 Fiona Poyer1
Fiona Poyer1 Raúl Jimenez Heredia2,3,4
Raúl Jimenez Heredia2,3,4 Wolfgang Novak1
Wolfgang Novak1 Petra Zeitlhofer2,5
Petra Zeitlhofer2,5 Karin Nebral2,5
Karin Nebral2,5 Michael N. Dworzak1,2
Michael N. Dworzak1,2 Oskar A. Haas1,2,5*†
Oskar A. Haas1,2,5*† Kaan Boztug1,2,3,4*†
Kaan Boztug1,2,3,4*† Leo Kager1,2*†
Leo Kager1,2*†- 1St. Anna Children’s Hospital, Department of Pediatrics and Adolescent Medicine, Medical University of Vienna, Vienna, Austria
- 2St. Anna Children’s Cancer Research Institute (CCRI), Vienna, Austria
- 3Ludwig Boltzmann Institute for Rare and Undiagnosed Diseases, Vienna, Austria
- 4Center for Molecular Medicine Center for Molecular Medicine (CeMM) Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria
- 5Labdia, Labordiagnostik, Vienna, Austria
We report the case of a male Pakistani patient with a pathogenic homozygous loss of function variant in the non-homologous end-joining factor 1 (NHEJ1) gene. The growth retarded and microcephalic boy with clinodactyly of both hands and hyperpigmentation of the skin suffered from recurrent respiratory infections. He was five and a half years old when he came to our attention with refractory cytopenia and monosomy 7. Hematopoietic stem cell transplantation was considered but not feasible because there was no suitable donor available. Monosomy 7 was not detected anymore in subsequent bone marrow biopsies that were repeated in yearly intervals. Instead, seven and a half years later, a novel clone with a del(20q) appeared and steadily increased thereafter. In parallel, the patient’s blood count, which had remained stable for over 20 years without necessitating any specific therapeutic interventions, improved gradually and the erythropoiesis-associated dysplasia resolved.
Introduction
Inherited bone marrow failure (IBMF) syndromes are genetically heterogeneous hematopoietic stem cell disorders that impede the adequate production of one or more blood cell lineages and consequently predispose affected individuals to the development of myelodysplastic syndromes (MDS) as well as myeloid malignancies (1–3). The responsible genetic defects often produce recognizable syndromes, whose main features are microcephaly, growth retardation as well as inconsistent other physical malformations and organ abnormalities. Such germline alterations comprise pathogenic variants in genes that encode for transcription factors such as GATA2, RUNX1 and ETV6, products that are involved in telomere maintenance (dyskeratosis congenita), ribosomal biogenesis (Blackfan-Diamond anemia) and maturation (Shwachman-Diamond syndrome; SDS), DNA maintenance and repair (Fanconi anemia), protein folding and trafficking (severe congenital neutropenia) as well as in the regulation of cell proliferation and apoptosis (SAMD9/9L) (3–9). In many instances characteristic clinical, laboratory and hematologic parameters alone will already suffice to identify the respective syndrome. Nevertheless, the large number of not only genes but also of possible types of pathogenic variants that need to be considered even in such well-defined syndromes requires a thorough molecular genetic clarification to obtain a precise diagnosis. In case of overlapping symptoms and/or if no pathogenic sequence abnormalities are found, an even broader screening approach with whole exome sequencing may be necessary to identify rarer or even previously not considered causes of such diseases.
Herein we report the extraordinary disease development in a patient with distinctive yet originally difficult to interpret phenotypic features, in whom, after a long and challenging diagnostic odyssey, we finally succeeded to secure a homozygous pathogenic variant in the NHEJ1 gene as the responsible germ line defect.
Case Report
The now 26-year-old patient [case #17; Table 2 in our previous publication (10)] is the son of consanguineous Pakistani parents, who were healthy and had normal blood counts. One sister was healthy, a second sister suffered from hepatitis C, but had normal blood counts. He had been prone to infections, primarily recurrent bronchitis, since birth and was first seen in our clinic when he was five and a half years old with fever and coughing. He was growth retarded and had a microcephaly (<3rd percentile), clinodactyly of both hands and hyperpigmented skin above the knees and elbows. Apart from his microcephaly, imaging of the head, the thorax and the abdomen remained inconspicuous. Screening for causative infectious agents, including Mycobacterium tuberculosis, was unrewarding. Immunological analyses that were performed during the course of the disease revealed IgA deficiency and B-lymphocytopenia (Details are provided in Supplementary Table 1). Hematologic analysis at first presentation revealed a white blood cell count of 1.47 x109/L with an absolute neutrophil count of 0.76 x109/L and an absolute lymphocyte count of 0.47 x109/L, a hemoglobin level of 9.2 g/dL, MCV of 75fl, and a platelet count of 109 x109/L. Data on the long-term course of hematological parameters are provided in Supplementary Figure 1. A bone marrow (BM) examination disclosed reduced cellularity of all three lineages with an erythropoiesis-restricted dysplasia that, together with the presence of a fluorescence in situ hybridization (FISH)-verified monosomy 7 in 18% of the analyzed nuclei, was consistent with the diagnosis of a hypocellular myelodysplastic syndrome in form of a refractory cytopenia (11, 12). Although the appearance of monosomy 7 in IBMF is strongly indicative of an underlying SAMD9/9L or GATA2 deficiency, these disease-promoting germline causes were not known at that time (6, 13, 14).
We discounted the most likely causes of the patient’s problems, namely Fanconi anemia, Nijmegen breakage syndrome and dyskeratosis congenita with an originally negative diepoxybutane (DEB) breakage analysis in the one, molecular testing in the other and based on clinical parameters in the latter, respectively. Another DEB test that was performed at the age of 14 years showed an elevated chromosome breakage (4.78, normal <0.6) but without the pathognomonic Fanconi anemia-specific chromatid exchange figures. Moreover, a cell cycle analysis of the patient’s skin fibroblasts, which was kindly performed in the Department of Human Genetics, University of Würzburg, Würzburg, Germany, lacked the otherwise typical G2 cell cycle blockage (15). Normal pancreatic laboratory parameters also excluded a less likely Shwachman-Diamond syndrome and lymphocyte immunophenotyping the presence of paroxysmal nocturnal hemoglobinuria clones that are seen in up to 38% of refractory cytopenia cases (16, 17). Since the appearance of monosomy 7 in patients with such syndromic features and pancytopenia often precedes and forecasts the transformation into myeloid malignancy, the current EWOG-MDS 2006 protocol (NCT00662090) recommends as the treatment of choice to perform a hematopoietic stem cell transplantation (HSCT), which was not feasible in this case because we did not find a suitable donor. However, contrary to all odds, his blood counts remained stable at this low level over the following years, and he did not even require any transfusions. He had only a mild obstructive ventilation disorder and continued to suffer from recurrent but well-manageable pulmonary infections. Although we did not find the clone with the monosomy 7 anymore in the follow-up BM examination three months later, another clone with a deletion of the long arm of chromosome 20, del(20q), emerged seven and a half years later, when the patient was 13 years old. The respective FISH analyses were performed with a del(20q)-specific dual-color probe set (leicabiosystems.com). Subsequent BM examinations that were executed in yearly intervals thereafter showed that this abnormal clone steadily increased from originally 15% to 90% within the following six years and then dropped again to 68% a year later, at which point the erythropoiesis-restricted dysplasia also had resolved. Nonetheless, even two years later, we found that 34% of the peripheral blood cells still descended from this del(20q) clone, even though the patient was in good clinical condition and with a notably improved white blood cell count of 3.06 x109/L, an absolute neutrophil count of 1.27 x109/L, an absolute lymphocyte count of 1.53 x109/L, a hemoglobin level of 11.6 g/dL and a platelet count of 82 x109/L. The hematological parameters, however, display a fluctuating pattern during course (Supplementary Figure 1).
