Abstract
Systemic lupus erythematosus (SLE) is a heterogeneous disease characterized by the production of abnormal autoantibodies and immune complexes that can affect the organ and organ systems, particularly the kidneys and the cardiovascular system. Emerging evidence suggests that dysregulated lipid metabolism, especially in key effector cells, such as T cells, B cells, and innate immune cells, exerts complex effects on the pathogenesis and progression of SLE. Beyond their important roles as membrane components and energy storage, different lipids can also modulate different cellular processes, such as proliferation, differentiation, and survival. In this review, we summarize altered lipid metabolism and the associated mechanisms involved in the pathogenesis and progression of SLE. Furthermore, we discuss the recent progress in the role of lipid metabolism as a potential therapeutic target in SLE.
Introduction
Systemic lupus erythematosus (SLE) is a complicated autoimmune disease characterized by various immune defects and chronic inflammation affecting multiple tissues and organs, and with a remarkably high morbidity and mortality rate (1). Immunological abnormalities, including the production of abnormal autoantibodies directly against various components, deposition of complement-fixing immune complexes, and dysregulation of immune cells, are the major hallmarks of SLE pathogenesis. Different types of immunocytes such as dendritic cells (DCs), macrophages, and neutrophils have been implicated as key players in SLE (2). However, the complex mechanisms underlying this systemic autoimmune disease remains to be elucidated (3, 4).
Although the exact mechanisms of autoimmune responses in SLE are not well understood, rapid development of technologies offers opportunities to examine various aspects of the diseases, providing novel insights into their pathogenesis. Lipidomics, as an independent branch of metabolomics, can be used to identify temporal and spatial changes in the lipid profile, thus, unravelling the complicated etiologies at the molecular level (5–7). Multiple changes in the lipid profiles have been documented in various metabolomic studies. Lipids are composed of various types of water-insoluble molecules, including cholesterol, fatty acids, oxysterols, triglycerides, and sphingolipids, which are essential components of biological membranes and organelles (8, 9). Regulation of lipid metabolism, including lipid uptake, lipogenesis, lipolysis, and its mediators, is important for cellular homeostasis. In addition to serving as building blocks of membranes, lipids also function as sources of energy, as well as signaling molecules or secondary messengers and participate in the modulation of diverse pathophysiological processes, including cell proliferation, differentiation, migration, activation, and survival (10). Thus, imbalances in lipid homeostasis can result in toxicity and contribute to SLE (11).
Growing evidence suggests lipid-mediated activities in immunocytes and the critical role of lipid metabolic dysregulation in the pathogenesis and progression of SLE. However, the mechanisms through which lipid metabolism affects the immune system remain unclear. In this review, we summarize the growing advances in lipid metabolism in SLE that could facilitate the development of novel therapeutic targets.
Lipid Metabolism
Lipid metabolism, including anabolism and catabolism, is essential for almost every aspect of cellular funtioning (Figure 1). The fatty acid (FA) and cholesterol metabolic pathways are the most important pathways in lipid metabolism. A wide range of lipids, such as FAs, cholesterol, triglycerides, and steroids, participate in lipid synthesis critical for the cell (12). FAs are indispensable for lipid biosynthesis, the process of which involves multiple lipogenic enzymes as shown in Table 1, such as ATP citrate lyase, fatty acid synthase (FASN), acetyl-CoA carboxylase (ACC), and 3-hydroxy-3-methylglutaryl CoA reductase (HMGCR). Cholesterol, together with newly synthesized phospholipids and glycosphingolipids (GSLs), are critical components of the cell membrane microdomains called lipid rafts (45). These lipid rafts can coordinate interactions between key signaling molecules in order to regulate downstream transduction events, including modulation of antigen receptor signaling-mediated responses and the phosphoinositide 3-kinase (PI3K) pathway in SLE (46, 47). Liver X receptors α (LXRα) and β (LXRβ) are members of the nuclear receptor superfamily that are implicated in regulating lipid metabolism, including cholesterol, FAs, and phospholipids (48, 49). Furthermore, LXR maintains steady-state levels of various lipids by regulating the expression of sterol regulatory element-binding protein (SREBP), which is critical for lipogenesis (13, 14).
Figure 1

A simplified schematic representation of lipid metabolism. Lipid metabolic networks mainly involve catabolism (fatty acid oxidation (FAO)), anabolism (de novo lipogenesis) and storage as lipid droplets (LDs). Lipid raft are microdomains of the plasma membrane enriched in sphingolipids and cholesterol. The uptake of various lipids undergoes a series of processes and oxidation in the mitochondrion, which leads to the production of energy (substantial ATP) and ROS. Acetyl CoA, the end product of FAO pathway, can then either enter TCA cycle or synthesize multiple lipids, such as cholesterol and fatty acids. Lipids or derivatives of the lipid metabolism can activate transcription factors, including PPAR, LXR, then regulate activities of transporters to modulate downstream signals. Dyslipidemia, the disruption of lipid metabolism, is mainly characterized by elevated plasma levels of LDL, TG), TC), and reduced HDL.
Table 1
| Protein | Abbreviation | Functions in lipid metabolism | Participation in SLE | References |
|---|---|---|---|---|
| Sterol regulatory element-binding protein | SREBP | A lipogenetic enzyme | coordination of lipid synthesis and relevant receptors in cells | (13, 14) |
| Acetyl CoA | A lipid mediator | Alters lipid profiles and drives the TCA cycle for energy production | (15–17) | |
| Peroxisome proliferator-activated receptor | PPAR | A transcription factor control genes involved in lipid metabolism | Affects immunocytes differentiation and functions, including production of cytokines and antibodies, modulates inflammatory signals | (18–23) |
| Acetyl-CoA carboxylase | ACC | A rate-limiting in de novo lipogenesis and connects energy metabolism | Associated with differentiation and responses of T cells | (17, 24–27) |
| Fatty acid synthase | FASN | A lipogenetic enzyme | Associated with death and differentiation of immunocytes as well as cytokines production, such as IFN-γ | (28, 29) |
| AMP-activated protein kinase | AMPK | A key signal in lipid metabolic pathways | Modulates inflammatory and immune responses | (30, 31) |
| mammalian Target of rapamycin | mTOR | A key signal in lipid metabolic pathways | Important for the proliferation, activation and differentiation of immunocytes, associated with disease activity as well | (32–36) |
| Liver X receptor | LXR | A nuclear receptor controls cellular lipid metabolism | Facilitates macrophage cholesterol efflux, affects acquired immune responses, its polymorphisms are related to SLE patients | (37–41) |
| ATP-binding cassette transporters A1 and G1 | ABCA1/G1 | Mediation of the cholesterol efflux | Associated with functions of dendritic cells | (42) |
| Glutathione peroxidase 4 | GPX4 | Associated with lipid peroxidation and ferroptosis | Downregulated in neutrophils and associated with disease activity | (43, 44) |
Key genes involved in lipid metabolism of SLE.
In addition to lipid synthesis, lipid degradation is an important part of lipid metabolism, which is essential to sustain life. Lipolysis, the catabolic branch of the FA cycle, mainly refers to the process by which long-chain FAs are metabolized into acetyl-CoA (15). FAs or their derivatives can bind to and activate certain transcription factors. One of the most well-known transcription factors known to play a role in lipid metabolism is the peroxisome proliferator-activated receptor (PPAR) family, which consists mainly of PPARα, PPARγ, and PPARδ, also known as PPARβ (18). The accumulation of cellular lipids contributes to the activation of these transcription factors, including PPARγ and LXRs, and regulates the activities of transporters in order to modulate the efflux of free cholesterol and scavenger receptors. FAs generated by lipolysis are broken down through the fatty acid β-oxidation pathway in mitochondria, leading to the production of energy (ATP) and reactive oxygen species (ROS) (50). Fatty acid oxidation (FAO) is a mitochondrial aerobic process that converts FAs into multiple lipid mediators such as acetyl-CoA, which drives the tricarboxylic acid (TCA) cycle for energy production (16).
Lipid droplets (LDs) are ubiquitous organelles of lipid storage that serve as centers for lipid metabolism. The storage of neutral lipids in LDs provides energy and lipids for membrane lipid synthesis, which is critical for cell proliferation and remodeling (51). Moreover, LDs can prevent the accumulation of toxic lipids in the endoplasmic reticulum (52). The endoplasmic reticulum is an important organelle that participates in multiple physiological processes, including lipid synthesis (53). Lipid metabolism is an intricate process that generates multiple biological mediators. Many of these molecules are bioactive lipids involved in multiple signaling networks, participating in the development and progression of many inflammatory and autoimmune disorders, including SLE (54).
It is now known that lipidomics (also designated lipid profiling) can provided further insights into lipid metabolism that are beneficial to the advancement of medical research (55). In addition, lipoproteins, a group of biochemical assemblies that contain lipids and proteins, can serve as enzymes, transporters, and antigens capable of regulating multiple cellular activities. Distinct apolipoproteins regulate the metabolism of lipoproteins by participating in the transport and redistribution of lipids among cells and organs (56).
Dyslipidemia in SLE
Dyslipidemia refers to the disruption of lipid metabolism and is mainly characterized by elevated plasma levels of low-density lipoprotein (LDL), triglyceride (TG), and total cholesterol (TC) levels, as well as reduced high-density lipoprotein (HDL) levels. Dyslipidemia is believed to be involved in disease pathogenesis, especially SLE (57). Cumulative evidence strongly supports the hypothesis that patients with SLE are prone to cardiovascular complications (58). The prevalence of ischemic heart disease in SLE patients ranges from 3.8 to 16%, which is close to 10-fold higher than that observed in the general population, and 50-fold higher in young women of reproductive age (59). The prevalence of dyslipidemia is significantly higher in a retrospective study of patients with SLE than that in otherwise healthy people (60). The prevalence of dyslipidemia ranges from 68-100% among adults with SLE (61). In the Systemic Lupus International Collaborating Clinic cohort study of newly diagnosed patients with SLE (n=198), the prevalence of hypercholesterolemia was 36% (60). A 6-year population-based cohort study demonstrated that children with SLE were more susceptible to heart failure and dyslipidemia than those without SLE (62). Additionally, a cross-sectional controlled study which evaluated 33 adolescent girls with juvenile SLE found nearly half patients had dyslipidemia, including decreased HDL-c concentrations; besides, juvenile patients showed decreased concentrations of apolipoprotein A-1 (ApoA-1) and ratio of LDL/apolipoprotein B (ApoB), all of which could increase atherosclerotic risk (63). Dyslipidemia had a higher prevalence among young female patients with SLE than among controls; this was characterized by decreased TC, LDL-c, HDL-c, ApoA, and ApoB. Univariate correlational analyses exhibited complicated correlations between serum levels of certain lipids and the SLE disease activity (64). A study recruited 71 female SLE patients and found that the plasma TG level is an independent predictor of carotid atherosclerosis in women with SLE (65). In addition, dyslipidemia could affect the prognosis of SLE patients not only through CVD-related disorders but also injuries to other organs, such as lupus nephritis (LN) (66). Aberrant lipoproteins are strongly associated with abnormal renal function in chronic kidney diseases (67). Dyslipidemia is frequent and more serious in LN patients than in those with a similar degree of other chronic kidney diseases (68). High frequency of lipid abnormalities, including increased levels of TG (58.5%), TC (55.4%), LDL-c (30.8%), and lower levels of HDL (21.5%), which were strongly related to their proteinuria were found in patients with LN (69). Dyslipidemia, such as elevated TC levels, not only reflects the activity but also the severity of renal injury (70, 71). Overall, dyslipidemia as a significant risk factor can result in renal failure and higher mortality in patients with LN (70). Notably, systemic corticosteroids when used at high doses or for a prolonged duration, lead to adverse events such as immunosuppression and dyslipidemia.
