 Jia Yang
Jia Yang Yuan Hu
Yuan Hu Binjing Zhang
Binjing Zhang Xiao Liang
Xiao Liang Xin Li
Xin Li- 1Department of Gynecology and Obstetrics and Pediatric Nephrology Nursing, Development and Related Disease of Women and Children Key Laboratory of Sichuan Province, Key Laboratory of Birth Defects and Related Diseases of Women and Children, Ministry of Education, West China Second Hospital, Sichuan University, Chengdu, China
- 2State Key Laboratory of Oral Disease, National Clinical Research Center for Oral Diseases, West China Hospital of Stomatology, Sichuan University, Chengdu, China
Inflammation has emerged as a key player in regulating cancer initiation, progression, and therapeutics, acting as a double edged sword either facilitating cancer progression and therapeutic resistance or inducing anti-tumor immune responses. Accumulating evidence has linked the epigenetic modifications of histones to inflammation and cancer, and histone modifications-based strategies have shown promising therapeutic potentials against cancer. The jumonji C domain-containing (JMJD) family histone demethylases have exhibited multiple regulator functions in inflammatory processes and cancer development, and a number of therapeutic strategies targeting JMJD histone demethylases to modulate inflammatory cells and their products have been successfully evaluated in clinical or preclinical tumor models. This review summarizes current understanding of the functional roles and mechanisms of JMJD histone demethylases in crosstalk between inflammation and cancer, and highlights recent clinical and preclinical progress on harnessing the JMJD histone demethylases to regulate cancer-related inflammation for future cancer therapeutics.
Introduction
Inflammatory cells and inflammatory factors are important components of the tumor microenvironment (1–3). In solid tumors, hypoxia and acidic stress in tumor microenvironmental niches induces apoptosis of the fast-growing tumor cells, releasing cell fragments and chemokines and leading to inflammatory cell infiltration and secretion of inflammatory factors. Furthermore, extrinsic factors such as systematic diseases, infections, and smoking are also able to evoke and enhance tumor-associated inflammatory response processes. Accumulating evidence has suggested that both tumor-extrinsic and -intrinsic inflammation are closely linked to tumorigenesis, development, and therapy resistance (4–7). Recently, epigenetics, particularly the Jumonji C domain-containing (JMJD) protein family-mediated histone methylation mechanisms, have been identified as one of the most important mechanisms for regulating the occurrence and progression of malignant tumors. In addition, a number of JMJD proteins-targeted therapeutic strategies aiming to modulate inflammatory cells and their products have been developed in clinical or preclinical tumor models. In this review, we summarize the roles and mechanisms of JMJD protein family in cancer-related inflammation, and discuss the anti-cancer therapeutic approaches by modulating JMJD proteins-mediated inflammation.
JMJD Histone Demethylases in Cancer-Related Inflammation
Crosstalk Between Cancer and Inflammation
In 1800, Galenus was the first to hypothesize a correlation between inflammation and tumors, suggesting that tumors can arise in inflamed and damaged tissues (8). In the 19th century, researchers found that tumors usually occurred in chronic inflammation sites, and there were inflammatory cells in tumor biopsy tissues (9). This is the first human evidence supporting the relationship between cancer and inflammation, and these findings have been confirmed by epidemiological research where researchers found that individuals with chronic inflammation were more likely to develop cancer.
Indeed, it is estimated that 15-20% of cancers worldwide are associated with underlying infection and inflammation. Tumor-related inflammation can be divided into the following categories according to the inducement, mechanism, result and intensity: inflammation caused by microbial infection, inflammation caused by environmental exposure, inflammation secondary to tumor, and inflammation secondary to tumor treatment. Importantly, tumor-related chronic inflammation has been shown to be an important trigger factor that increases the risks of cancer and promotes the cancer progression (4). For example, inflammation caused by microbial infection, including persistent helicobacter pylori infection, is associated with gastric cancer and gastric mucosa-associated lymphoid tissue lymphoma. In addition, Hepatitis B or C virus infection increases the risk of hepatocellular carcinoma, and schistosome or bacillus infection has been associated with bladder cancer and colon cancer, respectively (10). Furthermore, inflammation caused by environmental exposure, including particulate irritants such as grass smoke, can lead to chronic obstructive pulmonary disease which has a high risk for lung cancer (11). Inhalation of asbestos or silica particles can also cause lung cancer, and the tumorigenicity of these particles is attributed to the inflammatory effect induced by inflammasome-produced interleukin (IL)-1β cytokine (12). Not surprisingly, administration of non-steroidal anti-inflammatory drugs has been reported to significantly reduce the morbidity and mortality of multiple types of cancer (13–15).
In addition to its role in cancer initiation, the inflammatory responses were also shown to play an important role in cancer progression and metastasis. It is well-established that except for the cancer cells themselves, the stroma, blood vessels, and infiltrating inflammatory cells in tumor microenvironment are also involved in the regulation of inflammation and cancer. Tumor-related inflammatory molecules are known to mainly include transcription factors (such as NF-κB, as well as the signal transduction activation transcription factor STAT3 and HIF-1α), chemotactic factors, cytokines (such as IL-1β, IL-6, IL-23 and TNF-α), cyclooxygenase, and inducible nitric oxide synthase (iNOS), etc. (16). Studies have shown that NF-κB and STAT3 transcription factors are the main regulators for promoting the production of pro-inflammatory cytokines as well as the persistence of important mediators of chronic inflammation, and thus involved in the occurrence and development of tumors (17–20). Furthermore, interactions between inflammatory cells, cancer cells and stromal cells within the tumor microenvironment exist through mutual induction, receptor regulation and biological effects (21, 22). In addition, there are a large number of active substances which act through their own functional properties or mediated signaling pathways to participate in the cellular biological events necessary for tumorigenesis and cancer development. Certain cytokines are known to activate a variety of signaling molecules controlling a wide range of cellular processes, and thus linking the inflammatory microenvironment with biological behaviors of tumor cells to precede cancer development. For example, IL-6, IL-8, IL-10, IL-18, TGF-β and other cytokines act as indispensable factors to promote inflammatory responses for cancer progression (23). Studies have also shown that IL-6 is known to have a direct effect on both tumor cells and tumor inflammatory microenvironment, and is associated with several tumor types, including lymphoma, mesothelioma, lung, breast, pancreatic, colon, prostate, and stomach cancer (24–27). It has been demonstrated that epigenetic machineries are involved in regulating the secretion of inflammatory cytokines or the activation of signaling pathways. Taking transforming growth factor TGF-β as an example, the binding of TGF-β and corresponding cell membrane surface receptor triggers the transactivation of Smad protein, mediating upregulated or downregulated expression of the downstream genes involved in cancer promotion or inhibition (28, 29). Abnormal DNA methylation of tumor cells has also been found in a variety of tumor tissues, which can down-regulate or inhibit TGF-β-mediated signaling pathway by modulating TGF-β and its receptors (28). These results highlight the important roles of pro-inflammatory cytokines in tumor inflammatory microenvironment and cancer progression.
Therefore, inflammation, especially chronic inflammation, plays an important role in cancer initiation and progression. In the process of tumor formation, inflammatory reactions promote tumor growth, produce a microenvironment conducive to tumor growth, induce gene mutations, and promote angiogenesis. In the later stage of tumor development, inflammatory mediators affect tumor cells promote tumor growth, angiogenesis, and metastasis by modulating biological behaviors of tumor cells.
Histone Methylation and Demethylation in Cancer-Related Inflammation
Epigenetics refers to the stable and heritable changes in gene expression or cell phenotype caused by reversibly modifying nucleotide or chromosome without changing DNA sequences. The concept of epigenetics was first proposed by Waddington, a British scientist, in 1942. Epigenetics refers to the change of gene expression level and function without the change of DNA sequence, and produce heritable phenotype. Epigenetics can be divided into two categories according to their action mechanisms to regulate gene expression, including the DNA methylation, histone modification, chromatin remodeling act by selective transcription and expression regulation of genes, as well as the non-coding RNAs act by post-transcriptional regulation of genes (30).
Histones are highly conserved proteins composed of H1, H3, H2A, H2B and H4, which form nucleosomes together with DNA (30, 31). As one of the basic structural components of chromosomes, histone is not only an important part of the DNA that participates in the composition of genetic material, but also plays an indispensable role in the regulation of gene expression. Histone modifications mainly include the following categories: phosphorylation (serine and threonine residues), methylation (arginine and lysine residues), acetylation (lysine residues), ubiquitination, glycosylation, ADP-ribosylation (lysine residues), ubiquitination and proline isomerization, etc. (32, 33). Chromatin is divided into heterochromatin and euchromatin according to the interaction of different modification types of histone tail residues with specific proteins (33). Histone modifications have been found to not only reversibly promote or inhibit the transcription and expression levels of corresponding genes, but also participate in several cellular processes such as cell proliferation, differentiation, and functions (34). The modifications of histones at individual sites occur following the corresponding reactions mediated by specific enzymes, and the modification status is dynamically regulated by the corresponding enzymes in the reaction. Histone methylation and demethylation, mainly occur on lysine (K) and arginine (R) residues of the histones, are one of the most studied epigenetic patterns in the tumor field (35–38). Histone methylation is the methylation of lysine (k) or arginine (s) residues at the N-terminal of H3 and H4 histones. Its function is to form and maintain the structure of heterochromatin, genomic imprinting, DNA repair, inactivation and transcription of X chromatin (39, 40). Histone methylation is mainly catalyzed by histone methyltransferase (HMT). Histone methylation can be involved in the maintenance of chromatin structure and the silencing or activation of gene expression. Histone demethylase, LSD1 can be roughly divided into two families: LSD (Lysine-specific Demethylase) and JMJD (JmjC Domain-containing Family). LSD1 can specifically remove the mono-dimethylation of H3K4 and H3K9. JmjC family can remove the modification of lysine trimethylation. In general, methylation of the K9, K27, and K20 residues of histone H3 normally induces heterochromatin formation and silencing of related genes, while methylation of the K4 residue of histone H3, K36 and K79 in turn regulates gene expression (39). Abundant experimental evidence has demonstrated that abnormal histone methylation regulates the biological functions of tumor cells, interfere with the immune response of tumor cells, and even the occurrence and development of certain tumors (41, 42). Taking the polarization process of tumor-associated macrophages (TAMs) as an example, it is currently believed that the transformation of TAMs from M1 type to M2 type is one of the important markers of tumor malignancy and development (43). It was found that DNA methyltransferase 3B (DNMT3b) was highly expressed in M1-type polarized macrophages, while it was lowly expressed in M2-type polarized macrophages, suggesting that abnormal methylation is involved in the transformation process of TAMs from M1-type to M2-type (44). Besides, the aberrant modification of histone H3K27 was also involved in the signaling pathways responsible for M2 polarization (45). Furthermore, several biological characteristics of tumor microenvironment, such as hypoxia, low pH, high interstitial hydraulic pressure, production of cytokines and proteolytic enzymes, and persistence of inflammatory response, are also important mechanisms for modulating inflammation-related histone methylation machinery during the occurrence and development of tumors.
JMJD Family Histone Demethylases
The JMJD protein family is composed of 33 human members. The defining element of this protein family is the Jumonji C (JmjC) domain with a length of approximately 170 amino acids, containing an iconic HX (D/E) domain (46, 47). In addition, the JmjC domain also contains 2-oxoglutarate (2OG)/α-ketoglutarate binding sites similar to the catalytic domains of other 2OG-dependent oxygenases (48). This leads to the proved for the first time that the catalytic activity of the JmjC domain exists for HIF1-α (hypoxia inducible factor 1 subunit alpha inhibitor), indicating that it can hydroxylate asparagine residues (49, 50). Based on the structure and function relationships, such structural features confer JMJD proteins the catalytic activity to hydroxylate methylated lysine residues and then demethylate them (51). The histone lysine demethylase activity has been proven to exist in some members of JMJD proteins by further experimental studies (52, 53), leading to certain JMJD family proteins being renamed as lysine demethylases (KDMs). Several members of the JMJD protein family, such as JMJD2B, JMJD2C, JMJD2D, JMJD3, JMJD5, JMJD6, and JMJD8, have been shown to be closely involved in the development and progression of chronic diseases. Proteins containing JMJD have relatively small molecular weights and perform different functions by hydroxylating proteins and RNA, and these proteins are mostly epigenetic regulators mediating demethylation of the histone proteins.
