Lung Airways and Parenchyma. Mucosal Immunol (2018) 11(4):1071–8. doi: 10.1038/s41385-018-0003-x
189. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The Hallmarks of Aging. Cell (2013) 153(6):1194–217. doi: 10.1016/j.cell.2013.05.039
190. Nguyen THO, McAuley JL, Kim Y, Zheng MZM, Gherardin NA, Godfrey DI, et al. Influenza, But Not SARS-CoV-2, Infection Induces a Rapid Interferon Response That Wanes With Age and Diminished Tissue-Resident Memory CD8+ T Cells. Clin Trans Immunol (2021) 10(1):e1242. doi: 10.1002/cti2.1242
191. Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D. T Cell Memory. Resident Memory CD8 T Cells Trigger Protective Innate and Adaptive Immune Responses. Science (2014) 346(6205):98–101. doi: 10.1126/science.1254536
192. Schenkel JM, Fraser KA, Vezys V, Masopust D. Sensing and Alarm Function of Resident Memory CD8+ T Cells. Nat Immunol (2013) 14(5):509–13. doi: 10.1038/ni.2568
193. Naylor K, Li G, Vallejo AN, Lee W-W, Koetz K, Bryl E, et al. The Influence of Age on T Cell Generation and TCR Diversity. J Immunol (2005) 174(11):7446–52. doi: 10.4049/jimmunol.174.11.7446
194. Pizzolla A, Nguyen TH, Sant S, Jaffar J, Loudovaris T, Mannering SI, et al. Influenza-Specific Lung-Resident Memory T Cells are Proliferative and Polyfunctional and Maintain Diverse TCR Profiles. J Clin Invest (2018) 128(2):721–33. doi: 10.1172/JCI96957
195. Turner DL, Bickham KL, Thome JJ, Kim CY, D’Ovidio F, Wherry EJ, et al. Lung Niches for the Generation and Maintenance of Tissue-Resident Memory T Cells. Mucosal Immunol (2014) 7(3):501–10. doi: 10.1038/mi.2013.67
196. Li C, Zhu B, Son YM, Wang Z, Jiang L, Xiang M, et al. The Transcription Factor Bhlhe40 Programs Mitochondrial Regulation of Resident CD8+ T Cell Fitness and Functionality. Immunity (2019) 51(3):491–507. doi: 10.1016/j.immuni.2019.08.013
197. MacIver NJ, Michalek RD, Rathmell JC. Metabolic Regulation of T Lymphocytes. Annu Rev Immunol (2013) 31:259–83. doi: 10.1146/annurev-immunol-032712-095956
198. Powell JD, Pollizzi K. Fueling Memories. Immunity (2012) 36(1):3–5. doi: 10.1016/j.immuni.2012.01.003
199. Saravia J, Raynor JL, Chapman NM, Lim SA, Chi H. Signaling Networks in Immunometabolism. Cell Res (2020) 30(4):328–42. doi: 10.1038/s41422-020-0301-1
200. O’Sullivan D. The Metabolic Spectrum of Memory T Cells. Immunol Cell Biol (2019) 97(7):636–46. doi: 10.1111/imcb.12274
201. Proietti M, Perruzza L, Scribano D, Pellegrini G, D’Antuono R, Strati F, et al. ATP Released by Intestinal Bacteria Limits the Generation of Protective IgA Against Enteropathogens. Nat Commun (2019) 10(1):1–11. doi: 10.1038/s41467-018-08156-z
202. Taheri M, Sarani H, Moazeni-Roodi A, Naderi M, Hashemi M. Association Between P2X7 Polymorphisms and Susceptibility to Tuberculosis: An Updated Meta-Analysis of Case-Control Studies. Medicina (2019) 55(6):298. doi: 10.3390/medicina55060298
203. Vardam-Kaur T, Sun J, da Silva HB. Metabolic Regulation of Tissue-Resident Memory CD8+ T Cells. Curr Opin Pharmacol (2021) 57:117–24. doi: 10.1016/j.coph.2021.02.004
204. Sun X, Zhou R, Lei Y, Hu J, Li X. The Ligand-Gated Ion Channel P2X7 Receptor Mediates NLRP3/caspase-1-Mediated Pyroptosis in Cerebral Cortical Neurons of Juvenile Rats With Sepsis. Brain Res (2020) 1748:147109. doi: 10.1016/j.brainres.2020.147109
205. Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 Receptor in Infection and Inflammation. Immunity (2017) 47(1):15–31. doi: 10.1016/j.immuni.2017.06.020
206. Stark R, Wesselink TH, Behr FM, Kragten NAM, Arens R, Koch-Nolte F, et al. T RM Maintenance is Regulated by Tissue Damage via P2RX7. Sci Immunol (2018) 3(30):eaau1022. doi: 10.1126/sciimmunol.aau1022
207. Fahrer AM, Konigshofer Y, Kerr EM, Ghandour G, Mack DH, Davis MM, et al. Attributes of γδ Intraepithelial Lymphocytes as Suggested by Their Transcriptional Profile. Proc Natl Acad Sci (2001) 98(18):10261–6. doi: 10.1073/pnas.171320798
208. Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, et al. Runx3 Programs CD8+ T Cell Residency in non-Lymphoid Tissues and Tumours. Nature (2017) 552(7684):253–7. doi: 10.1038/nature24993
209. Mackay LK, Minnich M, Kragten NAM, Liao Y, Nota B, Seillet C, et al. Hobit and Blimp1 Instruct a Universal Transcriptional Program of Tissue Residency in Lymphocytes. Science (2016) 352(6284):459–63. doi: 10.1126/science.aad2035
210. Kragten NAM, Behr FM, Vieira Braga FA, Remmerswaal EBM, Wesselink TH, Oja AE, et al. Blimp-1 Induces and Hobit Maintains the Cytotoxic Mediator Granzyme B in CD8 T Cells. Eur J Immunol (2018) 48(10):1644–62. doi: 10.1002/eji.201847771
211. Cheuk S, Schlums H, Sérézal IG, Martini E, Chiang SC, Marquardt N, et al. CD49a Expression Defines Tissue-Resident CD8+ T Cells Poised for Cytotoxic Function in Human Skin. Immunity (2017) 46(2):287–300. doi: 10.1016/j.immuni.2017.01.009
212. Li G, Larregina AT, Domsic RT, Stolz DB, Medsger TA Jr., Lafyatis R, et al. Skin-Resident Effector Memory CD8+ Cd28–T Cells Exhibit a Profibrotic Phenotype in Patients With Systemic Sclerosis. J Invest Dermatol (2017) 137(5):1042–50. doi: 10.1016/j.jid.2016.11.037
213. Huseby ES, Huseby PG, Shah S, Smith R, Stadinski BD. Pathogenic CD8 T Cells in Multiple Sclerosis and its Experimental Models. Front Immunol (2012) 3:64. doi: 10.3389/fimmu.2012.00064
214. Sasaki K, Bean A, Shah S, Schutten E, Huseby PG, Peters B, et al. Relapsing–remitting Central Nervous System Autoimmunity Mediated by GFAP-Specific CD8 T Cells. J Immunol (2014) 192(7):3029–42. doi: 10.4049/jimmunol.1302911
215. Machado-Santos J, Saji E, Tröscher AR, Paunovic M, Liblau R, Gabriely G, et al. The Compartmentalized Inflammatory Response in the Multiple Sclerosis Brain is Composed of Tissue-Resident CD8+ T Lymphocytes and B Cells. Brain (2018) 141(7):2066–82. doi: 10.1093/brain/awy151
216. Kuric E, Seiron P, Krogvold L, Edwin B, Buanes T, Hanssen KF, et al. Demonstration of Tissue Resident Memory CD8 T Cells in Insulitic Lesions in Adult Patients With Recent-Onset Type 1 Diabetes. Am J Pathol (2017) 187(3):581–8. doi: 10.1016/j.ajpath.2016.11.002
217. Hand TW, Cui W, Jung YW, Sefik E, Joshi NS, Chandele A, et al. Differential Effects of STAT5 and PI3K/AKT Signaling on Effector and Memory CD8 T-Cell Survival. Proc Natl Acad Sci (2010) 107(38):16601–6. doi: 10.1073/pnas.1003457107
218. Chetoui N, Boisvert M, Gendron S, Aoudjit F. Interleukin-7 Promotes the Survival of Human CD4+ Effector/Memory T Cells by Up-Regulating Bcl-2 Proteins and Activating the JAK/STAT Signalling Pathway. Immunology (2010) 130(3):418–26. doi: 10.1111/j.1365-2567.2009.03244.x
