 Naomi Benne
Naomi Benne Daniëlle ter Braake
Daniëlle ter Braake Arie Jan Stoppelenburg
Arie Jan Stoppelenburg Femke Broere
Femke Broere- 1Department of Biomolecular Health Sciences, Faculty of Veterinary Medicine, Utrecht University, Utrecht, Netherlands
- 2Department of Rheumatology, University Medical Center Utrecht, Utrecht, Netherlands
- 3Department of Clinical Immunology, University Medical Center Utrecht, Utrecht, Netherlands
- 4Department of Clinical Sciences, Faculty of Veterinary Medicine, Utrecht University, Utrecht, Netherlands
Autoimmune diseases affect many people worldwide. Current treatment modalities focus on the reduction of disease symptoms using anti-inflammatory drugs which can lead to side effects due to systemic immune suppression. Restoration of immune tolerance by down-regulating auto-reactive cells in an antigen-specific manner is currently the “holy grail” for the treatment of autoimmune diseases. A promising strategy is the use of nanoparticles that can deliver antigens to antigen-presenting cells which in turn can enhance antigen-specific regulatory T cells. In this review, we highlight some promising cell targets (e.g. liver sinusoidal endothelial cells and splenic marginal zone macrophages) for exploiting natural immune tolerance processes, and several strategies by which antigen-carrying nanoparticles can target these cells. We also discuss how nanoparticles carrying immunomodulators may be able to activate tolerance in other antigen-presenting cell types. Finally, we discuss some important aspects that must be taken into account when translating data from animal studies to patients.
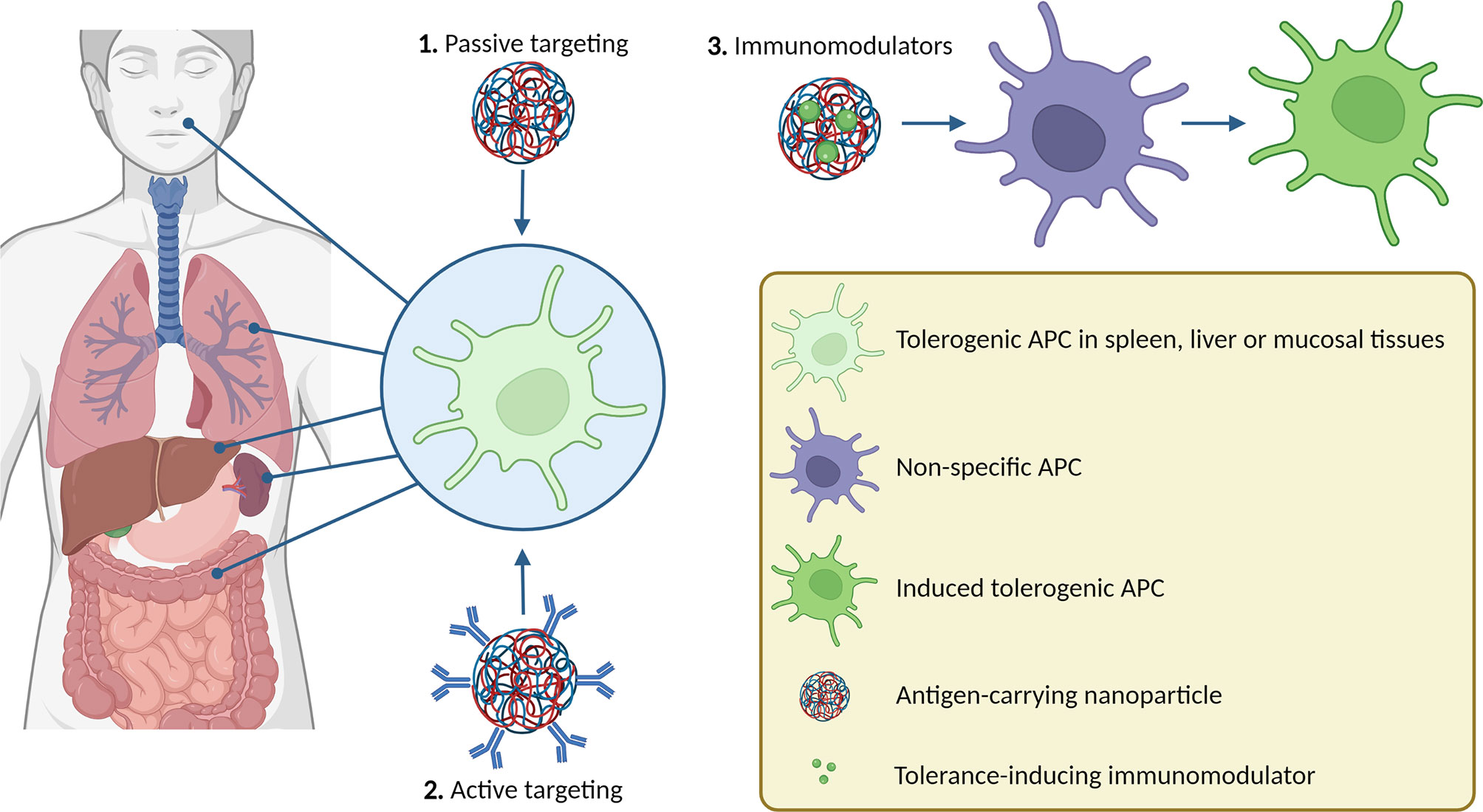
Graphical Abstract
1 Introduction
1.1 General
Our immune system has evolved to distinguish non-self from self-antigens to protect us against pathogens while also maintaining tissue homeostasis. Immune cells of both the innate and the adaptive immune systems are involved in this complex protection against potentially harmful intruders. The process of combating exogenous antigens by these immune cells is a powerful effector mechanism. However, when directed toward our own cells, tissues, or commensal microbes, this mechanism could be very destructive (1). To maintain immune homeostasis and prevent tissue damage, the immune system must be able to distinguish innocuous endogenous antigens from potentially harmful exogenous antigens (2). This mechanism is known as self-tolerance and is maintained by specialized immune cell subsets such as tolerogenic antigen-presenting cells (APCs) and regulatory T cells (Tregs) (3). In autoimmunity, Tregs are the most well-studied cell type and the focus of this review. Tregs can be derived from the thymus (4) or peripherally induced from effector T cell populations (5). Tregs are typically defined as CD45RO+TCR+CD4+CD25hiFoxp3+CD127- cells in humans, and CD4+CD25+Foxp3+ in mice (6), but can also express the suppressive receptors CTLA-4, PD-1, TIGIT, and GITR, among others (7, 8). Other suppressive T cell populations that do not express Foxp3 include T regulatory 1 (Tr1) cells, which secrete high levels of IL-10 and express CD49b and LAG-3 in humans and mice (9), and T helper 3 (Th3) cells, which secrete high levels of TGF-β (10). Apart from T cells, B cells also play an important role in maintaining immune homeostasis, including regulatory B cells (Bregs) (11), of which there are also several subtypes, as reviewed elsewhere (12). Immune tolerance can be disrupted due to multifactorial causes (13). This can prompt an overactive inflammatory response towards (auto)antigens (14), resulting in the development of autoimmune diseases. Current treatment modalities focus on the reduction of disease symptoms using anti-inflammatory drugs (e.g. corticosteroids) or biologicals (e.g. tumor necrosis factor (TNF)-α inhibitors) (15). Long-term use of these drugs coincides with unwanted side effects such as increased susceptibility to opportunistic infections and tumors (16). Significant research has focused on improving the targeting of these drugs, thereby reducing the dosage, and limiting off-target effects [as reviewed by Fang et al. (17)]. Although these therapies have improved patient care over the last decades, they fail to cure patients that suffer from autoimmune diseases, necessitating lifelong therapy.
Restoring the immune balance by down-regulating auto-reactive cells and enhancing Treg function is a promising strategy to treat autoimmune diseases. Importantly, this could result in long-lasting medication-free disease remission (18). Unfortunately, injection of free antigen is unlikely to result in sufficient accumulation in the specific cells that can induce tolerance. Besides, many autoantigens are poorly soluble, and due to their size or charge can bring about unwanted immunogenic effects if administered freely (19). A very promising approach is the use of APC-like nanoparticles (20), as reviewed elsewhere (21). Another strategy is to develop nanoparticulate delivery systems to facilitate the delivery of antigens to APCs in vivo.
Nanoparticles are drug delivery systems ranging from 1 to 1000 nm in size (22). These particles protect their cargo from enzymatic degradation and can be designed to accommodate for an antigen’s size, charge, and solubility, and (passively) target specific immune cells. This allows for higher therapeutic efficacy, reduces the required dose, and minimizes off-target effects or side effects associated with high doses or the intrinsic properties of the free antigen (23). Nanoparticles can target organs or cells depending on their physicochemical properties (e.g. size, charge, and rigidity) (24–27), or by the incorporation of targeting moieties, such as antibodies (28). Nanoparticles can be made of different materials, including polymers (29), metals (30), lipids (31), proteins (32), or a combination ft he above. Some of these materials have intrinsic immunomodulatory effects (33), which makes them interesting for use in immunotherapy. Furthermore, nanoparticles can carry both an antigen and an immunomodulator to induce tolerogenic phenotypes in APCs in vivo. In this review, we highlight some promising cell targets and several strategies by which antigen-carrying nanoparticles, with or without immunomodulators can target these cells.
1.2 Cellular Targets
Immune tolerance to circulating antigens is maintained by the spleen and liver (34, 35). These organs are responsible for filtering the blood and contain many specialized cell types that are involved in the clearance of apoptotic cells. This process is termed efferocytosis and is vital for immune homeostasis; the dysregulation of this process has been implicated in several autoimmune diseases (36, 37). Some APC subsets such as plasmacytoid dendritic cells (pDCs) express CD36 and CD61, which are efferocytic scavenger receptors involved in immune regulation (38). Cells expressing these receptors often show a decrease in the surface expression of the co-stimulatory molecules CD40 and CD86 and it has been shown that efferocytosis by DC subsets leads to a suppressive phenotype (39, 40). Furthermore, deficiency in scavenger receptor function has been described to be involved in the development of autoimmunity (41), and activation of the efferocytic receptor MER protected mice against the development of arthritis in collagen-induced arthritis and KRN serum transfer mouse models (42). In a study by Watkins et al., the importance of efferocytosis in immune tolerance was demonstrated by directly targeting antigens towards apoptotic erythrocytes. When these erythrocytes were efferocytosed in the spleen, the antigen was presented in a tolerogenic manner and induced long-lasting antigen-specific T cell anergy (43). In another study, it was shown that human apoptotic cells derived from peripheral blood mononuclear cells have low expression of HLA-DR and CD86, produce the anti-inflammatory cytokines IL-10 and TGF-β, and expressed Fas and caspase-3. In a mixed lymphocyte reaction, the apoptotic cells greatly reduced the proliferation of T cells as compared to non-apoptotic cells and reduced the expression of CD25, CD45RO, and OX40 on proliferated T cells. These apoptotic cells inhibited allogeneic immune responses in humanized non-obese diabetic (NOD)/severe combined immune deficiency (SCID)/γC mice (44). Furthermore, i.v. injection of apoptotic cells that expressed a MOG peptide prevented the development of EAE in mice. This was shown to be due to the accumulation of the apoptotic cells in the splenic marginal zone and antigen-specific T cell unresponsiveness, as measured by reduced proliferation of T cells and reduced production of IFNγ and IL-17 by T cells. Unfortunately, injection of the apoptotic cells after MOG immunization did not affect disease progression (45). Splenectomy abolishes the immune-suppressing effects of apoptotic cell-mimicking liposomes (35). These studies show that when antigen is taken up in the context of efferocytosis, it leads to immune suppression and that the spleen is vital in this process. The splenic marginal zone contains several cell subtypes such as marginal zone macrophages (MZMs), B cells, and DCs (46). For instance, MZMs expressing the macrophage receptor with a collagenous structure (MARCO) (47) and macrophages expressing CD169/sialic acid-binding immunoglobulin-type lectin-1 (Siglec-1) (48) can induce tolerance. Depletion of Siglec-1+ macrophages from the spleens in mice susceptible to EAE abrogated the protective effects of MOG-apoptotic cells (45). Siglec-1 is important for cell-cell contact and binds to CD8α+ DCs, which suggests that the tolerogenic immune effects of Siglec-1+ macrophages may be mediated through marginal zone DCs (49). CD8α+CD103+ DCs in the marginal zone of the spleen efficiently efferocytose apoptotic cells from the blood, and subsequently migrate to splenic T cells to present antigens in a tolerogenic fashion (50). Indeed, injected apoptotic cells are filtered by CD8α+ DCs in WT mice and lead to tolerance, while Siglec-1 depletion leads to uptake by CD8α-CD11b+ DCs instead (45).
