 Christian Gerecke
Christian Gerecke Caue Egea Rodrigues
Caue Egea Rodrigues Burkhard Kleuser
Burkhard Kleuser
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 21 March 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.861351
This article is part of the Research TopicEpigenetics of the Immune Component of InflammationView all 43 articles
Ten-eleven translocation proteins (TET1-3) are dioxygenases that oxidize 5-methyldeoxycytosine, thus taking part in passive and active demethylation. TETs have shown to be involved in immune cell development, affecting from self-renewal of stem cells and lineage commitment to terminal differentiation. In fact, dysfunction of TET proteins have been vastly associated with both myeloid and lymphoid leukemias. Recently, there has been accumulating evidence suggesting that TETs regulate immune cell function during innate and adaptive immune responses, thereby modulating inflammation. In this work, we pursue to review the current and recent evidence on the mechanistic aspects by which TETs regulate immune cell maturation and function. We will also discuss the complex interplay of TET expression and activity by several factors to modulate a multitude of inflammatory processes. Thus, modulating TET enzymes could be a novel pharmacological approach to target inflammation-related diseases and myeloid and lymphoid leukemias, when their activity is dysregulated.
Epigenetic modifications of DNA are essential for control of gene expression in cells. The 5´-methylation of cytosine in CpG-dinucleotides is one of the best-studied and most frequently observed epigenetic regulation element in mammalian cells. It plays a pivotal role in the establishment, maintenance, and persistence of gene expression patterns, contributing to nearly all cellular processes (1). Therefore, gene expression, which is modulated by epigenetic modifications, is placed by specific enzymes (“writers”), and recognized by effector proteins (“readers”). However, most of epigenetic marks are reversible, and various enzymes (“erasers”) remove these marks (2, 3). DNA methylation on the fifth carbon of cytosine in the CpG dinucleotide is carried out by “writer” DNA-methyltransferases (DNMTs), including DNMT1, DNMT3A, DNMT3B (whereas DNMT2 is considered a RNA-methyltransferase (4, 5), and is involved in different biological roles of various genomic regions (6). The DNA methyltransferases (DNMTs) convert cytidine to 5-methyl-2’-deoxycytidine (5-mdC) by transfer of a methyl group from S-adenyl methionine (SAM), which usually occurs on sites where a guanine nucleotide follows cytosine (CpG sites) in the DNA sequence (7). DNMT1 maintains the methylation pattern in DNA by binding to newly synthesized DNA and methylating it, copying the original methylation pattern. In contrast, DNMT3A and DNMT3B introduce methyl groups to DNA de novo. The maintenance of a balance between DNA hypomethylation and hypermethylation is crucial for physiological processes in the cell. Genome-wide DNA hypomethylation is associated with chromosomal instability, while loci-specific aberrant DNA hypermethylation in CpG islands of gene promoter regions can lead to gene silencing (8). This may result in facilitated tumorigenesis and cancer progression.
In the genome, 5-mdC are unequally distributed throughout the genome because the modified cytosine itself is mutagenic. It can undergo spontaneous hydrolytic deamination to cause C → T transitions, which in turn leads to severe DNA sequence changes (1). Nevertheless, approximately 70 - 80 % of CpGs are in a methylated status and therefore associated with transcriptional repression (9). However, in gene promoters CpGs are frequently unmethylated in order to control the transcriptional activity of the respective gene. Mostly, CpGs are clustered in CpG islands with a high content of cytosine and guanine base pairs (9). It has become clear that aberrant patterns of DNA methylation can lead to severe alterations in gene expression and be a fundamental part of carcinogenesis. Therefore, loss of normal DNA methylation occurs in DNA repetitive elements like silenced transposable elements (1). In contrast, aberrant DNA hypermethylation can be considered as a crucial event for the initiation and progression of cancer (10). This, in turn, can facilitate tumorigenesis and cancer progression due to decreased expression of e.g. tumor suppressor genes and disruption of cell cycle regulation, apoptosis, and/or DNA repair.
Once considered as non-reversible, recent technological advancements enabled great improvement in knowledge of DNA methylation dynamics and the possibilities of DNA demethylation processes (6). Although DNA demethylation via DNA replication-dependent passive dilution was known for several decades, the replication-independent active demethylation through the activity of the “eraser” enzymes ten-eleven translocation (TET) dioxygenases (TET1, TET2 and TET3) has just recently been described (11, 12). The TET enzyme family belongs to the α-ketoglutarate (αKG) dependent dioxygenases (αKGD) superfamily (synonymous to αKG is 2-oxoglutarate). In mammals, TETs catalyze the oxidation of 5-mdC to 5-hydroxymethyl-2’-deoxycytidine (5-hmdC), 5−formyl-2’-deoxycytidine (5-fdC) and 5−carboxyl-2’-deoxycytidine (5-cadC), facilitating replication-independent DNA demethylation via thymine DNA glycosylase (TDG) base excision repair of 5-fdC and 5-cadC (11, 12). The reaction mechanism is presented in Figure 1. However, more than a mere intermediate of DNA demethylation, 5-hmdC is a stable epigenetic modification distributed in a cell-, and tissue-type specific pattern and specially enriched at enhancers and gene bodies (13, 14). The TET-mediated conversion of 5-mdC to 5-hmdC at promoters, and subsequent DNA demethylation appears to be highly associated with transcriptional gene reactivation in several contexts (15, 16). As TET proteins and their products, i.e., oxidized cytosine bases, are involved in the maintenance of several biological processes, there has been accumulating evidence that they play a complex role in the regulation of inflammatory responses.
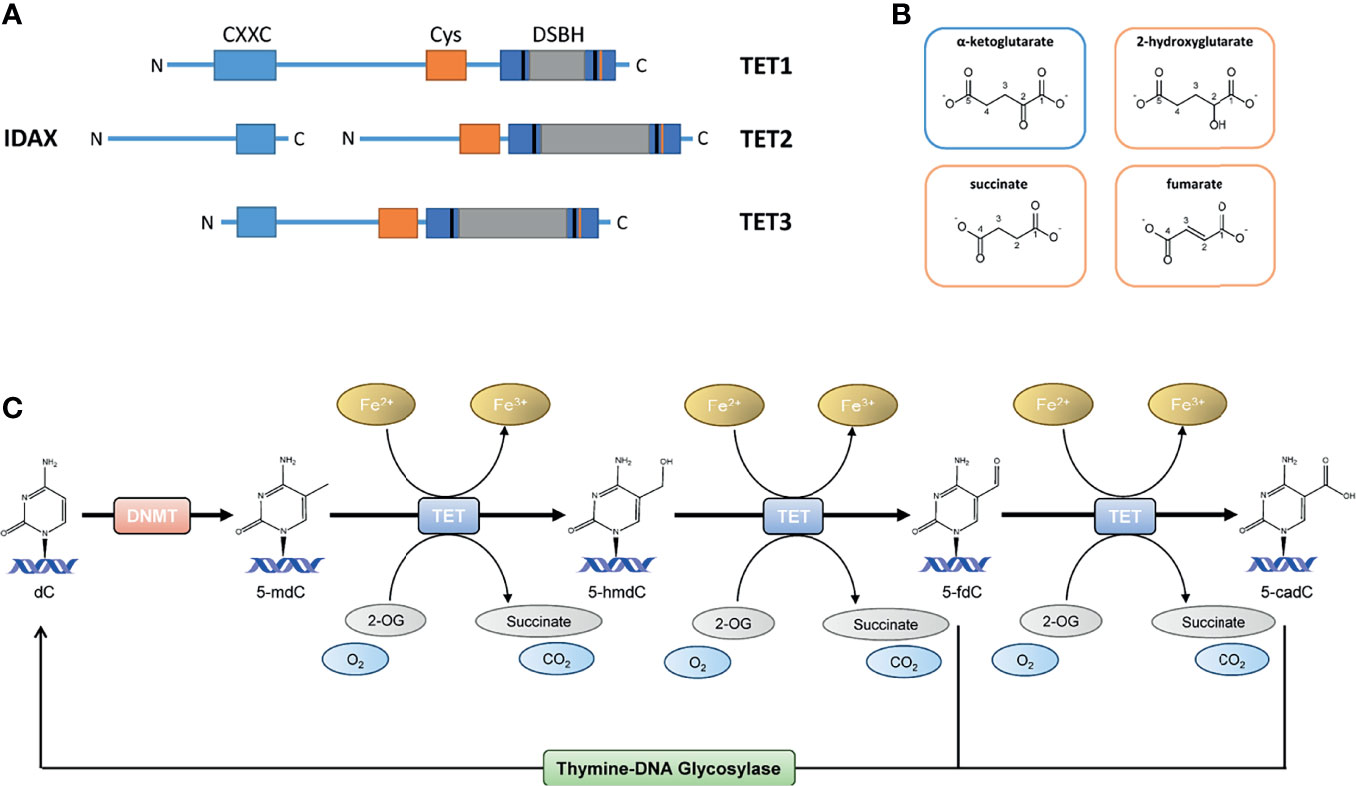
Figure 1 (A) The TET protein structures; full-length TET1 consists of 2039 aa, TET2 of 1921 aa and TET3 of 1803 aa. TET1, TET2 and TET3 have a C-terminal catalytic domain and contains a cysteine-rich domain and a double-stranded β-helix (DSBH) domain. A large low-complexity insert is found within the DSHB domain that may have regulatory roles via post-translational modifications. The N-terminal parts of TET1 and TET3 contain a DNA-binding CXXC domain. TET2 is lacking the CXXC domain, which CXXC finger protein 4 (IDAX) provides. (B) Structural similarities between the TET cosubstrate α-ketoglutarate (αKG) and the TET inhibiting molecules 2-hydroxyglutarate (2-HG), succinate and fumarate. (C) The TET enzyme family belongs to the αKG dependent dioxygenases (αKGD) superfamily. In mammals, TETs catalyze the oxidation of 5-methyl-2’-deoxycytidine (5-mdC) to 5-hydroxymethyl-2’-deoxycytidine (5-hmdC), 5−formyl-2’-deoxycytidine (5-fdC) and 5−carboxyl-2’-deoxycytidine (5-cadC), facilitating replication-independent DNA demethylation via thymine DNA glycosylase (TDG) base excision repair of 5-fdC and 5-cadC.
The TET enzymes are differentially expressed in several tissues during development and can regulate several conserved signaling pathways, which involve important transcription factors as Wingless (WNT), Notch, Sonic Hedgehog (SHH) and Transforming Growth Factor Beta (TGFB). Thus, the normal function of TET enzymes is fundamental for normal embryonic development and TET deficiency in animal models has shown to delay cell differentiation and result in dysregulated expression of genes involved in these signaling pathways. Consequently, the absence of TETs results in central nervous system defects and retinal deformity (17–22). Furthermore, TET deficiency plays a pivotal role in the initiation and progression of several malignancies and other aberrations.
In this review, we focus on highlighting the current understanding and emerging concepts in the mechanisms through which TET proteins and their products modulate inflammation in immune and non-immune cells, including also relevant aspects of the regulation of myeloid and lymphoid immune cell development, differentiation, and function. Finally, we present future potential perspectives of how these findings could pave the way to prevent and treat inflammation-related diseases. We outline current understanding of the roles of TET proteins in regulating adaptive and innate immune system and the crucial role in epigenetic modulation by promoting DNA demethylation, and producing oxidized products that affects these cells lineage and function. Additionally, we summarize how aberrant DNA-methylation plays a key role in proper hematopoietic stem and progenitor cells (HSPC) self-renewal and lineage differentiation dysregulation and can lead to aberrant stem cell function and cellular transformation.
Members of TET dioxygenase family are capable of oxidizing methylated 2’-deoxycytidines to 5-hmdC, 5-fdC and 5-cadC, which are eventually replaced by unmodified 2’-deoxycytidines because of TDG-mediated base excision repair (23–25). TET proteins belong to αKGD, employing Fe(II) as metal cofactor and αKG as cosubstrate (26, 27). The αKGD family has approximately 70 members in mammals. In addition to TETs, there exist numerous histone lysine demethylases, prolyl 4-hydroxylases that modify the hypoxia-inducible factor (HIF-P4Hs) or collagens (collagen P4Hs), the hypoxia-inducible factor asparagine hydroxylase FIH (factor inhibiting HIF) and FTO (fat mass and obesity-associated protein), the first identified RNA demethylase (15). The αKGDs, including TET enzymes, share the same reaction mechanism and cofactors, but their substrates vary from DNA to RNA, proteins and fatty acids. Besides Fe(II) and αKG, αKGDs require molecular oxygen (28, 29). The cofactors are coordinated at the active site by conserved residues, Fe(II) by two histidines and an aspartate and αKG by a positively charged arginine in TETs (Figure 2). The catalytic domains possess a double-stranded β-helix structure known as a jellyroll. Following cofactor and substrate binding, the molecular oxygen oxidizes Fe(II), inducing substrate oxidation and decarboxylation of succinate and CO2 (Figure 1C). The human TETs are large proteins, full-length TET1 being composed of 2039 amino acids (aa), TET2 of 1921 aa and TET3 of 1803 aa (23). The aminoterminal parts of TET1 and TET3 contain a DNA-binding CXXC domain (23), whereas this is lacking in TET2 but mediated by CXXC finger protein 4 (IDAX) (30). The catalytic domains are in the C-termini and contain a cysteine-rich domain and a double-stranded β-helix domain (Figure 1A). A large low-complexity insert is found within the double-stranded β-helix domain that may have regulatory roles via post-translational modifications. The human TETs are widely expressed, while experimental data suggest that TET1 is preferentially expressed in embryonic stem cells (ESCs) whereas TET2 and TET3 are expressed in many tissues and have overlapping expression profiles (23). The catalytic activity of the TETs is strongly dependent on Fe(II) and αKG (15, 29). From a therapeutic point of view, a limited number of known TET enzyme activators can significantly increase genome-wide 5-hmdC levels. Vitamin C is one of the best-known substrates of the TET enzymes as well as a potent antioxidant and reducing agent (31). It has been proposed that vitamin C is responsible for the restoration of TET enzyme catalytic activity through the reduction of Fe(III) to Fe(II) (32, 33). This hypothesis has, however, been contradicted by other studies in which other strong reducing agents were unable to enhance the TET-mediated hydroxylation of 5-mdC (34–36). The mechanism by which vitamin C contributes to increased levels of TET-mediated hydroxylation of 5-mdC is, therefore, presently unclear. Additionally, retinol is proved to increase TET2 expression in naïve ESCs (37). Besides, TET enzymatic activity is strongly dependent on the availability of reaction cofactors, including Fe(II), O2 and αKG (15). However, an activity suppression can occur via product inhibition of the αKG-metabolite succinate (Figure 1B). In this context, it is of great interest that although the oncometabolite 2-hydroxyglutarate (2-HG) has structural similarities to αKG, unlike this natural substrate, it inhibits TET enzymatic activity (Figure 1B) (15, 38–42). Indeed, the oncometabolite 2-HG is produced due to mutations of the isocitrate dehydrogenases genes IDH1 and IDH2, which play a central role in the citric cycle. The resulted dysfunctional enzymes exhibit a neomorphic gain of function, which leads to a decreased amount of the essential TET cosubstrate αKG as it is irreversibly converted to the oncometabolite 2-HG. Certainly, a specific CpG island methylator phenotype in gliomas and colorectal cancers that is characterized by DNA hypermethylation has been associated with IDH mutations, which has led to several speculations that IDH mutations contribute to tumorigenesis via altered epigenetic regulation (43–45).
