 Muzi Ouyang
Muzi Ouyang Changmeng Yu
Changmeng Yu Xiaolian Deng
Xiaolian Deng Yingyi Zhang
Yingyi Zhang Xudong Zhang1
Xudong Zhang1 Fangfang Duan
Fangfang Duan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 01 April 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.861559
This article is part of the Research TopicEpigenetics of the Immune Component of InflammationView all 43 articles
Cancer cells, as well as surrounding stromal and inflammatory cells, form an inflammatory tumor microenvironment (TME) to promote all stages of carcinogenesis. As an emerging post-translational modification (PTM) of serine and threonine residues of proteins, O-linked-N-Acetylglucosaminylation (O-GlcNAcylation) regulates diverse cancer-relevant processes, such as signal transduction, transcription, cell division, metabolism and cytoskeletal regulation. Recent studies suggest that O-GlcNAcylation regulates the development, maturation and functions of immune cells. However, the role of protein O-GlcNAcylation in cancer-associated inflammation has been less explored. This review summarizes the current understanding of the influence of protein O-GlcNAcylation on cancer-associated inflammation and the mechanisms whereby O-GlcNAc-mediated inflammation regulates tumor progression. This will provide a theoretical basis for further development of anti-cancer therapies.
The enzymatic process wherein carbohydrate moieties (referred to as glycans) get covalent attachment to proteins and lipids is defined as glycosylation. Protein glycosylation encompasses N-glycans, O-glycans, and proteoglycans. Until 1984, a novel form of protein O-glycosylation termed as O-linked β-N-Acetylglucosaminylation (O-GlcNAcylation) was firstly discovered by Hart and Torres (1). Unlike other types of protein glycosylation which glycosylate proteins either secreting from the cell, or residing in the extracellular plasma membrane, O-GlcNAcylation is an intracellular O-glycosylation, occurring in the nucleocytoplasmic and mitochondrial compartments. O-GlcNAcylation mediates the addition of a single monosaccharide, N-acetylglucosamine (GlcNAc), onto the hydroxyl groups of the amino acid serine or threonine residues of proteins to form a β-glycosidic bond. The donor substrate for protein O-GlcNAcylation is uridine diphosphate N-Acetylglucosamine (UDP-GlcNAc), which is the end product of the hexosamine biosynthetic pathway (HBP). As HBP integrates intermediate byproducts from lipids, amino acid and nucleotide metabolic pathways, O-GlcNAcylation is nutrition sensitive and is greatly impacted by the metabolism reprogramming in the tumor microenvironment.
To date, approximately 3000 human proteins have been confirmed to be O-GlcNAcylated (2). As one type of reversible protein post-translational modification, O-GlcNAcylation modulates protein functions mainly by regulating their enzyme activity, subcellular localization, protein stability, transcriptional activity and interaction with other proteins (3). Dysregulation in O-GlcNAc cycling has been implicated in the progression of various chronic human diseases including aging, obesity and diabetes, cardiovascular disease, neurodegenerative disorders, and carcinogenesis (4). Imbalanced levels of O-GlcNAcylation have been found in different kinds of cancers and contributing to various hallmarks of cancers such as tumor growth, metastasis, angiogenesis, cancer stem-like potential and metabolic reprogramming. Recent studies have identified O-GlcNAcylation is essential for the proliferation of corticotropic tumor cells (5). Increased O-GlcNAcylation together with mTOR pathways activation cooperate to increase Fatty acid synthase (FASN) expression to promote tumorigenesis of hepatic tumors (6). Moreover, O-GlcNAcylated MORC family CW-type zinc finger 2 (MORC2) at Thr556 is required for transcriptional activation of TGF-β1 to facilitate breast cancer cell migration and invasion (7). Inhibition of O-GlcNAcylation attenuates breast cancer stem-like cells (CSCs) potential (8). And genetic ablation of receptor for activated C kinase 1 (RACK1) O-GlcNAcylation at Ser122 dramatically suppressed angiogenesis in hepatocellular carcinoma (HCC) (9). Besides, β-catenin O-GlcNAcylation is accompanied by its nuclear translocation, thus enhancing the transcription of CEMIP to promote colorectal cancer metastasis via inducing glutamine metabolic reprogramming (10).
More than a century, accumulating evidence shows close association between chronic inflammation and increased risk of cancer progression and malignant development. Cancer-associated inflammation provides an immunosuppressive environment which aid the metabolic deregulations of the cancer cells, thus favoring tumor growth and metastasis (11). Moreover, metabolism reprogramming in tumor cells can also help sustain a chronic inflammation status (12). In comparation to normal cells, cancer cells have an altered cell metabolism to meet increased biosynthetic demand and energy requirements for tumor growth. Cancer cells took up high quantities of glucose and glutamine to sustain pools of various carbon intermediates. These metabolic changes accelerate the production of several molecules, such as lactate, reactive oxygen species (ROS) which simultaneously aid an inflammatory milieu (13). If the inflammation is unregulated and sustains to be chronic, various inflammatory cells will infiltrate and become active in the tumor microenvironment. These cells produce inflammatory mediators, such as growth factors, cytokines and chemokines and to stimulate tumor initiation and malignant growth.
Here, we focus on the latest research linking O-GlcNAcylation and cancer-associated inflammation and outline the underlying mechanisms of how O-GlcNAcylation drives cancer inflammation in the tumor microenvironment and promote cancer progression.
Before 1983, people never expected to find O-glycosylation occurring in the nucleocytoplasmic and mitochondrial compartments. O-GlcNAcylation was firstly reported in 1984 by Hart and Torres and they identified that the majority of O-glycosidically linked GlcNAc monosaccharide was inside the cells (1, 14). Their discovery was fascinating at that time. Primarily, O-GlcNAcylation was the first identified O-glycosylation which glycosylated proteins both in the nucleus and cytoplasm. Furthermore, similar to phosphorylation, O-GlcNAcylation was in a glycosylation and de-glycosylation dynamic equilibrium and was not as stable as proteoglycans in the extracellular matrix (15).
Additionally, there are several characteristics of this kind of post-translational modification. Initially, O-GlcNAcylation can only occur within the nucleocytoplasmic and mitochondrial compartments. Secondly, unlike “traditional glycosylation”, such as asparagine-linked or mucin-type O-glycosylation which can get extended into numerous different structures (16, 17), O-GlcNAcylation don’t get elongated to highly branched complicated structures (18). Instead, only one single GlcNAc was attached to the serine/threonine (S/T) residues on proteins by an O-linked β-glycosidic bond. Moreover, O-GlcNAcylation is a reversible and dynamic modification and mostly has a reciprocal relationship with phosphorylation. Numerous studies revealed O-GlcNAcylation occurs sequentially or reciprocally with phosphorylation on the same or nearby residues of numerous proteins. Here are some examples. For sequential crosstalk, Liu et al. determined that O-GlcNAcylation of Y-box binding protein 1(YB-1) at Thr126 was dependent on its phosphorylation at Ser102 to enhance YB-1 transcriptional activities in HepG2 cells (19). Tao and colleagues revealed that the TAK1 binding protein 3 (TAB3) was O-GlcNAcylated at Ser408 in triple negative breast cancer (TNBC), which was required for its Thr404 phosphorylation (20). In addition, Thr13 O-GlcNAcylation of MEK2 specifically enhanced its Thr394 phosphorylation together with downstream ERK1/2 activation to promote proliferation and migration of breast cancer cells (21). Recent study also identified Ser15 phosphorylation and Ser430 O-GlcNAcylation of PYGL were mutually reinforced under glucagon conditions in HCT116 cells (22). For reciprocal crosstalk, O-GlcNAcylation occupies the same site or adjacent positions to compete with phosphorylation and are capable to inhibit protein phosphorylation and vice-versa. Lei et al. determined O-GlcNAcylation of PFKFB3 could compete phosphorylation at the same Ser172 residue under hypoxic conditions in pancreatic cancer (23). Li et al. discovered Yes-associated protein (YAP) Ser109 O-GlcNAcylation promoted the malignant phenotypes in papillary thyroid cancer (PTC) cells by inducing YAP Ser127 dephosphorylation and activation (24). In NF-κB signaling, phosphorylation of p65 at Thr308 may prevent O-GlcNAcylation of p65 at Thr305 (25). Furthermore, O-GlcNAcylation/Phosphorylation crosstalk can even occur whereby the two PTMs are situated at quite distal sites, such as O-GlcNAcylation of endothelial nitric oxide synthase (eNOS) at Ser615 could interfere its phosphorylation at Ser1177 (26). LATS2 O-GlcNAcylation at Thr168 and Thr436 inhibited its phosphorylation at Ser872 and Thr1041, which has a decisive effect on LATS2 activation (27). Considering all of the above mentioned, this kind of O-GlcNAcylation/Phosphorylation relationship was termed “yin-yang model” (28). Moreover, apart from phosphorylation, O-GlcNAcylation has interplay with other posttranslational modifications, such as ubiquitination, acetylation and methylation (29–31).
Last but not least, another characteristic of O-GlcNAcylation is that in contrast to phosphorylation where there exist numerous kinases or phosphatases mediating to add or remove phosphorylation, the addition and removal of O-GlcNAcylation to target proteins are regulated by one single O-linked-β-N-acetylglucosamine transferase (O-GlcNAc transferase, OGT) and one single β-N-Acetylglucosaminidase (O-GlcNAcase, OGA), respectively. OGT uses UDP-GlcNAc to catalyze O-GlcNAc addition while OGA modulates O-GlcNAc removal from target proteins. Both OGT and OGA are highly conserved across evolution (32, 33). In mice, deletion of OGT is lethal at the embryonic level and conditional disruption of the OGA gene causes perinatal lethality, indicating the key role of O-GlcNAcylation in regulating fundamental cellular biological processes (34, 35).
One hallmark of cancer progression is altered metabolic state, shifting from oxidative phosphorylation to aerobic glycolysis which is termed as Warburg effect, characterized by large scale of glucose uptake (36). This phenomenon not only promote rapid ATP synthesis, but also help sustained production of glycolytic carbon intermediates required for the increased biosynthetic demands needed by the rapidly dividing cancer cells (36). The abundance of glucose in cancer cells primarily enters glycolysis, and also increases flux to glucose branched pathways, such as the hexosamine biosynthetic pathway (HBP).
HBP integrates various metabolic inputs, such as glucose, amino acid, fatty acid, nucleotide, to promote the synthesis of UDP-GlcNAc (37) (Figure 1). UDP-N-Acetlyglucosamine (UDP-GlcNAc) is the end product of HBP and is also the substrate for O-GlcNAcylation. As HBP integrates different metabolic intermediates fluxing to UDP-GlcNAc, O-GlcNAcylation act as a “nutritional sensor” (38, 39). Therefore, O-GlcNAcylation has been considered to serve in the regulation of cellular signaling, transcription in the cancer cells, as well as the adjacent stromal and inflammatory cells in response to nutrients and stresses in the TME. The signaling pathways activated in these cells in TME convert environmental cues into intracellular events, such as immune cell activation and inflammation to form an immunosuppressive inflammatory TME to promote cancer progression.
