 Ellen McKenna1,2
Ellen McKenna1,2 Richard Wubben3
Richard Wubben3 Johana M. Isaza-Correa1,2
Johana M. Isaza-Correa1,2 Ashanty M. Melo1,2
Ashanty M. Melo1,2 Aisling Ui Mhaonaigh4
Aisling Ui Mhaonaigh4 Niall Conlon5
Niall Conlon5 James S. O’Donnell6
James S. O’Donnell6 Clíona Ní Cheallaigh7,8
Clíona Ní Cheallaigh7,8 Tim Hurley1,2,9,10
Tim Hurley1,2,9,10 Nigel J. Stevenson3,11
Nigel J. Stevenson3,11 Mark A. Little4,6
Mark A. Little4,6 Eleanor J. Molloy1,2,9,10,12,13*
Eleanor J. Molloy1,2,9,10,12,13*- 1Discipline of Paediatrics, Dublin Trinity College, The University of Dublin, Dublin, Ireland
- 2Paediatric Research Laboratory, Trinity Translational Medicine Institute (TTMI), St James’ Hospital, Dublin, Ireland
- 3Viral Immunology Group, School of Biochemistry and Immunology, Trinity Biomedical Sciences Institute, Dublin, Ireland
- 4Trinity Health Kidney Centre, Trinity Translational Medicine Institute (TTMI), Trinity College Dublin, Dublin, Ireland
- 5Department of Immunology, St James’ Hospital, Trinity College Dublin, Dublin, Ireland
- 6Irish Centre for Vascular Biology, Dublin, Ireland
- 7Department of Clinical Medicine, Trinity Centre for Health Science, Trinity College Dublin, Dublin, Ireland
- 8Department of Infectious Diseases, St James’s Hospital, Dublin, Ireland
- 9Neonatology, Coombe Women and Infant’s University Hospital, Dublin, Ireland
- 10National Children’s Research Centre, Children’s Hospital Ireland (CHI) at Crumlin, Dublin, Ireland
- 11Viral Immunology Group, Royal College of Surgeons in Ireland - Medical College of Bahrain, Al Muharraq, Bahrain
- 12Neonatology, Children’s Hospital Ireland (CHI) at Crumlin, Dublin, Ireland
- 13Paediatrics, Children’s Hospital Ireland (CHI) at Tallaght, Tallaght University Hospital, Dublin, Ireland
Unusually for a viral infection, the immunological phenotype of severe COVID-19 is characterised by a depleted lymphocyte and elevated neutrophil count, with the neutrophil-to-lymphocyte ratio correlating with disease severity. Neutrophils are the most abundant immune cell in the bloodstream and comprise different subpopulations with pleiotropic actions that are vital for host immunity. Unique neutrophil subpopulations vary in their capacity to mount antimicrobial responses, including NETosis (the generation of neutrophil extracellular traps), degranulation and de novo production of cytokines and chemokines. These processes play a role in antiviral immunity, but may also contribute to the local and systemic tissue damage seen in acute SARS-CoV-2 infection. Neutrophils also contribute to complications of COVID-19 such as thrombosis, acute respiratory distress syndrome and multisystem inflammatory disease in children. In this Progress review, we discuss the anti-viral and pathological roles of neutrophils in SARS-CoV-2 infection, and potential therapeutic strategies for COVID-19 that target neutrophil-mediated inflammatory responses.
Introduction
Neutrophils are the first responders to infection and extravasate rapidly from the blood vessels into tissue. They are the most abundant leukocyte in blood, with about 1011 neutrophils produced by the bone marrow each day, representing 40-60% of circulating immune cells in healthy adults (1). Neutrophils kill pathogens using oxidative burst, degranulation, phagocytosis and the release of neutrophil extracellular traps (NETs) (2, 3). Their role is most prominent in bacterial infection but they can also contribute to antiviral immunity.
Severe disease in COVID-19 is associated to increased neutrophil-to-lymphocyte ratio and high expression of neutrophil-related cytokines IL-8 and IL-6 in serum, and neutrophilia has been described as a predictor of poor outcome (4–14). Peripheral blood neutrophil counts in patients with COVID-19, although not as elevated as bacterial pneumonia, are higher in severe COVID-19 compared with mild cases and most other viral infections (4, 15). Neutrophils are associated with the development of thrombosis and pulmonary infiltrates found in post-mortem samples following severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (16–18). In this Progress review, we focus on emerging data on the roles of neutrophils in the pathogenesis and response to SARS-CoV-2.
Neutrophils in COVID-19
An altered neutrophil-to-lymphocyte ratio occurs in many conditions such as cancer, cardiovascular disease, sepsis and inflammatory disorders, including Systemic lupus erythematosus (SLE) and psoriasis (19). Patients with COVID-19 with severe disease had significantly higher absolute neutrophil counts (8) similar to the neutrophilia in both Severe Acute Respiratory Syndrome (SARS) and Middle East Respiratory Syndrome (MERS) (20). The limited antiviral response in COVID-19 may exacerbate neutrophil infiltration, resulting in exuberant inflammation (21).
A small gene ontology (GO) analysis of COVID-19 infected cells indicated that neutrophil activation and degranulation are the most activated cellular immune processes in COVID-19, but did not play a role in the antibody-mediated elimination of SARS-CoV-2 in a passive immunisation model (22). Neutrophils contribute to hypersensitivity pneumonitis in SARS-CoV-2 infection and altered neutrophil immunometabolism, with accumulation of succinate correlating with disease severity (21). A rat coronavirus (RCoV) model demonstrated that neutrophils produce cytokines and chemokines in response to alveolar epithelial cell infection with SARS-CoV-2, resulting in an inflammatory response which contributes to lung injury (23).
Neutrophil Extracellular Traps
Neutrophil extracellular traps (NETs) are web-like chromatin structures released by neutrophils to degrade virulence factors and kill bacteria. Once unregulated in sepsis or severe COVID-19, they induce multiple organ damage, including arterial hypotension, hypoxemia, coagulopathy, renal, neurological, and hepatic dysfunction as consequence of a NETs-associated cytokine storm (24–26). Silva et al. found that gasdermin inhibition with disulfiram or genic deletion decreases NETs formation with reduced multiple organ dysfunction and mortality in a sepsis model (27). NETs concentration was markedly increased in the tracheal aspirate and plasma of patients hospitalised with COVID-19 as well as in SARS-CoV-2-infected lung airways and alveoli, with spontaneous NETs production from their neutrophils (13, 28–32). SARS-CoV-2 can directly induce healthy neutrophils to release NETs in vitro, which increase pulmonary epithelium cell death (28). NETs also appear to drive neuroinflammation in Ischemic Brain Damage (IBD) and IBD following COVID-19, by affecting the blood-brain barrier, promoting thrombosis, and by inducing neuronal damage through extruded NETs components, NETs-IL-1 loop and IL-17 cascades (33, 34), making them a promising target for therapy.
The first step in NETosis is cellular activation via pattern recognition receptors (PRR) such as Toll-like receptors 4 (TLR4), TLR7 and TLR8 in viral infections (24, 35, 36). Reactive oxygen species (ROS) are subsequently produced, resulting in the activation of protein arginase deiminase 4 (PAD4) which is responsible for chromatin decondensation (24, 37). Neutrophil elastase (NE), a granule protein, induces neutrophil nuclear membrane break down while granule protein gasdermin D facilitates pore formation in the cell membrane and mediates release of NETs into the extracellular space (Figure 1) (24, 31). NETs do play a role in viral clearance, but excessive NETs production exacerbates inflammation in acute respiratory distress syndrome (ARDS) and contributes to microvascular thrombosis (Figure 1) (38). These is potentially related to over-activation of the Stimulator of interferon genes (STING) pathway through cyclic GMP-AMP synthase (cGAS) in phagosomes, and by SARS-CoV-2 infection itself through Angiotensin-Converting Enzyme 2 (ACE2)-angiotensin II (39, 40). Pharmacological activation of the STING pathway may also regulate the effects of SARS-CoV-2 infection (41). NETs can also have different proteins cargo associated to their deoxyribonucleic acid (DNA), citrullinated histone 3 (cit-H3), NE, and myeloperoxidase (MPO) structure which can influence the type of immune response triggered (42). Severe COVID-19 patients were shown to have higher expression of the alarmin nuclear protein High mobility group box 1 (HMGB1), antiviral molecules like ISG-15 and LL-37, or functionally active tissue factor (TF) as protein cargo in NETs, produced mostly by normal density granulocytes (NDG) (43, 44). These cargo molecules induced thrombogenic activity and differential cytokines expression (43, 44).
