 Jeong-Yeon Mun1
Jeong-Yeon Mun1- 1Department of Biomedical Sciences, College of Natural Science, Dong-A University, Busan, South Korea
- 2Department of Health Sciences, The Graduate School of Dong-A University, Busan, South Korea
- 3College of Korean Medicine, Woosuk University, Jeonju, South Korea
The tumor microenvironment (TME) plays a critical role in tumorigenesis and is comprised of different components, including tumor cells, stromal cells, and immune cells. Among them, the relationship between each mediator involved in the construction of the TME can be understood by focusing on the secreting or expressing factors from each cells. Therefore, understanding the various interactions between each cellular component of the TME is necessary for precise therapeutic approaches. In carcinoma, stromal cells are well known to influence extracellular matrix (ECM) formation and tumor progression through multiple mediators. Immune cells respond to tumor cells by causing cytotoxicity or inflammatory responses. However, they are involved in tumor escape through immunoregulatory mechanisms. In general, anti-cancer therapy has mainly been focused on cancer cells themselves or the interactions between cancer cells and specific cell components. However, cancer cells directly or indirectly influence other TME partners, and members such as stromal cells and immune cells also participate in TME organization through their mutual communication. In this review, we summarized the relationship between stromal cells and immune cells in the TME and discussed the positive and negative relationships from the point of view of tumor development for use in research applications and therapeutic strategies.
Introduction
The tumor microenvironment (TME), a highly heterogeneous environment composed of many different types of cells and many molecules produced or released by tumor cells, stromal cells, and immune cells, is now widely recognized (1). The TME is composed of cellular components such as cancer-associated fibroblasts (CAFs), tumor-endothelial cells (TECs), cancer-associated adipocytes (CAAs), mesenchymal stem cells (MSCs), T cells, B cells, natural killer (NK) cells, and tumor-associated macrophages (TAMs) (2). In addition, the TME is rich in hypoxic, acidic, and immune/inflammatory cells known to play important roles in tumor development, growth, progression, and resistance to treatment (3–6). In the past, cancer therapies were designed to target cancer cells directly. Recently, they are designed to destroy networks formed by tumors. Immunotherapy, in addition to surgery, chemotherapy, and radiation therapy, has emerged as a breakthrough treatment modality for cancer patients (7). Besides therapies that directly target tumor cells, promising therapies targeting stromal cells present in the TME are also attracting attention. Targeting immunomodulatory pathways in the TME is considered a central step in cancer treatment (8–10). Indeed, the TME is currently in the spotlight as a new target for cancer therapeutics with many ongoing studies.
Treatments for cancer patients include surgical resection and chemotherapy or radiation therapy (11). With recent advances in onco-immunological studies, the use of immune checkpoint inhibitors (ICIs) taking advantage of the antitumor activity within the TME is considered as an effective therapeutic modality for cancer patients. Among them, inhibitors of the cytotoxic T lymphocyte antigen-4 (CTLA-4) blockade have shown remarkable efficacy in clinical trials. Both ipilimumab and tremelimumab are human antibodies to CTLA-4 (12, 13). In recent studies, both drugs have achieved considerable clinical success for patients with advanced malignant melanoma. A phase 3 study has shown that ipilimumab, a CTLA-4 blockade, can significantly improve the overall survival of melanoma patients. Based on results of these studies, ipilimumab, a T-cell potentiator, is considered useful as a treatment for advanced melanoma patients (14, 15). In addition, it has been suggested that PD-1/PD-L1 expression plays an important role in immune evasion by cells other than tumor cells present in the TME. Nivolumab is an ICI targeting PD-1/PD-L1. It is being used to treat melanoma, non-small cell lung cancer, kidney cancer, hepatocellular carcinoma, urinary tract cancer, gastric cancer, and triple-negative breast cancer (TNBC) (16–18). In addition to immunotherapy-based ipilimumab and nivolumab, a variety of FDA-approved ICI treatments such as cemiplimab and pembrolizumab are also being actively implemented for cancer treatment (19).
The expression of a modified T-cell receptor or chimeric antigen receptor (CAR) to enhance antigen specificity can be engineered using a patient’s own T cells. Thus, it is possible to efficiently generate tumor-targeting T cells by solving the fundamental problem without requiring the patient to activate de novo T cells (20, 21). Clinical trials conducted on patients with acute lymphocytic leukemia have shown a complete recovery in up to 92% of patients with very meaningful results (20). CAR-T cell therapy is being actively implemented for treating large B-cell lymphoma and B-cell precursor acute lymphoblastic leukemia (22, 23). Although this CAR-T cell therapy has achieved clinical success in some hematologic cancer patients (24), it has two main problems that need improvement. First, it is very difficult to obtain enough T cells from cancer patients to isolate CAR-T cells because of lymphocytopenia due to prior treatments. Second, there is not enough time to implement CAR-T cell therapy for rapidly advancing cancers (25). Tumor-infiltrating lymphocyte (TIL) therapy can be proposed as a form of therapy to solve these problems. TIL therapy is a treatment involves removing T cells infiltrating a patient’s tumor, proliferating them to a large amount in the laboratory, and injecting them back into the patient to help the patient’s immune system kill cancer cells (26). It has the advantage of being able to locate and destroy the patient’s tumor directly because it secures T cells that have already penetrated the patient’s tumor and reinjects them into the patient. Activated NK cells can also directly lyse tumor cells by releasing cytotoxic granules (including perforin and granzymes) in a manner similar to that of activated cytotoxic T cells. Since NK cells can eliminate tumors, immunotherapy based on NK cells has been developed. It is currently being used strategically. In addition, CAR-NK cells, like CAR-T cells, are genetically modified to express CARs that can recognize specific antigens that are characteristically overexpressed by target cells. Preclinical studies of CAR-NK cells have been performed on hematologic cancers and some solid cancers (27). These CAR-NK cells, along with T cell-based therapies, could be proposed as an improved therapy for solid tumors.
Strategies using drugs that can specifically target stromal cells within the TME have been proposed to further enhance patient survival and therapeutic effects. Therapeutic drugs that target stromal cells within the TME are under investigation (28). These stromal cells are important components of the TME as they express specific markers that can be targeted for tumor treatment (29). Of note, treatment with microsomal prostaglandin E synthase-1 inhibitor compound III targeting CAF-derived prostaglandin E2 (PGE2) can reduce tumor growth, suppress CAF migration/infiltration, and increase M1 macrophage ratio in neuroblastoma tumor studies (30). Furthermore, studies using a mouse model of cholangiocarcinoma induction have confirmed that navitoclax (BCl-2 inhibitor) treatment can induce CAF apoptosis, reduce the expression of tenascin C, and suppress tumor growth. This suggests that navitoclax may strategically target and destroy CAFs within TMEs to attack tumors (31). As an inhibitor of fibroblast growth factor receptors (FGFR), PD173704 can reduce the growth of both CAFs and endothelial cells (ECs), thereby inhibiting stromal cell-mediated FGFR pathway in a co-cultured environment of head and neck squamous cell carcinoma (HNSCC) cells. These inhibitor molecules can inhibit tumor cell growth, thus playing a crucial role in tumor-matrix interaction. They have been proposed as potential therapeutic agents for HNSCC (32). Phosphodiesterase (PDE), a class of enzymes that can hydrolyze cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP), are composed of 11 different subtypes. A lung cancer study has shown that phosphodiesterase-4 (PDE4) as a type of PDE can promote lung cancer proliferation and angiogenesis by having a crosstalk with hypoxia-inducible transcription factors (HIFs) factor (33). CC-5079 is an analog of these PDE4 inhibitors. It can inhibit the proliferation and migration of fibroblasts, bladder cells, and EC cells, stimulate mitogen-activated protein kinase phosphatase 1 (MKP1) expression, and inhibit micro-angiogenesis through its upregulation. It has been demonstrated that CC-5079 is a candidate for treating colon cancer by targeting ECs within the TME (34). However, further studies on the detailed mechanism of action of CC-5079 are needed. Although the TME component targeting therapy such as immune cell targeting therapy and stromal targeting therapy has been applied to treat cancer patients in various studies mentioned above, it has some limitations because the TME is heterogeneous, showing different characteristics in each patient (29).
Although the role of the TME in cancer processes has been extensively investigated, its contribution to the crosstalk between stromal cells and immune cells, which are components of the TME based on the tumor, is only partially known. Previous therapeutic approaches have focused on cancer cells and established relationships between cancer cells and immune cells or stromal cells. Based on an understanding of the relationship, it is more effective to consider treatment methods in a direction that can restore the relationship between stromal cells and cancer cells and the self-dependent role in the TME by understanding the relationship between cells in the TME. Communication between TME constituent cells should be understood and therapeutically targeted. Characteristic parts of cells must be considered. In this respect, unlike cancer cells, the somatic cells that make up our body are controllable. The need to find ways to overcome cancer treatment by properly regulating the communication between immune cells has emerged. In particular, exploring interactions and intercellular relationships in the TME will improve our understanding of cancer treatment (35, 36). Therefore, in this review, we described and summarized interactions, roles, and importance of the relationship between stromal cells and immune cells within the TME.
The Pro-Tumorigenic Effect of the Relationship Between Stromal Cells and Immune Cells in the TME
CAFs Interact With Myeloid Cell-Derived Immune Cells in the TME to Enhance Tumorigenesis and Immune Evasion
CAFs are the most dominant cell type in the TME. They are known to crosstalk with immune cells (37). CAFs may play a pivotal role in tumor development and survival as they are involved in TME composition and participate in mechanisms that promote tumor growth and invasion and subvert defense system (38–40). Recently, studies have attempted to clarify this point. CAFs can secrete components such as cytokines and chemokines known to be continuously activated in the TME through various signaling mechanisms and function as primary immunosuppressive mediators (41, 42). CAF-derived cytokines and chemokines are attracting attention not only for their roles in tumor progression, but also for their ability to regulate the recruitment and function of immune cells.
TAMs are macrophages that participate in the formation of the TME by producing cytokines, chemokines, and growth factors. TAMs are divided into two types, M1 and M2 macrophages (43, 44). Characteristically, M1 macrophages produce large amounts of pro-inflammatory cytokines and regulate the Th1 antitumor immune response, whereas M2 macrophages play an important role in tumor progression. Gokyavuz et al. have shown that CAFs can secrete monocyte chemotactic protein-1 (MCP-1) and stromal cell-derived factor-1 (SDF-1) and effectively recruit monocytes (45). MCP-1 is a chemokine that contributes to the recruitment of monocytes to the site of an inflammatory response. It is expressed in a variety of cancer types such as prostate and ovarian cancers (46, 47). SDF-1 is expressed in stromal fibroblasts in organs including the brain, breast, and lung. It is involved in cancer survival, proliferation, and metastasis (48, 49). Specifically, monocytes recruited by CAFs via MCP-1 and SDF-1 can reduce the secretion of pro-inflammatory cytokine interleukin (IL)-12 and increase the production of anti-inflammatory cytokine IL-10. CAF-educated monocytes can increase the motility and invasiveness of breast cancer cells and the expression of epithelial-mesenchymal transition (EMT)-related genes and vimentin proteins, eventually exerting their immunosuppressive role in breast cancer (45). In prostate cancer, CAFs can also promote the differentiation to the M2 macrophage phenotype via SDF-1. Analysis of patients with prostate cancer has confirmed a clear increase in the M2/M1 macrophage ratio, which is correlated with clinical prognosis. Thus, CAFs and M2-polarized macrophages actively contribute to the promotion of the invasive ability of prostate cancer cells. They are correlated with the aggressiveness of cancer via the infiltration of M2 macrophage (50). Chemokine (C-C motif) ligand 2 (CCL2; MCP-1) secreted by CAF can induce blood monocyte recruitment and differentiate into TAMs in breast cancer (51). Additional research has confirmed that the increase in monocyte migration to breast tumor spheroids is associated with CAF-derived CCL2 and that the CCR2A/2B pathway and the CCL2 receptor play important roles in monocyte recruitment (52). These findings suggest that CAF-derived factors can affect a wide range of processes, leading to monocyte recruitment and M2 macrophage differentiation.
