
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 17 May 2022
Sec. Primary Immunodeficiencies
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.869033
This article is part of the Research TopicDeciphering the Landscape of Immunohematology: Enhancing our Understanding and Management of Hematological Disorders Through Advances in Immunology and GeneticsView all 15 articles
 Maurizio Miano1*†
Maurizio Miano1*† Daniela Guardo1†
Daniela Guardo1† Alice Grossi2
Alice Grossi2 Elena Palmisani1
Elena Palmisani1 Francesca Fioredda1
Francesca Fioredda1 Paola Terranova1Enrico Cappelli1Michela Lupia1
Paola Terranova1Enrico Cappelli1Michela Lupia1 Monica Traverso3
Monica Traverso3 Gianluca Dell’Orso4Fabio Corsolini5
Gianluca Dell’Orso4Fabio Corsolini5 Andrea Beccaria1Marina Lanciotti1
Andrea Beccaria1Marina Lanciotti1 Isabella Ceccherini2‡
Isabella Ceccherini2‡ Carlo Dufour1‡
Carlo Dufour1‡Background: Evans syndrome (ES) is a rare disorder classically defined as the simultaneous or sequential presence of autoimmune haemolytic anaemia and immune thrombocytopenia, but it has also been described as the presence of at least two autoimmune cytopenias. Recent reports have shown that ES is often a manifestation of an underlying inborn error of immunity (IEI) that can benefit from specific treatments.
Aims: The aim of this study is to investigate the clinical and immunological characteristics and the underlying genetic background of a single-centre cohort of patients with ES.
Methods: Data were obtained from a retrospective chart review of patients with a diagnosis of ES followed in our centre. Genetic studies were performed with NGS analysis of 315 genes related to both haematological and immunological disorders, in particular IEI.
Results: Between 1985 and 2020, 40 patients (23 men, 17 women) with a median age at onset of 6 years (range 0–16) were studied. ES was concomitant and sequential in 18 (45%) and 22 (55%) patients, respectively. Nine of the 40 (8%) patients had a positive family history of autoimmunity. Other abnormal immunological features and signs of lymphoproliferation were present in 24/40 (60%) and 27/40 (67%) of cases, respectively. Seventeen out of 40 (42%) children fit the ALPS diagnostic criteria. The remaining 21 (42%) and 2 (5%) were classified as having an ALPS-like and an idiopathic disease, respectively. Eighteen patients (45%) were found to have an underlying genetic defect on genes FAS, CASP10, TNFSF13B, LRBA, CTLA4, STAT3, IKBGK, CARD11, ADA2, and LIG4. No significant differences were noted between patients with or without variant and between subjects with classical ES and the ones with other forms of multilineage cytopenias.
Conclusions: This study shows that nearly half of patients with ES have a genetic background being in most cases secondary to IEI, and therefore, a molecular evaluation should be offered to all patients.
Evans syndrome (ES) is a rare disorder classically defined by the concomitant or sequential presence of autoimmune haemolytic anaemia (AIHA) and immune thrombocytopenia (ITP) (1–3), but it is also described as cytopenia due to the immune-mediated destruction of at least two blood cells lineages (4–6). It can be either idiopathic or secondary to other conditions, such as infections, inborn errors of immunity (IEI), autoimmune and rheumatologic diseases, malignancies, and drugs.
In paediatric age, IEI and in particular primary immuno-regulatory disorders (PIRDs) play a relevant role in the development of autoimmune cytopenias (7–9). In the first reported paediatric cohort of ES, about 10% of patients were identified as having IEI (10). Since then, the increased use of techniques like next-generation sequencing (NGS) and whole exome sequence (WES) revealed closer relationships between ES and PIRDS as autoimmune lymphoproliferative syndrome (ALPS), ALPS-like disorders, and common variable immunodeficiency (CVID) that often present with autoimmune cytopenias (AC) (10–13), and outlined the role of variants on the clinical phenotype of ES. This was clearly shown by two recent studies. The first found seven pathogenic variants on CTLA4, LRBA, STAT3, and KRAS genes in a cohort of 18 children with ES (14), whereas the second identified an underlying genetic defect in 65% of cases and showed that patients carrying variants displayed a more severe disease and required more lines of treatment versus the ones without (15). Similar findings came from another multicentre study on 60 children where underlying immune dysregulation was detected in 42% of cases (6). This is relevant because the detection of specific monogenic defects may not only address a correct diagnosis, but also enable the use of targeted therapies (4, 16–18).
However, in the above mentioned large, multicentre studies, genetic analysis was not offered to all eligible patients but was performed according to the request of the attending physicians. In addition, financial access to molecular analysis may have somehow affected the prevalence and the type of underlying disorders (6, 14, 15). Moreover, the issue of the potential genetic and immunological differences between classical ES (association of AIHA and ITP) and other forms of multilineage cytopenia was not addressed.
The aim of this study is to evaluate the genetic background and the clinical/immunological features of a single-centre cohort of paediatric patients with ES and other multilineage cytopenias.
The clinical charts of all paediatric patients affected with classical ES and other multilineage cytopenias referred to our Unit between 1985 and 2020, identified via a clinical database, were reviewed.