Low HDL levels are one of the most common dyslipidemia observed in patients with SLE (72). It is a consensus that HDL-c, which can be routinely detected in clinical practice, is considered synonymous with HDL particle levels. Lupus HDL promotes pro-inflammatory responses through activation of the nuclear factor kappa B (NF-κB) (73). Serum cholesterol efflux capacity is impaired in SLE, with a specific mechanism pattern in each disease, independent of serum HDL levels (74). Another study found a higher prevalence of dyslipidemia in patients with SLE (46.2%) than that in controls (19.2%); besides, the patient group had higher Cys levels and lower HDL-c levels compared with the control (75). ApoA-1 is the main protein fraction of HDL and can stabilize paraoxonase-1 (PON-1), which protects LDL from oxidation (76). The ratio of baseline ApoB : ApoA-1 has been shown to correlate negatively with a low disease activity, accordingly, positively with the SLE disease activity index (SLEDAI) over a 5-year follow-up period (77). Elevated levels of antibodies against HDL and ApoA1 have been reported in patients with cardiovascular disease (CVD), and are significantly higher in patients with SLE, the levels of which were negatively correlated with the activity of paraoxonase (PON) (78). Moreover, PON1 arylesterase activity and total HDL antioxidant capacity are significantly decreased in patients with SLE compared with that in controls. All HDL sub-fraction levels and HDL antioxidant abilitiy were positively correlated with PON1 arylesterase activity and negatively correlated with disease activity as well as the examined inflammatory markers of interest, such as hsCRP and IL-6 (72). In autoimmune mouse models, autoantibodies against ApoA-1 contribute to a reduction in HDL-c levels and a lower PON1 activity independent of hepatic HDL synthesis (79). Additionally, studies have found negative correlations between serum HDL-c levels and disease activity. Two dyslipidemia patterns are evident in pediatric active SLE, characterized by elevated TG and low HDL levels (80). Patients with active disease show lower levels of HDL, which can be elevated after the use of prednisone and hydroxychloroquine (81). A study aimed to identify potential biomarkers of disease activity by analyzing the proteome of HDL particles in patients with SLE, the authors found that the plasma gelsolin levels decreased significantly in patients and were significantly associated with HDL-c levels, especially when patients developed a clinical flare, suggesting an association between dyslipidemia and disease activity (82).
A cohort study reported that, in pediatric SLE patients, higher LDL levels were associated with disease activity (83). Ricardo et al. obtained circulating LDL particles from SLE patients and found that LDL from a flare was more atherogenic and induced greater endothelial cell migration than LDL from inactive patients (84). Lipids are highly susceptible to oxidation, and products of lipid peroxidation are potential biomarkers suggestive of oxidative stress condition in SLE (85). The oxidative modification of LDL is an earlier event in atherosclerosis. Oxidized LDL (ox-LDL) is a common lipid peroxidation marker, represents various modifications of lipids by lipid peroxidation. Patients with SLE (especially those with CVD) show more oxidized epitopes on LDL than controls, and anticardiolipin antibodies in these patients recognize epitopes produced during lipid peroxidation (86, 87). Moreover, cross-reactivity was found between antiphospholipid antibodies, which are closely related to thrombosis, and antibodies to ox-LDL in SLE (88). More specifically, an increase in plasma levels of L5 (a sub-fraction of electronegative LDL), but not total LDL concentrations, may contribute to early vascular aging and premature atherosclerosis in SLE patients (89). SLE patients had higher levels of circulating ox-LDL, and patients with dyslipidemia were given a higher cumulative prednisolone dose than controls (90). Taken together, higher LDL and ox-LDL levels are positively correlated with a higher risk of cardiovascular complications in patients with SLE. An amount of HDL is dysfunctional and can not inhibit the oxidation of LDL in SLE patients.
Of note, as a conventional treatment in SLE, corticosteroids, particularly when used at high doses or prolonged duration, leads to adverse events such as immunosuppression and dyslipidemia. Corticosteroid-treated female patients with SLE have manifestations of higher levels of plasma ApoB as well as hyperlipidemia including VLDL, TG and LDL (91). While, among juvenile SLE, the cumulative corticosteroid dose was associated with a decrease in HDL-c (92). Interestingly, glucocorticoids regulate both lipid synthesis and degradation under different circumstances. The metabolic functions of glucocorticoid are mediated by the glucocorticoid receptor, a nuclear hormone receptor expressed various kinds of cells (93). Many mediators of the glucocorticoid receptor have been identified to regulate the lipid metabolism. Moreover, mechanisms of dyslipidemia induced by steroids are complex, including hepatic insulin resistance, higher levels of lipogenesis enzymes such as acetyl-CoA carboxylase and free fatty acid synthetase, lower levels of lipoprotein lipase and LDL receptor (94, 95). Therefore, if hormones are needed in long-term or large amount for SLE treatment, monitoring lipid profiles in blood are recommended. Notably, as the cornerstone in the treatment of SLE, antimalarials- hydroxychloroquine was demonstrated in a longitudinal study to exert favorable effects on lipids in SLE since the 3-month of hydroxychloroquine induced a significant reduction of TC and LDL, which determined an obvious decrease in the frequency of dyslipidemia (96). Similarly, a meta-analysis quantitively measured the beneficial effect of hydroxychloroquine on serum LDL levels in patients with SLE, and pooled data showed that hydroxychloroquine could lead to a reduction of mean LDL levels by 24.397 mg/dL (P = 0.002) (97). The effects of antimalarials on lipid profiles partially may because of an overall reduction in hepatic lipid synthesis (98). Besides, there is evidence that the lower levels of LDL induced by chloroquine in SLE are associated with the upregulation of LDL receptors (99). Hence, the benefits of hydroxychloroquine treatment in SLE not only because of its direct effects on disease activity but also its indirect effects on lipids to protect against dyslipidemia, especially those with higher cardiovascular risk or treated with glucocorticoids (100). While, dyslipidemia in SLE could also be induced by other immunosuppressant agents. Calcineurin inhibitors (e.g., tacrolimus and cyclosporine) widely used as immunosuppressants in autoimmune diseases especially in lupus nephritis, have side effects of hyperlipidemia as well (101). Specifically, cyclosporine could lead to hyperlipidemia including increased levels of TG, TC, LDL and Apo-B, partially through inhibiting certain hydroxylases and lipoprotein lipase (102). Besides, cyclosporine downregulates the LDL receptors and disturbs their binding of LDL-c, resulting in a decrease in cholesterol and LDL clearance respectively (95). What’s more, a novel calcineurin inhibitor, voclosporin, was approved for the treatment of adult patients with active lupus nephritis by the US Food and Drug Administration last year, exerts a more beneficial effect on lipid profiles such as lower levels of cholesterol and LDL than tacrolimus (103, 104). Meanwhile, another widely used immunosuppressant, cyclophosphamide can also reduce serum levels of LDL and VLDL in lupus nephritis patients (105). Overall, further explorations are needed to measure clinical benefits and lipid metabolic effects of these common drugs used in clinical practice.
Lipid Metabolism in Immunocytes in SLE
Current evidence suggests that lipids and lipid metabolites play an important role in the immune system by directly acting on immune cells through indirect modulation of antigen presentation and cytokine production (106). In view of the emerging role of lipid metabolism in autoimmune disorders, an increasing number of studies are exploring lipids in immunocytes in SLE (Figure 2). Aberrant immunity with inflammatory mediators and dysfunctional immune cells, especially the loss of T- and B-cell tolerance to self-antigens, are the major characteristics of SLE pathogenesis (107, 108). It has been reported that compared with healthy controls, lipidomics analysis of peripheral blood mononuclear cells from SLE patients revealed altered lipid metabolism, including significantly increased lysophospholipids, decreased plasmalogens, and altered phosphatidylserines (109).
Figure 2
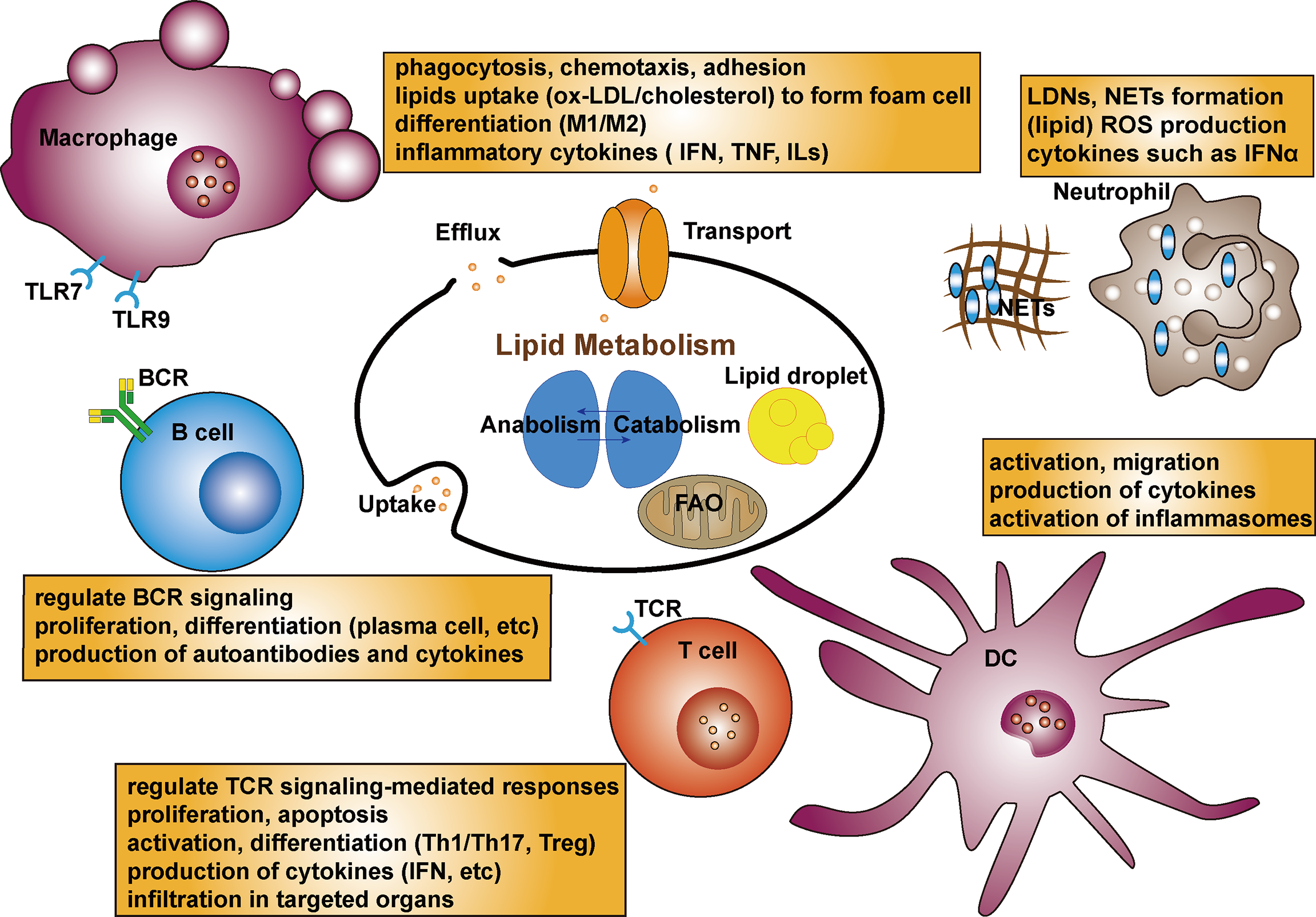
Schematic diagram of the lipid metabolism in immunocytes in SLE. Lipid rafts can regulate TCR signaling-mediated responses. Lipid metabolism as an important facilitator of T cell differentiation, can activate CD4+T cells then differentiate into Th or Treg cells. Lipid biosynthesis including cholesterol and fatty acid is critical for proliferation, apoptosis and production of cytokines in T cells. Dyslipidemia can promote the autoantibodies in B cells. Lipid rafts also regulate BCR signaling. Lipids composition could affect B cell proliferation, differentiation and production of autoantibodies as well as interleukins. Fatty acids can modulate the process of macrophage differentiation as well as adhesion on endothelial cells and their chemotaxis. The uptake of ox-LDL by macrophages uptake by the macrophage is the initial step to foam cell formation. Targeting receptors associated lipid metabolism can affect differentiation of monocytes and production of proinflammatory cytokines, which are related to TLR7 and TLR9 expression. Lipid profiles promote activation and migration of DCs. LDLs promote the production of IL-6 in DCs. Besides, cholesterol-enriched DCs show augmented cytokine secretion and activation of inflammasome. A subset of lupus proinflammatory neutrophils (LDGs) increase and promote risk of atherosclerosis. Oxidation of cholesterol and phospholipids can lead to lipid peroxidation, and higher level of ROS production with enhanced catalase activity of neutrophils are seen in SLE. NETs stimulate production of IFNs and proinflammatory cytokines. NETs possess active oxidant-generating enzymes, and modify HDL.
