 Jianan Zhao1,2,3
Jianan Zhao1,2,3 Kai Wei1,2,3
Kai Wei1,2,3 Cen Chang1,2,3
Cen Chang1,2,3 Lingxia Xu1,2,3
Lingxia Xu1,2,3 Ping Jiang1,2,3
Ping Jiang1,2,3 Shicheng Guo4,5*
Shicheng Guo4,5* Steven J. Schrodi4,5*
Steven J. Schrodi4,5* Dongyi He1,2,3,6*
Dongyi He1,2,3,6*- 1Guanghua Clinical Medical College, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 2Department of Rheumatology, Shanghai Guanghua Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 3Institute of Arthritis Research in Integrative Medicine, Shanghai Academy of Traditional Chinese Medicine, Shanghai, China
- 4Computation and Informatics in Biology and Medicine, University of Wisconsin-Madison, Madison, WI, United States
- 5Department of Medical Genetics, School of Medicine and Public Health, University of Wisconsin-Madison, Madison, WI, United States
- 6Arthritis Institute of Integrated Traditional and Western medicine, Shanghai Chinese Medicine Research Institute, Shanghai, China
Rheumatoid arthritis (RA) is an autoimmune disease that can cause joint damage and disability. Epigenetic variation, especially DNA methylation, has been shown to be involved in almost all the stages of the pathology of RA, from autoantibody production to various self-effector T cells and the defects of protective T cells that can lead to chronic inflammation and erosion of bones and joints. Given the critical role of T cells in the pathology of RA, the regulatory functions of DNA methylation in T cell biology remain unclear. In this review, we elaborate on the relationship between RA pathogenesis and DNA methylation in the context of different T cell populations. We summarize the relevant methylation events in T cell development, differentiation, and T cell-related genes in disease prediction and drug efficacy. Understanding the epigenetic regulation of T cells has the potential to profoundly translate preclinical results into clinical practice and provide a framework for the development of novel, individualized RA therapeutics.
Introduction
Rheumatoid arthritis (RA) is a chronic and systemic inflammatory illness that causes persistent synovial inflammation and bone and joint damage. RA increases the risk of various diseases such as respiratory infections, osteoporosis, cardiovascular disease, urinary tract disease, and cancer (1). The incidence in women is higher than that in men. RA has a somewhat bimodal distribution of age of onset, with the largest fraction initially being diagnosed in the 20-40s, and then a smaller fraction being diagnosed roughly in the 60s, and it can appear at any stage (1). Typically, numerous autoimmune antibodies and cytokines are present in the pre-onset stage, and this molecular signature further advances with the clinical onset of synovial hyperplasia and bone/joint damage. In the absence of effective therapy, the disease typically progresses, resulting in impairment of both physical and mental health (2).
Many factors influence RA, including heredity, metabolism, environmental factors, and microbiota– and involve a complex interplay of genetic predisposition, activation of a variety of immune cells, and imbalance of pro- and anti-inflammatory mechanisms. This pathobiological immune state ultimately drives the failure to regulate immune tolerance, causing the development of autoimmune diseases targeting the synovium (3–5) (see Figure 1). Current research has discovered that numerous gene variants contribute to illness development, implying that each susceptible gene variant contributes to the disease to varying degrees (6). In addition to gene variations, the interaction between the environment and genes influences gene expression, which is heavily influenced by epigenetic modifications. This includes DNA methylation, histone post-translational modification, miRNA, and, eventually, immune system regulation (6). DNA methylation refers to the addition of a methyl group to cytosine guanine (CG) dinucleotide (CpG). Two-thirds of CpGs in the human genome can be methylated (7). Under normal conditions, the 5-methylcytosine of CpG islands in DNA methylation prevents transcription factor complexes from binding to DNA through multiple processes, resulting in the inhibition of gene expression. On the other hand, DNA hypermethylation of gene promoters can enhance downstream gene transcription through a variety of mechanisms. Promoter hypermethylation, for example, blocks transcription inhibitors from binding to DNA (8). Hypermethylated advocates engage with enhancers to attract transcriptional activators and improve downstream gene transcription. Hypermethylation also promotes gene expression by binding to enhancers instead of insulators. Furthermore, in mammals, the hypermethylated promoter stimulates transcription of the first promoter of multiple variable promoters, resulting in increased target gene transcript output and decreased alternative spliceosome content (8).
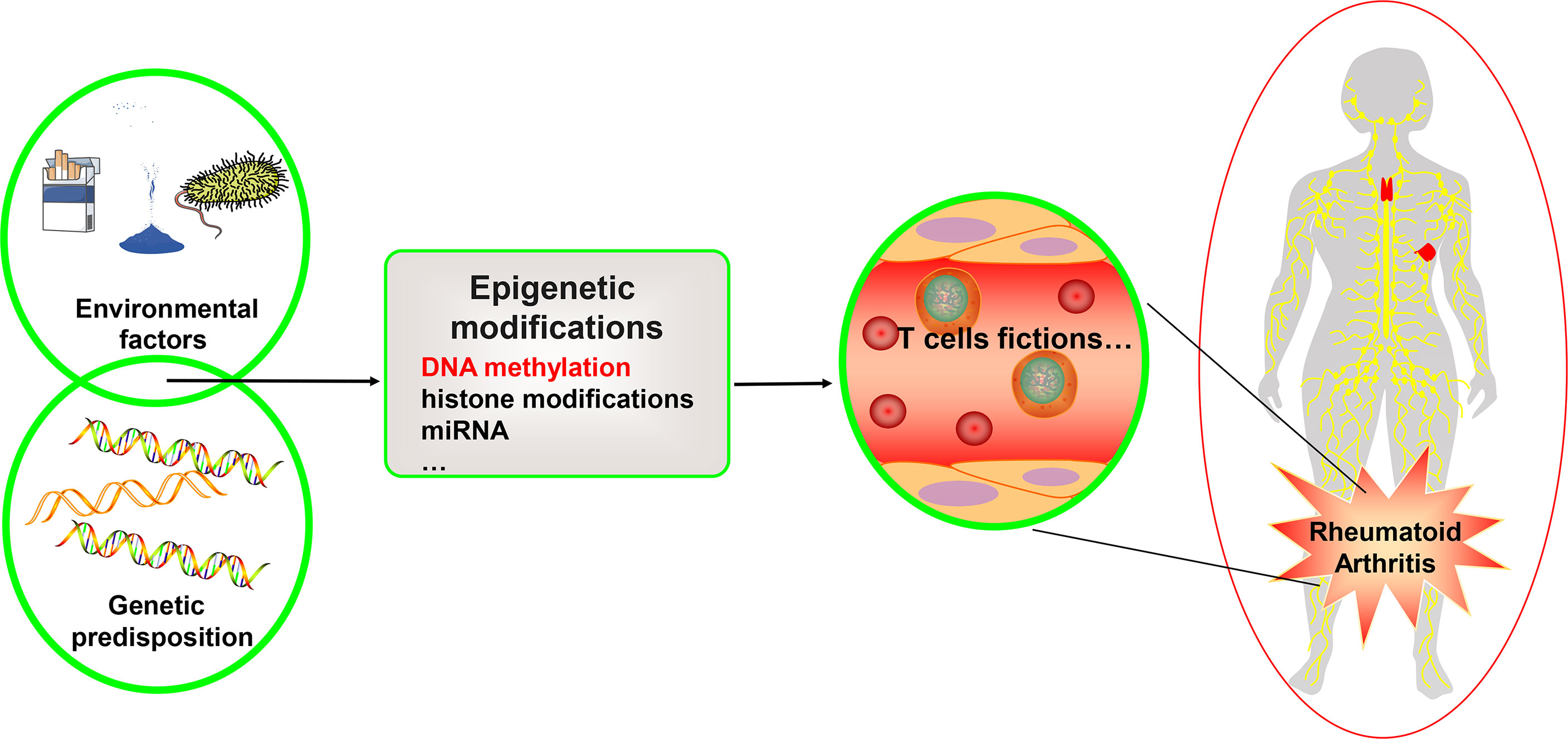
Figure 1 Genetics, environment, and DNA methylation are interrelated in T cell function for rheumatoid arthritis. Rheumatoid arthritis is a heritable autoimmune disease. Some gene variants can increase risk of disease. Environmental factors interact with the genetic background and can affect the function of the T cell subset, thus affecting rheumatoid arthritis through different epigenetic mechanisms, including DNA methylation, histone modifications, and microRNAs. We focus on the DNA methylation landscape in T cells.
T cells primarily engage with antigen-presenting cells and mix with antigen peptides provided by human leukocyte antigen (HLA) to create complexes that further mediate immunological responses (9). DNA methylation can influence various degrees of gene expression or selective splicing, both of which are associated with RA (10). The biological roles of the majority of the differential genes were shown to be associated with T cell development and activation by assessing gene expression and DNA methylation of CD4+ T cells in RA patients and healthy individuals (11). Many differentially methylated sites are found in T cell regulatory regions. Most gene variants were moderately or strongly correlated with methylation quantitative trait loci (meQTLs). Differentially variable positions (DVPs) are a type of CpG site. By studying the methylation of whole-genome DNA in monozygotic twins with discordant conditions, it was found that 1171 RA-related DVPs, of which 763 DVPs are in a highly variable state in RA twins, were mainly enriched in gene bodies and the 3’UTR of genes. Of the 763 DVPs, 563 were hypomethylated (12). Pathway analysis revealed that two of the top five were linked to T cells and cytokines (12). This suggests that the differential expression of RA genes may be partly driven by DNA methylation and are linked to variants of disease susceptibility genes that contribute to disease progression (11). In addition, there are several aberrant T-cell patterns in RA patients. In RA patients, both CD4+ naïve T cells and memory T cells exhibit premature aging and excessive telomere degradation (13). Furthermore, the ataxia telangiectasia mutated defect of T cells in RA patients leads to the destruction of the DNA repair mechanism (14). It enhances the sensitivity of T cells to apoptosis, while the apoptosis attrition of naive T cells ultimately accelerates immune senescence and the proliferation of autoimmune T cells (14). DNA methylation may cause aberrant activation and premature aging of T cells, contributing to atypical T-cell patterns in RA. In this article, we summarize and discuss the relationship between T cell DNA methylation and the pathological mechanism of RA in order to provide a theoretical foundation and reference for the development of novel clinical diagnosis and treatment programs from an epigenetic perspective (See Table 1).

Table 1 Relationship between T cell DNA methylation-related genes and rheumatoid arthritis.
The Potential Connection Between DNA Methylation of CD4+ T Cells and Rheumatoid Arthritis
Several intriguing results were discovered using transcriptomic data, methylation data, and bioinformatics to analyze the differential genes and differentially methylated genes of diverse immune cells in RA (54). First, the presence of anti-lymphocyte antibodies may have resulted in a considerable reduction in the total number of T and B cells in the peripheral blood of patients with RA compared to healthy controls (54, 55). Second, RA patient CD4+ T cells exhibit hypermethylated genes, including JUN, STAT1, PTEN, and CD44, compared to healthy controls, whereas hypomethylated genes include KRAS and ALB. Additionally, the expression levels of STAT5B, SOCS3, JUN, STAT1, KRAS, ALB, and CD44 were increased in RA. PTEN, FGFR2, and DICER1 expression levels were reduced. Gene ontology (GO) enrichment studies of differentially methylated genes are connected to T cell biological processes, suggesting that the regulation of CD4+ cells via DNA methylation plays an important role in RA (54). Finally, the analysis of the differentially methylated genes in the Kyoto Encyclopedia of Genes and Genomes (KEGG) showed that it was mainly related to thyroid hormone and estrogen signaling, hepatitis B virus (HBV), and the mitogen-activated protein kinase (MAPK) pathway (54). The results of the KEGG pathway analysis suggested various potential connections between gene DNA methylation and RA. Patients with RA have a higher prevalence of thyroid disease (56, 57). It often shows a certain degree of hypothyroidism at onset, which is used as a sign of autoimmune thyroiditis (58). RA and HBV are interconnected and have been widely studied (59). For example, RA patients receiving tofacitinib can induce HBV reactivation in HBsAg+ and HBsAg-/HBcAb+ patients (60), and HBV exposure correlates with rheumatoid factor (RF) (61). The increased incidence of RA in postmenopausal women may be related to the lack of estrogen. Studies have found that estrogen can promote the degradation of acid-sensitive ion channel 1a protein and inhibit the cytotoxicity of acidosis through autophagy-lysosome-dependent pathways, thereby protecting chondrocytes (62, 63).