AIHA was defined as the presence of haemolytic anaemia, a positive DAT, and the absence of other hereditary or acquired causes of haemolysis (16–18). AIN was defined as neutropenia due to the presence of indirect anti-neutrophil antibodies (19). ITP was defined as isolated thrombocytopenia (peripheral blood platelet count < 100,000 × 109/l) in the absence of other causes or disorders that may be associated (20, 21).
Patients presenting with the association of AIHA and ITP with or without autoimmune neutropenia (AIN) were defined as having a classical ES. Children suffering from AIHA and AIN or ITP and AIN were considered as having a multilineage autoimmune cytopenia (MAC). ALPS was defined according to the revised diagnostic criteria by Oliveira et al. (22) which needs the presence of two required criteria in addition to a primary or secondary accessory criterion to state a definitive and probable diagnosis, respectively. Patients with both definitive and probable diagnoses were considered in the ALPS group of our cohort. Patients who did not completely fulfil the ALPS diagnosis but, in addition to cytopenia, presented with at least one required or primary additional criterion of ALPS diagnostic criteria were classified as having an ALPS-like disorder. Patients without diagnostic criteria of ALPS, ALPS-like, or any other underlying systemic disorder were considered as having a primary disease. Lymphoproliferation was defined as the presence of chronic (>6 months), non-malignant, non-infectious lymphadenopathy, hepatomegaly, or splenomegaly. Other immune abnormalities were defined as the presence of any of the following: inflammatory bowel diseases, autoimmune hepatitis, autoimmune thyroiditis, celiac disease, and auto-antibody positivity (ANA, ENA, ASMA, ASCA, ANCA, anti-ADAMTS13, anti-parietal, anti-dsDNA, anti-SSA/Ro, or LAC).
Data on demographics, clinical features, laboratory and immunological findings, management, and outcome were collected.
All adult subjects provided written informed consent to participate to this study, while parental consent was obtained for children, as approved by the Istituto Gaslini Ethical Committee. The study and all analyses conformed to the 1975 Declaration of Helsinki. Novel variants reported here for the first time have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and assigned accession number SCV001424053-SCV001424130.
Peripheral lymphocyte subsets were evaluated from whole blood using an eight-colour immunostaining panel (lyse and wash procedure), a FACSCanto II flow cytometer (BD, Franklin Lakes, NJ, USA) equipped with three lasers (blue, red, violet), FACSDiva™ software (BD), and a large panel of RUO monoclonal antibodies and fluorochromes variously combined (all BD). CD3+CD4-CD8-TCRαβ+ T cells (double-negative T cells, DNTs) were calculated on total lymphocytes. An in vitro FAS-induced apoptosis test was considered pathological when positive in two separate assays.
DNA was isolated from peripheral blood samples of patients and their parents, when available, and tested for a selected list of genes through an NGS-based gene panel already reported (23). Sample library preparation and sequencing, and successive bioinformatics analyses, including variant annotation and interpretation, were carried out as described in Grossi et al. (23). The effect of variants was classified according with American College of Medical Genetics ACMG criteria (24) which are implemented in the VarSome database (www.varsome.com). In particular, the family segregation, population frequencies, functional prediction, and zygosity were taken into consideration.
Relevant variants were confirmed by polymerase chain reaction (PCR) amplification and direct Sanger sequencing of the corresponding DNA segments. X-inactivation analysis was performed using peripheral blood genomic DNA undigested or digested with restriction endonucleases sensitive to cytosine methylation (HpaII). PCR was carried out using two primers flanking the STR in the HUMARA gene. The PCR products were run on ABI PRISM 3130 (Applied Biosystems, Foster City, CA, USA). The ratio of active/inactive X chromosome was determined as described by Bolduc et al. (25).
Continuous variables were described as the median (range), and categorical variables were described as number (percentage). Quantitative variables were analysed using the χ2 test or Fisher exact test for small quantities. Differences between variables of various groups were considered significant when the P value was ≤ 0.05.
Forty patients (23 men, 17 women) with a median age at onset of 6 years (range 0–16) were studied. Twenty-two (55%) and 18/40 (45%) presented with ES and MAC, respectively. Seventeen out of 40 (43%) children met the ALPS diagnostic criteria. The remaining 18 (45%), 3 (7%), and 2 (5%) were classified as having an ALPS-like phenotype, CVID, and idiopathic cytopenia, respectively. Nine of the 40 (8%) patients had a positive family history of autoimmunity. Other immune abnormalities and signs of lymphoproliferation were present in 24/40 (60%) and 27/40 (67%) of cases, respectively. All patients but one required second- or further-line treatments which included mycophenolate mofetil (MMF) and sirolimus or both in 27 (67%), 18 (45%), and 14 (35%) cases, respectively.
The classical ES and MAC groups did not differ for any of the tested variables including family history of autoimmunity ALPS and ALPS-like/CVID phenotype with the exception of other immune abnormalities that were significantly more frequent (P = 0.03) in patients with MAC over those with classical ES. A trend of females to prevail in classical ES vs. MAC, without reaching statistical significance, was also observed (Table 1).

Table 1 Clinical and immunological differences according to type of cytopenia.
