
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 28 March 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.855978
This article is part of the Research Topic Insights in Cancer Immunity and Immunotherapy: 2021 View all 9 articles
 Michal A. Rahat1,2*
Michal A. Rahat1,2*Solid tumors metastasize very early in their development, and once the metastatic cell is lodged in a remote organ, it can proliferate to generate a metastatic lesion or remain dormant for long periods. Dormant cells represent a real risk for future tumor recurrence, but because they are typically undetectable and insensitive to current modalities of treatment, it is difficult to treat them in time. We describe the metastatic cascade, which is the process that allows tumor cells to detach from the primary tumor, migrate in the tissue, intravasate and extravasate the lymphatics or a blood vessel, adhere to a remote tissue and eventually outgrow. We focus on the critical enabling role of the interactions between tumor cells and immune cells, especially macrophages, in driving the metastatic cascade, and on those stages that can potentially be targeted. In order to prevent the metastatic cascade and tumor recurrence, we would need to target a molecule that is involved in all of the steps of the process, and evidence is brought to suggest that CD147/EMMPRIN is such a protein and that targeting it blocks metastasis and prevents tumor recurrence.
Recurrence of cancer due to a metastatic spread to remote organs is responsible for the vast majority of cancer-related deaths, despite successful treatment of the primary tumor (1). The metastatic cells are plastic, shift between different states and are often more resistant to different modalities of existing treatments (2), and therefore, present a great therapeutic challenge. Solid tumors can metastasize to a regional lymph node or to a secondary organ via lymphatics or blood vessels very early in their development, even before diagnosis of the primary tumor (3, 4). Metastasis is a multi-step process that begins with a change in the epithelial phenotype of the cells, a process known as the Epithelial-To-Mesenchymal Transition (EMT). EMT promotes the detachment of some tumor cells, increases their motility and changes their phenotype into a mesenchymal, spindle-like morphology (5). This increases their invasiveness and ability to resist different drugs (6, 7). The increased motility allows a cell to pave its way through the extracellular matrix (ECM), intravasate into a lymphatic or blood vessel to become a circulating tumor cell (CTC), and then extravasate in a regional lymph node or a distant tissue as a disseminated tumor cell (DTC) (8). However, while every metastasizing cell must undergo EMT, it is important to remember that EMT is not a binary process, and cancer cells are found in many hybrid forms along the epithelial/mesenchymal axis (hybrid E/M cells), having both epithelial and mesenchymal features that endow them with the plasticity to adjust to different microenvironments (5, 9). The DTCs that are lodged in the remote organ, either as single cells or as clusters of a few cells, may remain in a latent or quiescent state for years or decades (dormancy), undetected by imaging techniques, until they suddenly start proliferating (colonization). At this stage they undergo the inverse process to EMT - Mesenchymal to Epithelial Transition (MET), that allows their rapid outgrowth (the metastatic outbreak), ultimately generating the metastatic lesion (10).
This multi-step process, known as the metastatic cascade (11), is highly inefficient, and only few of the CTCs that embark on this journey will eventually be implanted in a secondary organ (about 0.01%) and become DTCs (12, 13). Dormant cells have been reported in many types of cancer (e.g., melanoma, lung cancer, glioblastoma, breast cancer, multiple myeloma, leukemia, colorectal, prostate, skin and gastric cancer), and the combination of their early dissemination and resistance to common modalities of treatment increases the risk of tumor recurrence (14, 15). Hence, a major therapeutic goal is to find successful therapies directed against different steps of the metastatic cascade, thus preventing recurrence and metastatic outbreak.
The EMT process initiates the metastatic cascade, and renders tumor cells more resistant to different therapies (16, 17), such as chemotherapies (18), targeted therapies (19) and immunotherapies (20). The EMT process has been linked to resistance to therapy, and several mechanisms were described, some of which are presented here. EMT depends on the induction of the EMT transcription factors (EMT-TFs), particularly members of the Snail, Twist1, and Zeb families. In addition to regulating the epithelial or mesenchymal phenotypes, these EMT-TFs also regulate the expression of ABC transporter genes and genes involved in drug-induced apoptosis, thus contributing to acquired drug resistance (17, 19). For example, high Snail levels induce the activity of the Multidrug Resistance Protein 1 (MDR1)/P-Glycoprotein, thus allowing better transport of drugs outside the cells and improving cell resistance (21), Twist1 suppresses the expression of the pro-apoptotic protein BIM (BCL2L11) to mediate resistance to tyrosine kinase inhibitor erlotinib (22), and Zeb1 enhanced the expression of Ataxia-Telangiectasia Mutated (ATM) kinase that promotes the DNA damage repair machinery and confer resistance to agents such as epirubicin (23). The pan-resistance in dormant cells, which inherently exhibit reduced metabolism and arrested proliferation that endows them with some resistance to therapy, is also partially attributed to low-level expression of E-cadherin that upon ligation provides signals that activate ERK and PI3K signaling pathways to ensure survival of DTCs. When these cells are exposed to chemotherapies these signals are amplified leading to drug resistance (2, 15, 24). To promote chemoresistance, extracellular vesicles (Evs) were shown to directly sequester drugs, either by up-taking them and secreting them outside the cells, or by exhibiting receptors or decoy receptors that bind therapeutic antibodies (e.g., with rituximab and CD20 or Herceptin and Her2). EVs can also horizontally transfer proteins, mRNAs, miRNAs or LncRNAs that express or regulate the expression of transporters and transcription factors, from chemo-resistant cells to chemo-sensitive cells or from stroma and immune cells to tumor cells promoting acquired resistance [reviewed in (25)]. The EMT process also promotes the recruitment of macrophages, MDSCs and fibroblasts to the tumor microenvironment (TME), and the tumor cells then interact with them and reprogram them in situ (26) to become tumor-associated macrophages (TAMs) and cancer-associated fibroblasts (CAFs) that secrete pro-inflammatory and pro-angiogenic mediators that affect drug resistance as well (16, 19). For example, IL-6 and Oncostatin-M have been implicated in the resistance to cisplatin and gemcitabine, respectively (27, 28). The suppressive microenvironment and increased IFNγ secretion that leads to increased PD-L1 expression on tumor cells and macrophages, inhibit T cells and NK cells and promote immune evasion of the DTCs (29, 30). Lastly, elevated deposition of ECM proteins and expression of integrins, increasing density and stiffness, was associated with increased drug resistance (31, 32).
Targeting the EMT process may enable us to reverse these effects and reduce metastasis. Different approaches to targeting EMT include inhibiting signals that promote it (e.g., TGFβ, HGF), targeting stromal cells that are needed to activate the EMT program, targeting specific EMT transcription factors or targeting the microRNAs that regulate them. Such approaches are extensively reviewed elsewhere (33), but although they may inhibit the metastatic cascade and reduce metastasis, it is not clear how they might affect dormant DTCs that have already disseminated to remote organs. However, dormant DTCs have already undergone EMT and activated dormancy programs (see below) that endow them with increased survival and pan-resistance to different therapies (34), suggesting that eradicating these dormant cells will prove challenging. Another option of targeting CTCs on route to the metastatic site was proposed. CTCs may be detected using liquid biopsies, but their presence after successful treatment or primary tumor resection, might indicate that an already existing dormant tumor sheds its cells (35), and therefore, they are not an obvious target.
There are three strategies to inhibit the outgrowth of dormant DTCs: a) kill DTCs directly, b) awaken dormant cells and simultaneously target them with therapies designed against proliferating cells (e.g., chemotherapies), or c) impose and maintain their dormant state indefinitely, or at least for a long time (36). Killing dormant DTCs or awakening them are strategies that should take into account their inherent resistance to cytotoxic drugs, targeted therapies and even immunotherapies (37, 38). Therefore, if any cells survive treatment, they are likely to become even more aggressive, and promoting their exit from dormancy might expedite their uncontrollable proliferation and enhance tumor relapse. The option of maintaining dormancy for long periods allows tumor cells to remain viable, and risks the possibility that eventually they will escape dormancy, suggesting that treatment should be given for life, thus increasing its potential toxicity and costs.
A closer look at dormancy shows that while most colonizing DTCs undergo apoptosis and die, and a few immediately proliferate to generate macro-metastases in a favorable pre-metastatic niche, some DTCs activate survival programs and enter the dormant state. These cells exhibit growth arrest and will remain suspended in the G0 phase (cellular dormancy), allowing them to re-enter cell cycle once conditions are more favorable (3, 12). Other DTCs form small cellular clusters that balance proliferation and apoptosis, remaining dormant either because pro-angiogenic signals are lacking (angiogenic dormancy) or because of the pressure exerted by the immune system (immune-mediated dormancy) drives apoptosis of the cells that is balanced by their proliferation (3, 12). The triggers that DTCs encounter in the metastatic microenvironment (MME) that determine their entry into or escape from a dormant state can be divided into cell-intrinsic and cell-extrinsic mechanisms, and both are not yet fully elucidated.
Cell-intrinsic mechanisms include metabolic pathways, autophagy, epigenetic mechanisms, and genetic mutations that directly or indirectly regulate the cell cycle machinery and proliferation. Experiments in vitro demonstrated that tumor cells can enter dormancy to survive deprivation of glucose or growth factors (e.g., IGF-2, PDGF) (37). This triggers arrest of proliferation and reduced expression of proteins involved in the regulation of cell cycle (e.g., c-Myc, Cyclin D1), or increased expression of the Cyclin Dependent Kinases (CDKs) inhibitors (e.g., p16, p21, p27) (39). Metabolic pathways respond to stress signaling, and reduction in PI3K signaling or increased autophagy has been linked to dormancy (40). An increase in the ratio between phosphorylated p38 and ERK MAPKs, reflecting stress-induced p38 activation, has often been used as a marker of dormancy, as was the increased expression of the orphan nuclear receptor (NR2F1) which induces SOX9 and RARβ, that in turn, elevate the CDK inhibitors p16 and p27 (3, 39, 41). Therapies that target specific elements in those pathways could be employed to induce dormancy. For example, genetic ablation of the FBXW7 protein that is part of an E3 ligase complex, promotes degradation of Cyclin E and c-Myc (42), whereas inhibitors of autophagy (e.g., hydroxychloroquine) inhibit proliferation and reduce DTCs survival (43). Such approaches are reviewed in (3, 39, 40).
