 Matthijs Luxen
Matthijs Luxen Matijs van Meurs
Matijs van Meurs Grietje Molema
Grietje Molema
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 13 May 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.867625
This article is part of the Research TopicSepsis: Studying the Immune System to Highlight Biomarkers for Diagnosis, Prognosis and Personalized TreatmentsView all 10 articles
Sepsis is a devastating clinical condition that can lead to multiple organ failure and death. Despite advancements in our understanding of molecular mechanisms underlying sepsis and sepsis-associated multiple organ failure, no effective therapeutic treatment to directly counteract it has yet been established. The endothelium is considered to play an important role in sepsis. This review highlights a number of signal transduction pathways involved in endothelial inflammatory activation and dysregulated endothelial barrier function in response to sepsis conditions. Within these pathways – NF-κB, Rac1/RhoA GTPases, AP-1, APC/S1P, Angpt/Tie2, and VEGF/VEGFR2 – we focus on the role of kinases and phosphatases as potential druggable targets for therapeutic intervention. Animal studies and clinical trials that have been conducted for this purpose are discussed, highlighting reasons why they might not have resulted in the expected outcomes, and which lessons can be learned from this. Lastly, opportunities and challenges that sepsis and sepsis-associated multiple organ failure research are currently facing are presented, including recommendations on improved experimental design to increase the translational power of preclinical research to the clinic.
Sepsis is a complex condition that involves a dysregulated host response to infection. Sepsis is the leading cause of multiple organ failure in patients treated in the intensive care unit, and sepsis-associated mortality is high (1). Our understanding of the underlying processes involved in the onset and progression of sepsis and sepsis-associated multiple organ failure (sepsis-MOF) has increased considerably in the past decades. However, this has not yet resulted in effective drug treatment regimens specifically targeting sepsis-related pathophysiological processes. At present, clinicians merely have infection source control, organ support machines, and antibiotics at their disposal to treat their patients. Therefore, the need for an effective drug treatment for sepsis and sepsis-MOF remains critically high.
The endothelium forms the inner lining of all blood vessels in the body. Since the early 2000s, especially the microvascular endothelium is considered a major player in the pathophysiology of sepsis and sepsis-MOF (2, 3). During sepsis, endothelial cells (EC) are activated by blood-derived stimuli, which can induce microvascular leakage, microvascular inflammation, and vascular hypocontractility, thereby contributing to septic shock and multiple organ failure (4–6). The intracellular signalling pathways that control endothelial activation in response to sepsis are numerous and display a high degree of interconnectedness. This means that different signal transduction pathways involved in the endothelial response to sepsis can share intracellular signalling axes. A corollary to this with regard to therapeutic strategy design is that pharmacological inhibition of one pathway may not be effective, as other pathways are activated unimpededly. Hence, therapeutic targeting of a single intracellular pathway in EC out of a myriad of potential targets is unlikely to improve sepsis outcome.
The aim of this review is to untangle the complexity of six signal transduction pathways that have been described to be involved in EC responses to sepsis conditions, namely NF-κB, Rac1/RhoA GTPases, AP-1, APC/S1P, Angpt/Tie2, and VEGF/VEGFR21. The role of kinases and phosphatases in these pathways will be emphasised throughout this review, as they hold promise as drug targets. The challenges for implementation of kinase inhibitor drug therapy to sepsis patients will also be addressed. The main proteins involved in the activation and downstream signalling of each pathway will be discussed, followed by a description of their engagement in sepsis animal studies, and of clinical trials in sepsis patients that intervene in the pathways of interest. An overview of all pathways will display their interconnectedness and show similarities and differences in the outcomes of their downstream signalling cascades. Discussing the molecular mechanisms of endothelial activation in sepsis in the light of clinical sepsis trials will help explain why some clinical trials failed, while animal studies reported potential benefit. By acknowledging and further mapping this complexity, different subsets of sepsis patients may be identified to define which patient groups may benefit from which treatment.
Once sepsis-induced hypotension can no longer be counteracted by fluid resuscitation and development of multiple organ failure occurs, the condition has developed into septic shock (1). The combination of both pro- and anti-inflammatory responses observed in sepsis is necessary for clearance of infection and tissue recovery, but an imbalance between both is thought to lead to organ injury and secondary infections (4, 7, 8). A substantial number of factors influence the course of disease progression, including the identity of the invading pathogen, the site of infection, the responses of (micro)vascular beds in organs, influence of comorbidities, and overall heterogeneity between patients (8).
Despite the huge demand for therapies to improve sepsis and sepsis-MOF outcome, and despite decades of research focused on drug development, clinicians currently lack pharmacological treatment options to directly target the processes that contribute to sepsis and sepsis-MOF. Close to 50 million sepsis cases are reported annually worldwide, of which 11 million patients do not survive their ailment (9). Sepsis represents one of the largest burdens to global health, with sepsis-related deaths representing 19.7% of deaths globally (9). Sepsis patients are prone to developing organ failure during the course of their disease, with an estimated 1 in every 3 patients developing acute kidney injury (10). This is associated with an increased likelihood of dying compared to patients suffering from acute kidney injury from other causes (11). Even patients that do survive carry an increased risk of developing chronic kidney disease, hereby also posing a huge socio-economic burden to society (10). In conjunction with the grim prospects for patients suffering from sepsis, this underscores the urgent need for novel and effective treatments for sepsis and sepsis-MOF.
In sepsis, EC are believed to play a role in many of the occurring pathophysiological processes in response to blood-derived stimuli. Activated EC facilitate leukocyte recruitment and extravasation into the underlying tissue, where they can cause damage to the tissue. In addition, endothelial inflammatory activation induces loss of endothelial barrier function, which can lead to increased microvascular leakage and subsequent tissue hypoxia and organ failure. While under normal conditions these endothelial processes aid clearance of infections and maintenance of homeostasis, exaggerated inflammatory conditions in sepsis overturn the delicate balance between functional and dysfunctional endothelial responses. Dysfunctional EC in sepsis are associated with glycocalyx shedding, decreased organ perfusion and oxygenation, and exacerbated microvascular permeability (2, 12, 13), which indicate an inability to maintain tissue homeostasis. In addition, EC play crucial roles in blood coagulation and thrombus formation, which can become dysregulated during sepsis. Albeit important, altered coagulation during sepsis is not extensively discussed in this review, yet excellent reviews on this topic are available (14, 15).
The various blood vessel segments that constitute the circulatory system - arteries, arterioles, capillaries, post-capillary venules, and veins – all have important roles in response to sepsis that vary from one another based on the type of segment. While modulation of blood flow, blood pressure, and shear stress is primarily controlled via the smooth muscle cells in the walls of arteries and smaller arterioles, capillaries and veins contribute little to nothing to these effects. Influx of leukocytes, on the other hand, is especially prominent in post-capillary venules (16), with the exception of lung where leukocyte transmigration primarily takes place in the alveolar capillaries (17). The capillaries and post-capillary venules are of particular interest with respect to sepsis, as they represent the major sites of microvascular leakage (18). Thus, EC from all segments of the circulatory system engage in the response of the body to sepsis, though in each segment they fulfil a different role. Care should be taken regarding the fact that not in all blood vessels nor in all organs similar responses are observed (6).
An integral question in microvascular EC research is if, and to what extent, EC in these different (micro)vascular segments respond to bloodborne stimuli. This depends on the presence of receptors, which may differ between different microvascular segments (19). Furthermore, even if receptors are expressed by EC at similar levels, downstream signalling may differ due to different expression levels of e.g. kinases or phosphatases (6, 20), though further research is required to characterise these differential expression patterns in EC in vivo. These differences may not only exist between different blood vessel segments, but can also exist between similar segments in different organs, e.g. pulmonary capillaries versus renal capillaries (21). Furthermore, variation in expression levels of molecules exists within different microvascular segments of one organ, as illustrated for kinase vascular endothelial growth factor receptor 2 (VEGFR2) in mouse kidney. While in glomeruli, which are specialised capillary networks responsible for filtering the blood, VEGFR2 expression was high, low expression was detected in arterioles and post-capillary venules (22). Organ-specific differences in EC responses to stimuli was illustrated in a study in which mice were exposed to the pro-inflammatory Gram-negative bacterium-derived lipopolysaccharide (LPS). While VEGFR2 expression in kidney had not changed 24 h after LPS administration, VEGFR2 expression in lung significantly decreased already 4 h after LPS administration (23). It is conceivable that the differential expression of receptors and molecules involved in signal transduction by EC in different organs and in different microvascular compartments underlies differential responses of organs in sepsis. By creating a framework of activation status of signal transduction pathways, basal gene/protein expression patterns, and differential changes in gene/protein expression in time in response to sepsis in microvascular compartments in different organs, pathways of interest can be further investigated for their responses to drug intervention studies.
Animal models often used to study the complex pathophysiology of sepsis include endotoxemia following injection with LPS, and cecal ligation and puncture (CLP)-induced polymicrobial sepsis. Endotoxemia results in a high peak of pro-inflammatory cytokines that is resolved relatively rapidly. CLP, on the other hand, is considered the gold standard among sepsis models, as it introduces an active site of bacterial infection in the abdomen. This mimics the course of human sepsis with regards to cytokine response and the development of protracted systemic inflammation more closely (24). LPS is an important component of the inflammatory response following CLP-sepsis, and is systemically elevated as early as 1 h after CLP surgery, though the inflammatory response to both endotoxemia and CLP consists of additional pro-inflammatory stimuli besides LPS (24, 25). Animal responses can be evaluated by e.g. pro-inflammatory cytokine levels and markers of endothelial inflammatory activation in the blood, immune cell infiltration into the tissue, circulating markers of organ function and organ damage, and structural integrity of organs assessed by histology.
During the pro-inflammatory phase of sepsis, EC become activated by exogenous agents such as LPS, and by endogenously produced cytokines and chemokines including tumour necrosis factor alpha (TNFα), interleukin (IL)-1β, and IL-6 (24, 26). In EC, LPS is recognised by both Toll-like receptor 4 (TLR4), an important part of the innate immune response against invading pathogens (27), and by retinoic acid-inducible gene I (RIG-I) (28). Immune cells in the host also respond to LPS by producing a plethora of pro-inflammatory cytokines, which can activate EC (29, 30). TNFα is recognised by EC via TNFα receptor type 1 (TNFR1) (31), IL-1β via a heterodimer of IL-1 receptor 1 and IL-1 receptor accessory protein (32, 33), and IL-6 by binding of IL-6/(soluble) IL-6 receptor complex to gp130 glycoprotein on the endothelial cell surface (34).
EC enter a state of inflammatory activation after stimulation by these pro-inflammatory mediators. This activated state is characterised by increased expression of adhesion molecules including E-selectin and vascular cell adhesion molecule 1 (VCAM-1), which facilitate rolling, adhesion, and transmigration of leukocytes through the endothelium into the underlying tissue. EC also start to produce cytokines and chemokines (22), including IL-6 and monocyte chemoattractant protein 1 (MCP-1), to attract circulating immune cells and signal to other EC (35–39). In addition, Weibel-Palade bodies, which are endothelial granules that contain P-selectin, IL-8, angiopoietin 2 (Angpt2), and von Willebrand Factor (vWF), rapidly release their content into the circulation in response to pro-inflammatory stimuli (40, 41). Endothelial inflammatory activation is also associated with proteolytic cleavage of the extracellular part of transmembrane proteins from the endothelial cell surface, resulting in circulating proteins. An example of this is vascular endothelial cadherin (VE-cadherin), the main structural unit of adherens junctions. Adherens junctions link neighbouring EC and are important contributors to endothelial barrier function, as their structural components serve to prevent free transport of molecules and cells from the blood into the underlying tissue (42, 43). Studies monitoring endothelial dysfunction frequently assess plasma levels of proteins including soluble VE-cadherin (sVE-cadherin), sE-selectin, or vWF to gain insight in the activation status of the endothelium. Similarly, plasma levels of pro-inflammatory cytokines can be monitored in time as determinants of sepsis progression. For instance, elevated levels of Angpt2 and VEGF, which play a role in signal transduction by endothelial-restricted receptor kinases Tie2 and VEGFR2 (discussed in more detail later), have both been associated with more severe disease progression and worsened clinical outcomes in sepsis patients (44, 45).
The following sections will discuss in greater detail six signal transduction pathways that have been described to be involved in EC responses to sepsis in vivo or sepsis conditions in vitro. Per pathway, the main molecules involved in the signal transduction cascade will be discussed, followed by a description of sepsis animal studies that investigate the pathway. Finally, an overview will be provided of clinical trials in sepsis patients intervening in the described pathways.
Endothelial inflammatory activation in sepsis can occur via recognition of bacterial products such as LPS by TLR4 and RIG-I, or through pro-inflammatory cytokines including TNFα produced by immune cells. TNFα is recognised by EC via TNFR1 and TNFα receptor type 2 (TNFR2), though the role of endothelial TNFR2 signalling is incompletely understood (31). Activation of TLR4/RIG-I by LPS as well as activation of TNFR1 by TNFα triggers translocation of NF-κB to the nucleus (31, 46, 47), where it induces transcription of pro-inflammatory genes (48) (Figure 1). In the absence of pro-inflammatory stimuli, NF-κB is retained in the cytoplasm via interaction with inhibitor of κB (IκB). This interaction conceals the nuclear localisation site of NF-κB, hereby preventing its translocation to the nucleus (49). IκB subtype IκBα can be phosphorylated by IκB kinase (IKK), which is a downstream responder of TNFR1/TLR4/RIG-I (28, 50, 51). This causes IκBα to dissociate from NF-κB, after which it is degraded in proteasomes. The pro-coagulatory protein thrombin also activates IKK in a RhoA-dependent manner (52), which will be discussed in more detail later.
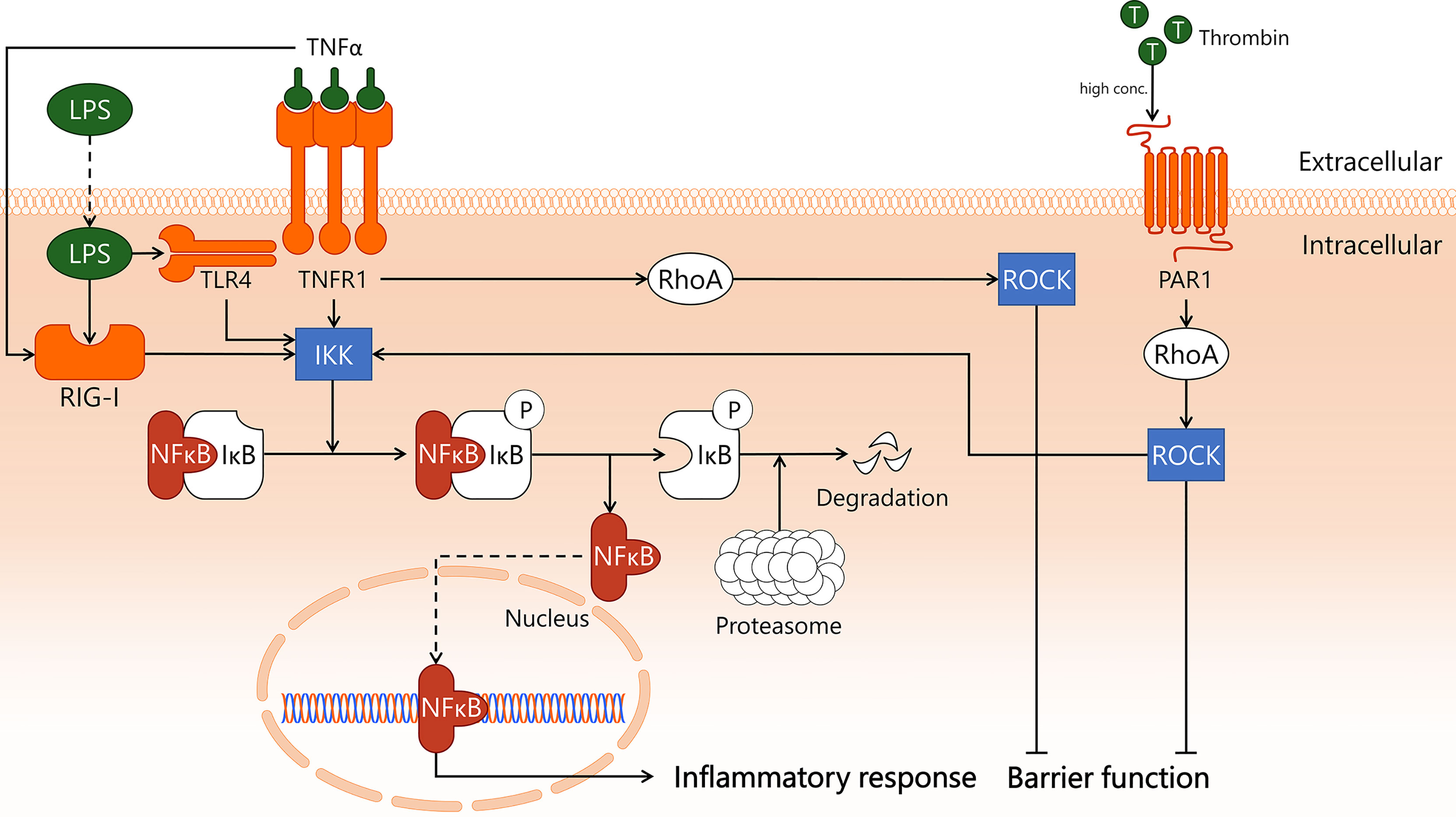
Figure 1 Transcription factor NF-κB induces endothelial inflammation during sepsis. In sepsis, endothelial cells are exposed to pro-inflammatory cytokine TNFα and Gram-negative bacterial product LPS, which incite pro-inflammatory responses. TNFα binds to its receptor TNFR1, activating both IKK and RhoA. TNFα also activates intracellular receptor RIG-I through unknown mechanisms. LPS is taken up by the cell, where it is recognised by both TLR4 and RIG-I, which activate IKK. High levels of thrombin induce PAR1-dependent activation of RhoA, which also activates IKK. In quiescence, IκB masks the nuclear translocation signal of NF-κB, hereby retaining it in the cytosol. Upon activation of IKK, IκB is phosphorylated and dissociates from NF-κB. While IκB is then enzymatically degraded by proteasomes, freed NF-κB translocates to the nucleus to incite an inflammatory response. In addition, the endothelial barrier function is negatively affected, following TNFR1- and PAR1-dependent activation of RhoA. IKK, IκB kinase; IκB, inhibitor of κB; LPS, lipopolysaccharide; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; PAR1, protease-activated receptor 1; RhoA, Ras homolog gene family, member A; RIG-I, retinoic acid-inducible gene I; ROCK, Rho-associated protein kinase; T, thrombin; TLR4, Toll-like receptor 4; TNFR1, tumor necrosis factor alpha receptor 1; TNFα, tumor necrosis factor alpha.
If translocation of NF-κB to the nucleus can somehow be inhibited, a major molecular pathway of pro-inflammatory activation of EC in sepsis is interrupted (53). In mouse CLP-sepsis, treatment with IKK inhibitor IKK 16, a 2-benzamido-pyrimidine, decreased the activity of the IKK complex (54), reduced IκBα phosphorylation, and inhibited nuclear translocation of NF-κB (55). IKK inhibition attenuated sepsis-associated dysfunction of the heart, based on improved cardiac function, and the kidney, based on normalisation of serum creatinine levels (55). Interestingly, IKK inhibition in mouse endotoxemia resulted in a ~30% decrease in lung neutrophil-associated myeloperoxidase activity, whereas in CLP this treatment resulted in a complete abrogation of lung myeloperoxidase activity (55). While no data were provided on which cell types responded to the inhibitor, it would be of interest to expand on this study by localising cells affected by NF-κB blockade, and to distil the role of EC therein.
Integrity of the endothelial barrier is important in homeostasis. In sepsis, disruption of endothelial barrier function leads to microvascular leakage and oedema formation, which is associated with worsened patient survival (43). EC can form intercellular protein-protein connections between adjacent cells, which are crucial in maintaining endothelial barrier function (56). The shape of EC can be altered by changing the properties of the cytoskeleton, thereby affecting cell-cell interactions, which in turn may compromise the integrity of the endothelial barrier. As actin filaments in EC are actively involved in barrier disruptive cytoskeleton rearrangements in sepsis (57, 58), the following section will discuss the signal transduction pathways underlying these processes.
Special complexes of actin bundles and myosin called stress fibres are regulated by a small GTPase called Ras homolog gene family, member A (RhoA), most notably via activation of Rho-associated protein kinase (Rho kinase), also known as ROCK (59). Stress fibre formation and contraction, as occurs in sepsis conditions, is a result of phosphorylation of the myosin light chain (MLC) (60, 61). The subsequent increase in tension on myosin-associated actin leads to altered cell structure, disrupted endothelial barrier, and increased microvascular permeability (62, 63). The balance between MLC phosphorylation and dephosphorylation is mainly regulated via MLC phosphatase and MLC kinase. ROCK reduces MLC phosphatase activity through phosphorylation of MLC phosphatase at an inhibitory site, as a consequence of which MLC dephosphorylation is prevented and stress fibre formation and contraction is stimulated (64, 65). Another small GTPase, Ras-related C3 botulinum toxin substrate 1 or Rac1, has opposing effects on endothelial barrier function compared to RhoA. Rac1 decreases MLC kinase activity through its effector protein p21-activated kinase (PAK) (66), which reduces MLC phosphorylation levels and consequently prohibits stress fibre formation and contraction (67, 68). Rac1 further promotes endothelial barrier function through the assembly of cortical actin, which stabilises the cytoskeleton (69, 70). Rac1 and RhoA also control the activity of one another, with Rac1 inhibiting RhoA (71), and RhoA inhibiting Rac1 via ROCK (72), thereby making the molecular balance between protection and disruption of endothelial barrier function even more intricate (Figure 2).
