
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol. , 08 April 2022
Sec. Primary Immunodeficiencies
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.881206
 Moisés Labrador-Horrillo1†
Moisés Labrador-Horrillo1† Clara Franco-Jarava2,3,4,5†
Clara Franco-Jarava2,3,4,5† Marina Garcia-Prat5,6,7
Marina Garcia-Prat5,6,7 Alba Parra-Martínez5,6,7
Alba Parra-Martínez5,6,7 María Antolín8Sandra Salgado-Perandrés2,3,5Aina Aguiló-Cucurull2,3,5
María Antolín8Sandra Salgado-Perandrés2,3,5Aina Aguiló-Cucurull2,3,5 Mónica Martinez-Gallo2,3,4,5*
Mónica Martinez-Gallo2,3,4,5* Roger Colobran2,3,4,5,8*
Roger Colobran2,3,4,5,8*SASH3 is a lymphoid-specific adaptor protein. In a recent study, SASH3 deficiency was described as a novel X-linked combined immunodeficiency with immune dysregulation, associated with impaired TCR signaling and thymocyte survival in humans. The small number of patients reported to date showed recurrent sinopulmonary, cutaneous and mucosal infections, and autoimmune cytopenia. Here we describe an adult patient previously diagnosed with common variable immunodeficiency (CVID) due to low IgG and IgM levels and recurrent upper tract infections. Two separate, severe viral infections drew our attention and pointed to an underlying T cell defect: severe varicella zoster virus (VZV) infection at the age of 4 years and bilateral pneumonia due type A influenza infection at the age of 38. Genetic testing using an NGS-based custom-targeted gene panel revealed a novel hemizygous loss-of-function variant in the SASH3 gene (c.505C>T/p.Gln169*). The patient’s immunological phenotype included marked B cell lymphopenia with reduced pre-switch and switch memory B cells, decreased CD4+ and CD8+ naïve T cells, elevated CD4+ and CD8+ TEMRA cells, and abnormal T cell activation and proliferation. The patient showed a suboptimal response to Streptococcus pneumoniae (polysaccharide) vaccine, and a normal response to Haemophilus influenzae type B (conjugate) vaccine and SARS-CoV-2 (RNA) vaccine. In summary, our patient has a combined immunodeficiency, although he presented with a phenotype resembling CVID. Two severe episodes of viral infection alerted us to a possible T-cell defect, and genetic testing led to SASH3 deficiency. Our patient displays a milder phenotype than has been reported previously in these patients, thus expanding the clinical spectrum of this recently identified inborn error of immunity.
Common variable immunodeficiency (CVID) is the most frequently diagnosed primary immunodeficiency in adults. It is characterized by overt hypogammaglobulinemia (low IgG and IgA levels, with or without IgM), and a poor or absent antibody response to infection and immunization. The clinical manifestations of CVID are heterogeneous, and include recurrent respiratory tract infections and other complications related to immune dysregulation, such as gastrointestinal, autoimmune, and lymphoproliferative disorders. Genetically, the heterogeneity is even greater, and during the last decade the increasing use of next-generation sequencing (NGS) technology has accelerated the discovery of novel genes associated with a CVID phenotype, via autosomal dominant or recessive inheritance. The immunological consequences of some of these genetic defects clearly go beyond B cell involvement, and this has progressively faded out the boundaries between CVID and combined immunodeficiency (CID). One example is NFKB1 haploinsufficiency, identified in 2015 and considered the most common monogenic cause of CVID (1, 2). Several reports provide evidence that NFKB1 haploinsufficiency also underlies a variable type of CID affecting both the B and T lymphocyte compartments, with a broadened spectrum of disease manifestations, including Epstein Barr virus (EBV)-induced lymphoproliferative disease (3–5). A similar situation is seen in LRBA deficiency and CTL4 haploinsufficiency. These two genetic defects have been repeatedly identified in patients initially diagnosed as having CVID and they are usually included in the gene list to be tested in CVID patients (6–8). However, both disorders display a complex variety of clinical and immunologic abnormalities, in most cases involving T cell defects (9).
Because of the specificity and the clinical consequences of these and other genetic defects associated with CVID, once the genetic diagnosis has been established in a patient previously considered to have CVID, the diagnosis should be changed to the respective genetic cause (eg, to NFKB1 deficiency or LRBA deficiency) (PMID:34153571). Still, it is important to keep in mind the various genetic etiologies that can underlie the CVID diagnosis.