Lipid Metabolism in Lymphocytes in SLE
T cells, especially CD4+T cells, are important in driving B cell production of abnormal autoantibodies and are central to SLE pathogenesis (110). Abnormal lipid metabolism can impair T-cell function in SLE. Lipid rafts can regulate T-cell antigen receptor (TCR) signaling-mediated responses in T cells. Lipid raft analysis demonstrated that cholesterol efflux mediated by HDL and ApoA-1 could link immunity and cardiovascular protection. ApoA-1 and HDL decreased cholesterol and major histocompatibility class II protein content, indicating that cholesterol efflux from antigen-presenting cells to HDL and ApoA-1 could block antigen presentation and T-cell activation by decreasing lipid raft assembly (111). A study indicated that increased FcR and activated Syk kinase in SLE T cells have more ganglioside-containing lipid rafts; meanwhile, cross-linking of lipid rafts could promote earlier and stronger calcium responses (112). Therefore, changes in lipid rafts can strengthen and promote T-cell activities in SLE. Furthermore, aggregation of lipid rafts and rewiring of the CD3 complex grants T cells a lower activation threshold and distorts downstream signalings, consequently directing the abnormal expression of adhesion molecules in aggregated lipid rafts to relavant targeted organs (113). Treatment targeting lipid raft-associated signaling has been demonstrated to restore defects in T cell activation, and reduce the production of pro-inflammatory cytokines in lupus auto-reactive T cells, which provides a rationale for further research (114). Glycosphingolipids (GSLs), which are composed of a ceramide backbone, are enriched in lipid rafts. Increased GSL synthesis is a feature of T cells in SLE and is closely related to TCR activation. CD4+ T cells from patients with SLE show an aberrant profile of lipid raft-associated GSLs compared with that from volunteers, and elevated GSLs are suggested to be associated with overexpression of LXRβ, a nuclear receptor mentioned before affects acquired immune responses as well (115). In addition to its influence on CD4+ T cell signaling and function, the inhibition of GSL biosynthesis in vitro can decrease the production of anti-dsDNA antibody by activated B cells. Similarly, decreasing the levels of GSLs in CD4+ T cells led to a moderation of the T cell response toward activation; concomitantly, dramatic decreasing IL-2 production and T-cell proliferation have been observed as well (116). In lymphocytes, the synthesis of GSLs is mainly controlled by the transcription factor Friend leukemia integration 1 (Fli1), which can affect the function of immune cells and implicated in the pathogenesis of autoimmune disorders (117). A significant decrease in the number of CD4+T cells was observed in the kidneys of MRL/lpr Fli1(+/-) mice compared with that of Fli1(+/+) nephritic mice; reducing Fli1 in lupus-prone mice would protect against lupus nephritis partially by regulating CXCR3 expression on T cells and reducing T-cell infiltration of kidneys (118).
Lipid metabolism is increasingly recognized as an important facilitator of T cell differentiation. Activated CD4+T cells can differentiate into T helper (Th) cells or inducible T regulatory (Treg) cell subsets with distinct immunological functions. Lipid biosynthesis, including cholesterol and fatty acids, is critical for the proliferation and differentiation of T cells, especially a subset of Th17 cells (119). Th17 cells are featured by the expression of their signature cytokine IL-17A and the master transcription factor retinoid-related orphan receptor (120). Acetyl-CoA is an important metabolite involved in the synthesis, degradation, and remodeling of various lipids, thus altering lipid profiles. Acetyl-CoA carboxylase 1 (ACC1) associates key energy metabolism with lipogenesis and is a rate-limiting enzyme involved in de novo lipogenesis through the carboxylation of acetyl-CoA (17). Most de novo synthesized fatty acids are incorporated into phospholipids or localized to lipid rafts to be implicated in membrane-related activities, and some are stored in the form of lipid droplets to buffer excess lipids. T cells deficient in specific ACC1 with impaired lipid synthesis show reduced potential for differentiation of naive CD4+T cells into both Th1 and Th17 cells and higher proportions of Treg cells (24). Regulation of de novo fatty acid synthesis by targeting ACC1 in T cells could be therapeutic for autoimmune and inflammatory diseases (25, 26). Mice with a deletion of ACC1 specifically in T cells, failed to respond efficiently and were susceptible to infection, suggesting the critical role of ACC1-dependent fatty acid synthesis in T cells to fight exogenous infection (27). Voss et al. explored the effects of lipid metabolism on apoptosis sensitivity in T cells, showing that inhibition of FASN protected human CD4+T cells from restimulation-induced cell death (RICD), the process of which relied on sphingolipid synthesis and FAO (28). FASN, downstream of ACC, is a key lipid metabolic regulator for the generation of inflammatory Th17 cells, and inhibition of FASN contributes to the production of IFN-γ by Th17 cells (29). Studies in both human and murine models have indicated the essential role of Th17 cells in SLE pathogenesis; thus, targeting mediators implicated in the regulation of Th17 would be meaningful (121). The CD95 ligand (CD95L) is expressed by various immunocytes and can trigger apoptosis. Metalloprotease-cleaved CD95L (cl-CD95L) is implicated in homeostasis of the immune system (122). Th17 cells stimulated with cl-CD95L could produce sphingosine 1-phosphate (S1P), a potent lipid mediator distributed to HDL, through which Th17 cells transmigrate across the endothelial barrier; thus, exploring the CD95 relevant signaling pathway could be therapeutic for SLE patients (123, 124). Inhibiting FA synthesis in memory CD4+T cells of SLE patients decreased interferon-γ production and increased Foxp3 expression in T-bet+Foxp3+ cells (125).
Oxidative stress is augmented in patients with SLE, as indicated by increased products of lipid peroxidation such as malonaldehyde (MDA) and decreased antioxidants such as glutathione, which are implicated in the regulation of the immune system (126). AMP-activated protein kinase (AMPK), an important regulator in metabolism, is a rate-limiting enzyme in promoting FAO, while AMPK-dependent ACC phosphorylation can inhibit de novo lipogenesis (30). Treg cells activate AMPK and are dependent on lipid oxidation (31). In T cells, the dysfunction of mitochondrion promotes lipid hydroperoxides release, which spread oxidative stress to other organelles and peripheral blood (127). A case-control study enrolled 204 SLE patients and 256 healthy volunteers and measured their lipid peroxides and found that increased peroxides were associated with a Th1 and Th17 immune shift (128). Moreover, AKT/mTOR signaling is important in SLE and is activated in T cells of patients (32). It is known that in activated immune cells, mTOR promotes lipid synthesis and is critical for the differentiation of CD4+T and CD8+T cells (33). Recently, a cohort of juvenile-onset SLE patients found that patients with a higher ratio of ApoB : ApoA1 exhibited higher frequencies of CD8+T cells; besides, a CD8+T-cell transcriptomic profile enriched in genes related to atherosclerosis, including those related to interferon signaling (77). Invariant natural killer T (iNKT) cells, which exhibit defective autoimmunity can promote atherosclerosis through CD1d-mediated lipid antigen presentation, and link lipids with immune responses in SLE patients with CVD (129). The inhibitory receptor B and T lymphocyte attenuator (BTLA) can negatively modulate immune responses. The BTLA signaling pathway is altered in SLE T cells, and the impaired capacity of BTLA can be corrected by normalizing the lipid metabolism in lupus CD4+ T cells (130).
Emerging evidence suggests that B cells in autoimmunity are accompanied by significant shifts in metabolic processes, thus determining the mechanisms of this new area of active study.
In animal models of SLE, hyperlipidemia promoted the production of autoantibodies in B cells by inducing autoimmune CXCR3+ follicular helper T cells (131). However, studies involving the role of lipid metabolism in B cells in SLE patients is limited relative to T cells. Defective control of cellular signals in lymphocytes, such as B-cell receptor (BCR) signaling, could directly result in autoimmunity in SLE (132). Assessment of B cells from patients with SLE showed larger stained lipid rafts, and lower molecular weight isoform of CD45 in lipid rafts compared with healthy controls; the abnormal CD45 translocation is associated with the dynamics of Lyn (a negative regulator of BCR signaling) in lipid rafts and BCR–antigen contact regions (133). Similarly, purified lipid raft signaling domains in B cells collected from a series of SLE patients showed lower Lyn levels and abnormal translocation to lipid rafts, and these changes in Lyn were in relation to increased spontaneous proliferation, production of anti-dsDNA auto-antibodies and cytokines (134). Apart from the importance of LXRs in lipid metabolism, they can modulate functions of immunocytes as well (37). Pharmacological activation of LXRs ameliorates disease activity in lupus-prone mice (38). The authors found that a cholesterol-containing diet in ApoE/LXRβ-deficient mice would result in lupus-like disease, and defects of LXRβ in CD11c+cells led to B-cell expansion (39). In addition to T cells, over-activation of mTOR signaling in B cells also relates to plasmablast counts and SLE activity (34). Evidence suggests that the mTORC1 pathway is prominently activated in lupus Atypical memory B cells (AtMs), and blocking mTORC1 signaling significantly reduces the terminal differentiation of AtMs (35). Mechanisms of mTOR signaling are complicated by B cell formation, activation, and differentiation in SLE. Lupus-prone mice treated with mTOR inhibitors showed a reduction in B cells differentiation into germinal centers and plasma cells, and lower disease activity (36). Recently, a study suggested a molecular link between the dysregulation of lipid metabolism and the pathogenesis of lupus (135). The authors collected B cells from patients with lupus and found that inositol-requiring enzyme 1α was positively correlated with the amount of B cell lipid deposition, demonstrating that this enzyme could control plasma cell differentiation by modulating the coenzymes associated with fatty acid synthesis. Fatty acid composition can affect B-cell differentiation into autoantibody-producing plasmablasts in autoimmunity. Alterations in the activation of immunocytes caused by omega-3 fatty acids (a family of polyunsaturated fatty acids, PUFAs) have been described for many years (136). Supplementation with omega-3 FAs could reduce autoantibody production and immunocomplex deposition, and hinder interferon and chemokine gene expression in lupus (137, 138). Consistently, dietary short-chain FAs can regulate B cell differentiation, which critically underpins effective T-dependent and T-independent antibody responses in lupus-prone mice (139). Overall, these findings support the hypothesis that dietary supplementation with specific fatty acids can attenuate lupus disease activity by affecting B cells.
Lipid Metabolism in Innate Immune Cells in SLE
Macrophages are also important in the regulation of SLE by engulfing various lipids through phagocytosis. Nitro-fatty acids, reactive lipid species produced by metabolic and inflammatory reactions, can modulate immune cell functions, thus exerting an influence on various pathologies, particularly inflammation (140). Moreover, nitro-fatty acids can modulate the process of macrophage differentiation and adhesion to endothelial cells and their chemotaxis (141). Notably, the ox-LDL uptake through macrophage scavenger receptors is a key initial process in the formation of foam cells, which is a critical precursor to atherosclerosis (142). Interferon-α priming can enhance the uptake of ox-LDL in macrophages, which promotes the formation of foam cells in vitro, and monocytes from patients with SLE display higher lipid uptake (143). In a lupus-prone mouse model, FcgRIIB knockout (KO) mice, dysregulation of lipid metabolism in macrophages was found to be responsible for lipopolysaccharide (LPS) tolerance, such as higher levels of phosphatidylethanolamine (PE), a class of phospholipids in biological membranes found in their macrophages (144). Exploring approaches to regulate lipid metabolism is a potential strategy to harness LPS tolerance in lupus. Transcriptional regulation through PPARγ is essential for changes in lipid metabolism during monocyte differentiation into macrophages. Blocking PPARγ-dependent signaling during monocyte differentiation could result in a macrophage phenotype characterized by attenuated reduced inflammatory responses to oxidized lipoproteins and saturated FAs, which could benefit patients with cardiometabolic disorders (145). Agents targeting PPARγ can promote the differentiation of monocytes towards an M2 phenotype and ameliorate the condition of SLE patients (19). The enzyme 12/15-PG (12/15-LO) oxygenates free and phospholipid-bound PUFAs. The expression of 12/15-LO is induced by the stimulation with some interleukins and is restricted to certain macrophages. It has been revealed that defective 12/15-LO activity led to an abnormal monocyte phagocytosis, subsequent antigen presentation of aberrant antigens derived from apoptosis, and a lupus-like autoimmune disease (146). Defects in eliminating dying cells contribute to the accumulation and secondary necrosis of apoptotic cells, which release signals such as S1P to induce phagocytic removal. S1P, a mediator produced from membrane sphingolipids, could regulate trafficking of adaptive immunocytes (147). Dying cell-derived S1P activates erythropoietin (EPO) signaling in macrophages, then enhances clearance of dead cells through PPARγ signaling. Consistently, macrophage-specific Epor(-/-) mice could develop lupus-prone models, the condition of which was improved by interference with S1P-EPO signaling (148). Liver X receptors (LXRs) play pivotal roles in the transcriptional control of cholesterol and lipid metabolism; its isotype, LXRα is extensively expressed in macrophages. Several animal models of atherosclerosis have demonstrated that stimulating LXRs could reduce disease development, which could be attributed in part to the capability of LXRs to facilitate macrophage cholesterol efflux (40). LXRα gene promoter polymorphisms are reported to be related to Korean SLE patients (41). A study assessed cytokine expression related to LXRα polymorphisms in monocyte-derived macrophages obtained from SLE patients and found that higher levels of pro-inflammatory cytokines are associated with the expression of Toll-like receptors (149). Overall, LXRs play complicated roles in macrophages and can connect the inflammatory response and lipid metabolism in SLE. Furthermore, it has been reported that in lupus-prone mice, injuries of end-organs could be rescued by an agonist of LXR (150). Hence, future investigations focusing on the role of LXRs in pharmacotherapy of patients with SLE are warranted.