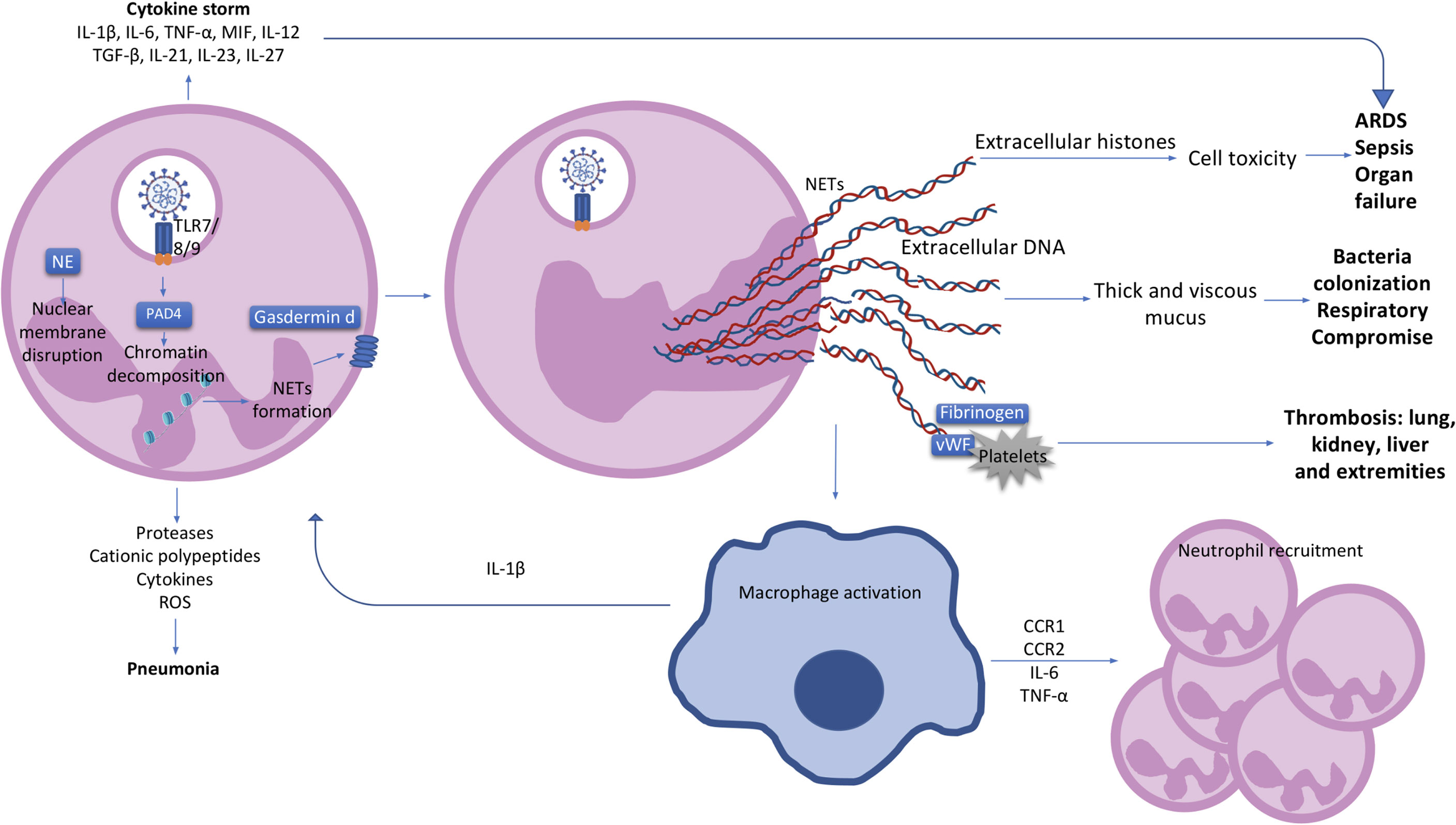
Figure 1 The neutrophil and clinical characteristics of COVID-19 patients. Activated neutrophils can produce cytokines such as IL-1β, IL-6, TNF-α, MIF, IL-12, TGF-β, IL-21, IL-23 and IL-27, contributing to a cytokine storm and the further development of ARDS and organ failure in COVID-19 patients. Pneumonia in COVID-19 patients is most likely to be caused by the production of proteases, cationic polypeptides, cytokines and ROS by neutrophils. Upon SARS-CoV-2 recognition by TLR7/8/9, protein arginase deiminase 4 (PAD4) is activated, which induces chromatin decondensation through histones citrullination and consequently NETs formation. Neutrophil nuclear membrane is disrupted by neutrophil elastase (NE) and gasdermin D which facilitates the formation of a pore in the neutrophil cell membrane and mediates release of the contents of NETs into the extracellular space. NETs induce macrophage activation and IL-1β production resulting in a positive loop with neutrophils and NETs formation. Macrophages also secrete CCR1, CCR2, IL-6 and TNF-α leading to further neutrophil recruitment. Extracellular histones presented in NETs causes cell cytotoxicity contributing with ARDS, sepsis and organ failure observed in COVID-19 patients. Extracellular DNA induces thick and viscous mucus production allowing bacteria colonization and respiratory failure. NETs also interact with fibrinogen, VWF and platelets causing thrombosis in several organs such as lung, kidney, liver and extremities. IL, interleukin; TNF, tumour necrosis factor; MIF, macrophage migration inhibitory factor; ARDS, acute respiratory distress syndrome; ROS, reactive oxygen species; PAD4, protein arginase deiminase 4; NETs, neutrophil extracellular trap; NE, neutrophil elastase; CCR, chemokine receptor; DNA, deoxyribonucleic acid; VWF, von Willebrand factor.
Inflammasome Activation in COVID-19
COVID-19 is characterised by a cytokine storm and the Pyrin domain containing 3 (NLRP3) inflammasome has been implicated. The inflammasomes are molecular mechanism involving multiprotein complexes which regulate the production of pro-inflammatory cytokines. NLRP3, a member of the nucleotide oligomerization domain (NOD)-like receptor (NLR) family, is present in neutrophils (17). After NLRP3 activation, pro-caspase 1 is cleaved to the active form caspase 1, leading to the cleavage of pro-inflammatory pro-IL-1β and pro-IL-18 into the active forms (Figure 2) (45). Single-stranded ribonucleic acid (ssRNA) viruses, such as SARS-CoV-2, induce Nuclear factor kappa B (NF-κB) activation and the further production of pro-IL-1β and pro-IL-18 (45, 46). Simultaneously, ROS and Adenosine 5’-triphosphate (ATP) produced by mitochondria trigger NLRP3 inflammasome assembly (46). Active NLRP3 inflammasome is present in peripheral blood mononuclear cells (PBMCs) and post-mortem tissues of COVID-19 patients, and high expression of its derived products such as Casp1p20 and IL-18 were seen to correlate with disease severity and poor clinical outcome (47). NLRP3 inflammasome activation has also been described in neutrophils of severe COVID-19 patients (48). Aymonnier et al. found that neutrophils from COVID-19 patients with respiratory failure demonstrated NLRP3 inflammasome molecule Apoptosis-associated speck-like protein containing a CARD (ASC) specks, and their early formation in NETosis. In patients with severe COVID-19 neutrophils with intact multilobulated nuclei, ASC specks formation and histone H3 citrullination was elevated (48). In a murine model they also showed transient presence of ASC specks at the microtubule organizing center, before nuclear rounding, early in NETosis (48). In addition, SARS-CoV-2 has been shown to directly activate the NLRP3 inflammasome through viroporin protein 3a, which most likely acts by the formation of K+ and Ca+ channels (49). Such direct activation of the inflammasome leads to the production of IL-1β and IL-18, perpetuating inflammation and resulting in further neutrophil activation (50). NLRP3 inflammasome activation in the blood of patients reveals an impaired immature neutrophil response in severe COVID-19. Inflammasome signature analysis in circulating myeloid cells allows COVID-19 patients to be stratified and predicts evolution of disease severity (51).
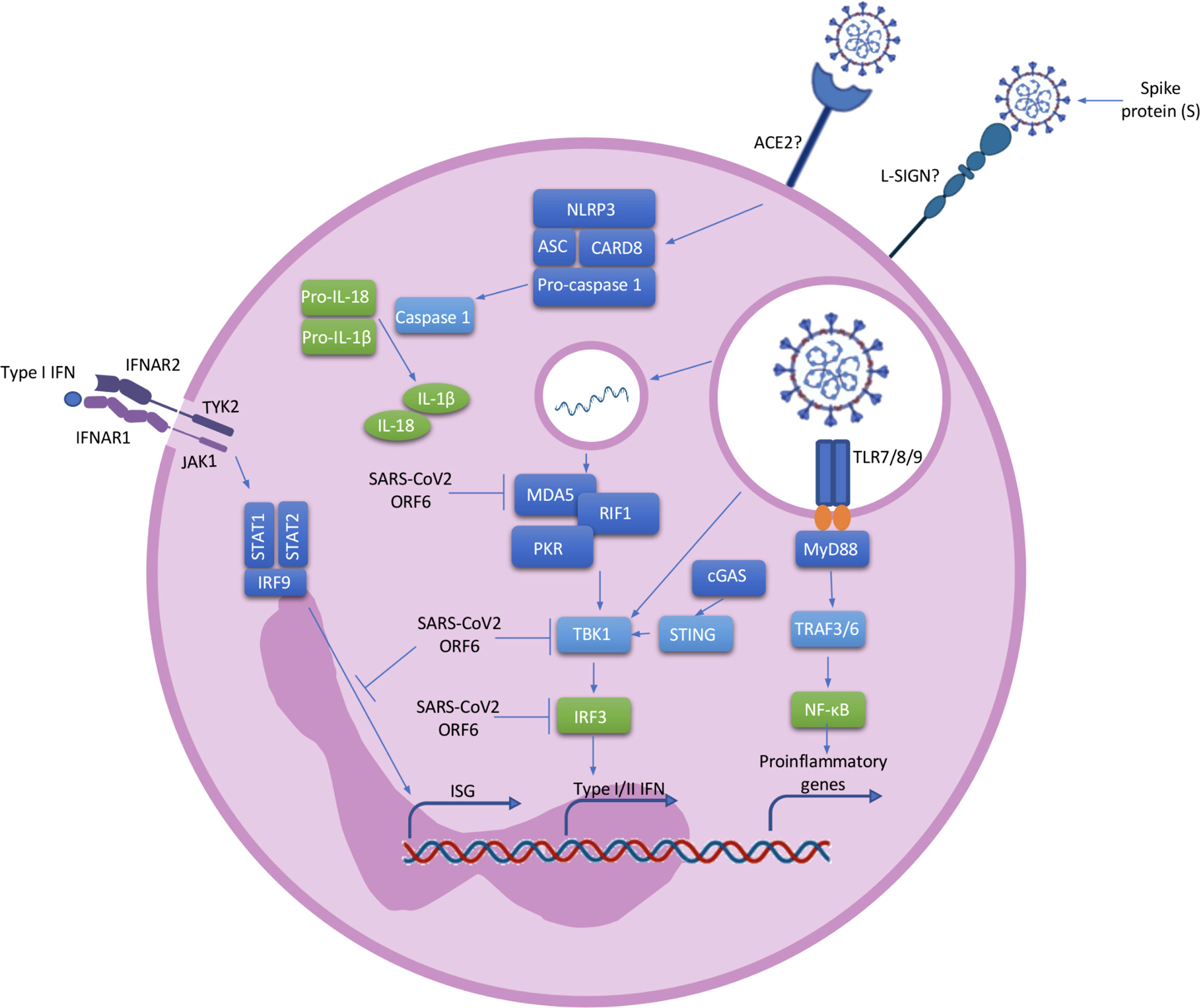
Figure 2 Neutrophil activation. ACE2 or L-SING receptors on neutrophils most likely recognise SARS-CoV2 via a spike (S) protein on its surface. Once the virus enters the cell, ssRNA viruses such as SARS-CoV-2 are recognised by TLR 7/8/9 which induce the activation of the MyD88 pathway. MyD88 activates TRAF3 and TRAF6 which result in the transcription of NF-κB and IRF7 associated genes. The activated NF-κB pathway leads to the transcriptional induction of proinflammatory cytokines, chemokines and additional inflammatory mediators in neutrophils. In addition, cytosolic viral RNA recruiting MDA5, RIF1 and PKR lead to the activation of TBK1 and the further activation of IRF3 resulting in the transcription of type I/II IFN genes. The positive stimulatory loop by type I IFN induces the production of more IFNs through the JAK/STAT pathway and the induction of Interferon Stimulated Genes (ISG). At the same time, SARS-CoV-2 possess ORF6, an accessory protein antagonist of IFNs by the inhibition of MDA5, TBK1, IRF3 and IRF9. ssRNA viruses also cause the recruitment of the NLRP3 inflammasome complex and the further activation of pro-caspase-1 resulting in the cleavage of pro-IL-1β and pro-IL-18 into the active forms. ACE2, angiotensin-Converting Enzyme 2; L-SING, L-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin/CD209L; RNA, ribonucleic acid; ssRNA, Single-stranded RNA; TLR, toll-like receptor; MyD88, myeloid differentiation primary response 88; TRAF, tumor necrosis factor receptor (TNF-R)-associated factor; NF-κB, nuclear factor kappa B; IRF, Interferon Regulatory Factor; MDA, melanoma differentiation-associated protein; RIF, Replication Timing Regulatory Factor; PKR, protein kinase R; TBK, TANK Binding Kinase; IFN, interferon; ISG, Interferon Stimulated Genes; JAK-STAT, janus kinase; ORF, open Reading Frame; NLRP3, nod like receptor family, pyrin domain containing 3; IL, interleukin.
