 John C. Flickinger Jr1
John C. Flickinger Jr1 Jagmohan Singh1
Jagmohan Singh1 Yanki Yarman1
Yanki Yarman1 Robert D. Carlson1
Robert D. Carlson1 Joshua R. Barton1
Joshua R. Barton1 Scott A. Waldman1,2
Scott A. Waldman1,2 Adam E. Snook1,2,3*
Adam E. Snook1,2,3*- 1Department of Pharmacology & Experimental Therapeutics, Thomas Jefferson University, Philadelphia, PA, United States
- 2Sidney Kimmel Cancer Center, Philadelphia, PA, United States
- 3Department of Microbiology & Immunology, Thomas Jefferson University, Philadelphia, PA, United States
The Gram-positive bacterium Listeria monocytogenes (Lm) is an emerging platform for cancer immunotherapy. To date, over 30 clinical trials have been initiated testing Lm cancer vaccines across a wide variety of cancers, including lung, cervical, colorectal, and pancreatic. Here, we assessed the immunogenicity of an Lm vaccine against the colorectal tumor antigen GUCY2C (Lm-GUCY2C). Surprisingly, Lm-GUCY2C vaccination did not prime naïve GUCY2C-specific CD8+ T-cell responses towards the dominant H-2Kd-restricted epitope, GUCY2C254-262. However, Lm-GUCY2C produced robust CD8+ T-cell responses towards Lm-derived peptides suggesting that GUCY2C254-262 peptide may be subdominant to Lm-derived peptides. Indeed, incorporating immunogenic Lm peptides into an adenovirus-based GUCY2C vaccine previously shown to induce robust GUCY2C254-262 immunity completely suppressed GUCY2C254-262 responses. Comparison of immunogenic Lm-derived peptides to GUCY2C254-262 revealed that Lm-derived peptides form highly stable peptide-MHC complexes with H-2Kd compared to GUCY2C254-262 peptide. Moreover, amino acid substitution at a critical anchoring residue for H-2Kd binding, producing GUCY2CF255Y, significantly improved stability with H-2Kd and rescued GUCY2C254-262 immunogenicity in the context of Lm vaccination. Collectively, these studies suggest that Lm antigens may compete with and suppress the immunogenicity of target vaccine antigens and that use of altered peptide ligands with enhanced peptide-MHC stability may be necessary to elicit robust immune responses. These studies suggest that optimizing target antigen competitiveness with Lm antigens or alternative immunization regimen strategies, such as prime-boost, may be required to maximize the clinical utility of Lm-based vaccines.
Introduction
Due to the unprecedented success of immune checkpoint inhibitors (ICIs) and adoptive cell therapy, immunotherapy has established itself as a pillar of cancer management alongside chemotherapy, surgery, targeted therapies, and radiotherapy (1, 2). While ICIs have been practice-changing, only 20% of patients respond to ICI therapy (3, 4). Notably, responsiveness to ICI therapy is associated with immunologically “hot” tumors characterized by significant immune cell infiltration (5, 6). In this context, there has been renewed interest in utilizing cancer vaccines as agents that expand tumor-specific T cells and promote T-cell infiltration into tumor microenvironments, working synergistically with ICI therapy (3, 4, 7). However, methods of cancer vaccination are highly variable, including peptide, nucleic acid, microbial-based, and others, with no consensus on optimal vaccine platforms (8). Therefore, a greater understanding of vaccine vector biology is urgently needed.
The Gram-positive bacterium Listeria monocytogenes (Lm) is an emerging platform for cancer immunotherapy. Lm is an attractive vector due to its tropism for antigen-presenting cells, leading to potent CD8+ T-cell immunity (9) and its ability to engage multiple aspects of the innate immune system and remodel immunosuppressive microenvironments (10, 11). Thus, multiple recombinant Lm vaccines secreting tumor antigens capable of inducing antitumor immunity have been explored (12). Moreover, attenuated Lm strains with favorable safety profiles have been developed for clinical testing. To date, over 30 clinical trials have been initiated testing attenuated Lm-based vaccines for cancers (13), originating from lung (14), cervix (15), colorectum (16), pancreas (17), and others.
Here, we examined the immunogenicity of an Lm-based vaccine expressing the colorectal cancer antigen guanylyl cyclase C (Lm-GUCY2C). Surprisingly, Lm-GUCY2C failed to prime GUCY2C-specific immune responses in mice, despite generating robust Lm-specific immunity. Studies revealed competition with immunodominant Lm-derived CD8+ T-cell epitopes as the underlying mechanism, which could be reversed by enhancing the MHC-binding affinity of GUCY2C-derived CD8+ T-cell epitopes. These studies reveal important mechanisms restricting the efficacy of Lm-based vaccines and novel approaches to Lm design and use to enhance that efficacy, particularly in the context of self/tumor-associated antigens.
Materials and Methods
Vaccines and Peptides
The live-attenuated double-deleted (LADD) strain of Listeria monocytogenes (Lm) containing deletions in virulence factors internalin B and actA (ΔactAΔinlB) (18) was obtained from ATCC and served as the parental strain for all Lm vaccines in this study. Recombinant Lm-GUCY2C and Lm-LacZ were generated by gene synthesis of the codon-optimized mouse GUCY2C extracellular domain (GUCY2C23-429) or β-galactosidase618-1024, respectively, in-frame with a modified version of the first 100 amino acids of actA protein (called ActAN100* throughout) and the Syn18x5 enhancer sequence under control of the actA promoter (19). All other Lm constructs were similarly cloned. The genetic sequence was cloned into the pPL2 plasmid (kindly provided by Richard Calendar, UC Berkeley) and integrated into the Lm chromosome as previously described (20). Successful integration was confirmed by DNA sanger sequencing. Recombinant Lm was grown in brain-heart infusion (BHI) broth (Fisher Scientific) to OD600 of about 1.0 and stored as aliquots at -80°C until the day of vaccination (21). For in vitro validation studies, the mouse macrophage cell line J774A.1 cultured in DMEM supplemented with 10% FBS was infected at a 10:1 multiplicity of infection with control or GUCY2C Lm. After a 1 h incubation at 37°C, cells were washed 2x in PBS, resuspended in media containing 10 ug/mL gentamicin to eliminate free extracellular bacteria, and incubated an additional 5 h at 37°C. For immunofluorescence studies, Lm was labeled prior to infection by incubating with 2 mM CellTracker Red CMPTPX dye for 10 min at 37°C, and GUCY2C protein was stained using the anti-GUCY2C monoclonal antibody MS20 (22) followed by incubation with a peroxidase-conjugated 2° antibody for subsequent tyramide-FITC amplification. For western blot studies, protein was extracted from cells using M-PER reagent (Pierce) supplemented with protease inhibitors. GUCY2C protein was stained using MS20 (22) and p60 was stained using the anti-p60 monoclonal antibody p6017 (AdipoGen).
Replication-deficient adenovirus serotype 5 (Ad5) expressing mouse GUCY2C1-429 fused to the influenza HA107-119 CD4+ T-cell epitope known as S1 (Ad5-GUCY2C) was used as a positive control for generating GUCY2C-specific CD8+ T-cell responses (23). The adenoviral vaccine used in this study was produced by the Baylor College of Medicine in the Cell and Gene Therapy Vector Development Lab and certified to be negative for replication-competent adenovirus, mycoplasma, and host cell DNA contamination. All peptides used for experiments were custom synthesized by ThermoFisher Scientific and purified by HPLC to >95% purity.
Mice and Immunizations
Studies employed BALB/cJ mice (Jackson Laboratories). For Lm vaccinations, Lm aliquots were thawed on the day of use, incubated at 37°C for 60 min in BHI broth, washed 2x in PBS, and resuspended to a concentration of 5x107 colony-forming units (CFU)/mL in PBS. Mice were immunized intraperitoneally (i.p.) with 107 CFU of recombinant Lm vaccine. For Ad5 vaccinations, mice were immunized intramuscularly (i.m.) with 1010 vp of Ad5-GUCY2C delivered as two 50 uL injections, one in each hind limb. All studies employed a single administration of Lm or Ad5 priming followed by Lm boosting (prime-boost studies indicated in Figure 2). For priming experiments, mice were sacrificed 7 d following Lm vaccination. For prime-boost immunizations, Ad5 and Lm vaccines were administered 21 d apart, and animals were euthanized 6 d after final vaccination.
IFNγ ELISpot Assay
Enzyme-Linked Immunospot Assay (ELISpot) was performed using a mouse interferon-γ (IFNγ) single color ELISpot kit (Cellular Technology Limited) according to the manufacturer’s protocol. The evening before an experiment, 96-well plates were coated and incubated overnight at 4°C with IFNγ capture antibody. After splenocytes were isolated from immunized mice on the following day, plates were washed with PBS. Splenocytes were then plated in triplicate in a 0.1% DMSO solution of CTL-TEST medium (Cellular Technology Limited) with 10 ug/mL of peptide and incubated at 37°C for 24 h. For TCR avidity studies, splenocytes were pulsed with decreasing concentrations of GUCY2C254-262 peptide (10 ug/mL to 3 pg/mL) (23–25). The next day, splenocytes were removed, and development reagents were added to detect IFNγ-producing spot-forming cells (SFCs). The number of SFCs/well was determined using the SmartCount and Autogate functions of an ImmunoSpot S6 Universal Analyzer (Cellular Technology Limited). Peptide-specific responses were calculated by subtracting mean spot counts of 0.1% DMSO wells from peptide-pulsed wells.
MHC Class I Stability Assay
The TAP-deficient cell line RMA-S expressing the MHC class I molecule H-2Kd (26) was kindly provided by Dr. Sean Murphy (University of Washington) and was used for peptide-MHC stability experiments. As previously described (27), RMA-S-H-2Kd cells were incubated overnight at 26°C with 30 ug/mL of each peptide. In the morning, cells were incubated 2 h at 37°C, washed 3x with PBS to remove unbound peptide, and incubated an additional 2, 4, or 6 h at 37°C. Cells were then stained with anti-H-2Kd-PE antibody (Invitrogen, Clone SF1-1.1.1), and surface H-2Kd was quantified as mean fluorescence intensity (MFI) by flow cytometry. The percent change in surface peptide-H-2Kd complexes was calculated using the formula, (28). The t1/2 for the peptide-MHC complex was calculated by nonlinear regression.