Dendritic cells (DCs) are known for their ability to link innate and adaptive immune processes, and their uncontrolled activation drives autoimmunity (151). DCs can sense cytokines, danger signals, and lipid species such as saturated fatty acids and ox-LDL. Altered lipid concentrations or stimulation by lipids can modulate the functions of DCs (152). The direct administration of LDLs and ox-LDL to DCs could promote the production of IL-6, which in turn would enhance susceptibility to autoimmune diseases by regulating pathogenic autoimmune Th17 polarization and inflammatory functions (153). Lipid profiles that reflect atherosclerosis have been proven to lead to local activation of murine DCs in the skin, promote dermal inflammation, and induce lymph node hypertrophy (154). Cholesterol efflux mediated by HDL is dependent on the ATP-binding cassette transporters A1 and G1 (ABCA1/G1). Notably, mice deficient in ABCA1/G1 in DCs develops autoimmune nephritis; DC specific deficiency of ABCA1/G1 enhances the activation of T cells, as well as the polarization of Th1 and Th17 cells; cholesterol-enriched DCs shows augmented cytokine secretion and activation of inflammasomes (42). Increased expression of type 1 interferon (IFN)-regulated genes is considered a hallmark of SLE, and human plasmacytoid DCs are important in SLE pathogenesis through enhanced IFNα (155). Mitochondrial mitochondrial import of pyruvate and FA synthesis are important to the increased FAO in IFN-activated plasmacytoid DCs, and these changes in lipid metabolism are PPARα-dependent and critical to cell and cytokine functions (20). Given the importance of IFNs in SLE, there is a clinical need to explore approaches such as metabolic targeting as a potential adjunct to conventional treatment strategies.
Neutrophils can interact with various immunocytes and are implicated in the pathogenesis of inflammatory and autoimmune diseases (156). A distinct subset of lupus proinflammatory neutrophils (LDGs) has been found to increase the risk of CVD in SLE (157). The noncalcified plaque burden enhances the prevalence of high-risk plaques in some inflammatory conditions. Accordingly, the noncalcified plaque burden was found to be elevated in SLE, and was associated with LDGs and cholesterol efflux capacity. Neutrophils can undermine the function of HDL, thereby promoting atherogenesis (158). Substantial interest has been given to study the involvement of heightened levels of oxidative stress in SLE, which contributes to dysregulated immune system. In one study, a cafeteria diet-fed lupus-prone mice showed increased cholesterol levels, and neutrophils exhibited enhanced ROS production capacity, which may contribute to accelerated SLE onset (159). Moreover, SLE patients showed increased ROS production and enhanced catalase activity in neutrophils. Neutrophils are characterized by lower levels of Malondialdehyde (MDA), a lipid peroxidation biomarker, and high levels of protein oxidation (160). The oxidation of cholesterol and phospholipids containing PUFA chains can contribute to the lipid peroxidation, in which lipid hydroperoxides are critical intermediates. Mechanistically, inhibition of glutathione peroxidase 4 (GPX4) results in lipid peroxidation and induction of ferroptosis, a regulated cell death featured by the aggregation of lipid ROS dependent on the iron (43).
Recently, a study found that mice with neutrophil-specific GPX4 haploinsufficiency recapitulated the critical clinical manifestation of SLE patients, and the disease activity in mouse models could be obviously ameliorated by a specific ferroptosis inhibitor (44). Current evidence suggests that neutrophils in patients show the ability to form neutrophil extracellular traps (NETs), which could stimulate the production of IFNs and multiple cytokines. NETs possess oxidant-generating enzymes such as myeloperoxidase. Experiments confirmed that active oxidant-generating enzymes within NETs, such as MPO, could modify HDL and render lipoprotein proatherogenic (161). Taken together, these findings suggest an emerging role for NET-derived lipid oxidation in SLE-relevant atherosclerosis, which could be targeted in future therapies.
Targeting Lipid Metabolism Associated with Immunity for SLE Treatment
Current drugs used for SLE treatment include various immunosuppressive agents that can cause adverse reactions. As lipid-associated metabolic dysregulation has been implicated in the pathogenesis and progression of SLE and/or complications, including lupus nephritis and cardiovascular diseases, targeting lipid metabolism is a potential adjunct treatment in these situations. Notably, steroids, which are widely used in patients with SLE, can alter metabolite profiles and increase cardiovascular risk. Glucocorticoids have been reported to exhibit metabolic effects by increasing the production of leptin, which may activate mTOR (162). Immunosuppressive drug, mycophenolate mofetil (MMF), could prevent SLE-associated CVD by inhibiting CD4+ T cell activation and infiltration into atherosclerotic lesions, which was accompanied by a decline in IgG1 serum titers to ox-LDL (163). Therefore, treatment with MMF alone or in combination with statins may benefit patients with SLE and atherosclerosis. The B cell-activating factor (BAFF) receptor pathway is of importance for the promotion of B-cell differentiation and survival of plasma cells. Overexpression of BAFF in the model of transgenic mouse increases lupus-like autoantibodies in B cells (164). In 2011, the US Food and Drug Administration approved an antibody against soluble BAFF as a treatment for patients with SLE. A study demonstrated that inhibiting the BAFF receptor ablated B-2 cells and reduced the frequency of mouse atherosclerosis (165). BAFF blockade could be expected to lower the cardiovascular risk in patients with SLE. IFN, low-density neutrophils (LDNs), and NETs are now considered as potential key players in SLE-associated vascular damage. The Janus kinase (JAK) inhibitor tofacitinib has been demonstrated to improve immunity and vascular disorders in lupus-prone models by exerting effects on immune cells and cytokines (166). Recently, a phase 1 double-blind randomized trial of tofacitinib demonstrated to improve HDL-c levels, particle numbers, and cholesterol efflux capacity. The use of tofacitinib significantly reduced the type I IFN gene signature, levels of LDNs and circulating NETs, which consequently ameliorated SLE and its associated vascular dysfunction (167). In addition, baricitinib, another JAK inhibitor that controls intracellular signaling induced by cytokines such as IL-6 and IFN, was proven more effective in ameliorating clinical manifestations of SLE patients than placebo in a phase IIb clinical trial (168). Considering the role of PPARγ in the pathogenesis of SLE, agents targeting PPARγ may serve as adjuvants for the treatment of SLE. Pioglitazone, a PPARγ agonist, regulates CD4+T cell function and modulates the differentiation of monocytes and monocyte-derived macrophages, as previously reported in lupus (19, 21). Moreover, another PPARγ agonist, rosiglitazone, downregulates the production of autoantibodies and prevents atherosclerosis and lupus nephritis (22). The disruption of CD40 signaling influences atherogenesis in hyperlipidemic mice (169). PPARγ modulates inflammatory signals induced by the activation of CD40, which complicates the mechanisms of PPARγ in SLE (23). Combined with glucocorticoids, rosiglitazone induced stable tolerogenic dendritic cells in monocytes derived from patients with SLE (170). Taken together, PPAR-γ agonists could be further explored as potential treatments for SLE. Statins exert beneficial vascular effects in SLE through mechanisms including inhibition of cholesterol biosynthesis and immunomodulatory functions. Monocytes collected from patients with SLE were treated in vitro with fluvastatin, and cells showed altered mitochondrial membrane potential and higher oxidative stress, and in vivo fluvastatin treatment improved the condition of patients with reduced lipid levels and vascular inflammation (171). Antiphospholipid antibodies (aPL) (occur in 30–40% of patients with SLE) are associated with increased risk of atherosclerosis as well. A prospective open-label pilot study demonstrated that some biomarkers, including TNF-α and soluble CD40L, upregulated in aPL-positive patients can be reversibly reduced by fluvastatin (172). Lipidomics analysis of SLE showed that alterations in lipids in peripheral blood mononuclear cells could be improved after treatment with an antioxidant in vitro, and the levels of pro-inflammatory cytokines as well as IgG autoantibodies were significantly reduced after treatment in vivo (83).
Omega-3 fatty acids, which was proven to reduce SLE activity, should also be considered in addition to the conventional regimens used for SLE (173). It is emerging that the excessive release of STING-dependent type I IFNs is implicated in certain interferonopathies, including SLE. Nitro-fatty acids can affect the release of type I IFN by modifying STING-associated signaling. Besides, endogenously formed nitro-fatty acids can serve as inhibitors of the STING-IFN axis. Further research on the novel roles of these lipids in the treatment of SLE is needed (174). Overall, understanding the mechanisms of lipid metabolism in SLE, particularly in pathogenic immunocytes, offers an opportunity to develop novel adjunct therapies to ameliorate progression of lupus and its related complications.
Conclusion and Future Perspectives
In this review, we illustrated a complicated lipid metabolic network in SLE. This highlights that lipid synthesis, oxidation, degradation, metabolites, and related signaling processes are implicated in immunocyte activation, differentiation, and immune responses. Further investigation is required to develop new approaches targeting lipid metabolism to treat patients with SLE. Lipogenesis is a major target for treatment because cells rely on lipids that provide energy and related signaling for their basic activities. Moreover, immunocytes rely on lipids to exert their specific functions in response to stimuli; thus, balancing the effects of lipid metabolism on immunocytes and the internal environment is necessary to explore adjunct targets. Nevertheless, certain questions and challenges, such as metabolism being tissue-or organ-specific, and many aspects of lipid metabolic changes may even be unique to distinct immunocyte types, not to mention their sophisticated complexity at the biochemical level, remains to be addressed Although there are promises for clinical translation, the results of the emerging therapies are variable. Moreover, the mechanisms of lipid metabolism acting directly or indirectly on SLE are complex, and multiple factors have been implicated. The involvement of lipids in the immune system, especially among different cell populations, emphasizes the need to study the effects of interfering with lipid metabolism in suitable in vivo models, particularly in combination with conventional immunosuppression therapies. Rational strategies for SLE therapies should be further explored owing to the availability of agents targeting various parts of lipid metabolism. Lipidomics, an approach to provide global profiles of lipids, has improved significantly with the advancement of next-generation mass spectrometry instruments, and bioinformatics; all of which promoted lipid biology to the forefront of metabolism studies (175). Insights into the regulation of lipid metabolism may provide novel therapeutic strategies for SLE treatment.
Funding
This work was supported by National Natural Science Foundation of China (81788101, 81630044), Chinese Academy of Medical Science Innovation Fund for Medical Sciences (CIFMS) (2021-1-I2M-017, 2021-1-I2M-047, 2021-1-I2M-040, 2021-1-I2M-016, and 2021-1-I2M-026), and Capital’s Funds for Health Improvement and Research (2020-2-4019).
