
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 10 May 2022
Sec. Molecular Innate Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.866330
This article is part of the Research Topic Current Challenges in Complement Diagnostics View all 27 articles
 Yuzhou Zhang
Yuzhou Zhang Renee X. Goodfellow
Renee X. Goodfellow Nicolo Ghiringhelli Borsa
Nicolo Ghiringhelli Borsa Hannah C. DunlopStephen A. PrestiNicole C. Meyer
Hannah C. DunlopStephen A. PrestiNicole C. Meyer Dingwu Shao
Dingwu Shao Sarah M. Roberts
Sarah M. Roberts Michael B. Jones
Michael B. Jones Gabriella R. PitcherAmanda O. Taylor
Gabriella R. PitcherAmanda O. Taylor Carla M. Nester
Carla M. Nester Richard J. H. Smith*
Richard J. H. Smith*C3 glomerulopathy (C3G) and atypical hemolytic uremic syndrome (aHUS) are two rare diseases caused by dysregulated activity of the alternative pathway of complement secondary to the presence of genetic and/or acquired factors. Complement factor I (FI) is a serine protease that downregulates complement activity in the fluid phase and/or on cell surfaces in conjunction with one of its cofactors, factor H (FH), complement receptor 1 (CR1/CD35), C4 binding protein (C4BP) or membrane cofactor protein (MCP/CD46). Because altered FI activity is causally related to the pathogenesis of C3G and aHUS, we sought to test functional activity of select CFI missense variants in these two patient cohorts. We identified 65 patients (16, C3G; 48, aHUS; 1 with both) with at least one rare variant in CFI (defined as a MAF < 0.1%). Eight C3G and eleven aHUS patients also carried rare variants in either another complement gene, ADAMTS13 or THBD. We performed comprehensive complement analyses including biomarker profiling, pathway activity and autoantibody testing, and developed a novel FI functional assay, which we completed on 40 patients. Seventy-eight percent of rare CFI variants (31/40) were associated with FI protein levels below the 25th percentile; in 22 cases, FI levels were below the lower limit of normal (type 1 variants). Of the remaining nine variants, which associated with normal FI levels, two variants reduced FI activity (type 2 variants). No patients carried currently known autoantibodies (including FH autoantibodies and nephritic factors). We noted that while rare variants in CFI predispose to complement-mediated diseases, phenotypes are strongly contingent on the associated genetic background. As a general rule, in isolation, a rare CFI variant most frequently leads to aHUS, with the co-inheritance of a CD46 loss-of-function variant driving the onset of aHUS to the younger age group. In comparison, co-inheritance of a gain-of-function variant in C3 alters the phenotype to C3G. Defects in CFH (variants or fusion genes) are seen with both C3G and aHUS. This variability underscores the complexity and multifactorial nature of these two complement-mediated renal diseases.
The complement cascade is the cornerstone of the innate defense system. It is the first line of defense against foreign and altered host cells, it activates and potentiates adaptive immunity, and it provides integrated crosstalk with other pathways including the coagulation pathway (1). Activation of complement is triggered via one of three pathways–the classical (CP), lectin (LP) and alternative (AP)–all of which converge on C3bBb, a C3 convertase that cleaves C3 to C3b, to amplify the complement response and initiate the terminal pathway with ultimate formation of the membrane-bound lytic unit membrane attack complex (C5b-9). The entire cascade is tightly regulated by several mechanisms involving soluble and membrane-bound regulators such as factor H (FH), factor I (FI) and membrane cofactor protein (MCP/CD46) (2).
The primary function of FI is to downregulate complement activity by proteolytic inactivation of C3b to iC3b and C4b to iC4b in the presence of one of its four co-factors, FH, CR1, C4BP and MCP. FI-mediated regulation is especially important in controlling activity of the AP as this pathway is constitutively active. FI is synthesized by the liver as a 66 kDa single chain peptide that undergoes posttranslational glycosylation (adding 22 kDa of glycans) and protein cleavage to remove the signal sequence and four internal amino acids (RRKR, residues 336 to 339) to generate a mature protein with a heavy chain (50 kDa, Lys19–Ile335) and a light chain (38 kDa, Ile340–Val583) linked by a single disulfide bond (Cys255-Cys471) (3, 4). The five recognizable domains include a membrane-attack complex (FIMAC) domain, scavenger receptor cysteine-rich (SRCR) domain, two low-density lipoprotein receptor domains (LDLR1 and LDLR2) and, after a C-terminal linker on the heavy chain, the catalytic or serine protease (SP) domain with its catalytic triad His380-Asp429-Ser525 on the light chain (Figure 1) (5).
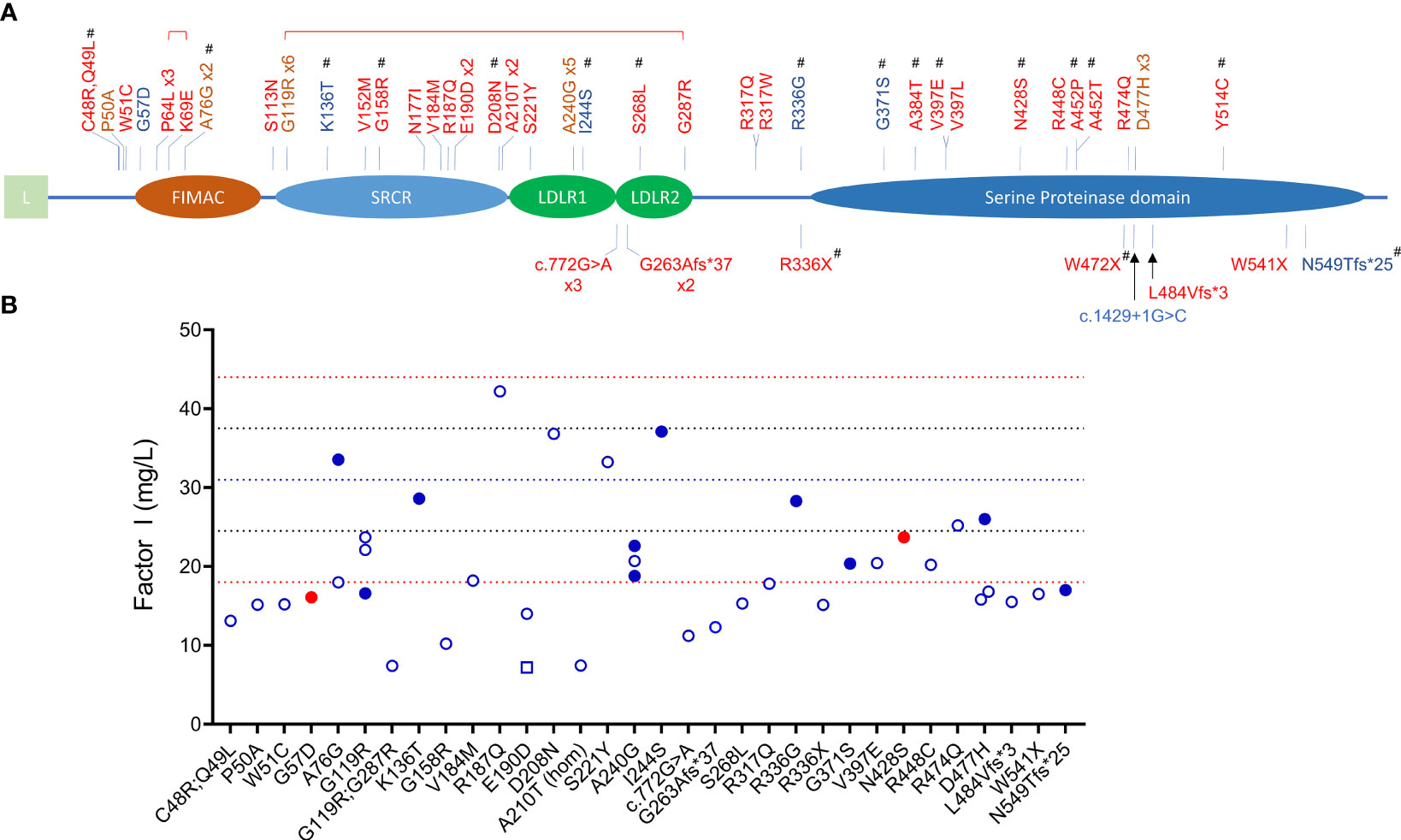
Figure 1 CFI variants identified in patients with C3G and aHUS. (A) Schematic view of factor I and the 45 rare variants (MAF < 0.1%) identified in 65 patients (red, aHUS; blue, C3G; brown, both C3G and aHUS). All variants were identified in heterozygosis except one homozygote (p.Ala210Thr) and two compound heterozygotes (brackets above variants indicate compound heterozygosity). Novel variants are marker with #. (L, leading sequence; FIMAC, Factor I Membrane Attack Complex; SRCR, Scavenger Receptor Cysteine-Rich; LDLR, Low-Density Lipoprotein Receptor). (B) FI levels in 40 patients (open circles, aHUS; filled blue circles, C3GN; filled red circles, DDD; open square, C3G/aHUS; dashed lines, reference values: red, upper and lower limits of normal; blue, percentile lines).
