 Stinne R. Greisen
Stinne R. Greisen Maithri Aspari
Maithri Aspari Bent Deleuran
Bent Deleuran- 1Department of Biomedicine, Aarhus University, Aarhus, Denmark
- 2Department of Rheumatology, Aarhus University Hospital, Aarhus, Denmark
Immune checkpoint receptors are key players in regulating the immune response. They are responsible for both generating an immune response sufficient to kill invading pathogens, balancing the same response, and protecting against tissue destruction or the development of autoimmune events. The central role of the co-inhibitory receptors also referred to as inhibitory immune checkpoints, including PD-1 and CTLA-4 has become especially evident with the cancer treatments targeting these receptors. Blocking these pathways enhances the immune activity, resulting in both an increased chance of cancer clearance, at the same time induction of immune-related adverse events (irAE). Some of these irAE progress into actual autoimmune diseases with autoantibodies and symptoms, undistinguished from the naturally occurring diseases. This review will take advantage of the lessons learned from immune checkpoint blockade and relate this knowledge to our understanding of the same pathways in naturally occurring autoimmune diseases, mainly focusing on rheumatic diseases.
Introduction: Balancing Health and Disease
The immune system is unique when it comes to its ability to protect our body and maintain homeostasis. It can both defend us against foreign pathogens and at the same time recognize and accept self-antigens. This equilibrium is also essential in avoiding cancer and controlling aging. Owing to the complexity of this system, we seem to be only scratching the surface in our understanding of the plethora of factors involved in this balanced regulation.
For T cells, the evolvement of a healthy immune system starts in the thymus where CD4 and CD8 T cells undergo positive and negative selection, to ensure an optimal reactivity to foreign antigens, and high tolerance towards self (1). Once out of the thymus, these cells will use an array of mechanisms when encountering a foreign antigen. The innate immune system is the first line of defense, subsequently resulting in the activation and recruitment of T cells and finally initiating an adaptive immune response. For antigen presenting cells to successfully activate a T cell, the T cell will need a second signal through an immune checkpoint, or co-receptor, which will be upregulated upon T cell receptor signaling or by cytokines (2, 3). The activation of co-receptors can either lead to increased activation of the T cell or inhibit the activation resulting in a dampened response or even anergy. The outcome from the balanced signaling between the multiple co-receptors thus determines the fate of an antigen-activated T cell. Often, the presentation of a high-affinity antigen that is significantly different from self, will result in an upregulation of the co-stimulatory receptors (CSR), like CD28 or 4-1BB, leading to full activation of the T cell (4). If the presented antigen resembles self or is of low affinity, upregulation of co-inhibitory receptors (CIR), including programmed death-1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) will be more pronounced (5, 6). These pathways will cause downregulation of the T cell activation, including diminished cytokine production, less proliferation, and reduced motility (7). Failure in balancing this second signal may cause chronic infections, cancers, or autoimmune diseases. The innate immune system can also initiate a chronic inflammatory condition, often referred to as autoinflammation. This topic is, however, beyond the scope of this review (8). In the following, we will focus on the CIR, their ligands, and their role in the development of autoimmune diseases. We consider this with the lessons learned from cancer treatment using immune checkpoint inhibitors (ICI), resulting in various inflammatory conditions, referred to as immune-related adverse events (irAE).
Results: The First 10 Years of Immune Checkpoint Inhibitor Therapy
One of the central features in cancer development is the escape of immune surveillance. One mechanism exploited by cancer cells is the upregulation of ligands for the CIR, causing infiltrating immune cells to be shut down, thus avoiding immune mediated killing (9). Immune checkpoint inhibitors block CIR or their ligands. The first drug on the market targeted CTLA-4 (10, 11) and has in conjunction with ICI towards the PD-1 pathway, revolutionized cancer treatment. As a result of the increased immune activation, irAE and reduced cancer burden often goes hand-in-hand. However, these drugs provide valuable insight into the understanding of the role of CIR in the development of autoimmunity. In some cases, irAE become chronic conditions, closely resembling an autoimmune disease. Presentation with a full-blown debut of an autoimmune disease, even with autoantibodies is described (12, 13). IrAE are seen with a large variation in intensity, but are very common and affect 50%-70% of patients in monotherapy and more than 90% in combination therapy (12). Often, combination therapy targets both CTLA-4 and PD-1, which makes it difficult to pinpoint a specific clinical manifestation of one or the other. Despite several years in use, it remains difficult to predict which patients will respond to the treatment, and the search for prognostic biomarkers is ongoing (14). The development and treatment of irAE are often the governing steps for patients in ICI treatment. In patients with known systemic immunological disorders, ICI treatment is still limited to very few trials. Worsening, or flare in their disease seems to closely follow tumor regression (15). Considering the occurrence of irAE, it is appealing that the CIR pathways play a prominent role in the development of autoimmune diseases (Figure 1).
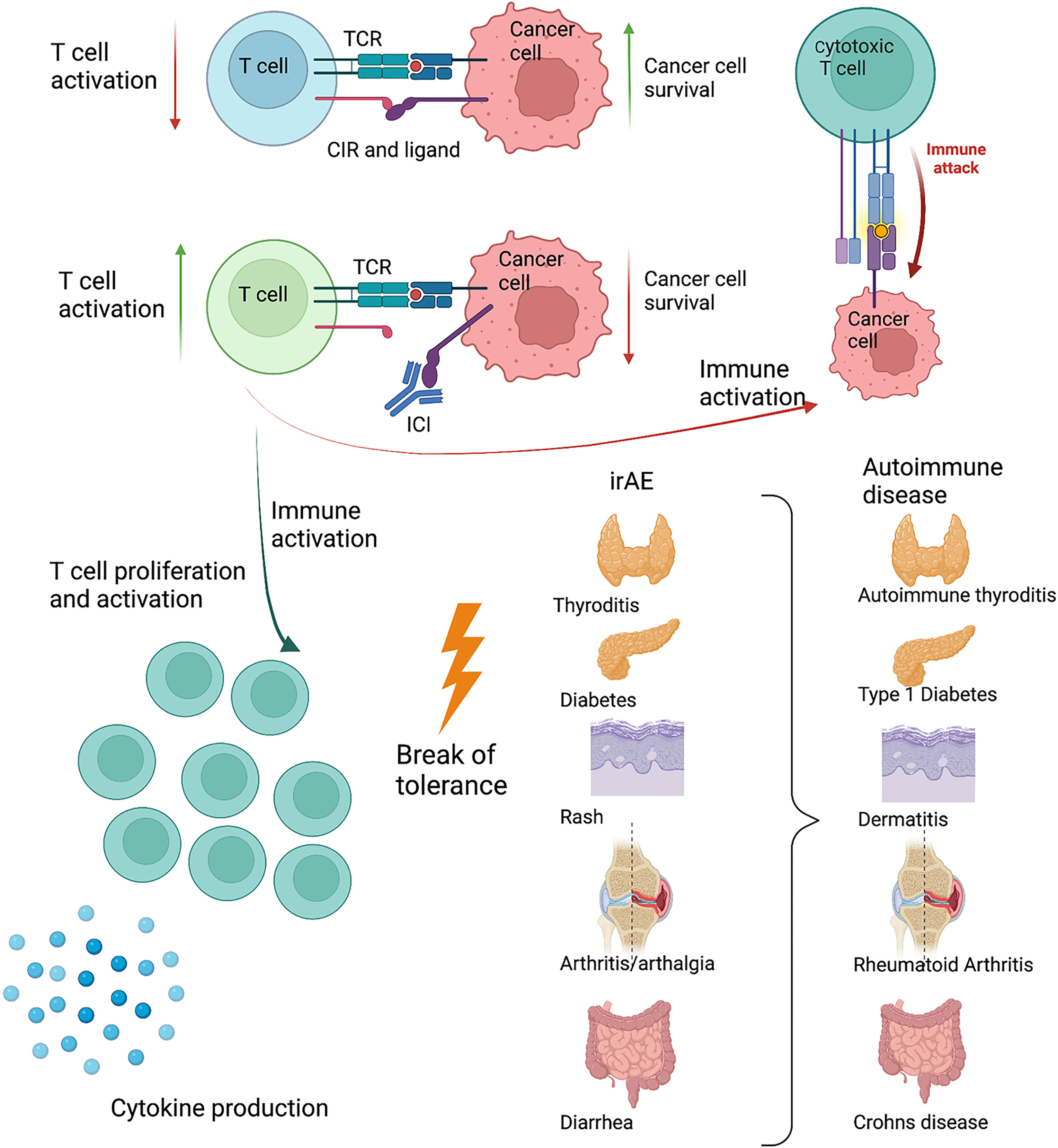
Figure 1 Schematic drawing of the interaction between an activated T cell and a cancer cell. The cancer cell exploits the PD-1 pathway, reducing T cell activity. When blocking the PD-1 pathway with antibodies, the T cell becomes sufficiently activated to kill the cancer cell. As a response to increased T cell activation and inflammation, immune related adverse events (irAE) develop. These target different organs and resembles known autoimmune diseases present in the same organ. Created with BioRender.com.
