 Aaren Kettelhut1*
Aaren Kettelhut1* Emily Bowman1
Emily Bowman1 Janelle Gabriel1
Janelle Gabriel1 Brittany Hand1
Brittany Hand1 Namal P. M. Liyanage2,3
Namal P. M. Liyanage2,3 Manjusha Kulkarni1Frances Avila-Soto1
Manjusha Kulkarni1Frances Avila-Soto1 Jordan E. Lake4†
Jordan E. Lake4† Nicholas T. Funderburg1†
Nicholas T. Funderburg1†- 1Department of Health and Rehabilitation Sciences, The Ohio State University, Columbus, OH, United States
- 2Department of Microbial Infection and Immunity, The Ohio State University, Columbus, OH, United States
- 3Department of Veterinary Biosciences, The Ohio State University, Columbus, OH, United States
- 4Department of Internal Medicine, University of Texas Health Science Center at Houston, Houston, TX, United States
Background: Transgender women (TW) are at increased risk for both human immunodeficiency virus (HIV) and cardiovascular disease (CVD). Antiretroviral therapy-treated HIV has been associated with a two-fold increased risk of CVD, potentially due to dysregulated Toll-like receptor (TLR)-induced immune activation. Use of estrogens in feminizing hormone therapy (FHT) may enhance inflammatory responses and the risk of cardiovascular mortality in TW. Despite this, the immunomodulatory effects of estrogen use in TW with HIV have been inadequately explored.
Methods: As an in vitro model for FHT, cryopreserved PBMCs (cryoPBMCs) from HIV negative (HIV-), HIV+ ART-suppressed (HIV+SP), and HIV+ ART-unsuppressed (HIV+USP) cisgender men were cultured overnight in the presence of 17-β estradiol or 17-α ethinylestradiol with and without the TLR4 agonist LPS or the TLR8 agonist ssPolyU. Monocyte activation (CD69, HLA-DR, CD38) was assessed by flow cytometry. Cytokine levels (IL-6, TNF-α, IL-1β, and IL-10) were measured in cell culture supernatants by Legendplex. Levels of phosphorylated TLR signaling molecules (JNK, MAPK p38) were assessed by Phosflow. Plasma levels of immune activation biomarkers (LPS-binding protein, monocyte activation markers sCD14 and sCD163, and inflammatory molecules IL-6 and TNF-α receptor I) were measured by ELISA.
Results: PBMCs from people with HIV (PWH) produced greater levels of inflammatory cytokines following exposure to LPS or ssPolyU compared to levels from cells of HIV- individuals. While estrogen exposure alone induced mild changes in immune activation, LPS-induced TLR4 activation was elevated with estrogen in cisgender men (CM) with HIV, increasing monocyte activation and inflammatory cytokine production (IL-6, TNF-α). Interestingly, testosterone inhibited LPS-induced cytokine production in CM regardless of HIV status. Plasma markers of immune activation and microbial translocation (e.g., sCD14, sCD163, LPS-binding protein) were generally higher in PWH compared to HIV- CM, and these markers were positively associated with in vitro responsiveness to estrogen and LPS in CM with HIV.
Conclusions: Our in vitro data suggest that estrogen exposure may enhance innate immune activation in PWH. Further examination is needed to fully understand the complex interactions of FHT, HIV, and CVD in TW, and determine optimal FHT regimens or supplementary treatments aimed at reducing excess immune activation.
Introduction
Transgender women (TW) are an underserved population in medicine. Treatment with gender-affirming, feminizing hormone therapy (FHT) using estrogen-based supplementation can significantly improve quality of life (1, 2). Despite the success of FHT, long-term estrogen use may contribute to increased prevalence of chronic co-morbidities including metabolic, pulmonary, cardiovascular, and immunologic complications (3–5).
Prevalence of human immunodeficiency virus (HIV) and cardiovascular disease (CVD) is higher in TW compared to the general population (6–11). TW have an approximately 3-fold greater risk of myocardial infarction (MI) compared to their cisgender counterparts (11). Additionally, MI prevalence is greater in TW receiving FHT compared to TW not on FHT (9). Studies have identified a 3-fold increased risk of cardiovascular mortality with estrogen use in TW (4). Despite these findings, mechanisms underlying FHT-associated CVD risk have been incompletely explored.
Increased CVD risk as a consequence of FHT may be particularly concerning in TW with HIV as HIV is linked to heightened CV morbidity and mortality (12). Globally, the prevalence of HIV in TW is 19% compared to < 1% in the general population; the CDC reports TW are 49 times more likely to live with HIV than cisgender women (CW) (6, 7). Current antiretroviral therapy (ART) regimens successfully manage HIV progression, however, persistent immune activation is a strong predictor of CVD in people with HIV (PWH) (13–16). Chronic immune activation may be driven by multiple factors, including the activation of Toll-like receptors (TLRs) by microbial products that translocate through damaged gut lumen or by products of low-level HIV replication (14–30). Increased monocyte and macrophage activation has been implicated in the pathogenesis of HIV-associated CVD (31–33). Exposure to bacterial (e.g., lipopolysaccharide (LPS) or flagellin) or viral products (e.g., single-stranded polyuridine (ssPolyU), imiquimod, or HIV-1) triggers cellular activation and production of pro-inflammatory mediators. Compared to those without HIV, exposure of myeloid cells from PWH to TLR ligands can result in elevated inflammatory cytokines levels including IL-6, a molecule linked to increased morbidity and mortality in PWH (34–36).
Several studies demonstrate that estrogen may enhance inflammatory responses to TLR ligands (37–41). We have measured cardiometabolic profiles and associated inflammatory biomarkers in cisgender men (CM) and TW and reported increased immune activation in the latter, regardless of HIV status. We also observed slight elevations in the CVD-associated molecules TNF-α receptor I (TNFRI), oxidized low-density lipoprotein (oxLDL), and human extracellular newly-identified receptor for advanced glycation end products binding protein (EN-RAGE) in TW on FHT compared to those off FHT (42). Further work is needed to elucidate the mechanisms that contribute to cardiometabolic risk in TW with HIV. Here, we explored the in vitro consequences of estrogen (17-β estradiol or 17-α ethinylestradiol) exposure to TLR-stimulated peripheral blood mononuclear cells (cryoPBMCs) from CM who were HIV negative (HIV-), HIV-positive with ART suppression (HIV+SP), or HIV-positive and ART-naïve/unsuppressed (HIV+USP) to test our hypothesis that estrogen can enhance TLR responsiveness in monocytes from PWH compared to those without HIV. While this work will be limited in its generalizability to TW, as these individuals may experience greater health disparities associated with CVD due to minority stress and marginalization when compared to CM (43), this work serves as a model to elucidate the immune modulatory effects of exogenous estrogen in those assigned male at birth with HIV.
Materials and Methods
Sample Collection and Cell Culture Methods
For HIV- donors, blood samples were collected by EDTA-containing Vacutainer tubes (BD Biosciences) from which PBMCs and plasma were isolated. PBMCs were freshly isolated by centrifugation over Ficoll-Hypaque and cryopreserved in freezing media made with 90% DMSO and 10% FBS at 1 ml per 1 x 107 cells. To isolate plasma, EDTA tubes were centrifuged for 10 minutes at 470 g.
CryoPBMCs and plasma from ART HIV+SP (viral load < 20 copies/mL) and HIV+USP (viral load > 10,000 copies/mL) PWH were supplied by the Center for AIDS Research Network of Integrated Clinical Systems repository (CFAR CNICS). All cryopreserved cells were thawed with pre-warmed phenol red free RPMI 1640 (Gibco) that contained 10% Human Male AB OTC Serum, heat inactivated (ATCC Lot #A18049). Trypan Blue staining (1:1) was used to assess viability of thawed PBMCs. Cells were rested overnight (1 x 106/ml) in 12 well culture plates before stimulating with 0.001% of ethanol (vehicle control), 1 μM of 17-β estradiol (Sigma-Aldrich), 1 μM of 17-α ethinylestradiol (Sigma-Aldrich), or 1 μM of testosterone (Sigma-Aldrich) in the absence or presence of 100 ng/ml lipopolysaccharide (Sigma-Aldrich) or 1 μg/ml ssPolyU (In vivogen) overnight in 37°C incubator with 5% CO2. All hormones were obtained in powder form and solubilized in 100% ethanol before filtering. Concentrations of hormones were selected based on initial dose response assays. For phosphoflow experiments, cells were exposed to 0.001% of ethanol or 1 μM of 17-β estradiol in the presence or absence of 100 ng/ml lipopolysaccharide for 20 minutes in 37°C incubator before cell collection.
Flow Cytometry
Cells were collected and supernatants stored in -80°C for cytokine analysis. Cells were washed in 1X Dulbecco Phosphate Buffered Saline (PBS; Gibco) followed by flow wash buffer. Cells were stained for 15 minutes at room temperature in the dark, washed, and fixed in 1% paraformaldehyde (Thermoscientific). Monocytes were identified by size, granularity, and surface expression of CD14 (anti-CD14 Pacific Blue or Pacific Blue isotype; BD Biosciences Cat# 558121, RRID : AB_397041 or BD Biosciences Cat# 558118, RRID : AB_397039, respectively). Monocyte activation markers were measured using anti-CD38 (phycoerythrin [PE]; BD Biosciences Cat# 555460, RRID : AB_395853), anti-CD69 (PE-Cyanine7 [PeCy7]; BD Biosciences Cat# 335792, RRID : AB_1937286), anti-HLADR (APC-Cyanine7 [APCCy7]; BD Biosciences Cat# 335814, RRID : AB_399991) or their respective isotypes (BD Biosciences Cat# 555749, RRID : AB_396091; BD Biosciences Cat# 348798, RRID : AB_400386) by Miltenyi MACSQuant Analyzer 10 flow cytometer (Miltenyi Bioscience MACSQuant Analyzer 10, RRID : SCR_020268) and MACSQuant analysis software (MACSQuantify, RRID : SCR_020943).
Phosphoflow Cytometry
Cells were collected and washed in PBS and flow wash buffer, similar to above protocol, then stained for surface expression of CD14 (anti-CD14 V500 or isotype; BD Biosciences Cat# 561392, RRID : AB_10611862 or BD Biosciences Cat# 561221, RRID : AB_10566127 respectively) for 20 minutes in the dark at 37°C. Monocyte intracellular expression of phosphorylated proteins were measured using an intracellular staining protocol. Cells were lysed at 37°C for 10 minutes with lyse/fix buffer (BD Phosflow), washed with PBS, and permeabilized at 4°C for 30 minutes with Perm Buffer III (BD Phosflow). Cells were washed 3 times in stain buffer (BD Phosflow) before staining in the dark at room temperature for 1 hour with anti-JNK (pT183/pY185) (PE; BD Biosciences Cat# 562480, RRID : AB_11153134) and anti-p38 MAPK (pT180/pY182) (peridinium-chlorophyl-protein Complex-Cyanine 5.5 [PerCP-Cy5.5]; BD Biosciences Cat# 560406, RRID : AB_164529). Cells were analyzed by the MACSQuant Analyzer 10 and analysis software. Monocytes were again identified by size, granularity, and surface expression of CD14.