By comparing the number of differentially methylated positions of memory CD4+ T cells and CD4+ naive T cells in RA patients, it was found that the number of the former was significantly greater than the latter. Most of the DNA methylation of differentially methylated positions (DMP) in RA patients with active disease is increased (64). Among the 101 RA genes recently identified (65), more than 30 overlapped with DMP (P=0.1 after 1000 permutations). One of these, UBASH3A, was found in CD4+ memory T cells. Its introns contain both single nucleotide polymorphisms (SNPs) that are correlated with RA risk r and two DMP and show a decrease in DNA methylation, which may increase gene expression (64). The protein encoded by UBASH3A, ubiquitin-associated and SH3 domain-containing A protein, primarily plays a role in the presentation of antigens to T cells and dampens T cell activity (41, 42). Therefore, the expression of UBASH3A may increase the expression of genes through DNA methylation to promote antigen presentation to autoimmune T cells. UBASH3A SNPs (rs1893592, rs3788013), rheumatoid factor, cyclic citrullinated peptide (anti-CCP), disease activity score in 28 joints (DAS28), and C-reactive protein (CRP) were all significantly correlated with RA susceptibility (66, 67). By studying peripheral blood samples from RA patients, researchers have found that CD4+ T memory cells have one hypermethylated CpG site, and CD4+ naïve T cells have 18 CpG sites; six are hypomethylated, and 12 are hypermethylated. The most important top 10 sites were located in TYK2, PRKAR1B, ABCC4, COMT, CAI2, MCF2L, GALNT9, C7orf50, and two intergenic regions (15). All 19 CpG sites were related to RA susceptibility. The hypomethylation status of CpG sites in the promoter regions of GATA3 and GATA3-AS1 (cg17566118 and cg15852223) was also found (15). The meQTL-mediated differentially methylated regions lead to the downregulation of GATA3 and CD83 expression (11). GATA3 is a transcription factor that drives the differentiation of anti-inflammatory Th2 cells, promotes the enhancement of the interleukin (IL)-4 promoter, and regulates the expression of the IL-4 gene. The decrease in its expression may further lead to an imbalance in anti-inflammatory mechanisms (48, 49). Oral administration of a-L-Guluronic acid, a non-steroidal anti-inflammatory drug (NSAID), can significantly increase the expression of IL-4 and GATA3 in peripheral blood mononuclear cells (PBMCs) and improve the symptoms of related diseases in RA patients (68). CD4+ T cells lacking CD83 exhibit enhanced pro-inflammatory differentiation ability under in vitro stimulation; for example, more interferon-gamma (IFN-γ) secretion and more Th1 and Th17 cell differentiation (31). The study detected genome-wide DNA methylation of CD4+ T cells from RA patients of Han Chinese ancestry. This reveals RA-related DNA methylation patterns in East Asian populations (18). There were 383 hypomethylation genes and 785 hypermethylated genes in CD4+ T cells. HDAC4, NXN, TBCD, and TMEM61 are the top four hypermethylated genes. HLA-DRB6, HLA-DQA1, HLA-E, ITIH3, TCN2, PRDM16, SLC1A5, and GALNT9 were hypomethylated. HLA-DQB1 is hypermethylated in the CpG island region, and the CpG framework is hypomethylated (18). In addition, the DNA methylation mechanism may also be a reason for the higher prevalence of RA in women. The hypomethylation status of CD40L in CD4+ T cells of female patients with RA is conducive to the expression of CD40L and partially explains the higher prevalence of RA in women (43). Consistent with this, the expression of 1,25(OH)2D3 and the vitamin D receptor (VDR) in the peripheral blood of RA patients is reduced. 1,25(OH)2D3 and VDR can inhibit the PKCδ/ERK pathway and promote DNA methylation of CD11a, CD70, and CD40L to inhibit CD4+ T cell activation (44).
In this section, we summarize the changes in CD4+ T cell-related differential DNA methylation, such as JUN, PTEN, and STAT1. Next, we explored the potential connection between its related genes and RA. Cell survival and death of CD4+ T cells play an essential role in RA (46). The upregulation of c-Jun expression can activate the MAPK signaling pathway and regulate cell proliferation, cell cycle, and apoptosis (20). Both STAT1 and STAT3 are related to IL-6 inflammatory factors and are involved in the course of RA (21). Gp130/STAT1 may increase the joint infiltration of T cells in antigen-induced arthritis (AIA) animal models (69). PTEN is mainly involved in cell proliferation and pro-inflammatory factors. Overexpression of PTEN can significantly reduce the expression of pro-inflammatory factors, chemokines, vascular cell adhesion factor-1, and vascular endothelial growth factor-α in AIA experimental animals. Consistent with the previous, the expression of PTEN is regulated by DNA methylation and is accompanied by changes in the AKT signal (22), and the methylation of PTEN has been shown to promote inflammation and activation of RA fibroblast-like synoviocyte (FLS) (23). Shikonin significantly reduces angiogenesis in RA. The mechanism may involve up-regulating the expression of PTEN, downregulating the expression of tumor necrosis factor-a (TNF-a), vascular endothelial growth factor 2, and IL-1β, and inhibiting the phosphorylation and gene expression of ERK1/2, JNK1/2, and P38 induced by TNF-α (70). In addition, PTEN is regulated by various miRNAs. Tocilizumab can regulate the proliferation of RA FLS and the production of pro-inflammatory factors via the MIR31HG/miR-214/PTEN/AKT axis (71). Therefore, PTEN may be a vital factor in inflammation in RA. CD44 is mainly used as a cell-surface adhesion molecule (72). The anti-CD44 antibody can significantly increase the incidence of experimental arthritis mice induced by type II collagen (24). Studies have shown that the preclinical stage of the disease in the K/BxN mouse model reduced CD4+ T lymphocytes. However, in the early clinical setting, most of the CD4+ T cells begin to express CD44, which is regarded as a sign of cell homeostasis expansion (73). Selenium supplementation can improve the joint pathological performance of CIA mouse models, reduce the number of CD4+CD44+ receptor activators of nuclear factor (NF)-κB ligand (RANKL) +T cells, and inhibit the expression of RANKL in mouse Th17 cells in vitro (74). Osteopontin in the synovial fluid of patients with RA induces Th17 differentiation and promotes the production of pro-inflammatory factor IL-17 through the IL-6/STAT3 pathway and CD44, CD29, and retinoic acid-related orphan receptor (ROR). The mechanism may be CD44 binding domain of CD4+ T cells induces H3 acetylation of the IL-17A promoter and promotes interaction with ROR (75). The overexpression of KRAS and BRAF in CD4+ T cells lowers the activation threshold of T cells to respond more efficiently to autoantigens in RA. This is related to the level of phosphorylated ERK (38).
The Effect of DNA Methylation on the Differentiation of CD4+ T Cells
T cells have cellular heterogeneity and can differentiate into different subsets under stimulation by different cytokines, such as Treg cells, Th1 cells, Th2 cells, Th17 cells, and T-follicular helper (Tfh) cells, which play different roles in RA (76). Th1 and Th17 cell subsets contribute to inflammation in RA. Many studies have shown that the inhibition of Th1 and Th17 cell subsets can alleviate RA (77–79). Th2 cell subsets can inhibit Th1 and Th17 cell differentiation and function by secreting IL-4 as a mechanism to antagonize inflammation (80). In early patients with RA, Th2 cells and their secreted cytokines are few, and the differentiation of CD4+ T cells is severely unbalanced, which significantly promotes the production of chronic inflammation (81). Studies have shown that T cells lacking DNA methyltransferase (DNMT) 3a are activated and produce IL-4 and IFNG site hypomethylation. When T cells with Th2 cell characteristics are biased to induce Th1 cells, they will express more IFN-γ, indicating that DNA methylation plays a vital role in controlling T cell differentiation according to the microenvironment (82). Further research shows that DNMT3a is an essential element that controls the differentiation of Th2 cells, expresses IL-13, and maintains the function of Th2 cells to inhibit inflammation (40). There is a substantial body of literature on how epigenetic mechanisms, including DNA methylation, affect the differentiation of lymphoid T cells (26, 83).
In response to IL-12 stimulation, CD4+ T cells differentiate into Th1 cells and produce IFN-γ. When activated for the first time, intracellular IFNG can be expressed under the stimulus of TCR signals and can bind to the transcription factor T-bet or STAT4. IL- 12 can also reactivate the IFNG of memory Th1 cells for a second time (84–86). Studies have shown that the hypomethylation pattern of the IFNG promoter region and the CNS-1 region may contribute to the differentiation of Th1 and establish a stable epigenetic imprint, which is conducive to the reactivation of Th1 and promotes inflammation (39). Many meaningful results have been obtained by detecting the differential DNA methylation sites of monocytes, memory CD4+ T cells, and CD4+ naïve T cells in patients with RA (87). First, 1047 and 913 differentially methylated CpGs were detected in CD4+ naïve T cells and memory CD4+ T cells, respectively. Second, CD4+ T cells in early RA patients seem to develop in the direction of Th17 cells. Differential methylation of TBX21 (T-bet in mice) may lead to a decrease in Th1 cell differentiation (85). At the same time, the IL-17 and IL-17R of CD4+naïve T cells undergo differential methylation modification, which tend to differentiate into pro-inflammatory Th17 cells and interact with TNF. Differential methylation of IFN-related genes affects gene expression (87). As previously reported, it may indicate the characteristics of chronic inflammation and predict the progression of arthritis in early RA (88, 89). Interestingly, when T cells were treated with DNA methyltransferase inhibitors, the expression of STAT4 was significantly upregulated. The shortening of the methylation site of the proximal regulatory element of its promoter greatly enhanced transcriptional activity, indicating that STAT4 in T cells is affected by DNA methylation regulation and is not regulated by promoter polymorphism (90). Decitabine, a DNA methylation inhibitor, can significantly improve the clinical symptoms of experimental arthritis mice induced by type II collagen, inhibit pro-inflammatory cytokines of Th1 and Th17 cells, reduce the production of anti-type II collagen antibodies (91), and promote the production of cytokines by Th2 cells (92). In addition, Tfh cells mainly promote B cell antibody maturation and development (93). The binding of BCL6, an essential molecule for Tfh cell differentiation, is associated with a decrease in 5-hydroxymethylcytosine (5hmC), implying a link between Tfh and DNA methylation (94). Hence, DNA methylation can regulate the differentiation of various T cell subsets via different mechanisms that affect the development of patients with RA.