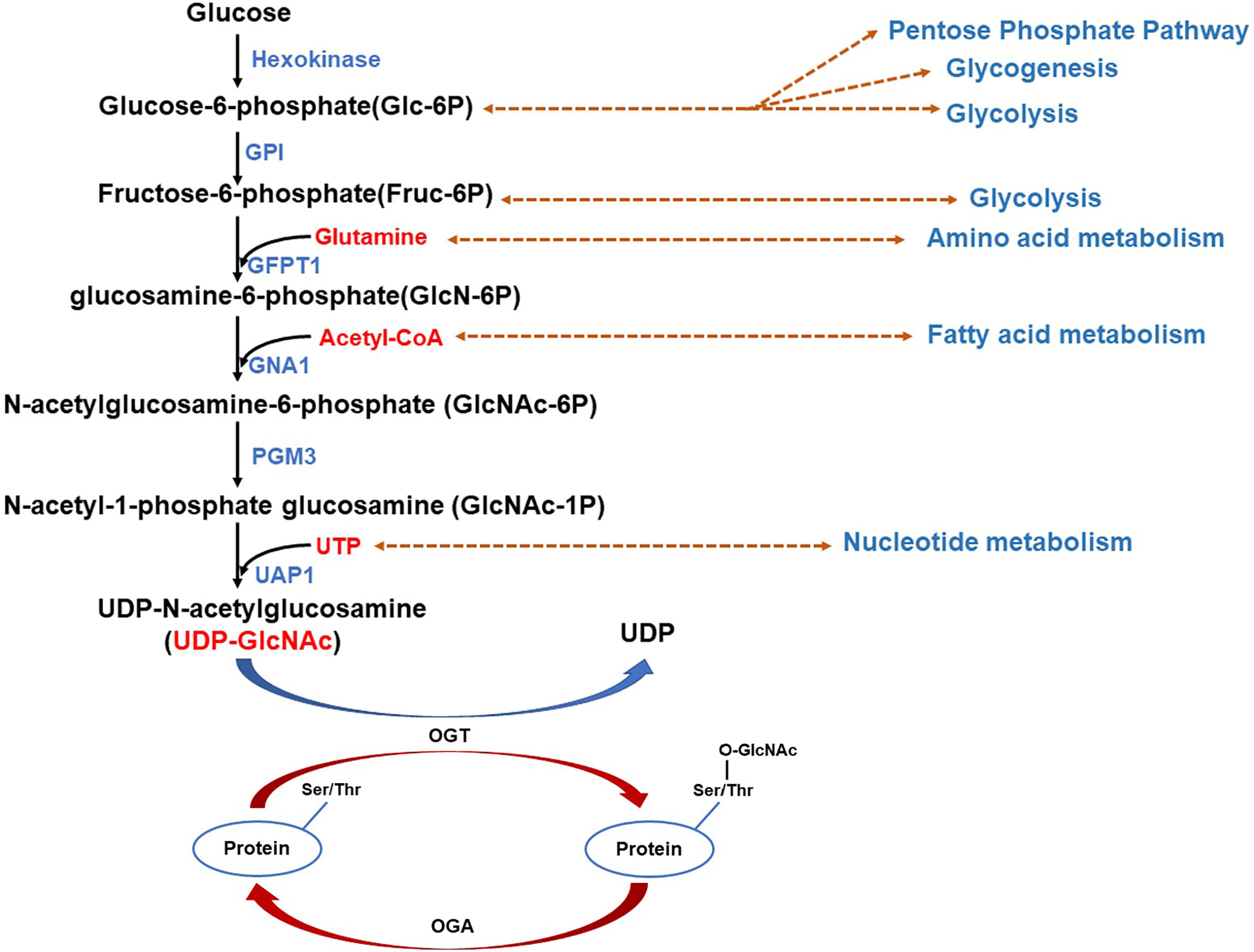
Figure 1 Schematic illustration of the hexosamine biosynthesis pathway (HBP) integrating different metabolic intermediates fluxing to UDP-N-Acetlyglucosamine (UDP-GlcNAc). Glucose is phosphorylated by hexokinase yielding glucose-6-phosphate (Glc-6P) which can be converted to fructose-6-phosphate (Fruc-6P) by glucose-6-phosphate isomerase (GPI). Both Glc-6P and Fruc-6P can be metabolized by glycolysis or the HBP. The rate-limiting enzyme of HBP: Glutamine-fructose 6-phosphate aminotransferase (GFPT1 or GFAT) mediates the diverge to HBP, conversing Fruc-6P to glucosamine-6-phosphate (GlcN-6P) from glutamine, the amide donor. Additionally, GFAT mRNA and protein expression was upregulated by saturated fatty acids (palmitate and stearate), whereas inhibition occurs owing to feedback from its enzymatic product GlcN-6P. Next, acetyl-CoA (AcCoA) and GlcN-6P are converted by D-glucosamine-6-phosphate N-Acetyltransferase (GNA1) to CoA and N-Acetylglucosamine-6-phosphate (GlcNAc-6P). After that, another enzyme, GlcNAc phosphomutase (PGM3), converts GlcNAc-6P to N-Acetyl-1-phosphate glucosamine (GlcNAc-1P), employing glucose-1,6-bisphosphate as a co-factor. The nucleoside (UTP) is then added to the sugar by UDP-N-Acetylhexosamine pyrophosphorylase (UAP1) yielding UDP-N-Acetlyglucosamine(UDP-GlcNAc). Glucose-6-phosphate isomerase (GPI); Glutamine-fructose 6-phosphate aminotransferase (GFPT1 or GFAT); D-glucosamine-6-phosphate N-Acetyltransferase (GNA1); GlcNAc phosphomutase (PGM3); Nucleoside (UTP); UDP-N-Acetylhexosamine pyrophosphorylase (UAP1).
The relationship between inflammation and cancer was firstly discovered in 1893 by Rudolf Virchow who found leucocytes in neoplastic tissues and make it clear that inflammation accompanies with cancer (40). Cancer-associated inflammation can fall into two categories: cancer extrinsic inflammation and cancer intrinsic inflammation. Cancer extrinsic inflammation is driven by chronic long-term inflammatory conditions which predispose to cancer (41). There are some risk factors accounting for the cancer extrinsic inflammation, composing of infections from bacterial and viral, obesity, autoimmune diseases, tobacco smoking, and excessive alcohol consumption. For example, infection with Helicobacter pylori positively correlates with tumorigenesis of gastric cancer and gastric mucosal lymphoma. Some autoimmune diseases, such as Crohn’s disease (CD) is associated with colon cancer progression. Around 15%–20% of all cancer cases show this kind of precancerous inflammatory conditions present before a malignant change occurs (41). All of these inflammatory conditions form a constant inflammatory state, causing precancerous inflammation lesions which will precede the development of cancer malignancy.
However, most cancers are not developed from long-lasting chronic inflammation (42). In contrast, cancer cells recruit immune cells and secrete inflammatory mediators to reshape the tumor microenvironment (TME) to promote cancer intrinsic inflammation (41). Cancer intrinsic inflammation or cancer-elicited inflammation is defined as inflammation driven by genetic events that cause neoplasia (39). It is elicited by genetic and/or epigenetic mutation (Oncogenes) that drives a tumor-promoting inflammatory milieu which involves the recruitment and activation of inflammatory cells. Cancer cells, as well as the adjacent stromal and inflammatory cells participate in well-orchestrated reciprocal interactions to form an inflammatory TME to promote cancer progression.
Both cancer extrinsic and intrinsic inflammation results in transcription factors and core signaling pathways activation to modulate the inflammatory response through soluble mediators (cytokines, chemokines) and cellular components (e.g. tumor-associated macrophages). During these processes, O-GlcNAcylation acts as a key orchestrator at the intersection of the intrinsic and extrinsic pathways to trigger immunosuppression in tumor microenvironment, thus favoring tumor tumorigenesis (Figure 2).
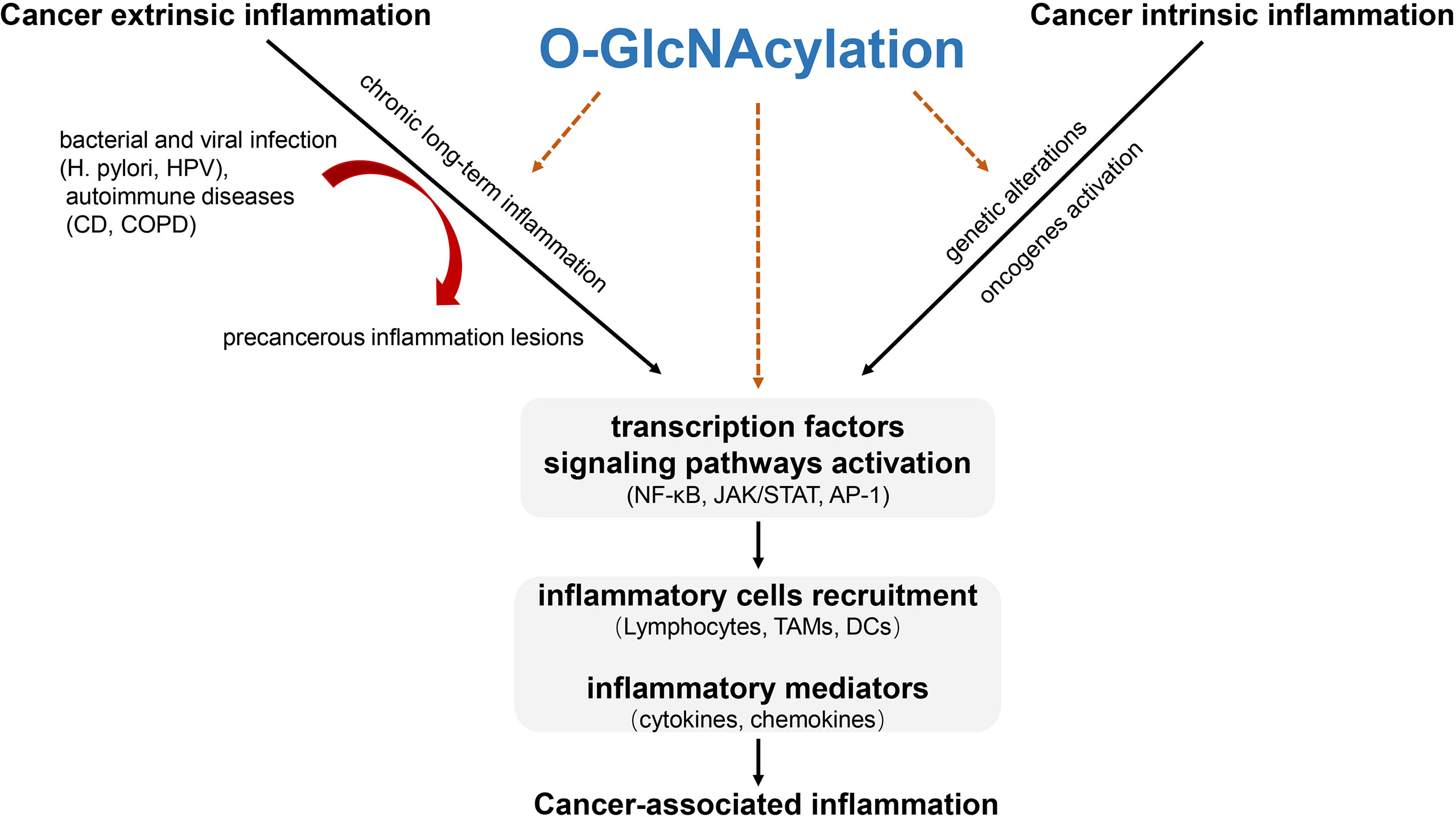
Figure 2 The role of O-GlcNAcylation involved in cancer-associated inflammation. Cancer-associated inflammation can fall into cancer extrinsic inflammation and cancer intrinsic inflammation. Cancer extrinsic inflammation is driven by long-lasting chronic inflammation to form precancerous inflammation lesions while cancer intrinsic inflammation is elicited by genetic and/or epigenetic mutation to form an inflammatory milieu. The two ways converge to promote transcription factors-mediated signaling pathways activation, thus facilitating inflammatory cells recruitment and inflammatory mediators’ production. O-GlcNAcylation promotes precancerous inflammation and acts as a key orchestrator at the intersection of the intrinsic and extrinsic inflammation through O-GlcNAcylating key transcription factors and functional proteins in inflammatory cells activation to trigger cancer-associated inflammation in tumor microenvironment. H. pylori, Helicobacter pylori; HPV, Human papillomavirus; CD, Crohn’s disease; COPD, Chronic obstructive pulmonary disease; TAMs, Tumor associated macrophages; DCs, Dendritic cells.