Many irAE tend to fade or disappear when the ICI treatment is ceased. These irAE thus deviate from what we normally see when examining an autoimmune disease. This supports a role for CIR serving to decrease immune activation, and their absence, or reduced function, leading to temporary symptoms or disease. Contrarily, upon considering the cases where the irAE progress into chronic diseases, these indicate that the dysfunction of CIR can induce a break in tolerance leading to autoimmunity (16). The factors determining these two different outcomes remain to be understood. More than 40% of the irAE develop into chronic conditions, often less responsive to steroids (17). The inflammatory conditions often present within the first 2-6 weeks of therapy, but may also arise already after the first treatment, or not until after several years of treatment (17, 18). Although autoimmune diseases are closely associated with HLA genotypes only a few studies have investigated the association between irAE and HLA type, and with no clear conclusion (19, 20).
It does occur that irAE are much more common than their “counter” autoimmune disease. One case is hypophysitis, an autoimmune reaction in the pituitary gland, very rare in its idiopathic form, but with an overall incidence after ICI treatment reported in a recent meta-analysis as 14%. This is a relatively common irAE, and also of significant severity, subsequently demanding life-long treatment as multiple hormonal axes are involved, and the damage is irreversible (21). Primary hypophysitis is not associated with systemic inflammation, and CIR have to our knowledge, not been investigated in the development of primary hypophysitis, where the mechanisms responsible for development are still largely unknown (22).
From a clinical viewpoint, it remains a challenge to treat patients with irAE especially if the symptoms become chronic, or hinder continued treatment of the oncologic disease. The first line of treatment is often corticosteroids, especially if these can be used locally (23, 24). However, systemic corticosteroid treatment results in both reduced antigen recognition and ability for cellular toxicity, especially ADCC. The use of systemic corticosteroid treatment thus raises a concern about the continued efficacy of the ICI to induce tumor eradication (25). This has resulted in a general consent to reducing corticosteroids to a minimum as quickly as possible, and a tendency to use disease-modifying anti-rheumatic drugs (DMARD) to reach control of the irAE. Anti-TNF antibodies and anti-IL-6R antibodies are the mainly used therapies, but large-scale and long-term studies are still not carried out (26, 27). Therefore, a better understanding of this area is needed, to improve treatment for both the cancer and the irAE.
The CD28/B7 Family
The CD28/B7 family of CSR and CIR is probably the best described among all co-receptors. CD28 was the first identified CSR and is crucial for optimal T cell activation (28). Signaling through the CD28 pathway induces complete T cell activation with increased motility, proliferation, and cytokine production including IL-2 and IFN-y. CD28 signals downstream through PI3K and PKC0, finally resulting in phosphorylation of AKT and NFkB. Mice lacking CD28 have a reduced immunoglobulin concentration and class switch, as well as decreased levels of IL-2. However, cytotoxic T cells do develop in these mice, and they do not succumb to infections (29). CD28 is necessary for the development of Tregs, but also favors the development of immune reactive T cells. Despite promising evidence (30), targeting CD28 in autoimmunity has not reached common clinical use, and especially for rheumatoid arthritis (RA), the focus has been on targeting CTLA-4, which also binds CD80, and competes with CD28 (31). This review will primarily focus on CIR, and the role of CD28 will not be discussed further. The two other two important members in the CD28/B7 family are CTLA-4 and PD-1, these being CIR (Figure 2).
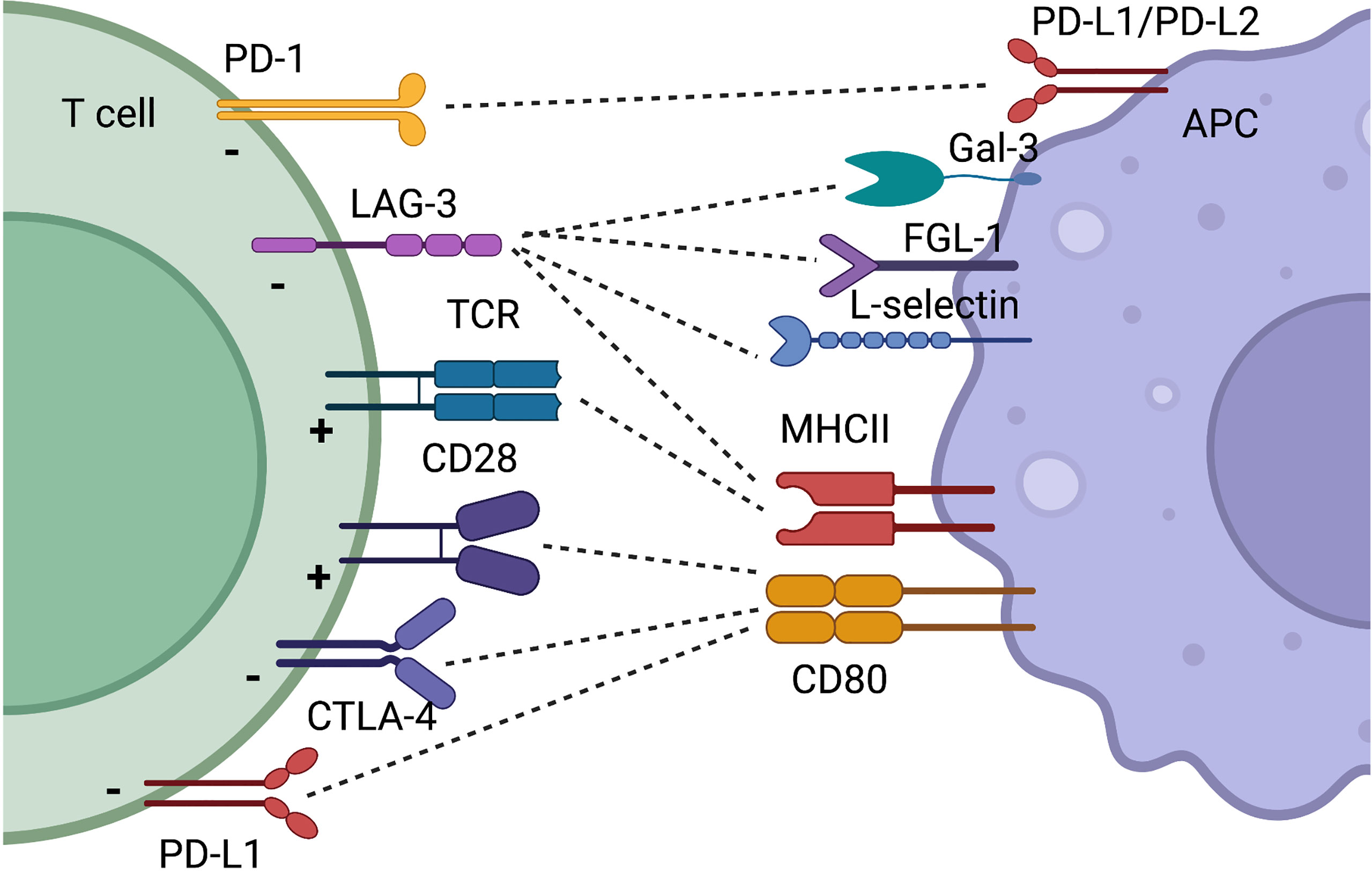
Figure 2 Simplified schematic illustration of CIR and CSR discussed in this review. Some receptors have binding partners which is illustrated by dotted lines. The function on the T cell by the receptor is indicated with a “+” for T cell activation and a “-” when T cell activity is decreased. Created with BioRender.com.
The PD-1 Pathway
PD-1 is a trans-membranous receptor mainly expressed by activated lymphocytes, however PD-1 expression has been described on several other cell types and in multiple tissues (32, 33). PD-1 has two ligands, PD-L1 and PD-L2 (34, 35). Engaging the PD-1 receptor causes phosphorylation of the ITSM motif and recruitment of SHP2, subsequently resulting in dephosphorylation of PI3K and AKT, eventually causing reduced activity in NFkB and thereof, a decreased T cell activation (3, 36, 37). Cancer cells utilize the expression of PD-L1 to silence inhibitory signals from cytotoxic T cells and other host immune cells. PD-1 or PD-L1 expression is often used as a marker (though poor) to identify tumors where treatment with ICI seems feasible (38). The PD-1 pathway not only signals through the PD-1 receptor; reverse signaling through PD-L1 is described, increasing cell survival and decreasing type 1 IFN responsiveness (39). This can be exploited by cancer cells, further supporting tumor growth and immune avoidance. PD-L1 reverse signaling is also crucial for chemokine-mediated dendritic cell migration from the skin to the lymph node (40). The role of reverse signaling in autoimmunity remains to be clarified.