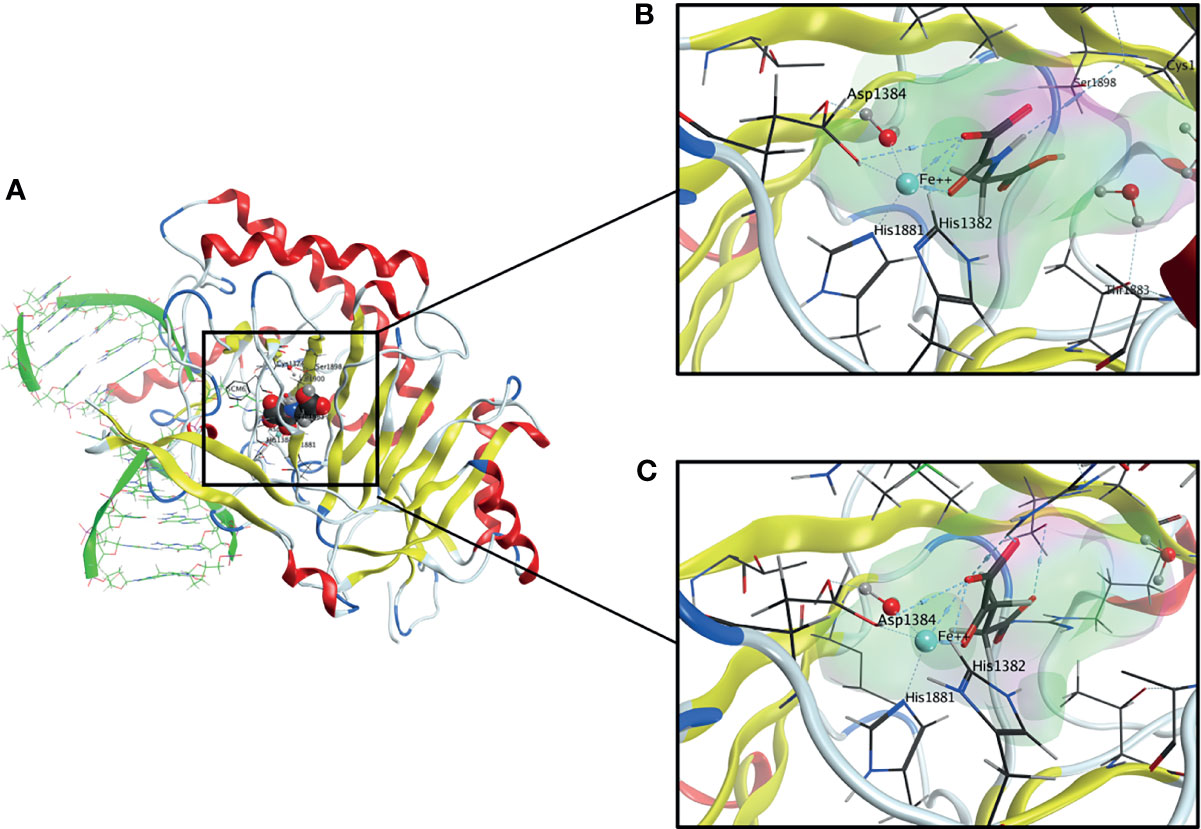
Figure 2 (A) Overall Structure of TET2-DNA Complex generated from pdb: 4NM6. The active site contains a highly conserved 2-His-1-carboxylate-amino acid residue triad motif in which the catalytically essential Fe(II) is fixated by two histidine residues and an aspartic acid residue. A water molecule is also an essential part of the complex. (B) The active site of TET2 containing N-Oxalylglycine (NOG). (C) The active site of TET2 containing the cosubstrate α-ketoglutarate (αKG).
Aberrant DNA hypermethylation has been associated with several types of cancer, including gliomas, acute myeloid leukemia (AML) and cancers of the lung, breast, ovaries, and colon (46–48). Additionally, a genome-wide decrease in 5-hmdC levels is considered an epigenetic hallmark in many cancers (29, 49) and several studies have highlighted the diagnostic and prognostic value of this mechanism. In hematological malignancies, TET1 was first identified as a gene fused to MLL (mixed-lineage leukemia) in an AML patient with a ten–eleven translocation (50); however, this translocation does not occur very frequently (51). Loss of function mutations in genes encoding TET enzymes occur frequently in hematopoietic malignancies, but rarely in solid tumors, which instead commonly have reduced enzymatic activity (Figure 3). The impairment of their expression and activity as defined by reduced 5-hmdC levels may be caused by other factors. Thus, in glioma, TET2 mutations have not been described; however, TET2 promoter methylation has been detected in 14% of low-grade glioma patients without IDH mutation (52, 53). Since promoter methylation is associated with transcriptional repression, this suggests a decreased TET2 expression in these patients, possibly leading to decreased levels of 5-hmdC. In addition, Müller et al. (54) showed a strong correlation between nuclear exclusion of TET1 and decreased levels in glioma. In their study, 61% of the tumor samples had decreased 5-hmdC levels. Of the 5-hmdC negative tumors, 70% showed nuclear exclusion of TET1, or no detectable TET1 protein, thereby demonstrating an additional mechanism that may lead to decreased 5-hmdC in glioma.
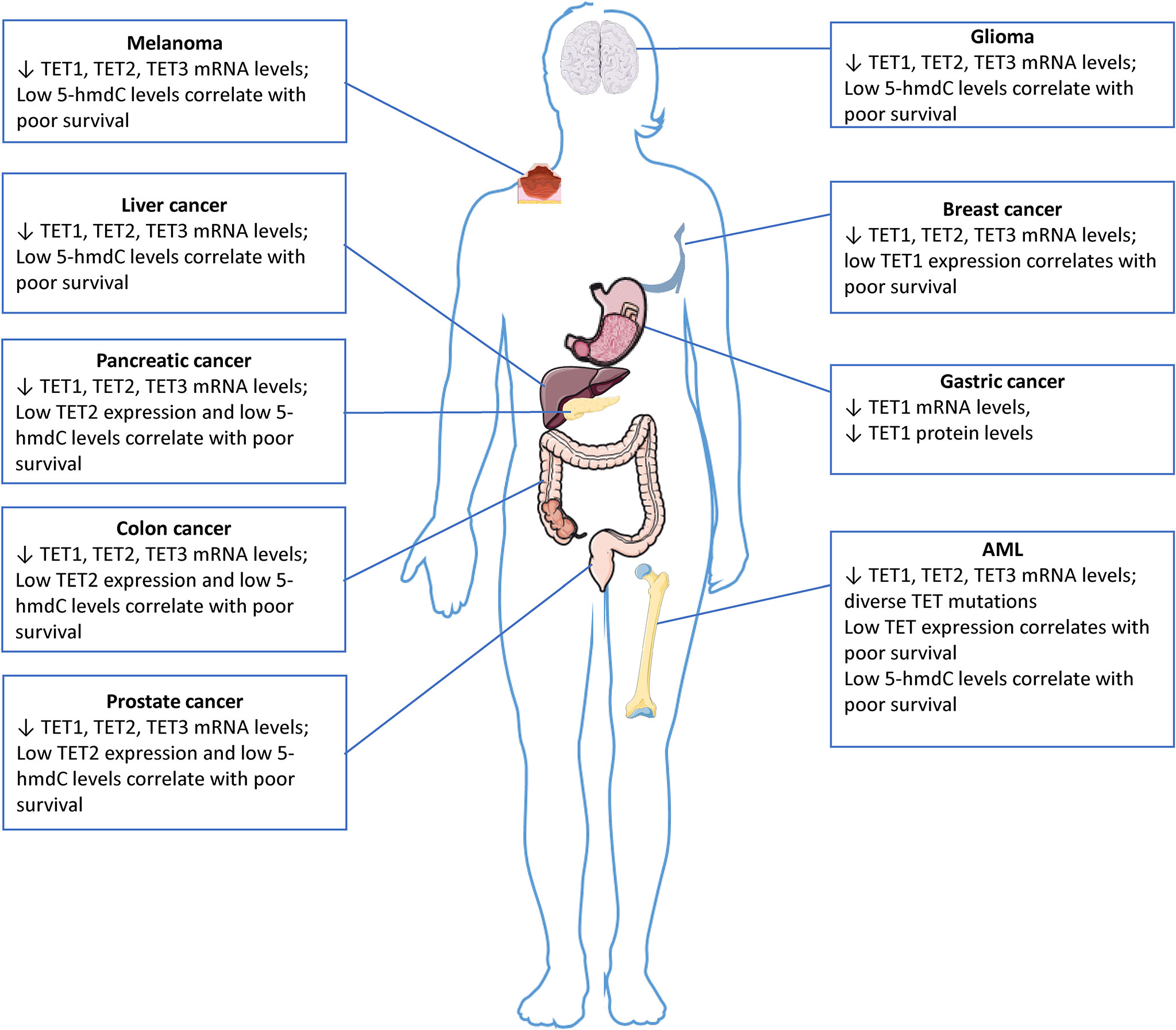
Figure 3 5-hmdC levels and TET expression in solid tumors and leukemia. The downregulation of TET gene expression, which is often associated with reduced 5-hmC levels, has been observed in numerous solid cancers. TET genes are rarely mutated in solid tumors, the impairment of their expression and activity as defined by reduced 5-hmC levels may be caused by other factors. The images were provided and adapted from Servier Medical Art (smart.servier.com).
TET proteins may also be post-transcriptionally down-regulated by microRNAs (miRNA) (55, 56). The miRNA miR-29 inhibits the translation of TET1 and TDG in lung cancer cells, in human dermal fibroblasts and vascular smooth muscle cells miR-29 inhibits formation of all TET isoforms resulting in effective 5-hmdC decrease (57, 58). Moreover, miR-26 and TET or TDG levels are inversely correlated during pancreatic progenitor cell differentiation (59). Additionally, in breast cancer the TET family is a target of miR-22. Here, it promotes invasiveness and metastasis by DNA methylation-dependent silencing of miR-200 through the direct targeting of TET proteins (56). Moreover, miR-22 is overexpressed and TET2 and miR-200 are downregulated in patients with gastric cancer, and the aberrant expression of both correlates with poor survival (60). Interestingly, in embryonal kidney cells the ancestor of the CXXC domain for TET2, IDAX, may stimulate caspase-dependent TET2 cleavage after targeting it to DNA (30). The interaction of TET proteins with OGT (O6-Guanine transferase) enables catalytic-independent gene expression regulation (61). Due to the common alteration of OGT in many cancers, it may affect the TET activity on oxidizing DNA, as well (62). Additionally, various TET-interacting proteins, such as EZH2 (Enhancer of zeste homolog 2), SIN3A (SIN3 transcription regulator family member A), the hematopoietic transcription factor SPI1 and EBF1 (B cell factor 1), may modify the actions of TET proteins; SPI1 (involved in the differentiation of B cells and myeloid lineages) is often deregulated in leukemia and potentially influences mRNA splicing (63). Another study found that TET2 mediates the IFN-γ/JAK/STAT signaling pathway to control chemokine and PD-L1 (Programmed cell death-ligand 1) expression, lymphocyte infiltration, and cancer immunity (64). Generally, reduced TET activity was associated with decreased Th1-type chemokines and tumor-infiltrating lymphocytes and the progression of human colon cancer. Deletion of TET2 in murine melanoma and colon tumor cells reduced chemokine expression and tumor-infiltrating lymphocytes, enabling tumors to evade antitumor immunity and to resist anti–PD-L1 therapy. Conversely, systematic injection of the TET activating compound vitamin C increased chemokines and tumor-infiltrating lymphocytes, leading to enhanced antitumor immunity and anti–PD-L1 efficacy and extended lifespan of tumor-bearing mice. It has been suggested, that TET activity could serve as a biomarker for predicting the efficacy and patient response to anti–PD-1/PD-L1 therapy. Additionally, stimulation of TET activity could serve as an adjuvant immunotherapy of solid tumors.
Especially hematological malignancies are often linked to loss of function mutations in TET genes (65), which leads to impaired cell differentiation and transformation (66, 67). Furthermore, four genes that play a role in the citric acid cycle, namely IDH1, IDH2, SDH (succinate dehydrogenase) and FH (fumarate hydratase), are frequently mutated in leukemias and various types of solid cancers (38, 68, 69). These genes are able to affect the activity of the TET proteins by changing the levels of metabolites that compete with the TET cofactor αKG. In humans, TET2 is one of the most frequently mutated genes in hematopoietic cancers of both myeloid and lymphoid origin (66). Human and murine hematopoietic progenitor and mature immune cells show a high TET2 expression and high levels of 5-hmdC (70). TET2 has been extensively shown to play an important role in the regulation of hematopoietic stem cell expansion, differentiation, and function (71). Loss of function TET2 mutations are often described as the first step in the multi-hit model of development of leukemia (72). Even before the discovery of the TET enzymatic function, deletions and somatic mutations in TET2 were identified across multiple exons in different types of blood cancers in humans (72). In myeloid leukemias, e.g., in ∼30% of cases of secondary AML, ∼17% of de novo AML, ∼30% of myelodysplastic syndromes (MDS), ∼50% of chronic myelomonocytic leukemias (CMMLs), and ∼20% of myeloproliferative neoplasms (MNP) show a mutated TET2. Additionally, TET2 is also found to be mutated in certain lymphoid leukemias, e.g., ∼17% of T-cell acute lymphoblastic leukemia (T-ALL), and in certain lymphomas, e.g., ∼33% of angioimmunoblastic T lymphomas, and in ∼12% of diffuse large B-cell lymphomas (73–75). TET2 mutations are shown to increase with ageing, being observed in peripheral blood cells of ∼5% - 10% of adults older than 65 years of age, and are one of the most commonly detected alterations in clonal hematopoiesis of indeterminate potential (CHIP). CHIP is a result from TET2-mutant hematopoietic stem cell proliferative advantage and is considered a preleukemic condition that accounts as a potential driver of myeloid dysfunction and development of inflammatory diseases such as type 2 diabetes, coronary heart disease, and ischemic stroke (76). Data from a recent meta-analysis has shown that in spite of the fact that TET2 mutations had no significant prognostic value on myelodysplastic syndromes, the response rates to hypomethylating agents were significantly different between patients with and without TET2 mutations (77). Both TET2-knockout murine and TET2-mutant human hematopoietic stem cells display resistance to colony-suppressive effects of TNF-α, therefore sustaining proliferative advantage in an inflammatory environment (78). In summary, the role of TET enzymes in the development of cancer has been examined and extensively proven (16, 66, 79) (Figure 3).
The cells of the innate immune system are often recognized as the first line of host defense against pathogens and injury, being fundamental not only for the initiation but also for the resolution of inflammatory processes. Here, myeloid cells, including macrophages, dendritic cells, neutrophils, mast cells and, with the involvement of epithelial and endothelial cells as well, are identified as important players in the recognition and response to potentially harmful external and internal stimuli. This system recognizes pathogens via a limited number of germline-encoded pattern-recognition receptors (PRRs), including toll-like receptors (TLRs), RIG-I-like receptors (RLRs) and the NOD-like receptors (NLRs), which initiate complex intracellular responses upon stimulation (80–85). All TET proteins (TET1-3) have been implicated in the differentiation and/or function of myeloid cells, regulating key aspects related to inflammation, which is illustrated in Figure 4.
