 Puja Bagri
Puja Bagri Varun C. Anipindi
Varun C. Anipindi Charu Kaushic
Charu Kaushic
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 14 April 2022
Sec. Mucosal Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.861444
Interleukin-17 (IL-17A) is a cytokine involved in a complex array of both protective and detrimental processes. Although early biological studies focused on the pro-inflammatory function of IL-17 in the context of autoimmune and inflammatory disorders, it has become increasingly evident that the roles of IL-17 are far more nuanced. Recent work has demonstrated that the functions of IL-17 are highly context- and tissue-dependent, and there is a fine balance between the pathogenic and protective functions of IL-17. This is especially evident in mucosal tissues such as the female reproductive tract, where IL-17 has been shown to play an important role in the immune response generated during fungal, bacterial and viral infections associated with protection, but also with inflammation. In this review, we discuss the evolving landscape of IL-17 biology within the context of the vaginal mucosa, focusing on key findings that highlight the importance of this cytokine in genital mucosal immunity.
Interleukin 17A (IL-17A; referred to as ‘IL-17’ here) was cloned in the early 1990s and initially known as cytotoxic T lymphocyte-associated antigen 8 (CTLA-8) (1). Although first recognized as a new cytokine in 1995, the importance of IL-17 remained obscure until almost a decade later, when it was discovered that a novel population of CD4+ T helper (Th) cells, subsequently named Th17 cells, were characterized by their secretion of IL-17 (2–4). Following this discovery, the role of IL-17 has been examined in the context of many disease models.
The IL-17 family of cytokines includes six similarly structured ligands (IL-17A to IL-17F), of which, IL-17A and IL-17F are most closely related functionally. IL-17 cytokines signal through a dimeric receptor composed of pairs of five subunits: IL-17RA through IL-17RE (5, 6). IL-17 and IL-17F exist either as homodimers or as a heterodimer, and signal through an obligate dimeric IL-17RA and IL-17RC receptor complex (7). There are two primary signalling pathways initiated upon IL-17 binding to its receptor complex [reviewed in (5, 8)] (Figure 1). The first is a canonical pathway, which leads to the activation of nuclear factor kappa B (NF-κB), mitogen-activated protein kinase (MAPK) and CCAAT-enhancer-binding protein (C/EBP) pathways, and results in transcriptional activation of downstream, pro-inflammatory target genes. The second, noncanonical pathway, leads to the stabilization of mRNA transcripts which encode for intrinsically unstable targets including cytokines and chemokines. Overall, at the transcriptional and post-transcriptional level, IL-17 enhances the production of several immune mediators including chemokines, cytokines, antimicrobial peptides (AMPs) and other primarily inflammatory effectors (9).
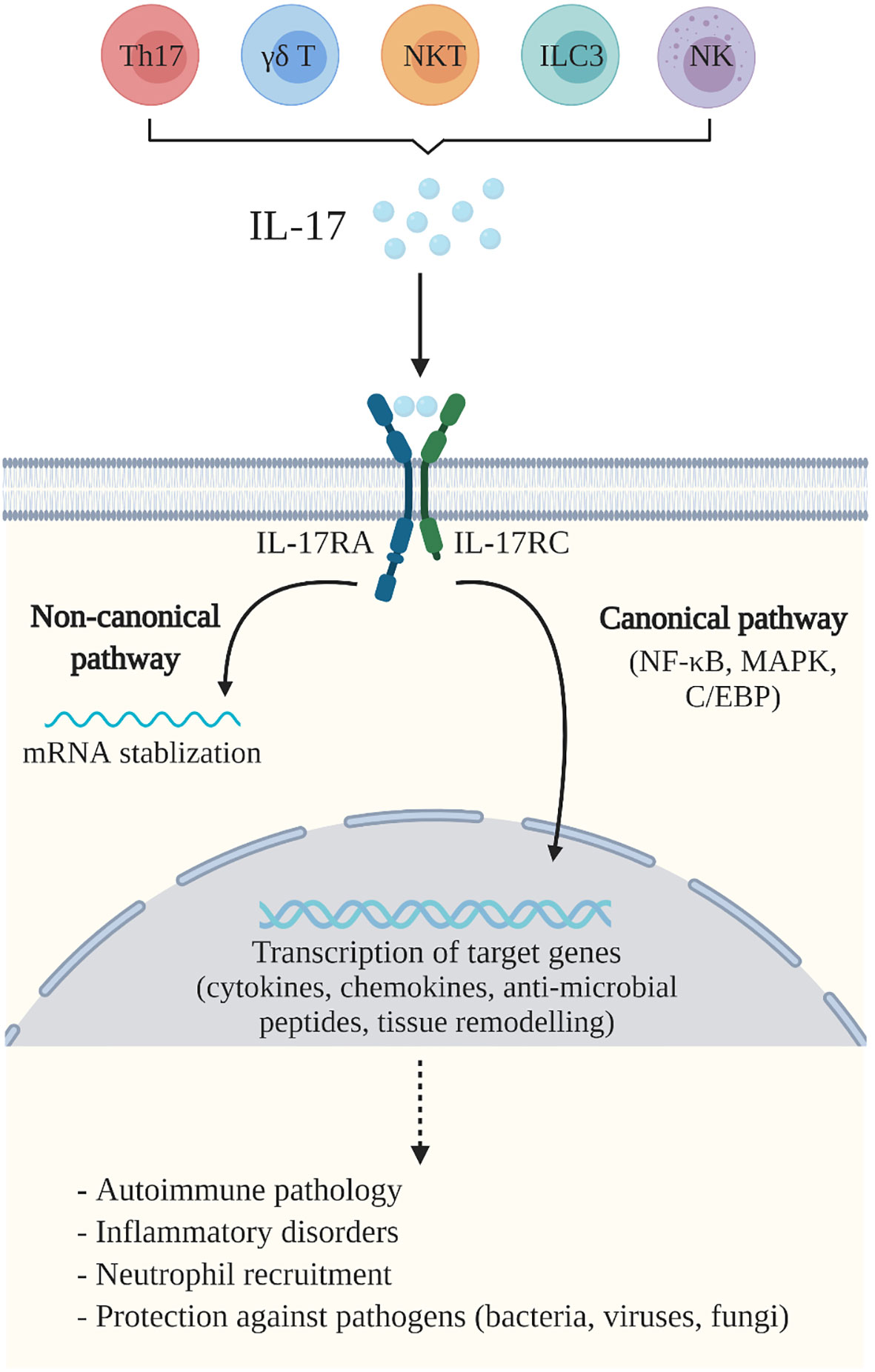
Figure 1 IL-17A-mediated immunity. IL-17A (IL-17) is produced by a variety of cells including Th17 cells, γδ T cells, natural killer (NK) cells, natural killer T (NKT) cells and group 3 innate lymphoid cells (ILC3). IL-17 binds to the IL-17 receptor (IL-17R)A and IL-17RC complex, expressed by a variety of cells including macrophages, fibroblasts, keratinocytes, and epithelial and endothelial cells. This initiates two primary IL-17 signalling pathways which mediate the essential functions of IL-17. The canonical pathway activates the nuclear factor kappa B (NF-κB), mitogen-activated protein kinase (MAPK) and CCAAT-enhancer-binding protein (C/EBP) pathways, that trigger transcriptional activation of downstream target genes, including pro-inflammatory cytokines, chemokines and anti-microbial peptides, as well as genes related to tissue remodeling. The non-canonical pathway leads to the stabilization of mRNA transcripts. Together, these pathways mediate immune responses which contribute to the pathogenesis of autoimmune and inflammatory diseases, neutrophil recruitment and are important for host defence against pathogens including bacteria, viruses and fungi. Created with BioRender.
IL-17 has several immunoregulatory functions, as detailed in a recent review (10). During infection, IL-17 is actively involved in neutrophil and monocyte recruitment through enhanced induction of various chemoattractants including CXCL1, CXCL2 and CXCL5 (11, 12). IL-17 is also known to induce granulocyte-colony-stimulating factor (G-CSF), which is involved in promoting the expansion and survival of neutrophils (13). Additionally, IL-17 plays an important role in maintaining epithelial barrier integrity by regulating the induction of AMPs during periods of homeostasis when the barrier is intact, and then by inducing immune mediators upon the loss of barrier integrity (14). Taken together, IL-17 is considered a key cytokine involved in the clearance of extracellular bacteria and fungi. However, in some cases aberrant IL-17 production can augment inflammation and cause tissue damage, which has been shown to occur in autoimmune or chronic inflammatory diseases such as rheumatoid arthritis (6).
The production of IL-17 is linked to several cellular sources. Traditionally, IL-17 is considered a cytokine secreted by Th17 cells, a subset of activated CD4+ T cells that secrete signature cytokines IL-17A and IL-17F, as well as IL-21 and IL-22 (12). Exposure of naïve CD4+ T cells to combinations of antigen-presenting cell (APC)-derived polarizing cytokines, including transforming growth factor-beta (TGF-β), IL-6, IL-21, IL-23 and IL-1β, leads to the differentiation of Th17 cells (15–18). Furthermore, Th17 cells are regulated by a master transcription factor, retinoic acid receptor-related orphan receptor gamma t (ROR-γt, encoded by Rorc), which induces the production of their signature cytokines (19). ROR-γt is also a major transcription factor associated with all subsets of IL-17-secreting cells, along with the transcription factor aryl hydrocarbon receptor (Ahr) (10).
In addition to Th17 cells, other cell types are also capable of producing IL-17. This includes innate and innate-like lymphocytes such as gamma-delta (γδ) T cells (20), natural killer (NK) cells, natural killer T (NKT) cells (21), lymphoid tissue inducer-like cells (LTi) and several populations of group 3 innate lymphoid (ILC3) cells (22, 23). These additional sources of IL-17 tend to accumulate at mucosal surfaces and have been shown to play an important role in the early immune response against pathogens, emphasizing the importance of IL-17 in innate immunity. Different pro-inflammatory cytokines including IL-1β and IL-23 are known to induce IL-17 production by these cells. Interestingly, γδ T cells in particular have been shown to be the primary source of IL-17 production in various settings of tissue homeostasis and infection [reviewed in (24)]. γδ T cells develop early in the fetus and provide immunity prior to the generation of adaptive immune responses (25). Unlike traditional T lymphocytes, γδ T cells can acquire their effector function during thymic development (25); thus, allowing them to produce baseline levels of IL-17 and/or respond early and more rapidly to pathogens compared to Th17 cells.