Both PD-1 and its ligands are present in soluble (s) forms. These have been investigated as biomarkers of inflammatory diseases and multiple cancers. Most evidence suggests that levels of the soluble receptors increase as a response to inflammation. The functional role, if any, still remains up for debate. The soluble forms have been suggested to both increase immune activity by blocking the PD-1 pathway and decrease immune activity by positively engaging the PD-1 pathway. The role of the soluble forms is further complicated when recombinant proteins are used as surrogates for the soluble forms in both in vivo and in vitro studies (41–44).
The PD-1 pathway is central in maintaining immunological balance (33) and knockout mice of the PD-1 receptor causes lupus-like disease at a late stage in life (45).
PD-1 inhibitors are better tolerated than CTLA-4 inhibitors, but irAE affecting the joints, lungs and thyroid are more common (12). Hypothyroidism is the most common endocrinopathy seen in 6-10% (46)whereas newly onset T1DM is seen in about 1% of patients. In the development of childhood T1DM, polymorphisms in the PD-1 receptor are associated with an increased risk of T1DM. Patients with newly onset T1DM have decreased numbers of circulating CD4+PD-1+ T cells, and PD-1 fails to upregulate on peripheral T cells (47). The PD-1 axis is only scarcely investigated thyroid autoimmunity, but polymorphism in the PD-1 gene is associated with an increased incidence of autoimmune thyroiditis (48). Increased numbers of PD-1 infiltrating T cells are also seen in autoimmune thyroiditis (49). With hypothyroidism being one of the most prominent irAE after PD-1 blockade, this pathway might be more important than previously considered in the development of thyroid autoimmunity.
Many patients also experience rheumatological toxicities and close to 40% of the patients develop signs of inflammatory joint pain (16). The rheumatological toxicities can develop into diseases with autoantibodies, and both RA, scleroderma-like disease (50, 51), polymyositis and Sjögrens are seen.
Especially blocking the PD-1 pathway can cause pneumonitis which is a severe irAE, with high mortality. Especially, does pre-existing pulmonary fibrosis increase the risk of anti-PD-1-related pneumonitis (52, 53).
In accordance with the development of rheumatological irAE the PD-1 pathway plays a significant role in several rheumatological diseases including; RA, lupus (SLE), polymyalgia, polymyositis, and SSc (54). In RA; we have reported PD-1 expression to be increased both in the synovium and in the peripheral circulation (55–57). The increased expression correlates with disease activity, probably representing an immune activation. Looking further into the cellular and peripheral populations, PD-1 expression is associated with a pathological T cell phenotype involved in B cell activation and plasma cell maturation, resulting in increased antibody production and disease severity (58, 59). However, PD-1 expression also characterize regulatory T cells (Treg) and Tex which both are associated with less immune activation, and a better prognosis in autoimmune diseases (32, 57).
Looking into SLE the picture remains complicated. Genetic variances in the PD-1 gene are considered related to the development of the disease (60), and PD-1 expression is upregulated and associated with disease severity (61, 62). Recent bioinformatic and multi-omics approaches have made it evident that PD-1 is expressed on multiple different cell types, which are not solemnly associated with immune down regulation (58). The distinction between the different cell types expressing PD-1 is especially important in the understanding of how the PD-1 pathway is associated with disease activity and severity in SLE, where it is demonstrated that the increased PD-1 expression is especially seen in T helper cell populations with more pro-inflammatory properties (63). This has become evident that especially T peripheral helper cells (Tph) cells are upregulated in SLE -supporting the close association with autoantibodies in this disease (64). Turning to Treg, the expression of PD-1 is not reported different in SLE patients, however, the expression of PD-L1 is suggested decreased in SLE Treg, and in accordance is negatively correlated to disease activity (65). Despite lupus-like disease being one of the clinical manifestations in PD-1 KO mice, SLE as an irAE is relatively rare (66). It could be hypothesized that diseases largely driven by auto antibodies will take a long time to develop. When more patients will initiate early treatment with ICI, potentially increasing both survival and treatment duration, SLE-like disease could become a more common irAE.
Systemic scleroderma (SSc) is also a rheumatic disease, where autoantibodies play a significant role. Again, few cases have been reported after ICI treatment (12, 67). In SSc, a dysfunctional PD-1 pathway has been correlated with disease outcomes and clinical parameters (68). Raised levels of sPD-1 and sPD-L2 has been associated with increased skin thickness. In SSc patients, the numbers of PD-1–expressing cells within the Treg cell subset and within γδT cells (69) were significantly increased compared to those of healthy subjects. Increased frequency of T lymphocytes co-expressing PD-1 and T cell immunoreceptor with Ig and ITIM domains (TIGIT) was also observed in SSc patients. This accumulated expression of multiple CIR is in accordance with the hallmarks of exhausted T cells (Tex).
In in vivo models of mice with Topoisomerase I(Top-I)–induced SSc, production of IL-10 by Top-I specific B cells in cultures with T cells and Top-I protein was significantly higher than that by conventional B cells, this effect could be overcome by injection with recombinant chimeric PD-1-Fc and PD-L2-Fc (68), thereby supporting activation of the PD-1 pathway. Thus, substantiating the regulatory effect of PD-1 in maintaining homeostasis. Suggesting that interaction of PD-1 and PD-L2 is required for the production of IL-10 by B cells during T cell–B cell autoantigen-specific cognate interactions in SSc.
Taken together, it is clear that signaling through the PD-1 receptor decreases immune activation, and supports immunological tolerance. However, PD-1 is expressed both by exhausted T cells, but also as a result of activation. Therefore, PD-1 can be expressed by T cell driving immune activation, but also by T cells participating in lowering the immune reaction. This adds to the complexity of the understanding of when and how PD-1 expressing T cells are associated with disease activity in inflammatory conditions.
CTLA-4
CTLA-4 is a transmembranous receptor expressed by activated T cells. Expression is upregulated upon T cell antigen encounter, and as for PD-1, low affinity antigens will induce a higher upregulation of CTLA-4. CTLA-4 binds to CD80/CD86 and competes with CD28 upon this binding, but with a higher avidity and affinity than CD28. A signal through CTLA-4 decreases T cell activation by the SHP2 and PP2A pathways, with many similarities to the PD-1 signaling pathway (70).
CTLA-4 is expressed by T cells, B cells, NK cells and regulatory T cells (Treg) (71, 72). T cells only express low levels of CTLA-4 on their surface, even when activated. CTLA-4 is mainly localized intracellular, where CTLA-4 is found in the golgi network, as well as in endosomes, secretory granules, and lysosomal vesicles from where it is circulated to the surface. This process is reviewed by Schneider et al (73).
CTLA-4 is also present in a soluble form, however, the importance of this is still not fully elucidated. Studies do support that sCTLA-4 plays an important role in keeping optimal immune surveillance (74).
In CTLA-4 knock-out mice, severe lymphoid proliferation and death appear, when the animals are 3-4 weeks old (75). Although no disease equivalent is known in humans, the importance of CTLA-4 in keeping tolerance is supported by fact that polymorphisms in the gene are associated with an increased risk of autoimmune disease. Importantly, these are not allocated to one disease but include T1DM, thyroiditis, Mb Addison, Crohn’s disease, RA, and multiple sclerosis, among many others (76). Engaging this pathway with CTLA-4:Ig it has been shown to be an effective treatment, highlighting that this pathway plays a role in maintaining immune reaction during autoimmune disease.
Antibodies targeting CTLA-4 are approved in multiple cancers including; metastatic melanoma, hepatocellular, renal carcinoma, mesothelioma, colon cancer, and non-small cell lung cancer, and more will be included in the near future. Monotherapy targeting CTLA-4 is associated with more severe irAE compared with monotherapy targeting the PD-1 pathway (12). This aligns with CTLA-4 KO mice having a much more severe phenotype than PD-1 KO mice (45, 75). The most common irAE from anti-CTLA-4 treatment are related to the gut and skin. This suggests CTLA-4 has a functional implication in preserving gut tolerance and supporting barrier functions (13, 77). Considering the evolving evidence of the major significance of the microbiota in the development of autoimmune diseases, including both rheumatic and gastrointestinal diseases, the role of CTLA-4, in combination with PD-1 is further highlighted by both their abilities to influence IgA synthesis and the microbiome (78, 79). Controversially, the CTLA-4:Ig fusion protein, Abatacept, was shown not to be clinically relevant in controlling Crohn’s Disease or ulcerative colitis in a clinical trial (80) . This does not exclude that CTLA-4 play an important role in keeping optimal conditions in the non-inflamed gut. This notion is supported by the observation that genetic changes in CTLA-4 is associated with an early onset of Crohn’s disease (81).
CTLA-4 functions in humans are elucidated by the in vivo use of CTLA-4-Ig fusion protein, used for the treatment of various immunological diseases. Abatacept was the first CTLA-4-Ig molecule to be approved for humans and has since been followed by Belatacept, with a higher affinity for CD86, making it 10-fold more potent in vitro, than Abatacept (82). These two CTLA-4-Ig molecules have been investigated in multiple autoimmune disorders, including RA, T1DM, diffuse cutaneous systemic sclerosis (dcSSc), psoriasis, and SLE. Both have also been tested, as inducers of tolerance to specific antigens upon organ transplantations. Especially Belatacept has shown to be efficacious in renal transplantation (83).
