 Ryu Watanabe
Ryu Watanabe Motomu Hashimoto
Motomu Hashimoto
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 29 July 2022
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.859502
This article is part of the Research Topic The Role of Monocytes/Macrophages in Autoimmunity and Autoinflammation View all 19 articles
Vasculitis is an autoimmune vascular inflammation with an unknown etiology and causes vessel wall destruction. Depending on the size of the blood vessels, it is classified as large, medium, and small vessel vasculitis. A wide variety of immune cells are involved in the pathogenesis of vasculitis. Among these immune cells, monocytes and macrophages are functionally characterized by their capacity for phagocytosis, antigen presentation, and cytokine/chemokine production. After a long debate, recent technological advances have revealed the cellular origin of tissue macrophages in the vessel wall. Tissue macrophages are mainly derived from embryonic progenitor cells under homeostatic conditions, whereas bone marrow-derived circulating monocytes are recruited under inflammatory conditions, and then differentiate into macrophages in the arterial wall. Such macrophages infiltrate into an otherwise immunoprotected vascular site, digest tissue matrix with abundant proteolytic enzymes, and further recruit inflammatory cells through cytokine/chemokine production. In this way, macrophages amplify the inflammatory cascade and eventually cause tissue destruction. Recent studies have also demonstrated that monocytes/macrophages can be divided into several subpopulations based on the cell surface markers and gene expression. In this review, the subpopulations of circulating monocytes and the ontogeny of tissue macrophages in the artery are discussed. We also update the immunopathology of large vessel vasculitis, with a special focus on giant cell arteritis, and outline how monocytes/macrophages participate in the disease process of vascular inflammation. Finally, we discuss limitations of the current research and provide future research perspectives, particularly in humans. Through these processes, we explore the possibility of therapeutic strategies targeting monocytes/macrophages in vasculitis.
Monocytes are circulating blood leukocytes that play important roles in the inflammatory response, and represent 10% of leukocytes in human blood (1). Monocytes are functionally characterized by their capacity for phagocytosis, antigen presentation, and cytokine/chemokine production, and originate in the bone marrow from a hematopoietic precursor which is common for several subsets of macrophages and dendritic cells (DCs). These cells are not only part of the innate immune system, but also the monocytic lineage that support the activation of the adaptive immune system by antigen presentation (2). Monocytes/macrophages are deeply involved in vascular inflammation including atherosclerosis and vasculitis as well.
Vasculitis is an autoimmune and/or autoinflammatory vascular inflammation and causes breakdown of the blood vessel walls. Based on the distribution of vessel involvement, it is classified as large, medium, and small vessel vasculitis (3). Large vessel vasculitis affects the aorta and its major branches and include giant cell arteritis (GCA) and Takayasu arteritis (TAK). The hallmark of the two diseases is granulomatous inflammation, which is primarily composed of CD4+ T cells and macrophages (4, 5). In GCA, name-giving multinucleated giant cells are often observed in the vascular tissue (Figure 1) and formed by Toll-like receptor (TLR) 2-induced fusion of macrophages (6). Thus, it is obvious that monocytes/macrophages are key players in the pathomechanisms of large vessel vasculitis. Since we have been working on the pathogenesis of GCA, this review will mainly focus on GCA.
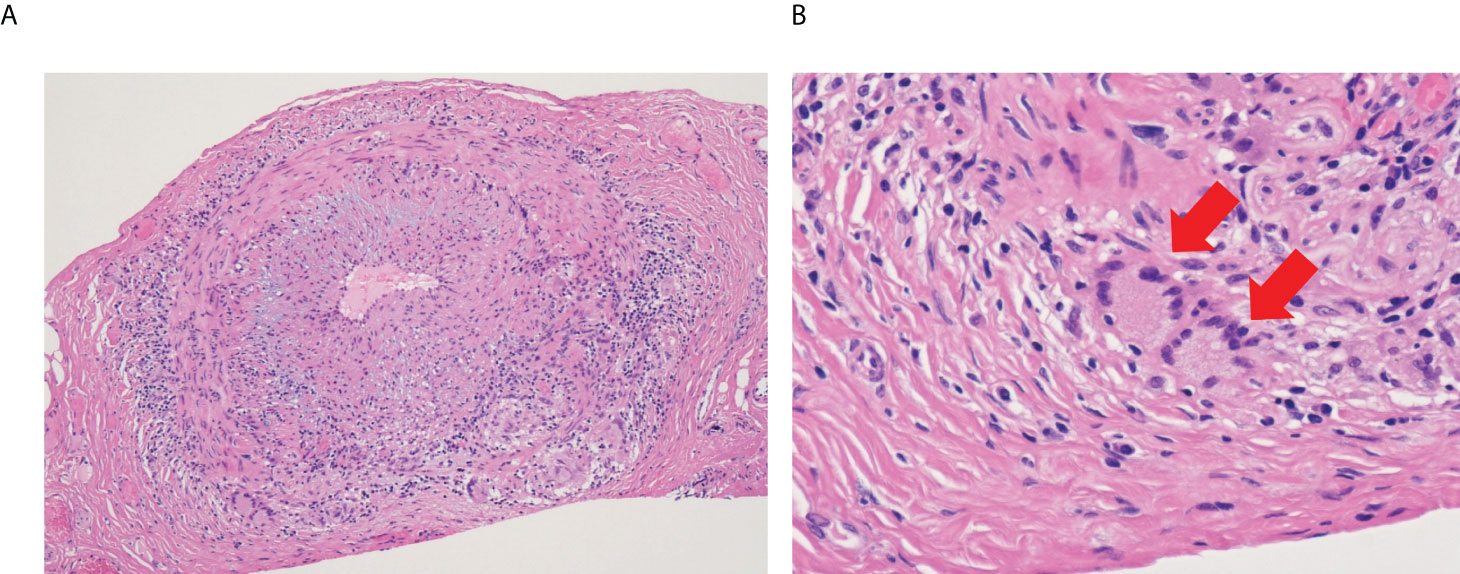
Figure 1 Microscopic image of giant cell arteritis. (A) Left temporal artery biopsy from 65-year-old woman with giant cell arteritis (Hematoxylin and eosin staining, x10). Lymphocytes and macrophages form granulomatous inflammation, and intimal hyperplasia causes narrowing of the blood lumen. (B) High power field image of the biopsy (Hematoxylin and eosin staining, x40). Red arrows show multinucleated giant cells.
The currently available treatments for GCA include glucocorticoids and tocilizumab (TCZ), an IL-6 receptor inhibitor. Even with the adequate use of glucocorticoids, inflammation of the temporal artery remains in about half of the patients after one year (7). Macrophages and giant cells also remain in one in four patients. On the other hand, TCZ reduces vascular inflammation detected by fluorodeoxyglucose-positron emission tomography (8) and flare-up of GCA (9). However, it is difficult to cure the disease, as shown by the flare-up after discontinuation of TCZ in most cases (8). Therefore, clinical unmet needs exist with the current therapies.
This review first summarizes the current knowledge of monocytes/macrophages subsets and the origin of tissue macrophages, particularly in the vascular tissue. Then, the pathogenic roles of monocytes/macrophages in the pathogenesis of large vessel vasculitis are presented. Finally, we discuss future perspectives for therapeutic options targeting monocytes/macrophages in large vessel vasculitis.
Monocytes differentiated from progenitor cells in the bone marrow reach the circulation. Currently, human circulating monocytes are divided into three subsets based on the expression of superficial CD14 (a cell co-receptor for lipopolysaccharide [LPS]) and CD16 (the low-affinity IgG receptor); “classical” CD14++CD16− monocytes (≥90%), “intermediate” CD14++CD16+ monocytes, and “non-classical” CD14+ CD16++ monocytes (10). These subsets are characterized by different levels of cell surface markers and chemokine receptors, but there appears to be a developmental relationship between these cells (from classical by intermediate to non-classical) (10). The classical monocytes are involved in a variety of immune response such as inflammation and tissue repair. The intermediate monocytes are characterized by the highest TLR2, TLR4, and human leukocyte antigen-D related expression among monocyte subsets, and have the highest antigen presenting ability. They have superior reactive oxygen species production and have a role in angiogenesis. The non-classical monocytes are called “patrolling” monocytes and have high ability to stimulate CD4+ T cells (11). The use of additional markers, such as C-C Chemokine Receptor 2 (CCR2) which is a key mediator of monocyte migration, for better delineation of monocyte subsets has been proposed (12), but its usefulness needs further study.
Conflicting data on cytokine production by the distinct monocyte subsets exist. We have previously reported that the intermediate monocytes treated with LPS produced the most IL-1β, IL-6, and TNFα (13). Wong et al. reported that non-classical monocytes produced the highest IL-1β and TNFα in response to LPS, but that equivalent amounts of IL-6 were secreted by the three subsets. (14). These inconsistencies are probably due to the different isolation methods used to purify the monocyte subsets (11).