RNA Isolation, cDNA Conversion, and Quantitative Polymerase Chain Reaction
Cells were stimulated as described above with 0.001% of ethanol, 1 μM of 17-β estradiol, or 1 μM of 17-α ethinylestradiol in the absence or presence of 100 ng/ml lipopolysaccharide or 1 μg/ml ssPolyU for 24 hours in the 37°C incubator. Cells were collected, washed in PBS, and placed in 1% 2-Mercaptoethanol (Sigma Aldrich) supplemented lysing buffer (Qiagen) and stored at -80°C overnight for optimal cell lysing. Cell supernatants were collected for cytokine analysis and stored at -80°C. RNA was extracted using the RNeasy Mini Kit (Qiagen) and RNase-Free DNase set (Qiagen). Extracted RNA was converted to cDNA using the iScript cDNA Synthesis Kit (Bio-rad) and S1000 thermal cycler (Bio-rad). Resultant cDNA along with IQ Sybr Green Supermix (Bio-rad) and relevant forward and reverse primers were used for qPCR analysis. This analysis assessed expression of cytokines IL6 (F: 5’CCAGGAGCCCAGCTATGAAC 3’, R: 5’CCCAGGGAGAAGGCAACTG 3’) and TNF-α (F: 5’GAGGCCAAGCCCTGGTATG 3’, R: 5’CGGGCCGATTGATCTCAGC 3’) utilizing MicroAmp Optical 6-well reaction plates and Real-Time PCR System Quant Studio 3 (Applied Biosystems; QuantStudio 3 Real Time PCR System, RRID : SCR_018712). Results were analyzed using QuantStudio design and analysis software.
MAPK p38 Inhibition
Cryopreserved PBMCs were rested overnight (1 x 106/ml) in 12 well culture plates before stimulating with 0.001% of ethanol (vehicle control) or 1 μM of 17-β estradiol (Sigma-Aldrich) in the absence or presence of 100 ng/ml lipopolysaccharide (Sigma-Aldrich) with or without 10 µM of p38 inhibitor SB203580 (Sigma-Aldrich) overnight in 37°C incubator with 5% CO2. Supernatants were collected for cytokine analysis and stored at -80°C.
Measurement of Cytokine Expression
Levels of inflammatory cytokines were measured in supernatants using the LEGENDplex HU Anti-virus response panel (13-plex) w/VbP (Biolegend). Supernatants collected from cells stimulated with ssPolyU or no stimulation were diluted 1:5 in provided assay buffer while those stimulated with LPS were diluted 1:100. LEGENDplex was run on MACSQuant 10 flow cytometer and analyzed by the LEGENDplex Data Analysis Software.
Plasma Biomarker Assessment
Plasma biomarker levels were measured by enzyme-linked immunosorbent assay (ELISA; R&D unless otherwise stated) using a SpectraMax 190 plate reader (SpectraMax 190 microplate reader, RRID : SCR_018932). These biomarkers include soluble CD14 (sCD14), soluble CD163 (sCD163), tumor necrosis factor receptor 1 (TNFR-1), high sensitivity interleukin 6 (HS IL6), LPS-binding protein (LBP, Hycult Biotech), and β D-glucan (BDG, MyBioSource). Data was collected in SoftMax Pro 7.0.2 (SoftMax Pro Data Acquisition and Analysis Software, RRID : SCR_014240).
Statistical Analysis
Statistical analysis for flow, gene expression, and cytokine analyses were performed in GraphPad Prism 8 (GraphPad Prism, RRID : SCR_002798) utilizing nonparametric paired and unpaired t tests along with nonparametric 1- way ANOVA analysis to compare data between our 3 donor groups with our different stimulations. Statistical analysis for ELISA and demographic data were also performed in GraphPad Prism 8 utilizing nonparametric 1- way ANOVA analysis to compare clinical and plasma biomarker data between donor groups. Spearman correlations were utilized to assess relationships between measured plasma biomarkers, plasma biomarkers and clinical data, and plasma biomarkers and changes in cytokine production levels. Correlation heatmaps were created with R software to assess relationships between clinical information, baseline immunoinflammatory profiles, and cytokine responses.
Results
Plasma Biomarkers of Immune Activation and Microbial Translocation are Elevated in PWH
Several studies, including our own, have reported increased levels of pro-inflammatory molecules in PWH and in TW on and off FHT compared to levels in CM, regardless of HIV status. Here, we explored the in vitro effects of estrogen on TLR-induced monocyte activation profiles in PWH. As a model for in vivo processes in TW initiating FHT, we utilized cryoPBMCs from 18 CM not known to have HIV and 29 CM with HIV, including individuals with HIV+SP (n=14) and HIV+USP (n=15) viral replication. Median viral loads (20 copies/mL and 98,138 copies/mL) and CD4+ T-cell counts (708 and 334 cells/μL) were significantly different in HIV+SP and HIV+USP participants (p<0.001). While median age was not significantly different among groups (HIV- 45.0; HIV+SP 50.5; HIV+USP 49.0, p>0.05), racial identification was different between HIV- individuals and PWH, with more White and Asian participants among the former group (p<0.01) (Table 1). Participants were approved for enrollment under the IRB protocol ID 2019H0113.
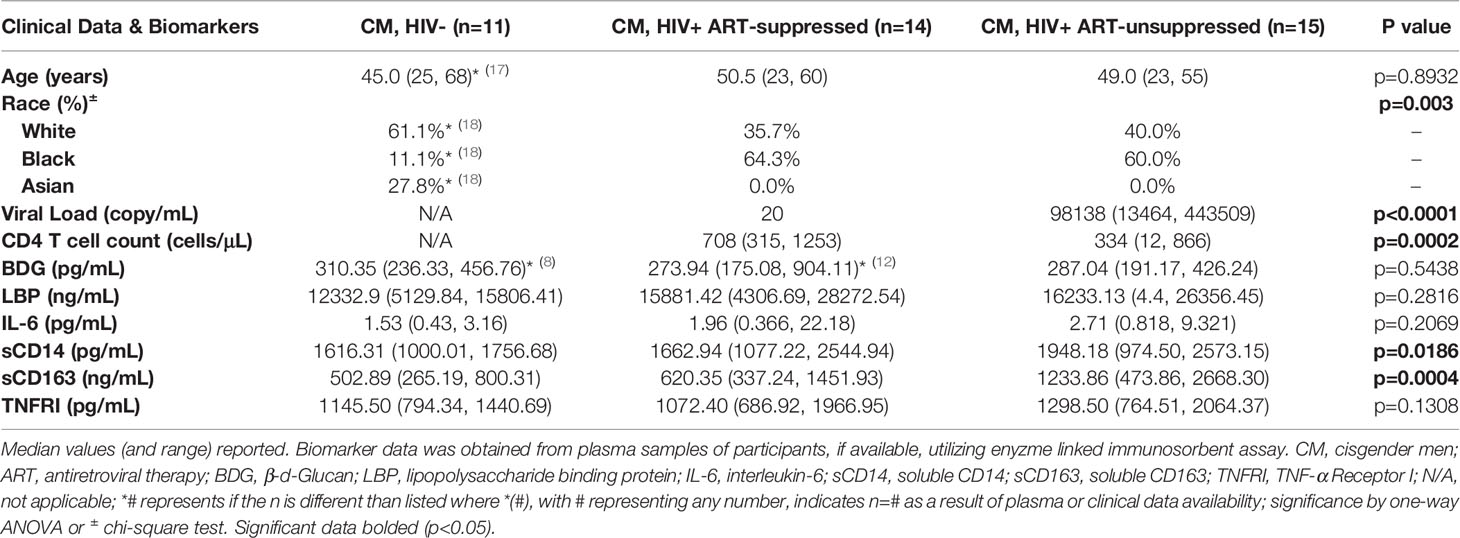
Table 1 Baseline clinical characteristics and biomarkers of immune activation and inflammation in plasma of HIV- and HIV+ antiretroviral therapy-suppressed and -unsuppressed donors.
Plasma biomarkers were measured to assess in vivo immune activation in participants with available samples (HIV- n=11, HIV+SP n=14, HIV HIV+USP n=15, Table 1), including potential drivers of inflammation in PWH (BDG and LBP), inflammatory molecules (IL-6, TNFRI), and monocyte activation markers (sCD14, sCD163). Of these, only CD14 and CD163 were significantly different between groups (p<0.05, p<0.001). Post hoc analysis revealed significant differences between HIV- and HIV+USP for both markers (CD14 p<0.05, CD163 p<0.001) and between HIV+SP and HIV+USP for CD163 (p<0.05). TNFRI was elevated in USP donors, while IL-6 and LBP increased in both groups with HIV compared to HN participants (p>0.05).
Antiretroviral Therapy Suppressed PWH Demonstrate Significant Positive Associations Between Markers of Immune Activation and LPS-Binding Protein
We assessed correlations among biomarkers and contributing mediators of immune activation in PWH to better understand activation profiles in participant groups. We found significant positive associations among LBP, IL-6, TNFRI, CD14, and CD163 in HIV+SP donors (p<0.05, Figure 1B). These associations, while not all statistically significant, remained in the HIV+USP group. Viral loads and CD4+ T cell counts were negatively associated in HIV+USP donors (p<0.05); however, viral loads were not significantly associated with markers of inflammation (Figure 1C). Fewer associations were seen in HIV- donors, with only one association reaching significance (p<0.05, Figure 1A). Age and LBP were significantly and negatively associated in HIV- donors, otherwise age was not significantly associated with immune activation in any participant group.