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Author contributions
YL and YS contributed to support the conception of the review. WS and PL contributed to writing the manuscript and making figures. JC, JM, and XZ read, discussed, and revised the manuscript. All authors listed have approved the submitted version and publication.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Glossary
| SLE | systemic lupus erythematosus |
| DCs | dendritic cells |
| FA | fatty acid |
| FAs | fatty acids |
| FASN | fatty acid synthase |
| ACC | acetyl-CoA carboxylase |
| HMGCR | 3-hydroxy-3-methylglutaryl CoA reductase |
| GSL | glycosphingolipid |
| PI3K | phosphoinositide 3-kinase |
| LXR | liver X receptor |
| SREBP | sterol regulatory element-binding protein |
| PPAR | peroxisome proliferator-activated receptor |
| ROS | reactive oxygen species |
| FAO | fatty acid oxidation |
| TCA | tricarboxylic acid |
| LDs | lipid droplets |
| LDL | low-density lipoprotein |
| TG | triglyceride |
| TC | total cholesterol |
| HDL | high-density lipoprotein |
| ApoA-1 | apolipoprotein A-1 |
| ApoB | apolipoprotein B |
| LN | lupus nephritis |
| NF-κB | the nuclear factor κB |
| PON-1 | paraoxonase-1 |
| SLEDAI | Systemic Lupus Erythematosus Disease Activity Index |
| CVD | cardiovascular disease |
| ox-LDL | oxidized LDL |
| TCR | T-cell antigen receptor |
| Fli1 | Friend leukaemia integration 1 |
| Th | T helper |
| T | Treg, T regulatory |
| RICD | restimulation-Induced Cell Death |
| S1P | sphingosine 1-phosphate |
| IFN-γ | interferon-γ |
| MDA | malonaldehyde |
| AMPK | AMP-activated protein kinase |
| iNKT | invariant natural killer T |
| BTLA | B and T lymphocyte attenuator |
| BCR | B cell receptor |
| AtMs | atypical memory B cells |
| PUFA | polyunsaturated fatty acids |
| EPO | erythropoietin |
| ABCA1/G1 | ATP Binding Cassette Transporters A1 and G1 |
| LDGs | low density granulocytes |
| GPX4 | glutathione peroxidase 4 |
| NETs | neutrophil extracellular traps |
| MMF | Mycophenolate mofetil |
| BAFF | B cell-activating factor |
| LDNs | low-density neutrophils |
| JAK | Janus Kinase |
| aPL | antiphospholipid antibodies |
References
1
Bernatsky S Boivin JF Joseph L Manzi S Ginzler E Gladman DD et al . Mortality in Systemic Lupus Erythematosus. Arthritis Rheum (2006) 54:2550–7. doi: 10.1002/art.21955
2
Pathak S Mohan C . Cellular And Molecular Pathogenesis of Systemic Lupus Erythematosus: Lessons From Animal Models. Arthritis Res Ther (2011) 13:241. doi: 10.1186/ar3465
3
Gergianaki I Bortoluzzi A Bertsias G . Update on the Epidemiology, Risk Factors, and Disease Outcomes of Systemic Lupus Erythematosus. Best Pract Res Clin Rheumatol (2018) 32:188–205. doi: 10.1016/j.berh.2018.09.004
4
Teruel M Alarcón-Riquelme ME . The Genetic Basis of Systemic Lupus Erythematosus: What Are the Risk Factors and What Have We Learned. J Autoimmun (2016) 74:161–75. doi: 10.1016/j.jaut.2016.08.001
5
Wang M Wang C Han RH Han X . Novel Advances in Shotgun Lipidomics for Biology and Medicine. Prog Lipid Res (2016) 61:83–108. doi: 10.1016/j.plipres.2015.12.002
6
Han X . Lipidomics for Studying Metabolism. Nat Rev Endocrinol (2016) 12:668–79. doi: 10.1038/nrendo.2016.98
7
Kang J Zhu L Lu J Zhang X . Application of Metabolomics in Autoimmune Diseases: Insight Into Biomarkers and Pathology. J Neuroimmunol (2015) 279:25–32. doi: 10.1016/j.jneuroim.2015.01.001
8
Holthuis JC Menon AK . Lipid Landscapes and Pipelines in Membrane Homeostasis. Nature (2014) 510:48–57. doi: 10.1038/nature13474
9
Torres M Parets S Fernández-Díaz J Beteta-Göbel R Rodríguez-Lorca R Román R et al . Lipids in Pathophysiology and Development of the Membrane Lipid Therapy: New Bioactive Lipids. Membranes (2021) 11:919. doi: 10.3390/membranes11120919
10
Santos AL Preta G . Lipids in the Cell: Organisation Regulates Function. Cell Mol Life Sci (2018) 75:1909–27. doi: 10.1007/s00018-018-2765-4
11
Zhang C Wang K Yang L Liu R Chu Y Qin X et al . Lipid Metabolism in Inflammation-Related Diseases. Analyst (2018) 143:4526–36. doi: 10.1039/c8an01046c
12
Howie D Ten Bokum A Necula AS Cobbold SP Waldmann H . The Role of Lipid Metabolism in T Lymphocyte Differentiation and Survival. Front Immunol (2017) 8:1949. doi: 10.3389/fimmu.2017.01949
13
Su W Peng J Li S Dai YB Wang CJ Xu H et al . Liver X Receptor α Induces 17β-Hydroxysteroid Dehydrogenase-13 Expression Through SREBP-1c. Am J Physiol Endocrinol Metab (2017) 312:E357–67. doi: 10.1152/ajpendo.00310.2016
14
Wagner BL Valledor AF Shao G Daige CL Bischoff ED Petrowski M et al . Promoter-Specific Roles For Liver X Receptor/Corepressor Complexes in the Regulation of ABCA1 and SREBP1 Gene Expression. Mol Cell Biol (2003) 23:5780–9. doi: 10.1128/mcb.23.16.5780-5789.2003
15
Zechner R Zimmermann R Eichmann TO Kohlwein SD Haemmerle G Lass A et al . FAT SIGNALS–Lipases and Lipolysis in Lipid Metabolism and Signaling. Cell Metab (2012) 15:279–91. doi: 10.1016/j.cmet.2011.12.018
16
Nowinski SM Solmonson A Rusin SF Maschek JA Bensard CL Fogarty S et al . Mitochondrial Fatty Acid Synthesis Coordinates Oxidative Metabolism in Mammalian Mitochondria. Elife (2020) 9:e58041. doi: 10.7554/eLife.58041
17
Wakil SJ Abu-Elheiga LA . Fatty Acid Metabolism: Target for Metabolic Syndrome. J Lipid Res (2009) 50 Suppl:S138–43. doi: 10.1194/jlr.R800079-JLR200
18
Lefterova MI Haakonsson AK Lazar MA Mandrup S . Pparγ and the Global Map of Adipogenesis and Beyond. Trends Endocrinol Metab (2014) 25:293–302. doi: 10.1016/j.tem.2014.04.001
19
Mohammadi S Saghaeian-Jazi M Sedighi S Memarian A . Immunomodulation in Systemic Lupus Erythematosus: Induction of M2 Population in Monocyte-Derived Macrophages by Pioglitazone. Lupus (2017) 26:1318–27. doi: 10.1177/0961203317701842
20
Wu D Sanin DE Everts B Chen Q Qiu J Buck MD et al . Type 1 Interferons Induce Changes in Core Metabolism That Are Critical for Immune Function. Immunity (2016) 44:1325–36. doi: 10.1016/j.immuni.2016.06.006
21
Zhao W Berthier CC Lewis EE McCune WJ Kretzler M Kaplan MJ . The Peroxisome-Proliferator Activated Receptor-γ Agonist Pioglitazone Modulates Aberrant T Cell Responses in Systemic Lupus Erythematosus. Clin Immunol (2013) 149:119–32. doi: 10.1016/j.clim.2013.07.002
22
Aprahamian T Bonegio RG Richez C Yasuda K Chiang LK Sato K et al . The Peroxisome Proliferator-Activated Receptor Gamma Agonist Rosiglitazone Ameliorates Murine Lupus by Induction of Adiponectin. J Immunol (2009) 182:340–6. doi: 10.4049/jimmunol.182.1.340
23
Oxer DS Godoy LC Borba E Lima-Salgado T Passos LA Laurindo I et al . Pparγ Expression is Increased in Systemic Lupus Erythematosus Patients and Represses CD40/CD40L Signaling Pathway. Lupus (2011) 10:575–87. doi: 10.1177/0961203310392419
24
Berod L Friedrich C Nandan A Freitag J Hagemann S Harmrolfs K et al . De Novo Fatty Acid Synthesis Controls the Fate Between Regulatory T and T Helper 17 Cells. Nat Med (2014) 20:1327–33. doi: 10.1038/nm.3704
25
Raha S Raud B Oberdörfer L Castro CN Schreder A Freitag J et al . Disruption of De Novo Fatty Acid Synthesis via Acetyl-CoA Carboxylase 1 Inhibition Prevents Acute Graft-Versus-Host Disease. Eur J Immunol (2016) 46:2233–8. doi: 10.1002/eji.201546152
26
Mamareli P Kruse F Lu CW Guderian M Floess S Rox K et al . Targeting Cellular Fatty Acid Synthesis Limits T Helper and Innate Lymphoid Cell Function During Intestinal Inflammation and Infection. Mucosal Immunol (2021) 14:164–76. doi: 10.1038/s41385-020-0285-7
27
Stüve P Minarrieta L Erdmann H Arnold-Schrauf C Swallow M Guderian M et al . De Novo Fatty Acid Synthesis During Mycobacterial Infection Is a Prerequisite for the Function of Highly Proliferative T Cells, But Not for Dendritic Cells or Macrophages. Front Immunol (2018) 9:495. doi: 10.3389/fimmu.2018.00495
28
Voss K Luthers CR Pohida K Snow AL . Fatty Acid Synthase Contributes to Restimulation-Induced Cell Death of Human CD4 T Cells. Front Mol Biosci (2019) 6:106. doi: 10.3389/fmolb.2019.00106
29
Young KE Flaherty S Woodman KM Sharma-Walia N Reynolds JM . Fatty Acid Synthase Regulates the Pathogenicity of Th17 Cells. J Leukoc Biol (2017) 102:1229–35. doi: 10.1189/jlb.3AB0417-159RR
30
Ma EH Poffenberger MC Wong AH Jones RG . The Role of AMPK in T Cell Metabolism and Function. Curr Opin Immunol (2017) 46:45–52. doi: 10.1016/j.coi.2017.04.004
31
Michalek RD Gerriets VA Jacobs SR Macintyre AN MacIver NJ Mason EF et al . Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4+ T Cell Subsets. J Immunol (2011) 186:3299–303. doi: 10.4049/jimmunol.1003613
32
Fernandez D Perl A . mTOR Signaling: A Central Pathway to Pathogenesis in Systemic Lupus Erythematosus? Discov Med (2010) 9:173–8.
33
Wahl DR Petersen B Warner R Richardson BC Glick GD Opipari AW . Characterization of the Metabolic Phenotype of Chronically Activated Lymphocytes. Lupus (2010) 19:1492–501. doi: 10.1177/0961203310373109
34
Torigoe M Iwata S Nakayamada S Sakata K Zhang M Hajime M et al . Metabolic Reprogramming Commits Differentiation of Human CD27(+)IgD(+) B Cells to Plasmablasts or CD27(-)IgD(-) Cells. J Immunol (2017) 199:425–34. doi: 10.4049/jimmunol.1601908
35
Wu C Fu Q Guo Q Chen S Goswami S Sun S et al . Lupus-Associated Atypical Memory B Cells are Mtorc1-Hyperactivated and Functionally Dysregulated. Ann Rheum Dis (2019) 78:1090–100. doi: 10.1136/annrheumdis-2019-215039
36
Lee SY Moon SJ Kim EK Seo HB Yang EJ Son HJ et al . Metformin Suppresses Systemic Autoimmunity in Roquin(san/san) Mice Through Inhibiting B Cell Differentiation Into Plasma Cells via Regulation of AMPK/mTOR/Stat3. J Immunol (2017) 198:2661–70. doi: 10.4049/jimmunol.1403088
37
Kidani Y Bensinger SJ . Liver X Receptor and Peroxisome Proliferator-Activated Receptor as Integrators of Lipid Homeostasis and Immunity. Immunol Rev (2012) 249:72–83. doi: 10.1111/j.1600-065X.2012.01153.x
38
N AG Bensinger SJ Hong C Beceiro S Bradley MN Zelcer N et al . Apoptotic Cells Promote Their Own Clearance and Immune Tolerance Through Activation of the Nuclear Receptor LXR. Immunity (2009) 31:245–58. doi: 10.1016/j.immuni.2009.06.018
39
Ito A Hong C Oka K Salazar JV Diehl C Witztum JL et al . Cholesterol Accumulation in CD11c(+) Immune Cells Is a Causal and Targetable Factor in Autoimmune Disease. Immunity (2016) 45:1311–26. doi: 10.1016/j.immuni.2016.11.008
40
Schulman IG . Liver X Receptors Link Lipid Metabolism and Inflammation. FEBS Lett (2017) 591:2978–91. doi: 10.1002/1873-3468.12702
41
Jeon JY Nam JY Kim HA Park YB Bae SC Suh CH . Liver X Receptors Alpha Gene (NR1H3) Promoter Polymorphisms Are Associated With Systemic Lupus Erythematosus in Koreans. Arthritis Res Ther (2014) 16:R112. doi: 10.1186/ar4563
42
Westerterp M Gautier EL Ganda A Molusky MM Wang W Fotakis P et al . Cholesterol Accumulation in Dendritic Cells Links the Inflammasome to Acquired Immunity. Cell Metab (2017) 25:1294–304.e6. doi: 10.1016/j.cmet.2017.04.005
43
Forcina GC Dixon SJ . GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics (2019) 19:e1800311. doi: 10.1002/pmic.201800311
44
Li P Jiang M Li K Li H Zhou Y Xiao X et al . Glutathione Peroxidase 4-Regulated Neutrophil Ferroptosis Induces Systemic Autoimmunity. Nat Immunol (2021) 22:1107–17. doi: 10.1038/s41590-021-00993-3
45
Simons K Toomre D . Lipid Rafts and Signal Transduction. Nat Rev Mol Cell Biol (2000) 1:31–9. doi: 10.1038/35036052
46
Jury EC Flores-Borja F Kabouridis PS . Lipid Rafts in T Cell Signalling and Disease. Semin Cell Dev Biol (2007) 18:608–15. doi: 10.1016/j.semcdb.2007.08.002
47
Waddington KE Jury EC . Manipulating Membrane Lipid Profiles to Restore T-Cell Function in Autoimmunity. Biochem Soc Trans (2015) 43:745–51. doi: 10.1042/bst20150111
48
Calkin AC Tontonoz P . Transcriptional Integration of Metabolism by the Nuclear Sterol-Activated Receptors LXR and FXR. Nat Rev Mol Cell Biol (2012) 13:213–24. doi: 10.1038/nrm3312
49
Hong C Tontonoz P . Liver X Receptors in Lipid Metabolism: Opportunities for Drug Discovery. Nat Rev Drug Discov (2014) 13:433–44. doi: 10.1038/nrd4280
50
Rosca MG Vazquez EJ Chen Q Kerner J Kern TS Hoppel CL . Oxidation of Fatty Fcids is the Source of Increased Mitochondrial Reactive Oxygen Species Production in Kidney Cortical Tubules in Early Diabetes. Diabetes (2012) 61:2074–83. doi: 10.2337/db11-1437
51
Walther TC Chung J Farese RV Jr . Lipid Droplet Biogenesis. Annu Rev Cell Dev Biol (2017) 33:491–510. doi: 10.1146/annurev-Cellbio-100616-060608
52
Kassan A Herms A Fernández-Vidal A Bosch M Schieber NL Reddy BJ et al . Acyl-CoA Synthetase 3 Promotes Lipid Droplet Bogenesis in ER Microdomains. J Cell Biol (2013) 203:985–1001. doi: 10.1083/jcb.201305142
53
Stevenson J Huang EY Olzmann JA . Endoplasmic Reticulum-Associated Degradation and Lipid Homeostasis. Annu Rev Nutr (2016) 36:511–42. doi: 10.1146/annurev-nutr-071715-051030
54
Wójcik P Gęgotek A Žarković N Skrzydlewska E . Oxidative Stress and Lipid Mediators Modulate Immune Cell Functions in Autoimmune Diseases. Int J Mol Sci (2021) 22(2):723. doi: 10.3390/ijms22020723
55
Wenk MR . The Emerging Field of Lipidomics. Nat Rev Drug Discovery (2005) 4:594–610. doi: 10.1038/nrd1776
56
Mahley RW Innerarity TL Rall SC Jr Weisgraber KH . Plasma Lipoproteins: Apolipoprotein Structure and Function. J Lipid Res (1984) 25:1277–94.