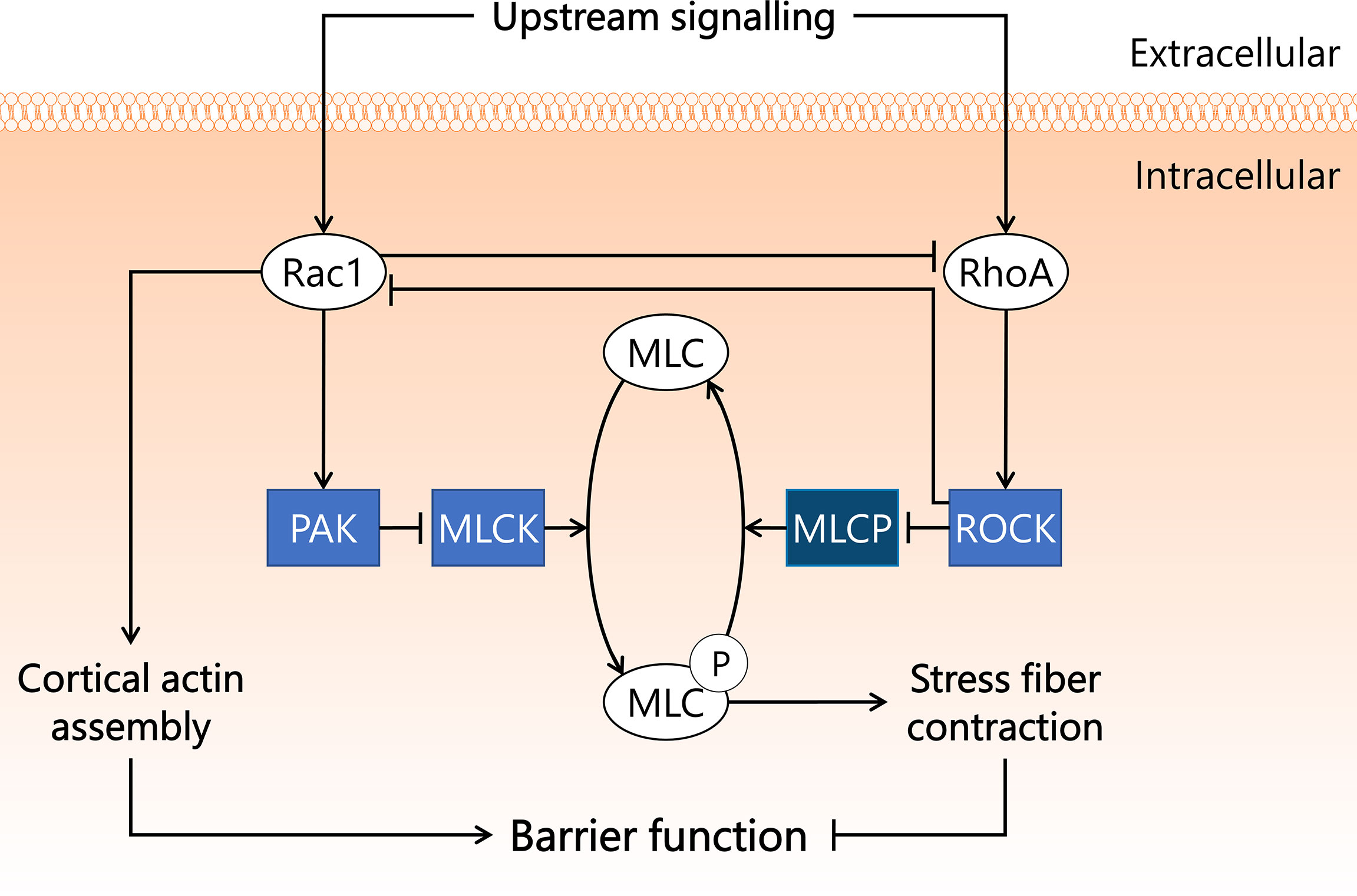
Figure 2 Rac1 and RhoA GTPases exert opposite effects on endothelial barrier function in sepsis. Endothelial barrier integrity is integral for maintaining blood pressure and preventing microvascular leakage and subsequent tissue hypoxia. Rac1 and RhoA GTPases both play important roles in endothelial barrier function, and are activated by a variety of upstream signals associated with sepsis. Rac1 inhibits RhoA and inhibits MLCK in a PAK-dependent manner. RhoA activates its effector kinase ROCK, which inhibits Rac1 activity and phosphorylates MLCP, resulting in inactivation. MLCK induces phosphorylation of MLC, a process reverted by MLCP. Upon phosphorylation, MLC induces the formation and contraction of stress fibres in the cell, leading to reduced cell-cell contacts and dysregulated endothelial barrier function. Activated Rac1 stimulates the assembly of cortical actin, which preserves endothelial barrier function. MLC, myosin light chain; MLCK, myosin light chain kinase; MLCP, myosin light chain phosphatase; PAK, p21-activated kinase; Rac1, Ras-related C3 botulinum toxin substrate 1; RhoA, Ras homolog gene family, member A; ROCK, Rho-associated protein kinase.
Several studies in rodent sepsis models showed involvement of Rac1 and/or RhoA GTPases in the development of sepsis-associated microvascular leakage and inflammation. In a study in which rats were treated with pharmacological ROCK inhibitor Y27632 at 2 mg/kg i.v. directly after CLP instalment, microvascular permeability (measured by FITC-BSA extravasation) decreased ~50% in lung and kidney 8 h after start of CLP compared to untreated rats (73). Another study in rats showed that pre-treatment with Y27632 at 1.5 mg/kg i.p. 20 min prior to CLP ameliorated both sepsis-induced acute lung injury and microvascular leakage in lung (74). Drug administration prior to sepsis onset in these studies does not allow translation of outcomes to therapeutic outcome expectations, as effects of drug pre-treatment are not representative of drug performance during intervention studies. These studies do, however, highlight the involvement of ROCK in the onset and early development of sepsis-induced microvascular permeability in lung and kidney. MicroRNAs (miR) are small non-coding RNAs that can bind to compatible sequences on target mRNAs, thereby sequestering them and preventing translation to protein. One week prior to CLP instalment, mice were injected with miR-539-5p agomir, which mimics the function of endogenous miR-539-5p and suppresses ROCK protein translation. Mice were then sacrificed 24 h after CLP surgery. Treatment with miR-539-5p agomir resulted in reduced pulmonary injury as assessed by histology, and reduced inflammation as assessed by IL-1β and IL-6 mRNA and protein levels in lung (75). The potential applicability of Y27632 and miR-539-5p as therapeutic strategy in sepsis remains to be established, as cell types affected by the treatment, and long-term effects on mortality were not reported. Importantly, ROCK fulfils a key role in maintaining basal endothelial barrier function, as was shown in in vitro experiments in human umbilical vein endothelial cells (76). More research is required to establish if in certain microvascular beds ROCK is crucial to maintain basal endothelial barrier function, to circumvent unwanted induction of microvascular leakage and inflammation following systemic ROCK inhibition.
Activator protein 1 (AP-1) is a transcription factor heterodimer involved in cell survival and induction of inflammation. The canonical AP-1 complex consists of cellular Fos proto-oncogene (c-Fos) and cellular Jun proto-oncogene (c-Jun), though over 20 members of the AP-1 transcription factor family are known to engage in AP-1 heterodimer formation (77, 78). It is unclear whether particular AP-1 transcription factor family members are cell type- or microvascular compartment-specific, and if so, what the implications are for AP-1-mediated gene transcription (79). The most well-described AP-1 transcription factor family members, c-Fos and c-Jun, can be phosphorylated, which leads to enhanced AP-1-dependent transcription rate of target genes (77, 78). c-Fos is phosphorylated by extracellular signal-regulated kinase (ERK) (80) and p38 mitogen-activated protein kinase (p38) (81), whereas c-Jun is phosphorylated by p38 (82) and c-Jun N-terminal kinase (JNK) (83). These three kinases – ERK, p38, and JNK – are all members of the mitogen-activated protein kinase family that engages in intracellular processes predominantly related to inflammation and cell survival. ERK, p38, and JNK contribute to the transcription and subsequent increase in protein levels of both c-Fos and c-Jun as well, and in this manner are involved in promoting AP-1 transcriptional activity at multiple levels (84–88). Furthermore, p38 has also been reported to lower the activity of both ERK (89) and JNK (87), although the latter interaction has not been confirmed to exist in EC. During endothelial inflammatory activation, the kinases ERK, p38, and JNK become activated by upstream signalling pathways initiated by LPS/TLR4 (90) and TNFα/TNFR1 complexes (91, 92). The PI3K/AKT signalling axis is also activated upon endothelial exposure to LPS (93) and TNFα (94). Furthermore, p38 and JNK are activated by Rac1 and RhoA through mechanisms that are not well described (95–98).
In comparison to NF-κB, AP-1 is less well described in relation to sepsis-related inflammatory activation of EC, and is instead primarily a topic of study in chronic inflammation and oncology (99, 100). As a result, various components of the signal transduction pathway displayed in Figure 3 represent state-of-the-art knowledge on AP-1 signalling in general, not in EC per se. Since in vitro studies in primary EC indicated that inhibition of the NF-κB pathway is insufficient to completely prevent an inflammatory response to e.g. LPS, a considerable portion of this response may well be linked to p38-dependent AP-1 activation (101, 102). The importance of AP-1 in sepsis was highlighted in a study using AP-1 decoy oligodeoxynucleotides (ODN), which showed improved survival in mouse CLP-sepsis (103). Furthermore, intervention treatment with the AP-1 decoy ODN 1 h after start of CLP resulted in decreased gene expression of pro-inflammatory cytokines (IL-1β, IL-6, TNFα, MCP-1) in lung and kidney, and a reduction in circulating IL-1β protein levels in blood 18 h after CLP. In addition, treatment with AP-1 decoy ODN induced a significant reduction of neutrophil influx in lung, kidney, and liver 18 h after start of CLP, and reduced sepsis-induced apoptosis in these same three organs (103). Treatment with c-Fos inhibitor T-5224 in mouse endotoxemia improved survival, and resulted in reduced serum blood urea nitrogen and creatinine levels, suggesting improved kidney function (104). Importantly, these studies did not report which cell types were affected by inhibiting AP-1 activity. Hence, the role of AP-1 in EC in sepsis in vivo requires further investigation.
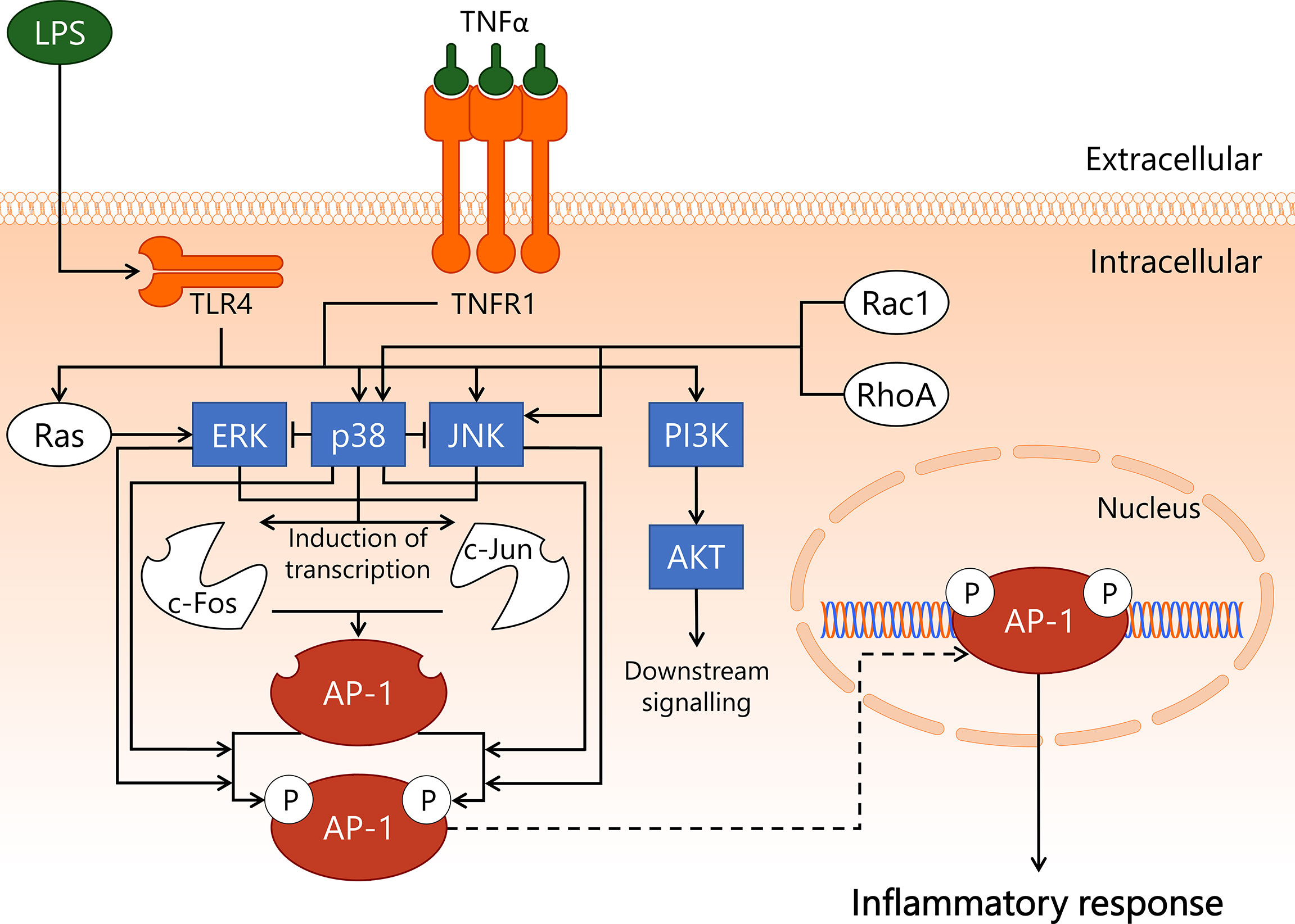
Figure 3 Activation of transcription factor AP-1 in endothelial cells during sepsis. During onset and development of sepsis, transcription factor AP-1 becomes activated in the endothelium. LPS/TLR4 and TNFα/TNFR1 promote activity of PI3K/AKT, Ras/ERK, p38, and JNK kinases, whereas p38 and JNK can also become activated by Rac1 or RhoA. ERK, p38, and JNK induce the transcription of the c-Fos and c-Jun proteins, which can form the AP-1 heterodimer. Phosphorylation of AP-1 enhances its transcriptional function. The c-Fos component of AP-1 is phosphorylated by ERK and p38, whereas the c-Jun component of AP-1 is phosphorylated by p38 and JNK. AP-1 induces transcription of genes mainly associated with inflammation and apoptosis. AKT, protein kinase B; AP-1, activator protein 1; c-Fos, cellular Fos proto-oncogene; c-Jun, cellular Jun proto-oncogene; ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase; LPS, lipopolysaccharide; p38, p38 mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinase; Rac1, Ras-related C3 botulinum toxin substrate 1; RhoA, Ras homolog gene family, member A; TLR4, Toll-like receptor 4; TNFR1, tumor necrosis factor alpha receptor 1; TNFα, tumor necrosis factor alpha.
A 2001 human endotoxemia study in which healthy volunteers received LPS via bolus i.v. injection reported absence of activated JNK in peripheral blood leukocytes, which made the authors conclude that JNK might not be an ideal target when attempting to abrogate inflammation (105). This conclusion does not take a contribution of non-leukocyte cell types to sepsis pathophysiology into account. Strikingly, pre-treatment with pharmacological JNK-inhibitor SP600125 4 h before CLP instalment in rats reduced lung injury (106), with SP600125 administration 1 h after CLP in mice resulting in decreased sepsis-induced lung and liver damage and significantly improved 5-day survival (107).
Also in the early 2000s, multiple studies were performed in which healthy volunteers were closely monitored after being treated with p38 inhibitor drugs followed by systemic LPS challenge. Beneficial effects of p38 inhibition compared to placebo included reduced circulating markers of endothelial inflammatory activation (108), reduced leukocyte activation and attenuated pro-inflammatory cytokine production (109), and maintenance of base level body temperature and heart rate (110). As the effect of p38 inhibition on the endothelium was not reported, the contribution of EC to the reported effects remains unclear. In part due to reported high levels of (hepato)toxicity following p38 inhibitor treatment (111), no follow up has taken place towards a successful therapeutic strategy revolving around p38 inhibition that benefits sepsis patients.
During sepsis, the balance between coagulation and anti-coagulation is skewed towards heightened coagulation due to increased production of pro-coagulatory factors and decreased production of anti-coagulatory factors, resulting in elevated blood viscosity (112). Sepsis-induced inflammatory activation of EC prompts vWF secretion, which results in microthrombi formation via recruitment and activation of platelets (15). Thrombus formation and altered coagulation in sepsis and the role of EC therein are reviewed in detail elsewhere (14, 15). Activated protein C (APC) is an important molecule in sepsis, as it has anti-inflammatory, anti-coagulatory, and anti-thrombotic properties (112, 113). Under homeostatic conditions, relatively high concentrations of APC (5-20 nM) stimulate protease-activated receptor 1 (PAR1) to elicit a protective effect on endothelial barrier function (114, 115). Additionally, activation of PAR1 by pro-coagulatory protein thrombin increases microvascular permeability via endothelial RhoA-dependent cytoskeleton rearrangement (116, 117). Hence, stimulation of the PAR1 receptor can have both protective and disruptive effects on endothelial barrier function, depending on its main agonist being either APC or thrombin, respectively (Figure 4). In sepsis, the APC:thrombin ratio is commonly decreased due to lowered levels of APC and/or elevated levels of thrombin (14), resulting in increased microvascular leakage following thrombin-dependent PAR1 stimulation (118). PAR1 involvement in human sepsis was investigated in a placebo-controlled study in which healthy volunteers received PAR1 inhibitor vorapaxar 24 h prior to LPS bolus infusion (119). Vorapaxar treatment was associated with reduced activation of coagulation, possibly via platelet-expressed PAR1 and decreased endothelial inflammatory activation in response to LPS as assessed by vWF and sE-selectin levels measured in blood. This outcome implies a predominantly harmful effect of PAR1 signalling in endotoxemia, with PAR1 inhibition resulting in reduced endothelial inflammatory activation (119).