Very recently, Delmonte et al. described a novel X-linked CID caused by SASH3 loss-of-function (LOF) mutations (10). The study included four unrelated patients with recurrent sinopulmonary, cutaneous and mucosal infections, and refractory autoimmune cytopenia. Although all patients were genetically diagnosed in the adulthood, their first clinical manifestations occurred in the childhood. The authors demonstrated that SASH3 deficiency cause a global defect on TCR signaling, which was already evident in T cell progenitors. This leads not only to a reduced numbers of CD3+ TCRαβ+ cells in the patients, but also to an enrichment of TCR clonotypes with molecular signatures of self-reactivity. Moreover, T cells exhibited functional defects as decreased proliferation and cell cycle progression and increased apoptosis in response to mitogens (10).
Here, we describe a patient initially diagnosed with CVID who presented a novel hemizygous LOF variant in the SASH3 gene. Our patient displays a milder phenotype than that of the reported patients, thus expanding the clinical spectrum of this novel inborn error of immunity (IEI).
A 43-year-old man, the only child of non-consanguineous parents, attended our allergy clinic with a history of peach allergy (oral allergy syndrome) from childhood. A few years before, the patient had been diagnosed of possible CVID in other center due to low IgG, low IgM, and recurrent upper tract infections. CT sinus scanning showed chronic rhinosinusitis that required 2 to 3 antibiotic courses per year during the following two years, without hospital admission. The chest CT study was normal. Immunoglobulin replacement therapy was not prescribed at that time. During the patient’s