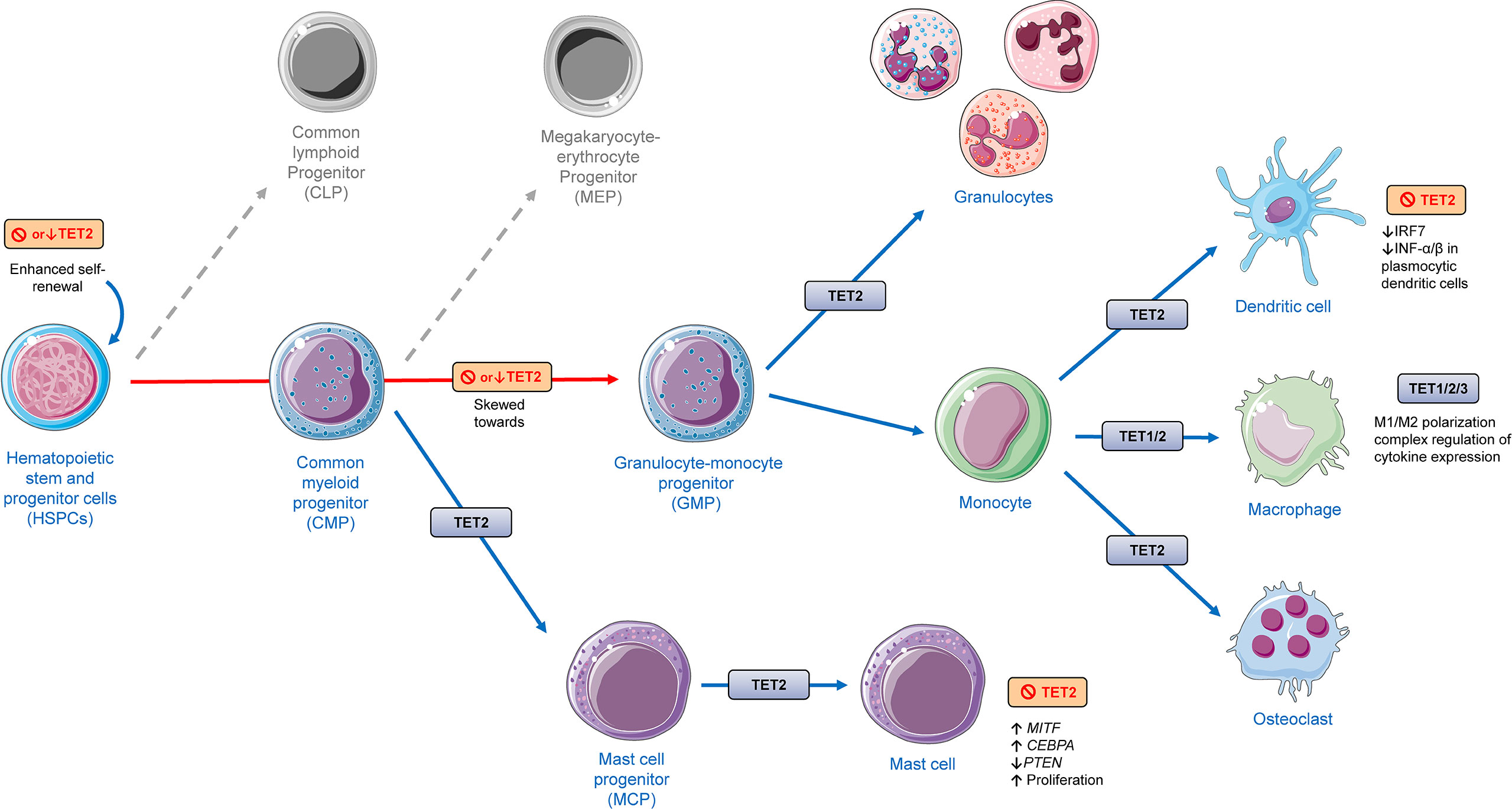
Figure 4 The role of TET proteins in myeloid cell lineage differentiation and function based on findings from different models presented in this review, including human and murine cells and animal experiments applying rodents. TET2 deletion has shown to promote increased hematopoietic stem and progenitor cells (HSPCs). TET2 has shown to be involved in different aspects of the differentiation of myeloid cells. Deletion or depletion of TET2 was shown to skew differentiation of HSPCs towards monocytic/granulocytic lineages. TET2 regulates the function of monocytic populations such as dendritic cells, macrophages, and osteoclasts. TET1/2 have been shown to play a role in monocyte to macrophage differentiation. All TET members (TET1/2/3) have been shown to have an effect in the function of macrophages, affecting their activation, polarization (M1/M2) and cytokine expression. TET2 was shown to regulate mast cell differentiation and function in both a catalytic-dependent and -independent manner. The cellular images were provided and adapted from Servier Medical Art (smart.servier.com).
TET1 has been suggested to play a role in the differentiation from monocytes to macrophages and to the activation of proinflammatory phenotype of macrophages. In the human monocytic leukemia cell line THP-1, TET1 mRNA is increased during macrophage differentiation induced by macrophage colony-stimulating factor (M-CSF). Moreover, 5-hmdC levels were shown to be enhanced on global levels and specifically in the TNF-α promoter during the differentiation of monocytes to macrophages. Importantly, CRISPR stable knockout of TET1 led to decreased expression of TNF-α and other pro-inflammatory cytokine genes, suggesting that TET1 is an important activator of the TNF-α gene in macrophages (86). In line with these findings, TET1 knockdown leads to inhibition of M1 macrophage polarization through the NF-κB (nuclear factor kappa B) signaling pathway. LPS/IFN-γ-stimulated macrophages were shown to display significant decrease in the production of proinflammatory markers such as IL-6, TNF-α, CCL2, and HLA-DR, when TET1 was down regulated (87). In a recently published study, TET1 has been shown to display a striking specificity in the regulation of gene expression in macrophages related to cell migration and trafficking. In LPS-stimulated macrophages, TET1 deletion repressed the expression of CXCL1 (C-X-C Motif Chemokine Ligand 1), a chemokine that regulates cell migration and recruitment, playing a role in neutrophil influx during lung inflammation, for example (88).
In primary murine bone-marrow-derived macrophages, TET3 was shown to inhibit type I IFN production after poly(I:C) stimulation or viral infection (89). Thus, this study showed that deletion of TET3 in macrophages elicited enhanced antiviral responses. Mechanistically, TET3 suppressed IFN-β production through the recruitment of the histone deacetylase 1 (HDAC1) to the IFNB1 promoter, a mechanism that is independent from its 5-mdC oxidative catalytic activity (89). Non-classical monocytes comprise around 2 - 11% of circulating monocytes and have distinguished expression of surface markers, transcriptomic and metabolic profiles in comparison to classical monocytes with complex functions in homeostasis control and in the pathophysiology of chronic inflammatory diseases (90, 91). Recently, TET3 has been shown to be involved in the control on the regulation of the repartition between subsets of classical and non-classical monocytes. In fact, TET3 knockout mice were shown to display an increased number of non-classical monocytes. In this context, it is of interest that non-classical monocyte numbers are significantly decreased in CMML patients (92).
There is an accumulating body of evidence that loss or depletion of TET2 leads to alterations in differentiation and function of different types of cells of myeloid lineage and their progenitors. Short hairpin RNA (shRNA)-mediated depletion of TET2 in murine bone marrow HSPCs has shown to skew their differentiation towards monocyte/macrophage lineages (93). Similarly, results from another working group shown that their TET2-knockout generated mouse model displayed increased Lin−Sca-1+c-Kit+ (LSK) cell pool with increased hematopoietic repopulating capacity and altered cell differentiation skewing toward monocytic/granulocytic lineages in an in vitro competitive reconstitution assay (94). Evidence from in vivo and in vitro experiments suggest that although TET2 loss does not affect markers of terminal macrophage differentiation, TET2 plays an important role in macrophage functional polarization (95). TET2 gene expression in murine macrophages has been shown to be induced by LPS treatment, an effect that was abolished by pretreatment with BAY 11–7082, an NF-κB inhibitor. Interestingly, here it was shown that unstimulated TET2–/– murine macrophages displayed increased gene expression of multiple proinflammatory cytokines and chemokines, while LPS-stimulated TET2–/– macrophages demonstrated impaired ability to resolve inflammation, expressing increased mRNA levels of IL1B, IL6, and ARG1 (Arginase 1) at later stages of LPS stimulation (95). Similar findings were observed with murine and human TET2–/– dendritic cells and macrophages. Furthermore, animal experiments demonstrated that TET2-deficient mice were more susceptible to colitis and to endotoxin shock in comparison to wild-type mice, displaying a more severe inflammatory phenotype and severe tissue damage (96). Mechanistic investigation revealed that TET2 interacts with IκBζ, a specific transcription factor for IL6, to promote IL6 transcription repression during inflammation resolution through the subsequent recruitment of HDAC2 (Histone Deacetylase 2) to the IL6 promoter, a mechanism that is independent from TET2 catalytic activity (96).
Evidence on the transcription repression of IL1B in macrophages mediated by TET2 via HDAC–mediated histone deacetylation of IL1B promoter has also been shown. Beyond its effect on the transcription of IL1B, TET2 deficiency in LPS/IFN-γ/ATP–treated macrophages was further shown to enhance the priming of NLRP3 inflammasome, promoting increased cleavage of pro–IL-1B to its active form. Moreover, results from animal experiments from this working group have revealed that TET2 loss of function in macrophages led to increased proatherogenic activity, suggesting a pivotal role of TET2-deficient macrophages in the acceleration of atherosclerosis that is associated with expansion of TET2-deficient HSPCs (97). In another study, TET2-deficiency in hematopoietic cells by both, specific ablation in myeloid cells and by partial reconstitution of the bone marrow with TET2-deficient HSPCs, led to exacerbated IL-1B, resulting in cardiac dysfunction with worse late-stage cardiac remodeling in two experimental murine models of heart failure (98). Consistently, findings from both studies have shown that the treatment with MCC950, a specific NLRP3 inflammasome inhibitor, was able abrogate the exacerbated IL-1B secretion, promoting an atheroprotective effect and hindering the development of heart failure in these murine models for clonal hematopoiesis associated with TET2 somatic mutations (97, 98).
Plasmacytoid dendritic cells express endosomal sensors TLR7/9 and have a massive capacity of producing INF, playing a fundamental role in both innate and adaptive immune responses against DNA and RNA viral infections. In order to elicit INF-α/β production, these cells activate upon stimulation important transcription factors such as IRF7 (interferon regulatory factor 7), NFκB and AP-1. TET2 was shown to be recruited by the zinc finger CXXC family epigenetic regulator CXXC5 to the CpG island containing promoter of IRF7, regulating its hypomethylation and thereby increasing its gene expression. In comparison to controls, TET deficient dendritic cells displayed decreased capacity to express IRF7. These findings suggest that along with CXXC5, TET2 is an important proinflammatory antiviral defense regulator of dendritic cells (99).
Recently, TET2 has also shown to be post-transcriptionally regulated during inflammatory processes affecting cytokine expression through macrophage activation via feedback regulatory mechanisms mediated by miRNAs (100, 101). Using different knockout and myeloid cell-specific transgenic mouse models, it has been shown that in LPS-activated bone-marrow derived macrophages, the miR-let-7a/let-7d/let-7f cluster (let-7adf) promotes IL-6 secretion by two different mechanisms. In the direct mechanism, let-7adf targets specifically TET2, thereby decreasing its expression at mRNA and protein levels. Indirectly, let-7adf has been suggested to regulate the Lin28a/SDHA axis in these LPS-activated macrophages (100). This miRNA cluster directly degradates TET2 mRNA and increases succinate levels, which inhibits TET enzymatic activity, as described above (102, 103).
Further, miR-125a-5p expression is upregulated in vascular endothelial cells treated with oxidized low-density lipoprotein. It targets the TET2 3′-untranslated region and so downregulates TET2 expression in a concentration-dependent manner. In direct consequence, these cells displayed decreased levels of 5-hmdC, mitochondrial dysfunction, increased reactive oxygen species, activation of NF-κB, NLRP3 inflammasome activation, increased release of IL-1B and IL-18 and cell death through pyroptosis (101). Interestingly, miR-125a-5p expression is enhanced in LPS-stimulated murine bone marrow-derived macrophages, suppressing the expression of the M1 phenotype while promoting the expression of the M2 phenotype (104). In contrast, another study has shown that LPS/IFN-γ-stimulated tumour-associated macrophages with increased expression of miR-125a-5p led to promotion of M1 polarization by targeting the factor FIH1 (inhibiting hypoxia-inducible factor-1 α) and inhibition of M2 polarization through targeting interferon regulatory factor 4 (IRF4) (105). Due to these contradictory results, further research needs to unveil possible mechanistic links between miR-125a-5p and TET2 to coordinate macrophage polarization.
TET2 has also shown to regulate mast cell differentiation, proliferation, and function (106–108) (Figure 4). TET2 loss of function in mast cells affects the 5-hmdC deposition and alters cytokine production. Cell culture experiments have shown that TET2−/− murine bone marrow progenitor cells displayed delayed differentiation to mast cells, while TET2−/− mast cells displayed altered 5-hmdC patterns, disrupted gene expression, decreased percentage of cells expressing IL-6, TNF-α, and IL-13 upon IgE and antigen stimulation, and marked increased proliferation. Interestingly, although vitamin C was able to enhance 5-hmdC levels and partially rescue the differential gene expression in TET2−/− mast cells, suggesting existing compensatory mechanisms, proliferation differences between TET2−/− and TET2+/+ cells remained unaffected (107). In another study, in comparison to controls, TET2–/– mice display increased numbers of immature promastocytes in the peritoneal cavity. RNA-sequencing analysis on bone marrow-derived mast cells derived from TET2–/– mice revealed transcriptional repression of genes required for mast cell differentiation, maturation, and function. TET2–/– mast cells displayed marked upregulation of genes involved in in inflammatory response, migration, growth, proliferation, and antiapoptotic mechanisms, whereas genes involved in negative regulation of cell proliferation and phosphorylation were downregulated. Importantly, TET2–/– mast cells display dysregulated expression of transcription factors, including MITF (Melanocyte Inducing Transcription Factor) and CEBPα (CCAAT/enhancer-binding protein alpha), and show reduced PTEN (Phosphatase and tensin homolog) expression because of PTEN promoter hypermethylation and hyperactivation of the PI3K/AKT/c-Myc pathway. Interestingly, PTEN expression and maturation was rescued in TET2–/– mast cells treated with the FDA-approved hypomethylating agent 5-azacytidine, whereas the co-treatment of these cells with 5-azacytidine and vitamin C led to a complete restoration of mast cell maturation and correction of hyperproliferation (106).
In mRNA oxidation-dependent manner, TET2 was shown to promote pathogen infection-induced myelopoiesis through ADAR1 (Adenosine deaminase acting on RNA)-mediated repression of SOCS3 (suppressor of cytokine signaling) expression at the post-transcription level. In mice experiments, TET2 regulated both, abdominal sepsis-induced emergency myelopoiesis and parasite-induced mast cell expansion. In comparison to controls, a TET2-deficient mice model of abdominal sepsis with acute mobilization and expansion of myeloid cells have shown to be protected from sepsis with lower mortality rates. Interestingly, while control mice developed neutrophilia and inflammatory monocytosis with increased serum levels of inflammatory cytokines (e.g., TNF-α, keratinocyte chemoattractant, macrophage inflammatory protein-1α), neutrophil and monocyte numbers were barely altered in TET2-deficient mice (108).