IL-17-mediated immunity has been demonstrated to play an important role in the immune response generated against pathogens in mucosal tissues including the female reproductive tract (FRT). The FRT is a unique mucosal site that is regulated by several factors in the microenvironment, including sex hormones and the vaginal microbiota [reviewed in (26–28)]. Importantly, the FRT is a critical site for enabling human reproductive success as well as protecting against sexually transmitted pathogens; thus, immune responses in this tissue must be well understood in order to help facilitate positive reproductive health. As such, we and others have focused on the role of this important immunoregulatory factor in the context of genital tract infections, and in this review, we will summarize and highlight the recent progress in the field of IL-17-mediated immunity during fungal, bacterial and viral infections in the FRT.
Candida species are part of the normal microflora of the FRT but can become pathogenic under certain circumstances (29). Vulvovaginal candidiasis (VVC) caused by Candida albicans (C. albicans), is an opportunistic fungal infection that affects approximately 75% of healthy women of reproductive age globally at least once during their lifetime (30). Furthermore, following primary infection with C. albicans, 5-10% of women will subsequently experience recurrent infection (31), which is defined as experiencing at least 3-4 episodes yearly (32). Thus, VVC infection is a very frequent and distressing condition that can significantly impact quality of life.
Both human and animal studies have examined the role of IL-17-mediated immunity during C. albicans infection. Animal models are commonly used to study VVC pathogenesis (33), and the mouse model closely recapitulates the human disease. Although mice are not naturally susceptible to vaginal infection with C. albicans, treatment with estradiol (E2) enables persistent infection to occur. In several studies, IL-17 was seen to play a clear protective role in several types of mucosal candidiasis (i.e. chronic mucocutaneous and oropharyngeal candidiasis) and help regulate antifungal immunity by upregulating pro-inflammatory cytokines, neutrophil-recruiting chemokines and AMPS (11, 34–36). The role of IL-17 during VVC, however, remains unclear. For example, patients who have genetic defects in IL-17 do not show increased susceptibility to VVC as they do to other mucocutaneous forms of candidiasis (37, 38). Similarly, animal studies have shown that in the absence of IL-17 and/or IL-17-mediated signalling, mice demonstrated worsened disease outcomes following both systemic (39) and oral infection with C. albicans (34), and this was related to impaired neutrophil recruitment and AMP production. However, unlike candidiasis infections at other mucosal sites, neutrophils have been shown to be more damaging than protective during VVC (29, 40) or they appear to have a limited impact on fungal infection in the FRT. For instance, Yano et al. (41) reported that in the absence of IL-17 and other Th17-related cytokines, mice had similar levels of fungal burden following vaginal C. albicans infection compared to wildtype (WT) mice, suggesting IL-17 was not involved in response to infection. In addition, recent findings from Peters et al. (42) demonstrated that mice lacking IL-17RA did not exhibit altered VVC susceptibility, regardless of E2 administration, further supporting the idea that the Th17/IL-17 axis plays no role in the immunopathogenesis of VVC. In contrast, Pietrella et al. (43) found that when Th17 differentiation was inhibited during VVC challenge, there was greater exacerbation of disease, along with significantly less production of protective AMPs. This suggests that IL-17 and AMPs play an important role in VVC immunity. Based on the limited studies conducted, it appears that instead of being neutrophil-dependent, protection against VVC is reliant on extrinsic factors such as AMPS and the maintenance of an intact epithelial barrier and a balanced vaginal microbiota (44). Overall, studies regarding the role of IL-17 during VVC are inconclusive, and further research is required to determine if the function of IL-17 is protective, as observed at other sites of candida infection.
Gonorrhea is an acute purulent genital tract infection caused by the Gram-negative bacterium, Neisseria gonorrhoeae (N. gonorrhoeae) (45). It is estimated that there are 78 million new cases of gonorrhea which occur globally each year (46), and due to the lack of a viable vaccine and the emergence of multi-drug resistant strains, N. gonorrhoeae is considered a serious infectious threat (46, 47). Rates of infection are higher in women compared to men, and gonorrhea infection can lead to further negative health outcomes in women including pelvic inflammatory disease (PID), infertility and ectopic pregnancy (48). Furthermore, if left untreated, infection with N. gonorrhoeae can enhance the transmission and acquisition of HIV by up to 5-fold (49).
N. gonorrhoeae is primarily an extracellular bacterium that induces a pro-inflammatory response consisting of the cytokines IL-6, IL-1β and tumor necrosis factor-alpha (TNF-α), as well as an influx of neutrophils (47). Interestingly, these cytokines are also linked to Th17 differentiation. As such, IL-17 levels have been reported to be elevated in individuals infected with N. gonorrhoeae (50, 51). The mouse model of gonorrhea, which requires mice to be treated with E2 prior to infection (52, 53), also demonstrates a strong Th17 response following infection. In this model, infection persists in rodents for about 10-20 days, after which the bacteria are cleared. Over the past several years, M.W. Russell and colleagues have extensively used the mouse model to better understand the role of IL-17 during gonorrheal infection. In initial studies, Feinen et al. (45) showed that IL-17 was critical for controlling early gonorrheal infection in vivo, as blocking IL-17 or preventing IL-17RA signalling in mice resulted in prolonged infection, along with significantly diminished neutrophil recruitment. Further, Russell and Feinen (54) reported that unlike IL-17, IL-22 does not appear to impact N. gonorrhoeae infection in vivo, suggesting IL-17 specifically, but not other Th17-related cytokines, is critical for bacterial clearance.
Interestingly, N. gonorrhoeae infection does not elicit a strong Th1 or Th2 response in mice (45) and although N. gonorrhoeae induces local inflammation, there is no acquired immunity or immunological memory established (53, 55). These findings show that primary infection does not lead to substantial or sustained antibody responses, and mice can be re-infected with the same strain of bacteria without displaying enhanced resistance such as elevated antibodies or enhanced CD4+ T cell responses (53). These observations closely reflect known features of the human immune response to uncomplicated N. gonorrhoeae infection, where there is limited humoral and T cell immunity, even with recurrent infection (56, 57). This led to the theory that N. gonorrhoeae selectively elicits Th17-dependent innate responses that it can overcome, including neutrophil recruitment and upregulation of AMP production, while suppressing Th1/Th2-driven adaptive immunity that may be able to protect against subsequent infection (45, 58–60). This would imply that Th17 immunity is manipulated by N. gonorrhoeae and used to evade host mechanisms of protection. In this regard, further work by Liu et al. has shown that the absence of protective Th1 responses can be attributed to the production of TGF-β, which occurs following N. gonorrhoeae infection and skews immune responses towards Th17-mediated immunity (59, 60). Together, these studies suggest that N. gonorrhoeae actually suppresses adaptive immunity by upregulating the production of immunosuppressive cytokines, TGF-β and IL-10. Liu et al. demonstrated that by blocking TGF-β, it is possible to reverse this host immune response and enable the development of protective anti-gonococcal immunity consisting of Th1-driven responses, the presence of anti-gonococcal IgG and IgA antibodies, establishment of immunological memory and enhanced clearance of bacteria (59, 60). Additional work is needed to understand how to better direct immunity during infection with N. gonorrhoeae to maximize protection, as well as to better understand how to leverage IL-17 immunity in this effort.
Chlamydia trachomatis (C. trachomatis) is an intracellular human bacterium that causes the most common bacterial sexually transmitted disease worldwide, with over 250 000 new infections contracted daily (61). Like many other STIs, women have disproportionately higher rates of chlamydia infection prevalence globally (62). Although antibiotics can be used to treat chlamydial infection, more than 70% of women show no signs or symptoms of active infection (63). As a result, untreated infections can cause significant reproductive tract pathology in women leading to the development of conditions such as PID, chronic pelvic pain and infertility. C. trachomatis is also the cause of preventable blindness (trachoma) in developing countries (64, 65).
Although IL-17 has been shown to be protective against extracellular pathogens, the role of IL-17 in protection against intracellular bacterial pathogens, including chlamydia, is less clear. Multiple mouse models for chlamydia have been developed over the past two decades and studies have provided extensive information regarding chlamydia pathogenesis, immune response and vaccine design. As with most animal models of infectious disease, there are differences between human and murine chlamydia infections which should be considered when extrapolating findings (66). For instance, similar to other models involving genital pathogens, the hormonal microenvironment of the murine FRT has to be manipulated in order for infection to occur. In the chlamydia models, mice require pre-treatment with progesterone to enhance susceptibility to infection. It is well known that changing the hormonal microenvironment alters the structural physiology of the FRT epithelium and may also influence the function of the immune cells present (67). Another difference is that in mice, disease develops after a single exposure to bacteria, after which infection is often cleared. This contrasts what occurs in humans, where secondary infections are often required to drive significant pathology and long-term chronic infections are common. Furthermore, different species of chlamydia display tropism for specific hosts (68). While human urogenital C. trachomatis strains can be used to infect mice, the most common murine model of genital chlamydia infection uses the mouse-adapted C. muridarum pathogen. Intravaginal infection with C. muridarum evades murine cell-autonomous immune mechanisms and establishes a self-resolving genital infection, whereas infection with human C. trachomatis is rapidly cleared from the murine FRT and fails to establish productive infection or induce pathogenic immune responses. As such, the C. muridarum genital infection model is more amenable to the study of immune mechanisms and appears to replicate many aspects of human infection. Studies have shown that C. muridarum first infects the vaginal and cervical epithelial cells, after which it ascends the reproductive tract and causes upper reproductive tract pathology, similar to chlamydia-associated disease sequelae observed in women infected with C. trachomatis (69, 70). The infection in mice is resolved after approximately 4-6 weeks and results in long-lived adaptive immunity that protects against re-infection (71). It has been shown that Th1 cells and interferon gamma (IFN-γ) are critical for protection against primary genital C. muridarum infection (72), while CD8+ T cell responses and antibody responses are important for protection against re-infection (73–78).