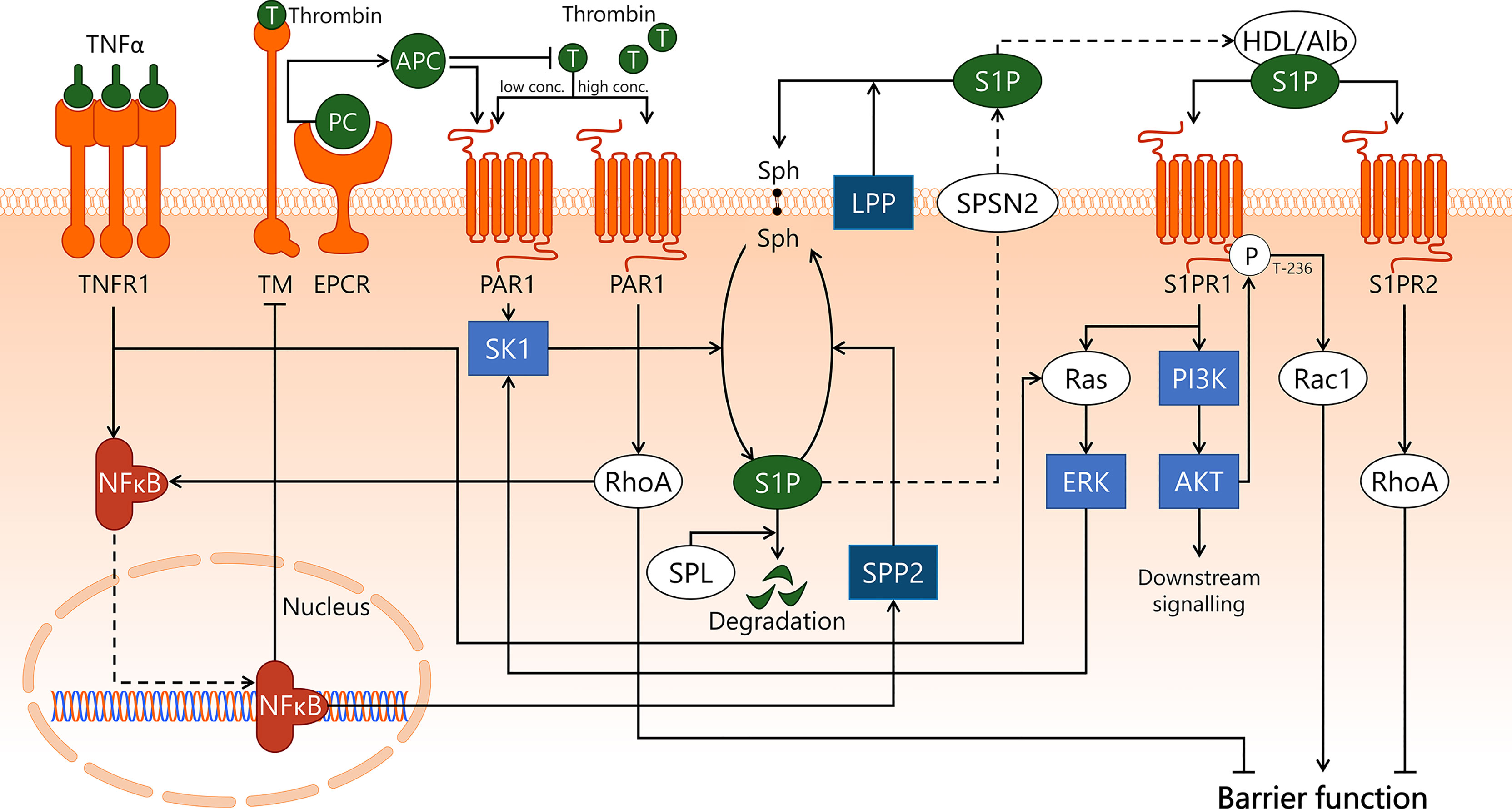
Figure 4 APC/S1P and associated signal transduction pathways in endothelial cells during sepsis. Sepsis pathophysiology is closely associated with microvascular leakage and aberrant coagulation. APC is involved in both processes, by promoting endothelial barrier function and by exerting anti-coagulatory effects. The conversion of protein C to APC by EPCR is stimulated upon association with thrombin/TM, after which APC inhibits thrombin formation. In sepsis-associated inflammation, pro-inflammatory cytokines such as TNFα are recognised by TNFR1 on the endothelial cell surface, leading to activation of Ras/ERK, and activation and subsequent translocation to the nucleus of transcription factor NF-κB. NF-κB induces downregulation of TM, hereby decreasing the turnover of PC to APC. APC and low concentrations of thrombin activate PAR1, which activates SK1. SK1 phosphorylates the lipid sphingosine to form S1P. This process can be reversed by SPP2, whose activity is increased upon NF-κB-induced transcription, and S1P can also be irreversibly degraded by SPL. S1P is transported out of the cell by SPNS2, where it is able to associate with carrier proteins such as albumin and HDL, or can be dephosphorylated by LPP. S1P can then activate S1PR1, leading to activation of Ras/ERK, which stimulates SK1 activity. S1PR1 activation also stimulates PI3K/AKT activity, which enables AKT to phosphorylate S1PR1 at the T-236 site, which is required for its ability to activate Rac1 and promote endothelial barrier function. Alternatively, a disruptive effect on the endothelial barrier function by RhoA is established both by activation of S1PR2 by S1P and through high concentrations of thrombin activating PAR1. In addition, specifically thrombin/PAR1-activated RhoA promotes NF-κB transcriptional activities. AKT, protein kinase B; Alb, albumin; APC, activated protein C; EPCR, endothelial protein C receptor; ERK, extracellular signal-regulated kinase; HDL, high-density lipoprotein; LPP, lipid phosphate phosphatase; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; PAR1, protease-activated receptor 1; PC, protein C; PI3K, phosphoinositide 3-kinase; Rac1, Ras-related C3 botulinum toxin substrate 1; Ras, Ras protein subfamily; RhoA, Ras homolog gene family, member A; S1P, sphingosine-1-phosphate; S1PR1, sphingosine-1-phosphate receptor 1; S1PR2, sphingosine-1-phosphate receptor 2; SK1, sphingosine kinase 1; Sph, sphingosine; SPL, sphingosine-1-phosphate lyase; SPNS2, Spinster homolog 2; SPP2, sphingosine-1-phosphate phosphatase 2; T, thrombin; TM, thrombomodulin; TNFR1, tumor necrosis factor alpha receptor 1; TNFα, tumor necrosis factor alpha.
Protein C is a zymogen that is primarily produced by the liver, which can be converted to APC by thrombin. In turn, APC inhibits thrombin formation, hereby forming a negative feedback loop that regulates thrombin levels (120). The conversion of protein C to APC is vastly accelerated in the presence of thrombomodulin (TM) and endothelial protein C receptor (EPCR) (121, 122), both of which are expressed on the surface of EC in dedicated microvascular segments (6, 112). The importance of EPCR was emphasised by a study in mouse endotoxemia, in which mice were genetically modified to express less than 10% EPCR protein compared to wildtype controls. Eighteen hours after administration of LPS, mice expressing low levels of EPCR had increased microvascular leakage compared to control mice, particularly in lung, kidney, and brain (123). The authors hypothesised that this outcome may be explained by reduced formation of APC following low EPCR expression, although evidence supporting this claim was not presented. Low APC levels may also be a result of loss of TM from the EC membrane, either due to NF-κB-mediated inhibition of expression or shedding from the endothelial cell surface (124–126). Sepsis-induced endothelial inflammatory activation can result in cleavage of TM from the endothelial membrane, resulting in elevated plasma levels of soluble TM (sTM). sTM is described as a potential endothelial damage marker with prognostic value for disease progression in sepsis (127–129). Interestingly, sTM partially retains its anti-coagulatory and anti-inflammatory functions, among others through continued conversion of protein C into APC and binding of thrombin (113, 130). Reduced expression of TM may be a crucial contributor to decrease APC levels in sepsis, and therefore a key process in sepsis-associated endothelial dysfunction, although further research on sTM is required to better understand its contribution to overall APC production.
A proposed mechanism underlying APC-dependent protection of endothelial barrier function involves the activation of sphingosine kinase 1 (SK1) by APC-dependent PAR1 signalling (114) (Figure 4). Intracellular SK1 phosphorylates sphingosine, a constituent of the cell membrane, to convert it into sphingosine-1-phosphate (S1P). S1P is transported out of the cell via mechanisms that are not fully understood, although non-ATP dependent transmembrane transporter Spinster homolog 2 is believed to play a role in the export (131). In the blood stream, S1P associates with carriers such as albumin and high-density lipoprotein (132). High-density lipoprotein contains a lipocalin called apolipoprotein M that is not only able to bind S1P, but also delivers it back to the endothelial cell surface to facilitate recognition by sphingosine-1-phosphate receptor 1 (S1PR1) (133). Activation of S1PR1 will lead to downstream signalling via the Ras protein subfamily (Ras)/ERK and phosphoinositide 3-kinase/protein kinase B (PI3K/AKT) signalling axes (134, 135). Activated AKT can then phosphorylate S1PR1 on its threonine-236 residue, which will lead to protection of the endothelial barrier function through activation of Rac1 (135). It was shown in EC in vitro that after inhibition of SK1 or S1PR1 prior to APC stimulation, this protective effect did not occur (114). This suggests that the protective effect of APC on endothelial barrier function is governed by PAR1, and dependent on both S1P formation and S1P-dependent activation of S1PR1. S1P also activates sphingosine-1-phosphate receptor 2 (S1PR2), which activates RhoA and causes disruption of the endothelial barrier function (136). The effect that will prevail – either S1PR1-induced barrier protection or S1PR2-induced barrier disruption – is believed to depend on expression levels of these S1PRs on the endothelial cell surface (137). Varying expression patterns of S1PRs are reported for different tissues (138), though distribution of microvascular S1PR expression is not well characterised. S1P availability is also dependent on its turnover rate, which occurs intracellularly either via cleavage by S1P lyases (139) or dephosphorylation by S1P phosphatases (140), or extracellularly via dephosphorylation by lipid phosphate phosphatases (141, 142). In human umbilical vein endothelial cells exposed to TNFα for 4 h, sphingosine-1-phosphate phosphatase 2 expression was induced up to 400 times, and its activity was increased (143). This illustrates involvement of pro-inflammatory signalling pathways in S1P turnover.
Various animal studies investigated the involvement of S1P and S1PRs in the onset and progression of sepsis. The 2-day and 10-day survival of S1PR2 KO mice was studied after intraperitoneal injection of E. coli (144) respectively induction of polymicrobial sepsis by CLP (145). Both studies found a significant increase in survival of S1PR2 KO mice compared to WT, and primarily attributed this finding to altered macrophage behaviour. Even though S1PR2 is abundantly expressed by macrophages, effects of genetic deletion on the behaviour of other cell types that also express S1PR2 – including EC – were not addressed. Treatment of mice with general S1PR agonist FTY720 and S1P lyase inhibitor 4-deoxypyridoxine before inducing sepsis by peritoneal contamination and infection (PCI) reduced microvascular leakage in lung and liver, and dampened IL-6, TNFα, and MCP-1 production. This indicates a positive effect of increased S1PR activation and elevated availability of S1P on sepsis-associated microvascular integrity loss and immunological response (146). It is difficult to identify the exact molecular mechanisms responsible for these outcomes, as all S1PRs are likely affected by this treatment.
Successful outcomes following treatment with S1P-system modifying drugs in animal sepsis models are not universal, as a study utilising a rat sepsis model called colon ascendens stent peritonitis (CASP) reported that treatment with S1PR1-agonist SEW2871 at 0.5 mg/kg i.v. 12 h after induction of sepsis did not improve microvascular leakage in mesenteric venules or serum creatinine levels in these animals. On the contrary, SEW2871 treatment resulted in bradyarrhythmia and cardiac arrest, which exacerbated mortality compared to untreated rats. The molecular mechanisms underlying these symptoms are speculated to be a result of S1PR1 internalisation in response to prolonged exposure to SEW2871 (147). In mouse CLP-sepsis, on the other hand, SEW2871 administration 6 h after start of CLP at 10 mg/kg i.p. exerted a protective effect on kidney function, as measured by reduced blood urea nitrogen and creatinine protein levels in serum, and kidney morphology 18 h after CLP (148). The differences in outcome may be explained by differences between the pathophysiology of CASP and CLP sepsis, which may affect the pharmacological effect of SEW2871. Furthermore, the timing and dosage between both studies also differed. SEW2871 administration at 0.5 mg/kg 12 h after sepsis induction failed to improve sepsis-induced leakage or kidney function, while 10 mg/kg 6 h after sepsis did improve kidney function. These results could indicate that S1PR1 is involved in early onset of sepsis, higher dosages of S1PR1 agonist are required to invoke protective effects, and that there is a limited window of treatment after sepsis onset for SEW2871 to attenuate sepsis-induced organ dysfunction.
Another endothelial protein that the sepsis field has turned its attention to, is receptor kinase tunica intima endothelial kinase 2 (Tek), also known as Tie2. Tie2 exhibits enriched endothelial expression, but is also present in a variety of other cell types (149). Two ligands play a key role in regulating Tie2 signalling. Under non-inflammatory conditions, binding of the agonist angiopoietin 1 (Angpt1) to Tie2 induces anti-inflammatory (150) and barrier protective effects (151, 152) (Figure 5). The second ligand is the antagonist Angpt2, which competitively binds to Tie2, thus inhibiting Angpt1-dependent protective signal transduction in EC (153). Activation of Tie2 leads to its internalisation and degradation, thereby reducing Tie2 availability at the cell surface (154). APC is also reported to induce Tie2 downstream signalling through functioning as an agonist for Tie2, and treatment with APC 5 min prior to LPS administration prevented LPS-induced microvascular leakage in lung and kidney in a Tie2-dependent manner (155). Interestingly, TM-facilitated formation of APC is inhibited by binding of both Angpt1 and Angpt2 to TM (156), suggesting involvement of angiopoietins in coagulation, and adding an additional connection between the Tie2 pathway and APC.
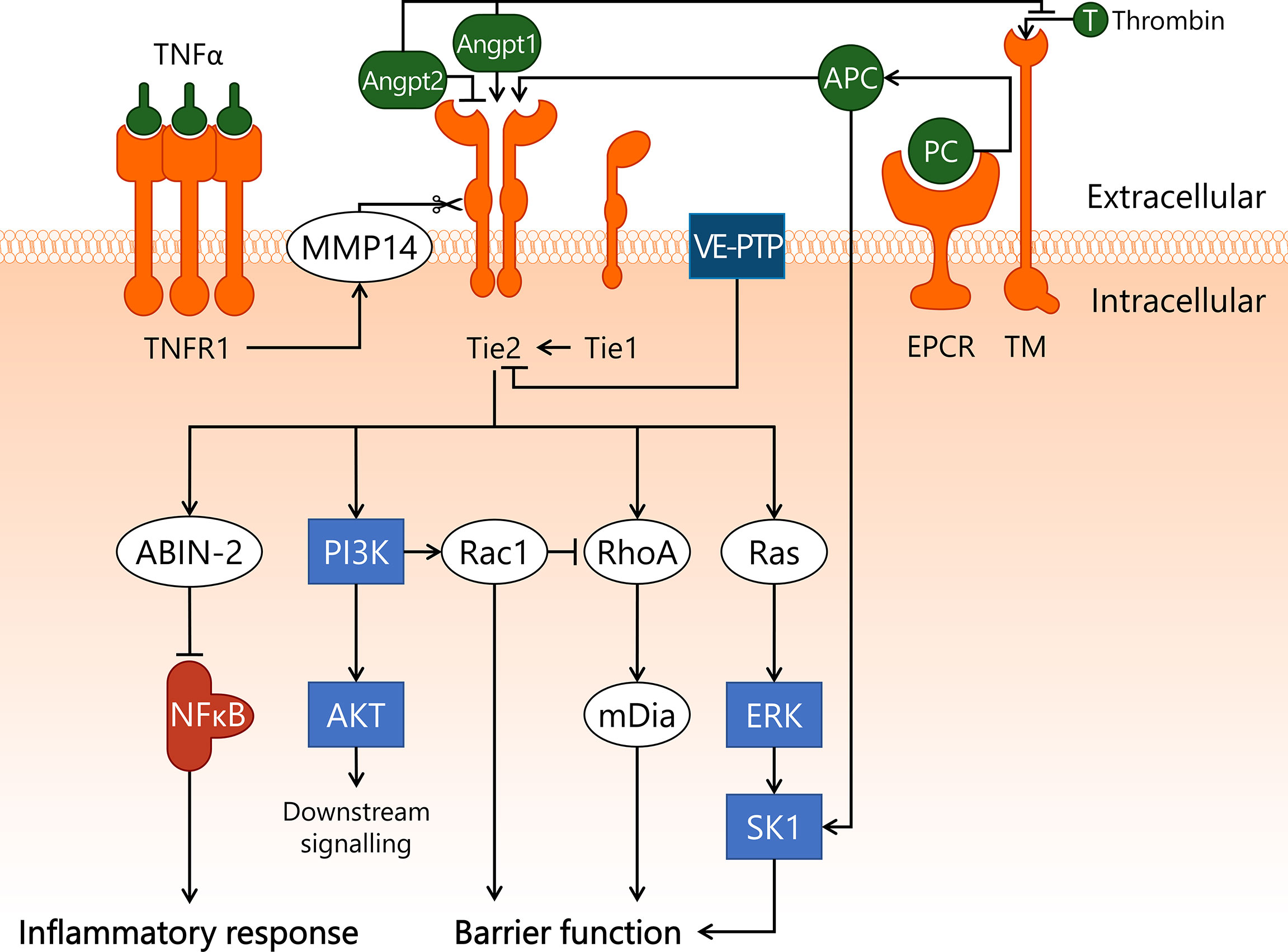
Figure 5 Endothelial Angpt/Tie2 signalling is compromised in sepsis. A key regulator of endothelial homeostasis – Tie2 – is heavily modulated in sepsis, which can result in endothelial dysfunction. Angpt1 and APC serve as agonists for Tie2, whereas Angpt2 competitively binds to Tie2, preventing its activation. Angpt1 and Angpt2 can also bind to TM, hereby inhibiting EPCR/TM/thrombin-dependent conversion of PC to APC. Tie2 activity is further modulated through association with Tie1, which lowers the activation threshold of Tie2 to promote its activation, and VE-PTP, which decreases Tie2 activation status through dephosphorylation. TNFα/TNRF1 promotes cleavage of the extracellular domain of Tie2 via MMP14. Upon activation, Tie2 exerts an anti-inflammatory effect by activating ABIN-2, which prevents nuclear translocation of NF-κB. In addition, Tie2 activates Ras/ERK/SK1 and PI3K/AKT. Finally, Tie2 has a protective effect on endothelial barrier function through PI3K-dependent activation of Rac1, and RhoA-dependent activation of mDia. ABIN-2, A20-binding inhibitor of NF-κB activation-2; AKT, protein kinase B; Angpt1, angiopoietin-1; Angpt2, angiopoietin-2; APC, activated protein C; ERK, extracellular signal-regulated kinase; mDia, mammalian Diaphanous-related formin Dia; MMP14, matrix metalloproteinase-14; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; PI3K, phosphoinositide 3-kinase; Rac1, Ras-related C3 botulinum toxin substrate 1; Ras, Ras protein subfamily; RhoA, Ras homolog gene family, member A; SK1, sphingosine kinase 1; T, thrombin; Tie1, tunica intima endothelial kinase 1; Tie2, tunica intima endothelial kinase 2; TM, thrombomodulin; TNFR1, tumor necrosis factor alpha receptor 1; TNFα, tumor necrosis factor alpha; VE-PTP, vascular endothelial protein tyrosine phosphatase.
The ability of Tie2 to initiate downstream signalling in response to its agonists is controlled by interactions with several associated regulatory proteins. Tie2 interacts with orphan receptor Tie1, which lowers the threshold of Tie2 activation under resting conditions (157, 158). This lowered activation threshold of Tie2 enables Angpt2 to function as a Tie2 agonist instead (157, 159), though the extent to which this interaction occurs in microvascular beds has not been reported. During inflammatory conditions Tie1 is cleaved, thereby reverting the lowered Tie2 activation threshold (157). Tie2 itself is also cleaved by matrix metalloproteinase-14 in response to inflammatory mediators such as TNFα, thereby inhibiting downstream signalling of Tie2 (160). Finally, Tie2 can be dephosphorylated by membrane-bound vascular endothelial protein tyrosine phosphatase (VE-PTP), leading to reduced Tie2-mediated signal transduction (161).
Downstream signalling of activated Tie2 includes multiple pathways and cellular processes. A Tie2-dependent anti-inflammatory effect is exerted through stimulation of A20-binding inhibitor of NF-κB activation-2 (ABIN-2) (150). ABIN-2 inhibits IKK, thereby preventing NF-κB translocation to the nucleus and interfering with inflammatory gene transcription (162). Tie2 also activates the PI3K/AKT pathway, after which PI3K stimulates Rac1. Rac1 then inhibits RhoA, resulting in protection of endothelial barrier function (71). Tie2 also activates mammalian Diaphanous-related formin Dia (mDia) in a RhoA-dependent manner (151). mDia exerts a protective effect on endothelial barrier function via an inhibitory interaction with the VEGFR2 pathway that will be discussed in more detail below. Finally, activation of Tie2 induces signalling through the Ras/ERK pathway (163), leading to activation of SK1 (164), thereby providing another connection between the Angpt/Tie2 pathway and the APC/S1P pathway.
Angpt/Tie2 signalling is disrupted during sepsis, due to elevated levels of Angpt2 (165). This results in increased endothelial inflammation and microvascular leakage (45, 166). Furthermore, in mouse endotoxemia Tie2 is downregulated in several organs, including brain, kidney and liver (167, 168). A study in renal biopsies of sepsis patients with renal dysfunction showed that mRNA levels of Tie2 were significantly decreased in renal biopsies of sepsis patients (169), though this effect was not reported in other studies (23, 170). Moreover, mRNA expression of both Angpt1 and Angpt2 were decreased in the kidney of sepsis patients (169). This indicates that therapeutic intervention in this pathway will require an approach that goes beyond merely altering the systemic Angpt1:Angpt2 ratio. Indeed, Angpt2 knockout failed to prevent kidney dysfunction in mouse endotoxemia, although a decrease in endothelial inflammatory activation was observed (168).
Angpt2-binding and Tie2-activating antibody (ABTAA), which induces oligomerisation of Angpt2 and subsequently activates Tie2, was used as intervention treatment in sepsis-associated microvascular dysfunction (171). In three different mouse sepsis models - CLP-sepsis, LPS endotoxemia, and S. aureus injection – ABTAA treatment 6 h and 18 h after sepsis induction improved survival rate. ABTAA was shown to reduce microvascular leakage in the lung assessed at 30 h after LPS injection. Furthermore, upon ABTAA administration 6 h after start of CLP, reduced heparanase protein expression was observed in lung and in glomeruli in the kidney 12 h post-CLP, indicating improved endothelial glycocalyx integrity. Finally, administration of ABTAA 6 h after the start of CLP lowered TNFα, IL-6, and Angpt2 protein levels in serum at 12 h, 18 h, and 24 h post-CLP (171). These anti-inflammatory capabilities of increased Tie2 activation likely contribute to normalisation of EC behaviour in sepsis. The above findings indicate that Tie2 signalling plays an important role in the onset and progression of various pathophysiological processes taking place during sepsis. Crucially, more research is required to elucidate which organs and which microvascular compartments are affected by altered Tie2 signalling during sepsis, before suitable targets within the Tie2 pathway can be identified for novel therapeutic strategies.
VEGF/VEGFR2 is a molecular signalling axis that controls microvascular permeability, by modifying endothelial behaviour in sepsis. Elevated plasma levels of VEGF have been reported in sepsis patients (172), which may lead to increased VEGF-dependent activation of VEGFR2. In mouse CLP-sepsis, treatment with anti-VEGF neutralising antibody bevacizumab at 0.1 mg/kg i.p. 1 h before CLP or 6 h after CLP improved survival rate (173), though these findings could not be reproduced in a later study in which bevacizumab was administered at 0.5 mg/kg i.p. immediately prior to CLP (174). Pre-treatment with bevacizumab 1 h before LPS injection i.p. in mice reduced microvascular permeability in kidney, lung, and spleen at 24 h after LPS administration (173). Pre-treatment with a neutralising antibody targeting both Angpt2 and VEGF (A2V) in mouse CLP-sepsis alleviated endothelial inflammatory activation in kidney as assessed by reduced intercellular adhesion molecule 1 (ICAM-1) protein expression in peritubular capillaries, reduced pulmonary permeability, and improved 5-day survival (175). These findings suggest that decreased interaction of VEGF and Angpt2 with their respective receptors results in survival benefit in this particular setting. However, it was not reported whether VEGF and Angpt2 plasma levels or VEGFR2 and Tie2 activation levels were affected in response to A2V treatment, which calls for further studies into the underlying molecular mechanisms of this treatment.