Functional Mechanisms of JMJD Histone Demethylases in Cancer-Related Inflammation
JMJD Histone Demethylases and Cancer-Related Inflammation
Methylation modifications have been demonstrated to regulate the expression and function of key signaling molecules released by inflammatory cells and play a key role in certain cancer types. As we all know, the target genes of HIF-1 α, a key factor involved in the serial effects of hypoxia on tumor cells, include certain members of the Jumonji structures (JMJD)-containing histone demethylases (54). Pollard et al. (55) has found that siRNA targeting HIF-1 α can significantly reduce the expression of JMJD2B and JMJD2C proteins, suggesting that hypoxia can induce the activation of histone demethylase and promote the process of histone demethylation. Studies have also shown that hypoxia can induce disturbance of other epigenetic mechanisms such as DNA methylation, histone modification (56) and miRNA expression profile change in tumor cells (57). Besides, tumor stromal cells, especially the immune cells infiltrated locally by tumor, are known to produce a large number of inflammatory cytokines in the tumor microenvironment, and thus making the tumor cells in a long-term inflammatory state (58). This persistent inflammatory response plays an important role in altering epigenetic modification status and disturbance during cancer development. Studies have found that stromal cells in the myeloma microenvironment could inhibit miRNA-15a/-16 expression in bone marrow cells by secreting high levels of the inflammatory factor IL-6, thus weakening the inhibitory effects on tumor cell proliferation and participating in the occurrence of drug resistance in myeloma. In turn, overexpression of inflammatory cytokines TNF-α and IL-6 can induce promoter methylation of signal transduction inhibitor 1 and down-regulate its expression, thus alleviating the inhibition of inflammatory response in tumors (59). In highly metastatic prostate cancer, inflammatory factor CXCL1 activates the NF-κB signaling pathway, deacetylates histone H3 and H4, and down-regulates the expression of extracellular matrix glycoprotein fibulin-D1 (59). Together, these studies suggest that inflammatory cytokines and chemokines could modulate epigenetic machineries to trigger remodeling of the tumor microenvironment, affecting cancer development.
JMJD histone demethylases are abnormally expressed in inflammatory conditions and are responsible for regulating the expression and secretion of inflammatory mediators. A number of studies have shown a significant link between inflammation and epigenome reprogramming brought about by the JMJD family, and the JMJD family has been shown to be critical in chronic inflammation and autoimmune diseases, including atherosclerosis (60). In addition, demethylation of H3K4me3 and H3K27 is necessary for M1 macrophages to initiate inflammatory cytokine production and M2 polarization (61). Considering their pro-inflammatory roles and the current evidence on inflammation-cancer transformation, we speculated that JMJD family histone demethylases might be a key node in the crosstalk between inflammation and cancer, and their related functional mechanisms and therapeutic potentials are discussed. Despite the large number of members of the JMJD family, we found that only a limited number of members are associated with inflammatory tumors and their mechanisms are relatively well studied. Next, we will focus on the association between JMJD2B, JMJD2C, JMJD2D, JMJD3 and JMJD5 with inflammatory tumors.
JMJD2B and Cancer-Related Inflammation
JMJD2B, also known as KDM4B, is a JmjC domain-containing histone demethylase belonged to the JMJD2 protein family. JMJD2B mainly targets the trimethyl of lysine 9 of histone H3 (H3K9me3) to catalyze its demethylation reaction. Such demethylation activity of JMJD2B has been found to play an important role in several biological processes, such as heterochromatin formation, X chromosome inactivation, gene transcription regulation, and stem cell differentiation (39, 60–64). Recently, JMJD2B has been recognized as a strong epigenetic modulator of inflammation (65), and have attracted much attention in the malignant progression of multiple types of cancer, such as breast cancer, gastric cancer, colorectal cancer, bladder cancer, and lung cancer (66–72).
As one of the most common malignant tumors worldwide, gastric cancer-related mortality ranked second amongst causes of death in cancer patients (73, 74). In detail, although the overall incidence of gastric cancer has fallen in the United States, its incidence in Asian countries is still on the high side (75). Among them, China is a high incidence area of gastric cancer, with an incidence of 34.6/100,000 and a mortality rate of 30.2/100,000 (76). Due to the insufficient early detection and diagnosis of gastric cancer, most patients have already entered the advanced or metastatic stage by the time of diagnosis. Besides, the currently available treatment options for advanced gastric cancer are still limited, surgery and combined chemotherapy have displayed insufficient effects on therapeutic effects, and the five-year survival among the gastric cancer patients remained very low (77). The age-standardized 5-year relative survival rate of gastric cancer patients in China was only 35.1% (78). Helicobacter pylori (H. pylori), a type of microaerobic Gram-negative bacteria that colonizes gastric mucosal epithelial cells, has been shown to be a risk factor for chronic gastritis, gastrointestinal ulcer, and gastric cancer (79). It has been classified as a class I carcinogen by the international agency for research on cancer (IARC) (80). Recent studies have suggested that H. pylori affected the invasion and metastasis of the gastric cancer by upregulating the expression of epithelial-mesenchymal transition (EMT)-related factors Snail 1, Slug and vimentin, partly relying on gastrin and matrix metalloproteinase 7 (MMP7), and enhancing the expression of Heparin-binding EGF-like growth factor (HB-EGF), thus promoting the EMT process in gastric cancer (81). In addition, H. pylori activates multiple intracellular pathways in epithelial cells, such as the NF-κB, β-catenin pathway and PI3K/AKT pathway, which induces increased inflammatory cytokine production, altered apoptosis rate, and deregulated epithelial cell proliferation and differentiation, finally leading to the oncogenic transformation of early human gastric cancer (78, 82–88).
Histone modifications have been reported to be involved in immunosuppression, thereby providing a preferred microenvironment for infection with either bacteria or viruses and further development of human diseases, including cancer (89). As a risk factor associated with the occurrence and development of gastric cancer, H. pylori infection-induced epigenetic disturbances, especially the histone modification alterations, are closely involved in regulating the expression of genes crucial for tumorigenesis and cancer development (90, 91). As a histone demethylase, JMJD2B has been reported to possess oncogenic activities in multiple types of human cancers (66, 70). More specifically, JMJD2B plays an important in gastric cancer initiation and progression by promoting the proliferation, survival, invasion and metastasis of gastric cancer cells (92, 93).
It has been found that the mRNA and protein expression levels of JMJD2B in different gastric epithelial cells are significantly increased in response to H. pylori infection in a CagA-independent manner. Besides, it has been demonstrated that there is a potential binding site for β-catenin on the promoter region of JMJD2B (94). Indeed, amongst the multiple signaling pathways activated by H. pylori, β-catenin plays an important role in modulating dysregulated inflammation and gastric cancer progression (95). H. pylori infection induces the transcriptional activity of β-catenin, activates the nuclear LEF/TCF transcriptional factor, and regulates the expression levels of downstream target genes which are known to be involved in key cellular processes including cell cycle control, differentiation, and migration (96, 97). Meanwhile, in response to H. pylori infection, β-catenin has been shown to enhance the transcriptional activity of JMJD2B by recognizing the active region of the JMJD2B gene promoter (94). Besides, existing evidence has highlighted an interaction between JMJD2B and NF-κB in gastric cancer cells. It has been reported that H. pylori induces expression of cyclooxygenase 2 (COX-2) by activating the transcription factor NF-κB, which is a key regulator of immune and inflammatory responses, and thus further modulating the inflammatory response and contributing to the carcinogenesis of gastric cancer (98). Upon exposure to H. pylori infection, an increased binding of JMJD2B with NF-κB to COX-2 promoter has been found to be coupled with the decrease of H3K9me3 and increase of H3K9me2 levels (94), and it can be concluded that JMJD2B could facilitate H. pylori-induced COX-2 transcription. Indeed, multiple studies have demonstrated that COX-2 is involved in H. pylori-induced inflammatory response and tumorigenesis (99, 100). Furthermore, an overexpressed expression pattern of COX-2 has been detected in primary gastric cancer, and has been shown to be closely related to the occurrence and distal metastasis of gastric cancer (101–104). Hence, the β-catenin-JMJD2B-COX-2 signaling cascade described above is a crucial JMJD2B-dependent pathogenic mechanism responsible for the H. pylori-induced initiation and development of gastric cancer (Figure 1).
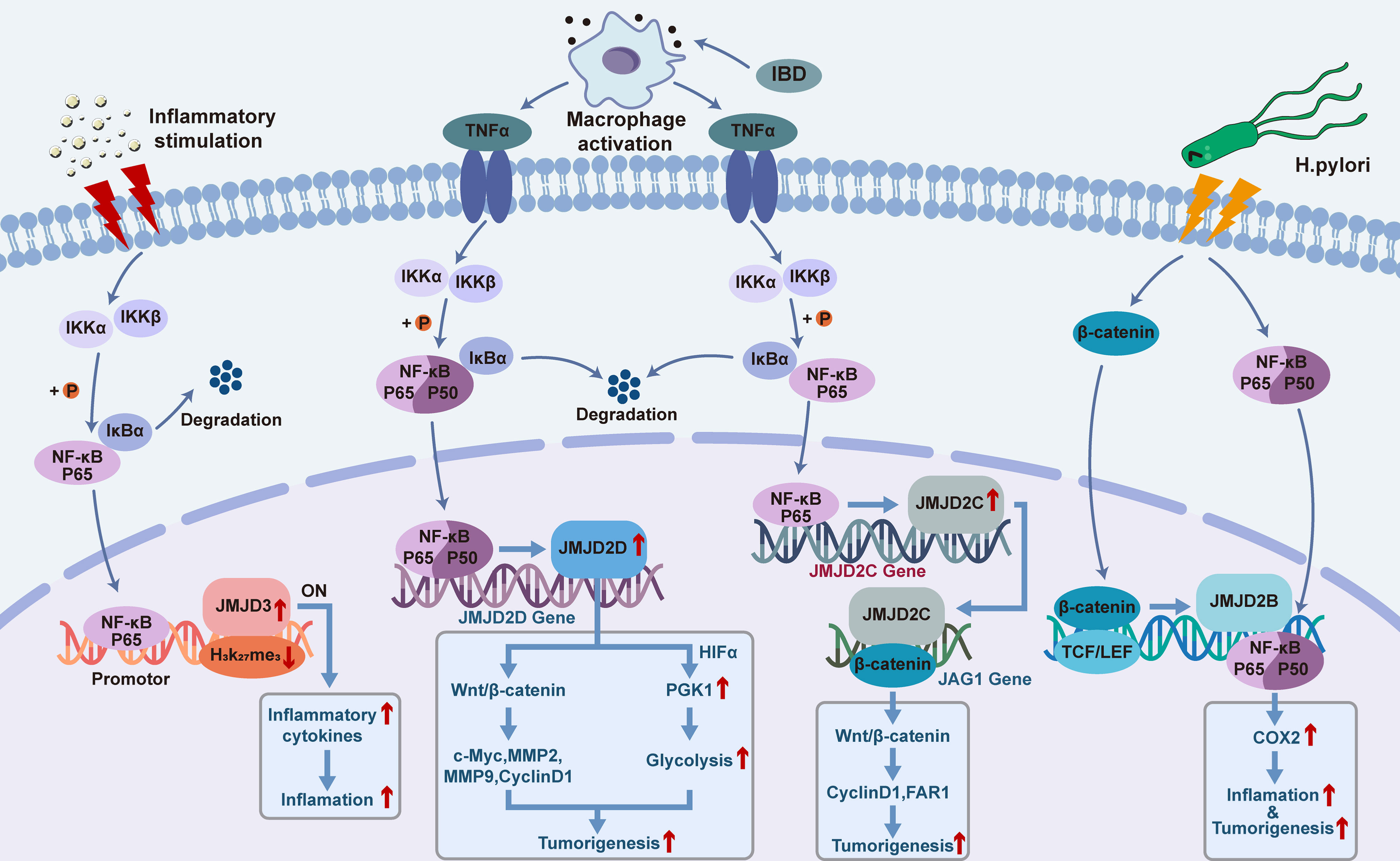
Figure 1 Functional roles and mechanisms of JMJD2B/C/D and JMJD3 in the crosstalk between inflammation and cancer.
Besides, JMJD2B has also been associated with gastric cancer through other mechanisms. When exposed to hypoxia and radiation, histone H3 methylation of gastric adenocarcinoma cells was in a state of dynamic transformation, and JMJD2B-mediated modification mechanisms function to enhance the expression of cyclin A1 (CCNA1) and further facilitate the growth of gastric adenocarcinoma cells under hypoxia and radiation stress (67). These results suggested that histone modifications catalyzed by JMJD2B played an important role in the pathogenesis of gastric cancer under environmental stress, and this protein could become a potential target for the future development of treatment options for gastric cancer.
JMJD2C and Cancer-Related Inflammation
JMJD2C gene is located in the p24.1 region of human chromosome 9. The total length of JMJD2C gene consists of 4239 bases, 13 different transcription products, including 22 exons and 3 promoters, forming a total of 6 variants. JMJD2C protein contains 1 JmjC, 1 JmjN, 2 phD-like zinc knuckle structures and 2 Tudor regions (105–107). In 2000, Y Ang et al. identified JMJD2C in the esophageal squamous cell carcinoma (ESCC) cells by comparative genomic hybridization, and named it as gene amplified in squamous cell carcinoma 1 (GASC1) (108). Subsequently, GASC1 was incorporated into the JMJD2 family by Katoh et al. (106), and is now also known as lysine specific demethylase 4C (KDM4C). JMJD2C can specifically remove lysine methyl groups from histone H3K9me3, H3K9me2, H3K36me3 and H3K36me2 through hydroxylation, thus participating in heterochromatin formation, gene imprinting, X chromosome inactivation, and gene transcription regulation (109). Up to now, studies on JMJD2C have attracted much attention on the following hot spots: its roles in controlling the stem cell fate (110–113), its involvement in the tumorigenesis and cancer development, as well as its potential for the development of novel anti-cancer drugs (105, 114, 115). Indeed, expression of JMJD2C has been found to be upregulated in breast cancer, gastric cancer, colorectal cancer, osteosarcoma, and other tumor types (116, 117).