Among various cancer types, pancreatic cancer has a low response to immunotherapy. Thus, it is necessary to understand the antitumor immune response in pancreatic stromal cells. Stromal cells in the pancreatic cancer microenvironment can generate numerous factors that support the growth and survival of tumor cells. They are being studied to increase the understanding and relevance of immune cells (53). Pancreatic stellate cells (PSCs) are resident cells in the pancreas under quiescent conditions. They are isolated from the human pancreas as fibroblast-like cells (54). These cells display some characteristics of activated myofibroblasts such as α-smooth muscle actin (α-SMA) expression and ECM proteins synthesis (55). Much evidence has confirmed the importance of PSCs in pancreatic ductal adenocarcinoma (PDAC) development. Importantly, myeloid-derived suppressor cells (MDSCs) are identified as immature myeloid cells that can induce immunosuppression, mediate multiple signaling pathways, and interact with immune cells and mediators (56, 57). It has been shown that MDSCs can promote cancer progression, angiogenesis, and metastasis and disrupt the efficacy of therapeutic agents (58, 59). In particular, the number of MDSCs is highly correlated with the staging of pancreatic cancer patients, indicating that an increase in MDSC levels could be an indicator of disease progression (60). Thus, MDSCs could be targeted in the treatment of PDAC patients. Mace et al. have reported that soluble factor IL-6 generated from patient PSCs with confirmed phenotypic α-smooth muscle actin and glial fibrillary acidic protein expression can promote the differentiation of MDSCs (61). PSCs in PDAC can modulate MDSC differentiation via IL-6 and STAT3 signal transduction pathways (61). Targeting PSCs in the TME by promoting the generation of immunosuppressive cells that suppress the innate or acquired immune response against pancreatic cancer may reduce MDSC levels, thereby enhancing the effectiveness of immunotherapy. Dendritic cells (DCs) represent immune cells that link innate and adaptive immunity. They are derived from hematopoietic bone marrow precursor cells (62). DCs are essential antigen-presenting cells (APCs) that can induce the activation of naïve T cells. Levels of peripheral blood DCs are decreased in cancer patients than in normal controls (63–65). By expressing high levels of immunoregulatory cytokines, they can induce the differentiation of regulatory T cells (Tregs) and help tumor cells evade the immune response. Cheng et al. have shown that CAF-derived IL-6 can induce the activation of the STAT3 pathway, leading to immunosuppressive cell types (66). Especially, STAT3 activation by CAF-derived IL-6 plays a pivotal role in indoleamine-2,3-dioxygenase (IDO) production and induced regulatory DC recruitment (66). Cheng et al. have evaluated CAF-regulated neutrophil function and the activation of hepatocellular carcinoma (HCC) via the IL-6/STAT3/PD-L1 signaling cascade (67). They found that IL-6 derived from CAFs could recruit PD-L1+ neutrophils and impair T-cell function via PD1/PD-L1 signaling (67). Cho et al. have reported that IL-6 and granulocyte-macrophage colony-stimulating factor (GM-CSF) released from cancer cell-activated CAFs through co-culture of monocytes and CAFs can increase TAM infiltration and metastasis and direct monocytes to differentiate into M2 TAMs (68). Thus, the development of inhibitors or neutralizing antibodies targeting IL-6, IDO, GM-CSF, and STAT3 may lead to a new cancer immunotherapeutic approach that can induce tumor immune evasion of CAFs via the cell network in the TME. In the TME, secreted chemokines including IL-8 play important roles in tumorigenesis. These chemokines could be secreted by CAF (69). According to a study on colorectal cancer-derived CAFs isolated from human colorectal cancer tissue, the CAF-derived IL-8 can be secreted to attract monocytes and promote polarization from anti-tumorigenic/pro-inflammatory M1 macrophages to pro-tumorigenic/anti-inflammatory M2 (70). These findings indicate that CAFs can also inhibit NK cell function and promote CRC cell progression by increasing TAM infiltration.
CAFs Interact With Lymphocytes in the TME to Enhance Tumorigenesis and Progression
Transforming growth factor-β (TGF-β) is a representative cytokine secreted from CAF with a strong immunomodulatory function. It is involved in the suppression of immune responses. It also plays an important role in tumor initiation, invasion, and metastasis (71–73). CAFs-derived TGF-β can regulate various types of immune cells through the paracrine signaling pathway with growth factors and cytokines. TGF-β can suppress IL-2 production and T cell proliferation. It plays an important role in CD4+CD25+ Treg production and function (74). Furthermore, it has been proposed that transcriptional inactivation of TGF-β by suppressing T-box expressed in T cells (T-bet) and GATA-binding protein 3 (GATA-3) expression can control the differentiation of murine CD4+ T cells (75, 76). According to immunological studies conducted in HCC cells, IL-10, TGF-β, and IL-4 secreted by stromal cells in the TME can increase polarization to M2 macrophages. It is known that M2 macrophages can enhance tissue remodeling, angiogenesis, tumor progression, and ultimately IL-10, increasing PD-L1 and human leukocyte antigen (HLA)-DR expression to induce immunosuppression (77–79).
Since stromal cells in the TME express cytokines and immunological factors, it is critical to consider stromal cells to rescue tumor-reactive CD8+ T cells from immune evasion mechanisms (80). According to a study by Lakins et al., CAF-educated T cells can induce death of T cells by PD-L2 and Fas ligand (FasL) engagement in the lung tumor stroma (81). These results indicate that CAFs can enhance tumor viability by inducing the dysfunction of encountered CD8+ T cells, suggesting that CAFs can directly contribute to the pro-tumor T cell immune response.
Wu et al. have investigated the heterogeneity of stromal cell population of TNBC patients using single-cell RNA sequencing technology and confirmed the existence of an inflammatory CAF (iCAF) subpopulation (82). These iCAFs showed upregulation of CXCL12 (SDF-1)-CXCR4 chemoattractant pathway genes and suggested a strong association of CD8+ T cell dysfunction with iCAF presence and exclusion (82). In breast cancer studies, four different CAF subsets of human breast cancer have been identified, among which the CAF subset 1 is characterized as immunosuppressive cells. CAF subset 1 can secrete CXCL12 to attract T cells, increase the survival of CD4+CD25+ T cells, and promote differentiation into CD25highFOXP3high Treg cells via B7H3, CD73, and DPP4 (83, 84). These studies indicate that the CAF-S1 fibroblast subset contributes to immunosuppression in breast cancer. Takahashi et al. have shown that IL-6, CXCL8, tumor necrosis factor (TNF), and vascular endothelial growth factor (VEGF)-α are detected more in CAFs than in normal fibroblasts in the TME of HNSCC. These CAFs can modulate effector T cell function by expressing co-regulatory molecules B7H1 and B7DC and enhance T cell apoptosis and the induction of Treg cells (85). Thus, CAFs have been implicated in the proliferation of CD4+FOXP3+ Treg cells, tumor progression, angiogenesis, and metastasis (86, 87). These cells play an important role in angiogenesis, invasion, and metastasis. They contribute to the immunosuppressive process that promotes tumor evasion by the T cell network within the TME of HNSCC. Moreover, CAFs can release different factors including chemokines, cytokines, and growth factors that can promote immunosuppression through recruitment of immunosuppressive cells such as Tregs and myeloid cells, upregulation of immune checkpoint molecules on T cells, and regulation of T cell migration (88). Notably, in PDACs, T cells can be inhibited by tumor stroma, suggesting that CAF-derived factors may regulate T cell function and phenotype (89–91). Most T cells in the TME are exhausted, leading to cancer immune evasion. Restoring exhausted T cells appears to be an excellent strategy in cancer immunotherapeutic therapy (92). PD-1, a critical inhibitory receptor regulating T cell exhaustion, can attenuate its ability to clear cancer via high expression in T cells (91). Reversing this T cell exhaustion represents a major strategy for cancer treatment (93).
Candidate factors that can interfere with signal transduction between CAF and NK cells have been suggested as potential strategies for cancer treatments (94). NK cells are cells that can produce cytokines to communicate with other cells. They have the ability to kill tumor cells (95, 96). NK cell activation releases perforin, granzymes, inflammatory cytokines, and chemokines toward target cells (97). PGE2 is a major product of arachidonic metabolism. It is synthesized via cyclooxygenase-2 (COX2) and prostaglandin synthase pathways (98). According to Balsamo et al., PGE2 released by melanoma-derived fibroblasts can suppress the expression of NK receptors, perforins, and granzymes, meaning that they can interfere with the phenotype and function of NK cells (99, 100). PGE2 secreted from HCC-associated fibroblasts can inhibit the expression of NK receptors exhibiting immune function, including NKp30, NKp44, and NKG2D, thus impairing NK cell-mediated cytolytic activity (101). In addition, it has been confirmed that blocking the activity of PGE2 and IDO in activated HCC-related fibroblasts can restore the function of NK cells and promote the progression of HCC (102). Thus, metastatic melanoma-derived fibroblasts and HCC-associated fibroblasts can release PGE2 and IDO to affect NK cell function and exert strong immunosuppressive activity (99–101).
TECs, CAAs, and MSCs Interact With Immune Cells in the TME to Induce Immune Evasion and Reduce Anti-Tumor Functions
Endothelial cells (ECs) are a major type of cells found inside the lining of blood vessels, lymph vessels, and heart. They fulfill many physiological processes in the body (103). ECs are included in stromal cells. They represent an important interface between tissue and blood (104). TECs found in most tumors can also form an essential vascular inner layer in tumors (105). TECs play an important role in orchestrating the TME. TECs are known to be particularly important for T cell recruitment and activation. Previous studies have shown that the interaction between T cells and ECs plays an important role in the regulation of the immune system during chronic inflammation (106). TECs in the tumor microenvironment are particularly relevant to circulating immune cells. They may influence anti-tumor cell immune responses. TECs are APCs with an inhibitory activity. They express MHC class II and PD-L1. They can impair the production of pro-inflammatory cytokines including IL-2, TNF-α, and IFN-γ in CD8+ T cells (107, 108). TECs are also known to play a critical role in tumor cell growth and invasion (109). Vascular cell adhesion molecule 1 (VCAM1) induction in endothelial cells can regulate tumor progression, provide angiogenic factors, promote neutrophil infiltration and tumor cell adhesion to the endothelium, and promote metastasis by sustaining vascular Notch1 signaling (110).
In gliomas, penetration of T cells into tumor tissue as TILs is extremely low. Although this feature is controversial for the correlation with TIL-induced tumor prognosis in glioma, it has a useful aspect in the evaluation of factors that reduce the presence of T cells in tumor tissues. Moreover, FasL is well-known as a pro-apoptotic cell surface protein that plays an important role in confirming T cell depletion in tumor tissues. This has been demonstrated by flow cytometric analysis, showing that FasL levels expressed in the endothelium of brain tumors are inversely correlated with the CD8+/CD4+ TIL ratio (111). That is, FasL expression indicates not only the CD8+/CD4+ TIL ratio, but also the tumor contribution by immune avoidance in brain tumors due to decreased T cell presence. In melanoma, it has been confirmed that TECs are APCs that express MHC class II and PD-L1 with an inhibitory activity. These TECs can inhibit the proliferation of CD8+ T cells via inhibitory cytokines including IL-10 and TGF-β. They can also attenuate the antitumor effect of antigen-specific CD8+ T cells. Experimental results have shown that TECs can induce immune responses of tumor antigen-specific CD8+ T cells through the PD-1/PD-L1 pathway and evade tumor immunity by regulating immunosuppressive CD4+ T cells in an antigen-specific manner (112). Common lymphatic endothelial and vascular endothelial receptor (CLEVER-1/stabilin-1) are also expressed on lymphatic vessels, high endothelial venules, and non-continuous endothelium (113). CLEVER-1/stabilin-1 identified in HCC endothelium can recruit FOXP3+ Treg cells (114). These findings make it possible to confirm the tumorigenic effect of TECs under the influence of various mechanisms and factors.