Genetic analysis was performed in all 40 patients, and variants were found in 18 (45%) of them. Variants were pathogenic/likely pathogenic in 13 and of unknown significance in 5 subjects. Patient carrying variants were not differently distributed in the classical ES and MAC groups. All variants defined as pathogenic were previously described or functionally validated (26, 27). Of note, they were all related to PIRDs and IEI. In particular, in 5/18 (28%) patients the gene was involved in the pathogenesis of ALPS (4 FAS, 1CASP10) and in 7/18 (39%) subject variants were implicated in ALPS-like disorders (4 TNFSF13B, 1 LRBA, 1 CTLA4, 1 STAT3). The remaining 6/18 cases (33%) were found to have an impairment in genes related to other IEI. Overall, genetic variants were found in 10/18 (55%) of patients with ALPS. Tables 2, 3 show the clinical/immunological details and the molecular results of patients carrying genetic variants, and the characteristics of the remaining cases are reported in Table 4.
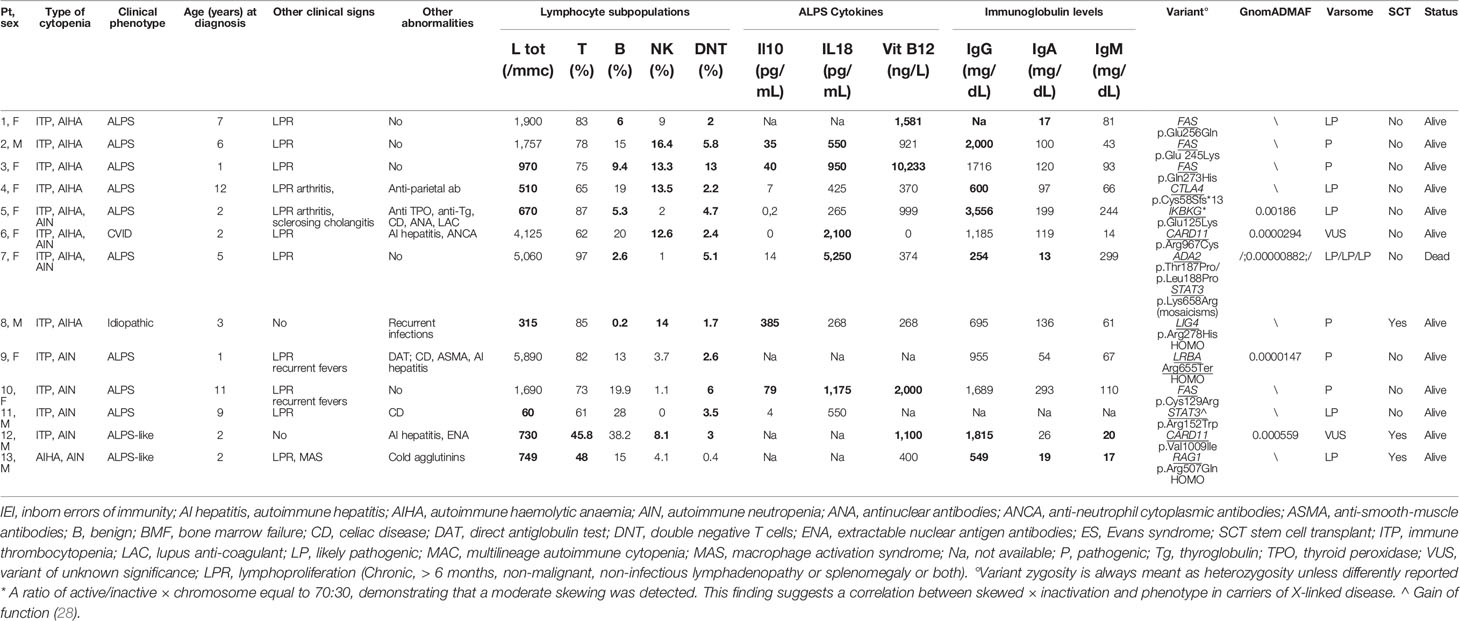
Table 2 Clinical/immunological characteristics of patients carrying pathogenic/likely pathogenic variants related t o IEI (abnormal results in bold).
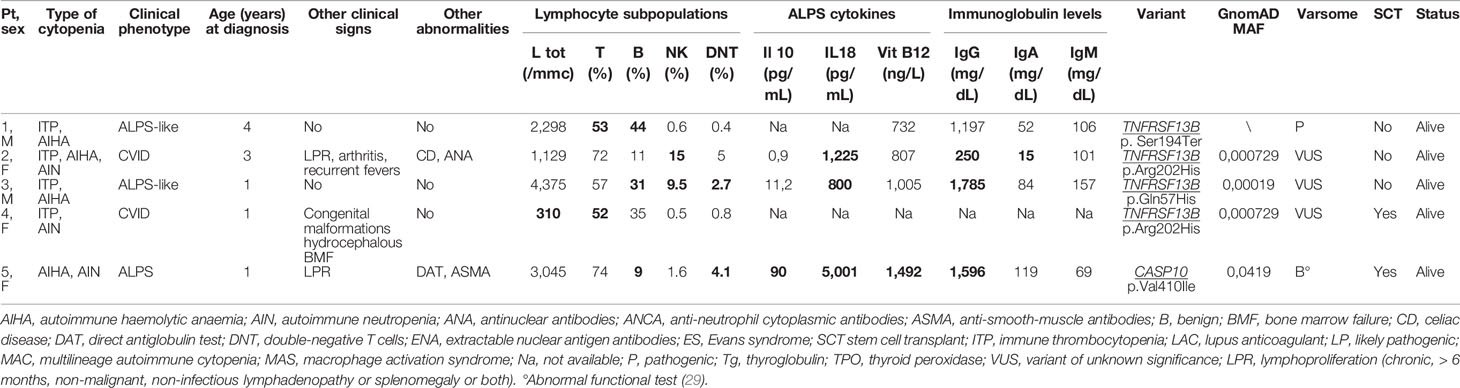
Table 3 Clinical/immunological characteristics of patients carrying pathogenic or of unknown significance variants related to risk factors for immune-dysregulation (abnormal results in bold).