57
Wang Y Yu H He J . Role of Dyslipidemia in Accelerating Inflammation, Autoimmunity, and Atherosclerosis in Systemic Lupus Erythematosus and Other Autoimmune Diseases. Discov Med (2020) 30:49–56.
58
Liu Y Kaplan MJ . Cardiovascular Disease in Systemic Lupus Erythematosus: An Update. Curr Opin Rheumatol (2018) 30:441–8. doi: 10.1097/bor.0000000000000528
59
Giannelou M Mavragani CP . Cardiovascular Disease in Systemic Lupus Erythematosus: A Comprehensive Update. J Autoimmun (2017) 82:1–12. doi: 10.1016/j.jaut.2017.05.008
60
Urowitz MB Gladman D Ibañez D Fortin P Sanchez-Guerrero J Bae S et al . Clinical Manifestations and Coronary Artery Disease Risk Factors at Diagnosis of Systemic Lupus Erythematosus: Data From an International Inception Cohort. Lupus (2007) 16:731–5. doi: 10.1177/0961203307081113
61
Castro LL Lanna CCD Ribeiro ALP Telles RW . Recognition And Control of Hypertension, Diabetes, and Dyslipidemia in Patients With Sytemic Lupus Erythematosus. Clin Rheumatol (2018) 37:2693–8. doi: 10.1007/s10067-018-4169-0
62
Chan PC Yu CH Yeh KW Horng JT Huang JL . Comorbidities of Pediatric Systemic Lupus Erythematosus: A 6-Year Nationwide Population-Based Study. J Microbiol Immunol Infect (2016) 49:257–63. doi: 10.1016/j.jmii.2014.05.001
63
Machado D Sarni RO Abad TT Silva SG Khazaal EJ Hix S et al . Lipid Profile Among Girls With Systemic Lupus Erythematosus. Rheumatol Int (2017) 37:43–8. doi: 10.1007/s00296-015-3393-z
64
Zhou B Xia Y She J . Dysregulated Serum Lipid Profile And Its Correlation to Disease Activity in Young Female Adults Diagnosed With Systemic Lupus Erythematosus: A Cross-Sectional Study. Lipids Health Dis (2020) 19:40. doi: 10.1186/s12944-020-01232-8
65
Fanlo-Maresma M Candás-Estébanez B Esteve-Luque V Padró-Miquel A Escrihuela-Vidal F Carratini-Moraes M et al . Asymptomatic Carotid Atherosclerosis Cardiovascular Risk Factors and Common Hypertriglyceridemia Genetic Variants in Patients With Systemic Erythematosus Lupus. Clin Med (2021) 10:2218. doi: 10.3390/jcm10102218
66
Tselios K Koumaras C Gladman DD Urowitz MB . Dyslipidemia in Systemic Lupus Erythematosus: Just Another Comorbidity? Semin Arthritis Rheum (2016) 45:604–10. doi: 10.1016/j.semarthrit.2015.10.010
67
Stangl S Kollerits B Lamina C Meisinger C Huth C Stöckl A et al . Association Between Apolipoprotein A-IV Concentrations and Chronic Kidney Disease in Two Large Population-Based Cohorts: Results From the KORA Studies. J Intern Med (2015) 278:410–23. doi: 10.1111/joim.12380
68
Chong YB Yap DY Tang CS Chan TM . Dyslipidaemia in Patients With Lupus Nephritis. Nephrology (2011) 16:511–7. doi: 10.1111/j.1440-1797.2011.01456.x
69
Sajjad S Farman S Saeed MA Ahmad NM Butt BA . Frequency of Dyslipidemia in Patients With Lupus Nephritis. Pak J Med Sci (2017) 33:358–62. doi: 10.12669/pjms.332.12410
70
Reich HN Gladman DD Urowitz MB Bargman JM Hladunewich MA Lou W et al . Persistent Proteinuria and Dyslipidemia Increase the Risk of Progressive Chronic Kidney Disease in Lupus Erythematosus. Kidney Int (2011) 79:914–20. doi: 10.1038/ki.2010.525
71
Kochan Z Szupryczynska N Malgorzewicz S Karbowska J . Dietary Lipids and Dyslipidemia in Chronic Kidney Disease. Nutrients (2021) 13:3138. doi: 10.3390/nu13093138
72
Gaál K Tarr T Lőrincz H Borbás V Seres I Harangi M et al . High-Density Lipopoprotein Antioxidant Capacity, Subpopulation Distribution and Paraoxonase-1 Activity in Patients With Systemic Lupus Erythematosus. Lipids Health Dis (2016) 15:60. doi: 10.1186/s12944-016-0229-0
73
Smith CK Seto NL Vivekanandan-Giri A Yuan W Playford MP Manna Z et al . Lupus High-Density Lipoprotein Induces Proinflammatory Responses in Macrophages by Binding Lectin-Like Oxidised Low-Density Lipoprotein Receptor 1 and Failing to Promote Activating Transcription Factor 3 Activity. Ann Rheum Dis (2017) 76:602–11. doi: 10.1136/annrheumdis-2016-209683
74
Ronda N Favari E Borghi MO Ingegnoli F Gerosa M Chighizola C et al . Impaired Serum Cholesterol Efflux Capacity in Rheumatoid Arthritis and Systemic Lupus Erythematosus. Ann Rheum Dis (2014) 73:609–15. doi: 10.1136/annrheumdis-2012-202914
75
Ortiz TT Terreri MT Caetano M Souza FS D'Almeida V Sarni RO et al . Dyslipidemia in Pediatric Systemic Lupus Erythematosus: The Relationship With Disease Activity and Plasma Homocysteine and Cysteine Concentrations. Ann Nutr Metab (2013) 63:77–82. doi: 10.1159/000351076
76
Vuilleumier N Charbonney E Fontao L Alvarez M Turck N Sanchez JC et al . Anti-(Apolipoprotein A-1) IgGs Are Associated With High Levels of Oxidized Low-Density Lipoprotein in Acute Coronary Syndrome. Clin Sci (2008) 115:25–33. doi: 10.1042/cs20070325
77
Robinson GA Waddington KE Coelewij L Peng J Naja M Wincup C et al . Increased Apolipoprotein-B:A1 Ratio Predicts Cardiometabolic Risk in Patients With Juvenile Onset SLE. EBioMedicine (2021) 65:103243. doi: 10.1016/j.ebiom.2021.103243
78
Narshi CB Giles IP Rahman A . The Endothelium: An Interface Between Autoimmunity and Atherosclerosis in Systemic Lupus Erythematosus? Lupus (2011) 20:5–13. doi: 10.1177/0961203310382429
79
Srivastava R Yu S Parks BW Black LL Kabarowski JH . Autoimmune-Mediated Reduction of High-Density Lipoprotein-Cholesterol and Paraoxonase 1 Activity in Systemic Lupus Erythematosus-Prone Gld Mice. Arthritis Rheum (2011) 63:201–11. doi: 10.1002/art.27764
80
Ardoin SP Sandborg C Schanberg LE . Management of Dyslipidemia in Children and Adolescents With Systemic Lupus Erythematosus. Lupus (2007) 16:618–26. doi: 10.1177/0961203307079566
81
Cardoso CR Signorelli FV Papi JA Salles GF . Prevalence and Factors Associated With Dyslipoproteinemias in Brazilian Systemic Lupus Erythematosus Patients. Rheumatol Int (2008) 28:323–7. doi: 10.1007/s00296-007-0447-x
82
Parra S Heras M Herrero P Amigó N Garcés E Girona J et al . Gelsolin: A New Biomarker of Disease Activity in SLE Patients Associated With HDL-C. Rheumatology (2020) 59:650–61. doi: 10.1093/rheumatology/kez293
83
Ardoin SP Schanberg LE Sandborg C Yow E Barnhart HX Mieszkalski K et al . Laboratory Markers of Cardiovascular Risk in Pediatric SLE: The APPLE Baseline Cohort. Lupus (2010) 19:1315–25. doi: 10.1177/0961203310373937
84
Rodríguez-Calvo R Guardiola M Oliva I Arrando H Arranz I Ferré A et al . Low-Density Lipoprotein From Active SLE Patients is More Atherogenic to Endothelial Cells Than Low-Density Lipoprotein From the Same Patients During Remission. Rheumatology (2021) 60:866–71. doi: 10.1093/rheumatology/keaa380
85
Spengler MI Svetaz MJ Leroux MB Bertoluzzo SM Parente FM Bosch P . Lipid Peroxidation Affects Red Blood Cells Membrane Properties in Patients With Systemic Lupus Erythematosus. Clin Hemorheol Microcirc (2014) 58:489–95. doi: 10.3233/ch-131716
86
Matsuura E Hughes GR Khamashta MA . Oxidation of LDL and Its Clinical Implication. Autoimmun Rev (2008) 7:558–66. doi: 10.1016/j.autrev.2008.04.018
87
Frostegård J Svenungsson E Wu R Gunnarsson I Lundberg IE Klareskog L et al . Lipid Peroxidation Is Enhanced in Patients With Systemic Lupus Erythematosus and Is Associated With Arterial and Renal Disease Manifestations. Arthritis Rheum (2005) 52:192–200. doi: 10.1002/art.20780
88
Vaarala O Alfthan G Jauhiainen M Leirisalo-Repo M Aho K Palosuo T . Crossreaction Between Antibodies to Oxidised Low-Density Lipoprotein and to Cardiolipin in Systemic Lupus Erythematosus. Lancet (1993) 341:923–5. doi: 10.1016/0140-6736(93)91213-6
89
Chan HC Chan HC Liang CJ Lee HC Su H Lee AS et al . Role of Low-Density Lipoprotein in Early Vascular Aging Associated With Systemic Lupus Erythematosus. Arthritis Rheumatol (2020) 72:972–84. doi: 10.1002/art.41213
90
Svenungsson E Jensen-Urstad K Heimbürger M Silveira A Hamsten A de Faire U et al . Risk Factors for Cardiovascular Disease in Systemic Lupus Erythematosus. Circulation (2001) 104:1887–93. doi: 10.1161/hc4101.097518
91
Ettinger WH Jr Hazzard WR . Elevated Apolipoprotein-B Levels in Corticosteroid-Treated Patients With Systemic Lupus Erythematosus. J Clin Endocrinol Metab (1988) 67:425–8. doi: 10.1210/jcem-67-3-425
92
Rodrigues WDR Sarni ROS Abad TTO Silva S Souza FIS Len CA et al . Lipid Profile of Pediatric Patients With Chronic Rheumatic Diseases - A Retrospective Analysis. Rev Assoc Med Bras (2020) 66:1093–9. doi: 10.1590/1806-9282.66.8.1093
93
John K Marino JS Sanchez ER Hinds TD Jr . The Glucocorticoid Receptor: Cause of or Cure for Obesity? Am J Physiol Endocrinol Metab (2016) 310:E249–57. doi: 10.1152/ajpendo.00478.2015
94
Strohmayer EA Krakoff LR . Glucocorticoids and Cardiovascular Risk Factors. Endocrinol Metab Clin North Am (2011) 40:409–17. doi: 10.1016/j.ecl.2011.01.011
95
Chakkera HA Sharif A Kaplan B . Negative Cardiovascular Consequences of Small Molecule Immunosuppressants. Clin Pharmacol Ther (2017) 102:269–76. doi: 10.1002/cpt.738
96
Cairoli E Rebella M Danese N Garra V Borba EF . Hydroxychloroquine Reduces Low-Density Lipoprotein Cholesterol Levels in Systemic Lupus Erythematosus: A Longitudinal Evaluation of the Lipid-Lowering Effect. Lupus (2012) 21:1178–82. doi: 10.1177/0961203312450084
97
Babary H Liu X Ayatollahi Y Chen XP Doo L Uppaluru LK et al . Favorable Effects of Hydroxychloroquine on Serum Low Density Lipid in Patients With Systemic Lupus Erythematosus: A Systematic Review and Meta-Analysis. Int J Rheum Dis (2018) 21:84–92. doi: 10.1111/1756-185X.13159
98
Beynen AC van der Molen AJ Geelen MJ . Inhibition of Hepatic Cholesterol Biosynthesis by Chloroquine. Lipids (1981) 16:472–4. doi: 10.1007/bf02535017
99
Sachet JC Borba EF Bonfá E Vinagre CG Silva VM Maranhão RC . Chloroquine Increases Low-Density Lipoprotein Removal From Plasma in Systemic Lupus Patients. Lupus (2007) 16:273–8. doi: 10.1177/09612033070160040901
100
Rahman P Gladman DD Urowitz MB Yuen K Hallett D Bruce IN . The Cholesterol Lowering Effect of Antimalarial Drugs Is Enhanced in Patients With Lupus Taking Corticosteroid Drugs. J Rheumatol (1999) 26:325–30.