The liver microenvironment also promotes tolerance (51) and human DCs derived from the liver are more suppressive than blood-derived DCs (52). The liver contains a multitude of cells that are inherently suppressive (34). For example, liver sinusoidal epithelial cells (LSECs) have been shown to induce differentiation of T cells to both Foxp3+ Tregs and Foxp3-LAG3+ Tr1 cells (53). Furthermore, Kupffer cells in the liver express low levels of co-stimulatory molecules and suppress T cell responses (54).
Finally, it is well known that oral and mucosal antigen application favors tolerance induction. In a collagen-induced arthritis mouse model, multiple oral administrations of type II collagen significantly reduced the severity of the disease (55), and in a human trial in rheumatoid arthritis patients, oral administration of type II collagen significantly reduced joint swelling and pain (56). Oral tolerance induction is independent of apoptotic pathways as splenectomy does not abrogate oral tolerance (57). The mesenteric lymph nodes are important for oral tolerance induction, as it was found that transplantation of peripheral lymph nodes into the gut mesenteries in mice did not allow for the induction of oral tolerance (58). This points to the importance of the microenvironment created by stromal cells in these lymphoid structures that facilitates tolerance induction. In mice removal of the superficial cervical and internal jugular lymph nodes which drain the nasal mucosa abrogated nasal tolerance induction (59). Specifically, it has been shown that CD11b+ DCs are important for oral tolerance in a collagen-induced arthritis model (60).
APCs involved in mucosal tolerance are mainly macrophages and DCs in the lungs (61), along with B cells, DCs, and macrophages in the nasal- and gut-associated lymphoid tissues (62–64). Other DCs involved in nasal and oral tolerance express the inhibitory Fc receptor for IgG IIB (FcγRIIB) (65, 66). FcγRIIB plays a key role in DC uptake, processing, and presentation of antigens (67) and a loss of this receptor has been shown to induce autoimmunity in mice (68). Another group of immune-suppressing mucosal DCs are CD103+ DCs, which can induce antigen-specific Foxp3+ Tregs (69, 70). Collectively, these studies show that there are several subtypes of APCs in the liver, spleen, and oral and mucosal lymphoid tissues which are specialized to induce tolerance and are therefore attractive for targeting nanoparticles. We will highlight several strategies by which this targeting can be achieved.
2 Passive Targeting
2.1 Physicochemical Properties
2.1.1 Charge
One of the great advantages of nanoparticles is that their physicochemical properties can be optimized to the application. Depending on their physicochemical properties, nanoparticles can elicit different immune responses. In the case of pro-inflammatory responses, this is generally achieved by mimicking pathogens (41). However, for tolerance induction, an attractive strategy would be to mimic apoptotic cells. Such apoptotic-like particles would efficiently be taken up directly or via the protein corona and processed through the tolerance-promoting effector mechanisms in the spleen and liver. Nanoparticle charge is one of the easiest and most effective ways to achieve this. When cells undergo apoptosis, they express the negatively charged phospholipid phosphatidylserine (PS) on their surface (71), which is recognized by receptors on efferocytes, such as stabilin-2 (72), TIM-4 (73), and CD300f (74, 75), as shown in Figure 1. The role of PS in apoptosis is extensively reviewed by Birge et al. and several types of nanoparticles containing PS have taken advantage of this pathway to induce tolerance in autoimmune models (76, 77). Unfortunately, empty PS liposomes have been shown to induce non-specific immune tolerance which may hamper clinical application (35, 78). Anionic phosphatidylglycerol (PG)-containing liposomes encapsulating an atherosclerosis-specific peptide significantly reduced disease progression in an atherosclerotic mouse model. However, the same liposomes without antigen did not have any effect on the disease, meaning that this effect was antigen-specific. The PG-liposomes were more effective at inducing antigen-specific Treg responses than PS-liposomes, even though both had similar surface charges (79). This was hypothesized to be due to the formation of a protein corona, specifically C1q binding to the PG-liposomes, which has been shown to have a tolerogenic effect intricately linked to the clearance of apoptotic cells by binding to scavenger receptors such as class F scavenger receptor (SR-F1) (41, 75, 80, 81). In subsequent studies, it was observed that the liposomes are selectively taken up by APCs in the liver and spleen (unpublished data). In another study, anionic poly(ethylene-co-maleic acid)-poly(lactic-co-glycolic acid) (PEMA-PLGA) nanoparticles encapsulating EAE peptides upregulated PD-L1 expression on liver CD103+ DCs and Kupffer cells. This in turn led to antigen-specific lymphocyte unresponsiveness, as measured by reduced proliferation of lymphocytes and reduced production of IL-17, GM-CSF, and IFNγ. The nanoparticles could both prevent and treat EAE in mice. This effect was even observed after splenectomy, underlining the importance of the liver in this model (82). Interestingly, protection against EAE was induced after i.v. injection, and to a lesser extent i.p. injection of nanoparticles, but not after oral or s.c. administration. This was hypothesized to be because these nanoparticles need to travel to the liver and spleen to exert their effects (83).
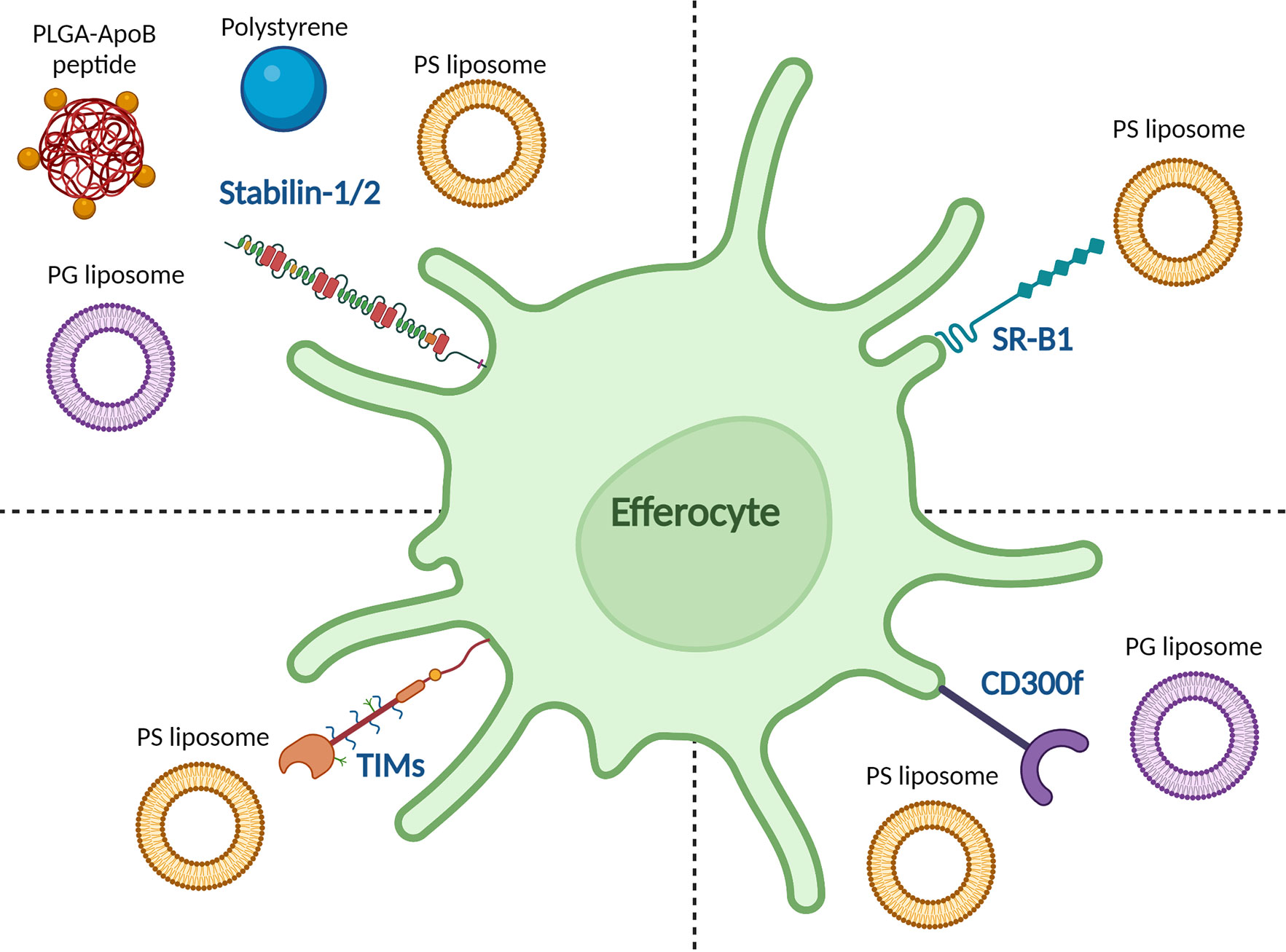
Figure 1 Reported binding of tolerance-inducing nanoparticles to efferocytic receptors on APCs. The stabilin-1/2 receptor was reported to bind to anionic polystyrene beads, apolipoprotein B (ApoB)-peptide-functionalized poly(lactic-co-glycolic acid) (PLGA) nanoparticles, and anionic phosphatidylserine (PS) and phosphatidylglycerol (PG) liposomes. The T cell/transmembrane, immunoglobulin, and mucin (TIM) receptor family recognize PS liposomes as does scavenger receptor class B type 1 (SR-B1). CD300f recognizes both PS and PG liposomes.
Splenic macrophages can also be targeted by negatively charged particles (47). PLGA particles coupled to encephalitogenic peptides were localized to the splenic marginal zone after i.v. injection and prevented and treated EAE in mice (84). i.v. injection of polystyrene beads coupled to zinc transporter 8- and islet-specific glucose-6-phosphatase catalytic subunit-related protein-derived peptides could induce antigen-specific tolerance in HLA-A2.1 transgenic mice (HHD) and prevent the development of diabetes in humanized NOD.β2m null HHD mice. The nanoparticles could also suppress antigen-specific CD8+ T cell responses in PBMCs from individuals with type 1 diabetes. Specifically, Siglec-1+ macrophages and marginal zone DCs produced CCL22 upon nanoparticle uptake, which mediated Foxp3+ Treg and CD103+ DC chemotaxis via CCR4 (85).
LSECs can also be targeted by negatively charged particles since stabilin receptors on LSECs preferentially bind negatively charged particles (86, 87). For instance, Carambia et al. designed anionic iron oxide nanoparticles with a poly(maleic anhydride-alt-1-octadecene)-coat. The particles were rapidly cleared from the plasma after i.v. injection and accumulated together with their antigen load in LSECs which led to an increase in Foxp3+ Tregs. The nanoparticles induced long-lasting protection to EAE and could even cure EAE in mice. The authors point out that in-depth analysis of the phenotype of nanoparticle-induced Tregs was lacking, as it was not possible to separate nanoparticle-induced Tregs from endogenously induced Tregs (88). Similarly, Saito et al. showed that PEMA-coated polylactide (PLA) nanoparticles accumulated in the liver and spleen after i.v. injection and were mainly associated with Kupffer cells and LSECs in the liver, which showed a reduction in CD86 expression. Ex vivo restimulation of splenocytes with the relevant antigen (PLP139-151) resulted in reduced production of IFNγ, IL-17, GM-CSF, TNFα, IL-2 and increased IL-10 in mice immunized with PLP nanoparticles as compared to mice injected with OVA nanoparticles. The PLP nanoparticles prevented EAE in mice (89).
The nanoparticle charge also influences targeting in mucosal tissues. Fromen et al. developed anionic and cationic rod-shaped nanoparticles of 80 nm x 320 nm in size, functionalized to ovalbumin. After delivery into the lungs of mice by orotracheal instillation, anionic nanoparticles were immunologically inert, while the cationic particles were pro-inflammatory, as demonstrated by higher gene expression of Ccl2, Il-10, Il-2, IL-6, Cxcl10, Ifnγ and Il-12β in homogenized lung cells. This was hypothesized to be because the anionic nanoparticles were taken up by alveolar macrophages, which maintain tissue homeostasis, while the cationic nanoparticles were taken up by lung DCs (90).