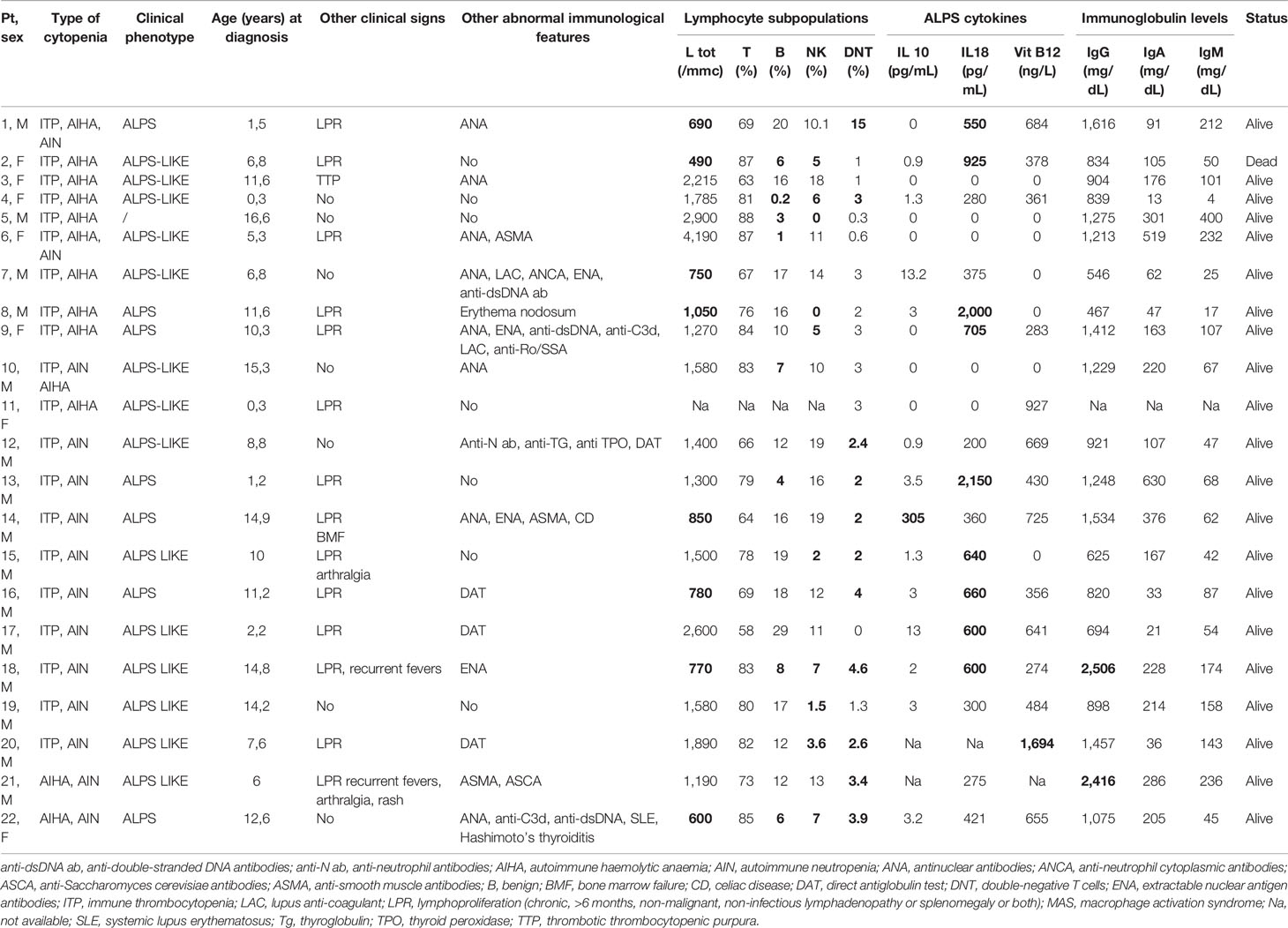
Table 4 Clinical/immunological characteristics of patients without genetic variant (abnormal results in bold).
In order to see whether there were differences between the patients carrying variants versus those who did not, we divided the whole cohort in these two groups. We did not find any significant difference for any of the tested variables (Table 5). However, although not reaching statistical significance, a greater need of second-line therapies was observed in the patients with variants (14/18, 78%) compared with the ones without (14/22, 64%). In line with this, patients carrying variants were also the only ones who required further treatment with stem cell transplantation (SCT): five of them were transplanted from haploidentical (3), sibling (1), and unrelated (1) donor, respectively, and are all alive and well at a median of 4.6 years after the procedure. Two patients (5%) died due to complication of the diseases. The median follow-up was 6.9 years (0,3–35).
