 Ying Li1
Ying Li1 Muhuo Ji
Muhuo Ji Jianjun Yang
Jianjun Yang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 06 May 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.855006
Sepsis is recognized as a life-threatening multi-organ dysfunction resulting from a dysregulated host response to infection. Although the incidence and mortality of sepsis decrease significantly due to timely implementation of anti-infective and support therapies, accumulating evidence suggests that a great proportion of survivors suffer from long-term cognitive impairment after hospital discharge, leading to decreased life quality and substantial caregiving burdens for family members. Several mechanisms have been proposed for long-term cognitive impairment after sepsis, which are not mutually exclusive, including blood-brain barrier disruption, neuroinflammation, neurotransmitter dysfunction, and neuronal loss. Targeting these critical processes might be effective in preventing and treating long-term cognitive impairment. However, future in-depth studies are required to facilitate preventive and/or treatment strategies for long-term cognitive impairment after sepsis.
The central nervous system (CNS) is one of the most vulnerable organs affected by sepsis (1, 2).
Accumulating evidence has suggested that sepsis survivors display long-term neurological sequelae. In particular, sepsis is associated with a 3-fold increase in the prevalence of cognitive impairment (1, 2), which mainly involves declarative memory, working memory, processing speed, and executive function. Generally, the degree of cognitive impairment is affected by poorer pre-sepsis health status, severity of sepsis, and quality of hospital treatment (1). However, in clinical practice, various risk factors also influence the occurrence of long-term cognitive impairment after sepsis. For instance, increased antibiotic treatment latency, delirium, dependence on mechanical ventilation, and long hospitalizations are associated with poor cognitive performance after discharge (3–6). Several animal models mimicking sepsis, including lipopolysaccharide (LPS) injection, cecal slurry injection, or cecal ligation puncture (CLP), have been widely applied in numerous preclinical studies. Nevertheless, the exact pathogenesis of long-term cognitive impairment after sepsis is still poorly understood. Here, we review multiple mechanisms underlying long-term cognitive impairment after sepsis. It is anticipated that future in-depth studies will pave the way for the development of preventive and/or treatment strategies for long-term cognitive impairment after sepsis.
Recently, the involvement of BBB dysregulation in cognitive impairment after sepsis has been a focus in this field (7, 8). The BBB is a tightly sealed interface between the peripheral circulation and the neuronal cells that is composed of capillary wall endothelial cells, pericytes, and astrocytes. Furthermore, the major components of the BBB and other central nervous system (CNS) cell types form a dynamic structure called the neurovascular unit, which is also critical for normal cognitive function (9). An intact BBB is essential for keeping pathogens, pro-inflammatory signals, and neurotoxic substances outside the brain (10). However, in the sepsis animal model induced by LPS injection, the function of pericytes and endothelial cells is significantly disrupted, leading to BBB damage and cognitive dysfunction (11, 12). Also, in patients suffering from critical illness, markers of BBB injury, such as S100β, are elevated and negatively associated with global cognition at 3 and 12 months after hospital discharge (13). At the molecular level, unlike peripheral endothelium, the BBB endothelium is sealed by tight junctions (TJs), restricting the paracellular routes for diffusion of polar molecules and macromolecules (14). Claudins and occludins are principal TJ proteins bridging adjacent endothelial cells while zonula occludens family (ZO-1, -2, and -3) are scaffolding proteins that provide structural adaptors of TJs (10). In vitro studies revealed decreased claudin 5, occludins, and ZO-1 levels in response to LPS stimulation in a concentration-dependent manner (15–18). A recent human autopsy study showed that claudins and occludins were lost in the cerebral endothelium of patients with fetal sepsis (19). Adherens junctions (AJs), another critical aspect of the BBB, connect the actin cytoskeleton of neighboring endothelial cells and thus strengthen the endothelial integrity of the BBB (20). Importantly, AJs modulate the passage of inflammatory cells such as lymphocytes, monocytes, and neutrophils in systemic inflammation (10). AJs are mainly composed of vascular endothelial cadherin and platelet endothelial cell adhesion molecule-1, the dysregulation of which are responsible for BBB hyperpermeability under inflammatory conditions (21, 22). Gap junctions enable exchange of electrical and metabolic signals between endothelial cells and astrocytes (23), which are dynamically changed in response to extracellular stimulations (24).
Many agents are effective in maintaining BBB integrity and have drawn increasing attention for use in preventing long-term cognitive impairment after sepsis. Biologically, hydrogen (H2) is a selective antioxidant that has been tested for the treatment of graft-versus-host disease, aplastic anemia, and hemorrhage (25). Its low molecular weight makes it easy for H2 to diffuse the BBB (26). Yu and colleagues (27) confirmed that immediate inhalation of 2% H2 after CLP decreased escape latency of mice in the Morris water maze (MWM). Intriguingly, H2 had no protective effect when nuclear factor erythroid-2-related factor 2 (Nrf2) was knocked out in the septic mouse model, indicating that Nrf2 is involved in its protective mechanism. The same group demonstrated improvement of spatial memory in the MWM after H2 inhalation in a chronic sepsis model of intraperitoneal injection of human stool suspension (28). The impaired BBB was again restored. Importantly, H2 treatment significantly upregulated Nrf2 expression, which further validated their previous findings. It is worth mentioning that the disrupted CA1 structure was rearranged and the lost pyramidal neurons were replenished in the CA1 area of the hippocampus after H2 treatment. However, their studies covered only a time window of 10 to 14 days after sepsis. Maintenance of BBB integrity and other beneficial effects of H2 in a longer period need to be verified in the future.
Neuroinflammation is a major component of the etiology of numerous neurological and neurodegenerative diseases (29). During sepsis, following the propagation of peripheral inflammation, the pro-inflammatory signals reach the brain via impaired BBB segments, and eventually trigger neuroinflammation, which is primarily characterized by the activation of microglia. In general, activated microglia have two distinct phenotypes, a pro-inflammatory M1 phenotype and an anti-inflammatory M2 phenotype. In both septic animals and patients, the activation of M1 microglia is consistently detected and widely investigated (30–32). It has been shown that inhibition of microglial activation with minocycline is sufficient to prevent long-term memory impairment of mice undergoing CLP procedure (33). In detail, microglia are resident macrophages of the brain parenchyma and are endowed with a host of membrane pattern recognition receptors for recognizing different pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) generated during sepsis (34). It is worth mentioning that one of the most important DAMPs is high‐mobility group box 1 (HMGB1), which is released by innate immune cells beyond LPS stimulation. Significantly, its persistent high serum level is involved in the pathogenesis of long-term memory impairments of sepsis survivors (35). Among various pattern recognition receptors, Toll-like receptors (TLRs) play unique roles in innate immunity in sepsis by specifically recognizing LPS and other PAMPs or DAMPs. Following the recognition of PAMPs and DAMPs by TLRs, multiple intracellular signaling pathways are activated, which lead to the activation of IκB kinase (IKK). IKK is then ubiquitinated and degraded. Afterwards, the nuclear localization sequences on NF-κB protein are exposed, freeing NF-κB dimer to translocate into the nucleus, where it binds to its consensus sequence on the promoter regions or enhancer regions of targeted genes and initiates the pro-inflammatory gene expression (36–38). Importantly, microglia express most TLRs in the brain, among which TLR2 and TLR4 have been regarded as the main ones involved in neuroinflammation (39, 40). It has been demonstrated that anti-TLR2 and anti-TLR4 treatment can prevent LPS-induced NF-κB activation (41). However, more studies are needed to provide mechanistic insight into how anti-TLR could be effective for avoiding long-term cognitive impairment after sepsis. Additionally, when the NF-κB signaling was activated by microRNA-301b, microglial activation in the hippocampus was then induced and excessive TNF-α and IL-Iβ were secreted, which eventually led to the disruption of spatial learning and memory (42). On the contrary, prophylactic inhibition of NF-κB by deleting IKKβ in microglia attenuated its activation and reduced the pro-inflammatory cytokines such as IL1β, IL6, and TNF-α (43). Therefore, NF-κB should be a potential target for modulating microglia activation status in sepsis.
The imbalance of kynurenine pathway (KP) of tryptophan metabolism in the CNS is another possible mechanism underlying microglia-related long-term cognitive impairment after sepsis. In the beginning of the KP, tryptophan in the brain is metabolized to kynurenine by 2,3-dioxygenase (IDO). Then, the KP segregates into two major branches. In the “neuroprotective” branch of KP, kynurenine is converted to kynurenic acid (KA) mainly in astrocytes. KA is an endogenous antagonist of the N-methyl-D-aspartate receptor (NMDAR) and α7 nicotinic acetylcholine receptor (α7nAChR), both of which are highly implicated in cognitive function. While in the “neurotoxic” branch, kynurenine is converted to quinolinic acid (QA) predominantly in activated microglia. QA is an NMDAR agonist and can also inhibit reuptake of glutamate by astrocytes, leading to excitotoxicity and neuroinflammation (44). In the context of sepsis-induced neuroinflammation, pro-inflammatory cytokines robustly upregulate the expression and activity of brain IDO, resulting in more production of kynurenine. Meanwhile, those cytokines also promote the diversion of the KP towards QA production from kynurenine. As a result, QA is excessively generated by microglia, leading to brain injury and cognitive impairment (45). Vitamin B6 acts as an important cofactor of the critical enzymes in the KP. Danielski et al. revealed that immediate administration of Vitamin B6 after CLP improved non-associative memory of mice in the object recognition test 10 days later (46). Thus, maintaining the homeostasis of the KP plays a key role in neuroinflammation modulation and may also be a possible therapeutic strategy.
The activation of microglia not only results in the activation of transcription factor NF-κB and subsequent pro-inflammatory cascades, but also induces over-generation of reactive oxidative species (ROS). The production of ROS in microglia is mostly mediated by NADPH oxidase (NOX) (47). Immunohistochemistry and single-cell RNA-sec study indicates that NOX2 is the most common NOX isotype in human and rodent microglia (48, 49). NOX2 activation is associated with oxidant production in sepsis while acute pharmacological inhibition of NOX2 with low dose apocynin could reduce oxidative stress and prevent spatial memory impairment in the MWM 15 days after sepsis (50). Notably, apocynin can eliminate memory deficits in the inhibitory avoidance test (50). Similarly, our previous study suggested that 10 consecutive days of apocynin treatment after CLP rescued associative memory as measured by freezing time in the fear conditioning test (51). Vitamin C has long been viewed as an antioxidant and has been applied to treat infection, cancer, and even COVID-19 most recently (52–55). In sepsis, high doses of Vitamin C 24 hours after CLP protected septic animals, including reduced cerebral inflammation, oxidative injury, and subsequent improved spatial memory in the MWM (56). Dimethyl fumarate is usually prescribed for relapsing multiple sclerosis and psoriasis. Its effect in cellular antioxidation and detoxification has attracted increasing attention (57). Zarbato and colleagues reported that dimethyl fumarate reduced ROS almost to the control level in a CLP rat model and reversed memory deficits in the object recognition task 10 days after surgery (58). In addition, antioxidant therapy is also helpful in ameliorating cognitive dysfunction in direct cerebral infection caused by microbials (59, 60). In the future, large-scale clinical studies are warranted to verify the potential of antioxidants for preventing long-term cognitive impairment after sepsis.