Tumor Studies
The murine CT26 colorectal cancer cell line expressing mouse GUCY2C and luciferase (23) was used for in vivo tumor studies. Seven days after final immunizations, mice received 5x105 CT26 cells via i.v. tail vein injection to model metastatic colorectal cancer recurrence in the lungs. Tumor burden was quantified by subcutaneous injection of 3.75 mg of D-luciferin potassium salt (Gold Biotechnologies) in PBS solution. Following an 8 min incubation, mice were imaged with a ten-second exposure using a Caliper IVIS Lumina XR imaging station (PerkinElmer). Total radiance (photons/second) was quantified by Living Image In Vivo Imaging Software (PerkinElmer).
Results
GUCY2C254-262 Is Subdominant to Lm-Derived Epitopes
Lm-GUCY2C was produced using a construct composed of the actA promoter, an enhancer, and a fusion protein of a modified version of the first 100 amino acids of actA (ActAN100*) and residues 23-429 of murine GUCY2C (Figure 1A and Supplementary Figure 1). Lm-GUCY2C successfully produces GUCY2C protein upon infection of J744A.1 macrophages, detected in the supernatant (Figure 1A) and in the cells (Figure 1B). However, while Ad5-GUCY2C induced robust GUCY2C-specific CD8+ T-cell responses, Lm-GUCY2C vaccination failed to prime CD8+ T cell responses to GUCY2C (Figure 1C). In contrast, Lm-GUCY2C induced robust Lm-specific CD8+ T-cell responses against multiple H-2Kd-restricted Lm epitopes derived from Listeriolysin-O (LLO), p60, and metalloprotease (Mpl) antigens (Figure 1D). Moreover, Lm-GUCY2C successfully boosted GUCY2C-specific CD8+ T-cell responses that were primed 21 days earlier with Ad5-GUCY2C (Figure 1E). Furthermore, in the context of a multi-epitope Lm vaccine that included epitopes from GUCY2C, β-galactosidase, and Ad5, robust responses were produced against the highly immunogenic foreign antigens β-galactosidase and Ad5, but not the self-antigen GUCY2C (Figure 1F). Collectively, these data suggest that the only GUCY2C CD8+ T-cell epitope in BALB/c mice (GUCY2C254-262) (29) is processed and presented upon Lm-GUCY2C administration but is unable to prime naive GUCY2C254-262-specific CD8+ T-cell responses. In the context of primarily T cells with weak TCR-peptide-MHC interactions escaping self-tolerance (30, 31), we hypothesized that the GUCY2C254-262 epitope might be subdominant in the context of live Lm vaccine vectors. To confirm that Lm epitopes limit GUCY2C254-262 immunogenicity, we utilized the Ad5-GUCY2C vaccine template, which produces robust GUCY2C254-262 responses (29, 32–34), and created an Ad5 vaccine composed of GUCY2C254-262, the S1 CD4+ helper T-cell epitope used in Ad5-GUCY2C vaccines (35), and immunogenic epitopes from LLO, p60 and Mpl (Figure 1G). Strikingly, this construct induced robust responses directed to the S1 CD4+ T-cell epitope and Lm-derived CD8+ T-cell epitopes but failed to induce responses to GUCY2C254-262 (Figure 1G).
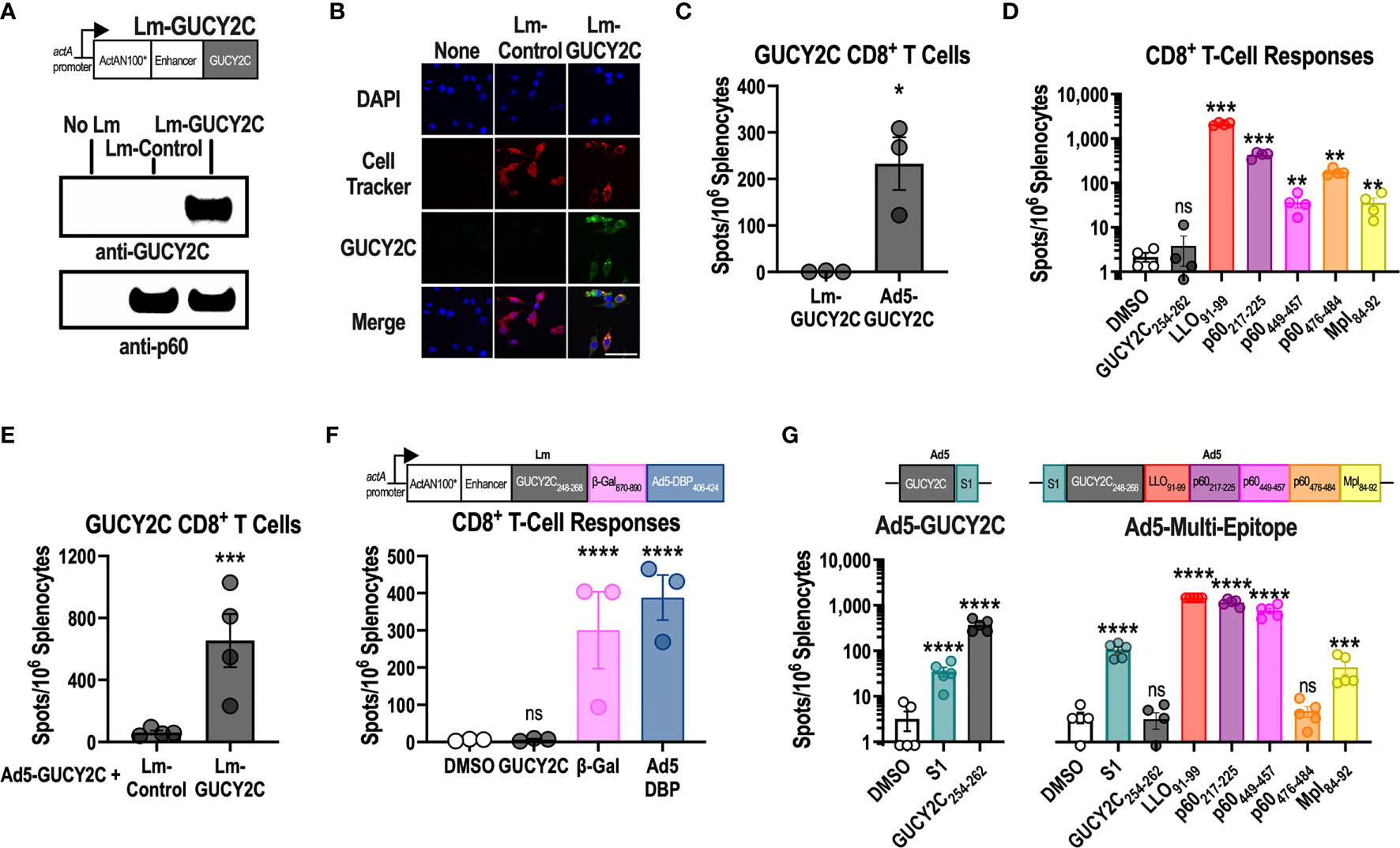
Figure 1 GUCY2C254-262 peptide is subdominant to Lm-derived peptides. (A) Lm-GUCY2C secretes a fusion protein comprised of ActAN100*, an enhancer sequence, and mouse GUCY2C23-429 under control of the actA promoter. (A, B) J774A.1 macrophages were uninfected or infected with Lm-Control or Lm-GUCY2C at a 10:1 MOI for 6 h at 37°C. GUCY2C fusion protein was detected by (A) western blot and (B) immunofluorescence. Scale bars in B are 50 um. (C, D) BALB/c mice (n=3-4/group) were immunized intraperitoneally (i.p.) with 107 colony-forming units (CFU) of recombinant Lm secreting GUCY2C (Lm-GUCY2C) or intramuscularly (i.m.) with 1010 vp of Ad5-GUCY2C. (E) Animals received 107 CFU of Lm-GUCY2C or Lm-Control 21 d after priming with 1010 vp of Ad5-GUCY2C. (C–E) Fourteen days after Ad5-GUCY2C immunization or 6-7 d after Lm administrations, mice were euthanized, and splenocytes were collected to quantify GUCY2C254-262 (C, E) or Lm-specific (D) T-cell responses, quantified by IFNγ ELISpot. (F) BALB/c mice (n=3) were immunized with an Lm containing a multi-epitope construct composed of GUCY2C, β-Gal, and Ad5 DBP epitopes (as depicted), and those responses were quantified 7 d later by IFNγ ELISpot. (G) BALB/c mice (n=5/group) were immunized with Ad5-GUCY2C (left) or a recombinant Ad5 containing S1, GUCY2C254-262, and the H-2Kd-restricted Lm epitopes as depicted (right). Responses were quantified 14 d later by IFNγ ELISpot. Statistical comparisons were made by unpaired T test (C, E) or one-way ANOVA with Bonferroni correction for multiple comparisons of epitope-specific response to control DMSO wells (D, F, G). Error bars indicate mean +/- SEM. Symbols indicate individual animals. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant.