Deficiency of FI leads to unchecked AP amplification and consumptive deficiency of complement components, such as C3, with various phenotypic consequences (6). Complete absence of FI, which is rare, results in severe and recurrent infections, glomerulonephritis and/or autoimmune disease (7–11). Partial FI deficiencies, which are much more common, lead to dysregulated C3 convertase activity and predispose to several complement-mediated diseases such as atypical hemolytic uremic syndrome (aHUS) (12–15), age-related macular degeneration (AMD) (16–18), and occasionally, C3 glomerulopathy (C3G) (19–22).
C3G and aHUS are rare renal diseases caused by dysregulated complement activity of the AP typically driven by genetic and/or acquired factors. In C3G, uncontrolled complement activity in the fluid phase and glomerular microenvironment leads to a characteristic pattern of glomerular injury defined by the predominant deposition of C3 fragments for which the disease is named (23–25). Based on electron microscopy, two major subclasses of C3G are recognized–C3 glomerulonephritis (C3GN) and dense deposit disease (DDD). Classic clinical findings of both subtypes are hematuria and proteinuria (26). aHUS is a form of complement-mediated thrombotic microangiopathy (TMA) characterized by microangiopathic hemolytic anemia, consumptive thrombocytopenia, and multisystem end organ involvement primarily affecting the kidney (27). In aHUS, dysregulated complement activity primarily occurs on cell surfaces, leading to local prothrombotic states, especially in the kidney.
Here, we reported 45 variants in CFI (MAF < 0.1%) identified in 65 patients with either C3G or aHUS. We completed detailed studies in 40 patients that included a novel FI functional assay and identified both type 1 and type 2 CFI variants.
Our C3G and TMA registry (C3G, n = 1048; TMA, n = 1468) was searched for patients who were evaluated between 2009-’21 and carried a CFI variant with a minor allele frequency (MAF) < 0.1%. Biomaterials included DNA (all patients) isolated from peripheral blood using established methods (28), and serum and plasma (most patients) collected at the time of disease onset (acute flare) in patients with aHUS or during ongoing disease (chronic period) in patients with C3G using a standard protocol (29) and stored as one-time-use aliquots at -80°C. The study was approved by the Institutional Review Board of Carver College of Medicine at the University of Iowa.
Genetic analysis was performed using a targeted sequencing panel, which captures relevant complement genes as well as THBD, ADAMTS13 and DGKE as we have described (30). In addition, the CFH-CFHR and CFI genomic regions were screened for copy number variation by multiplex ligation-dependent probe amplification (MLPA) using the MRC Holland SALSA kit and in-house designed probes.
Biomarker/functional testing was performed using a customized panel that includes ELISAs, radial immunodiffusion (RID), immunofixation electrophoresis (IFE), and hemolytic-based assays (29). Specifically, CP and AP activities were assessed using Quidel (San Diego, CA) and Wieslab kits (SVAR, Malmö, Sweden). Fluid-phase activity was also tested using IFE on a SPIFE machine (Helena Laboratories, Beaumont, TX). Serum levels of C3, C5, properdin, FH, FI and complement activation products C3c, Ba, Bb, sC5b-9 were measured using ELISA kits (Hycult Biotech, Wayne, PA or Quidel, San Diego, CA). C4, FB and FI were run by RID (The Binding Site, San Diego, CA). FH and FB autoantibody assays were done by ELISA against purified proteins FH and FB, respectively (Complement Tech, Tyler, TX). C3-, C4-, and C5-nephritic factors were detected by cell-based hemolytic methods (31).
To assess FI co-factor activity, we developed a novel FI functional assay using C3b-decorated sheep erythrocytes (C3b-ShE) prepared as previously described (32). To minimize the effect of endogenous FH, patient serum or plasma was diluted 1:32 in GVB-Mg2+ buffer (16). To assess FI function, 50 µL of diluted serum/plasma, 50 µL of C3b-ShE (1x109/mL) and 100 µL of co-factors were added to 200 µL of GVB-EDTA buffer. The co-factors [FH (Complement Tech, Tyler, TX), sCR1 (Celldex Therapeutics, New Haven, CT) or sMCP (Alexion Pharmaceuticals, Boston, MA)] were assayed at final concentrations of 50nM, 25nM and 50nM, respectively. The resulting mixture was incubated at 37°C for 15 minutes. After 3 washes, cells were resuspended in 50 µL of GVB-Mg2+ buffer and remaining C3b on ShE was titrated out by excess FB (5x of the amount at Z = 1) and FD (0.3 μg) in GVB-Mg2+ buffer to form C3 convertase at 30°C over a 5-minute period; the reaction was stopped by adding 300 µL of GVB-EDTA buffer. 50 μL of the mixture was transferred to an empty 96-well plate and hemolysis was induced by adding 50 μL of rat EDTA serum diluted (1:10) in GVB-EDTA buffer (as a source of C5-C9). After centrifugation at 1000 x g, cell-free supernatant was transferred to a flat bottom 96-well plate and absorbance was read at OD415. The percentage of hemolysis in each well was calculated using hypotonic lysis induced by water as 100%.
Serum or plasma (1:80 diluted) in Laemmli buffer with or without reducing reagent was separated on 4-15% polyacrylamide gels (Bio-Rad, Hercules, CA). After transferring, FI was visualized using rabbit monoclonal antibody to FI (ab278524, Abcam, Waltham, MA) followed by a secondary incubation and chemiluminescence.
Statistical analysis was performed using the Student’s t-test for two group comparisons, Fisher’s exact test for contingency tables, and Pearson correlation to measure linear relationships between variables using GraphPad Prism 8.2 (GraphPad Software, San Diego, CA). Error bars represent means ± SD. The number of samples and number of experimental repetitions are indicated in the figure legends. P < 0.05 was considered significant.
Sixty-five unrelated patients (sixteen C3G [ten, C3GN; six, DDD]; 48 aHUS; one C3G/aHUS) with rare variants in CFI were identified in our registry (Figure 1), which represents significant enrichment in our aHUS cohort as compared to the prevalence of rare variants with MAF < 0.1% reported in gnomAD (1.77% of 141433 individuals in gnomAD vs 1.62% of 1048 C3G patients (P > 0.05) and vs 3.34% of 1468 TMA patients (P < 0.0001)) (33). In addition, the prevalence of rare variants is significantly higher in aHUS as compared to C3G (P = 0.0078). Disease occurred at any age in both cohorts (median age, 27.5 vs 37, respectively), although more aHUS patients were diagnosed in early childhood (age < 5, six aHUS vs one C3G). In the aHUS cohort, there were also more females (female/male, 35/13), while in the C3G cohort, both genders were equally affected (male/female, 9/8) (Figure 2A). Demographic data and basic clinical information are shown in Table 1.