In addition to microglial activation, astroglial activation is also involved in neuroinflammation after sepsis. Like microglia, the astroglial pathology is also primarily mediated by TLRs. TLRs on astrocyte membranes are stimulated mainly through binding myeloid differentiation factor 88 or Toll/interferon-1 receptor domain-containing adaptor inducing interferon-β, followed by the induction of the NF-κB signaling as well (61). Moreover, S100B, a ligand of Receptor for Advanced Glycation End products (RAGE), is secreted by astrocytes in the CNS during sepsis, which is also recognized as a DAMP. High levels of S100β stimulates iNOS in astrocytes via activation of the RAGE/NF-κB pathway (62). Although the long-term cognition function was not assessed, Li and colleagues proved that inhibition of S100β/RAGE/NF-κB pathway reduced neuroinflammation, oxidative stress, and reactive gliosis in the hippocampus in the CLP sepsis model (63).
Activation of microglia and astroglia leads to a storm of pro-inflammatory cytokines including IL1-α, IL-1β, TNF-α, IL-6, IL-12, IL-23 inside the cerebral parenchyma (64, 65). On the other hand, the anti-inflammatory cytokines including IL-4 and IL-10 decrease considerably (31). IL-1β is one of the most relevant cytokines for mediating inflammatory injury in sepsis. After secretion, IL-1β causes synapse damage, contributing to short memory deficits in mice (66–68). Intriguingly, the application of IL-1Ra, a competitive inhibitor of IL-1β, can ameliorate synapse loss in cultured neurons in medium derived from cultured microglia with LPS stimulation (66). Moreover, intracisternal injection of IL-1Ra prevented non-associative memory and aversive memory deficits 10 days after CLP (69). These studies indicate that targeting IL-1β is sufficient to prevent long-term cognitive impairment after sepsis. TNF-α is released as a soluble cytokine primarily by microglia in the brain. It’s another crucial terminal product of the neuroinflammation cascade in sepsis. It has been suggested that systemic administration of TNF-α can induce cognitive impairment (70), while anti-TNF-α treatment yields favorable outcomes in diseases with cognitive decline due to neuroinflammation (71–73). In addition, TNF-α-modulating agents such as stains can prevent long-term spatial learning and memory impairment in the MWM in septic animals about 2 weeks after intraperitoneal injection of cecal material or CLP (74, 75). However, clinical trials using anti-TNF-α agents for preventing long-term cognitive impairment are still sparse.
Several other neuroinflammation-related mechanisms of long-term cognitive impairment after sepsis have been recently proposed. For example, neuroinflammation in sepsis is always accompanied by mitochondrial dysfunction which is associated with reduced ATP synthesis, over-production of ROS, and dysregulated apoptosis (76). Indeed, Manfredini et al. proved that activators of mitochondrial biogenesis such as rosiglitazone significantly improved impaired memory of rats 10 days after CLP. CD40-CD40 ligand pathway plays an important role in the neuroinflammation and oxidative stress in a series of disorders. Michels and colleagues concluded that long-term memory deficits identified by the inhibitory test and open-field task can be ameliorated by anti-CD40 treatment (77). Moreover, it has been demonstrated that fish oil-rich emulsion (78) and alpha-lipoic acid (79) may prevent long-term cognitive impairment by attenuating acute neuroinflammation in sepsis. Cerebral monocyte recruitment is another hallmark of neuroinflammation. Mouse monocytes can be divided into two distinct subsets: inflammatory monocytes with Ly6ChighCCR2highCX3CR1low expression and anti-inflammatory monocytes with Ly6ClowCCR2lowCX3CR1high (80). Inflammatory monocytes use chemokine receptor CCR2 for their adhesion to vesicular wall and infiltration into the brain (81). In a model of S. pneumoniae pneumonia-induced sepsis, intravital imaging of CCR2 reporter mice brain showed an increase in the rolling and adhesion of CCR2+ monocytes in the vasculature. This increasing recruitment of CCR2+ monocytes into the brain was followed by microglial activation and neuroinflammation 24 hours after infection. Meanwhile, spatial memory deficits, represented by less travel distance and less duration in the target quadrant in the MWM, appeared 2 weeks later and persisted for at least 9 weeks. Remarkably, anti-CCR2 treatment in the acute phase could abolish the rolling and adhesion of CCR2+ monocytes and thus prevent the long-term spatial memory deficits in the MWM (82). Taken together, intervention by inhibiting inflammatory reaction especially in the early phase should be considered in clinical practice and may serve as a possible therapeutic strategy.
Neurotransmitter dysfunction plays crucial roles in the pathophysiology of sepsis-associated cognitive decline (Figure 1). In the CNS, acetylcholine (ACh) modulates the state of a group of neurons in response to environmental stimulations. Specifically, cholinergic inputs from the medial septum innervate hippocampal circuits and thus regulate hippocampus-dependent cognitive function (83). In fact, acetylcholinesterase (AChE) inhibitors can ameliorate some cognitive symptoms of Alzheimer’s disease by blocking the breakdown of ACh in the brain (84). A significant increase of AChE activity was observed in septic patients experiencing cognitive dysfunction, suggesting that ACh is involved in sepsis-related cognitive impairment (85). In an animal study, the mice made more wrong choices in the 8-arm radial maze test after total recovery from sepsis induced by LPS, which was accompanied by substantial loss of cholinergic boutons in the parietal association and somatosensory cortical areas (86). α7nAChR is the most abundant nicotinic receptor of ACh expressed in the pyramidal neurons and GABAergic neurons in the hippocampus. In particular, α7nAChR plays a distinct role in modulating synaptic activity and plasticity due to its relatively high permeability to calcium. Therefore, α7nAChR agonist has been pursued as a potential therapeutic target for various diseases with cognitive impairments. Recently, selective agonism of α7nAChR has been demonstrated to reduce HMGB1 release and internalization, inhibit the NF-κB pathway (87, 88), attenuate neuroinflammation and significantly increase freezing time of mice in the contextual fear conditioning test (89). Conversely, another study suggests that inhibition of α7nAChR is sufficient to block the protective effects of dexmedetomidine in rescuing impaired cognition in endotoxemia (90). Recently, Tchessalova and Tronson developed a sub-chronic immune challenge model in which they performed intraperitoneal injection of LPS or Polyinosinic:polycytidylic acid intermittently. Using this model, they showed that female mice had significant impairments only in the object recognition, while male mice exhibited broader cognitive impairments in tests including the object recognition, and both context and tone fear conditioning several months later (91). With regard to sex difference, it has been shown that the dysregulated neuronal and synaptic genes in the hippocampus of old male mice recover slower from sepsis than old female mice in the CLP model (92). Interestingly, α7nAChR in the hippocampus of male mice was more vulnerable to sepsis than that of female mice, contributing to worse cognitive performance (93). In addition, genetic knockout of α7nAChRs only worsened the male’s performance in the spatial discrimination task (94). Taken together, α7nAChRs are implicated in the sex difference in susceptibility to long-term cognitive impairment after sepsis. Thus, these findings suggest that sex difference should be taken into consideration when designing α7nAChR-targeting medications for treating sepsis patients.
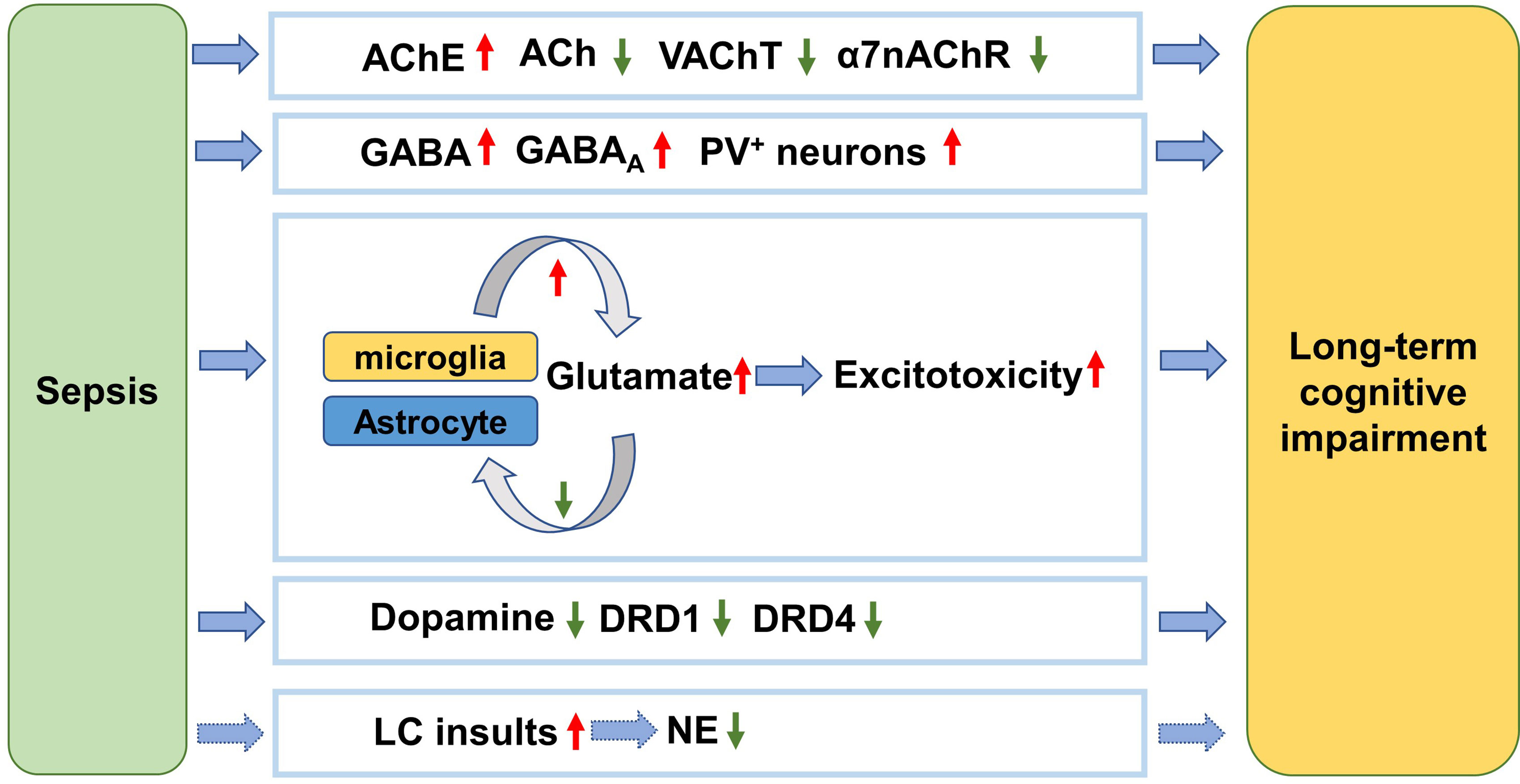
Figure 1 Neurotransmitter dysfunction mechanism underlying long-term cognitive impairment after sepsis. After sepsis, the AChE is activated and the ACh level is thus decreased. Meanwhile the cholinergic innervation is inhibited, as reflected by reduction of VAChT. The α7nAChRs are also downregulated. The suppression of ACh function is associated with cognitive impairment following sepsis. PV+ interneurons are activated after sepsis resulting in cognitive impairment. The activated microglia secret more glutamate while the damaged astrocytes reuptake less glutamate, causing excitotoxicity to the brain and cognitive impairment. The level of dopamine is decreased and the receptors are inhibited by sepsis, which is involved in cognitive impairment. The LC is the only source of NE in the brain. NE level decreases in response to sepsis, which contributes to cognitive impairment. AChE, acetylcholinesterase; ACh, acetylcholine; VAChT, vesicular acetylcholine transporter; α7nAChR, α7-nicotinic acetylcholine receptor; GABA, Gamma-aminobutyric acid; GABAA, GABA A receptor; PV, parvalbumin; DRD1, dopamine receptor D1; DRD4, dopamine receptor D4; LC, locus coeruleus; NE, norepinephrine.