The role of IL-17 during chlamydial infection has been extensively studied in the lungs, where it has been shown that in the absence of IL-17, there is greater replication of bacteria and decreased survival of infected mice (79). Further mechanistic studies have demonstrated that IL-17 appears to be necessary for DC priming of Th1 immunity in the lungs, without which there is compromised bacterial clearance (80). However, in the context of genital tract infection with C. muridarum, the role of IL-17 is less clear. For instance, some studies have shown that the importance of IL-17 in mediating protection against chlamydia is negligible. A study by Scurlock et al. (81) showed that although IL-17RA deficient (IL-17RA-/-) mice demonstrated reduced IFN-γ production in the lymph nodes and decreased neutrophil influx into the FRT, mice were still able to resolve primary C. muridarum infection normally and showed no differences in pathology compared to WT mice. Instead, both macrophage influx and TNF-α production was increased in the absence of IL-17, suggesting a compensatory mechanism to control infection. Likewise, Frazer et al. (82) showed that in the absence of IL-23, where chlamydia-specific Th17 responses were absent, mice exhibited normal susceptibility to genital infection and regular development of oviduct pathology.
In contrast, other findings suggest that IL-17 plays a pathogenic role during genital chlamydial infection. In a study by Andrew et al. (64), they found that the duration and magnitude of C. muridarum infection was significantly lower in IL-17 deficient (IL-17A-/-) mice compared to WT mice. In the absence of IL-17, they also noted decreased inflammatory pathology, which was related to significantly reduced recruitment of neutrophils and macrophages into the oviduct tissues. Interestingly, others have also shown that oviduct pathology observed following infection is associated with the infiltration of neutrophils into the FRT, as well as the production of inflammatory mediators and factors involved in tissue-remodeling such as matrix metalloproteases; all of which are mediated by IL-17 (83–86). Human studies have similarly associated greater neutrophil activation with increased disease progression in women infected with genital C. trachomatis (87) and suggested a role for IL-17 during infection, as cervical washes from infected women had 5-fold higher levels of IL-17 compared to uninfected controls (88). Altogether, studies regarding the importance of IL-17 during chlamydial infection are conflicting. This may be due to the fact that the strains of mice used in different studies vary, and it is well known that the mouse strain used for chlamydia infection may impact outcomes, including duration of infection, degree of upper genital tract infection, severity of infection-induced pathology and immune responses generated (71). As such, further work is required to better elucidate the role of IL-17 in the context of genital chlamydia infection.
HIV (human immunodeficiency virus) infection in the FRT leads to the rapid depletion of local CD4+ T cells, as well as viral dissemination throughout the body (89). It is a deadly virus if left untreated and can lead to the development of acquired immunodeficiency syndrome (AIDS). There are currently close to 38 million individuals living with HIV, with approximately 1.7 million new infections occurring each year (90). Women account for more than 50% of infections, and young women between the ages of 15–24 are particularly susceptible to infection (90). Although the risk of vaginal transmission is considered low, estimates indicate that 40% of HIV infections are initiated in the FRT (91). Additionally, infection with HIV is often associated with increased susceptibility to other sexually transmitted infections (STIs).
The role of IL-17 during HIV infection has not been comprehensively described, however, Th17 cells are important for HIV pathogenesis. HIV preferentially replicates in activated T cells, specifically CD4+ T cells (92), and the expression of the mucosal integrin α4β7 and HIV co-receptor CCR5 by activated T cells also increases susceptibility (93, 94). As such, Th17 cells, which are activated and terminally differentiated cells that express high levels of α4β7 and CCR5, are considered preferential target cells for HIV infection (95–100). In vitro and ex vivo studies using human cells and in vivo studies using a macaque model with the simian variant of the virus (SIV), have all shown that Th17 cells are target cells for HIV infection in mucosal tissues. For instance, Rodriguez-Garcia et al. (101) examined the phenotype and susceptibility of primary CD4+ T cells isolated from endometrium, endocervix, and ectocervix, to HIV infection ex vivo. They found that Th17 cells were the primary CD4+ T cell population which expressed HIV receptors CCR5 and CD90, and that these cells were the most susceptible to HIV infection in vitro. Likewise, several studies using samples collected from HIV-infected Kenyan women have also shown that Th17 cells are preferentially targeted by HIV, as Th17 cells were significantly depleted in infected individuals (93, 100, 102). Additionally, Boily-Larouche et al. (102) described a highly activated subset of CD4+ T cells in the FRT of HIV-infected female sex workers (FSWs) from Kenya, which expressed CD161 and differentiated into Th17 cells. These cells were found to express multiple HIV susceptibility markers and were severely depleted in HIV-infected FSWs, compared to uninfected FSWs. Similarly, McKinnon et al. (99) collected cervical cytobrush specimens from FSWs in Kenya and found that cervical IL-17+ CD4+ T cells preferentially co-expressed HIV receptors α4β7 and CCR5. Furthermore, these cervical Th17 cells were significantly depleted following HIV infection, suggesting they may serve as key target cells during HIV infection. Finally, in the macaque model of SIV infection, Stieh et al. (103) found that Th17 cells (CCR6+ CD4+) were also preferentially infected by SIV, and they showed that most SIV-infected cells expressed the master Th17 transcriptional regulator, ROR-γt.
Interestingly, Th17 and Th22 cells that make IL-17 and IL-122 play an important role in the renewal and maintenance of the mucosal epithelial barrier, which is known to be critical in protecting against HIV infection in both the gut and FRT (98, 104, 105). Several components of the mucosal epithelial barrier help mediate protection against HIV infection, including the physical composition of the barrier, the presence of immune cells, cytokines and antimicrobial factors, and the interactions between the barrier and the local microenvironment, including mucus and host microbiota (106). As such, the loss of Th17 cells may have a direct impact on the integrity of mucosal barrier and consequently impact further viral transmission and/or pathogenesis (107). This has been shown in the gut, where the depletion of Th17 and IL-22 producing T cells during chronic HIV infection has been associated with damage to the epithelial barrier and subsequent microbial translocation (108, 109). It is possible Th17 cells may be playing a similar protective role in the FRT during HIV infection, however more work is needed to elucidate this potential mechanism.
Herpes simplex virus type 2 (HSV-2), the primary virus causing genital herpes, is one of the most common STIs worldwide, with over 400 million individuals infected globally and approximately 17 million new infections occurring each year (110). Rates of infection are especially alarming in sub-Saharan Africa, where prevalence is as high as 80% amongst women between the ages of 15-49 (110). HSV-2 first infects genital epithelial cells, and then travels via retrograde transport along nerve axons to the dorsal root ganglia, where it establishes life-long latency (111). The neuronal cells act as a reservoir for the latent virus, which can be reactivated due to factors including stress and hormonal changes. Reactivation results in anterograde transport of the virus, resulting in productive replication in the FRT (112). Along with the development of painful genital ulcers, HSV-2 infection is associated with a 2- to 3-fold increased risk of HIV-1 acquisition and up to a 5-fold increase in transmission of HIV-1 (113, 114).
Unlike the role of IL-17 during bacterial and fungal infections at mucosal sites, very limited studies have examined the importance of IL-17 in viral infections, especially in the context of the FRT and HSV-2. As such, this has been an area of interest in our lab and we have focused extensively on the role of both innate and adaptive production of IL-17 in the FRT, along with the role it plays during viral infection with HSV-2. Additionally, we have examined how factors in the FRT microenvironment, such as female sex hormones and the vaginal microbiota, effect IL-17 production in the genital mucosa.
Our work over the past few years has provided significant insight regarding how IL-17 modulates critical anti-viral T cell responses in the FRT. While it is well established that anti-viral protection against HSV-2 in the mouse model of infection is largely mediated by Th1 responses (115–117), our findings are the first to show that Th17 responses are also important in the anti-viral immune response to HSV-2 infection (118–120). We directly examined the role of IL-17 in anti-viral protection against HSV-2 and found that compared to WT controls, IL-17A-/- mice immunized intravaginally or intranasally were more susceptible to HSV-2 challenge (119). IL-17A-/- mice had decreased survival, greater viral shedding, and more severe genital pathology post-challenge. Interestingly, we found that IL-17 played an important role in enhancing anti-viral Th1 responses in the FRT, as IL-17A-/- mice had impaired Th1 cells responses post-challenge (119) and mechanistically, this was associated with impaired Th1 priming by vaginal DCs (118). Taken together, these studies have demonstrated that IL-17 is critical for inducing an efficient Th1 immune response following HSV-2 immunization, resulting in effective protection against HSV-2.
While the presence of innate IL-17 has been implicated in the amplification of Th17 responses in other mucosal tissues (121–123), the significance of IL-17 produced by innate or innate-like sources in the FRT and its influence on Th17 immunity in the vaginal mucosa is less understood. Thus, we also investigated the how innate IL-17 in the FRT might induce adaptive Th17 responses (124). Our findings support results seen in other mucosa and demonstrate that innate IL-17 produced in the FRT is also important for inducing Th17 responses. Furthermore, our observation that vaginal DCs from IL-17A-/- mice produced lower amounts of IL-1β compared to WT DCs, suggested a mechanism in which innate IL-17 induces vaginal DCs to prime Th17 responses via IL-1β (124). Additionally, consistent with previous findings by Kim et al. (125), we showed that γδ+ cells are the primary source of innate IL-17 in the FRT under homeostatic conditions (124). We extended these findings to show that multiple factors found within the vaginal microenvironment, influence innate IL-17 production by γδ+ cells. We found that E2 treatment resulted in significantly greater proportions of IL-17-producing γδ+ T cells and that germ-free mice had significantly lower proportions (124). These results provide insight on how innate IL-17 can influence immune responses against infections in the FRT, as well as ways in which its production can be modulated.