RA is by far the disease with the highest number of reports, as Abatacept has been approved for the treatment of moderate to severe disease for several of years (84). Its efficacy is in line with anti-TNF-antibodies (85). Since anti-CTLA-4 antibodies are used to treat cancer, it is tempting to suspect that treatment of RA with Abatacept would increase the risk of developing malignancy. However, no clear picture emerges. Some studies have reported a small increased risk, others none (86–88).
This naturally led to the question of why treatment with CTLA-4:Ig in RA, does not result in a greater increase in their cancer risk when blocking CTLA-4 in cancer results in autoimmunity. RA is dominated by a high number of both Tex cells and senescent T cells (89, 90), similar to chronic infections diseases. In patients with chronic hepatitis C, antiviral treatment lead to a decreased number of Tex cells (91). Assuming that Abatacept will do the same in RA the risk of developing cancer will also decrease as Tex favors cancer development. Therefore, it is a balanced outcome of disease activity, disease length, and treatment, thus explaining the low or absent risk of developing cancer in the Abatacept studies. The understanding of CTLA-4’s role in RA mainly comes from studying the effects of Abatacept. One major point of action shown for Abatacept is the downregulation of cytokine production in co-cultures, resulting in significantly lower concentration of IL-2, TNF-a and IL-1b (92). Additionally, data suggest that macrophages shift from an M1 to an M2 phenotype, when cultured in the presence of Abatacept (93), favoring a less inflammatory environment.
Fully in line with CTLA-4 function, Abatacept treatment slightly decreases but does not completely inhibit a vaccination response (94, 95).
In SLE and other inflammatory connective tissue diseases, Abatacept has shown efficacy in animal models, but generally failed in clinical studies (96, 97). Since the first casuistic report of Abatacepts efficacy in SSc, a phase II study has supported CTLA-4s possible role in the treatment of this disease (98, 99). Evidence on CTLA-4’s role in initiating connective tissue diseases remains to be fully elucidated, and most evidence is only on the genetic polymorphisms of CTLA-4 being associated with an increased risk of connective tissue diseases (100).
LAG-3
Lymphocyte activation gene 3 (LAG-3) is still one of the less described CIR. LAG-3 is expressed upon T cell activation and has high homology with CD4. LAG-3 only binds to stabilized MHC class II molecules, suggesting that this inhibitory pathway exerts its functions when the T cells encounter antigens with a high specificity (101–103). Several other ligands for LAG-3 have been described. Among these, are Fibrinogen-like Protein 1 (FGL-1), L-selectin, and Galectin-3 (Gal-3) (104–106). As LAG-3 is also present in a soluble form, these multiple ligands and potential large multimeric formations suggest that the functionality of LAG-3 is complex. LAG-3 regulates T cell proliferation and homeostasis of both effector T cells and Tregs (107, 108).
Single LAG-3– and PD-1–deficient mice display minimal immunopathologic sequelae, double LAG-3/PD-1 knockout mice develop lethal systemic autoimmunity. Though mice lacking LAG-3 do not develop spontaneous autoimmune disease in non-autoimmune prone mouse strains, LAG-3 induced lethal myocarditis in BALB/c mice deficient for the gene encoding for PD-1. In addition, LAG-3 deficiency alone accelerated T1DM in nonobese diabetic(NOD) mice (10). Moreover, a cytotoxic LAG-3 Ab has been evaluated in a nonhuman primate model of delayed-type hypersensitivity (109). Contrarily, in a phase I clinical trial investigating psoriasis the depletion of LAG-3–positive T cells was linked to a reduced Th1-driven skin inflammation, lasting even in the absence of the depleting antibody (110).
The LAG-3 pathway is shown to be involved in the development of irAE such as colitis, RA, and diabetes (111, 112). Evidence from mouse studies suggests efficacy in cancer treatment, especially in combination with either anti-PD-1 or anti-CTLA-4 therapy. This has led to a recent study evaluating the effects of anti-LAG-3 antibodies in combination with anti-PD-1 antibodies, versus anti-PD-1 as monotherapy in patients with advanced melanoma (113). Here, addition of anti-LAG3 increased to the progression-free survival, but also increased the rate of irAE. With special relation to rheumatology, arthralgia was the most commonly increased irAE induced after targeting LAG-3. This supports LAG-3’s role in joint diseases. Currently, drugs that either augment LAG-3’s effects or deplete activated T cells that express LAG-3 are under development for chronic inflammatory diseases (114).
Despite emerging clinical trials with both agonistic and antagonistic antibodies targeting LAG-3, it is important to recognize that the knowledge of LAG-3 in relation to autoimmune disease is still rather limited. We have reported that LAG-3 could play a role in juvenile rheumatoid arthritis, supporting that LAG-3 could be important for immunoreactions and immune maturation during infancy (115).
Apart from the immune checkpoint molecules described here, many more are known, but few of these have reached the level of clinical studies. Among these are 4-1BB, TIGIT, TIM-3, B7-H3, B7-H4, ICOS to mention a few. The next decade will lead to new and interesting descriptions of effects and irAE that will help us to a better understanding of the immune system.
Conclusion
Integrating our knowledge of irAE provides a better understanding of the role of CIR in both the initiation and progression of inflammatory diseases. It is suggestive that CIR may participate in the break of tolerance, but also in keeping the diseases in a chronic state. Which additional factors are needed for a break of tolerance when CIR are blocked, remains to be understood. It has also become evident that not all autoimmune diseases are represented equally in the irAE spectrum. Apart from myositis and myocarditis, inflammatory connective tissue diseases like SLE and SSc are still not as common as arthritis and polymyalgia. Among endocrinopathies, the thyroid gland is much more commonly affected than the pancreas. The reasons for this could be plenty. One is the time perspective as ICI treatment has only been around for less than a decade, and both diabetes and connective tissue diseases may develop over a longer period of time. For connective tissue diseases, especially considering that autoantibodies are present years before the development of clinical symptoms (116–118), which suggests a prolonged period to induce the disease. Also, the presence of these autoantibodies may be crucial for initiating the disease, and targeting two CIR may not be enough for autoantibodies to develop. Future observational studies with long term follow-up could change this picture.
With ICI drugs coming to the market targeting different CIR, we might see a more specific organ preference, depending on the targeted CIR and thereby reflecting the pathology of the “natural” autoimmune disease. A blurred picture is starting to emerge, where we see a tendency for CTLA-4 blockade to induce gut affection more often than blockade of the PD-1 pathway. By contrast, PD-1 and LAG-3 blockade more often result in pneumonitis and joint affection (119).
Our knowledge of CIR role is mainly associated with adults. Our major expositor to different antigens is highest during the first part of life, supporting that CIR play distinct functions dependent on age. We do, however, still know very little of their function in the early stages of life.
Taken together, it is clear that we have only scratched the surface, when it comes to understanding the complexity immune checkpoint molecules have on development of diseases related to immunosurveillance.
Author Contributions
All authors contributed equally to searching and implementing relevant literature and editing the manuscript. SG prepared the initial draft and BD served as the senior and last author. All authors contributed to the article and approved the submitted version.
Funding
Funding was received from Aarhus University, Lundbeck Foundation (R-287-2018-1094) and Danish Rheumatism Association (A6443).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Thapa P, Farber DL. The Role of the Thymus in the Immune Response. Thorac Surg Clinics (2019) 29(2):123–31. doi: 10.1016/j.thorsurg.2018.12.001
2. Dubey C, Croft M, Swain SL. Costimulatory Requirements of Naive CD4+ T Cells. ICAM-1 or B7-1 can Costimulate Naive CD4 T Cell Activation But Both Are Required for Optimum Response. J Immunol (1995) 155(1):45–57.