In the hippocampus, GABAergic interneurons expressing parvalbumin regulate memory and learning (95, 96). Under LPS challenge, the number of parvalbumin-positive inhibitory synapses and frequency of miniature inhibitory postsynaptic currents obviously increased, indicating elevated GABAergic transmission of hippocampal neuronal circuits (97). Concomitantly, the declining cognitive function was identified by spontaneous alternation in the Y maze test, while selective blocking of GABAA receptor could reverse LPS-induced cognitive impairment (97). In contrast to GABA, glutamate is a prevalent excitatory neurotransmitter. In the septic brain, the activated microglia acquire an M1 phenotype and then produces excessive amounts of glutamate into the extracellular space, leading to exacerbated excitotoxicity to neuronal circuits and cognitive deficits. On the other hand, astrocytes are the primary glutamate buffering cells, maintaining the homeostasis of glutamate (98). However, astrocytes are damaged and lost during sepsis, presumably resulting in decreased glutamate uptake (99). Furthermore, astrocytes themselves also secrete glutamate and increase the excitability of adjacent neurons (100). Guanosine exerts neuroprotective effects probably by decreasing extracellular glutamate level and augment astrocytic glutamate uptake. Interestingly, guanosine was proven to eliminate the detrimental effects of CLP on rats’ memory function (101). These results suggest that glutamate dysregulation is involved in cognitive impairment following sepsis.
A recent study reported decreased dopamine levels in the hippocampus after CLP, which was associated with lower discrimination index in the object recognition test 12 days later and increasing escape latency ain the Barnes maze test 14 days later (102). However, intraperitoneal administration of L-dopa/Benserazide hydrochloride (L-DA) overtly reduced the percentage of activated microglia in hippocampus. Meanwhile, mRNA levels of pro-inflammatory cytokines such as TNF-α, IL-1α and IL-1β were significantly downregulated. Meaningfully, L-DA restored memory and learning function of septic mice in the object recognition test and the Barnes maze test. The authors also found that the L-DA’s inhibition of microglial activation was abolished when dopamine D1 receptor antagonist was added. However, it should be noted that L-DA was effective only when it was applied in the acute phase of sepsis (102). Moreover, significant dopamine D4 receptor loss was also reported in mice suffering from cognitive impairment in a CLP model while agonism of D4 was sufficient to rescue cognitive abnormalities (103).
Norepinephrine (NE) is another important neurotransmitter regulating cognition such as episodic memory and working memory (104). To date, there is sparse evidence that directly links NE dysregulation to long-term cognitive impairment after sepsis. In the CNS, most of the NE is released from the locus coeruleus (LC) neurons in the brain stem (105). LC neurons are vulnerable to toxins and inflammation due to their high energetic requirement (106). It can be assumed that overwhelming sepsis would injure LC neurons and thus cause NE deficits. Therefore, it’s worth testing NE modulators such as some antidepressants for preventing long-term cognitive impairment after sepsis in future (106).
Neuronal loss has been identified to be associated with long-term cognitive impairment in both septic animals and patients (Table 1). As for adult animals, Semmler et al. (86) showed that rats supposed to recover from sepsis still had neuronal loss in the CA1 and CA2 of the hippocampus and the prefrontal cortex 12 weeks after LPS exposure. Meanwhile, the rats spent more time in the corners in the open field test and had less correct entries during training trials in the 8-arm radial maze test, indicating impaired cognitive function. In adult mice undergoing CLP, several studies reported progressive and irreversible neuronal loss in CA1 and CA3, which was linked to longer escape latency in the MWM (74, 107, 108). In neonatal rats treated with LPS, loss of dopaminergic neurons in mesencephalic substantia nigra was revealed when they showed long-term impairment in locomotor function and spatial memory (110). In neonatal mice, LPS-induced systemic inflammation resulted in significant cerebellar atrophy as well as neuronal loss in the CA3 and the pons at least for 9 days (99). Interestingly, it was also found that sepsis caused loss of neural stem cell in the DG of mice, which contributed to hippocampal neurogenesis and cognitive impairment (114). Besides direct neuronal loss, Semmler et al. (86) noted that LPS caused a reduction of cholinergic innervation in the parietal cortex. In addition, Huerta et al. (115) provided data showing that CLP induced substantial decline of dendritic spine density of excitatory neurons in the basolateral nucleus of the amygdala and granule cells in the dentate gyrus (DG). Consequently, the mice still had contextual fear memory impairment 78 to 176 days after CLP. Hence, it can be inferred that the loss of neuronal synapse due to sepsis may also be implicated in sustained cognitive impairment. Nevertheless, there are also inconsistent results. For instance, some other researchers argued that mice with long-term cognitive impairment had no significant hippocampal volume reduction in MRI or obvious changes of cellularity of the DG region (82). In the future, novel approaches for detection of neuronal loss locally and globally will broaden our understanding of its role in sepsis-induced long-term cognitive impairment.
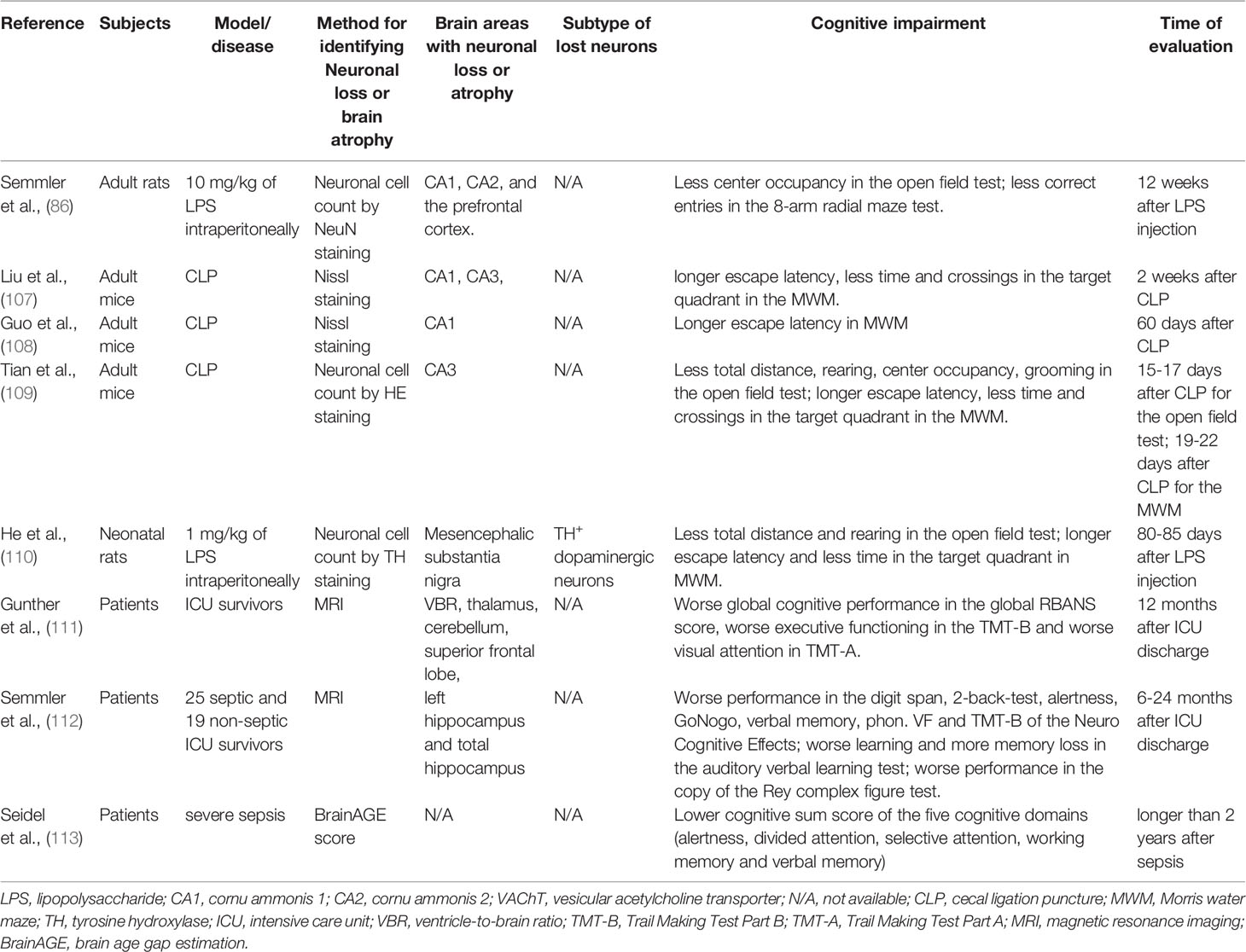
Table 1 Representative studies of neuronal loss mechanism underlying long-term cognitive impairment after sepsis.
As for septic patients, magnetic resonance imaging (MRI) technique has been the first choice for detecting brain atrophy due to neuronal loss. For example, a clinical study investigated the brain volume and cognition of sepsis patients. The results suggested that greater brain atrophy 3 months after sepsis could predict worse cognitive performance at 12 months (111). The affected areas included the frontal lobes, thalamus, and cerebellum. Another MRI study presented evidence of prominent atrophy of the left hippocampus in sepsis survivors who had permanent cognitive deficits in verbal learning and memory 6 to 24 months after discharge from the ICU (112). In patients with sepsis-induced brain dysfunction, Orhun and colleagues (116) found prominent atrophy of gay matter in several limbic structures including the temporal lobe and insula via a voxel-based morphometry analysis base on MRI. However, they (116) later identified atrophy of cerebral and cerebellar white matter, cerebral cortex, hippocampus, and amygdala whereas the brainstem, cerebellar cortex, and deep gray matter structures were relatively resistant to septic damage (117). The discrepancy of affected regions of the two studies might be attributed to the sepsis stages of patients, which indicated that the brain may undergo a dynamic alteration in response to sepsis. Based on structural MRI, the “brain age gap estimation” (BrainAGE) score reflects the age-specific grey matter atrophy of the whole brain (118). Seidel et al. (113) followed up 20 patients with severe sepsis and reported that patients suffered from cognitive impairment 2 years later or longer. It is worth mentioning that the severity of the patients’ cognitive impairment was closely associated with their BrainAGE scores (113).