The Defects of DNA Methylation in Treg Cells Promote Inflammation
Treg cells are defined as CD4+CD25+FOXP3+; their expression defects contribute to the production of autoimmunity because their functions can inhibit the proliferation and differentiation of autoimmune T cells (95). Treg cells also have cellular heterogeneity, mainly divided into resting-state CD45+FOXP3low Treg cells, activated CD45RA-FOXP3hi, and CD45- FOXP3lowTreg cells, and a subset of Treg cells expressing HLA-DR with early contact-dependent inhibition (96, 97). FOXP3 is the most commonly used marker for identifying Treg cells and is an essential transcription factor for the development and maintenance of suppressive function in Treg cells (26). There is a differential methylation region (DMR) upstream of the Foxp3 promoter in RA Treg cells, with enhancer activity sensitive to methylation-induced silencing. Both the expression of DMR and DNMT1/3 in RA Treg cells were downregulated, and the methylation of DMR was negatively correlated with the mRNA expression of Foxp3. RF-negative and-positive Treg cells also express Foxp3 differently. RF-positive Treg cells express lower Foxp3 levels and have a higher DMR methylation status (98). Tumor necrosis factor receptor 2 (TNFR2) is an important molecule that regulates Foxp3 expression. It can inhibit DNA methylation of the Foxp3 promoter by maintaining its expression in Treg cells. The lack of TNFR2 leads to the deterioration of experimental animal arthritis models, accompanied by a decrease in the number and a defect in the suppressive function of Treg cells. It leads Treg cells to inflammatory phenotypic differentiation of Th17 cells, which attempts to suppress inflammatory cells but fails (99). Consistent with this, the number of a subset of Treg cells expanded during the active stage of RA, similar to the pathogenic T cell phenotype (100). Similarly, the degree of methylation of Foxp3 in Treg cells of TNFR2 knockout mice increased. Under in vitro culture conditions, TNF upregulates Foxp3 expression in Treg cells through TNFR2 signaling, and TNFR2+ Treg cells increase in patients with RA receiving anti-TNF-α therapy for three months (101). Regular expression of CD83, SMAD7, and CTLA4 can maintain the functional integrity of Treg cells. Mice lacking CD83 cause abnormal Treg cell differentiation and promote inflammation (31, 32). The down-regulation of CD83 and SMAD7 may be caused by meQTL-mediated differentially methylated regions in RA (11). SMAD7 is significantly reduced in the synovial tissue of patients with RA, and the TGF-β/SMAD3 signal is enhanced considerably, further intensifying the pro-inflammatory response of Th1 and Th17 cells (102). Experimental arthritis mice lacking SMAD7 developed severe arthritis, including joint swelling, synovial hyperplasia, bone destruction, and immune cell infiltration. The mechanism may be related to the imbalance of inhibitory function in Treg cells, the inflammatory response of Th1 and Th17 cells, and the overactivation of the TGF-β/SMAD3/IL-6 pro-inflammatory signaling pathway (102). In addition, the Treg inhibitory function of patients with RA is impaired, and the expression of CTLA4 is downregulated, which may be due to methylation of the CTLA4 promoter. It causes Treg cells to fail to induce the expression of tryptophan-degrading enzyme indoleamine 2,3 -dioxygenase (IDO) and the activation of the immunomodulatory kynurenine pathway (103). Decitabine, a DNA methylation inhibitor, can induce the expansion of RA Treg cells through an IDO-dependent pathway and promote the apoptosis of Th1 and Th17 cells to improve RA (104). DNA methylation may affect Treg cell function, further influencing autoimmune T cells and inflammation in RA.
The DNA Methylation of T Cells as a Potential Biomarker for Rheumatoid Arthritis
The identification of DNA methylation in RA holds promise for augmenting and improving the clinical diagnosis process. At present, biomarkers for the diagnosis of RA include anti-citrullinated protein antibody (ACPA) and RF. The sensitivity is insufficient, and new markers are still needed to assist ACPA and RF, enabling more accurate diagnoses (105). The addition of other molecular tests to the markers of seropositivity offers an opportunity to increase the sensitivity of the combined test. Prime candidates for this expansion of the diagnostic utility of RA include genetic markers and differentially methylated regions. There are 1951 differentially methylated CpGs of T lymphocytes in patients with early RA, 60% of which were hypermethylated, representing 1216 genes. Of the top 15 genes, C6orf47, AGPAT9, DUSP22, SLC25A13, and SNORA15 were hypermethylated. MAGI2, C11orf58, POMZP3, ZNF708, and CLEC2L were hypomethylated (28). Similarly, the study identified 509 differentially methylated CpGs of T lymphocytes in RA. The top 15 with gene hypermethylation included DUSP22, C1orf106, ZFP57, ARSB, LSM5, and HIST1H4D, and hypomethylation included TMEM9, CCS, FBRSL1, GALNT9, PDHB, MGMT, DDO, KLF6, and RDH12 (29). DUSP22 has been shown many times to be DNA hypermethylated, which is closely related to the regulation of immunity and inflammation. Its reduced expression can promote CD4+ T cells to differentiate into Th1 and Th17 cells, thereby promoting inflammation (106). Similarly, the inhibition of DUSP22 promotes the activation, proliferation, and differentiation of CD4+T cells into Th1/Th17 cells, thus promoting an inflammatory response (107). In addition, serum DUSP22 levels were negatively correlated with ESR, CRP, and DAS28, and increased in patients with RA receiving DMARDs treatment (108). These results suggest that DUSP22 could potentially be used as a biomarker of RA. In addition, the detection of DNA methylation of the whole genome of CD4+ T cells revealed that the changes in the methylation levels of many genes are related to different aspects of RA. For example, OR5A2 (cg02981094), ALDH9A1 (cg03984859), and C5orf32 (cg02070114) are related to the course of RA. ZC3H11A (cg02337583) is related to RF. OAS2 (cg00085448) was related to the patient’s global assessment. C16orf71 (cg04705084) and LOC100129716 (cg00598143) were correlated with DAS28. SLC38A8 (cg01740650), C18orf19 (cg00448482), COL18A1 (cg04760448), BAT3 (cg05649229), and PLD3(cg07071106) were associated with erythrocyte sedimentation rate. HSPA12A (cg06942850) is associated with TJC (18). Further experiments are required to verify the accuracy of implementing these interesting results.
The Pharmacological Mechanism of Drugs for Rheumatoid Arthritis Involves the Regulation of DNA Methylation
Methotrexate (MTX) is a first-line drug for RA treatment. The recommended dose was 25 mg/week. It can be combined with glucocorticoids to achieve better clinical remission (109). DNA methylation analysis of CD4+ T cells purified from the peripheral blood of juvenile idiopathic arthritis revealed 145 differentially methylated sites. When removing four patients receiving MTX treatment, only 11 differentially methylated sites remained, strongly suggesting that the regulatory mechanism of MTX includes regulation of DNA methylation of CD4+ T cells (110). Studies have shown that MTX can affect one-carbon metabolism during the methyl transfer process of CpG island methylation through antifolate metabolism. One-carbon metabolism requires the participation of folic acid (111).. Through further gene pathway analysis of 11 differentially methylated sites, most of them are in the “cellular growth and proliferation, hematological system development and function, hematopoiesis” network centered on TNF-α (110). The study also found that methylated IL-32 and MRPL28 are attractive potential targets (110). The pro-inflammatory factor TNF-α induces the expression of IL-32, which together constitute a chronic autoinflammatory cycle. Anti-TNF-α treatment can significantly reduce IL-32 protein levels in the synovial membrane of patients with RA (45). MRPL28 is mainly involved in encoding the mitochondrial ribosomal protein L28, and mitochondrial dysfunction may cause CD4+T cells to be sensitive to multiple forms of cell death. Mitochondria are also involved in the energy metabolism of cells, suggesting that the methylation of CD4+ T cells can affect RA in part by regulating the function of mitochondria, which may be a potential mechanism for MTX treatment (46, 47). Significantly, MTX may affect gene expression by regulating the DNA methylation levels of multiple genes in T cells and further regulating multiple immune mechanisms to exert pharmacological effects. T cells in patients with RA who have just started receiving disease-modifying antirheumatic drug (DMARD) treatment show an overall DNA hypomethylation pattern. The expression of DNA methyltransferase 1 (DNMT1) is low, the expression of demethylases (ten-eleven translocation (TET)1, TET2, and TET3) is increased, and the expression of growth arrest and DNA-damage-inducible protein 45A (GADD45A) was downregulated. After MTX treatment, the overall DNA methylation level and DNMT1 levels increased (112). By comparing the methylation levels of whole blood samples of patients with RA who had a good or bad response to MTX therapy for weeks, it was found that the differential methylation sites related to MTX therapy were mainly near GATA3 (cg27427581), RPH3AL(cg21040096), and WDR27(cg09894276) (52). As mentioned above, GATA3 mainly promotes the differentiation of Th2 cells, indicating that MTX may partially regulate the methylation and gene expression of GATA3 to regulate the differentiation of CD4+ T cells to treat RA. Interestingly, the whole longitudinal genome of Japanese patients with RA receiving anti-TNF-α therapy showed that the gene expression of WDR27, MAP3K7, BACH2, and GFRA1 might be related to the therapeutic effect, further implying a correlation between DNA methylation, gene expression regulation, and various clinical therapies (53). As mentioned earlier, ALB in CD4+ T cells of patients with RA is hypomethylated (54). ALB is involved in the molecular mechanism of MTX treatment in RA (113). In addition, after receiving MTX treatment, the inhibitory function of RA Treg cells was restored, and the mechanism involved increased Foxp3 and CTLA-4 expression and reduced the methylation of the upstream enhancer of Foxp3 (27).
Biological agents are another treatment option for patients with RA who have a poor response to MTX treatment. TNF-α inhibitors are the most widely used. Various biological agents may exert pharmacological effects by regulating DNA methylation (109). By studying the differential gene expression and differential methylation of PBMCs, monocytes, and CD4+ T cells in patients with RA receiving adalimumab (ADA) or etanercept (ETN) before and after treatment, it was found that more than 100 differential genes had DNA methylation changes. Some CD4+T cells of patients with RA who respond to ADA therapy have the characteristics of upregulation of TNF signaling pathway-related genes, such as CTLA4, TNFSF13B, TNFRSF1B, TNFSF4, IRF1, and IL18R1. In CD4+ T cells of patients with RA responding to ETN therapy, Foxo signaling pathways, such as FOXO3, FOXO4, TGFBR1, and USP7, were upregulated. NOD-like receptor signaling pathways, such as GSDMD, RIPK3, CASP4, and JAK/STAT signaling pathways, such as CISH, SOCS2, and PIM1, were downregulated (114). In addition to anti-TNF-α therapy, the DNA methylation changes of IL-6 significantly affect the gene expression of early RA, which can promote inflammation through the JAK1/STAT3 pathway (87). The clearance of apoptotic cells by phagocytes has an immunosuppressive effect of preventing the induction of autoimmunity and inflammation. If it cannot be eliminated effectively, it may be harmful. The DNA of apoptotic T cells is demethylated, which may further promote inflammation and stimulate the production of pro-inflammatory factor IL-6 in macrophages by interacting with TLR in RA (50). This further suggests that the existing IL-6R therapy and JAK inhibitor therapy should be applied as soon as possible, and the mechanism may involve DNA methylation in RA. Some drugs or natural components could regulate DNA methylation or hydroxymethylation in diseases like cancer (115, 116). Furthermore, while some of these may have a therapeutic effect in RA, they may not be active directly in treating RA by modulating the DNA methylation status. We still believe some drugs or natural components have therapeutic potential for RA, given some association outcomes. Table 2 shows updates and summaries thereof. (See Table 2)
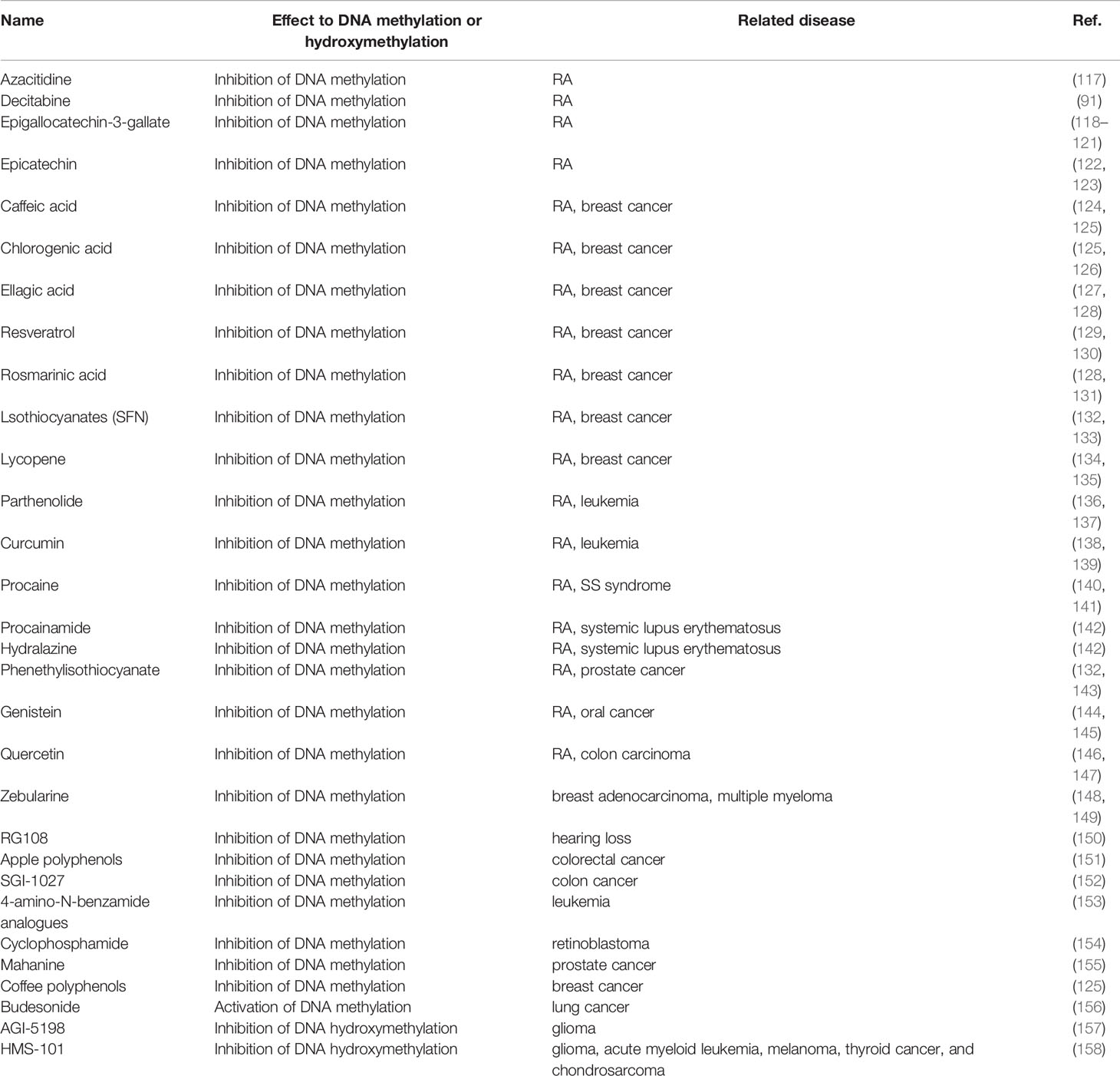
Table 2 Drugs or natural components that regulate DNA methylation or hydroxymethylation.