219. Grange M, Giordano M, Mas A, Roncagalli R, Firaguay G, Nunes JA, et al. Control of CD 8 T Cell Proliferation and Terminal Differentiation by Active STAT 5 and CDKN 2a/CDKN 2b. Immunology (2015) 145(4):543–57. doi: 10.1111/imm.12471
220. Quigley M, Huang X, Yang Y. Extent of Stimulation Controls the Formation of Memory CD8 T Cells. J Immunol (2007) 179(9):5768–77. doi: 10.4049/jimmunol.179.9.5768
221. Li Y, Shen C, Zhu B, Shi F, Eisen HN, Chen J. Persistent Antigen and Prolonged AKT–mTORC1 Activation Underlie Memory CD8 T Cell Impairment in the Absence of CD4 T Cells. J Immunol (2015) 195(4):1591–8. doi: 10.4049/jimmunol.1500451
222. Kim EH, Sullivan JA, Plisch EH, Tejera MM, Jatzek A, Choi KY, et al. Signal Integration by Akt Regulates CD8 T Cell Effector and Memory Differentiation. J Immunol (2012) 188(9):4305–14. doi: 10.4049/jimmunol.1103568
223. Ferreira C, Barros L, Baptista M, Blankenhaus B, Barros A, Figueiredo-Campos P, et al. Type 1 Treg Cells Promote the Generation of CD8+ Tissue-Resident Memory T Cells. Nat Immunol (2020) 21(7):766–76. doi: 10.1038/s41590-020-0674-9
224. Koch MA, Thomas KR, Perdue NR, Smigiel KS, Srivastava S, Campbell DJ. T-Bet+ Treg Cells Undergo Abortive Th1 Cell Differentiation Due to Impaired Expression of IL-12 Receptor β2. Immunity (2012) 37(3):501–10. doi: 10.1016/j.immuni.2012.05.031
225. Lommerts JE, Bekkenk MW, Luiten RM. Vitiligo Induced by Immune Checkpoint Inhibitors in Melanoma Patients: An Expert Opinion. Expert Opin Drug Safety (2021) 20(8):883–8. doi: 10.1080/14740338.2021.1915279
226. Stolley JM, Masopust D. Tissue-Resident Memory T Cells Live Off the Fat of the Land. Cell Res (2017) 27(7):847–8. doi: 10.1038/cr.2017.49
227. Jung JH, Rha MS, Sa M, Choi HK, Jeon JH, Seok H, et al. SARS-CoV-2-Specific T Cell Memory Is Sustained in COVID-19 Convalescent Patients for 10 Months with Successful Sevelopment of Stem Cell-Like Memory T Cells. Nat Commun (2021) 12(1):4043. doi: 10.1038/s41467-021-24377-1
228. Neidleman J, Luo X, George AF, McGregor M, Yang J, Yun C, et al. Distinctive Features of SARS-CoV-2-Specific T Cells Predict Recovery From Severe COVID-19. Cell Rep (2021) 36(3):109414. doi: 10.1016/j.celrep.2021.109414
229. Zhao Y, Kilian C, Turner J-E, Bosurgi L, Roedl K, Bartsch P, et al. Clonal Expansion and Activation of Tissue-Resident Memory-Like TH17 Cells Expressing GM-CSF in the Lungs of Patients With Severe COVID-19. Sci Immunol (2021) 6(56):eabf6692. doi: 10.1126/sciimmunol.abf6692
230. Roberts LM, Jessop F, Wehrly TD, Bosio CM. Cutting Edge: Lung-Resident T Cells Elicited by SARS-CoV-2 do Not Mediate Protection Against Secondary Infection. J Immunol (2021) 207(10):2399–404. doi: 10.4049/jimmunol.2100608
231. Goplen NP, Cheon IS, Sun J. Age-Related Dynamics of Lung-Resident Memory CD8+ T Cells in the Age of COVID-19. Front Immunol (2021) 12:323. doi: 10.3389/fimmu.2021.636118
232. Cheon IS, Li C, Son YM, Goplen NP, Wu Y, Cassmann T, et al. Immune Signatures Underlying Post-Acute COVID-19 Lung Sequelae. Sci Immunol (2021) 6(65):eabk1741. doi: 10.1126/sciimmunol.abk1741
233. Vahed SZ, Khatibi SMH, Ahmadian E, Ardalan M. Targeting Chronic COVID-19 Lung Injury; Tofacitinib can be Used Against Tissue-Resident Memory T Cells. Biomed Pharmacother (2022) 147:112614. doi: 10.1016/j.biopha.2022.112614