An expansion of intermediate monocytes has been implicated in various inflammatory diseases and vascular diseases such as atherosclerosis (15), coronary artery disease (13), and antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (16). It has been suggested that the ability of intermediate monocytes to present antigens and produce proinflammatory cytokines may be involved in the pathogenesis of such diseases.
Tissue macrophages are derived from embryonic or adult hematopoietic stem cell (HSC) progenitor cells under homeostatic conditions (17). Representatives of tissue macrophages are alveolar macrophages (lung), Kupffer cells (liver), osteoclast (bone), microglia (central nervous system), and so on. They are remarkably heterogenous in terms of their surface markers, transcriptome, and epigenomes (18). Monocyte-derived cells also contribute to the macrophage population in the tissues but are mostly associated with a response to inflammatory conditions. It seems more likely that local environmental imprinting is the critical determinant for macrophage identity and function, irrespective of their origin (18).
Recently, the ontogeny of arterial macrophages has been revealed by an elegant method combining the fate-mapping analysis and single-cell RNA sequencing (19). Yolk sac erythro-myeloid progenitors (EMPs) migrate to the arterial adventitia and give rise to adventitial macrophages. Surprisingly, with aging, these adventitial macrophages decline in numbers, and are not replenished by bone marrow-derived monocytes. During vascular inflammation, bone marrow-derived monocytes are recruited to the vascular site and differentiate into adventitial macrophages, while EMP-derived macrophages show self-renewal and contribute to tissue regeneration (19). It has been reported that, during infection, monocytes are educated to be tissue-specific in the bone marrow by signals produced at the site of inflammation (20), but it remains unclear whether this is the case during vascular inflammation.
The most well-described paradigm of macrophage polarization is the M1/M2 polarization axis. M1 and M2 macrophages are also referred to as classically or alternatively activated macrophages, respectively (21). M1 macrophages are activated by the microbial products and proinflammatory cytokines (IFN-γ and/or LPS or TNFα) and characterized by an excess production of proinflammatory cytokines (IL-1β, IL-6, IL-12, IL-23), chemokines, nitric oxide, and reactive oxygen intermediates. In contrast, M2 macrophages are activated by IL-4, IL-10, IL-13, and express mannose receptor (CD206), scavenger receptor A (CD204), and chemokine receptors. High levels of IL-10 are produced by M2 macrophages (22). M2 macrophages are further classified into M2a (IL4/IL-13-induced), M2b (LPS/immune complexes-induced), M2c (IL-10/TGFβ/glucocorticoids-induced), and M2d (tumor-associated factors-induced) macrophages (23, 24).
However, macrophage activation is not that simple. It should be noted that M1 and M2 macrophages are not completely distinct subsets, but they are often overlapping; for example, in atherosclerotic plaque, macrophages expressing both M1 and M2 markers do exist (25). Thus, consensus on how to define macrophage activation in vitro and in vivo has not yet been fully established. In this context, a group of scientists proposed the updated nomenclature for macrophage activation and polarization (26). In this proposal, they described a set of standards encompassing three principles—the source of macrophages, definition of the activators, and markers to describe macrophage activation—with the goal of unifying experimental standards (26). Technological advances, such as single cell RNA sequencing, may reveal further new macrophage subsets in the future (27).
An increased number of monocytes (monocytosis) is observed in the peripheral blood of active patients with GCA, and monocyte counts positively correlates with the C-reactive protein (CRP) levels (28). This observation is in line with the report that monocyte-derived macrophages are dominant among tissue macrophages during vascular inflammation (19). Subpopulation analysis using flow cytometry demonstrated that monocytosis in the peripheral blood was attributable to classical monocytes and slightly intermediate monocytes (29). Interestingly, treatment with corticosteroids suppress the numbers of intermediate and non-classical monocytes, but the number of classical monocytes is unaffected (29). It has been reported that glucocorticoid-induced depletion of non-classical monocytes is mediated by caspase-dependent apoptosis (30).
Although monocytes can differentiate into DCs, it has been reported that the number of circulating DCs were comparable between GCA patients and healthy individuals (31). Most quiescent tissues contain resident DC population, but during inflammation, monocyte-derived DCs compensate resident population in the tissue (32). However, it remains elusive whether this is the case in GCA.
It is no doubt that research on monocytes/macrophages in GCA has dramatically progressed since the discovery of IL-6 (33). IL-6 acts on hepatocytes to produce acute phase proteins such as CRP and serum amyloid A (34). It was found that plasma IL-6 levels reflect the disease activity of GCA (35). Although 60–80% of circulating monocytes in patients with GCA can produce IL-6, the major source of IL-6 production was activated macrophages in the vascular lesion (36). Tissue macrophages are activated by IFN-γ released from CD4+ T cells (4), and IL-6 shifts naïve CD4+ T cell differentiation towards Th17 cells, while inhibiting regulatory T cell (Treg) differentiation (37). Other proinflammatory cytokines, including IL-1β and TNFα, were also localized to tissue macrophages and giant cells (38).
Treatment with corticosteroids diminish IL-1β and IL-6 production from tissue macrophages (39). In contrast, IL-6 receptor inhibitor tocilizumab (TCZ) increases plasma IL-6 levels in patients with GCA (40). TCZ may have little direct effect on suppressing macrophage activation in the vascular tissue and/or block clearance of released IL-6 through IL-6 receptor. However, TCZ reduces relapse and has a steroid-tapering effect on GCA (9) maybe because it restores not only the number of Tregs but also the function of these cells (41–43). Thus, TCZ is widely recommended in the treatment guidelines (44, 45).
IL-12, which is produced by M1 macrophages, is a heterodimeric proinflammatory cytokine that favours the differentiation of Th1 cells (46). Recently, it has been reported that IL-12 promotes conversion from Th17 cells into IFN-γ-producing Th1-like cells, called “non-classic Th1 cells” (47, 48). This transformation is governed by the transcription factor Eomes (49). Indeed, IL-12 is highly enriched in the biopsy-positive temporal arteries (50); therefore, IFN-γ found in the vascular tissues may be derived from Th1 or non-classic Th1 cells. However, ustekinumab, an IL-12 and IL-23 inhibitor, failed to show its efficacy in the treatment of GCA (51).
Monocytes/macrophages and giant cells not only produce proinflammatory cytokines, but also contribute to tissue destruction. They produce excess proteolytic enzymes and proteinases such as collagenases, cathepsins, and matrix metalloproteinase (MMP)-2 and MMP-9, disrupt external and internal elastic membranes and cause vessel wall destruction (52). Our recent work has revealed that MMP-9-producing macrophages/giant cells are mainly located at the intima-media border and monocyte-derived macrophages from patients with GCA outperformed producing MMP-2 and MMP-9 compared with those from healthy donors (53). Since MMP-2 cleaves the propeptide from the pro-MMP-9 to release enzymatically active MMP-9, this combination of MMPs allows vascular lesions as an active MMP-9-rich environment. We further demonstrated that, using an artificial basement membrane system composed of collagen I and collage IV, MMP-9 released from the circulating monocytes degrades basement membrane and enables CD4+ T cell to invade into blood vessel. This study also showed that, using an experimental animal model of vasculitis, blocking MMP-9 was highly effective to protect vascular structure and homeostasis, suggesting that it may serve as a novel therapeutic option for large vessel vasculitis (53).
A recent report showed that most of the MMP-9-producing macrophages were CD206 positive and induced by granulocyte macrophage-colony stimulating factor (GM-CSF) (54). Generally, GM-CSF is considered to induce M1 phenotype in macrophages (55, 56), but it may induce M1 plus M2 phenotypes in GCA macrophages. GM-CSF, which is produced by macrophages, T cells, myofibroblasts, and endothelial cells in GCA-affected arteries (57), is expected to be a promising therapeutic target in GCA in recent years. Indeed, treatment of ex vivo cultured GCA arteries with the anti-GM-CSF receptor antagonist mavrilimumab successfully ameliorated vascular inflammation through reducing T cell and macrophage infiltration and neoangiogenesis (57). Among T cells, mavrilimumab specifically reduced Th1 cells, but not Th17 cells. In addition, in a phase 2, randomised, double-blind, placebo-controlled trial, mavrilimumab showed superiority to placebo in the analyses of time to flare and sustained remission for patients with GCA (58). Therefore, GM-CSF is not only a macrophage differentiation factor, but is also fundamentally involved in vascular inflammation.