Extrinsic factors in the MME can enforce DTCs dormancy. High levels of quiescent signals can be secreted from organ cells and resident immune cells, such as TGFβ2 and BMP7 in the bone, or BMP4 in the lung (12, 44–46). Thrombospondin-1 (Tsp-1) secreted from quiescent endothelial cells promotes dormancy, whereas in sprouting endothelial cells, TGFβ1 and Periostin secreted from endothelial tip cells promote tumor cell outgrowth (46). Low oxygen tensions (hypoxia) can impair the mTOR metabolic pathway to induce dormancy (47), or induce the expression of TGFβ2 and dormancy marker genes (48, 49). The composition of ECM proteins and the degree of tissue stiffness can also induce dormancy or outgrowth (44, 50, 51). For example, activation of β1-integrins promoted Src-FAK-ERK-MLCK signaling and escape from dormancy (52), and crosslinking of Collagen fibers by Zeb1-regulated Lysyl Oxidases that increase stiffness, promotes metastasis (53). In mouse syngeneic or xenograft models that were treated for oncogene inhibition (anti-Her2 and anti-ER treatments), primary tumors regressed leaving residual tumor cells locally or at remote sites, that exhibited reduced proliferation and a gene signature with reduced expression of Cyclins and CDKs, enhanced expression of ECM proteins (e.g., Fibronectin) and dormancy genes, such as Bhlhe41 (coding for DEC2), TGFβ2 and its receptor TGFβRIII, and Thsb1 (coding for Tsp-1). This gene signature was similar to dormancy induced by microenvironmental cues, and was different from genes expressed by primary tumor cells or recurrent metastatic tumor cells (38).
The MME consists of many factors arriving from systemic factors, extracellular vesicles (EVs) secreted from the primary tumor, soluble factors secreted from the resident tissue or immune cells, and from the DTCs themselves. These prepare the pre-metastatic niche (‘soil’) to receive the metastatic cells (‘seeds’) (54), determine organotropic metastasis (affinity of DTCs to specific organs) (55–57), and promote tumor cell-stroma interactions (56, 57) that eventually promote the escape of the DTCs from dormancy. However, the mechanisms that allow these myriad factors and pathways to interact and to determine dormancy or escape from it are only now beginning to be addressed. The reader is referred to some outstanding reviews that discuss these mechanisms and potential approaches to target them (3, 10, 34, 36, 39, 40, 45, 58, 59).
The central role tumor-associated macrophages (TAMs) play in promoting the primary tumor is now well recognized, but their role in metastasis is only now beginning to unfold. TAMs can be differently activated, and in the primary tumor they are often found in a mixture of both M2-activated (pro-tumoral, pro-angiogenic) and M1-activated (pro-inflammatory, anti-tumoral) macrophages. In the primary tumor, TAMs and myeloid-derived suppressor cells (MDSCs) secrete growth factors and pro-angiogenic factors that promote tumor progression, and express inhibitory receptors and cytokines that suppress cytotoxic T cells or lead to their exhaustion, thus contributing to evasion from immune recognition (60–64).
Crosstalk between specific TAMs subsets and tumor cells in specific niches in the primary tumor also help promote metastasis. Such interactions promote the EMT process and increase the ability of tumor cells to migrate and invade (65–67). Perivascular TAMs (identified as Tie2+VEGFA+MRC1+) promote angiogenesis and vascular permeability by enhancing VEGF secretion (68, 69), and assist in generating specialized temporal structures (called tumor microenvironment of metastasis-TMEM) that are based on direct cell-cell contacts with both tumor cells and endothelial cells. These TMEMs become the gateways which facilitate the intravasation of the metastatic cells (11, 70–72). Another subset of TAMs co-migrate along with the metastatic cells, secrete matrix metalloproteinases (MMPs) that degrade the ECM, and thus pave the way for the metastatic cells (54). TAMs can also utilize a myriad of mechanisms (e.g., secretion of IDO1, TGFβ1, PGE2 and IL-10 that promote Tregs, or expression of co-inhibitory receptors as PD-L1 that lead to T cell exhaustion) that provide local protection for the tumor cells against immune recognition along the metastatic cascade (71).
Metastasis-associated macrophages (MAMs) in the distant organs consist of either resident macrophages or blood monocytes-derived macrophages (BMDM) that are recruited into the site. The primary tumor can secrete soluble factors and exosomes that reprogram resident macrophages in distant organs to become MAMs that are predominantly M2-activated, recruit BMDMs and help generate the pre-metastatic niche (73). Soluble factors (e.g., VEGF, TGFβ, TNFα, CXCL12) secreted from the primary tumor can activate lung cells to express S100A8/S100A9 and MMP-9 (74), and circulating CD11b+ myeloid cells are recruited to the lung in a VEGFR1-dependent manner (75). These MAMs and recruited BMDMs secrete VEGF and IL-1β, and help promote extravasation of DTCs at the secondary organ and their seeding through cross-talk involving CD44, integrins and endothelial cell adhesion molecules (13). Different techniques used to deplete macrophages help exemplify their important role during DTCs extravasation, survival, seeding and initial growth in the lung or bone (76, 77). However, the role that TAMs play during colonization of DTCs and in the process of escape from dormancy and metastatic outgrowth is still not fully understood.
Limited evidence suggests that the immune system, particularly MAMs, is important during initiation of the MET process and colonization, as well as in the regulation of dormancy and the metastatic outbreak. The metastatic outbreak is associated with inflammation, as patients with chronic inflammatory diseases (e.g., obesity, asthma, rheumatoid arthritis, psoriasis), increased serum levels of pro-inflammatory cytokines (e.g., IL-6) and presence of inflammatory infiltrate have an increased risk of metastasis (13). Similarly, surgical resection of the primary tumor, which is followed by a healing response, can enhance the escape from dormancy (13, 78). The presence of macrophages and the factors they secrete (e.g., CCL2, CCL5, IL-8) could also promote the escape from dormancy by activating the ERK signaling pathway (79, 80). In addition, interactions mediated through the binding of VCAM-1 to α4 integrins on DTCs induce Akt activation and promote DTCs survival (81). TNFα-induced expression of the IL-35 receptor IL12Rβ2 already on 4T1 mammary cancer cells in the primary tumor, rendered these cells more susceptible to the effects of IL-35 secreted from MAMs upon arrival at the lung metastatic site. This induced the JAK2/STAT6/GATA3 signaling, initiated their MET process and enhanced their colonization (82). In bone metastases, secreted pro-inflammatory cytokines including Parathyroid hormone-releasing peptide (PTHrP) stimulate osteoclasts to secrete RANK-L to promote bone degradation, and this releases dormant cells from the control of the bone microenvironment and promotes their proliferation and escape from dormancy (83). However, not all pro-inflammatory signals induce metastatic outgrowth. For example, secretion of IFNβ and IFNγ from TAMs and lymphocytes was shown to induce dormancy in DTCs by activating the IDO1/AhR/p27-dependent pathway (84, 85), and chemotherapy-induced secretion of IFNβ enhanced immune dormancy in an IRF7-dependent manner, drastically reducing the presence of MDSCs in the primary tumor and in the lungs of syngeneic mice (86). Interactions between the Growth Arrest Specific 6 (GAS6) factor produced by osteoblasts and the Axl tyrosine kinase receptor expressed by disseminating prostate cancer cells, mediated the TGFβ2 signaling pathway and promoted dormancy of the tumor cells in the bone marrow niche (87).
Different methods used to deplete macrophages demonstrated the importance of TAMs and MAMs in metastasis. In mouse models, removal of the spleen, which acts as a reservoir of myeloid cells, reduces metastasis (88). A recent study showed that TAMs are critical in a model of breast-cancer bone metastasis, as their ablation or inhibition resulted in reduced proliferation and metastasis (77). These TAMs originated mostly from monocyte-derived macrophages and not from bone resident macrophages, and their activation depended on their high IL-4R expression (77).
ECM composition and density could also play a role in determining dormancy or escape from dormancy, and TAMs and MAMS have been shown to take part in regulating this effect (50). For example, depletion of myeloid cells prevented M-MDSCs-induced deposition of Versican. Versican was shown to attenuate Smad2 and Snail levels, thus inhibiting EMT and promoting MET at the metastatic site (89). Collaboration between TAMs and cancer-associated fibroblasts (CAFs) that was initiated by systemic inflammatory signals, resulted in production of high levels of ECM proteins, including collagen, fibronectin and laminin, which increased ECM stiffness and generated a fibrotic MME (24). These changes protected the emerging DTCs from cytotoxic drugs and targeted therapies (24, 32). Neutrophils (and some macrophages) are usually associated with Neutrophil Extracellular Traps (NETs) that contain among other proteins the Neutrophil Elastase and MMP-9. These enzymes can degrade and remodel Laminin-rich ECM, and the cleavage products activate the α3β1 integrin pathway to promote proliferation and escape from dormancy (90).
Overall, the majority of evidence so far suggest that TAMs and MAMs promote MET and the escape from dormancy, but our mechanistic knowledge about the specific contribution of TAMs to the final step of the metastatic outgrowth is still very limited and merits more investigations.