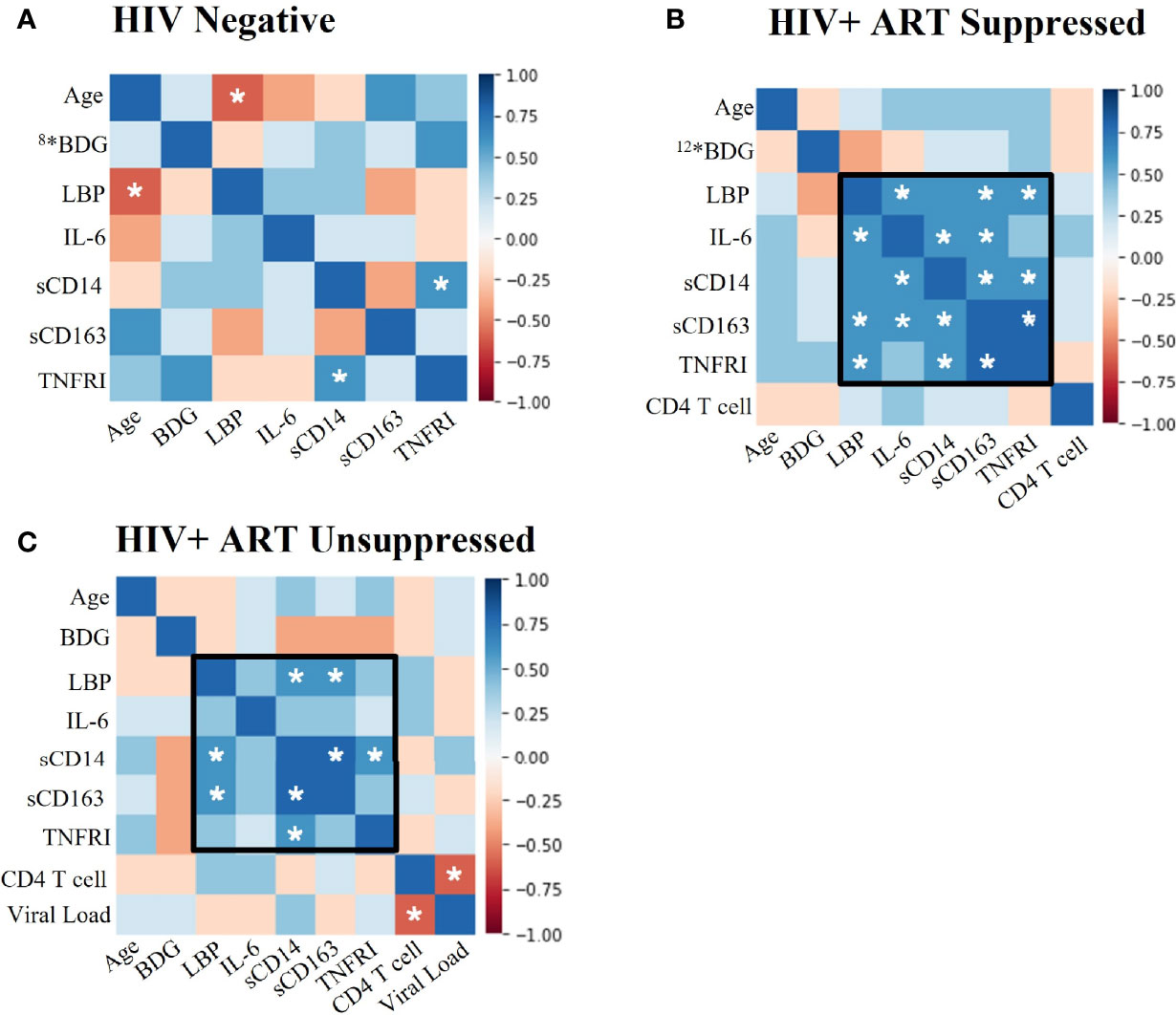
Figure 1 Plasma biomarkers show significant positive associations between LBP and immunoinflammatory markers in PWH. Spearman Rank Correlation between clinical and plasma biomarker data in (A) HIV negative (n=11), (B) HIV+ ART-suppressed (n=14), and (C) HIV+ ART-unsuppressed (n=15) donor groups. PWH, people with HIV; ART, antiretroviral therapy; BDG, β-d-Glucan; LBP, lipopolysaccharide binding protein; IL-6, interleukin-6; sCD14, soluble CD14; sCD163, soluble CD163; TNFRI, TNF-α Receptor I; *#, where # represents any number, indicates a different n than listed where #* is equivalent to n=# due to issues in plasma availability; white stars represent significance of p<0.05.
PWH Enhance Responses to Toll-Like Receptor Agonism
Alterations in TLR expression and function have been reported in PWH (36, 44, 45). We examined the effects of TLR ligands on monocyte activation markers (CD69, HLA-DR, CD38, n=10 all groups) and cytokine production (IL-6, TNF-α, IL-1β, IL-10, HIV- n=15, HIV+SP n=12, HIV HIV+USP n=15) from cryoPBMCs. The proportion of monocytes that expressed CD69 and the mean fluorescence intensities (MFIs) of HLA-DR and CD38 were increased in all groups following exposure to LPS or ssPolyU. TLR-induced expression of activation markers tended to be higher in PWH compared to expression on cells from people without HIV (Figures 2A–C). Exposure of cells to LPS or ssPolyU also increased production of inflammatory cytokines from all participant groups. Despite higher baseline cytokine production in HIV- participants, LPS exposure resulted in greater production of cytokines from the cells of PWH compared to cells from HIV- individuals (Figures 2D–G).
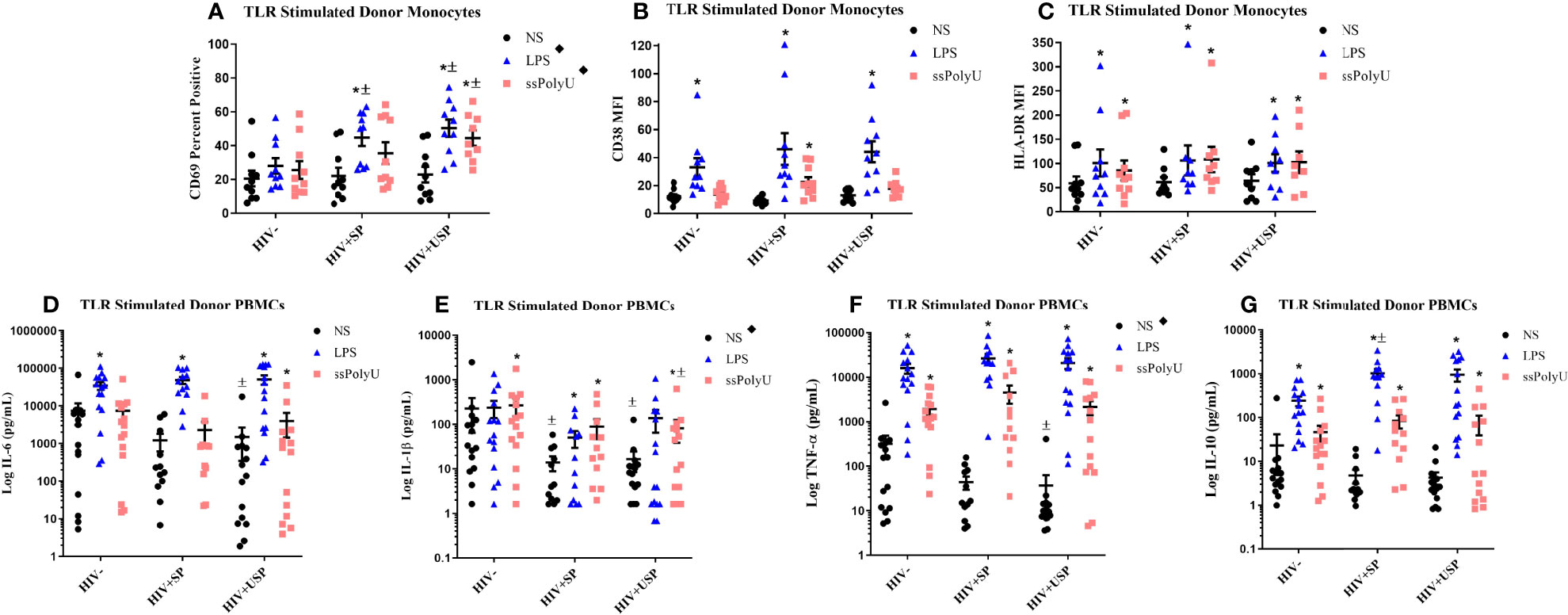
Figure 2 Toll-like receptor stimulation increases monocyte activation markers and cytokine production with greater increases in PWH. HIV+ ART-suppressed (HIV+SP), HIV+ ART-unsuppressed (HIV+USP), and HIV negative (HIV-) cryopreserved peripheral blood mononuclear cells (PBMCs) from CM were thawed and rested overnight in phenol red free RPMI supplemented with 10% Human AB Serum. Cells were treated overnight with no stimulation (NS), 100 ng/mL of TLR4 agonist LPS, or 1 μg/mL of TLR8 agonist ssPolyU. (A–C) Cells were collected and stained for CD14+ monocytes and analyzed for levels of relevant activation markers (CD69, CD38, HLA-DR) expressed as percent positive cells or mean fluorescent intensity (MFI) (n=10 all groups). (D–G) Supernatants were collected for protein analysis of inflammatory cytokines (IL-1β, IL-6, TNF-α, IL-10) by multiplex bead assay (HIV- n=15, HIV+SP n=12, HIV+USP n=15). Data is represented as mean and standard error of measure in (A–C) raw or (D–G) log format and was analyzed by *Wilcoxon signed rank test, ± Mann Whitney U, or ♦one-way ANOVA (*,±,♦p<0.05). CM, cisgender men; ART, antiretroviral therapy; LPS, lipopolysaccharide; ssPolyU, single stranded polyuridine; TLR, Toll-like receptor; NS, no stimulation; PWH, people with HIV; IL, interleukin; TNF, tumor necrosis factor.
17-β Estradiol Enhances LPS-Induced Activation Profiles in PWH
Estrogen may enhance TLR expression and function (37), therefore, we assessed alteration in cellular responses to LPS stimulation with estrogen treatment in our participants. Exposure of cells to either 17-β estradiol (17B) or 17-α ethinylestradiol (17A) alone inconsistently reduced expression of activation markers on monocytes from all participant groups (Supplemental Figures 1A–C). Exposure of cells from PWH to 17B, however, increased cytokine production, with significantly higher IL-6 production from HIV+SP versus HIV- cells (p<0.05, Supplemental Figures 1D–G).
Based on these results, and as 17B is the preferred estrogen for FHT (46), we focused on the effects of 17B in follow-up experiments. In combination with LPS, estrogen exposure typically enhanced monocyte activation marker expression in HIV+SP participants. HLA-DR expression levels were increased significantly in HIV+SP participants with the combination of estrogen and LPS compared to expression induced by LPS alone (p<0.05; Figure 3A). Cells from all groups increased cytokine production with combined treatment compared to LPS alone; IL-6 production was significantly increased with combined stimulation in HIV+SP PWH (p<0.05, Figure 3B). Greater production of IL-6 and TNF-α was seen in PWH compared to HIV- participants. Changes in mRNA expression of IL-6 and TNF-α reflected results seen at the protein level (Supplemental Figures 2–4). For comparison, we tested cryoPBMC responses to testosterone in the absence or presence of LPS. Combined treatment with LPS showed increases in activation marker expression on monocytes from all groups (Supplemental Figures 5A, B). Cytokine production tended to increase with testosterone alone - albeit to a lesser degree than estrogen alone for IL-6 and TNF-α – but testosterone decreased cytokine production when combined with LPS in all groups compared to levels from cells exposed to LPS alone (Supplemental Figures 5C, D).