As mentioned previously, the hormonal microenvironment can influence immune responses and disease outcomes in the FRT. For the past decade we have investigated the influence of E2 on immune responses in the FRT and found that E2 increases protection against HSV-2 infection, although the underlying immunological mechanisms remained unclear. Recently, we showed that better protection in E2-treated mice coincided with earlier recruitment and higher proportions of Th1 and Th17 cells in the FRT following either HSV-2 immunization (intravaginally or intranasally) (120) and/or challenge (118). This included greater establishment of tissue-resident memory CD4+ T cells in the FRT, which play a critical role against re-exposure to pathogens (120). Together with the reduced protection shown against HSV-2 challenge in E2-treated IL-17A-/- mice, these findings suggested that E2-mediated protection against HSV-2 is driven by the induction of Th17 responses, likely mediated by increased IL-1β production by vaginal CD11c+ DCs in the presence of E2. Thus, a novel and critical anti-viral role for IL-17 in the FRT has emerged through our work.
The understanding of IL-17 and its role in immune processes has advanced considerably over recent years. While IL-17 has been shown to play a key role in the maintenance of tissue integrity and the generation of protective immunity against extracellular pathogens such as bacteria and fungi, the pro-inflammatory nature of IL-17 has also been associated with excessive inflammation and immunopathology. Within the context of the FRT, findings from several research groups have demonstrated the importance of IL-17 function which goes beyond merely acting as an inflammatory cytokine. The relevance of IL-17 has been demonstrated in bacterial, fungal and viral infections within the FRT, where innate and adaptive production of IL-17 is involved in a variety of immunomodulatory processes including neutrophil recruitment, DC regulation and Th1 modulation (Figure 2). However, it has become increasingly clear that the overall function of IL-17 is highly contextual, depending on both the pathogen involved and the site of infection. For instance, studies examining C. albicans infection in the FRT have demonstrated that unlike the clear protective effects of IL-17 observed in chronic mucocutaneous and oropharyngeal candidiasis, the function of IL-17 during vulvovaginal candidiasis remains inconclusive. Similarly, while IL-17 has shown to be protective against chlamydial infection in the lungs, the role of IL-17 is less clear in the FRT. Some findings suggest IL-17 does not impact disease outcomes following genital chlamydial infection, while other studies have demonstrated a pathogenic role. This highlights the tissue-specific function of IL-17, as well as the necessity to further study the extent to which IL-17 is required in facilitating immunity during these infections. Additionally, some genital tract pathogens such as HIV and N. gonorrhoeae appear to leverage IL-17-mediated responses to their advantage by either targeting Th17 cells to promote infection or by using these responses to suppress the generation of protective adaptive immunity, respectively. Yet, our work has shown that IL-17 seems to have a broader protective role in the context of HSV-2 infection in the FRT. We have recently demonstrated that IL-17 modulates vaginal DCs to induce greater Th1 and Th17 immunity in the FRT and is also involved in increased establishment of memory T cells post-immunization, which results in greater protection against genital HSV-2 infection (Figure 3). Our studies have also emphasized the critical role of hormones in regulating levels of IL-17 within the reproductive mucosa. Altogether, these studies point to the need to better understand the dual role and balance between IL-17-mediated protection and pathology before considering therapeutically modulating the IL-17 pathway. Further research is needed to gain a better understanding of the underlying factors including the tissue microenvironment that regulate IL-17 and Th17 immune responses. Consequently, incentives directed towards developing vaccines or therapies for these pathogens must consider whether supporting or inhibiting IL-17 responses in the FRT will help shape the immune response towards a more robust protective phenotype that can help resist these pathogens.
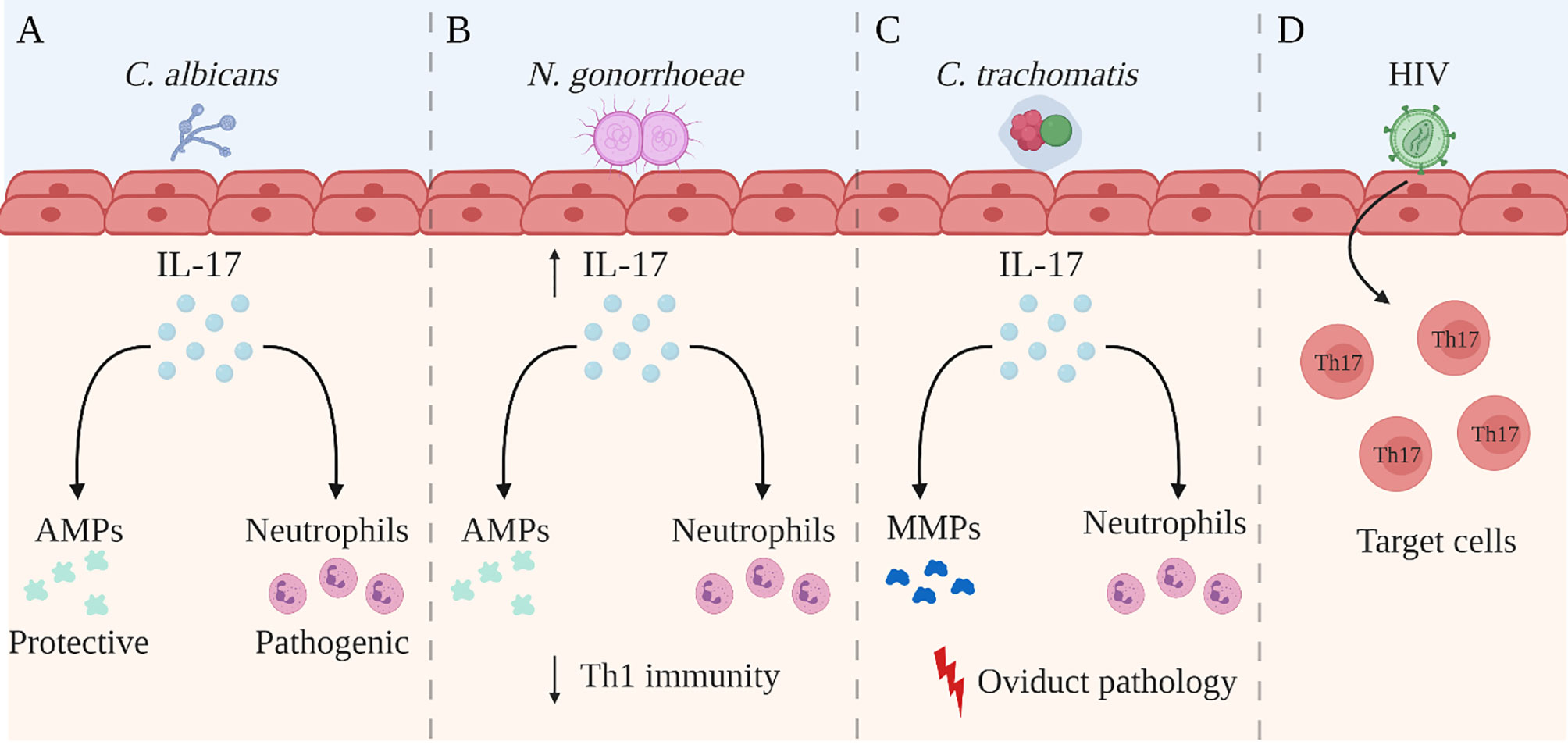
Figure 2 Summary of the role of IL-17 in the female reproductive tract. The role of IL-17A (IL-17) in the female reproductive tract (FRT) is disease-specific and is not yet completely understood. Studies have reported the following regarding the role of IL-17 during genital infection with Candida albicans (C. albicans), Neisseria gonorrhoea (N. gonorrhoeae), Chlamydia trachomatis (C. trachomatis), and human immunodeficiency virus (A) IL-17 is involved in recruiting neutrophils and inducing the production of anti-microbial peptides (AMPs) during C albicans infection. While AMPs have shown to be protective against C albicans infection in the FRT, neutrophils may be causing more damage than protection. (B) N. gonorrhoeae infection in the FRT selectively induces a robust Th17 response but reduced protective Th1 immunity, allowing evasion of host mechanisms of protection. However, the elevated IL-17 production has been shown to be important for controlling early gonococcal infection, as well as for recruiting AMPs and neutrophils. (C) During C trachomatis infection in the FRT, the recruitment of neutrophils and the production of matrix metalloproteases (MMPs) by IL-17 has been associated with greater oviduct pathology. (D) HIV preferentially infects activated Th17 cells which express CD4 and high levels of HIV co-receptors. Created with BioRender.