Epidemiological studies have attributed up to 25% of all cancers are related to chronic infection, termed as cancer extrinsic inflammation (43–45). Numerous studies identified that chronic inflammation is an vital risk factor for the development of cancer progression, such as Gastric cancer [Helicobacter pylori (H. pylori)] (46), Colorectal cancer [Inflammatory bowel disease (47)], Hepatocellular carcinoma [Hepatitis B/C virus (48)], Prostate cancer [or Prostatitis (49)], Pancreatic cancer [Pancreatitis (50)], Cervical cancer (Human papillomavirus (HPV) (51), Lung cancer [(Chronic obstructive pulmonary disease(COPD)] (52), Bladder cancer (Schistosoma haematobium) (53). All of these inflammatory conditions form a constant inflammatory state, causing precancerous inflammation lesions which will precede the development of cancer malignancy.
It has been shown that H. pylori infection creates an inflammatory environment, providing a favorable role in gastric cancer oncogenesis and is well known to be associated with the development of precancerous lesions such as chronic atrophic gastritis (AG) (54). Tae and colleagues determined both O-GlcNAcylation and OGT expression in the epithelium and mononuclear inflammatory cells from biopsied tissues of chronic gastritis (55). They also demonstrated that elevated expression of OGT and O-GlcNAcylation are found in H. pylori-infected chronic gastritis than those in chronic gastritis without H. pylori infection. Their findings demonstrated the importance of O-GlcNAcylation in the development of gastric cancer precancerous lesions.
Inflammation is also an established risk factor for colorectal cancer (CRC) (56). Ulcerative colitis (UC) and Crohn’s disease (CD) are common types of inflammatory bowel disease (IBD) which causes inflammation and irritation of the gastrointestinal tract, thus increasing risk for bowl cancer (57). Yang and colleagues discovered elevation of O-GlcNAcylation was found in dextran sulfate sodium (DSS)-induced colitis and increased O-GlcNAcylation promotes the progression of colitis-associated colorectal cancer (58). Elevated O-GlcNAc levels also showed in intestinal epithelial tissues of active CD patients and adherent-invasive Escherichia coli (AIEC) LF82-infected mice (59). Inhibition of O-GlcNAc protects mice from DSS (dextran sulfate sodium)- and AIEC LF82-induced intestinal inflammation. Moreover, gut microbiota also influences intestinal inflammatory physiology. The enzyme OGAs from gut microbiota help hydrolyse O-GlcNAcylated proteins in host cells, thus suppressing inflammatory response in the gut and protecting mice from chemically induced colonic inflammation (60). These results demonstrated O-GlcNAcylation provides a favorable role in IBD-induced chronic intestinal inflammation and inhibition of O-GlcNAc from gut microbiota can reverse colonic inflammation.
Chronic hepatitis B virus (HBV) infection contributes to at least 50% cases of Hepatocellular carcinoma (HCC) worldwide (61). HBV infection stimulates the host immune response and drives liver chronic necro-inflammation to promote hepatocarcinogenesis (62). Hu et al. revealed total O-GlcNAcylation levels got markedly higher regulation in liver tissues from patients with chronic hepatitis B (CHB) than in those from normal controls (63). HBV infection upregulated glucose transporter 1 (GLUT1) expression which facilitated glucose uptake, thus leading to an increase in protein O-GlcNAcylation. Pharmacological or transcriptional inhibition of HBP and O-GlcNAcylation enhanced HBV replication because O-GlcNAcylated SAMHD1 at Ser93 stabilizes SAMHD1 and enhances its antiviral activity. The findings reveal a link between HBP, O-GlcNAc modification, and host antiviral immune response against HBV.
Inflammation have been reported to promote the progression of benign prostatic hyperplasia (BPH) (64). Meta-Analysis suggested BPH is associated with an increased risk of prostate cancer, especially in Asian BPH patients (65). Gu et al. identified that elevated O-GlcNAcylation levels induces malignant transformation of nontumorigenic benign prostatic hyperplasia (BPH) cells, enhancing migratory and invasive ability through inhibiting the formation of the E-cadherin/catenin/cytoskeleton complex (66). O-GlcNAcylation is shown to be an inducer of prostate cancer progression.
Patients with acute pancreatitis (AP) characterized by inflammation had an increased risk of pancreatic cancer (67). Zhang et al. found that both OGT and O-GlcNAcylation were upregulated in acute pancreatitis cell model which was constructed with caerulein-stimulated AR42 J rat pancreatic acinar cells. Reducing the expression of OGT attenuated the severity of inflammation while O-GlcNAc upregulation increased the AP severity (68). In this AP model, both NF-κB subunit p65 and its upstream activating kinases IKKα were O-GlcNAcylated, which are responsible for the inflammatory NF-κB activation during acute pancreatitis. The results demonstrate that OGT-mediated O-GlcNAcylation promotes NF-κB-mediated inflammation in pancreatic acinar cells to promote the progression of AP.
Persistent genital high-risk human papillomavirus (HPV) infection accounts for about 99.7% of cervical cancer (69). Histological analysis revealed a higher degree of inflammation in biopsies of cervical mucosa from high risk (HR)−HPV−infected females, accompanied with an increased infiltration of neutrophils and lymphocytes into the epithelium, comparing with those who were uninfected (70). Cervical cancer can origin from persistent HPV lesions through the action of two HR HPV genes, E6 and E7 while HPV E6/E7 gene transcription can be upregulated by OGT though O-GlcNAc modification of HCF-1 in cervical cancer cells (71). Furthermore, levels of O-GlcNAc and OGT in HPV-associated cervical neoplasms were markedly increased relative to the normal cervix. Pharmacological inhibition of O-GlcNAcylation impaired HPV-mediated cervical carcinoma cells viability and transformation (72). Thus, O-GlcNAcylation serves as an essential regulator to facilitate HPV-associated malignancies.
Chronic obstructive pulmonary disease (COPD) is a characterized by persistent respiratory symptoms and enhanced inflammatory response (73). People with COPD have higher risk (4- to 6-fold) of developing lung cancer, ignore of the patients’ history, such as smoking history, age and sex (74, 75). HBP/O-GlcNAc modification activation is stimulated in human bronchial epithelial cells by FGF23 through the PLCγ signaling pathway, leading to NFAT activation and increased secretion of IL-6 which contribute to the progression of pathogenesis of chronic inflammatory airway diseases such as COPD (76). These findings revealed the link whereby FGF23 and the augmentation of O-GlcNAc levels regulate chronic airway inflammation.
Taken together, O-GlcNAcylation serve as a key regulator to facilitate the progression of these precancerous inflammation lesions, thus providing a tumor-supporting chronic immunosuppressive microenvironment for tumor initiation, growth and progression.
During both cancer intrinsic and extrinsic inflammation, a wide array of intracellular signaling transduction pathways are often dysregulated to stimulate malignant transformation. Transcription factors (TFs) (Nuclear factor-κB (NF-κB), STAT1/STAT3, HIFs, AP-1, and Nrf2) orchestrators various inflammation-related signaling pathways to modulate the inflammatory response, through inflammatory mediators (such as cytokines, chemokines) and immune cells infiltration (tumor-associated macrophages), thus promoting tumorigenesis. The following sections illustrate the role of O-GLCNAC in linking TFs with cancer-associated inflammation.
NF-κB activation plays central roles in inflammatory and immune responses (77). Nuclear factor-κB (NF-κB) consists of five master transcription factors, including p65 (RelA), RelB, c-Rel (Rel), p50/p105 (NF-κB1), and p52/p100 (NF-κB2) in mammals (78). In most cell types, NFκB is composed of p65 and p50 and is localized in the cytosol where it binds inhibitor (IκB). NF-κB can be activated by various stimuli which lead to the activation of the inhibitor of κB (IκB) kinase (IKK) complex. The activated IKK complex mediates phosphorylation of IκB for proteasomal degradation (79). Thus, the free NF-κB translocate from the cytoplasm to the nucleus, bind to DNA elements and activate the expression of target genes.
Studies revealed site specific O-GlcNAcylation is implicated in NF-κB pathway mediated inflammation. O-GlcNAcylation of NF-κB p65 on Thr352 decreases its binding to IκBα is required for transcriptional activity under hyperglycemic conditions (80). Yang et al. determined pancreatic ductal adenocarcinoma (PDAC) is supported by oncogenic NF-κB transcriptional activity and mutation of two p65 O-GlcNAc sites (T322A and T352A) attenuated the growth of PDAC (81). Apart from tumor growth, O-GlcNAcylation of NF-κB subunit p65 also promotes lung metastasis of cervical cancer cells via upregulating CXCR4 expression (82). Point mutated p65 at Thr322 or Thr352 in HeLa cells decreased CXCR4 expression compared to transfection with wild-type p65, indicating site specific p65 O-GlcNAcylation contributes to the regulation of CXCR4 expression in cervical cancer cells (82). Furthermore, O-GlcNAcylation upregulates matrix-metalloproteinases (MMPs) expression to enhance cholangiocarcinoma (CCA) cell migration and invasion via inducing NF-κB p65 nuclear translocation (83). However, whether up-regulation of MMPs mediated by O-GlcNAcylation in CCA is dependent on p65 O-GlcNAcylation at Thr322 and Thr352 is not investigated in this paper (83). In addition, there is some discrepancy between results from two studies on p65 O-GlcNAcylation in colon cancer progression. Yang et al. discovered p65 O-GlcNAcylation at Thr322 and Thr352 is required for transcriptional activation of p65 target genes (IL-6, TNF-α and MCP1) and promotes colonic inflammation and tumorigenesis (58). Nevertheless, Hirata and colleagues identified O-GlcNAcylation could protect from inflammation-induced colon carcinogenesis via suppressing NF-κB signaling (84). Their inconsistent findings could be from the difference of mouse model they used. Yang et al. implemented an Oga+/– mouse model while Hirata et al. utilized OGT-transgenic (OGT-Tg) mice. Besides subunit p65, O-GlcNAcylation of NF-κB subunit c-Rel can also get O-GlcNAcylated. O-GlcNAcylation of NF-κB subunit c-Rel at Ser350 was required for the DNA binding and transactivation. Blocking O-GlcNAcylation of this residue abrogated c-Rel-mediated expression of the cytokine-encoding genes IL2, IFNG, and CSF2 to promote T cell-mediated autoimmunity (85).
Moreover, the activation of NF-κB and IKK requires an upstream kinase complex consisting of TGFβ (transforming growth factor-beta)-activated protein kinase (TAK1) and adaptor proteins such as TAK1 binding protein 1 (TAB1), TAB2, TAB3 (86). Ser395 O-GlcNAcylation of TAB1 is essential for full TAK1 activation to induce NF-κB activation, thus favoring IL-6 and TNF-α production (87). Tao et al. revealed O-GlcNAcylation of TAB3 at Ser408 promotes triple negative breast cancer cell migration, invasion and is correlated with patient poor prognosis (20). The IKK complex is composed of two catalytic subunits, IKKα and IKKβ. O-GlcNAcylation of IKKβ sustained the TNFα-dependent IKKβ activation, thus stimulating NF-κB signaling and increased IKKβ expression is essential for cell viability in prostate cancer (88, 89). These studies collectively provide insights into the mechanism of O-GlcNAcylation regulating upstream signal transductors in NF-kB activation and suggests its important implications in tumor growth and metastasis.
Along with NF-κB, the Janus kinase/signal transduction and activator of transcription (JAK-STAT) signaling pathway is implicated in regulating cytokine-dependent inflammation and immunity in carcinogenesis (90). The binding of various ligands, usually cytokines, to cell-surface receptors cause the receptor to activate JAKs, which phosphorylate tyrosine residues on the receptor thereafter and recruit STATs proteins. Tyrosine-phosphorylated STATs dimerize and are then transported into the nucleus to transactivate target genes. There are four JAK proteins: JAK1, JAK2, JAK3, and TYK2. The STAT family comprises seven members: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6 (91). STAT3 acts as a key mediator of intestinal inflammation and tumorigenesis (92). Studies determined O-GlcNAcylation of STAT3 at Thr717 negatively regulates its phosphorylation in colon macrophages, which promote colonic inflammation and inflammation-driven tumorigenesis (92). O-GlcNAcylation of STAT5A at Thr92 accompanied enhanced STAT5 tyrosine phosphorylation, driving oncogenic transcription to induce myeloid transformation (93). Furthermore, STAT5 can directly regulate hypoxia inducible factor (HIF) 1β, whereas STAT3 directly controls HIF1α (94). HIF1α was reported to augment inflammation in a mice model of proximal colon cancer (95). Elevated O-GlcNAcylation stabilized HIF-1α levels, protecting breast cancer cells from ER stress-mediated apoptosis (96). These studies indicated O-GlcNAc-modified STATs proteins can regulate cancer-related inflammation independently or through modulating HIFs proteins production.