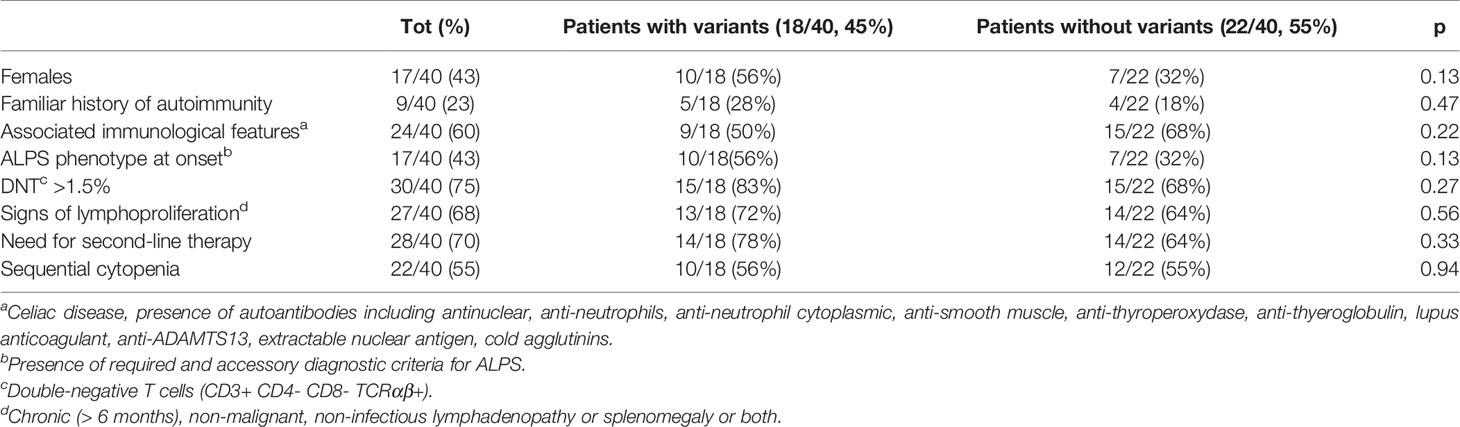
Table 5 Clinical and immunological differences according to variants.
Paediatric Evans syndrome is a very rare and challenging disease. To the best of our knowledge, this study reports on the largest monocentric cohort of children, homogenously screened with molecular analysis, and confirms the presence of underlying disorders in a considerable number of cases, highlighting the important role of genetic assessment in this setting of patients. This holds especially true in the case of IEI that are known to have overlapping phenotypes due to the incomplete penetrance of genetic defects and other still unknown epigenetic factors (9, 11).
Previous multicentric studies have already shown the presence of underlying immune dysregulation in most ES patients. However, in these studies, differently from ours in which all patients were genetically tested, patients underwent genetic analysis based on a prescription of the attending physician, on the availability of the diagnostic tools over the years, on financial access to analysis, and on the severity of the disease (6, 14, 15). This might have generated a selected cohort of patients that were more likely to show an underlying defect and might explain the lower detection rate in our patients (48%) compared to that of the larger multicentric French cohort (68%) (15). In addition, our results were obtained using NGS gene panel analysis, thus implying that a deeper investigation with WES or WGS (whole genome sequencing) might have increased the detection rate. Such further analysis should be taken into consideration in patients with negative NGS results, since the presence of other immuno-pathological manifestations in the majority of our patients is in keeping with the idea that both ES and MAC represent an epiphenomenon of some still unknown IEI.
As expected, most of the variants found in our cohort were involved in genes causing CVID, ALPS, or ALPS-like syndromes. Interestingly, patients carrying CTLA4 (27) and DADA2 (26) variants initially presented with an exclusive haematological phenotype which was followed by more typical signs of their diseases during adolescence/adulthood, highlighting that, in the paediatric age, cytopenia can be the first sign of a more complex disease which can show its complete phenotype later in life. In this respect, the DADA2 patient’s peripheral cytopenia worsened due to the occurrence of severe bone marrow failure. The coexistence of immune-mediated destruction of blood cells and marrow failure was also shown in both patients (Pt 9 and Pt 16) carrying two very rare variants of unknown significance on the CARD11 gene, described to be damaging in most scores and also included in the GUK (guanylate kinase-like) domain, critical for protein’s function, where other pathogenic variants have been reported. The phenomenon of the immune-mediated interplay between bone marrow and peripheral blood in the pathogenesis of cytopenia in IEI has already been highlighted by our group (30) and has clinical implications since these patients deserve a particular alert and specific follow-up.
The pathogenic variant IKBKG was found in a female presenting with isolated ES during childhood and showing further signs of immune dysregulation during adolescence. The abnormal chromosome X inactivation documented in our female patients carrying a X-linked disorder—known to be the cause of incontinentia pigmenti, ectodermal dysplasia, and immunodeficiency—may explain the milder phenotype. In addition, also the different degree of the protein impairment (NF-kappa-B essential modulator—NEMO), which may depend on the type of variant, may have contributed to the milder clinical issues (31, 32).
As expected, most patients showed higher levels of DNTs, which are well known to be raised not only in patients with ALPS but also in association with ALPS-like or CVID phenotypes (33). This indicates that increased DNTs may represent an important initial screening tool for patients with ES and MAC that can address subsequent specific immunological investigations (4, 34–36).