CD147 (also known as EMMPRIN or basigin) is a multifunctional transmembrane glycoprotein, with two extracellular Ig-like domains, that mediates interactions between tumor and stromal cells (91), and those interactions elevate the secretion of pro-angiogenic factors. CD147 is overexpressed in many types of tumors (91), and is elevated on metastatic cells (92–94) and in the serum of breast cancer patients with lymph node metastasis (95). CD147 is best known for its pro-angiogenic activity as an inducer of both VEGF and MMPs through homophilic interactions between tumor cells and stroma cells (mostly fibroblasts and macrophages). This homophilic interaction is localized to the N-terminus of the protein on the first of its two Ig-like domains (96, 97). Recently, we have identified a specific short epitope in the protein that is responsible for the induction of both VEGF and MMP-9 (98), and showed that CD147 is an important mediator of macrophage-tumor cell interactions, and its expression is increased when these cells are co-cultured in vitro (99). CD147 acts as a hub protein that binds to many protein partners and promotes the assembly of protein complexes (100), and therefore, is involved in mediating many functions (Table 1). For example, CD147 acts as a chaperone of the lactate transporters MCT-1 and MCT-4, to facilitate lactate efflux (126), which is critical in maintaining viability of tumor cells that rely mostly on glycolytic metabolism. CD147 also regulates Hyaluronan synthesis and can bind to the Hyaluronan receptor CD44, contributing to tumor cell invasiveness and chemoresistance (119, 135). Extracellular Cyclophilin, especially Cyclophilin A (CyPA) secreted by tumor cells, binds to its receptor CD147 to enhance tumor cell proliferation and survival and leukocyte chemotaxis and adhesion (105, 136). It was also suggested that by binding to CD147, CypA enhanced the localization of CD147 and CD44 complexes to lipid rafts and initiated a STAT3 signaling, thus activating Cyclin D1 and Survivin and promoting cell proliferation and survival (118). Additionally, CD147 has been linked to EMT, both as a mediator of TGFβ1 signaling leading to increased expression of the EMT transcription factors snail and slug (130, 131), and as a destabilizer of the interactions between E-cadherin and β-catenin (132). CD147 can also bind to the β1 integrins, especially α3β1 and α6β1 integrins, induce the FAK signaling pathway and promote invasion, MMPs secretion and metastasis (113, 137, 138). Invasiveness of malignant cells was associated with the increased expression of another binding partner - S100A9 (122, 139). Most importantly, CD147 activates the Wnt, the TGFβ1-Smad4, and/or the ERK1/2 pathways to enhance cell proliferation (101, 105, 140, 141), potentially implicating CD147 in the regulation of metastasis and escape from dormancy. Collectively, these studies suggest that CD147 acts as a scaffold protein that promotes the assembly of many proteins into one or more signaling complexes that enhances cell proliferation, metabolism, angiogenesis, invasiveness, EMT and survival. All these processes are necessary for tumor metastasis, and the increased proliferation and angiogenic switch promote the escape from cellular and angiogenic dormancy. These data place CD147 as an attractive candidate for targeting.
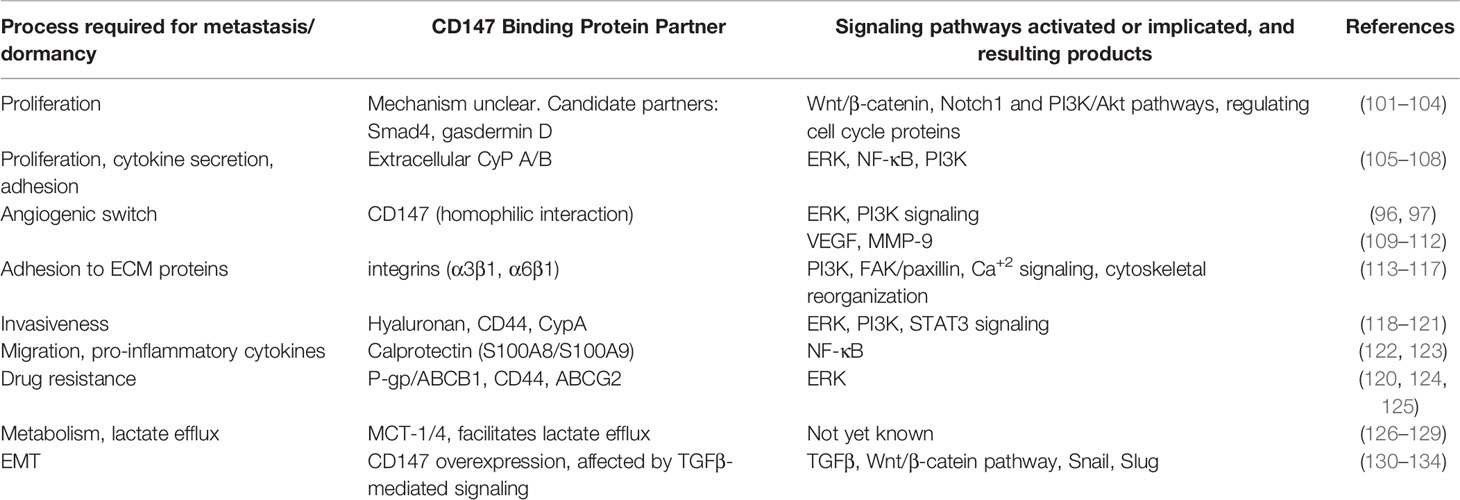
Table 1 The multifunctional CD147 protein acts as a hub protein that binds many protein partners, thus promoting processes that could lead to escape from dormancy.
Targeting CD147 by siRNA revealed much about the functions of the protein. Reduced CD147 expression inhibited the expression of MMP-9 and MMP-2 and markedly reduced the invasion of PC-3 prostate cancer cell line (142) and in the HCC cell line FHCC-98 (143). In the breast cancer MCF7 cell line it reduced both MMP-9 and VEGF expression, as well as cell proliferation, migration and invasion (93). Silencing CD147 in the leukemic U937 cell line inhibited cell viability and proliferation, and was associated with downregulation of Cyclin D1 and Cyclin E, and with increased sensitivity to the doxorubicin/Adriamycin (102). Silencing CD147 expression in the human malignant melanoma cell line A375 resulted in morphological changes, where adhesion to the collagen-coated plates, but not to fibronectin-, fibrinogen- or laminin-coated plates, was decreased, and integrin β1 expression was re-localized from the plasma membrane to the cytoplasm (114). In vivo, tumors generated with the HCC Hepa1-6 cell line that was silenced for CD147 expression, exhibited reduced tumor size and increased T cell infiltration and were sensitized to their cytotoxicity (144), suggesting that CD147 mediated escape from immune surveillance. Xenografts of the human HNSCC cell line FaDu cells demonstrated decreased tumor growth, reduced proliferation and reduced levels of MMP-9 and VEGF after CD147 was knocked down (145), and tumors generated with the mouse CD147-KD P388D1 cells resulted in smaller tumors, inhibited MMP-11 levels, and reduced ability to migrate to lymph nodes (146). These results implicate CD147 in the induction of MMPs, invasion, adhesion and proliferation, which are all critical during different stages of metastasis.
Most antibodies that were developed against CD147 remained in the pre-clinical stages. For example, a chimeric anti-CD147 antibody (CNTO3899) directed against the extracellular domains of CD147 inhibited tumor growth in a SCC-1 xenograft model of HNSCC, and even more so when combined with radiotherapy (147). Another murine monoclonal antibody (clone 1A6) that targeted human CD147 inhibited tumor growth in orthotopic model of human pancreatic cancer (148) and cutaneous squamous cell carcinoma (SCC) (149), and demonstrated reduced proliferation and migration of the tumor cell lines. Our group developed an antibody against the above-mentioned epitope in CD147, and demonstrated reduced tumor growth in two subcutaneous models of colon and renal cell carcinomas, and reduced metastasis in an orthotopic model of 4T1 mammary gland cancer (98). We also showed that the antibody reduced metastasis in a MDA-MB-231 orthotopic model (unpublished data). Moreover, active vaccination with a modified peptide against the same epitope resulted in inhibition of tumor growth and lung metastases, and prevented recurrence when challenged with repeated injections of the tumor cells (150). We showed that targeting CD147 with the antibody changed the TME by inducing a necroptotic death, which released dsRNA to the TME and shifted macrophage activation to promote anti-cancer immunity (151).
CD147 is highly expressed in hepatocellular carcinoma (HCC), which is a malignancy characterized by rapid progression, poor prognosis, and frequent tumor recurrence despite surgical treatment modalities (152, 153). In order to target CD147 in HCC, the anti-CD147 monoclonal antibody HAb18 was generated in mice immunized with hepatocellular carcinoma cells and cleaved with pepsin to remove the Fc fragment. The resulting F(ab’)2 fragment that targets the EC-1 extracellular domain of CD147 was conjugated to iodine (131I) and named Metuximab/Licartin (154, 155). In phase I/IIc clinical trials, radioimmunotherapy with 131I-metuximab was directed into the tumor and found to be safe and effective (154, 155), and subsequently was approved for clinical therapy of primary HCC by China State Food and Drug Administration. Overall, meta-analysis demonstrated that 131I-metuximab in combination with transcatheter arterial chemoembolization (TACE) or with radiofrequency ablation (RFA) exhibited high specificity and efficiency in killing tumor cells and extending survival (156–158). The use of Metuximab showed a decrease in recurrence and increased survival rate of treated patients after resection followed by orthotopic liver transplantation or in combination with radiofrequency ablation (152, 157), suggesting that the antibody can target residual metastatic cells. Mechanistically, it was suggested that the two components of 131I-metuximab - the radioactive moiety and the antibody fragment - synergize to kill HCC cells, inhibit their proliferation and reverse the EMT process (159). The same Chinese group later developed another human-mouse chimeric antibody that recognized the EC-2 domain of CD147 named metuzumab (160), and showed that it was efficient in inhibiting tumor growth in xenograft models of lung cancer human in SCID mice, and could sensitize tumor cells to chemotherapy (161). However, metuzumab has not yet reached clinical trials.