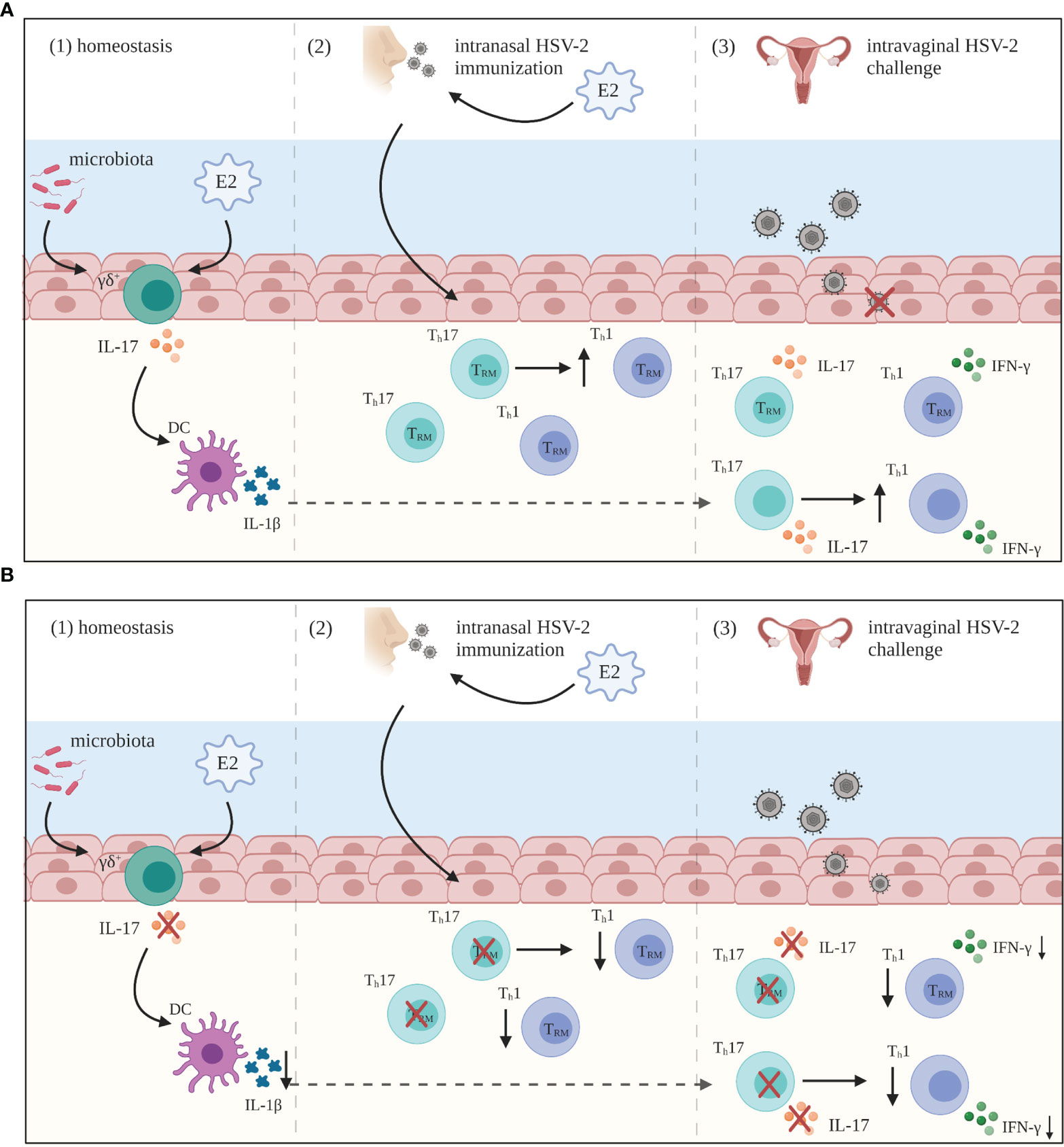
Figure 3 Summary of IL-17 mediated anti-HSV-2 immunity in the FRT. (A) (1) During periods of homeostasis, IL-17 is constitutively produced by innate lymphocyte populations, primarily gamma delta (γδ+) T cells, and regulated by estradiol (E2) and commensal microbiota. This innate IL-17 is important for the induction of Th17 responses primed by vaginal dendritic cells (DCs). (2) Following intranasal HSV-2 immunization under the influence of E2, IL-17 is important for the establishment of Th1 tissue-resident memory (TRM) cells in the female reproductive tract (FRT). (3) These Th17 and Th1 TRM cells are then able to protect against subsequent HSV-2 challenge. Additionally, IL-17 is critical for inducing IFN-γ+ CD4+ T cell recall responses in the FRT post-challenge. (B) In the absence of IL-17, there is reduced Th17 immunity generated (1), and overall anti-viral Th1 responses are significantly lowered both post-immunization (2) and post-challenge (3), even in the presence of E2. As a result, there is less protection generated against HSV-2 infection in the FRT. Created with BioRender.
The concept for this review was determined by CK. The first draft was written by PB. All authors participated in the editing and revision of the manuscript. PB provided the initial production of the figures and their revision. All authors participated in the final revision of the manuscript and approved the final version.
This research was funded by operating grants from the Canadian Institutes of Health Research (CIHR) to CK (FRN #93615) and an Applied HIV Research Chair Award to CK from the Ontario HIV Treatment Network (OHTN) (AHRC #779).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rouvier E, Luciani MF, Mattei MG, Denizot F, Golstein P. CTLA-8, Cloned From an Activated T Cell, Bearing AU-Rich Messenger RNA Instability Sequences, and Homologous to a Herpesvirus Saimiri Gene. J Immunol (1993) 150(12):5445–56.
2. Korn T, Oukka M, Kuchroo V, Bettelli E. Th17 Cells: Effector T Cells With Inflammatory Properties. Semin Immunol (2007) 19(6):362–71. doi: 10.1016/j.smim.2007.10.007
3. Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 Are Coexpressed by Th17 Cells and Cooperatively Enhance Expression of Antimicrobial Peptides. J Exp Med (2006) 203(10):2271–9. doi: 10.1084/jem.20061308
4. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A Distinct Lineage of CD4 T Cells Regulates Tissue Inflammation by Producing Interleukin 17. Nat Immunol (2005) 6(11):1133–41. doi: 10.1038/ni1261
5. Amatya N, Garg AV, Gaffen SL. IL-17 Signaling: The Yin and the Yang. Trends Immunol (2017) 38(5):310–22. doi: 10.1016/j.it.2017.01.006
6. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol (2009) 27:485–517. doi: 10.1146/annurev.immunol.021908.132710
7. Toy D, Kugler D, Wolfson M, Vanden Bos T, Gurgel J, Derry J, et al. Cutting Edge: Interleukin 17 Signals Through a Heteromeric Receptor Complex. J Immunol (2006) 177(1):36–9. doi: 10.4049/jimmunol.177.1.36
8. Monin L, Gaffen SL. Interleukin 17 Family Cytokines: Signaling Mechanisms, Biological Activities, and Therapeutic Implications. Cold Spring Harb Perspect Biol (2018) 10(4):a028522. doi: 10.1101/cshperspect.a028522
9. Onishi RM, Gaffen SL. Interleukin-17 and its Target Genes: Mechanisms of Interleukin-17 Function in Disease. Immunology (2010) 129(3):311–21. doi: 10.1111/j.1365-2567.2009.03240.x
10. Veldhoen M. Interleukin 17 Is a Chief Orchestrator of Immunity. Nat Immunol (2017) 18(6):612–21. doi: 10.1038/ni.3742
11. Conti HR, Gaffen SL. IL-17-Mediated Immunity to the Opportunistic Fungal Pathogen Candida Albicans. J Immunol (2015) 195(3):780–8. doi: 10.4049/jimmunol.1500909
12. Sandquist I, Kolls J. Update on Regulation and Effector Functions of Th17 Cells. F1000Res (2018) 7:205. doi: 10.12688/f1000research.13020.1
13. Parsonage G, Filer A, Bik M, Hardie D, Lax S, Howlett K, et al. Prolonged, Granulocyte-Macrophage Colony-Stimulating Factor-Dependent, Neutrophil Survival Following Rheumatoid Synovial Fibroblast Activation by IL-17 and TNFalpha. Arthritis Res Ther (2008) 10(2):R47. doi: 10.1186/ar2406
14. Pappu R, Rutz S, Ouyang W. Regulation of Epithelial Immunity by IL-17 Family Cytokines. Trends Immunol (2012) 33(7):343–9. doi: 10.1016/j.it.2012.02.008
15. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal Developmental Pathways for the Generation of Pathogenic Effector TH17 and Regulatory T Cells. Nature (2006) 441(7090):235–8. doi: 10.1038/nature04753
16. Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming Growth Factor-Beta Induces Development of the T(H)17 Lineage. Nature (2006) 441(7090):231–4. doi: 10.1038/nature04754
17. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the Context of an Inflammatory Cytokine Milieu Supports De Novo Differentiation of IL-17-Producing T Cells. Immunity (2006) 24(2):179–89. doi: 10.1016/j.immuni.2006.01.001
18. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 Programs T(H)-17 Cell Differentiation by Promoting Sequential Engagement of the IL-21 and IL-23 Pathways. Nat Immunol (2007) 8(9):967–74. doi: 10.1038/ni1488
19. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The Orphan Nuclear Receptor RORgammat Directs the Differentiation Program of Proinflammatory IL-17+ T Helper Cells. Cell (2006) 126(6):1121–33. doi: 10.1016/j.cell.2006.07.035
20. Cua DJ, Tato CM. Innate IL-17-Producing Cells: The Sentinels of the Immune System. Nat Rev Immunol (2010) 10(7):479–89. doi: 10.1038/nri2800
21. Kronenberg M. Toward an Understanding of NKT Cell Biology: Progress and Paradoxes. Annu Rev Immunol (2005) 23:877–900. doi: 10.1146/annurev.immunol.23.021704.115742
22. Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 Immune Axis: From Mechanisms to Therapeutic Testing. Nat Rev Immunol (2014) 14(9):585–600. doi: 10.1038/nri3707
23. Villanova F, Flutter B, Tosi I, Grys K, Sreeneebus H, Perera GK, et al. Characterization of Innate Lymphoid Cells in Human Skin and Blood Demonstrates Increase of NKp44+ ILC3 in Psoriasis. J Invest Dermatol (2014) 134(4):984–91. doi: 10.1038/jid.2013.477
24. Papotto PH, Ribot JC, Silva-Santos B. IL-17(+) γδ T Cells as Kick-Starters of Inflammation. Nat Immunol (2017) 18(6):604–11. doi: 10.1038/ni.3726
25. Sinkora M, Sinkorova J, Holtmeier W. Development of Gammadelta Thymocyte Subsets During Prenatal and Postnatal Ontogeny. Immunology (2005) 115(4):544–55. doi: 10.1111/j.1365-2567.2005.02194.x
26. Brotman RM, Ravel J, Bavoil PM, Gravitt PE, Ghanem KG. Microbiome, Sex Hormones, and Immune Responses in the Reproductive Tract: Challenges for Vaccine Development Against Sexually Transmitted Infections. Vaccine (2014) 32(14):1543–52. doi: 10.1016/j.vaccine.2013.10.010
27. Wessels JM, Felker AM, Dupont HA, Kaushic C. The Relationship Between Sex Hormones, the Vaginal Microbiome and Immunity in HIV-1 Susceptibility in Women. Dis Model Mech (2018) 11(9):dmm035147. doi: 10.1242/dmm.035147