B cells and plasma cells belong classically to cells of the adaptative immune system and play an important role in acute and chronic inflammatory processes, being involved in pathophysiological mechanisms of several inflammatory and autoimmune diseases due to their ability to drive or suppress inflammation (109–111). TET enzymes affect different aspects of B cell development, differentiation, and function, which is presented in Figure 5. Recent sequencing approaches of different human B cell subpopulations revealed intense alterations of the epigenomic landscape during B cell differentiation, characterized by progressive demethylation resulting in hypomethylation at later stages (112). While TET1 has been identified as a tumor suppressor of B cell lymphoma and its loss in HSPCs promotes differentiation bias towards B cell lymphopoiesis, its expression was shown to be decreased during B lineage commitment (113). The mRNA levels of TET2 and TET3, on the other hand, were observed to progressively increase during B cell development (114). In mice, TET2 and TET3 conditional knockout at early stages of B cell development has been shown to largely impede lineage-specific programmed demethylation events (115). Interestingly, TET2 and TET3 were shown to cooperate with B linage-specific transcription factors to promote 5-mdC oxidation and demethylation, thereby improving chromatin accessibility at B cell enhancers. Loss of both, TET2 and TET3 in mice has shown to lead to blockade of pro- to pre-B cell transition in the bone marrow, decreased IRF4 expression at mRNA and protein levels and impaired the germline transcription and rearrangement of the Ig light chain (Igκ) locus (116).
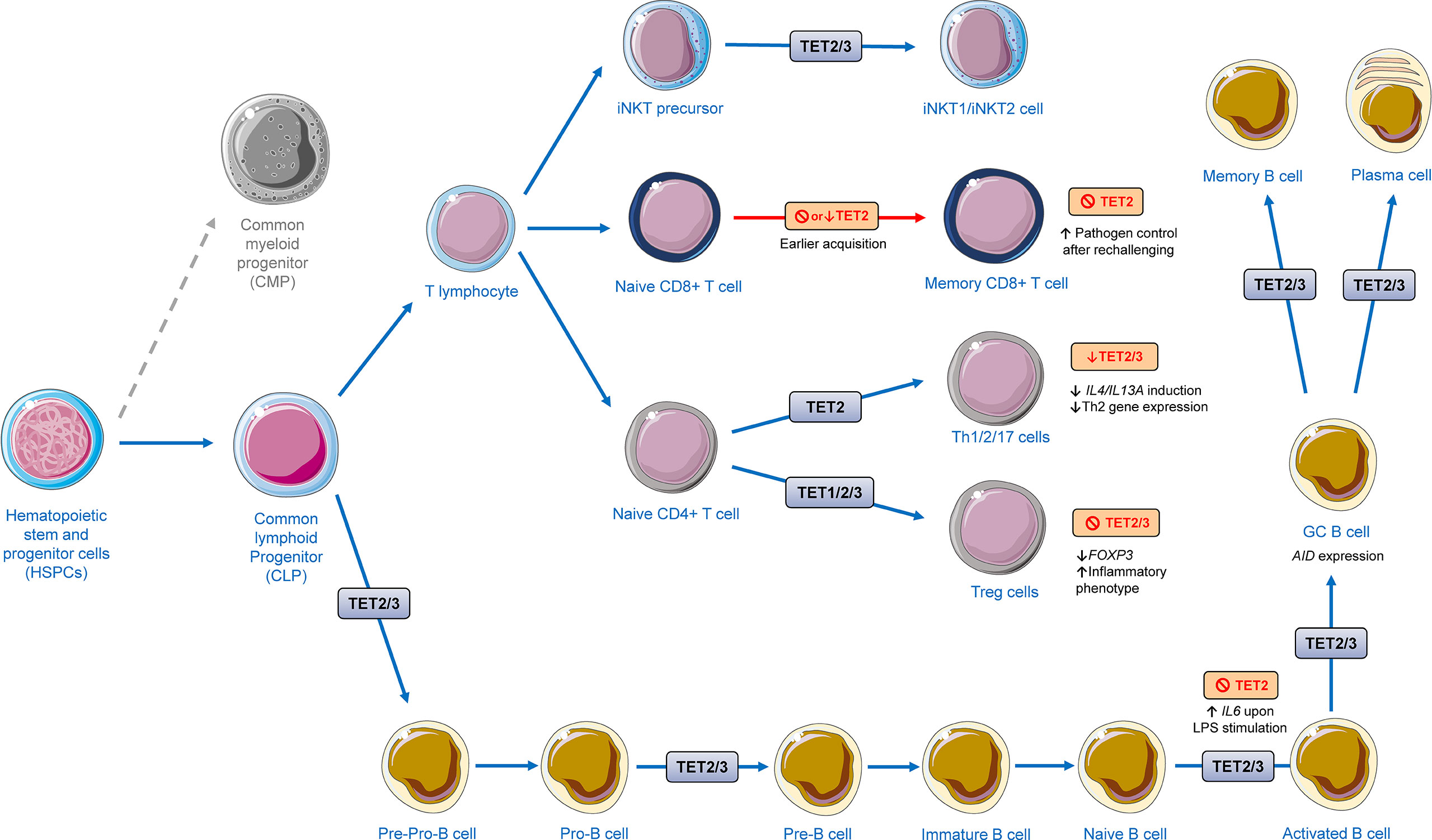
Figure 5 Regulation of lymphoid development and function by TET proteins. The presented scheme summarizes data from in vitro and murine models highlighted in this review. TET proteins have a great impact on the development of different lymphoid cells. TET2/3 were shown to regulate invariant natural killer T (iNKT) cells maturation and development and differentiation into iNKT1 and iNKT2. Ablation or depletion of TET2 has been shown to lead to early acquisition of memory CD8+ T cell without disrupting effector function after acute viral infection. TET2 has shown to play a role in differentiation of T helper (Th) cells. Knockdown of TET2/3 gives rise to the decreased responses of IL-4/13A induction against exogenous soluble antigen stimulation, leading to a restrained expression of Th2-related genes. TET1/2/3 regulate the stability of the regulatory T cells (Treg cells). TET2/3 deletion leads to decreased FOXP3 expression and a shift to an inflammatory phenotype. During B cell differentiation, TET2 and TET3 orchestrate B cell maturation and function. The cellular images were provided and adapted from Servier Medical Art (smart.servier.com).
Antigenic activation of B cells results in the formation of dynamic germinal centers (GC). Here, B cells clonally expand and undergo activation induced cytidine deaminase (AID)-induced hypermutations that are essential for shaping antibody affinity and diversity. Upon GC exit, B cells differentiate into antibody-secreting plasma cells and memory B cells (117). Both TET2 and TET3 have shown to play a role in antibody class switch recombination (118). In mice experiments, TET2 deletion alone was enough to disrupt transit of B cells through GCs, leading to GC hyperplasia, impairment of class switch recombination, and blockade of plasma cell differentiation. Remarkably, in a cohort of 128 patients with diffuse large B-cell lymphomas, TET2 loss of function mutations was found in ∼12% of the cases (119). In TET2 knockout mice, AID-induced demethylation was impaired and an aberrant DNA hypermethylation of regulatory elements was observed with potential disturbance on the binding of important transcription factors, such as transcription factors involved in exit from the GC reaction and B cell receptor, antigen presentation, and CD40 pathways. Importantly, hypermethylation signatures of murine GC B cells were found to be significantly reflected in primary cells from TET2 mutant patients with large B-cell lymphomas (120). It has been shown that TET is required for optimal AID expression, and both TET2 and TET3 were shown to guide the GC exit of B cells to antibody secreting plasma cells (114). Mechanistically, TET2 and TET3 enhance the expression of AID by depositing 5-hmdC and facilitating DNA demethylation at TET2-responsive enhancer elements located within the AID superenhancer, thereby maintaining chromatin accessibility and promoting gene expression (121).
TET2-deficient B cells were shown to express higher level of IL-6 but not TNF-α in response to LPS stimulation. These findings further demonstrate the general characteristics of TET2 in repressing IL6 transcription in B cells and indicate that TET2 may exert its broad repression of proinflammatory genes in different types of immune cells (96). TET2 and TET3 have also been implicated in the regulation of key aspects to prevent autoimmune inflammatory processes. The expression of the co-stimulatory molecule CD86 was shown to be upregulated on TET2- and TET3-deficient B cells, which displayed a permissive chromatin state at the CD86 locus as a result of decreased accumulation of HDACs. Dysregulated CD86 expression plays a role to the induction of autoimmune inflammatory processes, contributing significantly to aberrant activation of T and B cells in vitro and in vivo (122). Further research of how TETs regulate inflammation and autoimmune-related processes in B and plasma cells might reveal novel potential pharmacological targets and strategies to modulate autoimmune and chronic inflammatory diseases.
T cells, cells that are generally seem as members of the adaptive immune system, have shown to play a prominent role in acute and chronic inflammatory processes (123, 124). Genome-wide mapping of 5-hmdC levels in T cells has revealed dynamic changes during sequential steps of lineage commitment in the thymus and the periphery. During the process of T-cell development and differentiation, with 5-hmdC enrichment in gene bodies of certain genes of developing T cells being strongly positive correlated with respective gene expression (125). TET2 and TET3 are expressed at high levels in thymocytes and peripheral T cells, and are responsible for the majority of 5-hmdC modifications in these sets of T cells (Figure 5) (126–129). It has been shown, that the deletion of both, TET2 and TET3, in murine T cells caused a massive lymphoproliferative phenotype with enlarged spleen and lymph nodes. The TET2/TET3 double knockout mice did not live longer than 8 weeks due to decreased thymic cellularity, lower number of CD4+/CD8+ double positive cells, and an increased percentage of CD4+ and CD8+ single positive cells, including phenotypes reminiscent of thymic atrophy induced by stress or inflammation (130).
Depending on several signals, naïve CD4+ T cells can differentiate into multiple lineages, including T helper cells like, Th1, Th2, Th17, follicular T helper cells (Tfh), and regulatory T cells (Treg). These differentiated cells are characterized into these five major subsets based on their expression of signature cytokines and lineage-specific master transcription factors and play a critical role in coordinating actions of other immune and non-immune cells during inflammatory processes (131). CD4+ T cells homeostasis is important for healthy immune responses, but also plays a role in the pathogenesis of immune-mediated inflammatory diseases such as multiple sclerosis (MS), psoriasis, and asthma (132). TET2 has been shown to be more expressed in TCR (T-cell receptor)-activated Th cell subsets in comparison to TET1 and TET3. TET2 recruitment to the signature cytokine loci was dependent on lineage-specific transcription factors, regulating differentiation of Th1 and Th17. Under Th1 cell polarization in vitro, TET2−/− T cells displayed a marked decreased expression of IFN-γ at mRNA and protein levels, whereas TET2−/− Th17 cells displayed a decrease in the expression of IL-17 on mRNA and protein levels in comparison to controls. Using a mice model of myelin oligodendrocyte glycoprotein peptide-induced experimental autoimmune encephalomyelitis, TET2 expression has been shown to play a role in suppression of disease severity by regulating the expression of IL10, IL17, and IFNG in Th cell subsets (133). Importantly, IL10 expression is considered to be generally an anti-inflammatory cytokine that plays a fundamental role in preventing inflammatory and autoimmune diseases (134).
Treg cells are known for their anti-inflammatory properties, promoting tissue repair, and suppressing effector T cell proliferation (135–138). Under specific conditions, however, these cells may lose suppressive properties, releasing proinflammatory cytokines and consequently contributing to inflammation-related diseases such as in certain autoimmune diseases (139). The transcription factor Forkhead box P3 (FOXP3) is crucial for Treg cells and its expression is imperative and sufficient for their suppressive activity. A decrease of FOXP3 expression can shift the function of Treg cells, leading to expression of proinflammatory phenotypes with increased secretion IFN-γ and IL-17 (140). TET proteins have been shown to mediate the increase of 5-hmdC and loss of 5-mdC in Treg cell–specific hypomethylated regions, including conserved noncoding sequence elements (CNS) 1 and 2, intronic cis-regulatory elements in the FOXP3 locus. Moreover, TET2/TET3 deficient mice displayed markedly compromised stability of FOXP3 expression. Vitamin C potentiates TET activity thereby promoting FOXP3 expression stability (141). Treg cell specific demethylation region (TSDR) demethylation such as the demethylation of CNS2 has been shown to be required for a Treg immuno-suppressive phenotype induction in experiments that applied CRISPR-dCas9-TET1-mediated targeted DNA demethylation technology (142).. During thymic Treg development, IL-2 has been shown to be required to maintain increased expression levels of TET2, whereas TET2 downregulation was shown to prevent Treg cell specific demethylation region (TSDR) (143). In contrast, IL-6 has been shown to induce the expression of pro-inflammatory cytokines in Foxp3+ Treg cells. IL-6 signaling has been shown to induce Dnmt3a expression and activation, leading to DNA methylation of CNS2 and Foxp3 downregulation (144). Decreased TET2 and TET3 was shown to compromise physiological Treg function in animal experiments in which mice lacking TET2 and TET3 in Treg cells develop inflammatory disease with splenomegaly and leukocyte infiltration into the lung. Treg cells from these mice displayed dysregulation of Treg signature genes while also upregulating genes involved in cell cycle, DNA damage, and cancer, acquiring ultimately an inflammatory phenotype (145). Recently, miR142-3p has been shown to target TET2, impairing Treg cell differentiation and stability in models of type 1 diabetes, contributing to immune activation and progression during islet autoimmunity. Thus, inhibition of miR142-3p improved islet autoimmunity in animal experiments (146).
CD8+ T cells play an important role in the defense against pathogens and are also potentially important in defense against cancers (147). TET2 also regulates CD8+ T cell differentiation and function. Concomitant IL-12 and TCR-mediated stimulation of human naïve CD8+ T cells leads to TET2-mediated demethylation of the IFNG- promoter eliciting an increase in IFN-γ expression (148). Interestingly, in a murine model of acute viral infection, TET2 loss led to early acquisition of a memory CD8+ T cells without disrupting antigen-driven cell expansion or effector function after acute viral infection, with TET2-deficient memory CD8+ T cells displaying superior pathogen control after rechallenging. TET2 loss in these cells elicited altered DNA methylation patterns with the majority of differently methylated regions located in introns and coding sequences of genes involved in cellular growth, proliferation, development, death, and survival (149). Furthermore, concomitant deletion of TET2 and TET3 in mouse CD4+/CD8+ double-positive thymocytes have also shown to dysregulate development and proliferation of invariant natural killer T cells, suggesting that TET2 and TET3 carry proper development and maturation of these cells by suppressing aberrant proliferation mediated by TCR (130).
Altogether, there is data that demonstrates that TETs play an important role in different subset populations of T lymphocytes, affecting their differentiation and function. Future target research to understand mechanisms by which TETs regulate function of these cells during inflammatory processes in inflammation-related diseases might lead to the finding of new venues for inflammation-related disease prevention and treatment.
Neuroinflammation is an important defense mechanism of the central nervous system against pathogenic and or infectious insults that has also been identified as a common etiopathogenic factor involved in several central nervous system disorders, such as MS, depression, ischemic brain injury, Alzheimer’s disease, and Parkinson’s disease, Amyotrophic Lateral Sclerosis, and Huntington’s disease (150). Microglial cells, astrocytes, oligodendrocytes, infiltrating myeloid cells are the main reactive cellular components involved in neuroinflammation (151). Microglia are the resident macrophages of the central nervous system, playing a fundamental role in the immune responses in the brain thereby preserving the integrity of neuronal circuits. Remarkably, microglia can exhibit different phenotypes upon different stimuli (152). For example, microglia are classically activated in response to LPS or exposure to pro-inflammatory cytokines (e.g., IFN-γ, TNF-α), displaying a M1 proinflammatory phenotype (153), whereas in response to IL-4 exposure, microglia acquire complex antiinflammatory properties, altering their expression of cytokines, surface markers, phagocytosis capacity, and displaying enhanced potential to induce proliferation of T cells with a regulatory signature (154). Several epigenetic mechanisms, including histone modifications, DNA methylation, and expression of non-coding RNAs (ncRNAs) have been shown to modulate the alteration of microglia phenotypes, allowing these cells to display the ability to adapt and respond to the microenvironment and signal activation (155).