Conclusion
There are several unresolved issues regarding the role of DNA methylation in RA. First, a cell-specific investigation of the link between DNA methylation and RA is required to fully understand the impact of differential methylation on RA pathophysiology. Many studies have focused solely on the DNA methylation properties of the total cell population, ignoring the variability of cells within distinct cell subsets. There may be several mechanisms by which DNA methylation yields effects, and these effects may be substantially different across cell types. As a result, cell purification is required to properly evaluate DNA methylation and its link with gene expression and regulation of biological functions. There are some shortcomings, including the fact that there is little direct research-based evidence about other T cell subsets (CD8+ T cells, gamma delta T cells, and MAIT T cells) related to DNA methylation regulation. It is undeniable that T cells are heterogenous cell populations. Studying and clarifying the correlation between the DNA methylation patterns of other T cell subsets and RA is very important, and is a potential future research direction. Second, whether DNA methylation begins before the commencement of the disease or varies continually over the course of the ailment requires more well-designed clinical studies for sample collection to clarify its function. The detection of DNA methylation before illness onset can assist in identifying high-risk patients, allowing us to realize the objective of precision medicine. Finally, a distinction should be made between cell-specific alterations in DNA methylation in early and late RA. When comparing various study findings, we should keep the distinctions between chips and detecting systems in mind. Additionally, we mapped the protein interaction network based on the genes that undergo DNA methylation in T cells discussed in this article. Some of the proteins have interactions, potentially focusing on future research for further exploration (see Figure 2). In conclusion, integrating cell-specific DNA methylation data with other data, such as primary clinical data, gene expression data, proteomics data, and metabolomics data, would enable us to deliver a more powerful RA-related clinical treatment choice in the future.
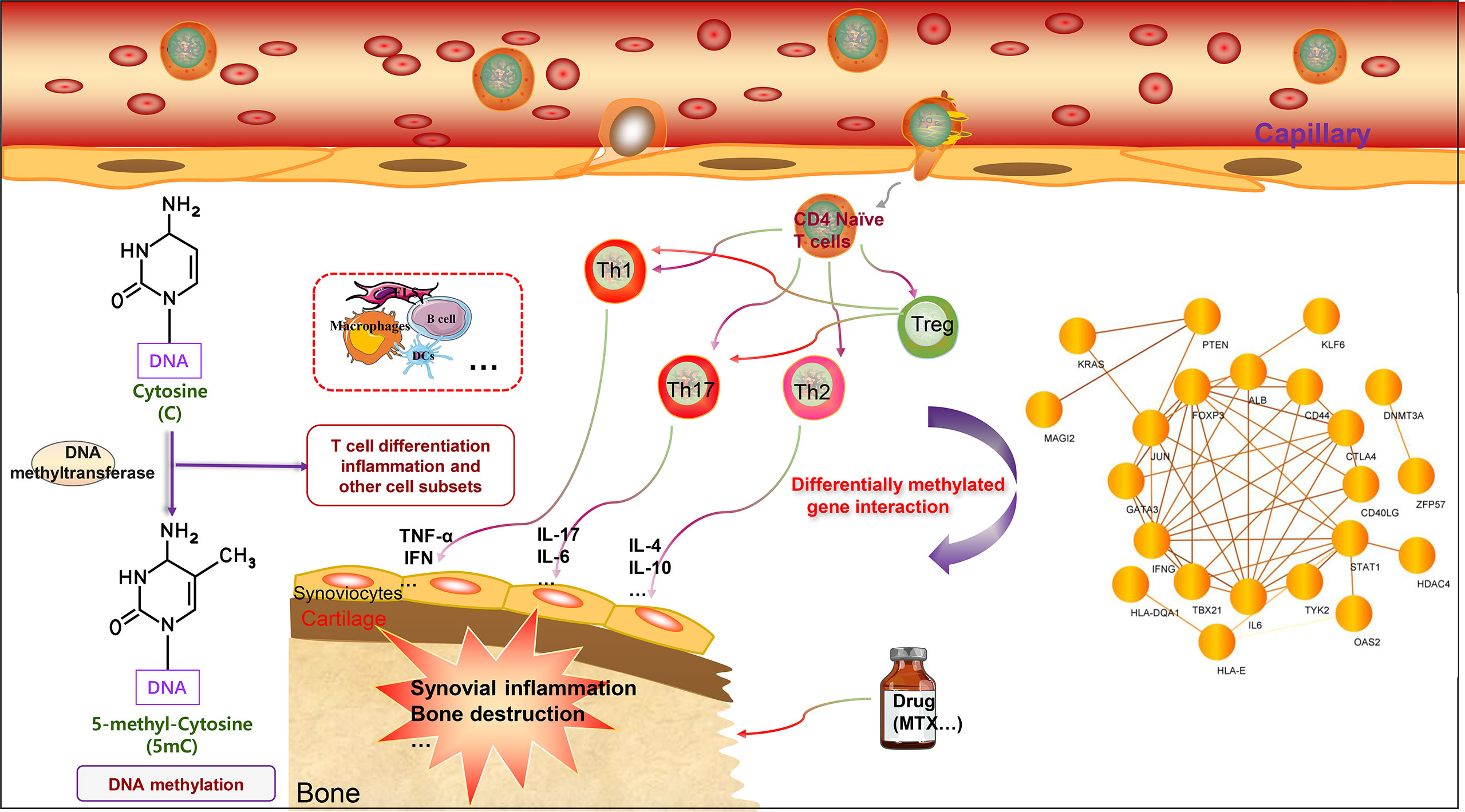
Figure 2 DNA methylation of T cell-related genes affects rheumatoid arthritis. The main characteristics of RA include chronic synovial inflammation and bone destruction. T cells can differentiate into different cell subgroups according to the intracellular microenvironment, including Treg cells, Th1 cells, Th2 cells, and Th17 cells. Th1 and Th17 cells can promote inflammation by secreting different pro-inflammatory factors, including TNF-α, IL-1, IL-6, IL-17, and IFN-γ. Th2 cells mainly secrete cytokines that inhibit inflammation, including IL-4 and IL-10. Treg cells specifically regulate the function, inhibit the differentiation of Th1 and Th17 cells, and thus inhibit inflammation. T cell-related genes can undergo DNA methylation modification to affect gene expression, further regulate T cell function, and affect the progression of RA. DNA methylation genes can also potentially serve as biomarkers for RA and predict drug efficacy. Interestingly, the DNA methylated genes related to T cells can form a network, and there are interactions worthy of further study. In addition, the methylation patterns of multiple immune cells present in RA warrant further investigation.
Author Contributions
JZ is responsible for the collection, collation and writing of the original manuscript. KW, CC, LX and PJ is responsible for the collection of the original manuscript. SG, SS, and DH are responsible for the revision and review of the manuscript. All authors reviewed and accepted with the final version.
Funding
This work was funded by the National Natural Science Funds of China (82074234), Shanghai Chinese Medicine Development Office,National Administration of Traditional Chinese Medicine,Regional Chinese Medicine (Specialist) Diagnosis and Treatment Center Construction Project-Rheumatology,State Administration of Traditional Chinese Medicine, National TCM Evidence-Based Medicine Research and Construction Project, Basic TCM Evidence-Based Capacity Development Program, Shanghai Municipal Health Commission, East China Region based Chinese and Western Medicine Joint Disease Specialist Alliance.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
RA, rheumatoid arthritis; CpG, cytosine guanine (CG) dinucleotide; HLA, human leukocyte antigen; hTERT, human telomerase reverse transcriptase; DVPs, differentially variable positions; KEGG, the Kyoto Encyclopedia of Genes and Genomes; GO, gene ontology; MAPK, the mitogen-activated protein kinase; HBV, hepatitis B virtus; RF, rheumatoid factor; DMP, differentially methylated positions; anti-CCP, cyclic citrullinated peptides; DAS28, disease activity score in 28 joints; CRP, C-reactive protein; SNPs, single nucleotide polymorphisms; NSAID, non-steroidal anti-inflammatory drug; PBMCs, peripheral blood mononuclear cells; IL, interleukin; IFN-γ, interferon-gamma; VDR, vitamin D receptor; FLS, fibroblast-like synoviocyte; TNF-a, tumor necrosis factor-a; RANKL, receptor activator of nuclear factor (NF)−κB ligand; Tfh, T-follicular helper; DNMT, DNA methyltransferase; TNFR2, tumor necrosis factor receptor 2; IDO, indoleamine 2,3 –dioxygenase; MTX, methotrexate; DMARDs, disease-modifying antirheumatic drugs; DNMT1, DNA methyltrasnferase 1; TET1, ten-eleven translocation 1; GADD45A, DNA-damage-inducible protein 45A; ADA, adalimumab; ETN, etanercept; ACPA, anti-citrullinated protein antibody.
References
1. Sparks J. Rheumatoid Arthritis. Ann Intern Med (2019) 170(1):ITC1–ITC16. doi: 10.7326/aitc201901010
2. Weyand CM, Goronzy JJ. The Immunology of Rheumatoid Arthritis. Nat Immunol (2021) 22(1):10–8. doi: 10.1038/s41590-020-00816-x
3. Catrina A, Joshua V, Klareskog L, Malmström V. Mechanisms Involved in Triggering Rheumatoid Arthritis. Immunol Rev (2016) 269(1):162–74. doi: 10.1111/imr.12379
4. Tan E, Smolen J. Historical Observations Contributing Insights on Etiopathogenesis of Rheumatoid Arthritis and Role of Rheumatoid Factor. J Exp Med (2016) 213(10):1937–50. doi: 10.1084/jem.20160792
5. McInnes I, Schett G. Pathogenetic Insights From the Treatment of Rheumatoid Arthritis. Lancet (London) (2017) 389(10086):2328–37. doi: 10.1016/s0140-6736(17)31472-1
6. Ballestar E. Epigenetic Alterations in Autoimmune Rheumatic Diseases. Nat Rev Rheumatol (2011) 7(5):263–71. doi: 10.1038/nrrheum.2011.16
7. Bird A. DNA Methylation Patterns and Epigenetic Memory. Genes Dev (2002) 16(1):6–21. doi: 10.1101/gad.947102
8. Smith J, Sen S, Weeks R, Eccles M, Chatterjee A. Promoter DNA Hypermethylation and Paradoxical Gene Activation. Trends Cancer (2020) 6(5):392–406. doi: 10.1016/j.trecan.2020.02.007
9. Cope A, Schulze-Koops H, Aringer M. The Central Role of T Cells in Rheumatoid Arthritis. Clin Exp Rheumatol (2007) 25(5 Suppl 46):S4–11.