Enhancing GUCY2C254-262 Quantity, Degradation, or Processing Does Not Overcome Immunodominance
During microbial infections, antigens are degraded into short peptide fragments and loaded onto MHC molecules for presentation to T cells (36). Interestingly, while a single bacterium expresses thousands of potentially antigenic proteins (37), the immune system concentrates T-cell responses towards only a few “dominant” antigenic sequences. In contrast, sequences that induce T-cell responses to a lesser, or undetectable, degree are termed “subdominant.” Because GUCY2C254-262 is subdominant to Lm epitopes in Lm-GUCY2C, we hypothesized that it might be possible to rescue Lm-GUCY2C immunogenicity by increasing the relative quantity of GUCY2C epitopes and produced a variety of Lm vaccines expressing GUCY2C in different contexts to test that hypothesis (Figure 2A). It has been reported that increasing the degradation rate of an antigen can enhance its immunogenicity (38–40). Thus, we mutated the N-terminal residue following the actA signal sequence (A30R) to destabilize the actA-GUCY2C fusion protein or incorporated ubiquitin into our actA-GUCY2C fusion protein (Ub-GUCY2C) to promote its degradation (41). We then cloned these constructs into Lm, generating the vaccines Lm-GUCY2CA30R and Lm-Ub-GUCY2C, respectively (Figure 2A). While both constructs resulted in increased degradation and much lower steady-state levels of GUCY2C (Figure 2A), vaccination with these constructs did not improve GUCY2C254-262 immunogenicity (Figure 2B). Recently, a group developing an Lm vaccine against the EGFRvIII variant reported that EGFRvIII immunogenicity was enhanced when repeating copies of epitope were encoded and flanked by sequences predicted to facilitate proteasomal cleavage (42). To similarly increase the copy number and improve processing of GUCY2C254-262 peptide, we used a similar design with 5 copies flanked by sequences designed to facilitate antigen processing (Lm-GUCY2Cx5; Figure 2C). However, Lm-GUCY2Cx5 similarly failed to induce GUCY2C254-262-specific CD8+ T-cell responses (Figure 2C). Finally, we altered the promoter and fusion protein by which GUCY2C is expressed. Lm vaccines advanced into clinical trials have predominantly expressed vaccine antigen under the hly promoter fused to LLO or under the actA promoter fused to actA (13). Importantly, antigen expression kinetics during infection also influences antigen immunogenicity (43), and hly and actA promoters are distinctly regulated (44). Moreover, fusion to LLO also has been reported to enhance subdominant epitope immunogenicity in some cases (45). Thus, we fused GUCY2C to LLO and expressed it under the hly promoter (Lm-LLO-GUCY2C; Figure 2D); however, Lm-LLO-GUCY2C also failed to prime GUCY2C-specific CD8+ T-cell responses (Figure 2D). Notably, while these Lm-GUCY2C variants were unable to prime responses, all were capable of boosting GUCY2C-specific memory CD8+ T-cell responses in mice primed with Ad5-GUCY2C (Figure 2E, F). Finally, recent studies report that subdominant T-cell clones express high levels of PD-1 upon activation, resulting in cell death, and that administration of PD-1-blocking antibody during vaccination significantly improves the expansion of subdominant T-cell clones (46, 47). However, treatment with anti-PD-1 antibody during Lm-GUCY2C immunization did not produce GUCY2C-specific CD8+ T-cell priming (Supplementary Figure 2).
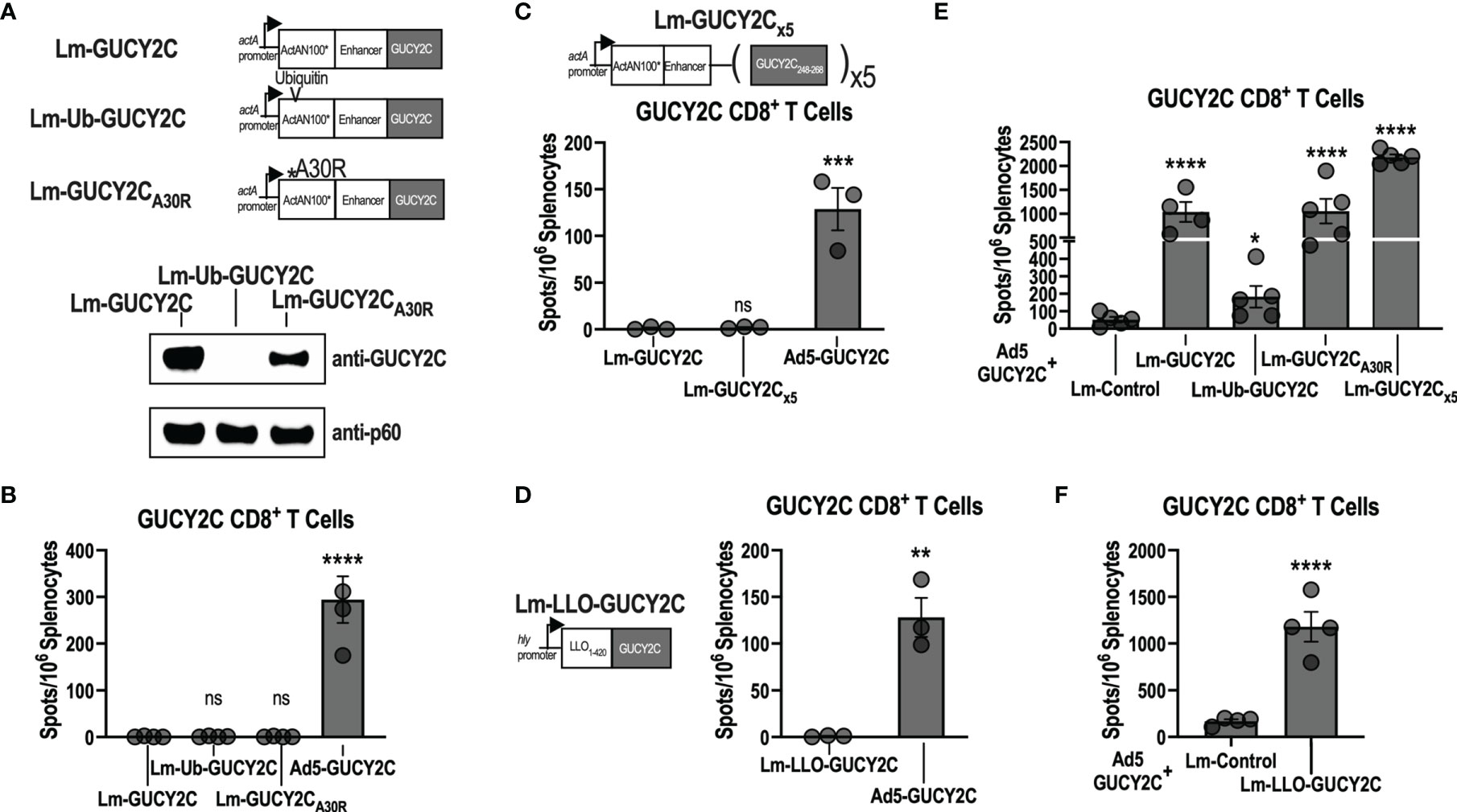
Figure 2 Lm-GUCY2C vaccines designed to enhance antigen processing do not improve GUCY2C254-262 immunogenicity. (A) Lm-GUCY2C used in Figure 1 secretes a fusion protein consisting of ActAN100* combined with an enhancer sequence and GUCY2C. Lm-GUCY2CA30R contains a point mutation at the N-terminal residue following the signal sequence of actA, and Lm-Ub-GUCY2C contains ubiquitin protein following the signal sequence within ActAN100* to enhance degradation, confirmed by western blot analysis. (B) BALB/c mice (n=3-4/group) were immunized i.p. with 107 CFU of Lm vaccines or 1010 vp of Ad5-GUCY2C vaccine and euthanized 7 or 14 d later, respectively, to quantify GUCY2C-specific CD8+ T-cell responses by IFNγ ELISpot. (C) To enhance the quantity of GUCY2C254-262 peptide and improve antigen processing, Lm-GUCY2Cx5 secretes a fusion protein containing five repeating copies of GUCY2C248-268 separated by sequences predicted to facilitate proteasomal cleavage. BALB/c mice (n=3-4/group) were immunized i.p. with 107 CFU of Lm-GUCY2Cx5 vaccine or 1010 vp of Ad5-GUCY2C vaccine and euthanized 7 or 14 d later, respectively, to quantify GUCY2C-specific CD8+ T-cell responses by IFNγ ELISpot. (D) Lm-LLO-GUCY2C secretes a fusion protein comprised of a truncated LLO protein fused to GUCY2C under control of the hly promoter. BALB/c mice (n=3/group) were immunized i.p with 107 CFU of Lm-LLO-GUCY2C or i.m. with 1010 vp of Ad5-GUCY2C and GUCY2C-specific CD8+ T-cell responses were quantified by IFNγ ELISpot 7 or 14 d later, respectively. (E, F) BALB/c mice (n=4-5/group) were immunized i.m. with 1010 vp of Ad5-GUCY2C vaccine on day 0 followed by 107 CFU of Lm vaccines i.p. on day 21, including Lm constructs designed to enhance epitope presentation employed in (A–C) and the LLO-based construct employed in (D). Six days after Lm boosting, GUCY2C-specific CD8+ T-cell responses were quantified by IFNγ ELISpot. Statistical comparisons were made to Lm-GUCY2C using one-way ANOVA with Bonferroni correction for multiple comparisons (B, C, E) or unpaired T test (D, F). Error bars indicate mean +/- SEM. Symbols indicate individual animals. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant.