Colorectal cancer, a common malignant tumor of the digestive tract system, ranks fifth in cancer incidence and mortality among China, and third in terms of incidence and mortality among all cancers in the United States (118). Interestingly, the incidence and mortality of males are significantly higher than that of females in China, while displaying basically the same in the United States (118, 119). Colorectal cancer develops slowly and is mostly transformed from chronic intestinal diseases. A large amount of evidence has indicated that colorectal cancer can be triggered by various factors, including gene mutation, epigenetic change, chronic inflammation, diet, and lifestyle (120). More specifically, multiple factors, including genetic factors (121, 122), family history (123), medical conditions such as chronic inflammatory bowel disease (124), type 2 diabetes (125), and previous cancer history of abdominal or pelvic radiation (126–129), have been reported to be potential risk factors of colorectal cancer in developing countries.
Currently, it is believed that colorectal cancer originates from colon cancer stem cells, and Notch and Wnt signaling pathways have been reported to be involved in the growth of the latter (130, 131). It has been shown that JMJD2C is involved in the interaction between Notch and Wnt pathways to promote the formation of stem cell-like spheres in colorectal cancer (132). In 2013, Yamamoto et al. evaluated the expression of JMJD2C gene in colorectal cancer cell line and found that JMJD2C was the downstream gene of β-catenin in Wnt pathway (132). Mechanistically, the expression level of JMJD2C protein was increased under the modulation of Wnt/β-catenin signaling pathway, and the protein product subsequently recruited β-catenin and co-located at the Jagged1 (JAG1) promoter for transactivation, thus enhancing the activation of Notch pathway and promoting the development of colorectal cancer (132). Kim et al. showed that JMJD2C gene was highly expressed in colorectal cancer and was necessary for the maximum growth of human colorectal carcinoma HCT-116 cell lines (133). It has also been reported that JMJD2C forms a complex with β-catenin and regulates the expression of several key downstream effectors of β-catenin signaling pathway. In addition, JMJD2C was found to upregulate the expression levels of cyclinD1 and proto-oncogene FAR1 to stimulate the proliferation of colon cancer cells (133). Besides, JMJD2C could positively regulate the anti-apoptotic factor BCL-2 to maintain the survival of cancer cells (133) (Figure 1).
JMJD2D and Cancer-Related Inflammation
JMJD2D (also known as KDM4D), a protein containing 523 amino acids with a molecular weight of 60 kDa, also belongs to the JMJD2 subfamily of the JMJD family histone demethylases while its structure is different from that of JMJD2A-2C (134). In details, the JmjN domain is located at 17-59 amino acids of the N terminus and acts to regulate gene transcription, while the JmjC domain is located at 146-312 amino acids of the N terminus and functions to catalyze the demethylation reaction (105). This protein has been reported to play an important role in many biological processes. For example, it has been reported that interactions between JMJD2D and androgen receptor (AR) enhance the transcription activity of androgen-dependent genes and regulate the activation of androgen signaling (135). In addition, JMJD2D was found to be recruited to the DNA damage sites in a poly (ADP-ribose) polymerase 1 (PARP 1)-dependent manner, contributing to the DNA double-strand break repair process (136). JMJD2D could also promote the initiation complex formation and regulate DNA replication (137), it can function as an RNA binding protein to increase its chromatin-binding capability and thus enhance the H3K9me3 demethylation activity (138). Besides, JMJD2D has been shown to bind with p53 to enhance the transcriptional activity of P21 (139).
Studies have revealed that JMJD2D can promote the growth and metastasis of colorectal cancer by enhancing the Wnt pathway and glycolysis, while inhibiting JMJD2D can prevent the occurrence and development of colorectal cancer (140) (Figure 1). And, overexpression of JMJD2D could activate the Wnt/β-catenin signaling pathway, causing upregulation of the downstream target genes of Wnt signaling such as c-myc, cyclin D1, MMP2, and MMP9 to promote the development of colorectal cancer (141). Under the condition of inflammatory bowel disease (IBD)-induced colon injury and intestinal bacterial translocation, activation of macrophages and secretion of inflammatory cytokine TNF-α trigger the activation of NF-κB signaling, leading to enhanced expression of JMJD2D in the colon epithelial cells. Meanwhile, JMJD2D has been shown to increase the transcriptional activity of phosphoglycerate Kinase 1 (PGK1) by interacting with HIF1-α, thus enhancing glycolysis to facilitate the progression of colorectal cancer. Further investigations on the functional role of JMJD2D histone demethylase activity in patients with colorectal cancer are imperative.
JMJD3 and Cancer-Related Inflammation
JMJD3, also known as KDM6B, is another member of the JMJD family with H3K27me3 histone demethylase activity (142). The human JMJD3 gene is located at 17p13.1 and encodes a protein consisting of 1682 amino acids with a molecular weight of 176632 Da. JMJD3 is located in both cytoplasm and nucleus, and the JmjC domain is indispensable for its H3K27me3 demethylation activity (143–145). It has been reported that this protein is involved in multiple cellular processes, including tumorigenesis and stem cell fate decision (143–146). Although JMJD3 is well-known for enzymatic activity to catalyze the conversion of H3K27me3 and H3K27me2 to H2K27me1, JMJD3 has been demonstrated to affect the proliferation of cancer cells and stem cells in either demethylation-dependent or -independent manner (147, 148). And, JMJD3 has been found to have both pro-cancer and anti-cancer properties by modulating the senescence and apoptosis of cancer cells (147).
JMJD3 plays an important role in the regulation of inflammation by affecting the polarization of macrophages, the differentiation of T cells, and the secretion of inflammatory factors. For example, it has been reported that serum amyloid A could upregulate JMJD3 via toll-like receptor (TLR) 2 and TLR4 pathways, thereby activating the secretion of proinflammatory cytokines of macrophages such as IL-1β, IL-8, and TNF-α (149). Besides, JMJD3 has been shown to mediate lipopolysaccharide-induced inflammation by directly targeting and promoting the transcription of important inflammatory factors such as TNF-α, IL-6, IL-1β, COX-2, and ICAM-1 (150).
As mentioned above, NF-κB is a key coordinator of innate immunity and inflammation, and an important endogenous tumor promoter (19). It has been demonstrated that in tumor initiating cells, tumor cells, or inflammatory cells, NF-κB might function by acting as the downstream of microbial or tissue damage perception through the TLR signaling pathway as well as the signaling pathways mediated by the inflammatory cytokines TNF-α and IL-1β. In tumor cells, epithelial cells and inflammatory cells that are at risk of carcinogen transformation, NF-κB activates the prostaglandin synthesis pathway (such as COX-2), iNOS, and angiogenic factors. Besides, another important functional mechanism of NF-κB in tumor cells is to promote cell survival by modulating the expression levels of anti-apoptotic genes such as BCL-2. Furthermore, more and more studies have indicated the inter-connections and compensatory pathways between NF-κB and HIF1-α system (18, 151, 152), linking innate immunity with hypoxic response. And, NF-κB has been associated with the occurrence and development of tumors due to its regulatory effects on tumor-related inflammation in inflamed tissues (153, 154). Importantly, the inflammatory responses mediated by NF-κB have been closely associated with the pathogenesis of multiple tumor types. For example, NF-κB acts as a transcription factor to promote the expression of inflammatory cytokines IL-6 and IL-15, contributing to the development of colon cancer (155–158). NF-κB promotes the expression of inflammatory cytokines such as IL-17, IL-8 and TNF-α and facilitates the development of prostate cancer (159, 160). Moreover, NF-κB induces lymphatic carcinoma by promoting the expression of IL-6 and inflammatory COX-2, and activated NF-κB promotes the expression of inflammatory factors such as IL-22, TNF-α and iNOS to benefit the progression of hepatocellular carcinoma (161). Although many experimental and clinical studies have indicated that inflammation has tumor precursor activity, some evidence does not fit this general pattern. For example, obvious chronic inflammatory reactions such as psoriasis are not correlated with an increased risk of skin cancer (162). On the other hand, the presence of inflammatory cells is associated with a better prognosis in certain tumors or tumor subgroups as exemplified by eosinophils in colon tumors, and TAM in breast tumors and pancreatic tumor subgroups. These observations have suggested that inflammatory responses might also be tumor-suppressive (163). Indeed, it has been shown that properly activated macrophages, a typical component of cancer-related inflammation, can kill tumor cells and trigger a cancer-destructive inflammatory response surrounding the blood vessel wall, although their tumor-promoting properties are dominate in most cases (163). It is worth noting that there is evidence showing that NF-κB is important in determining the balance between tumorigenicity and anti-tumor properties of macrophages (164, 165), and modulating the function of NF-κB might be promising to improve the efficiency of cancer immunotherapy. Das et al. have reported that both NF-κB and JMJD3 were significantly upregulated in LPS-induced macrophage cell lines in vitro (166). With use of JMJD3 gene knockout and stable transfection approaches, action mechanisms of the NF-κB-JMJD3 signal axis in monocytes were investigated; as a results, it was revealed that the deletion of JMJD3 resulted in enrichment of H3K27me3 in promoter regions of NF-κB-related inflammatory target genes such as monocyte chemoattractant protein-1 (MCP-1) and IL-1β, and inhibited the transcription and expression of genes associated with inflammation (166). These results suggested that JMJD3 in the NF-κB-JMJD3 signaling axis could enhance the expression of NF-κB-related inflammatory genes by regulating H3K27me3 methylation levels. In mechanism, the activated NF-κB-JMJD3 signaling pathway recruits upregulated NF-κB/P65 and JMJD3 to H3K27ME3-rich transcription initiation regions, and the H3K27me3 conformation changes through the demethylation of Jmjd3, thus exposing the target gene transcription initiation binding site. The binding of the donor transcription factor-κB initiates the transcription process, resulting in the release of inflammatory mediators and adhesion molecules, contributing to the development of many chronic inflammation and inflammation-related cancers (Figure 1). Moreover, it has been reported that JMJD3 can not only regulate the expression of some inflammatory factors, but also bind to the promoters of some oncogenes or tumor suppressor genes, and play different roles in different stages of the disease development. For example, it has been well-known that functional loss of Ink4B/p15-ARF/p14-INK4A/p16 is an important inducing factor for tumorigenesis in multiple cancer types, and JMJD3 has been reported to promote the expression of tumor suppressors p16 and P14 (167). Studies have shown that JMJD3 is involved in inducing polarization of M2-like macrophages and thus participates in tumor progression (168). Taken together, considering the promoting roles of inflammatory factors in cancer (169), it might be reasonable to speculate that blocking JMJD3 may serve as an entry point for targeted therapy of chronic inflammation or even as a novel antitumor target for inflammation-related cancers.
JMJD5 and Cancer-Related Inflammation
JMJD5 has two subtypes in human body, namely, subtype 1 and subtype 2 which containing 454 and 416 amino acid residues, respectively (170). Although JMJD5 (also known as KDM8) is a member of the JMJD family histone demethylases, this protein has been shown to function not only as a demethylase but also as a hydroxylase, playing an important role in cell cycle regulation, embryonic development, osteoclast differentiation, and circadian rhythm regulation (170, 171).
At present, although the pathogenesis of primary liver cancer is not clear, hepatitis B virus (HBV) infection and related immune response has been well-established to be involved in the pathogenesis of hepatocellular carcinoma (172). Kouwaki et al. has shown that JMJD5 could promote HBV replication through Gly135 interaction with HBV X protein (HBX), and thus JMJD5 is a novel HBX-binding protein responsible for the regulation of HBV replication (173). Although HBx has been confirmed to be localized in the cytoplasm and nucleus (174), JMJD5 is clearly localized in the nucleus, while co-expression of JMJD5 and HBX resulted in partial translocation of JMJD5 from the nucleus to the cytoplasm (173). To further confirm the role of JMJD5 and HBX interaction in HBV replication, researchers prepared JMJD5-knockout human “hemochromatotic” Huh7 cell lines and found that HBV replication was severely impaired in JMJD5 knockout cells (173). The interaction between JMJD5 and HBX was also found to be involved in post-transcriptional steps such as HBV capsid stabilization and aggregation. Gly135 of JMJD5 is a key amino acid residue that interacts with HBx, substitution of Gly135 by Glu could eliminate the repair effect of HBV replication by overexpressing JMJD5 in JMJD5-knockout cells (173). Moreover, expression of an enzyme deficient JMJD5 mutant that replaces His321 with Ala in the JmjC domain did not restore HBV replication in JMJD5 knockout cells (173). These results suggest that both the association of JMJD5 with HBX and the hydroxylase activity of JMJD5 are necessary for effective HBV replication. Besides, it has been suggested that JMJD5 could inhibit the proliferation and cell cycle progression of hepatocellular carcinoma cells by regulating P21 (175, 176), and thus affecting the development of hepatocellular carcinoma (Figure 2).