Cancer recurrence remains a major problem in cancer treatment, and is attributed to the awakening of dormant DTCs in local or remote organs. The development of metastasis is a complex, multi-step process whose molecular mechanisms are not yet fully elucidated, which is intimately associated with macrophages and other immune cells. Therefore, finding proteins that are closely implicated in the decision of DTCs to escape dormancy might provide new therapeutic targets that will prevent tumor recurrence, and it is reasonable to consider proteins that are involved in these immune interactions.
CD147 had been implicated in driving the EMT process, and has been shown to take part in the regulation of angiogenesis, proliferation, migration, invasion, and chemoresistance, all of which are processes critically required by the DTCs that escapes dormancy. Evidence that directly place CD147 as a participant in the MET process are not yet available, but its involvement in proliferation certainly suggests that this is the case. CD147 is also a mediator of tumor cell-macrophage interactions, so targeting it might disrupt those interactions and delay or prevent the escape from dormancy. Thus, targeting CD147, alone as a monotherapy or better yet in combination with radiotherapy, chemotherapy, or other immunotherapies, could potentially inhibit or prevent all processes that are essential for the escape from dormancy simultaneously.
The author confirms being the sole contributor of this work and has approved it for publication.
MR is supported by the Israel Science Foundation (ISF) grant number 2607/20 and the Israel Cancer Association (ICA) grant number (20210046).
MR is an inventor of a patent (US Grant US9688732B2, EP application EP2833900A4) related to peptide vaccination approach against CD147 mentioned in the manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Riihimäki M, Thomsen H, Sundquist K, Sundquist J, Hemminki K. Clinical Landscape of Cancer Metastases. Cancer Med (2018) 7:5534–42. doi: 10.1002/cam4.1697
2. Wells A, Clark A, Bradshaw A, Ma B, Edington H. The Great Escape: How Metastases of Melanoma, and Other Carcinomas, Avoid Elimination. Exp Biol Med (2018) 243:1245–55. doi: 10.1177/1535370218820287
3. Sauer S, Reed DR, Ihnat M, Hurst RE, Warshawsky D, Barkan D. Innovative Approaches in the Battle Against Cancer Recurrence: Novel Strategies to Combat Dormant Disseminated Tumor Cells. Front Oncol (2021) 11:659963. doi: 10.3389/fonc.2021.659963
4. Eyles J, Puaux AL, Wang X, Toh B, Prakash C, Hong M, et al. Tumor Cells Disseminate Early, But Immunosurveillance Limits Metastatic Outgrowth, in a Mouse Model of Melanoma. J Clin Invest (2010) 120:2030–9. doi: 10.1172/JCI42002
5. Bornes L, Belthier G, Van Rheenen J. Epithelial-To-Mesenchymal Transition in the Light of Plasticity and Hybrid E/M States. J Clin Med (2021) 10:2403. doi: 10.3390/jcm10112403
6. Agliano A, Calvo A, Box C. The Challenge of Targeting Cancer Stem Cells to Halt Metastasis. Semin Cancer Biol (2017) 44:25–42. doi: 10.1016/j.semcancer.2017.03.003
7. La Porta CAM, Zapperi S. Complexity in Cancer Stem Cells and Tumor Evolution: Toward Precision Medicine. Semin Cancer Biol (2017) 44:3–9. doi: 10.1016/j.semcancer.2017.02.007
8. Zhang Y, Weinberg RA. Epithelial-To-Mesenchymal Transition in Cancer: Complexity and Opportunities EMT: A Naturally Occurring Transdifferentia-Tion Program. Front Med (2018) 12:1–13. doi: 10.1007/s11684-018-0656-6
9. Sinha D, Saha P, Samanta A, Bishayee A. Emerging Concepts of Hybrid Epithelial-to-Mesenchymal Transition in Cancer Progression. Biomolecules (2020) 10:1–22. doi: 10.3390/biom10111561
10. Klein CA. Cancer Progression and the Invisible Phase of Metastatic Colonization. Nat Rev Cancer (2020) 20:681–94. doi: 10.1038/s41568-020-00300-6
11. Swierczak A, Pollard JW. Myeloid Cells in Metastasis. Cold Spring Harb Perspect Biol (2020) 10:a038026 (22 pages). doi: 10.1101/cshperspect.a038026
12. Hen O, Barkan D. Dormant Disseminated Tumor Cells and Cancer Stem/Progenitor-Like Cells: Similarities and Opportunities. Semin Cancer Biol (2020) 60:157–65. doi: 10.1016/j.semcancer.2019.09.002
13. Majidpoor J, Mortezaee K. Steps in Metastasis: An Updated Review. Med Oncol (2021) 38:1–17. doi: 10.1007/s12032-020-01447-w
14. Basu S, Dong Y, Kumar R, Jeter C, Tang DG. Slow-Cycling (Dormant) Cancer Cells in Therapy Resistance, Cancer Relapse and Metastasis. Semin Cancer Biol (2022) 78:90–103. doi: 10.1016/j.semcancer.2021.04.021
15. Ma B, Wells A, Wei L, Zheng J. Prostate Cancer Liver Metastasis: Dormancy and Resistance to Therapy. Semin Cancer Biol (2021) 71:2–9. doi: 10.1016/j.semcancer.2020.07.004
16. De Las Rivas J, Brozovic A, Izraely S, Casas-Pais A, Witz IP, Figueroa A. Cancer Drug Resistance Induced by EMT: Novel Therapeutic Strategies. Arch Toxicol (2021) 95:2279–97. doi: 10.1007/s00204-021-03063-7
17. Seo J, Ha J, Kang E, Cho S. The Role of Epithelial–Mesenchymal Transition-Regulating Transcription Factors in Anti-Cancer Drug Resistance. Arch Pharm Res (2021) 44:281–92. doi: 10.1007/s12272-021-01321-x
18. Farino CJ, Pradhan S, Slater JH. The Influence of Matrix-Induced Dormancy on Metastatic Breast Cancer Chemoresistance. ACS Appl Bio Mater (2020) 3:5832–44. doi: 10.1021/acsabm.0c00549
19. Erin N, Grahovac J, Brozovic A, Efferth T. Tumor Microenvironment and Epithelial Mesenchymal Transition as Targets to Overcome Tumor Multidrug Resistance. Drug Resist Update (2020) 53:100715. doi: 10.1016/j.drup.2020.100715
20. Chae YK, Chang S, Ko T, Anker J, Agte S, Iams W, et al. Epithelial-Mesenchymal Transition (EMT) Signature Is Inversely Associated With T-Cell Infiltration in non-Small Cell Lung Cancer (NSCLC). Sci Rep (2018) 8:2–9. doi: 10.1038/s41598-018-21061-1
21. Tomono T, Yano K, Ogihara T. Snail-Induced Epithelial-To-Mesenchymal Transition Enhances P-Gp-Mediated Multidrug Resistance in HCC827 Cells. J Pharm Sci (2017) 106:2642–9. doi: 10.1016/j.xphs.2017.03.011
22. Yochum ZA, Cades J, Wang H, Chatterjee S, Simons BW, O’Brien JP, et al. Targeting the EMT Transcription Factor TWIST1 Overcomes Resistance to EGFR Inhibitors in EGFR-Mutant Non-Small-Cell Lung Cancer. Oncogene (2019) 38:656–70. doi: 10.1038/s41388-018-0482-y
23. Zhang X, Zhang Z, Zhang Q, Zhang Q, Sun P, Xiang R, et al. ZEB1 Confers Chemotherapeutic Resistance to Breast Cancer by Activating ATM Article. Cell Death Dis (2018) 9:57–72. doi: 10.1038/s41419-017-0087-3
24. Ma B, Wells A, Clark AM. The Pan-Therapeutic Resistance of Disseminated Tumor Cells: Role of Phenotypic Plasticity and the Metastatic Microenvironment. Semin Cancer Biol (2020) 60:138–47. doi: 10.1016/j.semcancer.2019.07.021
25. Fontana F, Carollo E, Melling GE, Carter DRF. Extracellular Vesicles: Emerging Modulators of Cancer Drug Resistance. Cancers (Basel) (2021) 13:1–16. doi: 10.3390/cancers13040749
26. Low-Marchelli JM, Ardi VC, Vizcarra EA, Van Rooijen N, Quigley JP, Yang J. Twist1 Induces CCL2 and Recruits Macrophages to Promote Angiogenesis. Cancer Res (2013) 73:662–71. doi: 10.1158/0008-5472.CAN-12-0653
27. Shintani Y, Fujiwara A, Kimura T, Kawamura T, Funaki S, Minami M, et al. IL-6 Secreted From Cancer-Associated Fibroblasts Mediates Chemoresistance in NSCLC by Increasing Epithelial-Mesenchymal Transition Signaling. J Thorac Oncol (2016) 11:1482–92. doi: 10.1016/j.jtho.2016.05.025
28. Smigiel JM, Parameswaran N, Jackson MW. Potent EMT and CSC Phenotypes Are Induced by Oncostatin-M in Pancreatic Cancer. Mol Cancer Res (2017) 15:478–88. doi: 10.1158/1541-7786.MCR-16-0337
29. Goddard ET, Bozic I, Riddell SR, Ghajar CM. Dormant Tumour Cells, Their Niches and the Influence of Immunity. Nat Cell Biol (2018) 20:1240–9. doi: 10.1038/s41556-018-0214-0
30. Aqbi HF, Wallace M, Sappal S, Payne KK, Manjili MH. IFN-γ Orchestrates Tumor Elimination, Tumor Dormancy, Tumor Escape, and Progression. J Leukoc Biol (2018) 103:1219–23. doi: 10.1002/JLB.5MIR0917-351R
31. Zeltz C, Primac I, Erusappan P, Alam J, Noel A, Gullberg D. Cancer-Associated Fibroblasts in Desmoplastic Tumors: Emerging Role of Integrins. Semin Cancer Biol (2020) 62:166–81. doi: 10.1016/j.semcancer.2019.08.004