In contrast, macrophage-colony stimulating factor (M-CSF), which is generally considered to induce M2 phenotype in macrophages (59, 60), is shown to skew macrophages into different phenotypes, namely folate receptor β (FRβ)-positive macrophages (54). M-CSF is mainly produced by CD206+/MMP-9+ macrophages at the intima-media borders. Collectively, it has been proposed that, at the initial stage of GCA, infiltrated monocytes from the vasa vasorum are primed by local GM-CSF and differentiate into CD206+/MMP-9+ macrophages. They migrate to media and media-intima junction and promote tissue destruction, while stimulating angiogenesis through IL-13Rα2 signaling (61). At the late stage of GCA, CD206+/MMP-9+ macrophages often fuse to form multinucleated giant cells and release M-CSF at the intima-media borders. Multiple cytokines (TNF, IL-6, IFN-γ, IL-4, etc) and TLR2 are thought to be involved in the formation of multinucleated giant cells, but the precise mechanism remains unclear (6). M-CSF-skewed FRβ+ macrophages produce high concentrations of growth factors that activate myofibroblasts, leading to luminal occlusion (54, 62) (Figure 2). Anti-M-CSF antibodies have not been tested in patients with GCA so far.
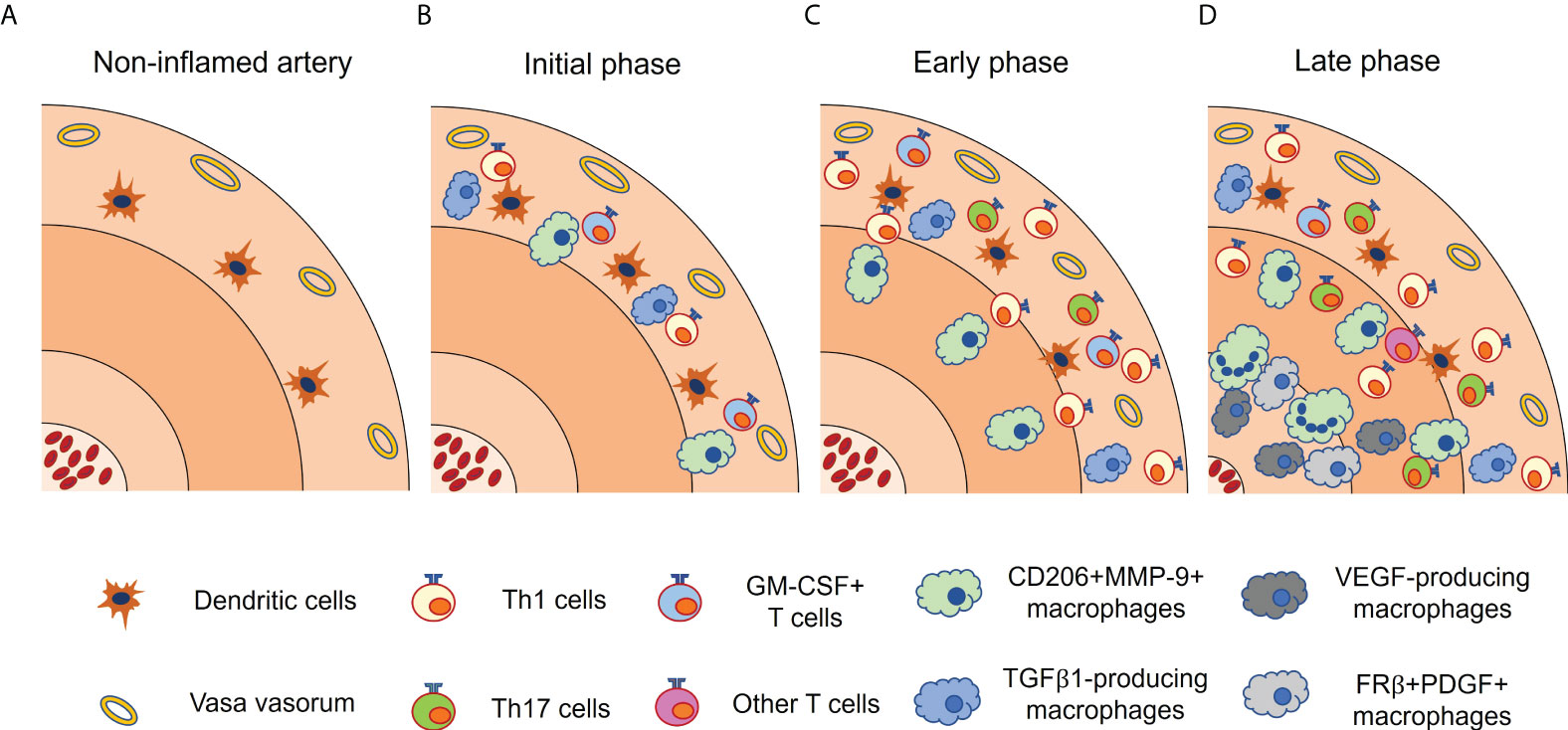
Figure 2 Functionally heterogenous macrophages in giant cell arteritis. Vascular lesion of giant cell arteritis contains a variety of macrophage subsets, each with a characteristic distribution. (A) In non-inflamed artery, vascular dendritic cells (vasDCs) reside in the media-adventitial border. (B) In the initial phase, vasDCs initiate inflammatory cascade, and recruits T cells and monocytes through chemokines. Infiltrated monocytes are differentiated into CD206+MMP-9+ macrophages by GM-CSF released from activated T cells. TGFβ1-producing macrophages are also present in the adventitia. (C) In the early phase, CD206+MMP-9+ macrophages migrate to the media and the media-intima border. Adventitial inflammation is increased. (D) In the late phase, CD206+MMP-9+ macrophages often fuse to form multinucleated giant cells and produce M-CSF, which gives rise to FRβ+ PDGF-producing macrophages at the media-intima border. Multiple cytokines and TLR2 are thought to be involved in the formation of multinucleated giant cells. VEGF-producing macrophages are preferentially located in the tunica media and intima. It should be noted that these macrophage subsets are not completely distinct. FRβ, folate receptor β; GM-CSF, granulocyte macrophage-colony stimulating factor; IFN, interferon; M-CSF, macrophage-colony stimulating factor; MMP, matrix metalloproteinase; PDGF, platelet-derived growth factor; TGFβ1, transforming growth factor β1; VEGF, vascular endothelial growth factor.
Tissue macrophages produce growth factors such as transforming growth factor β1 (TGFβ1), platelet-derived growth factors (PDGF), and fibroblast growth factors (FGF) (63, 64). These growth factors are considered to induce an excessive fibroproliferative response leading to luminal occlusion. TGFβ1-expressing macrophages coproduce IL-1β and IL-6 and exhibit a strong preference for localization in the adventitia. Although not clearly proven, given the cytokine profile and localization of TGFβ1-producing macrophages, it is likely that they emerge at the initial disease stage and are activated by IFN-γ released from Th1 cells (62) (Figure 2). In contrast, FRβ+ macrophages at the media-intima junction emerge at the late disease stage, and produce PDGF, which is closely associated with concentric intimal hyperplasia (54).
In addition, the number of newly formed blood vessel in the adventitia is associated with the production of vascular endothelial growth factor (VEGF), which is localized to tissue macrophages at the media-intima junction (65). VEGF production is augmented by IL-6 (66) and upregulates a NOTCH ligand, Jagged1, on the innermost microvascular endothelial cells. Jagged1 in turn stimulates NOTCH1 receptor on CD4+ T cell, skewing CD4+ T cell differentiation toward Th1 and Th17 (67). Therefore, anti-VEGF therapy may help to inhibit not only neoangiogenesis but also maldifferentiation of CD4+ T cells (68).
Alteration in systemic and local chemokine production and chemokine receptor expression has been reported (Figure 3). Among them, C-X-C motif Chemokine Ligand 9 (CXCL9), CXCL10, and CXCL11 levels are elevated in the serum of GCA patients (69). These chemokines are produced by tissue macrophages in response to IFN-γ (70) and recruit Th1 cells through C-X-C Motif Chemokine Receptor 3 (CXCR3). C-C motif Chemokine Ligand 3 (CCL3), CCL4, and CCL5 are also overproduced from macrophages and recruit T cells through CCR5 (71). Not only T cells but also monocytes are recruited by chemokines. For example, classical monocytes are recruited into vascular tissue by the CCL2-CCR2 axis (72), while non-classical monocytes depend on the CX3CL1-CX3CR1 axis (29). These observations suggest that tissue macrophages attract T cells, particularly Th1 cells, and monocytes/macrophages through multiple chemokines, amplifying vascular inflammation. In addition, a recent report has demonstrated that CXCL9 attracts CXCR3+ memory B cells, and CXCL13 recruits CXCR5+ memory B cells into vascular tissue, respectively (73). Notably, not only tissue macrophages but also vascular DCs produce chemokines such as CCL19 and CCL21, rendering vascular tissue as a chemokine-rich microenvironment (74). Further study is needed to test whether blocking the chemokines and chemokine receptors could have a therapeutic potential.