2.1.2 Other Physicochemical Properties
Apart from particle charge, the size of the nanoparticles can also determine their biodistribution and uptake by immune cells, as reviewed elsewhere (91). Specifically, concerning targeting tolerance-inducing cell subsets, it was shown that nanoparticles between 79 and 199 nm in size efficiently delivered siRNA to LSECs after i.v. injection, while 420 nm particles did not (92). Particle size is also important for mucosal tolerance induction; i.v. injection of 15 µm, but not 400 nm-sized PLGA nanoparticles led to the retention of these particles in the lungs. While both particles encapsulating PLP were able to prevent EAE development in a mouse model compared to untreated control, the larger particles were more effective (93). In addition, intratracheal administration of glycine-coated polystyrene nanoparticles of 50 nm in size, but not 500 nm were taken up by CD103+ DCs in the lungs (26, 94). Finally, 300 nm PLGA nanoparticles encapsulating type II collagen were found in the Peyer’s patches of mice after oral administration. The nanoparticles were able to significantly reduce arthritis scores in a collagen-induced arthritis mouse model. This coincided with reduced circulating anti-type II collagen IgG antibodies, reduced proliferation of draining lymph node lymphocytes after type II collagen restimulation, increased gene expression of Tgfβ in Peyer’s patches, and decreased gene expression of Tnfα in draining lymph nodes in mice immunized with antigen-loaded nanoparticles compared to placebo (55).
The rigidity of nanoparticles is gaining increasing attention as another important physicochemical parameter for tolerance induction (95). For instance, the FcγRIIB+ DCs involved in mucosal tolerance (65, 66) are sensitive to particle rigidity (96). Particle rigidity also influences whether circulating particles can reach sterically obscured cells (97) such as LSECs.
Interestingly, polyethylene glycol (PEG)ylation of PLGA nanoparticles can have profound effects on tolerance induction. Comparing PLGA-PEG to PLGA nanoparticles with identical size and charge, Li et al. show that PEGylation induced lower complement activation, neutrophil recruitment, and co-stimulatory molecule expression on DCs around the injection site after s.c. injection (98).
A unique challenge for the induction of mucosal tolerance is that many antigens are not mucoadhesive. Mucoadhesiveness of antigens is highly dependent on several factors such as hydrophilicity, molecular weight, charge, and chemical structure (99). Nanoparticles designed to be mucoadhesive can overcome this problem, and some of the properties that affect the mucoadhesion of formulations are charge, spreadability/rigidity, and ability to bind to the mucus substrate (100). For example, sublingual administration of ovalbumin adsorbed on mucoadhesive polymerized maltodextrin nanoparticles showed therapeutic tolerance in an ovalbumin-induced allergic mouse model, which was not observed with free ovalbumin. Unfortunately, cellular mechanisms were not reported in this study (101). Pulmonary administration of an EAE antigen with hyaluronic acid [a mucoadhesive (102)] abrogated EAE in mice (103), and peanut-induced anaphylaxis was only inhibited in mice receiving intranasal administration of a nanoemulsion with peanut extract, but not free extract (104). Finally, while only antigen-loaded PLGA nanoparticles increased Foxp3 gene expression in cervical lymph nodes and suppressed delayed-type hypersensitivity response in mice, intranasal administration of both antigen-loaded PLGA and PLGA-TMC (N-trimethyl chitosan, a mucoadhesive) nanoparticles suppressed proteoglycan-induced arthritis in an antigen-specific manner. The authors hypothesize that the discrepancy between the models could be explained by the chronic nature of the arthritis model compared to the delayed-type hypersensitivity model (105). Collectively, these studies show that the physicochemical properties of nanoparticles can be tuned to induce tolerance by targeting specific organs and/or cell subsets.
3 Active Targeting
Aside from targeting cell subsets via the physicochemical properties of nanoparticles, particles can be designed to actively target cells via the use of targeting moieties (106). The use of targeting antibodies for tolerance induction was extensively reviewed by Castenmiller et al. (28). Functionalization of PLGA nanoparticles with an ApoB peptide (a ligand for the stabilin-1 and stabilin-2 receptors expressed on LSECs) led to higher uptake by LSECs in vivo as compared to bare PLGA nanoparticles or mannan (another ligand for LSECs) nanoparticles. i.v. injection of the ApoB nanoparticles induced high TGF-β production by LSECs which coincided with an increase in Foxp3+ Tregs in the lungs. The nanoparticles could prevent and treat allergic symptoms in a pulmonary allergen sensitization model in mice (107). Another cell type, CD8+CD205+ DCs, can induce Foxp3+ Tregs from Foxp3- precursors in mice in the presence of a low dose of antigen (108). There is evidence that CD205 is important in the recognition of apoptotic cells (109). Targeting antigens to CD205+ cells in the spleen using anti-CD205 antibodies has shown to be effective at deletion of autoreactive CD8+ T cells (110), and at inducing tolerance in several autoimmune disease models (111, 112). Targeting Siglec-1 and other C-type lectin receptors can be achieved by glycosylated molecules (113), as reviewed elsewhere (114). Regarding mucosal tolerance, reports of specific targeting of antigens to CD103+ DCs using antibodies are divided amongst both pro-and anti-inflammatory responses, and co-administration of a pro-inflammatory adjuvant abrogates the tolerogenic effects of CD103+ DCs (115). Targeting FcγRIIB with specific antibodies has been used to induce tolerance (116, 117), but so far these targeting antibodies have not been combined with nanoparticles, although this approach would be promising. Using targeting moieties is an attractive strategy to target APC subsets, but care must be taken to avoid the production of anti-drug antibodies against the targeting ligand (118).
4 Immunomodulators
It may not always be necessary to target antigens to a specific APC subset. After systemic injection, APCs rapidly take up nanoparticles. If a nanoparticle encapsulates an antigen together with an immunomodulator, the immunomodulator can direct the APC towards a tolerogenic phenotype. Several studies demonstrate that co-encapsulation of an antigen with an immunomodulator (e.g., rapamycin, calcitriol, aryl hydrocarbon receptor ligands, or NF-κB inhibitors) can ameliorate autoimmunity, whilst free antigen does not (119–129). For example, s.c. injections of nanoparticles encapsulating MOG35-55 and dexamethasone significantly treated EAE in mice as compared to empty nanoparticles, dexamethasone nanoparticles, MOG35-55 nanoparticles, or free dexamethasone and MOG35-55. After ex vivo restimulation of splenocytes with MOG35-55 only spleens of mice that received the nanoparticles encapsulating MOG35-55 and dexamethasone had reduced IL-17 and GM-CSF production (130). In another study s.c. injection of retinoic acid/TGF-β/insulin peptide-encapsulating PLGA microparticles led to uptake by CD11c+ splenic DCs, and a significant increase in B220+CD19+CD1d+CD5+ Bregs, but not CD4+CD25+Foxp3+ Tregs in the mesenteric lymph nodes, as compared to control mice. These effects were acute (3 days after particle administration), and it is unknown how long-lasting they are. The particles could prevent diabetes in NOD mice, while the administration of free retinoic acid or TGF-β could not (131). There is also evidence that co-encapsulation is not always necessary. Lewis et al. prepared a mixture of distinct PLGA nano- and microparticles, namely vitamin D3 (1000 nm, phagocytosable), denatured human recombinant insulin (1000 nm), TGF-β1 (30 µm, non-phagocytosable), and GM-CSF (30 µm). When NOD mice were injected s.c. with the mix of particles, the phagocytosable particles were found in the paracortex of the draining lymph nodes and associated mainly with DCs. The particle-positive DCs expressed high PD-L1 and BTLA. In diabetic mice, the particle mixture enhanced Foxp3+ Tregs in the spleen and pancreatic lymph nodes and increased PD-1 on CD4+ and CD8+ T cells. Treatment with all particles administered s.c. significantly prevented and treated diabetes in NOD mice (132). All these studies demonstrate that the immunomodulator enables efficient tolerance induction even without active targeting specific organs or cellular subsets. This is underlined by the fact that these studies reported nanoparticle uptake by multiple subsets of DCs and macrophages in the spleen, lymph nodes, and liver, which gained a tolerogenic phenotype and were able to induce antigen-specific T cell tolerance.
While the current studies are very encouraging it is unclear whether immunomodulators that leak from formulations pose a safety concern by inducing non-specific immune suppression. Luo et al. proposed a nanoparticulate approach that could circumvent this problem. They prepared PLGA-PEG nanoparticles encapsulating the diabetes-specific peptide 2.5mi, together with a CRISPR-Cas9 plasmid and guide RNAs for CD80, CD86, and CD40. Upon i.v. administration, MHC-II+CD11c+ DCs in the lymph nodes, spleen, and blood took up these nanoparticles, and presented the peptide in the absence of costimulatory molecules, leading to an antigen-specific Foxp3+ Treg response. Only the complete formulation significantly reduced diabetes incidence in NOD/Ltj mice (133). In a similar but simpler approach, Krienke et al. made liposomes carrying mRNA coding for disease-relevant antigens that were specifically modified to suppress immune activation. The liposomes were taken up by different splenic CD11c+ DCs after i.v. injection. The DCs presented the antigen to T cells in the absence of costimulatory molecules which resulted in antigen-specific Foxp3+ Treg expansion and could prevent and treat EAE in mice. Specifically, MOG35-55–specific splenic CD4+ T cells from mice treated with the liposomes had high expression of inhibitory markers CD5, ICOS, LAG-3, PD-1, CTLA-4, TIGIT, and TIM-3 (134). These studies show that, if the nanoparticle is tolerance-inducing, either by the inclusion of immunomodulators or other methods, it can even skew pro-inflammatory APC subsets towards tolerance.
5 Translation to Human
While there are many promising pre-clinical studies with nanoparticles, translation of animal models to human patients is a difficult challenge. It is not fully understood why the translation often fails but can be due to multiple factors. For instance, the protein corona that forms around a nanoparticle after administration can differ between species, which may affect the stability, toxicity, and biodistribution of the nanoparticles (135, 136). Furthermore, there may be differences in phagocytosis of nanoparticles among species; comparing dogs, humans, and several strains of rats and mice, it was found that opsonization of dextran-coated iron oxide nanoparticles occurred mainly via the alternative complement pathway in humans, while in the other species this was dependent on Ca2+-sensitive pathways (137). Another study tested a wide range of lipid nanoparticles containing mRNA in mice with humanized livers, primatized livers, or “murinized” livers. It was found that mRNA delivery was more efficient to human hepatocytes and primate hepatocytes compared to murine hepatocytes and that there was a discrepancy between the most efficient lipid nanoparticles for delivery to murine vs. human hepatocytes, leading to false positives or negatives. Transcriptomic analysis revealed that in human hepatocytes clathrin-mediated endocytosis was increased while caveolin-mediated endocytosis was decreased after lipid nanoparticle administration, while in mice this was reversed (138). Finally, there are many different models for autoimmunity which have varying degrees of similarity to human patients, which can make the translation to humans difficult (6, 139). For translation from pre-clinical to clinical trials, humanized mice may be useful for studies with nanoparticles, and while some of the studies highlighted in this review use humanized mice (44, 85), this is not standard practice.
To further complicate translation, immune cell number, phenotype, and function can differ between healthy individuals and patients. For example, the DCs in the pancreatic lymph nodes of type I diabetes patients may have reduced tolerogenic function than those of healthy controls, and there were even differences in lymph node cell composition when correcting for sex and age (140). T cells, B cells, DCs, and other lymph node cells are also shown to be different in rheumatoid arthritis patients compared to healthy individuals (141–143). Furthermore, efferocytosis can be defective in patients with autoimmune diseases (144, 145) or obese individuals (146), so for these patients nanoparticles that aim to exploit efferocytosis may not be as effective. The tolerance-inducing strategy could be tailored towards each patient individually if the immune cell function of the patient was matched to the pathway by which the nanoparticles exert their immunomodulatory effects. Encouragingly, one study directly compared nanoparticles with a targeted approach (targeting to LSECs) to a general immune-suppressing approach (rapamycin nanoparticles) in a murine airway inflammation model, and found that both approaches similarly reduced antigen-specific allergic responses (147).