101
Mok CC . Towards New Avenues in the Management of Lupus Glomerulonephritis. Nat Rev Rheumatol (2016) 12:221–34. doi: 10.1038/nrrheum.2015.174
102
Farouk SS Rein JL . The Many Faces of Calcineurin Inhibitor Toxicity-What the FK? Adv Chronic Kidney Dis (2020) 27:56–66. doi: 10.1053/j.ackd.2019.08.006
103
Busque S Cantarovich M Mulgaonkar S Gaston R Gaber AO Mayo PR et al . The PROMISE Study: A Phase 2b Multicenter Study of Voclosporin (ISA247) Versus Tacrolimus in De Novo Kidney Transplantation. Am J Transplant (2011) 11:2675–84. doi: 10.1111/j.1600-6143.2011.03763.x
104
Rovin BH Teng YKO Ginzler EM Arriens C Caster DJ Romero-Diaz J et al . Efficacy and Safety of Voclosporin Versus Placebo for Lupus Nephritis (AURORA 1): A Double-Blind, Randomised, Multicentre, Placebo-Controlled, Phase 3 Trial. Lancet (2021) 397:2070–80. doi: 10.1016/S0140-6736(21)00578-X
105
Guleria A Phatak S Dubey D Kumar S Zanwar A Chaurasia S et al . NMR-Based Serum Metabolomics Reveals Reprogramming of Lipid Dysregulation Following Cyclophosphamide-Based Induction Therapy in Lupus Nephritis. J Proteome Res (2018) 17:2440–8. doi: 10.1021/acs.jproteome.8b00192
106
Cas MD Roda G Li F Secundo F . Functional Lipids in Autoimmune Inflammatory Diseases. Int J Mol Sci (2020) 21:3074. doi: 10.3390/ijms21093074
107
Katsuyama T Tsokos GC Moulton VR . Aberrant T Cell Signaling and Subsets in Systemic Lupus Erythematosus. Front Immunol (2018) 9:1088. doi: 10.3389/fimmu.2018.01088
108
Fillatreau S Manfroi B Dörner T . Toll-Like Receptor Signalling in B Cells During Systemic Lupus Erythematosus. Nat Rev Immunol (2021) 17:98–108. doi: 10.1038/s41584-020-00544-4
109
Hu C Zhang J Hong S Li H Lu L Xie G et al . Oxidative Stress-Induced Aberrant Lipid Metabolism is an Important Causal Factor for Dysfunction of Immunocytes From Patients With Systemic Lupus Erythematosus. Free Radic Biol Med (2021) 163:210–19. doi: 10.1016/j.freeradbiomed.2020.12.006
110
Chen PM Tsokos GC . T Cell Abnormalities in the Pathogenesis of Systemic Lupus Erythematosus: An Update. Curr Rheumatol Rep (2021) 23:12. doi: 10.1007/s11926-020-00978-5
111
Wang SH Yuan SG Peng DQ Zhao SP . HDL and ApoA-I Inhibit Antigen Presentation-Mediated T Cell Activation by Disrupting Lipid Rafts in Antigen Presenting Cells. Atherosclerosis (2012) 225:105–14. doi: 10.1016/j.atherosclerosis.2012.07.029
112
Krishnan S Nambiar MP Warke VG Fisher CU Mitchell J Delaney N et al . Alterations in Lipid Raft Composition and Dynamics Contribute to Abnormal T Cell Responses in Systemic Lupus Erythematosus. J Immunol (2004) 172:7821–31. doi: 10.4049/jimmunol.172.12.7821
113
Crispín JC Kyttaris VC Juang YT Tsokos GC . How Signaling and Gene Transcription Aberrations Dictate the Systemic Lupus Erythematosus T Cell Phenotype. Trends Immunol (2008) 29:110–5. doi: 10.1016/j.it.2007.12.003
114
Jury EC Isenberg DA Mauri C Ehrenstein MR . Atorvastatin Restores Lck Expression and Lipid Raft-Associated Signaling in T Cells From Patients With Systemic Lupus Erythematosus. J Immunol (2006) 177:7416–22. doi: 10.4049/jimmunol.177.10.7416
115
McDonald G Deepak S Miguel L Hall CJ Isenberg DA Magee AI et al . Normalizing Glycosphingolipids Restores Function in CD4+ T Cells From Lupus Patients. J Clin Invest (2014) 124:712–24. doi: 10.1172/jci69571
116
Zhu Y Gumlaw N Karman J Zhao H Zhang J Jiang JL et al . Lowering Glycosphingolipid Levels in CD4+ T Cells Attenuates T Cell Receptor Signaling, Cytokine Production, and Differentiation to the Th17 Lineage. J Biol Chem (2011) 286:14787–94. doi: 10.1074/jbc.M111.218610
117
He YS Yang XK Hu YQ Xiang K Pan HF . Emerging Role of Fli1 in Autoimmune Diseases. Int Immunopharmacol (2021) 90:107127. doi: 10.1016/j.intimp.2020.107127
118
Sundararaj KP Thiyagarajan T Molano I Basher F Powers TW Drake RR et al . FLI1 Levels Impact CXCR3 Expression and Renal Infiltration of T Cells and Renal Glycosphingolipid Metabolism in the MRL/lpr Lupus Mouse Strain. J Immunol (2015) 195:5551–60. doi: 10.4049/jimmunol.1500961
119
Hu X Wang Y Hao LY Liu X Lesch CA Sanchez BM et al . Sterol Metabolism Controls T(H)17 Differentiation by Generating Endogenous Rorγ Agonists. Nat Chem Biol (2015) 11:141–7. doi: 10.1038/nchembio.1714
120
Dolff S Witzke O Wilde B . Th17 Cells in Renal Inflammation and Autoimmunity. Autoimmun Rev (2019) 18:129–36. doi: 10.1016/j.autrev.2018.08.006
121
Shin MS Lee N Kang I . Effector T-Cell Subsets in Systemic Lupus Erythematosus: Update Focusing on Th17 Cells. Curr Opin Rheumatol (2011) 23:444–8. doi: 10.1097/BOR.0b013e328349a255
122
Guégan JP Legembre P . Nonapoptotic Functions of Fas/CD95 in the Immune Response. FEBS J (2018) 285:809–27. doi: 10.1111/febs.14292
123
Poissonnier A Sanséau D Le Gallo M Malleter M Levoin N Viel R et al . CD95-Mediated Calcium Signaling Promotes T Helper 17 Trafficking to Inflamed Organs in Lupus-Prone Mice. Immunity (2016) 45:209–23. doi: 10.1016/j.immuni.2016.06.028
124
Christoffersen C Obinata H Kumaraswamy SB Galvani S Ahnström J Sevvana M et al . Endothelium-Protective Sphingosine-1-Phosphate Provided by HDL-Associated Apolipoprotein M. Proc Natl Acad Sci U S A (2011) 108:9613–8. doi: 10.1073/pnas.1103187108
125
Iwata S Zhang M Hao H Trimova G Hajime M Miyazaki Y et al . Enhanced Fatty Acid Synthesis Leads to Subset Imbalance and IFN-γ Overproduction in T Helper 1 Cells. Front Immunol (2020) 11:593103:593103. doi: 10.3389/fimmu.2020.593103
126
Shah D Mahajan N Sah S Nath SK Paudyal B . Oxidative Stress and Its Biomarkers in Systemic Lupus Erythematosus. J BioMed Sci (2014) 21:23. doi: 10.1186/1423-0127-21-23
127
Perl A . Oxidative Stress in the Pathology and Treatment of Systemic Lupus Erythematosus. Nat Rev Rheumatol (2013) 9:674–86. doi: 10.1038/nrrheum.2013.147
128
Scavuzzi BM Simão ANC Iriyoda TMV Lozovoy MAB Stadtlober NP Franchi Santos L et al . Increased Lipid and Protein Oxidation and Lowered Anti-Oxidant Defenses in Systemic Lupus Erythematosus Are Associated With Severity of Illness, Autoimmunity, Increased Adhesion Molecules, and Th1 and Th17 Immune Shift. Immunol Res (2018) 66:158–71. doi: 10.1007/s12026-017-8960-9
129
Smith E Croca S Waddington KE Sofat R Griffin M Nicolaides A et al . Cross-Talk Between iNKT Cells and Monocytes Triggers an Atheroprotective Immune Response in SLE Patients With Asymptomatic Plaque. Sci Immunol (2016) 1:eaah4081. doi: 10.1126/sciimmunol.aah4081
130
Sawaf M Fauny JD Felten R Sagez F Gottenberg JE Dumortier H et al . Defective BTLA Functionality Is Rescued by Restoring Lipid Metabolism in Lupus CD4+ T Cells. JCI Insight (2018) 3:e99711. doi: 10.1172/jci.insight.99711
131
Ryu H Chung Y . Dyslipidemia Promotes Ferminal Center Reactions via IL-27. BMB Rep (2018) 51:371–2. doi: 10.5483/BMBRep.2018.51.8.171
132
Nashi E Wang Y Diamond B . The Role of B Cells in Lupus Pathogenesis. Int J Biochem Cell Biol (2010) 42:543–50. doi: 10.1016/j.biocel.2009.10.011
133
Flores-Borja F Kabouridis PS Jury EC Isenberg DA Mageed RA . Altered Lipid Raft-Associated Proximal Signaling and Translocation of CD45 Tyrosine Phosphatase in B Lymphocytes From Patients With Systemic Lupus Erythematosus. Arthritis Rheum (2007) 56:291–302. doi: 10.1002/art.22309
134
Flores-Borja F Kabouridis PS Jury EC Isenberg DA Mageed RA . Decreased Lyn Expression and Translocation to Lipid Raft Signaling Domains in B Lymphocytes From Patients With Systemic Lupus Erythematosus. Arthritis Rheum (2005) 52:3955–65. doi: 10.1002/art.21416
135
Zhang Y Gui M Wang Y Mani N Chaudhuri S Gao B et al . Inositol-Requiring Enzyme 1α-Mediated Synthesis of Monounsaturated Fatty Acids as a Driver of B Cell Differentiation and Lupus-Like Autoimmune Disease. Arthritis Rheumatol (2021) 73:2314–26. doi: 10.1002/art.41883
136
Gutiérrez S Svahn SL Johansson ME . Effects of Omega-3 Fatty Acids on Immune Cells. Int J Mol Sci (2019) 20:5028. doi: 10.3390/ijms20205028
137
Kobayashi A Ito A Shirakawa I Tamura A Tomono S Shindou H et al . Dietary Supplementation With Eicosapentaenoic Acid Inhibits Plasma Cell Differentiation and Attenuates Lupus Autoimmunity. Front Immunol (2021) 12:650856. doi: 10.3389/fimmu.2021.650856
138
Benninghoff AD Bates MA Chauhan PS Wierenga KA Gilley KN Holian A et al . Docosahexaenoic Acid Consumption Impedes Early Interferon- and Chemokine-Related Gene Expression While Suppressing Silica-Triggered Flaring of Murine Lupus. Front Immunol (2019) 10:2851. doi: 10.3389/fimmu.2019.02851
139
Sanchez HN Moroney JB Gan H Shen T Im JL Li T et al . B Cell-Intrinsic Epigenetic Modulation of Antibody Responses by Dietary Fiber-Derived Short-Chain Fatty Acids. Nat Commun (2020) 11:60. doi: 10.1038/s41467-019-13603-6
140
Trostchansky A Rubbo H . Nitrated Fatty Acids: Mechanisms of Formation, Chemical Characterization, and Biological Properties. Free Radic Biol Med (2008) 44:1887–96. doi: 10.1016/j.freeradbiomed.2008.03.006
141
Verescakova H Ambrozova G Kubala L Perecko T Koudelka A Vasicek O et al . Nitro-Oleic Acid Regulates Growth Factor-Induced Differentiation of Bone Marrow-Derived Macrophages. Free Radic Biol Med (2017) 104:10–9. doi: 10.1016/j.freeradbiomed.2017.01.003