clinical follow-up in our hospital, he received a diagnosis of nonspecific lipid transfer protein (nsLTP)-mediated peach allergy. Low IgG levels and absent IgM were confirmed (Table 1). A meticulous anamnesis revealed other infectious episodes before the CVID diagnosis, including a severe varicella zoster virus (VZV) infection at the age of 4 that required hospitalization. During adolescence, he experienced mild warts on the hands that did not require treatment and resolved in 3 to 4 years. The patient also mentioned bilateral pneumonia due to type A influenza infection that had required ICU admission (6 days) 5 years previously (at age 38 years). There was no family history of severe infections.
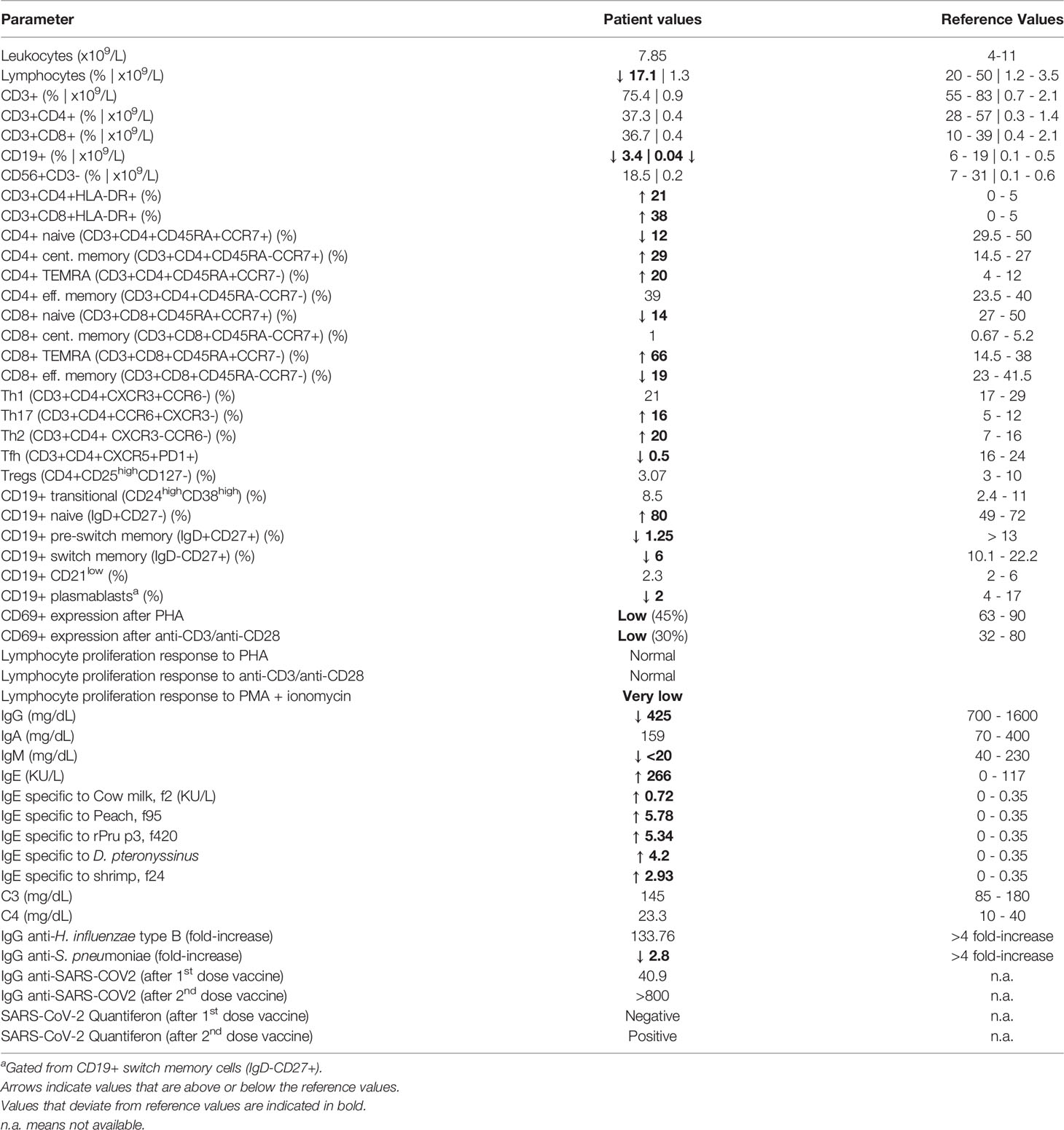
Table 1 Immunological parameters of SASH3-deficient patient.
Although the patient was initially diagnosed with CVID, the history of severe viral infections was an alert sign pointing to possible CID. He was tested using our NGS-based custom-targeted gene panel including 427 IEI-associated genes. We found a novel hemizygous variant in SASH3, consisting of a single nucleotide change (c.505C>T) leading to a premature STOP codon (p.Gln169*). This is a private variant (not present in the main population databases) and is predicted to cause LOF by truncating the SASH3 protein upstream of the SAM and SH3 domains. To date only three SASH3 variants have been reported, two nonsense and one missense, with p.Gln169* being the most N-terminal one (Figure 1A). The variant was confirmed by Sanger and family study, which showed that the mother is a heterozygous carrier (Figure 1B). Western blot of the patient’s PBMCs using a polyclonal antibody targeting the N-terminus of SASH3 showed complete absence of protein, thus confirming the LOF nature of the c.505C>T/p.Gln169* variant (Figure 1C).