Upon binding of VEGF, VEGFR2 will autophosphorylate several tyrosine sites in its intracellular domain to initiate downstream signalling via Src protein tyrosine kinase (Src) (176, 177) (Figure 6). VE-PTP can dephosphorylate these autophosphorylation sites, hereby reducing the ability of VEGFR2 to activate its downstream signalling targets (178). VE-PTP-induced dephosphorylation of VEGFR2 is dependent on Tie2 (178). In addition, Tie2 stimulates mDia to sequester Src (151), rendering Src unable to induce further downstream VEGFR2 signal transduction. Activated Src interacts with focal adhesion kinase 1 (FAK1), resulting in phosphorylation and increased activity of both Src and FAK1 (179). Both Src and FAK1 then activate the Ras/ERK signalling pathway (180). FAK1 is most well-known for its role in integrin signalling, and is implicated in endothelial inflammatory activation (181–183). In rat endotoxemia, knockdown of FAK1 by siRNA elicited a protective effect through a reduction of injurious inflammation-induced remodelling of lung tissue (184). FAK1 knockdown also inhibited LPS-induced inflammatory activation in human umbilical vein endothelial cells (183), as well as prevented injurious remodelling of cardiac tissue in rat endotoxemia (185). These studies underscore the role of FAK1 in some deleterious processes following endotoxemia-induced inflammation, yet much remains to be elucidated regarding the exact molecular involvement of FAK1 in inflammation in general and in sepsis in particular. Importantly, further identification of the cell types in which FAK1 contributes to these processes is required before therapeutic strategies and accompanying biomarker readouts can be designed.
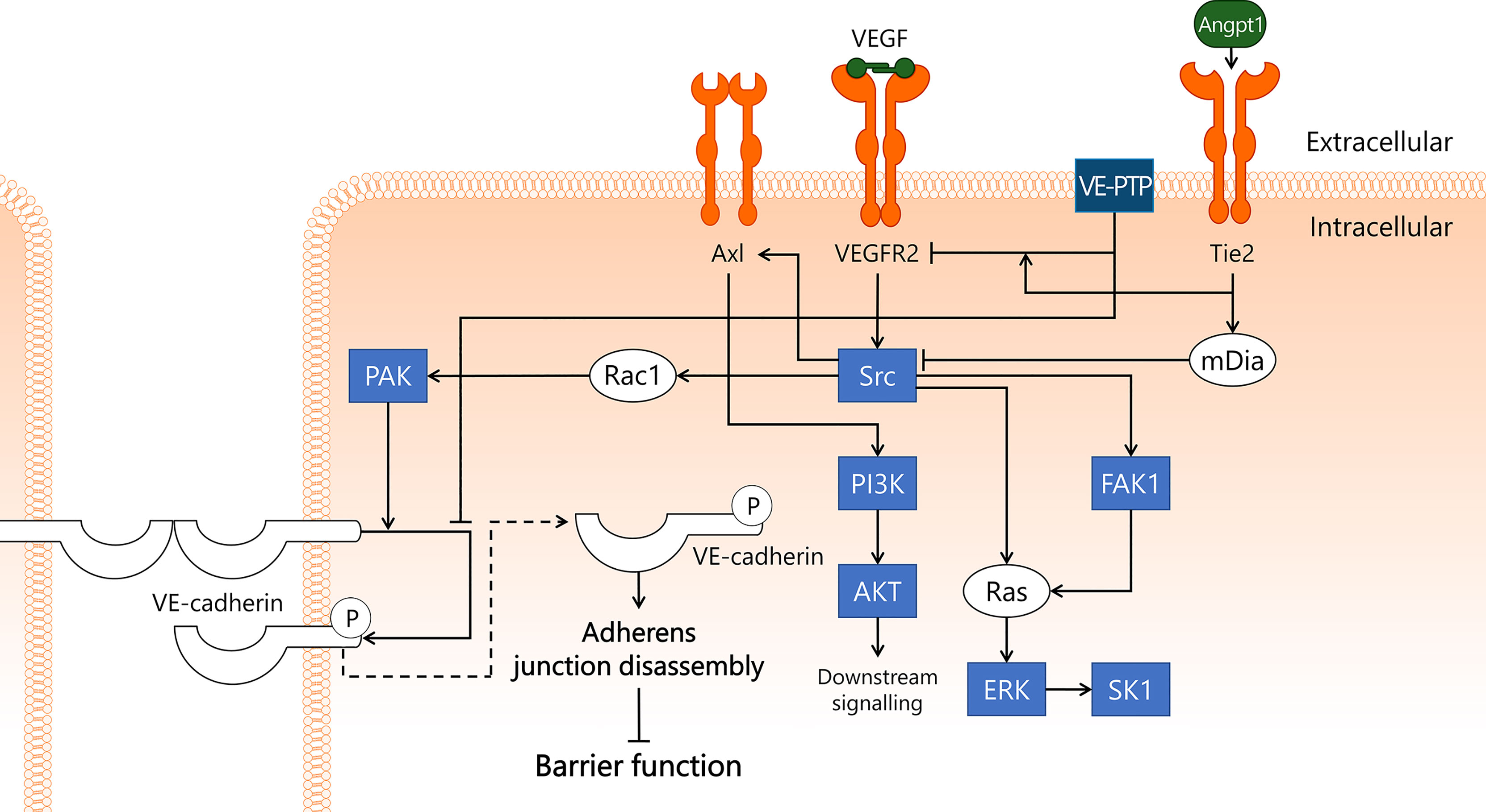
Figure 6 VEGF/VEGFR2 pathway disrupts endothelial barrier function in sepsis. VEGFR2 is well-known for its role in angiogenesis, but it is also thought to play a key role in sepsis-induced endothelial barrier dysfunction. VEGF binds to VEGFR2, leading to activation of Src kinase. Src then binds to and activates FAK1, which induces activation of Ras/ERK/SK1 through both Src and FAK1. In addition, Src activates PI3K/AKT via Axl, and Rac1/PAK. PAK phosphorylates VE-cadherin, leading to its internalisation and subsequent disassembly of adherens junctions, which results in disrupted endothelial barrier function. This process is inhibited by VE-PTP, which closely associates with VE-cadherin, hereby preventing its phosphorylation. VE-PTP also dephosphorylates VEGFR2, a process that is promoted by the vicinity of Tie2. Tie2 activation induces activation of mDia, which sequesters Src and prevents its involvement in the VEGF/VEGFR2 pathway. AKT, protein kinase B; Angpt1, angiopoietin-1; Axl, Axl receptor tyrosine kinase; ERK, extracellular signal-regulated kinase; FAK1, focal adhesion kinase 1; mDia, mammalian Diaphanous-related formin Dia; PAK, p21-activated kinase; PI3K, phosphoinositide 3-kinase; Rac1, Ras-related C3 botulinum toxin substrate 1; Ras, Ras protein subfamily; SK1, sphingosine kinase 1; Src, Src protein tyrosine kinase; Tie2, tunica intima endothelial kinase 2; VEGF, vascular endothelial growth factor; VEGFR2, vascular endothelial growth factor receptor 2; VE-cadherin, vascular endothelial cadherin; VE-PTP, vascular endothelial protein tyrosine phosphatase.
In addition to FAK1, Src activates transmembrane Axl receptor tyrosine kinase (Axl), which then leads to activation of the PI3K/AKT pathway (186). Axl can also be activated by its extracellular ligand, growth arrest specific 6 (Gas6), which is increased in sepsis in the blood and is suggested to play a role in systemic inflammation (187). A clinical study in sepsis patients reported that elevated Gas6 levels were associated with increased mortality (188). At this point, the exact role of Gas6 in sepsis is not well understood, nor is it known whether interactions with Axl in microvascular EC contribute to morbidity and/or mortality in sepsis patients.
Finally, Src plays a crucial role in VEGFR2-induced endothelial barrier dysfunction in conjunction with VE-cadherin, the main structural component of adherens junctions that is responsible for the physical interaction between neighbouring EC. Src induces activation of Rac1 and its effector protein PAK, which then phosphorylates VE-cadherin (176). This phosphorylation leads to internalisation of VE-cadherin, resulting in disassembly of the adherens junctions, loss of endothelial barrier function, and ultimately increased microvascular leakage (176, 189). In mice, stable VEGFR2/VE-cadherin complex formation in heart EC was disrupted in a Src-dependent fashion following VEGF injection (190). Internalisation of phosphorylated VE-cadherin may be prevented by dephosphorylation by VE-PTP (191). In this manner, VE-PTP contributes to adherens junction stability and endothelial barrier integrity. In addition to being internalised after phosphorylation, VE-cadherin can also be shed from the endothelial cell surface into the circulation. A study in sepsis patients reported elevated sVE-cadherin levels in a subset of patients that were suffering from acute kidney injury (192). There are also organ-specific variations in VE-cadherin expression in response to inflammatory stimuli, as VE-cadherin mRNA levels in the lung decreased significantly 4 h after i.p. LPS administration, whereas no changes were observed in the kidney (23). It remains to be determined whether loss of VE-cadherin in sepsis is occurring from the majority of EC, or only affects specific microvascular compartments, and whether sVE-cadherin exerts a signalling function through recognition by and interaction with EC. By quantifying VE-cadherin protein levels in different microvascular beds in organs during onset and development of sepsis to determine changes in expression levels in response to sepsis, or by studying the effects of exposing EC to sVE-cadherin, a deeper understanding of the role of VE-cadherin in sepsis can be obtained.
Several clinical trials in severe sepsis patients have been conducted to assess the effects of intervening in one of the discussed signal transduction pathways. Studied molecular targets include components of the NF-κB pathway (Figure 1). For example, blockade of TLR4 was employed in a clinical trial in which sepsis patients were treated with eritoran, a synthetic TLR4 antagonist that reduced LPS-dependent activation of TLR4. No effects on 28-day mortality were observed compared to patients who received placebo (193). TAK-242, also known as resatorvid, is a small molecule inhibitor of TLR4 that interacts with the intracellular domain of TLR4, thereby inhibiting the downstream signalling cascade initiated by bacterial LPS. TAK-242 treatment in sepsis patients did not affect plasma IL-6 levels, indicating no overall change in inflammatory status of the patients. Importantly, 28-day mortality was also not improved after drug treatment (194).
Another approach investigated blockade of TNFα to blunt the inflammatory response in sepsis patients. As a primary mediator of systemic inflammation in sepsis, its neutralisation could in theory prevent further downstream activation of harmful responses, as shown in baboons (195). This rationale was followed in a study in which a neutralising polyclonal anti-TNFα antibody fragment called AZD9773 was administered to patients with severe sepsis. AZD9773 effectively reduced circulating TNFα plasma levels, yet did not result in reduction of IL-6 plasma levels, nor did it improve 90-day survival of patients (196).
Taking out one major upstream component of an inflammatory signalling pathway to counteract the detrimental effects of sepsis on the microvasculature seems a straightforward manner to modulate microvascular inflammation and leakage. However, the clinical trials discussed above illustrate that this approach is not by definition effective in treating sepsis patients. This may be explained by the existence of compensatory molecular mechanisms, where alternative signal transduction pathways become activated to compensate for the lack of activation of the inhibited pathway. For instance, exposure of EC to LPS or TNFα both lead to activation and translocation of NF-κB. When TNFα-dependent activation of NF-κB is inhibited in sepsis patients, it is conceivable that this is insufficient to abolish inflammatory activation, as e.g. LPS-dependent endothelial inflammatory activation will still be possible. Similarly, when TLR4 is blocked with drugs like eritoran or TAK-242, the activation of RIG-I by LPS may remain unaffected or in theory could even increase to compensate for reduced TLR4 activation.
A major setback to the field of sepsis and sepsis-MOF research was the PROWESS-SHOCK clinical trial published in 2001. The overall aim was to improve sepsis patient outcome through administration of APC, which was hypothesised to prohibit sepsis-related coagulopathy and attenuate inflammatory activation. Sepsis patients were treated with recombinant human APC, also known as drotrecogin alfa (activated) (DrotAA, or Xigris). DrotAA was initially reported to reduce mortality in patients with severe sepsis (197), but in a later study no such evidence was found. Moreover, DrotAA caused bleeding complications due to its anti-coagulatory effects (198). The outcome of the PROWESS-SHOCK study has played a role in perpetuating the stigma surrounding sepsis research as “the graveyard for pharmaceutical companies” (199). This is a self-fulfilling prophecy, for a condition as complex and heterogeneous as sepsis will require large clinical studies with proper patient subtyping to establish treatment effectiveness, to increase our understanding of sepsis pathophysiology, and to identify biomarkers for different disease stages. When pharmaceutical companies are hesitant to invest in new sepsis research because it is deemed too high-risk, the progress of the sepsis research field as a whole will consequently be slowed down considerably.
The overall complexity and heterogeneous presentation of sepsis led to the identification of 4 distinct clinical phenotypes, including a phenotype characterised by abnormal coagulation (200). The SCARLET clinical trial investigated whether the anti-coagulatory capabilities of sTM (a.k.a. ART-123) could ameliorate the disseminated coagulation in patients suffering from sepsis-associated coagulopathy. However, treatment with recombinant human sTM did not reduce 28-day mortality (201). A follow-up clinical trial called SCARLET2 was later launched, focusing on severe sepsis patients with coagulopathy and dysfunction of at least one organ, but the study was withdrawn to accommodate changes in study design based on new results (NCT03517501).
While anti-VEGF neutralising antibody bevacizumab showed variable results in mouse sepsis (173, 174), a clinical trial was set up to test its effectiveness in sepsis patients, in particular in preventing development of sepsis-associated acute respiratory distress syndrome. This study was prematurely withdrawn before participants were enrolled (NCT01314066).
The above discussed clinical trials include interventions in the NF-κB, APC/S1P, and VEGF/VEGFR2 signalling pathways. No studies intervening in the AP-1, Rac1/RhoA GTPases, and Angpt/Tie2 signalling pathways have yet reached the stage of clinical trials. These pathways still represent promising therapeutic targets to ameliorate endothelial dysfunction in sepsis. Furthermore, it is important to note that this review focuses on the role of the endothelium in sepsis yet the discussed signalling pathways are not exclusive to EC. As a result, clinical trials intervening in these pathways likely affect the endothelium, but also a wide range of other cell types. For this reason, it is vital that markers for endothelial damage and/or inflammation are being investigated and reported, as it is one of few available tools to study EC responses in patients. In actuality, endothelial damage and inflammation markers are seldomly studied or reported in clinical trials, thereby obscuring the effects of these treatments on the endothelium.
The collective effort of in vitro studies, animal experiments, analyses of human samples obtained from patients, and clinical trials conducted in sepsis patients have contributed tremendously to our understanding of the cellular and molecular pathways in EC that contribute to the onset and progression of sepsis and sepsis-MOF. This review described six signal transduction pathways involved in endothelial activation in sepsis and in conditions mimicking sepsis, that play crucial roles in modulating coagulation, microvascular permeability, and leukocyte recruitment. An integrated schematic presentation of what we know so far about these pathways is represented in Figure 7 (relevant references are summarised in Supplementary Table 1), in which the sheer number of connections both within and between signal transduction pathways can be appreciated. We now face the challenge of not only expanding this knowledge further, but also to translate it into effective drug treatment strategies. In the clinical trials discussed, the underlying rationale often revolves around neutralisation of systemic pro-inflammatory mediators. Therapeutic regimens in clinical trials seldomly include pharmacological (kinase) inhibitor drugs, as summarised by Marshall in 2014 (202). This particular treatment strategy in sepsis is still in its infancy, and therapeutic targeting of kinases and phosphatases holds promise towards the development of successful treatment regimens for sepsis patients. This is spurred by technological advancements to analyse kinase and phosphatase activity, that now allow us to determine kinome and phosphatome profiles in cells and tissue during a particular condition (183, 203). This strategy reveals an unbiased selection of kinases and phosphatases with increased or decreased activity, and thereby will help identify potential targets for therapeutic intervention, as well as detecting potential side effects in non-diseased cells and tissues (204).
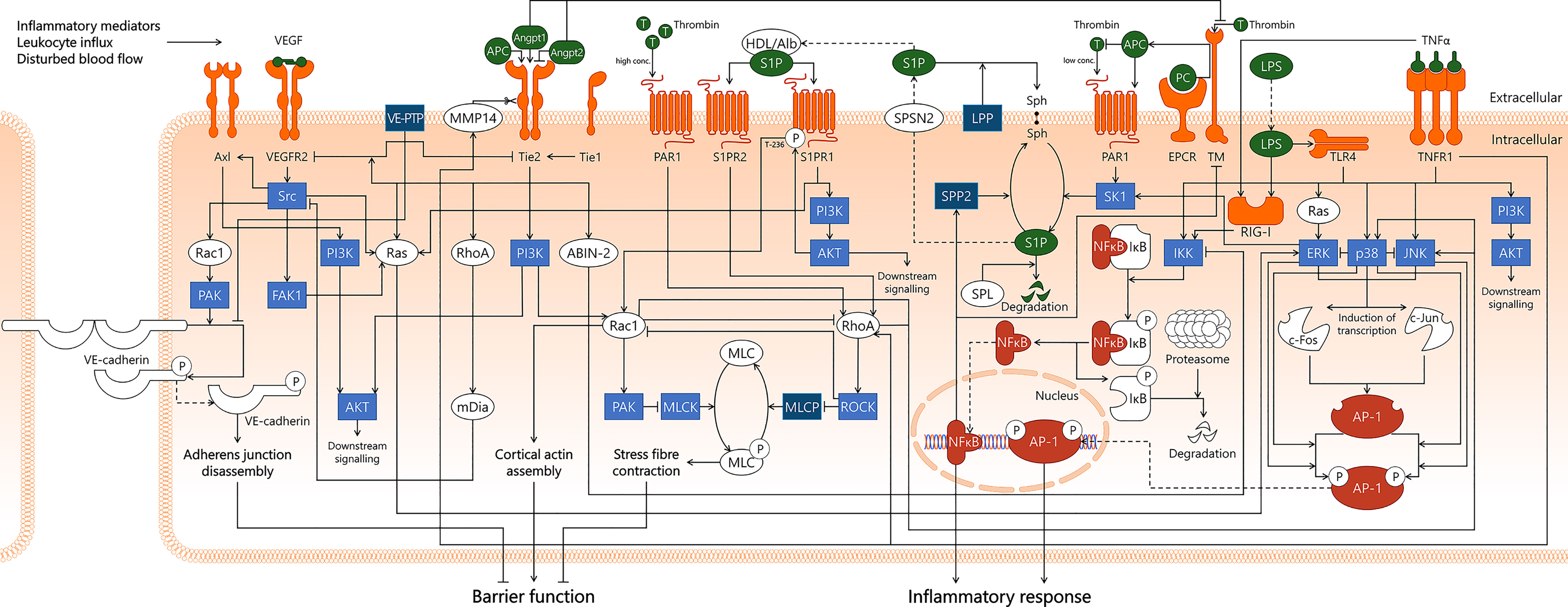
Figure 7 Integration of signal transduction pathways involved in endothelial responses in sepsis that affect endothelial barrier function and inflammatory responses. From left to right, this figure displays the VEGF/VEGFR2, Angpt/Tie2, Rac1/RhoA GTPases, APC/S1P, NF-κB, and AP-1 signal transduction pathways. This overview illustrates the complexity of the interactions of pathways involved in the endothelial response to sepsis, as well as displaying the numerous links that exist between different pathways. Coagulation-affecting pathways are not separately indicated for clarity reasons. ABIN-2, A20-binding inhibitor of NF-κB activation-2; AKT, protein kinase B; Alb, albumin; Angpt1, angiopoietin-1; Angpt2, angiopoietin-2; APC, activated protein C; AP-1, activator protein 1; Axl, Axl receptor tyrosine kinase; c-Fos, cellular Fos proto-oncogene; c-Jun, cellular Jun proto-oncogene; EPCR, endothelial protein C receptor; ERK, extracellular signal-regulated kinase; FAK1, focal adhesion kinase 1; HDL, high-density lipoprotein; IKK, IκB kinase; IκB, inhibitor of κB; JNK, c-Jun N-terminal kinase; LPP, lipid phosphate phosphatase; LPS, lipopolysaccharide; mDia, mammalian Diaphanous-related formin Dia; MLC, myosin light chain; MLCK, myosin light chain kinase; MLCP, myosin light chain phosphatase; MMP14, matrix metalloproteinase-14; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; p38, p38 mitogen-activated protein kinase; PAK, p21-activated kinase; PAR1, protease-activated receptor 1; PC, protein C; PI3K, phosphoinositide 3-kinase; Rac1, Ras-related C3 botulinum toxin substrate 1; Ras, Ras protein subfamily; RhoA, Ras homolog gene family, member A; RIG-I, retinoic acid-inducible gene I; ROCK, Rho-associated protein kinase; S1P, sphingosine-1-phosphate; S1PR1, sphingosine-1-phosphate receptor 1; S1PR2, sphingosine-1-phosphate receptor 2; SK1, sphingosine kinase 1; Sph, sphingosine; SPL, sphingosine-1-phosphate lyase; SPNS2, Spinster homolog 2; SPP2, sphingosine-1-phosphate phosphatase 2; Src, Src protein tyrosine kinase; T, thrombin; Tie1, tunica intima endothelial kinase 1; Tie2, tunica intima endothelial kinase 2; TLR4, Toll-like receptor 4; TM, thrombomodulin; TNFR1, tumor necrosis factor alpha receptor 1; TNFα, tumor necrosis factor alpha; VEGF, vascular endothelial growth factor; VEGFR2, vascular endothelial growth factor receptor 2; VE-cadherin, vascular endothelial cadherin; VE-PTP, vascular endothelial protein tyrosine phosphatase.
