 Fang Wei
Fang Wei Xiao-Xia Cheng
Xiao-Xia Cheng John Zhao Xue
John Zhao Xue Shao-An Xue
Shao-An Xue- Genetic Engineering Laboratory, School of Biological & Environmental Engineering, Xi’An University, Xi’An, China
Immunotherapy of cancer has made tremendous progress in recent years, as demonstrated by the remarkable clinical responses obtained from adoptive cell transfer (ACT) of patient-derived tumor infiltrating lymphocytes, chimeric antigen receptor (CAR)-modified T cells (CAR-T) and T cell receptor (TCR)-engineered T cells (TCR-T). TCR-T uses specific TCRS optimized for tumor engagement and can recognize epitopes derived from both cell-surface and intracellular targets, including tumor-associated antigens, cancer germline antigens, viral oncoproteins, and tumor-specific neoantigens (neoAgs) that are largely sequestered in the cytoplasm and nucleus of tumor cells. Moreover, as TCRS are naturally developed for sensitive antigen detection, they are able to recognize epitopes at far lower concentrations than required for CAR-T activation. Therefore, TCR-T holds great promise for the treatment of human cancers. In this focused review, we summarize basic, translational, and clinical insights into the challenges and opportunities of TCR-T. We review emerging strategies used in current ACT, point out limitations, and propose possible solutions. We highlight the importance of targeting tumor-specific neoAgs and outline a strategy of combining neoAg vaccines, checkpoint blockade therapy, and adoptive transfer of neoAg-specific TCR-T to produce a truly tumor-specific therapy, which is able to penetrate into solid tumors and resist the immunosuppressive tumor microenvironment. We believe such a combination approach should lead to a significant improvement in cancer immunotherapies, especially for solid tumors, and may provide a general strategy for the eradication of multiple cancers.
Introduction
Immunotherapy of cancer based on adoptive cell transfer (ACT) of T lymphocytes can be classified into three approaches. The first, tumor-infiltrating lymphocyte (TIL) therapy, harvests naturally occurring T cells that have already penetrated patient tumors, expands them ex vivo, and then re-infuses them into patients (1, 2). However, it is often very difficult to isolate tumor-specific TILs, which are not present in all patients or may generate too few cells for therapeutic efficacy.
The second approach, chimeric antigen receptor (CAR)-modified T cells (CAR-T), bypasses this problem by directly engineering T cells with known tumor-specific CARs. CARs are fusion molecules that link a single-chain antibody with T-cell activation signaling domains such as CD28-CD3ζ (3) or 4-1BB-CD3ζ (4). When a CAR is transduced into human T cells, the antibody fragment is expressed on the surface of the engineered T cells to recognize a tumor antigen expressed on tumor cells, while the CD28-CD3ζ or 4-1BB-CD3ζ domain delivers a stimulatory signal once the antibody binds to a tumor antigen, activating CAR-T cells to attack the tumor.
CAR-T cells are not restricted by MHC molecules, and thus one CAR-T construct can be used to treat any patient regardless of genetic background. Currently, the most widely used and successful CAR is the CD19-CAR, which recognizes the CD19 molecule expressed on the surface of B cells, thus can eliminate some B-cell-derived leukemias and lymphomas (5, 6), including complete response in nearly 90% of B-cell leukemia patients (5). However, antibody-based CARs can only recognize antigens expressed on the cell surface, and not intracellular antigens, limiting the number of targets and potential tumor types addressable by CAR-T therapy.
The third approach, T cell receptor (TCR)-engineered T cells (TCR-T), uses TCRS as found on native T cells to confer specificity, instead of antibody-based CARs. TCRS can be isolated from tumor-reactive T cells and further modified for enhanced expression and functions. TCRS can recognize both cell-surface and intracellular targets, these include neoantigens (neoAgs) that arise from mutations and are specific to tumor cells. The disadvantage of using TCR-T is that TCRS are restricted by MHC molecules, thus any given TCR can only be used to treat patients with the corresponding MHC genetic background. In the following sections, we describe the settings in which TCR-T may prove most effective.
TCR-T Immunotherapy for Cancer as a Complement to CAR-T
CAR-T has been most successful in hematological malignancies (7–10), with FDA-approved therapies targeting CD19 (Kymriah, Yescarta, Tecartus, Breyanzi) and BCMA (Abecma) as of December 2021. CD19-CAR-T can achieve complete response in nearly 90% of B cell leukemia patients, although 50% of patients may nonetheless relapse (11). One of the major reasons for this relapse is loss of surface expression of CD19 from tumor cells (12), thus evading recognition by CD19-CAR-T cells. These patients may no longer respond to CD19-CAR-T, although targeting different antigens may still be viable, in which case TCR-T may be used as a late-line option. For example, a recent study reported transfer of WT1-TCR-engineered donor T cells into AML patients at high risk of relapse following allogeneic stem cell transplantation, 12/12 treated patients achieved relapse-free survival (13), compared to 54% in a concurrent group of 88 similar high-risk patients, and WT1-TCR-T cells also showed prolonged persistence and maintenance of antigen-specific polyfunctional activity.
The greater opportunity for TCR-T may exist in solid tumors, where CAR-T has been less effective (14–16). The mechanisms behind these limitations are poorly understood and under active investigation. CAR-T recognition is limited to surface antigens, and moreover CAR-T activation requires higher concentration of target antigens (17, 18). This lower sensitivity helps avoid damage to normal tissues with low antigen expression (19), but conversely may be unsuitable for tumors with similarly low tumor antigen expression. For example, CAR-T specific for anaplastic lymphoma kinase (ALK) showed variable efficacy towards different cell lines depending on expression level of ALK (17). Recently, a study investigated B cell malignancies with up to 33-fold lower CD20 expression than healthy B cells – below the concentration required to activate CAR-T – but found that CD20-specific TCR-T clones with high avidity were able to overcome self-tolerance and eliminate these tumor cells (20). It is estimated that CAR-T cells need in the order of hundreds of target molecules to be activated (17, 18, 21), whereas TCR-T can be activated by a single target molecule (22).
Initial clinical studies of TCR-T in solid tumors have shown promising results (23, 24). An affinity-enhanced NYESO1-TCR achieved 45-55% clinical response rate in metastatic melanoma patients (25, 26), and 50-61% clinical response rate in metastatic synovial sarcoma patients (25–27). The same NYESO1-TCR achieved 80% clinical response rate in multiple myeloma patients without apparent side effects, including 70% complete response rate with median progression-free survival of 19 months (28). Recently, a phase 1 trial of TCR-T targeting HPV-16 E7 in metastatic HPV-associated epithelial cancers achieved 50% clinical response rate (6/12 patients), including 4/8 patients refractory to PD-1 blockade (29).
In recent years, neoAgs have been discovered as a class of immunogenic tumor-specific antigens that are derived from tumor-specific mutations of self-proteins (30–32) or from tumor-causing oncogenic viral proteins (33) in the estimated 15% of human cancers attributed to viruses (34). T cells specific for neoAgs and viral proteins would not have undergone central thymic tolerance selection, making it possible to isolate high-avidity T cell clones against these targets. These antigens are rarely expressed on the cell surface, and represent a therapeutic opportunity in solid tumors using TCR-T that CAR-T may be unable to match (35).
Despite these promising results (24–27, 29), several hurdles remain to be overcome to realize the true promise of TCR-T immunotherapy. In early clinical trials, some non-responder patients lacked in vivo persistence of the infused T cells (36, 37), suggesting that the transferred TCR-T cells need additional support to enhance their in vivo survival. Some patients with late relapse showed no evidence of T cell infiltration in the tumor, and moreover infused TCR-T cells face a hostile immunosuppressive tumor microenvironment (TME). In the following sections, we discuss current and future engineering strategies to address these challenges to deliver effective and long-lasting tumor control to a broad range of cancer patients.
Overcoming the Challenges of TCR-T Cancer Immunotherapy
Enhancing TCR Expression and Function Through Reducing TCR Mis-Pairing
TCR-T therapy relies on mRNA or viral transduction of tumor-reactive TCR genes to redirect T cell specificity towards tumor cells. However, using either mRNAs or randomly integrating viruses to deliver the exogenous TCR, leaves the endogenous TCR genes intact. Therefore, this could potentially result in some degree of mis-pairing between the introduced and endogenous TCR chains (38–40). Mis-pairing poses certain safety risks, as T cells expressing mis-paired TCRS may be auto-reactive against the patient’s MHC molecules. Indeed, a murine study showed that TCR mis-pairing in the context of adoptive transfer of TCR-gene-modified T cells combined with increased conditioning resulted in graft-versus-host disease (GvHD) and serious animal death (41). Similarly, an in vitro study of human EBV-transformed lymphoblastoid cell lines showed that mis-paired TCRS drove potentially dangerous off-target toxicity (42).
Several strategies have been explored to prevent TCR mis-pairing (43, 44). The interaction between TCRα and TCRβ chains is largely governed by the invariant Cα/Cβ-interface (45), enabling modification of this region to prevent pairing with endogenous TCRS. Reciprocal “knob-hole” amino acid changes in the center of the TCR C domains led to preferential pairing of the modified chains while disfavoring combinations with native TCR chains (46). Introduction of an additional inter-chain disulfide bond within the TCR Cα/Cβ-interface (47) also enhanced the pairing of the modified chains whilst reducing the efficiency of pairing with wild-type chains (48, 49). This preferential pairing of cysteine-modified TCR chains has accounted for improved TCR gene expression and enhanced antitumor activity of transduced T cells (50). Replacing the human TCR constant domains with whole (49, 51, 52) or partial (53, 54) murine sequences represents an alternative strategy to reduce unwanted mis-pairing, and can also increase the expression level of the introduced TCR genes (51). The enhanced expression of the human/murine hybrid TCR in human T cells may be partly due to the greater binding capacity of the murine TCR constant domains to human CD3 molecules when compared with human TCR constant domains (51). Finally, instead of using murine sequences, exchanging the human TCR constant domains Cα and Cβ with each other (domain swapping), or replacing Cα and Cβ with the corresponding γδ TCR constant domains, could also generate functional TCRS with reduced mis-pairing and improved safety profile (55). However, it is important to note that the individual TCR subfamily V-domains and even the antigen-binding CDR3 α/β-loops may also contribute to the interaction of TCR α and β chains (56, 57), and hence manipulation of the TCR constant domains can only partially reduce the frequency of mis-pairing, rather than eliminate the risk completely.
Another common approach to reduce mis-pairing is to generate a so-called single-chain TCR (Sc-TCR) by covalently linking the Vα and Vβ domains with a polylinker (PLK), resulting in a single polypeptide which will in theory inhibit mis-pairing through steric hindrance (58). T-cell-activation signaling upon antigen encounter is provided by fusion of CD3ζ onto the Cβ-chain within the Sc-TCR. Using this approach, Sebestyen et al. showed preferential pairing between CD3ζ-modified TCR α and β chains while reducing mis-pairing with unmodified TCR chains (59). To develop this concept further, Aggen et al. replaced the constant domains of the Sc-TCR construct with a CD28 or 4-1BB together with CD3ζ or LCK signaling domains (60). Although this strategy was able to reduce mis-pairing, activation of these T cells upon antigen encounter no longer followed natural TCR signaling pathways, but rather that of conventional CARs. Because CAR signaling is less efficient than that of the TCR complex (23, 61), Voss et al. developed an alternative Sc-TCR scaffold Vα-PLK-Vβ-Cβ plus Cα (62), relying on assembly with the native CD3 complex for more physiologic T-cell signaling. To stabilize the structure of the Sc-TCR, we introduced an extra new disulfide bond between the Vα and the polylinker, which strengthens the interaction between the Vα and Vβ domains, favoring surface expression of the Sc-TCR, while also greatly reduced TCR mis-pairing (63). One of the potential drawbacks of using this technology is that not all of the TCRS can form a stable Sc-TCR. According to our experience, some of the weak TCRS can not be expressed on the surface of T cells as a Sc-TCR due to the weak interaction between the Vα and Vβ domains. With such weak TCR, genetic engineering of certain frame work regions of the TCR may be able to help to resolve the problem (64).