Neutrophil Subsets in COVID-19
Heterogeneity within the neutrophil population during infection has been demonstrated in multiple diseases, and different subsets have defined roles in influencing the inflammatory response (38, 52). Neutrophil subsets varying in their density, maturity and expression of surface markers have been reported in COVID-19 (53, 54). Classically, in sepsis, immature neutrophils are released from the bone marrow and Carissimo et al. found increased immature neutrophils in whole blood that correlated with increased IL-6 and IP-10, and COVID-19 disease severity (55). The ratio of immature neutrophils to gamma delta (Vδ)2 T cells could predict severe COVID-19 (55). Additionally, a shift toward immature neutrophils as the driver of hyperinflammation is associated with severe COVID-19 disease (56).
Recently there has been a renewed interest in immunomodulatory neutrophil subsets, specifically in the field of cancer, SLE and sepsis, including low density granulocytes (LDGs) and myeloid derived suppressor cells (MDSCs) (38), but there is not a consensus on nomenclature and classification (57). MDSCs are a mixed population of mature and immature cells with differing immunomodulatory roles (58). There is a lack of clarity on the phenotypical and functional characteristics of MDSCs and their relationship to LDGs but their defining characteristic is suppression of the adaptive immune response (59). MDSC expansion is linked to G-CSF, a cytokine increased in the lungs of COVID-19 patients (60) and almost 90% of mononuclear cells in the severe disease cohort were MDSCs. Proportion of LDGs increases with disease severity in COVID-19 patients, as well as their production of NETs when compared to healthy controls (43, 61).
Morrisey et al. described a population of LDGs correlating with disease severity and hypercoagulable state in COVID-19 patients (53). A population of CD45+CD66b+CD16IntCD44lowCD11bInt LDGs was found in patients with severe disease, which displayed enhanced phagocytic capacity, spontaneous NETs formation and elevated cytokine production. Similarly, an immune-suppressive CD16bright/CD62Ldim neutrophil subtype was increased in patients developing pulmonary embolism (PE) on the day of ICU admission (54). Using whole blood transcriptomics analysis, increased NLRP3 inflammasome, monocytes and LDGs were found in the lungs of COVID-19 patients, and neutrophil activation-associated signatures correlated to disease severity (62). In COVID-19, immature neutrophils are expanded and show increased programmed death ligand (PD-L) 1, which suppresses T cells, and reduced oxidative burst functions with no change in phagocytosis in severe COVID-19 (63). Chevrier et al. found higher LDGs were present in COVID-19 patients early in the course of the disease and decreased in convalescence using mass cytometry and serum proteomics, but CD16low neutrophil population remained expanded over the disease course (64). COVID-19 induced-ARDS is associated with MDSC expansion, reduced lymphocyte function and arginine shortage, through increased arginase activity, therefore arginase supplementation may be therapeutic (65). Further study into the role of neutrophil subsets in COVID-19 is warranted, potentially as biomarkers of disease severity, or as new targets for therapeutic approaches.
Neutrophil Response to SARS-CoV-2
Does SARS-CoV-2 Actively Infect Neutrophils?
Although neutrophils express the L-SIGN and DC-SIGN C-type lectins receptors that have been suggested to act as entry receptors for SARS-CoV-2, there is conflicting evidence about active infection of neutrophils with the virus. In other ssRNA viruses such as West Nile and influenza virus neutrophils serve as an important viral reservoir and contain actively replicating virus, and studies with human immunodeficiency virus (HIV) and Respiratory syncytial virus (RSV) viral models suggest that neutrophils can internalise virus without productive infection (66). Neutrophils are important for viral detection and initiation of downstream effector immune pathways but the replicative ability of ssRNA virus SARS-CoV-2 within neutrophils is not known.
ACE2 is the primary cell entry receptor for SARS-CoV-2 and ACE2 deficiency is associated with worse outcomes in COVID-19 (67). Entry of SARS-CoV2 into the cells following membrane fusion majorly down-regulates ACE2 receptors, with loss of the catalytic effect of these receptors at the external site of the membrane (68). This induces increased pulmonary inflammation and coagulation due to enhanced and unopposed angiotensin II effects. ACE2 down-regulation induced by viral invasion may be especially detrimental in people with baseline ACE2 deficiency (68). Following viral entry, the additional ACE2 deficiency may exacerbate the dysregulation between ACE→Angiotensin II→AT1 receptor axis (potentially adverse) and the ACE2→Angiotensin→Mas receptor axis (negative regulator of angiotensin II activity, potentially protective recombinant ACE2) (68). Therefore, angiotensin and angiotensin II type 1 receptor blockers may be beneficial in patients with severe SARS-CoV-2 (68). However, two large cohort studies showed that angiotensin-converting enzyme inhibitors (ACEIs)/angiotensin receptor blockers (ARBs) use was not associated with increased SARS-CoV-2 infection, but was in fact associated with a lower risk of all-cause mortality in hospitalized patients (69, 70). Further studies are needed to test the protective effects of ACEIs/ARBs in COVID-19 (69, 70). NETs triggered by SARS-CoV-2 depend on ACE2, serine protease TMPRSS2, virus replication, and PAD-4 (28). ACE is important in neutrophil antibacterial activity. Veras at al found that NETosis was facilitated in neutrophils in patients with COVID-19 (28). Neutrophils express ACE2 similar to other immune cells and it is postulated that allows the virus-triggered cell activation and NETosis (28). Knockout of this gene in mice or treatment with an ACE inhibitor increased susceptible to bacterial infection by methicillin-resistant Staphylococcus aureus (MRSA). Mice overexpressing ACE in neutrophils have increased killing of MRSA Pseudomonas aeruginosa, and Klebsiella pneumoniae, with increased neutrophil production of reactive oxygen species (ROS) independent of the angiotensin II AT1 receptor (71).
Dysfunctional Neutrophil Activation in COVID-19
Neutrophils express all known Toll-like receptors (TLRs) with the exception of TLR3 (72). TLR7, TLR8 and TLR9 are involved in the detection of ssRNA viruses such as SARS-COV-2 (73). Activation of these receptors leads to downstream activation of NF-κB and interferon regulatory factor (IRF7), and the subsequent production of pro-inflammatory cytokines and chemokines in neutrophils (Figure 2) (74). In conjunction with neutrophils, these pro-inflammatory cytokines and chemokines drive the characteristic hyperinflammation and pulmonary infiltration seen in severe COVID-19 (74). Neutrophils also produce type 1 interferons (IFN-α/IFNβ) through the activation of IRF proteins (75) and this broad, but dysregulated, pro-inflammatory and antiviral response puts selective pressure on these highly pathogenic respiratory viruses. The host response to SARS-CoV-2 has also been broadly defined as a significantly depleted type 1 IFN response, with a consistent upregulation of chemotactic signals (CCL8, CCL2, CXCL2, CXCL8 and CXCL9), most of which are key mediators of neutrophil recruitment. Liao et al. found that in the lungs of patients with severe COVID-19, macrophages exacerbate inflammation by producing chemokines that recruit neutrophils to the site of infection through chemokine receptors CC-chemokine receptor 1 (CCR1) and C-X-C chemokine receptor type 2 (CXCR2) (57). Using a SARS-CoV-2 animal model early induction of CXCL9 and CCL8 was found consistent with observations in primary human bronchial epithelial cells infected with SARS-CoV-2. At day 7, despite waning levels of virus, elevated CCR5, CCL2, CXCL9 and IL-6 were found in the animal model, suggesting neutrophil-mediated inflammation may persist after the virus has been cleared (76). This may correlate with the clinical findings of persistent symptoms and fatigue with post-viral infection complications in some patients.
The loss of IFN signalling is vital to understanding why SARS-CoV-2 elicits such a potent inflammatory and neutrophilic chemotactic response. For instance, bats appear to limit the inflammatory and neutrophilic chemotactic response when infected with coronaviruses endemic in the bat population (77). Banerjee et al. have proposed that bats possess repressors of NF-κB signalling, a potent inductor of pro-inflammatory and chemotactic responses, allowing these strains of the viruses to become endemic in the population. However, unlike bats, humans lack this repressor activity rendering us susceptible to this uncontrollable neutrophil-mediated inflammatory response following viral infection (77).
Neutrophils and Thrombosis
Coagulation cascade activation is a common finding in patients with COVID-19 and is associated with disease severity (78). Elevated levels of fibrin D-dimer degradation products, a marker of fibrin degradation indicating overactive coagulation, correlates with a worse clinical outcome (79). High plasma levels of plasminogen activator (tPA) and plasminogen activator inhibitor-1 (PAI-1) in hospitalised COVID-19 patients had strong correlations with neutrophil counts and activation, and extremely high levels of tPA increasing fibrinolysis (80). Plasmatic matrix metalloproteinase-9 (MMP-9) was likewise increased in COVID-19 patients which induced platelet and neutrophil activation, and NETs formation in vitro (81). Post-mortem studies have consistently shown that micro-thrombi are present throughout the pulmonary vasculature (82). Collectively, these data suggest that coagulation activation and vasculopathy within the lungs (pulmonary intravascular coagulopathy [PIC]) plays a role in modulating COVID-19 pathogenesis (78). The biological mechanisms through which SARS-CoV-2 infection causes PIC within the lung blood vessels remain poorly understood (83). However, recent autopsy studies have reported significant endothelial cell (EC) damage, apoptosis, loss of tight junctions and separation from the basement membrane (84). Local inflammation and dysregulated pro-inflammatory cytokine generation within the lungs are a major factor as well as local hypoxia and complement activation, which significantly enhance procoagulant pathways and downregulate anticoagulant pathways in vivo. Moreover, ECs express the ACE2 receptor through which SARS-CoV-2 gains entry into cells, and electron microscopy studies have reported viral inclusion bodies within ECs.