Despite all of these challenges, there have been several successful phase 1 and phase 2 clinical trials using antigen-carrying nanoparticles. Lutterotti et al. coupled myelin-derived peptides to apoptotic PBMCs derived from MS patients. In this phase 1 trial, the cells were well-tolerated, and at a dose >1 x 109 cells there was a decrease in T cell proliferation in response to restimulation with the peptides (148). Another phase 1 clinical trial used CD206-targeting mannosylated liposomes carrying myelin-derived peptides for the treatment of MS. The liposomes were well-tolerated, and patients showed decreased serum concentrations of IL-7, IL-2, CCL2, and CCL4, while TNFα increased (149, 150). Finally, PLGA nanoparticles encapsulating gluten protein were tested in phase 1 and phase 2a trials in patients with celiac disease. The nanoparticles were well-tolerated and significantly reduced antigen-specific IFNγ production. After oral gluten challenge, the patients that received the nanoparticles showed reduced percentages of circulating CD4+CD38+α4β7+, CD8+CD38+αEβ7+, and γδ+CD38+αEβ7+ T cells compared to patients that received placebo (151). These promising results show that there are good prospects for antigen-carrying nanoparticles to treat autoimmunity in human patients.
6 Discussion
Current treatments of autoimmune diseases are focused on the management of symptoms using non-antigen-specific anti-inflammatory drugs. These drugs require life-long adherence and can be expensive, especially in the case of the biologicals, while potentially causing severe side effects (152). So far there is no cure for such diseases. Therefore, there is an urgent need to develop antigen-specific treatments for patients, which can induce long-lasting immune tolerance without causing general immune suppression. In this review, we have highlighted different strategies in which nanoparticles can be used to induce antigen-specific tolerance to treat autoimmune diseases and summarized these in Table 1. The most commonly used nanoparticles for passive targeting of antigens to APCs are polymeric nanoparticles and liposomes. The most well-studied biodegradable polymeric particles are (PEGylated) PLGA nanoparticles. Several promising studies also use PEMA coating to enhance the efferocytic effects. In studies that prevent and/or treat autoimmunity, the polymeric nanoparticles are anionic and generally around 300 nm in size. For liposomal nanoparticles that carry antigens, again anionic charge, provided by e.g. PG or PS is necessary for tolerance induction. The liposomes further generally contain the zwitterionic helper lipid PC and may contain cholesterol. They can range in size from around 150 nm up to 700 nm. On the other hand, mRNA requires cationic lipids for complexation, so for mRNA delivery, cationic lipids such as DOTMA and DOTAP are commonly used.
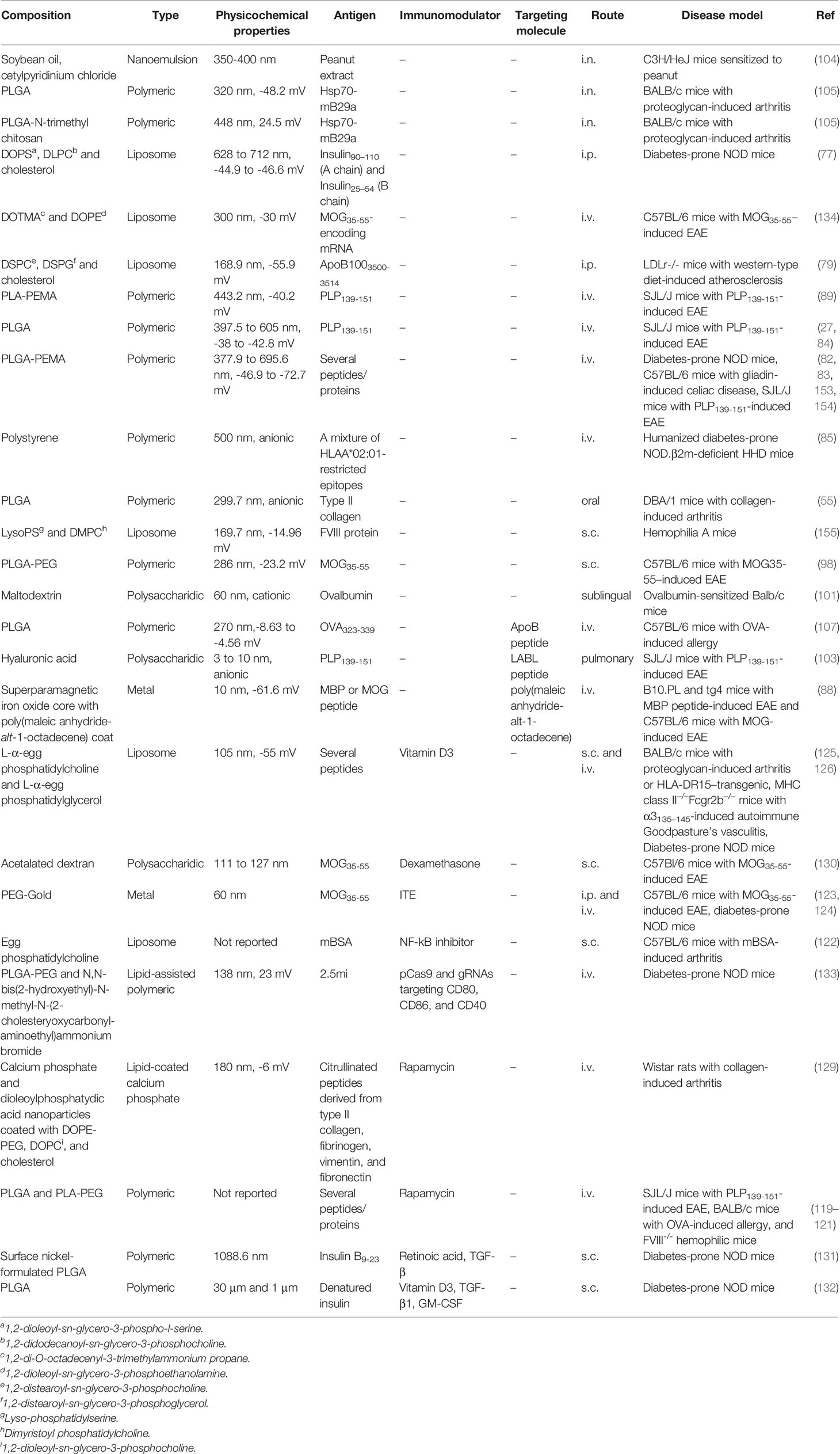
Table 1 Overview of antigen-carrying nanoparticles that resulted in immune suppression in autoimmune disease models.
Once a targeting molecule is included in the formulation, the physicochemical properties of the nanoparticles may be less important. Similarly, for nanoparticles that co-encapsulate antigens and immunomodulators, there is a wider range of materials and physicochemical properties that can be used, as targeting specific APC subsets is not the goal. Here, the main focus is on the stability of the particle and the successful encapsulation and retention of the cargo inside the particle.
There have been reports of tolerance induction using antigen-free nanoparticles (35, 78, 94), which can be unfavorable if it leads to general immune suppression. Furthermore, while the incorporation of immunomodulators into nanoparticles is effective, care should be taken that the immunomodulator does not leak from the nanoparticle before reaching the site of action. Therefore, nanoparticle immunotherapy should always be tested for non-antigen-specific effects, and in the case of co-encapsulation of an immunomodulator, in vivo stability of the immunomodulator should be evaluated. In the case of using targeting ligands on the surface of nanoparticles, there is a risk for the production of anti-drug antibodies against the targeting ligand, which can abrogate the targeting effects of the nanoparticle, or in severe cases even lead to an unwanted immune response (118). In this case, the prevention of the production of such antibodies e.g. by optimal dosing schemes needs to be taken into account (156).
Where reported in in vivo studies, we have outlined the biodistribution of nanoparticles, uptake by APC subsets, and immunological mechanisms for tolerance induction (summarized in Figure 2). However, not all studies have examined these aspects, and there is still much to be gained from research that describes well-characterized nanoparticles and their effects on the uptake by APC subsets and subsequent induced immune responses.
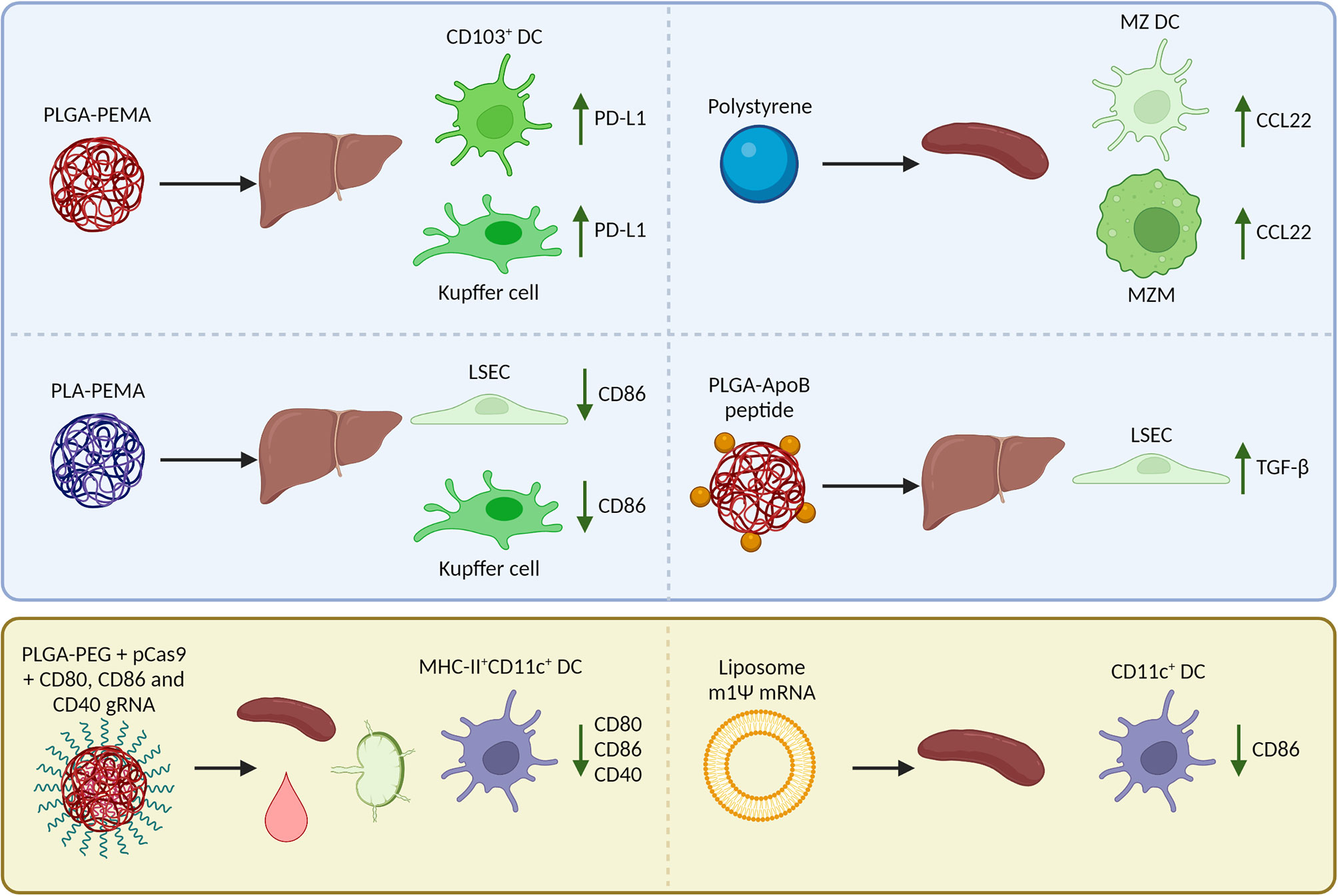
Figure 2 Reported biodistribution, cellular uptake, and Foxp3+ regulatory T cell (Treg)-inducing mechanisms in mice after i.v. injection of nanoparticles encapsulating an antigen (peptide/mRNA). The blue box (top) summarizes nanoparticles that target natural tolerogenic antigen-presenting cells in the liver and spleen including dendritic cells (DCs), macrophages, and liver sinusoidal endothelial cells (LSECs) to enhance tolerance. After uptake of nanoparticles, these cells upregulate inhibitory receptors, downregulate costimulatory molecules, produce chemokines that attract Tregs, or cytokines that induce Tregs. The yellow box (bottom) summarizes nanoparticles that are taken up by a wide range of APCs including DCs in the spleen, lymph nodes, and blood. The nanoparticles are designed to inhibit co-stimulation in target cells so that the antigen can be presented in a tolerogenic manner and induce Tregs. PLGA, poly (lactic-co-glycolic acid); PLA, polylactic acid; PEMA, poly(ethylene-co-maleic acid); ApoB, apolipoprotein B; PEG, polyethylene glycol; MZ, marginal zone; m1Ψ mRNA, 1 methylpseudouridine-modified messenger RNA; PD-L1, programmed death-ligand 1; CCL22, C-C motif chemokine 22; TGF-β, Transforming growth factor β.