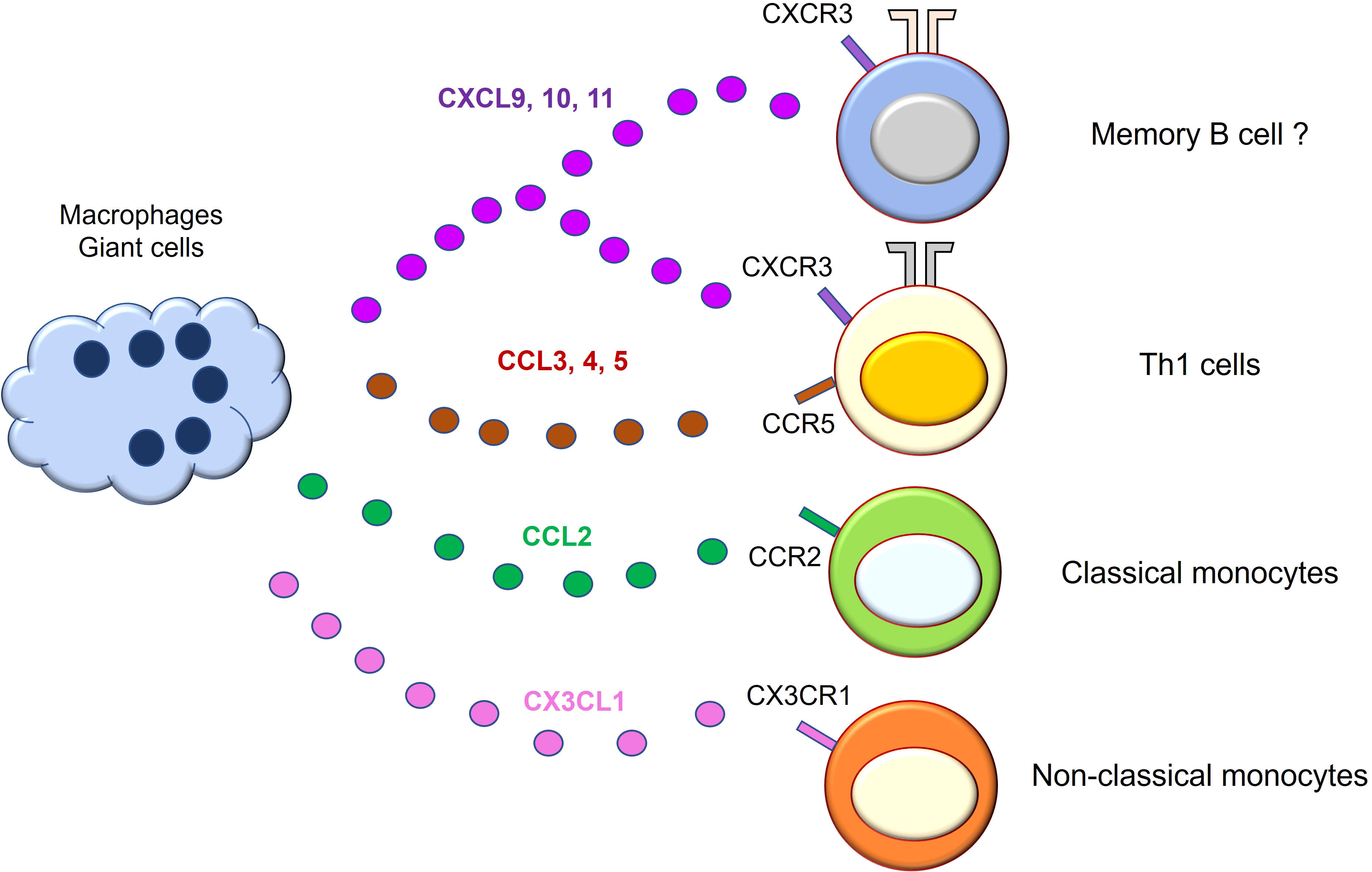
Figure 3 Macrophages/giant cells are professional chemokine producers in giant cell arteritis. Macrophages/giant cells in the vascular lesion of giant cell arteritis actively engage in chemokine production. The released chemokines amplify vascular inflammation by mobilizing cells that express the corresponding chemokine receptors. CXCL9, 10, and 11 recruit Th1 cells and memory B cells through CXCR3 receptor on the cell surface. CCL3, 4, and 5 recruit T cells expressing CCR5. CCL2 recruits CCR2-expressing classical monocytes. CX3CL1 mediates non-classical monocyte mobilization through CX3CR1 receptor. CCL, C-C motif Chemokine Ligand; CXCL, C-X-C motif Chemokine Ligand; CXCR, C-X-C Motif Chemokine Receptor.
Not only costimulatory molecules like CD28, but also coinhibitory molecules, such as programmed death 1 (PD-1), are expressed on T cell surface, and the clinical significance of blocking the PD-1/programmed death ligand 1 (PD-L1) interaction has become clear in cancer immunotherapy. Surprisingly, vascular DCs residing at the media-adventitial boarder have defective PD-L1 expression, which is critically involved in the pathomechanisms of GCA (75). PD-L1-deficient DCs have an increased potential to activate T cells and polarize naïve CD4+ T cell differentiation into Th1, Th17, and IL-21-producing T cells (76). Monocytes/macrophages derived from patients with GCA also had decreased expression of PD-L1 (70), although the significance of the deficient expression requires further elucidation. Taken together, PD-L1 immunoinhibitory mechanism to inhibit T cell hyperactivation is defective in myeloid lineage on vascular lesion in GCA. It is necessary to elucidate the mechanism of PD-L1 expression on vascular DCs and tissue macrophages. Also, testing the effect of PD-L1 signal-inducing agents, such as fusion proteins linking the extracellular domain of PD-L1 to the Fc portion of immunoglobulin (PD-L1 Fc), is warranted.
Unlike GCA, it is difficult to perform biopsies of affected lesions in TAK, and only specimens that have undergone surgery are used for research. In addition, large amounts of steroids are often administered prior to surgery, making it rare to obtain an active untreated vascular sample. Thus, the pathogenesis of TAK has not been fully elucidated. Although such bias is undeniable, M1 macrophages are dominant in aortic lesions of TAK (77, 78), which may be linked to excess IFN-γ produced by CD4+ T cells, CD8+ T cells, and natural killer cells (79). In vitro production of MMP-2 and MMP-9 in monocyte-derived macrophages is mildly increased compared with that from healthy donors (80). Steroid treatment transforms M1 macrophages into M2 macrophages and diminishes CCL2-expressing M1 macrophages (78). Thus, M2 macrophages dominate in treated aortic lesion and promote tissue remodeling with an excess fibrotic response.
Recently, single cell RNA sequencing was applied to examine the transcriptome of peripheral blood mononuclear cells from TAK patients (81). The study demonstrated that CD14+ monocytes were increased, and gene expressions involved in oxidative stress were enriched. These monocytes may serve as a reservoir of tissue macrophages.
We have reviewed the role of monocytes/macrophages in large vessel vasculitis, particularly in GCA. Of note, functionally distinct macrophage subsets have been increasingly identified in GCA (62), although there were no studies comparing monocytes/macrophages from cranial GCA and those from large vessel GCA. Also, it becomes clearer that circulating monocytes, rather than embryonic progenitor-derived macrophages, cause vascular inflammation by migrating and differentiating into the distinct subsets of macrophages, although it has not yet been fully investigated in human. In particular, since GCA only affects people over the age of 50, the number of tissue resident macrophages in the vasculature may be decreased. Furthermore, low grade inflammation caused by aging, which is called inflammaging, inevitably affects monocyte/macrophages, T cells, and vascular cells both in the circulation and in the vascular tissue (82, 83). Cellular senescence of immune cells is often linked to the senescence-associated secretory phenotype, which could be implicated in the pathomechanisms of GCA (62, 84).
Considering disease mechanisms mediated by monocytes/macrophages in GCA, inhibiting the migration of circulating monocytes or inhibiting their differentiation and function in the tissues may be therapeutic strategies targeting macrophages. The possible therapeutic options in GCA are summarized in Figure 4. Blockade of the chemokine and chemokine receptor interaction attracting circulating monocytes and T cells could be the preferential therapeutic option based on the pathomechanisms. However, it is speculated that by the time symptoms appear, a significant number of monocytes have already been recruited to the vascular tissues, and differentiated macrophages are refractory to the current therapies. Therefore, it is unclear to what extent chemokine blockade is effective. In fact, many attempts have been made to treat rheumatic diseases by blocking the chemokine-chemokine receptor interaction, but many of the results have been disappointing so far (85).