Neutrophils and platelets are key modulators of thrombosis. Significant NETosis is found in patients with severe COVID-19 and is important in thrombus aetiology (85). NETs can bind to platelets, triggering platelet activation, and through their citrullinated histone H3 (citH3) they can also interact with procoagulant von Willebrand factor (VWF) (85). In addition to their effects on primary hemostasis, NETs also enhance local thrombin generation. In particular, NETs initiate coagulation activation through the alternative contact pathway and trigger thrombin generation by enhancing the intrinsic tissue-factor dependent pathway. NETs have also been described to over-activate the STING pathway through the cGAS sensor in phagosomes (40). The over-activation of the STING-pathway increases hyper-coagulability via interferon-β and tissue factor, released by monocytes-macrophages, and can be inhibited upstream the STING-pathway by aspirin, intravenous immunoglobulins and Vitamin-D (40). NETs histones can activate platelets by stimulating platelet TLR4 and TLR2; neutrophils can bind to these active platelets through surface glycoprotein Ib to induce NETosis and, consequently, result in thrombosis (85). Platelet activation is associated with disease severity in COVID-19 (86). Finally, NETosis has potent pro-inflammatory effects on ECs, which serve to attenuate the normal ability of ECs to regulate procoagulant pathways (87, 88). NETs and thrombosis have been implicated in several disorders including cancer, SLE, rheumatoid arthritis (RA), atherosclerosis and ischemic stroke. NETs have been shown to invade microthrombi in septic patients and contribute to organ damage, hence it is likely that neutrophils are a mediator of organ dysfunction in COVID-19 (31).
Neutrophils and COVID-19 in Children
The severity of COVID-19 differs between age-groups, and children, especially neonates, exhibit milder disease with only a small proportion require intensive care with acute respiratory illness. There are many theories about this discrepancy, which is also seen with other similar viral illnesses, and the decreased expression of ACE2 and NETs formation may be contributory (89). However, a multisystem inflammatory disease in children (MIS-C) or paediatric multisystem inflammatory syndrome temporally associated with COVID-19 (PIMS-TS) has emerged in children, occurring weeks after the primary infection with SARS-Cov-2, that can lead to serious and life-threatening illness in previously healthy children (90). There is no internationally accepted single definition of MIS-C/PIMS-TS, but most case definitions require multi-organ dysfunction, systemic inflammation evidence of recent a SARS-CoV-2 infection, and the exclusion of other causes. The clinical presentation and laboratory findings in MIS-C are similar to Kawasaki’s disease and toxic shock syndrome, and considered to be a spectrum of disease (90).
Similar to adults with COVID-19, neutrophilia and lymphocytopenia are common in MIS-C. Neutrophils play a key functional role in Kawasaki disease with recent descriptions of NETosis and neutrophil activation in the form of CD11b and CD64 production (91). Neutrophil counts predict responsiveness of patients with Kawasaki disease to intravenous immunoglobulin therapy, also used in MIS-C (92, 93). Neutrophils activation marker Fc γ receptor I (FcγRI; CD64) was described to be highly expressed on neutrophils of treatment-naive MIS-C patients in acute phase compared with healthy controls (94). These patients also showed increase levels of the neutrophils chemoattractant cytokine IL-8 (94). Ramaswamy et al. talk of a potential myeloid dysfunction in MIS-C patients based on the high expression of alarmin-related S100A genes in neutrophils and monocytes, and the significant reduction in key antigen-presentation molecules such as HLA class II and CD86 (95). Additional research is required to fully understand the role of neutrophils in MIS-C and to determine whether treatments used in Kawasaki disease such as intravenous immunoglobulin therapy could also be used with MIS-C patients.
Therapeutic Targeting of Neutrophils
Targeting Cytokines
The efficacy of targeting cytokines produced by various immune cells, including neutrophils, is being explored in ongoing clinical trials. Neutrophils produce IL-6, and IL-6 inhibitor tocilizumab has been shown to decreases neutrophil survival and lipopolysaccharides (LPS)-induced oxidative burst, as well as neutrophil release from the bone marrow and lung demargination (96, 97). Tocilizumab has been approved by the United States Food and Drug Administration (FDA) for use in COVID-19 patients and decreased mortality, poor outcome and mechanical ventilation (98, 99). Clazakizumab also targets IL-6 and is currently being evaluated for safety in several clinical trials of patients with life-threatening COVID-19 (Table 1). The interleukin-6 receptor inhibitors (IL6ri) sarilumab or tocilizumab decreased intubation and mortality in a study including 255 patients with COVID-19 (100). Doxycycline (a tetracycline) reduces IL-6, IL-1β and TNF-α levels, however, doxycycline treatment did not have a significant clinical impact on time to recovery, hospital admissions or deaths related to COVID-19 in patients with high risk to adverse outcomes (101).
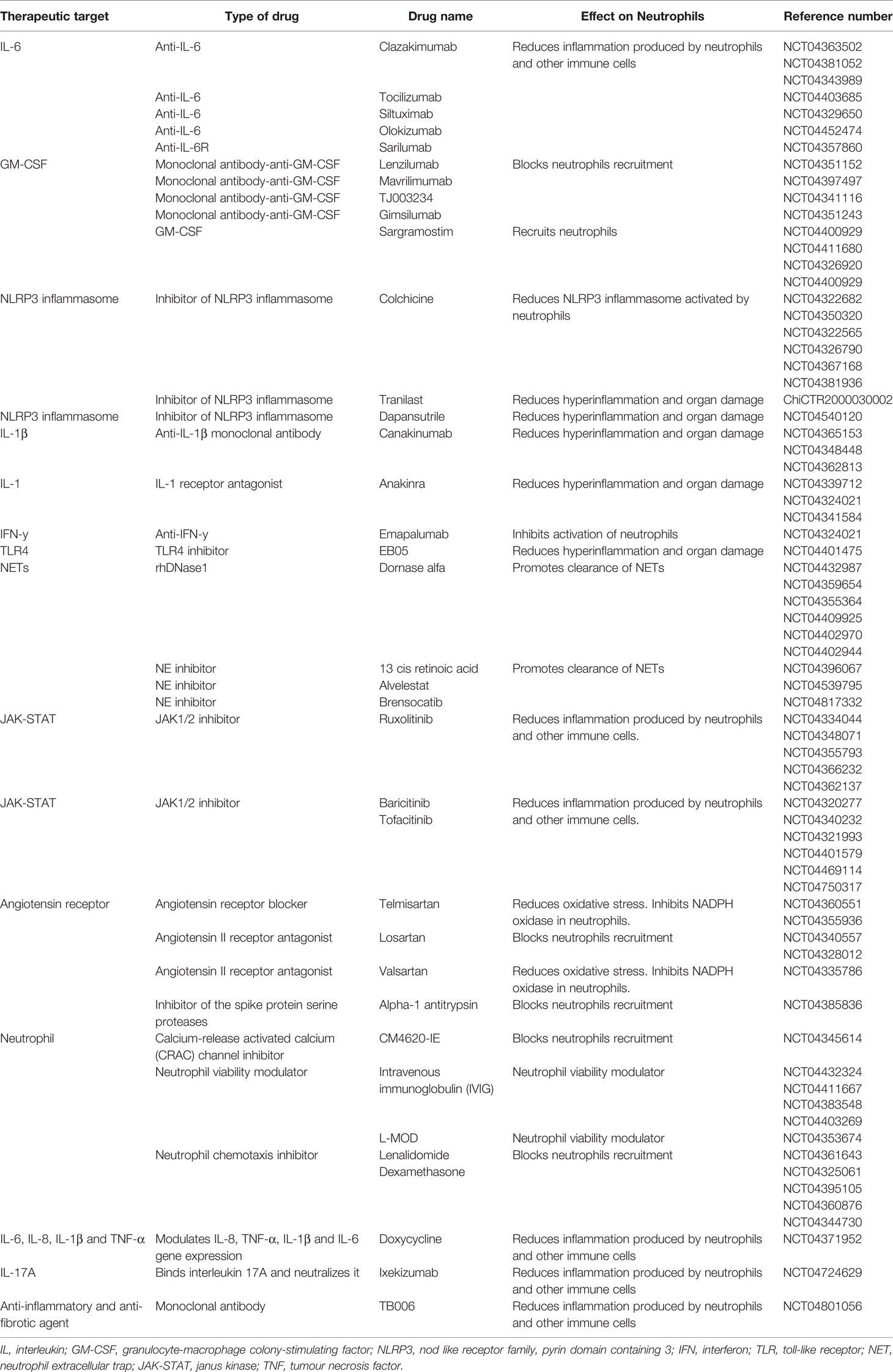
Table 1 Clinical trials therapeutically targeting neutrophils.
Granulocyte-macrophage colony-stimulating factor (GM-CSF) is involved in neutrophil recruitment, survival, IL-6 release and priming for NETosis (102, 103). Mavrilimumab, an anti-GM-CSF receptor-α monoclonal antibody, improved clinical outcomes in patients with COVID-19 pneumonia and systemic hyperinflammation (104). In contrast, sargramostim, a recombinant human GM-CSF is under investigation, to improve the immune response by recruiting neutrophils, dendritic cells and macrophages to fight the virus and to repair tissue damage (Table 1), although there may be significant risks including neurotoxicity (103). GM-CSF also induces the expansion of immunosuppressive MDSCs, which impair NK cells, CD8+ T cells and increase proliferation of immunosuppressive T regulatory (Treg) cells (105, 106). GM-CSF stimulates the expression of IL-1β, IL-6, TNFα and other pro-inflammatory cytokines and chemokines, therefore, its inhibition would more broadly dampen hyperinflammation than therapy for IL-6 alone. In patients with rheumatoid arthritis this strategy is used for those unresponsive to anti-TNF therapy or tocilizumab (106). Cytokine signalling pathways are targeted by using inhibitors of JAK1/JAK2, to potentially reduce inflammation (Table 1). Clinical trials using JAK1/JAKK2 inhibitor Baricitinib showed reduction in 30-day mortality in over 70s with moderate-to-severe COVID-19 pneumonia, and combined with Remdesivir decreased recovery time and reduced 28-day mortality, serious events and new infections (107, 108). Reduction in the risk of death or respiratory failure was also described in a clinical trial including 289 COVID-19 patients when comparing the effects of JAK inhibitor Tofacitinib with a placebo (109).