3. Croft M, Dubey C. Accessory Molecule and Costimulation Requirements for CD4 T Cell Response. Crit Rev Immunol (2017) 37(2-6):261–90. doi: 10.1615/CritRevImmunol.v37.i2-6.60
4. Laderach D, Movassagh M, Johnson A, Mittler RS, Galy A. 4-1bb Co-Stimulation Enhances Human CD8+ T Cell Priming by Augmenting the Proliferation and Survival of Effector CD8+ T Cells. Int Immunol (2002) 14(10):1155–67. doi: 10.1093/intimm/dxf080
5. Sharpe AH. Mechanisms of Costimulation. Immunol Rev (2009) 229(1):5–11. doi: 10.1111/j.1600-065X.2009.00784.x
6. Riella LV, Paterson AM, Sharpe AH, Chandraker A. Role of the PD-1 Pathway in the Immune Response. Am J Transplant (2012) 12(10):2575–87. doi: 10.1111/j.1600-6143.2012.04224.x
7. Murakami N, Riella LV. Co-Inhibitory Pathways and Their Importance in Immune Regulation. Transplantation (2014) 98(1):3–14. doi: 10.1097/TP.0000000000000169
8. Place DE, Kanneganti TD. The Innate Immune System and Cell Death in Autoinflammatory and Autoimmune Disease. Curr Opin Immunol (2020) 67:95–105. doi: 10.1016/j.coi.2020.10.013
9. Cha JH, Chan LC, Li CW, Hsu JL, Hung MC. Mechanisms Controlling PD-L1 Expression in Cancer. Mol Cell (2019) 76(3):359–70. doi: 10.1016/j.molcel.2019.09.030
10. Savoia P, Astrua C, Fava P. Ipilimumab (Anti-Ctla-4 Mab) in the Treatment of Metastatic Melanoma: Effectiveness and Toxicity Management. Hum Vaccines Immunother (2016) 12(5):1092–101. doi: 10.1080/21645515.2015.1129478
11. Leach DR, Krummel MF, Allison JP. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science (1996) 271(5256):1734–6. doi: 10.1126/science.271.5256.1734
12. Ramos-Casals M, Brahmer JR, Callahan MK, Flores-Chavez A, Keegan N, Khamashta MA, et al. Immune-Related Adverse Events of Checkpoint Inhibitors. Nat Rev Dis Primers (2020) 6(1):38. doi: 10.1038/s41572-020-0179-8
13. Ghosh N, Chan KK, Jivanelli B, Bass AR. Autoantibodies in Patients With Immune-Related Adverse Events From Checkpoint Inhibitors. JCR (2021) 28(2):1–8. doi: 10.1097/RHU.0000000000001777
14. Nakamura Y. Biomarkers for Immune Checkpoint Inhibitor-Mediated Tumor Response and Adverse Events. Front Med (Lausanne) (2019) 6:119. doi: 10.3389/fmed.2019.00119
15. Abdel-Wahab N, Shah M, Lopez-Olivo MA, Suarez-Almazor ME. Use of Immune Checkpoint Inhibitors in the Treatment of Patients With Cancer and Preexisting Autoimmune Disease: A Systematic Review. Ann Intern Med (2018) 168(2):121–30. doi: 10.7326/M17-2073
16. Johnson DB, Nebhan CA, Moslehi JJ, Balko JM. Immune-Checkpoint Inhibitors: Long-Term Implications of Toxicity. Nat Rev Clin Oncol (2022) 19(4):254–67. doi: 10.1038/s41571-022-00600-w
17. Patrinely JR Jr., Johnson R, Lawless AR, Bhave P, Sawyers A, Dimitrova M, et al. Chronic Immune-Related Adverse Events Following Adjuvant Anti-PD-1 Therapy for High-Risk Resected Melanoma. JAMA Oncol (2021) 7(5):744–8. doi: 10.1001/jamaoncol.2021.0051
18. Ghisoni E, Wicky A, Bouchaab H, Imbimbo M, Delyon J, Gautron Moura B, et al. Late-Onset and Long-Lasting Immune-Related Adverse Events From Immune Checkpoint-Inhibitors: An Overlooked Aspect in Immunotherapy. Eur J Cancer (2021) 149:153–64. doi: 10.1016/j.ejca.2021.03.010
19. Hasan Ali O, Berner F, Bomze D, Fassler M, Diem S, Cozzio A, et al. Human Leukocyte Antigen Variation is Associated With Adverse Events of Checkpoint Inhibitors. Eur J Cancer (2019) 107:8–14. doi: 10.1016/j.ejca.2018.11.009
20. Iafolla MAJ, Yang C, Chandran V, Pintilie M, Li Q, Bedard PL, et al. Predicting Toxicity and Response to Pembrolizumab Through Germline Genomic HLA Class 1 Analysis. JNCI Cancer Spectr (2021) 5(1):1–9. doi: 10.1093/jncics/pkaa115
21. Jessel S, Weiss SA, Austin M, Mahajan A, Etts K, Zhang L, et al. Immune Checkpoint Inhibitor-Induced Hypophysitis and Patterns of Loss of Pituitary Function. Front Oncol (2022) 12:836859. doi: 10.3389/fonc.2022.836859
22. Langlois F, Varlamov EV, Fleseriu M. Hypophysitis, the Growing Spectrum of a Rare Pituitary Disease. J Clin Endocrinol Metab (2022) 107(1):10–28. doi: 10.1210/clinem/dgab672
23. Naing A, Hajjar J, Gulley JL, Atkins MB, Ciliberto G, Meric-Bernstam F, et al. Strategies for Improving the Management of Immune-Related Adverse Events. J ImmunoTher Cancer (2020) 8(2):1–9. doi: 10.1136/jitc-2020-001754
24. Kumar V, Chaudhary N, Garg M, Floudas CS, Soni P, Chandra AB. Current Diagnosis and Management of Immune Related Adverse Events (irAEs) Induced by Immune Checkpoint Inhibitor Therapy. Front Pharmacol (2017) 8. doi: 10.3389/fphar.2017.00311
25. Petrelli F, Signorelli D, Ghidini M, Ghidini A, Pizzutilo EG, Ruggieri L, et al. Association of Steroids Use With Survival in Patients Treated With Immune Checkpoint Inhibitors. Cancers (Basel) (2020) 4:1–11.doi: 10.3390/cancers12030546
26. Chen AY, Wolchok JD, Bass AR. TNF in the Era of Immune Checkpoint Inhibitors: Friend or Foe? Nat Rev Rheumatol (2021) 17(4):213–23. doi: 10.1038/s41584-021-00584-4
27. Dimitriou F, Hogan S, Menzies AM, Dummer R, Long GV. IL-6 Blockade for Prophylaxis and Management of Immune-Related Adverse Events (irAEs) With Anti-PD-1 Based Immunotherapy. J Clin Oncol (2021) 39(15_suppl):214–24. doi: 10.1200/JCO.2021.39.15_suppl.9553
28. Linsley PS, Ledbetter JA. The Role of the CD28 Receptor During T Cell Responses to Antigen. Annu Rev Immunol (1993) 11:191–212. doi: 10.1146/annurev.iy.11.040193.001203
29. Shahinian A, Pfeffer K, Lee KP, Kundig TM, Kishihara K, Wakeham A, et al. Differential T Cell Costimulatory Requirements in CD28-Deficient Mice. Science (1993) 261(5121):609–12. doi: 10.1126/science.7688139
30. Vierboom MP, Breedveld E, Kap YS, Mary C, Poirier N, t Hart BA, et al. Clinical Efficacy of a New CD28-Targeting Antagonist of T Cell Co-Stimulation in a non-Human Primate Model of Collagen-Induced Arthritis. Clin Exp Immunol (2016) 183(3):405–18. doi: 10.1111/cei.12739
31. Porciello N, Kunkl M, Tuosto L. CD28 Between Tolerance and Autoimmunity: The Side Effects of Animal Models. F1000Res (2018) 7. doi: 10.12688/f1000research.14046.1
32. Sharpe AH, Pauken KE. The Diverse Functions of the PD1 Inhibitory Pathway. Nat Rev Immunol (2018) 18(3):153–67. doi: 10.1038/nri.2017.108
33. Keir ME, Freeman GJ, Sharpe AH. PD-1 Regulates Self-Reactive CD8 +T Cell Responses to Antigen in Lymph Nodes and Tissues. J Immunol (2007) 179(8):5064–70. doi: 10.4049/jimmunol.179.8.5064
34. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed Death-1 Ligand 1 Interacts Specifically With the B7-1 Costimulatory Molecule to Inhibit T Cell Responses. Immunity (2007) 27(1):111–22. doi: 10.1016/j.immuni.2007.05.016
35. Liang SC, Latchman YE, Buhlmann JE, Tomczak MF, Horwitz BH, Freeman GJ, et al. Regulation of PD-1, PD-L1, and PD-L2 Expression During Normal and Autoimmune Responses. Eur J Immunol (2003) 33(10):2706–16. doi: 10.1002/eji.200324228
36. Marasco M, Berteotti A, Weyershaeuser J, Thorausch N, Sikorska J, Krausze J, et al. Molecular Mechanism of SHP2 Activation by PD-1 Stimulation. Sci Adv (2020) 6(5):eaay4458. doi: 10.1126/sciadv.aay4458
37. Patsoukis N, Duke-Cohan JS, Chaudhri A, Aksoylar HI, Wang Q, Council A, et al. Interaction of SHP-2 SH2 Domains With PD-1 ITSM Induces PD-1 Dimerization and SHP-2 Activation. Commun Biol (2020) 3(1):1–13. doi: 10.1038/s42003-020-0845-0
38. Doroshow DB, Bhalla S, Beasley MB, Sholl LM, Kerr KM, Gnjatic S, et al. PD-L1 as a Biomarker of Response to Immune-Checkpoint Inhibitors. Nat Rev Clin Oncol. (2021) 18(6):345–62. . doi: 10.1038/s41571-021-00473-5
39. Gato-Canas M, Zuazo M, Arasanz H, Ibanez-Vea M, Lorenzo L, Fernandez-Hinojal G, et al. PDL1 Signals Through Conserved Sequence Motifs to Overcome Interferon-Mediated Cytotoxicity. Cell Rep (2017) 20(8):1818–29. doi: 10.1016/j.celrep.2017.07.075
40. Lucas ED, Schafer JB, Matsuda J, Kraus M, Burchill MA, Tamburini BAJ. PD-L1 Reverse Signaling in Dermal Dendritic Cells Promotes Dendritic Cell Migration Required for Skin Immunity. Cell Rep (2020) 33(2):108258. doi: 10.1016/j.celrep.2020.108258