Aside from TCR protein design, another way of reducing TCR mis-pairing and its related side effects is to knock out the endogenous TCRS via genetic engineering, which also reduces competition for CD3 binding from endogenous TCRS (65). Several strategies have been explored to achieve this goal, including the use of siRNAs (66, 67), zinc-finger nucleases (68), transcription activator-like effector nucleases (TALENs) (69, 70), and CRISPR/Cas9 technologies (71, 72). As the CRISPR/Cas9 has several advantages, including (i) simple and highly efficient editing, (ii) rapid and affordable manufacturing, (iii) versatile multiplex genome editing through simultaneously targeting several genes, and (iv) user-friendly and easily deliverable. Therefore, this CRISPR/Cas9 system holds great promise and may lead the way for future genetic engineering of T cells for cancer immunotherapy (73). The feasibility of genome editing using CRISPR/Cas9 targeting the TRAC and TRBC loci was recently demonstrated in primary T cells (71, 74). Using multiplex genome editing, the beta-2 microglobulin class I MHC and PD-1 genes can also be disrupted alongside the TCR α and β genes (75, 76). Removal of endogenous TCR and class I MHC eliminates allogeneic antigen recognition and reduces risk of GvHD and donor T cell rejection, generating allogeneic ‘universal’ T cells that can be infused in any recipient (77, 78), as opposed to autologous T cells that can only be re-infused into the donor patient. The immune checkpoint gene PD-1 is removed to enhance T cell activity, for which reason the T cell suppressor LAG-3 has also been knocked out to improve antitumor activity in vitro and in murine xenografts (79). We anticipate that other immune inhibitory receptors such as TGFβ receptors can also be disrupted to generate universal CAR-T and TCR-T cells with enhanced resistance to the inhibitory TME.
TCR-T cells have been generated by TCR transduction of T cells in which the endogenous TCR α chain (80), β chain (81), or both α and β chains (82) were removed using CRISPR/Cas9, or by orthotopic replacement of TCR-αβ chains with tumor reactive TCRS using CRISPR/Cas9 (83, 84), resulting in engineered TCR-T cells with enhanced TCR expression and prolonged control of tumor growth in preclinical murine models. However, for clinical use, the potential off-target toxicity of the CRISPR/Cas9 technology has to be taken into considerations (85). A recent study showed that DNA breaks introduced by sgRNA/Cas9 can lead to on-target mutagenesis, such as large deletions and genomic rearrangements at the targeted sites in mouse embryonic stem cells and in a human differentiated cell line (86). Therefore, strategies for improving safety of the CRISPR/Cas9 technology need to be put in place. To reduce the off-target toxicity, high-fidelity CRISPR–Cas9 nuclease has been developed (87), and a recent review has provided many strategies about how to refine the CRISPR/Cas9 technology for clinical applications (88). Recently, a phase 1 clinical trial was designed to test multiplex CRISPR-Cas9 gene editing of T cells from patients with advanced, refractory cancer (89), in which endogenous TCR α and β chains were removed to prevent mis-pairing, and PD-1 removed to avoid T cell exhaustion. The NYESO1-TCR engineered T cells persisted for up to 9 months and trafficked to tumor sites, demonstrating proof-of-concept for multiplex CRISPR gene editing in cell therapy. Another study applied CRISPR-edited T cells in patients with refractory non-small-cell lung cancer also concluded that clinical application of CRISPR–Cas9 gene-edited T cells is generally safe and feasible (90).
To achieve the best results of maximally reducing mis-pairing and enhancing expression and function of the tumor-specific TCR, we have recently combined multiple strategies by knocking out endogenous TCR using CRISPR/Cas9 together with transduction of a single-chain EBV-specific TCR (EBV-Sc-TCR) (91). This almost eliminated mis-pairing between the introduced EBV-Sc-TCR and endogenous TCR chains, and we further enhanced tumor-specific TCR expression, functional avidity, and IL-2 production by introducing an extra intra-chain disulfide bond between the Vα and the poly-linker (91).
Enhancing Persistence and Anti-Tumor Functions of the Genetically Engineered T Cells
T cell persistence is a fundamental requisite for durable immunosurveillance, as many clinical trials revealed that most non-responder patients showed no in vivo persistence of the infused tumor specific T cells (36, 37), and in contrast, patients who achieved complete response or relapse-free survival and tumor control showed robust proliferative capacity and long-term persistence of engineered T cells (13, 27). To maintain persistence of the transferred T cells, a variety of cytokines have been coadministered to support T cell survival and expansion. The standard ACT regimen comprises lymphodepletion with cytotoxic agents, including cyclophosphamide and fludarabine, followed by administration of recombinant human IL-2 after T cell transfer (92). Systemic delivery of IL-2, is known to expand T cells while maintaining functional activity (93), has achieved durable regression in some metastatic melanoma and renal cancer patients (94), is approved by the FDA, and is used in both CAR-T and TCR-T immunotherapy of cancers today. However, there is evidence suggests that IL-2 may preferentially expand CD4+ regulatory T (Treg) cells rather than tumor-killing CD8+ cytotoxic T cells (CTLs) (95, 96). Therefore, recent attention has focused on modifying the IL-2 molecule to preferentially bind and activate CD8+ CTLs over Treg cells (97). For example, a half-life-extended super mutant IL-2 conjugated to a tumor-targeting antibody allowed more efficient CTL stimulation and expansion in the TME, resulting in significantly improved complete response rate and lower tumor relapse in vivo (97).
IL-7, is a hematopoietic cytokine regulating multiple aspects of T cell biology (98), is essential for T cell survival and homeostatic proliferation, and promotes the survival of naïve and memory T cells by upregulating the antiapoptotic molecule Bcl-2 (99, 100). IL-7 supplementation improved the persistence and efficacy of transferred T cells, supporting its usage as an adjuvant for adoptive immunotherapy (101). When IL-7 was co-expressed in NKG2D-based CAR-T cells, it enhanced CAR-T persistence and expansion while inhibiting apoptosis and exhaustion (102). Similarly, IL-7 co-expression in GPC3-CAR-T cells improved CAR-T efficacy toward liver cancer (103).
IL-12 is a major contributor to effective anti-tumor immune responses (104), stimulating the effector functions of activated T cells and NK cells via induction of cytotoxic enzymes such as perforin and cytokines such as IFN-γ (104, 105). Cytotoxic enzymes can mediate direct killing of tumor cells (106), while production of IFNγ from NK cells as well as CD4+ and CD8+ T cells inhibits tumor growth (107, 108). IL-12 further modifies the TME through inhibition of Tregs (107), upregulating MHC class I presentation on tumor cells (109, 110), and converting immunosuppressive M2 macrophages into activated antitumor M1 macrophages (111). IL-12 also prevents the activation-induced cell death of naïve CD8+ T cells, favoring their survival and differentiation towards the effector phenotype to sustain anti-tumor activity against mouse models of melanoma (112).
These studies demonstrate that IL-12 is not only required for the activation of anti-tumor cytotoxic immune responses, but also directly relieves immune suppression (107). However, systemic administration of IL-12 is very toxic (113), severely limiting its utility in clinical applications. To minimize systemic exposure and potential toxicity while maintaining the beneficial effects of IL-12, several strategies have been explored, for example local delivery (114, 115), encapsulating IL-12 with nanoparticles or heparin (116, 117). Alternatively, deleting the N-terminal signal peptide of IL-12 or tethering IL-12 to the surface of TCR-engineered T cells via a membrane anchor prevents secretion (118, 119), thereby attenuating toxicity while improving antitumor efficacy. These treatment strategies may have broad applications to cellular therapy with TILs, CAR-T, and TCR-T cells. A recent multi-center phase 1 trial used a chemically activatable IL-12 gene delivered into the tumor site, where IL-12 expression triggered by the drug veledimex achieved conversion of an immunologically “cold” TME to an inflamed “hot” TME with increased influx of IFN-γ–producing T cells (120). IL-18 is another cytokine that shares biological effects with IL-12 but with reduced toxicity (121). Recent studies exploring IL-18 in the place of IL-12 suggest that CAR-T cells engineered to secrete IL-18 enhances CAR-T cell survival and antitumor activity both in vitro and in vivo by producing IFN-γ and several other cytokines, stimulating expansion of human CD4+ cells as well as activating the endogenous immune system in immunocompetent mice (122, 123).
IL-15 is known to stimulate the generation of stem cell memory T cells (Tscm) with potential to sustain durable T cell responses (124). Unlike IL-2, IL-15 does not bind to the IL-2Rα chain, and thus does not stimulate Tregs and may have a more selective effect. When compared with IL2, IL15 tend to enhance CAR-T cell antitumor activity by preserving their Tscm phenotype (125). A comparison of CAR-T cell expansion in the presence of IL-2, IL-15, or a combination of IL-15/IL-7, revealed that IL-15 best enhances CAR-T persistence and function in a mouse model of multiple myeloma (126). Preclinical observations strongly support the antitumor activity of IL-15 mediated by CD8+ T cells (127), and IL‐15 co-expression in CD19-CAR-T not only revealed a strong killing effect against leukemia cells, but most of the persistent T cells were phenotypically consistent with Tscm that drive long‐term persistence (128).
To improve IL-15 half-life and effectiveness in vivo, IL-15 was associated with IL-15-receptor-α to form a pre-bound IL-15/IL-15Rα dimer, which showed stronger antitumor activity than IL-15 monomer (129). Recently, subcutaneous injection of recombinant human IL15 was tested in patients with advanced solid tumors, although the treatment produced substantial increases in circulating NK and CD8+ T cells, nonetheless, no objective responses were observed (130). However, when IL-15/IL-15Rα sushi-domain was co-expressed on CD5-specific CAR-T cells, and tested in a patient with relapsed T-lymphoblastic lymphoma with CNS infiltration, a rapid ablation of the CNS lymphoblasts to undetectable levels within 4 weeks and disease remission was observed (131). To address IL-15-induced immune checkpoint activation, IL-15 can also be combined with anti-PD-(L)1 and anti-CTLA-4 antibodies (132).
IL-21 is a newly discovered member of the common γ-chain family of cytokines. Like IL-12 and IL-15, and in contrast to IL-2, IL-21 does not stimulate Tregs, instead, it inhibits Treg expansion through suppression of Foxp3, thus favoring the enrichment of antigen-specific CD8+ T cells (133). IL-21 facilitates the maturation and enhances the cytotoxicity of CD8+ T cells and NK cells, and promotes the differentiation of memory CD8+ T cells (134, 135). IL-21 synergizes when combined with IL-12 to further inhibit Tregs (136), and synergizes when combined with IL-15 to expand CD28-expressing antigen-specific CD8+ T cells (137, 138). Utilizing these characteristics, IL-21 performed much better than IL-2 or IL-15 during in vitro generation of antigen-specific CD8+ CTL and in an in vivo murine model of cancer immunotherapy (139, 140). In murine tumor models, intratumoral injection of IL-21 strongly inhibited tumor growth and increased the frequency of tumor-infiltrating CD8+ T cells and mice survival (141). In a phase 1/2 trial, 4 out of 4 leukemia patients who received WT1-specific CTL generated in the presence of IL-21 demonstrated both relapse-free survival without GvHD and did not need further anti-leukemic treatment (142).
