
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 04 May 2022
Sec. Primary Immunodeficiencies
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.856601
This article is part of the Research Topic Deciphering the Landscape of Immunohematology: Enhancing our Understanding and Management of Hematological Disorders Through Advances in Immunology and Genetics View all 15 articles
 Saniya Sharma*
Saniya Sharma* Rakesh Kumar Pilania
Rakesh Kumar Pilania Gummadi Anjani
Gummadi Anjani Murugan Sudhakar
Murugan Sudhakar Kanika Arora
Kanika Arora Rahul Tyagi
Rahul Tyagi Manpreet Dhaliwal
Manpreet Dhaliwal Pandiarajan Vignesh
Pandiarajan Vignesh Amit Rawat
Amit Rawat Surjit Singh
Surjit SinghInborn errors of immunity (IEIs) are a group of heterogeneous disorders characterized by a broad clinical spectrum of recurrent infections and immune dysregulation including autoimmunity and lymphoproliferation (LP). LP in the context of IEI may be the presenting feature of underlying immune disorder or may develop during the disease course. However, the correct diagnosis of LP in IEI as benign or malignant often poses a diagnostic dilemma due to the non-specific clinical features and overlapping morphological and immunophenotypic features which make it difficult to treat. There are morphological clues to LP associated with certain IEIs. A combination of ancillary techniques including EBV-associated markers, flow cytometry, and molecular assays may prove useful in establishing a correct diagnosis in an appropriate clinical setting. The present review attempts to provide comprehensive insight into benign and malignant LP, especially the pathogenesis, histological clues, diagnostic strategies, and treatment options in patients with IEIs.
Lymphoproliferation (LP) in inborn errors of immunity (IEI) refers to persistent polyclonal, oligoclonal, or monoclonal proliferation of lymphoid cells in the clinical setting of primary immunodeficiency or immune dysregulation (1, 2). The incidence of LP in IEI varies from 0.7 to 18% (3). Typically, LP occurs during disease evolution in a patient with underlying primary immunodeficiency. However, it is difficult to assess the cases with LP as the presenting feature of IEI, posing a diagnostic challenge as there are no guidelines on the diagnosis and management of such cases. Moreover, the diagnostic and therapeutic approach toward cases with non-malignant LP in IEI is not clear. The current review will attempt to summarize the clinicopathological aspects and diagnostic approach to LP in IEI with the aim to provide an insight into early diagnosis and timely management of these cases.
Patients with LP associated with IEI usually present at a younger age with frequent extranodal involvement as compared to their immunocompetent counterparts. Clinical symptoms are non-specific and mimic those of an infection, inflammation, or neoplasia. The clinical features are characterized by chronic or recurrent lymphadenopathy, hepatosplenomegaly, extranodal infiltration, and/or peripheral blood lymphocytosis (4). In certain immune disorders such as autoimmune lymphoproliferative syndrome (ALPS) and X-linked lymphoproliferation (XLP), LP is the predominant feature at disease onset. In a diagnosed case of IEI, it is often challenging to accurately define LP as benign or malignant as both have different therapeutic and prognostic implications. Besides, the clinical suspicion and diagnosis of immune disorder underlying LP is a mammoth task and requires expertise. The relevance of correctly diagnosing and treating benign LP in IEI lies in the fact that it may not only act as a precursor lesion of lymphoid malignancy but also may be associated with an increased risk of hemophagocytic lymphohistiocytosis (HLH) (5). Thus, in a given case an apparently innocuous LP may have a sinister connotation. Treatment and prognosis of LP in IEI depend upon the severity of the underlying immune defect and need to be assessed individually.
A recent nomenclature has attempted to classify LP in the setting of immunodeficiency based on the morphology of lesion, viral infection status (EBV or HHV-8), and the clinical immunodeficiency state (2). With respect to LP in IEI, the morphological spectrum chiefly encompasses Epstein-Barr virus (EBV)-associated B-cell LPDs similar to the histological lesions observed in post-transplant lymphoproliferative disorders (PTLD). These include B-cell lymphoid hyperplasias: follicular, plasmacytic, and infectious mononucleosis (IM)-like; polymorphous B-cell LP; indolent lymphomas; aggressive non-Hodgkin lymphomas (NHL); and classical Hodgkin lymphoma (HL)- like LP (2). In general, T/NK-cell LPs are rare in IEI, the majority being benign/reactive CD8+T-cell infiltrates. Certain morphological clues in tissue biopsies may help to predict the underlying immune disorder [Figure 1]. In a recent review investigating benign and malignant LP, distinct histopathological alterations in the distribution of CD4+ follicular helper T-cells, follicular dendritic cells, and mantle zone naïve B-cells correlate with the different patterns in the development of germinal centers (6). Nevertheless, achieving a specific diagnosis is often challenging owing to the significant degree of morphological and immunophenotypic overlap between the benign and malignant lesions such as polymorphous B-cell LP is a masquerader of Hodgkin lymphoma (HL) and benign CD8+T-cell/histiocytic infiltrates in common variable immunodeficiency (CVID) could be easily confused with mimicking T- large granular lymphocytic leukemia (T-LGLL) leading to overtreatment.
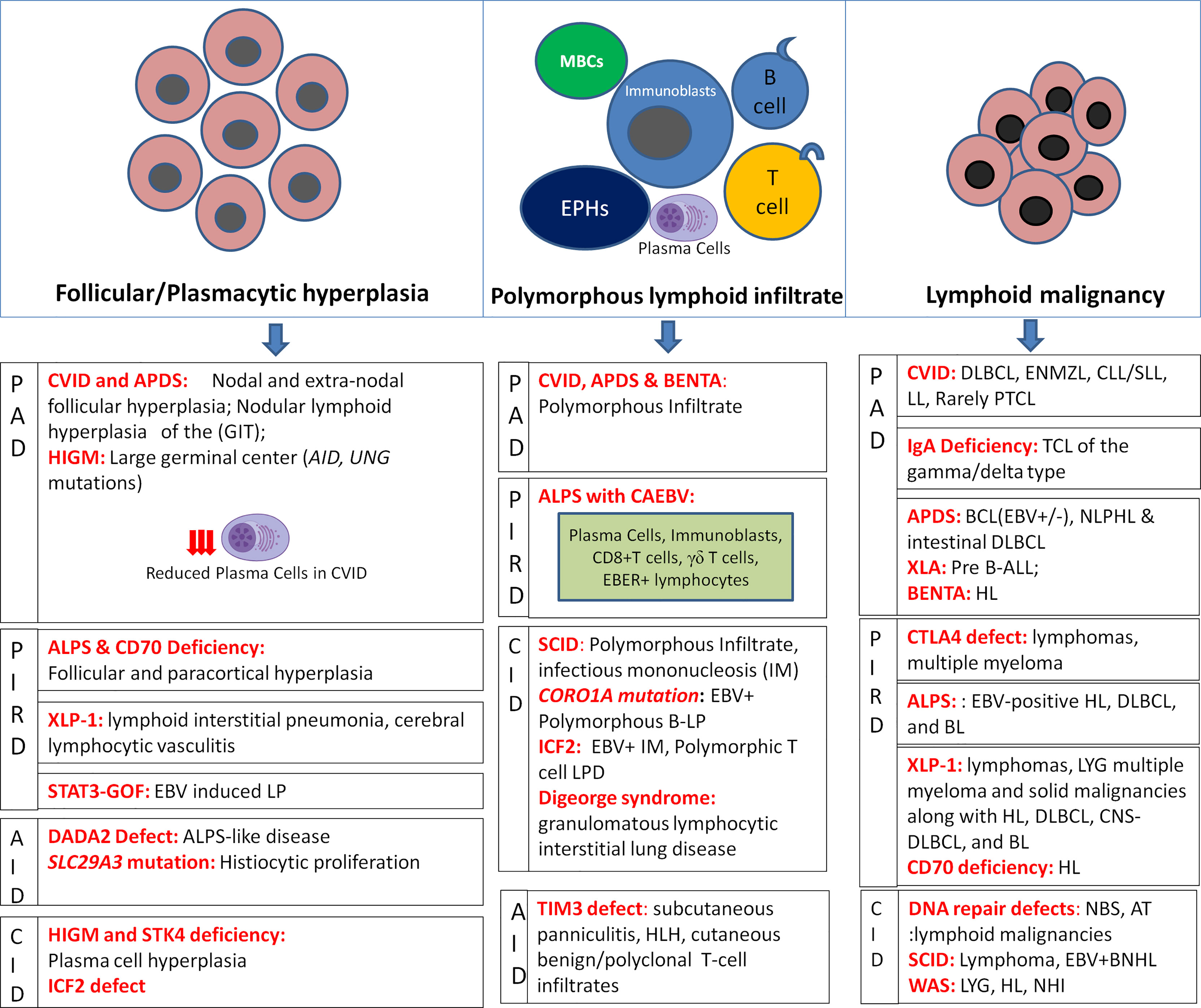
Figure 1 Histopathological alterations in LP associated with IEIs depicting hyperplasias, polymorphous lymphoid infiltrates, and lymphoid malignancies as per IEI disease phenotypes (MBCs, monocytoid B-cells; EPHCs, epithelioid histiocytes).