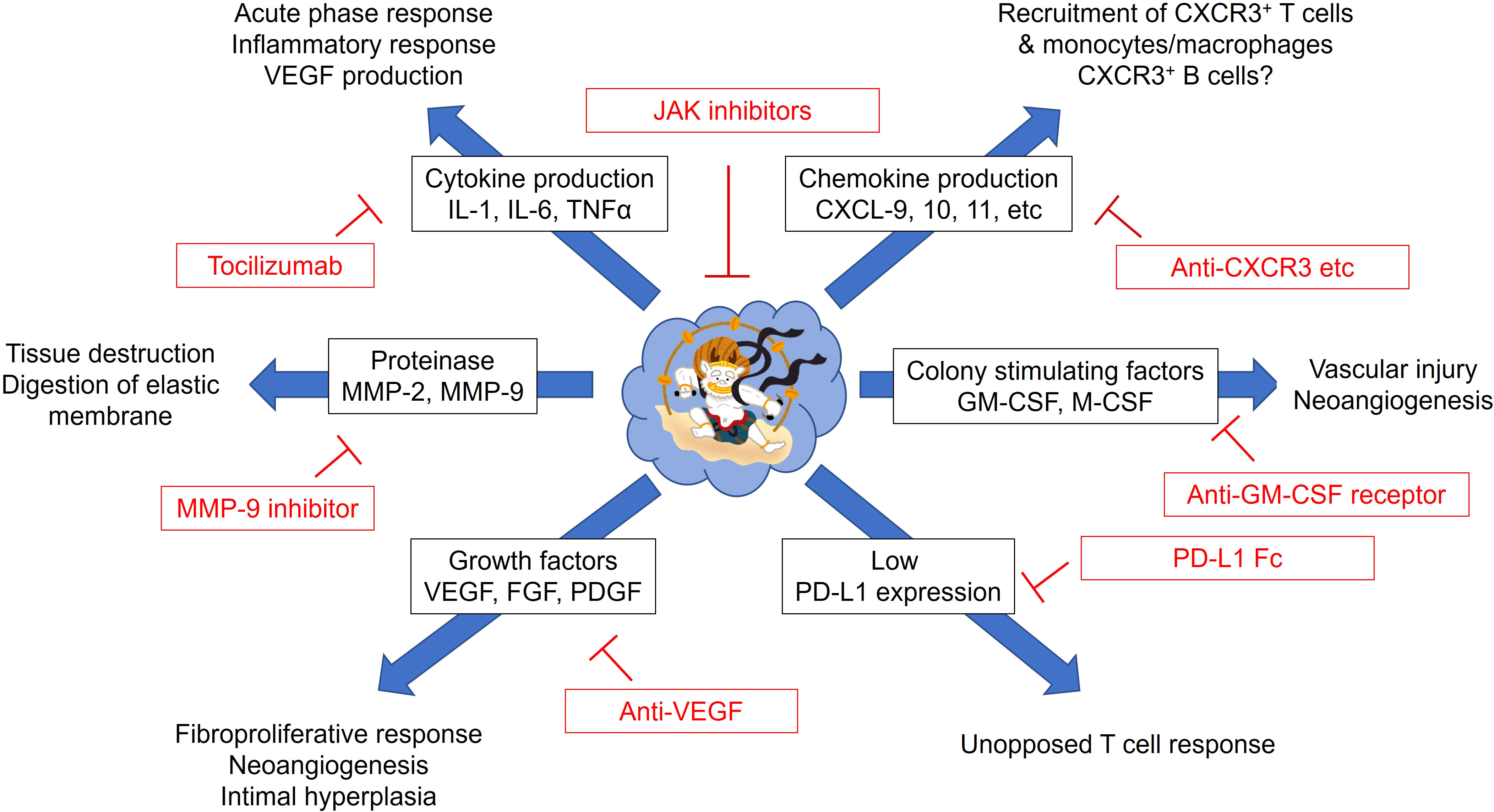
Figure 4 Possible therapeutic strategies for giant cell arteritis targeting monocytes and macrophages. Monocytes/macrophages from patients with giant cell arteritis have pleiotropic functions. Excess production of proinflammatory cytokines (IL-1β, IL-6, and TNFα), chemokines (CXCL9, 10, and 11), proteolytic enzymes (MMP-2 and MMP-9), colony stimulating factors (GM-CSF and M-CSF), growth factors (VEGF, FGF, PDGF) could be targeted by the corresponding inhibitors. Immune dysregulation by defective PD-L1 expression on monocytes/macrophages could be corrected by PD-L1 Fc. Janus kinase (JAK) inhibitors may directly suppress the function of monocytes/macrophages. CXCL, C-X-C motif Chemokine Ligand; CXCR, C-X-C Motif Chemokine Receptor; FGF, fibroblast growth factor; GM-CSF, granulocyte macrophage-colony stimulating factor; M-CSF, macrophage-colony stimulating factor; MMP, matrix metalloproteinase; PDGF, platelet-derived growth factor; PD-L1, programmed death ligand 1; VEGF, vascular endothelial growth factor.
Proinflammatory cytokines contribute profoundly to the exacerbation of vasculitis. The GiACTA trial have successfully demonstrated that blocking the IL-6 signal with TCZ suppresses flare of GCA and has steroid-sparing effect (9). However, as mentioned, recurrence after the discontinuation or even during TCZ therapy remains common. In addition, blocking TNFα with infliximab yielded disappointing results for GCA (86). Moreover, anakinra, an IL-1 receptor antagonist, has been shown the efficacy against GCA in case series (87), but its efficacy and safety have not fully been confirmed in the large-scale trials. Therefore, accumulating evidence shows that single cytokine inhibition may not be sufficient to completely diminish vascular inflammation.
As we have seen, treatment that suppresses a single therapeutic target, such as chemokines, proinflammatory cytokines, proteolytic enzymes, or growth factors, may not be sufficient for treating GCA. A combination of these or agents that inhibits the multiple cellular signaling, such as Janus kinase (JAK) inhibitors, may be effective (88). Multiple cytokines which are implicated in the pathomechanisms of GCA, such as IL-6, IFN-γ, IFN-α, GM-CSF, utilize the JAK-signal transducer and activator of transcription (STAT) pathway (89). Indeed, increased activities of the JAK-STAT pathway has been reported both in the vascular lesions and in circulating T cells (90, 91). In experimental animal model of large vessel vasculitis, JAK inhibitors not only reduced T cell infiltration and T cell-derived cytokine production, but also inhibited macrophage infiltration and growth factor production, resulting in reduced neoangiogenesis and intimal hyperplasia (90).
CD4+ T cells from patients with TAK are also dependent on the JAK-STAT pathway (92). In addition, genome-wide association study has demonstrated that IL-12B is an susceptibility gene in TAK (93) and risk allele of IL-12B was associated with vascular damage in TAK (94). Since IL-12 utilizes the JAK-STAT pathway as a downstream signaling, JAK inhibitors could be promising agents for TAK as well (92, 95).
Finally, PD-L1 deficiency seems not specific to GCA monocytes. Monocytes derived from ANCA-associated vasculitis have the same defect (96). Lower PD-L1 expression leads to increased stimulatory capacity of monocytes, thus leading to overactivation of CD4+ T cells. The defective PD-L1 expression was due to an enhanced lysosomal degradation of PD-L1 (96). As the efficacy of PD-L1 Fc has been shown in a mouse model of lupus (97), PD-L1 Fc may induce negative signals to overactivated T cells in vasculitis and ameliorate vascular inflammation. Alternatively, agents that inhibit PD-L1 degradation in lysosomes may have therapeutic potentials.
In conclusion, recent advances in the research have clarified the origin and various roles of monocytes/macrophages in vasculitis. Drugs that inhibit multiple therapeutic targets simultaneously, rather than a single target, or agents that block multiple cellular signaling may be effective; however, verification of the efficacy and the safety of such drugs is essential.
RW drafted the manuscript. MH revised and finalized the manuscript. All authors contributed to the article and approved the submitted version.
This work was in part supported by JSPS KAKENHI Grant Number 20K17418, a grant-in-aid of the Cardiovascular Research Fund, Tokyo, Japan and grant for Promoting Research and Survey in Rheumatic Diseases by Japan Rheumatism Foundation to RW.
We would like to thank Dr. Yuto Kaimi (Department of Pathology, Osaka Metropolitan University Graduate School of Medicine) for pathological analysis and Enago for the language review (https://www.enago.jp/).