32. Hanker AB, Estrada MV, Bianchini G, Moore PD, Zhao J, Cheng F, et al. Extracellular Matrix/Integrin Signaling Promotes Resistance to Combined Inhibition of HER2 and PI3K in HER2+ Breast Cancer. Cancer Res (2017) 77:3280–92. doi: 10.1158/0008-5472.CAN-16-2808
33. Liu QL, Luo M, Huang C, Chen HN, Zhou ZG. Epigenetic Regulation of Epithelial to Mesenchymal Transition in the Cancer Metastatic Cascade: Implications for Cancer Therapy. Front Oncol (2021) 11:657546. doi: 10.3389/fonc.2021.657546
34. Korentzelos D, Clark AM. Wells A. A Perspective on Therapeutic Pan-Resistance in Metastatic Cancer. Int J Mol Sci (2020) 21:7304–26. doi: 10.3390/ijms21197304
35. Meng S, Tripathy D, Frenkel EP, Shete S, Naftalis EZ, Huth JF, et al. Circulating Tumor Cells in Patients With Breast Cancer Dormancy. Clin Cancer Res (2004) 10:8152–62. doi: 10.1158/1078-0432.CCR-04-1110
36. Gomatou G, Syrigos N, Vathiotis IA, Kotteas EA. Tumor Dormancy: Implications for Invasion and Metastasis. Int J Mol Sci (2021) 22:4862–78. doi: 10.3390/ijms22094862
37. Nabil WNN, Xi Z, Song Z, Jin L, Zhang XD, Zhou H, et al. Towards a Framework for Better Understanding of Quiescent Cancer Cells. Cells (2021) 10:562–81. doi: 10.3390/cells10030562
38. Ruth JR, Pant DK, Pan Tc, Seidel HE, Baksh SC, Keister BA, et al. Cellular Dormancy in Minimal Residual Disease Following Targeted Therapy. Breast Cancer Res (2021) 23:1–23. doi: 10.1186/s13058-021-01416-9
39. Damen MPF, van Rheenen J, Scheele CLGJ. Targeting Dormant Tumor Cells to Prevent Cancer Recurrence. FEBS J (2021) 288:6286–303. doi: 10.1111/febs.15626
40. Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of Disseminated Cancer Cell Dormancy: An Awakening Field. Nat Rev Cancer (2014) 14:611–22. doi: 10.1038/nrc3793
41. Sosa MS, Parikh F, Maia AG, Estrada Y, Bosch A, Bragado P, et al. NR2F1 Controls Tumour Cell Dormancy via SOX9- and Rarβ-Driven Quiescence Programmes. Nat Commun (2015) 6:6170–84. doi: 10.1038/ncomms7170
42. Shimizu H, Takeishi S, Nakatsumi H, Nakayama KI. Prevention of Cancer Dormancy by Fbxw7 Ablation Eradicates Disseminated Tumor Cells. JCI Insight (2019) 4:e125138. doi: 10.1172/jci.insight.125138
43. Chen Y, Gibson SB. Three Dimensions of Autophagy in Regulating Tumor Growth: Cell Survival/Death, Cell Proliferation, and Tumor Dormancy. Biochim Biophys Acta - Mol Basis Dis (2021) 1867:166265. doi: 10.1016/j.bbadis.2021.166265
44. Fiore APZP, Ribeiro P de F, Bruni-Cardoso A. Sleeping Beauty and the Microenvironment Enchantment: Microenvironmental Regulation of the Proliferation-Quiescence Decision in Normal Tissues and in Cancer Development. Front Cell Dev Biol (2018) 6:59. doi: 10.3389/fcell.2018.00059
45. Prunier C, Baker D, ten Dijke P, Ritsma L. TGF-β Family Signaling Pathways in Cellular Dormancy. Trends Cancer (2019) 5:66–78. doi: 10.1016/j.trecan.2018.10.010
46. Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, et al. The Perivascular Niche Regulates Breast Tumour Dormancy. Nat Cell Biol (2013) 15:807–17. doi: 10.1038/ncb2767
47. Hoppe-Seyler K, Bossler F, Lohrey C, Bulkescher J, Rösl F, Jansen L, et al. Induction of Dormancy in Hypoxic Human Papillomavirus-Positive Cancer Cells. Proc Natl Acad Sci USA (2017) 114:E990–8. doi: 10.1073/pnas.1615758114
48. Fluegen G, Avivar-Valderas A, Wang Y, Padgen MR, Williams JK, Nobre AR, et al. Phenotypic Heterogeneity of Disseminated Tumour Cells Is Preset by Primary Tumour Hypoxic Microenvironments. Nat Cell Biol (2017) 19:120–32. doi: 10.1038/ncb3465
49. Bragado P, Estrada Y, Parikh F, Krause S, Capobianco C, Farina HG, et al. TGF-β2 Dictates Disseminated Tumour Cell Fate in Target Organs Through TGF-β-RIII and P38α/β Signalling. Nat Cell Biol (2013) 15:1351–61. doi: 10.1038/ncb2861
50. Di Martino JS, Akhter T, Bravo-Cordero JJ. Remodeling the ECM: Implications for Metastasis and Tumor Dormancy. Cancers (Basel) (2021) 13:4916. doi: 10.3390/cancers13194916
51. Coban B, Bergonzini C, Zweemer AJM, Danen EHJ. Metastasis: Crosstalk Between Tissue Mechanics and Tumour Cell Plasticity. Br J Cancer (2021) 124:49–57. doi: 10.1038/s41416-020-01150-7
52. Barkan D, El Touny LH, Michalowski AM, Smith JA, Chu I, Davis AS, et al. Metastatic Growth From Dormant Cells Induced by a Col-I-Enriched Fibrotic Environment. Cancer Res (2010) 70:5706–16. doi: 10.1158/0008-5472.CAN-09-2356
53. Peng DH, Ungewiss C, Tong P, Byers LA, Wang J, Canales JR, et al. ZEB1 Induces LOXL2-Mediated Collagen Stabilization and Deposition in the Extracellular Matrix to Drive Lung Cancer Invasion and Metastasis. Oncogene (2017) 36:1925–38. doi: 10.1038/onc.2016.358
54. Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. Pre-Metastatic Niches: Organ-Specific Homes for Metastases. Nat Rev Cancer (2017) 17:302–17. doi: 10.1038/nrc.2017.6
55. Grigoryeva ES, Savelieva OE, Popova NO, Cherdyntseva NV, Perelmuter VM. Do Tumor Exosome Integrins Alone Determine Organotropic Metastasis? Mol Biol Rep (2020) 47:8145–57. doi: 10.1007/s11033-020-05826-4
56. Izraely S, Witz IP. Site-Specific Metastasis: A Cooperation Between Cancer Cells and the Metastatic Microenvironment. Int J Cancer (2021) 148:1308–22. doi: 10.1002/ijc.33247
57. Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, et al. Tumour Exosome Integrins Determine Organotropic Metastasis. Nature (2015) 527:329–35. doi: 10.1038/nature15756
58. Jahanban-Esfahlan R, Seidi K, Manjili MH, Jahanban-Esfahlan A, Javaheri T, Zare P. Tumor Cell Dormancy: Threat or Opportunity in the Fight Against Cancer. Cancers (Basel) (2019) 11:1207. doi: 10.3390/cancers11081207
59. Gomis RR, Gawrzak S. Tumor Cell Dormancy. Mol Oncol (2017) 11:62–78. doi: 10.1016/j.molonc.2016.09.009
60. Coffelt SB, Hughes R, Lewis CE. Tumor-Associated Macrophages: Effectors of Angiogenesis and Tumor Progression. Biochim Biophys Acta (2009) 1796:11–8. doi: 10.1016/j.bbcan.2009.02.004
61. Solinas G, Germano G, Mantovani A, Allavena P. Tumor-Associated Macrophages (TAM) as Major Players of the Cancer-Related Inflammation. J Leukoc Biol (2009) 86:1065–73. doi: 10.1189/jlb.0609385
62. Balkwill FR, Mantovani A. Cancer-Related Inflammation: Common Themes and Therapeutic Opportunities. Semin Cancer Biol (2012) 22:33–40. doi: 10.1016/j.semcancer.2011.12.005
63. Bronte V. Tumor Cells Hijack Macrophages via Lactic Acid. Immunol Cell Biol (2014) 92:647–9. doi: 10.1038/icb.2014.67
64. Lewis CE, Harney AS, Pollard JW. The Multifaceted Role of Perivascular Macrophages in Tumors. Cancer Cell (2016) 30:365. doi: 10.1016/j.ccell.2016.07.009
65. She L, Qin Y, Wang J, Liu C, Zhu G, Li G, et al. Tumor-Associated Macrophages Derived CCL18 Promotes Metastasis in Squamous Cell Carcinoma of the Head and Neck. Cancer Cell Int (2018) 18:120. doi: 10.1186/s12935-018-0620-1
66. Chen Z, Dong F, Lu J, Wei L, Tian L, Ge H, et al. Polarized M2c Macrophages Have a Promoting Effect on the Epithelial-to-Mesenchymal Transition of Human Renal Tubular Epithelial Cells. Immunobiology (2018) 223:826–33. doi: 10.1016/j.imbio.2018.08.008
67. Zeng XY, Xie H, Yuan J, Jiang XY, Yong JH, Zeng D, et al. M2-Like Tumor-Associated Macrophages-Secreted EGF Promotes Epithelial Ovarian Cancer Metastasis via Activating EGFR-ERK Signaling and Suppressing lncRNA LIMT Expression. Cancer Biol Ther (2019) 20:956–66. doi: 10.1080/15384047.2018.1564567
68. Asiry S, Kim G, Filippou PS, Sanchez LR, Entenberg D, Marks DK, et al. The Cancer Cell Dissemination Machinery as an Immunosuppressive Niche: A New Obstacle Towards the Era of Cancer Immunotherapy. Front Immunol (2021) 12:654877. doi: 10.3389/fimmu.2021.654877
69. El-Kenawi A, Hänggi K, Ruffell B. The Immune Microenvironment and Cancer Metastasis. Cold Spring Harb Perspect Med (2020) 10:1–24. doi: 10.1101/cshperspect.a037424
70. Rohan TE, Xue X, Lin HM, D’Alfonso TM, Ginter PS, Oktay MH, et al. Tumor Microenvironment of Metastasis and Risk of Distant Metastasis of Breast Cancer. J Natl Cancer Inst (2014) 106:dju136. doi: 10.1093/jnci/dju136