CAAs play a central role in tumorigenesis, tumor growth, and metastasis (115). CAAs can help cancer cells by storing energy as triacylglycerol and directly providing lipids. They can release pro-inflammatory cytokines such as IL-8, CCL2, VEGF, TGF-β, and cathepsin S that can recruit bone marrow cells to the TME, thus regulating the differentiation of M2/MDSC and promoting the angiogenesis process (116–119). A breast cancer study by Arendt et al. has shown that adipocytes in human and mouse breast tissues can activate and recruit macrophages via the CCL-2/IL-1β/CXCL12 signaling pathway (117). These activated macrophages are sufficient to promote angiogenesis and accelerate stromal vascularization and breast cancer formation. Understanding major interactions between immune cells and CAAs in the TME can be an effective way to improve the effectiveness of existing therapies. PD-L1 can regulate anti-tumor immunity as described above. It is the main target of checkpoint-blocking immunotherapy. Previous studies on breast cancer have shown that the contribution of PD-L1 expression to adipogenesis remains an issue that merits further investigation. During the adipogenesis process, the expression of PD-L1 in primary human adipose stromal cells and adipocytes is highly induced. It is known that PD-L1 expression in breast cancer adipocytes is significantly elevated and that adipocyte-derived PD-L1 can inhibit the activity of important antitumor functions of CD8+ T cells (120). Fatty acids (FAs) as CAA-derived metabolites can influence immune cell homeostasis and differentiation (121), leading to immune evasion and tumor progression. Based on this, FAs are necessary for promoting proper differentiation of neutrophils. They can potentially promote the function of tumor-associated neutrophils (TANs). Besides FAs, CAA-derived IL-8 can recruit neutrophils to the TME. In PDAC, neutrophils are recruited by CAA-derived IL-1β to promote tumor progression and further activate PSCs (122).
In the TME, MSCs are multipotent stromal stem cells found in most cancers that play a central role in cancer cell growth, invasion, and metastasis by interacting with the tumor and immune cells in the TME (123, 124). MSC-derived TGF-β can increase the frequency of Treg cells, protect breast cancer cells, and support the growth of breast cancer (125). These MSCs can induce IL-10+ regulatory B (Breg) cells involved in the production of immunosuppressive environments through SDF-1α and CXCR7 (126). IL-10+ Breg cells are a subset of immunosuppressive cells. The frequency or the function of Breg cells is involved in the tumorigenesis of some cancers (127–130). It has been shown that PD-L1 in stromal cells in the TME can be induced by TNF-α signals to promote the progression of colorectal cancer by suppressing the antitumor immune response of CD8+ T cells (131). Recently, research and development for antitumor treatments targeting CAAs has progressed rapidly. Because CAAs can interact with TME component cells in a complex cell network, they can be combined with a variety of therapies, including targeted or immunotherapies, which can selectively eliminate tumor-promoting CAAs.
Immune Cells Contribute to Immune Suppression With Stromal Cells in the TME
In the TME formed by tumor cells, the interaction between immune cells and stromal cells plays an important role in both cancer progression and anticancer activity. Among various types of immune cells in the TME, neutrophils have received less attention than other immune cell types. IL-1β is a cytokine secreted by neutrophils that can activate ECs. Treatment with IL-1 receptor antagonists can reduce in vitro endothelial cell migration, where IL-1β secreted by Ly6G+ neutrophils can directly activate endothelial cells and MMP-9, thus effectively increasing metastasis capability (132, 133). This demonstrates the distinct metastatic role of Ly6G+ neutrophils and confirms that they are beneficial for promoting tumors (132, 134). These increases of MMPs not only contribute to local invasion and metastasis-related cascades, but also contribute to the intravascular invasion process (135–137). Further research is needed to determine the role of neutrophils in the TME of various cancer types.
Among various immune cells, TAMs exist in the vicinity of CAFs and constitute the most abundant innate immune cell type (138). TAMs are classified as pro-tumorigenic macrophages that can promote the development of malignant tumors and activate CAFs to aid in tumor progression (139, 140). Tokuda et al. have shown that osteopontin is a key molecule involved in cancer-CAF-TAM interactions and that increased osteopontin can promote malignant tumors (141). This characterizes the importance of cancer cell-TAM-CAF interactions in HCC. Lung cancer is developed in the region of major fibrosis. Particularly, idiopathic pulmonary fibrosis case has been reported to be associated with increased risk of lung cancer (142, 143). Wnt7a secreted by M2 macrophages in pulmonary fibrosis can interact with Frizzled-1 to activate Wnt/β-catenin signaling and promote differentiation of MSCs into myofibroblasts. In this way, myofibroblasts activated in idiopathic pulmonary fibrosis can lead to accumulation of ECM, resulting in an acute exacerbation of lung disease such as lung cancer (144, 145). In addition, TAMs can regulate communication with cancer cells and other stromal cells in the TME through nanovesicle secretion, which carries various molecules, including microRNAs (146). Exosomes containing microRNAs can affect cellular processes and promote tumor progression and angiogenesis. Macrophage-derived exosomes containing miR-155-5p and miR-221-5p can be transmitted to ECs via an E2F Transcription Factor 1 (E2F1)-dependent manner, thus promoting EC proliferation and PDAC growth (147, 148).
Human decidual NK cells are CD56superbrightCD16- that can increase tumor growth by angiogenic activity via the production of VEGF, placental growth factor, and IL-8 (149). In non-small cell lung cancer patients, pro-angiogenic factors such as VEGF and placental growth factor are released from NK cells and defined NK cell subsets to promote human umbilical vein endothelial cell migration and capillary-like structure formation (150, 151). Mast cells in the TME can regulate adaptive immunity to tumors. Recent studies have shown that the infiltration of mast cells into tumors can indicate a poor patient prognosis (152, 153). A study of neurofibromas by Yang et al. has shown that Nf1+/- mast cells can secrete TGF-β, thus promoting fibroblast proliferation via TGF-β (154). In a human prostate cancer microtissue model, mast cells can release tryptase to enhance CAF-induced transformation of epithelial cell morphology, thus playing an important role in prostate cancer progression (155). In the TME of PDAC, mast cells are essential for tumorigenesis. Mast cells can secrete cytokines IL-13 and tryptase and promote the proliferation of PSCs (156). This suggests that mast cell infiltration and activation can contribute to the formation of dense fibrotic stromal formation characteristics of PDACs and that mast cells can promote PSC proliferation in the TME. Thus, targeting mast cells could be a way to improve PDAC therapy. Table 1 describes tumorigenic effects of various mediators in TME on the relationship between stromal cells and immune cells.

Table 1 Tumorigenic effect of the relationship between stromal cells and immune cells in TME.
Anti-Tumorigenic Association Between Stromal Cells and Immune Cells in the TME
Crosstalk Between Stromal Cells and Immune Cells in the TME Induces Anti-Tumor Immunity
Although numerous studies have suggested that CAFs can exert a tumorigenic effect, other studies have suggested that they are also involved in tumor suppression. According to Ozdemir’s study, CAF-depleted tumors are associated with increased CTLA-4 expression with a reduced Teff/Treg cell ratio in the PDAC model. In this study, the cytotoxic Teff/Treg ratio was decreased in the myofibroblast-depleted tumor and associated with a significant elevation in CTLA-4. Thus, CAF depletion can induce immunosuppression, reduce survival, and further accelerate PDAC (157). Suppression of the checkpoint blockade using CAF depletion and anti-CTLA-4 antibody can improve mouse PDAC tumors and increase overall survival (157). CAF depletion has breakthrough efficacy with significant changes in major contributors to cancer development within the TME. McAndrews et al. have demonstrated that depletion of αSMA+ CAFs is associated with increased Lgr5+ cancer stem cells and the generation of an immunosuppressive TME with increased frequency of Foxp3+ Treg cells and suppression of CD8+ T cells. Thus, αSMA+ CAFs in CRC can promote an anti-tumor effect via BMP4/TGF-β signaling (158). Interestingly, infiltrated CD8+ T cell accumulation in non-small cell lung cancer with a high proportion of fibroblasts is associated with the expression of CCL19 identified in the lungs and tumors of patients (159). Fibroblasts expressing CCL19 can form perivascular niches and promote the accumulation of CD8+ T cells. These results can be proposed as a new marker for immune tumor treatment by targeting CAFs that produce CCL19 (159). A study by Kamata et al. using G12DKRAS and V600EBRAF-driven mouse models that develop lung adenocarcinoma and adenoma has confirmed that stanniocalcin1 secreted by tumor-associated fibroblasts can inhibit TAM differentiation. The secreted stanniocalcin1 can inhibit TAM differentiation by sequestering the binding of glucose regulatory protein 94 (GRP94), an autocrine macrophage differentiation-inducing factor, to the scavenger receptor (160).
A variety of cytokines have been shown to be either pro- or anti-inflammatory depending on the cell type and disease model. IL-33 is an inflammatory cytokine released during necrotic cell death (161). CAFs can release IL-33, induce metastasis via EMT, and promote cell migration and invasion. In cancer immune response, IL-33 exhibits both pro-tumoral functions and antitumor functions (162). IL-33 can also induce IL-33 gene expression in HNSCC cells through a positive feedback process (163). The production of IL-33 is responsible for the antitumor response as CD8+ T cell infiltration. In a colon cancer model, IL-33 can increase interferon (IFN)-γ production by tumor-invasive CD4+ and CD8+ T cells. This has been confirmed by the accumulation of infiltrated CD8+ T cells, which exerts an antitumor effect (164). These results demonstrate the synergistic increase in IFN-γ, and IL-12 release, along with increases in proliferation, infiltration, and the number of cytotoxic NK cells activated by IL-33 (165). IL-33 administered to a mouse breast cancer model can potently suppress lung metastasis and increase the number of NK cells recruited within the TME (166). Furthermore, it has been shown that NK cell depletion in IL-33/ST2-deficient mice is associated with tumor growth promotion (167).
Interestingly, inhibitor of κB kinase beta (IKKβ)-depletion in intestinal mesenchymal cells (IMCs) can decrease immune cells infiltration and the expression of several pro-inflammatory mediators. Supernatant of IKKβ-depleted IMCs mouse model also shows decreased secretion of chemokine MIP2, cytokines IL-6, TNF, FOX2, and MMP9 in organ culture. Therefore, IKKβ in IMCs of inflammation-associated colorectal cancer might have a tumor-suppressive effect (168).
Several studies have reported that the sirtuin 1 (SIRT1) signaling pathway can regulate vascular inflammation. The role and molecular interaction of SIRT1 and Toll-like receptor 2 (TLR2) in monocyte adhesion to the vascular endothelium have been found to be important. These results suggest potential therapeutic targets for a variety of vascular inflammation, including atherosclerosis (169–171), although these results are not about cancer. Recent studies have shown that extracellular vesicles (EVs) derived from immune cells can perform various roles in immune responses (172). EVs can deliver not only proteins, but also biomolecules such as nucleic acids and lipids. They play a very essential role in the cell-to-cell communication process. Activated CD8+ T cells in a mouse model can temporarily release cytotoxic EVs and prevent the progression, invasion, and metastasis of fibroblast stroma-mediated tumors (173).