The activator protein-1 (AP-1) family of transcription factors consist of multiple Jun (c-Jun, JunB, and JunD) and Fos (c-Fos, FosB, Fra1, and Fra2) members (97). Similar to NF-κB, AP-1 can also bind promoters of inflammatory mediators (IL6, IL8) to promote cancer-associated inflammation (98). Studies suggest that OGT plays an oncogenic role in non-alcoholic fatty liver disease-associated hepatocellular carcinoma (NAFLD-HCC) through activating JNK/c-Jun/AP-1 cascade by increasing p-JNK, p-c-Jun protein expression and AP-1 activation. In keeping with this, NF-κB cascade was activated and NF-κB DNA binding activity was enhanced to induce downstream genes (TNF-α) expression (99). As JNK and NF-κB signaling pathways are the major endoplasmic reticulum (ER) stress-related oncogenic signaling pathways, OGT acts as a mediator to induce ER stress to promote NAFLD-HCC. Mechanistically, OGT regulates lipid metabolism through increasing palmitic acid production and reactive oxygen species (ROS), thereby activating ER stress, and ER stress-related JNK/c-Jun/AP1 and NF-κB pathways. OGT may serve as a potential therapeutic target in NAFLD-HCC.
Taking together, these findings provide compelling evidence for cooperative relationship between O-GlcNAcylation and NF-κB, STATs, HIF-1, AP-1-mediated cancer-related signaling pathway.
A tumor contains not only a group of cancer cells, but also a heterogeneous collection of infiltrating and resident host cells, such as stromal fibroblasts, endothelial cells and immune cells like macrophages and lymphocytes and the non-cellular extracellular components such as cytokines, growth factors, collagen, fibronectin, laminin, among others (100, 101). A dynamic and reciprocal relationship develops between cancer cells and resident host cells of the tumor microenvironment to create an immunosuppressive environment to promote tumor growth and metastasis. Here we illustrate how O-GlcNAcylation regulates TME cells functions in tumor immune microenvironment and also summarize site-specific O-GlcNAcylation of functional proteins in regulating inflammation-related signaling pathways and stromal cells in TME (Figure 3).
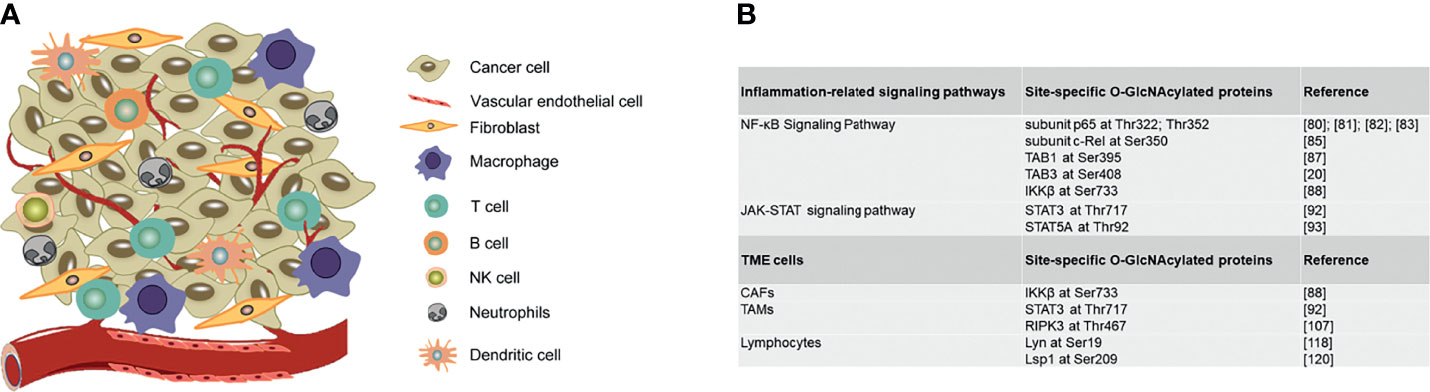
Figure 3 Site-specific O-GlcNAcylation of functional proteins in regulating inflammation-related signaling pathways and stromal cells in TME. (A) Graphic representation of TME cells. (B) Site-specific O-GlcNAcylated proteins in regulating inflammation-related signaling pathways and stromal cells in TME. TME, Tumor microenvironment; CAFs, Cancer-associated fibroblast; TAMs, Tumor associated macrophages.
To maintain vessel homeostasis, tumor endothelial cells (TECs) not only activate on environmental stressors, such as pro-angiogenic signals and hypoxia to initiate tumor angiogenesis, but also regulate peripheral immune cell trafficking into the tumor compartment (102). Hypoxia is a common physiological and pathophysiological occurrence in tumors and coexist with inflammation. Studies revealed hypoxia regulates vascular inflammatory response through upregulation of 26S proteasome activity and downregulate OGT expression in vascular endothelial cells (103). Mechanically, hypoxia generates reactive oxygen or nitrogen species (ROS/RNS) to enhanced 26S proteasome functionality, favoring E3 ubiquitin ligase β-TrCP1-mediated OGT degradation to facilitate vascular endothelial inflammation. Up-regulation of OGT can prevent this hypoxia-mediated inflammation in vascular endothelial cells, suggesting OGT may be targeted to treat diseases characterized by hypoxic inflammation (103).
Inflammation is often accompanied by the recruitment of fibroblasts and the induction of fibrosis. Cancer-associated fibroblast (CAFs) promote the deposition of collagen and various ECM components in the TME to facilitate tumorigenesis (104). Kawauchi and colleagues revealed that O-GlcNAcylation of IKKβ occurred in both p53-deficient mouse embryonic fibroblasts (MEFs) and transformed human fibroblasts (88). O-GlcNAcylation of IKKβ occurred at Ser733 sustained the TNFα-dependent IKKβ activity and enhance NF-κB activity. Thus, these results implicate O-GlcNAcylation of IKKβ as a central component linking glucose metabolism to IKK–NF-κB signaling pathway, providing a favorable role in inflammation-associated tumor development.
Tumor associated macrophages (TAMs) are derived from circulating monocytes and are divided into two subtypes: M1 and M2 macrophages. M1 macrophages are pro-inflammatory and can produce pro-inflammatory cytokines such as interleukin-1β (IL-1β), IL-6, IL-12, IL-23, and TNF-α while M2 macrophages are anti-inflammatory and produce anti-inflammatory cytokines such as IL-10 and TGF-β (105). O-GlcNAcylation promotes opposing effects in TAMs. In colorectal cancer, increased O-GlcNAcylation skewed macrophage polarization to a M2-like phenotype to enhance cancer progression and immune evasion (106). O-GlcNAcylation of STAT3 at Thr717 regulates its phosphorylation in colon macrophages negatively, which was accompanied by exacerbated colonic inflammation and inflammation-driven carcinogenesis (92). Nevertheless, a decreased HBP activity and protein O-GlcNAcylation was observed in LPS-stimulated macrophages where O-GlcNAcylation of RIPK3 at Thr467 suppresses RHIM-mediated RIPK3-RIPK1 interaction and downstream RIPK3 kinase activation, thus inhibiting inflammatory responses and inflammation-associated necroptosis (107). Moreover, LPS treatment increases the interaction of OGT with transcriptional corepressor mammalian Sin3A (mSin3A) to inhibit inducible nitric oxide synthase (iNOS) transcription while GlcN-induced hyper-O-GlcNAcylation inhibits LPS-driven activation of NF-κB and iNOS expression in RAW264.7 cells, which are monocyte/macrophage-like cells derived from BALB/c mice. These results indicate that OGT functions as a transcriptional repressor and exerts inhibitory effects on LPS-induced inflammation (108). Taken together, the above results collectively show O-GlcNAcylation appears to have opposite functions in inflammatory responses in macrophages. On some occasions, O-GlcNAcylation promotes inflammatory responses in macrophages, and in certain scenarios may has an anti-inflammatory function.
Tumors was infiltrated with diverse immune cells and there are three main types of lymphocytes: T cells, B cells, and Natural killer (NK) cells. Within the tumor microenvironment there are several distinct populations of T cells that influence tumorigenesis, comprising of cytotoxic (CD8+) T cells and helper (CD4+) T cells which could differentiate into Th1, Th2, Th17, and regulatory T cells (Tregs) Upon activation (109).
Following tumor infiltration, naive CD8+ T cells are differentiated into effector CD8+ T cells and further differentiated and activated into cytotoxic and memory CD8+ T cells (110). It has been reported that O-GlcNAcylated proteins enriched in Murine effector and memory-like CD8+ T cells. O-GlcNAcylation in effector CD8+ T cells promote transcription and translation essential for the regulation of fast cell proliferation, while protein O-GlcNAcylation in memory-like CD8+ T cells is involved in the transcription, mRNA processing, and translation during memory T cell formation (111). The main cluster of proteins identified O-GlcNAcylation between effector- and memory-like T cells consisted primarily of histones, identifying O-GlcNAc critical role as part of the “histone code” in T cell subgroups. Exosomes (30–100 nm small lipid bilayer extracellular vesicles) are secreted by various kinds of cells and play an important role in intercellular communication due to their ability to transfer proteins, lipids and nucleic acids to surrounding recipient cells. Numerous studies have demonstrated cancer-derived exosomes alter immune players in the TME to induce a pro-tumoral environment to facilitate tumor progression. Yuan et al. identified OGT in exosome from Esophageal carcinoma stem cells (ECSCs) promoted the expression of PD-1 in neighboring CD8+ T cells, thus favoring ECSCs the ability of immune escape (112). Moreover, O-GlcNAcylation also play an important role in the differentiation of Th cells. Elevated O-GlcNAcylation promotes increased IL-17A production in CD4+ T cells polarized to the Th17 lineage (113). Administration of Thiamet-G, which is an O-GlcNAcase inhibitor, increased the binding of transcription factor RORγt for commitment of the Th17 lineage, to IL-17 promoter and therefore promotes IL-17 production and Th17 differentiation (114). Subsequently, pro-inflammatory responses were enhanced by Th17 cells. In Treg cells, O-GlcNAcylation stabilizes FOXP3 and activates IL-2/STAT5 signaling, thus supporting Treg cells lineage stability and promoting the suppressive program of Treg cells (115). Collectively, O-GlcNAcylation is therefore shown to be an essential regulator in T cell development, transformation and differentiation. In addition, Loss of OGT blocked T cell progenitor renewal, malignant transformation and peripheral T cell clonal expansion (116).
B cells contribute to adaptive immune responses responsible for antibody production and promoting T cell activation via antigen presentation. Compared to T cells, relatively few B cells infiltrated in the tumor microenvironment. B cells are typically found in lymph nodes in close proximity to the TME (100). Following antigen engagement with B-cell receptors (BCRs), a series of signaling cascades involving the activation of spleen tyrosine kinase (Syk) and Lck/Yes-related Novel protein tyrosine kinase (Lyn) are triggered, which further activate downstream pathways, such as PLC-γ2/calcium/NFAT, IKK/NF-κB to induce transcription of genes that regulate the functions of activated B cells (117). Studies determined that O-GlcNAcylation of Lyn at Ser19 is detrimental for Lyn activation and Syk interaction in BCR-mediated B-cell activation (118). NF-κB and NFAT can be O-GlcNAcylated by OGT which mediates their translocation to the nucleus, sensitizing T and B lymphocytes toward activation (119). Lack of OGT in B-cell development not only inhibits activation of BCR signaling, but also enhances apoptosis of mature B cells, thus perturbing B-cell homeostasis. In addition, apoptosis initiates following BCR activation to eliminate auto-reactive B cells. O-GlcNAcylation of a pro-apoptotic regulator Lsp1 at Ser209 is required for the recruitment of its kinase, PKC-β1, to stimulate Ser243 phosphorylation, leading ERK activation and downregulation of BCL-2 and BCL-xL which initiate B-cell activation and apoptosis (120).