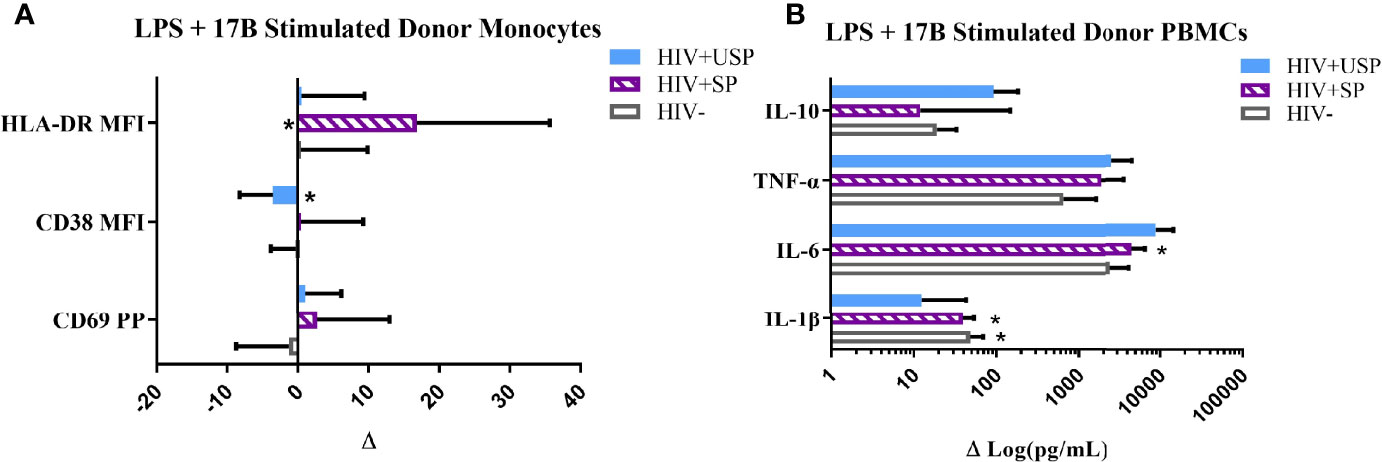
Figure 3 Estrogen differentially modulates monocyte activation and cytokine production with LPS stimulation. HIV+ ART-suppressed (HIV+SP), HIV+ ART-unsuppressed (HIV+USP), and HIV negative (HIV-) cryopreserved peripheral blood mononuclear cells (PBMCs) from CM were thawed and rested overnight in phenol red free RPMI supplemented with 10% Human AB Serum. Cells were treated overnight with 0.001% ethanol (vehicle) or 1 μM of either 17β estradiol (17B) (17A) +/- 100 ng/mL of LPS. (A) Cells were collected and stained for CD14+ monocytes and analyzed for levels of relevant activation markers (CD69, CD38, HLA-DR) expressed as percent positive cells or mean fluorescent intensity (MFI) (n=10). (B) Supernatants were collected for protein analysis of inflammatory cytokines (IL-1β, IL-6, TNF-α, IL-10) by multiplex bead assay (HIV- n=15, HIV+SP n=12, HIV+USP n=15). Data is normalized to LPS + vehicle and is represented as delta change (Δ) of marker or cytokine expression with mean and standard error of measure shown. Data in (B) represented in log format. Data was analyzed by *Wilcoxon signed rank test which assessed for significant differences in activation marker expression or cytokine production between LPS + vehicle and LPS + 17B stimulation for each donor group (*p<0.05). Mann Whitney U and one-way ANOVA analyses were also run, but no significant differences were found. CM, cisgender men; ART, antiretroviral therapy; LPS, lipopolysaccharide; IL, interleukin; TNF, tumor necrosis factor.
In Vivo Activation Profiles May Influence Ex Vivo Responsiveness to Estrogen and TLR Stimulation in PWH
We next examined relationships between in vivo profiles of inflammation and immune activation in our participant groups and the ex vivo responsiveness of PBMCs to TLR and estrogen stimulations. LPS-stimulated HIV- PBMCs showed negative associations between plasma IL-6 and LBP levels and ex vivo cytokine production (Figure 4A). Negative associations were also seen among several plasma biomarkers and LPS-induced cytokine production in our HIV+SP group (Figure 4B). In PWH, positive associations were observed between estrogen-induced cytokine production and participant activation profiles (Figures 4B, C). In those without HIV, however, estrogen-induced cytokine production had mixed associations with activation profiles. The relationships seen between estrogen-induced cytokine production and immune activation profiles remained consistent with the addition of LPS in both HIV- and HIV+SP participant groups. Combined LPS- and estrogen-induced cytokine production in HIV+USP participants no longer showed positive or significant associations with immune activation profiles.
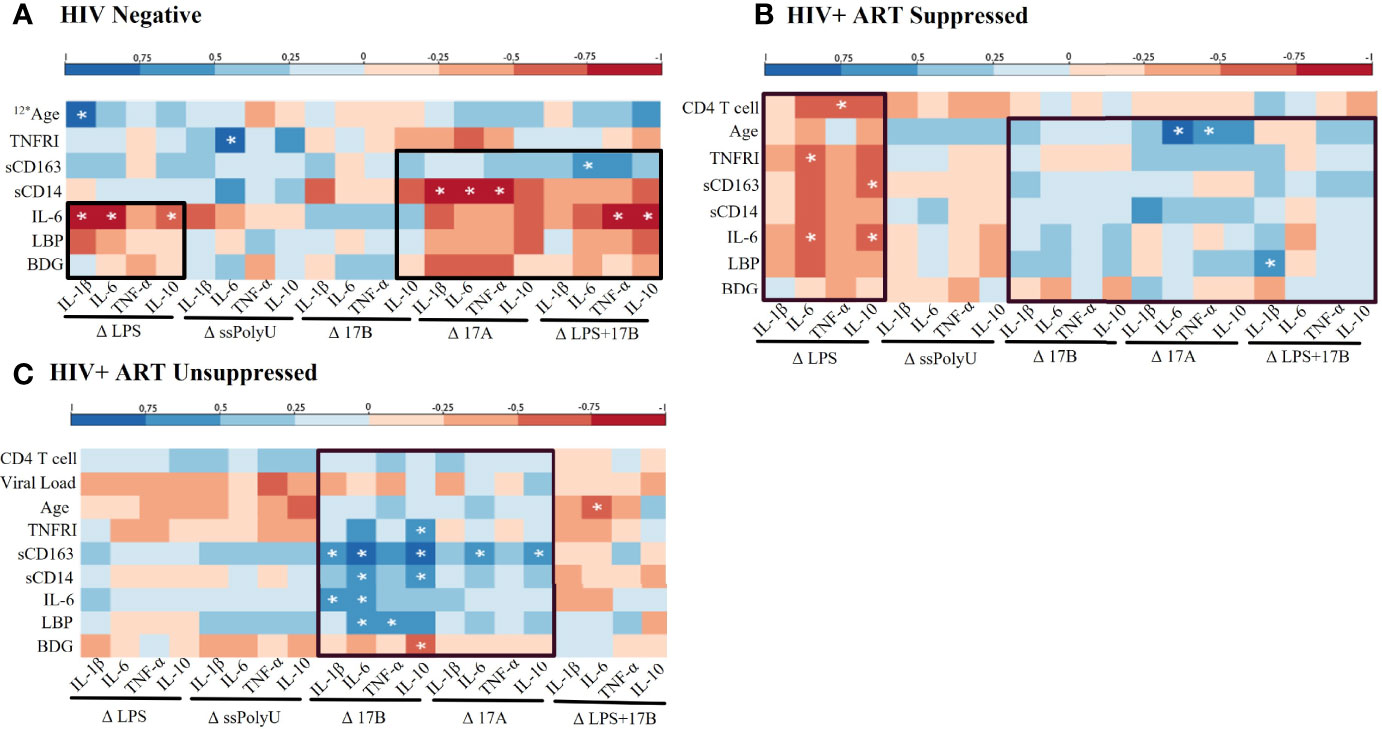
Figure 4 Baseline in vivo inflammatory profiles may play a role in response to in vitro stimulations. Spearman Rank Correlation between clinical or plasma biomarker data and delta change (Δ) in cytokine production, determined by ELISA or bead assay respectively, by TLR and/or estrogen stimulation in (A) HIV negative (n=8), (B) HIV+ ART-suppressed (n=12), and (C) HIV+ ART-unsuppressed (n=15) donor groups. ART, antiretroviral therapy; LPS, lipopolysaccharide; ssPolyU, single stranded polyuridine; 17B, 17β estradiol; 17A, 17α ethinylestradiol; BDG, β-d-Glucan; LBP, lipopolysaccharide binding protein; IL-6, interleukin-6; sCD14, soluble CD14; sCD163, soluble CD163; TNFRI, TNF-α Receptor I; IL, interleukin; TNF, tumor necrosis factor; ELISA, enyzme linked immunosorbent assay. #* represents different n than listed where #* is equivalent to n=# due to issues in plasma availability *p<0.05.
Inhibition of MAPK p38 Abates 17β Enhanced LPS-Induced Inflammatory Responses
Estrogen may modulate activation of intracellular signaling including the phosphorylation of MAPK p38 and JNK, molecules that influence inflammatory responses to TLR stimulation (47–49). We analyzed phosphorylation of these signaling molecules in the monocytes of our participant groups in the presence or absence of TLR4 stimulation. Although not statistically significant, HIV- individuals on average showed upregulation of both the proportion of monocytes that had phosphorylated JNK and MAPK p38, as well as the MFI of these phosphorylated molecules, while variable responses were seen in the cells of HIV+SP and HIV+USP participants exposed to LPS (Figure 5). Treatment with 17B alone or in combination with LPS showed significant alterations in the percent positive (PP) monocytes for phosphorylated JNK (pJNK) in HIV- and HIV+USP individuals, while minor changes were induced in HIV+SP participants. Phosphorylated MAPK p38 (pMAPK p38) showed decreased or little change in the PP monocytes with 17B treatment alone, however, the addition of 17B to LPS stimulation significantly increased pMAPK p38 in HIV+SP participants when compared to HIV- individuals (p<0.05, Figure 6). As previously demonstrated, 17B stimulation enhances LPS-induced production of IL-6 and TNF-α to a greater degree in PWH (Figure 3). To further understand the mechanism by which estrogen may enhance inflammatory responses and the importance of MAPK p38 signaling in PWH, we inhibited MAPK p38 activation with SB203580. Inhibition of MAPK p38 decreased LPS-induced cytokine production in both HIV- and HIV+ SP groups (Figure 7). Inhibition appeared to also reduce estrogen’s synergistic effects on IL-6 and TNF-α production with LPS stimulation, compared to production with LPS alone, in HIV+SP individuals (Figures 7C, D). The role of p38 activation in estrogen-induced cytokine production requires further study.