In platinum-based chemotherapy for ovarian cancer cells, IFN-γ-producing CD8+ T cells show altered glutathione (GSH)/cystine metabolism of fibroblasts and reduced fibroblast-mediated platinum resistance (174). Thus, IFN-γ-producing CD8+ T cells can eliminate chemoresistance of ovarian tumors and offer a combined treatment method that utilizes immune and stromal cell relationships in cancer treatment. In addition to effector T cells, other immune cells can also influence tumors by controlling fibroblast function. Interestingly, when DCs as potent APCs are fused with CAFs, they can stimulate T cells to attack cancer cells. These DC/CAF fusion cells can produce TNF-α, IL-1β, IL-6, and IL-12p70 and stimulate T cells to produce IFN-α and IFN-γ. T cells activated by the fusion of DCs and CAFs can induce a strong cytotoxic T cell response that has emerged as a new antitumor response through tumor growth inhibition (175). A multiple myeloma study has analyzed the activity of cytokine-stimulated NK cells on tumor-associated endothelial cells. IL15-activated-NK cells can enhance the killing of multiple myeloma patient’s endothelial cells via DNAX accessory molecule 1 (DNAM-1) (176). Table 2 describes anti-tumorigenic effects of various mediators in TME on the relationship between stromal cells and immune cells. It is necessary to understand the complexity between stromal cells and immune cells and carefully consider their targeting and use in drug development to overcome cancer treatment resistance.
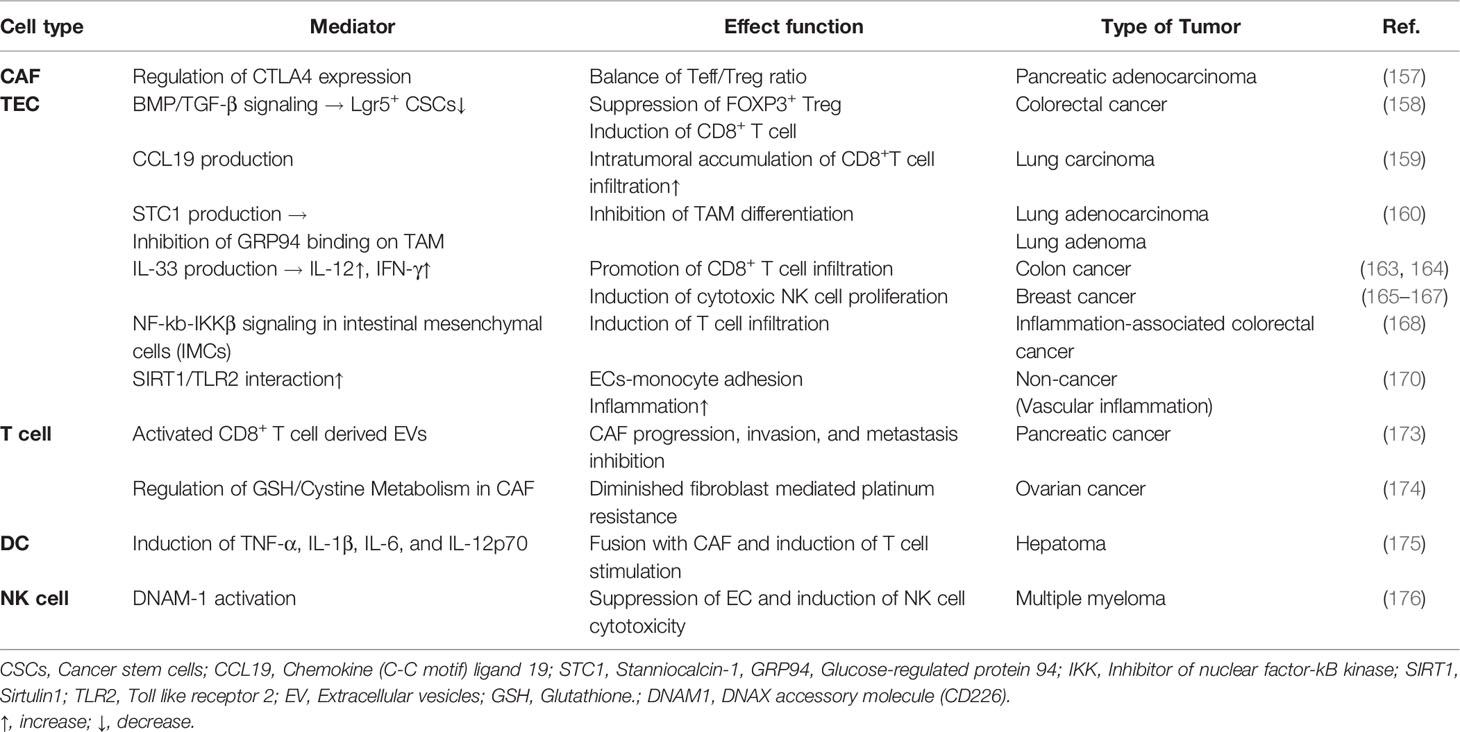
Table 2 Anti-tumorigenic effect of the relationship between stromal cells and immune cells in TME.
Conclusions and Prospection
Early studies have focused on “tumor cells” to explain anticancer drug resistance and progression of tumors by specific signaling mechanisms, rather than heterogenic TME characteristics. Successful cancer treatment strategies need to be developed based on understanding the relationship between stromal cells and immune cells in the TME. Recently, research on cancer cells has continued to transition toward studying the network of stromal cells and inflammatory immune cells in the TME. Ultimately, the organic relationship between stromal cells and immune cells present in the TME not only involves pro-tumorigenic and anti-tumorigenic cells, but also shows a mixed environment of these aspects in the TME (Figure 1).
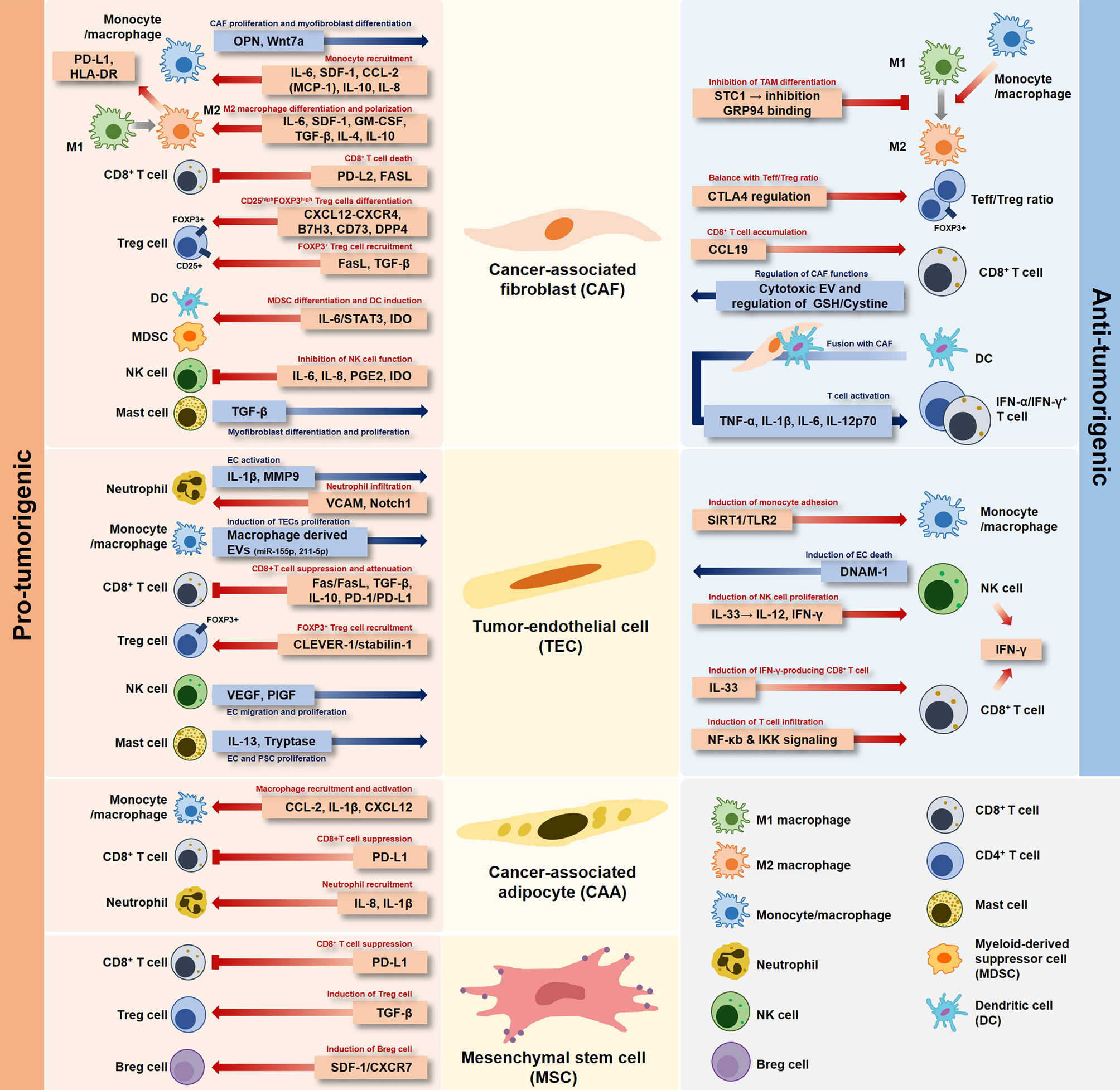
Figure 1 Dual role of stromal cells in the tumor microenvironment. Each stromal cell developed in the tumor-specific microenvironment has a pro-tumorigenic or anti-tumorigenic role depending on the interaction with immune cells.
Since the TME is heterogeneous, the relationship between stromal cells and immune cells, which are components of the TME, is complex. However, additional mechanisms that control the interactions between immune cells and stromal cells in the TME remain unknown. Therefore, rather than interpreting a one-way relationship between two cells, it is necessary to understand and approach two-way relationships. The relationship between immune cells and stromal cells in the TME merits further investigation.
In particular, the most interesting aspect of the two orientations proposed in this review is the association of stromal cells with immune cells in controlling tumor formation and development. The network requires more attention when developing drugs that target their relationships. Increasing the understanding of the network between stromal cells and immune cells within the TME will ultimately improve the development and efficacy of cancer therapies. Finally, understanding the complex interactions between stromal cells and immune cells within the TME is necessary to identify potential strategies for cancer treatment.
Author Contributions
HK contributed to study conception. J-YM, S-HL, and JL performed literature review and analysis and revised the manuscript. J-YM and HK drafted the manuscript, figures, and tables. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (NRF-2020R1C1C1003676).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Labani-Motlagh A, Ashja-Mahdavi M, Loskog A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front Immunol (2020) 11:940. doi: 10.3389/fimmu.2020.00940
2. Chen F, Zhuang XQ, Lin LY, Yu PF, Wang Y, Shi YF, et al. New Horizons in Tumor Microenvironment Biology: Challenges and Opportunities. BMC Med (2015) 13:45. doi: 10.1186/s12916-015-0278-7
3. Petrova V, Annicchiarico-Petruzzelli M, Melino G, Amelio I. The Hypoxic Tumour Microenvironment. Oncogenesis (2018) 7:10. doi: 10.1038/s41389-017-0011-9
4. Feng LZ, Dong ZL, Tao DL, Zhang YC, Liu Z. The Acidic Tumor Microenvironment: A Target for Smart Cancer Nano-Theranostics. Natl Sci Rev (2018) 5(2):269–86. doi: 10.1093/nsr/nwx062
5. Greten FR, Grivennikov SI. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity (2019) 51(1):27–41. doi: 10.1016/j.immuni.2019.06.025
6. Gonzalez H, Hagerling C, Werb Z. Roles of the Immune System in Cancer: From Tumor Initiation to Metastatic Progression. Gene Dev (2018) 32(19-20):1267–84. doi: 10.1101/gad.314617.118
7. Bondhopadhyay B, Sisodiya S, Chikara A, Khan A, Tanwar P, Afroze D, et al. Cancer Immunotherapy: A Promising Dawn in Cancer Research. Am J Blood Res (2020) 10(6):375–85.