Genetic Analyses
When our patient was 21 years old, we finally succeeded to identify the genetic cause of his physical and hematological problems with our next-generation targeted sequencing hematology panel. This approach uncovered a unique and hitherto undescribed pathogenic homozygous missense variant (NM_024782.2:c.236T>C, p.Leu79Pro; GRCh37) in exon 3 of the NHEJ1 (“non-homologous end-joining factor 1”, also known as XLF “XRCC4-like factor” or Cernunnos, OMIM *611290) gene on chromosome 2(q35) (10). The pathogenic variant had a CADD v.1.3 (“combined annotation dependent depletion”) score of 31 and was thus classified as being intolerable (Figure 1, left) (19). Segregation analysis confirmed the heterozygous carrier status of both parents (Figure 1, right).
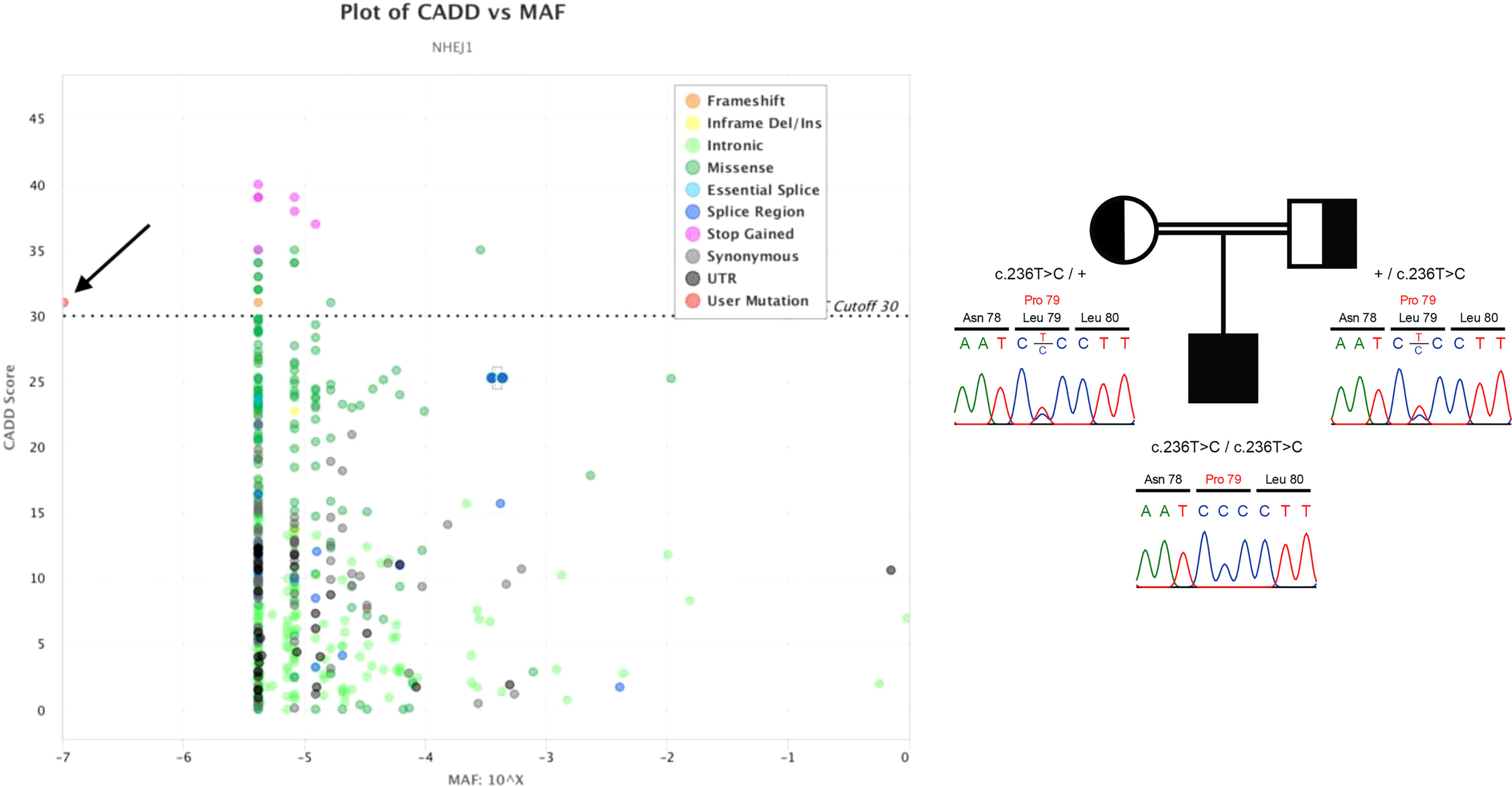
Figure 1 Left: CADD versus minor allele frequency (MAF) plot of all known NEHJ1 sequence variants visualized by PopViz (18). The horizontal axis shows the MAF scores and the vertical axis the CADD ones. The specific types of the various sequence variants, which were collected from the gnomAD r2.0.2 database (https://gnomad.broadinstitute.org/gene/ENSG00000187736?dataset=gnomad_r2_1), are color-coded, and the pathogenic variant of our patient is indicated by the black arrow. Right: The patient’s homozygous NEHJ1 gene pathogenic variant (NM_024782.2:c.236T>C, p.Leu79Pro) was inherited from his heterozygous parents.
At this time, immunophenotyping of peripheral blood cells revealed a selective CD19+ B-cell lymphopenia with a nadir of 0.02 x109/L. Immunoglobulin G and M levels (IgA deficiency) as well as the results of the stimulated T cell proliferation tests were normal, although the vaccine-dependent reactivity was moderately reduced. Since an ongoing telomere length shortening of hematopoietic stem cells had been previously reported to contribute to the development of cytopenia in NHEJ1-deficient cases, we had this parameter examined in the Department of Pediatric Hematology and Oncology, University of Freiburg, Germany (20, 21). Comparison of his telomere repeat copy number with that of reference samples confirmed that the telomeres in his peripheral blood cells were, with a ratio of 0.57, indeed severely shortened, namely to an extent that was below the first percentile (0.61) of a healthy control cohort (n=90).