Natural killer (NK) cells are a population of innate lymphoid cells involved in immunosurveillance of solid tumors. These cells possess powerful cytotoxic activity orchestrated by an intricate network of signals to control tumor growth and mediate anti-metastatic effect in tumors (121). In TME, high density of tumor-infiltrating NK cells predicts favorable prognosis in multiple human tumors (122, 123). Soluble HLA-G1α chain (sHLA-G1α chain) are secreted proteins to exhibit systemic immunoinhibitory functions in peripheral blood. It has been shown that O-GlcNAcylation decreased during NK cell cytotoxicity and that GST-sHLA-G1α chain could inhibit the decrease of O-GlcNAc level during the process to inhibit NK cell cytotoxicity, thus inducing immunotolerance (124). These findings suggest O-GlcNAcylation important implications in the cytotoxic signal transduction of NK cells. Upon activation, NK cells release cytotoxic granules containing perforin, cathepsins, and granzymes to directly lyse tumor cells. Cathepsin C can activate granzymes by proteolytic cleavage and granzymes can enter the target cell to induce apoptosis through various signaling pathways (125). Studies revealed Glucosamine (GlcN), an intermediate fluxing into HBP pathway to increase O-GlcNAcylation, could reduce NK cell cytotoxicity by altering the distribution of cathepsin C and E, indicating O-GlcNAcylation might play a role in regulating the cytotoxic activity of NK cells (126).
Dendritic cells (DCs) are a type of antigen-presenting cells which present antigens to T cells and mediate T cells activation to induce an antigen-specific immunotherapy response (127). DCs constitute a rare immune cell population within the tumor microenvironment but emerge as an essential antitumor component based on their ability to foster T cell immunity (128). Toll-like receptors (TLRs) are expressed on DCs, recognizing stimuli to activate DCs. After activation by TLRs, DCs upregulate glucose uptake with enhanced glycolytic rates (129). As HBP branches from glycolysis and forms UDP-GlcNAc to mediate protein O-GlcNAcylation, these studies point to the idea that O-GlcNAcylation might provide a favorable role in DCs activation and migration. Apart from glucose, amino acids in the environment of DCs play an important role in regulating their differentiation and activation. For example, intracellular glutamine (Gln) are enhanced in DCs after TLR receives a stimulation (127). As Gln is the substrate of HBP rate-limiting enzyme GFAT, these findings suggest O-GlcNAcylation may also involve DCs differentiation and activation.
Neutrophils are cells that carry out an innate immune response. For O-GlcNAcylation role in Neutrophils, increased O-GlcNAcylation promotes chemotaxis and cellular mobility of neutrophils (130). Studies demonstrate elevating O-GlcNAcylation levels by administration of PUGNAc, an OGA inhibitor, or GlcN activates protein Rac, an important small GTPase which plays an essential role for regulating neutrophil motility via activating P38 and p44/42 MAPK signaling (130).
Dysfunction in the O-GlcNAcylation has been associated with various chronic diseases, such as neurodegenerative diseases, diabetes and cancers (131). Thus, targeting the O-GlcNAc enzymes, OGT and OGA, or the specific O-GlcNAcylated proteins have therapeutic potential. In terms of OGA, many potent inhibitors have been developed to treat neurodegenerative disorders, such as Alzheimer’s disease (AD) since O-GlcNAcylation can compete with phosphorylation to reduce hyperphosphorylated tau aggregation in the disease background (132). Three OGA inhibitors have entered clinical trials: MK-8719 from Merck/Alectos, ASN-120290 from Asceneuron S.A., LY-3372689 from Eli Lilly, and ASN-120290 is scheduled for phase 2 clinical trial (132, 133). Due to complex expression and instability of isolated OGT, the discovery of potent OGT inhibitors is challenging. Several compounds have been developed to inhibit OGT for cancer therapy. Alloxan was the first OGT inhibitor reported but with off-target effects and general cellular toxicity (134). Pharmacological inhibition of OGT using Ac4-5SGlcNAc decreases aggressive phenotype of breast cancer cells (135). Another OGT inhibitor OSMI (OSMI-1 or OSMI-2) results in decreased tumor burden in pancreatic ductal adenocarcinoma (PDAC) and inhibit prostate cancer cells proliferation (136, 137). As both OGA and OGT are ubiquitously expressed in various tissues, therapeutic inhibition of OGT or OGA may cause global change in cellular biological process. As a result, studies aimed at obtaining specific inhibitor of key target O-GlcNAcylated proteins in a specific cancer should be an effective way for therapeutic intervention.
It is widely accepted that TME is a complex and dynamic environment, comprising of cancer cells, as well as the adjacent stromal and inflammatory cells. Due to the critical roles of the TME in regulating tumor progression, diverse cell types in TME represent targets for anticancer therapies, which is termed as TME-directed therapies, including immunotherapies (138). Cancer immunotherapy reactivates a patient’s immune system and prevent tumor immune escape. Immune checkpoint inhibitors (ICIs) are the most widely used clinically for cancer treatment. ICIs target immune checkpoint regulators to enhance anti-tumor immunity, including anti-cytotoxic T lymphocyte antigen-4 (CTLA-4), anti-programed cell death protein 1 (PD-1), and anti-PDL-1 (139). Ipilimumab (CTLA-4 inhibitor) is the first approved ICIs in 2011 and now used in the first-line setting to improve overall survival in patients with unresectable/metastatic melanoma (140). Another revolutionary immunotherapy for cancer is Chimeric antigen receptor (CAR)-T cell therapy, which genetically engineers T cells in vitro to possess the ability to specifically recognize and attack tumor cells (141). For anti-TAM cancer therapy, reprogramming M2 TAMs toward pro-inflammatory M1 phenotype is one strategy. Toll-like receptors (TLRs) are key players in M1 programming upon binding a ligand. Currently, Imiquimod (a TLR7 agonist) is now FDA-approved for topical administration in squamous and basal cell carcinoma (142).
However, there are still some challenges for cancer immunotherapy. For ICIs treatment, a large proportion of patients are either insensitive to ICIs or are burdened by adverse side effects. CAR-T cell therapy shows remarkable clinical responses in hematological malignancies, such as B cell leukemia or lymphoma but less efficacy in solid tumors and often cause severe toxicities, even tumor relapse (143). The therapeutic limits of immunotherapy are largely due to the network of the cells in the tumor microenvironments (TME) that operates in an immunosuppressive fashion to facilitate immune escape. Thus, an in-depth understanding of the formation of inflammatory milieu to overcome the immune suppressive TME, and finding new ways to sensitizing cancer immunotherapy are urgently needed.
Here, we focus on elucidating how O-GlcNAcylation regulates cancer-associated inflammation through identifying the role of O-GlcNAcylation in regulating precancerous inflammation lesions, inflammation-associated signaling pathways and stromal cells activation in TME. Metabolism reprogramming in TME helps sustain cancer-associated inflammation to form an inflammatory milieu to regulate cancer progression. O-GlcNAcylation acts as a metabolic sensor, linking nutrition homeostasis and cancer progression. Thus, understanding the role of O-GlcNAcylation in regulating cancer-associated inflammation and especially how site-specific O-GlcNAcylation of functional proteins in regulating inflammation-associated signaling pathways in TME is critical. Characterizing the significant roles of O-GlcNAcylation in cancer-associated inflammation may shed light on the development of new strategies targeting O-GlcNAcylated form of key proteins, thus boosting or rejuvenating immune responses against malignant cancers, and improving the therapeutic effect of cancer immunotherapy.
FD and XZ conceived and organized the manuscript. MO and CY wrote the manuscript. XD and YZ prepared the figures and contributed to the discussion. All authors have read and approved the final manuscript.
This work was supported by grants from the National Natural Science Foundation of China (32000543), Shenzhen Science and Technology Program (Grant No. 2021A22). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Torres CR, Hart GW. Topography and Polypeptide Distribution of Terminal N-Acetylglucosamine Residues on the Surfaces of Intact Lymphocytes. Evidence for O-linked GlcNAc. J Biol Chem (1984) 259:3308–17. doi: 10.1016/S0021-9258(17)43295-9
2. Ma J, Li Y, Hou C, Wu C. O-GlcNAcAtlas: A Database of Experimentally Identified O-GlcNAc Sites and Proteins. Glycobiology (2021) 31:719–23. doi: 10.1093/glycob/cwab003
3. Chatham JC, Zhang J, Wende AR. Role of O-Linked N-Acetylglucosamine Protein Modification in Cellular (Patho)Physiology. Physiol Rev (2021) 101:427–93. doi: 10.1152/physrev.00043.2019
4. Nie H, Yi W. O-GlcNAcylation, a Sweet Link to the Pathology of Diseases. J Zhejiang Univ Sci B (2019) 20:437–48. doi: 10.1631/jzus.B1900150
5. Massman LJ, Pereckas M, Zwagerman NT, Olivier-Van Stichelen S. O-GlcNAcylation Is Essential for Rapid Pomc Expression and Cell Proliferation in Corticotropic Tumor Cells. Endocrinology 162 (2021). doi: 10.1210/endocr/bqab178
6. Raab S, Gadault A, Very N, Decourcelle A, Baldini S, Schulz C, et al. Dual Regulation of Fatty Acid Synthase (FASN) Expression by O-GlcNAc Transferase (OGT) and mTOR Pathway in Proliferating Liver Cancer Cells. Cell Mol Life Sci (2021) 78:5397–413. doi: 10.1007/s00018-021-03857-z