TET2 has been observed to be upregulated in microglia cells upon TLR-mediated inflammatory stimuli through a NF-κB-dependent pathway. During inflammatory responses, TET2 was shown to alter transcription of important genes, leading to cellular metabolic reprogramming and expression of different inflammatory mediators. Moreover, in mice models for neuroinflammation and in samples from Alzheimer’s disease patients, TET2 expression was also shown to be increased. Collectively, these findings suggest that TET2 is involved in the inflammatory responses in microglia in vitro and in vivo (156). Contrastingly, TET2 depletion has also been associated with increased development of neuroinflammation in Alzheimer’s disease. A mouse model for Alzheimer’s disease indicated TET2 depletion resulted in increased amyloid-β plaque accumulation, microglia overgrowth and proinflammatory cytokine accumulation (e.g., IL-6, IL-1B, and TNF-α) (157).
TET2 has also been suggested to play a role in microglia proinflammatory activation in Parkinson’s disease. By applying a mice model of inflammation-mediated nigral neurodegeneration, it has been demonstrated that TET2 loss was accompanied by protection of nigral dopaminergic neurons. Thus, TET2 inactivation fully prevents nigral dopaminergic neuronal loss induced by previous inflammation. This is of interest as patients with Parkinson´s disease exhibit an epigenetic and transcriptional upregulation of TET2 (158).
Although astrocytes are essential for protection of neurons, there is growing evidence that astrocytes might also contribute to neuroinflammation when exposed to signals from damaged neurons, proinflammatory microglia and/or damaging insults (e.g., aggregated proteins, and environmental toxicants). Reactive astrocytes acquire the capacity to produce pro-inflammatory cytokines, such as IL-1B, and TNF-α, and ROS. DNA methylation and demethylation have been implicated in the differentiation and function of astrocytes (159, 160). For instance, vitamin C was shown to promote astrocyte differentiation by promoting TET-mediated DNA hydroxymethylation on astrocyte-specific genes (161). The activation of specific pathways including NF-κB and JAK/STAT pathways play an important role in the conversion of reactive astrocytes and in reactive astrogliosis (162).
As DNA demethylation of genes in the JAK-STAT pathway have shown to be important for enhanced activation of STATs for astrocyte differentiation (163) and TETs were shown to affect and be affected by the NF-κB pathway in other cell types (101, 156), it is likely that TETs might also play a role in the neuroinflammatory responses of astrocytes. Furthermore, TET activity seems to be important for generation of oligodendrocytes, which are the myelinating cells of the central nervous system. Oligodendrocyte pathology is therefore evident especially in MS. Oligodendrocytes are generated from oligodendrocyte progenitor cells (164) and all TET proteins (TET1, TET2, and TET3) were shown to play a role in the differentiation process with unique subcellular and temporal expression patterns (165). Moreover, DNA hydroxymethylation catalyzed by TET1 has been shown to be essential for myelin repair in young adults and defective in old mice (166). However, in MS remyelination is often incomplete. Interestingly, in patients with MS TET2 expression is significantly downregulated in peripheral blood mononuclear cells, which is associated with a decrease of 5-hmdC levels (167). Further studies are still needed to understand the complex role of TET enzymes in the pathophysiology of MS.
The loss of function of TETs, through either genetic mutations or catalytic inhibition, has shown a strong causal relationship with multiple malignancies (13, 65, 66). Based on recent discoveries, several new treatment options are now under consideration. Data from a recent meta-analysis has shown that, in spite TET2 mutations had no significant prognostic value on myelodysplastic syndromes, the response rates to hypomethylating agents were significantly different between patients with and without TET2 mutations (168). However, in patients with decreased 5-hmdC levels due to elevated miRNA or CXXC expression, TET proteins are still functional. These studies have already demonstrated that TET overexpression is able to rescue the phenotype caused by overexpression of TET-targeting miRNAs (55, 56). Moreover, the expression of TETs is shown to be regulated on several levels, i.e., at pre-transcriptional, pro-transcriptional (miRNAs), and post-translational levels and, as mentioned before in this review, its activity can be modulated by by re-inducing its activity or enhancing the remaining TET activity by certain compounds like vitamin C (Figure 6) (37). For instance, on pre-transcriptional level, TET1 promoter hypermethylation has been shown to be linked with downregulation of TET1 and breast cancer metastasis (169). Hydrogen sulfide derived from the metabolism of methionine has shown to lead to sulfhydration of the transcriptional activator nuclear transcription factor Y subunit beta (NFYB), resulting in increased transcription of TET1 and TET2 (170). Post-translational modification of TET enzymes, including O-linked-N-acetylglucosamine (O-GlcNAc) Transferase, phosphorylation, Poly-Adenosine Ribosylation (PARylation) and monoubiquitination have shown to significantly affect their enzymatic activity and stability (171). Although TET deficiency enhances cell survival and increases cell “stemness”, recent studies discuss the possibility of temporarily inhibiting TET activity in order to enhance immune responses. In chimeric antigen receptor T cells (CAR-T) are efficient therapeutic agent for B lymphocyte malignancies (172).. Here it has been shown, that TET2 elimination can change the epigenetic map of cells and promote the proliferation of CAR-T cells derived from a single cell clone, thereby promoting remission in leukemia patients and improving the efficacy of immunotherapy (172). Thus, TET2 deficiency facilitated differentiation and expansion of CD8+ T cells, which could provide protection against tumor and virus infiltration. Non-specific TET inhibitors as 2-hydroxyglutarate should be able to boost antigen-specific responses against tumours by themselves (118). Therefore, there are different potential strategies that could be used for modulating TET function in immune and non-immune cells, potentially furnishing future research with novel therapeutic venues involving TETs to target and ameliorate inflammation-related diseases. It is important to note, however, that these statements are highly exploratory and that the complexity of how TET enzymes regulate immune responses in a cell and tissue type-dependent manner represents a great obstacle for the exploration of these mechanisms for modulating inflammation and in the context inflammation-related diseases. Nevertheless, it appears highly dangerous to misregulate TET activity and 5-hmdC levels indiscriminately. Therefore, more investigations on how to target specific cell types and tissues are still needed. Summarily, intensive further research needs to be done applying different in vitro and in vivo models to elucidate the mechanisms underlying the complex regulation of the immune system performed by these enzymes and how their modulation might affect ultimately the immune responses.
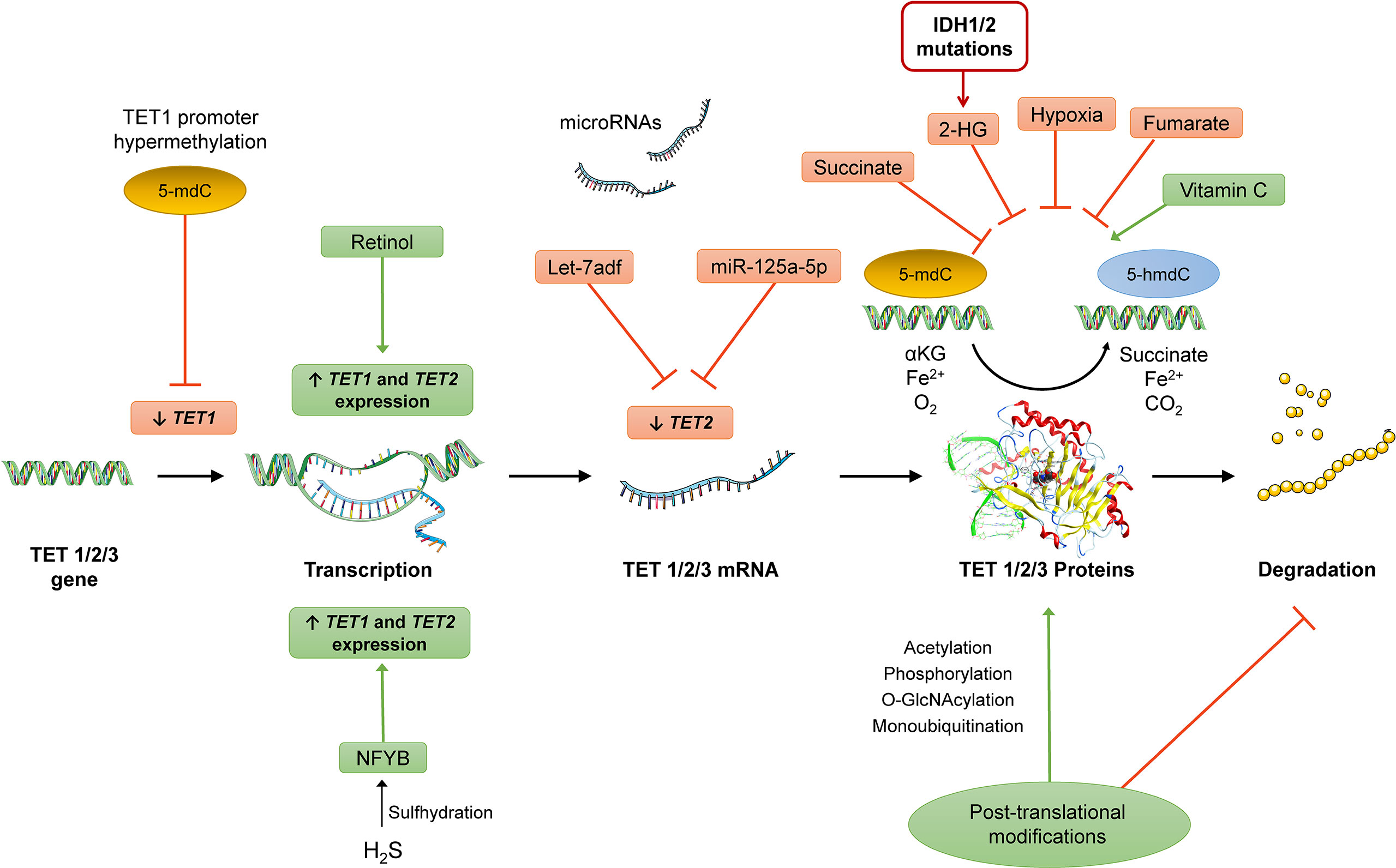
Figure 6 Potential targets for regulation of TET expression at pre-transcriptional (e.g., promoter hypermethylation of TET1l, pro-transcriptional (miRNAs), and post-translational levels (post-translational modifications that can affect TET enzymatic activity and/or stability) and induction/inhibition of TET activity. Different metabolic factors can affect TET enzymatic activity (e.g., 2-HG produced from mutated IDH1/2 enzymes, increased concentrations of succinate, and hypoxia conditions) and other compounds such as vitamin C and retinol were also shown to affect the activity of these enzymes. The nucleic acid images were provided and adapted from Servier Medical Art (smart.servier.com).