Many kinase inhibitors interact with the catalytic domain of a kinase, thereby rendering it unable to phosphorylate its substrate. Due to structural homology between kinases, kinase inhibitors tend to not only inhibit the activity of one kinase, but also alter the activity of other types of kinases (205). These so-called off-target effects are commonly seen as an impediment to a broader application of kinase inhibitor treatments, because it can lead to unwanted and unpredictable side-effects (206, 207). Moreover, in some instances the exact target(s) of kinase inhibitor drugs are poorly described. This is exemplified by a recent study revealing that kinase inhibitor OTS964 exerts its function not through its putative target PDZ-binding kinase, but rather via cyclin-dependent kinase 11 (208). The misidentification of the target of OTS964 likely stems from RNA interference promiscuity, an obstacle that is circumvented with recently developed CRISPR-based gene knockdown (208). In the development of new generation kinase inhibitor drugs, increasing emphasis is put on enhanced target specificity, while reducing off-target effects (209, 210). Although the reasoning behind this development is valid, it is likely not the way forward for the application of kinase inhibitor drugs for sepsis treatment. Keeping the considerable levels of redundancy between signalling pathways in mind, targeting a single kinase with a highly specific kinase inhibitor may potentially be ineffective, as related pathways will strive to compensate for the loss of activity of that particular kinase (211). Instead, kinase inhibitors with low(er) target specificity can target a wider group of kinases and might therefore be more effective in shutting down a combination of pathways without evoking compensation. This topic is reviewed in more detail elsewhere (209). If specific inhibition of a single kinase is preferred, it is convenient to also have drugs with higher levels of specificity at one’s disposal to satisfy both needs. The field of oncology has a long history with the development of and treatment with kinase inhibitors, which has led to an expanding number of kinase inhibitor drugs that are FDA/EMA-approved (212, 213). Whenever one of these drugs is found to improve the condition of sepsis patients, such repurposed drugs can complete mandatory intermediary steps more rapidly than newly developed drugs before allowing administration of this drug to sepsis patients (214). An example of a repurposed kinase inhibitor drug for sepsis is imatinib. In mouse CLP-sepsis this drug, developed as multi-target kinase inhibitor for anticancer treatment (215), attenuated renal microvascular leakage (216). Both repurposing as well as off-label use of kinase inhibitor drugs are becoming increasingly commonplace in oncology, which could serve as an example for drug development for sepsis (217, 218).
Another important consideration involves the position of the target kinase of choice in its respective signalling cascade. On the one hand, it could be effective to target a kinase that functions during the initiation of the sepsis-induced intracellular signalling cascade, e.g. tyrosine kinase receptors, which then inhibits the very start of the signal transduction pathway. On the other hand, a strategy relying on kinases that are further downstream in the signalling cascade has the benefit of only targeting components of the pathway that are deemed injurious. In this way, non-damaging or potential beneficial signalling pathways of the original stimulus remain unaffected, whilst only the harmful pathway is inhibited. Opting for targeting kinases at either the start or end of the signalling cascade will most likely depend on the existence of pathway redundancies, the target cell of interest, the stage of the target disease, and insights in the time frame in which a particular treatment can effectively block signalling in its target cells. During early sepsis, pro-inflammatory mediators will already have had the opportunity to initiate intracellular signalling in various cell types involved in sepsis pathophysiology. Inhibition of kinases after they have already initiated signalling is likely ineffective. Instead, a more effective approach could involve targeting kinases that become activated at later stages of sepsis, considering that it typically requires hours to days to develop clinical symptoms before sepsis can be diagnosed. This also highlights the need for reliable blood biomarkers to detect early sepsis, and to establish the stage of disease progression in patients. More research is required to identify windows of time within which therapeutic treatment with a particular drug of interest would have favourable effects for sepsis patients.
With regard to the administration of kinase inhibitor drugs to patients, another issue that needs to be addressed is the different cell types that are targeted by the treatment. For example, a certain kinase inhibitor treatment might prevent sepsis-induced dysfunction of EC in glomeruli of the kidney upon systemic administration. The drug will also have access to and thus affect cells other than glomerular endothelium. This can lead to altered kinase activity in other microvascular beds in the kidney and in other organs, as well as in non-endothelial cell types throughout the body. Even though some kinases and phosphatases are known to be restrictedly expressed by EC, the majority of kinases and phosphatases is expressed in a multitude of cell types. For that reason, systemic inhibition of kinases or phosphatases could lead to side-effects of the treatment that in a worst-case scenario completely counteract its beneficial effects. Deleterious side-effects of drugs may depend on the activation status of the targeted kinase in these non-target cell and tissue types, but this knowledge is currently lacking. Recent advances in kinome and phosphatome activity arrays (204), as well as phosphoproteomics (219), represent novel opportunities to contribute to our understanding of kinase activation status in different cell and tissue types, and may help predict side-effects of particular drug treatments.
Most research that strives to identify novel druggable targets in sepsis and sepsis-MOF follows the traditional pipeline of in vitro studies, followed by animal studies, and in some cases the study next advances to the stage of clinical trials (220). Even though this strategy has led to success in other fields (213), and has also contributed to our current understanding of sepsis, it has not yet led to the development of an effective therapeutic strategy to halt the development of multiple organ failure as a result of sepsis (221). One reason for this might lie in the complexity of sepsis, or that in vitro studies are generally a poor predictor of how cell types respond to a certain stimulus or drug in vivo. This is particularly true for EC, which exhibit unique gene expression profiles and behaviour when being in their microvascular environment, a feature that is lost when they are isolated and cultured (222). Immortalised endothelial cell lines are the farthest removed from mimicking the in vivo situation. This issue is somewhat salvaged by culturing primary EC (223), but also these cells quickly drift into a generic EC phenotype, leading to the loss of their unique in vivo characteristics. This loss of unique characteristics is likely a combination of the absence of flow in most in vitro settings, lack of interaction with supporting cells such as pericytes and smooth muscle cells, and lack of interaction with bloodborne cells (222).
To maximise the translational value of the available models, we propose that we depart from the linearity and unidirectionality of the traditional research pipeline and adopt an approach that facilitates going back and forth between in vitro, in vivo, ex vivo, and between animal models and patients. By no means does this suggest omitting any of the aforementioned steps, but it rather ensures that information on the involvement of a target molecule in the disease process is maximised to enable informed decision-making. These intermediary steps include organ-on-a-chip, organoids, ex vivo precision cut organ slices, and patient biopsies. In short, organ-on-a-chip are microdevices containing different cell types that can mimic key characteristics of particular organs in vitro (224). Organoids rely on self-organisation of the cells to form more complex 3D structures compared to organ-on-a-chip, which are also used to study particular aspects of organ behaviour in vitro (225). To identify the involvement of a target kinase in the development of sepsis in EC, organ-on-a-chip and organoids could be implemented to study the activity of the target kinase under homeostatic and pro-inflammatory conditions. This would increase our understanding of the various cell types within an organ that express the kinase of interest. Innovations to enable (vascular) flow in these models are under development (226), and vascular flow in kidney organoids has been described as a prerequisite for proper vascularisation to take place (227). Further developments are required for these models to become relevant tools to study endothelial heterogeneity, as the kidney cortex alone consists of distinct microvascular beds, i.e. arterioles, glomeruli, peritubular capillaries, and post-capillary venules (6), which represent a level of structural complexity not yet present in kidney organoids. Post-mortem organ biopsies of both sepsis and non-sepsis donors could help determine the presence of the target kinase in different organs using protein detection tools ranging from ELISA to proteomics. In addition, using kinase activity arrays (204) and phosphoproteomics (219) to compare kinase activation status in biopsies from both sepsis and non-sepsis donors could hint towards the involvement of the target kinase in sepsis.
Precision cut tissue slices (PCTS) represent small sections of viable human tissue, in which cell types can be studied while situated in their original pathophysiological niche (228, 229). Importantly, the retained endothelial heterogeneity of the microvascular compartments in these organs provides valuable information on microvascular compartment-specific responses to certain stimuli that would be unattainable in vitro (230). One could also imagine a setting in which PCTS from both humans and experimental animals are compared for their response to a particular kinase inhibitor drug. If the response of the human PCTS differs greatly from that of animal PCTS, continuation of this study in that particular animal model might not be worthwhile, as this indicates that the effects of drug treatment in animal will not be representative of effects in humans. In the case of biomarker development in conjunction with novel therapies, a biomarker of sepsis found in a human PCTS model can be validated in human patient blood or urine. If its presence is confirmed, it can be determined whether this is also the case in the animal model utilised for sepsis. If so, one can determine expression levels of the biomarker in e.g. mice upon treatment with a novel drug, and next translate this back to the human PCTS model. In this manner, there is constant crosstalk between models, and incongruencies caused by differences in models or differences between species are discovered at an early stage. These tools might offer earlier confirmation that a target and/or biomarker of interest truly is involved in the development of sepsis or indicate that pursuit of the target is not worthwhile. In the end, all experimental models come with their own advantages and disadvantages, and this realisation can help select combinations of models that will answer the research question at hand. The traditional research pipeline has not resulted in a drug to effectively treat sepsis, which can be viewed as a confirmation that in vitro and animal studies alone are poor predictors of pathophysiology in sepsis patients. Inclusion of organ-on-a-chip, organoids, precision cut organ slices, and patient biopsies when designing novel drug therapies will enable making better-informed decisions and will allow resources to be spent on more promising targets and biomarkers.
There are additional ways in which sepsis animal studies and clinical trials can be further optimised. For instance, many animal studies and clinical trials with sepsis patients tend to focus on a limited number of readouts, including IL-6 plasma levels as a parameter of inflammation, or biomarkers indicating dysfunction of a single organ. Sepsis is by definition a systemic disease, that concurrently induces dysfunction of multiple organs. Using an animal sepsis model to study the effects of an experimental drug only on e.g. sepsis-induced acute lung injury is an interesting quest in its own right but fails to acknowledge the fact that the drug might also influence the disease trajectory in organs that are not of primary interest. The drug might be reported as a promising perspective for sepsis patients because it reduced acute lung injury, while at the same time effects on e.g. kidney and liver were not reported, or vice versa. To gain a more complete overview of the effects that drugs have on sepsis-related pathophysiology, effects of drugs should be assessed in brain, heart, lung, liver, and kidney, as they are most commonly affected in sepsis. Such an approach would vastly increase the quantity and quality of information that is generated per animal. This is especially true for vascular research, because the extensive heterogeneity of EC between and within organs may underlie microvascular subsets responding divergently from one another (6). By studying and reporting the effects of drugs on all major organs, studies will become increasingly informative, while simultaneously enhancing the translatability of this translational research.
This ties in with the previously discussed PROWESS-SHOCK clinical trial, which eventually showed not to improve survival of sepsis patients despite promising preceding findings. Firstly, sepsis treatment should not negatively affect non-diseased tissue in such a way that it worsens the overall condition of the patient or studied animal. In the case of PROWESS-SHOCK, patients receiving treatment initially showed attenuated systemic inflammation and improved survival (197), positive effects that were not found in a later study (198). The reported lack of improved survival in the latter study cannot be fully explained, but complications caused by the effects of the drug on coagulation and subsequent bleeding complications may have played a role. Gaining an indication of whether the positive effects of a drug exceed the negative side-effects before entering clinical trials is exactly what the sepsis and sepsis-MOF research field is struggling with. The variation between and within different sepsis animal models (221, 231) only further adds to the already limited translatability of preclinical research to the clinical phase (220). In addition, coagulopathy exemplifies the differences in symptoms between human sepsis and animal models of sepsis, as it is a common condition in sepsis patients, yet many animal models of sepsis fail to produce symptoms mimicking sepsis-associated coagulopathy (232), thereby considerably reducing their translational value. Secondly, the PROWESS-SHOCK study forces us once more to acknowledge the extreme heterogeneity of sepsis patients, which is underscored by the recent description of the existence of four clinical sepsis phenotypes. These phenotypes are characterised based on a variety of host-response biomarkers, including markers of abnormal coagulation (200). Coagulopathy affects individual patients to different extents at different times (14). Therefore, a drug with coagulation-modulatory side-effects might be harmless or even beneficial for one patient, while being detrimental to a patient with a different clinical sepsis phenotype.
There are two final points that may improve the translatability of animal studies or clinical trials for sepsis. First of all, a shift from drug pre-treatment to drug intervention studies in animal models of polymicrobial sepsis will help assess drug effectiveness in a setting that is clinically more relevant. While pre-treatment with a drug of choice before inducing sepsis can indicate the involvement of the target under study in the onset and progression of sepsis, it does not provide any information on applicability of the drug for sepsis treatment. Hence, when performing animal studies with novel drugs aimed to ameliorate sepsis, intervention studies need to take up a considerable portion of the research, as these are better representatives of the performance of the drug in the complexity of the pathophysiological process. This is tying in with the second point, that deals with the importance of providing proof of the activity of the drug in its target tissue and its targeted cells. When a kinase inhibitor drug is administered in an animal sepsis model to ameliorate acute lung injury by targeting a kinase in lung EC, it is insufficient to simply report the extent of acute lung injury or show altered levels of soluble markers of endothelial dysfunction in plasma. Neither parameter provides the necessary proof that the pulmonary vasculature was indeed effectively targeted by the drug. Instead, one might consider quantification of inflammatory proteins expressed by target cells in the target tissue, in addition to quantifying activity levels of the target kinase of the inhibitor in EC. In addition, tools such as laser microdissection can be used to zoom in on individual microvascular compartments, for instance to study their unique gene expression patterns in both health and disease (22). Laser microdissected material is also compatible with (mi)RNA sequencing platforms, allowing for high-throughput characterisation of microvascular compartment-specific signatures (233). These types of analyses will create better insights into the molecular mechanisms of drugs and will enable identification of subsets of cells that are, or are not, affected by the drug treatment. Anything other than direct evidence is suboptimal, because ambiguity about the pharmacological mechanism of a drug at this phase is bound to lead to unforeseen side-effects of the drug at subsequent stages of development.
Despite considerable advances in our understanding of sepsis pathophysiology, clinicians and researchers have not yet been able to develop effective drug treatment regimens to prevent or treat sepsis and sepsis-MOF. In this review, we set out to highlight the role of endothelial cells in sepsis, as these represent an understudied cellular player in the pathophysiology of sepsis, and we provided a comprehensive overview of the major signal transduction pathways involved in sepsis-induced endothelial cell activation and dysfunction. We discussed animal studies and clinical trials intervening in these pathways, with emphasis on how the involvement of interconnected kinases and phosphatases form sepsis-associated intracellular signalling networks. Despite the potential that kinase (and phosphatase) inhibitor drugs hold as a treatment for sepsis, several challenges need to be overcome before their applicability for sepsis patients can be assessed. The molecular mechanisms involved in sepsis are so complex, that it seems unlikely that any approach targeting a single cell type or a single pathway will succeed. Yet through adopting a research design with which the amount of information obtained from a single study increases, while simultaneously enhancing the translatability of outcomes in the lab to clinical applications via intermediary models including organoids and precision cut tissue slices, the overarching goal of uncovering effective therapeutic treatments for sepsis and sepsis-MOF becomes within reach.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This project is co-financed by the Ministry of Economic Affairs and Climate Policy by means of the PPP-allowance made available by the Top Sector Life Sciences & Health to stimulate public-private partnerships (#6334, MM).