To reduce the toxicity and increase the half-life of IL-21, IL-21 has been conjugated to tumor-targeting antibodies such as anti-EGFR antibody (143), selectively expanding functional CTLs while restricting exhausted T cells in the TME. IL-21 upregulates perforin and granzyme expression in memory and effector CD8+ T cells (144), thus augments the antitumor activity of CD8+ T cells (145), consistent with the requirement of IL-21 for the long-term maintenance and function of CD8+ T cells (146). IL-21 fused to anti-PD-1 antibody stimulated generation of Tscm with enhanced cell proliferation and tumor-specific CD8+ T cells, outperforming anti-PD-1 antibody and IL-21 infused as separate treatments (147). These results demonstrated that IL-21 can be used alone or in combination with other cytokines to produce tumor-specific T cells with a memory phenotype, with enhanced persistence, proliferative capacity, and antitumor efficacy for adoptive cancer immunotherapies.
Endogenous immune cells can act as a “sink” for administered cytokines (148), thus the use of a lymphodepleting conditioning regimen prior to ACT helps to spare the limited cytokines for the transferred T cells. Moreover, conditioning can also eliminate immunosuppressive Tregs and MDSCs (149), further supporting the engraftment and expansion of engineered T cells and improving therapy persistence and efficacy (150, 151).
Finally, purposeful selection of T cell sub-populations is another way to enhance persistence and functionality of the adoptively transferred T cells. Less differentiated T cells such as Tscm and central memory (Tcm) cells are more effective than effector T cells when transferred into tumor-bearing mice (152), thus CAR modification of naïve T cells can generate antigen-specific Tscm and Tcm cells with long in vivo persistence which mediated robust, long-lasting antitumor responses (153, 154). To preserve this early differentiated T cell population, tumor-specific CTLs can be stimulated by a combination of IL-21 and anti-CD3/anti-CD28 antibody-conjugated microbeads or nanomatrices (155, 156). Addition of IL-21 alone or in combination with other cytokines (such as IL-7 and IL-15) into the expansion culture, can further support the enrichment and expansion of Tscm cells with superior antitumor activity (157), consistent with recent clinical data that WT1-TCR engineered T cells generated in the presence of IL-21 showed long-lasting persistence with superior anti-leukemia activity in humans (13, 142).
Enhancing the Homing and Penetration of Engineered T Cells Into Solid Tumors
To achieve tumor eradication, cancer-specific CTLs need to migrate and penetrate into solid tumors (158), driven by interactions between tumor-secreted chemokines and chemokine receptors expressed on CTLs (159–163). This process is rate-limited when CTLs express chemokine receptors at low density or that do not match the specific chemokines secreted by the tumors (164, 165). This creates an opportunity to engineer tumor-specific T cells’ chemokine receptors to match known chemokines or cytokines abundant in the TME, with encouraging results for enhanced CTL homing and antitumor efficacy.
The chemokines CCL2, CCL7, and CCL8 are expressed in many cancer types as well as cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), MDSCs, and mesenchymal stem cells found in the TME, which support the tumor growth and metastasis (164). All three of these chemokines are ligands for the CCR2 receptor, thus when CCR2b was transduced together with a CAR specific for GD2 into human T cells, these modified T cells showed enhanced trafficking with >10-fold improved homing to CCL2-secreting neuroblastoma, and significantly enhanced activity against neuroblastoma xenografts in vivo (166). The same approach, resulted in 12.5-fold increase in infiltration of CAR-T specific for mesothelin into established mouse tumors, and significantly enhanced antitumor activity and tumor eradication (167). When CCR2 was transduced together with a TCR specific for WT1 into CD3+ human T cells, double gene-modified CD3+ T cells demonstrated CCL2-tropic tumor trafficking and potentiated antitumor activity against WT1-expressing LK79 lung cancer cells both in vitro and in vivo (168). Similarly, transduction of CCR2 into TCR-T cells specific for the SV40 large T antigen, enhanced recruitment into CCL2-expressing metastatic prostate adenocarcinoma, and improved in vivo antitumor effect (169).
Multiple chemokine ligands for the CXCR2 receptor are expressed in many tumors (170), and also promote tumor initiation, proliferation, migration, metastasis, and immune invasion. Thus, CXCR2 has been explored intensively for cancer immunotherapy. Human hepatocellular carcinoma (HCC) tumor tissues and cell lines express several chemokine ligands for CXCR2, however, both human peripheral T cells and TILs of HCC lack expression of CXCR2. In a recent study (171), Liu et al. transduced human T cells with a GPC3-CAR together with CXCR2; compared with CAR-T cells without CXCR2, these cells exhibited identical cytotoxicity but significantly increased migration in vitro, as well as accelerated in vivo trafficking and tumor-specific accumulation in a xenograft tumor model. Similarly, CXCR2 enhanced trafficking and in vivo antitumor efficacy of CAR-T cells specific for integrin αvβ6 in advanced pancreatic and ovarian tumor xenograft models (172). In the TCR-T field, when CXCR2 was transduced into pmel-1 TCR transgenic T cells (173), or MAGE-A3-specific TCR-engineered T cells (174), the CXCR2-TCR-T cells showed increased in vivo homing, enhanced tumor infiltration, and preferential accumulation in tumor sites in mice, with enhanced survival and tumor regression compared with mice receiving control TCR-T cells. These results indicate that introduction of the CXCR2 gene into tumor-specific T cells can enhance their homing and localization to tumors and improve antitumor immune responses. CXCR2 has also been used to enhance the migration and homing of NK cells to CXCL5-expressing renal cell carcinomas (175). Recently, Jin et al. used radiation therapy to induce tumor secretion of IL-8 (CXCL8), and found that CD70-CAR-engineered T cells expressing either of the IL-8 receptors CXCR1 or CXCR2, showed enhanced migration and persistence, leading to complete tumor regression and immunologic memory in models of aggressive tumors, including glioblastoma, ovarian, and pancreatic cancers (176). Like radiation therapy, chemotherapy may also induce chemokine secretion from tumor cells, resulting in increased homing and infiltration of adoptively transferred T cells (177). These studies indicate that genetic engineering of tumor-specific T cells with chemokine receptors can be combined with conventional radiation and chemotherapy to enhance antitumor efficacy. CXCR1 has also been used to enhance migration and tumor infiltration of NK cells modified with a CAR specific for NKG2D (178).
Other chemokine receptors used in this way include CCR4 and CXCR4. Similarly, coexpression of CCR4 enhanced migration of CD30-specific CAR-T cells in response to CCL17 secreted by Hodgkin’s lymphoma in murine xenografts (179). CXCR4 has also been explored as a means of recruiting T cells into the bone marrow, whose microenvironment is suggested to improve memory T cell formation and self-renewal. Khan (180) et al. overexpressed CXCR4 in CD8+ T cells, observing enhanced migration toward CXCL12-expressing cells in the bone marrow, with enhanced memory differentiation, expansion, persistence, and antitumor function of adoptively transferred T cells. CXCR4 also enhanced migration of NK cells to bone marrow as a means of targeting bone-marrow-resident tumor cells such as leukemia (181). CXCR4-modified CAR-NK cells also significantly improved survival and tumor regression of mice bearing glioblastoma (182).
Aside from engineering T cells with chemokine receptors, chemokines can be directly introduced into tumors to enhance T cell recruitment. For example, intratumoral injection of CXCL2 plasmid DNA combined with inactivated Sendai virus envelope, suppressed the growth of murine breast cancers and inhibited lung metastasis through recruitment of CTLs and neutrophils, further enhanced with anti-PD-1 antibodies to inhibit T cell exhaustion (183). Chemokines can also be introduced into tumors through chemokine-armed oncolytic viruses, which simultaneously, replicate within and directly kill tumor cells to amplify antitumor efficacy (184). T cells can also be engineered to express both chemokines and cytokines to improve antitumor efficacy. For example, transduction of CAR-T cells to express IL-7 and the chemokine CCL-19, not only enhanced T cell survival, infiltration and accumulation in the tumor, but also achieved complete regression of pre-established solid tumors and prolonged mouse survival (185). When the chemokine CCL21 and IL7 was transduced into CAR-T cells, it significantly improved survival and infiltration of both CAR-T and dendritic cells in the tumor, leading to complete tumor remission (186).
Overcoming the Immunosuppressive Tumor Micro-Environment
Infiltration of genetically engineered T cells into the tumor is only the first step in fighting cancers. Tumor cells inhabit a heterogeneous microenvironment of infiltrating and resident host cells, secreted factors and extracellular matrix (187). Infiltrated cells include immune cells, such as T cells (TILs and Tregs), macrophages (M1 and M2), and MDSCs, and secreted factors include the immunosuppressive cytokines IL-10 and TGFβ. The TME also includes stromal cells such as CAFs and TAMs. These components can mutually interact to induce a supportive milieu for malignant cell growth, migration, and metastasis, that evades the immune system and tumor-specific CTLs (188, 189).
Most tumor stromal cells in the TME express the immunosuppressive checkpoint ligand PD-L1 (190–192), which can interact with PD-1 expressed on T cells, resulting in inhibition of antitumor function and exhaustion of adoptively transferred TILs (193), CAR-T (194) and TCR-T (195). This effect can be relieved via checkpoint blockade with anti-PD-1 (196–199) and anti-PD-L1 (200, 201) antibodies. CTLA-4 expressed on activated T cells have a similar effect, as CTLA-4 binds to CD80/86 on antigen-presenting cells with higher affinity in competition with the T cell costimulation molecule CD28, dampening antitumor immunity (202). Anti-CTLA4 antibodies both block the interaction between CTLA4 and CD80/86, and can also deplete Tregs (203), thus facilitate the costimulation and expansion of tumor-specific CTL with improved clinical benefits (204, 205).
Checkpoint inhibitors alone induce a response rate of approximately 20% of patients in one meta-analysis (206), and some responding patients will develop resistance (207). One important resistance mechanism is the upregulation of PD-L1 expression on tumor cells treated with immunotherapy, resulting in T cell exhaustion and relapse (207, 208). Immune checkpoint blockade is also associated with significant and in some cases life-threatening toxicity (209). An alternative approach to eliminating the immunosuppressive effect of PD-1 on tumor-specific CTLs uses CRISPR/Cas9 technology to remove PD-1 from CAR-T (210), and TCR-T cells (89). It is possible to go beyond PD-1-deletion by introducing a chimeric switch receptor (CSR) consisting of a PD-1 extracellular domain (PD1ex) and CD28 intracellular domain (CD28in). When this PD-1:CD28 CSR was transduced together with a CAR (211, 212) or TCR (213, 214) into T cells, the engineered CTLs still interact with PD-L1 on tumor cells, but delivers a costimulation signal via CD28 rather than an inhibitory signal. CAR-T cells generated using this strategy show increased cytokine production (211), enhanced killing ability, and increase in central memory T cells (212). Similarly,this PD-1:CD28 CSR enhanced TCR-T cells to increase cytokine production and cell proliferation in vitro and in vivo (213), and prevented PD-L1 upregulation and Th2 polarization in the TME (214). CSR TCR-T cells also synergized with anti-PD-L1 antibody to secrete more IFNγ compared with control TCR-T (214). Recently, this strategy has begun to be used in the clinic. In a CD-19 CAR-T study of relapsed/refractory diffuse large B-cell lymphoma (215), 6 patients who progressed following CD19-CAR-T therapy, were given CAR-T cells engineered with CSR PD1ex-CD28in, of which 3/6 patients achieved complete remission, and 1/6 achieved partial response. In another study, in relapsed/refractory PD-L1+ B-cell lymphoma (216), CSR-engineered CAR-T cells targeting CD19 showed superior T-cell proliferation, cytokine production, and cancer cell killing in vitro and in vivo. Among 17 treated adult patients, 10 patients had objective response (58.8%), including 7 with complete remission (41.2%). In both trials no severe neurologic toxicity or cytokine release syndrome was observed. Endogenous PD-1 was not depleted in these trials, thus we anticipate additional opportunity to enhance antitumor activity by combining CSR-engineered T cells with PD-1 knockout. The same CD28 CSR approach has also been applied to the immune checkpoint molecules TIGIT (T cell immunoreceptor with Ig and ITIM domains) and CTLA-4. Co-transduction of a TIGIT : CD28 CSR together with a tumor-specific TCR or CAR into human T-cells, drove enhanced cytokine production and superior anti-tumor function in a xenograft model of established human melanoma tumors (217). Transduction of a CTLA-4:CD28 CSR into tumor-specific T cells, resulted in elevated IFN-γ and IL-2 production and enhanced antitumor effect without systemic autoimmunity (218). A recent study engineered a CTLA4:CD28-CD3z CSR with the intracellular domains of both CD28 and CD3z, demonstrating increased cytokine production and cytotoxicity in vitro and in xenograft models (219). These engineered CAR-T cells were found to accumulate in tumors and to target MDSCs without severe GvHD or CRS (219).