10. Liu Y, Aryee M, Padyukov L, Fallin M, Hesselberg E, Runarsson A, et al. Epigenome-Wide Association Data Implicate DNA Methylation as an Intermediary of Genetic Risk in Rheumatoid Arthritis. Nat Biotechnol (2013) 31(2):142–7. doi: 10.1038/nbt.2487
11. Ha E, Bang S, Lim J, Yun J, Kim J, Bae J, et al. Genetic Variants Shape Rheumatoid Arthritis-Specific Transcriptomic Features in CD4 T Cells Through Differential DNA Methylation, Explaining a Substantial Proportion of Heritability. Ann Rheum Dis (2021) 80(7):876–83. doi: 10.1136/annrheumdis-2020-219152
12. Webster A, Plant D, Ecker S, Zufferey F, Bell J, Feber A, et al. Increased DNA Methylation Variability in Rheumatoid Arthritis-Discordant Monozygotic Twins. Genome Med (2018) 10(1):64. doi: 10.1186/s13073-018-0575-9
13. Fujii H, Shao L, Colmegna I, Goronzy J, Weyand C. Telomerase Insufficiency in Rheumatoid Arthritis. P Natl Acad Sci USA (2009) 106(11):4360–5. doi: 10.1073/pnas.0811332106
14. Shao L, Fujii H, Colmegna I, Oishi H, Goronzy J, Weyand C. Deficiency of the DNA Repair Enzyme ATM in Rheumatoid Arthritis. J Exp Med (2009) 206(6):1435–49. doi: 10.1084/jem.20082251
15. Rhead B, Holingue C, Cole M, Shao X, Quach H, Quach D, et al. Rheumatoid Arthritis Naive T Cells Share Hypermethylation Sites With Synoviocytes. Arthritis Rheumatol (2017) 69(3):550–9. doi: 10.1002/art.39952
16. Zhou L, Chen L, Wang Y, Huang J, Yang G, Tan Z, et al. NR1I2Impact of , Adenosine Triphosphate-Binding Cassette Transporters Genetic Polymorphisms on the Pharmacokinetics of Ginsenoside Compound K in Healthy Chinese Volunteers. J ginseng Res (2019) 43(3):460–74. doi: 10.1016/j.jgr.2018.04.003
17. Jiang J, Qiu YH, Peng YP, Wang JJ. Immunoregulatory Role of Endogenous Catecholamines Synthesized by Immune Cells. Sheng li Xue Bao [Acta Physiologica Sinica] (2006) 58(4):309–17.
18. Guo S, Zhu Q, Jiang T, Wang R, Shen Y, Zhu X, et al. Genome-Wide DNA Methylation Patterns in CD4+ T Cells From Chinese Han Patients With Rheumatoid Arthritis. Mod Rheumatol (2017) 27(3):441–7. doi: 10.1080/14397595.2016.1218595
19. Chang L, Kan L. Mesenchymal Stem Cell-Originated Exosomal Circular RNA Circfbxw7 Attenuates Cell Proliferation, Migration and Inflammation of Fibroblast-Like Synoviocytes by Targeting miR-216a-3p/HDAC4 in Rheumatoid Arthritis. J Inflammation Res (2021) 14:6157–71. doi: 10.2147/jir.S336099
20. Deng Y, Wang J, Huang M, Xu G, Wei W, Qin H. Inhibition of miR-148a-3p Resists Hepatocellular Carcinoma Progress of Hepatitis C Virus Infection Through Suppressing C-Jun and MAPK Pathway. J Cell Mol Med (2019) 23(2):1415–26. doi: 10.1111/jcmm.14045
21. Wang M, Wei J, Li H, Ouyang X, Sun X, Tang Y, et al. Leptin Upregulates Peripheral CD4CXCR5ICOS T Cells via Increased IL-6 in Rheumatoid Arthritis Patients. J Interferon Cytokine Res (2018) 38(2):86–92. doi: 10.1089/jir.2017.0031
22. Li X, Chen X, Bao J, Xu L, Zhang L, Huang C, et al. PTEN Negatively Regulates the Expression of Pro-Inflammatory Cytokines and Chemokines of Fibroblast-Like Synoviocytes in Adjuvant-Induced Arthritis. Artif Cells Nanomed Biotechnol (2019) 47(1):3687–96. doi: 10.1080/21691401.2019.1661849
23. Li X, Wu S, Yan Q, Wu Y, Chen H, Yin S, et al. PTEN Methylation Promotes Inflammation and Activation of Fibroblast-Like Synoviocytes in Rheumatoid Arthritis. Front Pharmacol (2021) 12:700373. doi: 10.3389/fphar.2021.700373
24. Zeidler A, Bräuer R, Thoss K, Bahnsen J, Heinrichs V, Jablonski-Westrich D, et al. Therapeutic Effects of Antibodies Against Adhesion Molecules in Murine Collagen Type II-Induced Arthritis. Autoimmunity (1995) 21(4):245–52. doi: 10.3109/08916939509001943
25. Kawashima M, Miossec P. mRNA Quantification of T-Bet, GATA-3, IFN-Gamma, and IL-4 Shows a Defective Th1 Immune Response in the Peripheral Blood From Rheumatoid Arthritis Patients: Link With Disease Activity. J Clin Immunol (2005) 25(3):209–14. doi: 10.1007/s10875-005-4092-4
26. Janson PC, Winerdal ME, Winqvist O. At the Crossroads of T Helper Lineage Commitment-Epigenetics Points the Way. Biochim Biophys Acta (2009) 1790(9):906–19. doi: 10.1016/j.bbagen.2008.12.003
27. Cribbs A, Kennedy A, Penn H, Amjadi P, Green P, Read J, et al. Methotrexate Restores Regulatory T Cell Function Through Demethylation of the FoxP3 Upstream Enhancer in Patients With Rheumatoid Arthritis. Arthritis Rheumatol (2015) 67(5):1182–92. doi: 10.1002/art.39031
28. Glossop J, Emes R, Nixon N, Packham J, Fryer A, Mattey D, et al. Genome-Wide Profiling in Treatment-Naive Early Rheumatoid Arthritis Reveals DNA Methylome Changes in T and B Lymphocytes. Epigenomics (2016) 8(2):209–24. doi: 10.2217/epi.15.103
29. Glossop JR, Emes RD, Nixon NB, Haworth KE, Packham JC, Dawes PT, et al. Genome-Wide DNA Methylation Profiling in Rheumatoid Arthritis Identifies Disease-Associated Methylation Changes That are Distinct to Individual T- and B-Lymphocyte Populations. Epigenetics (2014) 9(9):1228–37. doi: 10.4161/epi.29718
30. Yang M, Tao J, Wu H, Zhang L, Yao Y, Liu L, et al. Responses of Transgenic Melatonin-Enriched Goats on LPS Stimulation and the Proteogenomic Profiles of Their PBMCs. Int J Mol Sci (2018) 19(8):2406. doi: 10.3390/ijms19082406
31. Liedtke K, Alter C, Günther A, Hövelmeyer N, Klopfleisch R, Naumann R, et al. Endogenous CD83 Expression in CD4 Conventional T Cells Controls Inflammatory Immune Responses. J Immunol (2020) 204(12):3217–26. doi: 10.4049/jimmunol.2000042
32. Doebbeler M, Koenig C, Krzyzak L, Seitz C, Wild A, Ulas T, et al. CD83 Expression Is Essential for Treg Cell Differentiation and Stability. JCI Insight (2018) 3(11):e99712. doi: 10.1172/jci.insight.99712
33. Yang H, Chang L, Liang Y, Lin C, Wang P. A Genome-Wide Homozygosity Association Study Identifies Runs of Homozygosity Associated With Rheumatoid Arthritis in the Human Major Histocompatibility Complex. PloS One (2012) 7(4):e34840. doi: 10.1371/journal.pone.0034840
34. Hässler S, Bachelet D, Duhaze J, Szely N, Gleizes A, Hacein-Bey Abina S, et al. Clinicogenomic Factors of Biotherapy Immunogenicity in Autoimmune Disease: A Prospective Multicohort Study of the ABIRISK Consortium. PloS Med (2020) 17(10):e1003348. doi: 10.1371/journal.pmed.1003348
35. Iwaszko M, Świerkot J, Kolossa K, Jeka S, Wiland P, Bogunia-Kubik K. Polymorphisms Within the Human Leucocyte Antigen-E Gene and Their Associations With Susceptibility to Rheumatoid Arthritis as Well as Clinical Outcome of Anti-Tumour Necrosis Factor Therapy. Clin Exp Immunol (2015) 182(3):270–7. doi: 10.1111/cei.12696
36. Liao C, Chou P, Cheng C, Chang Y, Chi W, Tsai K, et al. Comparative Analysis of Novel Autoantibody Isotypes Against Citrullinated-Inter-Alpha-Trypsin Inhibitor Heavy Chain 3 (ITIH3)(542-556) Peptide in Serum From Taiwanese Females With Rheumatoid Arthritis, Primary Sjögren's Syndrome and Secondary Sjögren's Syndrome in Rheumatoid Arthritis. J Proteomics (2016) 141:1–11. doi: 10.1016/j.jprot.2016.03.031
37. Saeki N, Imai Y. Reprogramming of Synovial Macrophage Metabolism by Synovial Fibroblasts Under Inflammatory Conditions. Cell communication Signaling CCS (2020) 18(1):188. doi: 10.1186/s12964-020-00678-8
38. Singh K, Deshpande P, Li G, Yu M, Pryshchep S, Cavanagh M, et al. K-RAS GTPase- and B-RAF Kinase-Mediated T-Cell Tolerance Defects in Rheumatoid Arthritis. P Natl Acad Sci USA (2012) 109(25):E1629–37. doi: 10.1073/pnas.1117640109
39. Dong J, Chang H, Ivascu C, Qian Y, Rezai S, Okhrimenko A, et al. Loss of Methylation at the IFNG Promoter and CNS-1 Is Associated With the Development of Functional IFN-γ Memory in Human CD4(+) T Lymphocytes. Eur J Immunol (2013) 43(3):793–804. doi: 10.1002/eji.201242858
40. Yu Q, Zhou B, Zhang Y, Nguyen ET, Du J, Glosson NL, et al. DNA Methyltransferase 3a Limits the Expression of Interleukin-13 in T Helper 2 Cells and Allergic Airway Inflammation. Proc Natl Acad Sci USA (2012) 109(2):541–6. doi: 10.1073/pnas.1103803109
41. Tsygankov AY. Multidomain STS/TULA Proteins Are Novel Cellular Regulators. IUBMB Life (2008) 60(4):224–31. doi: 10.1002/iub.36
42. Call ME, Wucherpfennig KW. Molecular Mechanisms for the Assembly of the T Cell Receptor-CD3 Complex. Mol Immunol (2004) 40(18):1295–305. doi: 10.1016/j.molimm.2003.11.017
43. Liao J, Liang G, Xie S, Zhao H, Zuo X, Li F, et al. CD40L Demethylation in CD4(+) T Cells From Women With Rheumatoid Arthritis. Clin Immunol (2012) 145(1):13–8. doi: 10.1016/j.clim.2012.07.006
44. He X, Ding Y, Xiang W, Dang X. Roles of 1,25(OH)2D3 and Vitamin D Receptor in the Pathogenesis of Rheumatoid Arthritis and Systemic Lupus Erythematosus by Regulating the Activation of CD4+ T Cells and the Pkcδ/ERK Signaling Pathway. Cell Physiol Biochem (2016) 40:743–56. doi: 10.1159/000453135