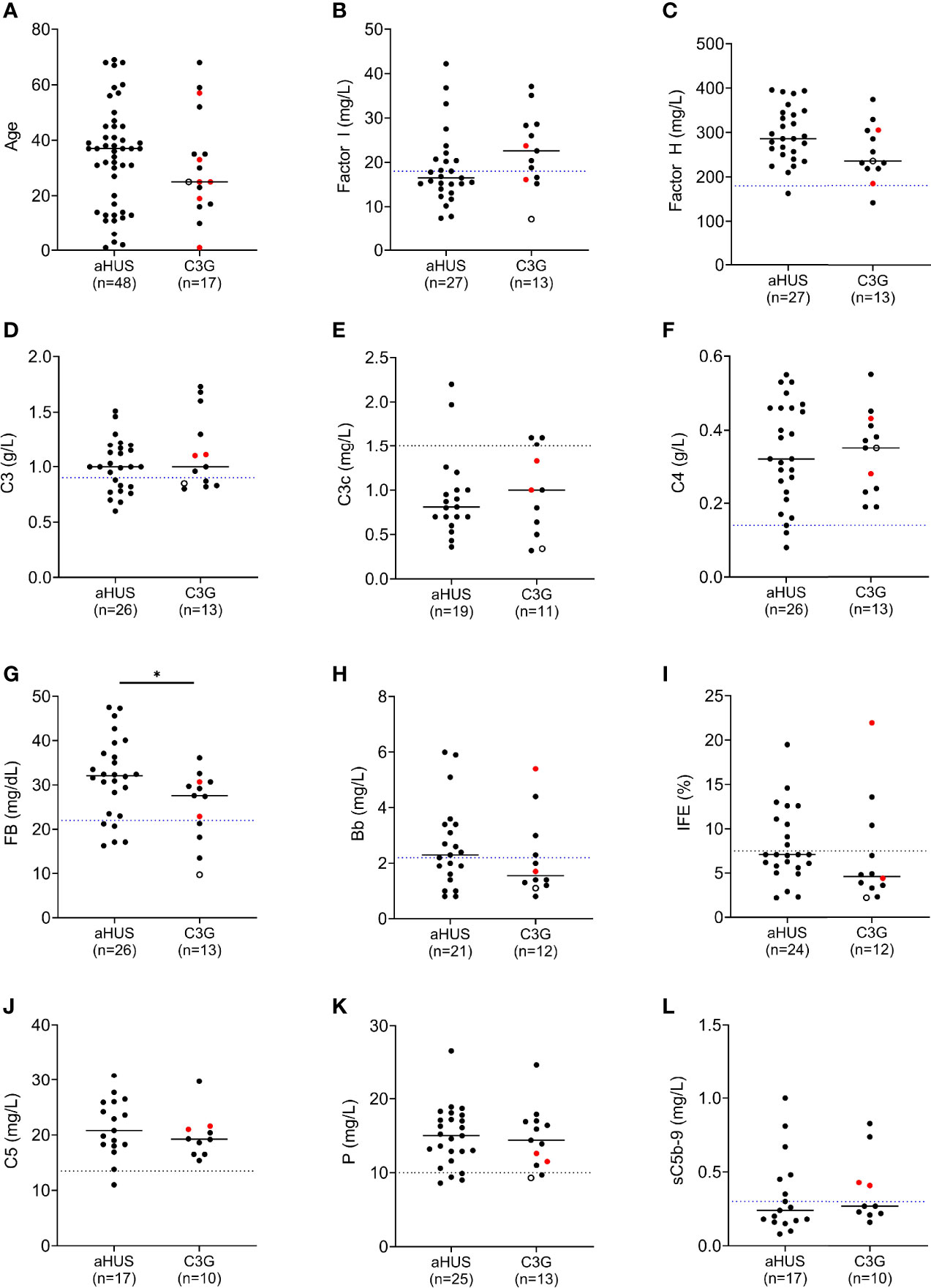
Figure 2 Complement biomarker profiling in aHUS and C3G patients. (A) Age distribution. (B) Factor I levels. (C) Factor H levels. (D–H) Biomarkers for alternative pathway (AP) activity including C3 and factor B, and associated activation products C3c and Bb. (I) AP fluid-phase activity by immunofixation eletrophoresis. (J–L) Biomarkers for terminal pathway activity including C5, properdin and soluble terminal complement complex (sC5b-9). (left column, aHUS; right column-red dots, DDD patients; black dots, C3GN patients; open circle, C3GN/aHUS patient; dashed line, lower limit of normal value for each assay; patients on eculizumab excluded from (J, L); *P < 0.05).
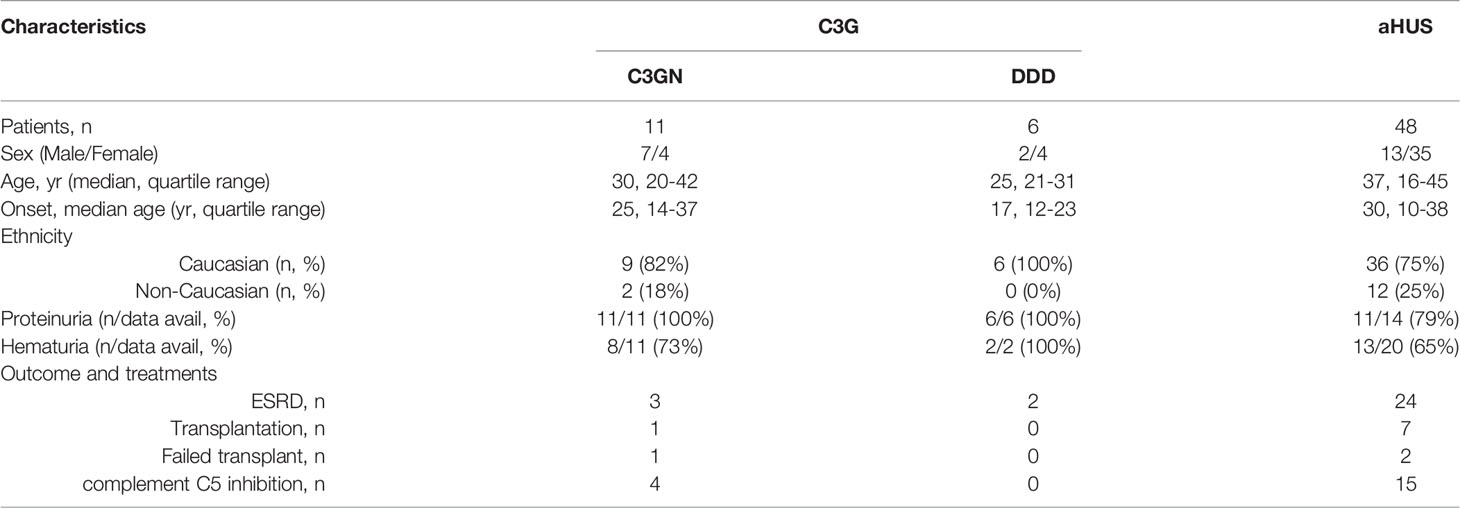
Table 1 Patient demographics.
Null alleles were found in nine of 48 (19%) patients with aHUS and two of 15 (13%) patients with C3G (Tables 2, 3). Three aHUS patients (#27, #28, #29) carried the same splice-region variant (c.772G>A, p.Ala258Thr; the last nucleotide of exon 5), which results in exon 5 skipping (34). Three patients (#35, #46, #48) carried nonsense mutations (p.Arg336Ter, p.Trp472Ter and p.Trp541Ter) and three patients (#31, #32, #47) had micro-deletions that result in frame shifts (two with c.786delA and one with c.1450_1454del). In the C3G cohort, two null alleles were identified–a microdeletion (c.1646delA; #58, C3GN) and a splice site variant (c.1429+1G>C; #64, DDD). Western blot confirmed the absence of detectable circulating truncated proteins in all patients with null alleles, suggesting that the transcribed mutant message undergoes nonsense-mediated decay. We also confirmed non-expression of the c.1429+1G>C allele on a western blot in a homozygote patient we are following for recurrent infections.
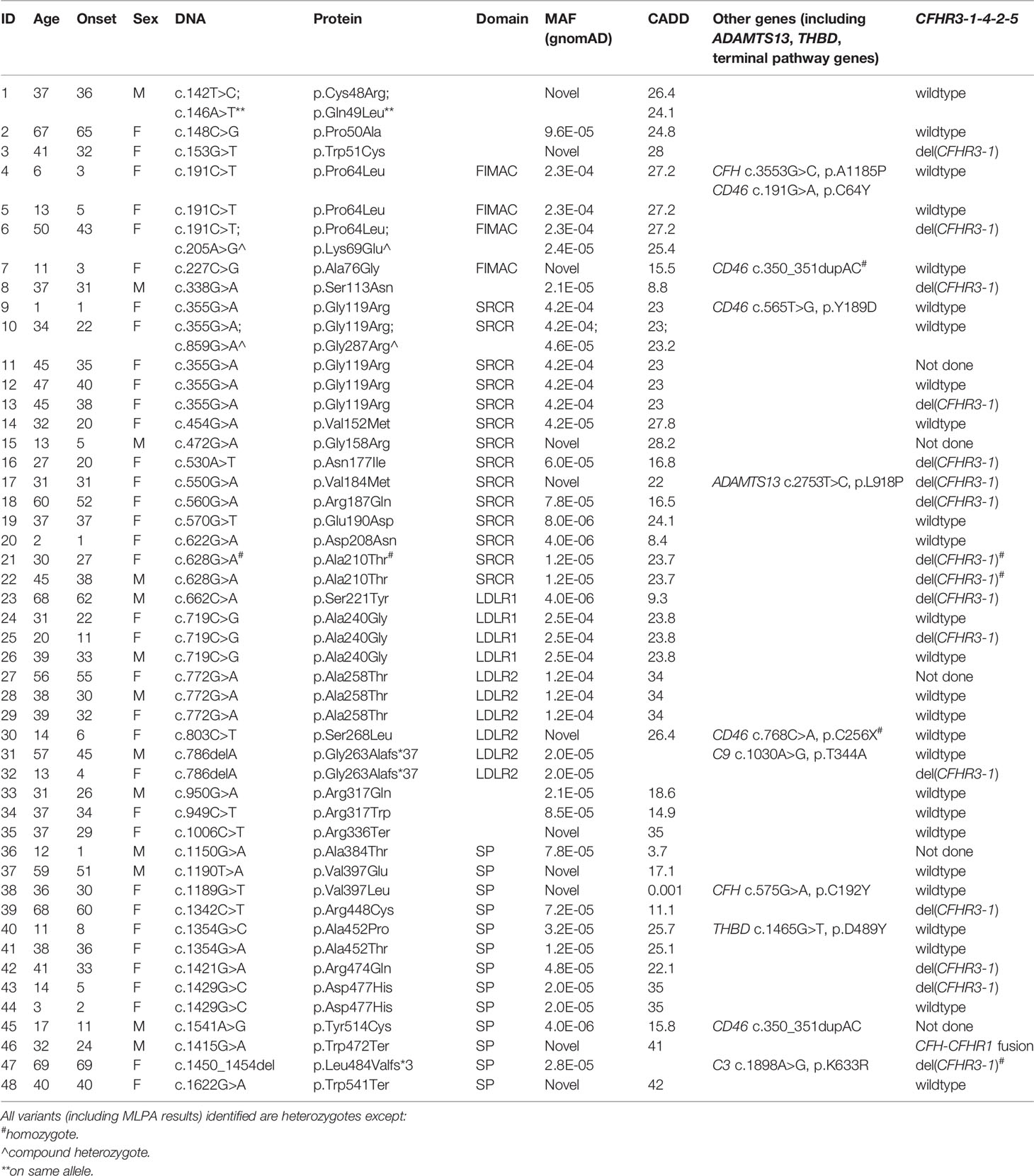
Table 2 Demographic and genetic data in patients with aHUS.