71. Sanchez LR, Borriello L, Entenberg D, Condeelis JS, Oktay MH, Karagiannis GS. The Emerging Roles of Macrophages in Cancer Metastasis and Response to Chemotherapy. J Leukoc Biol (2019) 106:259–74. doi: 10.1002/JLB.MR0218-056RR
72. Sharma VP, Tang B, Wang Y, Duran CL, Karagiannis GS, Xue EA, et al. Live Tumor Imaging Shows Macrophage Induction and TMEM-Mediated Enrichment of Cancer Stem Cells During Metastatic Dissemination. Nat Commun (2021) 12:1–24. doi: 10.1038/s41467-021-27308-2
73. Doak GR, Schwertfeger KL, Wood DK. Distant Relations: Macrophage Functions in the Metastatic Niche. Trends Cancer (2018) 4:445–59. doi: 10.1016/j.trecan.2018.03.011
74. Siakaeva E, Jablonska J. Pre-Metastatic Niche Formation by Neutrophils in Different Organs. Adv Exp Med Biol (2021) 1329:93–108. doi: 10.1007/978-3-030-73119-9_5
75. Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour-Mediated Upregulation of Chemoattractants and Recruitment of Myeloid Cells Predetermines Lung Metastasis. Nat Cell Biol (2006) 8:1369–75. doi: 10.1038/ncb1507
76. Qian B, Deng Y, Im JH, Muschel RJ, Zou Y, Li J, et al. istinct Macrophage Population Mediates Metastatic Breast Cancer Cell Extravasation, Establishment and Growth. PLoS One (2009) 4:16:e6562. doi: 10.1371/journal.pone.0006562
77. Ma RY, Zhang H, Li XF, Zhang CB, Selli C, Tagliavini G, et al. Monocyte-Derived Macrophages Promote Breast Cancer Bone Metastasis Outgrowth. J Exp Med (2020) 217:16:e20191820. doi: 10.1084/JEM.20191820
78. Krall JA, Reinhardt F, Mercury OA, Pattabiraman DR, Brooks MW, Dougan M, et al. The Systemic Response to Surgery Triggers the Outgrowth of Distant Immune-Controlled Tumors in Mouse Models of Dormancy. Sci Transl Med (2018) 10:1–12. doi: 10.1126/scitranslmed.aan3464
79. Baram T, Rubinstein-Achiasaf L, Ben-Yaakov H, Ben-Baruch A. Inflammation-Driven Breast Tumor Cell Plasticity: Stemness/EMT, Therapy Resistance and Dormancy. Front Oncol (2021) 10:614468:614468. doi: 10.3389/fonc.2020.614468
80. Khazali AS, Clark AM, Wells A. Inflammatory Cytokine IL-8/CXCL8 Promotes Tumour Escape From Hepatocyte-Induced Dormancy. Br J Cancer (2018) 118:566–76. doi: 10.1038/bjc.2017.414
81. Chen Q, Zhang XHF, Massagué J. Macrophage Binding to Receptor VCAM-1 Transmits Survival Signals in Breast Cancer Cells That Invade the Lungs. Cancer Cell (2011) 20:538–49. doi: 10.1016/j.ccr.2011.08.025
82. Lee CC, Lin JC, Hwang WL, Kuo YJ, Chen HK, Tai SK, et al. Macrophage-Secreted Interleukin-35 Regulates Cancer Cell Plasticity to Facilitate Metastatic Colonization. Nat Commun (2018) 9:3763. doi: 10.1038/s41467-018-06268-0
83. Ban J, Fock V, Aryee DNT, Kovar H. Mechanisms, Diagnosis and Treatment of Bone Metastases. Cells (2021) 10:1–30. doi: 10.3390/cells10112944
84. Liu Y, Lv J, Liu J, Liang X, Jin X, Xie J, et al. STAT3/p53 Pathway Activation Disrupts IFN-β-Induced Dormancy in Tumor-Repopulating Cells. J Clin Invest (2018) 128:1057–73. doi: 10.1172/JCI96329
85. Liu Y, Liang X, Yin X, Lv J, Tang K, Ma J, et al. Blockade of IDO-Kynurenine-AhR Metabolic Circuitry Abrogates IFN-γ-Induced Immunologic Dormancy of Tumor-Repopulating Cells. Nat Commun (2017) 8:1–15. doi: 10.1038/ncomms15207
86. Lan Q, Peyvandi S, Duffey N, Huang YT, Barras D, Held W, et al. Type I Interferon/IRF7 Axis Instigates Chemotherapy-Induced Immunological Dormancy in Breast Cancer. Oncogene (2019) 38:2814–29. doi: 10.1038/s41388-018-0624-2
87. Yumoto K, Eber MR, Wang J, Cackowski FC, Decker AM, Lee E, et al. Axl Is Required for TGF-β2-Induced Dormancy of Prostate Cancer Cells in the Bone Marrow. Sci Rep (2016) 6:1–16. doi: 10.1038/srep36520
88. Alečković M, McAllister SS, Polyak K. Metastasis as a Systemic Disease: Molecular Insights and Clinical Implications. Biochim Biophys Acta - Rev Cancer (2019) 1872:89–102. doi: 10.1016/j.bbcan.2019.06.002
89. Gao D, Joshi N, Choi H, Ryu S, Hahn M, Catena R, et al. Myeloid Progenitor Cells in the Premetastatic Lung Promote Metastases by Inducing Mesenchymal to Epithelial Transition. Cancer Res (2012) 72:1384–94. doi: 10.1158/0008-5472.CAN-11-2905
90. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil Extracellular Traps Produced During Inflammation Awaken Dormant Cancer Cells in Mice. Science (80-) (2018) 361:13:eaao4227. doi: 10.1126/science.aao4227
91. Grass GD, Toole BP. How, With Whom and When: An Overview of CD147-Mediated Regulatory Networks Influencing Matrix Metalloproteinase Activity. Biosci Rep (2016) 36:e00283–3. doi: 10.1042/BSR20150256
92. Liu N, Qi M, Li K, Zeng W, Li J, Yin M, et al. CD147 Regulates Melanoma Metastasis via the NFAT1-MMP-9 Pathway. Pigment Cell Melanoma Res (2020) 33:731–43. doi: 10.1111/pcmr.12886
93. Li F, Zhang J, Guo J, Jia Y, Han Y, Wang Z. RNA Interference Targeting CD147 Inhibits Metastasis and Invasion of Human Breast Cancer MCF-7 Cells by Downregulating MMP-9/VEGF Expression. Acta Biochim Biophys Sin (Shanghai) (2018) 50:676–84. doi: 10.1093/abbs/gmy062
94. Liu B, Wan Z, Sheng B, Lin Y, Fu T, Zeng Q, et al. Overexpression of EMMPRIN is Associated With Lymph Node Metastasis and Advanced Stage of Non-Small Cell Lung Cancer: A Retrospective Study. BMC Pulm Med (2017) 17:1–9. doi: 10.1186/s12890-017-0540-1
95. Kuang YH, Liu YJ, Tang LL, Wang SM, Yan GJ, Liao LQ. Plasma Soluble Cluster of Differentiation 147 Levels Are Increased in Breast Cancer Patients and Associated With Lymph Node Metastasis and Chemoresistance. Hong Kong Med J (2018) 24:252–60. doi: 10.12809/hkmj176865
96. Knutti N, Kuepper M, Friedrich K. Soluble Extracellular Matrix Metalloproteinase Inducer (EMMPRIN, EMN) Regulates Cancer-Related Cellular Functions by Homotypic Interactions With Surface CD147. FEBS J (2015) 282:4187–200. doi: 10.1111/febs.13414
97. Belton RJ, Chen L, Mesquita FS, Nowak RA. Basigin-2 Is a Cell Surface Receptor for Soluble Basigin Ligand. J Biol Chem (2008) 283:17805–14. doi: 10.1074/jbc.M801876200
98. Walter M, Simanovich E, Brod V, Lahat N, Bitterman H, Rahat MA. An Epitope-Specific Novel Anti-EMMPRIN Polyclonal Antibody Inhibits Tumor Progression. Oncoimmunology (2015) 5:12:e1078056. doi: 10.1080/2162402X.2015.1078056
99. Amit-Cohen BC, Rahat MM, Rahat MA. Tumor Cell-Macrophage Interactions Increase Angiogenesis Through Secretion of EMMPRIN. Front Physiol (2013) 4:178. doi: 10.3389/fphys.2013.00178
100. Grass GD, Tolliver LB, Bratoeva M, Toole BP. CD147, CD44, and the Epidermal Growth Factor Receptor (EGFR) Signaling Pathway Cooperate to Regulate Breast Epithelial Cell Invasiveness. J Biol Chem (2013) 288:26089–104. doi: 10.1074/jbc.M113.497685
101. Qin H, Rasul A, Li X, Masood M, Yang G, Wang N, et al. CD147-Induced Cell Proliferation Is Associated With Smad4 Signal Inhibition. Exp Cell Res (2017) 358:279–89. doi: 10.1016/j.yexcr.2017.07.003
102. Gao H, Jiang Q, Han Y, Peng J, Wang C. shRNA-Mediated EMMPRIN Silencing Inhibits Human Leukemic Monocyte Lymphoma U937 Cell Proliferation and Increases Chemosensitivity to Adriamycin. Cell Biochem Biophys (2014) 71:827–35. doi: 10.1007/s12013-014-0270-4
103. Yong Y, Zhang RY, Liu ZK, Wei D, Shang YK, Wu J, et al. Gamma-Secretase Complex-Dependent Intramembrane Proteolysis of CD147 Regulates the Notch1 Signaling Pathway in Hepatocellular Carcinoma. J Pathol (2019) 249:255–67. doi: 10.1002/path.5316
104. Peng J, Jiang H, Guo J, Huang J, Yuan Q, Xie J, et al. CD147 Expression Is Associated With Tumor Proliferation in Bladder Cancer via GSDMD. BioMed Res Int (2020) 20207:7638975. doi: 10.1155/2020/7638975
105. Sakamoto M, Miyagaki T, Kamijo H, Oka T, Boki H, Takahashi-shishido N, et al. CD147-Cyclophilin a Interactions Promote Proliferation and Survival of Cutaneous T-Cell Lymphoma. Int J Mol Sci (2021) 22:7889–902. doi: 10.3390/ijms22157889
106. Zhu D, Wang Z, Zhao JJ, Calimeri T, Meng J, Hideshima T, et al. The Cyclophilin A-CD147 Complex Promotes the Proliferation and Homing of Multiple Myeloma Cells. Nat Med (2015) 21:572–80. doi: 10.1038/nm.3867
107. Dawar FU, Xiong Y, Khattak MNK, Li J, Lin L, Mei J. Potential Role of Cyclophilin A in Regulating Cytokine Secretion. J Leukoc Biol (2017) 102:989–92. doi: 10.1189/jlb.3ru0317-090rr