45. Heinhuis B, Koenders M, van Riel P, van de Loo F, Dinarello C, Netea M, et al. Tumour Necrosis Factor Alpha-Driven IL-32 Expression in Rheumatoid Arthritis Synovial Tissue Amplifies an Inflammatory Cascade. Ann Rheum Dis (2011) 70(4):660–7. doi: 10.1136/ard.2010.139196
46. Zhao J, Jiang P, Guo S, Schrodi SJ, Apoptosis HD. Autophagy, NETosis, Necroptosis, and Pyroptosis Mediated Programmed Cell Death as Targets for Innovative Therapy in Rheumatoid Arthritis. Front Immunol (2021) 12:809806. doi: 10.3389/fimmu.2021.809806
47. Zhao J, Hu Y, Peng J. Targeting Programmed Cell Death in Metabolic Dysfunction-Associated Fatty Liver Disease (MAFLD): A Promising New Therapy. Cell Mol Biol Lett (2021) 26(1):17. doi: 10.1186/s11658-021-00254-z
48. Wan Y. GATA3: A Master of Many Trades in Immune Regulation. Trends Immunol (2014) 35(6):233–42. doi: 10.1016/j.it.2014.04.002
49. Ohmura K, Nguyen L, Locksley R, Mathis D, Benoist C. Interleukin-4 can be a Key Positive Regulator of Inflammatory Arthritis. Arthritis Rheum-US (2005) 52(6):1866–75. doi: 10.1002/art.21104
50. Notley C, Jordan C, McGovern J, Brown M, Ehrenstein M. DNA Methylation Governs the Dynamic Regulation of Inflammation by Apoptotic Cells During Efferocytosis. Sci Rep-UK (2017) 7:42204. doi: 10.1038/srep42204
51. Guo K, Major G, Foster H, Bassendine M, Collier J, Ross D, et al. Defective Repair of O6-Methylguanine-DNA in Primary Sjögren's Syndrome Patients Predisposed to Lymphoma. Ann Rheum Dis (1995) 54(3):229–32. doi: 10.1136/ard.54.3.229
52. Nair N, Plant D, Verstappen S, Isaacs J, Morgan A, Hyrich K, et al. Differential DNA Methylation Correlates With Response to Methotrexate in Rheumatoid Arthritis. Rheumatol (Oxford) (2020) 59(6):1364–71. doi: 10.1093/rheumatology/kez411
53. Honne K, Hallgrímsdóttir I, Wu C, Sebro R, Jewell N, Sakurai T, et al. A Longitudinal Genome-Wide Association Study of Anti-Tumor Necrosis Factor Response Among Japanese Patients With Rheumatoid Arthritis. Arthritis Res Ther (2016) 18:12. doi: 10.1186/s13075-016-0920-6
54. Fang Q, Li T, Chen P, Wu Y, Wang T, Mo L, et al. Comparative Analysis on Abnormal Methylome of Differentially Expressed Genes and Disease Pathways in the Immune Cells of RA and SLE. Front Immunol (2021) 12:668007. doi: 10.3389/fimmu.2021.668007
55. Chen Q, Ross A. Vitamin A and Immune Function: Retinoic Acid Modulates Population Dynamics in Antigen Receptor and CD38-Stimulated Splenic B Cells. P Natl Acad Sci USA (2005) 102(40):14142–9. doi: 10.1073/pnas.0505018102
56. Stagi S, Giani T, Simonini G, Falcini F. Thyroid Function, Autoimmune Thyroiditis and Coeliac Disease in Juvenile Idiopathic Arthritis. Rheumatol (Oxford) (2005) 44(4):517–20. doi: 10.1093/rheumatology/keh531
57. Li Q, Wang B, Mu K, Zhang J, Yang Y, Yao W, et al. Increased Risk of Thyroid Dysfunction Among Patients With Rheumatoid Arthritis. Front Endocrinol (Lausanne) (2018) 9:799. doi: 10.3389/fendo.2018.00799
58. Pörings A, Lowin T, Dufner B, Grifka J, Straub R. A Thyroid Hormone Network Exists in Synovial Fibroblasts of Rheumatoid Arthritis and Osteoarthritis Patients. Sci Rep-UK (2019) 9(1):13235. doi: 10.1038/s41598-019-49743-4
59. Hsu C, Lang H, Huang K, Lin H, Chen C. Association of Rheumatoid Arthritis and Hepatitis B Infection: A Nationwide Nested Case-Control Study From 1999 to 2009 in Taiwan. Medicine (2016) 95(18):e3551. doi: 10.1097/md.0000000000003551
60. Wang ST, Tseng CW, Hsu CW, Tung CH, Huang KY, Lu MC, et al. Reactivation of Hepatitis B Virus Infection in Patients With Rheumatoid Arthritis Receiving Tofacitinib. Int J Rheum Dis (2021) 24(11):1362–9. doi: 10.1111/1756-185x.14217
61. Generali E, De Santis M, Isailovic N, Palermo B, Guidelli G, Ceribelli A, et al. Rheumatoid Factor and Anti-Citrullinated Peptide Antibodies in the General Population: Hepatitis B and C Virus Association and 15-Year-Risk of Rheumatoid Arthritis. Clin Exp Rheumatol (2021) 39(1):38–43.
62. Su JW, Li SF, Tao JJ, Xu YY, Wang K, Qian XW, et al. Estrogen Protects Against Acidosis-Mediated Articular Chondrocyte Injury by Promoting ASIC1a Protein Degradation. Eur J Pharmacol (2021) 908:174381. doi: 10.1016/j.ejphar.2021.174381
63. Hang X, Zhang Z, Niu R, Wang C, Yao J, Xu Y, et al. Estrogen Protects Articular Cartilage by Downregulating ASIC1a in Rheumatoid Arthritis. J Inflammation Res (2021) 14:843–58. doi: 10.2147/jir.S295222
64. Guderud K, Sunde L, Flåm S, Mæhlen M, Mjaavatten M, Lillegraven S, et al. Rheumatoid Arthritis Patients, Both Newly Diagnosed and Methotrexate Treated, Show More DNA Methylation Differences in CD4 Memory Than in CD4 Naïve T Cells. Front Immunol (2020) 11:194. doi: 10.3389/fimmu.2020.00194
65. Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of Rheumatoid Arthritis Contributes to Biology and Drug Discovery. Nature (2014) 506(7488):376–81. doi: 10.1038/nature12873
66. Yang X, Liu J, Chen S, Li M, Zhang M, Leng R, et al. UBASH3A Gene Polymorphisms and Expression Profile in Rheumatoid Arthritis. Autoimmunity (2019) 52(1):21–6. doi: 10.1080/08916934.2019.1581773
67. Liu D, Liu J, Cui G, Yang H, Cao T, Wang L. UBASH3AEvaluation of the Association of and With Rheumatoid Arthritis and Disease Activity and Severity in Han Chinese. Oncotarget (2017) 8(61):103385–92. doi: 10.18632/oncotarget.21875
68. Khadem Azarian S, Jafarnezhad-Ansariha F, Nazeri S, Azizi G, Aghazadeh Z, Hosseinzadeh E, et al. Effects of Guluronic Acid, as a New NSAID With Immunomodulatory Properties on IL-17, Rorγt, IL-4 and GATA-3 Gene Expression in Rheumatoid Arthritis Patients. Immunopharmacol Immunotoxicol (2020) 42(1):22–7. doi: 10.1080/08923973.2019.1702053
69. Jones G, Greenhill C, Williams J, Nowell M, Williams A, Jenkins B, et al. Exacerbated Inflammatory Arthritis in Response to Hyperactive Gp130 Signalling Is Independent of IL-17a. Ann Rheum Dis (2013) 72(10):1738–42. doi: 10.1136/annrheumdis-2013-203771
70. Liu C, He L, Wang J, Wang Q, Sun C, Li Y, et al. Anti-Angiogenic Effect of Shikonin in Rheumatoid Arthritis by Downregulating PI3K/AKT and MAPKs Signaling Pathways. J Ethnopharmacol (2020) 260:113039. doi: 10.1016/j.jep.2020.113039
71. Cao L, Jiang H, Yang J, Mao J, Wei G, Meng X, et al. LncRNA MIR31HG Is Induced by Tocilizumab and Ameliorates Rheumatoid Arthritis Fibroblast-Like Synoviocyte-Mediated Inflammation via miR-214-PTEN-AKT Signaling Pathway. Aging (Albany NY) (2021) 13(21):24071–85. doi: 10.18632/aging.203644
72. Kurohori Y, Sato K, Suzuki S, Kashiwazaki S. Adhesion Molecule Expression on Peripheral Blood Mononuclear Cells in Rheumatoid Arthritis: Positive Correlation Between the Proportion of L-Selectin and Disease Activity. Clinl rheumatol (1995) 14(3):335–41. doi: 10.1007/bf02208350
73. Jang E, Kim H, Cho S, Paik D, Kim J, Lee S, et al. Prevention of Spontaneous Arthritis by Inhibiting Homeostatic Expansion of Autoreactive CD4+ T Cells in the K/BxN Mouse Model. Arthritis Rheum-US (2006) 54(2):492–8. doi: 10.1002/art.21567
74. Qin J, Huang X, Wang N, Zhou P, Zhang H, Chen Z, et al. Supranutritional Selenium Suppresses ROS-Induced Generation of RANKL-Expressing Osteoclastogenic CD4 T Cells and Ameliorates Rheumatoid Arthritis. Clin Transl Immunol (2021) 10(9):e1338. doi: 10.1002/cti2.1338
75. Chen G, Zhang X, Li R, Fang L, Niu X, Zheng Y, et al. Role of Osteopontin in Synovial Th17 Differentiation in Rheumatoid Arthritis. Arthritis Rheum-US (2010) 62(10):2900–8. doi: 10.1002/art.27603
76. Zhao J, Guo S, Schrodi S, He D. Molecular and Cellular Heterogeneity in Rheumatoid Arthritis: Mechanisms and Clinical Implications. Front Immunol (2021) 12:790122. doi: 10.3389/fimmu.2021.790122
77. Ye H, Fu D, Fang X, Xie Y, Zheng X, Fan W, et al. Casein Kinase II Exacerbates Rheumatoid Arthritis via Promoting Th1 and Th17 Cell Inflammatory Responses. Expert Opin Ther Targets (2021) 25(11):1017–24. doi: 10.1080/14728222.2021.2010190
78. Lin J, Tang J, Lin J, He Y, Yu Z, Jiang R, et al. YY1 Regulation by miR-124-3p Promotes Th17 Cell Pathogenicity Through Interaction With T-Bet in Rheumatoid Arthritis. JCI Insight (2021) 6(22):e149985. doi: 10.1172/jci.insight.149985
79. Li Y, Yang W, Wang F. The Relationship of Blood CDC42 Level With Th1 Cells, Th17 Cells, Inflammation Markers, Disease Risk/Activity, and Treatment Efficacy of Rheumatoid Arthritis. Ir J Med Sci (2021). doi: 10.1007/s11845-021-02858-y
80. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-Producing CD4+ Effector T Cells Develop via a Lineage Distinct From the T Helper Type 1 and 2 Lineages. Nat Immunol (2005) 6(11):1123–32. doi: 10.1038/ni1254
81. Skapenko A, Wendler J, Lipsky PE, Kalden JR, Schulze-Koops H. Altered Memory T Cell Differentiation in Patients With Early Rheumatoid Arthritis. J Immunol (1999) 163(1):491–9.