28. Wira CR, Rodriguez-Garcia M, Patel MV. The Role of Sex Hormones in Immune Protection of the Female Reproductive Tract. Nat Rev Immunol (2015) 15(4):217–30. doi: 10.1038/nri3819
29. Mengesha BG, Conti HR. The Role of IL-17 in Protection Against Mucosal Candida Infections. J Fungi (Basel) (2017) 3(4):52. doi: 10.3390/jof3040052
30. Sobel JD, Faro S, Force RW, Foxman B, Ledger WJ, Nyirjesy PR, et al. Vulvovaginal Candidiasis: Epidemiologic, Diagnostic, and Therapeutic Considerations. Am J Obstet Gynecol Am J Obstet Gynecol (1998) 178(2):203–11. doi: 10.1016/s0002-9378(98)80001-x
31. Fidel PL Jr., Sobel JD. Immunopathogenesis of Recurrent Vulvovaginal Candidiasis. Clin Microbiol Rev (1996) 9(3):335–48. doi: 10.1128/cmr.9.3.335
32. Magliani W, Conti S, Cassone A, De Bernardis F, Polonelli L. New Immunotherapeutic Strategies to Control Vaginal Candidiasis. Trends Mol Med (2002) 8(3):121–6. doi: 10.1016/s1471-4914(01)02268-7
33. Naglik JR, Fidel PL Jr., Odds FC. Animal Models of Mucosal Candida Infection. FEMS Microbiol Lett (2008) 283(2):129–39. doi: 10.1111/j.1574-6968.2008.01160.x
34. Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, et al. Th17 Cells and IL-17 Receptor Signaling Are Essential for Mucosal Host Defense Against Oral Candidiasis. J Exp Med (2009) 206(2):299–311. doi: 10.1084/jem.20081463
35. Eyerich K, Foerster S, Rombold S, Seidl HP, Behrendt H, Hofmann H, et al. Patients With Chronic Mucocutaneous Candidiasis Exhibit Reduced Production of Th17-Associated Cytokines IL-17 and IL-22. J Invest Dermatol (2008) 128(11):2640–5. doi: 10.1038/jid.2008.139
36. Pirofski LA, Casadevall A. Rethinking T Cell Immunity in Oropharyngeal Candidiasis. J Exp Med (2009) 206(2):269–73. doi: 10.1084/jem.20090093
37. Rosentul DC, Delsing CE, Jaeger M, Plantinga TS, Oosting M, Costantini I, et al. Gene Polymorphisms in Pattern Recognition Receptors and Susceptibility to Idiopathic Recurrent Vulvovaginal Candidiasis. Front Microbiol (2014) 5:483. doi: 10.3389/fmicb.2014.00483
38. Ferwerda B, Ferwerda G, Plantinga TS, Willment JA, van Spriel AB, Venselaar H, et al. Human Dectin-1 Deficiency and Mucocutaneous Fungal Infections. N Engl J Med (2009) 361(18):1760–7. doi: 10.1056/NEJMoa0901053
39. Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of Interleukin-17A for Systemic Anti-Candida Albicans Host Defense in Mice. J Infect Dis (2004) 190(3):624–31. doi: 10.1086/422329
40. Dongari-Bagtzoglou A, Villar CC, Kashleva H. Candida Albicans-Infected Oral Epithelial Cells Augment the Anti-Fungal Activity of Human Neutrophils In Vitro. Med Mycol (2005) 43(6):545–9. doi: 10.1080/13693780500064557
41. Yano J, Kolls JK, Happel KI, Wormley F, Wozniak KL, Fidel PL Jr. The Acute Neutrophil Response Mediated by S100 Alarmins During Vaginal Candida Infections is Independent of the Th17-Pathway. PloS One (2012) 7(9):e46311. doi: 10.1371/journal.pone.0046311
42. Peters BM, Coleman BM, Willems HME, Barker KS, Aggor FEY, Cipolla E, et al. The Interleukin (IL) 17r/IL-22r Signaling Axis Is Dispensable for Vulvovaginal Candidiasis Regardless of Estrogen Status. J Infect Dis (2020) 221(9):1554–63. doi: 10.1093/infdis/jiz649
43. Pietrella D, Rachini A, Pines M, Pandey N, Mosci P, Bistoni F, et al. Th17 Cells and IL-17 in Protective Immunity to Vaginal Candidiasis. PloS One (2011) 6(7):e22770. doi: 10.1371/journal.pone.0022770
44. Fidel PL Jr. History and Update on Host Defense Against Vaginal Candidiasis. Am J Reprod Immunol (2007) 57(1):2–12. doi: 10.1111/j.1600-0897.2006.00450.x
45. Feinen B, Jerse AE, Gaffen SL, Russell MW. Critical Role of Th17 Responses in a Murine Model of Neisseria Gonorrhoeae Genital Infection. Mucosal Immunol (2010) 3(3):312–21. doi: 10.1038/mi.2009.139
46. Wi T, Lahra MM, Ndowa F, Bala M, Dillon JR, Ramon-Pardo P, et al. Antimicrobial Resistance in Neisseria Gonorrhoeae: Global Surveillance and a Call for International Collaborative Action. PloS Med (2017) 14(7):e1002344. doi: 10.1371/journal.pmed.1002344
47. Stevens JS, Criss AK. Pathogenesis of Neisseria Gonorrhoeae in the Female Reproductive Tract: Neutrophilic Host Response, Sustained Infection, and Clinical Sequelae. Curr Opin Hematol (2018) 25(1):13–21. doi: 10.1097/MOH.0000000000000394
48. Mayor MT, Roett MA, Uduhiri KA. Diagnosis and Management of Gonococcal Infections. Am Fam Phys (2012) 86(10):931–8.
49. Lewis DA, Latif AS, Ndowa F. WHO Global Strategy for the Prevention and Control of Sexually Transmitted Infections: Time for Action. Sex Transm Infect (2007) 83(7):508–9. doi: 10.1136/sti.2007.028142
50. Gagliardi MC, Starnino S, Teloni R, Mariotti S, Dal Conte I, Di Carlo A, et al. Circulating Levels of Interleukin-17A and Interleukin-23 are Increased in Patients With Gonococcal Infection. FEMS Immunol Med Microbiol (2011) 61(1):129–32. doi: 10.1111/j.1574-695X.2010.00759.x
51. Masson L, Salkinder AL, Olivier AJ, McKinnon LR, Gamieldien H, Mlisana K, et al. Relationship Between Female Genital Tract Infections, Mucosal Interleukin-17 Production and Local T Helper Type 17 Cells. Immunology (2015) 146(4):557–67. doi: 10.1111/imm.12527
52. Jerse AE. Experimental Gonococcal Genital Tract Infection and Opacity Protein Expression in Estradiol-Treated Mice. Infect Immun (1999) 67(11):5699–708. doi: 10.1128/IAI.67.11.5699-5708.1999
53. Song W, Condron S, Mocca BT, Veit SJ, Hill D, Abbas A, et al. Local and Humoral Immune Responses Against Primary and Repeat Neisseria Gonorrhoeae Genital Tract Infections of 17beta-Estradiol-Treated Mice. Vaccine (2008) 26(45):5741–51. doi: 10.1016/j.vaccine.2008.08.020
54. Feinen B, Russell MW. Contrasting Roles of IL-22 and IL-17 in Murine Genital Tract Infection by Neisseria Gonorrhoeae. Front Immunol (2012) 3:11. doi: 10.3389/fimmu.2012.00011
55. Packiam M, Veit SJ, Anderson DJ, Ingalls RR, Jerse AE. Mouse Strain-Dependent Differences in Susceptibility to Neisseria Gonorrhoeae Infection and Induction of Innate Immune Responses. Infect Immun (2010) 78(1):433–40. doi: 10.1128/IAI.00711-09
56. Hedges SR, Mayo MS, Mestecky J, Hook EW 3rd, Russell MW. Limited Local and Systemic Antibody Responses to Neisseria Gonorrhoeae During Uncomplicated Genital Infections. Infect Immun (1999) 67(8):3937–46. doi: 10.1128/iai.67.8.3937-3946.1999
57. Hedges SR, Sibley DA, Mayo MS, Hook EW 3rd, Russell MW. Cytokine and Antibody Responses in Women Infected With Neisseria Gonorrhoeae: Effects of Concomitant Infections. J Infect Dis (1998) 178(3):742–51. doi: 10.1086/515372
58. Liu Y, Hammer LA, Liu W, Hobbs MM, Zielke RA, Sikora AE, et al. Experimental Vaccine Induces Th1-Driven Immune Responses and Resistance to Neisseria Gonorrhoeae Infection in a Murine Model. Mucosal Immunol (2017) 10(6):1594–608. doi: 10.1038/mi.2017.11
59. Liu Y, Islam EA, Jarvis GA, Gray-Owen SD, Russell MW. Neisseria Gonorrhoeae Selectively Suppresses the Development of Th1 and Th2 Cells, and Enhances Th17 Cell Responses, Through TGF-β-Dependent Mechanisms. Mucosal Immunol (2012) 5(3):320–31. doi: 10.1038/mi.2012.12