Accumulating evidence has provided mechanistic insights into neuronal loss in sepsis and subsequent long-term cognitive impairment. Apoptosis (119), autophagy (120), and pyroptosis (121) have been identified as possible mechanisms of neuronal loss in sepsis (Figure 2). In response to sepsis, mitochondrial dysfunction, and over-production of ROS, apoptosis and autophagy are stimulated to regulate the death of neuronal cells (122). Indeed, by consuming and recycling macromolecules and damaged organelles, autophagy restores physiological ROS level and maintains cellular homeostasis. Alternatively, when ROS is produced excessively, apoptosis will be initiated to clear damaged cells (123).
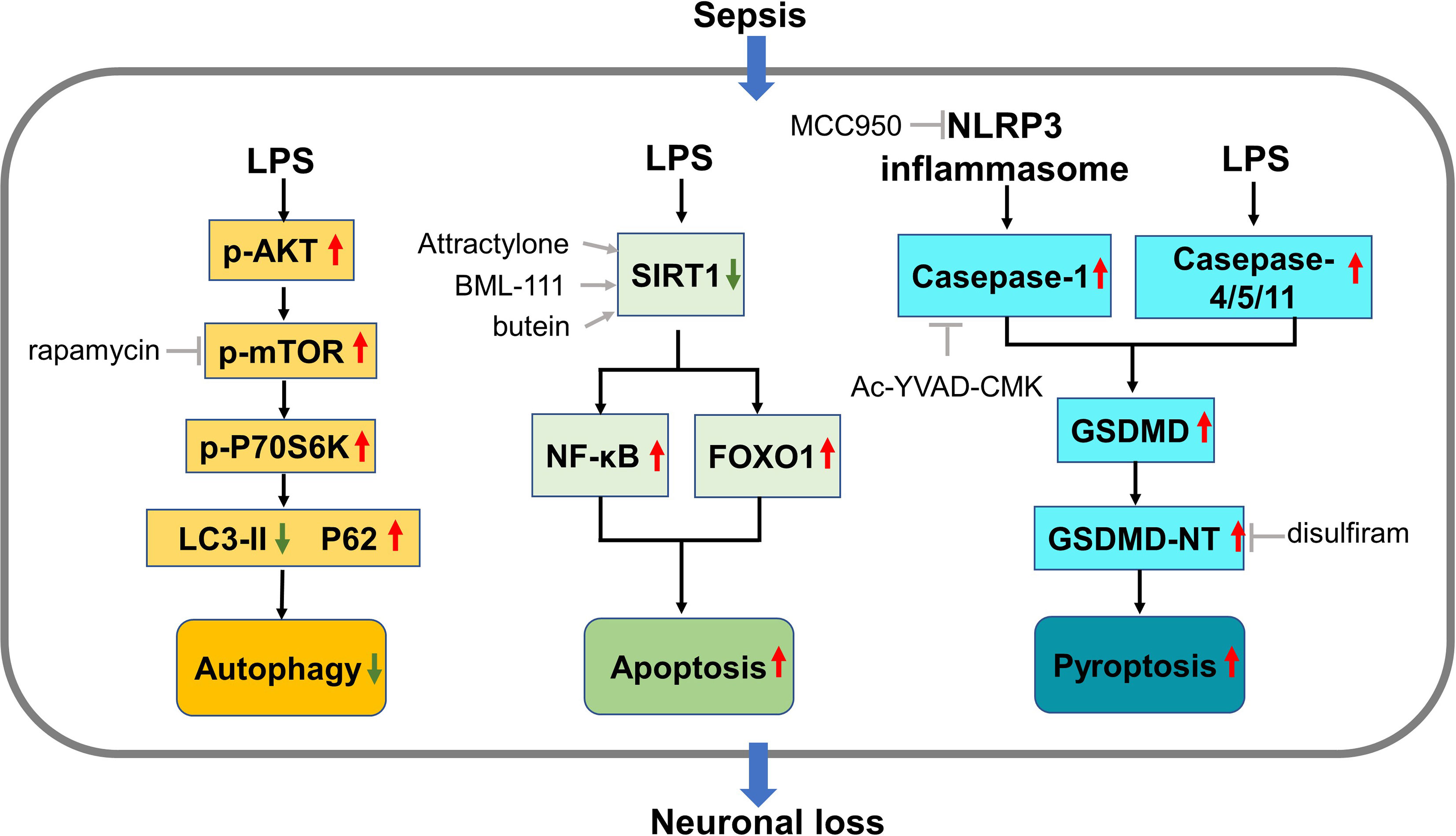
Figure 2 Mechanisms of neuronal loss in sepsis. During sepsis, the AKT/mTOR pathway is activated and inhibits the normal autophagy process in neurons, leading to autophagic neuronal death. Suppression of SIRT1 by LPS activates NF-κB and FOXO1, triggering neuron apoptosis. Inflammasome generated in sepsis activates casepase-1 while LPS activates casepase-4/5/11, both of which promotes the generation of GSDMD. Cleavage of GSDMD produces GSDMD-NT which forms membrane pores and lead to neuronal pyroptosis. LPS, lipopolysaccharides; mTOR, mammalian/mechanistic target of rapamycin; LC3, SIRT1, silent information regulator 1; FOXO1, forkhead box protein O1; GSDMD, gasdermin D; GSDMD-NT, GSDMD-N-terminal products.
It has been acknowledged that enhancing autophagy may prevent cognitive impairment in sepsis by reducing neuronal cell damage (124). One of the most widely investigated pathways implicated in this process is the AKT/mTOR pathway. Generally, it participates in the neuronal development, survival and functioning of mature neurons, and is implicated in neuronal death in brain injury and diseases (125). Specifically, this pathway plays an indispensable role in the initiation of autophagy (123). For example, when CLP caused neuronal loss and subsequent long-term cognitive impairment, it upregulated the phosphorylation of key components of the AKT/mTOR pathway in the hippocampal neurons of mice, which suggested the activation of this pathway and suppression of autophagy. Concomitantly, the autophagy markers such as P62 and LC3 were reduced by CLP, indicating suppression of autophagy. Intriguingly, administration of rapamycin, an inhibitor of the AKT/mTOR pathway, rescued spatial memory by restoring the autophagic activity (107). The same team later reported that, along with the long-term neuronal loss in the CA1 region, the Akt/mTOR signaling was increasingly activated over time (from 14 to 60 days) after CLP (108). However, rapamycin was only effective when it’s used at 14 days but not 60 days after CLP, indicating that the neuronal death is irreversible and timely intervention is necessary. Inconsistently, other authors pointed out that the Akt/mTOR signaling was suppressed in the CLP model (126). In addition, by restoring suppressed Akt/mTOR signaling, recombinant human erythropoietin exerted neuroprotective effect and rescued the spatial memory 1 week after CLP. Moreover, the beneficial outcomes of exogenous recombinant human erythropoietin were abolished by inhibiting this pathway by using rapamycin (126). These contradictory results suggest that researchers should deeply explore the complicated role of Akt/mTOR in autophagy in sepsis.
Apoptotic neurons are repeatedly observed in the hippocampus of CLP model, contributing to permanent neuronal loss and cognitive impairment (127–130). Moreover, pre-treatment with medications that inhibit caspase-dependent apoptosis can attenuate the cognitive dysfunction 7 days after CLP (129). Sirtuins are histone deacetylases implicated in numerous cellular processes. As a member of the sirtuin family, silent information regulator 1 (SIRT1) plays an important role in the modulation of apoptosis (131, 132). Intriguingly, several teams consistently revealed that SIRT-activating agents can alleviate long-term cognitive impairment after sepsis by reducing neuronal apoptosis (109, 130, 133).
Pyroptosis is a rapid form of programmed cell death mediated by gasdermin D (GSDMD). It can be categorized into a canonical pathway mediated by casepase-1 and a noncanonical pathway mediated by casepase-4/5/11. In the canonical pathway, inflammasomes generated in response to external stimuli promotes the maturation of casepase-1. Casepase-1 then promotes the maturation of IL-1β and IL-18. Meanwhile, Casepase-1 further cleaves GSDMD into N- and C-terminal components (GSDMD-NT and -CT). GSDMD-NT can insert into and form pores in plasma membranes, leading to lytic cell death and secretion of mature IL-1β and IL-18. Similarly, in the noncanonical pathway, cytosolic LPS is sensed by casepase-4/5/11, which are in turn activated and cleave GSDMD into GSDMD-NT. In sepsis, both pathways can be activated (134). The NOD-like receptor and pyrin domain containing 3 (NLRP3) is one of the most-explored inflammasomes involved in the canonical pathway (135). Yang et al. (136) found that prophylactic delivery of either NLRP3 inhibitor or casepase-1 inhibitor before CLP surgery protected CA1 neurons from degeneration and thus preserved learning and memory function of mice 2 weeks later. Even 3 months after LPS stimulation, the knockout of NLRP3 protected neuronal structure including dendritic arbors and spines as well as synaptic plasticity (137). A recent report evidenced that disulfiram, an alcohol deterrent, can interfere with GSDMD-mediated pore-forming, making it an attractive medication for treating sepsis-associated long-term cognitive impairment (138).
Like dementia, long-term cognitive impairment after sepsis is almost irreversible, which decreases life quality of patients and increases economic burden. When treating septic patients, clinicians should be alert to this sequela and act as early as possible. As for the mechanisms, long-term cognitive impairment is profoundly associated with a cascade of several detrimental events, including BBB disruption, neuroinflammation, neurotransmitter dysfunction, and neuronal loss (Figure 3). Based on these mechanisms, various interventions have been tested in numerous animal studies. The positive results indicate that targeting certain critical steps of those pathologic events will be promising for preventing and treating long-term cognitive impairment after sepsis. Despite significant advances having been made in our understanding of sepsis -related long-term cognitive impairment, several questions remain. Firstly, the complex interplay among these mechanisms should be taken into consideration when designing pre-clinical and clinical studies. Secondly, there has been some criticism about the translational value of current animal models of sepsis, particularly for the CLP model (139). Finally, there is currently no consensus on the timelines for long-term cognitive assessment after sepsis. Addressing these important issues will significantly improve our understanding of long-term cognitive impairment after sepsis.