Among the multiple immunosuppressive factors secreted within the TME, TGF-β plays a central role driving tumor signaling, remodeling, and metabolism (220). TGF-β is produced by many cell types including tumor cells, stromal cells and Tregs (221), and stimulates autocrine and paracrine signaling to promote angiogenesis (222), suppress CD8+ and Th1 anti-tumor responses (223), and induce epithelial-to−mesenchymal transition of neoplastic cells and thus facilitate tumor invasion (224). Recent clinical data associated patient non-responders to checkpoint blockade with TGF-β signaling (225). Therefore, blocking TGF-β signaling in the TME could potentiate antitumor responses. Indeed, one possible mechanism of anti-CTLA4 antibody therapy is the depletion of immunosuppressive TGF-β-producing Treg cells (203), thus facilitating the costimulation and expansion of tumor-specific CTL with improved clinical benefit (204, 205). To further improve on the efficacy of checkpoint inhibitor antibodies, bifunctional antibody-ligand traps comprising an antibody targeting CTLA-4 or PD-L1 fused to a TGFβ receptor II ectodomain (TGFβRIIecd) have been generated (226), in which TGFβRIIecd sequesters TGF-β secreted in the TME, while the checkpoint inhibitor antibody depletes Tregs and facilitates the CTL costimulation. This dual strategy may be more effective against cancers that are resistant to immune checkpoint inhibitors alone.
As an alternative to TGF-β sequestration, tumor-reactive T cells can be transduced with a dominant-negative TGFβ receptor-II (dnTGFβRII), generating TGF-β-resistant antitumor T cells (227). TCR-T cells expressing dnTGFβRII demonstrated complete tumor regression and prolonged survival in a mouse model of advanced and invasive prostate carcinoma. In a recent clinical study, patients with relapsed Hodgkin lymphoma were treated with EBV-specific T cells engineered to express dnTGFβRII, and 4/8 patients showed an objective clinical response (228), demonstrating that TGFβ-resistant tumor-specific T cells can persist safely in patients and potentially enhance the efficacy of T cell immunotherapy. To take advantage of the high concentration of TGF-β in the TME, a recent study fused the TGF-β receptor II (TGFβRII) extracellular domain to the intracellular domain of 4-1BB to convert the immunosuppressive effect of TGF-β into an immunostimulatory signal (229). The same cells were also transduced with a CAR-CD3ζ targeting the prostate-specific antigen and an inverted cytokine receptor consisting of the IL-4R extracellular domain fused to the IL-7R intracellular domain. Coexpression of these 3 transgenic receptors generated an additive effect with improved expansion, persistence, tumor lysis, and selective antitumor activity in vivo. Transduction of the TGFβRII-41BB CSR together with a NYESO1-specific TCR promoted abundant effector cytokine production in T cells, resulting in markedly enhanced tumor clearance in an in vivo solid tumor model (230). Like TGF-β, the Fas ligand-mediated T cell death signal that is highly expressed in the TME can also be converted into pro-survival signal via CSR, by fusing the Fas extracellular domain with the 4-1BB intracellular domain (231), resulting in engineered T cells with increased pro-survival signaling, proliferation, antitumor function, and enhanced in vivo efficacy against leukemia and pancreatic cancer mouse models. These studies clearly demonstrate the potential for using CSRs to convert the TME’s immunosuppressive signals into immunostimulatory signals in engineered antitumor T cells.
Enhancing Tumor-Specific Killing by Targeting Neoantigens
Over the past decade, many tumor-associated antigens (TAAs) have been discovered and investigated as targets for cancer immunotherapy. These include the cancer testis antigens (e.g., New York esophageal squamous cell carcinoma 1 (NY-ESO)-1) (25), melanoma-associated antigen (MAGE-A3 (174), MAGE-A4) (232), differentiation antigens (e.g., melanoma antigen recognized by T-cells 1 (MART-1) (233), tyrosinase/gp100 (234), overexpressed oncogenes (e.g., Wilms’ Tumor antigen 1 (WT1) (13, 235, 236), surviving (237), tumor suppressor genes (e.g., TP53) (238); and TAAs that are organ-specific (e.g., prostate-specific antigens) (239) or cell-type-specific antigens that are transiently expressed during differentiation (e.g., terminal deoxynucleotidyl transferase) (240), or normally expressed only during embryonic development (e.g., carcinoembryonic antigen) (241). While many of these have advanced to the clinic, the residual expression of many TAAs in normal tissues often leads to toxicity from TAA-targeted therapy (234, 242–244). There is therefore a need to target truly tumor-specific antigens. This is the basis for targeting antigens from oncogenic viruses, such as HPV (245), EBV (246) and HBV (247), which can potentially eradicate virus-induced cancer cells (248, 249).
While most cancers are not viral in origin, they share a hallmark of genomic instability (250), which often leads to the occurrence of a large number of mutations. Mutant amino acid coding sequences can be expressed, processed, and presented on the surface of tumor cells as cancer-specific neoAgs, and subsequently recognized by T cells. CTLs targeting neoAgs are less likely to react against normal tissues or face immune tolerance. Indeed, evidence from treatment of cancers with checkpoint blockade suggests that tumors with higher mutational burden are likely to respond to immunotherapy (251, 252). In a study of 266 cancer patients, responders to checkpoint blockade therapy more often had tumors harboring TILs (so-called ‘hot’ tumors), while non-responders had tumors with few TILs (‘cold’ tumors) (253). It is thought that tumors harboring more mutations generate more neoAgs, which can be recognized by neoAg-specific TILs (254). These TILs are frequently suppressed in the TME by immune checkpoint molecules such as CTLA-4 and PD-1/PD-L1, but can be reactivated following checkpoint blockade and thus able to induce tumor regression. As a result, cancers with high mutational load, such as melanoma and lung cancer, are more susceptible to checkpoint blockade therapies. There is also evidence that checkpoint blockade not only increases the number but also enhances the antitumor activity of neoAg-specific TILs (255).
Combining these clinical data, we hypothesize that immunotherapy through checkpoint blockade could be further augmented with neoantigen vaccines. Such vaccines could stimulate and amplify neoAg-specific TILs, which are released and reactivated upon checkpoint blockade to destroy tumor cells. Indeed, a personalized RNA-based vaccine was recently used to treat stage III and IV melanoma patients (256). All 13 patients developed T cell responses against multiple neo-epitopes, and each patient developed T cells against at least three mutations. Vaccination reduced rate of metastases and sustained progression-free survival in 8 patients. Notably, 1 patient showed complete response when the vaccination was combined with PD-1 blockade therapy. In another study, 4/6 neoAg-vaccinated patients showed no tumor recurrence at 25 months after treatment, while 2/6 patients with recurrent disease subsequently showed complete tumor regression following treatment with anti-PD-1 therapy (257). A phase Ib trial combining a personalized neoAg vaccine with PD-1 blockade found durable, neoAg-specific CD4+ and CD8+ T cell responses in all 82 treated patients, and T cells migrated to metastatic tumors and mediated tumor cell killing (258). Many clinical studies suggest that neoAg-specific T cells are the main mediators of tumor destruction in patients who responded to checkpoint blockade therapy (252, 259) or adoptive T cell transfer (260, 261). Both CD8+ and CD4+ neoAg-specific T cells contribute to lasting tumor clearance (262–265), and work continues to develop strategies to promote maximal cytotoxic T cell responses (266).
Summary and Future Perspective
The power of the human immune system in fighting cancer has been demonstrated by the adoptive transfer of TILs (1, 2) and CAR-T therapy for hematological malignancies (5, 6). However, the promise of adoptive cell therapy for solid tumors has not yet been fully realized (14, 15). TCR-T therapy holds a number of advantages over alternative strategies. TCRS can recognize epitopes derived from both surface and intracellular proteins, enabling detection of a much broader range of targets compared to CAR-T, including TAAs, cancer germline antigens, viral oncoproteins, and neoAgs. Moreover, TCRS have naturally developed to sensitively detect low epitope concentrations, down to as little as a single molecule. Recent clinical research on TCR-T has produced meaningful responses in a variety of cancers (24–27), and in some cases durable and curative responses in solid tumor patients (29, 262–265). However, as many of these studies mainly targeted TAAs, we envisage targeting tumor-specific neoAgs to produce more profound antitumor immune responses. To achieve the goal of complete eradication of solid tumors, several aspects need to be considered:
1. Targeting tumor-specific antigens, combining neoAg vaccines, checkpoint blockade therapy, and adoptive transfer of genetically engineered neoAg-specific T cells (Figure 1). While targeting neoAgs can achieve complete tumor regressions in some settings, combination strategies are likely to expand their utility and increase response rates. We propose to extend the concept of combining neoAg vaccines and checkpoint blockade therapy, together with the adoptive transfer of neoAg-specific T cells as a generalizable therapeutic strategy. This starts with high-throughput screening of large patient cohorts – encompassing multiple tumor types – to identify and collect a library of patient-derived neoAgs. This will enable vaccination using combinations of unique and shared cancer-specific neoAgs, to be tested in conjunction with checkpoint blockade therapies. In a second step, we propose to isolate neoAg-specific CTLs from responding patients, especially the CTLs specific for neoAgs representing driver mutations, to construct a TCR library that can be used to generate cell therapies for patients where vaccination is not effective (267). Challenges faced in this step include the limited availablity of patient derived materials and the low frequencies of neoAg-specific T cells from patients. To overcome these limitations, Stronen et al. developed a strategy for the induction and isolation of neoAg reactive T cells from healthy donor T cell repertoires (268), which contain higher frequency of neoAg reactive T cells that are not affected by the patient’s TME. Recently, this strategy has been optimized by Ali et al. into a standard protocol which will facilitate the isolation of neoAg specific T cells for cancer immunotherapy (269).
2. Eliminating TCR mis-pairing. Although neoAgs represent ideal targets for cancer immunotherapy, nonetheless, they are frequently ignored by patient’s TILs (268). Under such situation, adoptive transfer of high avidity neoAg-specific TCR-T cells will be a valuable supplementation to the neoAg vaccine and check point blockade therapies. However, mis-pairing of introduced TCR with endogenous TCRS could potentially cause auto-reactivity against patient’s MHC molecules, thus thoughtful innovations in TCR engineering technology could be incorporated. In this regard, genetic engineering of the TCR constant domains can be combined with design of a single-chain TCR, and the CRISPR/Cas9 genome editing can be used to orthotopically replace the endogenous TCR with tumor reactive TCR (83, 84). Through applying these recent innovative technologies in T cell engineering, TCR mis-pairing can be eliminated, while generating antitumor T cells with enhanced TCR expression and functions.