In severe combined immunodeficiency (SCID), loss of T/NK cell-mediated immune surveillance leads to uncontrolled EBV-driven B-cell LP that may progress into lymphoma (7). Homozygous and hypomorphic missense mutations in CORO1A may present as early-onset EBV+B-LP as a result of CD4+CD45RA+ T-cell lymphopenia and impaired invariant NKT-cell development and survival defects (8). Dedicator of cytokinesis-8 (DOCK8) deficiency/hyper IgE syndrome (HIES) leads to impaired NK-cell function and increased predisposition to EBV+ lymphomas (9). In hyper IgM syndrome (HIGM) with CD40LG/CD40 mutations, the defective antibody response to antigens results in the malignant transformation of B-cells (10). Serine/threonine-protein kinase 4 (STK4) deficiency, has been associated with nodal and extra-nodal EBV+ LP and B-cell lymphoma suggesting the role of EBV infection in inducing LP (11). Inducible tyrosine kinase (ITK) deficiency is associated with progressive CD4+T-cell and NKT-cell lymphopenia, and hypogammaglobulinemia resulting in EBV viremia and immune dysregulation leading to massive LP (12).
Histopathological changes in SCID comprise polymorphous LP characterized by systemic proliferation of highly polymorphous B-lymphoid cells showing plasmacytoid and immunoblastic differentiation that may also progress into HLH and EBV+B-NHL (1). EBV-encoded RNA (EBER)+ polymorphous CD20+ B-cell LP and DLBCL have been reported in patients with hypomorphic CORO1A mutations (8). DOCK8 deficiency has been reported to manifest as young-onset EBV+ and EBV- lymphomas and EBV+ pulmonary lymphomatoid granulomatosis (LYG) (9). In HIGM, the absence of germinal centers in lymph nodes and a massive extranodal accumulation of plasma cells that secrete IgM, particularly in GIT, to T-LGLL, HL, and EBV+B-NHL have been reported (1). STK4-deficiency is associated with plasma cell hyperplasia and EBV+ B-cell polymorphous LP, and B-cell lymphomas showing plasmacytic differentiation (11). ITK deficiency is chiefly associated with EBV+B-LPD, especially HL (12)
Wiskott-Aldrich syndrome (WAS) patients have an increased predisposition to develop EBV-driven LYG, NHL, and HL usually following a long duration of non-malignant LP due to defective immune surveillance against the virus-infected cells (13). Patients with DNA repair defects, Nijmegen breakage syndrome (NBS), and ataxia-telangiectasia (AT) show a high predisposition to lymphoid malignancies at a young age as a result of genomic instability, chromosomal abnormalities, combined B- and T-cell immunodeficiency, and radiation hypersensitivity (14, 15). Histologically, WAS is associated with EBV+ extranodal clonal angioinvasive B-LP (LYG), HL (nodular sclerosing and lymphocyte depleted), and B-NHL, particularly DLBCL (1). Notably, T-NHL, T-acute lymphoblastic leukemia/lymphoma (T-ALL/LBL), and clonal non-malignant T-cell proliferations are more common than B-NHLs in AT, and NBS (1). An increased propensity for T-prolymphocytic leukemia (T-PLL) has been reported in children with AT (1). Among B-cell lymphomas, classical HL, diffuse large B-cell lymphoma (DLBCL), and Burkitt lymphoma (BL) are commonly described (1). Immunodeficiency with centromeric instability and facial anomalies type 2 (ICF2) is a DNA-methylation disorder with a high susceptibility to EBV infection manifesting as EBV+ IM, HLH, chronic active EBV infection (CAEBV), HL, large B-cell lymphoma, and polymorphic non-clonal T-cell LPD (16). Patients with Di-George syndrome may present with CD8+ granulomatous cutaneous T-cell lymphoma and granulomatous lymphocytic interstitial lung disease (GLILD) characterized by ill-defined non-necrotizing lymphohistiocytic granulomas, CD20+ B-cell-rich lymphoid nodules, CD4+ T-cell-rich interstitial pneumonia, and peribronchiolar follicular hyperplasia with reduced regulatory T-cells (Tregs) (17). Homozygous Post-meiotic segregation 2 (PMS2) mutations, a DNA mismatch-repair defect is characterized chiefly by poor antibody responses and low B-cell number and is associated with B- and T-cell leukemia/lymphoma besides colorectal cancer and brain tumors (18). The other CIDs associated with increased risk of lymphomas include Bloom syndrome, ligase 1 and MCM4 deficiencies, and cartilage hair hypoplasia (19).