Epigenetic regulation plays an important role in modulating immune responses against infection or injury. Besides DNMT-mediated DNA-methylation, the TET enzymes are involved in immune cell development, affecting self-renewal of stem cells and lineage commitment to terminal differentiation. Thus, TETs and DNA-hydroxymethylation are important modulators of immune responses and pathogenesis of inflammatory diseases. Additionally, aberrant DNA-hydroxymethylation plays a key role in dysregulation of HSPC self-renewal and lineage differentiation and can lead to aberrant stem cell function and cellular transformation, leading to both myeloid and lymphoid leukemias. Data demonstrate that TET enzymes regulate a broad range of mechanisms in most of immune cells. Thus, they play an important role in different subset populations of T lymphocytes, affecting their differentiation and function. In addition, they regulate inflammation and autoimmune-related processes in B and plasma cells, as well. Furthermore, TET proteins are involved in the initiation and development of autoimmune diseases like MS and others. Here, the complex role of TET enzymes in the pathophysiology of this is still unclear. In this review, we focused on highlighting the current understanding and emerging concepts in the mechanisms through which TET proteins and their products modulate inflammation in immune and non-immune cells, including also relevant aspects of the regulation of myeloid and lymphoid immune cell development, differentiation, and function. Besides all these fundamental questions, modulating the activity of epigenetic regulating enzymes including TET proteins may be a promising way to alter and to achieve the desired magnitude and direction of immune responses.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
We acknowledge support by the Open Access Publication Fund of the Freie Universität Berlin. Dr. Christian Gerecke is funded by Deutsche Forschungsgemeinschaft (DFG - German Research Foundation) grant number GE 3321/2-1.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
2-HG, 2-hydroxyglutarate; αKG, α-ketoglutarate; αKGD, α-ketoglutarate dependent dioxygenases; 5-cadC, 5−carboxyl-2’-deoxycytidine; 5-fdC, 5-formyl-2’-deoxycytidine; 5-hmdC, 5-hydroxymethyl-2’-deoxycytidine; 5-mdC, 5-methyl-2’-deoxycytidine; aa, amino acid; ADAR1, adenosine deaminase acting on RNA 1; AID, activation induced DNA cytosine deaminase; AITL, angioimmunoblastic T cell lymphomas; AML, acute myeloid leukemia; AKT, Akt kinase; BER, base excision repair; CAR, chimeric antigen receptor; CD4/CD8, cluster of differentiation; CEBPα, CCAAT/enhancer-binding protein alpha; CHIP, clonal hematopoiesis of indeterminate potential; CMML, chronic myelomonocytic leukemia; c-myc, cellular myelocytomatosis; CpG, Cytosine Guanine dinucleotide; CXCL1, C-X-C motif chemokine ligand 1; CXXC, cysteine xx cysteine; DC, dendritic cells; DNMTs, DNA-Methyltransferase; FH, fumarate hydratase; FIH, factor inhibiting HIF; FOXP3, forkhead box P3; FTO, fat mass and obesity-associated protein; GATA3, GATA binding protein 3; GC, germinal centers; HDAC, histone deacetylase; HIF, hypoxia inducible factor; HSPC, hematopoietic stem and progenitor cells; IDAX, CXXC finger protein 4; IDH, isocitrate dehydrogenase; IFNB1, interferon beta 1; IFN-γ, interferon gamma; Igκ, Ig light chain; IKBZ, IkappaB zeta; IL, Interleukin; iNKT, invariant natural killer T cell; IRF4, interferon regulatory factor 4; IRF7, interferon regulatory factor 7; JAK, janus kinase; KLF13, Krueppel-like factor 13; let-7adf, miR-let-7a/let-7d/let-7f cluster; LPS, lipopolysaccharide; LSK, Lin−Sca-1+c-Kit+; MDS, myelodysplastic syndromes; MITF, melanocyte inducing transcription factor; MiRNA, microRNA; MLL, mixed-lineage leukemia 1; MNP, myeloproliferative neoplasms; MS, multiple sclerosis; ncRNAs, non-coding RNAs; NF-kB, nuclear factor kappa B; NLRs, NOD-like receptors; NLRP3, NLR family pyrin domain containing 3; NOD, nodulation factors; OGT, O6- guanine transferase; OPCs, oligodendrocyte precursor cells; P4H, prolyl-4-hydroxylase; PD-L1, programmed death-ligand 1; PI3K, phosphatidylinositol 3-kinase regulatory subunit alpha; PTEN, phosphatase and tensin homolog; RIG, retinoic acid inducible gene; RLRs, RIG-I-like receptors; ROS, reactive oxygen species; SDH, succinate dehydrogenase; shRNA, short hairpin RNA; Sin3A, SIN3 transcription regulator family member A; SOCS3, suppressor of cytokine signaling; STAT, signal transducer and activator of transcription; TBX21, T-box transcription factor TBX21; Tfh, follicular T helper cells; Th cell, T helper cell; Treg cell, regulatoric T cell; TDG, thymine DNA glycosylase; TET, ten eleven translocation protein; TLRs, toll-like receptors; TNFα, tumour necrosis factor alpha; ZBTB7b, zinc finger and BTB domain-containing protein 7B

1. Jones PA, Baylin SB. The Fundamental Role of Epigenetic Events in Cancer. Nat Rev Genet (2002) 3(6):415–28. doi: 10.1038/nrg816
2. Allis CD, Jenuwein T. The Molecular Hallmarks of Epigenetic Control. Nat Rev Genet (2016) 17(8):487–500. doi: 10.1038/nrg.2016.59
3. Rausch C, Hastert FD, Cardoso MC. DNA Modification Readers and Writers and Their Interplay. J Mol Biol (2019) 432(6):1731–46. doi: 10.1016/j.jmb.2019.12.018
4. Feinberg AP. Phenotypic Plasticity and the Epigenetics of Human Disease. Nature (2007) 447(7143):433–40. doi: 10.1038/nature05919
6. Greenberg MVC, Bourc’his D. The Diverse Roles of DNA Methylation in Mammalian Development and Disease. Nat Rev Mol Cell Biol (2019) 20(10):590–607. doi: 10.1038/s41580-019-0159-6
7. Moore LD, Le T, Fan G. DNA Methylation and its Basic Function. Neuropsychopharmacology (2013) 38(1):23–38. doi: 10.1038/npp.2012.112
8. Baylin SB, Belinsky SA, Herman JG. Aberrant Methylation of Gene Promoters in Cancer—Concepts, Misconcepts, and Promise. J Natl Cancer Inst (2000) 92(18):1460–1. doi: 10.1093/jnci/92.18.1460
9. Jones PA. Functions of DNA Methylation: Islands, Start Sites, Gene Bodies and Beyond. Nat Rev Genet (2012) 13(7):484–92. doi: 10.1038/nrg3230
10. Knudson AG Jr., Strong LC. Mutation and Cancer: A Model for Wilms’ Tumor of the Kidney. J Natl Cancer Inst (1972) 48(2):313–24. doi: 10.1093/jnci/48.2.313
11. Shen L, Song C-X, He C, Zhang Y. Mechanism and Function of Oxidative Reversal of DNA and RNA Methylation. Annu Rev Biochem (2014) 83:585–614. doi: 10.1146/annurev-biochem-060713-035513
12. Wu X, Zhang Y. TET-Mediated Active DNA Demethylation: Mechanism, Function and Beyond. Nat Rev Genet (2017) 18(9):517–34. doi: 10.1038/nrg.2017.33
13. Kohli RM, Zhang Y. TET Enzymes, TDG and the Dynamics of DNA Demethylation. Nature (2013) 502(7472):472–9. doi: 10.1038/nature12750
14. Yin X, Xu Y. Structure and Function of TET Enzymes. Adv Exp Med Biol (2016) 945:275–302. doi: 10.1007/978-3-319-43624-1_12
15. Koivunen P, Laukka T. The TET Enzymes. Cell Mol Life Sci (2018) 75(8):1339–48. doi: 10.1007/s00018-017-2721-8
16. Rasmussen KD, Helin K. Role of TET Enzymes in DNA Methylation, Development, and Cancer. Genes Dev (2016) 30(7):733–50. doi: 10.1101/gad.276568.115
17. Xu Q, Wang C, Zhou JX, Xu ZM, Gao J, Sui P, et al. Loss of TET Reprograms Wnt Signaling Through Impaired Demethylation to Promote Lung Cancer Development. Proc Natl Acad Sci U.S.A. (2022) 119(6):e2107599119. doi: 10.1073/pnas.2107599119
18. Wu MZ, Chen SF, Nieh S, Benner C, Ger LP, Jan CI, et al. Hypoxia Drives Breast Tumor Malignancy Through a TET-Tnfalpha-P38-MAPK Signaling Axis. Cancer Res (2015) 75(18):3912–24. doi: 10.1158/0008-5472.CAN-14-3208
19. Seritrakul P, Gross JM. Tet-Mediated DNA Hydroxymethylation Regulates Retinal Neurogenesis by Modulating Cell-Extrinsic Signaling Pathways. PloS Genet (2017) 13(9):e1006987. doi: 10.1371/journal.pgen.1006987
20. Sekita Y, Sugiura Y, Matsumoto A, Kawasaki Y, Akasaka K, Konno R, et al. AKT Signaling Is Associated With Epigenetic Reprogramming via the Upregulation of TET and its Cofactor, Alpha-Ketoglutarate During Ipsc Generation. Stem Cell Res Ther (2021) 12(1):510. doi: 10.1186/s13287-021-02578-1
21. Liu L, He X, Zhao M, Yang S, Wang S, Yu X, et al. Regulation of DNA Methylation and 2-OG/TET Signaling by Choline Alleviated Cardiac Hypertrophy in Spontaneously Hypertensive Rats. J Mol Cell Cardiol (2019) 128:26–37. doi: 10.1016/j.yjmcc.2019.01.011
22. Li X, Yue X, Pastor WA, Lin L, Georges R, Chavez L, et al. Tet Proteins Influence the Balance Between Neuroectodermal and Mesodermal Fate Choice by Inhibiting Wnt Signaling. Proc Natl Acad Sci U.S.A. (2016) 113(51):E8267–76. doi: 10.1073/pnas.1617802113
23. Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet Proteins in 5mc to 5hmc Conversion, ES-Cell Self-Renewal and Inner Cell Mass Specification. Nature (2010) 466(7310):1129–33. doi: 10.1038/nature09303
24. Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet Proteins can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science (2011) 333(6047):1300–3. doi: 10.1126/science.1210597
25. He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-Mediated Formation of 5-Carboxylcytosine and Its Excision by TDG in Mammalian DNA. Science (2011) 333(6047):1303–7. doi: 10.1126/science.1210944
26. Lio CJ, Yue X, Lopez-Moyado IF, Tahiliani M, Aravind L, Rao A. TET Methylcytosine Oxidases: New Insights From a Decade of Research. J Biosci (2020) 45. doi: 10.1007/s12038-019-9973-4
27. Loenarz C, Schofield CJ. Physiological and Biochemical Aspects of Hydroxylations and Demethylations Catalyzed by Human 2-Oxoglutarate Oxygenases. Trends Biochem Sci (2011) 36(1):7–18. doi: 10.1016/j.tibs.2010.07.002
28. Rose NR, McDonough MA, King ON, Kawamura A, Schofield CJ. Inhibition of 2-Oxoglutarate Dependent Oxygenases. Chem Soc Rev (2011) 40(8):4364–97. doi: 10.1039/c0cs00203h
29. Delatte B, Deplus R, Fuks F. Playing Tetris With DNA Modifications. EMBO J (2014) 33(11):1198–211. doi: 10.15252/embj.201488290
30. Ko M, An J, Bandukwala HS, Chavez L, Aijo T, Pastor WA, et al. Modulation of TET2 Expression and 5-Methylcytosine Oxidation by the CXXC Domain Protein IDAX. Nature (2013) 497(7447):122–6. doi: 10.1038/nature12052
31. Hore TA, von Meyenn F, Ravichandran M, Bachman M, Ficz G, Oxley D, et al. Retinol and Ascorbate Drive Erasure of Epigenetic Memory and Enhance Reprogramming to Naive Pluripotency by Complementary Mechanisms. Proc Natl Acad Sci U.S.A. (2016) 113(43):12202–7. doi: 10.1073/pnas.1608679113
32. Minor EA, Court BL, Young JI, Wang G. Ascorbate Induces Ten-Eleven Translocation (Tet) Methylcytosine Dioxygenase-Mediated Generation of 5-Hydroxymethylcytosine. J Biol Chem (2013) 288(19):13669–74. doi: 10.1074/jbc.C113.464800
33. Monfort A, Wutz A. Breathing-in Epigenetic Change With Vitamin C. EMBO Rep (2013) 14(4):337–46. doi: 10.1038/embor.2013.29
34. Blaschke K, Ebata KT, Karimi MM, Zepeda-Martínez JA, Goyal P, Mahapatra S, et al. Vitamin C Induces Tet-Dependent DNA Demethylation and a Blastocyst-Like State in ES Cells. Nature (2013) 500(7461):222–6. doi: 10.1038/nature12362
35. Coulter JB, O’Driscoll CM, Bressler JP. Hydroquinone Increases 5-Hydroxymethylcytosine Formation Through Ten Eleven Translocation 1 (TET1) 5-Methylcytosine Dioxygenase. J Biol Chem (2013) 288(40):28792–800. doi: 10.1074/jbc.M113.491365
36. Yin R, Mao SQ, Zhao B, Chong Z, Yang Y, Zhao C, et al. Ascorbic Acid Enhances Tet-Mediated 5-Methylcytosine Oxidation and Promotes DNA Demethylation in Mammals. J Am Chem Soc (2013) 135(28):10396–403. doi: 10.1021/ja4028346
37. Hore TA, von Meyenn F, Ravichandran M, Bachman M, Ficz G, Oxley D, et al. Retinol and Ascorbate Drive Erasure of Epigenetic Memory and Enhance Reprogramming to Naïve Pluripotency by Complementary Mechanisms. Proc Natl Acad Sci (2016) 113(43):12202–7. doi: 10.1073/pnas.1608679113
38. Losman J-A, Kaelin WG Jr. What a Difference a Hydroxyl Makes: Mutant IDH, (R)-2-Hydroxyglutarate, and Cancer. Genes Dev (2013) 27(8):836–52. doi: 10.1101/gad.217406.113
39. Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, et al. The Oncometabolite 2-Hydroxyglutarate Inhibits Histone Lysine Demethylases. EMBO Rep (2011) 12(5):463–9. doi: 10.1038/embor.2011.43
40. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-Associated IDH1 Mutations Produce 2-Hydroxyglutarate. Nature (2009) 462(7274):739–44. doi: 10.1038/nature08617
41. Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, et al. Transformation by the (R)-Enantiomer of 2-Hydroxyglutarate Linked to EGLN Activation. Nature (2012) 483(7390):484–8. doi: 10.1038/nature10898
42. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-Hydroxyglutarate is a Competitive Inhibitor of Alpha-Ketoglutarate-Dependent Dioxygenases. Cancer Cell (2011) 19(1):17–30. doi: 10.1016/j.ccr.2010.12.014
43. Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, et al. Identification of a Cpg Island Methylator Phenotype That Defines a Distinct Subgroup of Glioma. Cancer Cell (2010) 17(5):510–22. doi: 10.1016/j.ccr.2010.03.017
44. Whitehall VL, Dumenil TD, McKeone DM, Bond CE, Bettington ML, Buttenshaw RL, et al. Isocitrate Dehydrogenase 1 R132C Mutation Occurs Exclusively in Microsatellite Stable Colorectal Cancers With the Cpg Island Methylator Phenotype. Epigenetics (2014) 9(11):1454–60. doi: 10.4161/15592294.2014.971624
45. Laukka T, Mariani CJ, Ihantola T, Cao JZ, Hokkanen J, Kaelin WG Jr., et al. Fumarate and Succinate Regulate Expression of Hypoxia-Inducible Genes via TET Enzymes. J Biol Chem (2016) 291(8):4256–65. doi: 10.1074/jbc.M115.688762
46. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell (2010) 18(6):553–67. doi: 10.1016/j.ccr.2010.11.015
47. Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH Mutation Impairs Histone Demethylation and Results in a Block to Cell Differentiation. Nature (2012) 483(7390):474–8. doi: 10.1038/nature10860
48. Zheng S, Houseman EA, Morrison Z, Wrensch MR, Patoka JS, Ramos C, et al. DNA Hypermethylation Profiles Associated With Glioma Subtypes and EZH2 and IGFBP2 Mrna Expression. Neuro Oncol (2011) 13(3):280–9. doi: 10.1093/neuonc/noq190
49. Lian CG, Xu Y, Ceol C, Wu F, Larson A, Dresser K, et al. Loss of 5-Hydroxymethylcytosine is an Epigenetic Hallmark of Melanoma. Cell (2012) 150(6):1135–46. doi: 10.1016/j.cell.2012.07.033
50. Lorsbach RB, Moore J, Mathew S, Raimondi SC, Mukatira ST, Downing JR. TET1, a Member of a Novel Protein Family, is Fused to MLL in Acute Myeloid Leukemia Containing the T(10;11)(Q22;Q23). Leukemia (2003) 17(3):637–41. doi: 10.1038/sj.leu.2402834
51. Lee SG, Cho SY, Kim MJ, Oh SH, Cho EH, Lee S, et al. Genomic Breakpoints and Clinical Features of MLL-TET1 Rearrangement in Acute Leukemias. Haematologica (2013) 98(4):e55–7. doi: 10.3324/haematol.2012.076323
52. Kroeze LI, van der Reijden BA, Jansen JH. 5-Hydroxymethylcytosine: An Epigenetic Mark Frequently Deregulated in Cancer. Biochim Biophys Acta (2015) 1855(2):144–54. doi: 10.1016/j.bbcan.2015.01.001
53. Kim YH, Pierscianek D, Mittelbronn M, Vital A, Mariani L, Hasselblatt M, et al. TET2 Promoter Methylation in Low-Grade Diffuse Gliomas Lacking IDH1/2 Mutations. J Clin Pathol (2011) 64(10):850–2. doi: 10.1136/jclinpath-2011-200133
54. Muller T, Gessi M, Waha A, Isselstein LJ, Luxen D, Freihoff D, et al. Nuclear Exclusion of TET1 is Associated With Loss of 5-Hydroxymethylcytosine in IDH1 Wild-Type Gliomas. Am J Pathol (2012) 181(2):675–83. doi: 10.1016/j.ajpath.2012.04.017
55. Cheng J, Guo S, Chen S, Mastriano SJ, Liu C, D'Alessio AC, et al. An Extensive Network of TET2-Targeting Micrornas Regulates Malignant Hematopoiesis. Cell Rep (2013) 5(2):471–81. doi: 10.1016/j.celrep.2013.08.050
56. Song SJ, Poliseno L, Song MS, Ala U, Webster K, Ng C, et al. Micro RNA-Antagonism Regulates Breast Cancer Stemness and Metastasis via TET-Family-Dependent Chromatin Remodeling. Cell (2013) 154(2):311–24. doi: 10.1016/j.cell.2013.06.026
57. Morita S, Horii T, Kimura M, Ochiya T, Tajima S, Hatada I. Mir-29 Represses the Activities of DNA Methyltransferases and DNA Demethylases. Int J Mol Sci (2013) 14(7):14647–58. doi: 10.3390/ijms140714647
58. Wang Y, Zhang X, Li H, Yu J, Ren X. The Role of Mi RNA-29 Family in Cancer. Eur J Cell Biol (2013) 92(3):123–8. doi: 10.1016/j.ejcb.2012.11.004
59. Fu X, Jin L, Wang X, Luo A, Hu J, Zheng X, et al. Micro RNA-26a Targets Ten Eleven Translocation Enzymes and is Regulated During Pancreatic Cell Differentiation. Proc Natl Acad Sci U.S.A. (2013) 110(44):17892–7. doi: 10.1073/pnas.1317397110
60. Song F, Yang D, Liu B, Guo Y, Zheng H, Li L, et al. Integrated Micro RNA Network Analyses Identify a Poor-Prognosis Subtype of Gastric Cancer Characterized by the Mir-200 Family. Clin Cancer Res (2014) 20(4):878–89. doi: 10.1158/1078-0432.CCR-13-1844
61. Deplus R, Delatte B, Schwinn MK, Defrance M, Mendez J, Murphy N, et al. TET2 and TET3 Regulate Glcnacylation and H3K4 Methylation Through OGT and SET1/COMPASS. EMBO J (2013) 32(5):645–55. doi: 10.1038/emboj.2012.357
62. Ma Z, Vocadlo DJ, Vosseller K. Hyper-O-Glcnacylation is Anti-Apoptotic and Maintains Constitutive NF-Kappab Activity in Pancreatic Cancer Cells. J Biol Chem (2013) 288(21):15121–30. doi: 10.1074/jbc.M113.470047
63. Turkistany SA, DeKoter RP. The Transcription Factor PU.1 is a Critical Regulator of Cellular Communication in the Immune System. Arch Immunol Ther Exp (Warsz) (2011) 59(6):431–40. doi: 10.1007/s00005-011-0147-9
64. Xu YP, Lv L, Liu Y, Smith MD, Li WC, Tan XM, et al. Tumor Suppressor TET2 Promotes Cancer Immunity and Immunotherapy Efficacy. J Clin Invest (2019) 129(10):4316–31. doi: 10.1172/JCI129317
65. Huang Y, Rao A. Connections Between TET Proteins and Aberrant DNA Modification in Cancer. Trends Genet (2014) 30(10):464–74. doi: 10.1016/j.tig.2014.07.005
66. Ko M, An J, Pastor WA, Koralov SB, Rajewsky K, Rao A. TET Proteins and 5-Methylcytosine Oxidation in Hematological Cancers. Immunol Rev (2015) 263(1):6–21. doi: 10.1111/imr.12239
67. Lio CJ, Yuita H, Rao A. Dysregulation of the TET Family of Epigenetic Regulators in Lymphoid and Myeloid Malignancies. Blood (2019) 134(18):1487–97. doi: 10.1182/blood.2019791475
68. Rocquain J, Carbuccia N, Trouplin V, Raynaud S, Murati A, Nezri M, et al. Combined Mutations of ASXL1, CBL, FLT3, IDH1, IDH2, JAK2, KRAS, NPM1, NRAS, RUNX1, TET2 and WT1 Genes in Myelodysplastic Syndromes and Acute Myeloid Leukemias. BMC Cancer (2010) 10:401. doi: 10.1186/1471-2407-10-401
69. Ryan DG, Murphy MP, Frezza C, Prag HA, Chouchani ET, O'Neill LA, et al. Coupling Krebs Cycle Metabolites to Signalling in Immunity and Cancer. Nat Metab (2019) 1:16–33. doi: 10.1038/s42255-018-0014-7
70. Ko M, Rao A. TET2: Epigenetic Safeguard for HSC. Blood (2011) 118(17):4501–3. doi: 10.1182/blood-2011-08-373357
71. Jiang S. Tet2 at the Interface Between Cancer and Immunity. Commun Biol (2020) 3(1):667. doi: 10.1038/s42003-020-01391-5
72. Bowman RL, Levine RL. TET2 in Normal and Malignant Hematopoiesis. Cold Spring Harbor Perspect Med (2017) 7(8):a026518. doi: 10.1101/cshperspect.a026518
73. Pan F, Wingo TS, Zhao Z, Gao R, Makishima H, Qu G, et al. Tet2 Loss Leads to Hypermutagenicity in Haematopoietic Stem/Progenitor Cells. Nat Commun (2017) 8(1):15102. doi: 10.1038/ncomms15102
74. Lewis AC, Kats LM. Non-Genetic Heterogeneity, Altered Cell Fate and Differentiation Therapy. EMBO Mol Med (2021) 13(3):e12670. doi: 10.15252/emmm.202012670
75. Bensberg M, Rundquist O, Selimović A, Lagerwall C, Benson M, Gustafsson M, et al. TET2 as a Tumor Suppressor and Therapeutic Target in T-Cell Acute Lymphoblastic Leukemia. Proc Natl Acad Sci (2021) 118(34):e2110758118. doi: 10.1073/pnas.2110758118
76. Ferrone CK, Blydt-Hansen M, Rauh MJ. Age-Associated TET2 Mutations: Common Drivers of Myeloid Dysfunction, Cancer and Cardiovascular Disease. Int J Mol Sci (2020) 21(2):626. doi: 10.3390/ijms21020626
77. Lin Y, Lin Z, Cheng K, Fang Z, Li Z, Luo Y, et al. Prognostic Role of TET2 Deficiency in Myelodysplastic Syndromes: A Meta-Analysis. Oncotarget (2017) 8(26):43295–305. doi: 10.18632/oncotarget.17177
78. Abegunde SO, Buckstein R, Wells RA, Rauh MJ. An Inflammatory Environment Containing Tnfα Favors Tet2-Mutant Clonal Hematopoiesis. Exp Hematol (2018) 59:60–5. doi: 10.1016/j.exphem.2017.11.002
79. Bray JK, Dawlaty MM, Verma A, Maitra A. Roles and Regulations of TET Enzymes in Solid Tumors. Trends Cancer (2021) 7(7):635–46. doi: 10.1016/j.trecan.2020.12.011
80. Medzhitov R. Origin and Physiological Roles of Inflammation. Nature (2008) 454(7203):428–35. doi: 10.1038/nature07201
81. Akira S, Uematsu S, Takeuchi O. Pathogen Recognition and Innate Immunity. Cell (2006) 124(4):783–801. doi: 10.1016/j.cell.2006.02.015
82. Bassler K, Schulte-Schrepping J, Warnat-Herresthal S, Aschenbrenner AC, Schultze JL. The Myeloid Cell Compartment—Cell by Cell. Annu Rev Immunol (2019) 37(1):269–93. doi: 10.1146/annurev-immunol-042718-041728
83. Iqbal AJ, Fisher EA, Greaves DR. Inflammation-a Critical Appreciation of the Role of Myeloid Cells. Microbiol Spectr (2016) 4(5). doi: 10.1128/microbiolspec.MCHD-0027-2016
84. Nahrendorf M. Myeloid Cells in Cardiovascular Organs. J Intern Med (2019) 285(5):491–502. doi: 10.1111/joim.12844
85. O’Neill LA, Golenbock D, Bowie AG. The History of Toll-Like Receptors - Redefining Innate Immunity. Nat Rev Immunol (2013) 13(6):453–60. doi: 10.1038/nri3446
86. Sun F, Abreu-Rodriguez I, Ye S, Gay S, Distler O, Neidhart M, et al. TET1 Is an Important Transcriptional Activator of Tnfα Expression in Macrophages. PloS One (2019) 14(6):e0218551. doi: 10.1371/journal.pone.0218551
87. Huang Y, Tian C, Li Q, Xu Q. TET1 Knockdown Inhibits Porphyromonas Gingivalis LPS/IFN-γ-Induced M1 Macrophage Polarization Through the NF-κb Pathway in THP-1 Cells. Int J Mol Sci (2019) 20(8):2023. doi: 10.3390/ijms20082023
88. Yee KM, Shuai RW, Liu B, Huynh CA, Niu C, Lee HR, et al. TET1 Controls Cxcl1 Induction by DNA Demethylation and Promotes Neutrophil Recruitment During Acute Lung Injury. bioRxiv (2021) 2021:9.. doi: 10.1101/2021.09.07.459280
89. Xue S, Liu C, Sun X, Li W, Zhang C, Zhou X, et al. TET3 Inhibits Type I IFN Production Independent of DNA Demethylation. Cell Rep (2016) 16(4):1096–105. doi: 10.1016/j.celrep.2016.06.068
90. Sampath P, Moideen K, Ranganathan UD, Bethunaickan R. Monocyte Subsets: Phenotypes and Function in Tuberculosis Infection. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.01726
91. Kapellos TS, Bonaguro L, Gemünd I, Reusch N, Saglam A, Hinkley ER, et al. Human Monocyte Subsets and Phenotypes in Major Chronic Inflammatory Diseases. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.02035
92. Selimoglu-Buet D, Rivière J, Ghamlouch H, Bencheikh L, Lacout C, Morabito M, et al. A Mir-150/TET3 Pathway Regulates the Generation of Mouse and Human Non-Classical Monocyte Subset. Nat Commun (2018) 9(1):5455. doi: 10.1038/s41467-018-07801-x
93. Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, et al. Impaired Hydroxylation of 5-Methylcytosine in Myeloid Cancers With Mutant TET2. Nature (2010) 468(7325):839–43. doi: 10.1038/nature09586
94. Li Z, Cai X, Cai C-L, Wang J, Zhang W, Petersen BE, et al. Deletion of Tet2 in Mice Leads to Dysregulated Hematopoietic Stem Cells and Subsequent Development of Myeloid Malignancies. Blood (2011) 118(17):4509–18. doi: 10.1182/blood-2010-12-325241
95. Cull AH, Snetsinger B, Buckstein R, Wells RA, Rauh MJ. Tet2 Restrains Inflammatory Gene Expression in Macrophages. Exp Hematol (2017) 55:56–70.e13. doi: 10.1016/j.exphem.2017.08.001
96. Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, et al. Tet2 is Required to Resolve Inflammation by Recruiting Hdac2 to Specifically Repress IL-6. Nature (2015) 525(7569):389–93. doi: 10.1038/nature15252
97. Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, et al. Clonal Hematopoiesis Associated With TET2 Deficiency Accelerates Atherosclerosis Development in Mice. Sci (New York N.Y.) (2017) 355(6327):842–7. doi: 10.1126/science.aag1381
98. Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1β/Nlrp3 Inflammasome. J Am Coll Cardiol (2018) 71(8):875–86. doi: 10.1016/j.jacc.2017.12.037
99. Ma S, Wan X, Deng Z, Shi L, Hao C, Zhou Z, et al. Epigenetic Regulator CXXC5 Recruits DNA Demethylase Tet2 to Regulate TLR7/9-Elicited IFN Response in Pdcs. J Exp Med (2017) 214(5):1471–91. doi: 10.1084/jem.20161149
100. Jiang S, Yan W, Wang SE, Baltimore D. Dual Mechanisms of Posttranscriptional Regulation of Tet2 by Let-7 Microrna in Macrophages. Proc Natl Acad Sci (2019) 116(25):12416–21. doi: 10.1073/pnas.1811040116
101. Zhaolin Z, Jiaojiao C, Peng W, Yami L, Tingting Z, Jun T, et al. Oxldl Induces Vascular Endothelial Cell Pyroptosis Through Mir-125a-5p/TET2 Pathway. J Cell Physiol (2019) 234(5):7475–91. doi: 10.1002/jcp.27509
102. Laukka T, Mariani CJ, Ihantola T, Cao JZ, Hokkanen J, Kaelin WG Jr, et al. Fumarate and Succinate Regulate Expression of Hypoxia-Inducible Genes via TET Enzymes *. J Biol Chem (2016) 291(8):4256–65. doi: 10.1074/jbc.M115.688762
103. Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of α-KG-Dependent Histone and DNA Demethylases by Fumarate and Succinate That are Accumulated in Mutations of FH and SDH Tumor Suppressors. Genes Dev (2012) 26(12):1326–38. doi: 10.1101/gad.191056.112
104. Banerjee S, Cui H, Xie N, Tan Z, Yang S, Icyuz M, et al. Mir-125a-5p Regulates Differential Activation of Macrophages and Inflammation. J Biol Chem (2013) 288(49):35428–36. doi: 10.1074/jbc.M112.426866
105. Zhao JL, Huang F, He F, Gao CC, Liang SQ, Ma PF, et al. Forced Activation of Notch in Macrophages Represses Tumor Growth by Upregulating Mir-125a and Disabling Tumor-Associated Macrophages. Cancer Res (2016) 76(6):1403–15. doi: 10.1158/0008-5472.CAN-15-2019
106. Palam LR, Mali RS, Ramdas B, Srivatsan SN, Visconte V, Tiu RV, et al. Loss of Epigenetic Regulator TET2 and Oncogenic KIT Regulate Myeloid Cell Transformation via PI3K Pathway. JCI Insight (2018) 3(4):e94679. doi: 10.1172/jci.insight.94679
107. Montagner S, Leoni C, Emming S, Della Chiara G, Balestrieri C, Barozzi I, et al. TET2 Regulates Mast Cell Differentiation and Proliferation Through Catalytic and Non-Catalytic Activities. Cell Rep (2016) 15(7):1566–79. doi: 10.1016/j.celrep.2016.04.044
108. Shen Q, Zhang Q, Shi Y, Shi Q, Jiang Y, Gu Y, et al. Tet2 Promotes Pathogen Infection-Induced Myelopoiesis Through M RNA Oxidation. Nature (2018) 554(7690):123–7. doi: 10.1038/nature25434
109. Debes GF, McGettigan SE. Skin-Associated B Cells in Health and Inflammation. J Immunol (Baltimore Md.: 1950) (2019) 202(6):1659–66. doi: 10.4049/jimmunol.1801211
110. Cain D, Kondo M, Chen H, Kelsoe G. Effects of Acute and Chronic Inflammation on B-Cell Development and Differentiation. J Invest Dermatol (2009) 129(2):266–77. doi: 10.1038/jid.2008.286