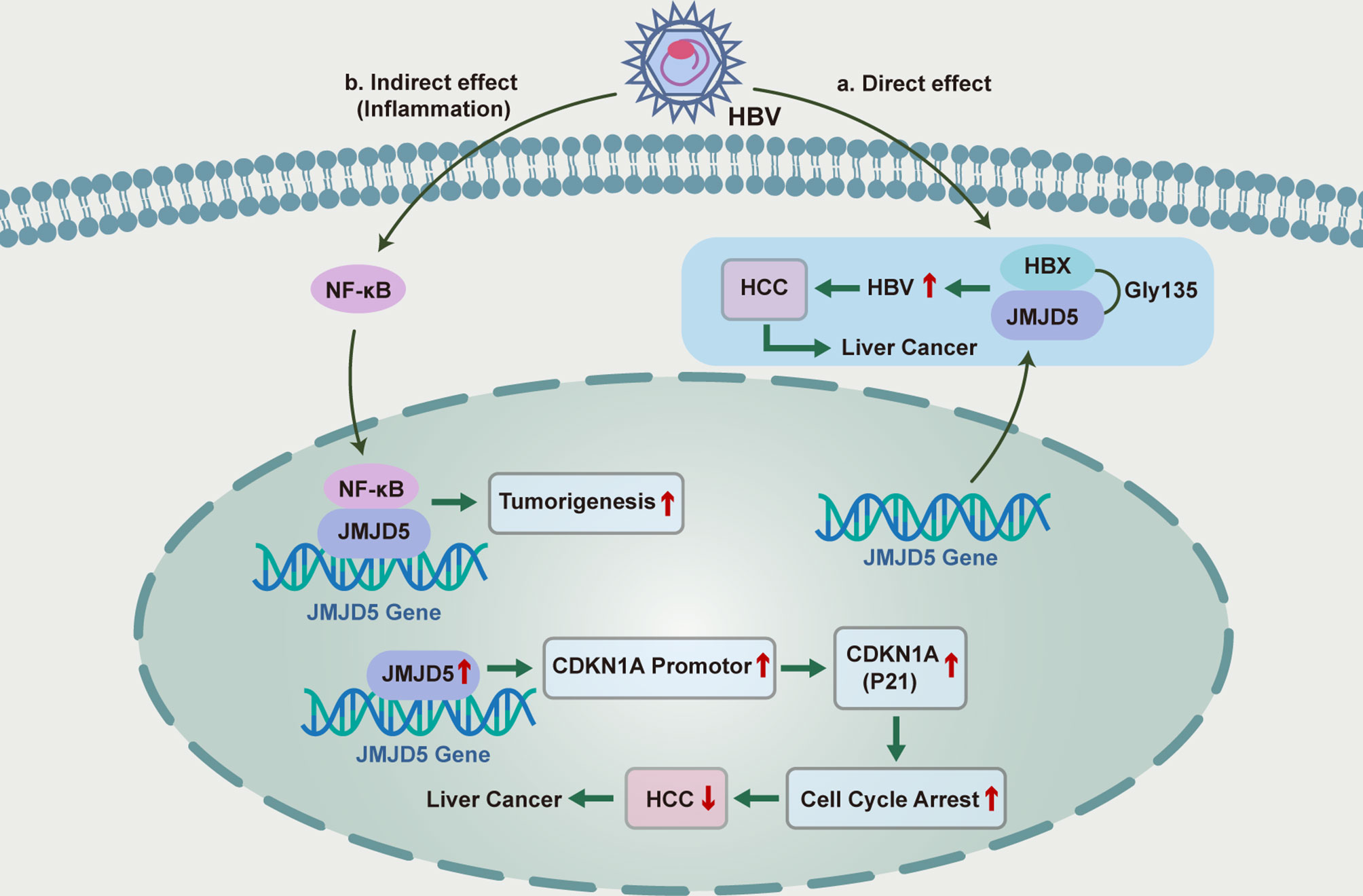
Figure 2 Functional roles and mechanisms of JMJD5 in the inflammatory regulation of HBC-induced hepatocellular carcinoma.
Targeting the JMJD Histone Demethylases for Anti-Cancer Therapies
Herein, we summarized the preclinical or clinical progress of anti-cancer drug development by targeting the JMJD histone demethylases mentioned above.
Pre-Clinical Trials Targeting the JMJD Histone Demethylases
With the gradual revelation of its role in tumorigenesis and development, JMJD3 has become a potential target in tumor therapy. At present, aiming at modulating the cancer promoting effect of JMJD3, specific small molecule inhibitors are synthesized to block the histone demethylation function of JMJD3 protein. For example, GSK-J4, a selective inhibitor of JMJD3 and UTX, has been proved to exert anticancer activity in a variety of tumor cell types, with its unique advantages of low action concentration, almost non-toxicity to normal cells, and reversible modification (177–179). As for colorectal cancer, Zhang J et al. found that GSK-J4 can strongly inhibit the characteristics of tumor initiation cells- and stem cells- related genes in colorectal cancer, reshape the epigenetic pattern, reduce the malignant phenotype of colorectal cancer cells, and sensitize them to chemotherapy (179). In the study of prostate cancer, Cao Z et al. found that GSK-J4 increased the H3K27me3 levels on cyclinD1 promoter by inhibiting JMJD3 expression, and thus inducing cell cycle arrest and tumor cell death (180). These findings suggest that GSK-J4 could be able to reverse JMJD3-modulated biological behaviors of tumor cells. Although targeting JMJD3 has good antitumor effect, the role of JMJD3 and UTX in the same kind of tumor is not exactly the same (177), and the potential side effects of GSK-J4 should be taken into careful consideration. Moreover, JMJD3 has antitumor effect in some tumors, and enhancing the expression and activity of JMJD3 by vitamin D has been shown therapeutic effects in certain tumors such as colon cancer. However, there are currently few known JMJD3 activators, and, more efforts are needed to make the therapeutic effect more specific and safe.
JIB-04 is a small molecule compound showing inhibitory effects on the histone demethylation activity of JMJD proteins, without affecting other α-ketoglutarate dependent hydroxylase or histone modification enzymes (181). The therapeutic effects of JIB-04 have been evaluated by cell viability assays in a large cohort of cells, including human lung cancer cell lines, primary or immortalized non-tumorigenic human bronchial epithelial cells, as well as prostate cancer cell line, primary prostate stromal cells, and prostate epithelial cells. In addition, colony formation studies in multiple cell lines also confirmed that JIB-04 effectively inhibited tumorigenesis by interfering with anchoring independent cell growth. As for in vivo studies, Wang et al. constructed two xenograft mouse models of H358 and A549 in their experiment (181). In all drug-treated animals, tumor growth rates were significantly reduced in the JIB-04 drug group compared to the carrier group. Significant reductions in final tumor weight were also observed in both models, with no effect on overall body weight or overall health. Histological evaluation of major organs revealed no abnormalities except increased liver weight and liver vacuoles. Taken together, these findings suggested that this compound could alter the transcriptional programs responsible for the growth of cancer cells but not normal cells, inducing cancer-specific cell death. The mechanism and target therapies of JMJD protein family in inflammatory tumor were summarized in Table 1.
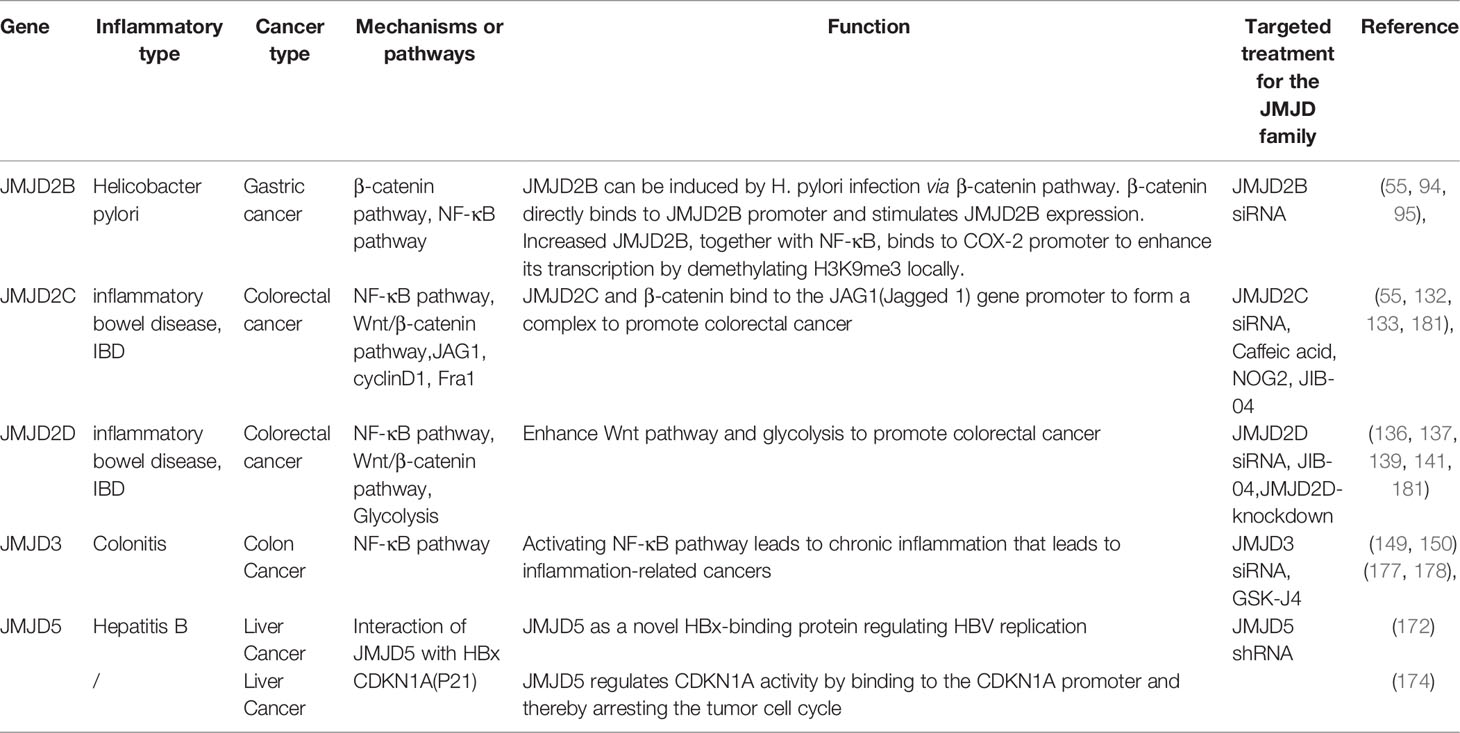
Table 1 The mechanism and target therapies of JMJD protein family in inflammatory tumor.
Clinical Trials Targeting the JMJD Histone Demethylases
So far, the only clinical trial of JMJD histone demethylases-based anti-cancer therapeutics is targeting JMJD2C. In view of the above, JMJD2C plays an important role in tumor development, and inhibition of this enzyme activity might hinder the proliferation and other biological behaviors of tumor cells (182). High expression of JMJD2C has been observed in a portion of clinical samples from patients with esophageal squamous cell carcinoma (ESCC), with its expression levels positively correlated with lymph nodes and tumor metastasis, as well as the low survival rate of ESCC (183). JMJD2C inhibitors available mainly include α-ketoglutaric acid analogues, such as N-oxalylglycine2 (NOG2), which can interfere with JmjC catalytic domain and affect JMJD demethylase activity through competitive antagonism against α-ketoglutaric acid (184, 185). Currently, a clinical trial evaluating caffeic acid as a JMJD2C modulator is being carried out in China, with the aim to investigate the efficacy and safety of caffeic acid in the treatment of advanced ESCC in China. Briefly, 240 patients with advanced ESCC will be randomly assigned to one of two groups: group A (caffeic acid treatment) or group B (placebo), followed for one year to determine the overall survival and progression-free survival of each group. Although the results of the clinical study have not yet been published, this trial might provide a paradigm for JMJD proteins-based precise targeted tumor therapy in the future. Considering the involvement of JMJD histone demethylases in cancer-related inflammation, targeting members of this protein family would contribute to the achievement of better outcomes for cancer therapeutics.
Conclusions and Perspectives
Cancer-associated inflammation is one of the key features of cancer. Several members of the JMJD family histone demethylases have been shown to be involved in inflammation and its regulation and play an important role in many factors affecting tumor biology. From the perspective of tumor development, epigenetic regulation occurs at different stages of tumorigenesis, progression and metastasis, and is one of the important “tools” for the “dialogue” between tumor cells and inflammatory system. Inflammation can induce new epigenetic modifications in tumor cells, which in turn remodels the inflammatory tumor microenvironment. As an important part of epigenetics, abnormal patterns of the JMJD family-mediated histone methylation have been identified in the crosstalk between tumor progression and inflammatory processes, affecting the occurrence and development of different types of tumors. The complex relationship among inflammation, tumor and epigenetics also provides novel opportunities for the development of cancer therapeutics. Actually, targeted regulation of epigenetics for anti-cancer treatment has gradually attracted attention of researchers.