In CVID, LP is one of the prominent clinical features accounting for approximately 20% of cases (20). Lymphadenopathy has been associated with the expansion of transitional B-cells (21). A recent report based on the United States Immunodeficiency Network (USIDNET) registry reported lymphoma in 8% of 1091 CVID patients (22). Lymphoma is the second major cause of mortality in CVID after chronic lung disease (23). B-cell lymphomas arise chiefly from germinal center-experienced mature B-cells that have undergone somatic hypermutation of Ig genes, correlating with a higher frequency of DLBCL, extranodal marginal zone lymphoma (ENMZL), and HL in CVID (24). Granulomatous CVID has been reported to involve every organ system occurring in 8-20% of CVID cases (25). The prevalence of LP in X-linked agammaglobulinemia (XLA) is extremely low (approximately 0.7%) in comparison to other IEIs (26). Nevertheless, the presence of lymphadenopathy in XLA is considered a matter of concern as it may harbor a lymphomatous process. Patients with selective IgA deficiency are usually asymptomatic, although may rarely develop infections, autoimmune disorders, and malignancies (27, 28). Considering the shared genetic basis between CVID and selective IgA deficiency, a higher incidence of malignancies has been observed, primarily gastrointestinal (29, 30). Activated phosphoinositide 3-kinase delta syndrome (APDS) is chiefly characterized by recurrent respiratory tract infections, CAEBV, immune dysregulation, and recurrent or persistent LP presenting as lymphadenopathy, splenomegaly, mucosal nodular lymphoid hyperplasia, and lymphoma (31). CAEBV results from impaired NK and CD8+ T cell-mediated killing of infected cells by EBV. Persistent EBV infection and B-cell proliferation due to constitutive activation of B-cell intrinsic PI3Kδ signaling contribute to lymphomagenesis in APDS (32). B-cell expansion with nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and T-cell anergy syndrome (BENTA) is a rare disorder featured by constitutive activation of the NF-κB signaling pathway leading to EBV-driven polyclonal B-cell LP manifesting as lymphocytosis, splenomegaly, and lymphadenopathy (33). NFKB1 deficiency is a clinically heterogeneous PAD with impaired function of B-cells with or without T-cell dysfunction and is characterized by recurrent infections, cytopenias, and EBV+ B-LP (34). Defects in activation-induced cytidine deaminase (AID) and uracil DNA glycosylase (UNG) in HIGM are frequently associated with lymphoid hyperplasia (35).
The morphological spectrum of LP in CVID is heterogeneous ranging from benign follicular hyperplasia and paracortical expansion with EBV-positive B-cells including large pleomorphic Reed-Sternberg (RS)-like cells to clonal but non-malignant B-cell nodular lymphoid hyperplasia of the gastrointestinal tract with a near-total absence of plasma cells to DLBCL, ENMZL, CLL/SLL, LPL, HL and rarely peripheral T-cell lymphoma (PTCL) (1). Notably, CD8+ cytotoxic T-cell proliferations are common in peripheral blood and hepatic sinusoids of CVID patients which is often difficult to differentiate from T-LGLL (36). Precursor B-ALL has been reported in XLA (37). Though rare, a few case reports have described T-cell lymphoma of the γδ type in selective IgA deficiency (28). APDS is frequently associated with non-malignant gastrointestinal and respiratory nodular mucosal lymphoid hyperplasia. These lesions show atypical follicular hyperplasia with disrupted and hyperplastic germinal centers, attenuated mantle zones, and perisinusoidal aggregates of monocytoid B-cells (31). There is hyperplasia of CD4+PD1+ follicular T-helper cells and PD1+CD57+CD8+ senescent T-cells and IgM+ plasma cells with reduced IgG+ plasma cells. EBV-associated oligoclonal polymorphous- B-cell LP has also been reported comprising polymorphous lymphoid infiltrate comprising of B- and T-cells, plasma cells, epithelioid histiocytes, and monocytoid B-cells. APDS shows an increased predisposition to EBV+/- B-cell lymphomas including intestinal DLBCL, and nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL) (32). A dual clinicopathological presentation of APDS with early-onset HLH followed by HL has also been reported (38). EBV+ polymorphous B-LPD and HL are described in BENTA disease (33). NFKB1-deficient patients may present with generalized lymphadenopathy with EBV+ reactive lymphoid hyperplasia of increasing clinical severity (34). AID and UNG mutated HIGM shows enlarged lymph nodes with large germinal centers (19, 35).
In ALPS, homozygous mutations in FAS and FASL genes are associated with impaired cytotoxicity and severely reduced activation-induced cell death (AICD) in B- and T-cells resulting in clinically severe disease and massive LP including lymphomagenesis (39). Besides ALPS, Caspase-8 mutations may also present as end-organ LP, granulomatous inflammation, mesenteric lymphadenopathy, and recurrent EBV infection (40). CD25, CD122, cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) deficiencies, and signal transducer and activator of transcription 3 (STAT-3) gain-of-function mutations lead to impaired Treg function leading to impaired suppression of effector T cells causing immune dysregulation with autoimmunity and may present with LP with recurrent EBV infections (19, 41). The estimated risk of malignancies in affected patients with CTLA-4 deficiency is approximately 12.9% (41). CTLA4 insufficiency and biallelic LRBA mutations share similar clinical features and are also associated with granulomatous lymphocytic interstitial lung disease (GLILD) (17, 42). Elevated STAT3 signaling leads to defects in phosphorylation of STAT5 and STAT1, impaired Treg function, enhanced T-helper cell-17 differentiation, LP, and systemic autoimmunity (19, 42). XLP1 and 2 are associated with EBV-driven HLH and LP due to severely reduced iNKT cells, although XLP1 shows an increased risk of lymphomagenesis (19). Another defect in CTP synthase 1 (CTPS1) also leads to fatal viral (EBV) infections which results in LP and non-Hodgkin B cell lymphoma (19, 43). Similarly, RAS guanyl-releasing protein 1 (RASGRP1) deficiency has been involved with T-cell lymphopenia and EBV-associated B-cell lymphoma (44). Accelerated loss of TCR repertoire diversity has been observed in these cases. Additionally, CD70, CD137(4-1BB), CARMIL2, PRKCD, and CD27 deficiencies and X-linked magnesium EBV and neoplasia (XMEN) exhibit unique susceptibility to EBV infection and associated LP as a result of impaired T-cell activation and proliferation (19). Impaired CD27-CD70 interactions lead to loss of CD4+ and CD8+ T-cell response, impaired EBV killing, and CAEBV with hypogammaglobulinemia (45). Interestingly, CD70 deficiency may clinically mimic periodic fever syndrome (45). Fanconi anemia-associated protein 24 (FAAP24) deficiency, a familial HLH syndrome, results in fatal EBV-driven LP due to the failure of cytotoxic CD8+T cells to kill EBV-infected B-cells (19).
Histopathological features in ALPS include marked proliferation of non-clonal CD4-/CD8-CD45RA+ CD57+TCRαβ+ cytotoxic (TIA+ and perforin+) double-negative T-cells (DNTs), involving peripheral blood, lymph nodes, liver, spleen, and extranodal sites, a few cases may show γδ-T-cell proliferation (36, 46). Follicular and paracortical hyperplasia and PTGC have been frequently described (1). HL, classical and NLPHL, DLBCL, Rosai-Dorfman disease (RDD), and rarely PTCL have been well-described in ALPS (1). Histopathological features of LP in ALPS may overlap with CAEBV, especially in those cases where classic ALPS morphology is not evident. In such cases, florid paracortical hyperplasia comprising plasma cells, immunoblasts, EBER+ lymphocytes, cytotoxic T-cells, and γδ-T-cells is the predominant finding (46). CTLA-4 deficient patients have an increased risk of solid and lymphoid malignancies including lymphomas and multiple myeloma. The reported cases are mostly EBV-positive HL, DLBCL, and BL (41). XLP1 associated LP ranges from non-malignant LP including lymphoid interstitial pneumonia, LYG, cerebral lymphocytic vasculitis, HLH, and severe IM, to malignant lesions like HL, DLBCL, CNS DLBCL, and BL (47). Patients with 4-1BB deficiency have been diagnosed with EBER+ HL and CD20-CD38+ DLBCL on lymph node biopsies (48). CD70 deficiency can show varied presentations ranging from EBV+IM, reactive follicular and paracortical hyperplasia to HL (45). CD27 deficiency manifests as EBV+HLH and lymphomas (19).