Lm-Derived Epitopes Exhibit Superior Peptide-MHC Stability Compared to GUCY2C254-262
In addition to antigen quantity (48), temporal expression (43), and degradation (49), competition between immunodominant and subdominant epitopes may reflect differing affinities for MHC class I and/or stabilities of the peptide-MHC complex (50). Thus, we compared the predicted binding affinity of GUCY2C254-262 for its cognate MHC class I molecule, H-2Kd, with that of all known H-2Kd-restricted Lm epitopes using NetMHCPan4.0 (Figure 3A). Notably, the predicted binding affinity of GUCY2C254-262 for H-2Kd was more than 15x lower than that of all Lm-derived peptides (Figure 3A). We next compared the stability of the GUCY2C254-262-H-2Kd complex to that of the Lm epitopes using the TAP-deficient cell line, RMA-S, stably expressing H-2Kd. In the absence of TAP, cytosolic peptides are not transported to the endoplasmic reticulum, resulting in empty MHC molecules that are highly unstable and rapidly internalized from the cell surface (51). However, the addition of exogenous peptide stabilizes MHC molecules on the cell surface, and the stability of different peptide-MHC complexes can be compared by the decay of the complex and reduced surface MHC levels (52). Thus, to assess the stability of the GUCY2C and Lm peptide-MHC complexes, we quantified the decay of surface peptide-H-2Kd over time (Figure 3B). As predicted from the in silico affinity algorithms (Figure 3A), GUCY2C254-262-H-2Kd complexes rapidly decayed from the cell surface compared to complexes formed with Lm epitopes (Figure 3B). Nonlinear regression analyses estimate the t1/2 for GUCYC254-262-H-2Kd complexes at 1.8 h, while the t1/2 for H-2Kd complexes with Lm epitopes ranged from 4.6 to 10.0 h (Figure 3C). Collectively, these data suggest that the GUCY2C254-262 epitope forms an unstable complex with its cognate MHC class I molecule, H-2Kd, compared to H-2Kd complexes formed with dominant Lm epitopes.
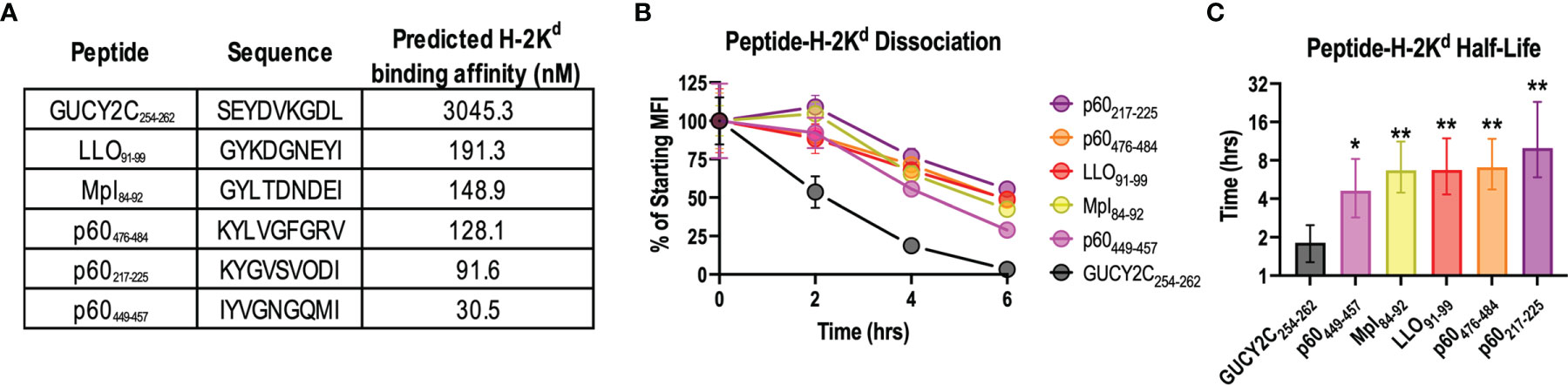
Figure 3 The GUCY2C254-262-H-2Kd complex is less stable than peptide-H-2Kd complexes derived from Lm epitopes. (A) The predicted binding affinity of GUCY2C254-262 peptide for H-2Kd was compared to that of all known Lm-specific H-2Kd-restricted epitopes using NetMHCPan4.0. (B, C) The stability of peptide-H-2Kd complexes was measured using the TAP-deficient cell line, RMA-S H-2Kd. (B) The normalized specific mean fluorescence intensities (MFI) are shown. Error bars indicate mean +/- SEM of technical replicates. (C) Half-lives (t1/2) were determined by nonlinear regression of (B). Error bars indicate computed t1/2 +/- 95% confidence intervals. Statistical comparisons were made to GUCY2C254-262 by extra sum-of-squares F Test with Bonferroni correction. Data are representative of two experiments. *P < 0.05; **P < 0.01.
F255Y Mutation of GUCY2C254-262 Enhances Stability of the GUCY2C254-262-H-2Kd Complex
The above data demonstrate that the GUCY2C254-262 epitope interacts weakly with H-2Kd compared to dominant Lm epitopes in the Lm-GUCY2C vaccine (Figure 3). Notably, the strength of the peptide-MHC interaction is most significantly impacted by a few amino acids at specific locations along the peptide (anchoring residues). At anchoring residues, amino acids from the peptide are partially or fully buried within the pockets of the MHC molecule, forcing significant peptide-MHC interactions at these locations (53). As a result, amino acids at these positions are highly conserved for each MHC allele. For the H-2Kd allele, anchoring residues are located at positions 2, 5, and 9 along the peptide (53). Employing the most conserved amino acid at each anchoring residue position for immunogenic H-2Kd epitopes (53), we generated three different GUCY2C254-262 peptide variants, each using the preferred amino acid at a given anchor, resulting in improved predicted affinity for H-2Kd (Figure 4A). To confirm that amino acid substitution at anchoring residues did not alter recognition by T-cells specific for the native GUCY2C254-262 epitope, splenocytes from mice primed against the native sequence (Ad5-GUCY2C) were employed in an ELISpot assay with native GUCY2C254-262, GUCY2CF255Y, GUCY2CV258S, or GUCY2CL262I point mutations (Figure 4B). Notably, GUCY2CV258S was poorly recognized by GUCY2C254-262-specific T-cells (Figure 4B) and was not explored further. However, GUCY2CF255Y and GUCY2CL262I peptide recognition by GUCY2C254-262-specific CD8+ T-cells was equivalent to recognition of the native GUCY2C254-262 peptide sequence (Figure 4B). Next, we assessed the stability of the GUCY2C254-262-H-2Kd complex between native, GUCY2CF255Y, and GUCY2CL262I sequences (Figures 4C, D). As expected, the GUCY2CF255Y-H-2Kd complex was significantly more stable than the native GUCY2C complex and shifted the half-life from 2.1 h to 8.3 h, respectively. However, the GUCY2CL262I-H-2Kd complex was less stable (1.0 h) than the native sequence. Thus, the GUCY2CF255Y peptide is recognized similarly to the native peptide sequence by T cells primed against native GUCY2C (Figure 4B) but forms significantly more stable complexes with H-2Kd (Figure 4C, D).
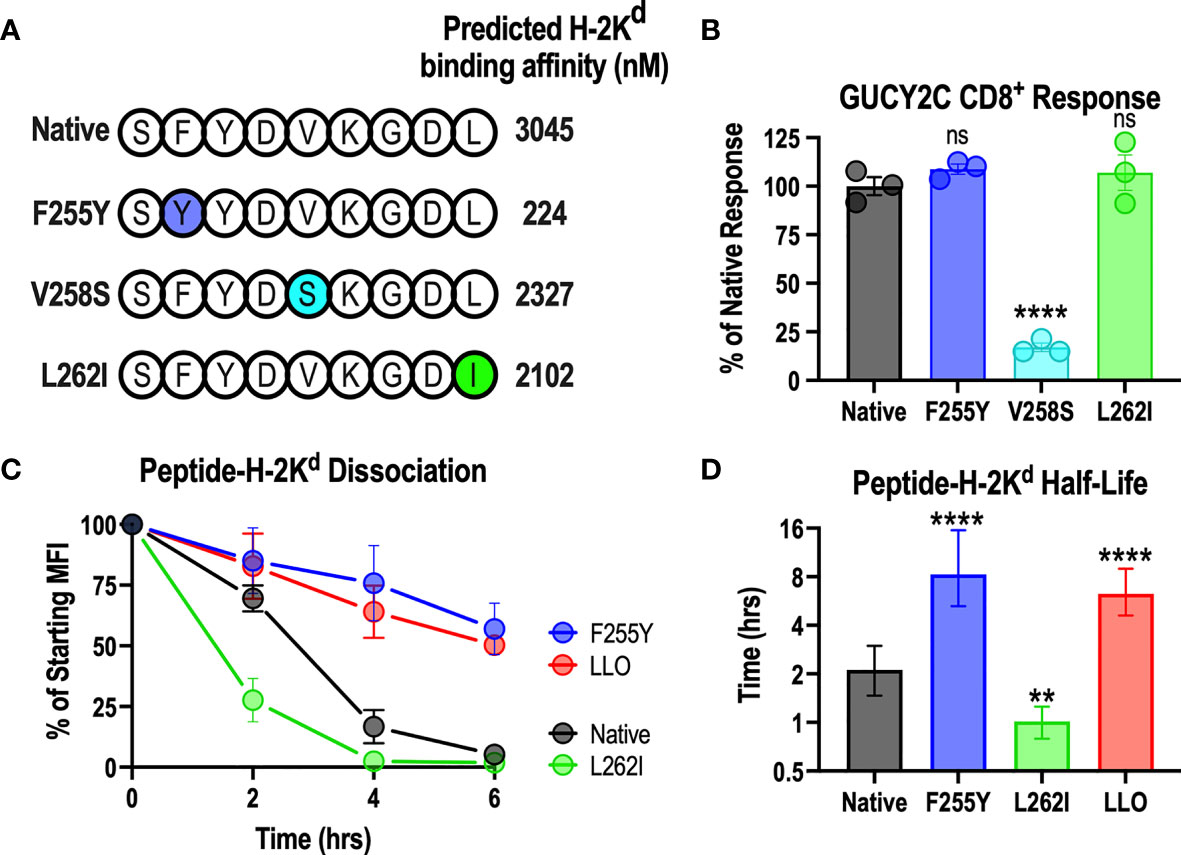
Figure 4 Anchoring residue modification improves the stability of the GUCY2C254-262-H-2Kd complex. (A) Shows the amino acid sequence of native and altered peptides predicted with NetMHCPan 4.0 to improve H-2Kd binding affinity. (B) Splenocytes from mice immunized against the native GUCY2C254-262 epitope with Ad5-GUCY2C were incubated overnight with native GUCY2C254-262 peptide or altered GUCY2C peptides, and the number of IFNγ-producing T cells were quantified by ELISpot (relative to recognition of the native GUCY2C254-262 peptide). Normalized spot counts were compared by one-way ANOVA to native GUCY2C254-262 peptide with Bonferroni correction for multiple comparisons. Error bars indicate mean +/- SEM of technical replicates. (C) The stability of the peptide-H-2Kd complexes was assessed using RMA-S H-Kd cells, and the normalized specific MFIs are shown. Error bars indicate mean +/- SEM of two experiments. (D) Half-lives (t1/2) were determined by nonlinear regression of (C). Error bars indicate computed t1/2 +/- 95% confidence intervals. Statistical comparisons were made to native GUCY2C by extra sum-of-squares F Test with Bonferroni correction. **P < 0.01; ****P < 0.0001; ns, not significant.