However, there are still problems to be solved when harnessing the histone demethylase activity of JMJD family proteins. First, histone demethylation activities have been shown to be partially responsible for the regulatory effects of JMJD histone demethylases on cancer-related inflammation, other involved mechanisms need to be further elucidated. Second, a large number of downstream targets of JMJD histone demethylases remain to be identified, which might make the therapeutic outcomes of targeted therapies unpredictable and induce treatment resistance. Third, there are only few specific small molecule compounds available to target the histone demethylation activity of JMJD proteins, and the crystal structure and structure-activity relationship of these proteins need to be clarified to facilitate high throughput drug screening in the future. The solution of the above problems could help us to have a deeper understanding on the crosstalk between inflammation and cancer, benefiting future development of personalized therapeutic strategies targeting the JMJD family histone demethylases.
Author Contributions
Designed and wrote the manuscript: JY and YH. Revised the content: XiaL, XinL and BZ. Modified the language: XiaL and XinL. Draw the images: JY. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by grants from the National Natural Science Foundation of China (No. 81903033), the National Natural Science Foundation of China (No. 81902662), the National Natural Science Foundation of China (No. 81821002), Sichuan Science and Technology Program 2021YJ0011. This study was also supported by the Basic research project of Nursing Department of West China Second Hospital (HLBKJ202126) and Foundation of Development and Related Diseases of Women and Children Key Laboratory of Sichuan Province (Grant No.2022003).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chou J, Shahi P, Werb Z. MicroRNA-Mediated Regulation of the Tumor Microenvironment. Cell Cycle (2013) 12(20):3262–71. doi: 10.4161/cc.26087
2. Naito Y, Yoshioka Y, Yamamoto Y, Ochiya T. How Cancer Cells Dictate Their Microenvironment: Present Roles of Extracellular Vesicles. Cell Mol Life Sci (2017) 74(4):697–713. doi: 10.1007/s00018-016-2346-3
3. Suzuki T, Minagawa S, Yamazaki T, Arai T, Kanai M, Shinjo S, et al. Loss of Hypoxia Inducible Factor-1alpha Aggravates Gammadelta T-Cell-Mediated Inflammation During Acetaminophen-Induced Liver Injury. Hepatol Commun (2018) 2(5):571–81. doi: 10.1002/hep4.1175
4. Balkwill F, Mantovani A. Inflammation and Cancer: Back to Virchow? Lancet (2001) 357(9255):539–45. doi: 10.1016/S0140-6736(00)04046-0
5. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science (2006) 313(5795):1960–4. doi: 10.1126/science.1129139
6. Whiteside TL. The Tumor Microenvironment and Its Role in Promoting Tumor Growth. Oncogene (2008) 27(45):5904–12. doi: 10.1038/onc.2008.271
7. Park MH, Hong JT. Roles of Nf-Kappab in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells (2016) 5(2):15. doi: 10.3390/cells5020015
8. Trinchieri G. Cancer and Inflammation: An Old Intuition With Rapidly Evolving New Concepts. Annu Rev Immunol (2012) 30:677–706. doi: 10.1146/annurev-immunol-020711-075008
9. Sansone P, Bromberg J. Environment, Inflammation, and Cancer. Curr Opin Genet Dev (2011) 21(1):80–5. doi: 10.1016/j.gde.2010.11.001
10. Hann HW, Coben R, Brown D, Needleman L, Rosato E, Min A, et al. A Long-Term Study of the Effects of Antiviral Therapy on Survival of Patients With Hbv-Associated Hepatocellular Carcinoma (Hcc) Following Local Tumor Ablation. Cancer Med (2014) 3(2):390–6. doi: 10.1002/cam4.197
11. Punturieri A, Szabo E, Croxton TL, Shapiro SD, Dubinett SM. Lung Cancer and Chronic Obstructive Pulmonary Disease: Needs and Opportunities for Integrated Research. J Natl Cancer Inst (2009) 101(8):554–9. doi: 10.1093/jnci/djp023
12. Takahashi K, Ishii Y. Asbestos and the Industrial Safety and Health Law-In Reference to the Ordinance on Prevention of Hazards Due to Specified Chemical Substances and the Ordinance on Prevention of Health Impairment Due to Asbestos. J UOEH (2013) 35(Suppl):121–6. doi: 10.7888/juoeh.35.121
13. Flossmann E, Rothwell PM, British Doctors Aspirin T, the UKTIAAT. Effect of Aspirin on Long-Term Risk of Colorectal Cancer: Consistent Evidence From Randomised and Observational Studies. Lancet (2007) 369(9573):1603–13. doi: 10.1016/S0140-6736(07)60747-8
14. Chan AT, Ogino S, Fuchs CS. Aspirin and the Risk of Colorectal Cancer in Relation to the Expression of Cox-2. N Engl J Med (2007) 356(21):2131–42. doi: 10.1056/NEJMoa067208
15. Koehne CH, Dubois RN. Cox-2 Inhibition and Colorectal Cancer. Semin Oncol (2004) 31(2 Suppl 7):12–21. doi: 10.1053/j.seminoncol.2004.03.041
16. Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. Il-6 and Stat3 Are Required for Survival of Intestinal Epithelial Cells and Development of Colitis-Associated Cancer. Cancer Cell (2009) 15(2):103–13. doi: 10.1016/j.ccr.2009.01.001
17. Karin M, Greten FR. Nf-Kappab: Linking Inflammation and Immunity to Cancer Development and Progression. Nat Rev Immunol (2005) 5(10):749–59. doi: 10.1038/nri1703
18. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. Nf-Kappab Links Innate Immunity to the Hypoxic Response Through Transcriptional Regulation of Hif-1alpha. Nature (2008) 453(7196):807–11. doi: 10.1038/nature06905
19. Karin M. Nuclear Factor-Kappab in Cancer Development and Progression. Nature (2006) 441(7092):431–6. doi: 10.1038/nature04870
20. Grivennikov SI, Karin M. Dangerous Liaisons: Stat3 and Nf-Kappab Collaboration and Crosstalk in Cancer. Cytokine Growth Factor Rev (2010) 21(1):11–9. doi: 10.1016/j.cytogfr.2009.11.005
21. van Kempen LC, Ruiter DJ, van Muijen GN, Coussens LM. The Tumor Microenvironment: A Critical Determinant of Neoplastic Evolution. Eur J Cell Biol (2003) 82(11):539–48. doi: 10.1078/0171-9335-00346
22. Atretkhany KN, Drutskaya MS, Nedospasov SA, Grivennikov SI, Kuprash DV. Chemokines, Cytokines and Exosomes Help Tumors to Shape Inflammatory Microenvironment. Pharmacol Ther (2016) 168:98–112. doi: 10.1016/j.pharmthera.2016.09.011
23. Lippitz BE. Cytokine Patterns in Patients With Cancer: A Systematic Review. Lancet Oncol (2013) 14(6):e218–28. doi: 10.1016/S1470-2045(12)70582-X
24. Wang X, Yang F, Xu G, Zhong S. The Roles of Il-6, Il-8 and Il-10 Gene Polymorphisms in Gastric Cancer: A Meta-Analysis. Cytokine (2018) 111:230–6. doi: 10.1016/j.cyto.2018.08.024
25. Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, et al. Activation of an Il6 Inflammatory Loop Mediates Trastuzumab Resistance in Her2+ Breast Cancer by Expanding the Cancer Stem Cell Population. Mol Cell (2012) 47(4):570–84. doi: 10.1016/j.molcel.2012.06.014
26. Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Kloppel G, et al. Stat3/Socs3 Activation by Il-6 Transsignaling Promotes Progression of Pancreatic Intraepithelial Neoplasia and Development of Pancreatic Cancer. Cancer Cell (2011) 19(4):456–69. doi: 10.1016/j.ccr.2011.03.009
27. Waldner MJ, Foersch S, Neurath MF. Interleukin-6–a Key Regulator of Colorectal Cancer Development. Int J Biol Sci (2012) 8(9):1248–53. doi: 10.7150/ijbs.4614
28. Butz H, Racz K, Hunyady L, Patocs A. Crosstalk Between Tgf-Beta Signaling and the Microrna Machinery. Trends Pharmacol Sci (2012) 33(7):382–93. doi: 10.1016/j.tips.2012.04.003
29. Sun X, Zhang J, Hou Z, Han Q, Zhang C, Tian Z. Mir-146a Is Directly Regulated by Stat3 in Human Hepatocellular Carcinoma Cells and Involved in Anti-Tumor Immune Suppression. Cell Cycle (2015) 14(2):243–52. doi: 10.4161/15384101.2014.977112
30. Allis CD, Jenuwein T. The Molecular Hallmarks of Epigenetic Control. Nat Rev Genet (2016) 17(8):487–500. doi: 10.1038/nrg.2016.59
31. Cantacorps L, Alfonso-Loeches S, Guerri C, Valverde O. Long-Term Epigenetic Changes in Offspring Mice Exposed to Alcohol During Gestation and Lactation. J Psychopharmacol (2019) 33(12):1562–72. doi: 10.1177/0269881119856001
32. Kouzarides T. Chromatin Modifications and Their Function. Cell (2007) 128(4):693–705. doi: 10.1016/j.cell.2007.02.005
33. Couture JF, Trievel RC. Histone-Modifying Enzymes: Encrypting an Enigmatic Epigenetic Code. Curr Opin Struct Biol (2006) 16(6):753–60. doi: 10.1016/j.sbi.2006.10.002
34. Lawrence M, Daujat S, Schneider R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet (2016) 32(1):42–56. doi: 10.1016/j.tig.2015.10.007
35. Heard E, Rougeulle C, Arnaud D, Avner P, Allis CD, Spector DL. Methylation of Histone H3 at Lys-9 Is an Early Mark on the X Chromosome During X Inactivation. Cell (2001) 107(6):727–38. doi: 10.1016/s0092-8674(01)00598-0
36. Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, et al. Role of Histone H3 Lysine 27 Methylation in X Inactivation. Science (2003) 300(5616):131–5. doi: 10.1126/science.1084274
37. Greer EL, Shi Y. Histone Methylation: A Dynamic Mark in Health, Disease and Inheritance. Nat Rev Genet (2012) 13(5):343–57. doi: 10.1038/nrg3173
38. Tokunaga R, Sakamoto Y, Nakagawa S, Miyake K, Izumi D, Kosumi K, et al. The Prognostic Significance of Histone Lysine Demethylase Jmjd3/Kdm6b in Colorectal Cancer. Ann Surg Oncol (2016) 23(2):678–85. doi: 10.1245/s10434-015-4879-3
39. Martin C, Zhang Y. The Diverse Functions of Histone Lysine Methylation. Nat Rev Mol Cell Biol (2005) 6(11):838–49. doi: 10.1038/nrm1761
40. Zhang Y, Reinberg D. Transcription Regulation by Histone Methylation: Interplay Between Different Covalent Modifications of the Core Histone Tails. Genes Dev (2001) 15(18):2343–60. doi: 10.1101/gad.927301
41. Hino S, Kohrogi K, Nakao M. Histone Demethylase Lsd1 Controls the Phenotypic Plasticity of Cancer Cells. Cancer Sci (2016) 107(9):1187–92. doi: 10.1111/cas.13004
42. Arcipowski KM, Martinez CA, Ntziachristos P. Histone Demethylases in Physiology and Cancer: A Tale of Two Enzymes, Jmjd3 and Utx. Curr Opin Genet Dev (2016) 36:59–67. doi: 10.1016/j.gde.2016.03.010
43. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage Polarization: Tumor-Associated Macrophages as a Paradigm for Polarized M2 Mononuclear Phagocytes. Trends Immunol (2002) 23(11):549–55. doi: 10.1016/s1471-4906(02)02302-5
44. Yang X, Wang X, Liu D, Yu L, Xue B, Shi H. Epigenetic Regulation of Macrophage Polarization by DNA Methyltransferase 3b. Mol Endocrinol (2014) 28(4):565–74. doi: 10.1210/me.2013-1293