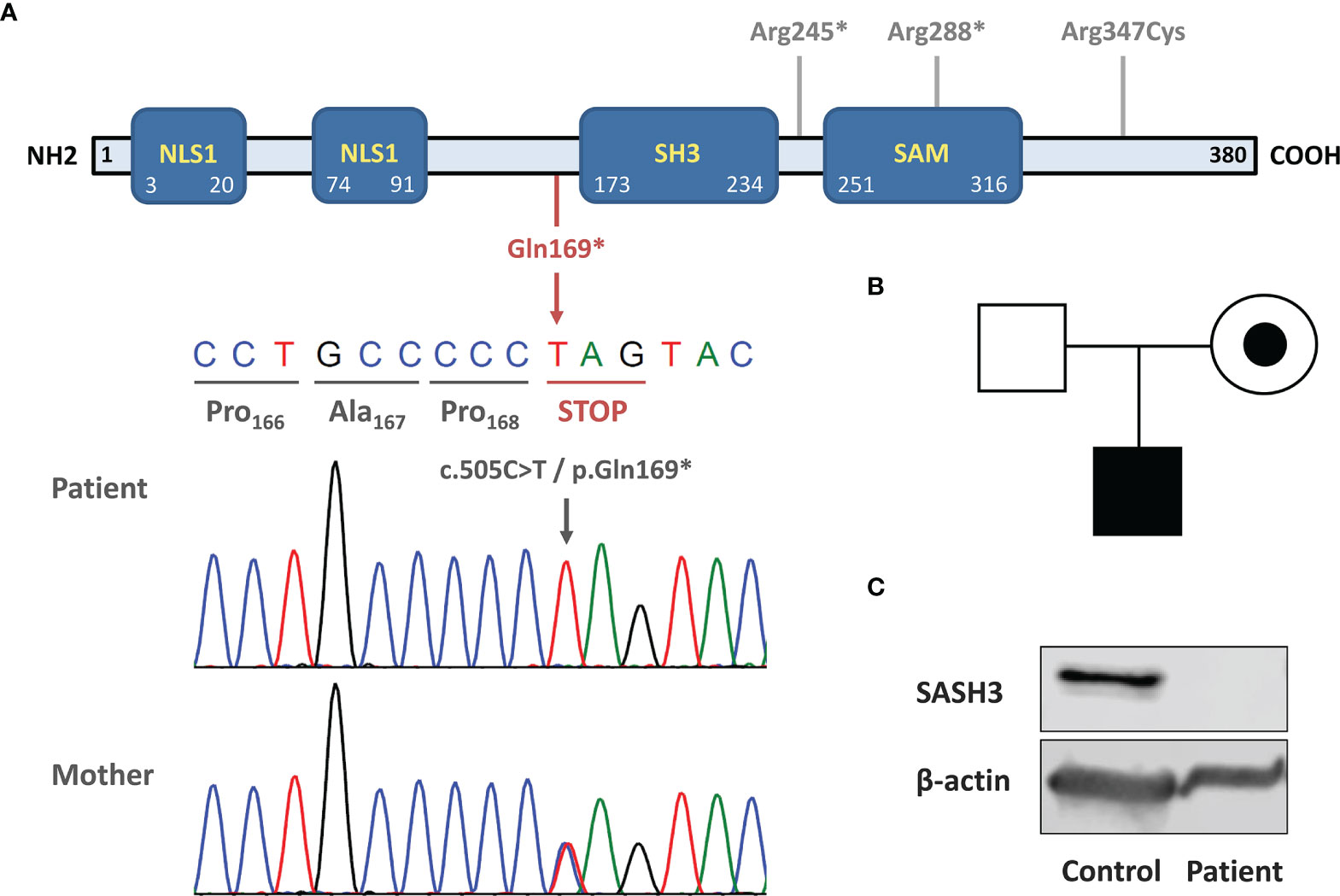
Figure 1 Novel loss-of-function variant causing SASH3 deficiency. (A) Schematic structure of SASH3 protein. The location of the c.505C>T/p.Gln169* variant is indicated below in red and Sanger sequencing confirmation is shown. The three other variants reported by Delmonte et al. are indicated in gray in the upper part. (B) Pedigree and familial segregation of the SASH3 variant. The patient was hemizygous and his mother, a heterozygous carrier. (C) Western blot showing absence of SASH3 protein in patient PBMCs. The immunogen recognized by the antibody used (SASH3 polyclonal antibody #PA5-70305, Termofisher Scientific) is located at the N-terminal region of human SASH3 protein, before the amino acid 169 (where the STOP codon is located).
After the genetic finding of SASH3 deficiency, recently reported as a new type of CID (10), we aimed to assess the patient’s immunological phenotype. White blood cell count was normal, but he had marked B cell lymphopenia both in percentage and absolute numbers (Table 1). Most B cells were naïve, with reductions in pre-switch and switch memory B cells (Figure 2A). Plasmablasts were decreased, whereas CD19+CD21low B cells (usually expanded in autoimmune conditions) were in the normal range.
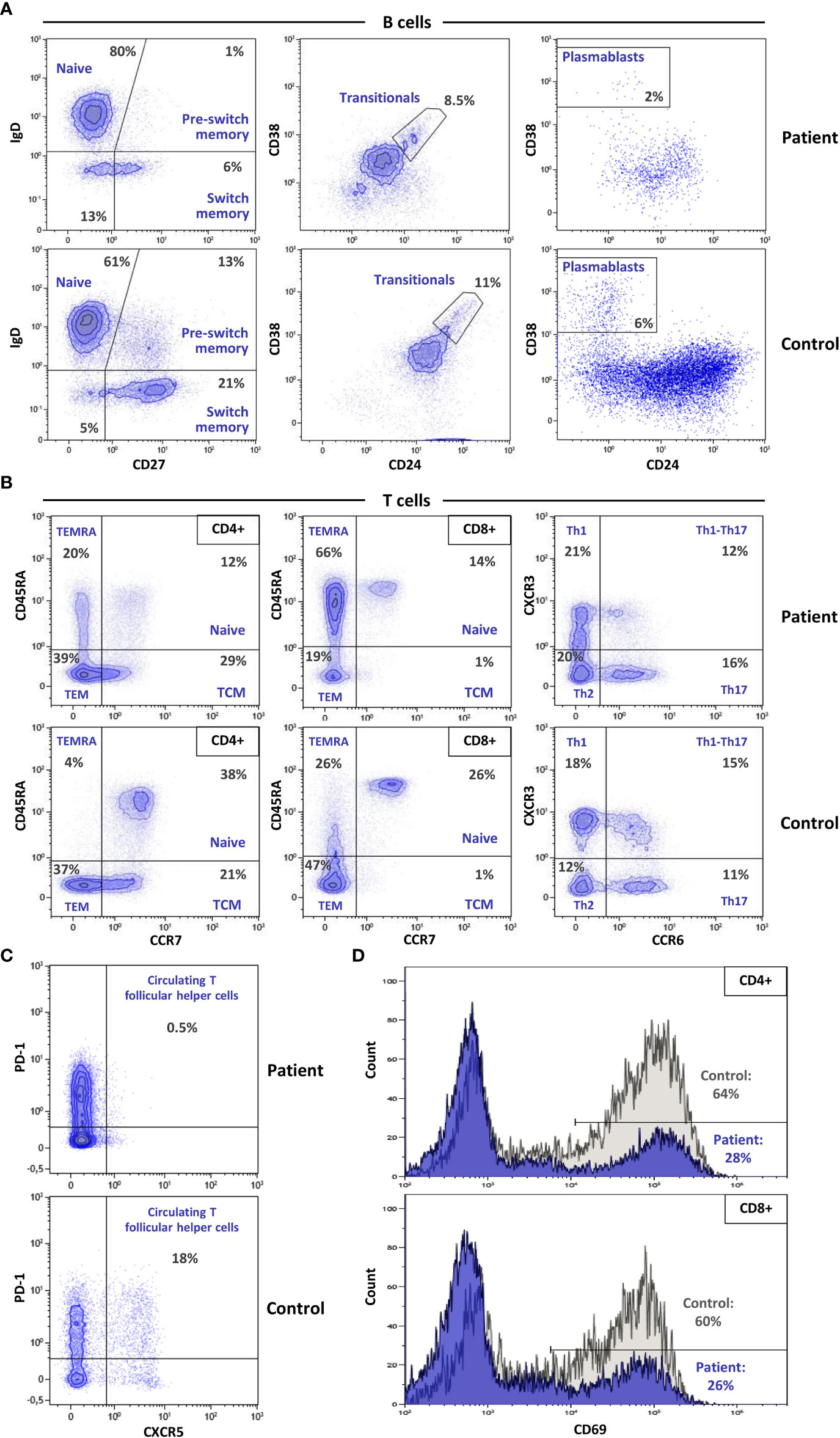
Figure 2 Multicolor flow cytometry evaluation of advanced lymphocyte subpopulations and T cell activation. (A) Dot plots illustrating the main differences in B cell subpopulations between the patient and a healthy control show a dramatic reduction in pre-switch, isotype