The authors declare that this review does not contain any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol (2009) 27:669–92. doi: 10.1146/annurev.immunol.021908.132557
2. Huber R, Pietsch D, Gunther J, Welz B, Vogt N, Brand K. Regulation of monocyte differentiation by specific signaling modules and associated transcription factor networks. Cell Mol Life Sci (2014) 71(1):63–92. doi: 10.1007/s00018-013-1322-4
3. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised international chapel hill consensus conference nomenclature of vasculitides. Arthritis Rheum (2013) 65(1):1–11. doi: 10.1002/art.37715
4. Weyand CM, Goronzy JJ. Immune mechanisms in medium and large-vessel vasculitis. Nat Rev Rheumatol (2013) 9(12):731–40. doi: 10.1038/nrrheum.2013.161
5. Watanabe R, Hosgur E, Zhang H, Wen Z, Berry G, Goronzy JJ, et al. Pro-inflammatory and anti-inflammatory T cells in giant cell arteritis. Joint Bone Spine (2017) 84(4):421–6. doi: 10.1016/j.jbspin.2016.07.005
6. Brooks PJ, Glogauer M, McCulloch CA. An overview of the derivation and function of multinucleated giant cells and their role in pathologic processes. Am J Pathol (2019) 189(6):1145–58. doi: 10.1016/j.ajpath.2019.02.006
7. Maleszewski JJ, Younge BR, Fritzlen JT, Hunder GG, Goronzy JJ, Warrington KJ, et al. Clinical and pathological evolution of giant cell arteritis: a prospective study of follow-up temporal artery biopsies in 40 treated patients. Mod Pathol (2017) 30(6):788–96. doi: 10.1038/modpathol.2017.10
8. Quinn KA, Dashora H, Novakovich E, Ahlman MA, Grayson PC. Use of 18F-fluorodeoxyglucose positron emission tomography to monitor tocilizumab effect on vascular inflammation in giant cell arteritis. Rheumatology (Oxford) (2021) 60(9):4384–9. doi: 10.1093/rheumatology/keaa894
9. Stone JH, Tuckwell K, Dimonaco S, Klearman M, Aringer M, Blockmans D, et al. Trial of tocilizumab in giant-cell arteritis. N Engl J Med (2017) 377(4):317–28. doi: 10.1056/NEJMoa1613849
10. Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, et al. Nomenclature of monocytes and dendritic cells in blood. Blood (2010) 116(16):e74–80. doi: 10.1182/blood-2010-02-258558
11. Ozanska A, Szymczak D, Rybka J. Pattern of human monocyte subpopulations in health and disease. Scand J Immunol (2020) 92(1):e12883. doi: 10.1111/sji.12883
12. Franca CN, Izar MCO, Hortencio MNS, do Amaral JB, Ferreira CES, Tuleta ID, et al. Monocyte subtypes and the CCR2 chemokine receptor in cardiovascular disease. Clin Sci (Lond) (2017) 131(12):1215–24. doi: 10.1042/CS20170009
13. Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med (2016) 213(3):337–54. doi: 10.1084/jem.20150900
14. Wong KL, Tai JJ, Wong WC, Han H, Sem X, Yeap WH, et al. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood (2011) 118(5):e16–31. doi: 10.1182/blood-2010-12-326355
15. Kapellos TS, Bonaguro L, Gemund I, Reusch N, Saglam A, Hinkley ER, et al. Human monocyte subsets and phenotypes in major chronic inflammatory diseases. Front Immunol (2019) 10:2035. doi: 10.3389/fimmu.2019.02035
16. Vegting Y, Vogt L, Anders HJ, de Winther MPJ, Bemelman FJ, Hilhorst ML. Monocytes and macrophages in ANCA-associated vasculitis. Autoimmun Rev (2021) 20(10):102911. doi: 10.1016/j.autrev.2021.102911
17. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol (2019) 19(6):369–82. doi: 10.1038/s41577-019-0127-6
18. Mowat AM, Scott CL, Bain CC. Barrier-tissue macrophages: functional adaptation to environmental challenges. Nat Med (2017) 23(11):1258–70. doi: 10.1038/nm.4430
19. Weinberger T, Esfandyari D, Messerer D, Percin G, Schleifer C, Thaler R, et al. Ontogeny of arterial macrophages defines their functions in homeostasis and inflammation. Nat Commun (2020) 11(1):4549. doi: 10.1038/s41467-020-18287-x
20. Askenase MH, Han SJ, Byrd AL, Morais da Fonseca D, Bouladoux N, Wilhelm C, et al. Bone-Marrow-Resident NK cells prime monocytes for regulatory function during infection. Immunity (2015) 42(6):1130–42. doi: 10.1016/j.immuni.2015.05.011
21. Gordon S. Alternative activation of macrophages. Nat Rev Immunol (2003) 3(1):23–35. doi: 10.1038/nri978
22. Labonte AC, Tosello-Trampont AC, Hahn YS. The role of macrophage polarization in infectious and inflammatory diseases. Mol Cells (2014) 37(4):275–85. doi: 10.14348/molcells.2014.2374
23. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep (2014) 6:13. doi: 10.12703/P6-13
24. Arora S, Dev K, Agarwal B, Das P, Syed MA. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology (2018) 223(4-5):383–96. doi: 10.1016/j.imbio.2017.11.001
25. Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR, et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res (2010) 107(6):737–46. doi: 10.1161/CIRCRESAHA.109.215715
26. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41(1):14–20. doi: 10.1016/j.immuni.2014.06.008
27. Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science (2017) 356(6335). doi: 10.1126/science.aah4573
28. van Sleen Y, Graver JC, Abdulahad WH, van der Geest KSM, Boots AMH, Sandovici M, et al. Leukocyte dynamics reveal a persistent myeloid dominance in giant cell arteritis and polymyalgia rheumatica. Front Immunol (2019) 10:1981. doi: 10.3389/fimmu.2019.01981
29. van Sleen Y, Wang Q, van der Geest KSM, Westra J, Abdulahad WH, Heeringa P, et al. Involvement of monocyte subsets in the immunopathology of giant cell arteritis. Sci Rep (2017) 7(1):6553. doi: 10.1038/s41598-017-06826-4
30. Dayyani F, Belge KU, Frankenberger M, Mack M, Berki T, Ziegler-Heitbrock L. Mechanism of glucocorticoid-induced depletion of human CD14+CD16+ monocytes. J Leukoc Biol (2003) 74(1):33–9. doi: 10.1189/jlb.1202612
31. Matsumoto K, Suzuki K, Yoshimoto K, Seki N, Tsujimoto H, Chiba K, et al. Significant association between clinical characteristics and changes in peripheral immuno-phenotype in large vessel vasculitis. Arthritis Res Ther (2019) 21(1):304. doi: 10.1186/s13075-019-2068-7
32. Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology (2018) 154(1):3–20. doi: 10.1111/imm.12888
33. Hirano T, Yasukawa K, Harada H, Taga T, Watanabe Y, Matsuda T, et al. Complementary DNA for a novel human interleukin (BSF-2) that induces b lymphocytes to produce immunoglobulin. Nature (1986) 324(6092):73–6. doi: 10.1038/324073a0
34. Castell JV, Gomez-Lechon MJ, David M, Andus T, Geiger T, Trullenque R, et al. Interleukin-6 is the major regulator of acute phase protein synthesis in adult human hepatocytes. FEBS Lett (1989) 242(2):237–9. doi: 10.1016/0014-5793(89)80476-4
35. Roche NE, Fulbright JW, Wagner AD, Hunder GG, Goronzy JJ, Weyand CM. Correlation of interleukin-6 production and disease activity in polymyalgia rheumatica and giant cell arteritis. Arthritis Rheum (1993) 36(9):1286–94. doi: 10.1002/art.1780360913
36. Wagner AD, Goronzy JJ, Weyand CM. Functional profile of tissue-infiltrating and circulating CD68+ cells in giant cell arteritis. evidence for two components of the disease. J Clin Invest (1994) 94(3):1134–40. doi: 10.1172/JCI117428
37. Kishimoto T, Kang S. IL-6 revisited: From rheumatoid arthritis to CAR T cell therapy and COVID-19. Annu Rev Immunol (2022) 40:323–48. doi: 10.1146/annurev-immunol-101220-023458
38. Field M, Cook A, Gallagher G. Immuno-localisation of tumour necrosis factor and its receptors in temporal arteritis. Rheumatol Int (1997) 17(3):113–8. doi: 10.1007/s002960050019