3. Maintaining long-lasting immunosurveillance against tumors and keeping patients in relapse-free survival. To achieve this goal, a fundamental requisite is persistence of genetically engineered T cells after ACT. Provision of cytokines can play important roles in supporting T cell survival and functions (99, 100, 137, 138), but is often associated with severe cytotoxicity if delivered systemically (113). Therefore, controlled and targeted delivery of cytokines through genetic engineering of tumor-specific T cells (116–119), can not only support T cell survival and generate long-term memory T cells (124, 138), but can also modify the TME to create an inflammatory environment (109, 111), and maintain a “hot” tumor milieu that self-sustains the antitumor immune responses (120).
4. Facilitating migration and penetration of genetically engineered T cells into the solid tumor (158). Genetic engineering of tumor-specific T cells with chemokine receptors that match chemokines secreted by the TME can be adopted to recruit T cells to the tumor sites (168, 174). For enhanced antitumor effect, chemokines and cytokines can be combined (185, 186), and introduced through oncolytic viruses or vaccine adjuvants (159, 184). Radiation and chemotherapy can further augment ACT by stimulating chemokine secretion from tumor cells, increasing homing and infiltration of adoptively transferred T cells into solid tumors with enhanced antitumor activity (176, 177).
5. Overcoming the immunosuppressive effects of the hostile TME and fully realizing the antitumor potential of engineered T cells. The TME represents a formidable hostile environment for antitumor T cells and favors tumor growth, metastasis, and immune evasion. The field has made advances in blocking the immunosuppressive factors of the TME (206), and developed innovative genetic engineering strategies to convert immunosuppressive ligands/factors into immunostimulatory signals (211–214, 229). These strategies can not only remodel the immunosuppressive network within the TME (230), and convert ‘cold’ tumors lacking TILs into ‘hot’ tumors with genetically engineered T cells, but can also enhance T cell co-stimulation and survival, and produce TME-resistant antitumor T cells (214, 231, 270). These TME-resistant T cells can be further expanded by neoAg vaccines, and their antitumor activity can be further enhanced by the checkpoint blockade therapies, and potentially lead to complete tumor eradication.
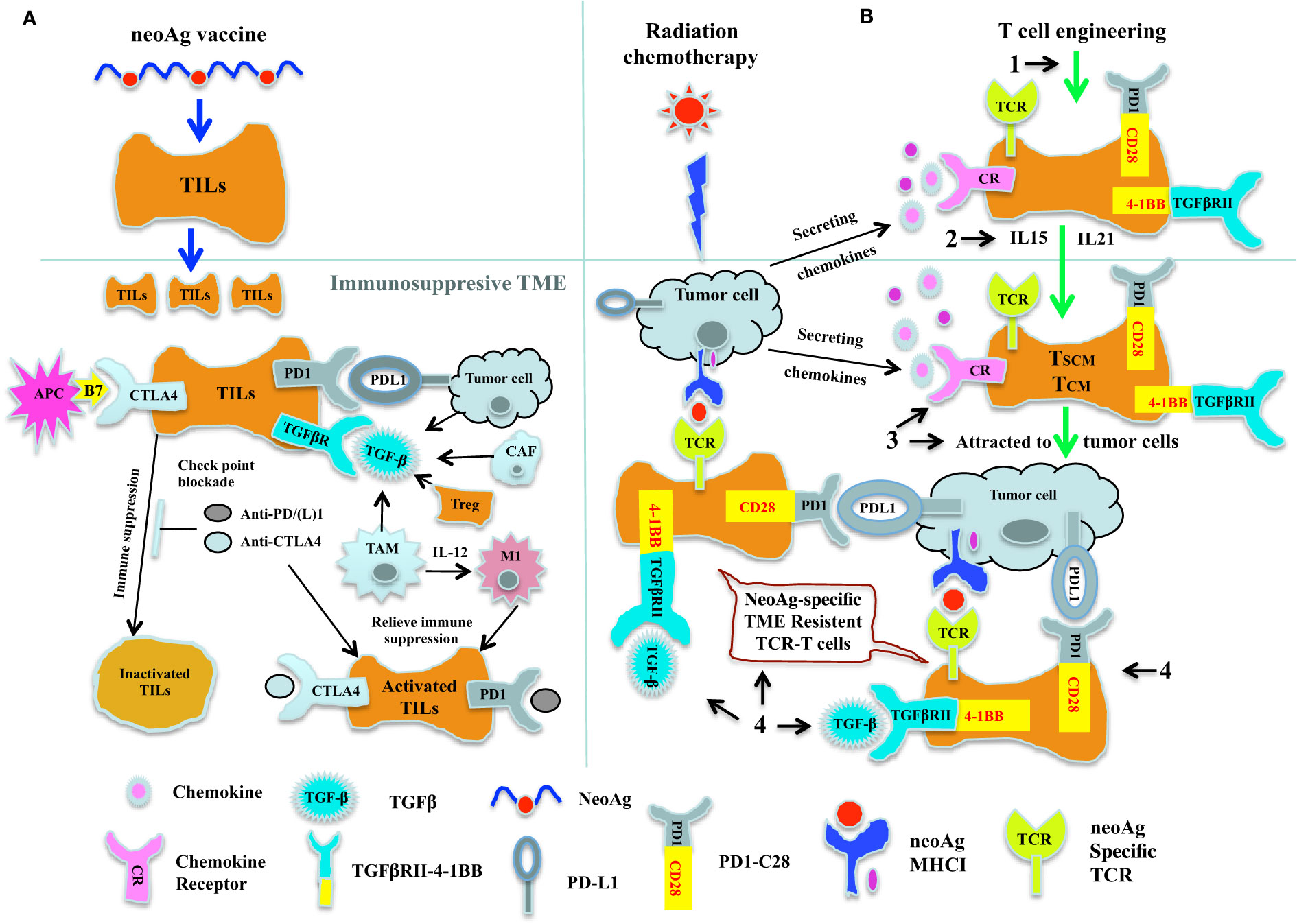
Figure 1 Cancer immunotherapy by combining neoAg vaccine and chekpoint blockade therapy together with adoptive transfer of neoAg-specific TCR-T. (A) Combination of neoAg vaccine and chekpoint blockade therapy. NeoAg vaccines can be used to stimulate and expand tumor reactive CTLs in the circulation system. When these expanded CTLs come into the TME, they will be inactivated by the check point molecules such as PDL1 and CTLA4, or by the immunosuppressive cytokine such as TGF-β (left). Using check point blockade therapies such as anti-PD(L)1 or anti-CTLA4 to block the check point interactions (middle) or using immunomodulatory cytokines such as IL-12 (right) to convert the tumor associated macrophage M2 into activated antitumor M1, these neoAg vaccine expanded CTLs can be reactivated and attack the tumor. (B) Genetically engineered TCR-T cells as a complement to neoAg vaccine and chekpoint blockade therapy. Radiation and chemotherapy can induce secretion of chemokines, which could potentially attract the tumor reactive CTLs to the tumor site. But more than offten, tumor reactive CTLs from patients are either too rare or with low avidity, thus could not control the tumor growth, as reflected by the fact that only a proportion of patients responded to neoAg vaccines or check point blockade therapies. Therefore, genetic engineering strategies could be used to complement the neoAg vaccines and check point blockade therapies. (1). By transducing patient’s T cells with neoAg-specific TCR, we could obtain truly tumor specific T cells. (2). Expanding these neoAg-specific TCR-T cells with IL-15 or IL-21, we could potentially acquire T cells with early differentiated phenotype of Tscm and Tcm. (3). By introducing chemokine receptor genes into these TCR modified T cells, these TCR-T cells could be attracted to the tumor site. (4). By expressing chimeric switch receptor (CSR) on these neoAg-specific TCR-T cells, the immunosuppressive effect of certain immune suppression factors such as PD-L1 or TGF-β within the TME could be potentially converted into immunostimulatory signals inside these TCR-T cells. Thus, with these innovative engineering strategies, we could not only obtain sufficient numbers of high avidity, early differentiated long lasting tumor reactive TCR-T cells, but these T cells could also be attracted and infiltrate into the solid tumor, and within the TME, these TCR-T cells would have the ability not only to resist the the immunosuppressive effect of the TME, but could also get stimulated and further expanded by neoAg vaccine and check point blockade therapies, and finally achieve the ultimate goal of destroying the tumor.
With the fast development and innovation of genetic engineering technologies, incorporating the aspects summarized above into the TCR-T therapeutics development, TCR-engineered T cells can be made truly tumor-specific and have the ability to migrate and penetrate into solid tumors, and become TME-resistant. TCR-T has potential to become a powerful tool for fighting cancers, especially solid tumors where other approaches have been less effective. By combining neoAg vaccines, checkpoint blockade therapy, and the adoptive transfer of neoAg-specific TCR-engineered T cells, we believe such a combination approach could lead to significant improvement in cancer immunotherapies, and this approach is scalable across different tumor types, and may provide a general strategy for the eradication of multiple cancers.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the National Natural Science Foundation of China (NSFC-81972883); Social Development & Scientific Technology Key Project of Shaanxi Province (2016SF-079); and Scientific Technology Program & Innovation Fund of Xi’An City Special Project (CXY1531WL12).
Conflict of Interest
SAX is an inventor on patents describing a Sc-TCR technology.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
CAR, Chimeric antigen receptor; CDR3, Complementarity-determining region 3; Cα: Constant alpha; Cβ: Constant beta; CRISPR, Clustered regularly interspaced short palindromic repeat; CRS: Cytokine release syndrome; CSR, Chimeric switch receptor; CTL, Cytotoxic T-Lymphocyte; CTLA4: Cytotoxic T-Lymphocyte Associated Antigen 4; EBV, Epstein-Barr Virus; GvHD, Graft-vs-host disease; HLA, Human leukocyte antigen; IFNγ, Interferon gamma; IL2, Interleukin 2; LCK, Lymphocyte-specific protein tyrosine kinase; mCα, Murine Constant alpha; MDSC, Myeloid-derived suppressor cells; MHC, Major histocompatibility complex; PBMC, Peripheral blood mononuclear cell; PLK, Polylinker; Sc, Single chain; Sc-TCR, Single-chain T-cell receptor; siRNA, Small interfering RNA; sgRNA, Single guide RNA; TAA, Tumor associated antigen; TALENs, Transcription activator-like effector nucleases; Tcm, Central memory T cells; TCR, T-cell receptor; TGF-β; Transforming growth factor-β; TNFα, Tumor necrosis factor-alpha; Treg, Regulatory T cells; TRAC, T cell receptor constant-alpha; TRBC, T cell receptor constant-beta; Tscm, Stem cell memory T cells; WT1, Wilms’ tumor 1.