41. Incorvaia L, Fanale D, Badalamenti G, Porta C, Olive D, De Luca I, et al. Baseline Plasma Levels of Soluble PD-1, PD-L1, and BTN3A1 Predict Response to Nivolumab Treatment in Patients With Metastatic Renal Cell Carcinoma: A Step Toward a Biomarker for Therapeutic Decisions. OncoImmunology (2020) 9(1):1832348. doi: 10.1080/2162402X.2020.1832348
42. Mildner F, Sopper S, Amann A, Pircher A, Pall G, Kock S, et al. Systematic Review: Soluble Immunological Biomarkers in Advanced non-Small-Cell Lung Cancer (NSCLC). Crit Rev Oncol/Hematol (2020) 153:102948–8. doi: 10.1016/j.critrevonc.2020.102948
43. Shi B, Du X, Wang Q, Chen Y, Zhang X. Increased PD-1 on CD4(+)CD28(-) T Cell and Soluble PD-1 Ligand-1 in Patients With T2DM: Association With Atherosclerotic Macrovascular Diseases. Metabolism (2013) 62(6):778–85. doi: 10.1016/j.metabol.2012.12.005
44. Bommarito D, Hall C, Taams LS, Corrigall VM. Inflammatory Cytokines Compromise Programmed Cell Death-1 (PD-1)-Mediated T Cell Suppression in Inflammatory Arthritis Through Up-Regulation of Soluble PD-1. Clin Exp Immunol (2017) 188(3):455–66. doi: 10.1111/cei.12949
45. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of Lupus-Like Autoimmune Diseases by Disruption of the PD-1 Gene Encoding an ITIM Motif-Carrying Immunoreceptor. Immunity (1999) 11(2):141–51. doi: 10.1016/S1074-7613(00)80089-8
46. Barroso-Sousa R, Barry WT, Garrido-Castro AC, Hodi FS, Min L, Krop IE, et al. Incidence of Endocrine Dysfunction Following the Use of Different Immune Checkpoint Inhibitor Regimens: A Systematic Review and Meta-Analysis. JAMA Oncol (2018) 4(2):173–82. doi: 10.1001/jamaoncol.2017.3064
47. Falcone M, Fousteri G. Role of the PD-1/PD-L1 Dyad in the Maintenance of Pancreatic Immune Tolerance for Prevention of Type 1 Diabetes. Front Endocrinol (2020) 11:1–11. doi: 10.3389/fendo.2020.00569
48. Hayashi M, Kouki T, Takasu N, Sunagawa S, Komiya I. Association of an a/C Single Nucleotide Polymorphism in Programmed Cell Death-Ligand 1 Gene With Graves' Disease in Japanese Patients. Eur J Endocrinol (2008) 158(6):817–22. doi: 10.1530/EJE-07-0649
49. Alvarez-Sierra D, Marin-Sanchez A, Ruiz-Blazquez P, de Jesus Gil C, Iglesias-Felip C, Gonzalez O, et al. Analysis of the PD-1/PD-L1 Axis in Human Autoimmune Thyroid Disease: Insights Into Pathogenesis and Clues to Immunotherapy Associated Thyroid Autoimmunity. J Autoimmun (2019) 103:102285. doi: 10.1016/j.jaut.2019.05.013
50. Barbosa NS, Wetter DA, Wieland CN, Shenoy NK, Markovic SN, Thanarajasingam U. Scleroderma Induced by Pembrolizumab: A Case Series. Mayo Clinic Proc (2017) 92(7):1158–63. doi: 10.1016/j.mayocp.2017.03.016
51. Terrier B, Humbert S, Preta LH, Delage L, Razanamahery J, Laurent-Roussel S, et al. Risk of Scleroderma According to the Type of Immune Checkpoint Inhibitors. Autoimmun Rev (2020) 19(8):102596–6. doi: 10.1016/j.autrev.2020.102596
52. Yamaguchi T, Shimizu J, Hasegawa T, Horio Y, Inaba Y, Yatabe Y, et al. Pre-Existing Pulmonary Fibrosis is a Risk Factor for Anti-PD-1-Related Pneumonitis in Patients With non-Small Cell Lung Cancer: A Retrospective Analysis. Lung Cancer (2018) 125:212–7. doi: 10.1016/j.lungcan.2018.10.001
53. Dobre IA, Frank AJ, D'Silva KM, Christiani DC, Okin D, Sharma A, et al. Outcomes of Patients With Interstitial Lung Disease Receiving Programmed Cell Death 1 Inhibitors: A Retrospective Case Series. Clin Lung Cancer (2021) 22(5):e738–e44. doi: 10.1016/j.cllc.2021.01.014
54. Zhang S, Wang L, Li M, Zhang F, Zeng X. The PD-1/PD-L Pathway in Rheumatic Diseases. J Formosan Med Assoc = Taiwan yi zhi (2021) 120(1 Pt 1):48–59. doi: 10.1016/j.jfma.2020.04.004
55. Greisen SR, Rasmussen TK, Stengaard-Pedersen K, Hetland ML, Horslev-Petersen K, Hvid M, et al. Increased Soluble Programmed Death-1 (sPD-1) is Associated With Disease Activity and Radiographic Progression in Early Rheumatoid Arthritis. Scandinavian J Rheumatol (2013) 43(2):101–8. doi: 10.3109/03009742.2013.823517
56. Greisen SR, Kragstrup TW, Thomsen JS, Horslev-Pedersen K, Hetland ML, Stengaard-Pedersen K, et al. The Programmed Death-1 Pathway Counter-Regulates Inflammation-Induced Osteoclast Activity in Clinical and Experimental Settings. Front Immunol (2022) 13:1–12. doi: 10.3389/fimmu.2022.773946
57. Greisen SR, Yan Y, Hansen AS, Veno MT, Nyengaard JR, Moestrup SK, et al. Extracellular Vesicles Transfer the Receptor Programmed Death-1 in Rheumatoid Arthritis. Front Immunol (2017) 8:e1694–14. doi: 10.3389/fimmu.2017.00851
58. Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically Expanded Peripheral T Helper Cell Subset Drives B Cells in Rheumatoid Arthritis. Nature (2017) 542(7639):110–4. doi: 10.1038/nature20810
59. Marks KE, Rao DA. T Peripheral Helper Cells in Autoimmune Diseases. Immunol Rev (2022) 307:1–12. doi: 10.1111/imr.13069
60. Curran CS, Gupta S, Sanz I, Sharon E. PD-1 Immunobiology in Systemic Lupus Erythematosus. J Autoimmun (2019) 97(3):1–9. doi: 10.1016/j.jaut.2018.10.025
61. Du Y, Nie L, Xu L, Wu X, Zhang S, Xue J. Serum Levels of Soluble Programmed Death-1 (sPD-1) and Soluble Programmed Death Ligand 1(sPD-L1) in Systemic Lupus Erythematosus: Association With Activity and Severity. Scandinavian J Immunol (2020) 92(1):1–11. doi: 10.1111/sji.12884
62. Jiao Q, Liu C, Yang Z, Ding Q, Wang M, Li M, et al. Upregulated PD-1 Expression is Associated With the Development of Systemic Lupus Erythematosus, But Not the PD-1.1 Allele of the PDCD1 Gene. Int J Genomics 2014 (2014) p:10–2. doi: 10.1155/2014/950903
63. Han L, Yang X, Yu Y, Wan W, Lv L, Zou H. Associations of Circulating CXCR3–PD-1+Cd4+T Cells With Disease Activity of Systemic Lupus Erythematosus. Modern Rheumatol (2019) 29(3):461–9. doi: 10.1080/14397595.2018.1469581
64. Lin J, Yu Y, Ma J, Ren C, Chen W. PD-1+CXCR5-Cd4+T Cells are Correlated With the Severity of Systemic Lupus Erythematosus. Rheumatol (United Kingdom) (2019) 58(12):2188–92. doi: 10.1093/rheumatology/kez228
65. Zhao L, Zhou X, Zhou X, Wang H, Gu L, Ke Y, et al. Low Expressions of PD-L1 and CTLA-4 by Induced CD4+CD25+ Foxp3+ Tregs in Patients With SLE and Their Correlation With the Disease Activity. Cytokine (2020) 133:155119–9. doi: 10.1016/j.cyto.2020.155119
66. Antonazzo IC, Poluzzi E, De Ponti F. Drug-Induced Systemic Lupus Erythematosus: Should Immune Checkpoint Inhibitors be Added to the Evolving List? Ann Rheum Dis (2021) 80(7):e120. doi: 10.1136/annrheumdis-2019-215819
67. Allenbach Y, Anquetil C, Manouchehri A, Benveniste O, Lambotte O, Lebrun-Vignes B, et al. Immune Checkpoint Inhibitor-Induced Myositis, the Earliest and Most Lethal Complication Among Rheumatic and Musculoskeletal Toxicities. Autoimmun Rev (2020) 19(8):102586. doi: 10.1016/j.autrev.2020.102586
68. Fukasawa T, Yoshizaki A, Ebata S, Nakamura K, Saigusa R, Miura S, et al. Contribution of Soluble Forms of Programmed Death 1 and Programmed Death Ligand 2 to Disease Severity and Progression in Systemic Sclerosis. Arthritis {\&} Rheumatol (2017) 69(9):1879–90. doi: 10.1002/art.40164
69. Fleury M, Belkina AC, Proctor EA, Zammitti C, Simms RW, Lauffenburger DA, et al. Increased Expression and Modulated Regulatory Activity of Coinhibitory Receptors PD-1, TIGIT, and TIM-3 in Lymphocytes From Patients With Systemic Sclerosis. Arthritis Rheumatol (2018) 70(4):566–77. doi: 10.1002/art.40399
70. Chuang E, Fisher TS, Morgan RW, Robbins MD, Duerr JM, Vander Heiden MG, et al. The CD28 and CTLA-4 Receptors Associate With the Serine/Threonine Phosphatase PP2A. Immunity (2000) 13(3):313–22. doi: 10.1016/S1074-7613(00)00031-5
71. Lindsten T, Lee KP, Harris ES, Petryniak B, Craighead N, Reynolds PJ, et al. Characterization of CTLA-4 Structure and Expression on Human T Cells. J Immunol (1993) 151(7):3489–99.