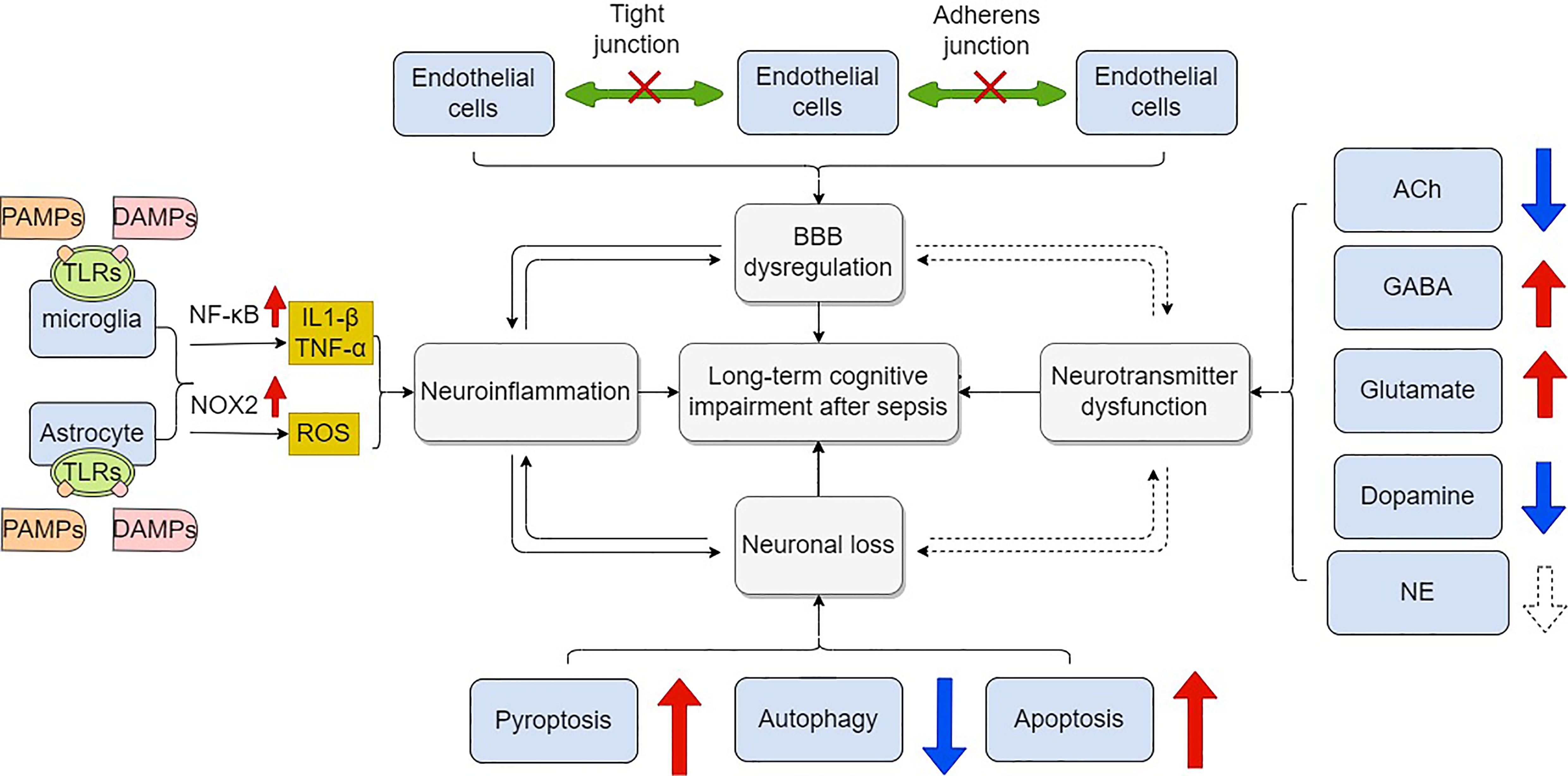
Figure 3 Mechanisms of long –term cognitive impairment after sepsis. Blood brain barrier is composed of endothelial cells, astrocytes and pericytes. These cells are connected by tight junctions, adherens junctions and gap junctions. Dysfunction of these components leads to increased permeability of BBB in response to sepsis, which facilitates long-term cognitive impairment. Microglia and astrocyte are over-activated mainly through the activation of NF-κB signaling when PAMPs and DAMPs including LPS by binding to their ligands (namely, TLRs) on cell membrane. Pro-inflammatory cytokines are subsequently over-produced while anti-inflammatory cytokines are down-regulated. Neuroinflammation further leads to cognitive impairment. Overproduction of ROS is medicated by NOX2 in activated microglia and astrocytes. Neuronal loss is common in sepsis. Apoptosis, autophagy, and pyroptosis are involved in the modulation of neuronal death. Finally, dysregulation of neurotransmitters including ACh, GABA, glutamate, dopamine and NE also contributes to cognitive impairment in sepsis. BBB, blood brain barrier; PAMPs, pathogen-associated molecular patterns; DAMPs, damage-associated molecular patterns; LPS, lipopolysaccharide; TLRs, toll like receptors; ROS, reactive oxidative species; NOX2, NADPH oxidase 2; ACh, acetylcholine; GABA, Gamma-aminobutyric acid; NE, norepinephrine.
YL drafted the first version of the manuscript. MJ revised the manuscript. JY approved the final manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the grants from the National Natural Science Foundation of China (81772126, 81971892, 81971020).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank Matthew Parry for proofreading the manuscript.
1. Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-Term Cognitive Impairment and Functional Disability Among Survivors of Severe Sepsis. Jama (2010) 304(16):1787–94. doi: 10.1001/jama.2010.1553
2. Prescott HC, Angus DC. Enhancing Recovery From Sepsis: A Review. Jama (2018) 319(1):62–75. doi: 10.1001/jama.2017.17687
3. Calsavara AJC, Costa PA, Nobre V, Teixeira AL. Factors Associated With Short and Long Term Cognitive Changes in Patients With Sepsis. Sci Rep (2018) 8(1):4509. doi: 10.1038/s41598-018-22754-3
4. Brummel NE, Bell SP, Girard TD, Pandharipande PP, Jackson JC, Morandi A, et al. Frailty and Subsequent Disability and Mortality Among Patients With Critical Illness. Am J Respir Crit Care Med (2017) 196(1):64–72. doi: 10.1164/rccm.201605-0939OC
5. Bruck E, Schandl A, Bottai M, Sackey P. The Impact of Sepsis, Delirium, and Psychological Distress on Self-Rated Cognitive Function in ICU Survivors-a Prospective Cohort Study. J Intensive Care (2018) 6:2. doi: 10.1186/s40560-017-0272-6
6. Rengel KF, Hayhurst CJ, Pandharipande PP, Hughes CG. Long-Term Cognitive and Functional Impairments After Critical Illness. Anesth Analg (2019) 128(4):772–80. doi: 10.1213/ANE.0000000000004066
7. Nwafor DC, Brichacek AL, Mohammad AS, Griffith J, Lucke-Wold BP, Benkovic SA, et al. Targeting the Blood-Brain Barrier to Prevent Sepsis-Associated Cognitive Impairment. J Cent Nerv Syst Dis (2019) 11:1179573519840652. doi: 10.1177/1179573519840652
8. Barichello T, Sayana P, Giridharan VV, Arumanayagam AS, Narendran B, Della Giustina A, et al. Long-Term Cognitive Outcomes After Sepsis: A Translational Systematic Review. Mol Neurobiol (2019) 56(1):186–251. doi: 10.1007/s12035-018-1048-2
9. Schaeffer S, Iadecola C. Revisiting the Neurovascular Unit. Nat Neurosci (2021) 24(9):1198–209. doi: 10.1038/s41593-021-00904-7
10. Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol Rev (2019) 99(1):21–78. doi: 10.1152/physrev.00050.2017
11. Zeng H, He X, Tuo Q-H, Liao D-F, Zhang G-Q, Chen J-X. LPS Causes Pericyte Loss and Microvascular Dysfunction via Disruption of Sirt3/angiopoietins/Tie-2 and HIF-2α/Notch3 Pathways. Sci Rep (2016) 6:20931–1. doi: 10.1038/srep20931
12. Ince C, Mayeux PR, Nguyen T, Gomez H, Kellum JA, Ospina-Tascon GA, et al. The Endothelium in Sepsis. Shock (Augusta Ga) (2016) 45(3):259–70. doi: 10.1097/SHK.0000000000000473
13. Hughes CG, Patel MB, Brummel NE, Thompson JL, McNeil JB, Pandharipande PP, et al. Relationships Between Markers of Neurologic and Endothelial Injury During Critical Illness and Long-Term Cognitive Impairment and Disability. Intensive Care Med (2018) 44(3):345–55. doi: 10.1007/s00134-018-5120-1
14. Lochhead JJ, Yang J, Ronaldson PT, Davis TP. Structure, Function, and Regulation of the Blood-Brain Barrier Tight Junction in Central Nervous System Disorders. Front Physiol (2020) 11:914. doi: 10.3389/fphys.2020.00914
15. Qin LH, Huang W, Mo XA, Chen YL, Wu XH. LPS Induces Occludin Dysregulation in Cerebral Microvascular Endothelial Cells via MAPK Signaling and Augmenting MMP-2 Levels. Oxid Med Cell Longev (2015) 2015:120641. doi: 10.1155/2015/120641
16. Hu M, Liu B. Resveratrol Attenuates Lipopolysaccharide-Induced Dysfunction of Blood-Brain Barrier in Endothelial Cells via AMPK Activation. Korean J Physiol Pharmacol (2016) 20(4):325–32. doi: 10.4196/kjpp.2016.20.4.325
17. Wu XX, Huang XL, Chen RR, Li T, Ye HJ, Xie W, et al. Paeoniflorin Prevents Intestinal Barrier Disruption and Inhibits Lipopolysaccharide (LPS)-Induced Inflammation in Caco-2 Cell Monolayers. Inflammation (2019) 42(6):2215–25. doi: 10.1007/s10753-019-01085-z
18. Sheth P, Delos Santos N, Seth A, LaRusso NF, Rao RK. Lipopolysaccharide Disrupts Tight Junctions in Cholangiocyte Monolayers by a C-Src-, TLR4-, and LBP-Dependent Mechanism. Am J Physiol Gastrointest Liver Physiol (2007) 293(1):G308–18. doi: 10.1152/ajpgi.00582.2006
19. Erikson K, Tuominen H, Vakkala M, Liisanantti JH, Karttunen T, Syrjala H, et al. Brain Tight Junction Protein Expression in Sepsis in an Autopsy Series. Crit Care (2020) 24(1):385. doi: 10.1186/s13054-020-03101-3
20. Stamatovic SM, Johnson AM, Keep RF, Andjelkovic AV. Junctional Proteins of the Blood-Brain Barrier: New Insights Into Function and Dysfunction. Tissue Barriers (2016) 4(1):e1154641. doi: 10.1080/21688370.2016.1154641
21. Yan Z, Wang ZG, Segev N, Hu S, Minshall RD, Dull RO, et al. Rab11a Mediates Vascular Endothelial-Cadherin Recycling and Controls Endothelial Barrier Function. Arterioscler Thromb Vasc Biol (2016) 36(2):339–49. doi: 10.1161/ATVBAHA.115.306549
22. Radeva MY, Waschke J. Mind the Gap: Mechanisms Regulating the Endothelial Barrier. Acta Physiol (Oxf) (2018) 222(1):e12860. doi: 10.1111/apha.12860
23. Totland MZ, Rasmussen NL, Knudsen LM, Leithe E. Regulation of Gap Junction Intercellular Communication by Connexin Ubiquitination: Physiological and Pathophysiological Implications. Cell Mol Life Sci CMLS (2020) 77(4):573–91. doi: 10.1007/s00018-019-03285-0
24. Goodenough DA, Paul DL. Gap Junctions. Cold Spring Harb Perspect Biol (2009) 1(1):a002576. doi: 10.1101/cshperspect.a002576
25. Qian L, Wu Z, Cen J, Pasca S, Tomuleasa C. Medical Application of Hydrogen in Hematological Diseases. Oxid Med Cell Longev (2019) 2019:3917393. doi: 10.1155/2019/3917393
26. Xuemei Ma FX. Molecular Hydrogen and its Potential Application in Therapy of Brain Disorders. Brain Disord Ther (2015) 04(01):154. doi: 10.4172/2168-975X.1000154
27. Yu Y, Feng J, Lian N, Yang M, Xie K, Wang G, et al. Hydrogen Gas Alleviates Blood-Brain Barrier Impairment and Cognitive Dysfunction of Septic Mice in an Nrf2-Dependent Pathway. Int Immunopharmacol (2020) 85:106585. doi: 10.1016/j.intimp.2020.106585
28. Jiang Y, Zhang K, Yu Y, Wang Y, Lian N, Xie K, et al. Molecular Hydrogen Alleviates Brain Injury and Cognitive Impairment in a Chronic Sequelae Model of Murine Polymicrobial Sepsis. Exp Brain Res (2020) 238(12):2897–908. doi: 10.1007/s00221-020-05950-4
29. Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell (2018) 173(5):1073–81. doi: 10.1016/j.cell.2018.05.003
30. Hoogland IC, Houbolt C, van Westerloo DJ, van Gool WA, van de Beek D. Systemic Inflammation and Microglial Activation: Systematic Review of Animal Experiments. J Neuroinflamm (2015) 12:114. doi: 10.1186/s12974-015-0332-6
31. Zhao J, Bi W, Xiao S, Lan X, Cheng X, Zhang J, et al. Neuroinflammation Induced by Lipopolysaccharide Causes Cognitive Impairment in Mice. Sci Rep (2019) 9(1):5790. doi: 10.1038/s41598-019-42286-8
32. Lemstra AW, Groen in't Woud JC, Hoozemans JJ, van Haastert ES, Rozemuller AJ, Eikelenboom P, et al. Microglia Activation in Sepsis: A Case-Control Study. J Neuroinflamm (2007) 4:4. doi: 10.1186/1742-2094-4-4
33. Michels M, Vieira AS, Vuolo F, Zapelini HG, Mendonça B, Mina F, et al. The Role of Microglia Activation in the Development of Sepsis-Induced Long-Term Cognitive Impairment. Brain Behav Immun (2015) 43:54–9. doi: 10.1016/j.bbi.2014.07.002
34. Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, et al. The Microglial Sensome Revealed by Direct RNA Sequencing. Nat Neurosci (2013) 16(12):1896–905. doi: 10.1038/nn.3554
35. Chavan SS, Huerta PT, Robbiati S, Valdes-Ferrer SI, Ochani M, Dancho M, et al. HMGB1 Mediates Cognitive Impairment in Sepsis Survivors. Mol Med (Cambridge Mass) (2012) 18(1):930–7. doi: 10.2119/molmed.2012.00195
36. Liu SF, Malik AB. NF-Kappa B Activation as a Pathological Mechanism of Septic Shock and Inflammation. Am J Physiol Lung Cell Mol Physiol (2006) 290(4):L622–45. doi: 10.1152/ajplung.00477.2005
37. Taniguchi K, Karin M. NF-Kappab, Inflammation, Immunity and Cancer: Coming of Age. Nat Rev Immunol (2018) 18(5):309–24. doi: 10.1038/nri.2017.142
38. Williams LM, Gilmore TD. Looking Down on NF-KappaB. Mol Cell Biol (2020) 40(15):e00104-20. doi: 10.1128/MCB.00104-20
39. Tsujimoto H, Ono S, Efron PA, Scumpia PO, Moldawer LL, Mochizuki H. Role of Toll-Like Receptors in the Development of Sepsis. Shock (Augusta Ga) (2008) 29(3):315–21. doi: 10.1097/SHK.0b013e318157ee55
40. Lorne E, Dupont H, Abraham E. Toll-Like Receptors 2 and 4: Initiators of non-Septic Inflammation in Critical Care Medicine? Intensive Care Med (2010) 36(11):1826–35. doi: 10.1007/s00134-010-1983-5
41. Lima CX, Souza DG, Amaral FA, Fagundes CT, Rodrigues IP, Alves-Filho JC, et al. Therapeutic Effects of Treatment With Anti-TLR2 and Anti-TLR4 Monoclonal Antibodies in Polymicrobial Sepsis. PloS One (2015) 10(7):e0132336. doi: 10.1371/journal.pone.0132336
42. Tang CZ, Zhang DF, Yang JT, Liu QH, Wang YR, Wang WS. Overexpression of microRNA-301b Accelerates Hippocampal Microglia Activation and Cognitive Impairment in Mice With Depressive-Like Behavior Through the NF-kappaB Signaling Pathway. Cell Death Dis (2019) 10(4):316. doi: 10.1038/s41419-019-1522-4
43. Zaghloul N, Kurepa D, Bader MY, Nagy N, Ahmed MN. Prophylactic Inhibition of NF-kappaB Expression in Microglia Leads to Attenuation of Hypoxic Ischemic Injury of the Immature Brain. J Neuroinflamm (2020) 17(1):365. doi: 10.1186/s12974-020-02031-9
44. Mithaiwala MN, Santana-Coelho D, Porter GA, O'Connor JC. Neuroinflammation and the Kynurenine Pathway in CNS Disease: Molecular Mechanisms and Therapeutic Implications. Cells (2021) 10(6):1548. doi: 10.3390/cells10061548
45. Verdonk F, Petit AC, Abdel-Ahad P, Vinckier F, Jouvion G, de Maricourt P, et al. Microglial Production of Quinolinic Acid as a Target and a Biomarker of the Antidepressant Effect of Ketamine. Brain Behav Immun (2019) 81:361–73. doi: 10.1016/j.bbi.2019.06.033
46. Danielski LG, Giustina AD, Goldim MP, Florentino D, Mathias K, Garbossa L, et al. Vitamin B(6) Reduces Neurochemical and Long-Term Cognitive Alterations After Polymicrobial Sepsis: Involvement of the Kynurenine Pathway Modulation. Mol Neurobiol (2018) 55(6):5255–68. doi: 10.1007/s12035-017-0706-0
47. Haslund-Vinding J, McBean G, Jaquet V, Vilhardt F. NADPH Oxidases in Oxidant Production by Microglia: Activating Receptors, Pharmacology and Association With Disease. Br J Pharmacol (2017) 174(12):1733–49. doi: 10.1111/bph.13425
48. Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J Neurosci (2014) 34(36):11929–47. doi: 10.1523/JNEUROSCI.1860-14.2014
49. Sorce S, Nuvolone M, Keller A, Falsig J, Varol A, Schwarz P, et al. The Role of the NADPH Oxidase NOX2 in Prion Pathogenesis. PloS Pathog (2014) 10(12):e1004531. doi: 10.1371/journal.ppat.1004531
50. Hernandes MS, D'Avila JC, Trevelin SC, Reis PA, Kinjo ER, Lopes LR, et al. The Role of Nox2-Derived ROS in the Development of Cognitive Impairment After Sepsis. J Neuroinflamm (2014) 11:36. doi: 10.1186/1742-2094-11-36
51. Ji MH, Qiu LL, Tang H, Ju LS, Sun XR, Zhang H, et al. Sepsis-Induced Selective Parvalbumin Interneuron Phenotype Loss and Cognitive Impairments may be Mediated by NADPH Oxidase 2 Activation in Mice. J Neuroinflamm (2015) 12:182. doi: 10.1186/s12974-015-0401-x
52. Padayatty SJ, Katz A, Wang Y, Eck P, Kwon O, Lee JH, et al. Vitamin C as an Antioxidant: Evaluation of its Role in Disease Prevention. J Am Coll Nutr (2003) 22(1):18–35. doi: 10.1080/07315724.2003.10719272
53. Ngo B, Van Riper JM, Cantley LC, Yun J. Targeting Cancer Vulnerabilities With High-Dose Vitamin C. Nat Rev Cancer (2019) 19(5):271–82. doi: 10.1038/s41568-019-0135-7
54. Carita AC, Fonseca-Santos B, Shultz JD, Michniak-Kohn B, Chorilli M, Leonardi GR. Vitamin C: One Compound, Several Uses. Advances for Delivery, Efficiency and Stability. Nanomedicine (2020) 24:102117. doi: 10.1016/j.nano.2019.102117
55. Cerullo G, Negro M, Parimbelli M, Pecoraro M, Perna S, Liguori G, et al. The Long History of Vitamin C: From Prevention of the Common Cold to Potential Aid in the Treatment of COVID-19. Front Immunol (2020) 11:574029. doi: 10.3389/fimmu.2020.574029
56. Zhang N, Zhao W, Hu ZJ, Ge SM, Huo Y, Liu LX, et al. Protective Effects and Mechanisms of High-Dose Vitamin C on Sepsis-Associated Cognitive Impairment in Rats. Sci Rep (2021) 11(1):14511. doi: 10.1038/s41598-021-93861-x
57. Saidu NEB, Kavian N, Leroy K, Jacob C, Nicco C, Batteux F, et al. Dimethyl Fumarate, a Two-Edged Drug: Current Status and Future Directions. Med Res Rev (2019) 39(5):1923–52. doi: 10.1002/med.21567
58. Zarbato GF, de Souza Goldim MP, Giustina AD, Danielski LG, Mathias K, Florentino D, et al. Dimethyl Fumarate Limits Neuroinflammation and Oxidative Stress and Improves Cognitive Impairment After Polymicrobial Sepsis. Neurotoxicity Res (2018) 34(3):418–30. doi: 10.1007/s12640-018-9900-8
59. Reis PA, Comim CM, Hermani F, Silva B, Barichello T, Portella AC, et al. Cognitive Dysfunction is Sustained After Rescue Therapy in Experimental Cerebral Malaria, and is Reduced by Additive Antioxidant Therapy. PloS Pathog (2010) 6(6):e1000963. doi: 10.1371/journal.ppat.1000963
60. Barichello T, Santos AL, Savi GD, Generoso JS, Otaran P, Michelon CM, et al. Antioxidant Treatment Prevents Cognitive Impairment and Oxidative Damage in Pneumococcal Meningitis Survivor Rats. Metab Brain Dis (2012) 27(4):587–93. doi: 10.1007/s11011-012-9315-9
61. Gorina R, Font-Nieves M, Márquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 Activation Induces a Proinflammatory Environment Through the Interplay Between MyD88-Dependent Nfκb Signaling, MAPK, and Jak1/Stat1 Pathways. Glia (2011) 59(2):242–55. doi: 10.1002/glia.21094
62. Angelo MF, Aguirre A, Aviles Reyes RX, Villarreal A, Lukin J, Melendez M, et al. The Proinflammatory RAGE/NF-KappaB Pathway is Involved in Neuronal Damage and Reactive Gliosis in a Model of Sleep Apnea by Intermittent Hypoxia. PloS One (2014) 9(9):e107901. doi: 10.1371/journal.pone.0107901