To determine the exact location and extension of the hematopoiesis-restricted del(20q), we performed a CytoScan™ HD array analysis. This array comprises 2,670,000 markers, including 750,000 single nucleotide polymorphism probes (Applied Biosystems™, Thermo Fisher Scientific, Waltham, MA, USA). We obtained the data from a commercial service provider and analyzed them in-house with the Chromosome Analysis Suite (ChAS; Applied Biosystems™, ThermoFisher Scientific) software package version 4.1 as described previously in detail (22). Overall, six percent of the autosomal genome was homozygous, indicating consanguinity in the family. In addition to the del(20q) we noted seven such consanguinity-associated homozygous regions that were larger than 3Mb, namely on chromosomes 2(q34-q37.2), which contained the NHEJ1 pathogenic variants, 3(p12.3-q13.31), 7(q34-q36.1), 8(q22.1-q22.3), 9(p21.3-q21.32), 14(q11.2-q12) and 21(q11.2-q21.3) as well as a unique 78kb duplication at chromosome 20(p11.2) that partially disrupted the GINS1 gene (Supplementary Table 2). Biallelic loss of function pathogenic variants in this gene cause another very rare, phenotypically very similar immunodeficiency syndrome (IMD55; OMIM #617827) with intrauterine growth retardation, chronic neutropenia, and natural killer cell deficiency (23). The 22,124 Mb large interstitial del(20q) encompassed nearly the entire long arm and removed 244 genes that are contained in this region (Figure 2). Of the three genes that are of specific relevance in the context of an acquired del(20q), two, namely the imprinted L3MBTL1 and SGK2 genes were deleted, whereas the EIF6 gene was not (Figure 2) (24–28).
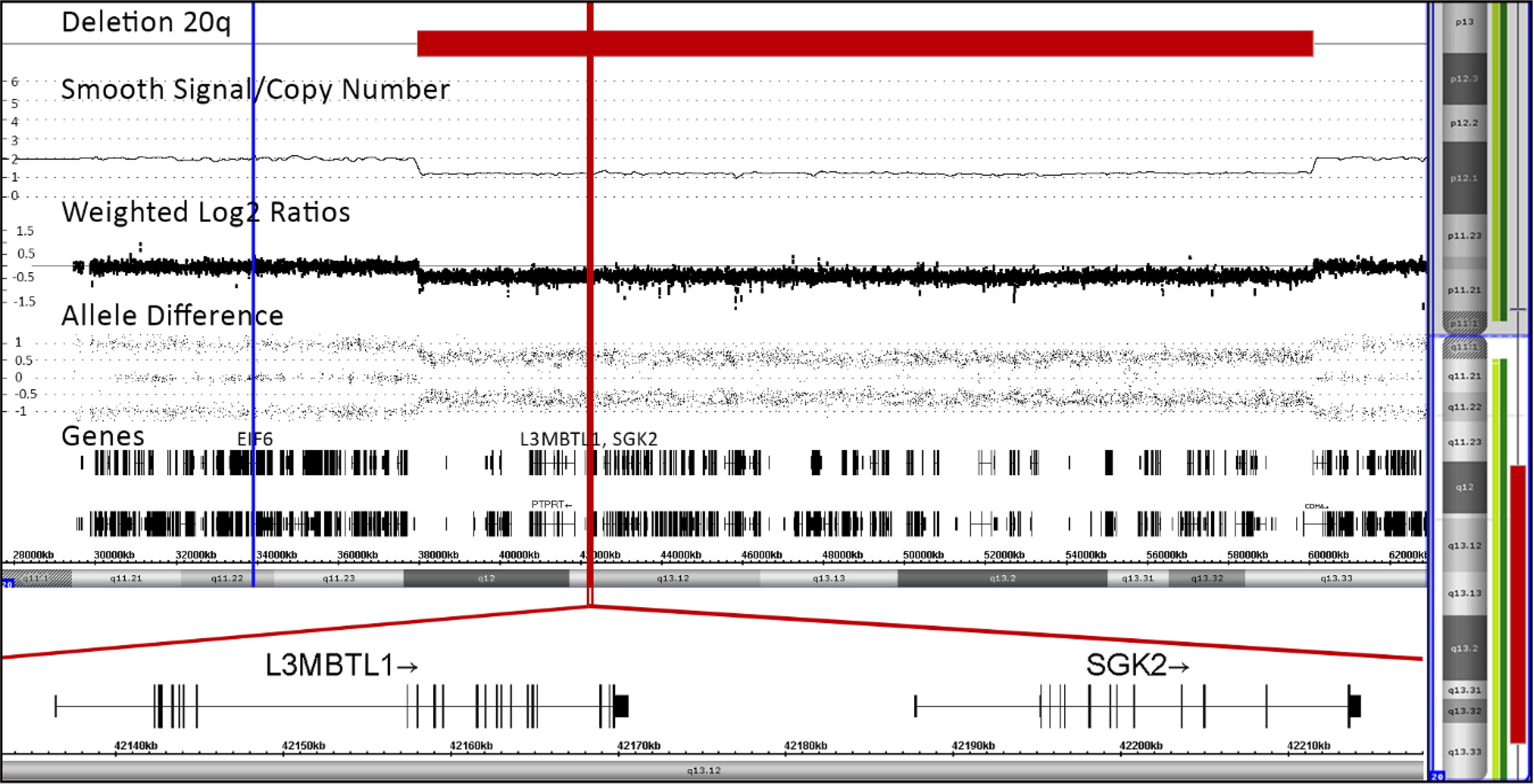
Figure 2 Array pattern of long arm of chromosome 20 showing the interstitial deletion with the following bordering coordinates: chr20:37948298-60071887 (hg19). The deletion extends over 22,124 Mb and encompasses 244 genes, a list of which is provided in the Supplementary Table 2. The location of the three potentially most relevant genes, EIF6, L3MBTL1 and SGK2, are indicated with blue and red lines, respectively (24–28). The orientation and detailed structure of the two imprinted ones in the deletion are shown in the blown-up section on the bottom part of the Figure.
Discussion
The case presented herein is only the second NHEJ1-deficient patient who reached adulthood without transplantation (29). Despite the early onset of a refractory cytopenia with a transient monosomy 7 and the emergence and subsequent expansion of a del(20q) clone many years later, his disease neither progressed into a bona fide myeloid malignancy nor did it require any specific therapeutic care during the now overall 20 year-long observation period. Given that even his genetic condition and his hematologic disease alone would have sufficed to transplant him, the lack of a suitable donor was in retrospect a stroke of luck for both the patient as well as the treating physicians (11, 14, 30, 31).