82. Gamper C, Agoston A, Nelson W, Powell J. Identification of DNA Methyltransferase 3a as a T Cell Receptor-Induced Regulator of Th1 and Th2 Differentiation. J Immunol (2009) 183(4):2267–76. doi: 10.4049/jimmunol.0802960
83. Wilson CB, Rowell E, Sekimata M. Epigenetic Control of T-Helper-Cell Differentiation. Nat Rev Immunol (2009) 9(2):91–105. doi: 10.1038/nri2487
84. Seder RA, Gazzinelli R, Sher A, Paul WE. Interleukin 12 Acts Directly on CD4+ T Cells to Enhance Priming for Interferon Gamma Production and Diminishes Interleukin 4 Inhibition of Such Priming. Proc Natl Acad Sci USA (1993) 90(21):10188–92. doi: 10.1073/pnas.90.21.10188
85. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A Novel Transcription Factor, T-Bet, Directs Th1 Lineage Commitment. Cell (2000) 100(6):655–69. doi: 10.1016/s0092-8674(00)80702-3
86. Thierfelder WE, van Deursen JM, Yamamoto K, Tripp RA, Sarawar SR, Carson RT, et al. Requirement for Stat4 in Interleukin-12-Mediated Responses of Natural Killer and T Cells. Nature (1996) 382(6587):171–4. doi: 10.1038/382171a0
87. Pitaksalee R, Burska A, Ajaib S, Rogers J, Parmar R, Mydlova K, et al. Differential CpG DNA Methylation in Peripheral Naïve CD4 T-Cells in Early Rheumatoid Arthritis Patients. Clin Epigenet (2020) 12(1):54. doi: 10.1186/s13148-020-00837-1
88. Lübbers J, Brink M, van de Stadt L, Vosslamber S, Wesseling J, van Schaardenburg D, et al. The Type I IFN Signature as a Biomarker of Preclinical Rheumatoid Arthritis. Ann Rheum Dis (2013) 72(5):776–80. doi: 10.1136/annrheumdis-2012-202753
89. van Baarsen LG, Bos WH, Rustenburg F, van der Pouw Kraan TC, Wolbink GJ, Dijkmans BA, et al. Gene Expression Profiling in Autoantibody-Positive Patients With Arthralgia Predicts Development of Arthritis. Arthritis Rheum (2010) 62(3):694–704. doi: 10.1002/art.27294
90. Shin H, Park H, Jeong S, Park H, Kim Y, Cho S, et al. STAT4 Expression in Human T Cells Is Regulated by DNA Methylation But Not by Promoter Polymorphism. J Immunol (2005) 175(11):7143–50. doi: 10.4049/jimmunol.175.11.7143
91. Petralia MC, Mazzon E, Basile MS, Cutuli M, Di Marco R, Scandurra F, et al. Effects of Treatment With the Hypomethylating Agent 5-Aza-2'-Deoxycytidine in Murine Type II Collagen-Induced Arthritis. Pharmaceuticals (Basel) (2019) 12(4):174. doi: 10.3390/ph12040174
92. Bird J, Brown D, Mullen A, Moskowitz N, Mahowald M, Sider J, et al. Helper T Cell Differentiation Is Controlled by the Cell Cycle. Immunity (1998) 9(2):229–37. doi: 10.1016/s1074-7613(00)80605-6
93. Qiu H, Wu H, Chan V, Lau C, Lu Q. Transcriptional and Epigenetic Regulation of Follicular T-Helper Cells and Their Role in Autoimmunity. Autoimmunity (2017) 50(2):71–81. doi: 10.1080/08916934.2017.1284821
94. Liu X, Lu H, Chen T, Nallaparaju KC, Yan X, Tanaka S, et al. Genome-Wide Analysis Identifies Bcl6-Controlled Regulatory Networks During T Follicular Helper Cell Differentiation. Cell Rep (2016) 14(7):1735–47. doi: 10.1016/j.celrep.2016.01.038
95. Buckner J. Mechanisms of Impaired Regulation by CD4(+)CD25(+)FOXP3(+) Regulatory T Cells in Human Autoimmune Diseases. Nat Rev Immunol (2010) 10(12):849–59. doi: 10.1038/nri2889
96. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional Delineation and Differentiation Dynamics of Human CD4+ T Cells Expressing the FoxP3 Transcription Factor. Immunity (2009) 30(6):899–911. doi: 10.1016/j.immuni.2009.03.019
97. Baecher-Allan C, Wolf E, Hafler D. MHC Class II Expression Identifies Functionally Distinct Human Regulatory T Cells. J Immunol (2006) 176(8):4622–31. doi: 10.4049/jimmunol.176.8.4622
98. Kennedy A, Schmidt E, Cribbs A, Penn H, Amjadi P, Syed K, et al. A Novel Upstream Enhancer of FOXP3, Sensitive to Methylation-Induced Silencing, Exhibits Dysregulated Methylation in Rheumatoid Arthritis Treg Cells. Eur J Immunol (2014) 44(10):2968–78. doi: 10.1002/eji.201444453
99. Tseng W, Huang Y, Clanchy F, McNamee K, Perocheau D, Ogbechi J, et al. Foxp3TNF Receptor 2 Signaling Prevents DNA Methylation at the Promoter and Prevents Pathogenic Conversion of Regulatory T Cells. P Natl Acad Sci USA (2019) 116(43):21666–72. doi: 10.1073/pnas.1909687116
100. Rossetti M, Spreafico R, Consolaro A, Leong J, Chua C, Massa M, et al. TCR Repertoire Sequencing Identifies Synovial Treg Cell Clonotypes in the Bloodstream During Active Inflammation in Human Arthritis. Ann Rheum Dis (2017) 76(2):435–41. doi: 10.1136/annrheumdis-2015-208992
101. Santinon F, Batignes M, Mebrek M, Biton J, Clavel G, Hervé R, et al. Involvement of Tumor Necrosis Factor Receptor Type II in FoxP3 Stability and as a Marker of Treg Cells Specifically Expanded by Anti-Tumor Necrosis Factor Treatments in Rheumatoid Arthritis. Arthritis Rheumatol (2020) 72(4):576–87. doi: 10.1002/art.41134
102. Zhou G, Sun X, Qin Q, Lv J, Cai Y, Wang M, et al. Loss of Smad7 Promotes Inflammation in Rheumatoid Arthritis. Front Immunol (2018) 9:2537. doi: 10.3389/fimmu.2018.02537
103. Cribbs A, Kennedy A, Penn H, Read J, Amjadi P, Green P, et al. Treg Cell Function in Rheumatoid Arthritis Is Compromised by Ctla-4 Promoter Methylation Resulting in a Failure to Activate the Indoleamine 2,3-Dioxygenase Pathway. Arthritis Rheumatol (2014) 66(9):2344–54. doi: 10.1002/art.38715
104. Huang YS, Tseng WY, Clanchy FIL, Topping LM, Ogbechi J, McNamee K, et al. Pharmacological Modulation of T Cell Immunity Results in Long-Term Remission of Autoimmune Arthritis. P Natl Acad Sci USA (2021) 118(19):e2100939118. doi: 10.1073/pnas.2100939118
105. Guo S, Xu L, Chang C, Zhang R, Jin Y, He D. Epigenetic Regulation Mediated by Methylation in the Pathogenesis and Precision Medicine of Rheumatoid Arthritis. Front Genet (2020) 11:811(811). doi: 10.3389/fgene.2020.00811
106. Shi X, Yang W, Wang N, Zhu J. Circulating JNK Pathway-Associated Phosphatase Level Correlates With Decreased Risk, Activity, Inflammation Level and Reduced Clinical Response to Tumor Necrosis Factor-α Inhibitor in Crohn Disease Patients. Med (Baltimore) (2019) 98(33):e16622. doi: 10.1097/md.0000000000016622
107. Zhou R, Chang Y, Liu J, Chen M, Wang H, Huang M, et al. JNK Pathway-Associated Phosphatase/DUSP22 Suppresses CD4(+) T-Cell Activation and Th1/Th17-Cell Differentiation and Negatively Correlates With Clinical Activity in Inflammatory Bowel Disease. Front Immunol (2017) 8:781. doi: 10.3389/fimmu.2017.00781
108. Song D, Zhu X, Wang F, Sun J. Longitudinal Monitor of Jun N-Terminal Kinase Pathway Associated Phosphatase Reflects Clinical Efficacy to Triple Conventional Disease-Modifying Anti-Rheumatic Drugs Treatment in Rheumatoid Arthritis Patients. Inflammopharmacology (2021) 29(4):1131–8. doi: 10.1007/s10787-021-00823-w
109. Aletaha D, Smolen JS. Diagnosis and Management of Rheumatoid Arthritis: A Review. JAMA (2018) 320(13):1360–72. doi: 10.1001/jama.2018.13103
110. Ellis JA, Munro JE, Chavez RA, Gordon L, Joo JE, Akikusa JD, et al. Genome-Scale Case-Control Analysis of CD4+ T-Cell DNA Methylation in Juvenile Idiopathic Arthritis Reveals Potential Targets Involved in Disease. Clin Epigenet (2012) 4(1):20. doi: 10.1186/1868-7083-4-20
111. Foley DL, Craig JM, Morley R, Olsson CA, Dwyer T, Smith K, et al. Prospects for Epigenetic Epidemiology. Am J Epidemiol (2009) 169(4):389–400. doi: 10.1093/aje/kwn380
112. de Andres M, Perez-Pampin E, Calaza M, Santaclara F, Ortea I, Gomez-Reino J, et al. Assessment of Global DNA Methylation in Peripheral Blood Cell Subpopulations of Early Rheumatoid Arthritis Before and After Methotrexate. Arthritis Res Ther (2015) 17:233. doi: 10.1186/s13075-015-0748-5
113. Wang Q, Fan Z, Li J, Fu L, Yan L, Yang B. Systematic Analysis of the Molecular Mechanisms of Methotrexate Therapy for Rheumatoid Arthritis Using Text Mining. Clin Exp Rheumatol (2021) 39(4):829–37.