switching, and plasmablast populations in the patient. (B) Advanced T lymphocyte analysis shows decreased naïve CD4 and CD8 populations, with a striking increase of lymphocytes with TEMRA phenotype. (C) Absence of cTfh cells in the patient vs. the control. (D) CD69 expression after activation with anti-CD3/CD28 in CD4 (upper) and CD8 (lower) lymphocytes, depicts decreased activation capacity in the SASH3-deficient patient.
T cell numbers were normal in both the CD4+ and CD8+ compartments, but analysis of T lymphocyte subset distributions demonstrated a marked decrease in CD4+ and CD8+ naïve T cells (Figure 2B; Table 1). Conversely, CD4+ and CD8+ TEMRA cells were elevated, more markedly in the case of CD8+ T cells, and were accompanied by an increase in HLA-DR expression. There was a complete absence of circulating T follicular helper (cTfh) cells (Figure 2C). Distributions of Th1, Th2, Th17, Tregs, and NK cells were in the normal range.
CD4+ and CD8+ T cells both showed low CD69 expression on stimulation with anti-CD3 and anti-CD28, indicating defective activation (Figure 2D). In vitro T-cell proliferation was also tested. Results were normal when using PHA and anti-CD3/anti-CD28 stimulation, but proliferation was very low in response to PMA + ionomycin (Table 1), indicating that distal intracellular signaling may have been affected.
Finally, vaccine responsiveness was evaluated. The patient showed a suboptimal response to Streptococcus pneumoniae (polysaccharide) vaccine and a normal response to Haemophilus influenzae type B (conjugate) vaccine. There was a good humoral and cellular response to SARS-CoV-2 vaccine (BNT162b2, Pfizer-BioNTech), especially after the second dose (Table 1).