References
1. Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer Regression and Autoimmunity in Patients After Clonal Repopulation With Antitumor Lymphocytes. Science (New York NY) (2002) 298(5594):850–4. doi: 10.1126/science.1076514
2. Rosenberg SA, Dudley ME. Cancer Regression in Patients With Metastatic Melanoma After the Transfer of Autologous Antitumor Lymphocytes. Proc Natl Acad Sci USA (2004) 101(Suppl 2):14639–45. doi: 10.1073/pnas.0405730101
3. Maher J, Brentjens RJ, Gunset G, Rivière I, Sadelain M. Human T-Lymphocyte Cytotoxicity and Proliferation Directed by a Single Chimeric TCRzeta/CD28 Receptor. Nat Biotechnol (2002) 20(1):70–5. doi: 10.1038/nbt0102-70
4. Song DG, Ye Q, Carpenito C, Poussin M, Wang LP, Ji C, et al. In Vivo Persistence, Tumor Localization, and Antitumor Activity of CAR-Engineered T Cells Is Enhanced by Costimulatory Signaling Through CD137 (4-1bb). Cancer Res (2011) 71(13):4617–27. doi: 10.1158/0008-5472.Can-11-0422
5. Anagnostou T, Riaz IB, Hashmi SK, Murad MH, Kenderian SS. Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy in Acute Lymphocytic Leukaemia: A Systematic Review and Meta-Analysis. Lancet Haematol (2020) 7(11):e816–e26. doi: 10.1016/s2352-3026(20)30277-5
6. Brudno JN, Kochenderfer JN. Chimeric Antigen Receptor T-Cell Therapies for Lymphoma. Nat Rev Clin Oncol (2018) 15(1):31–46. doi: 10.1038/nrclinonc.2017.128
7. Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T Cells With Chimeric Antigen Receptors Have Potent Antitumor Effects and Can Establish Memory in Patients With Advanced Leukemia. Sci Transl Med (2011) 3(95):95ra73. doi: 10.1126/scitranslmed.3002842
8. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric Antigen Receptor-Modified T Cells for Acute Lymphoid Leukemia. N Engl J Med (2013) 368(16):1509–18. doi: 10.1056/NEJMoa1215134
9. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults With B-Cell Lymphoblastic Leukemia. N Engl J Med (2018) 378(5):439–48. doi: 10.1056/NEJMoa1709866
10. Boyiadzis MM, Dhodapkar MV, Brentjens RJ, Kochenderfer JN, Neelapu SS, Maus MV, et al. Chimeric Antigen Receptor (Car) T Therapies for the Treatment of Hematologic Malignancies: Clinical Perspective and Significance. J Immunother Cancer (2018) 6(1):137. doi: 10.1186/s40425-018-0460-5
11. Ruella M, Maus MV. Catch Me If You Can: Leukemia Escape After CD19-Directed T Cell Immunotherapies. Comput Struct Biotechnol J (2016) 14:357–62. doi: 10.1016/j.csbj.2016.09.003
12. Orlando EJ, Han X, Tribouley C, Wood PA, Leary RJ, Riester M, et al. Genetic Mechanisms of Target Antigen Loss in CAR19 Therapy of Acute Lymphoblastic Leukemia. Nat Med (2018) 24(10):1504–6. doi: 10.1038/s41591-018-0146-z
13. Chapuis AG, Egan DN, Bar M, Schmitt TM, McAfee MS, Paulson KG, et al. T Cell Receptor Gene Therapy Targeting WT1 Prevents Acute Myeloid Leukemia Relapse Post-Transplant. Nat Med (2019) 25(7):1064–72. doi: 10.1038/s41591-019-0472-9
14. Newick K, O’Brien S, Moon E, Albelda SM. Car T Cell Therapy for Solid Tumors. Annu Rev Med (2017) 68:139–52. doi: 10.1146/annurev-med-062315-120245
15. D’Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M. Car-T Cells: The Long and Winding Road to Solid Tumors. Cell Death Dis (2018) 9(3):282. doi: 10.1038/s41419-018-0278-6
16. Zhang BL, Qin DY, Mo ZM, Li Y, Wei W, Wang YS, et al. Hurdles of CAR-T Cell-Based Cancer Immunotherapy Directed Against Solid Tumors. Sci China Life Sci (2016) 59(4):340–8. doi: 10.1007/s11427-016-5027-4
17. Walker AJ, Majzner RG, Zhang L, Wanhainen K, Long AH, Nguyen SM, et al. Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Mol Ther (2017) 25(9):2189–201. doi: 10.1016/j.ymthe.2017.06.008
18. Watanabe K, Terakura S, Martens AC, van Meerten T, Uchiyama S, Imai M, et al. Target Antigen Density Governs the Efficacy of Anti-CD20-CD28-CD3 Z Chimeric Antigen Receptor-Modified Effector CD8+ T Cells. J Immunol (2015) 194(3):911–20. doi: 10.4049/jimmunol.1402346
19. Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, et al. Tuning Sensitivity of CAR to Egfr Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res (2015) 75(17):3505–18. doi: 10.1158/0008-5472.Can-15-0139
20. Jahn L, van der Steen DM, Hagedoorn RS, Hombrink P, Kester MG, Schoonakker MP, et al. Generation of CD20-Specific TCRS for TCR Gene Therapy of CD20low B-Cell Malignancies Insusceptible to CD20-Targeting Antibodies. Oncotarget (2016) 7(47):77021–37. doi: 10.18632/oncotarget.12778
21. Watanabe K, Kuramitsu S, Posey AD Jr., June CH. Expanding the Therapeutic Window for CAR T Cell Therapy in Solid Tumors: The Knowns and Unknowns of CAR T Cell Biology. Front Immunol (2018) 9:2486. doi: 10.3389/fimmu.2018.02486
22. Huang J, Brameshuber M, Zeng X, Xie J, Li QJ, Chien YH, et al. A Single Peptide-Major Histocompatibility Complex Ligand Triggers Digital Cytokine Secretion in CD4(+) T Cells. Immunity (2013) 39(5):846–57. doi: 10.1016/j.immuni.2013.08.036
23. Chandran SS, Klebanoff CA. T Cell Receptor-Based Cancer Immunotherapy: Emerging Efficacy and Pathways of Resistance. Immunol Rev (2019) 290(1):127–47. doi: 10.1111/imr.12772
24. Tsimberidou AM, Van Morris K, Vo HH, Eck S, Lin YF, Rivas JM, et al. T-Cell Receptor-Based Therapy: An Innovative Therapeutic Approach for Solid Tumors. J Hematol Oncol (2021) 14(1):102. doi: 10.1186/s13045-021-01115-0
25. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor Regression in Patients With Metastatic Synovial Cell Sarcoma and Melanoma Using Genetically Engineered Lymphocytes Reactive With Ny-Eso-1. J Clin Oncol (2011) 29(7):917–24. doi: 10.1200/jco.2010.32.2537
26. Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, Feldman SA, et al. A Pilot Trial Using Lymphocytes Genetically Engineered With an Ny-Eso-1-Reactive T-Cell Receptor: Long-Term Follow-Up and Correlates With Response. Clin Cancer Res (2015) 21(5):1019–27. doi: 10.1158/1078-0432.Ccr-14-2708
27. D’Angelo SP, Melchiori L, Merchant MS, Bernstein D, Glod J, Kaplan R, et al. Antitumor Activity Associated With Prolonged Persistence of Adoptively Transferred Ny-Eso-1 (C259)T Cells in Synovial Sarcoma. Cancer Discov (2018) 8(8):944–57. doi: 10.1158/2159-8290.CD-17-1417
28. Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, et al. Ny-Eso-1-Specific TCR-Engineered T Cells Mediate Sustained Antigen-Specific Antitumor Effects in Myeloma. Nat Med (2015) 21(8):914–21. doi: 10.1038/nm.3910
29. Nagarsheth NB, Norberg SM, Sinkoe AL, Adhikary S, Meyer TJ, Lack JB, et al. TCR-Engineered T Cells Targeting E7 for Patients With Metastatic HPV-Associated Epithelial Cancers. Nat Med (2021) 27(3):419–25. doi: 10.1038/s41591-020-01225-1
30. Tran E, Robbins PF, Rosenberg SA. ‘Final Common Pathway’ of Human Cancer Immunotherapy: Targeting Random Somatic Mutations. Nat Immunol (2017) 18(3):255–62. doi: 10.1038/ni.3682
31. Vormehr M, Diken M, Boegel S, Kreiter S, Türeci Ö, Sahin U. Mutanome Directed Cancer Immunotherapy. Curr Opin Immunol (2016) 39:14–22. doi: 10.1016/j.coi.2015.12.001
32. Schumacher TN, Schreiber RD. Neoantigens in Cancer Immunotherapy. Science (New York NY) (2015) 348(6230):69–74. doi: 10.1126/science.aaa4971
34. zur Hausen H. Viruses in Human Cancers. Science (New York NY) (1991) 254(5035):1167–73. doi: 10.1126/science.1659743
35. Garber K. Driving T-Cell Immunotherapy to Solid Tumors. Nat Biotechnol (2018) 36(3):215–9. doi: 10.1038/nbt.4090
36. Michalek J, Kocak I, Fait V, Zaloudik J, Hajek R. Detection and Long-Term in Vivo Monitoring of Individual Tumor-Specific T Cell Clones in Patients With Metastatic Melanoma. J Immunol (2007) 178(11):6789–95. doi: 10.4049/jimmunol.178.11.6789
37. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of Response and Resistance to CD19 Chimeric Antigen Receptor (Car) T Cell Therapy of Chronic Lymphocytic Leukemia. Nat Med (2018) 24(5):563–71. doi: 10.1038/s41591-018-0010-1