111. Pioli PD. Plasma Cells, the Next Generation: Beyond Antibody Secretion. Front Immunol (2019) 10(2768). doi: 10.3389/fimmu.2019.02768
112. Kulis M, Merkel A, Heath S, Queirós AC, Schuyler RP, Castellano G, et al. Whole-Genome Fingerprint of the DNA Methylome During Human B Cell Differentiation. Nat Genet (2015) 47(7):746–56. doi: 10.1038/ng.3291
113. Cimmino L, Dawlaty MM, Ndiaye-Lobry D, Yap YS, Bakogianni S, Yu Y, et al. TET1 is a Tumor Suppressor of Hematopoietic Malignancy. Nat Immunol (2015) 16(6):653–62. doi: 10.1038/ni.3148
114. Schoeler K, Aufschnaiter A, Messner S, Derudder E, Herzog S, Villunger A, et al. TET Enzymes Control Antibody Production and Shape the Mutational Landscape in Germinal Centre B Cells. FEBS J (2019) 286(18):3566–81. doi: 10.1111/febs.14934
115. Orlanski S, Labi V, Reizel Y, Spiro A, Lichtenstein M, Levin-Klein R, et al. Tissue-Specific DNA Demethylation is Required for Proper B-Cell Differentiation and Function. Proc Natl Acad Sci (2016) 113(18):5018. doi: 10.1073/pnas.1604365113
116. Lio C-W, Zhang J, González-Avalos E, Hogan PG, Chang X, Rao A. TET2 and TET3 Cooperate With B-Lineage Transcription Factors to Regulate DNA Modification and Chromatin Accessibility. eLife (2016) 5:e18290. doi: 10.7554/eLife.18290.030
117. Mesin L, Ersching J, Victora GD. Germinal Center B Cell Dynamics. Immunity (2016) 45(3):471–82. doi: 10.1016/j.immuni.2016.09.001
118. Lio C-WJ, Rao A. TET Enzymes and 5hmc in Adaptive and Innate Immune Systems. Front Immunol (2019) 10(210). doi: 10.3389/fimmu.2019.00210
119. Dominguez PM, Ghamlouch H, Rosikiewicz W, Kumar P, Béguelin W, Fontán L, et al. TET2 Deficiency Causes Germinal Center Hyperplasia, Impairs Plasma Cell Differentiation, and Promotes B-Cell Lymphomagenesis. Cancer Discovery (2018) 8(12):1632–53. doi: 10.1158/2159-8290.CD-18-0657
120. Rosikiewicz W, Chen X, Dominguez PM, Ghamlouch H, Aoufouchi S, Bernard OA, et al. TET2 Deficiency Reprograms the Germinal Center B Cell Epigenome and Silences Genes Linked to Lymphomagenesis. Sci Adv (2020) 6(25):eaay5872–eaay5872. doi: 10.1126/sciadv.aay5872
121. Lio C-WJ, Shukla V, Samaniego-Castruita D, González-Avalos E, Chakraborty A, Yue X, et al. TET Enzymes Augment Activation-Induced Deaminase (AID) Expression via 5-Hydroxymethylcytosine Modifications at the Aicda Superenhancer. Sci Immunol (2019) 4(34):eaau7523. doi: 10.1126/sciimmunol.aau7523
122. Tanaka S, Ise W, Inoue T, Ito A, Ono C, Shima Y, et al. TET2 and TET3 in B Cells are Required to Repress CD86 and Prevent Autoimmunity. Nat Immunol (2020) 21(8):950–61. doi: 10.1038/s41590-020-0700-y
123. Cope AP. Studies of T-Cell Activation in Chronic Inflammation. Arthritis Res (2002) 4 Suppl 3(Suppl 3):S197–211. doi: 10.1186/ar557
124. Moro-García MA, Mayo JC, Sainz RM, Alonso-Arias R. Influence of Inflammation in the Process of T Lymphocyte Differentiation: Proliferative, Metabolic, and Oxidative Changes. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.00339
125. Tsagaratou A, Äijö T, Lio C-WJ, Yue X, Huang Y, Jacobsen SE, et al. Dissecting the Dynamic Changes of 5-Hydroxymethylcytosine in T-Cell Development and Differentiation. Proc Natl Acad Sci (2014) 111(32):E3306. doi: 10.1073/pnas.1412327111
126. Tsagaratou A. Deciphering the Multifaceted Roles of TET Proteins in T-Cell Lineage Specification and Malignant Transformation. Immunol Rev (2021) 300(1):22–36. doi: 10.1111/imr.12940
127. Tsagaratou A, Lio CJ, Yue X, Rao A. TET Methylcytosine Oxidases in T Cell and B Cell Development and Function. Front Immunol (2017) 8:220. doi: 10.3389/fimmu.2017.00220
128. Tsagaratou A, Rao A. TET Proteins and 5-Methylcytosine Oxidation in the Immune System. Cold Spring Harb Symp Quant Biol (2013) 78:1–10. doi: 10.1101/sqb.2013.78.020248
129. Tsiouplis NJ, Bailey DW, Chiou LF, Wissink FJ, Tsagaratou A. TET-Mediated Epigenetic Regulation in Immune Cell Development and Disease. Front Cell Dev Biol (2021) 8. doi: 10.3389/fcell.2020.623948
130. Tsagaratou A, Gonzalez-Avalos E, Rautio S, Scott-Browne JP, Togher S, Pastor WA, et al. TET Proteins Regulate the Lineage Specification and TCR-Mediated Expansion of Inkt Cells. Nat Immunol (2017) 18(1):45–53. doi: 10.1038/ni.3630
131. Zhu X, Zhu J. CD4 T Helper Cell Subsets and Related Human Immunological Disorders. Int J Mol Sci (2020) 21(21):8011. doi: 10.3390/ijms21218011
132. Hirahara K, Nakayama T. CD4+ T-Cell Subsets in Inflammatory Diseases: Beyond the Th1/Th2 Paradigm. Int Immunol (2016) 28(4):163–71. doi: 10.1093/intimm/dxw006
133. Ichiyama K, Chen T, Wang X, Yan X, Kim B-S, Tanaka S, et al. The Methylcytosine Dioxygenase Tet2 Promotes DNA Demethylation and Activation of Cytokine Gene Expression in T Cells. Immunity (2015) 42(4):613–26. doi: 10.1016/j.immuni.2015.03.005
134. Iyer SS, Cheng G. Role of Interleukin 10 Transcriptional Regulation in Inflammation and Autoimmune Disease. Crit Rev Immunol (2012) 32(1):23–63. doi: 10.1615/CritRevImmunol.v32.i1.30
135. Okeke EB, Uzonna JE. The Pivotal Role of Regulatory T Cells in the Regulation of Innate Immune Cells. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.00680
136. Shevyrev D, Tereshchenko V. Treg Heterogeneity, Function, and Homeostasis. Front Immunol (2020) 10. doi: 10.3389/fimmu.2019.03100
137. Dowling MR, Kan A, Heinzel S, Marchingo JM, Hodgkin PD, Hawkins ED. Regulatory T Cells Suppress Effector T Cell Proliferation by Limiting Division Destiny. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.02461
138. Lei H, Schmidt-Bleek K, Dienelt A, Reinke P, Volk H-D. Regulatory T Cell-Mediated Anti-Inflammatory Effects Promote Successful Tissue Repair in Both Indirect and Direct Manners. Front Pharmacol (2015) 6:184–4. doi: 10.3389/fphar.2015.00184
139. Gouirand V, Habrylo I, Rosenblum MD. Regulatory T Cells and Inflammatory Mediators in Autoimmune Disease. J Invest Dermatol (2021) 142(3):774–80. doi: 10.1016/j.jid.2021.05.010
140. Li Z, Li D, Tsun A, Li B. FOXP3+ Regulatory T Cells and Their Functional Regulation. Cell Mol Immunol (2015) 12(5):558–65. doi: 10.1038/cmi.2015.10
141. Yue X, Trifari S, Äijö T, Tsagaratou A, Pastor WA, Zepeda-Martínez JA, et al. Control of Foxp3 Stability Through Modulation of TET Activity. J Exp Med (2016) 213(3):377–97. doi: 10.1084/jem.20151438
142. Kressler C, Gasparoni G, Nordström K, Hamo D, Salhab A, Dimitropoulos C, et al. Targeted De-Methylation of the FOXP3-TSDR is Sufficient to Induce Physiological FOXP3 Expression But Not a Functional Treg Phenotype. Front Immunol (2021) 11. doi: 10.3389/fimmu.2020.609891
143. Nair VS, Oh KI. Down-Regulation of Tet2 Prevents TSDR Demethylation in IL2 Deficient Regulatory T Cells. Biochem Biophys Res Commun (2014) 450(1):918–24. doi: 10.1016/j.bbrc.2014.06.110
144. Garg G, Muschaweckh A, Moreno H, Vasanthakumar A, Floess S, Lepennetier G, et al. Blimp1 Prevents Methylation of Foxp3 and Loss of Regulatory T Cell Identity at Sites of Inflammation. Cell Rep (2019) 26(7):1854–1868.e5. doi: 10.1016/j.celrep.2019.01.070
145. Yue X, Lio C-WJ, Samaniego-Castruita D, Li X, Rao A. Loss of TET2 and TET3 in Regulatory T Cells Unleashes Effector Function. Nat Commun (2019) 10(1):2011. doi: 10.1038/s41467-019-09541-y
146. Scherm MG, Serr I, Zahm AM, Schug J, Bellusci S, Manfredini R, et al. Mi RNA142-3p Targets Tet2 and Impairs Treg Differentiation and Stability in Models of Type 1 Diabetes. Nat Commun (2019) 10(1):5697–7. doi: 10.1038/s41467-019-13587-3
147. Haring JS, Badovinac VP, Harty JT. Inflaming the CD8+ T Cell Response. Immunity (2006) 25(1):19–29. doi: 10.1016/j.immuni.2006.07.001
148. Zebley CC, Abdelsamed HA, Ghoneim HE, Alli S, Haydar D, Harris T, et al. IL-12 Signaling Promotes TET2-Mediated DNA Demethylation During CD8 T Cell Effector Differentiation. bioRxiv 2020: p (2020) 2020:11. doi: 10.2139/ssrn.3720007
149. Carty SA, Gohil M, Banks LB, Cotton RM, Johnson ME, Stelekati E, et al. The Loss of TET2 Promotes CD8(+) T Cell Memory Differentiation. J Immunol (Baltimore Md.: 1950) (2018) 200(1):82–91. doi: 10.4049/jimmunol.1700559
150. Mishra A, Bandopadhyay R, Singh PK, Mishra PS, Sharma N, Khurana N. Neuroinflammation in Neurological Disorders: Pharmacotherapeutic Targets From Bench to Bedside. Metab Brain Dis (2021) 36(7):1591–626. doi: 10.1007/s11011-021-00806-4
151. Ransohoff RM, Schafer D, Vincent A, Blachère NE, Bar-Or A. Neuroinflammation: Ways in Which the Immune System Affects the Brain. Neurotherapeutics: J Am Soc Exp Neurother (2015) 12(4):896–909. doi: 10.1007/s13311-015-0385-3
152. Saijo K, Glass CK. Microglial Cell Origin and Phenotypes in Health and Disease. Nat Rev Immunol (2011) 11(11):775–87. doi: 10.1038/nri3086
153. Lively S, Schlichter LC. Microglia Responses to Pro-Inflammatory Stimuli (LPS, Ifnγ+Tnfα) and Reprogramming by Resolving Cytokines (IL-4, IL-10). Front Cell Neurosci (2018) 12:215–5. doi: 10.3389/fncel.2018.00215
154. Zuiderwijk-Sick EA, van der Putten C, Timmerman R, Veth J, Pasini EM, van Straalen L, et al. Exposure of Microglia to Interleukin-4 Represses NF-κb-Dependent Transcription of Toll-Like Receptor-Induced Cytokines. Front Immunol (2021) 12(4743). doi: 10.3389/fimmu.2021.771453
155. Cheray M, Joseph B. Epigenetics Control Microglia Plasticity. Front Cell Neurosci (2018) 12(243). doi: 10.3389/fncel.2018.00243
156. Carrillo-Jimenez A, Deniz Ö, Niklison-Chirou MV, Ruiz R, Bezerra-Salomão K, Stratoulias V, et al. TET2 Regulates the Neuroinflammatory Response in Microglia. Cell Rep (2019) 29(3):697–713.e8. doi: 10.1016/j.celrep.2019.09.013
157. Li L, Qiu Y, Miao M, Liu Z, Li W, Zhu Y, et al. Reduction of Tet2 Exacerbates Early Stage Alzheimer’s Pathology and Cognitive Impairments in 2×Tg-AD Mice. Hum Mol Genet (2020) 29(11):1833–52. doi: 10.1093/hmg/ddz282
158. Marshall LL, Killinger BA, Ensink E, Li P, Li KX, Cui W, et al. Epigenomic Analysis of Parkinson’s Disease Neurons Identifies Tet2 Loss as Neuroprotective. Nat Neurosci (2020) 23(10):1203–14. doi: 10.1038/s41593-020-0690-y
159. Neal M, Richardson JR. Epigenetic Regulation of Astrocyte Function in Neuroinflammation and Neurodegeneration. Biochim Biophys Acta Mol basis Dis (2018) 1864(2):432–43. doi: 10.1016/j.bbadis.2017.11.004
160. Villarreal A, Vogel T. Different Flavors of Astrocytes: Revising the Origins of Astrocyte Diversity and Epigenetic Signatures to Understand Heterogeneity After Injury. Int J Mol Sci (2021) 22(13):6867. doi: 10.3390/ijms22136867
161. Kim J-H, Kim M, He X-B, Wulansari N, Yoon B-H, Bae D-H, et al. Vitamin C Promotes Astrocyte Differentiation Through Dna Hydroxymethylation. Stem Cells (2018) 36(10):1578–88. doi: 10.1002/stem.2886
162. Pavlou MAS, Grandbarbe L, Buckley NJ, Niclou SP, Michelucci A. Transcriptional and Epigenetic Mechanisms Underlying Astrocyte Identity. Prog Neurobiol (2019) 174:36–52. doi: 10.1016/j.pneurobio.2018.12.007
163. Fan G, Martinowich K, Chin MH, He F, Fouse SD, Hutnick L, et al. DNA Methylation Controls the Timing of Astrogliogenesis Through Regulation of JAK-STAT Signaling. Development (2005) 132(15):3345–56. doi: 10.1242/dev.01912
164. Kuhn S, Gritti L, Crooks D, Dombrowski Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells (2019) 8(11):1424. doi: 10.3390/cells8111424
165. Zhao X, Dai J, Ma Y, Mi Y, Cui D, Ju G, et al. Dynamics of Ten-Eleven Translocation Hydroxylase Family Proteins and 5-Hydroxymethylcytosine in Oligodendrocyte Differentiation. Glia (2014) 62(6):914–26. doi: 10.1002/glia.22649
166. Moyon S, Frawley R, Marechal D, Huang D, Marshall-Phelps KLH, Kegel L, et al. TET1-Mediated DNA Hydroxymethylation Regulates Adult Remyelination in Mice. Nat Commun (2021) 12(1):3359. doi: 10.1038/s41467-021-23735-3
167. Calabrese R, Valentini E, Ciccarone F, Guastafierro T, Bacalini MG, Ricigliano VA, et al. TET2 Gene Expression and 5-Hydroxymethylcytosine Level in Multiple Sclerosis Peripheral Blood Cells. Biochim Biophys Acta (2014) 1842(7):1130–6. doi: 10.1016/j.bbadis.2014.04.010
168. Guo Z, Zhang SK, Zou Z, Fan RH, Lyu XD. Prognostic Significance of TET2 Mutations in Myelodysplastic Syndromes: A Meta-Analysis. Leuk Res (2017) 58:102–7. doi: 10.1016/j.leukres.2017.03.013
169. Sang Y, Cheng C, Tang XF, Zhang MF, Lv XB. Hypermethylation of TET1 Promoter is a New Diagnosic Marker for Breast Cancer Metastasis. Asian Pac J Cancer Prev (2015) 16(3):1197–200. doi: 10.7314/APJCP.2015.16.3.1197
170. Oh SA, Li MO. Tets Link Hydrogen Sulfide to Immune Tolerance. Immunity (2015) 43(2):211–3. doi: 10.1016/j.immuni.2015.08.001
171. Arvinden VR, Deva Magendhra Rao AK, Rajkumar T, Mani S. Regulation and Functional Significance of 5-Hydroxymethylcytosine in Cancer. Epigenomes (2017) 1(3):19. doi: 10.3390/epigenomes1030019
Keywords: epigenetics, TETs, inflammation, DNA-methylation, DNA-hydroxymethylation, dioxygenases, immune cell regulation
Citation: Gerecke C, Egea Rodrigues C, Homann T and Kleuser B (2022) The Role of Ten-Eleven Translocation Proteins in Inflammation. Front. Immunol. 13:861351. doi: 10.3389/fimmu.2022.861351
Received: 24 January 2022; Accepted: 28 February 2022;
Published: 21 March 2022.
Edited by:
Hai-Jing Zhong, Jinan University, ChinaReviewed by:
Varun Sasidharan Nair, Helmholtz Association of German Research Centers (HZ), GermanyCopyright © 2022 Gerecke, Egea Rodrigues, Homann and Kleuser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Burkhard Kleuser, YnVya2hhcmQua2xldXNlckBmdS1iZXJsaW4uZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.