142
Moore KJ Sheedy FJ Fisher EA . Macrophages in Atherosclerosis: A Dynamic Balance. Nat Rev Immunol (2013) 13:709–21. doi: 10.1038/nri3520
143
Li J Fu Q Cui H Qu B Pan W Shen N et al . Interferon-α Priming Promotes Lipid Uptake and Macrophage-Derived Foam Cell Formation: A Novel Link Between Interferon-α and Atherosclerosis in Lupus. Arthritis Rheum (2011) 63:492–502. doi: 10.1002/art.30165
144
Jaroonwitchawan T Visitchanakun P Dang PC Ritprajak P Palaga T Leelahavanichkul A . Dysregulation of Lipid Metabolism in Macrophages is Responsible for Severe Endotoxin Tolerance in FcgRIIB-Deficient Lupus Mice. Front Immunol (2020) 11:959. doi: 10.3389/fimmu.2020.00959
145
Namgaladze D Kemmerer M von Knethen A Brüne B . AICAR Inhibits Pparγ During Monocyte Differentiation to Attenuate Inflammatory Responses to Atherogenic Lipids. Cardiovasc Res (2013) 98:479–87. doi: 10.1093/cvr/cvt073
146
Uderhardt S Herrmann M Oskolkova OV Aschermann S Bicker W Ipseiz N et al . 12/15-Lipoxygenase Orchestrates the Clearance of Apoptotic Cells and Maintains Immunologic Tolerance. Immunity (2012) 36:834–46. doi: 10.1016/j.immuni.2012.03.010
147
Cartier A Hla T . Sphingosine 1-Phosphate: Lipid Signaling in Pathology and Therapy. Science (2019) 366:eaar5551. doi: 10.1126/science.aar5551
148
Luo B Gan W Liu Z Shen Z Wang J Shi R et al . Erythropoeitin Signaling in Macrophages Promotes Dying Cell Clearance and Immune Tolerance. Immunity (2016) 44:287–302. doi: 10.1016/j.immuni.2016.01.002
149
Kim HA Baek WY Han MH Jung JY Suh CH . The Liver X Receptor is Upregulated in Monocyte-Derived Macrophages and Modulates Inflammatory Cytokines Based on Lxrα Polymorphism. Mediators Inflamm (2019) 2019:6217548. doi: 10.1155/2019/6217548
150
Han S Zhuang H Shumyak S Wu J Xie C Li H et al . Liver X Receptor Agonist Therapy Prevents Diffuse Alveolar Hemorrhage in Murine Lupus by Repolarizing Macrophages. Front Immunol (2018) 9:135. doi: 10.3389/fimmu.2018.00135
151
Banchereau J Steinman RM . Dendritic Cells and the Control of Immunity. Nature (1998) 392:245–52. doi: 10.1038/32588
152
Ibrahim J Nguyen AH Rehman A Ochi A Jamal M Graffeo CS et al . Dendritic Cell Populations With Different Concentrations of Lipid Regulate Tolerance and Immunity in Mouse and Human Liver. Gastroenterology (2012) 143:1061–72. doi: 10.1053/j.gastro.2012.06.003
153
Lim H Kim YU Sun H Lee JH Reynolds JM Hanabuchi S et al . Proatherogenic Conditions Promote Autoimmune T Helper 17 Cell Responses In Vivo. Immunity (2014) 40:153–65. doi: 10.1016/j.immuni.2013.11.021
154
Angeli V Llodrá J Rong JX Satoh K Ishii S Shimizu T et al . Dyslipidemia Associated With Atherosclerotic Disease Systemically Alters Dendritic Cell Mobilization. Immunity (2004) 21:561–74. doi: 10.1016/j.immuni.2004.09.003
155
Rönnblom L Eloranta ML . The Interferon Signature in Autoimmune Diseases. Curr Opin Rheumato (2013) 125:248–53. doi: 10.1097/BOR.0b013e32835c7e32
156
Nathan C . Neutrophils and Immunity: Challenges and Opportunities. Nat Rev Immunol (2006) 6:173–82. doi: 10.1038/nri1785
157
Denny MF Yalavarthi S Zhao W Thacker SG Anderson M Sandy AR et al . A Distinct Subset of Proinflammatory Neutrophils Isolated From Patients With Systemic Lupus Erythematosus Induces Vascular Damage and Synthesizes Type I IFNs. J Immunol (2010) 184:3284–97. doi: 10.4049/jimmunol.0902199
158
Carlucci PM Purmalek MM Dey AK Temesgen-Oyelakin Y Sakhardande S Joshi AA et al . Neutrophil Subsets and Their Gene Signature Associate With Vascular Inflammation and Coronary Atherosclerosis in Lupus. JCI Insight (2018) 3:e99276. doi: 10.1172/jci.insight.99276
159
Toller-Kawahisa JE Canicoba NC Venancio VP Kawahisa R Antunes LM Cunha TM et al . Systemic Lupus Erythematosus Onset in Lupus-Prone B6.MRL/lpr Mice Is Influenced by Weight Gain and Is Preceded by an Increase in Neutrophil Oxidative Burst Activity. Free Radic Biol Med (2015) 86:362–73. doi: 10.1016/j.freeradbiomed.2015.06.005
160
Elloumi N Ben Mansour R Marzouk S Mseddi M Fakhfakh R Gargouri B et al . Differential Reactive Oxygen Species Production of Neutrophils and Their Oxidative Damage in Patients With Active and Inactive Systemic Lupus Erythematosus. Immunol Lett (2017) 184:1–6. doi: 10.1016/j.imlet.2017.01.018
161
Smith CK Vivekanandan-Giri A Tang C Knight JS Mathew A Padilla RL et al . Neutrophil Extracellular Trap-Derived Enzymes Oxidize High-Density Lipoprotein: An Additional Proatherogenic Mechanism in Systemic Lupus Erythematosus. Arthritis Rheumatol (2014) 66:2532–44. doi: 10.1002/art.38703
162
Tanaka N Masuoka S Kusunoki N Nanki T Kawai S . Serum Resistin Level and Progression of Atherosclerosis During Glucocorticoid Therapy for Systemic Autoimmune Diseases. Metabolites (2016) 6:28. doi: 10.3390/metabo6030028
163
van Leuven SI Mendez-Fernandez YV Wilhelm AJ Wade NS Gabriel CL Kastelein JJ . Mycophenolate Mofetil But Not Atorvastatin Attenuates Atherosclerosis in Lupus-Prone LDLr(-/-) Mice. Ann Rheum Dis (2012) 71:408–14. doi: 10.1136/annrheumdis-2011-200071
164
Caro-Maldonado A Wang R Nichols AG Kuraoka M Milasta S Sun LD et al . Metabolic Reprogramming Is Required for Antibody Production That Is Suppressed in Anergic But Exaggerated in Chronically BAFF-Exposed B Cells. J Immunol (2014) 192:3626–36. doi: 10.4049/jimmunol.1302062
165
Tsiantoulas D Sage AP Göderle L Ozsvar-Kozma M Murphy D Porsch F et al . B Cell-Activating Factor Neutralization Aggravates Atherosclerosis. Circulation (2018) 138:2263–73. doi: 10.1161/circulationaha.117.032790
166
Furumoto Y Smith CK Blanco L Zhao W Brooks SR Thacker SG et al . Tofacitinib Ameliorates Murine Lupus and Its Associated Vascular Dysfunction. Arthritis Rheumatol (2017) 69:148–60. doi: 10.1002/art.39818
167
Hasni SA Gupta S Davis M Poncio E Temesgen-Oyelakin Y Carlucci PM et al . Phase 1 Double-Blind Randomized Safety Trial of the Janus Kinase Inhibitor Tofacitinib in Systemic Lupus Erythematosus. Nat Commun (2021) 12:3391. doi: 10.1038/s41467-021-23361-z
168
Wallace DJ Furie RA Tanaka Y Kalunian KC Mosca M Petri MA et al . Baricitinib for Systemic Lupus Erythematosus: A Double-Blind, Randomised, Placebo-Controlled, Phase 2 Trial. Lancet (2018) 392:222–31. doi: 10.1016/s0140-6736(18)31363-1
169
Mach F Schönbeck U Sukhova GK Atkinson E Libby P . Reduction of Atherosclerosis in Mice by Inhibition of CD40 Signaling. Nature (1998) 394:200–3. doi: 10.1038/28204
170
Obreque J Vega F Torres A Cuitino L Mackern-Oberti JP Viviani P et al . Autologous Tolerogenic Dendritic Cells Derived From Monocytes of Systemic Lupus Erythematosus Patients and Healthy Donors Show a Stable and Immunosuppressive Phenotype. Immunology (2017) 152:648–59. doi: 10.1111/imm.12806
171
Ruiz-Limon P Barbarroja N Perez-Sanchez C Aguirre MA Bertolaccini ML Khamashta MA et al . Atherosclerosis and Cardiovascular Disease in Systemic Lupus Erythematosus: Effects of In Vivo Statin Treatment. Ann Rheum Dis (2015) 74:1450–8. doi: 10.1136/annrheumdis-2013-204351
172
Erkan D Willis R Murthy VL Basra G Vega J Ruiz-Limón P et al . A Prospective Open-Label Pilot Study of Fluvastatin on Proinflammatory and Prothrombotic Biomarkers in Antiphospholipid Antibody Positive Patients. Ann Rheum Dis (2014) 73:1176–80. doi: 10.1136/annrheumdis-2013-203622
173
Duarte-García A Myasoedova E Karmacharya P Hocaoğlu M Murad MH Warrington KJ et al . Effect of Omega-3 Fatty Acids on Systemic Lupus Erythematosus Disease Activity: A Systematic Review and Meta-Analysis. Autoimmun Rev (2020) 19:102688. doi: 10.1016/j.autrev.2020.102688
174
Hansen AL Buchan GJ Rühl M Mukai K Salvatore SR Ogawa E et al . Nitro-Fatty Acids Are Formed in Response to Virus Infection and Are Potent Inhibitors of STING Palmitoylation and Signaling. Proc Natl Acad Sci U S A (2018) 115:E7768–75. doi: 10.1073/pnas.1806239115
175
Lv J Zhang L Yan F Wang X . Clinical Lipidomics: A New Way to Diagnose Human Diseases. Clin Transl Med (2018) 7:12. doi: 10.1186/s40169-018-0190-9
Summary
Keywords
systemic lupus erythematosus (SLE), dyslipidemia (DLP), lipid metabolism, immunocyte, autoimmunity
Citation
Sun W, Li P, Cai J, Ma J, Zhang X, Song Y and Liu Y (2022) Lipid Metabolism: Immune Regulation and Therapeutic Prospectives in Systemic Lupus Erythematosus. Front. Immunol. 13:860586. doi: 10.3389/fimmu.2022.860586
Received
23 January 2022
Accepted
28 February 2022
Published
18 March 2022
Volume
13 - 2022
Edited by
Hai-Feng Pan, Anhui Medical University, China
Reviewed by
Kongyang Ma, Sun Yat-sen University, China; Yafeng Li, The Fifth Hospital of Shanxi Medical University, China
Updates
Copyright
© 2022 Sun, Li, Cai, Ma, Zhang, Song and Liu.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yudong Liu, yudongliu1983@126.com; Yong Song, yong_song6310@yahoo.com
†These authors have contributed equally to this work
This article was submitted to Autoimmune and Autoinflammatory Disorders, a section of the journal Frontiers in Immunology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.