The NHEJ1 gene encodes one of the components of the principle nonhomologous end-joining repair pathway, whose other constituents are the products of LIG4 (encoding DNA ligase IV), PRKDC (encoding DNA-PKcs), DCLRE1C (encoding Artemis), and XRCC4 (encoding XRCC4) (32–35). This system is not only responsible for the repair of double strand breaks but also for the appropriate execution of V(D)J recombination (32–35). Thus, pathogenic variants in any of these genes increase the radiosensitivity of affected tissues and disturb V(D)J as well as class switch recombination processes. The ensuing problems produce developmental defects in form of a growth delay, microcephaly and dysmorphic facial features as well as various types of (severe) combined immunodeficiencies with differing degrees of B and T cell lymphocytopenia (9, 29, 32–37). In addition, such germline pathogenic variants also predispose affected individuals to the development of autoimmune diseases, lymphomas, bone marrow failure as well as lymphoid and occasionally also myeloid leukemias (9, 34, 38, 39). Although we did not perform any functional assays or radiosensitivity studies, the elevated chromosome breakage in the second DEB test at least provides some evidence that the double strand breakage repair was indeed impaired.
Since the overlapping actions of the PAXX and ATM gene products can to some extent compensate functional impairments of the NHEJ1 protein, one would not expect that it plays such a vital role as, for instance, that of the LIG4-encoded DNA ligase IV (34, 35, 40, 41). Nevertheless, NHEJ1 deficiencies still affect the respective repair and recombination processes quite profoundly, so that the ensuing clinical consequences usually resemble those of the otherwise more severe LIG4 defects (34). Less than 50 cases with bi-allelic NHEJ1 loss-of-function pathogenic variants have so far been documented in the literature (21, 29, 31, 32, 36, 37, 42–44). The heterogeneous phenotypes and variable clinical courses of patients with different but also identical pathogenic variants severely impede any attempts to establish an even only approximate genotype-phenotype relationship, not least also because the effects of the diverse pathogenic variants are also cell type-specific and differentiation stage-dependent (29, 42, 45). Cases in point are, for instance, the progressive lymphocytopenia as well as bone marrow aplasia in some NHEJ1-deficient individuals, which almost certainly can be put down to a premature aging of hematopoietic stem cells (21, 45, 46). This problem is most likely triggered by the inability of the affected stem cells to properly repair continuously accumulating double strand breaks as well as by a pathogenic variant-triggered decrease in telomerase activity that, as also seen in our patient, leads to a gradual loss of telomeres (21, 45, 46). We are aware of altogether five patients with such a bone marrow aplasia, all of whom were transplanted and, all but one, were alive at the time of reporting (21, 31, 32).
Abnormalities of chromosome 7 are the most common acquired genetic changes in childhood myelodysplastic syndromes. They comprise the loss of an entire copy as well as various structural abnormalities in form of deletions, translocations and isochromosomes of its long arm (14, 47). A monosomy 7 is seen in virtually all types of IBMF, but the frequency of its occurrence varies depending on the underlying primary germ line defect (14, 47). The two most common ones are the SAMD9/SAMD9L and GATA2 syndromes (6, 7, 14). Together they account for at least 50% of pediatric MDS with monosomy 7, although the disease emerges primarily in younger children in the former and primarily in adolescent ones in the latter (6, 7, 14). Moreover, monosomy 7 is also the most common alteration in patients with a hypocellular refractory cytopenia, although it is seen in only approximately nine percent of them (11, 12, 30). The only other case that is vaguely comparable to ours is one with a LIG4 germline pathogenic variant and an MDS-related deletion of 7q (48).
The emergence of a monosomy 7 in patients with a hypocellular refractory cytopenia usually concurs with a high probability of disease progression. Nevertheless, in some of the patients the abnormal clone may disappear again and thereby lead to a spontaneous improvement or even disease remission. In the meantime, such transient forms of monosomy 7 are well documented in the literature (13, 49–54). In case of SAMD9/9L-associated disorders, monosomy 7 always results from the nonrandom loss of the homologue that carries the respective SAMD9/9L germline defect (6, 7, 13, 14, 47, 55, 56). These clones may occasionally experience a spontaneous duplication of the remaining homologue that carries the wild-type SAMD9/9L, which will then promote the functional normalization of the bone marrow (6, 7, 13, 14). Since the NHEJ1 gene is located on chromosome 2 rather than on chromosome 7, we neither expected nor detected such a repair process-associated uniparental disomy 7 in the array analyses and conclude that the monosomic clone had no competitive advantage and simply got lost again (6, 7, 14, 51, 52).
The subsequent appearance and gradual increase of another clone with a del(20q), seven and a half years later, concurred with an improvement of his blood counts as well as with the continuous resolution of the erythropoietic dysplasia. A del(20q), either alone or in combination with other chromosome abnormalities, is seen in many different types of myeloid malignancies of all age groups (57, 58). Incidental observations in non-myeloid malignancies and unexplained cytopenia, however, prove that it is not always a bona fide indicator of malignancy and that in such instances the progression to MDS is extremely low (57–59). In children, a solitary del(20q) occurs in an age-dependent manner almost exclusively in those who suffer from a SDS (27, 28, 60–63). With a prevalence of 20% it is also the most common acquired abnormality followed by an isochromosome 7(q10), which is seen in 10% (63). Both these changes may occur either alone, simultaneously, sequentially, or even only transiently, but irrespective of the specific constellation, cases affected by either abnormality hardly ever progress into a genuine myeloid malignancy (28, 63, 64). The cytopenia of SDS patients with a del(20q) remains as stable and the dysplastic alterations as mild as the ones that we observed in our NEJH1-deficient case. The positive influence of a del(20q) on disease development has been put down to the facts that affected totipotent stem cells not only maintain their multipotential differentiation capacity but that, in addition, they also gain a selective advantage (60). However, the impact of such abnormal stem cells in individual settings is virtually impossible to predict, because their destiny is primarily governed by their competitive fitness, which in turn is to a large extent also influenced by a variety of individual host factors and, not least, of course by the type of the preexistent germline defect.
The three genes that are currently in the focus of interest in this context are L3MBTL1 and SGK2, which were lost, and EIF6, which was retained in our case (24, 26, 65). L3MBTL1 and SGK2 are two paternally expressed imprinted genes that encode a transcriptional repressor and a serine/threonine protein kinase, respectively. These genes are in the minimal commonly deleted region of 30 adult cases with a solitary del(20q), but also lost in SDS patients (26, 28, 57, 66). In vitro experiments revealed that their regulatory interactions vary in different hematopoietic lineages and successive stages of differentiation (25, 67, 68). Their coordinated silencing maintains megakaryopoiesis and enhances erythropoiesis but does apparently not equip affected stem and early progenitor cells with any selective advantage (25, 69). Attempts to associate the parental origin of the deletion with their anticipated allele-specific expression patterns produced no clear results and therefore also no coherent picture. Explanations for the various confusing discrepancies ranged from problems that may have arisen from inadequate clone sizes, from admixture of normal cells, from loss of imprinting and, most intriguing, also from the concurrence of two distinct clones, in which one lost the maternal and the other one the paternal allele (25, 26, 67, 69–71). The best clinical, albeit indirect evidence that a clonal del(20q) indeed enhances erythropoiesis derives from polycythemia cases (69). Another notable example relates to the observation that the hemoglobin concentrations and red blood cell counts of SDS patients with a del(20)(q) are higher than those without such a deletion (26).