72. Annunziato F, Cosmi L, Liotta F, Lazzeri E, Manetti R, Vanini V, et al. Phenotype, Localization, and Mechanism of Suppression of CD4+CD25+ Human Thymocytes. J Exp Med (2002) 196(3):379–87. doi: 10.1084/jem.20020110
73. Schneider H, Rudd CE. Diverse Mechanisms Regulate the Surface Expression of Immunotherapeutic Target CTLA-4. Front Immunol (2014) 5:1–10. doi: 10.3389/fimmu.2014.00619
74. Chen Z, Zhou F, Huang S, Jiang T, Chen L, Ge L, et al. Association of Cytotoxic T Lymphocyte Associated Antigen-4 Gene (Rs60872763) Polymorphism With Crohn's Disease and High Levels of Serum sCTLA-4 in Crohn's Disease. J Gastroenterol Hepatol (2011) 26(5):924–30. doi: 10.1111/j.1440-1746.2011.06662.x
75. Khattri R, Auger JA, Griffin MD, Sharpe AH, Bluestone JA. Lymphoproliferative Disorder in CTLA-4 Knockout Mice is Characterized by CD28-Regulated Activation of Th2 Responses. J Immunol (1999) 162(10):5784–91.
76. Gough SCL, Walker LSK, Sansom DM. CTLA4 Gene Polymorphism and Autoimmunity. Immunol Rev (2005) 204:102–15. doi: 10.1111/j.0105-2896.2005.00249.x
77. Dougan M. Checkpoint Blockade Toxicity and Immune Homeostasis in the Gastrointestinal Tract. Front Immunol (2017) 8:1547. doi: 10.3389/fimmu.2017.01547
78. Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, et al. Microbiome-Derived Inosine Modulates Response to Checkpoint Inhibitor Immunotherapy. Science (2020) 369(6509):1481–9. doi: 10.1126/science.abc3421
79. Kawamoto S, Tran TH, Maruya M, Suzuki K, Doi Y, Tsutsui Y, et al. The Inhibitory Receptor PD-1 Regulates IgA Selection and Bacterial Composition in the Gut. Science (2012) 336(6080):485–9. doi: 10.1126/science.1217718
80. Sandborn WJ, Colombel JF, Sands BE, Rutgeerts P, Targan SR, Panaccione R, et al. Abatacept for Crohn's Disease and Ulcerative Colitis. Gastroenterology (2012) 143(1):62–9 e4. doi: 10.1053/j.gastro.2012.04.010
81. Zeissig S, Petersen BS, Tomczak M, Melum E, Huc-Claustre E, Dougan SK, et al. Early-Onset Crohn's Disease and Autoimmunity Associated With a Variant in CTLA-4. Gut (2015) 64(12):1889–97. doi: 10.1136/gutjnl-2014-308541
82. Larsen CP, Pearson TC, Adams AB, Tso P, Shirasugi N, Strobert E, et al. Rational Development of LEA29Y (Belatacept), a High-Affinity Variant of CTLA4-Ig With Potent Immunosuppressive Properties. Am J Transplant (2005) 5(3):443–53. doi: 10.1111/j.1600-6143.2005.00749.x
83. Badell IR, Parsons RF, Karadkhele G, Cristea O, Mead S, Thomas S, et al. Every 2-Month Belatacept Maintenance Therapy in Kidney Transplant Recipients Greater Than 1-Year Posttransplant: A Randomized, Noninferiority Trial. Am J Transplant (2021) 21(9):3066–76. doi: 10.1111/ajt.16538
84. Kremer JM, Westhovens R, Leon M, Di Giorgio E, Alten R, Steinfeld S, et al. Treatment of Rheumatoid Arthritis by Selective Inhibition of T-Cell Activation With Fusion Protein CTLA4Ig. New Engl J Med (2003) 349(20):1907–15. doi: 10.1056/NEJMoa035075
85. Fleischmann R, Weinblatt ME, Schiff M, Khanna D, Maldonado MA, Nadkarni A, et al. Patient-Reported Outcomes From a Two-Year Head-To-Head Comparison of Subcutaneous Abatacept and Adalimumab for Rheumatoid Arthritis. Arthritis Care Res (2016) 68(7):907–13. doi: 10.1002/acr.22763
87. de Germay S, Bagheri H, Despas F, Rousseau V, Montastruc F. Abatacept in Rheumatoid Arthritis and the Risk of Cancer: A World Observational Post-Marketing Study. Rheumatology (2020) 59(9):228. doi: 10.1093/rheumatology/kez604
88. Huss V, Bower H, Wadstrom H, Frisell T, Askling J. Short- and Longer-Term Cancer Risks With Biologic and Targeted Synthetic Disease-Modifying Antirheumatic Drugs as Used Against Rheumatoid Arthritis in Clinical Practice. Rheumatology (2021) 61(5):1810–8. doi: 10.1093/rheumatology/keab570
89. Weyand CM, Yang Z, Goronzy JJ. T-Cell Aging in Rheumatoid Arthritis. Curr Opin Rheumatol (2014) 26(1):93–100. doi: 10.1097/BOR.0000000000000011
90. Miggelbrink AM, Jackson JD, Lorrey SJ, Srinivasan ES, Waibl-Polania J, Wilkinson DS, et al. CD4 T-Cell Exhaustion: Does it Exist and What are its Roles in Cancer? Clin Cancer Res (2021) 27(21):5742–52. doi: 10.1158/1078-0432.CCR-21-0206
91. Osuch S, Laskus T, Berak H, Perlejewski K, Metzner KJ, Paciorek M, et al. Decrease of T-Cells Exhaustion Markers Programmed Cell Death-1 and T-Cell Immunoglobulin and Mucin Domain-Containing Protein 3 and Plasma IL-10 Levels After Successful Treatment of Chronic Hepatitis C. Sci Rep (2020) 10(1):16060. doi: 10.1038/s41598-020-73137-6
92. Cutolo M, Soldano S, Montagna P, Sulli A, Seriolo B, Villaggio B, et al. CTLA4-Ig Interacts With Cultured Synovial Macrophages From Rheumatoid Arthritis Patients and Downregulates Cytokine Production. Arthritis Res Ther (2009) 11(6):R176. doi: 10.1186/ar2865
93. Cutolo M, Soldano S, Gotelli E, Montagna P, Campitiello R, Paolino S, et al. CTLA4-Ig Treatment Induces M1-M2 Shift in Cultured Monocyte-Derived Macrophages From Healthy Subjects and Rheumatoid Arthritis Patients. Arthritis Res Ther (2021) 23(1):306. doi: 10.1186/s13075-021-02691-9
94. Crnkic Kapetanovic M, Saxne T, Jonsson G, Truedsson L, Geborek P. Rituximab and Abatacept But Not Tocilizumab Impair Antibody Response to Pneumococcal Conjugate Vaccine in Patients With Rheumatoid Arthritis. Arthritis Res Ther (2013) 15(5):R171. doi: 10.1186/ar4358
95. Alten R, Bingham CO 3rd, Cohen SB, Curtis JR, Kelly S, Wong D, et al. Antibody Response to Pneumococcal and Influenza Vaccination in Patients With Rheumatoid Arthritis Receiving Abatacept. BMC Musculoskel Disord (2016) 17(1):231. doi: 10.1186/s12891-016-1082-z
96. Furie R, Nicholls K, Cheng TT, Houssiau F, Burgos-Vargas R, Chen SL, et al. Efficacy and Safety of Abatacept in Lupus Nephritis: A Twelve-Month, Randomized, Double-Blind Study. Arthritis Rheumatol (2014) 66(2):379–89. doi: 10.1002/art.38260
97. Merrill JT, Burgos-Vargas R, Westhovens R, Chalmers A, D'Cruz D, Wallace DJ, et al. The Efficacy and Safety of Abatacept in Patients With non-Life-Threatening Manifestations of Systemic Lupus Erythematosus: Results of a Twelve-Month, Multicenter, Exploratory, Phase IIb, Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheumatism (2010) 62(10):3077–87. doi: 10.1002/art.27601