Author Contributions
The manuscript was written by NB with extensive input and editing by all other authors. All authors contributed to the article and approved the submitted version.
Funding
This work was funded in part by the DC4Balance collaboration project. which is co-funded by the PPP Allowance made available by Health~Holland, Top Sector Life Sciences and Health, to the Dutch Cooperation of Health Foundations (SGF) and the Dutch Arthritis Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chaplin DD. Overview of the Immune Response. J Allergy Clin Immunol (2010) 125(2 Suppl 2):S3–23. doi: 10.1016/j.jaci.2009.12.980
2. Pozsgay J, Szekanecz Z, Sarmay G. Antigen-Specific Immunotherapies in Rheumatic Diseases. Nat Rev Rheumatol (2017) 13(9):525–37. doi: 10.1038/nrrheum.2017.107
3. Avdeeva A, Rubtsov Y, Dyikanov D, Popkova T, Nasonov E. Regulatory T Cells in Patients With Early Untreated Rheumatoid Arthritis: Phenotypic Changes in the Course of Methotrexate Treatment. Biochimie (2020) 174:9–17. doi: 10.1016/j.biochi.2020.03.014
4. Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, et al. Thymic Selection of CD4+CD25+ Regulatory T Cells Induced by an Agonist Self-Peptide. Nat Immunol (2001) 2(4):301–6. doi: 10.1038/86302
5. Schmitt EG, Williams CB. Generation and Function of Induced Regulatory T Cells. Front Immunol (2013) 4:152. doi: 10.3389/fimmu.2013.00152
6. Hahn SA, Bellinghausen I, Trinschek B, Becker C. Translating Treg Therapy in Humanized Mice. Front Immunol (2015) 6:623. doi: 10.3389/fimmu.2015.00623
7. Ohkura N, Sakaguchi S. Transcriptional and Epigenetic Basis of Treg Cell Development and Function: Its Genetic Anomalies or Variations in Autoimmune Diseases. Cell Res (2020) 30(6):465–74. doi: 10.1038/s41422-020-0324-7
8. Shevyrev D, Tereshchenko V. Treg Heterogeneity, Function, and Homeostasis. Front Immunol (2019) 10:3100. doi: 10.3389/fimmu.2019.03100
9. Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, et al. Coexpression of CD49b and LAG-3 Identifies Human and Mouse T Regulatory Type 1 Cells. Nat Med (2013) 19(6):739–46. doi: 10.1038/nm.3179
10. Carrier Y, Yuan J, Kuchroo VK, Weiner HL. Th3 Cells in Peripheral Tolerance. I. Induction of Foxp3-Positive Regulatory T Cells by Th3 Cells Derived From TGF-Beta T Cell-Transgenic Mice. J Immunol (2007) 178(1):179–85. doi: 10.4049/jimmunol.178.1.179
11. Ran Z, Yue-Bei L, Qiu-Ming Z, Huan Y. Regulatory B Cells and Its Role in Central Nervous System Inflammatory Demyelinating Diseases. Front Immunol (2020) 11:1884. doi: 10.3389/fimmu.2020.01884
12. Catalan D, Mansilla MA, Ferrier A, Soto L, Oleinika K, Aguillon JC, et al. Immunosuppressive Mechanisms of Regulatory B Cells. Front Immunol (2021) 12:611795. doi: 10.3389/fimmu.2021.611795
13. McInnes IB, Schett G. The Pathogenesis of Rheumatoid Arthritis. N Engl J Med (2011) 365(23):2205–19. doi: 10.1056/NEJMra1004965
14. Schinnerling K, Aguillon JC, Catalan D, Soto L. The Role of Interleukin-6 Signalling and Its Therapeutic Blockage in Skewing the T Cell Balance in Rheumatoid Arthritis. Clin Exp Immunol (2017) 189(1):12–20. doi: 10.1111/cei.12966
15. Kerschbaumer A, Sepriano A, Smolen JS, van der Heijde D, Dougados M, van Vollenhoven R, et al. Efficacy of Pharmacological Treatment in Rheumatoid Arthritis: A Systematic Literature Research Informing the 2019 Update of the EULAR Recommendations for Management of Rheumatoid Arthritis. Ann Rheum Dis (2020) 79(6):744–59. doi: 10.1136/annrheumdis-2019-216656
16. Wang W, Zhou H, Liu L. Side Effects of Methotrexate Therapy for Rheumatoid Arthritis: A Systematic Review. Eur J Med Chem (2018) 158:502–16. doi: 10.1016/j.ejmech.2018.09.027
17. Fang G, Zhang Q, Pang Y, Thu HE, Hussain Z. Nanomedicines for Improved Targetability to Inflamed Synovium for Treatment of Rheumatoid Arthritis: Multi-Functionalization as an Emerging Strategy to Optimize Therapeutic Efficacy. J Control Release (2019) 303:181–208. doi: 10.1016/j.jconrel.2019.04.027
18. Keijzer C, van der Zee R, van Eden W, Broere F. Treg Inducing Adjuvants for Therapeutic Vaccination Against Chronic Inflammatory Diseases. Front Immunol (2013) 4:245. doi: 10.3389/fimmu.2013.00245
19. Griffin JD, Song JY, Sestak JO, DeKosky BJ, Berkland CJ. Linking Autoantigen Properties to Mechanisms of Immunity. Adv Drug Deliv Rev (2020) 165-166:105–16. doi: 10.1016/j.addr.2020.04.005
20. Umeshappa CS, Singha S, Blanco J, Shao K, Nanjundappa RH, Yamanouchi J, et al. Suppression of a Broad Spectrum of Liver Autoimmune Pathologies by Single Peptide-MHC-Based Nanomedicines. Nat Commun (2019) 10(1):2150. doi: 10.1038/s41467-019-09893-5
21. Serra P, Santamaria P. Peptide-MHC-Based Nanomedicines for the Treatment of Autoimmunity: Engineering, Mechanisms, and Diseases. Front Immunol (2020) 11:621774. doi: 10.3389/fimmu.2020.621774
22. Smith DM, Simon JK, Baker JR Jr. Applications of Nanotechnology for Immunology. Nat Rev Immunol (2013) 13(8):592–605. doi: 10.1038/nri3488
23. Tibbitt MW, Dahlman JE, Langer R. Emerging Frontiers in Drug Delivery. J Am Chem Soc (2016) 138(3):704–17. doi: 10.1021/jacs.5b09974
24. Benne N, van Duijn J, Kuiper J, Jiskoot W, Slütter N. Orchestrating Immune Responses: How Size, Shape and Rigidity Affect the Immunogenicity of Particulate Vaccines. J Control Release (2016) 234:124–34. doi: 10.1016/j.jconrel.2016.05.033
25. Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, Bachmann MF. Nanoparticles Target Distinct Dendritic Cell Populations According to Their Size. Eur J Immunol (2008) 38(5):1404–13. doi: 10.1002/eji.200737984
26. Hardy CL, Lemasurier JS, Mohamud R, Yao J, Xiang SD, Rolland JM, et al. Differential Uptake of Nanoparticles and Microparticles by Pulmonary APC Subsets Induces Discrete Immunological Imprints. J Immunol (2013) 191(10):5278–90. doi: 10.4049/jimmunol.1203131
27. Pearson RM, Casey LM, Hughes KR, Wang LZ, North MG, Getts DR, et al. Controlled Delivery of Single or Multiple Antigens in Tolerogenic Nanoparticles Using Peptide-Polymer Bioconjugates. Mol Ther (2017) 25(7):1655–64. doi: 10.1016/j.ymthe.2017.04.015
28. Castenmiller C, Keumatio-Doungtsop BC, van Ree R, de Jong EC, van Kooyk Y. Tolerogenic Immunotherapy: Targeting DC Surface Receptors to Induce Antigen-Specific Tolerance. Front Immunol (2021) 12(422):643240. doi: 10.3389/fimmu.2021.643240
29. Rao JP, Geckeler KE. Polymer Nanoparticles: Preparation Techniques and Size-Control Parameters. Prog Polymer Sci (2011) 36(7):887–913. doi: 10.1016/j.progpolymsci.2011.01.001
30. Kelly KL, Coronado E, Zhao LL, Schatz GC. The Optical Properties of Metal Nanoparticles: The Influence of Size, Shape, and Dielectric Environment. J Phys Chem B (2003) 107(3):668–77. doi: 10.1021/jp026731y
31. Pattni BS, Chupin VV, Torchilin VP. New Developments in Liposomal Drug Delivery. Chem Rev (2015) 115(19):10938–66. doi: 10.1021/acs.chemrev.5b00046
32. Hawkins MJ, Soon-Shiong P, Desai N. Protein Nanoparticles as Drug Carriers in Clinical Medicine. Adv Drug Deliv Rev (2008) 60(8):876–85. doi: 10.1016/j.addr.2007.08.044
33. Song W, Musetti SN, Huang L. Nanomaterials for Cancer Immunotherapy. Biomaterials (2017) 148:16–30. doi: 10.1016/j.biomaterials.2017.09.017
34. Tiegs G, Lohse AW. Immune Tolerance: What Is Unique About the Liver. J Autoimmun (2010) 34(1):1–6. doi: 10.1016/j.jaut.2009.08.008
35. Hosseini H, Li H, Kanellakis P, Tay C, Cao A, Tipping P, et al. Phosphatidylserine Liposomes Mimic Apoptotic Cells to Attenuate Atherosclerosis by Expanding Polyreactive IgM Producing B1a Lymphocytes. Cardiovasc Res (2015) 106(3):443–52. doi: 10.1093/cvr/cvv037
36. Abdolmaleki F, Farahani N, Gheibi Hayat SM, Pirro M, Bianconi V, Barreto GE, et al. The Role of Efferocytosis in Autoimmune Diseases. Front Immunol (2018) 9(1645):1645. doi: 10.3389/fimmu.2018.01645
37. Doran AC, Yurdagul A Jr, Tabas I. Efferocytosis in Health and Disease. Nat Rev Immunol (2020) 20(4):254–67. doi: 10.1038/s41577-019-0240-6
38. Parcina M, Schiller M, Gierschke A, Heeg K, Bekeredjian-Ding I. PDC Expressing CD36, CD61 and IL-10 may Contribute to Propagation of Immune Tolerance. Autoimmunity (2009) 42(4):353–5. doi: 10.1080/08916930902831969
39. Pujol-Autonell I, Ampudia RM, Planas R, Marin-Gallen S, Carrascal J, Sanchez A, et al. Efferocytosis Promotes Suppressive Effects on Dendritic Cells Through Prostaglandin E2 Production in the Context of Autoimmunity. PloS One (2013) 8(5):e63296. doi: 10.1371/journal.pone.0063296
40. Ip WK, Lau YL. Distinct Maturation of, But Not Migration Between, Human Monocyte-Derived Dendritic Cells Upon Ingestion of Apoptotic Cells of Early or Late Phases. J Immunol (2004) 173(1):189–96. doi: 10.4049/jimmunol.173.1.189
41. Ramirez-Ortiz ZG, Pendergraft WF 3rd, Prasad A, Byrne MH, Iram T, Blanchette CJ, et al. The Scavenger Receptor SCARF1 Mediates the Clearance of Apoptotic Cells and Prevents Autoimmunity. Nat Immunol (2013) 14(9):917–26. doi: 10.1038/ni.2670
42. Waterborg CEJ, Beermann S, Broeren MGA, Bennink MB, Koenders MI, van Lent P, et al. Protective Role of the MER Tyrosine Kinase via Efferocytosis in Rheumatoid Arthritis Models. Front Immunol (2018) 9:742. doi: 10.3389/fimmu.2018.00742
43. Watkins EA, Antane JT, Roberts JL, Lorentz KM, Zuerndorfer S, Dunaif AC, et al. Persistent Antigen Exposure via the Eryptotic Pathway Drives Terminal T Cell Dysfunction. Sci Immunol (2021) 6(56):eabe1801. doi: 10.1126/sciimmunol.abe1801
44. Pilon C, Stehle T, Beldi-Ferchiou A, Matignon M, Thiolat A, Burlion A, et al. Human Apoptotic Cells, Generated by Extracorporeal Photopheresis, Modulate Allogeneic Immune Response. Front Immunol (2019) 10(2908):2908. doi: 10.3389/fimmu.2019.02908