GM is cofounder and CSO/CT of Vivomicx, which provides laser microdissection analysis services. Vivomicx financially contributes to the PPP-allowance.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank Jort Greefhorst for his contribution to the earliest iterations of the signal transduction pathway figures. The figures in this manuscript contain elements created by Motifolio (www.motifolio.com).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.867625/full#supplementary-material
ABIN-2: A20-binding inhibitor of NF-κB activation-2
AKT: protein kinase B
Angpt1: angiopoietin-1
Angpt2: angiopoietin-2
APC: activated protein C
AP-1: activator protein 1
Axl: Axl receptor tyrosine kinase
CASP: colon ascendens stent peritonitis
CLP: cecal ligation and puncture
c-Fos: cellular Fos proto-oncogene
c-Jun: cellular Jun proto-oncogene
EC: endothelial cell
EPCR: endothelial protein C receptor
ERK: extracellular signal-regulated kinase
FAK1: focal adhesion kinase 1
Gas6: growth arrest specific 6
HDL: high-density lipoprotein
ICAM-1: intercellular adhesion molecule 1
IKK: IκB kinase
IL-1β: interleukin 1 beta
IL-6: interleukin 6
IκB: inhibitor of κB
JNK: c-Jun N-terminal kinase
LPS: lipopolysaccharide
MCP-1: monocyte chemoattractant protein 1
mDia: mammalian Diaphanous-related formin Dia
miR: microRNA
MLC: myosin light chain
MLCK: myosin light chain kinase
MLCP: myosin light chain phosphatase
MMP14: matrix metalloproteinase-14
NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells
p38: p38 mitogen-activated protein kinase
PAK: p21-activated kinase
PAR1: protease-activated receptor 1
PCI: peritoneal contamination and infection
PI3K: phosphoinositide 3-kinase
Rac1: Ras-related C3 botulinum toxin substrate 1
Ras: Ras protein subfamily
RhoA: Ras homolog gene family, member A
RIG-I: retinoic acid-inducible gene I
ROCK: Rho-associated protein kinase
S1P: sphingosine-1-phosphate
S1PR1: sphingosine-1-phosphate receptor 1
S1PR2: sphingosine-1-phosphate receptor 2
sepsis-MOF: sepsis-associated multiple organ failure
SK1: sphingosine kinase 1
Src: Src protein tyrosine kinase
Tie1: tunica intima endothelial kinase 1
Tie2: tunica intima endothelial kinase 2
TLR4: Toll-like receptor 4
TM: thrombomodulin
TNFR1: tumour necrosis factor alpha receptor 1
TNFR2: tumour necrosis factor alpha receptor 2
TNFα: tumour necrosis factor alpha
VCAM-1: vascular endothelial adhesion molecule 1
VEGF: vascular endothelial growth factor
VEGFR2: vascular endothelial growth factor receptor 2
VE-cadherin: vascular endothelial cadherin
VE-PTP: vascular endothelial protein tyrosine phosphatase
vWF: von Willebrand Factor
1. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA (2016) 315(8):801. doi: 10.1001/jama.2016.0287
2. Aird WC. The Role of the Endothelium in Severe Sepsis and Multiple Organ Dysfunction Syndrome. Blood (2003) 101(10):3765–77. doi: 10.1182/blood-2002-06-1887
3. Verma SK, Molitoris BA. Renal Endothelial Injury and Microvascular Dysfunction in Acute Kidney Injury. Semin Nephrol (2015) 35(1):96–107. doi: 10.1016/j.semnephrol.2015.01.010
4. Angus DC, van der Poll T. Severe Sepsis and Septic Shock. N Engl J Med (2013) 369(9):840–51. doi: 10.1056/NEJMra1208623
5. Dellinger RP, Levy MM, Rhodes A, Annane D, Gerlach H, Opal SM, et al. Surviving Sepsis Campaign: International Guidelines for Management of Severe Sepsis and Septic Shock: 2012. Crit Care Med (2013) 41(2):580–637. doi: 10.1097/CCM.0b013e31827e83af
6. Molema G, Zijlstra JG, van Meurs M, Kamps JAAM. Renal Microvascular Endothelial Cell Responses in Sepsis-Induced Acute Kidney Injury. Nat Rev Nephrol (2022) 18(2):95–112. doi: 10.1038/s41581-021-00489-1
7. Chousterman BG, Swirski FK, Weber GF. Cytokine Storm and Sepsis Disease Pathogenesis. Semin Immunopathol (2017) 39(5):517–28. doi: 10.1007/s00281-017-0639-8
8. Gotts JE, Matthay MA. Sepsis: Pathophysiology and Clinical Management. BMJ (2016) 353:i1585. doi: 10.1136/bmj.i1585
9. Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, Regional, and National Sepsis Incidence and Mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet (2020) 395(10219):200–11. doi: 10.1016/S0140-6736(19)32989-7
10. Peerapornratana S, Manrique-Caballero CL, Gómez H, Kellum JA. Acute Kidney Injury From Sepsis: Current Concepts, Epidemiology, Pathophysiology, Prevention and Treatment. Kidney Int (2019) 96(5):1083–99. doi: 10.1016/j.kint.2019.05.026
11. Bagshaw SM, Uchino S, Bellomo R, Morimatsu H, Morgera S, Schetz M, et al. Septic Acute Kidney Injury in Critically Ill Patients: Clinical Characteristics and Outcomes. Clin J Am Soc Nephrol (2007) 2(3):431–9. doi: 10.2215/CJN.03681106
12. Joffre J, Hellman J, Ince C, Ait-Oufella H. Endothelial Responses in Sepsis. Am J Respir Crit Care Med (2020) 202(3):361–70. doi: 10.1164/rccm.201910-1911TR
13. Aird W. Endothelium as a Therapeutic Target in Sepsis. Curr Drug Targets (2007) 8(4):501–7. doi: 10.2174/138945007780362782
14. Levi M, van der Poll T. Coagulation and Sepsis. Thromb Res (2017) 149:38–44. doi: 10.1016/j.thromres.2016.11.007
15. Chang JC. Sepsis and Septic Shock: Endothelial Molecular Pathogenesis Associated With Vascular Microthrombotic Disease. Thromb J (2019) 17(1):10. doi: 10.1186/s12959-019-0198-4
16. Aird WC. Phenotypic Heterogeneity of the Endothelium. Circ Res (2007) 100(2):158–73. doi: 10.1161/01.RES.0000255691.76142.4a
17. Rossaint J, Zarbock A. Tissue-Specific Neutrophil Recruitment Into the Lung, Liver, and Kidney. J Innate Immun (2013) 5(4):348–57. doi: 10.1159/000345943
18. Aird WC. Endothelial Cell Heterogeneity. Cold Spring Harb Perspect Med (2012) 2(1):a006429. doi: 10.1101/cshperspect.a006429
19. Molema G. Heterogeneity in Endothelial Responsiveness to Cytokines, Molecular Causes, and Pharmacological Consequences. Semin Thromb Hemost (2010) 36(03):246–64. doi: 10.1055/s-0030-1253448
20. Moldobaeva A, Baek A, Wagner EM. MIP-2 Causes Differential Activation of RhoA in Mouse Aortic Versus Pulmonary Artery Endothelial Cells. Microvasc Res (2008) 75(1):53–8. doi: 10.1016/j.mvr.2007.06.007
21. Langenkamp E, Molema G. Microvascular Endothelial Cell Heterogeneity: General Concepts and Pharmacological Consequences for Anti-Angiogenic Therapy of Cancer. Cell Tissue Res (2009) 335(1):205–22. doi: 10.1007/s00441-008-0642-4
22. Yan R, Van Meurs M, Popa ER, Li R, Zwiers PJ, Zijlstra JG, et al. Early Heterogenic Response of Renal Microvasculature to Hemorrhagic Shock/Resuscitation and the Influence of NF-κb Pathway Blockade. Shock (2019) 51(2):200–12. doi: 10.1097/SHK.0000000000001126
23. Aslan A, Van Meurs M, Moser J, Popa ER, Jongman RM, Zwiers PJ, et al. Organ-Specific Differences in Endothelial Permeability-Regulating Molecular Responses in Mouse and Human Sepsis. Shock (2017) 48(1):69–77. doi: 10.1097/SHK.0000000000000841
24. Seemann S, Zohles F, Lupp A. Comprehensive Comparison of Three Different Animal Models for Systemic Inflammation. J BioMed Sci (2017) 24(1):60. doi: 10.1186/s12929-017-0370-8
25. Villa P, Sartor G, Angelini M, Sironi M, Conni M, Gnocchi P, et al. Pattern of Cytokines and Pharmacomodulation in Sepsis Induced by Cecal Ligation and Puncture Compared With That Induced by Endotoxin. Clin Diagn Lab Immunol (1995) 2(5):549–53. doi: 10.1128/cdli.2.5.549-553.1995
26. Cavaillon J, Adib-Conquy M, Fitting C, Adrie C, Payen D. Cytokine Cascade in Sepsis. Scand J Infect Dis (2003) 35(9):535–44. doi: 10.1080/00365540310015935
27. Akira S, Takeda K, Kaisho T. Toll-Like Receptors: Critical Proteins Linking Innate and Acquired Immunity. Nat Immunol (2001) 2(8):675–80. doi: 10.1038/90609
28. Moser J, Heeringa P, Jongman RM, Zwiers PJ, Niemarkt AE, Yan R, et al. Intracellular RIG-I Signaling Regulates TLR4-Independent Endothelial Inflammatory Responses to Endotoxin. J Immunol (2016) 196(11):4681–91. doi: 10.4049/jimmunol.1501819
29. Cohen J. The Immunopathogenesis of Sepsis. Nature (2002) 420(6917):885–91. doi: 10.1038/nature01326
30. Le KTT, Chu X, Jaeger M, Plantinga JA, Matzaraki V, Withoff S, et al. Leukocyte-Released Mediators in Response to Both Bacterial and Fungal Infections Trigger IFN Pathways, Independent of IL-1 and TNF-α, in Endothelial Cells. Front Immunol (2019) 10:1–13. doi: 10.3389/fimmu.2019.02508
31. Zhou Z, Connell MC, MacEwan DJ. TNFR1-Induced NF-κb, But Not ERK, P38mapk or JNK Activation, Mediates TNF-Induced ICAM-1 and VCAM-1 Expression on Endothelial Cells. Cell Signal (2007) 19(6):1238–48. doi: 10.1016/j.cellsig.2006.12.013
32. Peters VA, Joesting JJ, Freund GG. IL-1 Receptor 2 (IL-1R2) and Its Role in Immune Regulation. Brain Behav Immun (2013) 32:1–8. doi: 10.1016/j.bbi.2012.11.006
33. Budamagunta V, Manohar-Sindhu S, Yang Y, He Y, Traktuev DO, Foster TC, et al. Senescence-Associated Hyper-Activation to Inflammatory Stimuli in vitro. Aging (Albany NY) (2021) 13(15):19088–107. doi: 10.18632/aging.203396
34. Kang S, Kishimoto T. Interplay Between Interleukin-6 Signaling and the Vascular Endothelium in Cytokine Storms. Exp Mol Med (2021) 53(7):1116–23. doi: 10.1038/s12276-021-00649-0
35. Blecharz-Lang KG, Wagner J, Fries A, Nieminen-Kelhä M, Rösner J, Schneider UC, et al. Interleukin 6-Mediated Endothelial Barrier Disturbances Can Be Attenuated by Blockade of the IL6 Receptor Expressed in Brain Microvascular Endothelial Cells. Transl Stroke Res (2018) 9(6):631–42. doi: 10.1007/s12975-018-0614-2
36. Dzenko KA, Song L, Ge S, Kuziel WA, Pachter JS. CCR2 Expression by Brain Microvascular Endothelial Cells Is Critical for Macrophage Transendothelial Migration in Response to CCL2. Microvasc Res (2005) 70(1–2):53–64. doi: 10.1016/j.mvr.2005.04.005
37. Salcedo R, Ponce ML, Young HA, Wasserman K, Ward JM, Kleinman HK, et al. Human Endothelial Cells Express CCR2 and Respond to MCP-1: Direct Role of MCP-1 in Angiogenesis and Tumor Progression. Blood (2000) 96(1):34–40. doi: 10.1182/blood.V96.1.34.013a49_34_40
38. Zegeye MM, Lindkvist M, Fälker K, Kumawat AK, Paramel G, Grenegård M, et al. Activation of the JAK/STAT3 and PI3K/AKT Pathways are Crucial for IL-6 Trans-Signaling-Mediated Pro-Inflammatory Response in Human Vascular Endothelial Cells. Cell Commun Signal (2018) 16(1):55. doi: 10.1186/s12964-018-0268-4
39. Wang Y, Liu Q, Liu T, Zheng Q, Xu X, Liu X, et al. Early Plasma Monocyte Chemoattractant Protein 1 Predicts the Development of Sepsis in Trauma Patients. Med (Baltimore) (2018) 97(14):e0356. doi: 10.1097/MD.0000000000010356
40. McCormack JJ, Lopes da Silva M, Ferraro F, Patella F, Cutler DF. Weibel–Palade Bodies at a Glance. J Cell Sci (2017) 130(21):3611–7. doi: 10.1242/jcs.208033
41. Rondaij MG, Bierings R, Kragt A, Van Mourik JA, Voorberg J. Dynamics and Plasticity of Weibel-Palade Bodies in Endothelial Cells. Arterioscler Thromb Vasc Biol (2006) 26(5):1002–7. doi: 10.1161/01.ATV.0000209501.56852.6c
42. Radeva MY, Waschke J. Mind the Gap: Mechanisms Regulating the Endothelial Barrier. Acta Physiol (2018) 222(1):e12860. doi: 10.1111/apha.12860
43. Goldenberg NM, Steinberg BE, Slutsky AS, Lee WL. Broken Barriers: A New Take on Sepsis Pathogenesis. Sci Transl Med (2011) 3(88):1–7. doi: 10.1126/scitranslmed.3002011
44. Yano K, Liaw PC, Mullington JM, Shih S-C, Okada H, Bodyak N, et al. Vascular Endothelial Growth Factor Is an Important Determinant of Sepsis Morbidity and Mortality. J Exp Med (2006) 203(6):1447–58. doi: 10.1084/jem.20060375
45. Kumpers P, Van Meurs M, David S, Molema G, Bijzet J, Lukasz A, et al. Time Course of Angiopoietin-2 Release During Experimental Human Endotoxemia and Sepsis. Crit Care (2009) 13(3):R64. doi: 10.1186/cc7866
46. Akira S, Sato S. Toll-Like Receptors and Their Signaling Mechanisms. Scand J Infect Dis (2003) 35(9):555–62. doi: 10.1080/00365540310015683
47. Daun JM, Fenton MJ. Interleukin-1/Toll Receptor Family Members: Receptor Structure and Signal Transduction Pathways. J Interf Cytokine Res (2000) 20(10):843–55. doi: 10.1089/10799900050163217
48. Kisseleva T, Song L, Vorontchikhina M, Feirt N, Kitajewski J, Schindler C. NF-κb Regulation of Endothelial Cell Function During LPS-Induced Toxemia and Cancer. J Clin Invest (2006) 116(11):2955–63. doi: 10.1172/JCI27392
49. Jacobs MD, Harrison SC. Structure of an Iκbα/NF-κb Complex. Cell (1998) 95(6):749–58. doi: 10.1016/S0092-8674(00)81698-0
50. Zheng Z, Li Z, Chen S, Pan J, Ma X. Tetramethylpyrazine Attenuates TNF-α-Induced iNOS Expression in Human Endothelial Cells: Involvement of Syk-Mediated Activation of PI3K-IKK-Iκb Signaling Pathways. Exp Cell Res (2013) 319(14):2145–51. doi: 10.1016/j.yexcr.2013.05.018
51. Menden H, Tate E, Hogg N, Sampath V. LPS-Mediated Endothelial Activation in Pulmonary Endothelial Cells: Role of Nox2-Dependent IKK-β Phosphorylation. Am J Physiol Cell Mol Physiol (2013) 304(6):L445–55. doi: 10.1152/ajplung.00261.2012
52. Anwar KN, Fazal F, Malik AB, Rahman A. RhoA/Rho-Associated Kinase Pathway Selectively Regulates Thrombin-Induced Intercellular Adhesion Molecule-1 Expression in Endothelial Cells via Activation of Iκb Kinase β and Phosphorylation of RelA/P65. J Immunol (2004) 173(11):6965–72. doi: 10.4049/jimmunol.173.11.6965
53. Ye X, Ding J, Zhou X, Chen G, Liu SF. Divergent Roles of Endothelial NF-κb in Multiple Organ Injury and Bacterial Clearance in Mouse Models of Sepsis. J Exp Med (2008) 205(6):1303–15. doi: 10.1084/jem.20071393
54. Waelchli R, Bollbuck B, Bruns C, Buhl T, Eder J, Feifel R, et al. Design and Preparation of 2-Benzamido-Pyrimidines as Inhibitors of IKK. Bioorg Med Chem Lett (2006) 16(1):108–12. doi: 10.1016/j.bmcl.2005.09.035
55. Coldewey SM, Rogazzo M, Collino M, Patel NSA, Thiemermann C. Inhibition of Iκb Kinase Reduces the Multiple Organ Dysfunction Caused by Sepsis in the Mouse. Dis Model Mech (2013) 6(4):1031–42. doi: 10.1242/dmm.012435
56. Claesson-Welsh L, Dejana E, McDonald DM. Permeability of the Endothelial Barrier: Identifying and Reconciling Controversies. Trends Mol Med (2021) 27(4):314–31. doi: 10.1016/j.molmed.2020.11.006
57. Gröger M, Pasteiner W, Ignatyev G, Matt U, Knapp S, Atrasheuskaya A, et al. Peptide Bβ15-42 Preserves Endothelial Barrier Function in Shock. PloS One (2009) 4(4):e5391. doi: 10.1371/journal.pone.0005391 Bozza P, editor.
58. Hoang MV, Nagy JA, Senger DR. Active Rac1 Improves Pathologic VEGF Neovessel Architecture and Reduces Vascular Leak: Mechanistic Similarities With Angiopoietin-1. Blood (2011) 117(5):1751–60. doi: 10.1182/blood-2010-05-286831
59. Essler M, Amano M, Kruse H-J, Kaibuchi K, Weber PC, Aepfelbacher M. Thrombin Inactivates Myosin Light Chain Phosphatase via Rho and Its Target Rho Kinase in Human Endothelial Cells. J Biol Chem (1998) 273(34):21867–74. doi: 10.1074/jbc.273.34.21867
60. Marcos-Ramiro B, García-Weber D, Millán J. TNF-Induced Endothelial Barrier Disruption: Beyond Actin and Rho. Thromb Haemost (2014) 112(12):1088–102. doi: 10.1160/th14-04-0299
61. Schnoor M, García Ponce A, Vadillo E, Pelayo R, Rossaint J, Zarbock A. Actin Dynamics in the Regulation of Endothelial Barrier Functions and Neutrophil Recruitment During Endotoxemia and Sepsis. Cell Mol Life Sci (2017) 74(11):1985–97. doi: 10.1007/s00018-016-2449-x
62. Rigor RR, Shen Q, Pivetti CD, Wu MH, Yuan SY. Myosin Light Chain Kinase Signaling in Endothelial Barrier Dysfunction. Med Res Rev (2013) 33(5):911–33. doi: 10.1002/med.21270
63. García Ponce A, Citalán Madrid AF, Vargas Robles H, Chánez Paredes S, Nava P, Betanzos A, et al. Loss of Cortactin Causes Endothelial Barrier Dysfunction via Disturbed Adrenomedullin Secretion and Actomyosin Contractility. Sci Rep (2016) 6(1):29003. doi: 10.1038/srep29003
64. Khromov AS, Momotani K, Jin L, Artamonov MV, Shannon J, Eto M, et al. Molecular Mechanism of Telokin-Mediated Disinhibition of Myosin Light Chain Phosphatase and cAMP/cGMP-Induced Relaxation of Gastrointestinal Smooth Muscle. J Biol Chem (2012) 287(25):20975–85. doi: 10.1074/jbc.M112.341479
65. Nakai K, Suzuki Y, Kihira H, Wada H, Fujioka M, Ito M, et al. Regulation of Myosin Phosphatase Through Phosphorylation of the Myosin-Binding Subunit in Platelet Activation. Blood (1997) 90(10):3936–42. doi: 10.1182/blood.V90.10.3936
66. Knaus UG, Wang Y, Reilly AM, Warnock D, Jackson JH. Structural Requirements for PAK Activation by Rac GTPases. J Biol Chem (1998) 273(34):21512–8. doi: 10.1074/jbc.273.34.21512
67. Sanders LC, Matsumura F, Bokoch GM, De Lanerolle P. Inhibition of Myosin Light Chain Kinase by P21-Activated Kinase. Sci (80- ) (1999) 283(5410):2083–5. doi: 10.1126/science.283.5410.2083
68. Köster DV, Mayor S. Cortical Actin and the Plasma Membrane: Inextricably Intertwined. Curr Opin Cell Biol (2016) 38:81–9. doi: 10.1016/j.ceb.2016.02.021
69. Wojciak-Stothard B, Tsang LYF, Haworth SG. Rac and Rho Play Opposing Roles in the Regulation of Hypoxia/Reoxygenation-Induced Permeability Changes in Pulmonary Artery Endothelial Cells. Am J Physiol Cell Mol Physiol (2005) 288(4):L749–60. doi: 10.1152/ajplung.00361.2004
70. Fukuhara S, Sakurai A, Sano H, Yamagishi A, Somekawa S, Takakura N, et al. Cyclic AMP Potentiates Vascular Endothelial Cadherin-Mediated Cell-Cell Contact To Enhance Endothelial Barrier Function Through an Epac-Rap1 Signaling Pathway. Mol Cell Biol (2005) 25(1):136–46. doi: 10.1128/MCB.25.1.136-146.2005
71. Mammoto T, Parikh SM, Mammoto A, Gallagher D, Chan B, Mostoslavsky G, et al. Angiopoietin-1 Requires P190 RhoGAP to Protect Against Vascular Leakage in Vivo. J Biol Chem (2007) 282(33):23910–8. doi: 10.1074/jbc.M702169200
72. Ohta Y, Hartwig JH, Stossel TP. FilGAP, a Rho- and ROCK-Regulated GAP for Rac Binds Filamin A to Control Actin Remodelling. Nat Cell Biol (2006) 8(8):803–14. doi: 10.1038/ncb1437
73. Zhang J, Yang G, Zhu Y, Peng X, Li T, Liu L. Role of Connexin 43 in Vascular Hyperpermeability and Relationship to Rock1-MLC 20 Pathway in Septic Rats. Am J Physiol Cell Mol Physiol (2015) 309(11):L1323–32. doi: 10.1152/ajplung.00016.2015
74. Cinel I, Ark M, Dellinger P, Karabacak T, Tamer L, Cinel L, et al. Involvement of Rho Kinase (ROCK) in Sepsis-Induced Acute Lung Injury. J Thorac Dis (2012) 4(1):30–9. doi: 10.3978/j.issn.2072-1439.2010.08.04
75. Meng L, Cao H, Wan C, Jiang L. MiR-539-5p Alleviates Sepsis-Induced Acute Lung Injury by Targeting ROCK1. Folia Histochem Cytobiol (2020) 57(4):168–78. doi: 10.5603/FHC.a2019.0019
76. Van Nieuw Amerongen GP, Beckers CML, Achekar ID, Zeeman S, Musters RJP, Van Hinsbergh VWM. Involvement of Rho Kinase in Endothelial Barrier Maintenance. Arterioscler Thromb Vasc Biol (2007) 27(11):2332–9. doi: 10.1161/ATVBAHA.107.152322
77. Eferl R, Wagner EF. AP-1: A Double-Edged Sword in Tumorigenesis. Nat Rev Cancer (2003) 3(11):859–68. doi: 10.1038/nrc1209
78. Garces De Los Fayos Alonso I, Liang H-C, Turner S, Lagger S, Merkel O, Kenner L. The Role of Activator Protein-1 (AP-1) Family Members in CD30-Positive Lymphomas. Cancers (Basel) (2018) 10(4):93. doi: 10.3390/cancers10040093
79. Matsuda N. Alert Cell Strategy in SIRS-Induced Vasculitis: Sepsis and Endothelial Cells. J Intensive Care (2016) 4(1):21. doi: 10.1186/s40560-016-0147-2
80. Monje P, Hernández-Losa J, Lyons RJ, Castellone MD, Gutkind JS. Regulation of the Transcriptional Activity of C-Fos by ERK. J Biol Chem (2005) 280(42):35081–4. doi: 10.1074/jbc.C500353200
81. Tanos T, Marinissen MJ, Leskow FC, Hochbaum D, Martinetto H, Gutkind JS, et al. Phosphorylation of C-Fos by Members of the P38 MAPK Family. J Biol Chem (2005) 280(19):18842–52. doi: 10.1074/jbc.M500620200
82. Humar M, Loop T, Schmidt R, Hoetzel A, Roesslein M, Andriopoulos N, et al. The Mitogen-Activated Protein Kinase P38 Regulates Activator Protein 1 by Direct Phosphorylation of C-Jun. Int J Biochem Cell Biol (2007) 39(12):2278–88. doi: 10.1016/j.biocel.2007.06.013
83. Morton S, Davis RJ, McLaren A, Cohen P. A Reinvestigation of the Multisite Phosphorylation of the Transcription Factor C-Jun. EMBO J (2003) 22(15):3876–86. doi: 10.1093/emboj/cdg388
84. Wang Y, Prywes R. Activation of the C-Fos Enhancer by the Erk MAP Kinase Pathway Through Two Sequence Elements: The C-Fos AP-1 and P62tcf Sites. Oncogene (2000) 19(11):1379–85. doi: 10.1038/sj.onc.1203443
85. Whitmarsh AJ, Yang SH, Su MS, Sharrocks AD, Davis RJ. Role of P38 and JNK Mitogen-Activated Protein Kinases in the Activation of Ternary Complex Factors. Mol Cell Biol (1997) 17(5):2360–71. doi: 10.1128/MCB.17.5.2360
86. Cavigelli M, Dolfi F, Claret FX, Karin M. Induction of C-Fos Expression Through JNK-Mediated TCF/Elk-1 Phosphorylation. EMBO J (1995) 14(23):5957–64. doi: 10.1002/j.1460-2075.1995.tb00284.x
87. Kayahara M, Wang X, Tournier C. Selective Regulation of C- Jun Gene Expression by Mitogen-Activated Protein Kinases via the 12- O -Tetradecanoylphorbol-13-Acetate- Responsive Element and Myocyte Enhancer Factor 2 Binding Sites. Mol Cell Biol (2005) 25(9):3784–92. doi: 10.1128/MCB.25.9.3784-3792.2005
88. Gratton J-P, Morales-Ruiz M, Kureishi Y, Fulton D, Walsh K, Sessa WC. Akt Down-Regulation of P38 Signaling Provides a Novel Mechanism of Vascular Endothelial Growth Factor-Mediated Cytoprotection in Endothelial Cells. J Biol Chem (2001) 276(32):30359–65. doi: 10.1074/jbc.M009698200
89. Abdel-Malak NA, Srikant CB, Kristof AS, Magder SA, Di Battista JA, Hussain SNA. Angiopoietin-1 Promotes Endothelial Cell Proliferation and Migration Through AP-1–Dependent Autocrine Production of Interleukin-8. Blood (2008) 111(8):4145–54. doi: 10.1182/blood-2007-08-110338
90. Jersmann HPA, Hii CST, Ferrante JV, Ferrante A. Bacterial Lipopolysaccharide and Tumor Necrosis Factor Alpha Synergistically Increase Expression of Human Endothelial Adhesion Molecules Through Activation of NF-κb and P38 Mitogen-Activated Protein Kinase Signaling Pathways. Infect Immun (2001) 69(3):1273–9. doi: 10.1128/IAI.69.3.1273-1279.2001 Moore RN, editor.