8. Pitt JM, Marabelle A, Eggermont A, Soria JC, Kroemer G, Zitvogel L. Targeting the Tumor Microenvironment: Removing Obstruction to Anticancer Immune Responses and Immunotherapy. Ann Oncol (2016) 27(8):1482–92. doi: 10.1093/annonc/mdw168
9. Klemm F, Joyce JA. Microenvironmental Regulation of Therapeutic Response in Cancer. Trends Cell Biol (2015) 25(4):198–213. doi: 10.1016/j.tcb.2014.11.006
10. Gajewski TF. Failure at the Effector Phase: Immune Barriers at the Level of the Melanoma Tumor Microenvironment. Clin Cancer Res (2007) 13(18):5256–61. doi: 10.1158/1078-0432.Ccr-07-0892
11. Baskar R, Lee KA, Yeo R, Yeoh KW. Cancer and Radiation Therapy: Current Advances and Future Directions. Int J Med Sci (2012) 9(3):193–9. doi: 10.7150/ijms.3635
12. Weber JS, O’Day S, Urba W, Powderly J, Nichol G, Yellin M, et al. Phase I/II Study of Ipilimumab for Patients With Metastatic Melanoma. J Clin Oncol (2008) 26(36):5950–6. doi: 10.1200/jco.2008.16.1927
13. Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, et al. Cancer Regression and Autoimmunity Induced by Cytotoxic T Lymphocyte-Associated Antigen 4 Blockade in Patients With Metastatic Melanoma. Proc Natl Acad Sci USA (2003) 100(14):8372–7. doi: 10.1073/pnas.1533209100
14. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved Survival With Ipilimumab in Patients With Metastatic Melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
15. Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, et al. Survival, Durable Tumor Remission, and Long-Term Safety in Patients With Advanced Melanoma Receiving Nivolumab. J Clin Oncol (2014) 32(10):1020–30. doi: 10.1200/jco.2013.53.0105
16. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab Plus Ipilimumab in Advanced Melanoma. N Engl J Med (2013) 369(2):122–33. doi: 10.1056/NEJMoa1302369
17. Hellmann MD, Callahan MK, Awad MM, Calvo E, Ascierto PA, Atmaca A, et al. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination With Ipilimumab in Small-Cell Lung Cancer. Cancer Cell (2018) 33(5):853–61.e4. doi: 10.1016/j.ccell.2018.04.001
18. Yau T, Hsu C, Kim TY, Choo SP, Kang YK, Hou MM, et al. Nivolumab in Advanced Hepatocellular Carcinoma: Sorafenib-Experienced Asian Cohort Analysis. J Hepatol (2019) 71(3):543–52. doi: 10.1016/j.jhep.2019.05.014
19. Vaddepally RK, Kharel P, Pandey R, Garje R, Chandra AB. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors Per NCCN Guidelines With the Level of Evidence. Cancers (Basel) (2020) 12(3):738. doi: 10.3390/cancers12030738
20. Androulla MN, Lefkothea PC. CAR T-Cell Therapy: A New Era in Cancer Immunotherapy. Curr Pharm Biotechnol (2018) 19(1):5–18. doi: 10.2174/1389201019666180418095526
21. Barrett DM, Grupp SA, June CH. Chimeric Antigen Receptor- and TCR-Modified T Cells Enter Main Street and Wall Street. J Immunol (2015) 195(3):755–61. doi: 10.4049/jimmunol.1500751
22. Vitale C, Strati P. CAR T-Cell Therapy for B-Cell non-Hodgkin Lymphoma and Chronic Lymphocytic Leukemia: Clinical Trials and Real-World Experiences. Front Oncol (2020) 10:849. doi: 10.3389/fonc.2020.00849
23. Sheykhhasan M, Manoochehri H, Dama P. Use of CAR T-Cell for Acute Lymphoblastic Leukemia (ALL) Treatment: A Review Study. Cancer Gene Ther (2022). doi: 10.1038/s41417-021-00418-1
24. Holstein SA, Lunning MA. CAR T-Cell Therapy in Hematologic Malignancies: A Voyage in Progress. Clin Pharmacol Ther (2020) 107(1):112–22. doi: 10.1002/cpt.1674
25. Sterner RC, Sterner RM. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J (2021) 11(4):69. doi: 10.1038/s41408-021-00459-7
26. Yu P, Fu YX. Tumor-Infiltrating T Lymphocytes: Friends or Foes? Lab Invest (2006) 86(3):231–45. doi: 10.1038/labinvest.3700389
27. Parihar R, Rivas C, Huynh M, Omer B, Lapteva N, Metelitsa LS, et al. NK Cells Expressing a Chimeric Activating Receptor Eliminate MDSCs and Rescue Impaired CAR-T Cell Activity Against Solid Tumors. Cancer Immunol Res (2019) 7(3):363–75. doi: 10.1158/2326-6066.Cir-18-0572
28. Zhong SW, Jeong JH, Chen ZK, Chen ZH, Luo JL. Targeting Tumor Microenvironment by Small-Molecule Inhibitors. Transl Oncol (2020) 13(1):57–69. doi: 10.1016/j.tranon.2019.10.001
29. Baghba R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M, et al. Tumor Microenvironment Complexity and Therapeutic Implications at a Glance. Cell Commun Signal (2020) 18(1):59. doi: 10.1186/s12964-020-0530-4
30. Kock A, Larsson K, Bergqvist F, Eissler N, Elfman LHM, Raouf J, et al. Inhibition of Microsomal Prostaglandin E Synthase-1 in Cancer-Associated Fibroblasts Suppresses Neuroblastoma Tumor Growth. Ebiomedicine (2018) 32:84–92. doi: 10.1016/j.ebiom.2018.05.008
31. Mertens JC, Fingas CD, Christensen JD, Smoot RL, Bronk SF, Werneburg NW, et al. Therapeutic Effects of Deleting Cancer-Associated Fibroblasts in Cholangiocarcinoma. Cancer Res (2013) 73(2):897–907. doi: 10.1158/0008-5472.Can-12-2130
32. Sweeny L, Liu ZY, Lancaster W, Hart J, Hartman YE, Rosenthal EL. Inhibition of Fibroblasts Reduced Head and Neck Cancer Growth by Targeting Fibroblast Growth Factor Receptor. Laryngoscope (2012) 122(7):1539–44. doi: 10.1002/lary.23266
33. Semenza GL. Regulation of Physiological Responses to Continuous and Intermittent Hypoxia by Hypoxia-Inducible Factor 1. Exp Physiol (2006) 91(5):803–6. doi: 10.1113/expphysiol.2006.033498
34. Vu HN, Miller WJ, O’Connor SA, He M, Schafer PH, Payvandi F, et al. CC-5079: A Small Molecule With MKP1, Antiangiogenic, and Antitumor Activity. J Surg Res (2010) 164(1):116–25. doi: 10.1016/j.jss.2009.01.031
35. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-Related Inflammation. Nature (2008) 454(7203):436–44. doi: 10.1038/nature07205
36. Grivennikov SI, Greten FR, Karin M. Immunity, Inflammation, and Cancer. Cell (2010) 140(6):883–99. doi: 10.1016/j.cell.2010.01.025
37. Mao XQ, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk Between Cancer-Associated Fibroblasts and Immune Cells in the Tumor Microenvironment: New Findings and Future Perspectives. Mol Cancer (2021) 20(1):131. doi: 10.1186/s12943-021-01428-1
38. Zitvogel L, Tesniere A, Kroemer G. Cancer Despite Immunosurveillance: Immunoselection and Immunosubversion. Nat Rev Immunol (2006) 6(10):715–27. doi: 10.1038/nri1936
39. Mueller MM, Fusenig NE. Friends or Foes - Bipolar Effects of the Tumour Stroma in Cancer. Nat Rev Cancer (2004) 4(11):839–49. doi: 10.1038/nrc1477
40. Kalluri R, Zeisberg M. Fibroblasts in Cancer. Nat Rev Cancer (2006) 6(5):392–401. doi: 10.1038/nrc1877
41. An YY, Liu FT, Chen Y, Yang Q. Crosstalk Between Cancer-Associated Fibroblasts and Immune Cells in Cancer. J Cell Mol Med (2020) 24(1):13–24. doi: 10.1111/jcmm.14745
42. Monteran L, Erez N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front Immunol (2019) 10:1835. doi: 10.3389/fimmu.2019.01835
43. Tamura R, Tanaka T, Yamamoto Y, Akasaki Y, Sasaki H. Dual Role of Macrophage in Tumor Immunity. Immunotherapy (2018) 10(10):899–909. doi: 10.2217/imt-2018-0006
44. Pan Y, Yu Y, Wang X, Zhang T. Tumor-Associated Macrophages in Tumor Immunity. Front Immunol (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
45. Gokyavuz B, Gunaydin G, Gedik ME, Kosemehmetoglu K, Karakoc D, Ozgur F, et al. Cancer Associated Fibroblasts Sculpt Tumour Microenvironment by Recruiting Monocytes and Inducing Immunosuppressive PD-1(+) TAMs. Sci Rep (2019) 9(1):3172. doi: 10.1038/s41598-019-39553-z
46. Zhang J, Lu Y, Pienta KJ. Multiple Roles of Chemokine (C-C Motif) Ligand 2 in Promoting Prostate Cancer Growth. J Natl Cancer Inst (2010) 102(8):522–8. doi: 10.1093/jnci/djq044
47. Furukawa S, Soeda S, Kiko Y, Suzuki O, Hashimoto Y, Watanabe T, et al. MCP-1 Promotes Invasion and Adhesion of Human Ovarian Cancer Cells. Anticancer Res (2013) 33(11):4785–90.