39. Brack A, Rittner HL, Younge BR, Kaltschmidt C, Weyand CM, Goronzy JJ. Glucocorticoid-mediated repression of cytokine gene transcription in human arteritis-SCID chimeras. J Clin Invest (1997) 99(12):2842–50. doi: 10.1172/JCI119477
40. Berger CT, Rebholz-Chaves B, Recher M, Manigold T, Daikeler T. Serial IL-6 measurements in patients with tocilizumab-treated large-vessel vasculitis detect infections and may predict early relapses. Ann Rheum Dis (2019) 78(7):1012–4. doi: 10.1136/annrheumdis-2018-214704
41. Miyabe C, Miyabe Y, Strle K, Kim ND, Stone JH, Luster AD, et al. An expanded population of pathogenic regulatory T cells in giant cell arteritis is abrogated by IL-6 blockade therapy. Ann Rheum Dis (2017) 76(5):898–905. doi: 10.1136/annrheumdis-2016-210070
42. Samson M, Greigert H, Ciudad M, Gerard C, Ghesquiere T, Trad M, et al. Improvement of treg immune response after treatment with tocilizumab in giant cell arteritis. Clin Transl Immunol (2021) 10(9):e1332. doi: 10.1002/cti2.1332
43. Adriawan IR, Atschekzei F, Dittrich-Breiholz O, Garantziotis P, Hirsch S, Risser LM, et al. Novel aspects of regulatory T cell dysfunction as a therapeutic target in giant cell arteritis. Ann Rheum Dis (2022) 81(1):124–31. doi: 10.1136/annrheumdis-2021-220955
44. Turesson C, Borjesson O, Larsson K, Mohammad AJ, Knight A. Swedish Society of rheumatology 2018 guidelines for investigation, treatment, and follow-up of giant cell arteritis. Scand J Rheumatol (2019) 48(4):259–65. doi: 10.1080/03009742.2019.1571223
45. Hellmich B, Agueda A, Monti S, Buttgereit F, de Boysson H, Brouwer E, et al. 2018 update of the EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis (2020) 79(1):19–30. doi: 10.1136/annrheumdis-2019-215672
46. Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol (2003) 3(2):133–46. doi: 10.1038/nri1001
47. Mazzoni A, Santarlasci V, Maggi L, Capone M, Rossi MC, Querci V, et al. Demethylation of the RORC2 and IL17A in human CD4+ T lymphocytes defines Th17 origin of nonclassic Th1 cells. J Immunol (2015) 194(7):3116–26. doi: 10.4049/jimmunol.1401303
48. Stadhouders R, Lubberts E, Hendriks RW. A cellular and molecular view of T helper 17 cell plasticity in autoimmunity. J Autoimmun (2018) 87:1–15. doi: 10.1016/j.jaut.2017.12.007
49. Mazzoni A, Maggi L, Siracusa F, Ramazzotti M, Rossi MC, Santarlasci V, et al. Eomes controls the development of Th17-derived (non-classic) Th1 cells during chronic inflammation. Eur J Immunol (2019) 49(1):79–95. doi: 10.1002/eji.201847677
50. Conway R, O'Neill L, McCarthy GM, Murphy CC, Fabre A, Kennedy S, et al. Interleukin 12 and interleukin 23 play key pathogenic roles in inflammatory and proliferative pathways in giant cell arteritis. Ann Rheum Dis (2018) 77(12):1815–24. doi: 10.1136/annrheumdis-2018-213488
51. Matza MA, Fernandes AD, Stone JH, Unizony SH. Ustekinumab for the treatment of giant cell arteritis. Arthritis Care Res (Hoboken) (2021) 73(6):893–7. doi: 10.1002/acr.24200
52. Rodriguez-Pla A, Bosch-Gil JA, Rossello-Urgell J, Huguet-Redecilla P, Stone JH, Vilardell-Tarres M. Metalloproteinase-2 and -9 in giant cell arteritis: involvement in vascular remodeling. Circulation (2005) 112(2):264–9. doi: 10.1161/CIRCULATIONAHA.104.520114
53. Watanabe R, Maeda T, Zhang H, Berry GJ, Zeisbrich M, Brockett R, et al. MMP (Matrix metalloprotease)-9-Producing monocytes enable T cells to invade the vessel wall and cause vasculitis. Circ Res (2018) 123(6):700–15. doi: 10.1161/circresaha.118.313206
54. Jiemy WF, van Sleen Y, van der Geest KS, Ten Berge HA, Abdulahad WH, Sandovici M, et al. Distinct macrophage phenotypes skewed by local granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) are associated with tissue destruction and intimal hyperplasia in giant cell arteritis. Clin Transl Immunol (2020) 9(9):e1164. doi: 10.1002/cti2.1164
55. Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD. Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J Immunol (2007) 178(8):5245–52. doi: 10.4049/jimmunol.178.8.5245
56. Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol (2008) 8(7):533–44. doi: 10.1038/nri2356
57. Corbera-Bellalta M, Alba-Rovira R, Muralidharan S, Espigol-Frigole G, Rios-Garces R, Marco-Hernandez J, et al. Blocking GM-CSF receptor alpha with mavrilimumab reduces infiltrating cells, pro-inflammatory markers and neoangiogenesis in ex vivo cultured arteries from patients with giant cell arteritis. Ann Rheum Dis (2022) 81(4):524–36. doi: 10.1136/annrheumdis-2021-220873
58. Cid MC, Unizony SH, Blockmans D, Brouwer E, Dagna L, Dasgupta B, et al. Efficacy and safety of mavrilimumab in giant cell arteritis: a phase 2, randomised, double-blind, placebo-controlled trial. Ann Rheum Dis (2022) 81(5):653–61. doi: 10.1136/annrheumdis-2021-221865
59. Mia S, Warnecke A, Zhang XM, Malmstrom V, Harris RA. An optimized protocol for human M2 macrophages using m-CSF and IL-4/IL-10/TGF-beta yields a dominant immunosuppressive phenotype. Scand J Immunol (2014) 79(5):305–14. doi: 10.1111/sji.12162
60. Buchacher T, Ohradanova-Repic A, Stockinger H, Fischer MB, Weber V. M2 polarization of human macrophages favors survival of the intracellular pathogen chlamydia pneumoniae. PloS One (2015) 10(11):e0143593. doi: 10.1371/journal.pone.0143593
61. van Sleen Y, Jiemy WF, Pringle S, van der Geest KSM, Abdulahad WH, Sandovici M, et al. A distinct macrophage subset mediating tissue destruction and neovascularization in giant cell arteritis: Implication of the YKL-40/Interleukin-13 receptor alpha2 axis. Arthritis Rheumatol (2021) 73(12):2327–37. doi: 10.1002/art.41887
62. Esen I, Jiemy WF, van Sleen Y, van der Geest KSM, Sandovici M, Heeringa P, et al. Functionally heterogenous macrophage subsets in the pathogenesis of giant cell arteritis: Novel targets for disease monitoring and treatment. J Clin Med (2021) 10(21):4958. doi: 10.3390/jcm10214958
63. Weyand CM, Wagner AD, Bjornsson J, Goronzy JJ. Correlation of the topographical arrangement and the functional pattern of tissue-infiltrating macrophages in giant cell arteritis. J Clin Invest (1996) 98(7):1642–9. doi: 10.1172/JCI118959
64. Kaiser M, Weyand CM, Bjornsson J, Goronzy JJ. Platelet-derived growth factor, intimal hyperplasia, and ischemic complications in giant cell arteritis. Arthritis Rheum (1998) 41(4):623–33. doi: 10.1002/1529-0131(199804)41:4<623::AID-ART9>3.0.CO;2-6
65. Kaiser M, Younge B, Bjornsson J, Goronzy JJ, Weyand CM. Formation of new vasa vasorum in vasculitis. production of angiogenic cytokines by multinucleated giant cells. Am J Pathol (1999) 155(3):765–74. doi: 10.1016/S0002-9440(10)65175-9
66. O'Neill L, McCormick J, Gao W, Veale DJ, McCarthy GM, Murphy CC, et al. Interleukin-6 does not upregulate pro-inflammatory cytokine expression in an ex vivo model of giant cell arteritis. Rheumatol Adv Pract (2019) 3(1):rkz011. doi: 10.1093/rap/rkz011
67. Wen Z, Shen Y, Berry G, Shahram F, Li Y, Watanabe R, et al. The microvascular niche instructs T cells in large vessel vasculitis via the VEGF-Jagged1-Notch pathway. Sci Transl Med (2017) 9(399):eaal3322. doi: 10.1126/scitranslmed.aal3322