7. Liu YY, Liu HY, Yu TJ, Lu Q, Zhang FL, Liu GY, et al. O-GlcNAcylation of MORC2 at Threonine 556 by OGT Couples TGF-Beta Signaling to Breast Cancer Progression. Cell Death Differ (2022). doi: 10.1038/s41418-021-00901-0
8. Akella NM, Le Minh G, Ciraku L, Mukherjee A, Bacigalupa ZA, Mukhopadhyay D, et al. O-GlcNAc Transferase Regulates Cancer Stem-Like Potential of Breast Cancer Cells. Mol Cancer Res (2020) 18:585–98. doi: 10.1158/1541-7786.MCR-19-0732
9. Duan F, Wu H, Jia D, Wu W, Ren S, Wang L, et al. O-GlcNAcylation of RACK1 Promotes Hepatocellular Carcinogenesis. J Hepatol (2018) 68:1191–202. doi: 10.1016/j.jhep.2018.02.003
10. Hua Q, Zhang B, Xu G, Wang L, Wang H, Lin Z, et al. CEMIP, a Novel Adaptor Protein of OGT, Promotes Colorectal Cancer Metastasis Through Glutamine Metabolic Reprogramming via Reciprocal Regulation of Beta-Catenin. Oncogene (2021) 40:6443–55. doi: 10.1038/s41388-021-02023-w
11. Gentric G, Mieulet V, Mechta-Grigoriou F. Heterogeneity in Cancer Metabolism: New Concepts in an Old Field. Antioxid Redox Signal (2017) 26:462–85. doi: 10.1089/ars.2016.6750
12. Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, et al. Normalization of CD4+ T Cell Metabolism Reverses Lupus. Sci Transl Med (2015) 7:274ra18. doi: 10.1126/scitranslmed.aaa0835
13. Neagu M, Constantin C, Popescu ID, Zipeto D, Tzanakakis G, Nikitovic D, et al. Inflammation and Metabolism in Cancer Cell-Mitochondria Key Player. Front Oncol (2019) 9:348. doi: 10.3389/fonc.2019.00348
14. Schwein PA, Woo CM. The O-GlcNAc Modification on Kinases. ACS Chem Biol (2020) 15:602–17. doi: 10.1021/acschembio.9b01015
15. Yang X, Qian K. Protein O-GlcNAcylation: Emerging Mechanisms and Functions. Nat Rev Mol Cell Biol (2017) 18:452–65. doi: 10.1038/nrm.2017.22
16. Pinho SS, Reis CA. Glycosylation in Cancer: Mechanisms and Clinical Implications. Nat Rev Cancer (2015) 15:540–55. doi: 10.1038/nrc3982
17. Brockhausen I, Yang J, Lehotay M, Ogata S, Itzkowitz S. Pathways of Mucin O-Glycosylation in Normal and Malignant Rat Colonic Epithelial Cells Reveal a Mechanism for Cancer-Associated Sialyl-Tn Antigen Expression. Biol Chem (2001) 382:219–32. doi: 10.1515/BC.2001.029
18. Hart GW. Three Decades of Research on O-GlcNAcylation - A Major Nutrient Sensor That Regulates Signaling, Transcription and Cellular Metabolism. Front Endocrinol (Lausanne) (2014) 5:183. doi: 10.3389/fendo.2014.00183
19. Liu Q, Tao T, Liu F, Ni R, Lu C, Shen A. Hyper-O-GlcNAcylation of YB-1 Affects Ser102 Phosphorylation and Promotes Cell Proliferation in Hepatocellular Carcinoma. Exp Cell Res (2016) 349:230–8. doi: 10.1016/j.yexcr.2016.10.011
20. Tao T, He Z, Shao Z, Lu H. TAB3 O-GlcNAcylation Promotes Metastasis of Triple Negative Breast Cancer. Oncotarget (2016) 7:22807–18. doi: 10.18632/oncotarget.8182
21. Xu Y, Sheng X, Zhao T, Zhang L, Ruan Y, Lu H. O-GlcNAcylation of MEK2 Promotes the Proliferation and Migration of Breast Cancer Cells. Glycobiology (2021) 31:571–81. doi: 10.1093/glycob/cwaa103
22. Chen YF, Zhu JJ, Li J, Ye XS. O-GlcNAcylation Increases PYGL Activity by Promoting Phosphorylation. Glycobiology (2021) 32(2):101–9. doi: 10.1093/glycob/cwab114
23. Lei Y, Chen T, Li Y, Shang M, Zhang Y, Jin Y, et al. O-GlcNAcylation of PFKFB3 Is Required for Tumor Cell Proliferation Under Hypoxia. Oncogenesis (2020) 9:21. doi: 10.1038/s41389-020-0208-1
24. Li X, Wu Z, He J, Jin Y, Chu C, Cao Y, et al. OGT Regulated O-GlcNAcylation Promotes Papillary Thyroid Cancer Malignancy via Activating YAP. Oncogene (2021) 40:4859–71. doi: 10.1038/s41388-021-01901-7
25. Ma Z, Chalkley RJ, Vosseller K. Hyper-O-GlcNAcylation Activates Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells (NF-Kappab) Signaling Through Interplay With Phosphorylation and Acetylation. J Biol Chem (2017) 292:9150–63. doi: 10.1074/jbc.M116.766568
26. Aulak KS, Barnes JW, Tian L, Mellor NE, Haque MM, Willard B, et al. Specific O-GlcNAc Modification at Ser-615 Modulates eNOS Function. Redox Biol (2020) 36:101625. doi: 10.1016/j.redox.2020.101625
27. Kim E, Kang JG, Kang MJ, Park JH, Kim YJ, Kweon TH, et al. O-GlcNAcylation on LATS2 Disrupts the Hippo Pathway by Inhibiting Its Activity. Proc Natl Acad Sci USA (2020) 117:14259–69. doi: 10.1073/pnas.1913469117
28. Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross Talk Between O-GlcNAcylation and Phosphorylation: Roles in Signaling, Transcription, and Chronic Disease. Annu Rev Biochem (2011) 80:825–58. doi: 10.1146/annurev-biochem-060608-102511
29. Huang H, Wu Q, Guo X, Huang T, Xie X, Wang L, et al. O-GlcNAcylation Promotes the Migratory Ability of Hepatocellular Carcinoma Cells via Regulating FOXA2 Stability and Transcriptional Activity. J Cell Physiol (2021) 236:7491–503. doi: 10.1002/jcp.30385
30. Zhu Y, Hart GW. Nutrient Regulation of the Flow of Genetic Information by O-GlcNAcylation. Biochem Soc Trans (2021) 49:867–80. doi: 10.1042/BST20200769
31. Lin AP, Qiu Z, Ethiraj P, Sasi B, Jaafar C, Rakheja D, et al. MYC, Mitochondrial Metabolism and O-GlcNAcylation Converge to Modulate the Activity and Subcellular Localization of DNA and RNA Demethylases. Leukemia (2022). doi: 10.1038/s41375-021-01489-7
32. Kim JG, Nonneman D, Kim DW, Shin S, Rohrer GA. Polymorphism in the Intron 20 of Porcine O-Linked N-Acetylglucosamine Transferase. Asian-Australas J Anim Sci (2017) 30:1086–92. doi: 10.5713/ajas.17.0143
33. Keembiyehetty C, Love DC, Harwood KR, Gavrilova O, Comly ME, Hanover JA. Conditional Knock-Out Reveals a Requirement for O-Linked N-Acetylglucosaminase (O-GlcNAcase) in Metabolic Homeostasis. J Biol Chem (2015) 290:7097–113. doi: 10.1074/jbc.M114.617779
34. Urso SJ, Comly M, Hanover JA, Lamitina T. The O-GlcNAc Transferase OGT Is a Conserved and Essential Regulator of the Cellular and Organismal Response to Hypertonic Stress. PloS Genet (2020) 16:e1008821. doi: 10.1371/journal.pgen.1008821
35. Parker MP, Peterson KR, Slawson C. O-GlcNAcylation and O-GlcNAc Cycling Regulate Gene Transcription: Emerging Roles in Cancer. Cancers (Basel) (2021) 13(7):1666. doi: 10.3390/cancers13071666
36. Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab (2016) 23:27–47. doi: 10.1016/j.cmet.2015.12.006
37. Slawson C, Copeland RJ, Hart GW. O-GlcNAc Signaling: A Metabolic Link Between Diabetes and Cancer? Trends Biochem Sci (2010) 35:547–55. doi: 10.1016/j.tibs.2010.04.005
38. Whelan SA, Hart GW. Proteomic Approaches to Analyze the Dynamic Relationships Between Nucleocytoplasmic Protein Glycosylation and Phosphorylation. Circ Res (2003) 93:1047–58. doi: 10.1161/01.RES.0000103190.20260.37
39. Decourcelle A, Leprince D, Dehennaut V. Regulation of Polycomb Repression by O-GlcNAcylation: Linking Nutrition to Epigenetic Reprogramming in Embryonic Development and Cancer. Front Endocrinol (Lausanne) (2019) 10:117. doi: 10.3389/fendo.2019.00117
40. Balkwill F, Mantovani A. Inflammation and Cancer: Back to Virchow? Lancet (2001) 357:539–45. doi: 10.1016/S0140-6736(00)04046-0
41. Del Prete A, Allavena P, Santoro G, Fumarulo R, Corsi MM, Mantovani A. Molecular Pathways in Cancer-Related Inflammation. Biochem Med (Zagreb) (2011) 21:264–75. doi: 10.11613/BM.2011.036
42. Greten FR, Grivennikov SI. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity (2019) 51:27–41. doi: 10.1016/j.immuni.2019.06.025
43. Wu Y, Antony S, Meitzler JL, Doroshow JH. Molecular Mechanisms Underlying Chronic Inflammation-Associated Cancers. Cancer Lett (2014) 345:164–73. doi: 10.1016/j.canlet.2013.08.014
44. Hussain SP, Harris CC. Inflammation and Cancer: An Ancient Link With Novel Potentials. Int J Cancer (2007) 121:2373–80. doi: 10.1002/ijc.23173
45. Vendramini-Costa DB, Carvalho JE. Molecular Link Mechanisms Between Inflammation and Cancer. Curr Pharm Des (2012) 18:3831–52. doi: 10.2174/138161212802083707
46. McLean MH, El-Omar EM. Genetics of Inflammation in the Gastrointestinal Tract and How It Can Cause Cancer. Recent Results Cancer Res (2011) 185:173–83. doi: 10.1007/978-3-642-03503-6_11
47. O'Connor PM, Lapointe TK, Beck PL, Buret AG. Mechanisms by Which Inflammation may Increase Intestinal Cancer Risk in Inflammatory Bowel Disease. Inflamm Bowel Dis (2010) 16:1411–20. doi: 10.1002/ibd.21217
48. Ringelhan M, Pfister D, O'Connor T, Pikarsky E, Heikenwalder M. The Immunology of Hepatocellular Carcinoma. Nat Immunol (2018) 19:222–32. doi: 10.1038/s41590-018-0044-z
49. Cai T, Santi R, Tamanini I, Galli IC, Perletti G, Bjerklund Johansen TE, et al. Current Knowledge of the Potential Links Between Inflammation and Prostate Cancer. Int J Mol Sci (2019) 20(15):3833. doi: 10.3390/ijms20153833
50. Garcea G, Dennison AR, Steward WP, Berry DP. Role of Inflammation in Pancreatic Carcinogenesis and the Implications for Future Therapy. Pancreatology (2005) 5:514–29. doi: 10.1159/000087493
51. Franco EL, Rohan TE, Villa LL. Epidemiologic Evidence and Human Papillomavirus Infection as a Necessary Cause of Cervical Cancer. J Natl Cancer Inst (1999) 91:506–11. doi: 10.1093/jnci/91.6.506
52. Punturieri A, Szabo E, Croxton TL, Shapiro SD, Dubinett SM. Lung Cancer and Chronic Obstructive Pulmonary Disease: Needs and Opportunities for Integrated Research. J Natl Cancer Inst (2009) 101:554–9. doi: 10.1093/jnci/djp023
53. Porta C, Riboldi E, Sica A. Mechanisms Linking Pathogens-Associated Inflammation and Cancer. Cancer Lett (2011) 305:250–62. doi: 10.1016/j.canlet.2010.10.012
54. Watari J, Chen N, Amenta PS, Fukui H, Oshima T, Tomita T, et al. Helicobacter Pylori Associated Chronic Gastritis, Clinical Syndromes, Precancerous Lesions, and Pathogenesis of Gastric Cancer Development. World J Gastroenterol (2014) 20:5461–73. doi: 10.3748/wjg.v20.i18.5461