45. Takeuch O, Akira S. Epigenetic Control of Macrophage Polarization. Eur J Immunol (2011) 41(9):2490–3. doi: 10.1002/eji.201141792
46. Kooistra SM, Helin K. Molecular Mechanisms and Potential Functions of Histone Demethylases. Nat Rev Mol Cell Biol (2012) 13(5):297–311. doi: 10.1038/nrm3327
47. Markolovic S, Leissing TM, Chowdhury R, Wilkins SE, Lu X, Schofield CJ. Structure-Function Relationships of Human Jmjc Oxygenases-Demethylases Versus Hydroxylases. Curr Opin Struct Biol (2016) 41:62–72. doi: 10.1016/j.sbi.2016.05.013
48. Markolovic S, Wilkins SE, Schofield CJ. Protein Hydroxylation Catalyzed by 2-Oxoglutarate-Dependent Oxygenases. J Biol Chem (2015) 290(34):20712–22. doi: 10.1074/jbc.R115.662627
49. Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. Fih-1 Is an Asparaginyl Hydroxylase Enzyme That Regulates the Transcriptional Activity of Hypoxia-Inducible Factor. Genes Dev (2002) 16(12):1466–71. doi: 10.1101/gad.991402
50. McNeill LA, Hewitson KS, Claridge TD, Seibel JF, Horsfall LE, Schofield CJ. Hypoxia-Inducible Factor Asparaginyl Hydroxylase (Fih-1) Catalyses Hydroxylation at the Beta-Carbon of Asparagine-803. Biochem J (2002) 367(Pt 3):571–5. doi: 10.1042/BJ20021162
51. Trewick SC, McLaughlin PJ, Allshire RC. Methylation: Lost in Hydroxylation? EMBO Rep (2005) 6(4):315–20. doi: 10.1038/sj.embor.7400379
52. Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, et al. Histone Demethylation by a Family of Jmjc Domain-Containing Proteins. Nature (2006) 439(7078):811–6. doi: 10.1038/nature04433
53. Black JC, Van Rechem C, Whetstine JR. Histone Lysine Methylation Dynamics: Establishment, Regulation, and Biological Impact. Mol Cell (2012) 48(4):491–507. doi: 10.1016/j.molcel.2012.11.006
54. Xia X, Lemieux ME, Li W, Carroll JS, Brown M, Liu XS, et al. Integrative Analysis of Hif Binding and Transactivation Reveals Its Role in Maintaining Histone Methylation Homeostasis. Proc Natl Acad Sci USA (2009) 106(11):4260–5. doi: 10.1073/pnas.0810067106
55. Pollard PJ, Loenarz C, Mole DR, McDonough MA, Gleadle JM, Schofield CJ, et al. Regulation of Jumonji-Domain-Containing Histone Demethylases by Hypoxia-Inducible Factor (Hif)-1alpha. Biochem J (2008) 416(3):387–94. doi: 10.1042/BJ20081238
56. Scanlon SE, Glazer PM. Multifaceted Control of DNA Repair Pathways by the Hypoxic Tumor Microenvironment. DNA Repair (Amst) (2015) 32:180–9. doi: 10.1016/j.dnarep.2015.04.030
57. Hua Z, Lv Q, Ye W, Wong CK, Cai G, Gu D, et al. Mirna-Directed Regulation of Vegf and Other Angiogenic Factors Under Hypoxia. PloS One (2006) 1(1):e116. doi: 10.1371/journal.pone.0000116
58. Lu H, Ouyang W, Huang C. Inflammation, a Key Event in Cancer Development. Mol Cancer Res (2006) 4(4):221–33. doi: 10.1158/1541-7786.MCR-05-0261
59. Yasmin R, Siraj S, Hassan A, Khan AR, Abbasi R, Ahmad N. Epigenetic Regulation of Inflammatory Cytokines and Associated Genes in Human Malignancies. Mediators Inflammation (2015) 2015:201703. doi: 10.1155/2015/201703
60. De Santa F, Narang V, Yap ZH, Tusi BK, Burgold T, Austenaa L, et al. Jmjd3 Contributes to the Control of Gene Expression in Lps-Activated Macrophages. EMBO J (2009) 28(21):3341–52. doi: 10.1038/emboj.2009.271
61. Raghuraman S, Donkin I, Versteyhe S, Barres R, Simar D. The Emerging Role of Epigenetics in Inflammation and Immunometabolism. Trends Endocrinol Metab (2016) 27(11):782–95. doi: 10.1016/j.tem.2016.06.008
62. Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of Lsd1 Histone Demethylase Activity by Its Associated Factors. Mol Cell (2005) 19(6):857–64. doi: 10.1016/j.molcel.2005.08.027
63. Fodor BD, Kubicek S, Yonezawa M, O'Sullivan RJ, Sengupta R, Perez-Burgos L, et al. Jmjd2b Antagonizes H3k9 Trimethylation at Pericentric Heterochromatin in Mammalian Cells. Genes Dev (2006) 20(12):1557–62. doi: 10.1101/gad.388206
64. Ye L, Fan Z, Yu B, Chang J, Al Hezaimi K, Zhou X, et al. Histone Demethylases Kdm4b and Kdm6b Promotes Osteogenic Differentiation of Human Mscs. Cell Stem Cell (2012) 11(1):50–61. doi: 10.1016/j.stem.2012.04.009
65. Das ND, Choi MR, Jung KH, Park JH, Lee HT, Das A, et al. Functional Analysis of Histone Demethylase Jmjd2b on Lipopolysaccharide-Treated Murine Neural Stem Cells (Nscs). Neurotox Res (2013) 23(2):154–65. doi: 10.1007/s12640-012-9346-3
66. Yang J, Jubb AM, Pike L, Buffa FM, Turley H, Baban D, et al. The Histone Demethylase Jmjd2b Is Regulated by Estrogen Receptor Alpha and Hypoxia, and Is a Key Mediator of Estrogen Induced Growth. Cancer Res (2010) 70(16):6456–66. doi: 10.1158/0008-5472.CAN-10-0413
67. Kim JG, Yi JM, Park SJ, Kim JS, Son TG, Yang K, et al. Histone Demethylase Jmjd2b-Mediated Cell Proliferation Regulated by Hypoxia and Radiation in Gastric Cancer Cell. Biochim Biophys Acta (2012) 1819(11-12):1200–7. doi: 10.1016/j.bbagrm.2012.10.001
68. Liu Y, Zheng P, Liu Y, Ji T, Liu X, Yao S, et al. An Epigenetic Role for Prl-3 as a Regulator of H3k9 Methylation in Colorectal Cancer. Gut (2013) 62(4):571–81. doi: 10.1136/gutjnl-2011-301059
69. Kawazu M, Saso K, Tong KI, McQuire T, Goto K, Son DO, et al. Histone Demethylase Jmjd2b Functions as a Co-Factor of Estrogen Receptor in Breast Cancer Proliferation and Mammary Gland Development. PloS One (2011) 6(3):e17830. doi: 10.1371/journal.pone.0017830
70. Shi L, Sun L, Li Q, Liang J, Yu W, Yi X, et al. Histone Demethylase Jmjd2b Coordinates H3k4/H3k9 Methylation and Promotes Hormonally Responsive Breast Carcinogenesis. Proc Natl Acad Sci USA (2011) 108(18):7541–6. doi: 10.1073/pnas.1017374108
71. Toyokawa G, Cho HS, Iwai Y, Yoshimatsu M, Takawa M, Hayami S, et al. The Histone Demethylase Jmjd2b Plays an Essential Role in Human Carcinogenesis Through Positive Regulation of Cyclin-Dependent Kinase 6. Cancer Prev Res (Phila) (2011) 4(12):2051–61. doi: 10.1158/1940-6207.CAPR-11-0290
72. Fu L, Chen L, Yang J, Ye T, Chen Y, Fang J. Hif-1alpha-Induced Histone Demethylase Jmjd2b Contributes to the Malignant Phenotype of Colorectal Cancer Cells Via an Epigenetic Mechanism. Carcinogenesis (2012) 33(9):1664–73. doi: 10.1093/carcin/bgs217
73. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: Globocan Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2018) 68(6):394–424. doi: 10.3322/caac.21492
74. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: Globocan Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
75. Naylor GM, Gotoda T, Dixon M, Shimoda T, Gatta L, Owen R, et al. Why Does Japan Have a High Incidence of Gastric Cancer? Comparison of Gastritis Between Uk and Japanese Patients. Gut (2006) 55(11):1545–52. doi: 10.1136/gut.2005.080358
76. Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Piñeros M, et al. Estimating the Global Cancer Incidence and Mortality in 2018: GLOBOCAN Sources and Methods. Int J Cancer. (2019) 144(8):1941–53. doi: 10.1002/ijc.31937
77. Van Cutsem E, Sagaert X, Topal B, Haustermans K, Prenen H. Gastric Cancer. Lancet (2016) 388(10060):2654–64. doi: 10.1016/S0140-6736(16)30354-3
78. Allemani C, Matsuda T, Di Carlo V, Harewood R, Matz M, Nikšić M, et al. Global Surveillance of Trends in Cancer Survival 2000-14 (CONCORD-3): Analysis of Individual Records for 37513025 Patients Diagnosed With One of 18 Cancers From 322 Population-Based Registries in 71 Countries. Lancet (2018) 391(10125):1023–75. doi: 10.1016/S0140-6736(17)33326-3
79. Peek RM Jr, Crabtree JE. Helicobacter Infection and Gastric Neoplasia. J Pathol (2006) 208(2):233–48. doi: 10.1002/path.1868
80. Houghton J, Wang TC. Helicobacter Pylori and Gastric Cancer: A New Paradigm for Inflammation-Associated Epithelial Cancers. Gastroenterology (2005) 128(6):1567–78. doi: 10.1053/j.gastro.2005.03.037
81. Yin Y, Grabowska AM, Clarke PA, Whelband E, Robinson K, Argent RH, et al. Helicobacter Pylori Potentiates Epithelial:Mesenchymal Transition in Gastric Cancer: Links to Soluble Hb-Egf, Gastrin and Matrix Metalloproteinase-7. Gut (2010) 59(8):1037–45. doi: 10.1136/gut.2009.199794
82. Ernst PB, Peura DA, Crowe SE. The Translation of Helicobacter Pylori Basic Research to Patient Care. Gastroenterology (2006) 130(1):188–206; quiz 12-3. doi: 10.1053/j.gastro.2005.06.032
83. Tabassam FH, Graham DY, Yamaoka Y. Helicobacter Pylori Activate Epidermal Growth Factor Receptor- and Phosphatidylinositol 3-Oh Kinase-Dependent Akt and Glycogen Synthase Kinase 3beta Phosphorylation. Cell Microbiol (2009) 11(1):70–82. doi: 10.1111/j.1462-5822.2008.01237.x
84. Nagy TA, Frey MR, Yan F, Israel DA, Polk DB, Peek RM Jr.. Helicobacter Pylori Regulates Cellular Migration and Apoptosis by Activation of Phosphatidylinositol 3-Kinase Signaling. J Infect Dis (2009) 199(5):641–51. doi: 10.1086/596660
85. Bronte-Tinkew DM, Terebiznik M, Franco A, Ang M, Ahn D, Mimuro H, et al. Helicobacter Pylori Cytotoxin-Associated Gene a Activates the Signal Transducer and Activator of Transcription 3 Pathway In Vitro and In Vivo. Cancer Res (2009) 69(2):632–9. doi: 10.1158/0008-5472.CAN-08-1191
86. Maeda S, Akanuma M, Mitsuno Y, Hirata Y, Ogura K, Yoshida H, et al. Distinct Mechanism of Helicobacter Pylori-Mediated Nf-Kappa B Activation Between Gastric Cancer Cells and Monocytic Cells. J Biol Chem (2001) 276(48):44856–64. doi: 10.1074/jbc.M105381200
87. Franco AT, Israel DA, Washington MK, Krishna U, Fox JG, Rogers AB, et al. Activation of Beta-Catenin by Carcinogenic Helicobacter Pylori. Proc Natl Acad Sci USA (2005) 102(30):10646–51. doi: 10.1073/pnas.0504927102
88. Cho SO, Lim JW, Kim KH, Kim H. Involvement of Ras and Ap-1 in Helicobacter Pylori-Induced Expression of Cox-2 and Inos in Gastric Epithelial Ags Cells. Dig Dis Sci (2010) 55(4):988–96. doi: 10.1007/s10620-009-0828-y
89. Bhavsar AP, Guttman JA, Finlay BB. Manipulation of Host-Cell Pathways by Bacterial Pathogens. Nature (2007) 449(7164):827–34. doi: 10.1038/nature06247
90. Nardone G, Compare D, De Colibus P, de Nucci G, Rocco A. Helicobacter Pylori and Epigenetic Mechanisms Underlying Gastric Carcinogenesis. Dig Dis (2007) 25(3):225–9. doi: 10.1159/000103890
91. Ding SZ, Goldberg JB, Hatakeyama M. Helicobacter Pylori Infection, Oncogenic Pathways and Epigenetic Mechanisms in Gastric Carcinogenesis. Future Oncol (2010) 6(5):851–62. doi: 10.2217/fon.10.37
92. Li W, Zhao L, Zang W, Liu Z, Chen L, Liu T, et al. Histone Demethylase Jmjd2b Is Required for Tumor Cell Proliferation and Survival and Is Overexpressed in Gastric Cancer. Biochem Biophys Res Commun (2011) 416(3-4):372–8. doi: 10.1016/j.bbrc.2011.11.045