SASH3 (also known as SH3-containing lymphocyte protein, SLY1) is a lymphoid-specific adaptor protein. SASH3 deficiency, recently described by Delmonte et al., is a novel combined immunodeficiency with immune dysregulation, associated with impaired TCR signaling and thymocyte survival in humans (10). These authors identified four unrelated males carrying three novel deleterious hemizygous LOF variants in the SASH3 gene. The present study is the second to date describing a patient with SASH3 deficiency. Our patient carried the c.505C>T/p.Gln169* variant, which truncates SASH3 protein upstream of the SAM and SH3 domains and leads to a complete absence of the protein.
Comparison of the four patients reported by Delmonte et al. and our patient shows some similarities and differences (Table 2). At first glimpse, our patient experienced a less severe clinical course. He was diagnosed with possible CVID at the age of 41 years due to low IgG and IgM levels and recurrent upper tract infections (with a normal chest CT scan). But up to now, he has not shown autoimmune cytopenia or other forms of autoimmunity, and he has no relevant skin manifestations (severe warts or infections) or signs of lymphoproliferation. However, the history of two pertinent viral infections, severe VZV infection in childhood and bilateral pneumonia due type A influenza infection at the age of 38 years, indicated a possible underlying T cell defect. Indeed, as in the previously reported cases, our patient showed a marked decrease in CD4 and CD8 naïve T cells with defective T cell activation and proliferation. Strikingly, whereas the four patients described by Delmonte et al. had severe NK cell lymphopenia, our patient had normal NK counts (Table 2).
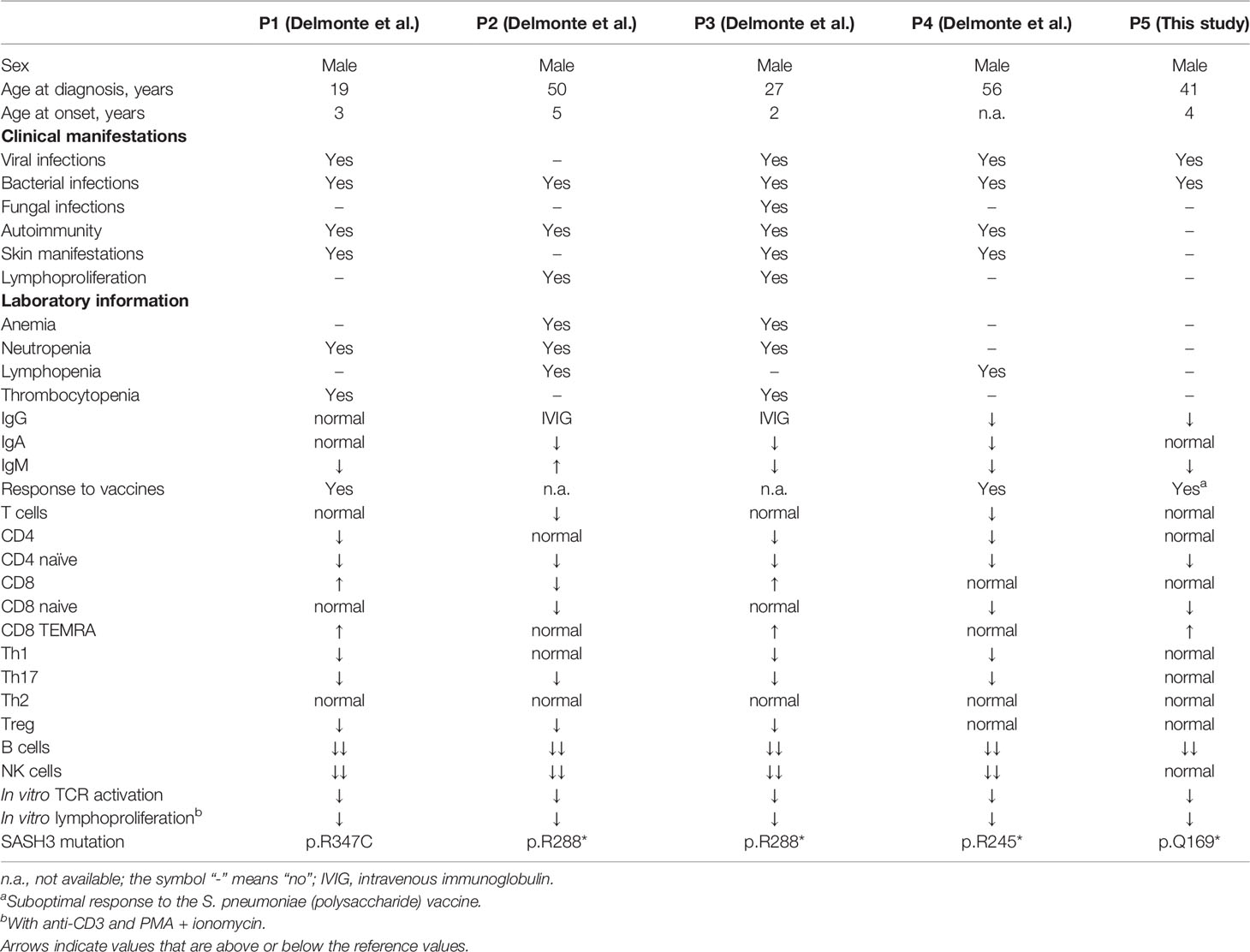
Table 2 Clinical and immunological features of all reported SASH3 deficient patients.