55. Jang TJ, Kim UJ. O-GlcNAcylation Is Associated With the Development and Progression of Gastric Carcinoma. Pathol Res Pract (2016) 212:622–30. doi: 10.1016/j.prp.2016.04.002
56. Itzkowitz SH, Yio X. Inflammation and Cancer IV. Colorectal Cancer in Inflammatory Bowel Disease: The Role of Inflammation. Am J Physiol Gastrointest Liver Physiol (2004) 287:G7–17. doi: 10.1152/ajpgi.00079.2004
58. Yang YR, Kim DH, Seo YK, Park D, Jang HJ, Choi SY, et al. Elevated O-GlcNAcylation Promotes Colonic Inflammation and Tumorigenesis by Modulating NF-KappaB Signaling. Oncotarget (2015) 6:12529–42. doi: 10.18632/oncotarget.3725
59. Sun QH, Wang YS, Liu G, Zhou HL, Jian YP, Liu MD, et al. Enhanced O-Linked Glcnacylation in Crohn's Disease Promotes Intestinal Inflammation. EBioMedicine (2020) 53:102693. doi: 10.1016/j.ebiom.2020.102693
60. He X, Gao J, Peng L, Hu T, Wan Y, Zhou M, et al. Bacterial O-GlcNAcase Genes Abundance Decreases in Ulcerative Colitis Patients and Its Administration Ameliorates Colitis in Mice. Gut (2021) 70:1872–83. doi: 10.1136/gutjnl-2020-322468
61. Xie Y. Hepatitis B Virus-Associated Hepatocellular Carcinoma. Adv Exp Med Biol (2017) 1018:11–21. doi: 10.1007/978-981-10-5765-6_2
62. Chen Y, Tian Z. HBV-Induced Immune Imbalance in the Development of HCC. Front Immunol (2019) 10:2048. doi: 10.3389/fimmu.2019.02048
63. Hu J, Gao Q, Yang Y, Xia J, Zhang W, Chen Y, et al. Hexosamine Biosynthetic Pathway Promotes the Antiviral Activity of SAMHD1 by Enhancing O-GlcNAc Transferase-Mediated Protein O-GlcNAcylation. Theranostics (2021) 11:805–23. doi: 10.7150/thno.50230
64. Song H, Shen Q, Hu S, Jin J. The Role of Macrophage Migration Inhibitory Factor in Promoting Benign Prostatic Hyperplasia Epithelial Cell Growth by Modulating COX-2 and P53 Signaling. Biol Open (2020) 9(11):bio053447. doi: 10.1242/bio.053447
65. Dai X, Fang X, Ma Y, Xianyu J. Benign Prostatic Hyperplasia and the Risk of Prostate Cancer and Bladder Cancer: A Meta-Analysis of Observational Studies. Med (Baltimore) (2016) 95:e3493. doi: 10.1097/MD.0000000000003493
66. Gu Y, Gao J, Han C, Zhang X, Liu H, Ma L, et al. O-GlcNAcylation Is Increased in Prostate Cancer Tissues and Enhances Malignancy of Prostate Cancer Cells. Mol Med Rep (2014) 10:897–904. doi: 10.3892/mmr.2014.2269
67. Kirkegard J, Cronin-Fenton D, Heide-Jorgensen U, Mortensen FV. Acute Pancreatitis and Pancreatic Cancer Risk: A Nationwide Matched-Cohort Study in Denmark. Gastroenterology (2018) 154:1729–36. doi: 10.1053/j.gastro.2018.02.011
68. Zhang D, Cai Y, Chen M, Gao L, Shen Y, Huang Z. OGT-Mediated O-GlcNAcylation Promotes NF-kappaB Activation and Inflammation in Acute Pancreatitis. Inflamm Res (2015) 64:943–52. doi: 10.1007/s00011-015-0877-y
69. Okunade KS. Human Papillomavirus and Cervical Cancer. J Obstet Gynaecol (2020) 40:602–8. doi: 10.1080/01443615.2019.1634030
70. Fernandes JV, DE Medeiros Fernades TA, DE Azevedo JC, Cobucci RN, DE Carvalho MG, Andrade VS, et al. Link Between Chronic Inflammation and Human Papillomavirus-Induced Carcinogenesis (Review). Oncol Lett (2015) 9:1015–26. doi: 10.3892/ol.2015.2884
71. Kim M, Kim YS, Kim H, Kang MY, Park J, Lee DH, et al. O-Linked N-Acetylglucosamine Transferase Promotes Cervical Cancer Tumorigenesis Through Human Papillomaviruses E6 and E7 Oncogenes. Oncotarget (2016) 7:44596–607. doi: 10.18632/oncotarget.10112
72. Zeng Q, Zhao RX, Chen J, Li Y, Li XD, Liu XL, et al. O-Linked GlcNAcylation Elevated by HPV E6 Mediates Viral Oncogenesis. Proc Natl Acad Sci USA (2016) 113:9333–8. doi: 10.1073/pnas.1606801113
73. Singh D, Agusti A, Anzueto A, Barnes PJ, Bourbeau J, Celli BR, et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease: The GOLD Science Committee Report 2019. Eur Respir J (2019) 53(5):1900164. doi: 10.1183/13993003.00164-2019
74. Gonzalez J, Marin M, Sanchez-Salcedo P, Zulueta JJ. Lung Cancer Screening in Patients With Chronic Obstructive Pulmonary Disease. Ann Transl Med (2016) 4:160. doi: 10.21037/atm.2016.03.57
75. Parris BA, O'Farrell HE, Fong KM, Yang IA. Chronic Obstructive Pulmonary Disease (COPD) and Lung Cancer: Common Pathways for Pathogenesis. J Thorac Dis (2019) 11:S2155–72. doi: 10.21037/jtd.2019.10.54
76. Krick S, Helton ES, Hutcheson SB, Blumhof S, Garth JM, Denson RS, et al. FGF23 Induction of O-Linked N-Acetylglucosamine Regulates IL-6 Secretion in Human Bronchial Epithelial Cells. Front Endocrinol (Lausanne) (2018) 9:708. doi: 10.3389/fendo.2018.00708
77. Hayden MS, Ghosh S. Signaling to NF-Kappab. Genes Dev (2004) 18:2195–224. doi: 10.1101/gad.1228704
78. Xia Y, Shen S, Verma IM. NF-Kappab, an Active Player in Human Cancers. Cancer Immunol Res (2014) 2:823–30. doi: 10.1158/2326-6066.CIR-14-0112
79. Liu F, Xia Y, Parker AS, Verma IM. IKK Biology. Immunol Rev (2012) 246:239–53. doi: 10.1111/j.1600-065X.2012.01107.x
80. Yang WH, Park SY, Nam HW, Kim DH, Kang JG, Kang ES, et al. NFKappaB Activation Is Associated With Its O-GlcNAcylation State Under Hyperglycemic Conditions. Proc Natl Acad Sci USA (2008) 105:17345–50. doi: 10.1073/pnas.0806198105
81. Ma Z, Vocadlo DJ, Vosseller K. Hyper-O-GlcNAcylation Is Anti-Apoptotic and Maintains Constitutive NF-KappaB Activity in Pancreatic Cancer Cells. J Biol Chem (2013) 288:15121–30. doi: 10.1074/jbc.M113.470047
82. Ali A, Kim SH, Kim MJ, Choi MY, Kang SS, Cho GJ, et al. O-GlcNAcylation of NF-KappaB Promotes Lung Metastasis of Cervical Cancer Cells via Upregulation of CXCR4 Expression. Mol Cells (2017) 40:476–84. doi: 10.14348/molcells.2017.2309
83. Phoomak C, Vaeteewoottacharn K, Sawanyawisuth K, Seubwai W, Wongkham C, Silsirivanit A, et al. Mechanistic Insights of O-GlcNAcylation That Promote Progression of Cholangiocarcinoma Cells via Nuclear Translocation of NF-Kappab. Sci Rep (2016) 6:27853. doi: 10.1038/srep27853
84. Hirata Y, Nakagawa T, Moriwaki K, Koubayashi E, Kakimoto K, Takeuchi T, et al. Augmented O-GlcNAcylation Alleviates Inflammation-Mediated Colon Carcinogenesis via Suppression of Acute Inflammation. J Clin Biochem Nutr (2018) 62:221–9. doi: 10.3164/jcbn.17-106
85. Ramakrishnan P, Clark PM, Mason DE, Peters EC, Hsieh-Wilson LC, Baltimore D. Activation of the Transcriptional Function of the NF-KappaB Protein C-Rel by O-GlcNAc Glycosylation. Sci Signal (2013) 6:ra75. doi: 10.1126/scisignal.2004097. Erratum in: Sci Signal. 2014 Apr 29;7(323):er3
86. Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, et al. TAB2 and TAB3 Activate the NF-KappaB Pathway Through Binding to Polyubiquitin Chains. Mol Cell (2004) 15:535–48. doi: 10.1016/j.molcel.2004.08.008
87. Pathak S, Borodkin VS, Albarbarawi O, Campbell DG, Ibrahim A, van Aalten DM. O-GlcNAcylation of TAB1 Modulates TAK1-Mediated Cytokine Release. EMBO J (2012) 31:1394–404. doi: 10.1038/emboj.2012.8
88. Kawauchi K, Araki K, Tobiume K, Tanaka N. Loss of P53 Enhances Catalytic Activity of IKKbeta Through O-Linked Beta-N-Acetyl Glucosamine Modification. Proc Natl Acad Sci USA (2009) 106:3431–6. doi: 10.1073/pnas.0813210106
89. Pflueger D, Terry S, Sboner A, Habegger L, Esgueva R, Lin PC, et al. Discovery of Non-ETS Gene Fusions in Human Prostate Cancer Using Next-Generation RNA Sequencing. Genome Res (2011) 21:56–67. doi: 10.1101/gr.110684.110
90. Yu H, Pardoll D, Jove R. STATs in Cancer Inflammation and Immunity: A Leading Role for STAT3. Nat Rev Cancer (2009) 9:798–809. doi: 10.1038/nrc2734
91. Hu X, Li J, Fu M, Zhao X, Wang W. The JAK/STAT Signaling Pathway: From Bench to Clinic. Signal Transduct Target Ther (2021) 6:402. doi: 10.1038/s41392-021-00791-1
92. Li X, Zhang Z, Li L, Gong W, Lazenby AJ, Swanson BJ, et al. Myeloid-Derived Cullin 3 Promotes STAT3 Phosphorylation by Inhibiting OGT Expression and Protects Against Intestinal Inflammation. J Exp Med (2017) 214:1093–109. doi: 10.1084/jem.20161105
93. Freund P, Kerenyi MA, Hager M, Wagner T, Wingelhofer B, Pham HTT, et al. O-GlcNAcylation of STAT5 Controls Tyrosine Phosphorylation and Oncogenic Transcription in STAT5-Dependent Malignancies. Leukemia (2017) 31:2132–42. doi: 10.1038/leu.2017.4
94. Rauth M, Freund P, Orlova A, Grunert S, Tasic N, Han X, et al. Cell Metabolism Control Through O-GlcNAcylation of STAT5: A Full or Empty Fuel Tank Makes a Big Difference for Cancer Cell Growth and Survival. Int J Mol Sci (2019) 20(5):1028. doi: 10.3390/ijms20051028
95. Mladenova DN, Dahlstrom JE, Tran PN, Benthani F, Bean EG, Ng I, et al. HIF1alpha Deficiency Reduces Inflammation in a Mouse Model of Proximal Colon Cancer. Dis Model Mech (2015) 8:1093–103. doi: 10.1242/dmm.019000
96. Ferrer CM, Lynch TP, Sodi VL, Falcone JN, Schwab LP, Peacock DL, et al. O-GlcNAcylation Regulates Cancer Metabolism and Survival Stress Signaling via Regulation of the HIF-1 Pathway. Mol Cell (2014) 54:820–31. doi: 10.1016/j.molcel.2014.04.026
97. Qiao Y, He H, Jonsson P, Sinha I, Zhao C, Dahlman-Wright K. AP-1 Is a Key Regulator of Proinflammatory Cytokine TNFalpha-Mediated Triple-Negative Breast Cancer Progression. J Biol Chem (2016) 291:5068–79. doi: 10.1074/jbc.M115.702571
98. Chang WT, Hong MY, Chen CL, Hwang CY, Tsai CC, Chuang CC. Mutant Glucocorticoid Receptor Binding Elements on the Interleukin-6 Promoter Regulate Dexamethasone Effects. BMC Immunol (2021) 22:24. doi: 10.1186/s12865-021-00413-z
99. Xu W, Zhang X, Wu JL, Fu L, Liu K, Liu D, et al. O-GlcNAc Transferase Promotes Fatty Liver-Associated Liver Cancer Through Inducing Palmitic Acid and Activating Endoplasmic Reticulum Stress. J Hepatol (2017) 67:310–20. doi: 10.1016/j.jhep.2017.03.017
100. Anderson NM, Simon MC. The Tumor Microenvironment. Curr Biol (2020) 30:R921–5. doi: 10.1016/j.cub.2020.06.081
101. Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M, et al. Tumor Microenvironment Complexity and Therapeutic Implications at a Glance. Cell Commun Signal (2020) 18:59. doi: 10.1186/s12964-020-0530-4