48. Lippitz BE. Cytokine Patterns in Patients With Cancer: A Systematic Review. Lancet Oncol (2013) 14(6):e218–28. doi: 10.1016/S1470-2045(12)70582-X
49. Domanska UM, Kruizinga RC, Nagengast WB, Timmer-Bosscha H, Huls G, de Vries EG, et al. A Review on CXCR4/CXCL12 Axis in Oncology: No Place to Hide. Eur J Cancer (2013) 49(1):219–30. doi: 10.1016/j.ejca.2012.05.005
50. Comito G, Giannoni E, Segura CP, Barcellos-de-Souza P, Raspollini MR, Baroni G, et al. Cancer-Associated Fibroblasts and M2-Polarized Macrophages Synergize During Prostate Carcinoma Progression. Oncogene (2014) 33(19):2423–31. doi: 10.1038/onc.2013.191
51. Silzle T, Kreutz M, Dobler MA, Brockhoff G, Knuechel R, Kunz-Schughart LA. Tumor-Associated Fibroblasts Recruit Blood Monocytes Into Tumor Tissue. Eur J Immunol (2003) 33(5):1311–20. doi: 10.1002/eji.200323057
52. Ksiazkiewicz M, Gottfried E, Kreutz M, Mack M, Hofstaedter F, Kunz-Schughart LA. Importance of CCL2-CCR2A/2B Signaling for Monocyte Migration Into Spheroids of Breast Cancer-Derived Fibroblasts. Immunobiology (2010) 215(9-10):737–47. doi: 10.1016/j.imbio.2010.05.019
53. Liu XM, Xu J, Zhang B, Liu J, Liang C, Meng QC, et al. The Reciprocal Regulation Between Host Tissue and Immune Cells in Pancreatic Ductal Adenocarcinoma: New Insights and Therapeutic Implications. Mol Cancer (2019) 18(1):184. doi: 10.1186/s12943-019-1117-9
54. Saotome T, Inoue H, Fujimiya M, Fujiyama Y, Bamba T. Morphological and Immunocytochemical Identification of Periacinar Fibroblast-Like Cells Derived From Human Pancreatic Acini. Pancreas (1997) 14(4):373–82. doi: 10.1097/00006676-199705000-00008
55. Jaster R. Molecular Regulation of Pancreatic Stellate Cell Function. Mol Cancer (2004) 3:26. doi: 10.1186/1476-4598-3-26
56. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated Regulation of Myeloid Cells by Tumours. Nat Rev Immunol (2012) 12(4):253–68. doi: 10.1038/nri3175
57. Gabrilovich DI, Nagaraj S. Myeloid-Derived Suppressor Cells as Regulators of the Immune System. Nat Rev Immunol (2009) 9(3):162–74. doi: 10.1038/nri2506
58. Bruno A, Mortara L, Baci D, Noonan DM, Albini A. Myeloid Derived Suppressor Cells Interactions With Natural Killer Cells and Pro-Angiogenic Activities: Roles in Tumor Progression. Front Immunol (2019) 10:771. doi: 10.3389/fimmu.2019.00771
59. Markowitz J, Brooks TR, Duggan MC, Paul BK, Pan XL, Wei L, et al. Patients With Pancreatic Adenocarcinoma Exhibit Elevated Levels of Myeloid-Derived Suppressor Cells Upon Progression of Disease. Cancer Immunol Immun (2015) 64(2):149–59. doi: 10.1007/s00262-014-1618-8
60. Porembka MR, Mitchem JB, Belt BA, Hsieh CS, Lee HM, Herndon J, et al. Pancreatic Adenocarcinoma Induces Bone Marrow Mobilization of Myeloid-Derived Suppressor Cells Which Promote Primary Tumor Growth. Cancer Immunol Immun (2012) 61(9):1373–85. doi: 10.1007/s00262-011-1178-0
61. Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, et al. Pancreatic Cancer-Associated Stellate Cells Promote Differentiation of Myeloid-Derived Suppressor Cells in a STAT3-Dependent Manner. Cancer Res (2013) 73(10):3007–18. doi: 10.1158/0008-5472.Can-12-4601
62. Patente TA, Pinho MP, Oliveira AA, Evangelista GCM, Bergami-Santos PC, Barbuto JAM. Human Dendritic Cells: Their Heterogeneity and Clinical Application Potential in Cancer Immunotherapy. Front Immunol (2018) 9:3176. doi: 10.3389/fimmu.2018.03176
63. Ninomiya T, Akbar SM, Masumoto T, Horiike N, Onji M. Dendritic Cells With Immature Phenotype and Defective Function in the Peripheral Blood From Patients With Hepatocellular Carcinoma. J Hepatol (1999) 31(2):323–31. doi: 10.1016/s0168-8278(99)80231-1
64. Maecker B, Mougiakakos D, Zimmermann M, Behrens M, Hollander S, Schrauder A, et al. Dendritic Cell Deficiencies in Pediatric Acute Lymphoblastic Leukemia Patients. Leukemia (2006) 20(4):645–9. doi: 10.1038/sj.leu.2404146
65. Wojas K, Tabarkiewicz J, Jankiewicz M, Rolinski J. Dendritic Cells in Peripheral Blood of Patients With Breast and Lung Cancer–A Pilot Study. Folia Histochem Cytobiol (2004) 42(1):45–8.
66. Cheng JT, Deng YN, Yi HM, Wang GY, Fu BS, Chen WJ, et al. Hepatic Carcinoma-Associated Fibroblasts Induce IDO-Producing Regulatory Dendritic Cells Through IL-6-Mediated STAT3 Activation. Oncogenesis (2016) 5:e198. doi: 10.1038/oncsis.2016.7
67. Cheng Y, Li H, Deng Y, Tai Y, Zeng K, Zhang Y, et al. Cancer-Associated Fibroblasts Induce PDL1+ Neutrophils Through the IL6-STAT3 Pathway That Foster Immune Suppression in Hepatocellular Carcinoma. Cell Death Dis (2018) 9(4):422. doi: 10.1038/s41419-018-0458-4
68. Cho H, Seo Y, Loke KM, Kim SW, Oh SM, Kim JH, et al. Cancer-Stimulated CAFs Enhance Monocyte Differentiation and Protumoral TAM Activation via IL6 and GM-CSF Secretion. Clin Cancer Res (2018) 24(21):5407–21. doi: 10.1158/1078-0432.CCR-18-0125
69. Ji Z, Tian W, Gao W, Zang R, Wang H, Yang G. Cancer-Associated Fibroblast-Derived Interleukin-8 Promotes Ovarian Cancer Cell Stemness and Malignancy Through the Notch3-Mediated Signaling. Front Cell Dev Biol (2021) 9:684505. doi: 10.3389/fcell.2021.684505
70. Zhang RS, Qi F, Zhao F, Li G, Shao SL, Zhang XC, et al. Cancer-Associated Fibroblasts Enhance Tumor-Associated Macrophages Enrichment and Suppress NK Cells Function in Colorectal Cancer. Cell Death Dis (2019) 10(4):273. doi: 10.1038/s41419-019-1435-2
71. Massague J, Blain SW, Lo RS. TGF Beta Signaling in Growth Control, Cancer, and Heritable Disorders. Cell (2000) 103(2):295–309. doi: 10.1016/S0092-8674(00)00121-5
72. Erdogan B, Webb DJ. Cancer-Associated Fibroblasts Modulate Growth Factor Signaling and Extracellular Matrix Remodeling to Regulate Tumor Metastasis. Biochem Soc T (2017) 45:229–36. doi: 10.1042/Bst20160387
73. Welters MJP, Santegoets SJ, van der Burg SH. The Tumor Microenvironment and Immunotherapy of Oropharyngeal Squamous Cell Carcinoma. Front Oncol (2020) 10:545385. doi: 10.3389/fonc.2020.545385
74. Chen WJ, Jin WW, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of Peripheral CD4(+)CD25(-) Naive T Cells to CD4(+)CD25(+) Regulatory T Cells by TGF-Beta Induction of Transcription Factor Foxp3. J Exp Med (2003) 198(12):1875–86. doi: 10.1084/jem.20030152
75. Gorelik L, Constant S, Flavell RA. Mechanism of Transforming Growth Factor Beta-Induced Inhibition of T Helper Type 1 Differentiation. J Exp Med (2002) 195(11):1499–505. doi: 10.1084/jem.20012076
76. Gorelik L, Fields PE, Flavell RA. Cutting Edge: TGF-Beta Inhibits Th Type 2 Development Through Inhibition of GATA-3 Expression. J Immunol (2000) 165(9):4773–7. doi: 10.4049/jimmunol.165.9.4773
77. Huang Y, Ge WH, Zhou JR, Gao BQ, Qian XH, Wang WL. The Role of Tumor Associated Macrophages in Hepatocellular Carcinoma. J Cancer (2021) 12(5):1284–94. doi: 10.7150/jca.51346
78. Chen YB, Song YC, Du W, Gong LL, Chang HC, Zou ZZ. Tumor-Associated Macrophages: An Accomplice in Solid Tumor Progression. J Biomed Sci (2019) 26(1):78. doi: 10.1186/s12929-019-0568-z
79. Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, et al. Activated Monocytes in Peritumoral Stroma of Hepatocellular Carcinoma Foster Immune Privilege and Disease Progression Through PD-L1. J Exp Med (2009) 206(6):1327–37. doi: 10.1084/jem.20082173
80. Xie Q, Ding J, Chen Y. Role of CD8(+) T Lymphocyte Cells: Interplay With Stromal Cells in Tumor Microenvironment. Acta Pharm Sin B (2021) 11(6):1365–78. doi: 10.1016/j.apsb.2021.03.027
81. Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-Associated Fibroblasts Induce Antigen-Specific Deletion of CD8(+) T Cells to Protect Tumour Cells. Nat Commun (2018) 9:948. doi: 10.1038/s41467-018-03347-0
82. Wu SZ, Roden DL, Wang CF, Holliday H, Harvey K, Cazet AS, et al. Stromal Cell Diversity Associated With Immune Evasion in Human Triple-Negative Breast Cancer. EMBO J (2020) 39(19):e104063. doi: 10.15252/embj.2019104063
83. Givel AM, Kieffer Y, Scholer-Dahirel A, Sirven P, Cardon M, Pelon F, et al. Mir200-Regulated CXCL12beta Promotes Fibroblast Heterogeneity and Immunosuppression in Ovarian Cancers. Nat Commun (2018) 9(1):1056. doi: 10.1038/s41467-018-03348-z
84. Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell (2018) 33(3):463–79.e10. doi: 10.1016/j.ccell.2018.01.011
85. Takahashi H, Sakakura K, Kawabata-Iwakawa R, Rokudai S, Toyoda M, Nishiyama M, et al. Immunosuppressive Activity of Cancer-Associated Fibroblasts in Head and Neck Squamous Cell Carcinoma. Cancer Immunol Immunother (2015) 64(11):1407–17. doi: 10.1007/s00262-015-1742-0
86. Rasanen K, Vaheri A. Activation of Fibroblasts in Cancer Stroma. Exp Cell Res (2010) 316(17):2713–22. doi: 10.1016/j.yexcr.2010.04.032
87. Ostman A, Augsten M. Cancer-Associated Fibroblasts and Tumor Growth - Bystanders Turning Into Key Players. Curr Opin Genet Dev (2009) 19(1):67–73. doi: 10.1016/j.gde.2009.01.003
88. Gorchs L, Kaipe H. Interactions Between Cancer-Associated Fibroblasts and T Cells in the Pancreatic Tumor Microenvironment and the Role of Chemokines. Cancers (Basel) (2021) 13(12):2995. doi: 10.3390/cancers13122995
89. Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, et al. Activated Pancreatic Stellate Cells Sequester CD8+ T Cells to Reduce Their Infiltration of the Juxtatumoral Compartment of Pancreatic Ductal Adenocarcinoma. Gastroenterology (2013) 145(5):1121–32. doi: 10.1053/j.gastro.2013.07.025
90. Hartmann N, Giese NA, Giese T, Poschke I, Offringa R, Werner J, et al. Prevailing Role of Contact Guidance in Intrastromal T-Cell Trapping in Human Pancreatic Cancer. Clin Cancer Res (2014) 20(13):3422–33. doi: 10.1158/1078-0432.CCR-13-2972
91. Gorchs L, Fernandez Moro C, Bankhead P, Kern KP, Sadeak I, Meng Q, et al. Human Pancreatic Carcinoma-Associated Fibroblasts Promote Expression of Co-Inhibitory Markers on CD4(+) and CD8(+) T-Cells. Front Immunol (2019) 10:847. doi: 10.3389/fimmu.2019.00847