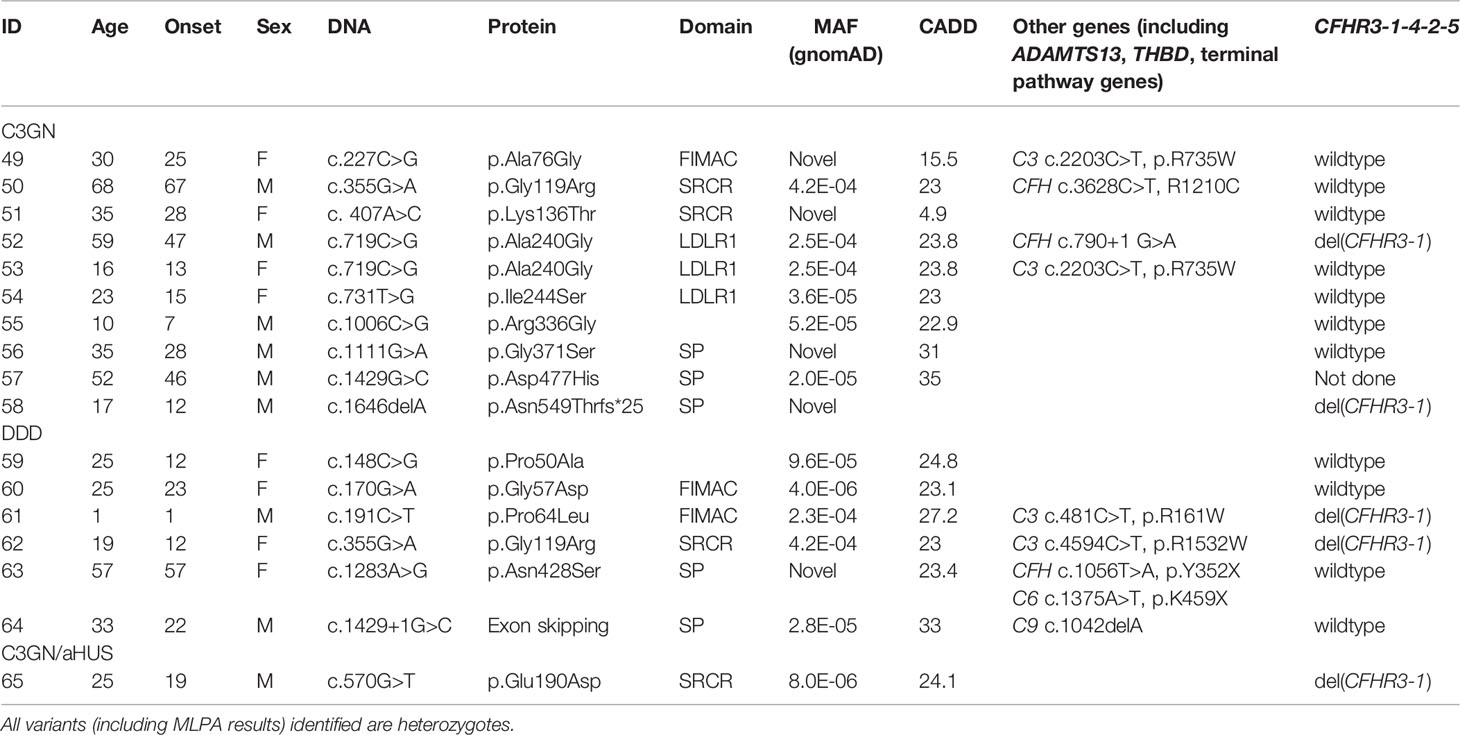
Table 3 Demographic and genetic data in patients with C3G.
Most variants (64/65) were found in heterozygosis, although we identified one aHUS patient homozygous for p.Ala210Thr (#21) and two who were compound heterozygotes (p.Pro64Leu/p.Lys69Glu, #6; p.Gly119Arg/p.Gly287Arg, #10). One aHUS patient carried two variants on the same allele (p.Cys48Arg/p.Gln49Leu, #1) (Table 2). No C3G patients carried a second rare variant in CFI and no large genomic deletions in the CFI region were identified (Tables 3).
Genetic variants (pathogenic or likely pathogenic, MAF < 0.1%) in other complement or TMA genes were identified in ten patients with aHUS and eight with C3G. One aHUS patient (#46) carried a CFH-CFHR1 fusion gene identified by MLPA (Table 2). In addition, two DDD patients carried a null allele in a terminal pathway gene (#63, #64) (Table 3).
Homozygous deletions of CFHR3-CHR1 were found in 3/60 (5%) patients, however none was positive for factor H autoantibodies (FHAAs). All patients were also negative for FBAAs and all C3G patients were negative for C3-, C4- and C5-nephritic factors.
FI levels were measured in 40 patients (12 C3G; 27 aHUS and 1 C3G/aHUS) (Figure 2B and Supplementary Tables 1, 2). In 31 of 40 (78%) patients, these levels were in the lowest quartile of the normal reference range (24.5 mg/L, normal reference mean = 31, standard deviation = 6.5). The lowest FI levels were found in two aHUS patients with biallelic variants. Excluding these two patients, FI levels were higher in C3G patients as compared to aHUS patients, however the difference was not statistically significant (median C3G vs aHUS, 24 vs 19.4, P = 0.09). FH levels showed the reverse trend and tended to be lower in C3G patients, although this comparison also did not reach statistical significance (Figure 2C, P = 0.08).
Several serum complement biomarkers were abnormal, including C3, which was low in four of twelve (33%) C3G and nine of 27 (33%) aHUS patients (Figure 2D), consistent with increased C3 convertase activity. In both cohorts, elevation in C3c occurred in a few patients while C4 was normal in most patients (Figures 2E, F). FB was low in three of twelve (25%) patients with C3G and five of 26 (19%) aHUS patients (Figure 2G), while Bb was high in four of eleven (36%) C3G and twelve of 21 (57%) aHUS patients (Figure 2H). Overall, the levels of FI showed moderate correlation with plasma C3 and factor B levels (Pearson’s r correlation = 0.39 and 0.36; P = 0.014 and 0.026, respectively). These findings also reflect increased convertase activity associated with a decrease in FI. Ba was elevated in most patients (Supplementary Tables 1, 2) however this biomarker does not accurately reflect complement dysregulation but rather the degree of renal injury (35, 36). The positive IFE in three of eleven (27%) C3G and nine of 24 (38%) aHUS patients recapitulated dysregulated upstream complement activity (Figure 2I). Of terminal pathway biomarkers (Figures 2J–L), sC5b-9 was elevated in six of seventeen (35%) aHUS and four of ten (40%) C3G patients not on Eculizumab (Figure 2L).