Patients with T-cell immunoglobulin and mucin domain-containing protein 3 (TIM3) deficiency usually present with subcutaneous panniculitis, HLH, cutaneous benign/polyclonal T-cell infiltrates, and panniculitis-like T-cell lymphoma of subcutaneous tissue and mesenteric fat (49). Deficiency of adenosine deaminase 2 (DADA2) is a recently described autoinflammatory disorder that is caused by bi-allelic mutations in the CECR1 gene encoding adenosine deaminase 2 (ADA2). Around 30% of cases present with splenomegaly and 10% present with lymphadenopathy. DADA2 may have varied clinical presentations ranging from vasculopathy, chronic liver disease, immune cytopenias, immunodeficiency, hypogammaglobulinemia, and LP (50). LP in DADA2 includes T-LGLL, ALPS-like disease, and HL. Germline mutation in the SLC29A3 gene causes histiocytosis-lymphadenopathy plus syndrome which is characterized by abnormal histiocytic proliferation and accumulation in lymph nodes, liver, spleen, GIT, CNS, skin, and kidneys causing organ damage (19).
Somatic activating mutations in NRAS and KRAS genes may produce an ALPS-like phenotype, Ras-associated autoimmune leukoproliferative disorder (RALD) that is featured by chronic non-malignant LP, autoimmune cytopenias, monocytosis, and hypergammaglobulinemia. LP is usually indolent, although myeloid malignancies have been reported in a few cases (19, 51).
Histopathological changes of LP often correlate with the underlying IEI. However, the pathologist should be aware of the potential diagnostic pitfalls while analyzing tissue biopsies, especially CD8+ cytotoxic T-cell infiltrates and polymorphous B-cell LP that could easily be mistaken for lymphoma leading to aggressive therapy. It is critical to understand that in IEIs, a major proportion of LP is constituted by polyclonal/oligoclonal, benign/reactive lymphoid infiltrates and every case is not a lymphoma, although the risk of malignant transformation is higher as compared to immunocompetent individuals. Detection of EBV encoded small RNA by in-situ hybridization (EBER-ISH), EBV encoded nucleic antigen (EBNA) and latent membrane protein (LMP) in lymphoid cells correlates well with EBV-infection and LP. EBV infection may cause downregulation of B-cell markers (CD20, CD79b, and CD19) and upregulation of CD30 (1). Immunohistochemistry (IHC), although used routinely for the detection of B-, T- cell, histiocytes, and plasma cell lineage antigens, should be interpreted with caution and in an appropriate clinical context. The tumor cells express high levels of programmed death-ligand 1 (PDL1) in CVID associated EBV+ DLBCL and HL (24). Immune checkpoint inhibitors are used to block this immune checkpoint protein, PDL1 expressed by tumor cells in HL to augment the T-cell mediated immune response. Although rare, it is difficult to diagnose T-cell lymphomas in IEIs other than DNA-repair defects owing to a high frequency of reactive/non-malignant T-cell LP. In such cases, a comprehensive approach including clinical phenotype, size of lymph node/lesion, architectural effacement, cellular/nuclear atypia, TCR gene rearrangements, chromosomal aberrations, and immunophenotyping should be followed (36).
Flow cytometric in situ hybridization (Flow-FISH) is a powerful diagnostic tool for EBV+LP as it can quantify and phenotypically characterize the EBER-positive lymphocytes (52). In CVID, flow cytometry-based detection of an increased proportion of CD21low B-cells and transitional B-cells (CD38+IgM+) and decreased proportion of class-switched memory B-cells correlates with splenomegaly, granulomatous inflammation, and lymphadenopathy (53). Several fluorescent-labeled antibodies against B/T/NK-cell lineage markers are routinely used to differentiate between reactive versus neoplastic populations. Kappa/lambda ratio (>3 or <0.5) is a more reliable indicator of the clonal process, although the complete absence of light chain expression has been described (54). T-cell receptor-Vβ (TCR-Vβ) repertoire assay is used to assess clonality in suspected T-cell LP covering almost 70% of the TCR repertoire. It is recommended for peripheral blood samples only as it gives inconclusive results with tissue samples and bone marrow aspirates (54). Nevertheless, flow cytometry is an invaluable and robust test with an accuracy of 70-90% in LPDs (54) [Supplementary Table 1].
Molecular assays used to assess clonality in suspected LP include Southern blot hybridization (SBH), restriction fragment length polymorphism (RFLP), and polymerase chain reaction (PCR)-based assays like fragment length analysis using capillary electrophoresis (Gene scan) and next-generation sequencing (NGS). These assays are largely based on detecting clonal Ig/TCR gene rearrangements that are random recombination events between one of several V, (D), and J segments generating unique exon sequences encoding antigen-bearing sites on Ig or TCR molecules for each lymphocyte. These events occur during the initial B- and T- cell development and impart diversity to Ig/TCR molecules (up to 1012) expressed by each lymphocyte. When identical sequences are shared by cells, it represents the clonal nature of that population, and identification of such homogeneous/clonal or heterogeneous/polyclonal populations forms the basis of clonality testing in suspected LP. A standardized and optimized multiplex PCR has been developed that targets nearly all Ig/TCR (IgH, IgK, TCRγ, TCRβ, and incomplete IgH D-J and TCRβ D-J) rearrangements by fragment length analysis (55). Due to the remarkably increased rates of clonality detection in B- and T-cell malignancies, these multiplex PCR assays have become gold-standard for clonality testing in LP. The interpretation is based on the identification of specific patterns of peaks generated by Genescan classified as clonal, polyclonal, pseudoclonal, multiple products, and non-evaluable (55). Although applicable in more than 95% of cases for routine clonality diagnostics across multiple centers, these assays have their own pitfalls, particularly when low-intensity clonal signals are generated (55). Moreover, they do not provide information about the clone-specific sequence and need to be run in duplicate for reproducible results. With the advent of NGS technology, Ig/TCR rearrangements may be targeted with higher sensitivity and improved clonality detection rate. The advantages of NGS over other molecular assays include a) improved detection of clonal populations in clinical samples with lymphoid cells undergoing somatic hypermutation b) the patient-specific index clone could be accurately sequenced and monitored for clonal evolution c) the entire spectrum of gene rearrangements in a sample could be visualized simultaneously, and d) clonal association between different lesions may be determined (56). Amplicon-based and targeted NGS-based strategies have been established to detect rearrangements and translocations including IGH, IGK, IGL, TRA, TRB, TRG, and TRD genetic loci in clinical samples (57). The targeted NGS panels are more economical, with less sample requirement, and multiplexing in a single assay, and enable uniform reporting of results along with the potential to detect copy number variations (CNV), translocations, somatic mutations, and indels associated with LPDs, thus achieving a sensitivity of 99.6% (57).
Despite the availability and recent advancements in diagnostic techniques, many patients still remain undiagnosed and are either undertreated or overtreated. Being a complex disorder, it is important to understand the clinical phenotype and pathophysiology of LP in IEIs. We attempt to propose a diagnostic algorithm of investigating patients with LP with regard to specific IEIs (Figure 2).

Figure 2 Diagnostic algorithm of investigations for LP in IEI according to clinical phenotype (FCM, flow cytometry; CBC, complete blood count; PBF, peripheral blood film; CXR, chest X-ray; CT, computed tomography; HPE, histopathological examination; NGS, next-generation sequencing; PCR, polymerase chain reaction; IHC, immunohistochemistry; EBER-ISH, Epstein-Barr virus-encoded RNA- in-situ hybridization; MLPA, multiplex ligation-dependent probe amplification; FISH, fluorescence in-situ hybridization).