No clinical and immunological differences were noted comparing patients with or without a genetic diagnosis. Similarly, apart from a slight—although not significant—female predominance in patients with ES and a statistically significant higher presence of immune abnormalities in MAC cases, we did not notice other differences between both groups. This strengthens the concept that multilineage cytopenia can be an epiphenomenon of an underlying disorder, regardless of the involved cell line. Nonetheless, the several additional manifestations of autoimmunity and of immune dysregulation noted in patients with MAC may reflect the more heterogeneous genetic background we found in this group.
As already reported in the French cohort (15), most patients needed second- or further-line immunosuppressive therapies which, in most cases, were successful. In fact, treatments as mycophenolate mofetil and sirolimus, well known to be effective in autoimmune cytopenias (37, 38), represent an appropriate approach for patients non-responding to steroids. Nonetheless, the identification of specific molecular defects, following a proper genetic screening, may lead to the administration of targeted therapies, as in the case of one patient of our cohort, affected with CTLA4 haploinsufficiency, who was successfully treated with abatacept (27). Few non-responding patients—all with a demonstrated underlying defect—successfully underwent SCT (39).
In conclusion, both ES and MAC should be considered an epiphenomenon of underlying IEI which are detected in about half of patients. Therefore, in these cases, genetic screening has to be considered a fundamental step of the diagnostic work-up that should be offered to all patients who may potentially benefit from specific follow-up and treatments.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found as follows: https://www.ncbi.nlm.nih.gov/clinvar/, SCV001424053-SCV001424130.
The studies involving human participants were reviewed and approved by Comitato etico Regione Liguria. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
MM and DG designed the research and wrote the paper. AG, MaL, MT, and IC performed the genetic analysis. PT, EC, and MiL performed the laboratory assays and functional studies. EP, FF, GO, and AB contributed the clinical data. CD coordinated the research and revised the manuscript. All authors contributed to the article and approved the submitted version.
We acknowledge ERG S.p.A., Rimorchiatori Riuniti (Genoa), Cambiaso Risso Marine (Genoa), Saar Depositi Oleari Portuali (Genoa), ONLUS Nicola Ferrari, and Ministero della Salute-Ricerca corrente 2021 for supporting the activity of Hematology Unit of IRCCS Istituto Giannina Gaslini.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Dr. Ubaldo Rosati and Dr. Cristina Arduino are acknowledged for supporting the research activity of Heamtology Unit.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.869033/full#supplementary-material
1. Wang WC. Evans Syndrome in Childhood: Pathophysiology, Clinical Course, and Treatment. Am J Pediatr Hematol Oncol (1988) 10:330–8. doi: 10.1097/00043426-198824000-00013
2. Mathew P, Chen G, Wang W. Evans Syndrome: Results of a National Survey. J Pediatr Hematol Oncol (1997) 19:433–7. doi: 10.1097/00043426-199709000-00005
3. Savaşan S, Warrier I, Ravindranath Y. The Spectrum of Evans' Syndrome. Arch Dis Child (1997) 77:245–8. doi: 10.1136/adc.77.3.245
4. Miano M. How I Manage Evans Syndrome and AIHA Cases in Children. Br J Haematol (2016) 172:524–34. doi: 10.1111/bjh.13866
5. Rivalta B, Zama D, Pancaldi G, Facchini E, Cantarini ME, Miniaci A, et al. Evans Syndrome in Childhood: Long Term Follow-Up and the Evolution in Primary Immunodeficiency or Rheumatological Disease. Front Pediatr (2019) 7:304. doi: 10.3389/fped.2019.00304
6. Grimes AB, Kim TO, Kirk SE, Flanagan J, Lambert MP, Grace RF, et al. Refractory Autoimmune Cytopenias in Pediatric Evans Syndrome With Underlying Systemic Immune Dysregulation. Eur J Haematol (2021) 106:783–7. doi: 10.1111/ejh.13600
7. Walter JE, Farmer JR, Foldvari Z, Torgerson TR, Cooper MA. Mechanism-Based Strategies for the Management of Autoimmunity and Immune Dysregulation in Primary Immunodeficiencies. J Allergy Clin Immunol Pract (2016) 4:1089–100. doi: 10.1016/j.jaip.2016.08.004
8. Walter JE, Ayala IA, Milojevic D. Autoimmunity as a Continuum in Primary Immunodeficiency. Curr Opin Pediatr (2019) 31:851–62. doi: 10.1097/MOP.0000000000000833