GUCY2CF255Y Mutation Rescues Lm-GUCY2C Immunogenicity and Antitumor Efficacy
Having demonstrated that GUCY2C254-262 peptide interacts weakly with H-2Kd compared to peptides from Lm (Figure 3) and that GUCY2CF255Y modification significantly improves the stability of the peptide-MHC complex (Figure 4), we hypothesized that improved stability of the GUCY2CF255Y peptide with H-2Kd may elevate GUCY2C254-262 within the Lm immunodominance hierarchy. Thus, we cloned new Lm vaccines identical to Lm-GUCY2C but containing the GUCY2CF255Y (Lm-GUCY2CF255Y) or GUCY2CL262I (Lm-GUCY2CL262I) modifications and assessed their ability to induce responses recognizing the native GUCY2C254-262 epitope by IFNy ELISpot (Figure 5A). Indeed, improving the stability of the GUCY2C254-262-H-2Kd complex with GUCY2CF255Y (Figure 4) rescued GUCY2C immunogenicity following Lm-GUCY2CF255Y vaccination (Figure 5A). Similarly, Lm-GUCY2CL262I failed to rescue responses (Figure 5A), reflecting worsened stability (Figure 4). Given that altering epitope residues may impact T-cell recognition (54), we next examined cross-reactivity of responses elicited to native and mutated GUCY2CF255Y, as well as the avidity of the vaccine-induced T-cell pools for native GUCY2C254-262. Notably, T-cells from mice immunized with native Ad5-GUCY2C or Lm-GUCY2CF255Y were equally cross-reactive to native and GUCY2CF255Y peptides (Figure 5B). Moreover, the avidity of T-cell pools induced by Ad5-GUCY2C and Lm-GUCY2CF255Y immunization were identical for native GUCY2C254-262 epitope (Figure 5C). Together, these data suggest that the GUCY2C254-262-reactive CD8+ T-cell pool is similar in mice primed against the native GUCY2C and GUCY2CF255Y sequences. Finally, we confirmed that Lm-GUCY2CF255Y induces antitumor immunity against colorectal cancer cells expressing native GUCY2C (Figures 5D–F). Mice were immunized with Lm-Control, Lm-GUCY2C, Lm-GUCY2CL262I, or Lm-GUCY2CF255Y in a model of recurrent, metastatic colorectal cancer using the CT26 cell line expressing GUCY2C and firefly luciferase. As expected, the priming of GUCY2C254-262-specific CD8+ T cells by Lm-GUCY2CF255Y reduced metastatic tumor burden (Figures 5D, E) and improved survival (Figure 5F), in striking contrast to Lm-GUCY2C and Lm-GUCY2CL262I, reflecting the immunodominance of Lm epitopes in those vaccines.
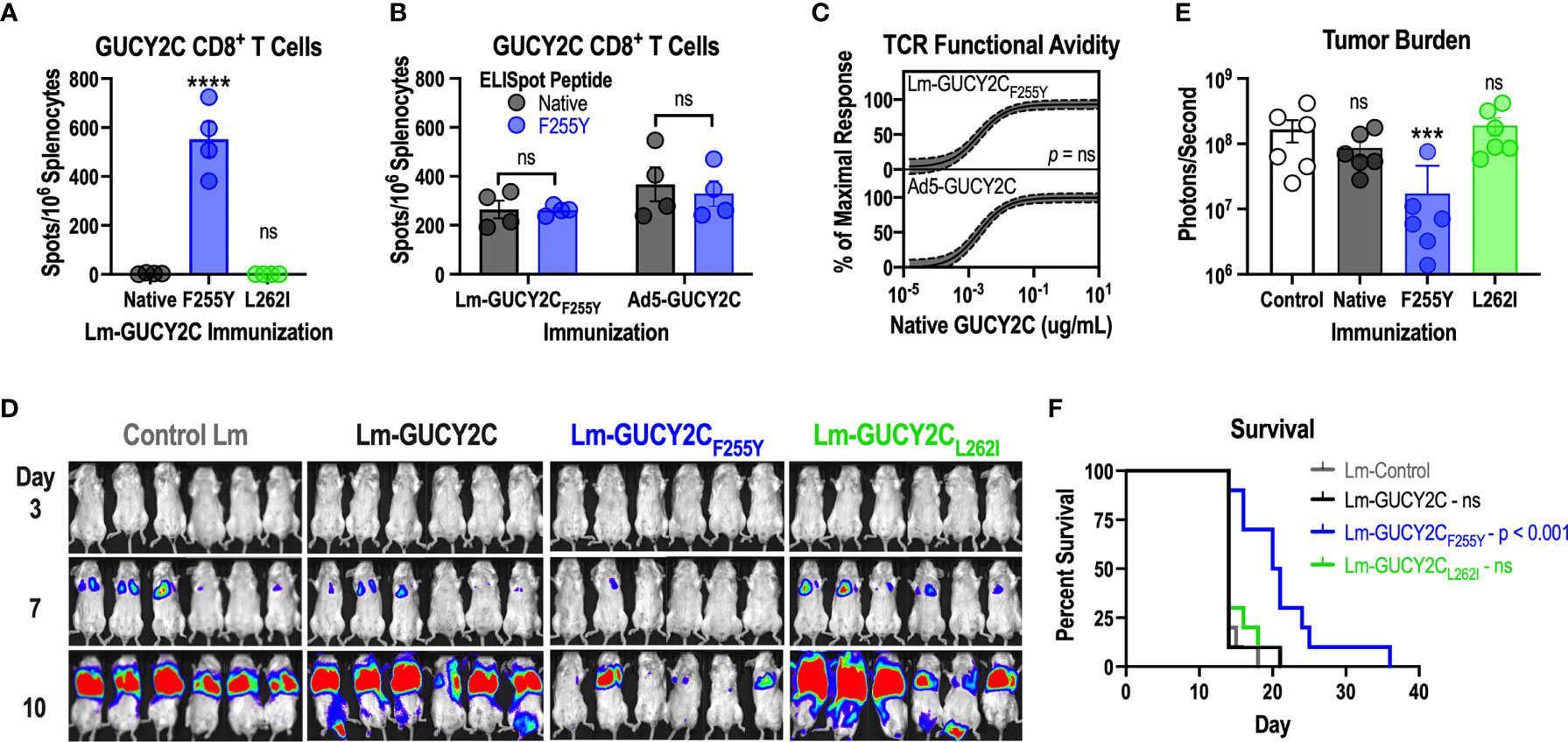
Figure 5 Lm-GUCY2CF255Y primes GUCY2C-specific CD8+ T-cell responses and antitumor immunity. (A) BALB/c mice (n=4/group) were immunized i.p. with 107 CFU of Lm secreting native GUCY2C or GUCY2C with altered GUCY2C254-262 epitopes, and GUCY2C254-262-specific T-cell responses were quantified 7 d later by IFNγ ELISpot. (B, C) BALB/c mice (n=4/group) were immunized i.p. with 107 CFU of Lm-GUCY2CF255Y or i.m. with 1010 vp of Ad5-GUCY2C and euthanized 7 or 14 d later, respectively. (B) Splenocytes from immunized mice were pulsed with native GUCY2C254-262 peptide or GUCY2CF255Y peptide to assess the magnitude of responses by IFNγ ELISpot. (C) Splenocytes from immunized mice were pulsed with decreasing concentrations of native GUCY2C254-262 peptide to assess functional TCR avidity. Nonlinear regression (solid line) of GUCY2C-specific CD8+ T-cell avidity is depicted with 95% confidence intervals (dashed lines). Statistical comparison was made by extra sum-of-squares F Test. (D–F) BALB/c mice (n=10/group) were immunized i.p. with 107 CFU control, GUCY2C, GUCY2CL262I, or GUCY2CF255Y Lm. Seven days later, mice were challenged with the CT26 colorectal cancer cell line expressing GUCY2C and luciferase. (D) Tumor burden was monitored by bioluminescence imaging and quantified on day 7 (E). (F) Survival was monitored longitudinally. Data were analyzed by one-way (A, E) or two-way (B) ANOVA with Bonferroni correction for multiple comparisons to compare 1 or 2 variables, respectively. Error bars indicate mean +/- SEM. Survival (F) was analyzed by the Mantel-Cox log rank test with all immunized groups compared to control with Bonferroni correction. Symbols indicate individual animals. ***P < 0.001; ****P < 0.0001; ns, not significant.
Discussion
The broader vaccine field, as well as cancer vaccines specifically, employs a wide variety of vaccination platforms. Indeed, a recent review assessing clinical trials testing personalized cancer vaccines between 2017 and 2020 found that methods of cancer vaccination are highly variable and noted that a “…lack of consensus on the best approach to the systematic testing of antigen and vaccine efficacy was evident” (8). Of 23 trials analyzed, 6 utilized mRNA, 6 utilized peptide, 5 utilized Listeria monocytogenes, 3 utilized DNA, 2 utilized mRNA in combination with a viral vector, and 1 utilized yeast. These observations underscore the necessity to evaluate current methods to define optimal methods of cancer vaccination.