EIF6, the retained gene, encodes the eukaryotic translation initiation factor 6, which is an essential ribosome chaperone protein (65, 72). To allow the formation of the mature 80S ribosome, this factor must first be released from the pre-60S ribosome subunit by the SBDS protein. Thus, EIF6 inactivating point mutations or deletions become only relevant in patients with preexisting SBDS mutations, in whom they will help to reestablish a normal SBDS : EIF6 protein ratio, which improves the maturation and translational capacity of the ribosomes and consequently also enhance the competitive fitness of the affected hematopoietic cells (65, 72). Deletions of the EIF6 locus in other forms of myeloid malignancies might therefore be functionally irrelevant and merely coincidental.
Taken together, the indolent development of a chromosomally abnormal refractory cytopenia in our patient with an already preexistent severe DNA repair defect is quite remarkable but not a unique phenomenon. Such observations reinforce the growing awareness that the emergence of abnormal clones in IBMF syndromes is not deterministic of malignant transformation. Instead, as we show herein, it can also stabilize and improve the disease process in a quite unexpected manner. The big challenge that derives from this insight is now the need to delineate harmless or even favorable abnormalities from undisputable malignant ones. The definition of even only approximate distinguishing criteria will significantly help to advance clinical decision processes, especially whether and for how long one can rely on a “watch and wait” strategy or whether at all and when one should pursue a more aggressive treatment, such as stem cell transplantation (65, 73–75).
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
Conceptualization and supervision: LK, KB, OAH; manuscript drafting and writing: FP, OAH; methodology: RJH, PZ, KN; data analysis and interpretation, RJH, PZ, KN, OAH, KB; provision of clinical information and patient care: FP, WN, MND, LK; illustrations: RJH, PZ, KN. All authors have read and agreed to the published version of the manuscript.
Conflict of Interest
Authors PZ, KN and OAH were employed by company Labdia, Labordiagnostik.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the patient and his family for their consent to use the information provided herein and to publish their case. The genetic laboratory part was supported by the “Österreichische Kinderkrebsforschung” and a charitable donation of the Kapsch group (http://www.kapsch.net/). We also acknowledge the networking support by the COST Action CA16223 LEukaemia GENe Discovery by data sharing, mining and collaboration (LEGEND) as well as by the IBFM Leukemia & Lymphoma Genetic Predisposition Committee.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.869047/full#supplementary-material
References
1. Kennedy AL, Shimamura A. Genetic Predisposition to MDS: Clinical Features and Clonal Evolution. Blood (2019) 133(10):1071–85. doi: 10.1182/blood-2018-10-844662
2. Sperling AS, Gibson CJ, Ebert BL. The Genetics of Myelodysplastic Syndrome: From Clonal Haematopoiesis to Secondary Leukaemia. Nat Rev Cancer (2017) 17(1):5–19. doi: 10.1038/nrc.2016.112
3. Wilson DB, Link DC, Mason PJ, Bessler M. Inherited Bone Marrow Failure Syndromes in Adolescents and Young Adults. Ann Med (2014) 46(6):353–63. doi: 10.3109/07853890.2014.915579
4. Haas OA. Primary Immunodeficiency and Cancer Predisposition Revisited: Embedding Two Closely Related Concepts Into an Integrative Conceptual Framework. Front Immunol (2019) 12(9):3136. doi: 10.3389/fimmu.2018.03136
5. Oved JH, Babushok DV, Lambert MP, Wolfset N, Kowalska MA, Poncz M, et al. Human Mutational Constraint as a Tool to Understand Biology of Rare and Emerging Bone Marrow Failure Syndromes. Blood Adv (2020) 4(20):5232–45. doi: 10.1182/bloodadvances.2020002687
6. Sahoo SS, Kozyra EJ, Wlodarski MW. Germline Predisposition in Myeloid Neoplasms: Unique Genetic and Clinical Features of GATA2 Deficiency and SAMD9/SAMD9L Syndromes. Best Pract Res Clin Haematol (2020) 33(3):101197. doi: 10.1016/j.beha.2020.101197
7. Sahoo SS, Pastor VB, Goodings C, Voss RK, Kozyra EJ, Szvetnik A, et al. Clinical Evolution, Genetic Landscape and Trajectories of Clonal Hematopoiesis in SAMD9/SAMD9L Syndromes. Nat Med (2021) 27(10):1806–17. doi: 10.1038/s41591-021-01511-6
8. Moreno OM, Paredes AC, Suarez-Obando F, Rojas A. An Update on Fanconi Anemia: Clinical, Cytogenetic and Molecular Approaches (Review). BioMed Rep (2021) 15(3):74. doi: 10.3892/br.2021.1450
9. Sharma R, Lewis S, Wlodarski MW. DNA Repair Syndromes and Cancer: Insights Into Genetics and Phenotype Patterns. Front Pediatr (2020) 23(8):570084. doi: 10.3389/fped.2020.570084
10. Kager L, Jimenez Heredia R, Hirschmugl T, Dmytrus J, Krolo A, Muller H, et al. Targeted Mutation Screening of 292 Candidate Genes in 38 Children With Inborn Haematological Cytopenias Efficiently Identifies Novel Disease-Causing Mutations. Br J Haematol (2018) 182(2):251–8. doi: 10.1111/bjh.15389
11. Kardos G, Baumann I, Passmore SJ, Locatelli F, Hasle H, Schultz KR, et al. Refractory Anemia in Childhood: A Retrospective Analysis of 67 Patients With Particular Reference to Monosomy 7. Blood (2003) 102(6):1997–2003. doi: 10.1182/blood-2002-11-3444
12. Niemeyer CM, Baumann I. Classification of Childhood Aplastic Anemia and Myelodysplastic Syndrome. Hematol Am Soc Hematol Educ Program (2011) 2011:84–9. doi: 10.1182/asheducation-2011.1.84