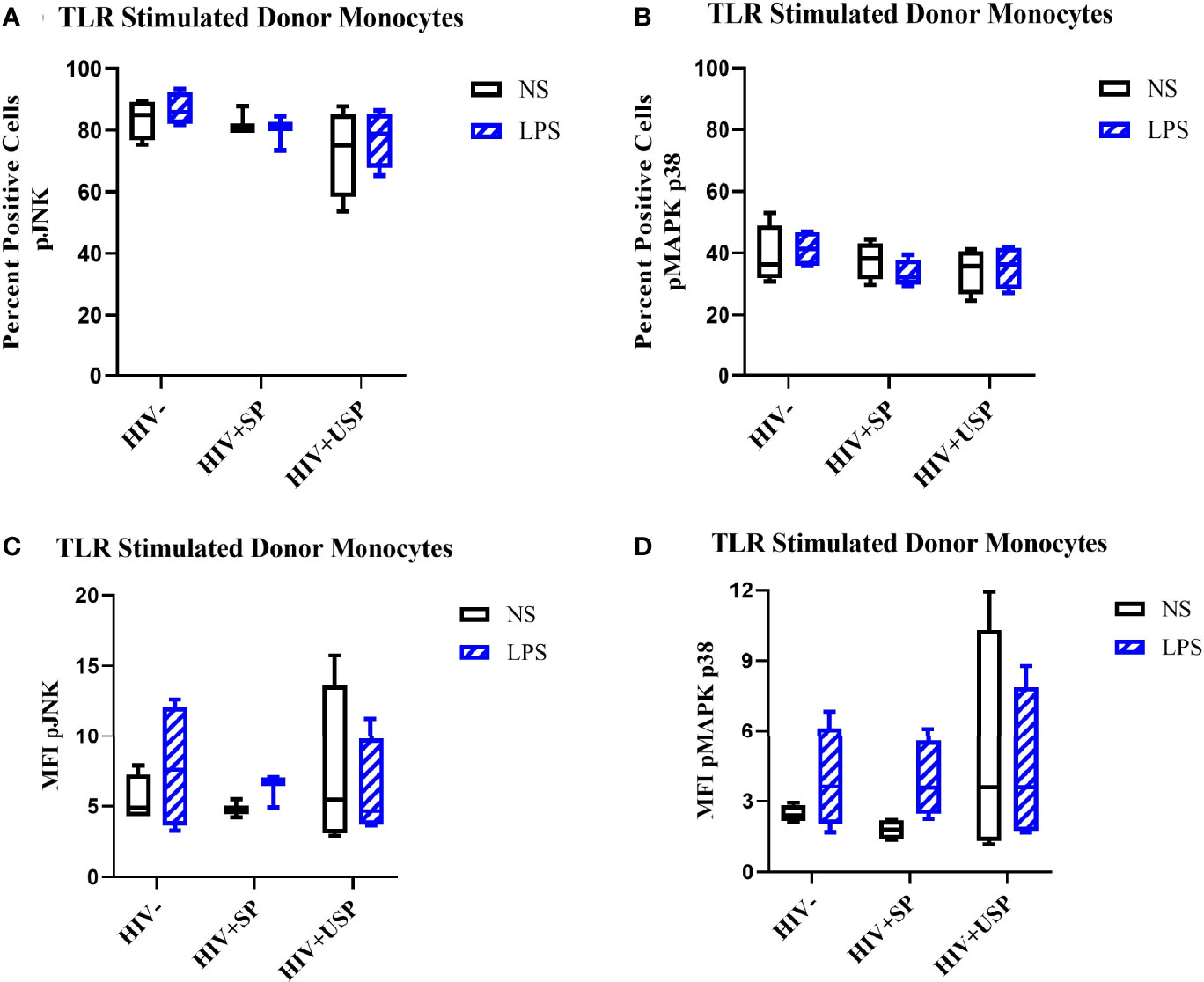
Figure 5 LPS increases phosphorylation levels of JNK and p38 in HIV- and HIV+USP, but not HIV+SP, participants. HIV+ ART-suppressed (HIV+SP), HIV+ ART-unsuppressed (HIV+USP), and HIV negative (HIV-) cryopreserved peripheral blood mononuclear cells (PBMCs) from CM were thawed and rested overnight in phenol red free RPMI supplemented with 10% Human AB Serum. Cells were treated for 20 minutes with no stimulation or 100 ng/mL of LPS. Cells were collected and stained for CD14+ monocytes and analyzed for intracellular levels of (A, C) phosphorylated c-Jun NH2-terminal kinase (n=4 all except HIV+SP n=3) or (B, D) mitogen activated protein kinase mammalian p38 (n=4 all) (pJNK or pMAPK p38 respectively) expressed as (A, B) percent positive cells or (C, D) MFI. Data is represented as mean and standard error of measure and was analyzed by Wilcoxon signed rank test, Mann Whitney U, or one-way ANOVA although no significant differences were found. CM, cisgender men; ART, antiretroviral therapy; LPS, lipopolysaccharide; TLR, Toll-like receptor; NS, no stimulation; MFI, mean fluorescent intensity.
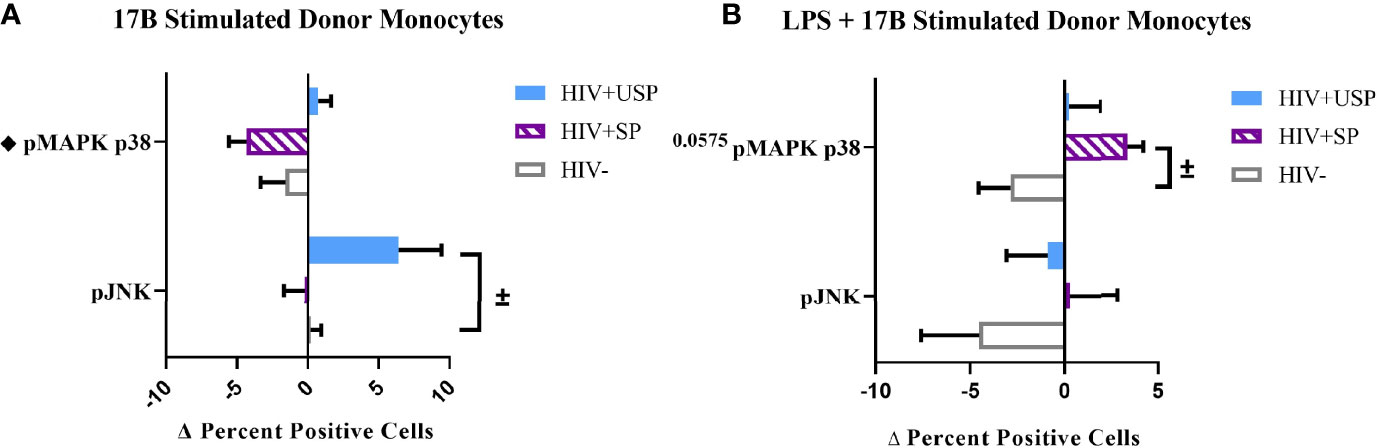
Figure 6 Phosphorylation of MAPK p38 in HIV+SP participants is upregulated with 17B treatment in the presence of LPS, but not alone. HIV+ ART-suppressed (HIV+SP), HIV+ ART-unsuppressed (HIV+USP), and HIV negative (HIV-) cryopreserved peripheral blood mononuclear cells (PBMCs) from CM were thawed and rested overnight in phenol red free RPMI supplemented with 10% Human AB Serum. Cells were treated for 20 minutes with 0.001% ethanol (vehicle) or 1 μM of either 17β estradiol (17B) (A) alone (n=4 all except HIV+SP n=3) or (B) with 100 ng/mL of LPS (n=4 all). Cells were collected and stained for CD14+ monocytes and analyzed for intracellular levels of phosphorylated c-Jun NH2-terminal kinase or mitogen-activated protein kinase mammalian p38 (pJNK or pMAPK p38) expressed as delta change (Δ) in percent positive cells. Data is represented as mean and standard error of measure and normalized to vehicle control (A) alone or (B) with 100 ng/mL of LPS. Data was analyzed by ±Mann Whitney U or ♦one-way ANOVA (±,♦p<0.05). CM, cisgender men; ART, antiretroviral therapy; LPS, lipopolysaccharide; TLR, Toll-like receptor.
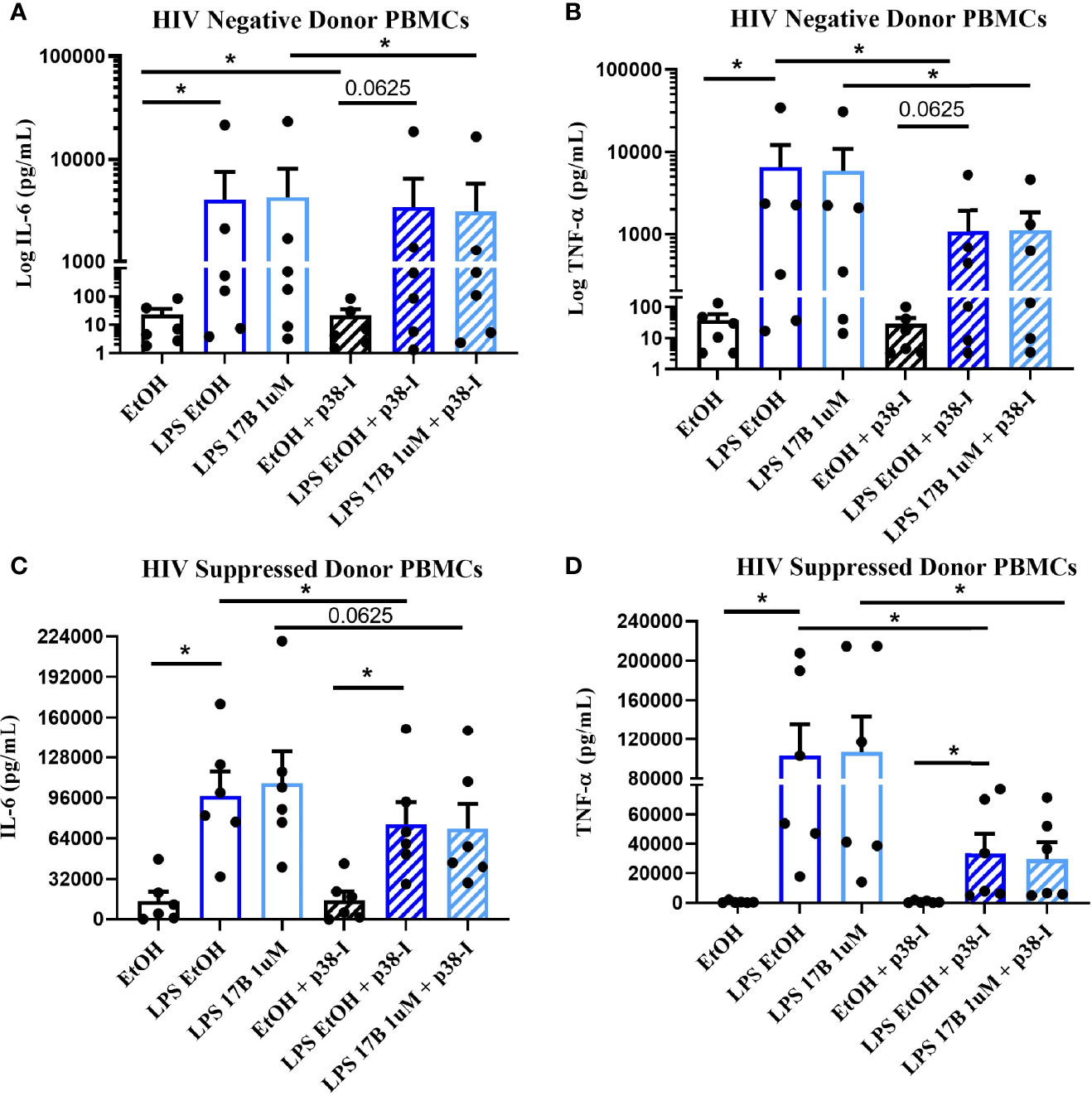
Figure 7 Inhibition of MAPK p38 in the presence of LPS and 17B may downregulate IL-6 and TNF-α production in ART-suppressed participants. 6 HIV+ ART-suppressed and HIV negative cryopreserved peripheral blood mononuclear cells (PBMCs) from CM were thawed and rested overnight in phenol red free RPMI supplemented with 10% Human AB Serum. Cells were treated overnight with 0.001% ethanol (vehicle) or 1 μM of 17β estradiol (17B) +/- 100 ng/mL of LPS in the presence or absence of MAPK p38 inhibitor SB203580 10 µM (p38-I). Supernatants were collected for protein analysis of inflammatory cytokines (A, C) IL-6 or (B, D) TNF-α by multiplex bead assay. Data presented in (A, B) log or (C, D) raw format and was analyzed by *Wilcoxon signed rank test. CM, cisgender men; ART, antiretroviral therapy; LPS, lipopolysaccharide; IL, interleukin; EtOH, ethanol; TNF, tumor necrosis factor.