114. Tao W, Concepcion A, Vianen M, Marijnissen A, Lafeber F, Radstake T, et al. Multiomics and Machine Learning Accurately Predict Clinical Response to Adalimumab and Etanercept Therapy in Patients With Rheumatoid Arthritis. Arthritis Rheumatol (2021) 73(2):212–22. doi: 10.1002/art.41516
115. Ciechomska M, Roszkowski L, Maslinski W. DNA Methylation as a Future Therapeutic and Diagnostic Target in Rheumatoid Arthritis. Cells (2019) 8(9):953. doi: 10.3390/cells8090953
116. Jha M, Aggarwal R, Jha A, Shrivastava A. Natural Compounds: DNA Methyltransferase Inhibitors in Oral Squamous Cell Carcinoma. Appl Biochem Biotechnol (2015) 177(3):577–94. doi: 10.1007/s12010-015-1768-y
117. Sun ZH, Liu YH, Liu JD, Xu DD, Li XF, Meng XM, et al. MeCP2 Regulates PTCH1 Expression Through DNA Methylation in Rheumatoid Arthritis. Inflammation (2017) 40(5):1497–508. doi: 10.1007/s10753-017-0591-8
118. Tasneem S, Liu B, Li B, Choudhary MI, Wang W. Molecular Pharmacology of Inflammation: Medicinal Plants as Anti-Inflammatory Agents. Pharmacol Res (2019) 139:126–40. doi: 10.1016/j.phrs.2018.11.001
119. Karatas A, Dagli AF, Orhan C, Gencoglu H, Ozgen M, Sahin N, et al. Epigallocatechin 3-Gallate Attenuates Arthritis by Regulating Nrf2, HO-1, and Cytokine Levels in an Experimental Arthritis Model. Biotechnol Appl Biochem (2020) 67(3):317–22. doi: 10.1002/bab.1860
120. Lee SY, Jung YO, Ryu JG, Oh HJ, Son HJ, Lee SH, et al. Epigallocatechin-3-Gallate Ameliorates Autoimmune Arthritis by Reciprocal Regulation of T Helper-17 Regulatory T Cells and Inhibition of Osteoclastogenesis by Inhibiting STAT3 Signaling. J Leukoc Biol (2016) 100(3):559–68. doi: 10.1189/jlb.3A0514-261RR
121. Singh AK, Umar S, Riegsecker S, Chourasia M, Ahmed S. Regulation of Transforming Growth Factor β-Activated Kinase Activation by Epigallocatechin-3-Gallate in Rheumatoid Arthritis Synovial Fibroblasts: Suppression of K(63) -Linked Autoubiquitination of Tumor Necrosis Factor Receptor-Associated Factor 6. Arthritis Rheumatol (2016) 68(2):347–58. doi: 10.1002/art.39447
122. Hsiao HB, Wu JB, Lin WC. Anti-Arthritic and Anti-Inflammatory Effects of (-)-Epicatechin-3-O-β-D-Allopyranoside, a Constituent of Davallia Formosana. Phytomedicine (2019) 52:12–22. doi: 10.1016/j.phymed.2018.09.192
123. Lee WJ, Shim JY, Zhu BT. Mechanisms for the Inhibition of DNA Methyltransferases by Tea Catechins and Bioflavonoids. Mol Pharmacol (2005) 68(4):1018–30. doi: 10.1124/mol.104.008367
124. Wang W, Sun W, Jin L. Caffeic Acid Alleviates Inflammatory Response in Rheumatoid Arthritis Fibroblast-Like Synoviocytes by Inhibiting Phosphorylation of Iκb Kinase α/β and Iκbα. Int Immunopharmacol (2017) 48:61–6. doi: 10.1016/j.intimp.2017.04.025
125. Lee WJ, Zhu BT. Inhibition of DNA Methylation by Caffeic Acid and Chlorogenic Acid, Two Common Catechol-Containing Coffee Polyphenols. Carcinogenesis (2006) 27(2):269–77. doi: 10.1093/carcin/bgi206
126. Chauhan PS, Satti NK, Sharma P, Sharma VK, Suri KA, Bani S. Differential Effects of Chlorogenic Acid on Various Immunological Parameters Relevant to Rheumatoid Arthritis. Phytother Res (2012) 26(8):1156–65. doi: 10.1002/ptr.3684
127. Arab HH, Gad AM, Fikry EM, Eid AH. Ellagic Acid Attenuates Testicular Disruption in Rheumatoid Arthritis via Targeting Inflammatory Signals, Oxidative Perturbations and Apoptosis. Life Sci (2019) 239:117012. doi: 10.1016/j.lfs.2019.117012
128. Paluszczak J, Krajka-Kuźniak V, Baer-Dubowska W. The Effect of Dietary Polyphenols on the Epigenetic Regulation of Gene Expression in MCF7 Breast Cancer Cells. Toxicol Lett (2010) 192(2):119–25. doi: 10.1016/j.toxlet.2009.10.010
129. Yang G, Chang CC, Yang Y, Yuan L, Xu L, Ho CT, et al. Resveratrol Alleviates Rheumatoid Arthritis via Reducing ROS and Inflammation, Inhibiting MAPK Signaling Pathways, and Suppressing Angiogenesis. J Agric Food Chem (2018) 66(49):12953–60. doi: 10.1021/acs.jafc.8b05047
130. Papoutsis AJ, Borg JL, Selmin OI, Romagnolo DF. BRCA-1 Promoter Hypermethylation and Silencing Induced by the Aromatic Hydrocarbon Receptor-Ligand TCDD are Prevented by Resveratrol in MCF-7 Cells. J Nutr Biochem (2012) 23(10):1324–32. doi: 10.1016/j.jnutbio.2011.08.001
131. Hur YG, Suh CH, Kim S, Won J. Rosmarinic Acid Induces Apoptosis of Activated T Cells From Rheumatoid Arthritis Patients via Mitochondrial Pathway. J Clin Immunol (2007) 27(1):36–45. doi: 10.1007/s10875-006-9057-8
132. Mohanty S, Sahoo AK, Konkimalla VB, Pal A, Si SC. Naringin in Combination With Isothiocyanates as Liposomal Formulations Potentiates the Anti-Inflammatory Activity in Different Acute and Chronic Animal Models of Rheumatoid Arthritis. ACS Omega (2020) 5(43):28319–32. doi: 10.1021/acsomega.0c04300
133. Meeran SM, Patel SN, Tollefsbol TO. Sulforaphane Causes Epigenetic Repression of hTERT Expression in Human Breast Cancer Cell Lines. PloS One (2010) 5(7):e11457. doi: 10.1371/journal.pone.0011457
134. Moia VM, Leal Portilho F, Almeida Pádua T, Barbosa Corrêa L, Ricci-Junior E, Cruz Rosas E, et al. Lycopene Used as Anti-Inflammatory Nanodrug for the Treatment of Rheumathoid Arthritis: Animal Assay, Pharmacokinetics, ABC Transporter and Tissue Deposition. Colloids Surf B Biointerfaces (2020) 188:110814. doi: 10.1016/j.colsurfb.2020.110814
135. King-Batoon A, Leszczynska JM, Klein CB. Modulation of Gene Methylation by Genistein or Lycopene in Breast Cancer Cells. Environ Mol Mutagen (2008) 49(1):36–45. doi: 10.1002/em.20363
136. Williams B, Lees F, Tsangari H, Hutchinson MR, Perilli E, Crotti TN. Assessing the Effects of Parthenolide on Inflammation, Bone Loss, and Glial Cells Within a Collagen Antibody-Induced Arthritis Mouse Model. Mediators Inflammation (2020) 2020:6245798. doi: 10.1155/2020/6245798
137. Liu Z, Liu S, Xie Z, Pavlovicz RE, Wu J, Chen P, et al. Modulation of DNA Methylation by a Sesquiterpene Lactone Parthenolide. J Pharmacol Exp Ther (2009) 329(2):505–14. doi: 10.1124/jpet.108.147934
138. Mohammadian Haftcheshmeh S, Khosrojerdi A, Aliabadi A, Lotfi S, Mohammadi A, Momtazi-Borojeni AA. Immunomodulatory Effects of Curcumin in Rheumatoid Arthritis: Evidence From Molecular Mechanisms to Clinical Outcomes. Rev Physiol Biochem Pharmacol (2021) 179:1–29. doi: 10.1007/112_2020_54
139. Liu Z, Xie Z, Jones W, Pavlovicz RE, Liu S, Yu J, et al. Curcumin Is a Potent DNA Hypomethylation Agent. Bioorg Med Chem Lett (2009) 19(3):706–9. doi: 10.1016/j.bmcl.2008.12.041
140. Wu F, Wang J, Sun J, Shen L, Liu M, Zhao E. Procaine Stimulates Aquaporin−5 Expression in Human Salivary Gland Ductal Cells via the Suppression of DNA Methyltransferase−1. Mol Med Rep (2018) 17(6):7996–8002. doi: 10.3892/mmr.2018.8821
141. Fearnley GR, Lackner R, Meanock RI, Bywaters EG. Pilot Study of Intra-Articular Procaine and Hydrocortisone Acetate in Rheumatoid Arthritis. Ann Rheum Dis (1956) 15(2):134–9. doi: 10.1136/ard.15.2.134
142. Richardson B, Scheinbart L, Strahler J, Gross L, Hanash S, Johnson M. Evidence for Impaired T Cell DNA Methylation in Systemic Lupus Erythematosus and Rheumatoid Arthritis. Arthritis Rheum (1990) 33(11):1665–73. doi: 10.1002/art.1780331109
143. Wang LG, Beklemisheva A, Liu XM, Ferrari AC, Feng J, Chiao JW. Dual Action on Promoter Demethylation and Chromatin by an Isothiocyanate Restored GSTP1 Silenced in Prostate Cancer. Mol Carcinog (2007) 46(1):24–31. doi: 10.1002/mc.20258
144. Li J, Gang D, Yu X, Hu Y, Yue Y, Cheng W, et al. Genistein: The Potential for Efficacy in Rheumatoid Arthritis. Clin Rheumatol (2013) 32(5):535–40. doi: 10.1007/s10067-012-2148-4
145. Fang MZ, Chen D, Sun Y, Jin Z, Christman JK, Yang CS. Reversal of Hypermethylation and Reactivation of P16ink4a, RARbeta, and MGMT Genes by Genistein and Other Isoflavones From Soy. Clin Cancer Res (2005) 11(19 Pt 1):7033–41. doi: 10.1158/1078-0432.Ccr-05-0406
146. Yuan K, Zhu Q, Lu Q, Jiang H, Zhu M, Li X, et al. Quercetin Alleviates Rheumatoid Arthritis by Inhibiting Neutrophil Inflammatory Activities. J Nutr Biochem (2020) 84:108454. doi: 10.1016/j.jnutbio.2020.108454
147. Tan S, Wang C, Lu C, Zhao B, Cui Y, Shi X, et al. Quercetin Is Able to Demethylate the P16ink4a Gene Promoter. Chemotherapy (2009) 55(1):6–10. doi: 10.1159/000166383
148. Yap ZH, Kong WY, Azeez AR, Fang CM, Ngai SC. Anti-Cancer Effects of Epigenetics Drugs Scriptaid and Zebularine in Human Breast Adenocarcinoma Cells. Anticancer Agents Med Chem (2021). doi: 10.2174/1871520621666210608103251
149. Krzeminski P, García-Sanz R, Gutiérrez NC. Zebularine-Induced Myeloma Cell Death Is Accompanied by Decreased C-Myc Expression. Cell Oncol (Dordr) (2020) 43(4):743–50. doi: 10.1007/s13402-020-00516-6
150. Zheng Z, Zeng S, Liu C, Li W, Zhao L, Cai C, et al. The DNA Methylation Inhibitor RG108 Protects Against Noise-Induced Hearing Loss. Cell Biol Toxicol (2021) 37(5):751–71. doi: 10.1007/s10565-021-09596-y
151. Fini L, Selgrad M, Fogliano V, Graziani G, Romano M, Hotchkiss E, et al. Annurca Apple Polyphenols Have Potent Demethylating Activity and Can Reactivate Silenced Tumor Suppressor Genes in Colorectal Cancer Cells. J Nutr (2007) 137(12):2622–8. doi: 10.1093/jn/137.12.2622
152. Datta J, Ghoshal K, Denny WA, Gamage SA, Brooke DG, Phiasivongsa P, et al. A New Class of Quinoline-Based DNA Hypomethylating Agents Reactivates Tumor Suppressor Genes by Blocking DNA Methyltransferase 1 Activity and Inducing its Degradation. Cancer Res (2009) 69(10):4277–85. doi: 10.1158/0008-5472.Can-08-3669
153. Rilova E, Erdmann A, Gros C, Masson V, Aussagues Y, Poughon-Cassabois V, et al. Design, Synthesis and Biological Evaluation of 4-Amino-N- (4-Aminophenyl)Benzamide Analogues of Quinoline-Based SGI-1027 as Inhibitors of DNA Methylation. ChemMedChem (2014) 9(3):590–601. doi: 10.1002/cmdc.201300420
154. Jin L, Ma X, Lei X, Tong J, Wang R. Cyclophosphamide Inhibits Pax5 Methylation to Regulate the Growth of Retinoblastoma via the Notch1 Pathway. Hum Exp Toxicol (2021) 40(12_suppl):S497–s508. doi: 10.1177/09603271211051601
155. Jagadeesh S, Sinha S, Pal BC, Bhattacharya S, Banerjee PP. Mahanine Reverses an Epigenetically Silenced Tumor Suppressor Gene RASSF1A in Human Prostate Cancer Cells. Biochem Biophys Res Commun (2007) 362(1):212–7. doi: 10.1016/j.bbrc.2007.08.005
156. Pereira MA, Tao L, Liu Y, Li L, Steele VE, Lubet RA. Modulation by Budesonide of DNA Methylation and mRNA Expression in Mouse Lung Tumors. Int J Cancer (2007) 120(5):1150–3. doi: 10.1002/ijc.22468
157. Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, et al. An Inhibitor of Mutant IDH1 Delays Growth and Promotes Differentiation of Glioma Cells. Science (2013) 340(6132):626–30. doi: 10.1126/science.1236062
Keywords: rheumatoid arthritis, DNA methylation, T-cell, precision medicine, therapeutic target
Citation: Zhao J, Wei K, Chang C, Xu L, Jiang P, Guo S, Schrodi SJ and He D (2022) DNA Methylation of T Lymphocytes as a Therapeutic Target: Implications for Rheumatoid Arthritis Etiology. Front. Immunol. 13:863703. doi: 10.3389/fimmu.2022.863703
Received: 27 January 2022; Accepted: 14 February 2022;
Published: 03 March 2022.
Edited by:
Haitao Wang, Center for Cancer Research, National Cancer Institute (NIH), United StatesReviewed by:
Xinwei Wu, National Institutes of Health (NIH), United StatesLaila A. Damiati, University of Jeddah, Saudi Arabia
Guangchun Han, University of Texas MD Anderson Cancer Center, United States
Copyright © 2022 Zhao, Wei, Chang, Xu, Jiang, Guo, Schrodi and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steven J. Schrodi, U2Nocm9kaUB3aXNjLmVkdQ==; Shicheng Guo, U2hpY2hlbmcuR3VvQHdpc2MuZWR1; Dongyi He, ZG9uZ3lpaGVAbWVkbWFpbC5jb20uY24=