In nine patients [four aHUS patients (p.Arg187Gln, p.Asp208Asn, p.Ser221Tyr, p.Arg474Gln); five C3G patients (p.Ala76Gly, p.Lys136Thr, p.Ile244Ser, p.Arg336Gly, p.Asp477His)], FI levels were above the lowest quartile and in these patients, we evaluated FI co-factor activity. Two type 2 variants were identified–p.Arg474Gln and p.Arg336Gly–both of which reduced C3b cleavage activity with all three cofactors (FH, sCR1 and MCP) (Figures 3A–C). Western blotting showed that p.Arg336Gly circulates as unprocessed single chain (Figure 3D). No functional impairment of FI activity was detectable with the other variants.
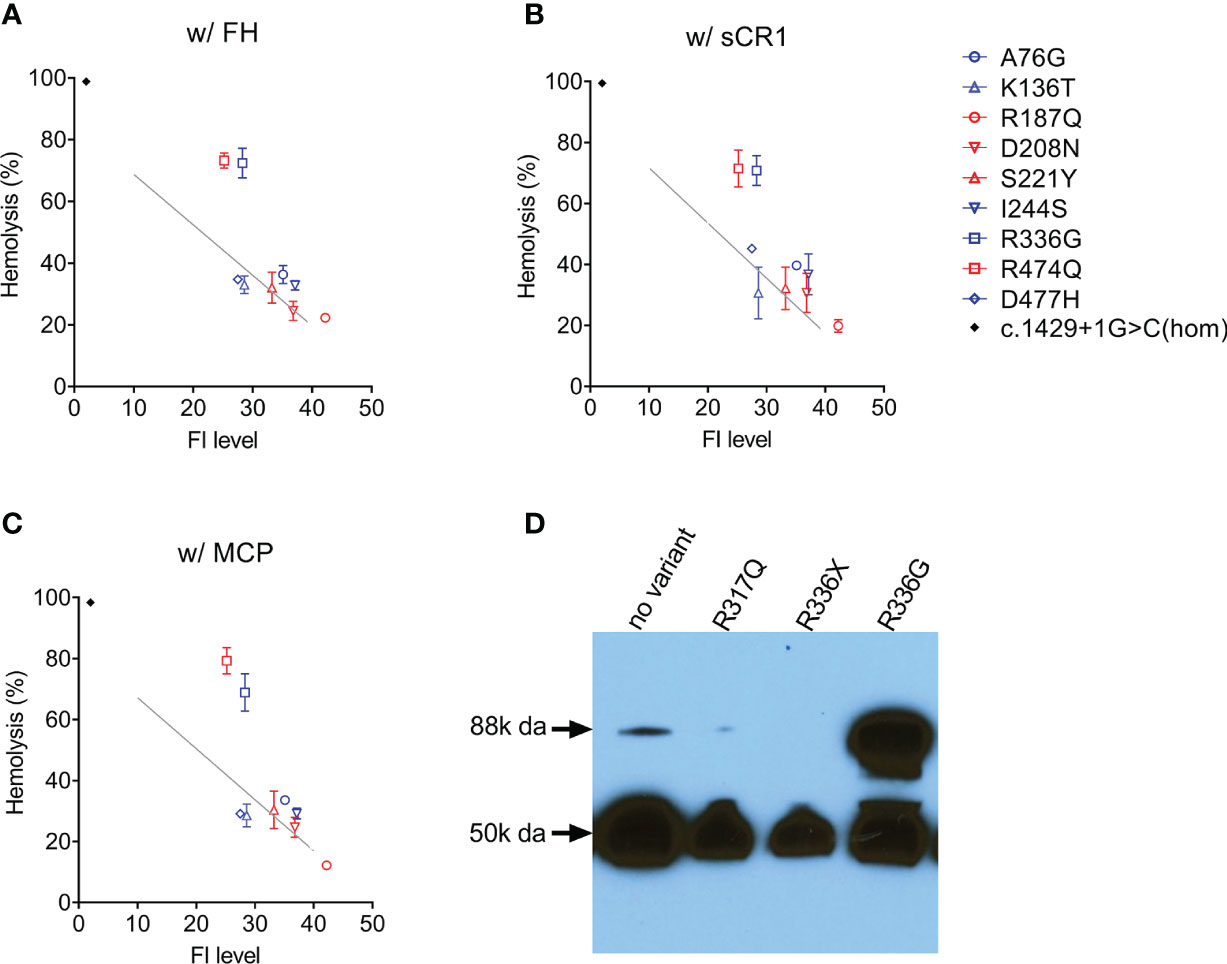
Figure 3 Assessing FI function. In the presence of a cofactor (A) FH 50 nM; (B) sCR1 25nM; (C) MCP 50nM, C3b cleavage activity by FI in patient serum (diluted 1:32) is measured using C3b-decorated sheep erythrocytes. Non-cleaved C3b is developed by the addition of both FB/FD to form C3 convertase and rat-EDTA serum as a source of C5-C9. Percent hemolysis is calculated and plotted as a function of FI concentration determined by ELISA. The grey line in each figure serves as a normal reference line calculated from a normal sample (FI concentration, 40 mg/L) with serial dilutions. Serum from a CFI c.1429+1G>C homozygote (FI is undetectable) serves as a positive control (black dot). (D) Circulating FI by a Western blot. Patient sample (1:80 diluted in PBS) with reducing reagent separated on a 4-15% polyacrylamide gel and transferred. FI was visualized by an antibody specific to the heavy chain (50k Da). CFI p.Arg336Gly results in an unprocessed single chain (88k Da).
Sixty-five unrelated patients [sixteen C3G (ten, C3GN; six, DDD); 48 aHUS; one C3G/aHUS] with rare variants in CFI were identified in our registry, which represents significant enrichment in our aHUS cohort as compared to the prevalence of rare CFI variants reported in gnomAD (1.77% of 141433 individuals in gnomAD vs 1.62% of 1048 C3G patients (P > 0.05) and vs 3.34% of 1468 TMA patients (P < 0.0001)) (33). Eighteen of these variants are novel and three (p.Val184Met, p.Arg448Cys, p.Trp541Ter) are reported for the first time in an aHUS cohort [previously associated only with AMD (18, 37–39)]. In 40 patients, we completed serological and functional studies to facilitate the classification of these variants based on ACMG guidelines.
Low FI levels have a variety of phenotypic consequences with the most extreme, complete CFI deficiency, leading to uncontrolled AP activity and C3 consumption (7). As a consequence of C3 consumption, severe and recurrent pyogenic infections with encapsulated organisms such as Streptococcus pneumoniae, Haemophilus influenzae and Neisseria meningitidis develop, driven by defective opsonization, immune adherence and phagocytosis (40). Whilst we have not seen complete FI deficiency in our C3G and aHUS cohorts, we have identified several patients with low or borderline low FI levels (16-24.5 mg/L, normal range 18 – 44) with the presenting phenotype typically driven by other acquired or genetic factors. For example, in isolation a rare CFI variant most frequently leads to aHUS, however the co-inheritance of a CD46 loss-of-function pathogenic variant drives the onset of aHUS to the younger age group (#4, #7, #9, #30, #45) while a gain-of-function pathogenic variant in C3 alters the phenotype to C3G (4 of 15 vs 1 of 48, C3G vs aHUS, P = 0.0097). Defects in CFH (variants or fusion genes) are seen with both C3G and aHUS (3 of 15 vs 2 of 48, C3G vs aHUS, P = 0.0828).
Autoantibodies to complement components (such as FHAAs, FBAAs, C3-, C4-, C5-nephritic factors) also play an important role in the pathogenesis of C3G and aHUS (23, 41), and while none of the patients in this study was co-positive for autoantibodies, we did not consider this finding surprising. With respect to aHUS, the prevalence of FHAAs is generally low, and when identified, they are typically seen in the pre-teenage years in children who are homozygous for the deletion of CFHR3-CFHR1. This genotype was identified only three times in our aHUS cohort, all in adults. With respect to C3G, about 60% of patients are positive for genetic mutations and/or autoantibodies as drivers of disease (19, 29). Of this 60%, ~10% are co-positive for both genetic drivers and autoantibodies, ~70% have only autoantibodies, and ~20% have only genetic variants. In our C3G cohort, half had additional genetic variants contributing to the complement dysregulation.
In 27 patients in our aHUS cohort, we measured FI levels and identified 18 type 1 variants (low expression) and five variants with limited expression (within 1st quartile, Figure 1B). The remaining 4 variants–p.Arg187Gln, p.Asp208Asn, p.Ser221Tyr and p.Arg474Gln–were associated with normal FI levels (above the lowest quartile). Of these, p.Asp208Asn is a novel variant, while p.Arg187Gln, p.Ser221Tyr and p.Arg474Gln have been reported. Although no data are provided for p.Arg187Gln, p.Ser221Tyr was associated with normal FI levels in two AMD studies (16, 18). p.Arg474Gln has also been reported in AMD cohorts with normal FI levels (18, 42, 43) and in an aHUS cohort with low FI levels, although the aHUS patient also carried another type 1 variant p.Ala258Thr (44). Our functional data show that p.Arg474Gln affects FI cofactor activity with all three cofactors (FH, MCP, sCR1) (Figure 3), making p.Arg474Gln a type 2 pathogenic variant. No functional defects were observed with p.Arg187Gln, p.Asp208Asn and p.Ser221Tyr, and based on these data, these three variants should be classified as likely benign.