91. Surapisitchat J, Hoefen RJ, Pi X, Yoshizumi M, Yan C, Berk BC. Fluid Shear Stress Inhibits TNF- Activation of JNK But Not ERK1/2 or P38 in Human Umbilical Vein Endothelial Cells: Inhibitory Crosstalk Among MAPK Family Members. Proc Natl Acad Sci (2001) 98(11):6476–81. doi: 10.1073/pnas.101134098
92. Xu XS, Vanderziel C, Bennett CF, Monia BP. A Role for c-Raf Kinase and Ha-Ras in Cytokine-mediated Induction of Cell Adhesion Molecules. J Biol Chem (1998) 273(50):33230–8. doi: 10.1074/jbc.273.50.33230
93. Xu H, Song J, Gao X, Xu Z, Xu X, Xia Y, et al. Paeoniflorin Attenuates Lipopolysaccharide-Induced Permeability of Endothelial Cells: Involvements of F-Actin Expression and Phosphorylations of PI3K/Akt and PKC. Inflammation (2013) 36(1):216–25. doi: 10.1007/s10753-012-9537-3
94. Lo H, Lai T, Li C, Wu W. TNF-α Induces CXCL1 Chemokine Expression and Release in Human Vascular Endothelial Cells In Vitro via Two Distinct Signaling Pathways. Acta Pharmacol Sin (2014) 35(3):339–50. doi: 10.1038/aps.2013.182
95. Yamauchi J, Kaziro Y, Itoh H. Differential Regulation of Mitogen-Activated Protein Kinase Kinase 4 (MKK4) and 7 (MKK7) by Signaling From G Protein βγ Subunit in Human Embryonal Kidney 293 Cells. J Biol Chem (1999) 274(4):1957–65. doi: 10.1074/jbc.274.4.1957
96. Yamauchi J, Tsujimoto G, Kaziro Y, Itoh H. Parallel Regulation of Mitogen-Activated Protein Kinase Kinase 3 (MKK3) and MKK6 in Gq-Signaling Cascade. J Biol Chem (2001) 276(26):23362–72. doi: 10.1074/jbc.M011752200
97. Guo F, Zhou Z, Dou Y, Tang J, Gao C, Huan J. GEF-H1/RhoA Signalling Pathway Mediates Lipopolysaccharide-Induced Intercellular Adhesion Molecular-1 Expression in Endothelial Cells via Activation of P38 and NF-κb. Cytokine (2012) 57(3):417–28. doi: 10.1016/j.cyto.2011.12.009
98. Peng J, He F, Zhang C, Deng X, Yin F. Protein Kinase C-α Signals P115RhoGEF Phosphorylation and RhoA Activation in TNF-α-Induced Mouse Brain Microvascular Endothelial Cell Barrier Dysfunction. J Neuroinflamm (2011) 8(1):28. doi: 10.1186/1742-2094-8-28
99. Yeung YT, Aziz F, Guerrero-Castilla A, Arguelles S. Signaling Pathways in Inflammation and Anti-Inflammatory Therapies. Curr Pharm Des (2018) 24(14):1449–84. doi: 10.2174/1381612824666180327165604
100. Trop-Steinberg S, Azar Y. AP-1 Expression and its Clinical Relevance in Immune Disorders and Cancer. Am J Med Sci (2017) 353(5):474–83. doi: 10.1016/j.amjms.2017.01.019
101. Dayang E-Z, Plantinga J, Ter Ellen B, Van Meurs M, Molema G, Moser J. Identification of LPS-Activated Endothelial Subpopulations With Distinct Inflammatory Phenotypes and Regulatory Signaling Mechanisms. Front Immunol (2019) 10:1–12. doi: 10.3389/fimmu.2019.01169
102. Kułdo JM, Westra J, Ásgeirsdóttir SA, Kok RJ, Oosterhuis K, Rots MG, et al. Differential Effects of NF-κb and P38 MAPK Inhibitors and Combinations Thereof on TNF-α- and IL-1β-Induced Proinflammatory Status of Endothelial Cells In Vitro. Am J Physiol Physiol (2005) 289(5):C1229–39. doi: 10.1152/ajpcell.00620.2004
103. Imaizumi T, Matsuda N, Tomita K, Palikhe S, Ohashi W, Hattori K, et al. Activator Protein-1 Decoy Oligodeoxynucleotide Transfection Is Beneficial in Reducing Organ Injury and Mortality in Septic Mice. Crit Care Med (2018) 46(5):e435–42. doi: 10.1097/CCM.0000000000003009
104. Ishida M, Ueki M, Morishita J, Ueno M, Shiozawa S, Maekawa N. T-5224, a Selective Inhibitor of C-Fos/activator Protein-1, Improves Survival by Inhibiting Serum High Mobility Group Box-1 in Lethal Lipopolysaccharide-Induced Acute Kidney Injury Model. J Intensive Care (2015) 3(1):49. doi: 10.1186/s40560-015-0115-2
105. Van Den Blink B, Branger J, Weijer S, Van Deventer SJH, Van Der Poll T, Peppelenbosch MP. Human Endotoxemia Activates P38 MAP Kinase and P42/44 MAP Kinase, But Not C-Jun N-Terminal Kinase. Mol Med (2001) 7(11):755–60. doi: 10.1007/BF03401965
106. Fang W, Cai S-X, Wang C-L, Sun X-X, Li K, Yan X-W, et al. Modulation of Mitogen-Activated Protein Kinase Attenuates Sepsis-Induced Acute Lung Injury in Acute Respiratory Distress Syndrome Rats. Mol Med Rep (2017) 16(6):9652–8. doi: 10.3892/mmr.2017.7811
107. Pizzino G, Bitto A, Pallio G, Irrera N, Galfo F, Interdonato M, et al. Blockade of the JNK Signalling as a Rational Therapeutic Approach to Modulate the Early and Late Steps of the Inflammatory Cascade in Polymicrobial Sepsis. Mediators Inflamm (2015) 2015:1–7. doi: 10.1155/2015/591572
108. Fijen JW, Tulleken JE, Muller Kobold AC, De Boer P, Van der Werf TS, Ligtenberg JJM, et al. Inhibition of P38 Mitogen-Activated Protein Kinase: Dose-Dependent Suppression of Leukocyte and Endothelial Response After Endotoxin Challenge in Humans*. Crit Care Med (2002) 30(4):841–5. doi: 10.1097/00003246-200204000-00021
109. Branger J, Van den Blink B, Weijer S, Madwed J, Bos CL, Gupta A, et al. Anti-Inflammatory Effects of a P38 Mitogen-Activated Protein Kinase Inhibitor During Human Endotoxemia. J Immunol (2002) 168(8):4070–7. doi: 10.4049/jimmunol.168.8.4070
110. Fijen JW, Zijlstra JG, De Boer P, Spanjersberg R, Cohen Tervaert JW, Van Der Werf TS, et al. Suppression of the Clinical and Cytokine Response to Endotoxin by RWJ-67657, a P38 Mitogen-Activated Protein-Kinase Inhibitor, in Healthy Human Volunteers. Clin Exp Immunol (2001) 124(1):16–20. doi: 10.1046/j.1365-2249.2001.01485.x
111. Coulthard LR, White DE, Jones DL, McDermott MF, Burchill SA. P38mapk: Stress Responses From Molecular Mechanisms to Therapeutics. Trends Mol Med (2009) 15(8):369–79. doi: 10.1016/j.molmed.2009.06.005
112. Levi M, Van der Poll T. Inflammation and Coagulation. Crit Care Med (2010) 38(SUPPL. 2):S26–34. doi: 10.1097/CCM.0b013e3181c98d21
114. Feistritzer C, Riewald M. Endothelial Barrier Protection by Activated Protein C Through PAR1-Dependent Sphingosine 1–Phosphate Receptor-1 Crossactivation. Blood (2005) 105(8):3178–84. doi: 10.1182/blood-2004-10-3985
115. Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of Endothelial Cell Protease Activated Receptor 1 by the Protein C Pathway. Sci (80- ) (2002) 296(5574):1880–2. doi: 10.1126/science.1071699
116. Adyshev DM, Dudek SM, Moldobaeva N, Kim K, Ma S-F, Kasa A, et al. Ezrin/radixin/moesin Proteins Differentially Regulate Endothelial Hyperpermeability After Thrombin. Am J Physiol Cell Mol Physiol (2013) 305(3):L240–55. doi: 10.1152/ajplung.00355.2012
117. Vouret-Craviari V, Grall D, Van Obberghen-Schilling E. Modulation of Rho GTPase Activity in Endothelial Cells by Selective Proteinase-Activated Receptor (PAR) Agonists. J Thromb Haemost (2003) 1(5):1103–11. doi: 10.1046/j.1538-7836.2003.00238.x
118. Li Z, Yin M, Zhang H, Ni W, Pierce RW, Zhou HJ, et al. BMX Represses Thrombin-PAR1–Mediated Endothelial Permeability and Vascular Leakage During Early Sepsis. Circ Res (2020) 126(4):471–85. doi: 10.1161/CIRCRESAHA.119.315769
119. Schoergenhofer C, Schwameis M, Gelbenegger G, Buchtele N, Thaler B, Mussbacher M, et al. Inhibition of Protease-Activated Receptor (PAR1) Reduces Activation of the Endothelium, Coagulation, Fibrinolysis and Inflammation During Human Endotoxemia. Thromb Haemost (2018) 118(07):1176–84. doi: 10.1055/s-0038-1655767
120. Nicolaes GAF, Thomassen MCLGD, Tans G, Rosing J, Hemker HC. Effect of Activated Protein C on Thrombin Generation and on the Thrombin Potential in Plasma of Normal and APC-Resistant Individuals. Blood Coagul Fibrinolysis (1997) 8(1):28–38. doi: 10.1097/00001721-199701000-00006
121. Stearns-Kurosawa DJ, Kurosawa S, Mollica JS, Ferrell GL, Esmon CT. The Endothelial Cell Protein C Receptor Augments Protein C Activation by the Thrombin-Thrombomodulin Complex. Proc Natl Acad Sci (1996) 93(19):10212–6. doi: 10.1073/pnas.93.19.10212
122. Taylor FB, Peer GT, Lockhart MS, Ferrell G, Esmon CT. Endothelial Cell Protein C Receptor Plays an Important Role in Protein C Activation In Vivo. Blood (2001) 97(6):1685–8. doi: 10.1182/blood.V97.6.1685
123. Von Drygalski A, Furlan-Freguia C, Ruf W, Griffin JH, Mosnier LO. Organ-Specific Protection Against Lipopolysaccharide-Induced Vascular Leak Is Dependent on the Endothelial Protein C Receptor. Arterioscler Thromb Vasc Biol (2013) 33(4):769–76. doi: 10.1161/ATVBAHA.112.301082
124. Moore KL, Esmon CT, Esmon NL. Tumor Necrosis Factor Leads to the Internalization and Degradation of Thrombomodulin From the Surface of Bovine Aortic Endothelial Cells in Culture. Blood (1989) 73(1):159–65. doi: 10.1182/blood.V73.1.159.159
125. Lin P-Y, Shen H-C, Chen C-J, Wu S-E, Kao H-L, Huang J-H, et al. The Inhibition in Tumor Necrosis Factor-α-Induced Attenuation in Endothelial Thrombomodulin Expression by Carvedilol Is Mediated by Nuclear Factor-κb and Reactive Oxygen Species. J Thromb Thrombolysis (2010) 29(1):52–9. doi: 10.1007/s11239-009-0318-2
126. Sohn RH, Deming CB, Johns DC, Champion HC, Bian C, Gardner K, et al. Regulation of Endothelial Thrombomodulin Expression by Inflammatory Cytokines is Mediated by Activation of Nuclear Factor-Kappa B. Blood (2005) 105(10):3910–7. doi: 10.1182/blood-2004-03-0928
127. Yin Q, Liu B, Chen Y, Zhao Y, Li C. The Role of Soluble Thrombomodulin in the Risk Stratification and Prognosis Evaluation of Septic Patients in the Emergency Department. Thromb Res (2013) 132(4):471–6. doi: 10.1016/j.thromres.2013.08.011
128. Lin J-J, Hsiao H-J, Chan O-W, Wang Y, Hsia S-H, Chiu C-H. Increased Serum Thrombomodulin Level Is Associated With Disease Severity and Mortality in Pediatric Sepsis. PloS One (2017) 12(8):e0182324. doi: 10.1371/journal.pone.0182324 Cox D, editor.
129. Zhang J, Xue M, Chen Y, Liu C, Kuang Z, Mu S, et al. Identification of Soluble Thrombomodulin and Tissue Plasminogen Activator-Inhibitor Complex as Biomarkers for Prognosis and Early Evaluation of Septic Shock and Sepsis-Induced Disseminated Intravascular Coagulation. Ann Palliat Med (2021) 10(10):10170–84. doi: 10.21037/apm-21-2222
130. Loghmani H, Conway EM. Exploring Traditional and Nontraditional Roles for Thrombomodulin. Blood (2018) 132(2):148–58. doi: 10.1182/blood-2017-12-768994
131. Hisano Y, Kobayashi N, Yamaguchi A, Nishi T. Mouse SPNS2 Functions as a Sphingosine-1-Phosphate Transporter in Vascular Endothelial Cells. PloS One (2012) 7(6):e38941. doi: 10.1371/journal.pone.0038941 Holowka D, editor.
132. Aoki S, Yatomi Y, Ohta M, Osada M, Kazama F, Satoh K, et al. Sphingosine 1-Phosphate–Related Metabolism in the Blood Vessel. J Biochem (2005) 138(1):47–55. doi: 10.1093/jb/mvi100
133. Christoffersen C, Obinata H, Kumaraswamy SB, Galvani S, Ahnstrom J, Sevvana M, et al. Endothelium-Protective Sphingosine-1-Phosphate Provided by HDL-Associated Apolipoprotein M. Proc Natl Acad Sci (2011) 108(23):9613–8. doi: 10.1073/pnas.1103187108
134. Mullershausen F, Zecri F, Cetin C, Billich A, Guerini D, Seuwen K. Persistent Signaling Induced by FTY720-Phosphate Is Mediated by Internalized S1P1 Receptors. Nat Chem Biol (2009) 5(6):428–34. doi: 10.1038/nchembio.173
135. Lee M-J, Thangada S, Paik J-H, Sapkota GP, Ancellin N, Chae S-S, et al. Akt-Mediated Phosphorylation of the G Protein-Coupled Receptor EDG-1 Is Required for Endothelial Cell Chemotaxis. Mol Cell (2001) 8(3):693–704. doi: 10.1016/S1097-2765(01)00324-0
136. Li Q, Chen B, Zeng C, Fan A, Yuan Y, Guo X, et al. Differential Activation of Receptors and Signal Pathways Upon Stimulation by Different Doses of Sphingosine-1-Phosphate in Endothelial Cells. Exp Physiol (2015) 100(1):95–107. doi: 10.1113/expphysiol.2014.082149
137. Sanchez T. Sphingosine-1-Phosphate Signaling in Endothelial Disorders. Curr Atheroscler Rep (2016) 18(6):31. doi: 10.1007/s11883-016-0586-1
138. Wang C, Mao J, Redfield S, Mo Y, Lage JM, Zhou X. Systemic Distribution, Subcellular Localization and Differential Expression of Sphingosine-1-Phosphate Receptors in Benign and Malignant Human Tissues. Exp Mol Pathol (2014) 97(2):259–65. doi: 10.1016/j.yexmp.2014.07.013
139. Bandhuvula P, Tam YY, Oskouian B, Saba JD. The Immune Modulator FTY720 Inhibits Sphingosine-1-Phosphate Lyase Activity. J Biol Chem (2005) 280(40):33697–700. doi: 10.1074/jbc.C500294200
140. Ogawa C, Kihara A, Gokoh M, Igarashi Y. Identification and Characterization of a Novel Human Sphingosine-1-Phosphate Phosphohydrolase, Hspp2. J Biol Chem (2003) 278(2):1268–72. doi: 10.1074/jbc.M209514200
141. Jasinska R, Zhang QX, Pilquil C, Singh I, Xu J, Dewald J, et al. Lipid Phosphate Phosphohydrolase-1 Degrades Exogenous Glycerolipid and Sphingolipid Phosphate Esters. Biochem J (1999) 340( Pt 3):677–86. doi: 10.1042/bj3400677
142. Nishi T, Kobayashi N, Hisano Y, Kawahara A, Yamaguchi A. Molecular and Physiological Functions of Sphingosine 1-Phosphate Transporters. Biochim Biophys Acta (2014) 1841(5):759–65. doi: 10.1016/j.bbalip.2013.07.012
143. Mechtcheriakova D, Wlachos A, Sobanov J, Kopp T, Reuschel R, Bornancin F, et al. Sphingosine 1-Phosphate Phosphatase 2 is Induced During Inflammatory Responses. Cell Signal (2007) 19(4):748–60. doi: 10.1016/j.cellsig.2006.09.004
144. Song F, Hou J, Chen Z, Cheng B, Lei R, Cui P, et al. Sphingosine-1-Phosphate Receptor 2 Signaling Promotes Caspase-11–Dependent Macrophage Pyroptosis and Worsens Escherichia Coli Sepsis Outcome. Anesthesiology (2018) 129(2):311–20. doi: 10.1097/ALN.0000000000002196
145. Hou J, Chen Q, Zhang K, Cheng B, Xie G, Wu X, et al. Sphingosine 1-Phosphate Receptor 2 Signaling Suppresses Macrophage Phagocytosis and Impairs Host Defense Against Sepsis. Anesthesiology (2015) 123(2):409–22. doi: 10.1097/ALN.0000000000000725
146. Hemdan NYA, Weigel C, Reimann C-M, Gräler MH. Modulating Sphingosine 1-Phosphate Signaling With DOP or FTY720 Alleviates Vascular and Immune Defects in Mouse Sepsis. Eur J Immunol (2016) 46(12):2767–77. doi: 10.1002/eji.201646417
147. Flemming S, Burkard N, Meir M, Schick MA, Germer C-T, Schlegel N. Sphingosine-1-Phosphate Receptor-1 Agonist Sew2871 Causes Severe Cardiac Side Effects and Does Not Improve Microvascular Barrier Breakdown in Sepsis. Shock (2018) 49(1):71–81. doi: 10.1097/SHK.0000000000000908
148. Wang Z, Sims CR, Patil NK, Gokden N, Mayeux PR. Pharmacologic Targeting of Sphingosine-1-Phosphate Receptor 1 Improves the Renal Microcirculation During Sepsis in the Mouse. J Pharmacol Exp Ther (2015) 352(1):61–6. doi: 10.1124/jpet.114.219394
149. Makinde T, Agrawal DK. Intra and Extravascular Transmembrane Signalling of Angiopoietin-1-Tie2 Receptor in Health and Disease. J Cell Mol Med (2008) 12(3):810–28. doi: 10.1111/j.1582-4934.2008.00254.x
150. Hughes DP, Marron MB, Brindle NPJ. The Antiinflammatory Endothelial Tyrosine Kinase Tie2 Interacts With a Novel Nuclear Factor-κb Inhibitor ABIN-2. Circ Res (2003) 92(6):630–6. doi: 10.1161/01.RES.0000063422.38690.DC
151. Gavard J, Patel V, Gutkind JS. Angiopoietin-1 Prevents VEGF-Induced Endothelial Permeability by Sequestering Src Through Mdia. Dev Cell (2008) 14(1):25–36. doi: 10.1016/j.devcel.2007.10.019
152. Yun J-H, Han MH, Jeong H-S, Lee D-H, Cho C-H. Angiopoietin 1 Attenuates Interleukin-6-Induced Endothelial Cell Permeability Through SHP-1. Biochem Biophys Res Commun (2019) 518(2):286–93. doi: 10.1016/j.bbrc.2019.08.048
153. Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, et al. Angiopoietin-2, a Natural Antagonist for Tie2 That Disrupts In Vivo Angiogenesis. Science (1997) 277(5322):55–60. doi: 10.1126/science.277.5322.55
154. Bogdanovic E, Nguyen VPKH, Dumont DJ. Activation of Tie2 by Angiopoietin-1 and Angiopoietin-2 Results in Their Release and Receptor Internalization. J Cell Sci (2006) 119(17):3551–60. doi: 10.1242/jcs.03077
155. Minhas N, Xue M, Jackson CJ. Activated Protein C Binds Directly to Tie2: Possible Beneficial Effects on Endothelial Barrier Function. Cell Mol Life Sci (2017) 74(10):1895–906. doi: 10.1007/s00018-016-2440-6
156. Daly C, Qian X, Castanaro C, Pasnikowski E, Jiang X, Thomson BR, et al. Angiopoietins Bind Thrombomodulin and Inhibit its Function as a Thrombin Cofactor. Sci Rep (2018) 8(1):505. doi: 10.1038/s41598-017-18912-8
157. Korhonen EA, Lampinen A, Giri H, Anisimov A, Kim M, Allen B, et al. Tie1 Controls Angiopoietin Function in Vascular Remodeling and Inflammation. J Clin Invest (2016) 126(9):3495–510. doi: 10.1172/JCI84923
158. Mueller SB, Kontos CD. Tie1: An Orphan Receptor Provides Context for Angiopoietin-2/Tie2 Signaling. J Clin Invest (2016) 126(9):3188–91. doi: 10.1172/JCI89963
159. Yuan HT, Khankin EV, Karumanchi SA, Parikh SM. Angiopoietin 2 Is a Partial Agonist/Antagonist of Tie2 Signaling in the Endothelium. Mol Cell Biol (2009) 29(8):2011–22. doi: 10.1128/MCB.01472-08
160. Thamm K, Schrimpf C, Retzlaff J, Idowu TO, Van Meurs M, Zijlstra JG, et al. Molecular Regulation of Acute Tie2 Suppression in Sepsis. Crit Care Med (2018) 46(9):e928–36. doi: 10.1097/CCM.0000000000003269
161. Souma T, Thomson BR, Heinen S, Anna Carota I, Yamaguchi S, Onay T, et al. Context-Dependent Functions of Angiopoietin 2 Are Determined by the Endothelial Phosphatase VEPTP. Proc Natl Acad Sci (2018) 115(6):1298–303. doi: 10.1073/pnas.1714446115
162. Liu W-K, Yen P-F, Chien C-Y, Fann M-J, Su J-Y, Chou C-K. The Inhibitor ABIN-2 Disrupts the Interaction of Receptor-Interacting Protein With the Kinase Subunit IKKgamma to Block Activation of the Transcription Factor NF-kappaB and Potentiate Apoptosis. Biochem J (2004) 378(Pt 3):867–76. doi: 10.1042/BJ20031736
163. Kim I, Ryu YS, Kwak HJ, Ahn SY, Oh J, Yancopoulos GD, et al. EphB Ligand, Ephrinb2, Suppresses the VEGF- and Angiopoietin-1-Induced Ras/mitogen-Activated Protein Kinase Pathway in Venous Endothelial Cells. FASEB J (2002) 16(9):1126–8. doi: 10.1096/fj.01-0805fje
164. Pitson SM, Moretti PAB, Zebol JR, Lynn HE, Xia P, Vadas MA, et al. Activation of Sphingosine Kinase 1 by ERK1/2-Mediated Phosphorylation. EMBO J (2003) 22(20):5491–500. doi: 10.1093/emboj/cdg540
165. David S, Mukherjee A, Ghosh CC, Yano M, Khankin EV, Wenger JB, et al. Angiopoietin-2 may Contribute to Multiple Organ Dysfunction and Death in Sepsis*. Crit Care Med (2012) 40(11):3034–41. doi: 10.1097/CCM.0b013e31825fdc31
166. Augustin HG, Young Koh G, Thurston G, Alitalo K. Control of Vascular Morphogenesis and Homeostasis Through the Angiopoietin–Tie System. Nat Rev Mol Cell Biol (2009) 10(3):165–77. doi: 10.1038/nrm2639
167. Van Meurs M, Kurniati NF, Wulfert FM, Asgeirsdottir SA, De Graaf IA, Satchell SC, et al. Shock-Induced Stress Induces Loss of Microvascular Endothelial Tie2 in the Kidney Which Is Not Associated With Reduced Glomerular Barrier Function. Am J Physiol Physiol (2009) 297(2):F272–81. doi: 10.1152/ajprenal.00137.2009
168. Kurniati NF, van Meurs M, vom Hagen F, Jongman RM, Moser J, Zwiers PJ, et al. Pleiotropic Effects of Angiopoietin-2 Deficiency Do Not Protect Mice Against Endotoxin-Induced Acute Kidney Injury. Nephrol Dial Transplant (2013) 28(3):567–75. doi: 10.1093/ndt/gfs336
169. Aslan A, Jongman RM, Moser J, Stegeman CA, van Goor H, Diepstra A, et al. The Renal Angiopoietin/Tie2 System in Lethal Human Sepsis. Crit Care (2014) 18(2):423. doi: 10.1186/cc13806
170. Volbeda M, Jou-Valencia D, van den Heuvel MC, Knoester M, Zwiers PJ, Pillay J, et al. Comparison of Renal Histopathology and Gene Expression Profiles Between Severe COVID-19 and Bacterial Sepsis in Critically Ill Patients. Crit Care (2021) 25(1):202. doi: 10.1186/s13054-021-03631-4
171. Han S, Lee S-J, Kim KE, Lee HS, Oh N, Park I, et al. Amelioration of Sepsis by TIE2 Activation–Induced Vascular Protection. Sci Transl Med (2016) 8(335):1–12. doi: 10.1126/scitranslmed.aad9260
172. van der Flier M, van Leeuwen HJ, van Kessel KP, Kimpen JL, Hoepelman AI, Geelen SP. Plasma Vascular Endothelial Growth Factor in Severe Sepsis. Shock (2005) 23(1):35–8. doi: 10.1097/01.shk.0000150728.91155.41
173. Jeong SJ, Han SH, Kim CO, Choi JY, Kim JM. Anti-Vascular Endothelial Growth Factor Antibody Attenuates Inflammation and Decreases Mortality in an Experimental Model of Severe Sepsis. Crit Care (2013) 17(3):R97. doi: 10.1186/cc12742
174. Besnier E, Brakenhielm E, Richard V, Tamion F. Does Anti-VEGF Bevacizumab Improve Survival in Experimental Sepsis? Crit Care (2017) 21(1):163. doi: 10.1186/s13054-017-1734-x
175. Hauschildt J, Schrimpf C, Thamm K, Retzlaff J, Idowu TO, von Kaisenberg C, et al. Dual Pharmacological Inhibition of Angiopoietin-2 and VEGF-A in Murine Experimental Sepsis. J Vasc Res (2020) 57(1):34–45. doi: 10.1159/000503787
176. Gavard J, Gutkind JS. VEGF Controls Endothelial-Cell Permeability by Promoting the β-Arrestin-Dependent Endocytosis of VE-Cadherin. Nat Cell Biol (2006) 8(11):1223–34. doi: 10.1038/ncb1486
177. Lee M, Choy WC, Abid MR. Direct Sensing of Endothelial Oxidants by Vascular Endothelial Growth Factor Receptor-2 and C-Src. PloS One (2011) 6(12):e28454. doi: 10.1371/journal.pone.0028454 Pal S, editor.