234. Jung JH, Rha M-S, Sa M, Choi HK, Jeon JH, Seok H, et al. SARS-CoV-2-Specific T Cell Memory is Sustained in COVID-19 Convalescent Patients for 10 Months With Successful Development of Stem Cell-Like Memory T Cells. Nat Commun (2021) 12(1):4043. doi: 10.1038/s41467-021-24377-1
235. Guerrera G, Picozza M, D’Orso S, Placido R, Pirronello M, Verdiani A, et al. BNT162b2 Vaccination Induces Durable SARS-CoV-2–Specific T Cells With a Stem Cell Memory Phenotype. Sci Immunol 6(66):eabl5344. doi: 10.1126/sciimmunol.abl5344
236. Azgomi MS, La Manna MP, Badami GD, Ragonese P, Trizzino A, Dieli F, et al. A Rapid and Simple Multiparameter Assay to Quantify Spike-Specific CD4 and CD8 T Cells After SARS-CoV-2 Vaccination: A Preliminary Report. Biomedicines (2021) 9(11). doi: 10.3390/biomedicines9111576
237. Brehm MA, Daniels KA, Welsh RM. Rapid Production of TNF-α Following TCR Engagement of Naive CD8 T Cells. J Immunol (2005) 175(8):5043–9. doi: 10.4049/jimmunol.175.8.5043
238. Arunachalam PS, Wimmers F, Mok CKP, Perera RAPM, Scott M, Hagan T, et al. Systems Biological Assessment of Immunity to Mild Versus Severe COVID-19 Infection in Humans. Science (2020) 369(6508):1210–20. doi: 10.1126/science.abc6261
239. Filbin MR, Mehta A, Schneider AM, Kays KR, Guess JR, Gentili M, et al. Longitudinal Proteomic Analysis of Severe COVID-19 Reveals Survival-Associated Signatures, Tissue-Specific Cell Death, and Cell-Cell Interactions. Cell Rep Med (2021) 2(5):100287. doi: 10.1016/j.xcrm.2021.100287
240. Thwaites RS, Sanchez Sevilla Uruchurtu A, Siggins MK, Liew F, Russell CD, Moore SC, et al. Inflammatory Profiles Across the Spectrum of Disease Reveal a Distinct Role for GM-CSF in Severe COVID-19. Sci Immunol (2021) 6(57):eabg9873. doi: 10.1126/sciimmunol.abg9873
241. Fox CJ, Hammerman PS, Thompson CB. Fuel Feeds Function: Energy Metabolism and the T-Cell Response. Nat Rev Immunol (2005) 5(11):844–52. doi: 10.1038/nri1710
242. Bordbar A, Mo ML, Nakayasu ES, Schrimpe-Rutledge AC, Kim YM, Metz TO, et al. Model-Driven Multi-Omic Data Analysis Elucidates Metabolic Immunomodulators of Macrophage Activation. Mol Syst Biol (2012) 8(1):558. doi: 10.1038/msb.2012.21
243. O’Neill LAJ, Hardie DG. Metabolism of Inflammation Limited by AMPK and Pseudo-Starvation. Nature (2013) 493(7432):346–55. doi: 10.1038/nature11862
Keywords: CD8 TRM cells, CD8 TSCM cells, metabolism, differentiation, infectious diseases
Citation: La Manna MP, Shekarkar Azgomi M, Tamburini B, Badami GD, Mohammadnezhad L, Dieli F and Caccamo N (2022) Phenotypic and Immunometabolic Aspects on Stem Cell Memory and Resident Memory CD8+ T Cells. Front. Immunol. 13:884148. doi: 10.3389/fimmu.2022.884148
Received: 25 February 2022; Accepted: 19 May 2022;
Published: 17 June 2022.
Edited by:
Loretta Tuosto, Sapienza University of Rome, ItalyReviewed by:
Claudio Procaccini, Consiglio Nazionale Delle Ricerche (CNR), ItalyRajko Reljic, St George’s, University of London, United Kingdom
Copyright © 2022 La Manna, Shekarkar Azgomi, Tamburini, Badami, Mohammadnezhad, Dieli and Caccamo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nadia Caccamo, bmFkaWEuY2FjY2Ftb0B1bmlwYS5pdA==
†These authors share first authorship
‡These authors share last authorship