92. Jiang Y, Li Y, Zhu B. T-Cell Exhaustion in the Tumor Microenvironment. Cell Death Dis (2015) 6:e1792. doi: 10.1038/cddis.2015.162
93. Zhang Z, Liu S, Zhang B, Qiao L, Zhang Y, Zhang Y. T Cell Dysfunction and Exhaustion in Cancer. Front Cell Dev Biol (2020) 8:17. doi: 10.3389/fcell.2020.00017
94. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or Adaptive Immunity? The Example of Natural Killer Cells. Science (2011) 331(6013):44–9. doi: 10.1126/science.1198687
95. Cooper MA, Fehniger TA, Caligiuri MA. The Biology of Human Natural Killer-Cell Subsets. Trends Immunol (2001) 22(11):633–40. doi: 10.1016/S1471-4906(01)02060-9
96. Lopez-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of Metastasis by NK Cells. Cancer Cell (2017) 32(2):135–54. doi: 10.1016/j.ccell.2017.06.009
97. Topham NJ, Hewitt EW. Natural Killer Cell Cytotoxicity: How do They Pull the Trigger? Immunology (2009) 128(1):7–15. doi: 10.1111/j.1365-2567.2009.03123.x
98. Brown JR, DuBois RN. COX-2: A Molecular Target for Colorectal Cancer Prevention. J Clin Oncol (2005) 23(12):2840–55. doi: 10.1200/JCO.2005.09.051
99. Balsamo M, Scordamaglia F, Pietra G, Manzini C, Cantoni C, Boitano M, et al. Melanoma-Associated Fibroblasts Modulate NK Cell Phenotype and Antitumor Cytotoxicity. Proc Natl Acad Sci USA (2009) 106(49):20847–52. doi: 10.1073/pnas.0906481106
100. Pietra G, Manzini C, Rivara S, Vitale M, Cantoni C, Petretto A, et al. Melanoma Cells Inhibit Natural Killer Cell Function by Modulating the Expression of Activating Receptors and Cytolytic Activity. Cancer Res (2012) 72(6):1407–15. doi: 10.1158/0008-5472.Can-11-2544
101. Li T, Yang Y, Hua X, Wang G, Liu W, Jia C, et al. Hepatocellular Carcinoma-Associated Fibroblasts Trigger NK Cell Dysfunction via PGE2 and IDO. Cancer Lett (2012) 318(2):154–61. doi: 10.1016/j.canlet.2011.12.020
102. Asghar K, Farooq A, Zulfiqar B, Rashid MU. Indoleamine 2,3-Dioxygenase: As a Potential Prognostic Marker and Immunotherapeutic Target for Hepatocellular Carcinoma. World J Gastroentero (2017) 23(13):2286–93. doi: 10.3748/wjg.v23.i13.2286
103. Mai J, Virtue A, Shen J, Wang H, Yang XF. An Evolving New Paradigm: Endothelial Cells–Conditional Innate Immune Cells. J Hematol Oncol (2013) 6:61. doi: 10.1186/1756-8722-6-61
104. Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C, Marini FC. Tumor-Associated Stromal Cells as Key Contributors to the Tumor Microenvironment. Breast Cancer Res (2016) 18(1):84. doi: 10.1186/s13058-016-0740-2
105. Salazar N, Zabel BA. Support of Tumor Endothelial Cells by Chemokine Receptors. Front Immunol (2019) 10:147. doi: 10.3389/fimmu.2019.00147
106. Certo M, Elkafrawy H, Pucino V, Cucchi D, Cheung KCP, Mauro C. Endothelial Cell and T-Cell Crosstalk: Targeting Metabolism as a Therapeutic Approach in Chronic Inflammation. Br J Pharmacol (2021) 178(10):2041–59. doi: 10.1111/bph.15002
107. Nagl L, Horvath L, Pircher A, Wolf D. Tumor Endothelial Cells (TECs) as Potential Immune Directors of the Tumor Microenvironment - New Findings and Future Perspectives. Front Cell Dev Biol (2020) 8:766. doi: 10.3389/fcell.2020.00766
108. Hirosue S, Vokali E, Raghavan VR, Rincon-Restrepo M, Lund AW, Corthesy-Henrioud P, et al. Steady-State Antigen Scavenging, Cross-Presentation, and CD8+ T Cell Priming: A New Role for Lymphatic Endothelial Cells. J Immunol (2014) 192(11):5002–11. doi: 10.4049/jimmunol.1302492
109. Maishi N, Hida K. Tumor Endothelial Cells Accelerate Tumor Metastasis. Cancer Sci (2017) 108(10):1921–6. doi: 10.1111/cas.13336
110. Wieland E, Rodriguez-Vita J, Liebler SS, Mogler C, Moll I, Herberich SE, et al. Endothelial Notch1 Activity Facilitates Metastasis. Cancer Cell (2017) 31(3):355–67. doi: 10.1016/j.ccell.2017.01.007
111. Yu JS, Lee PK, Ehtesham M, Samoto K, Black KL, Wheeler CJ. Intratumoral T Cell Subset Ratios and Fas Ligand Expression on Brain Tumor Endothelium. J Neuro-Oncol (2003) 64(1):55–61. doi: 10.1023/A:1024933925645
112. Taguchi K, Onoe T, Yoshida T, Yamashita Y, Tanaka Y, Ohdan H. Tumor Endothelial Cell-Mediated Antigen-Specific T-Cell Suppression via the PD-1/PD-L1 Pathway. Mol Cancer Res (2020) 18(9):1427–40. doi: 10.1158/1541-7786.Mcr-19-0897
113. Politz O, Gratchev A, McCourt PA, Schledzewski K, Guillot P, Johansson S, et al. Stabilin-1 and -2 Constitute a Novel Family of Fasciclin-Like Hyaluronan Receptor Homologues. Biochem J (2002) 362(Pt 1):155–64. doi: 10.1042/0264-6021:3620155
114. Shetty S, Weston CJ, Oo YH, Westerlund N, Stamataki Z, Youster J, et al. Common Lymphatic Endothelial and Vascular Endothelial Receptor-1 Mediates the Transmigration of Regulatory T Cells Across Human Hepatic Sinusoidal Endothelium. J Immunol (2011) 186(7):4147–55. doi: 10.4049/jimmunol.1002961
115. Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, et al. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res (2011) 71(7):2455–65. doi: 10.1158/0008-5472.CAN-10-3323
116. Fain JN. Release of Interleukins and Other Inflammatory Cytokines by Human Adipose Tissue is Enhanced in Obesity and Primarily Due to the Nonfat Cells. Vit Horm (2006) 74:443–77. doi: 10.1016/S0083-6729(06)74018-3.
117. Arendt LM, McCready J, Keller PJ, Baker DD, Naber SP, Seewaldt V, et al. Obesity Promotes Breast Cancer by CCL2-Mediated Macrophage Recruitment and Angiogenesis. Cancer Res (2013) 73(19):6080–93. doi: 10.1158/0008-5472.Can-13-0926
118. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity Is Associated With Macrophage Accumulation in Adipose Tissue. J Clin Invest (2003) 112(12):1796–808. doi: 10.1172/Jci200319246
119. Piya MK, McTernan PG, Kumar S. Adipokine Inflammation and Insulin Resistance: The Role of Glucose, Lipids and Endotoxin. J Endocrinol (2013) 216(1):T1–T15. doi: 10.1530/Joe-12-0498
120. Wu BG, Sun XJ, Gupta HB, Yuan B, Li JW, Ge F, et al. Adipose PD-L1 Modulates PD-1/PD-L1 Checkpoint Blockade Immunotherapy Efficacy in Breast Cancer. Oncoimmunology (2018) 7(11):e1500107. doi: 10.1080/2162402x.2018.1500107
121. Kedia-Mehta N, Finlay DK. Competition for Nutrients and its Role in Controlling Immune Responses. Nat Commun (2019) 10(1):2123. doi: 10.1038/s41467-019-10015-4
122. Vazquez Rodriguez G, Abrahamsson A, Jensen LDE, Dabrosin C. Adipocytes Promote Early Steps of Breast Cancer Cell Dissemination via Interleukin-8. Front Immunol (2018) 9:1767. doi: 10.3389/fimmu.2018.01767
123. Zhao SM, Wehner R, Bornhauser M, Wassmuth R, Bachmann M, Schmitz M. Immunomodulatory Properties of Mesenchymal Stromal Cells and Their Therapeutic Consequences for Immune-Mediated Disorders. Stem Cells Dev (2010) 19(5):607–14. doi: 10.1089/scd.2009.0345
124. Trivanovic D, Krstic J, Djordjevic IO, Mojsilovic S, Santibanez JF, Bugarski D, et al. The Roles of Mesenchymal Stromal/Stem Cells in Tumor Microenvironment Associated With Inflammation. Mediat Inflamm (2016) 2016:14. doi: 10.1155/2016/7314016
125. Patel SA, Meyer JR, Greco SJ, Corcoran KE, Bryan M, Rameshwar P. Mesenchymal Stem Cells Protect Breast Cancer Cells Through Regulatory T Cells: Role of Mesenchymal Stem Cell-Derived TGF-Beta. J Immunol (2010) 184(10):5885–94. doi: 10.4049/jimmunol.0903143
126. Qin Y, Zhou ZH, Zhang F, Wang Y, Shen B, Liu Y, et al. Induction of Regulatory B-Cells by Mesenchymal Stem Cells is Affected by SDF-1 Alpha-CXCR7. Cell Physiol Biochem (2015) 37(1):117–30. doi: 10.1159/000430338
127. Zhou X, Su YX, Lao XM, Liang YJ, Liao GQ. CD19(+)IL-10(+) Regulatory B Cells Affect Survival of Tongue Squamous Cell Carcinoma Patients and Induce Resting CD4(+) T Cells to CD4(+)Foxp3(+) Regulatory T Cells. Oral Oncol (2016) 53:27–35. doi: 10.1016/j.oraloncology.2015.11.003
128. Ammirante M, Luo JL, Grivennikov S, Nedospasov S, Karin M. B-Cell-Derived Lymphotoxin Promotes Castration-Resistant Prostate Cancer. Nature (2010) 464(7286):302–U187. doi: 10.1038/nature08782
129. Schioppa T, Moore R, Thompson RG, Rosser EC, Kulbe H, Nedospasov S, et al. B Regulatory Cells and the Tumor-Promoting Actions of TNF-Alpha During Squamous Carcinogenesis. Proc Natl Acad Sci USA (2011) 108(26):10662–7. doi: 10.1073/pnas.1100994108
130. Olkhanud PB, Damdinsuren B, Bodogai M, Gress RE, Sen R, Wejksza K, et al. Tumor-Evoked Regulatory B Cells Promote Breast Cancer Metastasis by Converting Resting CD4(+) T Cells to T-Regulatory Cells. Cancer Res (2011) 71(10):3505–15. doi: 10.1158/0008-5472.Can-10-4316
131. O’Malley G, Treacy O, Lynch K, Naicker SD, Leonard NA, Lohan P. Stromal Cell PD-L1 Inhibits CD8(+) T-Cell Antitumor Immune Responses and Promotes Colon Cancer (Vol 62018). Cancer Immunol Res (2019) 7(1):162–:pg 1426. doi: 10.1158/2326-6066.Cir-18-0889
132. Kowanetz M, Wu XM, Lee J, Tan M, Hagenbeek T, Qu XP, et al. Granulocyte-Colony Stimulating Factor Promotes Lung Metastasis Through Mobilization of Ly6G+Ly6C+ Granulocytes. Proc Natl Acad Sci USA (2010) 107(50):21248–55. doi: 10.1073/pnas.1015855107
133. Yan HH, Pickup M, Pang YL, Gorska AE, Li ZY, Chytil A, et al. Gr-1+CD11b+ Myeloid Cells Tip the Balance of Immune Protection to Tumor Promotion in the Premetastatic Lung. Cancer Res (2010) 70(15):6139–49. doi: 10.1158/0008-5472.Can-10-0706
134. Nozawa H, Chiu C, Hanahan D. Infiltrating Neutrophils Mediate the Initial Angiogenic Switch in a Mouse Model of Multistage Carcinogenesis. Proc Natl Acad Sci USA (2006) 103(33):12493–8. doi: 10.1073/pnas.0601807103
135. Ardi VC, Kupriyanova TA, Deryugina EI, Quigley JP. Human Neutrophils Uniquely Release TIMP-Free MMP-9 to Provide a Potent Catalytic Stimulator of Angiogenesis. Proc Natl Acad Sci USA (2007) 104(51):20262–7. doi: 10.1073/pnas.0706438104