178. Hayashi M, Majumdar A, Li X, Adler J, Sun Z, Vertuani S, et al. VE-PTP Regulates VEGFR2 Activity in Stalk Cells to Establish Endothelial Cell Polarity and Lumen Formation. Nat Commun (2013) 4(1):1672. doi: 10.1038/ncomms2683
179. Calalb MB, Polte TR, Hanks SK. Tyrosine Phosphorylation of Focal Adhesion Kinase at Sites in the Catalytic Domain Regulates Kinase Activity: A Role for Src Family Kinases. Mol Cell Biol (1995) 15(2):954–63. doi: 10.1128/MCB.15.2.954
180. Hood JD, Frausto R, Kiosses WB, Schwartz MA, Cheresh DA. Differential αv Integrin–Mediated Ras-ERK Signaling During Two Pathways of Angiogenesis. J Cell Biol (2003) 162(5):933–43. doi: 10.1083/jcb.200304105
181. Lim S-T, Miller NLG, Chen XL, Tancioni I, Walsh CT, Lawson C, et al. Nuclear-Localized Focal Adhesion Kinase Regulates Inflammatory VCAM-1 Expression. J Cell Biol (2012) 197(7):907–19. doi: 10.1083/jcb.201109067
182. Murphy JM, Jeong K, Rodriguez YAR, Kim J-H, Ahn E-YE, Lim S-TS. FAK and Pyk2 Activity Promote TNF-α and IL-1β-Mediated Pro-Inflammatory Gene Expression and Vascular Inflammation. Sci Rep (2019) 9(1):7617. doi: 10.1038/s41598-019-44098-2
183. Dayang E-Z, Luxen M, Kuiper T, Yan R, Rangarajan S, Van Meurs M, et al. Pharmacological Inhibition of Focal Adhesion Kinase 1 (FAK1) and Anaplastic Lymphoma Kinase (ALK) Identified via Kinome Profile Analysis Attenuates Lipopolysaccharide-Induced Endothelial Inflammatory Activation. BioMed Pharmacother (2021) 133:111073. doi: 10.1016/j.biopha.2020.111073
184. Petroni RC, Teodoro WR, Guido MC, Barbeiro HV, Abatepaulo F, Theobaldo MC, et al. Role of Focal Adhesion Kinase in Lung Remodeling of Endotoxemic Rats. Shock (2012) 37(5):524–30. doi: 10.1097/SHK.0b013e31824c7665
185. Guido MC, Clemente CF, Moretti AI, Barbeiro HV, Debbas V, Caldini EG, et al. Small Interfering RNA Targeting Focal Adhesion Kinase Prevents Cardiac Dysfunction in Endotoxemia. Shock (2012) 37(1):77–84. doi: 10.1097/SHK.0b013e31823532ec
186. Ruan G-X, Kazlauskas A. Axl Is Essential for VEGF-A-Dependent Activation of PI3K/Akt. EMBO J (2012) 31(7):1692–703. doi: 10.1038/emboj.2012.21
187. Ekman C, Linder A, Åkesson P, Dahlbäck B. Plasma Concentrations of Gas6 (Growth Arrest Specific Protein 6) and its Soluble Tyrosine Kinase Receptor Saxl In Sepsis and Systemic Inflammatory Response Syndromes. Crit Care (2010) 14(4):R158. doi: 10.1186/cc9233
188. Stalder G, Que YA, Calzavarini S, Burnier L, Kosinski C, Ballabeni P, et al. Study of Early Elevated Gas6 Plasma Level as a Predictor of Mortality in a Prospective Cohort of Patients With Sepsis. PloS One (2016) 11(10):e0163542. doi: 10.1371/journal.pone.0163542 Garcia De Frutos P, editor.
189. Smith RO, Ninchoji T, Gordon E, André H, Dejana E, Vestweber D, et al. Vascular Permeability in Retinopathy Is Regulated by VEGFR2 Y949 Signaling to VE-Cadherin. Elife (2020) 9:1–20. doi: 10.7554/eLife.54056
190. Weis S, Shintani S, Weber A, Kirchmair R, Wood M, Cravens A, et al. Src Blockade Stabilizes a Flk/cadherin Complex, Reducing Edema and Tissue Injury Following Myocardial Infarction. J Clin Invest (2004) 113(6):885–94. doi: 10.1172/JCI200420702
191. Nawroth R, Poell G, Ranft A, Kloep S, Samulowitz U, Fachinger G, et al. VE-PTP and VE-Cadherin Ectodomains Interact to Facilitate Regulation of Phosphorylation and Cell Contacts. EMBO J (2002) 21(18):4885–95. doi: 10.1093/emboj/cdf497
192. Yu W-K, McNeil JB, Wickersham NE, Shaver CM, Bastarache JA, Ware LB. Vascular Endothelial Cadherin Shedding Is More Severe in Sepsis Patients With Severe Acute Kidney Injury. Crit Care (2019) 23(1):18. doi: 10.1186/s13054-019-2315-y
193. Opal SM, Laterre P-F, Francois B, LaRosa SP, Angus DC, Mira J-P, et al. Effect of Eritoran, an Antagonist of MD2-TLR4, on Mortality in Patients With Severe Sepsis. JAMA (2013) 309(11):1154. doi: 10.1001/jama.2013.2194
194. Rice TW, Wheeler AP, Bernard GR, Vincent J-L, Angus DC, Aikawa N, et al. A Randomized, Double-Blind, Placebo-Controlled Trial of TAK-242 for the Treatment of Severe Sepsis*. Crit Care Med (2010) 38(8):1685–94. doi: 10.1097/CCM.0b013e3181e7c5c9
195. Tracey KJ, Fong Y, Hesse DG, Manogue KR, Lee AT, Kuo GC, et al. Anti-Cachectin/TNF Monoclonal Antibodies Prevent Septic Shock During Lethal Bacteraemia. Nature (1987) 330(6149):662–4. doi: 10.1038/330662a0
196. Bernard GR, Francois B, Mira J-P, Vincent J-L, Dellinger RP, Russell JA, et al. Evaluating the Efficacy and Safety of Two Doses of the Polyclonal Anti-Tumor Necrosis Factor-α Fragment Antibody AZD9773 in Adult Patients With Severe Sepsis and/or Septic Shock. Crit Care Med (2014) 42(3):504–11. doi: 10.1097/CCM.0000000000000043
197. Bernard GR, Vincent J-L, Laterre P-F, LaRosa SP, Dhainaut J-F, Lopez-Rodriguez A, et al. Efficacy and Safety of Recombinant Human Activated Protein C for Severe Sepsis. N Engl J Med (2001) 344(10):699–709. doi: 10.1056/NEJM200103083441001
198. Ranieri VM, Thompson BT, Barie PS, Dhainaut J-F, Douglas IS, Finfer S, et al. Drotrecogin Alfa (Activated) in Adults With Septic Shock. N Engl J Med (2012) 366(22):2055–64. doi: 10.1056/NEJMoa1202290
199. Riedemann NC, Guo R-F, Ward PA. Novel Strategies for the Treatment of Sepsis. Nat Med (2003) 9(5):517–24. doi: 10.1038/nm0503-517
200. Seymour CW, Kennedy JN, Wang S, Chang C-CH, Elliott CF, Xu Z, et al. Derivation, Validation, and Potential Treatment Implications of Novel Clinical Phenotypes for Sepsis. JAMA (2019) 321(20):2003. doi: 10.1001/jama.2019.5791
201. Vincent J-L, Francois B, Zabolotskikh I, Daga MK, Lascarrou J-B, Kirov MY, et al. Effect of a Recombinant Human Soluble Thrombomodulin on Mortality in Patients With Sepsis-Associated Coagulopathy. JAMA (2019) 321(20):1993. doi: 10.1001/jama.2019.5358
202. Marshall JC. Why Have Clinical Trials in Sepsis Failed? Trends Mol Med (2014) 20(4):195–203. doi: 10.1016/j.molmed.2014.01.007
203. De Borst MH, Diks SH, Bolbrinker J, Schellings MW, Van Dalen MBA, Peppelenbosch MP, et al. Profiling of the Renal Kinome: A Novel Tool to Identify Protein Kinases Involved in Angiotensin II-Dependent Hypertensive Renal Damage. Am J Physiol Physiol (2007) 293(1):F428–37. doi: 10.1152/ajprenal.00367.2006
204. Radu M, Chernoff J. Recent Advances in Methods to Assess the Activity of the Kinome. F1000Research (2017) 6(0):1004. doi: 10.12688/f1000research.10962.1
205. Wilson LJ, Linley A, Hammond DE, Hood FE, Coulson JM, MacEwan DJ, et al. New Perspectives, Opportunities, and Challenges in Exploring the Human Protein Kinome. Cancer Res (2018) 78(1):15–29. doi: 10.1158/0008-5472.CAN-17-2291
206. Paech F, Bouitbir J, Krähenbühl S. Hepatocellular Toxicity Associated With Tyrosine Kinase Inhibitors: Mitochondrial Damage and Inhibition of Glycolysis. Front Pharmacol (2017) 8:1–13. doi: 10.3389/fphar.2017.00367
207. Booth L, Poklepovic A, Dent P. Not the Comfy Chair! Cancer Drugs That Act Against Multiple Active Sites. Expert Opin Ther Targets (2019) 23(11):893–901. doi: 10.1080/14728222.2019.1691526
208. Lin A, Giuliano CJ, Palladino A, John KM, Abramowicz C, Lou YM, et al. Off-Target Toxicity Is a Common Mechanism of Action of Cancer Drugs Undergoing Clinical Trials. Sci Transl Med (2019) 11(509):eaaw8412. doi: 10.1126/scitranslmed.aaw8412
209. Cohen P, Cross D, Jänne PA. Kinase Drug Discovery 20 Years After Imatinib: Progress and Future Directions. Nat Rev Drug Discov (2021) 20(7):551–69. doi: 10.1038/s41573-021-00195-4
210. Ferguson FM, Gray NS. Kinase Inhibitors: The Road Ahead. Nat Rev Drug Discov (2018) 17(5):353–77. doi: 10.1038/nrd.2018.21
211. Hammaker D, Firestein GS. “Go Upstream, Young Man”: Lessons Learned From the P38 Saga. Ann Rheum Dis (2010) 69(Suppl 1):i77–82. doi: 10.1136/ard.2009.119479
213. Mullard A. 2019 FDA Drug Approvals. Nat Rev Drug Discov (2020) 19(2):79–84. doi: 10.1038/d41573-020-00001-7
214. Pushpakom S, Iorio F, Eyers PA, Escott KJ, Hopper S, Wells A, et al. Drug Repurposing: Progress, Challenges and Recommendations. Nat Rev Drug Discov (2019) 18(1):41–58. doi: 10.1038/nrd.2018.168
215. Waller CF. Imatinib Mesylate. In: Recent Results in Cancer Research. Cham: Springer International Publishing (2018). p. 1–27. doi: 10.1007/978-3-319-91439-8_1
216. Aman J, Van Bezu J, Damanafshan A, Huveneers S, Eringa EC, Vogel SM, et al. Effective Treatment of Edema and Endothelial Barrier Dysfunction With Imatinib. Circulation (2012) 126(23):2728–38. doi: 10.1161/CIRCULATIONAHA.112.134304
217. Sleire L, Førde HE, Netland IA, Leiss L, Skeie BS, Enger PØ. Drug Repurposing in Cancer. Pharmacol Res (2017) 124:74–91. doi: 10.1016/j.phrs.2017.07.013
218. Levêque D. Off-Label Use of Targeted Therapies in Oncology. World J Clin Oncol (2016) 7(2):253. doi: 10.5306/wjco.v7.i2.253
219. Ochoa D, Jarnuczak AF, Viéitez C, Gehre M, Soucheray M, Mateus A, et al. The Functional Landscape of the Human Phosphoproteome. Nat Biotechnol (2020) 38(3):365–73. doi: 10.1038/s41587-019-0344-3
220. Seyhan AA. Lost in Translation: The Valley of Death Across Preclinical and Clinical Divide – Identification of Problems and Overcoming Obstacles. Transl Med Commun (2019) 4(1):18. doi: 10.1186/s41231-019-0050-7
221. Lewis AJ, Rosengart MR. Bench-To-Bedside: A Translational Perspective on Murine Models of Sepsis. Surg Infect (Larchmt) (2018) 19(2):137–41. doi: 10.1089/sur.2017.308
222. Cleuren ACA, Van der Ent MA, Jiang H, Hunker KL, Yee A, Siemieniak DR, et al. The In Vivo Endothelial Cell Translatome Is Highly Heterogeneous Across Vascular Beds. Proc Natl Acad Sci (2019) 116(47):23618–24. doi: 10.1073/pnas.1912409116
223. Deng L, Pollmeier L, Zhou Q, Bergemann S, Bode C, Hein L, et al. Gene Expression in Immortalized Versus Primary Isolated Cardiac Endothelial Cells. Sci Rep (2020) 10(1):2241. doi: 10.1038/s41598-020-59213-x
224. Low LA, Mummery C, Berridge BR, Austin CP, Tagle DA. Organs-On-Chips: Into the Next Decade. Nat Rev Drug Discov (2021) 20(5):345–61. doi: 10.1038/s41573-020-0079-3
225. Hofer M, Lutolf MP. Engineering Organoids. Nat Rev Mater (2021) 6(5):402–20. doi: 10.1038/s41578-021-00279-y
226. Wu Q, Liu J, Wang X, Feng L, Wu J, Zhu X, et al. Organ-On-a-Chip: Recent Breakthroughs and Future Prospects. BioMed Eng Online (2020) 19(1):9. doi: 10.1186/s12938-020-0752-0
227. Nishinakamura R. Human Kidney Organoids: Progress and Remaining Challenges. Nat Rev Nephrol (2019) 15(10):613–24. doi: 10.1038/s41581-019-0176-x
228. Bigaeva E, Gore E, Simon E, Zwick M, Oldenburger A, De Jong KP, et al. Transcriptomic Characterization of Culture-Associated Changes in Murine and Human Precision-Cut Tissue Slices. Arch Toxicol (2019) 93(12):3549–83. doi: 10.1007/s00204-019-02611-6
229. Majorova D, Atkins E, Martineau H, Vokral I, Oosterhuis D, Olinga P, et al. Use of Precision-Cut Tissue Slices as a Translational Model to Study Host-Pathogen Interaction. Front Vet Sci (2021) 8:1–7. doi: 10.3389/fvets.2021.686088
230. Ásgeirsdóttir SA, Talman EG, De Graaf IA, Kamps JAAM, Satchell SC, Mathieson PW, et al. Targeted Transfection Increases siRNA Uptake and Gene Silencing of Primary Endothelial Cells In Vitro — A Quantitative Study. J Control Release (2010) 141(2):241–51. doi: 10.1016/j.jconrel.2009.09.008
231. Lewis AJ, Yuan D, Zhang X, Angus DC, Rosengart MR, Seymour CW. Use of Biotelemetry to Define Physiology-Based Deterioration Thresholds in a Murine Cecal Ligation and Puncture Model of Sepsis. Crit Care Med (2016) 44(6):e420–31. doi: 10.1097/CCM.0000000000001615
232. Berthelsen LO, Kristensen AT, Tranholm M. Animal Models of DIC and Their Relevance to Human DIC: A Systematic Review. Thromb Res (2011) 128(2):103–16. doi: 10.1016/j.thromres.2010.12.002
Keywords: endothelial cells (EC), sepsis, sepsis-induced organ injury, sepsis-induced multiple organ failure, drug treatment, signal transduction, kinases and phosphatases
Citation: Luxen M, van Meurs M and Molema G (2022) Unlocking the Untapped Potential of Endothelial Kinase and Phosphatase Involvement in Sepsis for Drug Treatment Design. Front. Immunol. 13:867625. doi: 10.3389/fimmu.2022.867625
Received: 01 February 2022; Accepted: 28 March 2022;
Published: 13 May 2022.
Edited by:
Bernahrd Ryffel, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Johanna Hol Fosse, Norwegian Veterinary Institute (NVI), NorwayCopyright © 2022 Luxen, van Meurs and Molema. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthijs Luxen, bS5sdXhlbkB1bWNnLm5s
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.