Discussion
Transgender women are a clinically unique population who are especially vulnerable to both HIV and CVD. As HIV-induced chronic immune activation and inflammation can contribute to CVD risk, and estrogen exposure may exacerbate inflammatory responses, research is needed to elucidate the effects of FHT in TW with HIV.
ART reduces AIDS morbidity and mortality in PWH. Despite its ability to suppress viral replication to undetectable levels and allow restoration of peripheral CD4+ T cell counts in most individuals, gut permeability is often persistently altered in PWH due to incomplete restoration of CD4+ T cells in the gut mucosa. LPS, a bacterial product which can translocate through the gut lining, may drive HIV-induced inflammatory states through activation of myeloid cells by TLR4 ligation (50). As expected, levels of LPS-binding protein, monocyte activation markers, and inflammatory molecules tended to be increased in PWH compared to levels in people without HIV (51–53). In this study, significant positive correlations among markers of monocyte activation, inflammation, and our marker of microbial translocation were identified within PWH, relationships that were not seen among the HIV- group, signaling that microbial translocation may play an influential role in altering immune activation in this cohort.
The expression and function of TLRs are thought to be altered in PWH due to chronic exposure to bacterial and viral products from microbial translocation, HIV replication, or co-infections. In this cohort, exposure of PBMCs to either TLR4 or TLR8 agonists, LPS and ssPolyU, increased inflammatory cytokine production and monocyte activation marker expression regardless of HIV serostatus. TLR-induced activation was notably higher in PWH despite reduced baseline levels. Altered immune responses to microbial products may have arisen in these individuals after long-term exposure to chronic inflammatory stimuli prompting epigenetic or metabolic reprogramming of innate immune cell responses, a phenomenon known as trained immunity. In PWH, trained immunity may optimize inflammatory responses to successive pathogens; in the setting of chronic TLR activation, however, a persistently elevated inflammatory response may promote pathways associated with cardiometabolic risk (54, 55). Additional studies are needed to further assess the role of trained immunity in chronic HIV infection.
Several in vivo markers of monocyte activation and inflammation were inversely related to levels of cytokine production following in vitro LPS exposure among ART-suppressed PWH in our cohort. LPS desensitization, or reduced responsiveness to repeated exposure, in myeloid cells is an adaptive response to limit inflammation during chronic LPS exposure (56). Similar trends existed in HIV- individuals with elevated immune activation and LBP compared to those with reduced profiles of immune activation. While similar relationships were not consistently seen in HIV+USP individuals, there may be a unique interplay between high levels of HIV replication and products of microbial translocation that should be considered. Viral and bacterial products may trigger independent immune pathways and may differentially alter functional responses to TLR stimuli.
Chronic exposure to inflammatory stimuli in vivo may play a role in producing altered functional responses to TLR ligation. Studies of endogenous estrogen in human and animal models suggest estrogen modulates inflammation and TLR responsiveness (37). We hypothesized that estrogen exposure to HIV- cells would result in downregulation of immune activation, as estrogen may play an atheroprotective role, but these effects could be reversed in PWH due to functional reprogramming of cells after exposure to chronic inflammatory environments in vivo. While estrogen had mild effects on cellular activation in all donor groups, the directionality of the response to estrogen differed by HIV serostatus, with increases in inflammatory cytokines in PWH. Interestingly, estrogen’s atheroprotective role in HIV- individuals may be reflected by the reduced cytokine production in those with greater in vivo activation profiles. In contrast, PWH tended to increase inflammatory cytokines in response to estrogen in individuals with greater in vivo immune activation, indicating a potential shift in the cellular response to estrogen with concurrent HIV infection.
Studies of HIV in CW have revealed faster progression to AIDS compared to men with HIV and similar viral loads, as well as altered levels of plasma markers of microbial and immune activation after treatment among CW versus CM with HIV (57, 58). These differences may be explained by estrogen’s ability to modulate TLR-mediated pro-inflammatory pathways, which contribute to chronic immune activation associated with CV morbidity and mortality (57–61). The addition of 17-β estradiol tended to enhance LPS-induced expression of the activation marker HLA-DR on monocytes, and production of cytokines IL-6 and TNF-α from the PBMCs of PWH regardless of ART status; enhancement of these molecules by the addition of estrogen was reduced in those without HIV. Among HIV- individuals, increased immune activation was inversely associated with LPS-induced cytokine production following estrogen exposure, while cytokine production was directly related to in vivo immune activation in PWH. In the latter group, this effect differs from the indirect relationship seen with LPS alone indicating the presence of estrogen may alter functional desensitization to LPS in suppressed individuals with greater activation profiles. Previous cross-sectional biomarker data identified increased levels of inflammatory molecules associated with CVD risk, such as EN-RAGE, oxLDL, and TNFRI, in TW both on and off FHT compared to CM (42). In the presence of estrogen, elevated immune activation profiles in TW with HIV may indicate risk for enhanced inflammatory responses associated with CVD.
Estrogen can modulate the phosphorylation, and activation, of important intracellular signaling molecules in the TLR pathway including JNK and MAPK p38 (48, 49). Both of these kinases can induce upregulation of transcription factors associated with immune activation. While phosphorylation of JNK seemed to play a minor role in either HIV- or suppressed donors, phosphorylation of MAPK p38 in the presence of LPS stimulation was induced by 17-β estradiol in only ART-suppressed individuals. This may suggest that estrogen can upregulate activation of important TLR4 signaling molecules, and subsequent inflammation, during suppressive ART, but not among HIV- individuals. Inhibition of MAPK p38 normalized LPS and estrogen-induced cytokine production to levels seen with LPS alone in HIV+ SP, suggesting that MAPK p38 may play a partial role in additive effects of estrogen and LPS on cytokine production. While MAPK p38 phosphorylation in ART-suppressed individuals may be one mechanism by which estrogen alters HIV-induced inflammatory pathways, estrogen’s effects are likely complex and multifactorial, as many factors are hypothesized to influence the effects of endogenous estrogen in humans. Multiple cellular receptors have been identified for estrogen, but their functions are incompletely understood. Further research is needed to unravel the intersections among estrogen, TLRs, and drivers of inflammation in PWH.
Limitations exist in this study, including the small number of participants. As an exploratory study, these numbers were satisfactory in revealing trends that require further elucidation. Another limitation of this study was the use of cryopreserved PBMCs, as baseline activation of cells may be elevated, although cell viability was not a concern. To mitigate this issue, cells were rested overnight with supplemented media and human serum. A dose-response study was utilized to select a concentration for estrogen. While the dose chosen was greater than physiological estrogen levels, higher levels of estrogen are associated with more anti-inflammatory responses (37) furthering our hypothesis that estrogen use in biological males may have altered effects on inflammation. Due to limitations in human subject enrollment from COVID-19 restrictions, we utilized cells from CM to best replicate the biological environment present in TW pre-FHT. While a convenient solution, studies would better represent this population with cells from TW, as these individuals have increased risk for co-morbidities such as obesity, diabetes, lipid dysregulation, and CVD regardless of HIV. Further study is also needed to understand the full effects of FHT, as FHT can utilize both estrogens and anti-androgens that block the effects of testosterone. We report here that testosterone may play a role in lowering LPS-induced inflammation in PWH. Despite these factors, our findings demonstrate the need for clinical studies aimed at understanding the mechanistic role of estrogen to reduce CV mortality in TW with HIV.
Overall, this work suggests complex interactions may exist among HIV, CVD, and FHT in TW. As PWH are living longer, both increased frequency and severity of multiple end-organ diseases, such as CVDs, have been uncovered. While estrogen is a known modulator of TLR expression and function, the intensified responses to TLR activation with estrogen treatment in PWH cannot be fully explained by this phenomenon when compared to responses in HIV- individuals. This work demonstrates a unique synergistic effect of microbial-and-estrogen-induced immune activation in PWH independent of the increased activation seen with TLR ligation or estrogen treatment alone. Our findings may reflect alterations in cellular response due to chronic inflammatory stimuli that result in enhanced inflammatory pathways associated with CV morbidity.
To appropriately address clinical disparities in TW, current research assessing treatments aimed to reduce pivotal drivers of inflammation and downstream immune signaling underlying HIV-induced co-morbidities should be considered. As previously reviewed, studies range from altering the microbiota in the gut to reduce translocation of microbial products, to blocking the actions of important inflammatory molecules associated with HIV morbidity and mortality such as IL-6 (62). A multi-pronged approach with ART may be a beneficial route to provide equitable care. By understanding the mechanism behind estrogen-induced modulation of TLR pathways, we may identify important clinical indications for additional therapies in TW on FHT or identify alternative estrogen-like drugs designed to reduce deleterious immune effects in TW with HIV.