63. Huang L, Zhang L, Liu Z, Zhao S, Xu D, Li L, et al. Pentamidine Protects Mice From Cecal Ligation and Puncture-Induced Brain Damage via Inhibiting S100B/RAGE/NF-Kappab. Biochem Biophys Res Commun (2019) 517(2):221–6. doi: 10.1016/j.bbrc.2019.07.045
64. Moraes CA, Zaverucha-do-Valle C, Fleurance R, Sharshar T, Bozza FA, d'Avila JC. Neuroinflammation in Sepsis: Molecular Pathways of Microglia Activation. Pharm (Basel) (2021) 14(5):416. doi: 10.3390/ph14050416
65. Shulyatnikova T, Verkhratsky A. Astroglia in Sepsis Associated Encephalopathy. Neurochem Res (2020) 45(1):83–99. doi: 10.1007/s11064-019-02743-2
66. Moraes CA, Santos G, de Sampaio e Spohr TC, D'Avila JC, Lima FR, Benjamim CF, et al. Activated Microglia-Induced Deficits in Excitatory Synapses Through IL-1β: Implications for Cognitive Impairment in Sepsis. Mol Neurobiol (2015) 52(1):653–63. doi: 10.1007/s12035-014-8868-5
67. Mishra A, Kim HJ, Shin AH, Thayer SA. Synapse Loss Induced by Interleukin-1beta Requires Pre- and Post-Synaptic Mechanisms. J Neuroimmune Pharmacol (2012) 7(3):571–8. doi: 10.1007/s11481-012-9342-7
68. Kim HJ, Shin AH, Thayer SA. Activation of Cannabinoid Type 2 Receptors Inhibits HIV-1 Envelope Glycoprotein Gp120-Induced Synapse Loss. Mol Pharmacol (2011) 80(3):357–66. doi: 10.1124/mol.111.071647
69. Mina F, Comim CM, Dominguini D, Cassol OJ Jr, Dall Igna DM, Ferreira GK, et al. Il1-β Involvement in Cognitive Impairment After Sepsis. Mol Neurobiol (2014) 49(2):1069–76. doi: 10.1007/s12035-013-8581-9
70. Hennessy E, Gormley S, Lopez-Rodriguez AB, Murray C, Murray C, Cunningham C. Systemic TNF-α Produces Acute Cognitive Dysfunction and Exaggerated Sickness Behavior When Superimposed Upon Progressive Neurodegeneration. Brain Behav Immun (2017) 59:233–44. doi: 10.1016/j.bbi.2016.09.011
71. Terrando N, Monaco C, Ma D, Foxwell BM, Feldmann M, Maze M. Tumor Necrosis Factor-Alpha Triggers a Cytokine Cascade Yielding Postoperative Cognitive Decline. Proc Natl Acad Sci USA (2010) 107(47):20518–22. doi: 10.1073/pnas.1014557107
72. Shamim D, Laskowski M. Inhibition of Inflammation Mediated Through the Tumor Necrosis Factor Alpha Biochemical Pathway Can Lead to Favorable Outcomes in Alzheimer Disease. J Cent Nerv Syst Dis (2017) 9:1179573517722512. doi: 10.1177/1179573517722512
73. Bortolato B, Carvalho AF, Soczynska JK, Perini GI, McIntyre RS. The Involvement of TNF-α in Cognitive Dysfunction Associated With Major Depressive Disorder: An Opportunity for Domain Specific Treatments. Curr Neuropharmacol (2015) 13(5):558–76. doi: 10.2174/1570159X13666150630171433
74. Tian J, Tai Y, Shi M, Zhao C, Xu W, Ge X, et al. Atorvastatin Relieves Cognitive Disorder After Sepsis Through Reverting Inflammatory Cytokines, Oxidative Stress, and Neuronal Apoptosis in Hippocampus. Cell Mol Neurobiol (2020) 40(4):521–30. doi: 10.1007/s10571-019-00750-z
75. Reis PA, Alexandre PCB, D'Avila JC, Siqueira LD, Antunes B, Estato V, et al. Statins Prevent Cognitive Impairment After Sepsis by Reverting Neuroinflammation, and Microcirculatory/Endothelial Dysfunction. Brain Behav Immun (2017) 60:293–303. doi: 10.1016/j.bbi.2016.11.006
76. Manfredini A, Constantino L, Pinto MC, Michels M, Burger H, Kist LW, et al. Mitochondrial Dysfunction is Associated With Long-Term Cognitive Impairment in an Animal Sepsis Model. Clin Sci (London Engl 1979) (2019) 133(18):1993–2004. doi: 10.1042/CS20190351
77. Michels M, Danieslki LG, Vieira A, Florentino D, Dall'Igna D, Galant L, et al. CD40-CD40 Ligand Pathway is a Major Component of Acute Neuroinflammation and Contributes to Long-Term Cognitive Dysfunction After Sepsis. Mol Med (Cambridge Mass) (2015) 21(1):219–26. doi: 10.2119/molmed.2015.00070
78. Della Giustina A, Goldim MP, Danielski LG, Florentino D, Garbossa L, Joaquim L, et al. Fish Oil-Rich Lipid Emulsion Modulates Neuroinflammation and Prevents Long-Term Cognitive Dysfunction After Sepsis. Nutr (Burbank Los Angeles Cty Calif) (2020) 70:110417. doi: 10.1016/j.nut.2018.12.003
79. Della Giustina A, Goldim MP, Danielski LG, Florentino D, Mathias K, Garbossa L, et al. Alpha-Lipoic Acid Attenuates Acute Neuroinflammation and Long-Term Cognitive Impairment After Polymicrobial Sepsis. Neurochem Int (2017) 108:436–47. doi: 10.1016/j.neuint.2017.06.003
80. Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of Monocytes, Macrophages, and Dendritic Cells. Science (2010) 327(5966):656–61. doi: 10.1126/science.1178331
81. Xu J, Ganguly A, Zhao J, Ivey M, Lopez R, Osterholzer JJ, et al. CCR2 Signaling Promotes Brain Infiltration of Inflammatory Monocytes and Contributes to Neuropathology During Cryptococcal Meningoencephalitis. mBio (2021) 12(4):e0107621. doi: 10.1128/mBio.01076-21
82. Andonegui G, Zelinski EL, Schubert CL, Knight D, Craig LA, Winston BW, et al. Targeting Inflammatory Monocytes in Sepsis-Associated Encephalopathy and Long-Term Cognitive Impairment. JCI Insight (2018) 3(9):e99364. doi: 10.1172/jci.insight.99364
83. Haam J, Yakel JL. Cholinergic Modulation of the Hippocampal Region and Memory Function. J Neurochem (2017) 142 Suppl 2:111–21. doi: 10.1111/jnc.14052
84. Hung SY, Fu WM. Drug Candidates in Clinical Trials for Alzheimer's Disease. J BioMed Sci (2017) 24(1):47. doi: 10.1186/s12929-017-0355-7
85. Zujalovic B, Mayer B, Hafner S, Balling F, Barth E. AChE-Activity in Critically Ill Patients With Suspected Septic Encephalopathy: A Prospective, Single-Centre Study. BMC Anesthesiol (2020) 20(1):287. doi: 10.1186/s12871-020-01204-6
86. Semmler A, Frisch C, Debeir T, Ramanathan M, Okulla T, Klockgether T, et al. Long-Term Cognitive Impairment, Neuronal Loss and Reduced Cortical Cholinergic Innervation After Recovery From Sepsis in a Rodent Model. Exp Neurol (2007) 204(2):733–40. doi: 10.1016/j.expneurol.2007.01.003
87. Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, et al. Cholinergic Agonists Inhibit HMGB1 Release and Improve Survival in Experimental Sepsis. Nat Med (2004) 10(11):1216–21. doi: 10.1038/nm1124
88. Yang H, Liu H, Zeng Q, Imperato GH, Addorisio ME, Li J, et al. Inhibition of HMGB1/RAGE-Mediated Endocytosis by HMGB1 Antagonist Box A, Anti-HMGB1 Antibodies, and Cholinergic Agonists Suppresses Inflammation. Mol Med (Cambridge Mass) (2019) 25(1):13. doi: 10.1186/s10020-019-0081-6
89. Terrando N, Yang T, Ryu JK, Newton PT, Monaco C, Feldmann M, et al. Stimulation of the Alpha7 Nicotinic Acetylcholine Receptor Protects Against Neuroinflammation After Tibia Fracture and Endotoxemia in Mice. Mol Med (Cambridge Mass) (2015) 20:667–75. doi: 10.2119/molmed.2014.00143
90. Zhang X, Li Z, Sun X, Jin F, Liu J, Li J. Role of α7 Nicotinic Acetylcholine Receptor in Attenuation of Endotoxin Induced Delirium With Dexmedetomidine in Mice. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue (2016) 28(2):127–33. doi: 10.3760/cma.j.issn.2095-4352.2016.02.009
91. Tchessalova D, Tronson NC. Memory Deficits in Males and Females Long After Subchronic Immune Challenge. Neurobiol Learn Mem (2019) 158:60–72. doi: 10.1016/j.nlm.2019.01.003
92. Barter J, Kumar A, Stortz JA, Hollen M, Nacionales D, Efron PA, et al. Age and Sex Influence the Hippocampal Response and Recovery Following Sepsis. Mol Neurobiol (2019) 56(12):8557–72. doi: 10.1007/s12035-019-01681-y
93. Li Y, Du L, Guo X, Zhang X. Cognitive Impairments Relate to the Expression of α7 Nicotinic Acetylcholinergic Receptor (α7nachr) and the Proportion of M1/M2 Microglia in the Hippocampus of Male Mice With Sepsis-Associated Encephalopathy. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi = Chin J Cell Mol Immunol (2021) 37(11):961–6.