NLRP3 inflammasome activation in neutrophils is implicated with pulmonary inflammation and inhibition with MCC950 inhibited IL-1β in the lungs of cystic fibrosis mice (110). Tranilast is the first NLRP3 inflammasome inhibitor in clinical trials in the Chinese Clinical Trial Registry. Interleukin-1 blockade with canakinumab treatment increases neutrophil apoptosis and decreases pro-inflammatory signalling in the IL-1β pathway using gene expression and pathway data (111). Canakinumab, is another FDA approved drug under investigation in clinical trials, and may help reduce respiratory and cardiac damage. Colchicine targets the neutrophil and monocyte NLRP3 inflammasome, hence attenuating activation of IL-1β (112). However, no significant differences were seen in primary (disease progression or mortality) or secondary (time to discharge, proportion of patients discharged, time in Intensive Care unit (ICU) or duration of hospitalisation) outcomes in two separated clinical trials comparing patients who were given colchicine to placebo/usual care treated patients (113, 114). Anakinra (commercially known as Kineret) is an FDA approved human IL-1RA (inflammasome-regulated immune response inhibitor of IL-1) which may reduce hyperinflammation and organ damage (Table 1) (112). Clinical trials using anakinra as treatment for COVID-19 have reported conflicting results. One study described lower risk of clinical progression in patients who received anakinra compared to placebo, while other study reports no effect of anakinra treatment on in-hospital mortality or days of organ support (115–117). However, the European Medicines Agency (EMA) recommended the use of anakinra in December 2021, specifically for COVID-19 adult patients at risk of developing severe respiratory failure or with pneumonia requiring supplemental oxygen (118).
Intravenous Immunoglobulin (IVIG) and Corticosteroids
IVIG are purified IgG made from a pool of plasma from healthy donors (119) and modulate neutrophil viability through agonistic antibodies anti-Fas and Siglec-9 (120). It may also decrease neutrophil activation and NETs formation and mitigate vascular injury (121). IVIG has been tested in clinical trials in patients with COVID-19 (Table 1) and has shown to have therapeutic value (121). Similar positive effects of IVIG have been described in children with Kawasaki’s disease and MIS-C. However, ambiguity exists about dose dependent pro/anti-inflammatory effects as high dose IVIG is anti-inflammatory while a lower dose is considered pro-inflammatory (122). The widespread utility of this therapy may be precluded by plasma shortage, as it is also use as treatment in immunodeficiencies and inflammatory disorders. Treatment of healthy neutrophils with IVIG decreased NETosis and ROS production but enhanced phagocytosis (122).
The efficacy of treating COVID-19 patients with corticosteroids remains controversial. Lomas et al. have demonstrated that dexamethasone can inhibit neutrophil chemotaxis in vitro and in vivo (123). A variety of studies hypothesise that this anti-inflammatory drug may be effective in reducing ARDS and respiratory failure in COVID-19 patients (Table 1). The randomised evaluation of COVID-19 therapy (RECOVERY) trial in hospitalized COVID-19 patients found that treatment with dexamethasone results in a lower 28-day mortality for patients receiving oxygen only or ventilation, though no explanation of the mechanism for this was provided (124). Neutrophil-to-Lymphocyte ratio was reduced in patients treated with corticosteroids for COVID-19.
Targeting NETs
The targeting of neutrophil extracellular traps with dornase alfa, a human recombinant deoxyribonuclease (DNAse) enzyme, degrades DNA and promotes the clearance of NETs and has been used in patients with cystic fibrosis (125). Several studies are investigating the use of dornase alfa to improve pulmonary function in severe COVID-19 with ARDS (Table 1) (125). Similarly, all-trans retinoic acid, an inhibitor of NE (granular component involved in NETosis), is also being explored to improve lung injury in COVID-19 patients. COVID-19 is associated with a significant neutrophil NETs burden and targeting NETs-driven IL-1 signalling, using the IL-1 receptor antagonist, decreased NETosis and may modulate inflammation.
Conclusion
The clinical syndrome of severe COVID-19 has several unique features, including, unusually for a viral infection, an increased neutrophil-lymphocyte ratio. Neutrophils play a role in viral clearance in terms of NETs and the production of IFN. However, neutrophils can have detrimental effects by aiding the pathogenesis of SARS-CoV-2 and exacerbating complications of COVID-19 such as ARDS, thrombosis and MIS-C. Understanding the role of neutrophils in the pathogenesis of severe COVID-19 may lead to identification of key therapeutic targets and/or biomarkers for early identification of patients who may benefit from immunomodulatory agents to control hyperinflammation and reduce mortality rates.
Author Contributions
EM and AMM have performed literature research, designed the review layout, wrote, and revised the review. RW, AUM, NC, JO’D, CNC, TH, and NS have performed literature research, wrote, and revised the review. JI-C designed the review layout, and revised the review. ML has performed literature research, designed the review layout, wrote, and revised the review. EJM has performed literature research, designed the review layout, wrote, and revised the review. All authors agree to be accountable for the content of the work. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Rosales C. Neutrophil: A Cell With Many Roles in Inflammation or Several Cell Types? Front Physiol (2018) 9:113. doi: 10.3389/fphys.2018.00113
2. Lacy P. Mechanisms of Degranulation in Neutrophils. Allergy Asthma Clin Immunol (2006) 2:98–108. doi: 10.1186/1710-1492-2-3-98
3. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss D, et al. Neutrophil Extracellular Traps Kill Bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
4. Wan S, Xiang Y, Fang W, Zheng Y, Li B, Hu Y, et al. Clinical Features and Treatment of COVID-19 Patients in Northeast Chongqing. J Med Virol (2020) 92:797–806. doi: 10.1002/jmv.25783
5. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical Features of Patients Infected With 2019 Novel Coronavirus in Wuhan, China. Lancet (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
6. Liu J, Liu Y, Xiang P, Pu L, Xiong H, Li C, et al. Neutrophil-To-Lymphocyte Ratio Predicts Critical Illness Patients With 2019 Coronavirus Disease in the Early Stage. J Transl Med (2020) 18:206. doi: 10.1186/s12967-020-02374-0
7. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA (2020) 323:1061–9. doi: 10.1001/jama.2020.1585
8. Itelman E, Wasserstrum Y, Segev A, Avaky C, Negru L, Cohen D, et al. Clinical Characterization of 162 COVID-19 Patients in Israel: Preliminary Report From a Large Tertiary Center. Isr Med Assoc J (2020) 22:271–4.
9. Zeng Z, Feng S, Chen G, Wu J. Predictive Value of the Neutrophil to Lymphocyte Ratio for Disease Deterioration and Serious Adverse Outcomes in Patients With COVID-19: A Prospective Cohort Study. BMC Infect Dis (2021) 21:80. doi: 10.1186/s12879-021-05796-3
10. Seyit M, Avci E, Nar R, Senol H, Yilmaz A, Ozen M, et al. Neutrophil to Lymphocyte Ratio, Lymphocyte to Monocyte Ratio and Platelet to Lymphocyte Ratio to Predict the Severity of COVID-19. Am J Emerg Med (2021) 40:110–4. doi: 10.1016/j.ajem.2020.11.058
11. Moradi E, Teimouri A, Rezaee R, Morovatdar N, Foroughian M, Layegh P. Increased Age, Neutrophil-to-Lymphocyte Ratio (NLR) and White Blood Cells Count Are Associated With Higher COVID-19 Mortality. Am J Emerg Med (2021) 40:11–4. doi: 10.1016/j.ajem.2020.12.003
12. Li L, Li J, Gao M, Fan H, Wang Y, Xu X, et al. Interleukin-8 as a Biomarker for Disease Prognosis of Coronavirus Disease-2019 Patients. Front Immunol (2021) 11:602395. doi: 10.3389/fimmu.2020.602395
13. Masso-Silva J, Moshensky A, Lam M, Odish M, Patel A, Xu L, et al. Increased Peripheral Blood Neutrophil Activation Phenotypes and Neutrophil Extracellular Trap Formation in Critically Ill Coronavirus Disease 2019 (COVID-19) Patients: A Case Series and Review of the Literature. Clin Infect Dis (2022) 74:479–89. doi: 10.1093/cid/ciab437
14. Ma A, Zhang L, Ye X, Chen J, Yu J, Zhuang L, et al. High Levels of Circulating IL-8 and Soluble IL-2r Are Associated With Prolonged Illness in Patients With Severe COVID-19. Front Immunol (2021) 12:626235. doi: 10.3389/fimmu.2021.626235