Contributions
Estrogen has been recognized as an important modulator of immune activation including alterations in inflammation and Toll-like receptor function in innate immune cells. Mechanisms by which estrogen exerts these effects are incompletely understood and may depend on estrogen concentration, cell type, and cellular environments. While the use of estrogen in feminizing hormone therapy can improve quality of life in transgender women, research is needed to understand the side effects of long-term high dose usage. This is particularly concerning in the setting of HIV-associated chronic inflammation as Toll-like receptor 4 activation is linked to elevated systemic lipopolysaccharide and inflammation. Based on current literature, we propose that estrogen may upregulate immune activation of myeloid cells in people with HIV as a result of Toll-like receptor functional changes. The elevated immune activation seen here has been associated with increased risk for co-morbidities such as cardiovascular disease and higher mortality rates in people with HIV. As estrogen use in transgender women has been linked to three-fold increased risk of cardiovascular mortality, these results may provide a potential mechanism by which estrogen exerts deleterious effects and potential biomarkers that may indicate a need for additional immunotherapies or alternative estrogen-based therapies in people with HIV.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Institutional Review Board at Ohio State Wexner Medical Center. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
All authors contributed to experimental design, data analysis, and writing of the manuscript. AK and EB: performed experiments. BH: provided analytical expertise.
Funding
Research reported in our publication was supported by the National Institutes of Health supported by the National Institute of Allergic and Infectious Disease of the National Institutes of Health under grant numbers P30AI161943, 5R21AI143452 to JEL. This work was also supported, in part, by the National Center for Advancing Translational Sciences of the National Institutes of Health under Grant Numbers TL1TR002735 & UL1TR001450.
Author Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest
NF has served as a consultant for Gilead.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the Center for AIDS Research Network of Integrated Clinical Systems repository (CFAR CNICS) at Case Western Reserve University, Cleveland, OH, for supplying cryopreserved peripheral blood mononuclear cells from HIV-positive participants and relevant clinical and demographic information for our study. We also acknowledge the contributions provided by Jiao Yu at Case Western Reserve University for their design and creation of the correlation heatmaps seen here.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.879600/full#supplementary-material
Abbreviations
TW, Transgender women; CM, Cisgender men; CW, Cisgender women; FHT, Feminizing hormone therapy; 17B, 17-β estradiol; 17A, 17-α ethinylestradiol; HIV-, HIV negative; HIV+SP, HIV+ Suppressed; HIV+USP, HIV+ Unsuppressed.
References
1. Nguyen HB, Chavez AM, Lipner E, Hantsoo L, Kornfield SL, Davies RD, et al. Gender-Affirming Hormone Use in Transgender Individuals: Impact on Behavioral Health and Cognition. Curr Psychiatry Rep (2018) 20(12):110. doi: 10.1007/s11920-018-0973-0
2. Tangpricha V, Safer JD. Transgender Women: Evaluation and Management. (2021). Available from: https://www.uptodate.com/contents/transgender-women-evaluation-and-management.
3. de Blok CJM, Wiepjes CM, van Velzen DM, Staphorsius AS, Nota NM, Gooren LJG, et al. Mortality Trends Over Five Decades in Adult Transgender People Receiving Hormone Treatment: A Report From the Amsterdam Cohort of Gender Dysphoria. Lancet Diabetes Endocrinol (2021) 9(10):663–70. doi: 10.1016/s2213-8587(21)00185-6
4. Asscheman H, Giltay EJ, Megens JA, de Ronde WP, van Trotsenburg MA, Gooren LJ. A Long-Term Follow-Up Study of Mortality in Transsexuals Receiving Treatment With Cross-Sex Hormones. Eur J Endocrinol (2011) 164(4):635–42. doi: 10.1530/EJE-10-1038
5. Bretherton I, Spanos C, Leemaqz SY, Premaratne G, Grossmann M, Zajac JD, et al. Insulin Resistance in Transgender Individuals Correlates With Android Fat Mass. Ther Adv Endocrinol Metab (2021) 12:2042018820985681. doi: 10.1177/2042018820985681
6. Baral SD, Poteat T, Strömdahl S, Wirtz AL, Guadamuz TE, Beyrer C. Worldwide Burden of HIV in Transgender Women: A Systematic Review and Meta-Analysis. Lancet Infect Dis (2013) 13(3):214–22. doi: 10.1016/s1473-3099(12)70315-8
7. Silva-Santisteban A, Raymond HF, Salazar X, Villayzan J, Leon S, McFarland W, et al. Understanding the HIV/AIDS Epidemic in Transgender Women of Lima, Peru: Results From a Sero-Epidemiologic Study Using Respondent Driven Sampling. AIDS Behav (2012) 16(4):872–81. doi: 10.1007/s10461-011-0053-5
8. Wilson R, Spiers A, Ewan J, Johnson P, Jenkins C, Carr S. Effects of High Dose Oestrogen Therapy on Circulating Inflammatory Markers. Maturitas (2009) 62(3):281–6. doi: 10.1016/j.maturitas.2009.01.009
9. Wierckx K, Elaut E, Declercq E, Heylens G, De Cuypere G, Taes Y, et al. Prevalence of Cardiovascular Disease and Cancer During Cross-Sex Hormone Therapy in a Large Cohort of Trans Persons: A Case-Control Study. Eur J Endocrinol (2013) 169(4):471–8. doi: 10.1530/EJE-13-0493
10. Gooren LJ, Wierckx K, Giltay EJ. Cardiovascular Disease in Transsexual Persons Treated With Cross-Sex Hormones: Reversal of the Traditional Sex Difference in Cardiovascular Disease Pattern. Eur J Endocrinol (2014) 170(6):809–19. doi: 10.1530/EJE-14-0011
11. Nokoff NJ, Scarbro S, Juarez-Colunga E, Moreau KL, Kempe A. Health and Cardiometabolic Disease in Transgender Adults in the United States: Behavioral Risk Factor Surveillance System 2015. J Endocr Soc (2018) 2(4):349–60. doi: 10.1210/js.2017-00465
12. Shah ASV, Stelzle D, Lee KK, Beck EJ, Alam S, Clifford S, et al. Global Burden of Atherosclerotic Cardiovascular Disease in People Living With HIV: Systematic Review and Meta-Analysis. Circulation (2018) 138(11):1100–12. doi: 10.1161/CIRCULATIONAHA.117.033369
13. Hemkens LG, Bucher HC. HIV Infection and Cardiovascular Disease. Eur Heart J (2014) 35(21):1373–81. doi: 10.1093/eurheartj/eht528
14. Martinez-Picado J, Deeks SG. Persistent HIV-1 Replication During Antiretroviral Therapy. Curr Opin HIV AIDS (2016) 11(4):417–23. doi: 10.1097/COH.0000000000000287
15. Appay V, Sauce D. Immune Activation and Inflammation in HIV-1 Infection: Causes and Consequences. J Pathol (2008) 214(2):231–41. doi: 10.1002/path.2276
16. Kelesidis T, Kendall MA, Yang OO, Hodis HN, Currier JS. Biomarkers of Microbial Translocation and Macrophage Activation: Association With Progression of Subclinical Atherosclerosis in HIV-1 Infection. J Infect Dis (2012) 206(10):1558–67. doi: 10.1093/infdis/jis545
17. Ross AC, Rizk N, O'Riordan MA, Dogra V, El-Bejjani D, Storer N, et al. Relationship Between Inflammatory Markers, Endothelial Activation Markers, and Carotid Intima-Media Thickness in HIV-Infected Patients Receiving Antiretroviral Therapy. Clin Infect Dis (2009) 49(7):1119–27. doi: 10.1086/605578
18. Currier JS, Taylor A, Boyd F, Dezii CM, Kawabata H, Burtcel B, et al. Coronary Heart Disease in HIV-Infected Individuals. J Acquir Immune Defic Syndr (2003) 33(4):506–12. doi: 10.1097/00126334-200308010-00012
19. So-Armah KA, Tate JP, Chang CH, Butt AA, Gerschenson M, Gibert CL, et al. Do Biomarkers of Inflammation, Monocyte Activation, and Altered Coagulation Explain Excess Mortality Between HIV Infected and Uninfected People? J Acquir Immune Defic Syndr (2016) 72(2):206–13. doi: 10.1097/QAI.0000000000000954
20. McKibben RA, Margolick JB, Grinspoon S, Li X, Palella FJ Jr, Kingsley LA, et al. Elevated Levels of Monocyte Activation Markers Are Associated With Subclinical Atherosclerosis in Men With and Those Without HIV Infection. J Infect Dis (2015) 211(8):1219–28. doi: 10.1093/infdis/jiu594
21. Longenecker CT, Jiang Y, Orringer CE, Gilkeson RC, Debanne S, Funderburg NT, et al. Soluble CD14 Is Independently Associated With Coronary Calcification and Extent of Subclinical Vascular Disease in Treated HIV Infection. AIDS (2014) 28(7):969–77. doi: 10.1097/QAD.0000000000000158
22. Triant VA, Lee H, Hadigan C, Grinspoon SK. Increased Acute Myocardial Infarction Rates and Cardiovascular Risk Factors Among Patients With Human Immunodeficiency Virus Disease. J Clin Endocrinol Metab (2007) 92(7):2506–12. doi: 10.1210/jc.2006-2190
23. Zicari S, Sessa L, Cotugno N, Ruggiero A, Morrocchi E, Concato C, et al. Immune Activation, Inflammation, and Non-AIDS Co-Morbidities in HIV-Infected Patients Under Long-Term ART. Viruses (2019) 11(3):200. doi: 10.3390/v11030200
24. Dirajlal-Fargo S, El-Kamari V, Weiner L, Shan L, Sattar A, Kulkarni M, et al. Altered Intestinal Permeability and Fungal Translocation in Ugandan Children With Human Immunodeficiency Virus. Clin Infect Dis (2020) 70(11):2413–22. doi: 10.1093/cid/ciz561
25. Dirajlal-Fargo S, Albar Z, Bowman E, Labbato D, Sattar A, Karungi C, et al. Increased Monocyte and T-Cell Activation in Treated HIV+ Ugandan Children: Associations With Gut Alteration and HIV Factors. AIDS (2020) 34(7):1009–18. doi: 10.1097/QAD.0000000000002505