In that context, studies here examining a GUCY2C-directed Lm vaccine reveal a significant limitation of this vaccine platform. Notably, while Lm-GUCY2C induces robust Lm-specific CD8+ T-cell responses, it cannot prime GUCY2C-specific CD8+ T-cell responses (Figure 1B). In contrast, Lm-GUCY2C generates potent secondary expansion of GUCY2C-specific memory CD8+ T-cells (Figure 1D), suggesting that the primary GUCY2C254-262 epitope is immunologically active and presented by MHC but is subdominant to Lm epitopes, depending on the context (primary versus secondary GUCY2C exposure). Alternatively, this may reflect low abundance of GUCY2C254-262 epitope, falling below levels needed for priming, but above levels needed for boosting. The lack of priming with Lm-GUCY2C variants that enhance epitope abundance (Figure 2) suggest that abundance alone doesn’t explain its failure to prime. Instead, this appears to reflect the poor stability of the GUCY2C254-262-H-2Kd complex compared to Lm-derived peptide-H-2Kd complexes (>5-fold faster dissociation from MHC; Figure 3), which can be reversed by an altered peptide-ligand with enhanced peptide-MHC stability (Figure 4), Lm-GUCY2CF255Y, rescuing immunogenicity and antitumor efficacy (Figure 5). Collectively, these findings suggest that Lm-derived peptides may act as a significant competitive barrier to target antigen presentation and immunogenicity, limiting the effectiveness of Lm-based vaccines.
In the context of Lm-based cancer vaccines, there are multiple reports of limited immunogenicity when using Lm vaccines to prime CD8+ T-cell responses against cancer antigens. For example, Lm vaccines against the tumor-associated antigens PAP (55) and mesothelin (56) had poor immunogenicity alone but were more effective as booster vaccines in the context of a heterologous prime-boost immunization regimen, similar to observations with GUCY2C (Figure 1) (57). However, Lm vaccines alone against HER2 (58) and PSA (59) tumor antigens induce robust antitumor immunity. Interestingly, those studies employed foreign antigens (human HER2 or human PSA) in mice, which are not limited by self-tolerance mechanisms. Moreover, Lm-based immunization with HER2 in rat HER2 transgenic mice (60) or immunization with HPV-16 E7 in HPV16 E6/E7 transgenic mice (61) did successfully induce responses, despite limitations imposed by self-tolerance. Collectively, these studies suggest that the immunogenicity of Lm vaccines is likely dependent on the vaccine antigen and context and that weak antigens (some self-antigens like GUCY2C and some foreign antigens) may be more susceptible to competition with Lm-derived peptides than other antigens. Notably, a recent clinical trial testing an Lm-derived vaccine against mesothelin reported that T-cell responses towards mesothelin were inconsistent and lower in magnitude than LLO (14). Similarly, Lm expressing 4 prostate cancer antigens induced significantly lower and inconsistent responses to the prostate antigens than LLO in patients, resulting in early study termination (62). Together, these observations suggest that competition between immunodominant Lm antigens and vaccine antigens is conserved in humans.
Interestingly, Ad5-based vaccines expressing GUCY2C are highly capable of priming GUCY2C-specific immunity, despite containing viral peptides that could theoretically compete with GUCY2C epitopes for presentation. However, competition between Ad5 and GUCY2C peptides is likely significantly less than the competition between Lm and GUCY2C peptides for at least two reasons. First, the Ad5 genome contains roughly 40 protein-coding genes (63), while the Lm genome encompasses over 2800 proteins (64), creating significantly more opportunities for immunodominance over GUCY2C. Second, despite being attenuated to limit inter-cellular migration and pathogenicity, Lm vaccines are replication-competent. Thus, the synthesis and presentation of GUCY2C antigen is concurrent with the production and presentation of Lm antigens in infected APCs. In contrast, Ad5 vectors utilized clinically and in the present studies are replication-deficient, and target antigens are encoded within the episomal Ad5 genomic DNA. Thus, during the course of infection, the quantity of Ad5 epitope presentation is predicted to peak at injection and diminish over time as the administered vector is cleared, while GUCY2C presentation is initially absent until it is de novo synthesized by the host cell over a much more extended period. The lag time between Ad5 exposure and the synthesis of target antigen by the host cell may, therefore, allow for temporal separation between presentation of GUCY2C and Ad5 epitopes – a working hypothesis supported by previous studies revealing the ability of GUCY2C-conjugated, but not Ad5 structural, antigens to provide CD4+ T-cell help to GUCY2C-specific CD8+ T cells (35). Thus, a surplus of Lm antigens, in addition to a lack of temporal separation between Lm and target antigen presentation, may operate to restrict vaccine-directed immune responses in the context of Lm vaccines.
This is the first study to demonstrate that Lm-derived peptides may interfere with the immunogenicity of target vaccine antigens. These studies carry significant implications for designing and implementing Lm-based cancer vaccines. Specifically, Lm vaccines may be inferior at priming vaccine-specific responses than other vaccine platforms. While use of altered peptide ligands with enhanced peptide-MHC stability could ameliorate this drawback, the identification and validation of altered peptide ligands for relevant patient HLA alleles is a cumbersome endeavor. Therefore, using other vaccine vectors for priming immune responses may be more practical. Alternatively, Lm engineering approaches, such as episomal plasmid-based transgene expression (45, 58, 59, 65) may produce higher antigen expression than the approach used here, in which a single chromosomal copy of GUCY2C is created (12). Despite the limitation identified here, Lm vaccines may be particularly useful in a different context. Specifically, multiple studies have demonstrated the ability of Lm vaccines to boost memory CD8+ T-cell responses (55, 56). Additionally, preclinical and clinical studies suggest that Lm vaccines transform tumor microenvironments by increasing CD8+ T-cell infiltration (66), decreasing immunosuppressive regulatory T cells (10, 65) and myeloid-derived suppressor cells (11, 65), and repolarizing tumor-associated macrophages from M2 to M1 phenotypes (10, 66, 67). While the capabilities of Lm to boost CD8+ T-cell responses and remodel immunosuppressive tumor microenvironments may be a unique advantage compared to other platforms, the clinical significance of these findings and the extent to which other vaccine platforms compare has yet to be fully investigated. Moreover, any superiorities of Lm-based vaccines must also be weighed against the potential risks and toxicities associated with using a live bacterial platform. Indeed, despite the numerous successes developing vaccines to elicit protective antibody responses, often with attenuated or inactivated pathogens, very few vaccines that elicit protective CD8⁺ T-cell responses have been clinically successful. Continued investigation of currently available vaccine vectors and development of novel vaccine approaches and combinations are necessary to elicit robust CD8⁺ T-cell responses against cancer and intractable pathogens to address this unmet need.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by Thomas Jefferson University Institutional Animal Care and Use Committee (IACUC).
Author Contributions
These investigations were conducted, supporting methodologies were developed, and data were acquired and validated by JF, JS, YY, RC, and JB. Data were visualized, and the original draft was prepared, by JF. The project was conceptualized, data were reviewed, and the manuscript was reviewed and edited by all authors. AS provided administration and acquisition of financial support for the project leading to this publication. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants to SW from NIH (1R01 CA204881, 1R01 CA206026), the Department of Defense Congressionally Directed Medical Research Program W81XWH-17-PRCRP-TTSA, The Courtney Ann Diacont Memorial Foundation, and Targeted Diagnostic & Therapeutics, Inc; and to AS from the Department of Defense Congressionally Directed Medical Research Program W81XWH-19-1-0263, W81XWH-19-1-0067, and W81XWH-17-1-0299, and David and Lorraine Swoyer. The funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication. JF was supported by the Alfred W. and Mignon Dubbs Fellowship Fund and a PhRMA Foundation Pre-Doctoral Fellowship in Pharmacology/Toxicology. RC was supported by the T32 Training Program in Cancer Biology (5T32 CA236736-02). JB was supported by an NIH Ruth Kirschstein Individual Predoctoral MD/PhD Fellowship (F30 DK127639). SW is the Samuel M.V. Hamilton Professor of Thomas Jefferson University. Research reported in this publication utilized the Flow Cytometry and Lab Animal Shared Resources of the Sidney Kimmel Cancer Center at Jefferson Health and was supported by the National Cancer Institute of the National Institutes of Health under Award Number 5P30 CA056036-20. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Conflict of Interest