45. Miyake Y, Asano K, Kaise H, Uemura M, Nakayama M, Tanaka M. Critical Role of Macrophages in the Marginal Zone in the Suppression of Immune Responses to Apoptotic Cell-Associated Antigens. J Clin Invest (2007) 117(8):2268–78. doi: 10.1172/JCI31990
46. Bronte V, Pittet MJ. The Spleen in Local and Systemic Regulation of Immunity. Immunity (2013) 39(5):806–18. doi: 10.1016/j.immuni.2013.10.010
47. McGaha TL, Chen Y, Ravishankar B, van Rooijen N, Karlsson MC. Marginal Zone Macrophages Suppress Innate and Adaptive Immunity to Apoptotic Cells in the Spleen. Blood (2011) 117(20):5403–12. doi: 10.1182/blood-2010-11-320028
48. Ravishankar B, Shinde R, Liu H, Chaudhary K, Bradley J, Lemos HP, et al. Marginal Zone CD169+ Macrophages Coordinate Apoptotic Cell-Driven Cellular Recruitment and Tolerance. Proc Natl Acad Sci USA (2014) 111(11):4215–20. doi: 10.1073/pnas.1320924111
49. van Dinther D, Veninga H, Iborra S, Borg EGF, Hoogterp L, Olesek K, et al. Functional CD169 on Macrophages Mediates Interaction With Dendritic Cells for CD8(+) T Cell Cross-Priming. Cell Rep (2018) 22(6):1484–95. doi: 10.1016/j.celrep.2018.01.021
50. Qiu CH, Miyake Y, Kaise H, Kitamura H, Ohara O, Tanaka M. Novel Subset of CD8α+ Dendritic Cells Localized in the Marginal Zone Is Responsible for Tolerance to Cell-Associated Antigens. J Immunol (2009) 182(7):4127–36. doi: 10.4049/jimmunol.0803364
51. Xia S, Guo Z, Xu X, Yi H, Wang Q, Cao X. Hepatic Microenvironment Programs Hematopoietic Progenitor Differentiation Into Regulatory Dendritic Cells, Maintaining Liver Tolerance. Blood (2008) 112(8):3175–85. doi: 10.1182/blood-2008-05-159921
52. Bamboat ZM, Stableford JA, Plitas G, Burt BM, Nguyen HM, Welles AP, et al. Human Liver Dendritic Cells Promote T Cell Hyporesponsiveness. J Immunol (2009) 182(4):1901–11. doi: 10.4049/jimmunol.0803404
53. Xu X, Jin R, Li M, Wang K, Zhang S, Hao J, et al. Liver Sinusoidal Endothelial Cells Induce Tolerance of Autoreactive CD4+ Recent Thymic Emigrants. Sci Rep (2016) 6(1):19861. doi: 10.1038/srep19861
54. You Q, Cheng L, Kedl RM, Ju C. Mechanism of T Cell Tolerance Induction by Murine Hepatic Kupffer Cells. Hepatology (2008) 48(3):978–90. doi: 10.1002/hep.22395
55. Kim WU, Lee WK, Ryoo JW, Kim SH, Kim J, Youn J, et al. Suppression of Collagen-Induced Arthritis by Single Administration of Poly(Lactic-Co-Glycolic Acid) Nanoparticles Entrapping Type II Collagen: A Novel Treatment Strategy for Induction of Oral Tolerance. Arthritis Rheum (2002) 46(4):1109–20. doi: 10.1002/art.10198
56. Trentham DE, Dynesius-Trentham RA, Orav EJ, Combitchi D, Lorenzo C, Sewell KL, et al. Effects of Oral Administration of Type II Collagen on Rheumatoid Arthritis. Science (1993) 261(5129):1727–30. doi: 10.1126/science.8378772
57. Buettner M, Bornemann M, Bode U. Skin Tolerance Is Supported by the Spleen. Scand J Immunol (2013) 77(4):238–45. doi: 10.1111/sji.12034
58. Hammerschmidt SI, Ahrendt M, Bode U, Wahl B, Kremmer E, Forster R, et al. Stromal Mesenteric Lymph Node Cells Are Essential for the Generation of Gut-Homing T Cells In Vivo. J Exp Med (2008) 205(11):2483–90. doi: 10.1084/jem.20080039
59. Wolvers DAW, Coenen-de Roo CJJ, Mebius RE, van der Cammen MJF, Tirion F, Miltenburg AMM, et al. Intranasally Induced Immunological Tolerance Is Determined by Characteristics of the Draining Lymph Nodes: Studies With OVA and Human Cartilage Gp-39. J Immunol (1999) 162(4):1994–8.
60. Min SY, Park KS, Cho ML, Kang JW, Cho YG, Hwang SY, et al. Antigen-Induced, Tolerogenic CD11c+,CD11b+ Dendritic Cells Are Abundant in Peyer’s Patches During the Induction of Oral Tolerance to Type II Collagen and Suppress Experimental Collagen-Induced Arthritis. Arthritis Rheum (2006) 54(3):887–98. doi: 10.1002/art.21647
61. Suzuki T, Chow CW, Downey GP. Role of Innate Immune Cells and Their Products in Lung Immunopathology. Int J Biochem Cell Biol (2008) 40(6-7):1348–61. doi: 10.1016/j.biocel.2008.01.003
62. Takaki H, Ichimiya S, Matsumoto M, Seya T. Mucosal Immune Response in Nasal-Associated Lymphoid Tissue Upon Intranasal Administration by Adjuvants. J Innate Immun (2018) 10(5-6):515–21. doi: 10.1159/000489405
63. Wawrzyniak M, O’Mahony L, Akdis M. Role of Regulatory Cells in Oral Tolerance. Allergy Asthma Immunol Res (2017) 9(2):107–15. doi: 10.4168/aair.2017.9.2.107
64. Fujihashi K, Dohi T, Rennert PD, Yamamoto M, Koga T, Kiyono H, et al. Peyer’s Patches Are Required for Oral Tolerance to Proteins. Proc Natl Acad Sci USA (2001) 98(6):3310–5. doi: 10.1073/pnas.061412598
65. Samsom JN, van Berkel LA, van Helvoort JM, Unger WW, Jansen W, Thepen T, et al. Fc Gamma RIIB Regulates Nasal and Oral Tolerance: A Role for Dendritic Cells. J Immunol (2005) 174(9):5279–87. doi: 10.4049/jimmunol.174.9.5279
66. van Montfoort N, t Hoen PA, Mangsbo SM, Camps MG, Boross P, Melief CJ, et al. Fcgamma Receptor IIb Strongly Regulates Fcgamma Receptor-Facilitated T Cell Activation by Dendritic Cells. J Immunol (2012) 189(1):92–101. doi: 10.4049/jimmunol.1103703
67. Guilliams M, Bruhns P, Saeys Y, Hammad H, Lambrecht BN. The Function of Fcgamma Receptors in Dendritic Cells and Macrophages. Nat Rev Immunol (2014) 14(2):94–108. doi: 10.1038/nri3582
68. McGaha TL, Karlsson MC, Ravetch JV. FcgammaRIIB Deficiency Leads to Autoimmunity and a Defective Response to Apoptosis in Mrl-MpJ Mice. J Immunol (2008) 180(8):5670–9. doi: 10.4049/jimmunol.180.8.5670
69. Khare A, Krishnamoorthy N, Oriss TB, Fei M, Ray P, Ray A. Cutting Edge: Inhaled Antigen Upregulates Retinaldehyde Dehydrogenase in Lung CD103+ But Not Plasmacytoid Dendritic Cells to Induce Foxp3 De Novo in CD4+ T Cells and Promote Airway Tolerance. J Immunol (2013) 191(1):25–9. doi: 10.4049/jimmunol.1300193
70. Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, et al. A Functionally Specialized Population of Mucosal CD103+ DCs Induces Foxp3+ Regulatory T Cells via a TGF-Beta and Retinoic Acid-Dependent Mechanism. J Exp Med (2007) 204(8):1757–64. doi: 10.1084/jem.20070590
71. Huynh M-LN, Fadok VA, Henson PM. Phosphatidylserine-Dependent Ingestion of Apoptotic Cells Promotes TGF-β1 Secretion and the Resolution of Inflammation. J Clin Invest (2002) 109(1):41–50. doi: 10.1172/JCI0211638
72. Park SY, Jung MY, Kim HJ, Lee SJ, Kim SY, Lee BH, et al. Rapid Cell Corpse Clearance by Stabilin-2, a Membrane Phosphatidylserine Receptor. Cell Death Differ (2008) 15(1):192–201. doi: 10.1038/sj.cdd.4402242
73. Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a Phosphatidylserine Receptor. Nature (2007) 450(7168):435–9. doi: 10.1038/nature06307
74. Choi SC, Simhadri VR, Tian L, Gil-Krzewska A, Krzewski K, Borrego F, et al. Cutting Edge: Mouse CD300f (CMRF-35-Like Molecule-1) Recognizes Outer Membrane-Exposed Phosphatidylserine and Can Promote Phagocytosis. J Immunol (2011) 187(7):3483–7. doi: 10.4049/jimmunol.1101549
75. Nagata S, Hanayama R, Kawane K. Autoimmunity and the Clearance of Dead Cells. Cell (2010) 140(5):619–30. doi: 10.1016/j.cell.2010.02.014
76. Rodriguez-Fernandez S, Pujol-Autonell I, Brianso F, Perna-Barrull D, Cano-Sarabia M, Garcia-Jimeno S, et al. Phosphatidylserine-Liposomes Promote Tolerogenic Features on Dendritic Cells in Human Type 1 Diabetes by Apoptotic Mimicry. Front Immunol (2018) 9:253. doi: 10.3389/fimmu.2018.00253
77. Villalba A, Rodriguez-Fernandez S, Ampudia RM, Cano-Sarabia M, Perna-Barrull D, Bertran-Cobo C, et al. Preclinical Evaluation of Antigen-Specific Nanotherapy Based on Phosphatidylserine-Liposomes for Type 1 Diabetes. Artif Cells Nanomed Biotechnol (2020) 48(1):77–83. doi: 10.1080/21691401.2019.1699812
78. Ramos GC, Fernandes D, Charao CT, Souza DG, Teixeira MM, Assreuy J. Apoptotic Mimicry: Phosphatidylserine Liposomes Reduce Inflammation Through Activation of Peroxisome Proliferator-Activated Receptors (PPARs) In Vivo. Br J Pharmacol (2007) 151(6):844–50. doi: 10.1038/sj.bjp.0707302
79. Benne N, van Duijn J, Lozano Vigario F, Leboux RJT, van Veelen P, Kuiper J, et al. Anionic 1,2-Distearoyl-Sn-Glycero-3-Phosphoglycerol (DSPG) Liposomes Induce Antigen-Specific Regulatory T Cells and Prevent Atherosclerosis in Mice. J Control Release (2018) 291:135–46. doi: 10.1016/j.jconrel.2018.10.028
80. Clarke EV, Weist BM, Walsh CM, Tenner AJ. Complement Protein C1q Bound to Apoptotic Cells Suppresses Human Macrophage and Dendritic Cell-Mediated Th17 and Th1 T Cell Subset Proliferation. J Leukoc Biol (2015) 97(1):147–60. doi: 10.1189/jlb.3A0614-278R
81. Wicker-Planquart C, Dufour S, Tacnet-Delorme P, Bally I, Delneste Y, Frachet P, et al. Molecular and Cellular Interactions of Scavenger Receptor SR-F1 With Complement C1q Provide Insights Into Its Role in the Clearance of Apoptotic Cells. Front Immunol (2020) 11:544. doi: 10.3389/fimmu.2020.00544
82. McCarthy DP, Yap JW, Harp CT, Song WK, Chen J, Pearson RM, et al. An Antigen-Encapsulating Nanoparticle Platform for TH1/17 Immune Tolerance Therapy. Nanomedicine (2017) 13(1):191–200. doi: 10.1016/j.nano.2016.09.007
83. Hunter Z, McCarthy DP, Yap WT, Harp CT, Getts DR, Shea LD, et al. A Biodegradable Nanoparticle Platform for the Induction of Antigen-Specific Immune Tolerance for Treatment of Autoimmune Disease. ACS Nano (2014) 8(3):2148–60. doi: 10.1021/nn405033r
84. Getts DR, Martin AJ, McCarthy DP, Terry RL, Hunter ZN, Yap WT, et al. Microparticles Bearing Encephalitogenic Peptides Induce T-Cell Tolerance and Ameliorate Experimental Autoimmune Encephalomyelitis. Nat Biotechnol (2012) 30(12):1217–24. doi: 10.1038/nbt.2434