Low FI levels (< 25%) were found in six C3GN patients and one DDD patient (#60). Of the six C3GN patients, two CFI variants were identified in each of two patients (p.Gly119Arg, #50 and #62; p.Ala240Gly, #52 and #53), with the remaining two patients carrying truncating variants (p.Asn549Thrfs*25, #58; c.1429+1G>C, #64). Interestingly, p.Gly119Arg and p.Ala240Gly were also identified in aHUS patients (p.Gly119Arg, #9, #10, #11, #12, #13; p.Ala240Gly, #24, #25, #26) and are well-documented type 1 variants (18, 21, 45–48). p.Gly119Arg and p.Ala240Gly are also present in the general population with the highest minor allele frequencies in Europeans (MAF = 0.000852 and 0.000093, respectively). These data suggest that carrying p.Gly119Arg or p.Ala240Gly is a risk factor for complement-mediated disease depending, in part, on the expressivity of the other CFI allele and/or the presence of mutations in other genes. For example, one aHUS patient (#10) had extremely low FI levels as a result of compound heterozygosis (p.Gly119Arg and p.Gly287Arg on opposite alleles), while all C3G patients carrying either the p.Gly119Arg allele or the p.Ala240Gly allele had a second pathogenic or likely pathogenic variant in CFH or C3 (#50, #52, #53, #62; Table 3).
Similarly, p.Pro64Leu, a variant that has been documented in AMD patients with low FI expression (16, 18), was identified in three aHUS patients (one CFI compound het) and one DDD patient (#4, #5, #6 and #61). The DDD patient also carried a gain-of-functions variant in C3, p.Arg161Trp, which has been shown to increase C3b affinity for factor B and reduce binding to MCP, although FH-mediated regulation is unchanged (14, 49, 50).
With two other variants [p.Ala76Gly (novel), p.Asp477His] identified in both C3GN and aHUS patients (#49 vs #7 and #57 vs #43 and #44, C3G vs aHUS), we noticed much higher FI levels in C3G as compared to aHUS patients. In these five patients, all FI results were confirmed by another technique (RID) and Western blotting, and large genomic deletions were ruled out by MLPA. In addition, FI functional assays showed normal C3b cleavage in all patients. Further research is required to understand this variability. We classified both variants as VUSs (variant of unknown significance).
We completed functional studies on all C3G patients with normal FI levels (p.Lys136Thr, p.Ile244Ser, p.Arg336Gly, p.Asn428Ser) and observed significantly impaired FI function (C3b cleavage) with all cofactors (FH, MCP, sCR1) for only p.Arg336Gly (Figures 3A–C). Because maturation of FI requires a proteolytic process that removes four amino acids (Arg-Arg-Lys-Arg) at residues 336-339, FI p.Gly336 circulates as an unprocessed pro-peptide without functional activity (Figure 3D). This finding has also been reported for p.Arg339Lys and p.Arg339Glu in aHUS patients (21, 44) and p.Arg339Gln in AMD patients (18, 38, 43). Common to all these mutations is the removal of a positive charged residue from the consensus sequence R-x-K/R-R. Therefore, we would expect individuals carrying these variants to have an identifiable circulating unprocessed single peptide and these variants should be classified as likely pathogenic.
Finally, when measuring FI levels, it should be remembered that as an acute-phase protein its serum concentration increases non-specifically in response to many cytokines (17). In our experience, in the acute phase, FI levels can be elevated ~25% above baseline, however this increase does not occur in isolation but rather in conjunction with an increase in other complement biomarkers, especially FB, C4 and FH (Figure 4). To ensure that the impact of a rare hypomorphic allele variant is not masked by the normal allele, serial testing of complement biomarkers is advisable. Anti-C5 therapy with eculizumab does not impact FI levels although it does elevate plasma C5, suppress sC5b-9, and abolish activity of the classical and alternative pathways. And finally, we found that the prevalence of rare CFI variants is significantly higher in aHUS as compared to C3G (P = 0.0078). While this finding suggests that partial FI deficiency might have less impact on C3G, precisely how a relative deficiency in FI contributes to the underlying pathophysiology of these two diseases requires further study.
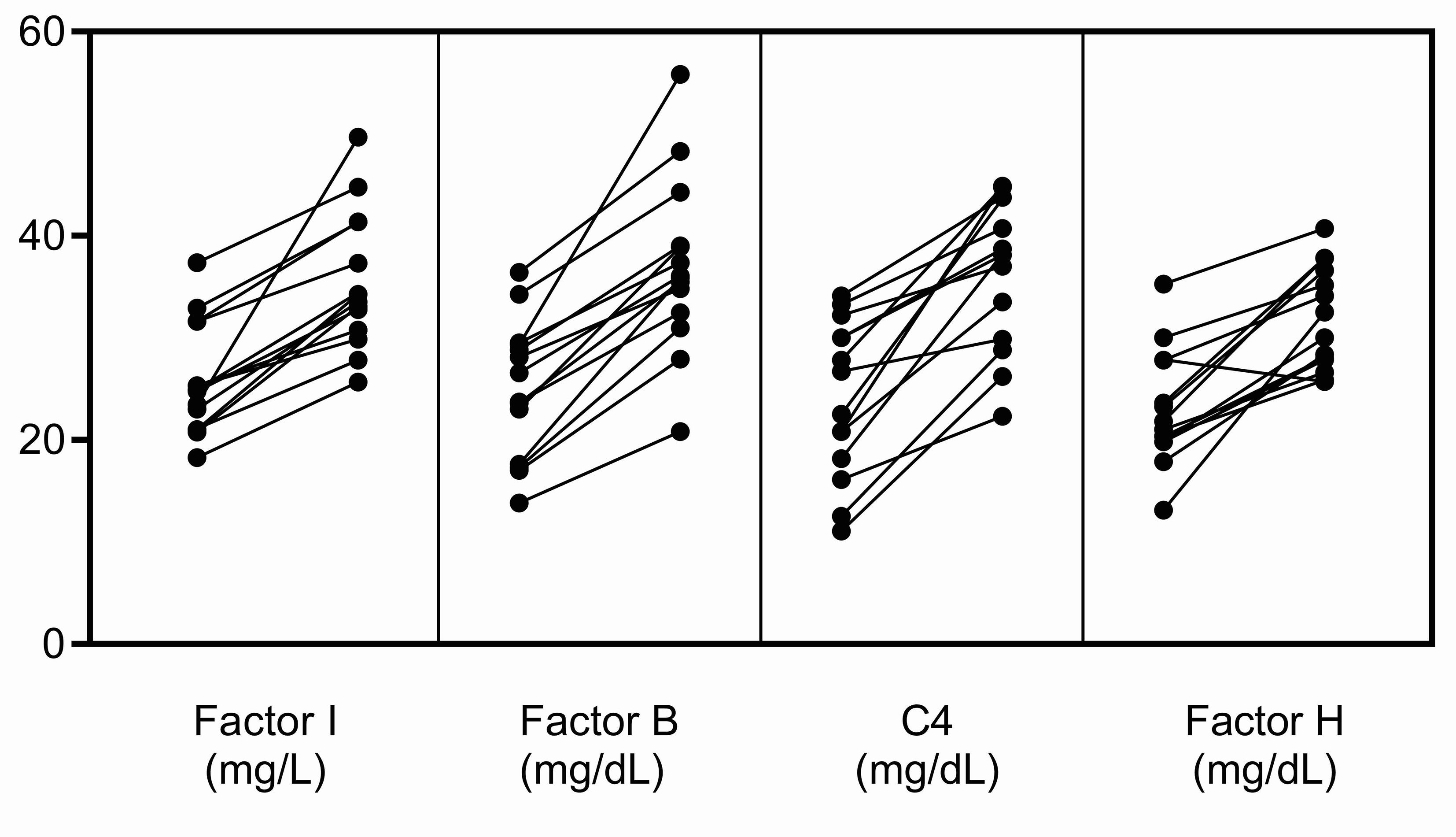
Figure 4 Increases in FI, FB, C4 and FH in the acute phase. These complement proteins are positive acute-phase reactants, and their levels were significantly increased in the acute phase in fourteen patients in our C3G and aHUS registries. The increase in FI does not occur in isolation but rather in conjunction with increases in other complement biomarkers, especially FB, C4 and FH. This pattern of increase was not observed in the patients described in this study (left, complement levels in remission; right, complement levels in the acute phase).