Improved survival with allogeneic reduced-intensity conditioning-hematopoietic stem cell transplant (RIC-HSCT) has been achieved in patients with IEI and B-cell lymphoma. These patients are treated with conventional chemotherapy and anti-CD20 monoclonal antibody (Rituximab) to achieve remission prior to the transplant. EBV infection in the pre-and post-transplant setting mandates initiation of therapy [Rituximab or EBV-cytotoxic T-lymphocytes (EBV-CTLs)] (58). HSCT is the definitive mode of therapy for many IEIs with LP including SCID and WAS. Apart from this, there are targeted therapies for some of the IEIs. Though management of CVID relies on long-term immunoglobulin replacement therapy (IVIg) and antibiotic prophylaxis, surveillance for complications is important (59). Follow-up for complications including malignancies, lymphoma, and non-neoplastic LP is important followed by appropriate therapy (22, 60).
Children with APDS show a significant reduction in infections on IVIg. HSCT with medium- or reduced-intensity conditioning has shown effective results (31, 61, 62) Autoimmune manifestations of APDS require immunosuppressive therapy (63). Rituximab has proven beneficial in the management of non-neoplastic LP. Inhibition of the downstream mTOR pathway by Sirolimus or Rapamycin decreases both non-neoplastic and neoplastic lymphoproliferation including regression of cutaneous T-cell lymphoma (31). In the European society for immunodeficiencies (ESID APDS) registry, a significant benefit (19/25) in the non-neoplastic LPD was seen on therapy with Sirolimus (64). Also, direct inhibition of the activated PI3Kδ by selective PI3Kδ inhibitors such as Leniolisib has shown a decrease in lymph node and spleen size in APDS (65).
In ALPS, treatment depends on manifestations and complications. Autoimmune manifestations, especially cytopenias require immunosuppressive therapy with corticosteroids and IVIg followed by steroid-sparing agents (66). The two most common first-line steroid-sparing agents used currently are mTOR inhibitor (Sirolimus) and Mycophenolate mofetil (MMF) (66–68). Long-term studies have shown durable improvement in lymphadenopathy and splenomegaly within 3 months of initiating sirolimus (69, 70). Patients with lymphadenopathy need to be closely monitored for lymphoma development. Severe and refractory cases require HSCT.
The definitive treatment of XLP is HSCT. However, therapy needs to be tailored according to the symptoms and infection profile. IVIg replacement therapy is essential in almost all patients to reduce infection risk and treat hypogammaglobulinemia. EBV+ patients need therapy with Rituximab. HLH needs aggressive therapy with high dose IVIg, Rituximab +/- HLH protocol. The presence of lymphoma needs surgery and chemoradiation with standard protocol followed by HSCT (71, 72).
In DADA2, management of vasculopathy and stroke is well-established with the use of anti-TNF therapy, however other manifestations including LP, cytopenias, and malignancy need immunosuppressive therapy. Treatment of these includes aggressive chemoradiation with IVIg therapy and TNF blockers to prevent disease progression on case to case basis (73). HSCT is the definitive therapy for the immunological, vascular, and hematological phenotype (74).
No clear treatment protocols for BENTA have been available as of now, however, patients receive multiple therapeutic modalities in the form of steroids, Rituximab, and Sirolimus with variable benefits. Long-term surveillance is important due to the increased risk of B-cell malignancy (75).
STAT3 gain-of-function mutations manifest with a variety of clinical symptoms including lymphoproliferation, and multiorgan autoimmunity. Patients have been treated with Azathioprine and MMF with poor response. Later, IL-6 blockers and HSCT were also tried (76, 77). Recently, the upstream inhibitors, such as Tocilizumab (anti-IL-6 receptor monoclonal antibody) and Janus kinase (JAK) inhibitors are available (78, 79).
In the patients with IEI, benign/reactive LP must be thoroughly investigated and monitored closely for malignant transformation. Any early-onset nodal or extranodal LP in an appropriate clinical context must be investigated for an underlying IEI. EBV testing must be done routinely in all patients. Flow cytometry and molecular genetics should be used in conjunction with morphology, especially in challenging cases.
SSh designed and supervised the manuscript. SSh, RP, GA, MS, KA, RT, MD, and PV reviewed the literature and wrote the manuscript. AR and SSi reviewed and edited the draft. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.856601/full#supplementary-material
1. Van Krieken JH, Onciu M, Elenitoba-Johnson KSJ, Jaffe ES. Lymphoproliferative Diseases Assocated With Primary Immune Disorders. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. Lyon: IARC (2008). p. 336–9.
2. Natkunam Y, Gratzinger D, Chadburn A, Goodlad JR, Chan JKC, Said J, et al. Immunodeficiency-Associated Lymphoproliferative Disorders: Time for Reappraisal? Blood (2018) 132(18):1871–8. doi: 10.1182/blood-2018-04-842559
3. Migliavacca M, Assanelli A, Ponzoni M, Pajno R, Barzaghi F, Giglio F, et al. First Occurrence of Plasmablastic Lymphoma in Adenosine Deaminase-Deficient Severe Combined Immunodeficiency Disease Patient and Review of the Literature. Front Immunol (2018) 9:113. doi: 10.3389/fimmu.2018.00113
4. Costagliola G, Consolini R. Lymphadenopathy at the Crossroad Between Immunodeficiency and Autoinflammation: An Intriguing Challenge. Clin Exp Immunol (2021) 205(3):288–305. doi: 10.1111/cei.13620
5. Forbes LR, Eckstein OS, Gulati N, Peckham-Gregory EC, Ozuah NW, Lubega J, et al. Genetic Errors of Immunity Distinguish Pediatric Nonmalignant Lymphoproliferative Disorders. J Allergy Clin Immunol (2021) 149(2):758–66. doi: 10.1016/j.jaci.2021.07.015. S0091-6749(21)01135-0.
6. Gars E, Butzmann A, Ohgami R, Balakrishna JP, O'Malley DP. The Life and Death of the Germinal Center. Ann Diagn Pathol (2020) 44:151421. doi: 10.1016/j.anndiagpath.2019.151421
7. Ratech H, Hirschhorn R, Greco MA. Pathologic Findings in Adenosine Deaminase Deficient-Severe Combined Immunodeficiency. II. Thymus, Spleen, Lymph Node, and Gastrointestinal Tract Lymphoid Tissue Alterations. Am J Pathol (1989) 135(6):1145–56.