85. Xu X, Bian L, Shen M, Li X, Zhu J, Chen S, et al. Multipeptide-Coupled Nanoparticles Induce Tolerance in ‘Humanised’ HLA-Transgenic Mice and Inhibit Diabetogenic CD8(+) T Cell Responses in Type 1 Diabetes. Diabetologia (2017) 60(12):2418–31. doi: 10.1007/s00125-017-4419-8
86. Campbell F, Bos FL, Sieber S, Arias-Alpizar G, Koch BE, Huwyler J, et al. Directing Nanoparticle Biodistribution Through Evasion and Exploitation of Stab2-Dependent Nanoparticle Uptake. ACS Nano (2018) 12(3):2138–50. doi: 10.1021/acsnano.7b06995
87. Arias-Alpizar G, Koch B, Hamelmann NM, Neustrup MA, Paulusse JMJ, Jiskoot W, et al. Stabilin-1 Is Required for the Endothelial Clearance of Small Anionic Nanoparticles. Nanomedicine (2021) 34:102395. doi: 10.1016/j.nano.2021.102395
88. Carambia A, Freund V, Schwinge D, Bruns OT, Salmen SC, Ittrich H, et al. Nanoparticle-Based Autoantigen Delivery to Treg-Inducing Liver Sinusoidal Endothelial Cells Enables Control of Autoimmunity in Mice. J Hepatol (2015) 62(6):1349–56. doi: 10.1016/j.jhep.2015.01.006
89. Saito E, Kuo R, Kramer KR, Gohel N, Giles DA, Moore BB, et al. Design of Biodegradable Nanoparticles to Modulate Phenotypes of Antigen-Presenting Cells for Antigen-Specific Treatment of Autoimmune Disease. Biomaterials (2019) 222:119432. doi: 10.1016/j.biomaterials.2019.119432
90. Fromen CA, Rahhal TB, Robbins GR, Kai MP, Shen TW, Luft JC, et al. Nanoparticle Surface Charge Impacts Distribution, Uptake and Lymph Node Trafficking by Pulmonary Antigen-Presenting Cells. Nanomedicine (2016) 12(3):677–87. doi: 10.1016/j.nano.2015.11.002
91. Hoshyar N, Gray S, Han H, Bao G. The Effect of Nanoparticle Size on In Vivo Pharmacokinetics and Cellular Interaction. Nanomed (Lond) (2016) 11(6):673–92. doi: 10.2217/nnm.16.5
92. Sato Y, Hatakeyama H, Hyodo M, Harashima H. Relationship Between the Physicochemical Properties of Lipid Nanoparticles and the Quality of siRNA Delivery to Liver Cells. Mol Ther (2016) 24(4):788–95. doi: 10.1038/mt.2015.222
93. Saito E, Gurczynski SJ, Kramer KR, Wilke CA, Miller SD, Moore BB, et al. Modulating Lung Immune Cells by Pulmonary Delivery of Antigen-Specific Nanoparticles to Treat Autoimmune Disease. Sci Adv (2020) 6(42):9317–33. doi: 10.1126/sciadv.abc9317
94. Mohamud R, LeMasurier JS, Boer JC, Sieow JL, Rolland JM, O'Hehir RE, et al. Synthetic Nanoparticles That Promote Tumor Necrosis Factor Receptor 2 Expressing Regulatory T Cells in the Lung and Resistance to Allergic Airways Inflammation. Front Immunol (2017) 8:1812. doi: 10.3389/fimmu.2017.01812
95. Benne N, Leboux RJT, Glandrup M, van Duijn J, Lozano Vigario F, Neustrup MA, et al. Atomic Force Microscopy Measurements of Anionic Liposomes Reveal the Effect of Liposomal Rigidity on Antigen-Specific Regulatory T Cell Responses. J Control Release (2020) 318:246–55. doi: 10.1016/j.jconrel.2019.12.003
96. Beningo KA, Wang YL. Fc-Receptor-Mediated Phagocytosis Is Regulated by Mechanical Properties of the Target. J Cell Sci (2002) 115(4):849–56. doi: 10.1242/jcs.115.4.849
97. Myerson JW, Braender B, McPherson O, Glassman PM, Kiseleva RY, Shuvaev VV, et al. Flexible Nanoparticles Reach Sterically Obscured Endothelial Targets Inaccessible to Rigid Nanoparticles. Adv Mater (2018) 30(32):e1802373. doi: 10.1002/adma.201802373
98. Li PY, Bearoff F, Zhu P, Fan Z, Zhu Y, Fan M, et al. PEGylation Enables Subcutaneously Administered Nanoparticles to Induce Antigen-Specific Immune Tolerance. J Control Release (2021) 331:164–75. doi: 10.1016/j.jconrel.2021.01.013
99. Shaikh R, Raj Singh TR, Garland MJ, Woolfson AD, Donnelly RF. Mucoadhesive Drug Delivery Systems. J Pharm Bioallied Sci (2011) 3(1):89–100. doi: 10.4103/0975-7406.76478
100. Boddupalli BM, Mohammed ZN, Nath RA, Banji D. Mucoadhesive Drug Delivery System: An Overview. J Adv Pharm Technol Res (2010) 1(4):381–7. doi: 10.4103/0110-5558.76436
101. Razafindratsita A, Saint-Lu N, Mascarell L, Berjont N, Bardon T, Betbeder D, et al. Improvement of Sublingual Immunotherapy Efficacy With a Mucoadhesive Allergen Formulation. J Allergy Clin Immunol (2007) 120(2):278–85. doi: 10.1016/j.jaci.2007.04.009
102. Sandri G, Rossi S, Ferrari F, Bonferoni MC, Zerrouk N, Caramella C. Mucoadhesive and Penetration Enhancement Properties of Three Grades of Hyaluronic Acid Using Porcine Buccal and Vaginal Tissue, Caco-2 Cell Lines, and Rat Jejunum. J Pharm Pharmacol (2004) 56(9):1083–90. doi: 10.1211/0022357044085
103. Thati S, Kuehl C, Hartwell B, Sestak J, Siahaan T, Forrest ML, et al. Routes of Administration and Dose Optimization of Soluble Antigen Arrays in Mice With Experimental Autoimmune Encephalomyelitis. J Pharm Sci (2015) 104(2):714–21. doi: 10.1002/jps.24272
104. O’Konek JJ, Landers JJ, Janczak KW, Goel RR, Mondrusov AM, Wong PT, et al. Nanoemulsion Adjuvant-Driven Redirection of TH2 Immunity Inhibits Allergic Reactions in Murine Models of Peanut Allergy. J Allergy Clin Immunol (2018) 141(6):2121–31. doi: 10.1016/j.jaci.2018.01.042
105. Keijzer C, Slutter B, van der Zee R, Jiskoot W, van Eden W, Broere F. PLGA, PLGA-TMC and TMC-TPP Nanoparticles Differentially Modulate the Outcome of Nasal Vaccination by Inducing Tolerance or Enhancing Humoral Immunity. PloS One (2011) 6(11):e26684. doi: 10.1371/journal.pone.0026684
106. Bottger R, Pauli G, Chao PH, Al Fayez N, Hohenwarter L, Li SD. Lipid-Based Nanoparticle Technologies for Liver Targeting. Adv Drug Deliv Rev (2020) 154-155:79–101. doi: 10.1016/j.addr.2020.06.017
107. Liu Q, Wang X, Liu X, Kumar S, Gochman G, Ji Y, et al. Use of Polymeric Nanoparticle Platform Targeting the Liver To Induce Treg-Mediated Antigen-Specific Immune Tolerance in a Pulmonary Allergen Sensitization Model. ACS Nano (2019) 13(4):4778–94. doi: 10.1021/acsnano.9b01444
108. Yamazaki S, Dudziak D, Heidkamp GF, Fiorese C, Bonito AJ, Inaba K, et al. CD8+ CD205+ Splenic Dendritic Cells Are Specialized to Induce Foxp3+ Regulatory T Cells. J Immunol (2008) 181(10):6923–33. doi: 10.4049/jimmunol.181.10.6923
109. Shrimpton RE, Butler M, Morel AS, Eren E, Hue SS, Ritter MA. CD205 (DEC-205): A Recognition Receptor for Apoptotic and Necrotic Self. Mol Immunol (2009) 46(6):1229–39. doi: 10.1016/j.molimm.2008.11.016
110. Mukhopadhaya A, Hanafusa T, Jarchum I, Chen YG, Iwai Y, Serreze DV, et al. Selective Delivery of Beta Cell Antigen to Dendritic Cells In Vivo Leads to Deletion and Tolerance of Autoreactive CD8+ T Cells in NOD Mice. Proc Natl Acad Sci USA (2008) 105(17):6374–9. doi: 10.1073/pnas.0802644105
111. Spiering R, Margry B, Keijzer C, Petzold C, Hoek A, Wagenaar-Hilbers J, et al. DEC205+ Dendritic Cell-Targeted Tolerogenic Vaccination Promotes Immune Tolerance in Experimental Autoimmune Arthritis. J Immunol (2015) 194(10):4804–13. doi: 10.4049/jimmunol.1400986
112. Wadwa M, Klopfleisch R, Buer J, Westendorf A. Targeting Antigens to Dec-205 on Dendritic Cells Induces Immune Protection in Experimental Colitis in Mice. Eur J Microbiol Immunol (Bp) (2016) 6(1):1–8. doi: 10.1556/1886.2015.00048
113. Nijen Twilhaar MK, Czentner L, Grabowska J, Affandi AJ, Lau CYJ, Olesek K, et al. Optimization of Liposomes for Antigen Targeting to Splenic CD169(+) Macrophages. Pharmaceutics (2020) 12(12):1138. doi: 10.3390/pharmaceutics12121138
114. Busold S, Nagy NA, Tas SW, van Ree R, de Jong EC, Geijtenbeek TBH. Various Tastes of Sugar: The Potential of Glycosylation in Targeting and Modulating Human Immunity via C-Type Lectin Receptors. Front Immunol (2020) 11:134. doi: 10.3389/fimmu.2020.00134
115. Semmrich M, Plantinga M, Svensson-Frej M, Uronen-Hansson H, Gustafsson T, Mowat AM, et al. Directed Antigen Targeting In Vivo Identifies a Role for CD103+ Dendritic Cells in Both Tolerogenic and Immunogenic T-Cell Responses. Mucosal Immunol (2012) 5(2):150–60. doi: 10.1038/mi.2011.61
116. Veri MC, Gorlatov S, Li H, Burke S, Johnson S, Stavenhagen J, et al. Monoclonal Antibodies Capable of Discriminating the Human Inhibitory Fcgamma-Receptor IIB (CD32B) From the Activating Fcgamma-Receptor IIA (CD32A): Biochemical, Biological and Functional Characterization. Immunology (2007) 121(3):392–404. doi: 10.1111/j.1365-2567.2007.02588.x
117. Horton HM, Chu SY, Ortiz EC, Pong E, Cemerski S, Leung IW, et al. Antibody-Mediated Coengagement of FcgammaRIIb and B Cell Receptor Complex Suppresses Humoral Immunity in Systemic Lupus Erythematosus. J Immunol (2011) 186(7):4223–33. doi: 10.4049/jimmunol.1003412
118. Baker MP, Reynolds HM, Lumicisi B, Bryson CJ. Immunogenicity of Protein Therapeutics: The Key Causes, Consequences and Challenges. Self Nonself (2010) 1(4):314–22. doi: 10.4161/self.1.4.13904
119. Maldonado RA, LaMothe RA, Ferrari JD, Zhang AH, Rossi RJ, Kolte PN, et al. Polymeric Synthetic Nanoparticles for the Induction of Antigen-Specific Immunological Tolerance. Proc Natl Acad Sci USA (2015) 112(2):E156–65. doi: 10.1073/pnas.1408686111
120. LaMothe RA, Kolte PN, Vo T, Ferrari JD, Gelsinger TC, Wong J, et al. Tolerogenic Nanoparticles Induce Antigen-Specific Regulatory T Cells and Provide Therapeutic Efficacy and Transferrable Tolerance Against Experimental Autoimmune Encephalomyelitis. Front Immunol (2018) 9:281. doi: 10.3389/fimmu.2018.00281
121. Zhang AH, Rossi RJ, Yoon J, Wang H, Scott DW. Tolerogenic Nanoparticles to Induce Immunologic Tolerance: Prevention and Reversal of FVIII Inhibitor Formation. Cell Immunol (2016) 301:74–81. doi: 10.1016/j.cellimm.2015.11.004
122. Capini C, Jaturanpinyo M, Chang HI, Mutalik S, McNally A, Street S, et al. Antigen-Specific Suppression of Inflammatory Arthritis Using Liposomes. J Immunol (2009) 182(6):3556–65. doi: 10.4049/jimmunol.0802972