68. Watanabe R, Goronzy JJ, Berry G, Liao YJ, Weyand CM. Giant cell arteritis: From pathogenesis to therapeutic management. Curr Treatm Opt Rheumatol (2016) 2(2):126–37. doi: 10.1007/s40674-016-0043-x
69. Baldini M, Maugeri N, Ramirez GA, Giacomassi C, Castiglioni A, Prieto-Gonzalez S, et al. Selective up-regulation of the soluble pattern-recognition receptor pentraxin 3 and of vascular endothelial growth factor in giant cell arteritis: relevance for recent optic nerve ischemia. Arthritis Rheum (2012) 64(3):854–65. doi: 10.1002/art.33411
70. Watanabe R, Hilhorst M, Zhang H, Zeisbrich M, Berry GJ, Wallis BB, et al. Glucose metabolism controls disease-specific signatures of macrophage effector functions. JCI Insight (2018) 3(20):e123047. doi: 10.1172/jci.insight.123047
71. Corbera-Bellalta M, Planas-Rigol E, Lozano E, Terrades-Garcia N, Alba MA, Prieto-Gonzalez S, et al. Blocking interferon gamma reduces expression of chemokines CXCL9, CXCL10 and CXCL11 and decreases macrophage infiltration in ex vivo cultured arteries from patients with giant cell arteritis. Ann Rheum Dis (2016) 75(6):1177–86. doi: 10.1136/annrheumdis-2015-208371
72. Cid MC, Hoffman MP, Hernandez-Rodriguez J, Segarra M, Elkin M, Sanchez M, et al. Association between increased CCL2 (MCP-1) expression in lesions and persistence of disease activity in giant-cell arteritis. Rheumatology (Oxford) (2006) 45(11):1356–63. doi: 10.1093/rheumatology/kel128
73. Graver JC, Abdulahad W, van der Geest KSM, Heeringa P, Boots AMH, Brouwer E, et al. Association of the CXCL9-CXCR3 and CXCL13-CXCR5 axes with b-cell trafficking in giant cell arteritis and polymyalgia rheumatica. J Autoimmun (2021) 123:102684. doi: 10.1016/j.jaut.2021.102684
74. Ma-Krupa W, Jeon MS, Spoerl S, Tedder TF, Goronzy JJ, Weyand CM. Activation of arterial wall dendritic cells and breakdown of self-tolerance in giant cell arteritis. J Exp Med (2004) 199(2):173–83. doi: 10.1084/jem.20030850
75. Zhang H, Watanabe R, Berry GJ, Vaglio A, Liao YJ, Warrington KJ, et al. Immunoinhibitory checkpoint deficiency in medium and large vessel vasculitis. Proc Natl Acad Sci U S A (2017) 114(6):E970–e979. doi: 10.1073/pnas.1616848114
76. Watanabe R, Zhang H, Berry G, Goronzy JJ, Weyand CM. Immune checkpoint dysfunction in large and medium vessel vasculitis. Am J Physiol Heart Circ Physiol (2017) 312(5):H1052–h1059. doi: 10.1152/ajpheart.00024.2017
77. Dos Santos JP, Artigiani Neto R, Mangueira CLP, Filippi RZ, Gutierrez PS, Westra J, et al. Associations between clinical features and therapy with macrophage subpopulations and T cells in inflammatory lesions in the aorta from patients with takayasu arteritis. Clin Exp Immunol (2020) 202(3):384–93. doi: 10.1111/cei.13489
78. Kong X, Xu M, Cui X, Ma L, Cheng H, Hou J, et al. Potential role of macrophage phenotypes and CCL2 in the pathogenesis of takayasu arteritis. Front Immunol (2021) 12:646516. doi: 10.3389/fimmu.2021.646516
79. Watanabe R, Berry GJ, Liang DH, Goronzy JJ, Weyand CM. Pathogenesis of giant cell arteritis and takayasu arteritis-similarities and differences. Curr Rheumatol Rep (2020) 22(10):68. doi: 10.1007/s11926-020-00948-x
80. Weyand CM, Watanabe R, Zhang H, Akiyama M, Berry GJ, Goronzy JJ. Cytokines, growth factors and proteases in medium and large vessel vasculitis. Clin Immunol (2019) 206:33–41. doi: 10.1016/j.clim.2019.02.007
81. Qing G, Zhiyuan W, Jinge Y, Yuqing M, Zuoguan C, Yongpeng D, et al. Single-cell RNA sequencing revealed CD14(+) monocytes increased in patients with takayasu's arteritis requiring surgical management. Front Cell Dev Biol (2021) 9:761300. doi: 10.3389/fcell.2021.761300
82. Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol (2018) 15(9):505–22. doi: 10.1038/s41569-018-0064-2
83. Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol (2018) 14(10):576–90. doi: 10.1038/s41574-018-0059-4
84. Watanabe R, Hashimoto M. Aging-related vascular inflammation: Giant cell arteritis and neurological disorders. Front Aging Neurosci (2022) 14:843305. doi: 10.3389/fnagi.2022.843305
85. Miyabe Y, Lian J, Miyabe C, Luster AD. Chemokines in rheumatic diseases: pathogenic role and therapeutic implications. Nat Rev Rheumatol (2019) 15(12):731–46. doi: 10.1038/s41584-019-0323-6
86. Hoffman GS, Cid MC, Rendt-Zagar KE, Merkel PA, Weyand CM, Stone JH, et al. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med (2007) 146(9):621–30. doi: 10.7326/0003-4819-146-9-200705010-00004
87. Deshayes S, Ly KH, Rieu V, Maigne G, Martin Silva N, Manrique A, et al. Steroid-sparing effect of anakinra in giant-cell arteritis: a case series with clinical, biological and iconographic long-term assessments. Rheumatology (Oxford) (2021) 61(1):400–6. doi: 10.1093/rheumatology/keab280
88. Watanabe R, Berry GJ, Liang DH, Goronzy JJ, Weyand CM. Cellular signaling pathways in medium and Large vessel vasculitis. Front Immunol (2020) 11:587089. doi: 10.3389/fimmu.2020.587089
89. Watanabe R, Hashimoto M. Perspectives of JAK inhibitors for Large vessel vasculitis. Front Immunol (2022) 13:881705. doi: 10.3389/fimmu.2022.881705
90. Zhang H, Watanabe R, Berry GJ, Tian L, Goronzy JJ, Weyand CM. Inhibition of JAK-STAT signaling suppresses pathogenic immune responses in medium and Large vessel vasculitis. Circulation (2018) 137(18):1934–48. doi: 10.1161/circulationaha.117.030423
91. Vieira M, Regnier P, Maciejewski-Duval A, Le Joncour A, Darasse-Jeze G, Rosenzwajg M, et al. Interferon signature in giant cell arteritis aortitis. J Autoimmun (2022) 127:102796. doi: 10.1016/j.jaut.2022.102796
92. Regnier P, Le Joncour A, Maciejewski-Duval A, Desbois AC, Comarmond C, Rosenzwajg M, et al. Targeting JAK/STAT pathway in takayasu's arteritis. Ann Rheum Dis (2020) 79(7):951–9. doi: 10.1136/annrheumdis-2019-216900
93. Terao C, Yoshifuji H, Matsumura T, Naruse TK, Ishii T, Nakaoka Y, et al. Genetic determinants and an epistasis of LILRA3 and HLA-B*52 in takayasu arteritis. Proc Natl Acad Sci U S A (2018) 115(51):13045–50. doi: 10.1073/pnas.1808850115
94. Kadoba K, Watanabe R, Iwasaki T, Nakajima T, Kitagori K, Akizuki S, et al. A susceptibility locus in the IL12B but not LILRA3 region is associated with vascular damage in takayasu arteritis. Sci Rep (2021) 11(1):13667. doi: 10.1038/s41598-021-93213-9
95. Watanabe R. JAK inhibitors as promising agents for refractory takayasu arteritis. Ann Rheum Dis (2020) 81(4):e67. doi: 10.1136/annrheumdis-2020-217577
96. Zeisbrich M, Chevalier N, Sehnert B, Rizzi M, Venhoff N, Thiel J, et al. CMTM6-deficient monocytes in ANCA-associated vasculitis fail to present the immune checkpoint PD-L1. Front Immunol (2021) 12:673912. doi: 10.3389/fimmu.2021.673912
Keywords: giant cell arteritis, large vessel vasculitis, macrophages, monocytes, takayasu arteritis, vasculitis
Citation: Watanabe R and Hashimoto M (2022) Pathogenic role of monocytes/macrophages in large vessel vasculitis. Front. Immunol. 13:859502. doi: 10.3389/fimmu.2022.859502
Received: 21 January 2022; Accepted: 11 July 2022;
Published: 29 July 2022.
Edited by:
Keishi Fujio, The University of Tokyo, JapanReviewed by:
Hiroaki Niiro, Kyushu University, JapanCopyright © 2022 Watanabe and Hashimoto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ryu Watanabe, ZG9jdG9yd2F0YW5hYmVyeXVAeWFob28uY28uanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.