8. Moshous D, Martin E, Carpentier W, Lim A, Callebaut I, Canioni D, et al. Whole-Exome Sequencing Identifies Coronin-1A Deficiency in 3 Siblings With Immunodeficiency and EBV-Associated B-Cell Lymphoproliferation. J Allergy Clin Immunol (2013) 131(6):1594–603. doi: 10.1016/j.jaci.2013.01.042
9. Dimitriades VR, Devlin V, Pittaluga S, Su HC, Holland SM, Wilson W, et al. DOCK 8 Deficiency, EBV+ Lymphomatoid Granulomatosis, and Intrafamilial Variation in Presentation. Front Pediatr (2017) 5:38. doi: 10.3389/fped.2017.00038
10. Fuentes-Páez G, Saornil MA, Herreras JM, Alonso-Ballesteros M, Sánchez PS, García-Tejeiro M. CHARGE Association, Hyper-Immunoglobulin M Syndrome, and Conjunctival MALT Lymphoma. Cornea (2007) 26(7):864–7. doi: 10.1097/ICO.0b013e31806c77d6
11. Saglam A, Cagdas D, Aydin B, Keles S, Reisli I, Arslankoz S, et al. STK4 Deficiency and EBV-Associated Lymphoproliferative Disorders, Emphasis on Histomorphology, and Review of Literature. Virchows Arch (2022) 480(2):393–401. doi: 10.1007/s00428-021-03147-w
12. Ghosh S, Bienemann K, Boztug K, Borkhardt A. Interleukin-2-Inducible T-Cell Kinase (ITK) Deficiency - Clinical and Molecular Aspects. J Clin Immunol (2014) 34(8):892–9. doi: 10.1007/s10875-014-0110-8
13. Du S, Scuderi R, Malicki DM, Willert J, Bastian J, Weidner N. Hodgkin's and Non-Hodgkin's Lymphomas Occurring in Two Brothers With Wiskott-Aldrich Syndrome and Review of the Literature. Pediatr Dev Pathol (2011) 14(1):64–70. doi: 10.2350/10-01-0787-CR.1
14. Gładkowska-Dura M, Dzierzanowska-Fangrat K, Dura WT, van Krieken JH, Chrzanowska KH, van Dongen JJ, et al. Unique Morphological Spectrum of Lymphomas in Nijmegen Breakage Syndrome (NBS) Patients With High Frequency of Consecutive Lymphoma Formation. J Pathol (2008) 216(3):337–44. doi: 10.1002/path.2418
15. Taylor AM, Metcalfe JA, Thick J, Mak YF. Leukemia and Lymphoma in Ataxia Telangiectasia. Blood (1996) 87(2):423–38. doi: 10.1182/blood.V87.2.423.bloodjournal872423
16. Padeira GL, Araújo C, Cordeiro AI, Freixo J, Martins CG, Neves JF. Case Report: Primary Immunodeficiencies, Massive EBV+ T-Cell Lympoproliferation Leading to the Diagnosis of ICF2 Syndrome. Front Immunol (2021) 12:654167. doi: 10.3389/fimmu.2021.654167
17. Sood AK, Funkhouser W, Handly B, Weston B, Wu EY. Granulomatous-Lymphocytic Interstitial Lung Disease in 22q11.2 Deletion Syndrome: A Case Report and Literature Review. Curr Allergy Asthma Rep (2018) 18(3):14. doi: 10.1007/s11882-018-0769-7
18. De Vos M, Hayward BE, Charlton R, Taylor GR, Glaser AW, Picton S, et al. PMS2 Mutations in Childhood Cancer. J Natl Cancer Inst (2006) 98(5):358–61. doi: 10.1093/jnci/djj073
19. Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J Clin Immunol (2020) 40(1):66–81. doi: 10.1007/s10875-020-00758-x
20. Gompels MM, Hodges E, Lock RJ, Angus B, White H, Larkin A, et al. Lymphoproliferative Disease in Antibody Deficiency: A Multi-Center Study. Clin Exp Immunol (2003) 134(2):314–20. doi: 10.1046/j.1365-2249.2003.02253
21. Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass Trial: Defining Subgroups in Common Variable Immunodeficiency. Blood (2008) 111(1):77–85. doi: 10.1182/blood-2007-06-091744
22. Yakaboski E, Fuleihan RL, Sullivan KE, Cunningham-Rundles C, Feuille E. Lymphoproliferative Disease in CVID: A Report of Types and Frequencies From a US Patient Registry. J Clin Immunol (2020) 40(3):524–30. doi: 10.1007/s10875-020-00769-8
23. Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and Mortality in Common Variable Immune Deficiency Over 4 Decades. Blood (2012) 119(7):1650–7. doi: 10.1182/blood-2011-09-377945
24. Wehr C, Houet L, Unger S, Kindle G, Goldacker S, Grimbacher B, et al. Altered Spectrum of Lymphoid Neoplasms in a Single-Center Cohort of Common Variable Immunodeficiency With Immune Dysregulation. J Clin Immunol (2021) 41(6):1250–65. doi: 10.1007/s10875-021-01016-4
25. Bonilla FA, Barlan I, Chapel H, Costa-Carvalho BT, Cunningham-Rundles C, de la Morena MT. International Consensus Document (ICON): Common Variable Immunodeficiency Disorders. J Allergy Clin Immunol Pract (2016) 4(1):38–59. doi: 10.1016/j.jaip.2015.07.025
26. Oertel SH, Riess H. Immunosurveillance, Immunodeficiency and LPs. Recent Results Cancer Res (2002) 159:1–8. doi: 10.1007/978-3-642-56352-2_1
27. Wobser M, Kerstan A, Kneitz H, Goebeler M, Kunzmann V, Rosenwald A, et al. Primary Cutaneous Marginal Zone Lymphoma With Sequential Development of Nodal Marginal Zone Lymphoma in a Patient With Selective Immunoglobulin A Deficiency. J Cutan Pathol (2013) 40(12):1035–41. doi: 10.1111/cup.12230
28. Ott MM, Ott G, Klinker H, Trunk MJ, Katzenberger T, Müller-Hermelink HK. Abdominal T-Cell non-Hodgkin's Lymphoma of the Gamma/Delta Type in a Patient With Selective Immunoglobulin A Deficiency. Am J Surg Pathol (1998) 22(4):500–6. doi: 10.1097/00000478-199804000-00017
29. Mellemkjaer L, Hammarstrom L, Andersen V, Yuen J, Heilmann C, Barington T, et al. Cancer Risk Among Patients With IgA Deficiency or Common Variable Immunodeficiency and Their Relatives: A Combined Danish and Swedish Study. Clin Exp Immunol (2002) 130(3):495–500. doi: 10.1046/j.1365-2249.2002.02004.x
30. Mortaz E, Tabarsi P, Mansouri D, Khosravi A, Garssen J, Velayati A, et al. Cancers Related to Immunodeficiencies: Update and Perspectives. Front Immunol (2016) 7:365. doi: 10.3389/fimmu.2016.00365
31. Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, et al. Clinical Spectrum and Features of Activated Phosphoinositide 3-Kinase δ Syndrome: A Large Patient Cohort Study. J Allergy Clin Immunol (2017) 139(2):597–606.e4. doi: 10.1016/j.jaci.2016.06.021
32. Rivalta B, Amodio D, Milito C, Chiriaco M, Di Cesare S, Giancotta C. Case Report: EBV Chronic Infection and Lymphoproliferation in Four APDS Patients: The Challenge of Proper Characterization, Therapy, and Follow-Up. Front Pediatr (2021) 9:703853. doi: 10.3389/fped.2021.703853