Regarding B cell status, our patient showed marked B cell lymphopenia with very low pre-switch and low switch memory B cells and plasmablasts, in accordance with findings in the other reported patients. The response to various vaccines was normal in all patients tested, although our patient had a suboptimal response to a polysaccharide vaccine (S. pneumoniae). Remarkably, our patient showed a complete absence of cTfh cells, a feature that had not been evaluated in the previous patients. cTfh cells are peripheral counterparts of conventional Tfh cells that are predominantly located in secondary lymphoid tissues and play a crucial role in T cell-dependent humoral immunity and the proper response to most vaccines. Why the response to vaccines was essentially normal in the absence of cTfh is challenging to explain, but the exact correlation between cTfh and successful antibody response remains controversial (11).
Our patient has an oral allergy syndrome with elevated total and allergen-specific IgE that was not described in the previously reported patients (10). It is difficult to establish whether this allergy is related with the SASH3 deficiency or is an independent phenomenon. Nonetheless, oral allergy syndrome is not rare and is likely not a distinctive feature of SASH3 deficiency.
The five SASH3 deficient patients reported to date recapitulate many of the immunological defects described in the two different Sly1 deficient mice models generated (Sly1Δ/Δ and Sly1-/-) (12, 13). In both models it has been shown that: (1) absence of SLY1 (SASH3) protein affects CD4 T cell development, T-cell proliferation and cytokine production; (2) SLY1 is an anti-apoptotic factor required for thymocyte development and (3) antibody responses to T-dependent and T-independent antigens were impaired. The functional T cell defects identified in SASH3 deficient patients confirm the crucial role of this lymphocyte-specific factor for T cell development and function in humans.
In summary, our patient has a combined immunodeficiency due to SASH3 deficiency, although he presented with a phenotype initially resembling CVID. Two separate, severe viral episodes alerted us to an underlying T-cell defect. The most relevant difference compared with the patients described so far is the absence of evident immune dysregulation. Undoubtedly, future cases of SASH3 deficiency will help to better define the prevalence of the various clinical and cellular characteristics of this IEI. In the meantime, SASH3 deficiency should be considered as one of the monogenic defects that can occur in male patients presenting with a CVID-like phenotype.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by Ethics Review Board of Hospital Universitari Vall d’Hebron. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
ML-H provided patient care and collected clinical data. MG-P and AP-M performed laboratory analysis and western blot under the supervision of CF-J. SS-P performed flow cytometry analysis under the supervision of MM-G. AA-C performed the molecular experiments under the supervision of RC. MA and RC analyzed the NGS data and provided the genetic diagnosis. ML-H, CF-J, MM-G, and RC analyzed and interpreted the data and wrote the manuscript. MM-G and RC designed and supervised the project, provided resources and edited the manuscript. All co-authors reviewed, commented and approved the final version of the manuscript.
This study was funded by Instituto de Salud Carlos III, grants PI17/00660 and PI20/00761, cofinanced by the European Regional Development Fund (ERDF).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
This study was supported by the European Reference Network for Rare Immunodeficiency, Autoinflammatory and Autoimmune Diseases Network (ERN-RITA).