9. Fischer A, Provot J, Jais JP, Alcais A, Mahlaoui N. Autoimmune and Inflammatory Manifestations Occur Frequently in Patients With Primary Immunodeficiencies. J Allergy Clin Immunol (2017) 140:1388–93.e8. doi: 10.1016/j.jaci.2016.12.978
10. Aladjidi N, Fernandes H, Leblanc T, Vareliette A, Rieux-Laucat F, Bertrand Y, et al. Evans Syndrome in Children: Long-Term Outcome in a Prospective French National Observational Cohort. Front Pediatr (2015) 3:79. doi: 10.3389/fped.2015.00079
11. Schmidt RE, Grimbacher B, Witte T. Autoimmunity and Primary Immunodeficiency: Two Sides of the Same Coin? Nat Rev Rheumatol (2017) 14:7–18. doi: 10.1038/nrrheum.2017.198
12. Bulkhi AA, Dasso JF, Schuetz C, Walter JE. Approaches to Patients With Variants in RAG Genes: From Diagnosis to Timely Treatment. Expert Rev ClinImmunol (2019) 15:1033–46. doi: 10.1080/1744666X.2020.1670060
13. Dorna MB, Barbosa PFA, Rangel-Santos A, Csomos K, Ujhazi B, Dasso JF, et al. Combined Immunodeficiency With Late-Onset Progressive Hypogammaglobulinemia and Normal B Cell Count in a Patient With RAG2 Deficiency. Front Pediatr (2019) 7:122. doi: 10.3389/fped.2019.00122
14. Besnard C, Levy E, Aladjidi N, Stolzenberg MC, Magerus-Chatinet A, Alibeu O, et al. Pediatric-Onset Evans Syndrome: Heterogeneous Presentation and High Frequency of Monogenic Disorders Including LRBA and CTLA4 Mutations. Clin Immunol (2018) 188:52–7. doi: 10.1016/j.clim.2017.12.009
15. Hadjadj J, Aladjidi N, Fernandes H, Leverger G, Magérus-Chatinet A, Mazerolles F, et al. Pediatric Evans Syndrome Is Associated With a High Frequency of Potentially Damaging Variants in Immune Genes. Blood (2019) 134:9–21. doi: 10.1182/blood-2018-11-887141
16. Ladogana S, Maruzzi M, Samperi P, Perrotta S, Del Vecchio GC, Notarangelo LD. Diagnosis and Management of Newly Diagnosed Childhood Autoimmune Haemolytic Anaemia. Recommendations From the Red Cell Study Group of the Paediatric Haemato-Oncology Italian Association. Blood Transfus (2017) 15:259–67. doi: 10.2450/2016.0072-16
17. Ladogana S, Maruzzi M, Samperi P, Condorelli A, Casale M, Giordano P. Second-Line Therapy in Paediatric Warm Autoimmune Haemolytic Anaemia. Guidelines From the Associazione Italiana Onco-Ematologia Pediatrica (AIEOP). Blood Transfus (2018) 16:352–7. doi: 10.2450/2018.0024-18
18. Hill QA, Hill A, Berentsen S. Defining Autoimmune Hemolyticanemia: A Systematic Review of the Terminology Used for Diagnosis and Treatment. Blood Adv (2019) 3:1897–906. doi: 10.1182/bloodadvances.2019000036
19. Fioredda F, Calvillo M, Bonanomi S, Coliva T, Tucci F, Farruggia P, et al. Congenital and Acquired Neutropenia Consensus Guidelines on Diagnosis From the Neutropenia Committee of the Marrow Failure Syndrome Group of the AIEOP (Associazione Italiana Emato-Oncologia Pediatrica). Pediatr Blood Cancer (2011) 57:10–7. doi: 10.1002/pbc.23108
20. Rodeghiero F, Stasi R, Gernsheimer T, Michel M, Provan D, Arnold DM, et al. Standardization of Terminology, Definitions and Outcome Criteria in Immune Thrombocytopenic Purpura of Adults and Children: Report From an International Working Group. Blood (2009) 113:2386–93. doi: 10.1182/blood-2008-07-162503
21. Miano M, Ramenghi U, Russo G, Rubert L, Barone A, Tucci F. Mycophenolatemofetil for the Treatment of Children With Immune Thrombocytopenia and Evans Syndrome. A Retrospective Data Review From the Italian Association of Paediatric Haematology/Oncology. Br J Haematol (2016) 175:490–5. doi: 10.1111/bjh.14261
22. Oliveira JB, Bleesing JJ, Dianzani U, Fleisher TA, Jaffe ES, Lenardo MJ, et al. Revised Diagnostic Criteria and Classification for the Autoimmune Lymphoproliferative Syndrome (ALPS): Report From the 2009 NIH International Workshop. Blood (2010) 116:e35-40. doi: 10.1182/blood-2010-04-280347
23. Grossi A, Miano M, Lanciotti M, Fioredda F, Guardo D, Palmisani E, et al. Targeted NGS Yields Plentiful Ultra-Rare Variants in InbornErrors of ImmunityPatients. Genes (2021) 12:1299. doi: 10.3390/genes12091299
24. Sue R, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
25. Bolduc V, Chagnon P, Provost S, Dubé MP, Belisle C, Gingras M, et al. No Evidence That Skewing of X Chromosome Inactivation Patterns Is Transmitted to Offspring in Humans. J Clin Invest (2008) 118:333–41. doi: 10.1172/JCI33166
26. Dell'Orso G, Grossi A, Penco F, Caorsi R, Palmisani E, Terranova P, et al. Case Report: Deficiency of Adenosine Deaminase 2 Presenting With Overlapping Features of Autoimmune Lymphoproliferative Syndrome and Bone Marrow Failure. Front Immunol (2021) 12:754029. doi: 10.3389/fimmu.2021.754029