136. Kessenbrock K, Plaks V, Werb Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell (2010) 141(1):52–67. doi: 10.1016/j.cell.2010.03.015
137. Hiratsuka S, Nakamura K, Iwai S, Murakami M, Itoh T, Kijima H, et al. MMP9 Induction by Vascular Endothelial Growth Factor Receptor-1 is Involved in Lung-Specific Metastasis. Cancer Cell (2002) 2(4):289–300. doi: 10.1016/S1535-6108(02)00153-8
138. Gunaydin G. CAFs Interacting With TAMs in Tumor Microenvironment to Enhance Tumorigenesis and Immune Evasion. Front Oncol (2021) 11:668349. doi: 10.3389/fonc.2021.668349
139. Noy R, Pollard JW. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity (2014) 41(1):49–61. doi: 10.1016/j.immuni.2014.06.010
140. Ueshima E, Fujimori M, Kodama H, Felsen D, Chen J, Durack JC, et al. Macrophage-Secreted TGF-Beta1 Contributes to Fibroblast Activation and Ureteral Stricture After Ablation Injury. Am J Physiol Renal Physiol (2019) 317(7):F52–64. doi: 10.1152/ajprenal.00260.2018
141. Tokuda K, Morine Y, Miyazaki K, Yamada S, Saito Y, Nishi M, et al. The Interaction Between Cancer Associated Fibroblasts and Tumor Associated Macrophages via the Osteopontin Pathway in the Tumor Microenvironment of Hepatocellular Carcinoma. Oncotarget (2021) 12(4):333–43. doi: 10.18632/oncotarget.27881
142. Park J, Kim DS, Shim TS, Lim CM, Koh Y, Lee SD, et al. Lung Cancer in Patients With Idiopathic Pulmonary Fibrosis. Eur Respir J (2001) 17(6):1216–9. doi: 10.1183/09031936.01.99055301
143. Haddad R, Massaro D. Idiopathic Diffuse Interstitial Pulmonary Fibrosis (Fibrosing Alveolitis), Atypical Epithelial Proliferation and Lung Cancer. Am J Med (1968) 45(2):211–9. doi: 10.1016/0002-9343(68)90039-9
144. Hou J, Shi J, Chen L, Lv Z, Chen X, Cao H, et al. M2 Macrophages Promote Myofibroblast Differentiation of LR-MSCs and are Associated With Pulmonary Fibrogenesis. Cell Commun Signal (2018) 16(1):89. doi: 10.1186/s12964-018-0300-8
145. Samarelli AV, Masciale V, Aramini B, Colo GP, Tonelli R, Marchioni A, et al. Molecular Mechanisms and Cellular Contribution From Lung Fibrosis to Lung Cancer Development. Int J Mol Sci (2021) 22(22):12179. doi: 10.3390/ijms222212179
146. Mashouri L, Yousefi H, Aref AR, Ahadi AM, Molaei F, Alahari SK. Exosomes: Composition, Biogenesis, and Mechanisms in Cancer Metastasis and Drug Resistance. Mol Cancer (2019) 18(1):75. doi: 10.1186/s12943-019-0991-5
147. Yang YH, Guo ZY, Chen WW, Wang XF, Cao M, Han X, et al. M2 Macrophage-Derived Exosomes Promote Angiogenesis and Growth of Pancreatic Ductal Adenocarcinoma by Targeting E2f2. Mol Ther (2021) 29(3):1226–38. doi: 10.1016/j.ymthe.2020.11.024
148. Gil Z, Billan S. Crosstalk Between Macrophages and Endothelial Cells in the Tumor Microenvironment. Mol Ther (2021) 29(3):895–6. doi: 10.1016/j.ymthe.2021.02.002
149. Hanna J, Goldman-Wohl D, Hamani Y, Avraham I, Greenfield C, Natanson-Yaron S, et al. Decidual NK Cells Regulate Key Developmental Processes at the Human Fetal-Maternal Interface. Nat Med (2006) 12(9):1065–74. doi: 10.1038/nm1452
150. Bruno A, Focaccetti C, Pagani A, Imperatori AS, Spagnoletti M, Rotolo N, et al. The Proangiogenic Phenotype of Natural Killer Cells in Patients With Non-Small Cell Lung Cancer. Neoplasia (2013) 15(2):133–U91. doi: 10.1593/neo.121758
151. Bassani B, Baci D, Gallazzi M, Poggi A, Bruno A, Mortara L. Natural Killer Cells as Key Players of Tumor Progression and Angiogenesis: Old and Novel Tools to Divert Their Pro-Tumor Activities Into Potent Anti-Tumor Effects. Cancers (2019) 11(4):461. doi: 10.3390/cancers11040461
152. Cai SW, Yang SZ, Gao J, Pan K, Chen JY, Wang YL, et al. Prognostic Significance of Mast Cell Count Following Curative Resection for Pancreatic Ductal Adenocarcinoma. Surgery (2011) 149(4):576–84. doi: 10.1016/j.surg.2010.10.009
153. Chang DZ, Ma Y, Ji BA, Wang HM, Deng DF, Liu Y, et al. Mast Cells in Tumor Microenvironment Promotes the In Vivo Growth of Pancreatic Ductal Adenocarcinoma. Clin Cancer Res (2011) 17(22):7015–23. doi: 10.1158/1078-0432.Ccr-11-0607
154. Yang FC, Chen S, Clegg T, Li X, Morgan T, Estwick SA, et al. Nf1+/- Mast Cells Induce Neurofibroma Like Phenotypes Through Secreted TGF-Beta Signaling. Hum Mol Genet (2006) 15(16):2421–37. doi: 10.1093/hmg/ddl165
155. Pereira BA, Lister NL, Hashimoto K, Teng L, Flandes-Iparraguirre M, Eder A, et al. Tissue Engineered Human Prostate Microtissues Reveal Key Role of Mast Cell-Derived Tryptase in Potentiating Cancer-Associated Fibroblast (CAF)-Induced Morphometric Transition In Vitro. Biomaterials (2019) 197:72–85. doi: 10.1016/j.biomaterials.2018.12.030
156. Ma Y, Hwang RF, Logsdon CD, Ullrich SE. Dynamic Mast Cell-Stromal Cell Interactions Promote Growth of Pancreatic Cancer. Cancer Res (2013) 73(13):3927–37. doi: 10.1158/0008-5472.CAN-12-4479
157. Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng XF, Wu CC, Simpson TR, et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer With Reduced Survival. Cancer Cell (2014) 25(6):719–34. doi: 10.1016/j.ccr.2014.04.005
158. McAndrews KM, Vazquez-Arreguin K, Kwak C, Sugimoto H, Zheng X, Li B, et al. alphaSMA(+) Fibroblasts Suppress Lgr5(+) Cancer Stem Cells and Restrain Colorectal Cancer Progression. Oncogene (2021) 40(26):4440–52. doi: 10.1038/s41388-021-01866-7
159. Cheng HW, Onder L, Cupovic J, Boesch M, Novkovic M, Pikor N, et al. CCL19-Producing Fibroblastic Stromal Cells Restrain Lung Carcinoma Growth by Promoting Local Antitumor T-Cell Responses. J Allergy Clin Immun (2018) 142(4):1257–+. doi: 10.1016/j.jaci.2017.12.998
160. Kamata T, So TY, Ahmed Q, Giblett S, Patel B, Luo JL, et al. Fibroblast-Derived STC-1 Modulates Tumor-Associated Macrophages and Lung Adenocarcinoma Development. Cell Rep (2020) 31(12):107802. doi: 10.1016/j.celrep.2020.107802
161. Choi MR, Sosman JA, Zhang B. The Janus Face of IL-33 Signaling in Tumor Development and Immune Escape. Cancers (2021) 13(13):3281. doi: 10.3390/cancers13133281
162. Andreone S, Gambardella AR, Mancini J, Loffredo S, Marcella S, La Sorsa V, et al. Anti-Tumorigenic Activities of IL-33: A Mechanistic Insight. Front Immunol (2020) 11:571593. doi: 10.3389/fimmu.2020.571593
163. Wasmer MH, Krebs P. The Role of IL-33-Dependent Inflammation in the Tumor Microenvironment. Front Immunol (2017) 7:682. doi: 10.3389/fimmu.2016.00682
164. Xia YL, Ohno T, Nishii N, Bhingare A, Tachinami H, Kashima Y, et al. Endogenous IL-33 Exerts CD8(+) T Cell Antitumor Responses Overcoming Pro-Tumor Effects by Regulatory T Cells in a Colon Carcinoma Model. Biochem Bioph Res Co (2019) 518(2):331–6. doi: 10.1016/j.bbrc.2019.08.058
165. Gao X, Wang XF, Yang QT, Zhao X, Wen W, Li G, et al. Tumoral Expression of IL-33 Inhibits Tumor Growth and Modifies the Tumor Microenvironment Through CD8(+) T and NK Cells. J Immunol (2015) 194(1):438–45. doi: 10.4049/jimmunol.1401344
166. Qi L, Zhang QY, Miao YH, Kang WY, Tian ZG, Xu DM, et al. Interleukin-33 Activates and Recruits Natural Killer Cells to Inhibit Pulmonary Metastatic Cancer Development. Int J Cancer (2020) 146(5):1421–34. doi: 10.1002/ijc.32779
167. Jovanovic I, Radosavljevic G, Mitrovic M, Juranic VL, McKenzie ANJ, Arsenijevic N, et al. ST2 Deletion Enhances Innate and Acquired Immunity to Murine Mammary Carcinoma. Eur J Immunol (2011) 41(7):1902–12. doi: 10.1002/eji.201141417
168. Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, et al. IKKbeta Links Inflammation and Tumorigenesis in a Mouse Model of Colitis-Associated Cancer. Cell (2004) 118(3):285–96. doi: 10.1016/j.cell.2004.07.013
169. Qiang L, Sample A, Liu H, Wu X, He YY. Epidermal SIRT1 Regulates Inflammation, Cell Migration, and Wound Healing. Sci Rep (2017) 7(1):14110. doi: 10.1038/s41598-017-14371-3
170. Lee SJ, Baek SE, Jang MA, Kim CD. SIRT1 Inhibits Monocyte Adhesion to the Vascular Endothelium by Suppressing Mac-1 Expression on Monocytes. Exp Mol Med (2019) 51(4):1–12. doi: 10.1038/s12276-019-0239-x
171. El Assar M, Angulo J, Rodriguez-Manas L. Oxidative Stress and Vascular Inflammation in Aging. Free Radic Biol Med (2013) 65:380–401. doi: 10.1016/j.freeradbiomed.2013.07.003
172. Veerman RE, Akpinar GG, Eldh M, Gabrielsson S. Immune Cell-Derived Extracellular Vesicles - Functions and Therapeutic Applications. Trends Mol Med (2019) 25(5):382–94. doi: 10.1016/j.molmed.2019.02.003
173. Seo N, Shirakura Y, Tahara Y, Momose F, Harada N, Ikeda H, et al. Activated CD8(+) T Cell Extracellular Vesicles Prevent Tumour Progression by Targeting of Lesional Mesenchymal Cells. Nat Commun (2018) 9:435. doi: 10.1038/s41467-018-02865-1
174. Wang WM, Kryczek I, Dostal L, Lin H, Tan LJ, Zhao LL, et al. Effector T Cells Abrogate Stroma-Mediated Chemoresistance in Ovarian Cancer. Cell (2016) 165(5):1092–105. doi: 10.1016/j.cell.2016.04.009
175. Qian LW, Tang ZR, Yin SH, Mo FZ, Yang XM, Hou XQ, et al. Fusion of Dendritic Cells and Cancer-Associated Fibroblasts for Activation of Anti-Tumor Cytotoxic T Lymphocytes. J BioMed Nanotechnol (2018) 14(10):1826–35. doi: 10.1166/jbn.2018.2616
Keywords: tumor microenvironment, immune cells, stromal cells, cancer-associated fibroblast (CAF), tumor endothelial cell, cancer-associated adipocyte, T cell, NK cell
Citation: Mun J-Y, Leem S-H, Lee JH and Kim HS (2022) Dual Relationship Between Stromal Cells and Immune Cells in the Tumor Microenvironment. Front. Immunol. 13:864739. doi: 10.3389/fimmu.2022.864739
Received: 28 January 2022; Accepted: 15 March 2022;
Published: 06 April 2022.
Edited by:
Jaewoo Hong, Catholic University of Daegu, South KoreaReviewed by:
You-Sun Kim, Ajou University, South KoreaYoe-Sik Bae, Sungkyunkwan University, South Korea
Copyright © 2022 Mun, Leem, Lee and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hyuk Soon Kim, hskimxo@dau.ac.kr