13. Pastor VB, Sahoo SS, Boklan J, Schwabe GC, Saribeyoglu E, Strahm B, et al. Constitutional SAMD9L Mutations Cause Familial Myelodysplastic Syndrome and Transient Monosomy 7. Haematologica (2018) 103(3):427–37. doi: 10.3324/haematol.2017.180778
14. Wlodarski MW, Sahoo SS, Niemeyer CM. Monosomy 7 in Pediatric Myelodysplastic Syndromes. Hematol Oncol Clin North Am (2018) 32(4):729–43. doi: 10.1016/j.hoc.2018.04.007
15. Seyschab H, Friedl R, Sun Y, Schindler D, Hoehn H, Hentze S, et al. Comparative Evaluation of Diepoxybutane Sensitivity and Cell Cycle Blockage in the Diagnosis of Fanconi Anemia. Blood (1995) 85(8):2233–7. doi: 10.1182/blood.V85.8.2233.bloodjournal8582233
16. Aalbers AM, van der Velden VH, Yoshimi A, Fischer A, Noellke P, Zwaan CM, et al. The Clinical Relevance of Minor Paroxysmal Nocturnal Hemoglobinuria Clones in Refractory Cytopenia of Childhood: A Prospective Study by EWOG-MDS. Leukemia (2014) 28(1):189–92. doi: 10.1038/leu.2013.195
17. de Winter DTC, Langerak AW, Te Marvelde J, Dworzak MN, De Moerloose B, Stary J, et al. The Variable Biological Signature of Refractory Cytopenia of Childhood (RCC), a Retrospective EWOG-MDS Study. Leuk Res (2021) 108:106652. doi: 10.1016/j.leukres.2021.106652
18. Zhang P, Bigio B, Rapaport F, Zhang SY, Casanova JL, Abel L, et al. PopViz: A Webserver for Visualizing Minor Allele Frequencies and Damage Prediction Scores of Human Genetic Variations. Bioinformatics (2018) 34(24):4307–9. doi: 10.1093/bioinformatics/bty536
19. Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: Predicting the Deleteriousness of Variants Throughout the Human Genome. Nucleic Acids Res (2019) 47(D1):D886–94. doi: 10.1093/nar/gky1016
20. Cawthon RM. Telomere Length Measurement by a Novel Monochrome Multiplex Quantitative PCR Method. Nucleic Acids Res (2009) 37(3):e21. doi: 10.1093/nar/gkn1027
21. Carrillo J, Calvete O, Pintado-Berninches L, Manguan-Garcia C, Sevilla Navarro J, Arias-Salgado EG, et al. Mutations in XLF/NHEJ1/Cernunnos Gene Results in Downregulation of Telomerase Genes Expression and Telomere Shortening. Hum Mol Genet (2017) 26(10):1900–14. doi: 10.1093/hmg/ddx098
22. Abbasi MR, Nebral K, Haslinger S, Inthal A, Zeitlhofer P, Konig M, et al. Copy Number Changes and Allele Distribution Patterns of Chromosome 21 in B Cell Precursor Acute Lymphoblastic Leukemia. Cancers (Basel) (2021) 13(18):4597. doi: 10.3390/cancers13184597
23. Cottineau J, Kottemann MC, Lach FP, Kang YH, Vely F, Deenick EK, et al. Inherited GINS1 Deficiency Underlies Growth Retardation Along With Neutropenia and NK Cell Deficiency. J Clin Invest (2017) 127(5):1991–2006. doi: 10.1172/JCI90727
24. Pressato B, Valli R, Marletta C, Mare L, Montalbano G, Lo Curto F, et al. Deletion of Chromosome 20 in Bone Marrow of Patients With Shwachman-Diamond Syndrome, Loss of the EIF6 Gene and Benign Prognosis. Br J Haematol (2012) 157(4):503–5. doi: 10.1111/j.1365-2141.2012.09033.x
25. Aziz A, Baxter EJ, Edwards C, Cheong CY, Ito M, Bench A, et al. Cooperativity of Imprinted Genes Inactivated by Acquired Chromosome 20q Deletions. J Clin Invest (2013) 123(5):2169–82. doi: 10.1172/JCI66113
26. Nacci L, Valli R, Maria Pinto R, Zecca M, Cipolli M, Morini J, et al. Parental Origin of the Deletion Del(20q) in Shwachman-Diamond Patients and Loss of the Paternally Derived Allele of the Imprinted L3MBTL1 Gene. Genes Chromosomes Cancer (2017) 56(1):51–8. doi: 10.1002/gcc.22401
27. Bezzerri V, Cipolli M. Shwachman-Diamond Syndrome: Molecular Mechanisms and Current Perspectives. Mol Diagn Ther (2019) 23(2):281–90. doi: 10.1007/s40291-018-0368-2
28. Valli R, Minelli A, Galbiati M, D'Amico G, Frattini A, Montalbano G, et al. Shwachman-Diamond Syndrome With Clonal Interstitial Deletion of the Long Arm of Chromosome 20 in Bone Marrow: Haematological Features, Prognosis and Genomic Instability. Br J Haematol (2019) 184(6):974–81. doi: 10.1111/bjh.15729
29. Sheikh F, Hawwari A, Alhissi S, Al Gazlan S, Al Dhekri H, Rehan Khaliq AM, et al. Loss of NHEJ1 Protein Due to a Novel Splice Site Mutation in a Family Presenting With Combined Immunodeficiency, Microcephaly, and Growth Retardation and Literature Review. J Clin Immunol (2017) 37(6):575–81. doi: 10.1007/s10875-017-0423-5
30. Locatelli F, Strahm B. How I Treat Myelodysplastic Syndromes of Childhood. Blood (2018) 131(13):1406–14. doi: 10.1182/blood-2017-09-765214
31. Slack J, Albert MH, Balashov D, Belohradsky BH, Bertaina A, Bleesing J, et al. Outcome of Hematopoietic Cell Transplantation for DNA Double-Strand Break Repair Disorders. J Allergy Clin Immunol (2018) 141(1):322–8.e310. doi: 10.1016/j.jaci.2017.02.036
32. Buck D, Malivert L, de Chasseval R, Barraud A, Fondaneche MC, Sanal O, et al. Cernunnos, a Novel Nonhomologous End-Joining Factor, Is Mutated in Human Immunodeficiency With Microcephaly. Cell (2006) 124(2):287–99. doi: 10.1016/j.cell.2005.12.030
33. Du L, Peng R, Bjorkman A, Filipe de Miranda N, Rosner C, Kotnis A, et al. Cernunnos Influences Human Immunoglobulin Class Switch Recombination and may be Associated With B Cell Lymphomagenesis. J Exp Med (2012) 209(2):291–305. doi: 10.1084/jem.20110325
34. Woodbine L, Gennery AR, Jeggo PA. The Clinical Impact of Deficiency in DNA non-Homologous End-Joining. DNA Repair (Amst) (2014) 16:84–96. doi: 10.1016/j.dnarep.2014.02.011