123. Yeste A, Takenaka MC, Mascanfroni ID, Nadeau M, Kenison JE, Patel B, et al. Tolerogenic Nanoparticles Inhibit T Cell-Mediated Autoimmunity Through SOCS2. Sci Signal (2016) 9(433):ra61. doi: 10.1126/scisignal.aad0612
124. Yeste A, Nadeau M, Burns EJ, Weiner HL, Quintana FJ. Nanoparticle-Mediated Codelivery of Myelin Antigen and a Tolerogenic Small Molecule Suppresses Experimental Autoimmune Encephalomyelitis. Proc Natl Acad Sci USA (2012) 109(28):11270–5. doi: 10.1073/pnas.1120611109
125. Galea R, Nel HJ, Talekar M, Liu X, Ooi JD, Huynh M, et al. PD-L1- and Calcitriol-Dependent Liposomal Antigen-Specific Regulation of Systemic Inflammatory Autoimmune Disease. JCI Insight (2019) 4(18). doi: 10.1172/jci.insight.126025
126. Bergot AS, Buckle I, Cikaluru S, Naranjo JL, Wright CM, Zheng G, et al. Regulatory T Cells Induced by Single-Peptide Liposome Immunotherapy Suppress Islet-Specific T Cell Responses to Multiple Antigens and Protect From Autoimmune Diabetes. J Immunol (2020) 204(7):1787–97. doi: 10.4049/jimmunol.1901128
127. Sands RW, Tabansky I, Verbeke CS, Keskin D, Michel S, Stern J, et al. Steroid-Peptide Immunoconjugates for Attenuating T Cell Responses in an Experimental Autoimmune Encephalomyelitis Murine Model of Multiple Sclerosis. Bioconjug Chem (2020) 31(12):2779–88. doi: 10.1021/acs.bioconjchem.0c00582
128. Kenison JE, Jhaveri A, Li Z, Khadse N, Tjon E, Tezza S, et al. Tolerogenic Nanoparticles Suppress Central Nervous System Inflammation. Proc Natl Acad Sci USA (2020) 117(50):32017–28. doi: 10.1073/pnas.2016451117
129. Chen XY, Du GS, Bai ST, Liu DJ, Li CL, Hou YY, et al. Restoring Immunological Tolerance in Established Experimental Arthritis by Combinatorial Citrullinated Peptides and Immunomodulatory Signals. Nano Today (2021) 41:101307. doi: 10.1016/j.nantod.2021.101307
130. Peine KJ, Guerau-de-Arellano M, Lee P, Kanthamneni N, Severin M, Probst GD, et al. Treatment of Experimental Autoimmune Encephalomyelitis by Codelivery of Disease Associated Peptide and Dexamethasone in Acetalated Dextran Microparticles. Mol Pharm (2014) 11(3):828–35. doi: 10.1021/mp4005172
131. Phillips BE, Garciafigueroa Y, Engman C, Liu W, Wang Y, Lakomy RJ, et al. Arrest in the Progression of Type 1 Diabetes at the Mid-Stage of Insulitic Autoimmunity Using an Autoantigen-Decorated All-Trans Retinoic Acid and Transforming Growth Factor Beta-1 Single Microparticle Formulation. Front Immunol (2021) 12:586220. doi: 10.3389/fimmu.2021.586220
132. Lewis JS, Stewart JM, Marshall GP, Carstens MR, Zhang Y, Dolgova NV, et al. Dual-Sized Microparticle System for Generating Suppressive Dendritic Cells Prevents and Reverses Type 1 Diabetes in the Nonobese Diabetic Mouse Model. ACS Biomater Sci Eng (2019) 5(5):2631–46. doi: 10.1021/acsbiomaterials.9b00332
133. Luo YL, Liang LF, Gan YJ, Liu J, Zhang Y, Fan YN, et al. An All-In-One Nanomedicine Consisting of CRISPR-Cas9 and an Autoantigen Peptide for Restoring Specific Immune Tolerance. ACS Appl Mater Interfaces (2020) 12(43):48259–71. doi: 10.1021/acsami.0c10885
134. Krienke C, Kolb L, Diken E, Streuber M, Kirchhoff S, Bukur T, et al. A Noninflammatory mRNA Vaccine for Treatment of Experimental Autoimmune Encephalomyelitis. Science (2021) 371(6525):145–53. doi: 10.1126/science.aay3638
135. Solorio-Rodríguez A, Escamilla-Rivera V, Uribe-Ramírez M, Chagolla A, Winkler R, García-Cuellar CM, et al. A Comparison of the Human and Mouse Protein Corona Profiles of Functionalized SiO2 Nanocarriers. Nanoscale (2017) 9(36):13651–60. doi: 10.1039/C7NR04685E
136. Müller LK, Simon J, Rosenauer C, Mailänder V, Morsbach S, Landfester K. The Transferability From Animal Models to Humans: Challenges Regarding Aggregation and Protein Corona Formation of Nanoparticles. Biomacromolecules (2018) 19(2):374–85. doi: 10.1021/acs.biomac.7b01472
137. Li Y, Wang G, Griffin L, Banda NK, Saba LM, Groman EV, et al. Complement Opsonization of Nanoparticles: Differences Between Humans and Preclinical Species. J Control Release (2021) 338:548–56. doi: 10.1016/j.jconrel.2021.08.048
138. Hatit MZC, Lokugamage MP, Dobrowolski CN, Paunovska K, Ni H, Zhao K, et al. Species-Dependent In Vivo mRNA Delivery and Cellular Responses to Nanoparticles. Nat Nanotechnol (2022) 17(2):1–9. doi: 10.1038/s41565-021-01030-y
139. Alves da Costa T, Lang J, Torres RM, Pelanda R. The Development of Human Immune System Mice and Their Use to Study Tolerance and Autoimmunity. J Transl Autoimmun (2019) 2:100021. doi: 10.1016/j.jtauto.2019.100021
140. Postigo-Fernandez J, Farber DL, Creusot RJ. Phenotypic Alterations in Pancreatic Lymph Node Stromal Cells From Human Donors With Type 1 Diabetes and NOD Mice. Diabetologia (2019) 62(11):2040–51. doi: 10.1007/s00125-019-04984-w
141. van Baarsen LG, de Hair MJ, Ramwadhdoebe TH, Zijlstra IJ, Maas M, Gerlag DM, et al. The Cellular Composition of Lymph Nodes in the Earliest Phase of Inflammatory Arthritis. Ann Rheum Dis (2013) 72(8):1420–4. doi: 10.1136/annrheumdis-2012-202990
142. Hahnlein JS, Nadafi R, de Jong T, Ramwadhdoebe TH, Semmelink JF, Maijer KI, et al. Impaired Lymph Node Stromal Cell Function During the Earliest Phases of Rheumatoid Arthritis. Arthritis Res Ther (2018) 20(1):35. doi: 10.1186/s13075-018-1529-8
143. Ramwadhdoebe TH, Ramos MI, Maijer KI, van Lienden KP, Maas M, Gerlag DM, et al. Myeloid Dendritic Cells Are Enriched in Lymph Node Tissue of Early Rheumatoid Arthritis Patients But Not in At Risk Individuals. Cells (2019) 8(7):756. doi: 10.3390/cells8070756
144. Suresh Babu S, Thandavarayan RA, Joladarashi D, Jeyabal P, Krishnamurthy S, Bhimaraj A, et al. MicroRNA-126 Overexpression Rescues Diabetes-Induced Impairment in Efferocytosis of Apoptotic Cardiomyocytes. Sci Rep (2016) 6(1):36207. doi: 10.1038/srep36207
145. Gaipl US, Kuhn A, Sheriff A, Munoz LE, Franz S, Voll RE, et al. Clearance of Apoptotic Cells in Human SLE. Curr Dir Autoimmun (2006) 9:173–87. doi: 10.1159/000090781
146. Fernandez-Boyanapalli R, Goleva E, Kolakowski C, Min E, Day B, Leung DY, et al. Obesity Impairs Apoptotic Cell Clearance in Asthma. J Allergy Clin Immunol (2013) 131(4):1041–7, 1047 e1-3. doi: 10.1016/j.jaci.2012.09.028
147. Liu Q, Wang X, Liu X, Liao YP, Chang CH, Mei KC, et al. Antigen- and Epitope-Delivering Nanoparticles Targeting Liver Induce Comparable Immunotolerance in Allergic Airway Disease and Anaphylaxis as Nanoparticle-Delivering Pharmaceuticals. ACS Nano (2021) 15(1):1608–26. doi: 10.1021/acsnano.0c09206
148. Lutterotti A, Yousef S, Sputtek A, Sturner KH, Stellmann JP, Breiden P, et al. Antigen-Specific Tolerance by Autologous Myelin Peptide-Coupled Cells: A Phase 1 Trial in Multiple Sclerosis. Sci Transl Med (2013) 5(188):188ra75. doi: 10.1126/scitranslmed.3006168
149. Lomakin Y, Belogurov A Jr, Glagoleva I, Stepanov A, Zakharov K, Okunola J, et al. Administration of Myelin Basic Protein Peptides Encapsulated in Mannosylated Liposomes Normalizes Level of Serum TNF-α and IL-2 and Chemoattractants CCL2 and CCL4 in Multiple Sclerosis Patients. Mediators Inflammation (2016) 2016:2847232–2847232. doi: 10.1155/2016/2847232
150. Belogurov A Jr, Zakharov K, Lomakin Y, Surkov K, Avtushenko S, Kruglyakov P, et al. CD206-Targeted Liposomal Myelin Basic Protein Peptides in Patients With Multiple Sclerosis Resistant to First-Line Disease-Modifying Therapies: A First-In-Human, Proof-Of-Concept Dose-Escalation Study. Neurotherapeutics: J Am Soc Exp Neurother (2016) 13(4):895–904. doi: 10.1007/s13311-016-0448-0
151. Kelly CP, Murray JA, Leffler DA, Getts DR, Bledsoe AC, Smithson G, et al. TAK-101 Nanoparticles Induce Gluten-Specific Tolerance in Celiac Disease: A Randomized, Double-Blind, Placebo-Controlled Study. Gastroenterology (2021) 161(1):66–80 e8. doi: 10.1053/j.gastro.2021.03.014
152. Domogalla MP, Rostan PV, Raker VK, Steinbrink K. Tolerance Through Education: How Tolerogenic Dendritic Cells Shape Immunity. Front Immunol (2017) 8:1764. doi: 10.3389/fimmu.2017.01764
153. Prasad S, Neef T, Xu D, Podojil JR, Getts DR, Shea LD, et al. Tolerogenic Ag-PLG Nanoparticles Induce Tregs to Suppress Activated Diabetogenic CD4 and CD8 T Cells. J Autoimmun (2018) 89:112–24. doi: 10.1016/j.jaut.2017.12.010
154. Freitag TL, Podojil JR, Pearson RM, Fokta FJ, Sahl C, Messing M, et al. Gliadin Nanoparticles Induce Immune Tolerance to Gliadin in Mouse Models of Celiac Disease. Gastroenterology (2020) 158(6):1667–81.e12. doi: 10.1053/j.gastro.2020.01.045
155. Glassman FY, Balu-Iyer SV. Subcutaneous Administration of Lyso-Phosphatidylserine Nanoparticles Induces Immunological Tolerance Towards Factor VIII in a Hemophilia A Mouse Model. Int J Pharm (2018) 548(1):642–8. doi: 10.1016/j.ijpharm.2018.07.018
Keywords: nanoparticles, autoimmunity, tolerance, antigen, immunotherapy, Treg
Citation: Benne N, ter Braake D, Stoppelenburg AJ and Broere F (2022) Nanoparticles for Inducing Antigen-Specific T Cell Tolerance in Autoimmune Diseases. Front. Immunol. 13:864403. doi: 10.3389/fimmu.2022.864403
Received: 28 January 2022; Accepted: 28 February 2022;
Published: 22 March 2022.
Edited by:
Kutty Selva Nandakumar, Karolinska Institutet (KI), SwedenReviewed by:
Pau Serra Devecchi, Institut de Recerca Biomèdica August Pi i Sunyer (IDIBAPS), SpainJames Paulson, The Scripps Research Institute, United States
Joseph R. Podojil, Northwestern University, United States
Copyright © 2022 Benne, ter Braake, Stoppelenburg and Broere. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Naomi Benne, bi5iZW5uZUB1dS5ubA==; Femke Broere, Zi5icm9lcmVAdXUubmw=