33. Arjunaraja S, Angelus P, Su HC, Snow AL. Impaired Control of Epstein-Barr Virus Infection in B-Cell Expansion With NF-κb and T-Cell Anergy Disease. Front Immunol (2018) 9:198. doi: 10.3389/fimmu.2018.00198
34. Boztug H, Hirschmugl T, Holter W, Lakatos K, Kager L, Trapin D, et al. NF-κb1 Haploinsufficiency Causing Immunodeficiency and EBV-Driven Lymphoproliferation. J Clin Immunol (2016) 36(6):533–40. doi: 10.1007/s10875-016-0306-1
35. Etzioni A, Ochs H. The Hyper IgM Syndrome—An Evolving Story. Pediatr Res (2004) 56:519–25. doi: 10.1203/01.PDR.0000139318.65842.4A
36. Gratzinger D, Jaffe ES, Chadburn A, Chan JK, de Jong D, Goodlad JR, et al. Primary/Congenital Immunodeficiency: 2015 SH/EAHP Workshop Report-Part 5. Am J Clin Pathol (2017) 147(2):204–16. doi: 10.1093/ajcp/aqw215
37. Hoshino A, Okuno Y, Migita M, Ban H, Yang X, Kiyokawa N, et al. X-Linked Agammaglobulinemia Associated With B-Precursor Acute Lymphoblastic Leukemia. J Clin Immunol (2015) 35(2):108–11. doi: 10.1007/s10875-015-0127-7
38. Cansever M, Zietara N, Chiang SCC, Ozcan A, Yilmaz E, Karakukcu M, et al. A Rare Case of Activated Phosphoinositide 3-Kinase Delta Syndrome (APDS) Presenting With Hemophagocytosis Complicated With Hodgkin Lymphoma. J Pediatr Hematol Oncol (2020) 42(2):156–9. doi: 10.1097/MPH.0000000000001487
39. Ruiz-García R, Mora S, Lozano-Sánchez G, Martínez-Lostao L, Paz-Artal E, Ruiz-Contreras J. Decreased Activation-Induced Cell Death by EBV-Transformed B-Cells From a Patient With Autoimmune Lymphoproliferative Syndrome Caused by a Novel FASLG Mutation. Pediatr Res (2015) 78(6):603–8. doi: 10.1038/pr.2015.170
40. Kanderova V, Grombirikova H, Zentsova I, Reblova K, Klocperk A, Fejtkova M. Lymphoproliferation, Immunodeficiency and Early-Onset Inflammatory Bowel Disease Associated With a Novel Mutation in Caspase 8. Haematologica (2019) 104(1):e32–4. doi: 10.3324/haematol.2018.201673
41. Egg D, Schwab C, Gabrysch A, Arkwright PD, Cheesman E, Giulino-Roth L, et al. Increased Risk for Malignancies in 131 Affected CTLA4 Mutation Carriers. Front Immunol (2018) 9:2012. doi: 10.3389/fimmu.2018.02012
42. Schwab C, Gabrysch A, Olbrich P, Patiño V, Warnatz K, Wolff D, et al. Phenotype, Penetrance, and Treatment of 133 Cytotoxic T-Lymphocyte Antigen 4-Insufficient Subjects. J Allergy Clin Immunol (2018) 142(6):1932–46. doi: 10.1016/j.jaci.2018.02.055
43. Martin E, Palmic N, Sanquer S, Lenoir C, Hauck F, Mongellaz C, et al. CTP Synthase 1 Deficiency in Humans Reveals its Central Role in Lymphocyte Proliferation. Nature (2014) 510(7504):288–92. doi: 10.1038/nature13386
44. Somekh I, Marquardt B, Liu Y, Rohlfs M, Hollizeck S, Karakukcu M, et al. Novel Mutations in RASGRP1 are Associated With Immunodeficiency, Immune Dysregulation, and EBV-Induced Lymphoma. J Clin Immunol (2018) 38(6):699–710. doi: 10.1007/s10875-018-0533-8
45. Caorsi R, Rusmini M, Volpi S, Chiesa S, Pastorino C, Sementa AR, et al. CD70 Deficiency Due to a Novel Mutation in a Patient With Severe Chronic EBV Infection Presenting As a Periodic Fever. Front Immunol (2018) 8:2015. doi: 10.3389/fimmu.2017.02015
46. Szczawińska-Popłonyk A, Grześk E, Schwartzmann E, Materna-Kiryluk A, Małdyk J. Case Report: Autoimmune Lymphoproliferative Syndrome vs. Chronic Active Epstein-Barr Virus Infection in Children: A Diagnostic Challenge. Front Pediatr (2021) 9:798959. doi: 10.3389/fped.2021.798959
47. Jiang Y, Firan M, Nandiwada SL, Reyes A, Marsh RA, Vogel TP, et al. The Natural History of X-Linked Lymphoproliferative Disease (XLP1): Lessons From a Long-Term Survivor. Case Rep Immunol (2020) 2020:8841571. doi: 10.1155/2020/8841571
48. Alosaimi MF, Hoenig M, Jaber F, Platt CD, Jones J, Wallace J, et al. Immunodeficiency and EBV-Induced Lymphoproliferation Caused by 4-1BB Deficiency. J Allergy Clin Immunol (2019) 144(2):574–83.e5. doi: 10.1016/j.jaci.2019.03.002
49. Wegehaupt O, Groß M, Wehr C, Marks R, Schmitt-Graeff A, Uhl M, et al. TIM-3 Deficiency Presenting With Two Clonally Unrelated Episodes of Mesenteric and Subcutaneous Panniculitis-Like T-Cell Lymphoma and Hemophagocytic Lymphohistiocytosis. Pediatr Blood Cancer (2020) 67(6):e28302. doi: 10.1002/pbc.28302
50. Alabbas F, Elyamany G, Alsharif O, Hershfield M, Meyts I. Childhood Hodgkin Lymphoma: Think Dada2. J Clin Immunol (2019) 39(1):26–9. doi: 10.1007/s10875-019-0590-7
51. Neven Q, Boulanger C, Bruwier A, de Ville de Goyet M, Meyts I, Moens L, et al. Clinical Spectrum of Ras-Associated Autoimmune Leukoproliferative Disorder (RALD). J Clin Immunol (2021) 41(1):51–8. doi: 10.1007/s10875-020-00883-7
52. Kawabe S, Ito Y, Gotoh K, Kojima S, Matsumoto K, Kinoshita T, et al. Application of Flow Cytometric in Situ Hybridization Assay to Epstein-Barr Virus-Associated T/natural Killer Cell Lymphoproliferative Diseases. Cancer Sci (2012) 103(8):1481–8. doi: 10.1111/j.1349-7006.2012.02305.x
53. Knight V. The Utility of Flow Cytometry for the Diagnosis of Primary Immunodeficiencies. Int J Lab Hematol (2019) 41 Suppl 1:63–72. doi: 10.1111/ijlh.13010
54. Ribera J, Zamora L, Juncà J, Rodríguez I, Marcé S, Cabezón M, et al. Usefulness of IGH/TCR PCR Studies in Lymphoproliferative Disorders With Inconclusive Clonality by Flow Cytometry. Cytomet B Clin Cytom (2014) 86(1):25–31. doi: 10.1002/cyto.b.21118
55. Langerak AW, Groenen PJ, Brüggemann M, Beldjord K, Bellan C, Bonello L, et al. EuroClonality/BIOMED-2 Guidelines for Interpretation and Reporting of Ig/TCR Clonality Testing in Suspected LPs. Leukemia (2012) 26(10):2159–71. doi: 10.1038/leu.2012.246
56. Arcila ME, Yu W, Syed M, Kim H, Maciag L, Yao J, et al. Establishment of Immunoglobulin Heavy (IGH) Chain Clonality Testing by Next-Generation Sequencing for Routine Characterization of B-Cell and Plasma Cell Neoplasms. J Mol Diagn (2019) 21(2):330–42. doi: 10.1016/j.jmoldx.2018.10.008