38. Schumacher TN. T-Cell-Receptor Gene Therapy. Nat Rev Immunol (2002) 2(7):512–9. doi: 10.1038/nri841
39. Xue SA, Stauss HJ. Enhancing Immune Responses for Cancer Therapy. Cell Mol Immunol (2007) 4(3):173–84.
40. Schmitt TM, Ragnarsson GB, Greenberg PD. T Cell Receptor Gene Therapy for Cancer. Hum Gene Ther (2009) 20(11):1240–8. doi: 10.1089/hum.2009.146
41. Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, et al. Lethal Graft-Versus-Host Disease in Mouse Models of T Cell Receptor Gene Therapy. Nat Med (2010) 16(5):565–70. doi: 10.1038/nm.2128
42. van Loenen MM, de Boer R, Amir AL, Hagedoorn RS, Volbeda GL, Willemze R, et al. Mixed T Cell Receptor Dimers Harbor Potentially Harmful Neoreactivity. Proc Natl Acad Sci USA (2010) 107(24):10972–7. doi: 10.1073/pnas.1005802107
43. Sommermeyer D, Neudorfer J, Weinhold M, Leisegang M, Engels B, Noessner E, et al. Designer T Cells by T Cell Receptor Replacement. Eur J Immunol (2006) 36(11):3052–9. doi: 10.1002/eji.200636539
44. Govers C, Sebestyén Z, Coccoris M, Willemsen RA, Debets R. T Cell Receptor Gene Therapy: Strategies for Optimizing Transgenic TCR Pairing. Trends Mol Med (2010) 16(2):77–87. doi: 10.1016/j.molmed.2009.12.004
45. Garcia KC, Degano M, Stanfield RL, Brunmark A, Jackson MR, Peterson PA, et al. An Alphabeta T Cell Receptor Structure at 2.5 a and Its Orientation in the TCR-MHC Complex. Science (New York NY) (1996) 274(5285):209–19. doi: 10.1126/science.274.5285.209
46. Voss RH, Willemsen RA, Kuball J, Grabowski M, Engel R, Intan RS, et al. Molecular Design of the Calphabeta Interface Favors Specific Pairing of Introduced TCRalphabeta in Human T Cells. J Immunol (2008) 180(1):391–401. doi: 10.4049/jimmunol.180.1.391
47. Boulter JM, Glick M, Todorov PT, Baston E, Sami M, Rizkallah P, et al. Stable, Soluble T-Cell Receptor Molecules for Crystallization and Therapeutics. Protein Eng (2003) 16(9):707–11. doi: 10.1093/protein/gzg087
48. Kuball J, Dossett ML, Wolfl M, Ho WY, Voss RH, Fowler C, et al. Facilitating Matched Pairing and Expression of TCR Chains Introduced Into Human T Cells. Blood (2007) 109(6):2331–8. doi: 10.1182/blood-2006-05-023069
49. Thomas S, Xue SA, Cesco-Gaspere M, San José E, Hart DP, Wong V, et al. Targeting the Wilms Tumor Antigen 1 by TCR Gene Transfer: TCR Variants Improve Tetramer Binding But Not the Function of Gene Modified Human T Cells. J Immunol (2007) 179(9):5803–10. doi: 10.4049/jimmunol.179.9.5803
50. Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, Morgan RA. Enhanced Antitumor Activity of T Cells Engineered to Express T-Cell Receptors With a Second Disulfide Bond. Cancer Res (2007) 67(8):3898–903. doi: 10.1158/0008-5472.Can-06-3986
51. Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced Antitumor Activity of Murine-Human Hybrid T-Cell Receptor (TCR) in Human Lymphocytes Is Associated With Improved Pairing and TCR/CD3 Stability. Cancer Res (2006) 66(17):8878–86. doi: 10.1158/0008-5472.Can-06-1450
52. Hart DP, Xue SA, Thomas S, Cesco-Gaspere M, Tranter A, Willcox B, et al. Retroviral Transfer of a Dominant TCR Prevents Surface Expression of a Large Proportion of the Endogenous TCR Repertoire in Human T Cells. Gene Ther (2008) 15(8):625–31. doi: 10.1038/sj.gt.3303078
53. Bialer G, Horovitz-Fried M, Ya’acobi S, Morgan RA, Cohen CJ. Selected Murine Residues Endow Human TCR With Enhanced Tumor Recognition. J Immunol (2010) 184(11):6232–41. doi: 10.4049/jimmunol.0902047
54. Sommermeyer D, Uckert W. Minimal Amino Acid Exchange in Human TCR Constant Regions Fosters Improved Function of TCR Gene-Modified T Cells. J Immunol (2010) 184(11):6223–31. doi: 10.4049/jimmunol.0902055
55. Bethune MT, Gee MH, Bunse M, Lee MS, Gschweng EH, Pagadala MS, et al. Domain-Swapped T Cell Receptors Improve the Safety of TCR Gene Therapy. Elife (2016) 5:e19095. doi: 10.7554/eLife.19095
56. Richman SA, Aggen DH, Dossett ML, Donermeyer DL, Allen PM, Greenberg PD, et al. Structural Features of T Cell Receptor Variable Regions That Enhance Domain Stability and Enable Expression as Single-Chain Valphavbeta Fragments. Mol Immunol (2009) 46(5):902–16. doi: 10.1016/j.molimm.2008.09.021
57. Heemskerk MH, Hagedoorn RS, van der Hoorn MA, van der Veken LT, Hoogeboom M, Kester MG, et al. Efficiency of T-Cell Receptor Expression in Dual-Specific T Cells Is Controlled by the Intrinsic Qualities of the TCR Chains Within the TCR-CD3 Complex. Blood (2007) 109(1):235–43. doi: 10.1182/blood-2006-03-013318
58. Willemsen RA, Weijtens ME, Ronteltap C, Eshhar Z, Gratama JW, Chames P, et al. Grafting Primary Human T Lymphocytes With Cancer-Specific Chimeric Single Chain and Two Chain TCR. Gene Ther (2000) 7(16):1369–77. doi: 10.1038/sj.gt.3301253
59. Sebestyén Z, Schooten E, Sals T, Zaldivar I, San José E, Alarcón B, et al. Human TCR That Incorporate CD3zeta Induce Highly Preferred Pairing Between TCRalpha and Beta Chains Following Gene Transfer. J Immunol (2008) 180(11):7736–46. doi: 10.4049/jimmunol.180.11.7736
60. Aggen DH, Chervin AS, Schmitt TM, Engels B, Stone JD, Richman SA, et al. Single-Chain Vαvβ T-Cell Receptors Function Without Mispairing With Endogenous TCR Chains. Gene Ther (2012) 19(4):365–74. doi: 10.1038/gt.2011.104
61. Harris DT, Kranz DM. Adoptive T Cell Therapies: A Comparison of T Cell Receptors and Chimeric Antigen Receptors. Trends Pharmacol Sci (2016) 37(3):220–30. doi: 10.1016/j.tips.2015.11.004
62. Voss RH, Thomas S, Pfirschke C, Hauptrock B, Klobuch S, Kuball J, et al. Coexpression of the T-Cell Receptor Constant Alpha Domain Triggers Tumor Reactivity of Single-Chain TCR-Transduced Human T Cells. Blood (2010) 115(25):5154–63. doi: 10.1182/blood-2009-11-254078
63. Knies D, Klobuch S, Xue SA, Birtel M, Echchannaoui H, Yildiz O, et al. An Optimized Single Chain TCR Scaffold Relying on the Assembly With the Native CD3-Complex Prevents Residual Mispairing With Endogenous TCRS in Human T-Cells. Oncotarget (2016) 7(16):21199–221. doi: 10.18632/oncotarget.8385
64. Thomas S, Mohammed F, Reijmers RM, Woolston A, Stauss T, Kennedy A, et al. Framework Engineering to Produce Dominant T Cell Receptors With Enhanced Antigen-Specific Function. Nat Commun (2019) 10(1):4451. doi: 10.1038/s41467-019-12441-w
65. Ahmadi M, King JW, Xue SA, Voisine C, Holler A, Wright GP, et al. CD3 Limits the Efficacy of TCR Gene Therapy in Vivo. Blood (2011) 118(13):3528–37. doi: 10.1182/blood-2011-04-346338
66. Ochi T, Fujiwara H, Okamoto S, An J, Nagai K, Shirakata T, et al. Novel Adoptive T-Cell Immunotherapy Using a WT1-Specific TCR Vector Encoding Silencers for Endogenous TCRS Shows Marked Antileukemia Reactivity and Safety. Blood (2011) 118(6):1495–503. doi: 10.1182/blood-2011-02-337089
67. Bunse M, Bendle GM, Linnemann C, Bies L, Schulz S, Schumacher TN, et al. RNAI-Mediated TCR Knockdown Prevents Autoimmunity in Mice Caused by Mixed TCR Dimers Following TCR Gene Transfer. Mol Ther (2014) 22(11):1983–91. doi: 10.1038/mt.2014.142
68. Provasi E, Genovese P, Lombardo A, Magnani Z, Liu PQ, Reik A, et al. Editing T Cell Specificity Towards Leukemia by Zinc Finger Nucleases and Lentiviral Gene Transfer. Nat Med (2012) 18(5):807–15. doi: 10.1038/nm.2700
69. Berdien B, Mock U, Atanackovic D, Fehse B. Talen-Mediated Editing of Endogenous T-Cell Receptors Facilitates Efficient Reprogramming of T Lymphocytes by Lentiviral Gene Transfer. Gene Ther (2014) 21(6):539–48. doi: 10.1038/gt.2014.26
70. Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, et al. Molecular Remission of Infant B-All After Infusion of Universal Talen Gene-Edited CAR T Cells. Sci Transl Med (2017) 9(374). doi: 10.1126/scitranslmed.aaj2013
71. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the Trac Locus With CRISPR/Cas9 Enhances Tumour Rejection. Nature (2017) 543(7643):113–7. doi: 10.1038/nature21405
72. Gaj T, Gersbach CA, Barbas CF 3rd. Zfn, Talen, and CRISPR/Cas-Based Methods for Genome Engineering. Trends Biotechnol (2013) 31(7):397–405. doi: 10.1016/j.tibtech.2013.04.004
73. Knott GJ, Doudna JA. CRISPR-Cas Guides the Future of Genetic Engineering. Science (New York NY) (2018) 361(6405):866–9. doi: 10.1126/science.aat5011
74. Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, et al. Reprogramming Human T Cell Function and Specificity With Non-Viral Genome Targeting. Nature (2018) 559(7714):405–9. doi: 10.1038/s41586-018-0326-5
75. Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin Cancer Res (2017) 23(9):2255–66. doi: 10.1158/1078-0432.Ccr-16-1300
76. Ren J, Zhang X, Liu X, Fang C, Jiang S, June CH, et al. A Versatile System for Rapid Multiplex Genome-Edited CAR T Cell Generation. Oncotarget (2017) 8(10):17002–11. doi: 10.18632/oncotarget.15218
77. Georgiadis C, Preece R, Nickolay L, Etuk A, Petrova A, Ladon D, et al. Long Terminal Repeat CRISPR-CAR-Coupled “Universal” T Cells Mediate Potent Anti-Leukemic Effects. Mol Ther (2018) 26(5):1215–27. doi: 10.1016/j.ymthe.2018.02.025
78. Liu X, Zhang Y, Cheng C, Cheng AW, Zhang X, Li N, et al. CRISPR-Cas9-Mediated Multiplex Gene Editing in CAR-T Cells. Cell Res (2017) 27(1):154–7. doi: 10.1038/cr.2016.142
79. Zhang Y, Zhang X, Cheng C, Mu W, Liu X, Li N, et al. CRISPR-Cas9 Mediated Lag-3 Disruption in CAR-T Cells. Front Med (2017) 11(4):554–62. doi: 10.1007/s11684-017-0543-6
80. Mastaglio S, Genovese P, Magnani Z, Ruggiero E, Landoni E, Camisa B, et al. Ny-Eso-1 TCR Single Edited Stem and Central Memory T Cells to Treat Multiple Myeloma Without Graft-Versus-Host Disease. Blood (2017) 130(5):606–18. doi: 10.1182/blood-2016-08-732636
81. Legut M, Dolton G, Mian AA, Ottmann OG, Sewell AK. CRISPR-Mediated TCR Replacement Generates Superior Anticancer Transgenic T Cells. Blood (2018) 131(3):311–22. doi: 10.1182/blood-2017-05-787598
82. Morton LT, Reijmers RM, Wouters AK, Kweekel C, Remst DFG, Pothast CR, et al. Simultaneous Deletion of Endogenous TCRαβ for TCR Gene Therapy Creates an Improved and Safe Cellular Therapeutic. Mol Ther (2020) 28(1):64–74. doi: 10.1016/j.ymthe.2019.10.001