26. Medzhitov R. Toll-Like Receptors and Innate Immunity. Nat Rev Immunol (2001) 1(2):135–45. doi: 10.1038/35100529
27. Kawasaki T, Kawai T. Toll-Like Receptor Signaling Pathways. Front Immunol (2014) 5:461. doi: 10.3389/fimmu.2014.00461
28. El-Zayat SR, Sibaii H, Mannaa FA. Toll-Like Receptors Activation, Signaling, and Targeting: An Overview Vol. 43. Bulletin of the National Research Centre (2019). doi: 10.1186/s42269-019-0227-2
29. Park BS, Lee JO. Recognition of Lipopolysaccharide Pattern by TLR4 Complexes. Exp Mol Med (2013) 45:e66. doi: 10.1038/emm.2013.97
30. Bernard MA, Han X, Inderbitzin S, Agbim I, Zhao H, Koziel H, et al. HIV-Derived ssRNA Binds to TLR8 to Induce Inflammation-Driven Macrophage Foam Cell Formation. PloS One (2014) 9(8):e104039. doi: 10.1371/journal.pone.0104039
31. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in Atherosclerosis: A Dynamic Balance. Nat Rev Immunol (2013) 13(10):709–21. doi: 10.1038/nri3520
32. Woollard KJ, Geissmann F. Monocytes in Atherosclerosis: Subsets and Functions. Nat Rev Cardiol (2010) 7(2):77–86. doi: 10.1038/nrcardio.2009.228
33. Libby P. Inflammation in Atherosclerosis. Nature (2002) 420(6917):868–74. doi: 10.1038/nature01323
34. Borges AH, O'Connor JL, Phillips AN, Neaton JD, Grund B, Neuhaus J, et al. Interleukin 6 Is a Stronger Predictor of Clinical Events Than High-Sensitivity C-Reactive Protein or D-Dimer During HIV Infection. J Infect Dis (2016) 214(3):408–16. doi: 10.1093/infdis/jiw173
35. Grund B, Baker JV, Deeks SG, Wolfson J, Wentworth D, Cozzi-Lepri A, et al. Relevance of Interleukin-6 and D-Dimer for Serious Non-AIDS Morbidity and Death Among HIV-Positive Adults on Suppressive Antiretroviral Therapy. PloS One (2016) 11(5):e0155100. doi: 10.1371/journal.pone.0155100
36. Jalbert E, Crawford TQ, D'Antoni ML, Keating SM, Norris PJ, Nakamoto BK, et al. IL-1Beta Enriched Monocytes Mount Massive IL-6 Responses to Common Inflammatory Triggers Among Chronically HIV-1 Infected Adults on Stable Anti-Retroviral Therapy at Risk for Cardiovascular Disease. PloS One (2013) 8(9):e75500. doi: 10.1371/journal.pone.0075500
37. Klein SL, Flanagan KL. Sex Differences in Immune Responses. Nat Rev Immunol (2016) 16(10):626–38. doi: 10.1038/nri.2016.90
38. Kovats S. Estrogen Receptors Regulate Innate Immune Cells and Signaling Pathways. Cell Immunol (2015) 294(2):63–9. doi: 10.1016/j.cellimm.2015.01.018
39. Young NA, Wu LC, Burd CJ, Friedman AK, Kaffenberger BH, Rajaram MV, et al. Estrogen Modulation of Endosome-Associated Toll-Like Receptor 8: An IFNalpha-Independent Mechanism of Sex-Bias in Systemic Lupus Erythematosus. Clin Immunol (2014) 151(1):66–77. doi: 10.1016/j.clim.2014.01.006
40. Filardo EJ, Quinn JA, Frackelton AR Jr., Bland KI. Estrogen Action via the G Protein-Coupled Receptor, GPR30: Stimulation of Adenylyl Cyclase and cAMP-Mediated Attenuation of the Epidermal Growth Factor Receptor-to-MAPK Signaling Axis. Mol Endocrinol (2002) 16(1):70–84. doi: 10.1210/mend.16.1.0758
41. Kang L, Zhang X, Xie Y, Tu Y, Wang D, Liu Z, et al. Involvement of Estrogen Receptor Variant ER-Alpha36, Not GPR30, in Nongenomic Estrogen Signaling. Mol Endocrinol (2010) 24(4):709–21. doi: 10.1210/me.2009-0317
42. Lake JE, Wang R, Barrett B, Funderburg N, Bowman E, Debroy P, et al. Hormone Use and HIV Alter Cardiovascular Biomarker Profiles in Transgender Women [CROI Abstract 645]. In: Special Issue: Abstracts From the 2020 Conference on Retroviruses and Opportunistic Infections. Boston, Massachusetts: Top Antivir Med (2020). p. 483.
43. Streed CG Jr., Beach LB, Caceres BA, Dowshen NL, Moreau KL, Mukherjee M, et al. Assessing and Addressing Cardiovascular Health in People Who Are Transgender and Gender Diverse: A Scientific Statement From the American Heart Association. Circulation (2021) 144(6):e136–e48. doi: 10.1161/CIR.0000000000001003
44. Funderburg N, Lederman MM, Feng Z, Drage MG, Jadlowsky J, Harding CV, et al. Human -Defensin-3 Activates Professional Antigen-Presenting Cells via Toll-Like Receptors 1 and 2. Proc Natl Acad Sci USA (2007) 104(47):18631–5. doi: 10.1073/pnas.0702130104
45. Hernandez JC, Stevenson M, Latz E, Urcuqui-Inchima S. HIV Type 1 Infection Up-Regulates TLR2 and TLR4 Expression and Function In Vivo and In Vitro. AIDS Res Hum Retroviruses (2012) 28(10):1313–28. doi: 10.1089/aid.2011.0297
46. Goldstein Z, Khan M, Reisman T, Safer JD. Managing the Risk of Venous Thromboembolism in Transgender Adults Undergoing Hormone Therapy. J Blood Med (2019) 10:209–16. doi: 10.2147/JBM.S166780
47. Prossnitz ER, Arterburn JB, Sklar LA. GPR30: A G Protein-Coupled Receptor for Estrogen. Mol Cell Endocrinol (2007) 265-266:138–42. doi: 10.1016/j.mce.2006.12.010
48. Fardoun MM, Issa K, Maaliki D, Nasser SA, Baydoun E, Eid AH. Estrogen Increases Expression of Vascular Alpha 2C Adrenoceptor Through the cAMP/Epac/JNK/AP-1 Pathway and Potentiates Cold-Induced Vasoconstriction. Vascul Pharmacol (2020) 131:106690. doi: 10.1016/j.vph.2020.106690
49. Seval Y, Cakmak H, Kayisli UA, Arici A. Estrogen-Mediated Regulation of P38 Mitogen-Activated Protein Kinase in Human Endometrium. J Clin Endocrinol Metab (2006) 91(6):2349–57. doi: 10.1210/jc.2005-2132
50. Ramendra R, Isnard S, Mehraj V, Chen J, Zhang Y, Finkelman M, et al. Circulating LPS and (1–>3)-Beta-D-Glucan: A Folie a Deux Contributing to HIV-Associated Immune Activation. Front Immunol (2019) 10:465. doi: 10.3389/fimmu.2019.00465
51. Nystrom J, Stenkvist J, Haggblom A, Weiland O, Nowak P. Low Levels of Microbial Translocation Marker LBP Are Associated With Sustained Viral Response After Anti-HCV Treatment in HIV-1/HCV Co-Infected Patients. PloS One (2015) 10(3):e0118643. doi: 10.1371/journal.pone.0118643
52. Neuhaus J, Jacobs DR Jr., Baker JV, Calmy A, Duprez D, La Rosa A, et al. Markers of Inflammation, Coagulation, and Renal Function Are Elevated in Adults With HIV Infection. J Infect Dis (2010) 201(12):1788–95. doi: 10.1086/652749
53. Mendez-Lagares G, Romero-Sanchez MC, Ruiz-Mateos E, Genebat M, Ferrando-Martinez S, Munoz-Fernandez MA, et al. Long-Term Suppressive Combined Antiretroviral Treatment Does Not Normalize the Serum Level of Soluble CD14. J Infect Dis (2013) 207(8):1221–5. doi: 10.1093/infdis/jit025
54. Owen AM, Fults JB, Patil NK, Hernandez A, Bohannon JK. TLR Agonists as Mediators of Trained Immunity: Mechanistic Insight and Immunotherapeutic Potential to Combat Infection. Front Immunol (2020) 11:622614. doi: 10.3389/fimmu.2020.622614
55. van der Heijden WA, Van de Wijer L, Keramati F, Trypsteen W, Rutsaert S, Horst RT, et al. Chronic HIV Infection Induces Transcriptional and Functional Reprogramming of Innate Immune Cells. JCI Insight (2021) 6(7):e145928. doi: 10.1172/jci.insight.145928
56. Ruckdeschel K, Richter K. Lipopolysaccharide Desensitization of Macrophages Provides Protection Against Yersinia Enterocolitica-Induced Apoptosis. Infect Immun (2002) 70(9):5259–64. doi: 10.1128/IAI.70.9.5259-5264.2002
57. Krebs SJ, Slike BM, Sithinamsuwan P, Allen IE, Chalermchai T, Tipsuk S, et al. Sex Differences in Soluble Markers Vary Before and After the Initiation of Antiretroviral Therapy in Chronically HIV-Infected Individuals. AIDS (2016) 30(10):1533–42. doi: 10.1097/QAD.0000000000001096
58. Farzadegan H, Hoover DR, Astemborski J, Lyles CM, Margolick JB, Markham RB, et al. Sex Differences in HIV-1 Viral Load and Progression to AIDS. Lancet (1998) 352(9139):1510–4. doi: 10.1016/s0140-6736(98)02372-1
59. Hanna DB, Lin J, Post WS, Hodis HN, Xue X, Anastos K, et al. Association of Macrophage Inflammation Biomarkers With Progression of Subclinical Carotid Artery Atherosclerosis in HIV-Infected Women and Men. J Infect Dis (2017) 215(9):1352–61. doi: 10.1093/infdis/jix082
60. Meier A, Chang JJ, Chan ES, Pollard RB, Sidhu HK, Kulkarni S, et al. Sex Differences in the Toll-Like Receptor-Mediated Response of Plasmacytoid Dendritic Cells to HIV-1. Nat Med (2009) 15(8):955–9. doi: 10.1038/nm.2004
61. Fischer J, Jung N, Robinson N, Lehmann C. Sex Differences in Immune Responses to Infectious Diseases. Infection (2015) 43(4):399–403. doi: 10.1007/s15010-015-0791-9
Keywords: estrogen, inflammation, monocytes, toll-like receptor 4, human immunodeficiency virus, cardiovascular disease, transgender women
Citation: Kettelhut A, Bowman E, Gabriel J, Hand B, Liyanage NPM, Kulkarni M, Avila-Soto F, Lake JE and Funderburg NT (2022) Estrogen May Enhance Toll-Like Receptor 4-Induced Inflammatory Pathways in People With HIV: Implications for Transgender Women on Hormone Therapy. Front. Immunol. 13:879600. doi: 10.3389/fimmu.2022.879600
Received: 19 February 2022; Accepted: 03 May 2022;
Published: 03 June 2022.
Edited by:
Anthony Jaworowski, RMIT University, AustraliaReviewed by:
Thomas A. Angelovich, RMIT University, AustraliaMabel Toribio, Massachusetts General Hospital and Harvard Medical School, United States
Copyright © 2022 Kettelhut, Bowman, Gabriel, Hand, Liyanage, Kulkarni, Avila-Soto, Lake and Funderburg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aaren Kettelhut, YWFyZW4ua2V0dGVsaHV0QG9zdW1jLmVkdQ==
†These authors share last authorship