108. Li M, Zhai Q, Bharadwaj U, Wang H, Li F, Fisher WE, et al. Cyclophilin A Is Overexpressed in Human Pancreatic Cancer Cells and Stimulates Cell Proliferation Through CD147. Cancer (2006) 106:2284–94. doi: 10.1002/cncr.21862
109. Zong J, Li Y, Du D, Liu Y, Yin Y. CD147 Induces Up-Regulation of Vascular Endothelial Growth Factor in U937-Derived Foam Cells Through PI3K/AKT Pathway. Arch Biochem Biophys (2016) 609:31–8. doi: 10.1016/j.abb.2016.09.001
110. Wu J, Hao ZW, Zhao YX, Yang XM, Tang H, Zhang X, et al. Full-Length Soluble CD147 Promotes MMP-2 Expression and is a Potential Serological Marker in Detection of Hepatocellular Carcinoma. J Transl Med (2014) 12:1–13. doi: 10.1186/1479-5876-12-190
111. Tang Y, Nakada MT, Rafferty P, Laraio J, McCabe FL, Millar H, et al. Regulation of Vascular Endothelial Growth Factor Expression by EMMPRIN via the PI3K-Akt Signaling Pathway. Mol Cancer Res (2006) 4:371–7. doi: 10.1158/1541-7786.MCR-06-0042
112. Zhou J, Zhu P, Jiang JL, Zhang Q, Wu ZB, Yao XY, et al. Involvement of CD147 in Overexpression of MMP-2 and MMP-9 and Enhancement of Invasive Potential of PMA-Differentiated THP-1. BMC Cell Biol (2005) 6:25–35. doi: 10.1186/1471-2121-6-25
113. Li L, Dong X, Peng F, Shen L. Integrin β1 Regulates the Invasion and Radioresistance of Laryngeal Cancer Cells by Targeting CD147. Cancer Cell Int (2018) 18:1–11. doi: 10.1186/s12935-018-0578-z
114. Nishibaba R, Higashi Y, Su J, Furukawa T, Kawai K, Kanekura T. CD147-Targeting siRNA Inhibits Cell-Matrix Adhesion of Human Malignant Melanoma Cells by Phosphorylating Focal Adhesion Kinase. J Dermatol (2012) 39:63–7. doi: 10.1111/j.1346-8138.2011.01304.x
115. Tang J, Wu YM, Zhao P, Yang XM, Jiang JL, Chen ZN. Overexpression of HAb18G/CD147 Promotes Invasion and Metastasis via α3β1 Integrin Mediated FAK-Paxillin and FAK-PI3K-Ca2+ Pathways. Cell Mol Life Sci (2008) 65:2933–42. doi: 10.1007/s00018-008-8315-8
116. Dai JY, Dou KF, Wang CH, Zhao P, Lau WB, Tao L, et al. The Interaction of HAb18G/CD147 With Integrin α6β1 and Its Implications for the Invasion Potential of Human Hepatoma Cells. BMC Cancer (2009) 9:337. doi: 10.1186/1471-2407-9-337
117. Lu M, Wu J, Hao ZW, Shang YK, Xu J, Nan G, et al. Basolateral CD147 Induces Hepatocyte Polarity Loss by E-Cadherin Ubiquitination and Degradation in Hepatocellular Carcinoma Progress. Hepatology (2018) 68:317–32. doi: 10.1002/hep.29798
118. Li L, Tang W, Wu X, Karnak D, Meng X, Thompson R, et al. HAb18G/CD147 Promotes Pstat3-Mediated Pancreatic Cancer Development via CD44s. Clin Cancer Res (2013) 19:6703–15. doi: 10.1158/1078-0432.ccr-13-0621
119. Grass GD, Dai L, Qin Z, Parsons C, Toole BP. CD147: Regulator of Hyaluronan Signaling in Invasiveness and Chemoresistance. Adv Cancer Res (2014) 123:351–73. doi: 10.1016/B978-0-12-800092-2.00013-7
120. Zhou S, Liao L, Chen C, Zeng W, Liu S, Su J, et al. CD147 Mediates Chemoresistance in Breast Cancer via ABCG2 by Affecting Its Cellular Localization and Dimerization. Cancer Lett (2013) 337:285–92. doi: 10.1016/j.canlet.2013.04.025
121. Dai L, Guinea MC, Slomiany MG, Bratoeva M, Grass GD, Tolliver LB, et al. CD147-Dependent Heterogeneity in Malignant and Chemoresistant Properties of Cancer Cells. Am J Pathol (2013) 182:577–85. doi: 10.1016/j.ajpath.2012.10.011
122. Hibino T, Sakaguchi M, Miyamoto S, Yamamoto M, Motoyama A, Hosoi J, et al. S100A9 Is a Novel Ligand of EMMPRIN That Promotes Melanoma Metastasis. Cancer Res (2013) 73:172–83. doi: 10.1158/0008-5472.CAN-11-3843
123. Sakaguchi M, Yamamoto M, Miyai M, Maeda T, Hiruma J, Murata H, et al. Identification of an S100A8 Receptor Neuroplastin-β and Its Heterodimer Formation With EMMPRIN. J Invest Dermatol (2016) 136:2240–50. doi: 10.1016/j.jid.2016.06.617
124. Zhao S, Chen C, Liu S, Zeng W, Su J, Wu L, et al. CD147 Promotes MTX Resistance by Immune Cells Through Up-Regulating ABCG2 Expression and Function. J Dermatol Sci (2013) 70:182–9. doi: 10.1016/j.jdermsci.2013.02.005
125. Li QQ, Wang WJ, Xu J, Cao XX, Chen Q, Yang JM, et al. Involvement of CD147 in Regulation of Multidrug Resistance to P-Gp Substrate Drugs and In Vitro invasion in breast cancer cells. Cancer Sci (2007) 98:1064–9. doi: 10.1111/j.1349-7006.2007.00487.x
126. Li X, Yu X, Dai D, Song X, Xu W. The Altered Glucose Metabolism in Tumor and a Tumor Acidic Microenvironment Associated With Extracellular Matrix Metalloproteinase Inducer and Monocarboxylate Transporters. Oncotarget (2016) 7:23141–55. doi: 10.18632/oncotarget.8153
127. Schneiderhan W, Scheler M, Holzmann KH, Marx M, Gschwend JE, Bucholz M, et al. CD147 Silencing Inhibits Lactate Transport and Reduces Malignant Potential of Pancreatic Cancer Cells in In Vivo and In Vitro Models. Gut (2009) 58:1391–8. doi: 10.1136/gut.2009.181412
128. Pinheiro C, Longatto-Filho A, Pereira SMM, Etlinger D, Moreira MAR, Jubé LF, et al. Monocarboxylate Transporters 1 and 4 Are Associated With CD147 in Cervical Carcinoma. Dis Markers (2009) 26:97–103. doi: 10.3233/DMA-2009-0596
129. Kendrick AA, Schafer J, Dzieciatkowska M, Nemkov T, D’Alessandro A, Neelakantan D, et al. CD147: A Small Molecule Transporter Ancillary Protein at the Crossroad of Multiple Hallmarks of Cancer and Metabolic Reprogramming. Oncotarget (2017) 8:6742–62. doi: 10.18632/oncotarget.14272
130. Suzuki S, Toyoma S, Tsuji T, Kawasaki Y, Yamada T. CD147 Mediates Transforming Growth Factor−β1−Induced Epithelial−Mesenchymal Transition and Cell Invasion in Squamous Cell Carcinoma of the Tongue. Exp Ther Med (2019) 17:2855–60. doi: 10.3892/etm.2019.7230
131. Ru NY, Wu J, Chen ZN, Bian H. HAb18G/CD147 is Involved in TGF-β-Induced Epithelial-Mesenchymal Transition and Hepatocellular Carcinoma Invasion. Cell Biol Int (2015) 39:44–51. doi: 10.1002/cbin.10341
132. Wang C, Zhang J, Fok KL, Tsang LL, Ye M, Liu J, et al. CD147 Induces Epithelial-To-Mesenchymal Transition by Disassembling Cellular Apoptosis Susceptibility Protein/E-Cadherin/β-Catenin Complex in Human Endometriosis. Am J Pathol (2018) 188:1597–607. doi: 10.1016/j.ajpath.2018.03.004
133. Wu J, Lu M, Li Y, Shang YK, Wang SJ, Meng Y, et al. Regulation of a TGF-β1-CD147 Self-Sustaining Network in the Differentiation Plasticity of Hepatocellular Carcinoma Cells. Oncogene (2016) 35:5468–79. doi: 10.1038/onc.2016.89
134. Chen Y, Tan W, Wang C. Tumor-Associated Macrophage-Derived Cytokines Enhance Cancer Stem-Like Characteristics Through Epithelial–Mesenchymal Transition. Onco Targets Ther (2018) 11:3817–26. doi: 10.2147/OTT.S168317
135. Toole BP. The CD147-HYALURONAN Axis in Cancer. Anat Rec (2020) 303:1573–83. doi: 10.1002/ar.24147
136. Dawar FU, Wu J, Zhao L, Khattak MNK, Mei J, Lin L. Updates in Understanding the Role of Cyclophilin A in Leukocyte Chemotaxis. J Leukoc Biol (2017) 101:823–6. doi: 10.1189/jlb.3RU1116-477R
137. Cui J, Huang W, Wu B, Jin J, Jing L, Shi WP, et al. N-Glycosylation by N-Acetylglucosaminyltransferase V Enhances the Interaction of CD147/basigin With Integrin β1 and Promotes HCC Metastasis. J Pathol (2018) 245:41–52. doi: 10.1002/path.5054
138. Li Y, Wu J, Song F, Tang J, Wang SJ, Yu XL, et al. Extracellular Membrane-Proximal Domain of HAb18G/CD147 Binds to Metal Ion-Dependent Adhesion Site (MIDAS) Motif of Integrin ??1 to Modulate Malignant Properties of Hepatoma Cells. J Biol Chem (2012) 287:4759–72. doi: 10.1074/jbc.M111.277699
139. Suzuki S, Honda K, Nanjo H, Iikawa N, Tsuji T, Kawasaki Y, et al. CD147 Expression Correlates With Lymph Node Metastasis Int1-T2 Squamous Cell Carcinoma of the Tongue. Oncol Lett (2017) 14:4670–6. doi: 10.3892/ol.2017.6808
140. Sidhu SS, Nawroth R, Retz M, Lemjabbar-Alaoui H, Dasari V, Basbaum C. EMMPRIN Regulates the Canonical Wnt/B-Catenin Signaling Pathway, a Potential Role in Accelerating Lung Tumorigenesis. Oncogene (2010) 29:4145–56. doi: 10.1038/onc.2010.166
141. Zhang JJ, Song CG, Dai JM, Zhang XQ, Lin P, Li L, et al. Inhibition of Mu-Opioid Receptor Suppresses Proliferation of Hepatocellular Carcinoma Cells via CD147-P53-MAPK Cascade Signaling Pathway. Am J Transl Res (2021) 13:3967–86.