15. Zhao Y, Nie H, Hu K, Wu X, Zhang Y, Wang M, et al. Abnormal Immunity of Non-Survivors With COVID-19: Predictors for Mortality. Infect Dis Poverty (2020) 9:108. doi: 10.1186/s40249-020-00723-1
16. Zuo Y, Zuo M, Yalavarthi S, Gockman K, Madison J, Shi H, et al. Neutrophil Extracellular Traps and Thrombosis in COVID-19. J Thromb Thrombolysis (2021) 51:446–53. doi: 10.1007/s11239-020-02324-z
17. Blasco A, Coronado M, Hernández-Terciado F, Martín P, Royuela A, Ramil E, et al. Assessment of Neutrophil Extracellular Traps in Coronary Thrombus of a Case Series of Patients With COVID-19 and Myocardial Infarction. JAMA Cardiol (2020) 6:1–6. doi: 10.1001/jamacardio.2020.7308
18. Calabrese F, Pezzuto F, Fortarezza F, Hofman P, Kern I, Panizo A, et al. Pulmonary Pathology and COVID-19: Lessons From Autopsy. The Experience of European Pulmonary Pathologists. Virchows Arch (2020) 477:359–72. doi: 10.1007/s00428-020-02886-6
19. Soliman W, Sherif N, Ghanima I, El-Badawy M. Neutrophil to Lymphocyte and Platelet to Lymphocyte Ratios in Systemic Lupus Erythematosus: Relation With Disease Activity and Lupus Nephritis. Reumatol Clin (2020) 16:255–61. doi: 10.1016/j.reuma.2018.07.008
20. Leist S, Jensen K, Baric R, Sheahan T. Increasing the Translation of Mouse Models of MERS Coronavirus Pathogenesis Through Kinetic Hematological Analysis. PloS One (2019) 14:e0220126–e. doi: 10.1371/journal.pone.0220126
21. McElvaney O, McEvoy N, McElvaney O, Carroll T, Murphy M, Dunlea D, et al. Characterization of the Inflammatory Response to Severe COVID-19 Illness. Am J Respir Crit Care Med (2020) 202:812–21. doi: 10.1164/rccm.202005-1583OC
22. Hemmat N, Derakhshani A, Baghi H, Silvestris N, Baradaran B, De Summa S. Neutrophils, Crucial, or Harmful Immune Cells Involved in Coronavirus Infection: A Bioinformatics Study. Front Genet (2020) 11:641. doi: 10.3389/fgene.2020.00641
23. Haick A, Rzepka J, Brandon E, Balemba O, Miura T. Neutrophils are Needed for an Effective Immune Response Against Pulmonary Rat Coronavirus Infection, But Also Contribute to Pathology. J Gen Virol (2014) 95:578–90. doi: 10.1099/vir.0.061986-0
24. Thiam H, Wong S, Wagner D, Waterman C. Cellular Mechanisms of NETosis. Annu Rev Cell Dev Biol (2020) 36:191–218. doi: 10.1146/annurev-cellbio-020520-111016
25. Kumar S, Payal N, Srivastava V, Kaushik S, Saxena J, Jyoti A. Neutrophil Extracellular Traps and Organ Dysfunction in Sepsis. Clin Chim Acta (2021) 523:152–62. doi: 10.1016/j.cca.2021.09.012
26. Kumar S, Gupta E, Kaushik S, Srivastava V, Saxena J, Mehta S, et al. Quantification of NETs Formation in Neutrophil and its Correlation With the Severity of Sepsis and Organ Dysfunction. Clin Chim Acta (2019) 495:606–10. doi: 10.1016/j.cca.2019.06.008
27. Silva C, Wanderley C, Veras F, Sonego F, Nascimento D, Gonçalves A, et al. Gasdermin D Inhibition Prevents Multiple Organ Dysfunction During Sepsis by Blocking NET Formation. Blood (2021) 138:2702–13. doi: 10.1182/blood.2021011525
28. Veras F, Cornejo Pontelli M, Meirelles Silva C, Toller-Kawahisa J, de Lima M, Carvalho Nascimento D, et al. SARS-CoV-2-Triggered Neutrophil Extracellular Traps Mediate COVID-19 Pathology. J Exp Med (2020) 217:e20201129. doi: 10.1084/jem.20201129
29. Radermecker C, Detrembleur N, Guiot J, Cavalier E, Henket M, d'Emal C, et al. Neutrophil Extracellular Traps Infiltrate the Lung Airway, Interstitial, and Vascular Compartments in Severe COVID-19. J Exp Med (2020) 217:e20201012. doi: 10.1084/jem.20201012
30. Middleton E, He X, Denorme F, Campbell R, Ng D, Salvatore S, et al. Neutrophil Extracellular Traps Contribute to Immunothrombosis in COVID-19 Acute Respiratory Distress Syndrome. Blood (2020) 136:1169–79. doi: 10.1182/blood.2020007008
31. Barnes B, Adrover J, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford J, et al. Targeting Potential Drivers of COVID-19: Neutrophil Extracellular Traps. J Exp Med (2020) 217:e20200652. doi: 10.1084/jem.20200652
32. Ouwendijk W, Raadsen M, Kampen J, Verdijk R, von der Thusen J, Guo L, et al. Neutrophil Extracellular Traps Persist at High Levels in the Lower Respiratory Tract of Critically Ill COVID-19 Patients. J Infect Dis (2021) 223:jiab053. doi: 10.1093/infdis/jiab053
33. Li C, Xing Y, Zhang Y, Hua Y, Hu J, Bai Y. Neutrophil Extracellular Traps Exacerbate Ischemic Brain Damage. Mol Neurobiol (2022) 59:643–56. doi: 10.1007/s12035-021-02635-z
34. Pramitasuri T, Laksmidewi A, Putra I, Dalimartha F. Neutrophil Extracellular Traps in Coronavirus Disease-19-Associated Ischemic Stroke: A Novel Avenue in Neuroscience. Exp Neurobiol (2021) 30:1–12. doi: 10.5607/en20048
35. Drescher B, Bai F. Neutrophil in Viral Infections, Friend or Foe? Virus Res (2013) 171:1–7. doi: 10.1016/j.virusres.2012.11.002
36. Naumenko V, Turk M, Jenne C, Kim S. Neutrophils in Viral Infection. Cell Tissue Res (2018) 371:505–16. doi: 10.1007/s00441-017-2763-0
37. Hiroki C, Toller-Kawahisa J, Fumagalli M, Colon D, Figueiredo L, Fonseca B, et al. Neutrophil Extracellular Traps Effectively Control Acute Chikungunya Virus Infection. Front Immunol (2020) 10:3108. doi: 10.3389/fimmu.2019.03108
38. Darcy C, Minigo G, Piera K, Davis J, McNeil Y, Chen Y, et al. Neutrophils With Myeloid Derived Suppressor Function Deplete Arginine and Constrain T Cell Function in Septic Shock Patients. Crit Care (2014) 18:R163. doi: 10.1186/cc14003
39. Apel F, Andreeva L, Knackstedt L, Streeck R, Frese C, Goosmann C, et al. The Cytosolic DNA Sensor cGAS Recognizes Neutrophil Extracellular Traps. Sci Signal (2021) 14:eaax7942. doi: 10.1126/scisignal.aax7942
40. Berthelot J, Drouet L, Lioté F. Kawasaki-Like Diseases and Thrombotic Coagulopathy in COVID-19: Delayed Over-Activation of the STING Pathway? Emerg Microbes Infect (2020) 9:1514–22. doi: 10.1080/22221751.2020.1785336
41. Li M, Ferretti M, Ying B, Descamps H, Lee E, Dittmar M, et al. Pharmacological Activation of STING Blocks SARS-CoV-2 Infection. Sci Immunol (2021) 6:eabi9007. doi: 10.1126/sciimmunol.abi9007
42. Mitsios A, Arampatzioglou A, Arelaki S, Mitroulis I, Ritis K. NETopathies? Unraveling the Dark Side of Old Diseases Through Neutrophils. Front Immunol (2017) 7:678. doi: 10.3389/fimmu.2016.00678
43. Torres-Ruiz J, Absalón-Aguilar A, Nuñez-Aguirre M, Pérez-Fragoso A, Carrillo-Vázquez D, Maravillas-Montero J, et al. Neutrophil Extracellular Traps Contribute to COVID-19 Hyperinflammation and Humoral Autoimmunity. Cells (2021) 10:2545. doi: 10.3390/cells10102545
44. Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos D, Metallidis S, Rafailidis P, et al. Complement and Tissue Factor-Enriched Neutrophil Extracellular Traps Are Key Drivers in COVID-19 Immunothrombosis. J Clin Invest (2020) 130:6151–7. doi: 10.1172/JCI141374
45. Mankan A, Dau T, Jenne D, Hornung V. The NLRP3/ASC/Caspase-1 Axis Regulates IL-1β Processing in Neutrophils. Eur J Immunol (2012) 42:710–5. doi: 10.1002/eji.201141921
46. Hayward J, Mathur A, Ngo C, Man S. Cytosolic Recognition of Microbes and Pathogens: Inflammasomes in Action. Microbiol Mol Biol Rev (2018) 82:e00015–18. doi: 10.1128/mmbr.00015-18
47. Rodrigues T, de Sá K, Ishimoto A, Becerra A, Oliveira S, Almeida L, et al. Inflammasomes are Activated in Response to SARS-CoV-2 Infection and are Associated With COVID-19 Severity in Patients. J Exp Med (2021) 218:e20201707. doi: 10.1084/jem.20201707
48. Aymonnier K, Ng J, Fredenburgh L, Zambrano-Vera K, Münzer P, Gutch S, et al. Inflammasome Activation in Neutrophils of Patients With Severe COVID-19. Blood Adv (2022) 6:2001–13. doi: 10.1182/bloodadvances.2021005949
49. Chen I, Moriyama M, Chang M, Ichinohe T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front Microbiol (2019) 10:50. doi: 10.3389/fmicb.2019.00050
50. Mousavizadeh L, Ghasemi S. Genotype and Phenotype of COVID-19: Their Roles in Pathogenesis. J Microbiol Immunol Infect (2021) 54:159–63. doi: 10.1016/j.jmii.2020.03.022
51. Courjon J, Dufies O, Robert A, Bailly L, Torre C, Chirio D, et al. Heterogeneous NLRP3 Inflammasome Signature in Circulating Myeloid Cells as a Biomarker of COVID-19 Severity. Blood Adv (2021) 5:1523–34. doi: 10.1182/bloodadvances.2020003918
52. Ui Mhaonaigh A, Coughlan A, Dwivedi A, Hartnett J, Cabral J, Moran B, et al. Low Density Granulocytes in ANCA Vasculitis Are Heterogenous and Hypo-Responsive to Anti-Myeloperoxidase Antibodies. Front Immunol (2019) 10:2603. doi: 10.3389/fimmu.2019.02603
53. Morrissey S, Geller A, Hu X, Tier D, Cooke E, Ding C, et al. Emergence of Low-Density Inflammatory Neutrophils Correlates With Hypercoagulable State and Disease Severity in COVID-19 Patients. MedRxiv (2020), 20106724. doi: 10.1101/2020.05.22.20106724