60. Liu Y, Russell MW. Diversion of the Immune Response to Neisseria Gonorrhoeae From Th17 to Th1/Th2 by Treatment With Anti-Transforming Growth Factor Beta Antibody Generates Immunological Memory and Protective Immunity. mBio (2011) 2(3):e00095-11. doi: 10.1128/mBio.00095-11
61. Herring A, Ballard R, Mabey D, Peeling RW, Initiative WTSTDD. Evaluation of Rapid Diagnostic Tests: Chlamydia and Gonorrhoea. Nat Rev Microbiol (2006) 4(12 Suppl):S41–8. doi: 10.1038/nrmicro1562
62. Rowley J, Vander Hoorn S, Korenromp E, Low N, Unemo M, Abu-Raddad LJ, et al. Chlamydia, Gonorrhoea, Trichomoniasis and Syphilis: Global Prevalence and Incidence Estimates. Bull World Health Organ (2019) 97(8):548–62p. doi: 10.2471/blt.18.228486
63. Stamm WE. Chlamydia Trachomatis Infections: Progress and Problems. J Infect Dis (1999) 179 Suppl 2(Supplement_2):S380–3. doi: 10.1086/513844
64. Andrew DW, Cochrane M, Schripsema JH, Ramsey KH, Dando SJ, O'Meara CP, et al. The Duration of Chlamydia Muridarum Genital Tract Infection and Associated Chronic Pathological Changes are Reduced in IL-17 Knockout Mice But Protection is Not Increased Further by Immunization. PloS One (2013) 8(9):e76664. doi: 10.1371/journal.pone.0076664
65. Beagley KW, Timms P. Chlamydia Trachomatis Infection: Incidence, Health Costs and Prospects for Vaccine Development. J Reprod Immunol (2000) 48(1):47–68. doi: 10.1016/s0165-0378(00)00069-3
66. O'Meara CP, Andrew DW, Beagley KW. The Mouse Model of Chlamydia Genital Tract Infection: A Review of Infection, Disease, Immunity and Vaccine Development. Curr Mol Med (2014) 14(3):396–421. doi: 10.2174/15665240113136660078
67. Wira CR, Fahey JV, Rodriguez-Garcia M, Shen Z, Patel MV. Regulation of Mucosal Immunity in the Female Reproductive Tract: The Role of Sex Hormones in Immune Protection Against Sexually Transmitted Pathogens. Am J Reprod Immunol (2014) 72(2):236–58. doi: 10.1111/aji.12252
68. Dockterman J, Coers J. Immunopathogenesis of Genital Chlamydia Infection: Insights From Mouse Models. Pathog Dis (2021) 79(4):ftab012. doi: 10.1093/femspd/ftab012
69. Hafner L, Beagley K, Timms P. Chlamydia Trachomatis Infection: Host Immune Responses and Potential Vaccines. Mucosal Immunol (2008) 1(2):116–30. doi: 10.1038/mi.2007.19
70. Kinnunen A, Molander P, Laurila A, Rantala I, Morrison R, Lehtinen M, et al. Chlamydia Trachomatis Reactive T Lymphocytes From Upper Genital Tract Tissue Specimens. Hum Reprod (2000) 15(7):1484–9. doi: 10.1093/humrep/15.7.1484
71. Farris CM, Morrison RP. Vaccination Against Chlamydia Genital Infection Utilizing the Murine C. Muridarum Model. Infect Immun (2011) 79(3):986–96. doi: 10.1128/iai.00881-10
72. Morrison RP, Caldwell HD. Immunity to Murine Chlamydial Genital Infection. Infect Immun (2002) 70(6):2741–51. doi: 10.1128/IAI.70.6.2741-2751.2002
73. Lampe MF, Wilson CB, Bevan MJ, Starnbach MN. Gamma Interferon Production by Cytotoxic T Lymphocytes is Required for Resolution of Chlamydia Trachomatis Infection. Infect Immun (1998) 66(11):5457–61. doi: 10.1128/iai.66.11.5457-5461.1998
74. Li LX, McSorley SJ. B Cells Enhance Antigen-Specific CD4 T Cell Priming and Prevent Bacteria Dissemination Following Chlamydia Muridarum Genital Tract Infection. PloS Pathog (2013) 9(10):e1003707. doi: 10.1371/journal.ppat.1003707
75. Magee DM, Williams DM, Smith JG, Bleicker CA, Grubbs BG, Schachter J, et al. Role of CD8 T Cells in Primary Chlamydia Infection. Infect Immun (1995) 63(2):516–21. doi: 10.1128/iai.63.2.516-521.1995
76. Morrison SG, Morrison RP. A Predominant Role for Antibody in Acquired Immunity to Chlamydial Genital Tract Reinfection. J Immunol (2005) 175(11):7536–42. doi: 10.4049/jimmunol.175.11.7536
77. Ramsey KH, Soderberg LS, Rank RG. Resolution of Chlamydial Genital Infection in B-Cell-Deficient Mice and Immunity to Reinfection. Infect Immun (1988) 56(5):1320–5. doi: 10.1128/iai.56.5.1320-1325.1988
78. Su H, Feilzer K, Caldwell HD, Morrison RP. Chlamydia Trachomatis Genital Tract Infection of Antibody-Deficient Gene Knockout Mice. Infect Immun (1997) 65(6):1993–9. doi: 10.1128/iai.65.6.1993-1999.1997
79. Zhang X, Gao L, Lei L, Zhong Y, Dube P, Berton MT, et al. A MyD88-Dependent Early IL-17 Production Protects Mice Against Airway Infection With the Obligate Intracellular Pathogen Chlamydia Muridarum. J Immunol (2009) 183(2):1291–300. doi: 10.4049/jimmunol.0803075
80. Bai H, Gao X, Zhao L, Peng Y, Yang J, Qiao S, et al. Respective IL-17A Production by Gammadelta T and Th17 Cells and its Implication in Host Defense Against Chlamydial Lung Infection. Cell Mol Immunol (2017) 14(10):850–61. doi: 10.1038/cmi.2016.53
81. Scurlock AM, Frazer LC, Andrews CW Jr., O'Connell CM, Foote IP, Bailey SL, et al. Interleukin-17 Contributes to Generation of Th1 Immunity and Neutrophil Recruitment During Chlamydia Muridarum Genital Tract Infection But is Not Required for Macrophage Influx or Normal Resolution of Infection. Infect Immun (2011) 79(3):1349–62. doi: 10.1128/iai.00984-10
82. Frazer LC, Scurlock AM, Zurenski MA, Riley MM, Mintus M, Pociask DA, et al. IL-23 Induces IL-22 and IL-17 Production in Response to Chlamydia Muridarum Genital Tract Infection, But the Absence of These Cytokines Does Not Influence Disease Pathogenesis. Am J Reprod Immunol (2013) 70(6):472–84. doi: 10.1111/aji.12171
83. Darville T, Andrews CW Jr., Laffoon KK, Shymasani W, Kishen LR, Rank RG. Mouse Strain-Dependent Variation in the Course and Outcome of Chlamydial Genital Tract Infection is Associated With Differences in Host Response. Infect Immun (1997) 65(8):3065–73. doi: 10.1128/iai.65.8.3065-3073.1997
84. Darville T, O'Neill JM, Andrews CW Jr., Nagarajan UM, Stahl L, Ojcius DM. Toll-Like Receptor-2, But Not Toll-Like Receptor-4, is Essential for Development of Oviduct Pathology in Chlamydial Genital Tract Infection. J Immunol (2003) 171(11):6187–97. doi: 10.4049/jimmunol.171.11.6187
85. Ramsey KH, Sigar IM, Schripsema JH, Shaba N, Cohoon KP. Expression of Matrix Metalloproteinases Subsequent to Urogenital Chlamydia Muridarum Infection of Mice. Infect Immun (2005) 73(10):6962–73. doi: 10.1128/IAI.73.10.6962-6973.2005
86. Shah AA, Schripsema JH, Imtiaz MT, Sigar IM, Kasimos J, Matos PG, et al. Histopathologic Changes Related to Fibrotic Oviduct Occlusion After Genital Tract Infection of Mice With Chlamydia Muridarum. Sex Transm Dis (2005) 32(1):49–56. doi: 10.1097/01.olq.0000148299.14513.11
87. Wiesenfeld HC, Heine RP, Krohn MA, Hillier SL, Amortegui AA, Nicolazzo M, et al. Association Between Elevated Neutrophil Defensin Levels and Endometritis. J Infect Dis (2002) 186(6):792–7. doi: 10.1086/342417
88. Jha R, Srivastava P, Salhan S, Finckh A, Gabay C, Mittal A, et al. Spontaneous Secretion of Interleukin-17 and -22 by Human Cervical Cells in Chlamydia Trachomatis Infection. Microbes Infect (2011) 13(2):167–78. doi: 10.1016/j.micinf.2010.10.012
89. Haase AT. Early Events in Sexual Transmission of HIV and SIV and Opportunities for Interventions. Annu Rev Med (2011) 62:127–39. doi: 10.1146/annurev-med-080709-124959
90. World Health Organization HIV data and statistics 2020. Available at: https://www.who.int/hiv/data/en (Accessed 2022, Jan 14).