SW is a member of the Board and Chair of the Scientific Advisory Board of, and AS is a consultant for, Targeted Diagnostics & Therapeutics, Inc., which provided research funding that, in part, supported this work and has a license to commercialize inventions related to this work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Dr. Richard Calendar for providing the pPL2 integration vector used in cloning Lm-GUCY2C, Dr. Sean Murphy for providing the RMA-S H-2Kd cell line, and Dr. Laurence Eisenlohr for discussion of protein degradation for epitope presentation.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.855759/full#supplementary-material
References
1. Emens LA, Ascierto PA, Darcy PK, Demaria S, Eggermont AMM, Redmond WL, et al. Cancer Immunotherapy: Opportunities and Challenges in the Rapidly Evolving Clinical Landscape. Eur J Cancer (2017) 81:116–29. doi: 10.1016/j.ejca.2017.01.035
2. Hunter P. The Fourth Pillar: Despite Some Setbacks in the Clinic, Immunotherapy has Made Notable Progress Toward Becoming an Additional Therapeutic Option Against Cancer. EMBO Rep (2017) 18:1889–92. doi: 10.15252/embr.201745172
3. Popovic A, Jaffee EM, Zaidi N. Emerging Strategies for Combination Checkpoint Modulators in Cancer Immunotherapy. J Clin Invest (2018) 128:3209–18. doi: 10.1172/JCI120775
4. Curran MA, Glisson BS. New Hope for Therapeutic Cancer Vaccines in the Era of Immune Checkpoint Modulation. Annu Rev Med (2019) 70:409–24. doi: 10.1146/annurev-med-050217-121900
5. Zemek RM, De Jong E, Chin WL, Schuster IS, Fear VS, Casey TH, et al. Sensitization to Immune Checkpoint Blockade Through Activation of a STAT1/NK Axis in the Tumor Microenvironment. Sci Transl Med (2019) 11:eaav7816. doi: 10.1126/scitranslmed.aav7816
6. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 Blockade Induces Responses by Inhibiting Adaptive Immune Resistance. Nature (2014) 515:568–71. doi: 10.1038/nature13954
7. Bashir B, Flickinger JC, Snook AE. Vaccines and Immune Checkpoint Inhibitors: A Promising Combination Strategy in Gastrointestinal Cancers. Immunotherapy (2021) 13:561–4. doi: 10.2217/imt-2021-0012
8. Shemesh CS, Hsu JC, Hosseini I, Shen B-Q, Rotte A, Twomey P, et al. Personalized Cancer Vaccines: Clinical Landscape, Challenges, and Opportunities. Mol Ther (2021) 29:555–70. doi: 10.1016/j.ymthe.2020.09.038
9. Chávez-Arroyo A, Portnoy DA. Why is Listeria Monocytogenes Such a Potent Inducer of CD8+ T-Cells? Cell Microbiol (2020) 22:e13175. doi: 10.1111/cmi.13175
10. Deng W, Lira V, Hudson TE, Lemmens EE, Hanson WG, Flores R, et al. Recombinant Listeria Promotes Tumor Rejection by CD8+ T Cell-Dependent Remodeling of the Tumor Microenvironment. Proc Natl Acad Sci USA (2018) 115:8179–84. doi: 10.1073/pnas.1801910115
11. Wallecha A, Singh R, Malinina I. Listeria Monocytogenes (Lm)-LLO Immunotherapies Reduce the Immunosuppressive Activity of Myeloid-Derived Suppressor Cells and Regulatory T Cells in the Tumor Microenvironment. J Immunother (2013) 36:468–76. doi: 10.1097/CJI.0000000000000000
12. Wood LM, Paterson Y. Attenuated Listeria Monocytogenes: A Powerful and Versatile Vector for the Future of Tumor Immunotherapy. Front Cell Infect Microbiol (2014) 4:51. doi: 10.3389/fcimb.2014.00051
13. Flickinger JC, Rodeck U, Snook AE. Listeria Monocytogenes as a Vector for Cancer Immunotherapy: Current Understanding and Progress. Vaccines (Basel) (2018) 6:48. doi: 10.3390/vaccines6030048
14. Brahmer JR, Johnson ML, Cobo M, Viteri S, Sarto JC, Sukari A, et al. JNJ-64041757 (JNJ-757), A Live, Attenuated, Double-Deleted Listeria Monocytogenes-Based Immunotherapy, in Patients With Non-Small Cell Lung Cancer: Results From 2 Phase 1 Studies. JTO Clin Res Rep (2020) 100103. doi: 10.1016/j.jtocrr.2020.100103
15. Basu P, Mehta A, Jain M, Gupta S, Nagarkar RV, John S, et al. A Randomized Phase 2 Study of ADXS11-001 Listeria Monocytogenes-Listeriolysin O Immunotherapy With or Without Cisplatin in Treatment of Advanced Cervical Cancer. Int J Gynecol Cancer (2018) 28:764–72. doi: 10.1097/IGC.0000000000001235
16. Hecht JR, Goldman JW, Hayes S, Balli D, Princiotta MF, Dennie JG, et al. Abstract CT007: Safety and Immunogenicity of a Personalized Neoantigen- Listeria Vaccine in Cancer Patients. Clin Trials (American Assoc Cancer Research) (2019) 79:CT007–7. doi: 10.1158/1538-7445.AM2019-CT007
17. Tsujikawa T, Crocenzi T, Durham JN, Sugar EA, Wu AA, Onners B, et al. Evaluation of Cyclophosphamide/GVAX Pancreas Followed by Listeria-Mesothelin (CRS-207) With or Without Nivolumab in Patients With Pancreatic Cancer. Clin Cancer Res (2020) 26:3578–88. doi: 10.1158/1078-0432.CCR-19-3978
18. Brockstedt DG, Giedlin MA, Leong ML, Bahjat KS, Gao Y, Luckett W, et al. Listeria-Based Cancer Vaccines That Segregate Immunogenicity From Toxicity. Proc Natl Acad Sci USA (2004) 101:13832–7. doi: 10.1073/pnas.0406035101
19. Lauer PM, Hanson WG. Protein Expression Enhancer Sequences and Use Thereof. (2020). Available at: https://patents.google.com/patent/US10526609B2/en.
20. Lauer P, Chow MYN, Loessner MJ, Portnoy DA, Calendar R. Construction, Characterization, and Use of Two Listeria Monocytogenes Site-Specific Phage Integration Vectors. J Bacteriol (2002) 184:4177–86. doi: 10.1128/JB.184.15.4177-4186.2002
21. Jones GS, D’Orazio SEF. Listeria Monocytogenes: Cultivation and Laboratory Maintenance. Curr Protoc Microbiol (2013) 31:9B.2.1–7. doi: 10.1002/9780471729259.mc09b02s31
22. Marszalowicz GP, Snook AE, Magee MS, Merlino D, Berman-Booty LD, Waldman SA. GUCY2C Lysosomotropic Endocytosis Delivers Immunotoxin Therapy to Metastatic Colorectal Cancer. Oncotarget (2014) 5:9460–71. doi: 10.18632/oncotarget.2455
23. Flickinger JC, Singh J, Carlson R, Leong E, Baybutt TR, Barton J, et al. Chimeric Ad5.F35 Vector Evades Anti-Adenovirus Serotype 5 Neutralization Opposing GUCY2C-Targeted Antitumor Immunity. J Immunother Cancer (2020) 8:e001046. doi: 10.1136/jitc-2020-001046
24. Abraham TS, Flickinger JC, Waldman SA, Snook AE. TCR Retrogenic Mice as a Model to Map Self-Tolerance Mechanisms to the Cancer Mucosa Antigen GUCY2C. J Immunol (2019) 202:1301–10. doi: 10.4049/jimmunol.1801206
25. Xiang B, Baybutt TR, Berman-Booty L, Magee MS, Waldman SA, Alexeev VY, et al. Prime-Boost Immunization Eliminates Metastatic Colorectal Cancer by Producing High-Avidity Effector CD8+ T Cells. J Immunol (2017) 198:3507–14. doi: 10.4049/jimmunol.1502672
26. Müllbacher A, Lobigs M, Kos FJ, Langman R. Alloreactive Cytotoxic T-Cell Function, Peptide Nonspecific. Scand J Immunol (1999) 49:563–9. doi: 10.1046/j.1365-3083.1999.00568.x
27. Watson AM, Mylin LM, Thompson MM, Schell TD. Modification of a Tumor Antigen Determinant to Improve Peptide/MHC Stability Is Associated With Increased Immunogenicity and Cross-Priming a Larger Fraction of CD8+ T Cells. J Immunol (2012) 189:5549–60. doi: 10.4049/jimmunol.1102221
28. Chefalo PJ, Harding CV. Processing of Exogenous Antigens for Presentation by Class I MHC Molecules Involves Post-Golgi Peptide Exchange Influenced by Peptide-MHC Complex Stability and Acidic Ph. J Immunol (2003) 170:643. doi: 10.4049/jimmunol.170.1.643
29. Snook AE, Magee MS, Marszalowicz GP, Schulz S, Waldman SA. Epitope-Targeted Cytotoxic T Cells Mediate Lineage-Specific Antitumor Efficacy Induced by the Cancer Mucosa Antigen GUCY2C. Cancer Immunol Immunother (2012) 61:713–23. doi: 10.1007/s00262-011-1133-0
30. Enouz S, Carrié L, Merkler D, Bevan MJ, Zehn D. Autoreactive T Cells Bypass Negative Selection and Respond to Self-Antigen Stimulation During Infection. J Exp Med (2012) 209:1769–79. doi: 10.1084/jem.20120905
31. Fu G, Casas J, Rigaud S, Rybakin V, Lambolez F, Brzostek J, et al. Themis Sets the Signal Threshold for Positive and Negative Selection in T-Cell Development. Nature (2013) 504:441–5. doi: 10.1038/nature12718
32. Snook AE, Baybutt TR, Xiang B, Abraham TS, Flickinger JC, Hyslop T, et al. Split Tolerance Permits Safe Ad5-GUCY2C-PADRE Vaccine-Induced T-Cell Responses in Colon Cancer Patients. J Immunother Cancer (2019) 7:104. doi: 10.1186/s40425-019-0576-2