93. Zhao L, Li W, Zang W, Liu Z, Xu X, Yu H, et al. Jmjd2b Promotes Epithelial-Mesenchymal Transition by Cooperating With Beta-Catenin and Enhances Gastric Cancer Metastasis. Clin Cancer Res (2013) 19(23):6419–29. doi: 10.1158/1078-0432.CCR-13-0254
94. Han F, Ren J, Zhang J, Sun Y, Ma F, Liu Z, et al. Jmjd2b Is Required for Helicobacter Pylori-Induced Gastric Carcinogenesis Via Regulating Cox-2 Expression. Oncotarget (2016) 7(25):38626–37. doi: 10.18632/oncotarget.9573
96. Gnad T, Feoktistova M, Leverkus M, Lendeckel U, Naumann M. Helicobacter Pylori-Induced Activation of Beta-Catenin Involves Low Density Lipoprotein Receptor-Related Protein 6 and Dishevelled. Mol Cancer (2010) 9:31. doi: 10.1186/1476-4598-9-31
97. Brabletz T, Jung A, Dag S, Hlubek F, Kirchner T. Beta-Catenin Regulates the Expression of the Matrix Metalloproteinase-7 in Human Colorectal Cancer. Am J Pathol (1999) 155(4):1033–8. doi: 10.1016/s0002-9440(10)65204-2
98. Lin MT, Zuon CY, Chang CC, Chen ST, Chen CP, Lin BR, et al. Cyr61 Induces Gastric Cancer Cell Motility/Invasion Via Activation of the Integrin/Nuclear Factor-Kappab/Cyclooxygenase-2 Signaling Pathway. Clin Cancer Res (2005) 11(16):5809–20. doi: 10.1158/1078-0432.CCR-04-2639
99. Chang YJ, Wu MS, Lin JT, Chen CC. Helicobacter Pylori-Induced Invasion and Angiogenesis of Gastric Cells Is Mediated by Cyclooxygenase-2 Induction Through Tlr2/Tlr9 and Promoter Regulation. J Immunol (2005) 175(12):8242–52. doi: 10.4049/jimmunol.175.12.8242
100. Rodriguez C, Lopez P, Pozo M, Duce AM, Lopez-Pelaez M, Fernandez M, et al. Cox2 Expression and Erk1/Erk2 Activity Mediate Cot-Induced Cell Migration. Cell Signal (2008) 20(9):1625–31. doi: 10.1016/j.cellsig.2008.05.008
101. Thiel A, Mrena J, Ristimaki A. Cyclooxygenase-2 and Gastric Cancer. Cancer Metastasis Rev (2011) 30(3-4):387–95. doi: 10.1007/s10555-011-9312-1
102. Yamac D, Ayyildiz T, Coskun U, Akyurek N, Dursun A, Seckin S, et al. Cyclooxygenase-2 Expression and Its Association With Angiogenesis, Helicobacter Pylori, and Clinicopathologic Characteristics of Gastric Carcinoma. Pathol Res Pract (2008) 204(8):527–36. doi: 10.1016/j.prp.2008.01.002
103. Lim HY, Joo HJ, Choi JH, Yi JW, Yang MS, Cho DY, et al. Increased Expression of Cyclooxygenase-2 Protein in Human Gastric Carcinoma. Clin Cancer Res (2000) 6(2):519–25. doi: 10.1093/infdis/141.1.92
104. Sung JJ, Leung WK, Go MY, To KF, Cheng AS, Ng EK, et al. Cyclooxygenase-2 Expression in Helicobacter Pylori-Associated Premalignant and Malignant Gastric Lesions. Am J Pathol (2000) 157(3):729–35. doi: 10.1016/S0002-9440(10)64586-5
105. Berry WL, Janknecht R. Kdm4/Jmjd2 Histone Demethylases: Epigenetic Regulators in Cancer Cells. Cancer Res (2013) 73(10):2936–42. doi: 10.1158/0008-5472.CAN-12-4300
106. Katoh M, Katoh M. Identification and Characterization of Jmjd2 Family Genes in Silico. Int J Oncol (2004) 24(6):1623–8. doi: 10.3892/ijo.24.6.1623
107. Leurs U, Clausen RP, Kristensen JL, Lohse B. Inhibitor Scaffold for the Histone Lysine Demethylase Kdm4c (Jmjd2c). Bioorg Med Chem Lett (2012) 22(18):5811–3. doi: 10.1016/j.bmcl.2012.07.091
108. Yang ZQ, Imoto I, Fukuda Y, Pimkhaokham A, Shimada Y, Imamura M, et al. Identification of a Novel Gene, Gasc1, Within an Amplicon at 9p23-24 Frequently Detected in Esophageal Cancer Cell Lines. Cancer Res (2000) 60(17):4735–9. doi: 10.1046/j.1523-5394.2000.85010.x
109. Nighot P, Ma T. Endocytosis of Intestinal Tight Junction Proteins: In Time and Space. Inflammation Bowel Dis (2021) 27(2):283–90. doi: 10.1093/ibd/izaa141
110. Das PP, Shao Z, Beyaz S, Apostolou E, Pinello L, De Los Angeles A, et al. Distinct and Combinatorial Functions of Jmjd2b/Kdm4b and Jmjd2c/Kdm4c in Mouse Embryonic Stem Cell Identity. Mol Cell (2014) 53(1):32–48. doi: 10.1016/j.molcel.2013.11.011
111. Cascante A, Klum S, Biswas M, Antolin-Fontes B, Barnabe-Heider F, Hermanson O. Gene-Specific Methylation Control of H3k9 and H3k36 on Neurotrophic Bdnf Versus Astroglial Gfap Genes by Kdm4a/C Regulates Neural Stem Cell Differentiation. J Mol Biol (2014) 426(20):3467–77. doi: 10.1016/j.jmb.2014.04.008
112. Klose RJ, Kallin EM, Zhang Y. Jmjc-Domain-Containing Proteins and Histone Demethylation. Nat Rev Genet (2006) 7(9):715–27. doi: 10.1038/nrg1945
113. Loh YH, Zhang W, Chen X, George J, Ng HH. Jmjd1a and Jmjd2c Histone H3 Lys 9 Demethylases Regulate Self-Renewal in Embryonic Stem Cells. Genes Dev (2007) 21(20):2545–57. doi: 10.1101/gad.1588207
114. Ishimura A, Terashima M, Kimura H, Akagi K, Suzuki Y, Sugano S, et al. Jmjd2c Histone Demethylase Enhances the Expression of Mdm2 Oncogene. Biochem Biophys Res Commun (2009) 389(2):366–71. doi: 10.1016/j.bbrc.2009.08.155
115. Zhao E, Ding J, Xia Y, Liu M, Ye B, Choi JH, et al. Kdm4c and Atf4 Cooperate in Transcriptional Control of Amino Acid Metabolism. Cell Rep (2016) 14(3):506–19. doi: 10.1016/j.celrep.2015.12.053
116. Li X, Dong S. Histone Demethylase Jmjd2b and Jmjd2c Induce Fibroblast Growth Factor 2: Mediated Tumorigenesis of Osteosarcoma. Med Oncol (2015) 32(3):53. doi: 10.1007/s12032-015-0503-4
117. Luo W, Chang R, Zhong J, Pandey A, Semenza GL. Histone Demethylase Jmjd2c Is a Coactivator for Hypoxia-Inducible Factor 1 That Is Required for Breast Cancer Progression. Proc Natl Acad Sci USA (2012) 109(49):E3367-76. doi: 10.1073/pnas.1217394109
118. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2018. CA Cancer J Clin (2018) 68(1):7–30. doi: 10.3322/caac.21442
119. Chen W. Cancer Statistics: Updated Cancer Burden in China. Chin J Cancer Res (2015) 27(1):1. doi: 10.3978/j.issn.1000-9604.2015.02.07
120. Wang D, DuBois RN. The Role of Anti-Inflammatory Drugs in Colorectal Cancer. Annu Rev Med (2013) 64:131–44. doi: 10.1146/annurev-med-112211-154330
121. Wolf AMD, Fontham ETH, Church TR, Flowers CR, Guerra CE, LaMonte SJ, et al. Colorectal Cancer Screening for Average-Risk Adults: 2018 Guideline Update From the American Cancer Society. CA Cancer J Clin (2018) 68(4):250–81. doi: 10.3322/caac.21457
122. de la Chapelle A. Genetic Predisposition to Colorectal Cancer. Nat Rev Cancer (2004) 4(10):769–80. doi: 10.1038/nrc1453
123. Giglia MD, Chu DI. Familial Colorectal Cancer: Understanding the Alphabet Soup. Clin Colon Rectal Surg (2016) 29(3):185–95. doi: 10.1055/s-0036-1584290
124. Herszenyi L, Barabas L, Miheller P, Tulassay Z. Colorectal Cancer in Patients With Inflammatory Bowel Disease: The True Impact of the Risk. Dig Dis (2015) 33(1):52–7. doi: 10.1159/000368447
125. Peeters PJ, Bazelier MT, Leufkens HG, de Vries F, De Bruin ML. The Risk of Colorectal Cancer in Patients With Type 2 Diabetes: Associations With Treatment Stage and Obesity. Diabetes Care (2015) 38(3):495–502. doi: 10.2337/dc14-1175
126. Reulen RC, Frobisher C, Winter DL, Kelly J, Lancashire ER, Stiller CA, et al. Long-Term Risks of Subsequent Primary Neoplasms Among Survivors of Childhood Cancer. JAMA (2011) 305(22):2311–9. doi: 10.1001/jama.2011.747
127. Henderson TO, Oeffinger KC, Whitton J, Leisenring W, Neglia J, Meadows A, et al. Secondary Gastrointestinal Cancer in Childhood Cancer Survivors: A Cohort Study. Ann Intern Med (2012) 156(11):757–66, W-260. doi: 10.7326/0003-4819-156-11-201206050-00002
128. Baxter NN, Tepper JE, Durham SB, Rothenberger DA, Virnig BA. Increased Risk of Rectal Cancer After Prostate Radiation: A Population-Based Study. Gastroenterology (2005) 128(4):819–24. doi: 10.1053/j.gastro.2004.12.038
129. Travis LB, Fossa SD, Schonfeld SJ, McMaster ML, Lynch CF, Storm H, et al. Second Cancers Among 40,576 Testicular Cancer Patients: Focus on Long-Term Survivors. J Natl Cancer Inst (2005) 97(18):1354–65. doi: 10.1093/jnci/dji278
130. de Sousa EMF, Vermeulen L. Wnt Signaling in Cancer Stem Cell Biology. Cancers (Basel) (2016) 8(7):60. doi: 10.3390/cancers8070060
131. Srinivasan T, Walters J, Bu P, Than EB, Tung KL, Chen KY, et al. Notch Signaling Regulates Asymmetric Cell Fate of Fast- and Slow-Cycling Colon Cancer-Initiating Cells. Cancer Res (2016) 76(11):3411–21. doi: 10.1158/0008-5472.CAN-15-3198
132. Yamamoto S, Tateishi K, Kudo Y, Yamamoto K, Isagawa T, Nagae G, et al. Histone Demethylase Kdm4c Regulates Sphere Formation by Mediating the Cross Talk Between Wnt and Notch Pathways in Colonic Cancer Cells. Carcinogenesis (2013) 34(10):2380–8. doi: 10.1093/carcin/bgt174
133. Kim TD, Fuchs JR, Schwartz E, Abdelhamid D, Etter J, Berry WL, et al. Pro-Growth Role of the Jmjd2c Histone Demethylase in Hct-116 Colon Cancer Cells and Identification of Curcuminoids as Jmjd2 Inhibitors. Am J Transl Res (2014) 6(3):236–47. doi: 10.4999/uhod.14490
134. Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, et al. Reversal of Histone Lysine Trimethylation by the Jmjd2 Family of Histone Demethylases. Cell (2006) 125(3):467–81. doi: 10.1016/j.cell.2006.03.028
135. Shin S, Janknecht R. Activation of Androgen Receptor by Histone Demethylases Jmjd2a and Jmjd2d. Biochem Biophys Res Commun (2007) 359(3):742–6. doi: 10.1016/j.bbrc.2007.05.179
136. Khoury-Haddad H, Guttmann-Raviv N, Ipenberg I, Huggins D, Jeyasekharan AD, Ayoub N. Parp1-Dependent Recruitment O138f Kdm4d Histone Demethylase to DNA Damage Sites Promotes Double-Strand Break Repair. Proc Natl Acad Sci USA (2014) 111(7):E728-37. doi: 10.1073/pnas.1317585111
137. Wu R, Wang Z, Zhang H, Gan H, Zhang Z. H3k9me3 Demethylase Kdm4d Facilitates the Formation of Pre-Initiative Complex and Regulates DNA Replication. Nucleic Acids Res (2017) 45(1):169–80. doi: 10.1093/nar/gkw848
138. Zoabi M, Nadar-Ponniah PT, Khoury-Haddad H, Usaj M, Budowski-Tal I, Haran T, et al. Rna-Dependent Chromatin Localization of Kdm4d Lysine Demethylase Promotes H3k9me3 Demethylation. Nucleic Acids Res (2014) 42(21):13026–38. doi: 10.1093/nar/gku1021
139. Kim TD, Oh S, Shin S, Janknecht R. Regulation of Tumor Suppressor P53 and Hct116 Cell Physiology by Histone Demethylase Jmjd2d/Kdm4d. PloS One (2012) 7(4):e34618. doi: 10.1371/journal.pone.0034618
140. Zhuo M, Chen W, Shang S, Guo P, Peng K, Li M, et al. Inflammation-Induced Jmjd2d Promotes Colitis Recovery and Colon Tumorigenesis by Activating Hedgehog Signaling. Oncogene (2020) 39(16):3336–53. doi: 10.1038/s41388-020-1219-2
141. Logan CY, Nusse R. The Wnt Signaling Pathway in Development and Disease. Annu Rev Cell Dev Biol (2004) 20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126