91. Hladik F, McElrath MJ. Setting the Stage: Host Invasion by HIV. Nat Rev Immunol (2008) 8(6):447–57. doi: 10.1038/nri2302
92. Zhang Z, Schuler T, Zupancic M, Wietgrefe S, Staskus KA, Reimann KA, et al. Sexual Transmission and Propagation of SIV and HIV in Resting and Activated CD4+ T Cells. Science (1999) 286(5443):1353–7. doi: 10.1126/science.286.5443.1353
93. Chege D, Chai Y, Huibner S, Kain T, Wachihi C, Kimani M, et al. Blunted IL17/IL22 and Pro-Inflammatory Cytokine Responses in the Genital Tract and Blood of HIV-Exposed, Seronegative Female Sex Workers in Kenya. PloS One (2012) 7(8):e43670. doi: 10.1371/journal.pone.0043670
94. Cicala C, Martinelli E, McNally JP, Goode DJ, Gopaul R, Hiatt J, et al. The Integrin Alpha4beta7 Forms a Complex With Cell-Surface CD4 and Defines a T-Cell Subset That is Highly Susceptible to Infection by HIV-1. Proc Natl Acad Sci USA (2009) 106(49):20877–82. doi: 10.1073/pnas.0911796106
95. Cecchinato V, Franchini G. Th17 Cells in Pathogenic Simian Immunodeficiency Virus Infection of Macaques. Curr Opin HIV AIDS (2010) 5(2):141–5. doi: 10.1097/COH.0b013e32833653ec
96. El Hed A, Khaitan A, Kozhaya L, Manel N, Daskalakis D, Borkowsky W, et al. Susceptibility of Human Th17 Cells to Human Immunodeficiency Virus and Their Perturbation During Infection. J Infect Dis (2010) 201(6):843–54. doi: 10.1086/651021
97. Gosselin A, Monteiro P, Chomont N, Diaz-Griffero F, Said EA, Fonseca S, et al. Peripheral Blood CCR4+CCR6+ and CXCR3+CCR6+CD4+ T Cells are Highly Permissive to HIV-1 Infection. J Immunol (2010) 184(3):1604–16. doi: 10.4049/jimmunol.0903058
98. McKinnon LR, Kaul R. Quality and Quantity: Mucosal CD4+ T Cells and HIV Susceptibility. Curr Opin HIV AIDS (2012) 7(2):195–202. doi: 10.1097/COH.0b013e3283504941
99. McKinnon LR, Nyanga B, Chege D, Izulla P, Kimani M, Huibner S, et al. Characterization of a Human Cervical CD4+ T Cell Subset Coexpressing Multiple Markers of HIV Susceptibility. J Immunol (2011) 187(11):6032–42. doi: 10.4049/jimmunol.1101836
100. Zhang ZQ, Wietgrefe SW, Li Q, Shore MD, Duan L, Reilly C, et al. Roles of Substrate Availability and Infection of Resting and Activated CD4+ T Cells in Transmission and Acute Simian Immunodeficiency Virus Infection. Proc Natl Acad Sci USA (2004) 101(15):5640–5. doi: 10.1073/pnas.0308425101
101. Rodriguez-Garcia M, Barr FD, Crist SG, Fahey JV, Wira CR. Phenotype and Susceptibility to HIV Infection of CD4+ Th17 Cells in the Human Female Reproductive Tract. Mucosal Immunol (2014) 7(6):1375–85. doi: 10.1038/mi.2014.26
102. Boily-Larouche G, Omollo K, Cheruiyot J, Njoki J, Kimani M, Kimani J, et al. CD161 Identifies Polyfunctional Th1/Th17 Cells in the Genital Mucosa That Are Depleted in HIV-Infected Female Sex Workers From Nairobi, Kenya. Sci Rep (2017) 7(1):11123. doi: 10.1038/s41598-017-11706-y
103. Stieh DJ, Matias E, Xu H, Fought AJ, Blanchard JL, Marx PA, et al. Th17 Cells Are Preferentially Infected Very Early After Vaginal Transmission of SIV in Macaques. Cell Host Microbe (2016) 19(4):529–40. doi: 10.1016/j.chom.2016.03.005
104. Estes JD, Harris LD, Klatt NR, Tabb B, Pittaluga S, Paiardini M, et al. Damaged Intestinal Epithelial Integrity Linked to Microbial Translocation in Pathogenic Simian Immunodeficiency Virus Infections. PloS Pathog (2010) 6(8):e1001052. doi: 10.1371/journal.ppat.1001052
105. Nazli A, Chan O, Dobson-Belaire WN, Ouellet M, Tremblay MJ, Gray-Owen SD, et al. Exposure to HIV-1 Directly Impairs Mucosal Epithelial Barrier Integrity Allowing Microbial Translocation. PloS Pathog (2010) 6(4):e1000852. doi: 10.1371/journal.ppat.1000852
106. Burgener A, McGowan I, Klatt NR. HIV and Mucosal Barrier Interactions: Consequences for Transmission and Pathogenesis. Curr Opin Immunol (2015) 36:22–30. doi: 10.1016/j.coi.2015.06.004
107. Dandekar S, George MD, Bäumler AJ. Th17 Cells, HIV and the Gut Mucosal Barrier. Curr Opin HIV AIDS (2010) 5(2):173–8. doi: 10.1097/COH.0b013e328335eda3
108. Kim CJ, Nazli A, Rojas OL, Chege D, Alidina Z, Huibner S, et al. A Role for Mucosal IL-22 Production and Th22 Cells in HIV-Associated Mucosal Immunopathogenesis. Mucosal Immunol (2012) 5(6):670–80. doi: 10.1038/mi.2012.72
109. Klatt NR, Estes JD, Sun X, Ortiz AM, Barber JS, Harris LD, et al. Loss of Mucosal CD103+ DCs and IL-17+ and IL-22+ Lymphocytes Is Associated With Mucosal Damage in SIV Infection. Mucosal Immunol (2012) 5(6):646–57. doi: 10.1038/mi.2012.38
110. Looker KJ, Magaret AS, Turner KME, Vickerman P, Gottlieb SL, Newman LM. Global Estimates of Prevalent and Incident Herpes Simplex Virus Type 2 Infections in 2012. PloS One (2015) 10(1):e114989. doi: 10.1371/journal.pone.0114989
111. Cunningham AL, Diefenbach RJ, Miranda-Saksena M, Bosnjak L, Kim M, Jones C, et al. The Cycle of Human Herpes Simplex Virus Infection: Virus Transport and Immune Control. J Infect Dis (2006) 194 Suppl 1(Supplement_1):S11–8. doi: 10.1086/505359
112. Kimberlin DW, Rouse DJ. Clinical Practice. Genital Herpes. N Engl J Med (2004) 350(19):1970–7. doi: 10.1056/NEJMcp023065
113. Freeman EE, Weiss HA, Glynn JR, Cross PL, Whitworth JA, Hayes RJ. Herpes Simplex Virus 2 Infection Increases HIV Acquisition in Men and Women: Systematic Review and Meta-Analysis of Longitudinal Studies. AIDS (2006) 20(1):73–83. doi: 10.1097/01.aids.0000198081.09337.a7
114. Looker KJ, Garnett GP, Schmid GP. An Estimate of the Global Prevalence and Incidence of Herpes Simplex Virus Type 2 Infection. Bull World Health Organ (2008) 86(10):805–12. doi: 10.2471/blt.07.046128
115. Dobbs ME, Strasser JE, Chu CF, Chalk C, Milligan GN. Clearance of Herpes Simplex Virus Type 2 by CD8+ T Cells Requires Gamma Interferon and Either Perforin- or Fas-Mediated Cytolytic Mechanisms. J Virol (2005) 79(23):14546–54. doi: 10.1128/jvi.79.23.14546-14554.2005
116. Harandi AM, Svennerholm B, Holmgren J, Eriksson K. Differential Roles of B Cells and IFN-Gamma-Secreting CD4(+) T Cells in Innate and Adaptive Immune Control of Genital Herpes Simplex Virus Type 2 Infection in Mice. J Gen Virol (2001) 82(Pt 4):845–53. doi: 10.1099/0022-1317-82-4-845
117. Parr MB, Parr EL. Vaginal Immunity in the HSV-2 Mouse Model. Int Rev Immunol (2003) 22(1):43–63. doi: 10.1080/08830180305228
118. Anipindi VC, Bagri P, Roth K, Dizzell SE, Nguyen PV, Shaler CR, et al. Estradiol Enhances CD4+ T-Cell Anti-Viral Immunity by Priming Vaginal DCs to Induce Th17 Responses via an IL-1-Dependent Pathway. PloS Pathog (2016) 12(5):e1005589. doi: 10.1371/journal.ppat.1005589
119. Bagri P, Anipindi VC, Nguyen PV, Vitali D, Stampfli MR, Kaushic C. Novel Role for Interleukin-17 in Enhancing Type 1 Helper T Cell Immunity in the Female Genital Tract Following Mucosal Herpes Simplex Virus 2 Vaccination. J Virol (2017) 91(23):e01234-17. doi: 10.1128/JVI.01234-17
120. Bagri P, Ghasemi R, McGrath JJC, Thayaparan D, Yu E, Brooks AG, et al. Estradiol Enhances Antiviral CD4(+) Tissue-Resident Memory T Cell Responses Following Mucosal Herpes Simplex Virus 2 Vaccination Through an IL-17-Mediated Pathway. J Virol (2020) 95(1):e01206-20. doi: 10.1128/jvi.01206-20
121. Do JS, Visperas A, Dong C, Baldwin WM 3rd, Min B. Cutting Edge: Generation of Colitogenic Th17 CD4 T Cells is Enhanced by IL-17+ γδ T Cells. J Immunol (2011) 186(8):4546–50. doi: 10.4049/jimmunol.1004021
122. Do JS, Visperas A, O'Brien RL, Min B. CD4 T Cells Play Important Roles in Maintaining IL-17-Producing γδ T-Cell Subsets in Naive Animals. Immunol Cell Biol (2012) 90(4):396–403. doi: 10.1038/icb.2011.50
123. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 Induce Innate IL-17 Production From Gammadelta T Cells, Amplifying Th17 Responses and Autoimmunity. Immunity (2009) 31(2):331–41. doi: 10.1016/j.immuni.2009.08.001
124. Anipindi VC, Bagri P, Dizzell SE, Jiménez-Saiz R, Jordana M, Snider DP, et al. IL-17 Production by γδ(+) T Cells Is Critical for Inducing T(h)17 Responses in the Female Genital Tract and Regulated by Estradiol and Microbiota. Immunohorizons (2019) 3(7):317–30. doi: 10.4049/immunohorizons.1900040
Keywords: IL-17, FRT, infection, fungal, bacterial, HIV, HSV, mucosal
Citation: Bagri P, Anipindi VC and Kaushic C (2022) The Role of IL-17 During Infections in the Female Reproductive Tract. Front. Immunol. 13:861444. doi: 10.3389/fimmu.2022.861444
Received: 24 January 2022; Accepted: 22 March 2022;
Published: 14 April 2022.
Edited by:
Carmen Fernández, Stockholm University, SwedenReviewed by:
Thomas Tsutomu Murooka, University of Manitoba, CanadaCopyright © 2022 Bagri, Anipindi and Kaushic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charu Kaushic, a2F1c2hpY0BtY21hc3Rlci5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.