54. Spijkerman R, Jorritsma N, Bongers S, Bindels B, Jukema B, Hesselink L, et al. An Increase in CD62Ldim Neutrophils Precedes the Development of Pulmonary Embolisms in COVID- 19 Patients. Scand J Immunol (2021) 93:e13023. doi: 10.1111/sji.13023
55. Carissimo G, Xu W, Kwok I, Abdad M, Chan Y, Fong S, et al. Whole Blood Immunophenotyping Uncovers Immature Neutrophil-to-VD2 T-Cell Ratio as an Early Marker for Severe COVID-19. Nat Commun (2020) 11:5243. doi: 10.1038/s41467-020-19080-6
56. Parackova Z, Zentsova I, Bloomfield M, Vrabcova P, Smetanova J, Klocperk A, et al. Disharmonic Inflammatory Signatures in COVID-19: Augmented Neutrophils’ But Impaired Monocytes’ and Dendritic Cells’ Responsiveness. Cells (2020) 9:2206. doi: 10.3390/cells9102206
57. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-Cell Landscape of Bronchoalveolar Immune Cells in Patients With COVID-19. Nat Med (2020) 26:842–4. doi: 10.1038/s41591-020-0901-9
58. Sagiv J, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, et al. Phenotypic Diversity and Plasticity in Circulating Neutrophil Subpopulations in Cancer. Cell Rep (2015) 10:562–73. doi: 10.1016/j.celrep.2014.12.039
59. Bronte V, Brandau S, Chen S, Colombo M, Frey A, Greten T, et al. Recommendations for Myeloid-Derived Suppressor Cell Nomenclature and Characterization Standards. Nat Commun (2016) 7:12150. doi: 10.1038/ncomms12150
60. Wu D, Yang X. TH17 Responses in Cytokine Storm of COVID-19: An Emerging Target of JAK2 Inhibitor Fedratinib. J Microbiol Immunol Infect (2020) 53:368–70. doi: 10.1016/j.jmii.2020.03.005
61. Torres-Ruiz J, Pérez-Fragoso A, Maravillas-Montero J, Llorente L, Mejía-Domínguez N, Páez-Franco J, et al. Redefining COVID-19 Severity and Prognosis: The Role of Clinical and Immunobiotypes. Front Immunol (2021) 12:689966. doi: 10.3389/fimmu.2021.689966
62. Aschenbrenner A, Mouktaroudi M, Krämer B, Oestreich M, Antonakos N, Nuesch-Germano M, et al. Disease Severity-Specific Neutrophil Signatures in Blood Transcriptomes Stratify COVID-19 Patients. Genome Med (2021) 13:7. doi: 10.1186/s13073-020-00823-5
63. Schulte-Schrepping J, Reusch N, Paclik D, Baßler K, Schlickeiser S, Zhang B, et al. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell (2020) 182:1419–40.e23. doi: 10.1016/j.cell.2020.08.001
64. Chevrier S, Zurbuchen Y, Cervia C, Adamo S, Raeber M, de Souza N, et al. A Distinct Innate Immune Signature Marks Progression From Mild to Severe COVID-19. Cell Rep Med (2020) 2:100166. doi: 10.1016/j.xcrm.2020.100166
65. Reizine F, Lesouhaitier M, Gregoire M, Pinceaux K, Gacouin A, Maamar A, et al. SARS-CoV-2-Induced ARDS Associates With MDSC Expansion, Lymphocyte Dysfunction, and Arginine Shortage. J Clin Immunol (2021) 41:515–25. doi: 10.1007/s10875-020-00920-5
66. Muralidharan A, Reid S. Complex Roles of Neutrophils During Arboviral Infections. Cells (2021) 10:1324. doi: 10.3390/cells10061324
67. Ni W, Yang X, Yang D, Bao J, Li R, Xiao Y, et al. Role of Angiotensin-Converting Enzyme 2 (ACE2) in COVID-19. Crit Care (2020) 24:422. doi: 10.1186/s13054-020-03120-0
68. Verdecchia P, Cavallini C, Spanevello A, Angeli F. The Pivotal Link Between ACE2 Deficiency and SARS-CoV-2 Infection. Eur J Intern Med (2020) 76:14–20. doi: 10.1016/j.ejim.2020.04.037
69. Mehta N, Kalra A, Nowacki AS, Anjewierden S, Han Z, Bhat P, et al. Association of Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers With Testing Positive for Coronavirus Disease 2019 (COVID-19). JAMA Cardiol (2020) 5:1020–6. doi: 10.1001/jamacardio.2020.1855
70. Zhang P, Zhu L, Cai J, Lei F, Qin JJ, Xie J, et al. Association of Inpatient Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers With Mortality Among Patients With Hypertension Hospitalized With COVID-19. Circ Res (2020) 126:1671–81. doi: 10.1161/CIRCRESAHA.120.317134
71. Khan Z, Shen XZ, Bernstein EA, Giani JF, Eriguchi M, Zhao TV, et al. Angiotensin-Converting Enzyme Enhances the Oxidative Response and Bactericidal Activity of Neutrophils. Blood (2017) 130:328–39. doi: 10.1182/blood-2016-11-752006
72. Hayashi F, Means T, Luster A. Toll-Like Receptors Stimulate Human Neutrophil Function. Blood (2003) 102:2660–9. doi: 10.1182/blood-2003-04-1078
73. Vaure C, Liu Y. Comparative Review of Toll-Like Receptor 4 Expression and Functionality in Different Animal Species. Front Immunol (2014) 5:316. doi: 10.3389/fimmu.2014.00316
74. Kawasaki T, Kawai T. Toll-Like Receptor Signaling Pathways. Front Immunol (2014) 5:461. doi: 10.3389/fimmu.2014.00461
75. Rocha B, Marques P, de Souza Leoratti F, Caroline Junqueira C, Batista Pereira D, do Valle Antonelli L, et al. Type I Interferon Transcriptional Signature in Neutrophils and Low-Density Granulocytes Are Associated With Tissue Damage in Malaria. Cell Rep (2015) 13:2829–41. doi: 10.1016/j.celrep.2015.11.055
76. Blanco-Melo D, Nilsson-Payant B, Liu W, Uhl S, Hoagland D, Møller R, et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell (2020) 181:1036–45.e9. doi: 10.1016/j.cell.2020.04.026
77. Banerjee A, Rapin N, Bollinger T, Misra V. Lack of Inflammatory Gene Expression in Bats: A Unique Role for a Transcription Repressor. Sci Rep (2017) 7:2232. doi: 10.1038/s41598-017-01513-w
78. Fogarty H, Townsend L, Ni Cheallaigh C, Bergin C, Martin-Loeches I, Browne P, et al. COVID19 Coagulopathy in Caucasian Patients. Br J Haematol (2020) 189:1044–9. doi: 10.1111/bjh.16749
79. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical Course and Risk Factors for Mortality of Adult Inpatients With COVID-19 in Wuhan, China: A Retrospective Cohort Study. Lancet (2020) 395:1054–62. doi: 10.1016/s0140-6736(20)30566-3
80. Zuo Y, Warnock M, Harbaugh A, Yalavarthi S, Gockman K, Zuo M, et al. Plasma Tissue Plasminogen Activator and Plasminogen Activator Inhibitor-1 in Hospitalized COVID-19 Patients. Sci Rep (2021) 11:1580. doi: 10.1038/s41598-020-80010-z
81. Petito E, Falcinelli E, Paliani U, Cesari E, Vaudo G, Sebastiano M, et al. Neutrophil More Than Platelet Activation Associates With Thrombotic Complications in COVID-19 Patients. J Infect Dis (2020) 223:jiaa756. doi: 10.1093/infdis/jiaa756
82. Wichmann D, Sperhake J, Lütgehetmann M, Steurer S, Edler C, Heinemann A, et al. Autopsy Findings and Venous Thromboembolism in Patients With COVID-19: A Prospective Cohort Study. Ann Intern Med (2020) 173:268–77. doi: 10.7326/m20-2003
83. McGonagle D, O'Donnell J, Sharif K, Emery P, Bridgewood C. Immune Mechanisms of Pulmonary Intravascular Coagulopathy in COVID-19 Pneumonia. Lancet Rheumatol (2020) 2:e437–e45. doi: 10.1016/s2665-9913(20)30121-1
84. Ackermann M, Verleden S, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med (2020) 383:120–8. doi: 10.1056/NEJMoa2015432
85. Kapoor S, Opneja A, Nayak L. The Role of Neutrophils in Thrombosis. Thromb Res (2018) 170:87–96. doi: 10.1016/j.thromres.2018.08.005
86. Comer S, Cullivan S, Szklanna P, Weiss L, Cullen S, Kelliher S, et al. COVID-19 Induces a Hyperactive Phenotype in Circulating Platelets. PloS Biol (2021) 19:e3001109. doi: 10.1371/journal.pbio.3001109
87. Folco E, Mawson T, Vromman A, Bernardes-Souza B, Franck G, Persson O, et al. Neutrophil Extracellular Traps Induce Endothelial Cell Activation and Tissue Factor Production Through Interleukin-1α and Cathepsin G. Arterioscler Thromb Vasc Biol (2018) 38:1901–12. doi: 10.1161/ATVBAHA.118.311150
88. Qi H, Yang S, Zhang L. Neutrophil Extracellular Traps and Endothelial Dysfunction in Atherosclerosis and Thrombosis. Front Immunol (2017) 8:928. doi: 10.3389/fimmu.2017.00928
89. Yost CC, Cody MJ, Harris ES, Thornton NL, McInturff AM, Martinez ML, et al. Impaired Neutrophil Extracellular Trap (NET) Formation: A Novel Innate Immune Deficiency of Human Neonates. Blood (2009) 113:6419–27. doi: 10.1182/blood-2008-07-171629
90. Feldstein L, Rose E, Horwitz S, Collins J, Newhams M, Son M, et al. Multisystem Inflammatory Syndrome in U.S. Children and Adolescents. N Engl J Med (2020) 383:334–46. doi: 10.1056/NEJMoa2021680
91. Yoshida Y, Takeshita S, Kawamura Y, Kanai T, Tsujita Y, Nonoyama S, et al. Enhanced Formation of Neutrophil Extracellular Traps in Kawasaki Disease. Pediatr Res (2020) 87:998–1004. doi: 10.1038/s41390-019-0710-3
92. Muto T, Masuda Y, Numoto S, Kodama S, Yamakawa K, Takasu M, et al. White Blood Cell and Neutrophil Counts and Response to Intravenous Immunoglobulin in Kawasaki Disease. Global Pediatr Health (2019) 6:2333794X19884826. doi: 10.1177/2333794X19884826