In summary, rare variants in CFI play a causal role in C3G and aHUS although the clinically observed phenotype is strongly contingent on the associated genetic background. Functional testing should be considered to assess FI activity if a rare CFI variant is identified. Our results suggest that the majority of CFI missense variants with a MAF < 0.1% will be type 1 variants (~80%), although a small of type 2 variants (~5%) will also be identified.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Institutional Review Board of Carver College of Medicine at the University of Iowa. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
YZ and RS conceived of the study, designed experiments, and wrote the manuscript. RG, NGB, HD, SP, NM, DS, SR, MJ, GP, and AT performed the experiments. CN provided crucial conceptual input. All authors reviewed and approved the manuscript.
This work was supported in part by National Institute of Health R01 DK110023.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are grateful to all patients whose participation made this research possible.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.866330/full#supplementary-material
Supplementary Table 1 | Biomarker data in patients with aHUS.
Supplementary Table 2 | Biomarker data in patients with C3G.
1. Markiewski MM, Lambris JD. The Role of Complement in Inflammatory Diseases From Behind the Scenes Into the Spotlight. Am J Pathol (2007) 171(3):715–27. doi: 10.2353/ajpath.2007.070166
2. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement System Part I - Molecular Mechanisms of Activation and Regulation. Front Immunol (2015) 6:262. doi: 10.3389/fimmu.2015.00262
3. Tsiftsoglou SA, Arnold JN, Roversi P, Crispin MD, Radcliffe C, Lea SM, et al. Human Complement Factor I Glycosylation: Structural and Functional Characterisation of the N-Linked Oligosaccharides. Biochim Biophys Acta (2006) 1764(11):1757–66. doi: 10.1016/j.bbapap.2006.09.007
4. Goldberger G, Arnaout MA, Aden D, Kay R, Rits M, Colten HR. Biosynthesis and Postsynthetic Processing of Human C3b/C4b Inactivator (Factor I) in Three Hepatoma Cell Lines. J Biol Chem (1984) 259(10):6492–7. doi: 10.1016/S0021-9258(20)82168-1
5. Roversi P, Johnson S, Caesar JJ, McLean F, Leath KJ, Tsiftsoglou SA, et al. Structural Basis for Complement Factor I Control and Its Disease-Associated Sequence Polymorphisms. Proc Natl Acad Sci USA (2011) 108(31):12839–44. doi: 10.1073/pnas.1102167108
6. Nilsson SC, Sim RB, Lea SM, Fremeaux-Bacchi V, Blom AM. Complement Factor I in Health and Disease. Mol Immunol (2011) 48(14):1611–20. doi: 10.1016/j.molimm.2011.04.004
7. Naesens L, Smet J, Tavernier SJ, Schelstraete P, Hoste L, Lambrecht S, et al. Plasma C3d Levels as a Diagnostic Marker for Complete Complement Factor I Deficiency. J Allergy Clin Immunol (2021) 147(2):749–53 e2. doi: 10.1016/j.jaci.2020.08.014
8. Haerynck F, Stordeur P, Vandewalle J, Van Coster R, Bordon V, De Baets F, et al. Complete Factor I Deficiency Due to Dysfunctional Factor I With Recurrent Aseptic Meningo-Encephalitis. J Clin Immunol (2013) 33(8):1293–301. doi: 10.1007/s10875-013-9944-8
9. Nita IM, Genel F, Nilsson SC, Smart J, Truedsson L, Choo S, et al. Molecular Characterization of Two Novel Cases of Complete Complement Inhibitor Factor I Deficiency. Mol Immunol (2011) 48(8):1068–72. doi: 10.1016/j.molimm.2011.01.012
10. Nielsen HE, Christensen KC, Koch C, Thomsen BS, Heegaard NH, Tranum-Jensen J. Hereditary, Complete Deficiency of Complement Factor H Associated With Recurrent Meningococcal Disease. Scand J Immunol (1989) 30(6):711–8. doi: 10.1111/j.1365-3083.1989.tb02480.x
11. Nilsson SC, Trouw LA, Renault N, Miteva MA, Genel F, Zelazko M, et al. Genetic, Molecular and Functional Analyses of Complement Factor I Deficiency. Eur J Immunol (2009) 39(1):310–23. doi: 10.1002/eji.200838702
12. Bu F, Maga T, Meyer NC, Wang K, Thomas CP, Nester CM, et al. Comprehensive Genetic Analysis of Complement and Coagulation Genes in Atypical Hemolytic Uremic Syndrome. J Am Soc Nephrol (2014) 25(1):55–64. doi: 10.1681/ASN.2013050453
13. Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaime F, Dragon-Durey MA, Ngo S, et al. Genetics and Outcome of Atypical Hemolytic Uremic Syndrome: A Nationwide French Series Comparing Children and Adults. Clin J Am Soc Nephrol (2013) 8(4):554–62. doi: 10.2215/CJN.04760512
14. Bresin E, Rurali E, Caprioli J, Sanchez-Corral P, Fremeaux-Bacchi V, Rodriguez de Cordoba S, et al. Combined Complement Gene Mutations in Atypical Hemolytic Uremic Syndrome Influence Clinical Phenotype. J Am Soc Nephrol (2013) 24(3):475–86. doi: 10.1681/ASN.2012090884
15. Westra D, Vernon KA, Volokhina EB, Pickering MC, van de Kar NC, van den Heuvel LP. Atypical Hemolytic Uremic Syndrome and Genetic Aberrations in the Complement Factor H-Related 5 Gene. J Hum Genet (2012) 57(7):459–64. doi: 10.1038/jhg.2012.57
16. Java A, Baciu P, Widjajahakim R, Sung YJ, Yang J, Kavanagh D, et al. Functional Analysis of Rare Genetic Variants in Complement Factor I (Cfi) Using a Serum-Based Assay in Advanced Age-Related Macular Degeneration. Transl Vis Sci Technol (2020) 9(9):37. doi: 10.1167/tvst.9.9.37
17. Hallam TM, Marchbank KJ, Harris CL, Osmond C, Shuttleworth VG, Griffiths H, et al. Rare Genetic Variants in Complement Factor I Lead to Low Fi Plasma Levels Resulting in Increased Risk of Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci (2020) 61(6):18. doi: 10.1167/iovs.61.6.18
18. Kavanagh D, Yu Y, Schramm EC, Triebwasser M, Wagner EK, Raychaudhuri S, et al. Rare Genetic Variants in the Cfi Gene Are Associated With Advanced Age-Related Macular Degeneration and Commonly Result in Reduced Serum Factor I Levels. Hum Mol Genet (2015) 24(13):3861–70. doi: 10.1093/hmg/ddv091
19. Iatropoulos P, Noris M, Mele C, Piras R, Valoti E, Bresin E, et al. Complement Gene Variants Determine the Risk of Immunoglobulin-Associated Mpgn and C3 Glomerulopathy and Predict Long-Term Renal Outcome. Mol Immunol (2016) 71:131–42. doi: 10.1016/j.molimm.2016.01.010
20. Zhao W, Ding Y, Lu J, Zhang T, Chen D, Zhang H, et al. Genetic Analysis of the Complement Pathway in C3 Glomerulopathy. Nephrol Dial Transplant (2018) 33(11):1919–27. doi: 10.1093/ndt/gfy033
21. Osborne AJ, Breno M, Borsa NG, Bu F, Fremeaux-Bacchi V, Gale DP, et al. Statistical Validation of Rare Complement Variants Provides Insights Into the Molecular Basis of Atypical Hemolytic Uremic Syndrome and C3 Glomerulopathy. J Immunol (2018) 200(7):2464–78. doi: 10.4049/jimmunol.1701695
22. Servais A, Noel LH, Roumenina LT, Le Quintrec M, Ngo S, Dragon-Durey MA, et al. Acquired and Genetic Complement Abnormalities Play a Critical Role in Dense Deposit Disease and Other C3 Glomerulopathies. Kidney Int (2012) 82(4):454–64. doi: 10.1038/ki.2012.63
23. Smith RJH, Appel GB, Blom AM, Cook HT, D’Agati VD, Fakhouri F, et al. C3 Glomerulopathy - Understanding a Rare Complement-Driven Renal Disease. Nat Rev Nephrol (2019) 15(3):129–43. doi: 10.1038/s41581-018-0107-2