27. Mazzoni M, Dell'Orso G, Grossi A, Ceccherini I, Viola S, Terranova P, et al. Underlying CTLA4 Deficiency in a Patient With Juvenile Idiopathic Arthritis and Autoimmune Lymphoproliferative Syndrome Features Successfully Treated With Abatacept-A Case Report. J Pediatr Hematol Oncol (2021) 43:e1168-e1172. doi: 10.1097/MPH.0000000000002120
28. Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-Onset Lymphoproliferation and Autoimmunity Caused by Germline STAT3 Gain-of-Function Mutations. Blood (2015) 125(4):591–9. doi: 10.1182/blood-2014-09-602763
29. Miano M, Cappelli E, Pezzulla A, Venè R, Grossi A, Terranova P, et al. FAS-Mediated Apoptosis Impairment in Patients With ALPS/ALPS-Like Phenotype Carrying Variants on CASP10 Gene. Br J Haematol (2019) 187(4):502–8. doi: 10.1111/bjh.16098
30. Miano M, Grossi A, Dell'Orso G, Lanciotti M, Fioredda F, Palmisani E, et al. Genetic Screening of Children With Marrow Failure. The Role of Primary Immunodeficiencies. Am J Hematol (2021) 96:1077–86. doi: 10.1002/ajh.26242
31. Fusco F, Bardaro T, Fimiani G, Mercadante V, Miano MG, Falco G, et al. Molecular Analysis of the Genetic Defect in a Large Cohort of IP Patients and Identification of Novel NEMO Mutations Interfering With NF-kappaB Activation. Hum Mol Genet (2004) 13:1763–73. doi: 10.1093/hmg/ddh192
32. Fusco F, Pescatore A, Conte MI, Mirabelli P, Paciolla M, Esposito E, et al. EDA-ID and IP, Two Faces of the Same Coin: How the Same IKBKG/NEMO Mutation Affecting the NF-κb Pathway Can Cause Immunodeficiency and/or Inflammation. Int W RevImmunol (2015) 34:445–59. doi: 10.3109/08830185.2015.1055331
33. Palmisani E, Miano M, Micalizzi C, Calvillo M, Pierri F, Terranova P, et al. Clinical Features and Therapeutic Challenges of Cytopenias Belonging to Alps and Alps-Related (ARS) Phenotype. Br J Haematol (2019) 184:861–4. doi: 10.1111/bjh.15178
34. Cifaldi C, Brigida I, Barzaghi F, Zoccolillo M, Ferradini V, Petricone D, et al. Targeted NGS Platforms for Genetic Screening and Gene Discovery in Primary Immunodeficiencies. Front Immunol (2019) 10:316. doi: 10.3389/fimmu.2019.00316
35. Miano M, Madeo A, Cappelli E, Lanza F, Lanza T, Stroppiano M, et al. Defective FAS-Mediated Apoptosis and Immune Dysregulation in Gaucher Disease. J Allergy Clin Immunol Pract (2020) 8:3535–42. doi: 10.1016/j.jaip.2020.06.065
36. Mendonça LO, Matucci-Cerinic C, Terranova P, Casabona F, Bovis F, Caorsi R, et al. The Challenge of Early Diagnosis of Autoimmune Lymphoproliferative Syndrome in Children With Suspected Autoinflammatory/Autoimmune Disorders. Rheumatology (2021) 61(2):696–704. doi: 10.1093/rheumatology/keab361
37. Miano M, Scalzone M, Perri K, Palmisani E, Caviglia I, Micalizzi C, et al. Mycophenolatemofetil and Sirolimus as Second or Further Line Treatment in Children With Chronic Refractory Primitive or Secondary Autoimmune Cytopenias: A Single Centre Experience. Br J Haematol (2015) 171(2):247–53. doi: 10.1111/bjh.13533
38. Bride KL, Vincent T, Smith-Whitley K, Lambert MP, Bleesing JJ, Seif AE, et al. Sirolimus Is Effective in Relapsed/Refractory Autoimmune Cytopenias: Results of a Prospective Multi-Institutional Trial. Blood (2016) 127(1):17–28. doi: 10.1182/blood-2015-07-657981
Keywords: Evans syndrome, autoimmune cytopenias, inborn errors of immunity (IEI), immune dysregulation, autoimmune haemolytic anaemia (AIHA), ITP (idiopathic thrombocytopenic purpura), autoimmune neutropenia (AIN), ALPS (autoimmune lymphoproliferative syndrome)
Citation: Miano M, Guardo D, Grossi A, Palmisani E, Fioredda F, Terranova P, Cappelli E, Lupia M, Traverso M, Dell’Orso G, Corsolini F, Beccaria A, Lanciotti M, Ceccherini I and Dufour C (2022) Underlying Inborn Errors of Immunity in Patients With Evans Syndrome and Multilineage Cytopenias: A Single-Centre Analysis. Front. Immunol. 13:869033. doi: 10.3389/fimmu.2022.869033
Received: 03 February 2022; Accepted: 07 April 2022;
Published: 17 May 2022.
Edited by:
Shanmuganathan Chandrakasan, Emory University, United StatesReviewed by:
Nicola Wright, University of Calgary, CanadaCopyright © 2022 Miano, Guardo, Grossi, Palmisani, Fioredda, Terranova, Cappelli, Lupia, Traverso, Dell’Orso, Corsolini, Beccaria, Lanciotti, Ceccherini and Dufour. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maurizio Miano, bWF1cml6aW9taWFub0BnYXNsaW5pLm9yZw==
†These authors share first authorship
‡These authors share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.