35. Slatter MA, Gennery AR. Update on DNA-Double Strand Break Repair Defects in Combined Primary Immunodeficiency. Curr Allergy Asthma Rep (2020) 20(10):57. doi: 10.1007/s11882-020-00955-z
36. Dutrannoy V, Demuth I, Baumann U, Schindler D, Konrat K, Neitzel H, et al. Clinical Variability and Novel Mutations in the NHEJ1 Gene in Patients With a Nijmegen Breakage Syndrome-Like Phenotype. Hum Mutat (2010) 31(9):1059–68. doi: 10.1002/humu.21315
37. Esmaeilzadeh H, Bordbar MR, Hojaji Z, Habibzadeh P, Afshinfar D, Miryounesi M, et al. An Immunocompetent Patient With a Nonsense Mutation in NHEJ1 Gene. BMC Med Genet (2019) 20(1):45. doi: 10.1186/s12881-019-0784-0
38. Altmann T, Gennery AR. DNA Ligase IV Syndrome; a Review. Orphanet J Rare Dis (2016) 11(1):137. doi: 10.1186/s13023-016-0520-1
39. Staines Boone AT, Chinn IK, Alaez-Verson C, Yamazaki-Nakashimada MA, Carrillo-Sanchez K, Garcia-Cruz MLH, et al. Failing to Make Ends Meet: The Broad Clinical Spectrum of DNA Ligase IV Deficiency. Case Series and Review of the Literature. Front Pediatr (2018) 6:426. doi: 10.3389/fped.2018.00426
40. Kumar V, Alt FW, Frock RL. PAXX and XLF DNA Repair Factors are Functionally Redundant in Joining DNA Breaks in a G1-Arrested Progenitor B-Cell Line. Proc Natl Acad Sci USA (2016) 113(38):10619–24. doi: 10.1073/pnas.1611882113
41. Lescale C, Lenden Hasse H, Blackford AN, Balmus G, Bianchi JJ, Yu W, et al. Specific Roles of XRCC4 Paralogs PAXX and XLF During V(D)J Recombination. Cell Rep (2016) 16(11):2967–79. doi: 10.1016/j.celrep.2016.08.069
42. Recio MJ, Dominguez-Pinilla N, Perrig MS, Rodriguez Vigil-Iturrate C, Salmon-Rodriguez N, Martinez Faci C, et al. Extreme Phenotypes With Identical Mutations: Two Patients With Same Non-Sense NHEJ1 Homozygous Mutation. Front Immunol (2018) 9:2959. doi: 10.3389/fimmu.2018.02959
43. Firtina S, Yin Ng Y, Hatirnaz Ng O, Kiykim A, Aydiner E, Nepesov S, et al. Mutational Landscape of Severe Combined Immunodeficiency Patients From Turkey. Int J Immunogenet (2020) 47(6):529–38. doi: 10.1111/iji.12496
44. Vignesh P, Rawat A, Kumrah R, Singh A, Gummadi A, Sharma M, et al. Clinical, Immunological, and Molecular Features of Severe Combined Immune Deficiency: A Multi-Institutional Experience From India. Front Immunol (2020) 11:619146. doi: 10.3389/fimmu.2020.619146
45. Tilgner K, Neganova I, Singhapol C, Saretzki G, Al-Aama JY, Evans J, et al. Brief Report: A Human Induced Pluripotent Stem Cell Model of Cernunnos Deficiency Reveals an Important Role for XLF in the Survival of the Primitive Hematopoietic Progenitors. Stem Cells (2013) 31(9):2015–23. doi: 10.1002/stem.1456
46. Avagyan S, Churchill M, Yamamoto K, Crowe JL, Li C, Lee BJ, et al. Hematopoietic Stem Cell Dysfunction Underlies the Progressive Lymphocytopenia in XLF/Cernunnos Deficiency. Blood (2014) 124(10):1622–5. doi: 10.1182/blood-2014-05-574863
47. Schwartz JR, Ma J, Lamprecht T, Walsh M, Wang S, Bryant V, et al. The Genomic Landscape of Pediatric Myelodysplastic Syndromes. Nat Commun (2017) 8(1):1557. doi: 10.1038/s41467-017-01590-5
48. Zhang MY, Keel SB, Walsh T, Lee MK, Gulsuner S, Watts AC, et al. Genomic Analysis of Bone Marrow Failure and Myelodysplastic Syndromes Reveals Phenotypic and Diagnostic Complexity. Haematologica (2015) 100(1):42–8. doi: 10.3324/haematol.2014.113456
49. Bluteau O, Sebert M, Leblanc T, Peffault de Latour R, Quentin S, Lainey E, et al. A Landscape of Germ Line Mutations in a Cohort of Inherited Bone Marrow Failure Patients. Blood (2018) 131(7):717–32. doi: 10.1182/blood-2017-09-806489
50. Jawad MD, Go RS, Ketterling RP, Begna KH, Reichard KK, Shi M. Transient Monosomy 7 in a Chronic Myelogenous Leukemia Patient During Nilotinib Therapy: A Case Report. Clin Case Rep (2016) 4(3):282–6. doi: 10.1002/ccr3.506
51. Mantadakis E, Shannon KM, Singer DA, Finklestein J, Chan KW, Hilden JM, et al. Transient Monosomy 7: A Case Series in Children and Review of the Literature. Cancer (1999) 85(12):2655–61. doi: 10.1002/(sici)1097-0142(19990615)85:12<2655::aid-cncr23>3.0.co;2-w
52. Parker TM, Klaassen RJ, Johnston DL. Spontaneous Remission of Myelodysplastic Syndrome With Monosomy 7 in a Young Boy. Cancer Genet Cytogenet (2008) 182(2):122–5. doi: 10.1016/j.cancergencyto.2008.01.003
53. Sevilla J, Querol S, Molines A, Gonzalez-Vicent M, Balas A, Carrio A, et al. Transient Donor Cell-Derived Myelodysplastic Syndrome With Monosomy 7 After Unrelated Cord Blood Transplantation. Eur J Haematol (2006) 77(3):259–63. doi: 10.1111/j.1600-0609.2006.00716.x
54. Stieglitz E, Loh ML. Genetic Predispositions to Childhood Leukemia. Ther Adv Hematol (2013) 4(4):270–90. doi: 10.1177/2040620713498161
55. Buonocore F, Kuhnen P, Suntharalingham JP, Del Valle I, Digweed M, Stachelscheid H, et al. Somatic Mutations and Progressive Monosomy Modify SAMD9-Related Phenotypes in Humans. J Clin Invest (2017) 127(5):1700–13. doi: 10.1172/JCI91913
56. Tesi B, Davidsson J, Voss M, Rahikkala E, Holmes TD, Chiang SCC, et al. Gain-Of-Function SAMD9L Mutations Cause a Syndrome of Cytopenia, Immunodeficiency, MDS, and Neurological Symptoms. Blood (2017) 129(16):2266–79. doi: 10.1182/blood-2016-10-743302
57. Bacher U, Haferlach T, Schnittger S, Zenger M, Meggendorfer M, Jeromin S, et al. Investigation of 305 Patients With Myelodysplastic Syndromes and 20q Deletion for Associated Cytogenetic and Molecular Genetic Lesions and Their Prognostic Impact. Br J Haematol (2014) 164(6):822–33. doi: 10.1111/bjh.12710