24. Sethi S, Gamez JD, Vrana JA, Theis JD, Bergen HR 3rd, Zipfel PF, et al. Glomeruli of Dense Deposit Disease Contain Components of the Alternative and Terminal Complement Pathway. Kidney Int (2009) 75(9):952–60. doi: 10.1038/ki.2008.657
25. Hou J, Markowitz GS, Bomback AS, Appel GB, Herlitz LC, Barry Stokes M, et al. Toward a Working Definition of C3 Glomerulopathy by Immunofluorescence. Kidney Int (2014) 85(2):450–6. doi: 10.1038/ki.2013.340
26. Bomback AS, Appel GB. Pathogenesis of the C3 Glomerulopathies and Reclassification of PGNn. Nat Rev Nephrol (2012) 8(11):634–42. doi: 10.1038/nrneph.2012.213
27. Berger BE. Atypical Hemolytic Uremic Syndrome: A Syndrome in Need of Clarity. Clin Kidney J (2019) 12(3):338–47. doi: 10.1093/ckj/sfy066
28. Bu F, Borsa NG, Jones MB, Takanami E, Nishimura C, Hauer JJ, et al. High-Throughput Genetic Testing for Thrombotic Microangiopathies and C3 Glomerulopathies. J Am Soc Nephrol (2016) 27(4):1245–53. doi: 10.1681/ASN.2015040385
29. Zhang Y, Nester CM, Martin B, Skjoedt MO, Meyer NC, Shao D, et al. Defining the Complement Biomarker Profile of C3 Glomerulopathy. Clin J Am Soc Nephrol (2014) 9(11):1876–82. doi: 10.2215/CJN.01820214
30. Bu F, Zhang Y, Wang K, Borsa NG, Jones MB, Taylor AO, et al. Genetic Analysis of 400 Patients Refines Understanding and Implicates a New Gene in Atypical Hemolytic Uremic Syndrome. J Am Soc Nephrol (2018) 29(12):2809–19. doi: 10.1681/ASN.2018070759
31. Zhang Y, Meyer NC, Wang K, Nishimura C, Frees K, Jones M, et al. Causes of Alternative Pathway Dysregulation in Dense Deposit Disease. Clin J Am Soc Nephrol (2012) 7(2):265–74. doi: 10.2215/CJN.07900811
32. Martin Merinero H, Zhang Y, Arjona E, Del Angel G, Goodfellow R, Gomez-Rubio E, et al. Functional Characterization of 105 Factor H Variants Associated With Atypical Hus: Lessons for Variant Classification. Blood (2021) 138(22):2185–201. doi: 10.1182/blood.2021012037
33. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The Mutational Constraint Spectrum Quantified From Variation in 141,456 Humans. Nature (2020) 581(7809):434–43. doi: 10.1038/s41586-020-2308-7
34. Ponce-Castro IM, Gonzalez-Rubio C, Delgado-Cervino EM, Abarrategui-Garrido C, Fontan G, Sanchez-Corral P, et al. Molecular Characterization of Complement Factor I Deficiency in Two Spanish Families. Mol Immunol (2008) 45(10):2764–71. doi: 10.1016/j.molimm.2008.02.008
35. Kaiser JP, Oppermann M, Gotze O, Deppisch R, Gohl H, Asmus G, et al. Significant Reduction of Factor D and Immunosuppressive Complement Fragment Ba by Hemofiltration. Blood Purif (1995) 13(6):314–21. doi: 10.1159/000170215
36. Zhang Y, Keenan A, Dai DF, May KS, Anderson EE, Lindorfer MA, et al. C3(H2O) Prevents Rescue of Complement-Mediated C3 Glomerulopathy in Cfh-/- Cfd-/- Mice. JCI Insight (2020) 5(9):e135758. doi: 10.1172/jci.insight.135758
37. Tan PL, Garrett ME, Willer JR, Campochiaro PA, Campochiaro B, Zack DJ, et al. Systematic Functional Testing of Rare Variants: Contributions of Cfi to Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci (2017) 58(3):1570–6. doi: 10.1167/iovs.16-20867
38. Geerlings MJ, Volokhina EB, de Jong EK, van de Kar N, Pauper M, Hoyng CB, et al. Genotype-Phenotype Correlations of Low-Frequency Variants in the Complement System in Renal Disease and Age-Related Macular Degeneration. Clin Genet (2018) 94(3-4):330–8. doi: 10.1111/cge.13392
39. Seddon JM, Yu Y, Miller EC, Reynolds R, Tan PL, Gowrisankar S, et al. Rare Variants in Cfi, C3 and C9 Are Associated With High Risk of Advanced Age-Related Macular Degeneration. Nat Genet (2013) 45(11):1366–70. doi: 10.1038/ng.2741
40. Lambris JD. The Multifunctional Role of C3, the Third Component of Complement. Immunol Today (1988) 9(12):387–93. doi: 10.1016/0167-5699(88)91240-6
41. Zhang Y, Ghiringhelli Borsa N, Shao D, Dopler A, Jones MB, Meyer NC, et al. Factor H Autoantibodies and Complement-Mediated Diseases. Front Immunol (2020) 11:607211. doi: 10.3389/fimmu.2020.607211
42. de Jong S, Volokhina EB, de Breuk A, Nilsson SC, de Jong EK, van der Kar N, et al. Effect of Rare Coding Variants in the Cfi Gene on Factor I Expression Levels. Hum Mol Genet (2020) 29(14):2313–24. doi: 10.1093/hmg/ddaa114
43. Fritsche LG, Igl W, Bailey JN, Grassmann F, Sengupta S, Bragg-Gresham JL, et al. A Large Genome-Wide Association Study of Age-Related Macular Degeneration Highlights Contributions of Rare and Common Variants. Nat Genet (2016) 48(2):134–43. doi: 10.1038/ng.3448
44. Szarvas N, Szilagyi A, Csuka D, Takacs B, Rusai K, Muller T, et al. Genetic Analysis and Functional Characterization of Novel Mutations in a Series of Patients With Atypical Hemolytic Uremic Syndrome. Mol Immunol (2016) 71:10–22. doi: 10.1016/j.molimm.2016.01.003
45. Bienaime F, Dragon-Durey MA, Regnier CH, Nilsson SC, Kwan WH, Blouin J, et al. Mutations in Components of Complement Influence the Outcome of Factor I-Associated Atypical Hemolytic Uremic Syndrome. Kidney Int (2010) 77(4):339–49. doi: 10.1038/ki.2009.472
46. Maga TK, Nishimura CJ, Weaver AE, Frees KL, Smith RJ. Mutations in Alternative Pathway Complement Proteins in American Patients With Atypical Hemolytic Uremic Syndrome. Hum Mutat (2010) 31(6):E1445–60. doi: 10.1002/humu.21256
47. Fakhouri F, Roumenina L, Provot F, Sallee M, Caillard S, Couzi L, et al. Pregnancy-Associated Hemolytic Uremic Syndrome Revisited in the Era of Complement Gene Mutations. J Am Soc Nephrol (2010) 21(5):859–67. doi: 10.1681/ASN.2009070706
48. Garg N, Zhang Y, Nicholson-Weller A, Khankin EV, Borsa NG, Meyer NC, et al. C3 Glomerulonephritis Secondary to Mutations in Factors H and I: Rapid Recurrence in Deceased Donor Kidney Transplant Effectively Treated With Eculizumab. Nephrol Dial Transplant (2018) 33(12):2260–5. doi: 10.1093/ndt/gfx369
49. Martinez-Barricarte R, Heurich M, Lopez-Perrote A, Tortajada A, Pinto S, Lopez-Trascasa M, et al. The Molecular and Structural Bases for the Association of Complement C3 Mutations With Atypical Hemolytic Uremic Syndrome. Mol Immunol (2015) 66(2):263–73. doi: 10.1016/j.molimm.2015.03.248
Keywords: complement, factor I, C3 glomerulopathy, atypical hemolytic uremic syndrome, C3 glomerulonephritis, dense deposit disease
Citation: Zhang Y, Goodfellow RX, Ghiringhelli Borsa N, Dunlop HC, Presti SA, Meyer NC, Shao D, Roberts SM, Jones MB, Pitcher GR, Taylor AO, Nester CM and Smith RJH (2022) Complement Factor I Variants in Complement-Mediated Renal Diseases. Front. Immunol. 13:866330. doi: 10.3389/fimmu.2022.866330
Received: 31 January 2022; Accepted: 08 March 2022;
Published: 10 May 2022.
Edited by:
Erik J. M. Toonen, Hycult Biotech, NetherlandsReviewed by:
Elisabetta Valoti, Mario Negri Pharmacological Research Institute (IRCCS), ItalyCopyright © 2022 Zhang, Goodfellow, Ghiringhelli Borsa, Dunlop, Presti, Meyer, Shao, Roberts, Jones, Pitcher, Taylor, Nester and Smith. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard J. H. Smith, UmljaGFyZC1zbWl0aEB1aW93YS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.