98. Khanna D, Spino C, Johnson S, Chung L, Whitfield ML, Denton CP, et al. Abatacept in Early Diffuse Cutaneous Systemic Sclerosis: Results of a Phase II Investigator-Initiated, Multicenter, Double-Blind, Randomized, Placebo-Controlled Trial. Arthritis Rheumatol (2020) 72(1):125–36. doi: 10.1002/art.41055
99. Chung L, Spino C, McLain R, Johnson SR, Denton CP, Molitor JA, et al. Safety and Efficacy of Abatacept in Early Diffuse Cutaneous Systemic Sclerosis (ASSET): Open-Label Extension of a Phase 2, Double-Blind Randomised Trial. Lancet Rheumatol (2020) 2(12):e743–e53. doi: 10.1016/S2665-9913(20)30237-X
100. Kailashiya V, Sharma HB, Kailashiya J. Role of CTLA4 A49G Polymorphism in Systemic Lupus Erythematosus and its Geographical Distribution. J Clin Pathol (2019) 72(10):659–62. doi: 10.1136/jclinpath-2019-206013
101. Baixeras E, Huard B, Miossec C, Jitsukawa S, Martin M, Hercend T, et al. Characterization of the Lymphocyte Activation Gene 3-Encoded Protein. A New Ligand for Human Leukocy Antigen Class H Antigens. J Exp Med (1992) 176(2):327–37. doi: 10.1084/jem.176.2.327
102. Huard B, Mastrangeli R, Prigent P, Bruniquel D, Donini S, El-Tayar N, et al. Characterization of the Major Histocompatibility Complex Class II Binding Site on LAG-3 Protein. Proc Natl Acad Sci United States America (1997) 94(11):5744–9. doi: 10.1073/pnas.94.11.5744
103. Maruhashi T, Okazaki IM, Sugiura D, Takahashi S, Maeda TK, Shimizu K, et al. LAG-3 Inhibits the Activation of CD4 + T Cells That Recognize Stable pMHCII Through its Conformation-Dependent Recognition of pMHCII. Nat Immunol (2018) 19(12):1415–26. doi: 10.1038/s41590-018-0217-9
104. Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al. Fibrinogen-Like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell (2019) 176(1-2):334–347.e12. doi: 10.1016/j.cell.2018.11.010
105. Xu F, Liu J, Liu D, Liu B, Wang M, Hu Z, et al. LSECtin Expressed on Melanoma Cells Promotes Tumor Progression by Inhibiting Antitumor T-Cell Responses. Cancer Res (2014) 74(13):3418–28. doi: 10.1158/0008-5472.CAN-13-2690
106. Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, et al. Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8 T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol Res (2015) 3(4):412–23. doi: 10.1158/2326-6066.CIR-14-0150
107. Andrews LP, Marciscano AE, Drake CG, Vignali DA. LAG3 (CD223) as a Cancer Immunotherapy Target. Immunol Rev (2017) 276(1):80–96. doi: 10.1111/imr.12519
108. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-Inhibitory Receptors With Specialized Functions in Immune Regulation. Immunity (2016) 44(5):989–1004. doi: 10.1016/j.immuni.2016.05.001
109. Poirier N, Haudebourg T, Brignone C, Dilek N, Hervouet J, Minault D, et al. Antibody-Mediated Depletion of Lymphocyte-Activation Gene-3 (LAG-3(+) )-Activated T Lymphocytes Prevents Delayed-Type Hypersensitivity in non-Human Primates. Clin Exp Immunol (2011) 164(2):265–74. doi: 10.1111/j.1365-2249.2011.04329.x
110. Ellis J, JBM D, Srinivasan N, Barrett C, Hopkins TG, Richards A, et al. Depletion of LAG-3(+) T Cells Translated to Pharmacology and Improvement in Psoriasis Disease Activity: A Phase I Randomized Study of mAb Gsk2831781. Clin Pharmacol Ther (2021) 109(5):1293–303. doi: 10.1002/cpt.2091
111. Bauche D, Joyce-Shaikh B, Jain R, Grein J, Ku KS, Blumenschein WM, et al. LAG3(+) Regulatory T Cells Restrain Interleukin-23-Producing CX3CR1(+) Gut-Resident Macrophages During Group 3 Innate Lymphoid Cell-Driven Colitis. Immunity (2018) 49(2):342–352 e5. doi: 10.1016/j.immuni.2018.07.007
112. Bettini M, Szymczak-Workman AL, Forbes K, Castellaw AH, Selby M, Pan X, et al. Cutting Edge: Accelerated Autoimmune Diabetes in the Absence of LAG-3. J Immunol (2011) 187(7):3493–8. doi: 10.4049/jimmunol.1100714
113. Tawbi HA, Schadendorf D, Lipson EJ, Ascierto PA, Matamala L, Castillo Gutierrez E, et al. Relatlimab and Nivolumab Versus Nivolumab in Untreated Advanced Melanoma. New Engl J Med (2022) 386(1):24–34. doi: 10.1056/NEJMoa2109970
114. Angin M, Brignone C, Triebel F. A LAG-3-Specific Agonist Antibody for the Treatment of T Cell-Induced Autoimmune Diseases. J Immunol (2020) 204(4):810–8. doi: 10.4049/jimmunol.1900823
115. Sag E, Demir S, Aspari M, Nielsen MA, Skejo C, Hvid M, et al. Juvenile Idiopathic Arthritis: Lymphocyte Activation Gene-3 is a Central Immune Receptor in Children With Oligoarticular Subtypes. Pediatr Res (2021) 90(4):744–51. doi: 10.1038/s41390-021-01588-2
116. Eriksson C, Kokkonen H, Johansson M, Hallmans G, Wadell G, Rantapaa-Dahlqvist S. Autoantibodies Predate the Onset of Systemic Lupus Erythematosus in Northern Sweden. Arthritis Res Ther (2011) 13(1):1–8. doi: 10.1186/ar3258
117. Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, et al. Development of Autoantibodies Before the Clinical Onset of Systemic Lupus Erythematosus. New Engl J Med (2003) 349(16):1526–33. doi: 10.1056/NEJMoa021933
118. Jonsson R, Theander E, Sjostrom B, Brokstad K, Henriksson G. Autoantibodies Present Before Symptom Onset in Primary Sjögren Syndrome. JAMA (2013) 310(17):1854–5. doi: 10.1001/jama.2013.278448
119. Verspohl SH, Holderried T, Behning C, Brossart P, Schafer VS. Prevalence, Therapy and Tumour Response in Patients With Rheumatic Immune-Related Adverse Events Following Immune Checkpoint Inhibitor Therapy: A Single-Centre Analysis. Ther Adv Musculoskelet Dis (2021) 13:1759720X211006963. doi: 10.1177/1759720X211006963
Keywords: PD-1, checkpoint inhibition therapy, autoimmunity, co-inhibitory receptors, immune related adverse events, rheumatic diseases
Citation: Greisen SR, Aspari M and Deleuran B (2022) Co-Inhibitory Molecules – Their Role in Health and Autoimmunity; Highlighted by Immune Related Adverse Events. Front. Immunol. 13:883733. doi: 10.3389/fimmu.2022.883733
Received: 25 February 2022; Accepted: 10 May 2022;
Published: 16 June 2022.
Edited by:
Rizgar A. Mageed, Queen Mary University of London, United KingdomReviewed by:
Dawn Elaine Smilek, UCSF, United StatesGiulia Di Dalmazi, G. d’Annunzio University of Chieti and Pescara, Italy
Copyright © 2022 Greisen, Aspari and Deleuran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stinne R. Greisen, c3JnQGJpb21lZC5hdS5kaw==