58. Ravindran A, He R, Ketterling RP, Jawad MD, Chen D, Oliveira JL, et al. The Significance of Genetic Mutations and Their Prognostic Impact on Patients With Incidental Finding of Isolated Del(20q) in Bone Marrow Without Morphologic Evidence of a Myeloid Neoplasm. Blood Cancer J (2020) 10(1):7. doi: 10.1038/s41408-020-0275-8
59. Martin I, Villamon E, Abellan R, Calasanz MJ, Irigoyen A, Sanz G, et al. Myelodysplastic Syndromes With 20q Deletion: Incidence, Prognostic Value and Impact on Response to Azacitidine of ASXL1 Chromosomal Deletion and Genetic Mutations. Br J Haematol (2021) 194(4):708–17. doi: 10.1111/bjh.17675
60. Crescenzi B, La Starza R, Sambani C, Parcharidou A, Pierini V, Nofrini V, et al. Totipotent Stem Cells Bearing Del(20q) Maintain Multipotential Differentiation in Shwachman Diamond Syndrome. Br J Haematol (2009) 144(1):116–9. doi: 10.1111/j.1365-2141.2008.07448.x
61. Maserati E, Pressato B, Valli R, Minelli A, Sainati L, Patitucci F, et al. The Route to Development of Myelodysplastic Syndrome/Acute Myeloid Leukaemia in Shwachman-Diamond Syndrome: The Role of Ageing, Karyotype Instability, and Acquired Chromosome Anomalies. Br J Haematol (2009) 145(2):190–7. doi: 10.1111/j.1365-2141.2009.07611.x
62. Burroughs L, Woolfrey A, Shimamura A. Shwachman-Diamond Syndrome: A Review of the Clinical Presentation, Molecular Pathogenesis, Diagnosis, and Treatment. Hematol Oncol Clin North Am (2009) 23(2):233–48. doi: 10.1016/j.hoc.2009.01.007
63. Furutani E, Liu S, Galvin A, Steltz S, Malsch MM, Loveless SK, et al. Hematologic Complications With Age in Shwachman-Diamond Syndrome. Blood Adv (2021) 6(1):297-306. doi: 10.1182/bloodadvances.2021005539
64. Dror Y, Durie P, Ginzberg H, Herman R, Banerjee A, Champagne M, et al. Clonal Evolution in Marrows of Patients With Shwachman-Diamond Syndrome: A Prospective 5-Year Follow-Up Study. Exp Hematol (2002) 30(7):659–69. doi: 10.1016/s0301-472x(02)00815-9
65. Kennedy AL, Myers KC, Bowman J, Gibson CJ, Camarda ND, Furutani E, et al. Distinct Genetic Pathways Define Pre-Malignant Versus Compensatory Clonal Hematopoiesis in Shwachman-Diamond Syndrome. Nat Commun (2021) 12(1):1334. doi: 10.1038/s41467-021-21588-4
66. Khan AW, Kennedy A, Furutani E, Myers K, Frattini A, Acquati F, et al. The Frequent and Clinically Benign Anomalies of Chromosomes 7 and 20 in Shwachman-Diamond Syndrome may be Subject to Further Clonal Variations. Mol Cytogenet (2021) 14(1):54. doi: 10.1186/s13039-021-00575-w
67. Bench AJ, Li J, Huntly BJ, Delabesse E, Fourouclas N, Hunt AR, et al. Characterization of the Imprinted Polycomb Gene L3MBTL, a Candidate 20q Tumour Suppressor Gene, in Patients With Myeloid Malignancies. Br J Haematol (2004) 127(5):509–18. doi: 10.1111/j.1365-2141.2004.05278.x
68. Benetatos L, Vartholomatos G. Imprinted Genes in Myeloid Lineage Commitment in Normal and Malignant Hematopoiesis. Leukemia (2015) 29(6):1233–42. doi: 10.1038/leu.2015.47
69. Perna F, Gurvich N, Hoya-Arias R, Abdel-Wahab O, Levine RL, Asai T, et al. Depletion of L3MBTL1 Promotes the Erythroid Differentiation of Human Hematopoietic Progenitor Cells: Possible Role in 20q- Polycythemia Vera. Blood (2010) 116(15):2812–21. doi: 10.1182/blood-2010-02-270611
70. Li J, Bench AJ, Vassiliou GS, Fourouclas N, Ferguson-Smith AC, Green AR. Imprinting of the Human L3MBTL Gene, a Polycomb Family Member Located in a Region of Chromosome 20 Deleted in Human Myeloid Malignancies. Proc Natl Acad Sci USA (2004) 101(19):7341–6. doi: 10.1073/pnas.0308195101
71. Schaub FX, Jager R, Looser R, Hao-Shen H, Hermouet S, Girodon F, et al. Clonal Analysis of Deletions on Chromosome 20q and JAK2-V617F in MPD Suggests That Del20q Acts Independently and Is Not One of the Predisposing Mutations for JAK2-V617F. Blood (2009) 113(9):2022–7. doi: 10.1182/blood-2008-07-167056
72. Tan S, Kermasson L, Hilcenko C, Kargas V, Traynor D, Boukerrou AZ, et al. Somatic Genetic Rescue of a Germline Ribosome Assembly Defect. Nat Commun (2021) 12(1):5044. doi: 10.1038/s41467-021-24999-5
73. Tsai FD, Lindsley RC. Clonal Hematopoiesis in the Inherited Bone Marrow Failure Syndromes. Blood (2020) 136(14):1615–22. doi: 10.1182/blood.2019000990
74. Gutierrez-Rodrigues F, Sahoo SS, Wlodarski MW, Young NS. Somatic Mosaicism in Inherited Bone Marrow Failure Syndromes. Best Pract Res Clin Haematol (2021) 34(2):101279. doi: 10.1016/j.beha.2021.101279
Keywords: NHEJ1, refractory cytopenia, monosomy 7, del(20q), myelodysplastic syndrome, NHEJ1-deficiency
Citation: Poyer F, Jimenez Heredia R, Novak W, Zeitlhofer P, Nebral K, Dworzak MN, Haas OA, Boztug K and Kager L (2022) Case Report: Refractory Cytopenia With a Switch From a Transient Monosomy 7 to a Disease-Ameliorating del(20q) in a NHEJ1-Deficient Long-term Survivor. Front. Immunol. 13:869047. doi: 10.3389/fimmu.2022.869047
Received: 03 February 2022; Accepted: 20 May 2022;
Published: 24 June 2022.
Edited by:
David Buchbinder, Children’s Hospital of Orange County, United StatesReviewed by:
Masatoshi Takagi, Tokyo Medical and Dental University, JapanThomas F. Michniacki, University of Michigan, United States
Copyright © 2022 Poyer, Jimenez Heredia, Novak, Zeitlhofer, Nebral, Dworzak, Haas, Boztug and Kager. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oskar A. Haas, b3NrYXIuaGFhc0BsYWJkaWEuYXQ=; Kaan Boztug, a2Fhbi5ib3p0dWdAY2NyaS5hdA==; Leo Kager, bGVvLmthZ2VyQHN0YW5uYS5hdA==
†These authors have contributed equally to this work and share last authorship