33. Snook AE, Baybutt TR, Hyslop T, Waldman SA. Preclinical Evaluation of a Replication-Deficient Recombinant Adenovirus Serotype 5 Vaccine Expressing Guanylate Cyclase C and the PADRE T-Helper Epitope. Hum Gene Ther Methods (2016) 27:238–50. doi: 10.1089/hgtb.2016.114
34. Snook AE, Stafford BJ, Li P, Tan G, Huang L, Birbe R, et al. Guanylyl Cyclase C-Induced Immunotherapeutic Responses Opposing Tumor Metastases Without Autoimmunity. J Natl Cancer Inst (2008) 100:950–61. doi: 10.1093/jnci/djn178
35. Snook AE, Magee MS, Schulz S, Waldman SA. Selective Antigen-Specific CD4(+) T-Cell, But Not CD8(+) T- or B-Cell, Tolerance Corrupts Cancer Immunotherapy. Eur J Immunol (2014) 44:1956–66. doi: 10.1002/eji.201444539
36. Hewitt EW. The MHC Class I Antigen Presentation Pathway: Strategies for Viral Immune Evasion. Immunology (2003) 110:163–9. doi: 10.1046/j.1365-2567.2003.01738.x
37. Bailey TW, do Nascimento NC, Bhunia AK. Genome Sequence of Listeria Monocytogenes Strain F4244, a 4b Serotype. Genome Announc (2017) 5:e01324-17. doi: 10.1128/genomeA.01324-17
38. Andersson HA, Barry MA. Maximizing Antigen Targeting to the Proteasome for Gene-Based Vaccines. Mol Ther (2004) 10:432–46. doi: 10.1016/j.ymthe.2004.05.035
39. Rasmussen AB, Zocca MB, Bonefeld CM, von Essen M, Lauritsen JPH, Tomra S, et al. Proteasomal Targeting and Minigene Repetition Improve Cell-Surface Presentation of a Transfected, Modified Melanoma Tumour Antigen. Scand J Immunol (2004) 59:220–7. doi: 10.1111/j.0300-9475.2004.01374.x
40. Miyazawa M, Ojima T, Katsuda M, Nakamura M, Hayata K, Nakamori M, et al. Dendritic Cell Vaccine Transduced With Ubiquitin-Mesothelin Fusion Gene for Pancreatic Cancer. JCO (2016) 34:e14586–6. doi: 10.1200/JCO.2016.34.15_suppl.e14586
41. Moors MA, Auerbuch V, Portnoy DA. Stability of the Listeria Monocytogenes Acta Protein in Mammalian Cells is Regulated by the N-End Rule Pathway. Cell Microbiol (1999) 1:249–57. doi: 10.1046/j.1462-5822.1999.00020.x
42. Zebertavage L, Bambina S, Shugart J, Alice A, Zens KD, Lauer P, et al. Bahjat KS. A Microbial-Based Cancer Vaccine for Induction of Egfrviii-Specific CD8+ T Cells and Anti-Tumor Immunity. PloS One (2019) 14:e0209153. doi: 10.1371/journal.pone.0209153
43. Dekhtiarenko I, Jarvis MA, Ruzsics Z, Čičin-Šain L. The Context of Gene Expression Defines the Immunodominance Hierarchy of Cytomegalovirus Antigens. J Immunol (2013) 190:3399–409. doi: 10.4049/jimmunol.1203173
44. Moors MA, Levitt B, Youngman P, Portnoy DA. Expression of Listeriolysin O and Acta by Intracellular and Extracellular Listeria Monocytogenes. Infect Immun (1999) 67:131–9. doi: 10.1128/IAI.67.1.131-139.1999
45. Singh R, Dominiecki ME, Jaffee EM, Paterson Y. Fusion to Listeriolysin O and Delivery by Listeria Monocytogenes Enhances the Immunogenicity of HER-2/Neu and Reveals Subdominant Epitopes in the FVB/N Mouse. J Immunol (2005) 175:3663–73. doi: 10.4049/jimmunol.175.6.3663
46. Memarnejadian A, Meilleur CE, Shaler CR, Khazaie K, Bennink JR, Schell TD, et al. PD-1 Blockade Promotes Epitope Spreading in Anticancer CD8+ T Cell Responses by Preventing Fratricidal Death of Subdominant Clones to Relieve Immunodomination. J Immunol (2017) 199:3348–59. doi: 10.4049/jimmunol.1700643
47. Friedman J, Moore EC, Zolkind P, Robbins Y, Clavijo PE, Sun L, et al. Neoadjuvant PD-1 Immune Checkpoint Blockade Reverses Functional Immunodominance Among Tumor Antigen-Specific T Cells. Clin Cancer Res (2020) 26:679–89. doi: 10.1158/1078-0432.CCR-19-2209
48. La Gruta NL, Kedzierska K, Pang K, Webby R, Davenport M, Chen W, et al. A Virus-Specific CD8+ T Cell Immunodominance Hierarchy Determined by Antigen Dose and Precursor Frequencies. Proc Natl Acad Sci USA (2006) 103:994–9. doi: 10.1073/pnas.0510429103
49. Kim A, Boronina TN, Cole RN, Darrah E, Sadegh-Nasseri S. Distorted Immunodominance by Linker Sequences or Other Epitopes From a Second Protein Antigen During Antigen-Processing. Sci Rep (2017) 7:46418. doi: 10.1038/srep46418
50. Yewdell JW. Confronting Complexity: Real-World Immunodominance in Antiviral CD8+ T Cell Responses. Immunity (2006) 25:533–43. doi: 10.1016/j.immuni.2006.09.005
51. Ljunggren HG, Stam NJ, Ohlén C, Neefjes JJ, Höglund P, Heemels MT, et al. Empty MHC Class I Molecules Come Out in the Cold. Nature (1990) 346:476–80. doi: 10.1038/346476a0
52. Busch DH, Pamer EG. MHC Class I/Peptide Stability: Implications for Immunodominance, In Vitro Proliferation, and Diversity of Responding CTL. J Immunol (1998) 160:4441–8.
53. Mitaksov V, Fremont DH. Structural Definition of the H-2Kd Peptide-Binding Motif. J Biol Chem (2006) 281:10618–25. doi: 10.1074/jbc.M510511200
54. Clay TM, Custer MC, McKee MD, Parkhurst M, Robbins PF, Kerstann K, et al. Changes in the Fine Specificity of Gp100(209-217)-Reactive T Cells in Patients Following Vaccination With a Peptide Modified at an HLA-A2.1 Anchor Residue. J Immunol (1999) 162:1749–55.
55. Johnson LE, Brockstedt D, Leong M, Lauer P, Theisen E, Sauer J-D, et al. Heterologous Vaccination Targeting Prostatic Acid Phosphatase (PAP) Using DNA and Listeria Vaccines Elicits Superior Anti-Tumor Immunity Dependent on CD4+ T Cells Elicited by DNA Priming. Oncoimmunology (2018) 7:e1456603. doi: 10.1080/2162402X.2018.1456603
56. Le DT, Wang-Gillam A, Picozzi V, Greten TF, Crocenzi T, Springett G, et al. Safety and Survival With GVAX Pancreas Prime and Listeria Monocytogenes-Expressing Mesothelin (CRS-207) Boost Vaccines for Metastatic Pancreatic Cancer. J Clin Oncol (2015) 33:1325–33. doi: 10.1200/JCO.2014.57.4244
57. Flickinger J, Singh J, Carlson R, Barton J, Baybutt T, Rappaport J, et al. Prime-Boost Immunization With Chimeric Adenoviral (Ad5.F35) and Listeria Vectors Is a Safe and Effective Strategy for Cancer Immunotherapy. (2022).
58. Shahabi V, Seavey MM, Maciag PC, Rivera S, Wallecha A. Development of a Live and Highly Attenuated Listeria Monocytogenes-Based Vaccine for the Treatment of Her2/Neu-Overexpressing Cancers in Human. Cancer Gene Ther (2011) 18:53–62. doi: 10.1038/cgt.2010.48
59. Shahabi V, Reyes-Reyes M, Wallecha A, Rivera S, Paterson Y, Maciag P. Development of a Listeria Monocytogenes Based Vaccine Against Prostate Cancer. Cancer Immunol Immunother (2008) 57:1301–13. doi: 10.1007/s00262-008-0463-z
60. Singh R, Paterson Y. In the FVB/N HER-2/Neu Transgenic Mouse Both Peripheral and Central Tolerance Limit the Immune Response Targeting HER-2/Neu Induced by Listeria Monocytogenes-Based Vaccines. Cancer Immunol Immunother (2007) 56:927–38. doi: 10.1007/s00262-006-0237-4
61. Sewell DA, Pan ZK, Paterson Y. Listeria-Based HPV-16 E7 Vaccines Limit Autochthonous Tumor Growth in a Transgenic Mouse Model for HPV-16 Transformed Tumors. Vaccine (2008) 26:5315–20. doi: 10.1016/j.vaccine.2008.07.036
62. Drake CG, Pachynski RK, Subudhi SK, McNeel DG, Antonarakis ES, Bauer TM, et al. Safety and Preliminary Immunogenicity of JNJ-64041809, a Live-Attenuated, Double-Deleted Listeria Monocytogenes-Based Immunotherapy, in Metastatic Castration-Resistant Prostate Cancer. Prostate Cancer Prostatic Dis (2021). doi: 10.1038/s41391-021-00402-8
63. Saha B, Wong CM, Parks RJ. The Adenovirus Genome Contributes to the Structural Stability of the Virion. Viruses (2014) 6:3563–83. doi: 10.3390/v6093563
64. Glaser P, Frangeul L, Buchrieser C, Rusniok C, Amend A, Baquero F, et al. Comparative Genomics of Listeria Species. Science (2001) 294:849–52. doi: 10.1126/science.1063447
65. Mkrtichyan M, Chong N, Abu Eid R, Wallecha A, Singh R, Rothman J, et al. Anti-PD-1 Antibody Significantly Increases Therapeutic Efficacy of Listeria Monocytogenes (Lm)-LLO Immunotherapy. J Immunother Cancer (2013) 1:15. doi: 10.1186/2051-1426-1-15
66. Hassan R, Alley E, Kindler H, Antonia S, Jahan T, Honarmand S, et al. Clinical Response of Live-Attenuated, Listeria Monocytogenes Expressing Mesothelin (CRS-207) With Chemotherapy in Patients With Malignant Pleural Mesothelioma. Clin Cancer Res (2019) 25:5787–98. doi: 10.1158/1078-0432.CCR-19-0070
Keywords: listeria, vaccine, immunodominance, colorectal cancer, GUCY2C
Citation: Flickinger JC Jr, Singh J, Yarman Y, Carlson RD, Barton JR, Waldman SA and Snook AE (2022) T-Cell Responses to Immunodominant Listeria Epitopes Limit Vaccine-Directed Responses to the Colorectal Cancer Antigen, Guanylyl Cyclase C. Front. Immunol. 13:855759. doi: 10.3389/fimmu.2022.855759
Received: 15 January 2022; Accepted: 10 February 2022;
Published: 09 March 2022.
Edited by:
Markus Germann, University of Basel, SwitzerlandReviewed by:
Yvonne Paterson, University of Pennsylvania, United StatesEmanuele Sasso, University of Naples Federico II, Italy
Copyright © 2022 Flickinger, Singh, Yarman, Carlson, Barton, Waldman and Snook. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adam E. Snook, YWRhbS5zbm9va0BqZWZmZXJzb24uZWR1