102. Nagl L, Horvath L, Pircher A, Wolf D. Tumor Endothelial Cells (TECs) as Potential Immune Directors of the Tumor Microenvironment - New Findings and Future Perspectives. Front Cell Dev Biol (2020) 8:766. doi: 10.3389/fcell.2020.00766
103. Liu H, Wang Z, Yu S, Xu J. Proteasomal Degradation of O-GlcNAc Transferase Elevates Hypoxia-Induced Vascular Endothelial Inflammatory Responsedagger. Cardiovasc Res (2014) 103:131–9. doi: 10.1093/cvr/cvu116
104. Zhao H, Wu L, Yan G, Chen Y, Zhou M, Wu Y, et al. Inflammation and Tumor Progression: Signaling Pathways and Targeted Intervention. Signal Transduct Target Ther (2021) 6:263. doi: 10.1038/s41392-021-00658-5
105. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage Plasticity, Polarization, and Function in Health and Disease. J Cell Physiol (2018) 233:6425–40. doi: 10.1002/jcp.26429
106. Rodrigues Mantuano N, Stanczak MA, Oliveira IA, Kirchhammer N, Filardy AA, Monaco G, et al. Hyperglycemia Enhances Cancer Immune Evasion by Inducing Alternative Macrophage Polarization Through Increased O-GlcNAcylation. Cancer Immunol Res (2020) 8:1262–72. doi: 10.1158/2326-6066.CIR-19-0904
107. Li X, Gong W, Wang H, Li T, Attri KS, Lewis RE, et al. O-GlcNAc Transferase Suppresses Inflammation and Necroptosis by Targeting Receptor-Interacting Serine/Threonine-Protein Kinase 3. Immunity (2019) 50:576–590 e6. doi: 10.1016/j.immuni.2019.01.007
108. Hwang SY, Hwang JS, Kim SY, Han IO. O-GlcNAc Transferase Inhibits LPS-Mediated Expression of Inducible Nitric Oxide Synthase Through an Increased Interaction With Msin3a in RAW264.7 Cells. Am J Physiol Cell Physiol (2013) 305:C601–8. doi: 10.1152/ajpcell.00042.2013
109. Hendry S, Salgado R, Gevaert T, Russell PA, John T, Thapa B, et al. Fox, Assessing Tumor-Infiltrating Lymphocytes in Solid Tumors: A Practical Review for Pathologists and Proposal for a Standardized Method From the International Immunooncology Biomarkers Working Group: Part 1: Assessing the Host Immune Response, TILs in Invasive Breast Carcinoma and Ductal Carcinoma In Situ, Metastatic Tumor Deposits and Areas for Further Research. Adv Anat Pathol (2017) 24:235–51. doi: 10.1097/PAP.0000000000000162
110. Raskov H, Orhan A, Christensen JP, Gogenur I. Cytotoxic CD8(+) T Cells in Cancer and Cancer Immunotherapy. Br J Cancer (2021) 124:359–67. doi: 10.1038/s41416-020-01048-4
111. Lopez Aguilar A, Gao Y, Hou X, Lauvau G, Yates JR, Wu P. Profiling of Protein O-GlcNAcylation in Murine CD8(+) Effector- and Memory-Like T Cells. ACS Chem Biol (2017) 12:3031–8. doi: 10.1021/acschembio.7b00869
112. Yuan Y, Wang L, Ge D, Tan L, Cao B, Fan H, et al. Exosomal O-GlcNAc Transferase From Esophageal Carcinoma Stem Cell Promotes Cancer Immunosuppression Through Up-Regulation of PD-1 in CD8(+) T Cells. Cancer Lett (2021) 500:98–106. doi: 10.1016/j.canlet.2020.12.012
113. Machacek M, Saunders H, Zhang Z, Tan EP, Li J, Li T, et al. Elevated O-GlcNAcylation Enhances Pro-Inflammatory Th17 Function by Altering the Intracellular Lipid Microenvironment. J Biol Chem (2019) 294:8973–90. doi: 10.1074/jbc.RA119.008373
114. Chang YH, Weng CL, Lin KI. O-GlcNAcylation and Its Role in the Immune System. J BioMed Sci (2020) 27:57. doi: 10.1186/s12929-020-00648-9
115. Liu B, Salgado OC, Singh S, Hippen KL, Maynard JC, Burlingame AL, et al. The Lineage Stability and Suppressive Program of Regulatory T Cells Require Protein O-GlcNAcylation. Nat Commun (2019) 10:354. doi: 10.1038/s41467-019-08300-3
116. Swamy M, Pathak S, Grzes KM, Damerow S, Sinclair LV, van Aalten DM, et al. Glucose and Glutamine Fuel Protein O-GlcNAcylation to Control T Cell Self-Renewal and Malignancy. Nat Immunol (2016) 17:712–20. doi: 10.1038/ni.3439
117. Sun Y, Yang Y, Zhao Y, Li X, Zhang Y, Liu Z. The Role of the Tyrosine Kinase Lyn in Allergy and Cancer. Mol Immunol (2021) 131:121–6. doi: 10.1016/j.molimm.2020.12.028
118. Wu JL, Chiang MF, Hsu PH, Tsai DY, Hung KH, Wang YH, et al. O-GlcNAcylation Is Required for B Cell Homeostasis and Antibody Responses. Nat Commun (2017) 8:1854. doi: 10.1038/s41467-017-01677-z
119. Golks A, Tran TT, Goetschy JF, Guerini D. Requirement for O-Linked N-Acetylglucosaminyltransferase in Lymphocytes Activation. EMBO J (2007) 26:4368–79. doi: 10.1038/sj.emboj.7601845
120. Wu JL, Wu HY, Tsai DY, Chiang MF, Chen YJ, Gao S, et al. Temporal Regulation of Lsp1 O-GlcNAcylation and Phosphorylation During Apoptosis of Activated B Cells. Nat Commun (2016) 7:12526. doi: 10.1038/ncomms12526
121. Melaiu O, Lucarini V, Cifaldi L, Fruci D. Influence of the Tumor Microenvironment on NK Cell Function in Solid Tumors. Front Immunol (2019) 10:3038. doi: 10.3389/fimmu.2019.03038
122. Sun Y, Sedgwick AJ, Palarasah Y, Mangiola S, Barrow AD. A Transcriptional Signature of PDGF-DD Activated Natural Killer Cells Predicts More Favorable Prognosis in Low-Grade Glioma. Front Immunol (2021) 12:668391. doi: 10.3389/fimmu.2021.668391
123. Liu S, Galat V, Galat Y, Lee YKA, Wainwright D, Wu J. NK Cell-Based Cancer Immunotherapy: From Basic Biology to Clinical Development. J Hematol Oncol (2021) 14:7. doi: 10.1186/s13045-020-01014-w
124. Yao AY, Tang HY, Wang Y, Feng MF, Zhou RL. Inhibition of the Activating Signals in NK92 Cells by Recombinant GST-sHLA-G1a Chain. Cell Res (2004) 14:155–60. doi: 10.1038/sj.cr.7290215
125. Perisic Nanut M, Pecar Fonovic U, Jakos T, Kos J. The Role of Cysteine Peptidases in Hematopoietic Stem Cell Differentiation and Modulation of Immune System Function. Front Immunol (2021) 12:680279. doi: 10.3389/fimmu.2021.680279
126. Božič J, Stoka V, Dolenc I. Glucosamine Prevents Polarization of Cytotoxic Granules in NK-92 Cells by Disturbing FOXO1/ERK/paxillin Phosphorylation. PloS One (2018) 13:e0200757. doi: 10.1371/journal.pone.0200757
127. Peng X, He Y, Huang J, Tao Y, Liu S. Metabolism of Dendritic Cells in Tumor Microenvironment: For Immunotherapy. Front Immunol (2021) 12:613492. doi: 10.3389/fimmu.2021.613492
128. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic Cells in Cancer Immunology and Immunotherapy. Nat Rev Immunol (2020) 20:7–24. doi: 10.1038/s41577-019-0210-z
129. Brombacher EC, Everts B. Shaping of Dendritic Cell Function by the Metabolic Micro-Environment. Front Endocrinol (Lausanne) (2020) 11:555. doi: 10.3389/fendo.2020.00555
130. Kneass ZT, Marchase RB. Protein O-GlcNAc Modulates Motility-Associated Signaling Intermediates in Neutrophils. J Biol Chem (2005) 280:14579–85. doi: 10.1074/jbc.M414066200
131. Lee BE, Suh PG, Kim JI. O-GlcNAcylation in Health and Neurodegenerative Diseases. Exp Mol Med (2021) 53:1674–82. doi: 10.1038/s12276-021-00709-5
132. Bartolome-Nebreda JM, Trabanco AA, Velter AI, Buijnsters P. O-GlcNAcase Inhibitors as Potential Therapeutics for the Treatment of Alzheimer's Disease and Related Tauopathies: Analysis of the Patent Literature. Expert Opin Ther Pat (2021) 31:1117–54. doi: 10.1080/13543776.2021.1947242
133. Balsollier C, Pieters RJ, Anderluh M. Overview of the Assays to Probe O-Linked Beta-N-Acetylglucosamine Transferase Binding and Activity. Molecules (2021) 26(4):1037. doi: 10.3390/molecules26041037
134. Liu Y, Ren Y, Cao Y, Huang H, Wu Q, Li W, et al. Discovery of a Low Toxicity O-GlcNAc Transferase (OGT) Inhibitor by Structure-Based Virtual Screening of Natural Products. Sci Rep (2017) 7:12334. doi: 10.1038/s41598-017-12522-0
135. Sodi VL, Bacigalupa ZA, Ferrer CM, Lee JV, Gocal WA, Mukhopadhyay D, et al. Nutrient Sensor O-GlcNAc Transferase Controls Cancer Lipid Metabolism via SREBP-1 Regulation. Oncogene (2018) 37:924–34. doi: 10.1038/onc.2017.395
136. Sharma NS, Gupta VK, Dauer P, Kesh K, Hadad R, Giri B, et al. O-GlcNAc Modification of Sox2 Regulates Self-Renewal in Pancreatic Cancer by Promoting Its Stability. Theranostics (2019) 9:3410–24. doi: 10.7150/thno.32615
137. Wu D, Jin J, Qiu Z, Liu D, Luo H. Functional Analysis of O-GlcNAcylation in Cancer Metastasis. Front Oncol (2020) 10:585288. doi: 10.3389/fonc.2020.585288
138. Bejarano L, Jordao MJC, Joyce JA. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov (2021) 11:933–59. doi: 10.1158/2159-8290.CD-20-1808
139. Wang Y, Tong Z, Zhang W, Zhang W, Buzdin A, Mu X, et al. FDA-Approved and Emerging Next Generation Predictive Biomarkers for Immune Checkpoint Inhibitors in Cancer Patients. Front Oncol (2021) 11:683419. doi: 10.3389/fonc.2021.683419
140. Lisi L, Lacal PM, Martire M, Navarra P, Graziani G. Clinical Experience With CTLA-4 Blockade for Cancer Immunotherapy: From the Monospecific Monoclonal Antibody Ipilimumab to Probodies and Bispecific Molecules Targeting the Tumor Microenvironment. Pharmacol Res (2022) 175:105997. doi: 10.1016/j.phrs.2021.105997
141. Safarzadeh Kozani P, Safarzadeh Kozani P, Rahbarizadeh F, Khoshtinat Nikkhoi S. Strategies for Dodging the Obstacles in CAR T Cell Therapy. Front Oncol (2021) 11:627549. doi: 10.3389/fonc.2021.627549
142. Boutilier AJ, Elsawa SF. Macrophage Polarization States in the Tumor Microenvironment. Int J Mol Sci (2021) 22(13):6995. doi: 10.3390/ijms22136995
Keywords: O-GlcNAcylation, TME (tumor microenvironment), cancer inflammation, post-translational modification (PTM), HBP pathway
Citation: Ouyang M, Yu C, Deng X, Zhang Y, Zhang X and Duan F (2022) O-GlcNAcylation and Its Role in Cancer-Associated Inflammation. Front. Immunol. 13:861559. doi: 10.3389/fimmu.2022.861559
Received: 24 January 2022; Accepted: 14 March 2022;
Published: 01 April 2022.
Edited by:
Guan-Jun Yang, Ningbo University, ChinaReviewed by:
Lara Abramowitz, National Institute of Diabetes and Digestive and Kidney Diseases (NIH), United StatesCopyright © 2022 Ouyang, Yu, Deng, Zhang, Zhang and Duan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fangfang Duan, ZHVhbmZmM0BtYWlsLnN5c3UuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.