142. Wang L, Wu G, Yu L, Yuan J, Fang F, Zhai Z, et al. Inhibition of CD147 Expression Reduces Tumor Cell Invasion in Human Prostate Cancer Cell Line via RNA Interference. Cancer Biol Ther (2006) 5:608–14. doi: 10.4161/cbt.5.6.2661
143. Xu HY, Qian AR, Shang P, Xu J, Kong LM, Bian HJ, et al. siRNA Targeted Against HAb18G/CD147 Inhibits MMP-2 Secretion, Actin and FAK Expression in Hepatocellular Carcinoma Cell Line via ERK1/2 Pathway. Cancer Lett (2007) 247:336–44. doi: 10.1016/j.canlet.2006.05.017
144. Ren Yx, Wang Sj, Fan Jh, Sun Sj, Li X, Padhiar AA, et al. CD147 Stimulates Hepatoma Cells Escaping From Immune Surveillance of T Cells by Interaction With Cyclophilin A. BioMed Pharmacother (2016) 80:289–97. doi: 10.1016/j.biopha.2016.03.036
145. Newman JR, Bohannon Ia, Zhang W, Skipper JB, Grizzle WE, Rosenthal EL. Modulation of Tumor Cell Growth In Vivo by Extracellular Matrix Metalloprotease Inducer. Arch Otolaryngol - Head Neck Surg (2008) 134:1218–24. doi: 10.1001/archotol.134.11.1218.Modulation
146. Jia L, Wei W, Cao J, Xu H, Miao X, Zhang J. Silencing CD147 Inhibits Tumor Progression and Increases Chemosensitivity in Murine Lymphoid Neoplasm P388D1 Cells. Ann Hematol (2009) 88:753–60. doi: 10.1007/s00277-008-0678-2
147. Dean NR, Newman JR, Helman EE, Zhang W, Safavy S, Weeks DM, et al. Anti-EMMPRIN Monoclonal Antibody as a Novel Agent for Therapy of Head and Neck Cancer. Clin Cancer Res (2009) 15:4058–65. doi: 10.1158/1078-0432.CCR-09-0212
148. Sweeny L, Hartman YE, Zinn KR, Prudent JR, Marshall DJ, Shekhani MS, et al. A Novel Extracellular Drug Conjugate Significantly Inhibits Head and Neck Squamous Cell Carcinoma. Oral Oncol (2013) 49:991–7. doi: 10.1016/j.oraloncology.2013.07.006
149. Frederick JW, Sweeny L, Hartman Y, Rosenthal EL. Epidermal Growth Factor Receptor Inhibition by Anti-CD147 Therapy in Cutaneous Squamous Cell Carcinoma. Head Neck (2016) 38:247–52. doi: 10.1002/HED
150. Simanovich E, Brod V, Rahat MM, Drazdov E, Miriam W, Shakya J, et al. Inhibition of Tumor Growth and Metastasis by EMMPRIN Multiple Antigenic Peptide (MAP) Vaccination Is Mediated by Immune Modulationtle. Oncoimmunology (2016) 6:e1261778. doi: 10.1080/2162402X.2016.1261778
151. Hijaze N, Ledersnaider M, Simanovich E, Kassem S, Rahat MA. Inducing Regulated Necrosis and Shifting Macrophage Polarization With Anti-EMMPRIN Antibody (161-pAb) and Complement Factors. J Leukoc Biol (2020) 110:343–56. doi: 10.1002/JLB.3A0520-333R
152. Xu J, Shen ZY, Chen XG, Zhang Q, Bian HJ, Zhu P, et al. A Randomized Controlled Trial of Licartin for Preventing Hepatoma Recurrence After Liver Transplantation. Hepatology (2007) 45:269–76. doi: 10.1002/hep.21465
153. Vogel A, Saborowski A. Adjuvant 131I-Metuximab in Hepatocellular Carcinoma: A New Option for an Old Drug? Lancet Gastroenterol Hepatol (2020) 5:517–9. doi: 10.1016/S2468-1253(20)30004-2
154. Zhang Z, Bian H, Feng Q, Mi L, Mo T, Kuang A, et al. Biodistribution and Localization of Iodine-131-Labeled Metuximab KEY WORDS RIB. Cancer Biol Therher (2006) 5:318–22. doi: 10.4161/cbt.5.3.2431
155. Chen ZN, Mi L, Xu J, Song F, Zhang Q, Zhang Z, et al. Targeting Radioimmunotherapy of Hepatocellular Carcinoma With Iodine (131I) Metuximab Injection: Clinical Phase I/II Trials. Int J Radiat Oncol Biol Phys (2006) 65:435–44. doi: 10.1016/j.ijrobp.2005.12.034
156. He Q, Lu WS, Liu Y, Guan YS, Kuang AR. 131I-Labeled Metuximab Combined With Chemoembolization for Unresectable Hepatocellular Carcinoma. World J Gastroenterol (2013) 19:9104–10. doi: 10.3748/wjg.v19.i47.9104
157. Bian H, Zheng JS, Nan G, Li R, Chen C, Hu CX, et al. Randomized Trial of [131I] Metuximab in Treatment of Hepatocellular Carcinoma After Percutaneous Radiofrequency Ablation. J Natl Cancer Inst (2014) 106:1–5. doi: 10.1093/jnci/dju239
158. Fan W, Wu Y, Lu M, Yao W, Cui W, Zhao Y, et al. A Meta-Analysis of the Efficacy and Safety of Iodine [131I] Metuximab Infusion Combined With TACE for Treatment of Hepatocellular Carcinoma. Clin Res Hepatol Gastroenterol (2019) 43:451–9. doi: 10.1016/j.clinre.2018.09.006
159. Wu L, Sun B, Lin X, Liu C, Qian H, Chen L, et al. I131 Reinforces Antitumor Activity of Metuximab by Reversing Epithelial–Mesenchymal Transition via VEGFR-2 Signaling in Hepatocellular Carcinoma. Genes to Cells (2018) 23:35–45. doi: 10.1111/gtc.12545
160. Zhang Z, Zhang Y, Sun Q, Feng F, Huhe M, Mi L, et al. Preclinical Pharmacokinetics, Tolerability, and Pharmacodynamics of Metuzumab, a Novel CD147 Human-Mouse Chimeric and Glycoengineered Antibody. Mol Cancer Ther (2015) 14:162–73. doi: 10.1158/1535-7163.mct-14-0104
Keywords: metastatic cascade, epithelial-to-mesenchymal transition (EMT), disseminated tumor cell (DTC), dormancy, tumor-associated macrophages (TAMs), CD147/EMMPRIN
Citation: Rahat MA (2022) Mini-Review: Can the Metastatic Cascade Be Inhibited by Targeting CD147/EMMPRIN to Prevent Tumor Recurrence? Front. Immunol. 13:855978. doi: 10.3389/fimmu.2022.855978
Received: 16 January 2022; Accepted: 07 March 2022;
Published: 28 March 2022.
Edited by:
Catherine Sautes-Fridman, U1138 Centre de Recherche des Cordeliers (CRC) (INSERM), FranceReviewed by:
Su Yin Lim, Macquarie University, AustraliaCopyright © 2022 Rahat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michal A. Rahat, bXJhaGF0QHRlY2huaW9uLmFjLmls; cmFoYXRfbWlraUBjbGFsaXQub3JnLmls
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.