94. Otto SL, Yakel JL. The Alpha7 Nicotinic Acetylcholine Receptors Regulate Hippocampal Adult-Neurogenesis in a Sexually Dimorphic Fashion. Brain Struct Funct (2019) 224(2):829–46. doi: 10.1007/s00429-018-1799-6
95. Murray AJ, Sauer JF, Riedel G, McClure C, Ansel L, Cheyne L, et al. Parvalbumin-Positive CA1 Interneurons are Required for Spatial Working But Not for Reference Memory. Nat Neurosci (2011) 14(3):297–9. doi: 10.1038/nn.2751
96. Ognjanovski N, Schaeffer S, Wu J, Mofakham S, Maruyama D, Zochowski M, et al. Parvalbumin-Expressing Interneurons Coordinate Hippocampal Network Dynamics Required for Memory Consolidation. Nat Commun (2017) 8:15039. doi: 10.1038/ncomms15039
97. Ji MH, Zhang L, Mao MJ, Zhang H, Yang JJ, Qiu LL. Overinhibition Mediated by Parvalbumin Interneurons Might Contribute to Depression-Like Behavior and Working Memory Impairment Induced by Lipopolysaccharide Challenge. Behav Brain Res (2020) 383:112509. doi: 10.1016/j.bbr.2020.112509
98. Mahmoud S, Gharagozloo M, Simard C, Gris D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance Between Glutamate Uptake and Release. Cells (2019) 8(2):184. doi: 10.3390/cells8020184
99. Cardoso FL, Herz J, Fernandes A, Rocha J, Sepodes B, Brito MA, et al. Systemic Inflammation in Early Neonatal Mice Induces Transient and Lasting Neurodegenerative Effects. J Neuroinflamm (2015) 12:82. doi: 10.1186/s12974-015-0299-3
100. Hamilton NB, Attwell D. Do Astrocytes Really Exocytose Neurotransmitters? Nat Rev Neurosci (2010) 11(4):227–38. doi: 10.1038/nrn2803
101. Petronilho F, Perico SR, Vuolo F, Mina F, Constantino L, Comim CM, et al. Protective Effects of Guanosine Against Sepsis-Induced Damage in Rat Brain and Cognitive Impairment. Brain Behav Immun (2012) 26(6):904–10. doi: 10.1016/j.bbi.2012.03.007
102. Li F, Zhang B, Duan S, Qing W, Tan L, Chen S, et al. Small Dose of L-Dopa/Benserazide Hydrochloride Improved Sepsis-Induced Neuroinflammation and Long-Term Cognitive Dysfunction in Sepsis Mice. Brain Res (2020) 1737:146780. doi: 10.1016/j.brainres.2020.146780
103. Ji M, Li S, Zhang L, Gao Y, Zeng Q, Mao M, et al. Sepsis Induced Cognitive Impairments by Disrupting Hippocampal Parvalbumin Interneuron-Mediated Inhibitory Network via a D4-Receptor Mechanism. Aging (2020) 12(3):2471–84. doi: 10.18632/aging.102755
104. Sara SJ. The Locus Coeruleus and Noradrenergic Modulation of Cognition. Nat Rev Neurosci (2009) 10(3):211–23. doi: 10.1038/nrn2573
105. Schwarz LA, Luo L. Organization of the Locus Coeruleus-Norepinephrine System. Curr Biol (2015) 25(21):R1051–6. doi: 10.1016/j.cub.2015.09.039
106. Mather M, Harley CW. The Locus Coeruleus: Essential for Maintaining Cognitive Function and the Aging Brain. Trends Cognit Sci (2016) 20(3):214–26. doi: 10.1016/j.tics.2016.01.001
107. Liu W, Guo J, Mu J, Tian L, Zhou D. Rapamycin Protects Sepsis-Induced Cognitive Impairment in Mouse Hippocampus by Enhancing Autophagy. Cell Mol Neurobiol (2017) 37(7):1195–205. doi: 10.1007/s10571-016-0449-x
108. Guo JN, Tian LY, Liu WY, Mu J, Zhou D. Activation of the Akt/mTOR Signaling Pathway: A Potential Response to Long-Term Neuronal Loss in the Hippocampus After Sepsis. Neural Regener Res (2017) 12(11):1832–42. doi: 10.4103/1673-5374.219044
109. Tian M, Qingzhen L, Zhiyang Y, Chunlong C, Jiao D, Zhang L, et al. Attractylone Attenuates Sepsis-Associated Encephalopathy and Cognitive Dysfunction by Inhibiting Microglial Activation and Neuroinflammation. J Cell Biochem (2019) 120:7101–8.. doi: 10.1002/jcb.27983
110. He W, Jiang M, Mao P, Yan F. TLR4 Inhibition Ameliorates Mesencephalic Substantia Nigra Injury in Neonatal Rats Exposed to Lipopolysaccharide via Regulation of Neuro-Immunity. Brain Res Bull (2020) 165:90–6. doi: 10.1016/j.brainresbull.2020.09.012
111. Gunther ML, Morandi A, Krauskopf E, Pandharipande P, Girard TD, Jackson JC, et al. The Association Between Brain Volumes, Delirium Duration, and Cognitive Outcomes in Intensive Care Unit Survivors: The VISIONS Cohort Magnetic Resonance Imaging Study*. Crit Care Med (2012) 40(7):2022–32. doi: 10.1097/CCM.0b013e318250acc0
112. Semmler A, Widmann CN, Okulla T, Urbach H, Kaiser M, Widman G, et al. Persistent Cognitive Impairment, Hippocampal Atrophy and EEG Changes in Sepsis Survivors. J Neurol Neurosurg Psychiatry (2013) 84(1):62–9. doi: 10.1136/jnnp-2012-302883
113. Seidel G, Gaser C, Götz T, Günther A, Hamzei F. Accelerated Brain Ageing in Sepsis Survivors With Cognitive Long-Term Impairment. Eur J Neurosci (2020) 52(10):4395–402. doi: 10.1111/ejn.14850
114. Bluemel P, Wickel J, Grunewald B, Ceanga M, Keiner S, Witte OW, et al. Sepsis Promotes Gliogenesis and Depletes the Pool of Radial Glia Like Stem Cells in the Hippocampus. Exp Neurol (2021) 338:113591. doi: 10.1016/j.expneurol.2020.113591
115. Huerta PT, Robbiati S, Huerta TS, Sabharwal A, Berlin RA, Frankfurt M, et al. Preclinical Models of Overwhelming Sepsis Implicate the Neural System That Encodes Contextual Fear Memory. Mol Med (Cambridge Mass) (2016) 22:789–99. doi: 10.2119/molmed.2015.00201
116. Orhun G, Esen F, Ozcan PE, Sencer S, Bilgic B, Ulusoy C, et al. Neuroimaging Findings in Sepsis-Induced Brain Dysfunction: Association With Clinical and Laboratory Findings. Neurocrit Care (2019) 30(1):106–17. doi: 10.1007/s12028-018-0581-1
117. Orhun G, Tuzun E, Bilgic B, Ergin Ozcan P, Sencer S, Barburoglu M, et al. Brain Volume Changes in Patients With Acute Brain Dysfunction Due to Sepsis. Neurocrit Care (2020) 32(2):459–68. doi: 10.1007/s12028-019-00759-8
118. Franke K, Gaser C. Ten Years of BrainAGE as a Neuroimaging Biomarker of Brain Aging: What Insights Have We Gained? Front Neurol (2019) 10:789. doi: 10.3389/fneur.2019.00789
119. Orihuela CJ, Fillon S, Smith-Sielicki SH, El Kasmi KC, Gao G, Soulis K, et al. Cell Wall-Mediated Neuronal Damage in Early Sepsis. Infect Immun (2006) 74(7):3783–9. doi: 10.1128/IAI.00022-06
120. Gu M, Mei XL, Zhao YN. Sepsis and Cerebral Dysfunction: BBB Damage, Neuroinflammation, Oxidative Stress, Apoptosis and Autophagy as Key Mediators and the Potential Therapeutic Approaches. Neurotoxicity Res (2021) 39(2):489–503. doi: 10.1007/s12640-020-00270-5
121. Guo R, Wang H, Cui N. Autophagy Regulation on Pyroptosis: Mechanism and Medical Implication in Sepsis. Mediators Inflamm (2021) 2021:9925059. doi: 10.1155/2021/9925059
122. Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, et al. Autophagy and Apoptosis Dysfunction in Neurodegenerative Disorders. Prog Neurobiol (2014) 112:24–49. doi: 10.1016/j.pneurobio.2013.10.004
123. Cooper KF. Till Death Do Us Part: The Marriage of Autophagy and Apoptosis. Oxid Med Cell Longev (2018) 2018:4701275. doi: 10.1155/2018/4701275
124. Yin X, Xin H, Mao S, Wu G, Guo L. The Role of Autophagy in Sepsis: Protection and Injury to Organs. Front Physiol (2019) 10:1071. doi: 10.3389/fphys.2019.01071
125. Switon K, Kotulska K, Janusz-Kaminska A, Zmorzynska J, Jaworski J. Molecular Neurobiology of mTOR. Neuroscience (2017) 341:112–53. doi: 10.1016/j.neuroscience.2016.11.017
126. Wang GB, Ni YL, Zhou XP, Zhang WF. The AKT/mTOR Pathway Mediates Neuronal Protective Effects of Erythropoietin in Sepsis. Mol Cell Biochem (2014) 385(1-2):125–32. doi: 10.1007/s11010-013-1821-5
127. Comim CM, Barichello T, Grandgirard D, Dal-Pizzol F, Quevedo J, Leib SL. Caspase-3 Mediates in Part Hippocampal Apoptosis in Sepsis. Mol Neurobiol (2013) 47(1):394–8. doi: 10.1007/s12035-012-8354-x
128. Zhou RX, Li YY, Qu Y, Huang Q, Sun XM, Mu DZ, et al. Regulation of Hippocampal Neuronal Apoptosis and Autophagy in Mice With Sepsis-Associated Encephalopathy by Immunity-Related GTPase M1. CNS Neurosci Ther (2020) 26(2):177–88. doi: 10.1111/cns.13229
129. Hu Y, Huang M, Wang P, Xu Q, Zhang B. Ucf-101 Protects Against Cerebral Oxidative Injury and Cognitive Impairment in Septic Rat. Int Immunopharmacol (2013) 16(1):108–13. doi: 10.1016/j.intimp.2013.03.019
130. Pan S, Wu Y, Pei L, Li S, Song L, Xia H, et al. BML-111 Reduces Neuroinflammation and Cognitive Impairment in Mice With Sepsis via the SIRT1/NF-kappaB Signaling Pathway. Front Cell Neurosci (2018) 12:267. doi: 10.3389/fncel.2018.00267
131. Herskovits AZ, Guarente L. SIRT1 in Neurodevelopment and Brain Senescence. Neuron (2014) 81(3):471–83. doi: 10.1016/j.neuron.2014.01.028
132. Jiao F, Gong Z. The Beneficial Roles of SIRT1 in Neuroinflammation-Related Diseases. Oxid Med Cell Longevity (2020) 2020:1–19. doi: 10.1155/2020/6782872
133. Zhu Y, Wang K, Ma Z, Liu D, Yang Y, Sun M, et al. SIRT1 Activation by Butein Attenuates Sepsis-Induced Brain Injury in Mice Subjected to Cecal Ligation and Puncture via Alleviating Inflammatory and Oxidative Stress. Toxicol Appl Pharmacol (2019) 363:34–46. doi: 10.1016/j.taap.2018.10.013
134. Wang Y, Liu X, Wang Q, Yang X. Roles of the Pyroptosis Signaling Pathway in a Sepsis-Associated Encephalopathy Cell Model. J Int Med Res (2020) 48(8):300060520949767. doi: 10.1177/0300060520949767
135. Swanson KV, Deng M, Ting JP. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
136. Fu Q, Wu J, Zhou XY, Ji MH, Mao QH, Li Q, et al. NLRP3/Caspase-1 Pathway-Induced Pyroptosis Mediated Cognitive Deficits in a Mouse Model of Sepsis-Associated Encephalopathy. Inflammation (2019) 42(1):306–18. doi: 10.1007/s10753-018-0894-4
137. Beyer MMS, Lonnemann N, Remus A, Latz E, Heneka MT, Korte M. Enduring Changes in Neuronal Function Upon Systemic Inflammation Are NLRP3 Inflammasome Dependent. J Neurosci (2020) 40(28):5480–94. doi: 10.1523/JNEUROSCI.0200-20.2020
138. Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, et al. FDA-Approved Disulfiram Inhibits Pyroptosis by Blocking Gasdermin D Pore Formation. Nat Immunol (2020) 21(7):736–45. doi: 10.1038/s41590-020-0669-6
Keywords: sepsis, cognitive impairment, BBB dysregulation, neuroinflammation, neurotransmitter dysfunction, neuronal loss
Citation: Li Y, Ji M and Yang J (2022) Current Understanding of Long-Term Cognitive Impairment After Sepsis. Front. Immunol. 13:855006. doi: 10.3389/fimmu.2022.855006
Received: 14 January 2022; Accepted: 11 April 2022;
Published: 06 May 2022.
Edited by:
Keliang Xie, Tianjin Medical University, ChinaReviewed by:
Patricio Huerta, Feinstein Institute for Medical Research, United StatesCopyright © 2022 Li, Ji and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianjun Yang, eWp5YW5nampAMTI2LmNvbQ==; Muhuo Ji, amltdWh1bzIwMDlAc2luYS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.