57. Stewart JP, Gazdova J, Darzentas N, Wren D, Proszek P, Fazio G, et al. Validation of the EuroClonality-NGS DNA Capture Panel as an Integrated Genomic Tool for Lymphoproliferative Disorders. Blood Adv (2021) 5(16):3188–98. doi: 10.1182/bloodadvances.2020004056
58. Cohen JM, Sebire NJ, Harvey J, Gaspar HB, Cathy C, Jones A, et al. Successful Treatment of Lymphoproliferative Disease Complicating Primary Immunodeficiency/Immunodysregulatory Disorders With Reduced-Intensity Allogeneic Stem-Cell Transplantation. Blood (2007) 110(6):2209–14. doi: 10.1182/blood-2006-12-062174
59. Cunningham-Rundles C. How I Treat Common Variable Immune Deficiency. Blood (2010) 116(1):7–15. doi: 10.1182/blood-2010-01-254417
60. Gangemi S, Allegra A, Musolino C. Lymphoproliferative Disease and Cancer Among Patients With Common Variable Immunodeficiency. Leuk Res (2015) 39(4):389–96. doi: 10.1016/j.leukres.2015.02.002
61. Notarangelo LD. Hematopoietic Stem Cell Transplantation for Activated Phosphoinositide 3-Kinase δ Syndrome: Who, When, and How? J Allergy Clin Immunol (2019) 143(1):91–3. doi: 10.1016/j.jaci.2018.08.039
62. Nademi Z, Slatter MA, Dvorak CC, Neven B, Fischer A, Suarez F, et al. Hematopoietic Stem Cell Transplant in Patients With Activated PI3K Delta Syndrome. J Allergy Clin Immunol (2017) 139(3):1046–9. doi: 10.1016/j.jaci.2016.09.040
63. Coulter TI, Cant AJ. The Treatment of Activated Pi3kδ Syndrome. Front Immunol (2018) 9:2043. doi: 10.3389/fimmu.2018.02043
64. Maccari ME, Abolhassani H, Aghamohammadi A, Aiuti A, Aleinikova O, Bangs C, et al. Disease Evolution and Response to Rapamycin in Activated Phosphoinositide 3-Kinase δ Syndrome: The European Society for Immunodeficiencies-Activated Phosphoinositide 3-Kinase δ Syndrome Registry. Front Immunol (2018) 9:543. doi: 10.3389/fimmu.2018.00543
65. Rao VK, Webster S, Dalm VASH, Šedivá A, van Hagen PM, Holland S, et al. Effective “Activated PI3Kδ Syndrome”-Targeted Therapy With the PI3Kδ Inhibitor Leniolisib. Blood (2017) 130(21):2307–16. doi: 10.1182/blood-2017-08-801191
66. Rao VK, Oliveira JB. How I Treat Autoimmune Lymphoproliferative Syndrome. Blood (2011) 118(22):5741–51. doi: 10.1182/blood-2011-07-325217
67. Matson DR, Yang DT. Autoimmune Lymphoproliferative Syndrome: An Overview. Arch Pathol Lab Med (2019) 144(2):245–51. doi: 10.5858/arpa.2018-0190-RS
68. George LA, Teachey DT. Optimal Management of Autoimmune Lymphoproliferative Syndrome in Children. Paediatr Drugs (2016) 18(4):261–72. doi: 10.1007/s40272-016-0175-3
69. Bride KL, Vincent T, Smith-Whitley K, Lambert MP, Bleesing JJ, Seif AE, et al. Sirolimus is Effective in Relapsed/Refractory Autoimmune Cytopenias: Results of a Prospective Multi-Institutional Trial. Blood (2016) 127(1):17–28. doi: 10.1182/blood-2015-07-657981
70. Teachey DT, Greiner R, Seif A, Attiyeh E, Bleesing J, Choi J, et al. Treatment With Sirolimus Results in Complete Responses in Patients With Autoimmune Lymphoproliferative Syndrome. Br J Haematol (2009) 145(1):101–6. doi: 10.1111/j.1365-2141.2009.07595.x
71. Panchal N, Booth C, Cannons JL, Schwartzberg PL. X-Linked Lymphoproliferative Disease Type 1: A Clinical and Molecular Perspective. Front Immunol (2018) 9:666. doi: 10.3389/fimmu.2018.00666
72. Booth C, Gilmour KC, Veys P, Gennery AR, Slatter MA, Chapel H, et al. X-Linked Lymphoproliferative Disease Due to SAP/SH2D1A Deficiency: A Multicenter Study on the Manifestations, Management and Outcome of the Disease. Blood (2011) 117(1):53–62. doi: 10.1182/blood-2010-06-284935
73. Alabbas F, Alsharief O, Meyts I, Albatniji F, Hershfield M, Mansoor A, et al. Deficiency of Adenosine Deaminase 2 (DADA2) Presenting As Familial Hodgkin Lymphoma. Blood (2018) 132(Supplement 1):5373. doi: 10.1182/blood-2018-99-116431
74. Meyts I, Aksentijevich I. Deficiency of Adenosine Deaminase 2 (DADA2): Updates on the Phenotype, Genetics, Pathogenesis, and Treatment. J Clin Immunol (2018) 38(5):569–78. doi: 10.1007/s10875-018-0525-8
75. Gupta M, Aluri J, Desai M, Lokeshwar M, Taur P, Lenardo M, et al. Clinical, Immunological, and Molecular Findings in Four Cases of B Cell Expansion With NF-κb and T Cell Anergy Disease for the First Time From India. Front Immunol (2018) 9:1049. doi: 10.3389/fimmu.2018.01049
76. Giovannini-Chami L, Vogel TP, Forbes LR, Fabre A, Trojani M-C, Leroy S, et al. STAT3 Gain of Function: A New Aetiology of Severe Rheumatic Disease. Rheumatology (2019) 58(2):365–7. doi: 10.1093/rheumatology/key308
77. Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-Onset Lymphoproliferation and Autoimmunity Caused by Germline STAT3 Gain-of-Function Mutations. Blood (2015) 125(4):591–9. doi: 10.1182/blood-2014-09-602763
78. Weinreich MA, Vogel TP, Rao VK, Milner JD. Up, Down, and All Around: Diagnosis and Treatment of Novel STAT3 Variant. Front Pediatr (2017) 5:49. doi: 10.3389/fped.2017.00049
Keywords: immunodeficiency, inborn errors of immunity (IEI), Ig/TCR gene rearrangements, lymphoproliferation, lymphoma
Citation: Sharma S, Pilania RK, Anjani G, Sudhakar M, Arora K, Tyagi R, Dhaliwal M, Vignesh P, Rawat A and Singh S (2022) Lymphoproliferation in Inborn Errors of Immunity: The Eye Does Not See What the Mind Does Not Know. Front. Immunol. 13:856601. doi: 10.3389/fimmu.2022.856601
Received: 17 January 2022; Accepted: 11 April 2022;
Published: 04 May 2022.
Edited by:
Shanmuganathan Chandrakasan, Emory University, United StatesReviewed by:
Eleonora Gambineri, University of Florence, ItalyCopyright © 2022 Sharma, Pilania, Anjani, Sudhakar, Arora, Tyagi, Dhaliwal, Vignesh, Rawat and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saniya Sharma, ZHJzYW5peWEuc2hhcm1hQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.