1. Fliegauf M, Bryant VL, Frede N, Slade C, Woon S-T, Lehnert K, et al. Haploinsufficiency of the NF-κb1 Subunit P50 in Common Variable Immunodeficiency. Am J Hum Genet (2015) 97(3):389–403. doi: 10.1016/j.ajhg.2015.07.008
2. Tuijnenburg P, Lango Allen H, Burns SO, Greene D, Jansen MH, Staples E, et al. Loss-Of-Function Nuclear Factor κb Subunit 1 (NFKB1) Variants Are the Most Common Monogenic Cause of Common Variable Immunodeficiency in Europeans. J Allergy Clin Immunol (2018) 142(4):1285–96. doi: 10.1016/j.jaci.2018.01.039
3. Boztug H, Hirschmugl T, Holter W, Lakatos K, Kager L, Trapin D, et al. NF-κb1 Haploinsufficiency Causing Immunodeficiency and EBV-Driven Lymphoproliferation. J Clin Immunol (2016) 36(6):533–40. doi: 10.1007/s10875-016-0306-1
4. Hoeger B, Serwas NK, Boztug K. Human NF-κb1 Haploinsufficiency and Epstein-Barr Virus-Induced Disease-Molecular Mechanisms and Consequences. Front Immunol (2017) 8:1978. doi: 10.3389/fimmu.2017.01978
5. Dieli-Crimi R, Martínez-Gallo M, Franco-Jarava C, Antolin M, Blasco L, Paramonov I, et al. Th1-Skewed Profile and Excessive Production of Proinflammatory Cytokines in a NFKB1-Deficient Patient With CVID and Severe Gastrointestinal Manifestations. Clin Immunol (2018) 195:49–58. doi: 10.1016/j.clim.2018.07.015
6. Ameratunga R, Lehnert K, Woon S-T, Gillis D, Bryant VL, Slade CA, et al. Review: Diagnosing Common Variable Immunodeficiency Disorder in the Era of Genome Sequencing. Clin Rev Allergy Immunol (2018) 54(2):261–8. doi: 10.1007/s12016-017-8645-0
7. Maffucci P, Filion CA, Boisson B, Itan Y, Shang L, Casanova J-L, et al. Genetic Diagnosis Using Whole Exome Sequencing in Common Variable Immunodeficiency. Front Immunol (2016) 7:220. doi: 10.3389/fimmu.2016.00220
8. de Valles-Ibáñez G, Esteve-Solé A, Piquer M, González-Navarro EA, Hernandez-Rodriguez J, Laayouni H, et al. Evaluating the Genetics of Common Variable Immunodeficiency: Monogenetic Model and Beyond. Front Immunol (2018) 9:636. doi: 10.3389/fimmu.2018.00636
9. Gámez-Díaz L, August D, Stepensky P, Revel-Vilk S, Seidel MG, Noriko M, et al. The Extended Phenotype of LPS-Responsive Beige-Like Anchor Protein (LRBA) Deficiency. J Allergy Clin Immunol (2016) 137(1):223–30. doi: 10.1016/j.jaci.2015.09.025
10. Delmonte OM, Bergerson JRE, Kawai T, Kuehn HS, McDermott DH, Cortese I, et al. SASH3 Variants Cause a Novel Form of X-Linked Combined Immunodeficiency With Immune Dysregulation. Blood (2021) 138(12):1019–33. doi: 10.1182/blood.2020008629
11. Ma CS, Wong N, Rao G, Avery DT, Torpy J, Hambridge T, et al. Monogenic Mutations Differentially Affect the Quantity and Quality of T Follicular Helper Cells in Patients With Human Primary Immunodeficiencies. J Allergy Clin Immunol (2015) 136(4):993–1006.e1. doi: 10.1016/j.jaci.2015.05.036
12. Beer S, Scheikl T, Reis B, Hüser N, Pfeffer K, Holzmann B. Impaired Immune Responses and Prolonged Allograft Survival in Sly1 Mutant Mice. Mol Cell Biol (2005) 25(21):9646–60. doi: 10.1128/MCB.25.21.9646-9660.2005
Keywords: primary immunodeficiencies, inborn errors of immunity, SASH3 deficiency, common variable immunodeficiency, combined immunodeficiency, genetics, mutation
Citation: Labrador-Horrillo M, Franco-Jarava C, Garcia-Prat M, Parra-Martínez A, Antolín M, Salgado-Perandrés S, Aguiló-Cucurull A, Martinez-Gallo M and Colobran R (2022) Case Report: X-Linked SASH3 Deficiency Presenting as a Common Variable Immunodeficiency. Front. Immunol. 13:881206. doi: 10.3389/fimmu.2022.881206
Received: 22 February 2022; Accepted: 16 March 2022;
Published: 08 April 2022.
Edited by:
Antonio Condino-Neto, University of São Paulo, BrazilReviewed by:
Alexandra Freeman, National Institutes of Health (NIH), United StatesCopyright © 2022 Labrador-Horrillo, Franco-Jarava, Garcia-Prat, Parra-Martínez, Antolín, Salgado-Perandrés, Aguiló-Cucurull, Martinez-Gallo and Colobran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roger Colobran, cmNvbG9icmFuQHZoZWJyb24ubmV0; Mónica Martinez-Gallo, bW1hcnRpbmV6QHZoZWJyb24ubmV0
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.