93. Pouletty M, Borocco C, Ouldali N, Caseris M, Basmaci R, Lachaume N, et al. Paediatric Multisystem Inflammatory Syndrome Temporally Associated With SARS-CoV-2 Mimicking Kawasaki Disease (Kawa-COVID-19): A Multicentre Cohort. Ann Rheum Dis (2020) 79:999–1006. doi: 10.1136/annrheumdis-2020-217960
94. Carter M, Fish M, Jennings A, Doores K, Wellman P, Seow J, et al. Peripheral Immunophenotypes in Children With Multisystem Inflammatory Syndrome Associated With SARS-CoV-2 Infection. Nat Med (2020) 26:1701–7. doi: 10.1038/s41591-020-1054-6
95. Ramaswamy A, Brodsky N, Sumida T, Comi M, Asashima H, Hoehn K, et al. Immune Dysregulation and Autoreactivity Correlate With Disease Severity in SARS-CoV-2-Associated Multisystem Inflammatory Syndrome in Children. Immunity (2021) 54:1083–95. doi: 10.1016/j.immuni.2021.04.003
96. Gaber T, Hahne M, Strehl C, Hoff P, Dörffel Y, Feist E, et al. Disentangling the Effects of Tocilizumab on Neutrophil Survival and Function. Immunol Res (2016) 64:665–76. doi: 10.1007/s12026-015-8770-x
97. Lok LSC, Farahi N, Juss JK, Loutsios C, Solanki CK, Peters AM, et al. Effects of Tocilizumab on Neutrophil Function and Kinetics. Eur J Clin Invest (2017) 47:736–45. doi: 10.1111/eci.12799
98. Molloy EJ, Bearer CF. COVID-19 in Children and Altered Inflammatory Responses. Pediatr Res (2020) 88:340–1. doi: 10.1038/s41390-020-0881-y
99. Molloy EJ, Lavizzari A, Klingenberg C, Profit J, Zupancic JAF, Davis AS, et al. Neonates in the COVID-19 Pandemic. Pediatr Res (2021) 89:1038–40. doi: 10.1038/s41390-020-1096-y
100. Sinha P, Mostaghim A, Bielick CG, McLaughlin A, Hamer DH, Wetzler LM, et al. Early Administration of Interleukin-6 Inhibitors for Patients With Severe COVID-19 Disease is Associated With Decreased Intubation, Reduced Mortality, and Increased Discharge. Int J Infect Dis (2020) 99:28–33. doi: 10.1016/j.ijid.2020.07.023
101. Butler C, Yu L, Dorward J, Gbinigie O, Hayward G, Saville B, et al. Doxycycline for Community Treatment of Suspected COVID-19 in People at High Risk of Adverse Outcomes in the UK (PRINCIPLE): A Randomised, Controlled, Open-Label, Adaptive Platform Trial. Lancet Respir Med (2021) 9:1010–20. doi: 10.1016/S2213-2600(21)00310-6
102. Castellani S, D'Oria S, Diana A, Polizzi A, Di Gioia S, Mariggiò M, et al. G-CSF and GM-CSF Modify Neutrophil Functions at Concentrations Found in Cystic Fibrosis. Sci Rep (2019) 9:12937. doi: 10.1038/s41598-019-49419-z
103. Mehta P, Porter J, Manson J, Isaacs J, Openshaw P, McInnes I, et al. Therapeutic Blockade of Granulocyte Macrophage Colony-Stimulating Factor in COVID-19-Associated Hyperinflammation: Challenges and Opportunities. Lancet Respir Med (2020) 8:822–30. doi: 10.1016/s2213-2600(20)30267-8
104. De Luca G, Cavalli G, Campochiaro C, Della-Torre E, Angelillo P, Tomelleri A, et al. GM-CSF Blockade With Mavrilimumab in Severe COVID-19 Pneumonia and Systemic Hyperinflammation: A Single-Centre, Prospective Cohort Study. Lancet Rheumatol (2020) 2:e465–e73. doi: 10.1016/S2665-9913(20)30170-3
105. Tavakkoli M, Wilkins C, Mones J, Mauro M. A Novel Paradigm Between Leukocytosis, G-CSF Secretion, Neutrophil-To-Lymphocyte Ratio, Myeloid-Derived Suppressor Cells, and Prognosis in Non-Small Cell Lung Cancer. Front Oncol (2019) 9:295. doi: 10.3389/fonc.2019.00295
106. Lang FM, Lee KMC, Teijaro JR, Becher B, Hamilton JA. GM-CSF-Based Treatments in COVID-19: Reconciling Opposing Therapeutic Approaches. Nat Rev Immunol (2020) 20:507–14. doi: 10.1038/s41577-020-0357-7
107. Kalil A, Patterson T, Mehta A, Tomashek K, Wolfe C, Ghazaryan V, et al. Baricitinib Plus Remdesivir for Hospitalized Adults With Covid-19. N Engl J Med (2021) 384:795–807. doi: 10.1056/NEJMoa2031994
108. Abizanda P, Calbo Mayo J, Mas Romero M, Cortés Zamora E, Tabernero Sahuquillo M, Romero Rizos L, et al. Baricitinib Reduces 30-Day Mortality in Older Adults With Moderate-to-Severe COVID-19 Pneumonia. J Am Geriatr Soc (2021) 69:2752–8. doi: 10.1111/jgs.17357
109. Guimarães P, Quirk D, Furtado R, Maia L, Saraiva J, Antunes M, et al. Tofacitinib in Patients Hospitalized With Covid-19 Pneumonia. N Engl J Med (2021) 385:406–15. doi: 10.1056/NEJMoa2101643
110. McElvaney OJ, Zaslona Z, Becker-Flegler K, Palsson-McDermott EM, Boland F, Gunaratnam C, et al. Specific Inhibition of the NLRP3 Inflammasome as an Antiinflammatory Strategy in Cystic Fibrosis. Am J Respir Crit Care Med (2019) 200:1381–91. doi: 10.1164/rccm.201905-1013OC
111. Torene R, Nirmala N, Obici L, Cattalini M, Tormey V, Caorsi R, et al. Canakinumab Reverses Overexpression of Inflammatory Response Genes in Tumour Necrosis Factor Receptor-Associated Periodic Syndrome. Ann Rheum Dis (2017) 76:303–9. doi: 10.1136/annrheumdis-2016-209335
112. Wu R, Wang L, Kuo H, Shannar A, Peter R, Chou P, et al. An Update on Current Therapeutic Drugs Treating COVID-19. Curr Pharmacol Rep (2020) 6:56–70. doi: 10.1007/s40495-020-00216-7
113. Absalón-Aguilar A, Rull-Gabayet M, Pérez-Fragoso A, Mejía-Domínguez N, Núñez-Álvarez C, Kershenobich-Stalnikowitz D, et al. Colchicine Is Safe Though Ineffective in the Treatment of Severe COVID-19: A Randomized Clinical Trial (COLCHIVID). J Gen Intern Med (2022) 37:4–14. doi: 10.1007/s11606-021-07203-8
114. RECOVERY CG. Colchicine in Patients Admitted to Hospital With COVID-19 (RECOVERY): A Randomised, Controlled, Open-Label, Platform Trial. Lancet Respir Med (2021) 9:1419–26. doi: 10.1016/S2213-2600(21)00435-5
115. Kyriazopoulou E, Huet T, Cavalli G, Gori A, Kyprianou M, Pickkers P, et al. Effect of Anakinra on Mortality in Patients With COVID-19: A Systematic Review and Patient-Level Meta-Analysis. Lancet Rheumatol (2021) 3:e690–e7. doi: 10.1016/S2665-9913(21)00216-2
116. Kyriazopoulou E, Poulakou G, Milionis H, Metallidis S, Adamis G, Tsiakos K, et al. Early Treatment of COVID-19 With Anakinra Guided by Soluble Urokinase Plasminogen Receptor Plasma Levels: A Double-Blind, Randomized Controlled Phase 3 Trial. Nat Med (2021) 27:1752–60. doi: 10.1038/s41591-021-01499-z
117. Derde L, Gordon A, Mouncey P, Al-Beidh F, Rowan K, Nichol A, et al. Effectiveness of Tocilizumab, Sarilumab, and Anakinra for Critically Ill Patients With COVID-19 The REMAP-CAP COVID-19 Immune Modulation Therapy Domain Randomized Clinical Trial. medRxiv (2021). doi: 10.1101/2021.06.18.21259133
118. European Medicines Agency. EMA Recommends Approval for Use of Kineret in Adults With COVID-19 (2021). Available at: https://www.ema.europa.eu/en/news/ema-recommends-approval-use-kineret-adults-covid-19 (Accessed April 26, 2022).
119. Molloy E. The Doctor’s Dilemma: Lessons From GB Shaw in a Modern Pandemic COVID-19. Pediatr Res (2021) 89:701–3. doi: 10.1038/s41390-020-0927-1
120. Schneider C, Wicki S, Graeter S, Timcheva T, Keller C, Quast I, et al. IVIG Regulates the Survival of Human But Not Mouse Neutrophils. Sci Rep (2017) 7:1296. doi: 10.1038/s41598-017-01404-0
121. Sakoulas G, Geriak M, Kullar R, Greenwood K, Habib M, Vyas A, et al. Intravenous Immunoglobulin Plus Methylprednisolone Mitigate Respiratory Morbidity in Coronavirus Disease 2019. Crit Care Explor (2020) 2:e0280. doi: 10.1097/CCE.0000000000000280
122. Liu X, Cao W, Li T. High-Dose Intravenous Immunoglobulins in the Treatment of Severe Acute Viral Pneumonia: The Known Mechanisms and Clinical Effects. Front Immunol (2020) 11:1660. doi: 10.3389/fimmu.2020.01660
123. Lomas D, Ip M, Chamba A, Stockley R. The Effect OfIn Vitro AndIn Vivo Dexamethasone on Human Neutrophil Function. Agents Actions (1991) 33:279–85. doi: 10.1007/BF01986574
124. RECOVERY Collaborative Group, Horby P, Pessoa-Amorim G, Peto L, Brightling C, Sarkar R, et al. Tocilizumab in Patients Admitted to Hospital With COVID-19 (RECOVERY): Preliminary Results of a Randomised, Controlled, Open-Label, Platform Trial. Lancet (2021) 397:1637–45. doi: 10.1016/S0140-6736(21)00676-0
Keywords: neutrophil, COVID-19, SARS-CoV-2, innate immunity, inflammation
Citation: McKenna E, Wubben R, Isaza-Correa JM, Melo AM, Mhaonaigh AU, Conlon N, O’Donnell JS, Ní Cheallaigh C, Hurley T, Stevenson NJ, Little MA and Molloy EJ (2022) Neutrophils in COVID-19: Not Innocent Bystanders. Front. Immunol. 13:864387. doi: 10.3389/fimmu.2022.864387
Received: 28 January 2022; Accepted: 29 April 2022;
Published: 01 June 2022.
Edited by:
Antonio Condino-Neto, University of São Paulo, BrazilReviewed by:
José Jiram Torres-Ruiz, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán (INCMNSZ), MexicoJorge Masso-Silva, University of California, San Diego, United States
Copyright © 2022 McKenna, Wubben, Isaza-Correa, Melo, Mhaonaigh, Conlon, O’Donnell, Ní Cheallaigh, Hurley, Stevenson, Little and Molloy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eleanor J. Molloy, ZWxlYW5vci5tb2xsb3lAdGNkLmll