142. Lan F, Bayliss PE, Rinn JL, Whetstine JR, Wang JK, Chen S, et al. A Histone H3 Lysine 27 Demethylase Regulates Animal Posterior Development. Nature (2007) 449(7163):689–94. doi: 10.1038/nature06192
143. Xiang Y, Zhu Z, Han G, Lin H, Xu L, Chen CD. Jmjd3 Is a Histone H3k27 Demethylase. Cell Res (2007) 17(10):850–7. doi: 10.1038/cr.2007.83
144. Hong S, Cho YW, Yu LR, Yu H, Veenstra TD, Ge K. Identification of Jmjc Domain-Containing Utx and Jmjd3 as Histone H3 Lysine 27 Demethylases. Proc Natl Acad Sci USA (2007) 104(47):18439–44. doi: 10.1073/pnas.0707292104
145. Kamikawa YF, Donohoe ME. The Localization of Histone H3k27me3 Demethylase Jmjd3 Is Dynamically Regulated. Epigenetics (2014) 9(6):834–41. doi: 10.4161/epi.28524
146. Swigut T, Wysocka J. H3k27 Demethylases, at Long Last. Cell (2007) 131(1):29–32. doi: 10.1016/j.cell.2007.09.026
147. Lhuissier E, Aury-Landas J, Allas L, Boittin M, Boumediene K, Bauge C. Antiproliferative Effect of the Histone Demethylase Inhibitor Gsk-J4 in Chondrosarcomas. IUBMB Life (2019) 71(11):1711–9. doi: 10.1002/iub.2110
148. Sherry-Lynes MM, Sengupta S, Kulkarni S, Cochran BH. Regulation of the Jmjd3 (Kdm6b) Histone Demethylase in Glioblastoma Stem Cells by Stat3. PloS One (2017) 12(4):e0174775. doi: 10.1371/journal.pone.0174775
149. Yan Q, Sun L, Zhu Z, Wang L, Li S, Ye RD. Jmjd3-Mediated Epigenetic Regulation of Inflammatory Cytokine Gene Expression in Serum Amyloid a-Stimulated Macrophages. Cell Signal (2014) 26(9):1783–91. doi: 10.1016/j.cellsig.2014.03.025
150. Yu S, Chen X, Xiu M, He F, Xing J, Min D, et al. The Regulation of Jmjd3 Upon the Expression of Nf-Kappab Downstream Inflammatory Genes in Lps Activated Vascular Endothelial Cells. Biochem Biophys Res Commun (2017) 485(1):62–8. doi: 10.1016/j.bbrc.2017.02.020
151. Mizukami Y, Jo WS, Duerr EM, Gala M, Li J, Zhang X, et al. Induction of Interleukin-8 Preserves the Angiogenic Response in Hif-1alpha-Deficient Colon Cancer Cells. Nat Med (2005) 11(9):992–7. doi: 10.1038/nm1294
152. Carbia-Nagashima A, Gerez J, Perez-Castro C, Paez-Pereda M, Silberstein S, Stalla GK, et al. Rsume, A Small Rwd-Containing Protein, Enhances Sumo Conjugation and Stabilizes Hif-1alpha During Hypoxia. Cell (2007) 131(2):309–23. doi: 10.1016/j.cell.2007.07.044
153. Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, et al. Ikkbeta Links Inflammation and Tumorigenesis in a Mouse Model of Colitis-Associated Cancer. Cell (2004) 118(3):285–96. doi: 10.1016/j.cell.2004.07.013
154. Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. Nf-Kappab Functions as a Tumour Promoter in Inflammation-Associated Cancer. Nature (2004) 431(7007):461–6. doi: 10.1038/nature02924
155. Harpel K, Leung S, Rice PF, Jones M, Barton JK, Bommireddy R. Imaging Colon Cancer Development in Mice: Il-6 Deficiency Prevents Adenoma in Azoxymethane-Treated Smad3 Knockouts. Phys Med Biol (2016) 61(3):N60–9. doi: 10.1088/0031-9155/61/3/N60
156. Waldner MJ, Neurath MF. Master Regulator of Intestinal Disease: Il-6 in Chronic Inflammation and Cancer Development. Semin Immunol (2014) 26(1):75–9. doi: 10.1016/j.smim.2013.12.003
157. Chua AC, Klopcic BR, Ho DS, Fu SK, Forrest CH, Croft KD, et al. Dietary Iron Enhances Colonic Inflammation and Il-6/Il-11-Stat3 Signaling Promoting Colonic Tumor Development in Mice. PloS One (2013) 8(11):e78850. doi: 10.1371/journal.pone.0078850
158. Bahri R, Pateras IS, D'Orlando O, Goyeneche-Patino DA, Campbell M, Polansky JK, et al. Il-15 Suppresses Colitis-Associated Colon Carcinogenesis by Inducing Antitumor Immunity. Oncoimmunology (2015) 4(9):e1002721. doi: 10.1080/2162402X.2014.1002721
159. De Angulo A, Faris R, Daniel B, Jolly C, deGraffenried L. Age-Related Increase in Il-17 Activates Pro-Inflammatory Signaling in Prostate Cells. Prostate (2015) 75(5):449–62. doi: 10.1002/pros.22931
160. Neveu B, Moreel X, Deschenes-Rompre MP, Bergeron A, LaRue H, Ayari C, et al. Il-8 Secretion in Primary Cultures of Prostate Cells Is Associated With Prostate Cancer Aggressiveness. Res Rep Urol (2014) 6:27–34. doi: 10.2147/RRU.S58643
161. Radaeva S, Sun R, Pan HN, Hong F, Gao B. Interleukin 22 (Il-22) Plays a Protective Role in T Cell-Mediated Murine Hepatitis: Il-22 Is a Survival Factor for Hepatocytes Via Stat3 Activation. Hepatology (2004) 39(5):1332–42. doi: 10.1002/hep.20184
162. Nickoloff BJ, Ben-Neriah Y, Pikarsky E. Inflammation and Cancer: Is the Link as Simple as We Think? J Invest Dermatol (2005) 124(6):x–xiv. doi: 10.1111/j.0022-202X.2005.23724.x
163. Mantovani A, Bottazzi B, Colotta F, Sozzani S, Ruco L. The Origin and Function of Tumor-Associated Macrophages. Immunol Today (1992) 13(7):265–70. doi: 10.1016/0167-5699(92)90008-U
164. Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, et al. "Re-Educating" Tumor-Associated Macrophages by Targeting Nf-Kappab. J Exp Med (2008) 205(6):1261–8. doi: 10.1084/jem.20080108
165. Saccani A, Schioppa T, Porta C, Biswas SK, Nebuloni M, Vago L, et al. P50 Nuclear Factor-Kappab Overexpression in Tumor-Associated Macrophages Inhibits M1 Inflammatory Responses and Antitumor Resistance. Cancer Res (2006) 66(23):11432–40. doi: 10.1158/0008-5472.CAN-06-1867
166. Das ND, Jung KH, Choi MR, Yoon HS, Kim SH, Chai YG. Gene Networking and Inflammatory Pathway Analysis in a Jmjd3 Knockdown Human Monocytic Cell Line. Cell Biochem Funct (2012) 30(3):224–32. doi: 10.1002/cbf.1839
167. Kim WY, Sharpless NE. The Regulation of Ink4/Arf in Cancer and Aging. Cell (2006) 127(2):265–75. doi: 10.1016/j.cell.2006.10.003
168. Liang X, Luo M, Shao B, Yang JY, Tong A, Wang RB, et al. Phosphatidylserine Released From Apoptotic Cells in Tumor Induces M2-Like Macrophage Polarization Through the PSR-STAT3-JMJD3 Axis. Cancer Commun (Lond). (2022) 42(3):205–22. doi: 10.1002/cac2.12272
169. Fu XT, Dai Z, Song K, Zhang ZJ, Zhou ZJ, Zhou SL, et al. Macrophage-Secreted Il-8 Induces Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma Cells by Activating the Jak2/Stat3/Snail Pathway. Int J Oncol (2015) 46(2):587–96. doi: 10.3892/ijo.2014.2761
170. Wang H, Zhou X, Wu M, Wang C, Zhang X, Tao Y, et al. Structure of the Jmjc-Domain-Containing Protein Jmjd5. Acta Crystallogr D Biol Crystallogr (2013) 69(Pt 10):1911–20. doi: 10.1107/S0907444913016600
171. Del Rizzo PA, Krishnan S, Trievel RC. Crystal Structure and Functional Analysis of Jmjd5 Indicate an Alternate Specificity and Function. Mol Cell Biol (2012) 32(19):4044–52. doi: 10.1128/MCB.00513-12
172. Tu T, Buhler S, Bartenschlager R. Chronic Viral Hepatitis and Its Association With Liver Cancer. Biol Chem (2017) 398(8):817–37. doi: 10.1515/hsz-2017-0118
173. Kouwaki T, Okamoto T, Ito A, Sugiyama Y, Yamashita K, Suzuki T, et al. Hepatocyte Factor Jmjd5 Regulates Hepatitis B Virus Replication Through Interaction With Hbx. J Virol (2016) 90(7):3530–42. doi: 10.1128/JVI.02776-15
174. Henkler F, Hoare J, Waseem N, Goldin RD, McGarvey MJ, Koshy R, et al. Intracellular Localization of the Hepatitis B Virus Hbx Protein. J Gen Virol (2001) 82(Pt 4):871–82. doi: 10.1099/0022-1317-82-4-871
175. Wu BH, Chen H, Cai CM, Fang JZ, Wu CC, Huang LY, et al. Epigenetic Silencing of Jmjd5 Promotes the Proliferation of Hepatocellular Carcinoma Cells by Down-Regulating the Transcription of Cdkn1a 686. Oncotarget (2016) 7(6):6847–63. doi: 10.18632/oncotarget.6867
176. Ocker M, Bitar SA, Monteiro AC, Gali-Muhtasib H, Schneider-Stock R. Epigenetic Regulation of P21(Cip1/Waf1) in Human Cancer. Cancers (Basel) (2019) 11(9):1343. doi: 10.3390/cancers11091343
177. Ntziachristos P, Tsirigos A, Welstead GG, Trimarchi T, Bakogianni S, Xu L, et al. Contrasting Roles of Histone 3 Lysine 27 Demethylases in Acute Lymphoblastic Leukaemia. Nature (2014) 514(7523):513–7. doi: 10.1038/nature13605
178. Hsieh IY, He J, Wang L, Lin B, Liang Z, Lu B, et al. H3k27me3 Loss Plays a Vital Role in Cemip Mediated Carcinogenesis and Progression of Breast Cancer With Poor Prognosis. BioMed Pharmacother (2020) 123:109728. doi: 10.1016/j.biopha.2019.109728
179. Zhang J, Ying Y, Li M, Wang M, Huang X, Jia M, et al. Targeted Inhibition of Kdm6 Histone Demethylases Eradicates Tumor-Initiating Cells Via Enhancer Reprogramming in Colorectal Cancer. Theranostics (2020) 10(22):10016–30. doi: 10.7150/thno.47081
180. Cao Z, Shi X, Tian F, Fang Y, Wu JB, Mrdenovic S, et al. Kdm6b Is an Androgen Regulated Gene and Plays Oncogenic Roles by Demethylating H3k27me3 at Cyclin D1 Promoter in Prostate Cancer. Cell Death Dis (2021) 12(1):2. doi: 10.1038/s41419-020-03354-4
181. Wang L, Chang J, Varghese D, Dellinger M, Kumar S, Best AM, et al. A Small Molecule Modulates Jumonji Histone Demethylase Activity and Selectively Inhibits Cancer Growth. Nat Commun (2013) 4:2035. doi: 10.1038/ncomms3035
182. Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, et al. The Putative Oncogene Gasc1 Demethylates Tri- and Dimethylated Lysine 9 on Histone H3. Nature (2006) 442(7100):307–11. doi: 10.1038/nature04837
183. Rose NR, Woon EC, Kingham GL, King ON, Mecinovic J, Clifton IJ, et al. Selective Inhibitors of the Jmjd2 Histone Demethylases: Combined Nondenaturing Mass Spectrometric Screening and Crystallographic Approaches. J Med Chem (2010) 53(4):1810–8. doi: 10.1021/jm901680b
184. Sun LL, Holowatyj A, Xu XE, Wu JY, Wu ZY, Shen JH, et al. Histone Demethylase Gasc1, A Potential Prognostic and Predictive Marker in Esophageal Squamous Cell Carcinoma. Am J Cancer Res (2013) 3(5):509–17. doi: 10.1186/bcr3445
Keywords: cancer, inflammation, cancer therapeutics, JMJD family, histone demethylases
Citation: Yang J, Hu Y, Zhang B, Liang X and Li X (2022) The JMJD Family Histone Demethylases in Crosstalk Between Inflammation and Cancer. Front. Immunol. 13:881396. doi: 10.3389/fimmu.2022.881396
Received: 22 February 2022; Accepted: 23 March 2022;
Published: 26 April 2022.
Edited by:
Guan-Jun Yang, Ningbo University, ChinaReviewed by:
Yuanbin Song, Sun Yat-sen University Cancer Center, ChinaXingmei Duan, Sichuan Academy of Medical Sciences and Sichuan Provincial People’s Hospital, China
Zhuobin Liang, Shenzhen Bay Laboratory, China
Copyright © 2022 Yang, Hu, Zhang, Liang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao Liang, eGlhb2xpYW5nOTEwMUAxNjMuY29t; Xin Li, bGl4aW4wOTE0MDcxQDEyNi5jb20=
†These authors have contributed equally to this work