83. Schober K, Müller TR, Gökmen F, Grassmann S, Effenberger M, Poltorak M, et al. Orthotopic Replacement of T-Cell Receptor A- and B-Chains With Preservation of Near-Physiological T-Cell Function. Nat Biomed Eng (2019) 3(12):974–84. doi: 10.1038/s41551-019-0409-0
84. Schober K, Müller TR, Busch DH. Orthotopic T-Cell Receptor Replacement-An “Enabler” for TCR-Based Therapies. Cells (2020) 9(6). doi: 10.3390/cells9061367
85. Zhang XH, Tee LY, Wang XG, Huang QS, Yang SH. Off-Target Effects in CRISPR/Cas9-Mediated Genome Engineering. Mol Ther Nucleic Acids (2015) 4(11):e264. doi: 10.1038/mtna.2015.37
86. Kosicki M, Tomberg K, Bradley A. Repair of Double-Strand Breaks Induced by CRISPR-Cas9 Leads to Large Deletions and Complex Rearrangements. Nat Biotechnol (2018) 36(8):765–71. doi: 10.1038/nbt.4192
87. Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, et al. High-Fidelity CRISPR-Cas9 Nucleases With No Detectable Genome-Wide Off-Target Effects. Nature (2016) 529(7587):490–5. doi: 10.1038/nature16526
88. Hirakawa MP, Krishnakumar R, Timlin JA, Carney JP, Butler KS. Gene Editing and CRISPR in the Clinic: Current and Future Perspectives. Biosci Rep (2020) 40(4):BSR20200127. doi: 10.1042/bsr20200127
89. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. Crispr-Engineered T Cells in Patients With Refractory Cancer. Science (New York NY) (2020) 367(6481):eaba7365. doi: 10.1126/science.aba7365
90. Lu Y, Xue J, Deng T, Zhou X, Yu K, Deng L, et al. Safety and Feasibility of CRISPR-Edited T Cells in Patients With Refractory Non-Small-Cell Lung Cancer. Nat Med (2020) 26(5):732–40. doi: 10.1038/s41591-020-0840-5
91. Xue SA, Chen Y, Voss RH, Kisan V, Wang B, Chen KK, et al. Enhancing the Expression and Function of an Ebv-TCR on Engineered T Cells by Combining Sc-TCR Design With CRISPR Editing to Prevent Mispairing. Cell Mol Immunol (2020) 17(12):1275–7. doi: 10.1038/s41423-020-0396-9
92. Dutcher JP, Schwartzentruber DJ, Kaufman HL, Agarwala SS, Tarhini AA, Lowder JN, et al. High Dose Interleukin-2 (Aldesleukin) - Expert Consensus on Best Management Practices-2014. J Immunother Cancer (2014) 2(1):26. doi: 10.1186/s40425-014-0026-0
93. Rosenberg SA. Il-2: The First Effective Immunotherapy for Human Cancer. J Immunol (2014) 192(12):5451–8. doi: 10.4049/jimmunol.1490019
94. Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, et al. High-Dose Recombinant Interleukin 2 Therapy for Patients With Metastatic Melanoma: Analysis of 270 Patients Treated Between 1985 and 1993. J Clin Oncol (1999) 17(7):2105–16. doi: 10.1200/jco.1999.17.7.2105
95. Sim GC, Martin-Orozco N, Jin L, Yang Y, Wu S, Washington E, et al. Il-2 Therapy Promotes Suppressive Icos+ Treg Expansion in Melanoma Patients. J Clin Invest (2014) 124(1):99–110. doi: 10.1172/jci46266
96. Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An Essential Role for the Il-2 Receptor in T(Reg) Cell Function. Nat Immunol (2016) 17(11):1322–33. doi: 10.1038/ni.3540
97. Sun Z, Ren Z, Yang K, Liu Z, Cao S, Deng S, et al. A Next-Generation Tumor-Targeting Il-2 Preferentially Promotes Tumor-Infiltrating CD8(+) T-Cell Response and Effective Tumor Control. Nat Commun (2019) 10(1):3874. doi: 10.1038/s41467-019-11782-w
98. Bradley LM, Haynes L, Swain SL. Il-7: Maintaining T-Cell Memory and Achieving Homeostasis. Trends Immunol (2005) 26(3):172–6. doi: 10.1016/j.it.2005.01.004
99. Fry TJ, Mackall CL. The Many Faces of Il-7: From Lymphopoiesis to Peripheral T Cell Maintenance. J Immunol (2005) 174(11):6571–6. doi: 10.4049/jimmunol.174.11.6571
100. Rathmell JC, Farkash EA, Gao W, Thompson CB. Il-7 Enhances the Survival and Maintains the Size of Naive T Cells. J Immunol (2001) 167(12):6869–76. doi: 10.4049/jimmunol.167.12.6869
101. Ding ZC, Habtetsion T, Cao Y, Li T, Liu C, Kuczma M, et al. Adjuvant Il-7 Potentiates Adoptive T Cell Therapy by Amplifying and Sustaining Polyfunctional Antitumor CD4+ T Cells. Sci Rep (2017) 7(1):12168. doi: 10.1038/s41598-017-12488-z
102. He C, Zhou Y, Li Z, Farooq MA, Ajmal I, Zhang H, et al. Co-Expression of Il-7 Improves NKG2D-Based CAR T Cell Therapy on Prostate Cancer by Enhancing the Expansion and Inhibiting the Apoptosis and Exhaustion. Cancers (2020) 12(7):1969. doi: 10.3390/cancers12071969
103. Xiong X, Xi J, Liu Q, Wang C, Jiang Z, Yue SY, et al. Co-Expression of Il-7 and Ph20 Promote Anti-Gpc3 CAR-T Tumour Suppressor Activity in Vivo and in Vitro. Liver Int (2021) 41(5):1033–43. doi: 10.1111/liv.14771
104. Tugues S, Burkhard SH, Ohs I, Vrohlings M, Nussbaum K, Vom Berg J, et al. New Insights Into Il-12-Mediated Tumor Suppression. Cell Death Differ (2015) 22(2):237–46. doi: 10.1038/CDd.2014.134
105. Vignali DA, Kuchroo VK. Il-12 Family Cytokines: Immunological Playmakers. Nat Immunol (2012) 13(8):722–8. doi: 10.1038/ni.2366
106. Hashimoto W, Osaki T, Okamura H, Robbins PD, Kurimoto M, Nagata S, et al. Differential Antitumor Effects of Administration of Recombinant Il-18 or Recombinant Il-12 Are Mediated Primarily by Fas-Fas Ligand- and Perforin-Induced Tumor Apoptosis, Respectively. J Immunol (1999) 163(2):583–9.
107. Cao X, Leonard K, Collins LI, Cai SF, Mayer JC, Payton JE, et al. Interleukin 12 Stimulates Ifn-Gamma-Mediated Inhibition of Tumor-Induced Regulatory T-Cell Proliferation and Enhances Tumor Clearance. Cancer Res (2009) 69(22):8700–9. doi: 10.1158/0008-5472.Can-09-1145
108. Matsushita H, Hosoi A, Ueha S, Abe J, Fujieda N, Tomura M, et al. Cytotoxic T Lymphocytes Block Tumor Growth Both by Lytic Activity and Ifnγ-Dependent Cell-Cycle Arrest. Cancer Immunol Res (2015) 3(1):26–36. doi: 10.1158/2326-6066.Cir-14-0098
109. Kerkar SP, Goldszmid RS, Muranski P, Chinnasamy D, Yu Z, Reger RN, et al. Il-12 Triggers a Programmatic Change in Dysfunctional Myeloid-Derived Cells Within Mouse Tumors. J Clin Invest (2011) 121(12):4746–57. doi: 10.1172/jci58814
110. Suzuki S, Umezu Y, Saijo Y, Satoh G, Abe Y, Satoh K, et al. Exogenous Recombinant Human Il-12 Augments Mhc Class I Antigen Expression on Human Cancer Cells in Vitro. Tohoku J Exp Med (1998) 185(3):223–6. doi: 10.1620/tjem.185.223
111. Yu X, Wu B, Ma T, Lin Y, Cheng F, Xiong H, et al. Overexpression of Il-12 Reverses the Phenotype and Function of M2 Macrophages to M1 Macrophages. Int J Clin Exp Pathol (2016) 9(9):8963–72.
112. Díaz-Montero CM, El Naggar S, Al Khami A, El Naggar R, Montero AJ, Cole DJ, et al. Priming of Naive CD8+ T Cells in the Presence of Il-12 Selectively Enhances the Survival of CD8+CD62lhi Cells and Results in Superior Anti-Tumor Activity in a Tolerogenic Murine Model. Cancer Immunol Immunother (2008) 57(4):563–72. doi: 10.1007/s00262-007-0394-0
113. Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, et al. Effects of Single-Dose Interleukin-12 Exposure on Interleukin-12-Associated Toxicity and Interferon-Gamma Production. Blood (1997) 90(7):2541–8.
114. Chinnasamy D, Yu Z, Kerkar SP, Zhang L, Morgan RA, Restifo NP, et al. Local Delivery of Interleukin-12 Using T Cells Targeting VEGF Receptor-2 Eradicates Multiple Vascularized Tumors in Mice. Clin Cancer Res (2012) 18(6):1672–83. doi: 10.1158/1078-0432.Ccr-11-3050
115. Nguyen KG, Vrabel MR, Mantooth SM, Hopkins JJ, Wagner ES, Gabaldon TA, et al. Localized Interleukin-12 for Cancer Immunotherapy. Front Immunol (2020) 11:575597. doi: 10.3389/fimmu.2020.575597
116. Li J, Lin W, Chen H, Xu Z, Ye Y, Chen M. Dual-Target Il-12-Containing Nanoparticles Enhance T Cell Functions for Cancer Immunotherapy. Cell Immunol (2020) 349:104042. doi: 10.1016/j.cellimm.2020.104042
117. Hwang MP, Fecek RJ, Qin T, Storkus WJ, Wang Y. Single Injection of Il-12 Coacervate as an Effective Therapy Against B16-F10 Melanoma in Mice. J Control Rel (2020) 318:270–8. doi: 10.1016/j.jconrel.2019.12.035
118. Wang P, Li X, Wang J, Gao D, Li Y, Li H, et al. Re-Designing Interleukin-12 to Enhance Its Safety and Potential as an Anti-Tumor Immunotherapeutic Agent. Nat Commun (2017) 8(1):1395. doi: 10.1038/s41467-017-01385-8
119. Zhang L, Davies JS, Serna C, Yu Z, Restifo NP, Rosenberg SA, et al. Enhanced Efficacy and Limited Systemic Cytokine Exposure With Membrane-Anchored Interleukin-12 T-Cell Therapy in Murine Tumor Models. J Immunother Cancer (2020) 8(1):e000210. doi: 10.1136/jitc-2019-000210
120. Chiocca EA, Yu JS, Lukas RV, Solomon IH, Ligon KL, Nakashima H, et al. Regulatable Interleukin-12 Gene Therapy in Patients With Recurrent High-Grade Glioma: Results of a Phase 1 Trial. Sci Transl Med (2019) 11(505):eaaw5680. doi: 10.1126/scitranslmed.aaw5680
121. Robertson MJ, Mier JW, Logan T, Atkins M, Koon H, Koch KM, et al. Clinical and Biological Effects of Recombinant Human Interleukin-18 Administered by Intravenous Infusion to Patients With Advanced Cancer. Clin Cancer Res (2006) 12(14 Pt 1):4265–73. doi: 10.1158/1078-0432.Ccr-06-0121
122. Hu B, Ren J, Luo Y, Keith B, Young RM, Scholler J, et al. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting Il-18. Cell Rep (2017) 20(13):3025–33. doi: 10.1016/j.celrep.2017.09.002
123. Avanzi MP, Yeku O, Li X, Wijewarnasuriya DP, van Leeuwen DG, Cheung K, et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and Through Activation of the Endogenous Immune System. Cell Rep (2018) 23(7):2130–41. doi: 10.1016/j.celrep.2018.04.051
124. Pilipow K, Roberto A, Roederer M, Waldmann TA, Mavilio D, Lugli E. Il15 and T-Cell Stemness in T-Cell-Based Cancer Immunotherapy. Cancer Res (2015) 75(24):5187–93. doi: 10.1158/0008-5472.Can-15-1498
125. Alizadeh D, Wong RA, Yang X, Wang D, Pecoraro JR, Kuo CF, et al. Il15 Enhances CAR-T Cell Antitumor Activity by Reducing Mtorc1 Activity and Preserving Their Stem Cell Memory Phenotype. Cancer Immunol Res (2019) 7(5):759–72. doi: 10.1158/2326-6066.Cir-18-0466
126. Battram AM, Bachiller M, Lopez V, Fernández de Larrea C, Urbano-Ispizua A, Martín-Antonio B. Il-15 Enhances the Persistence and Function of Bcma-Targeting CAR-T Cells Compared to Il-2 or Il-15/Il-7 by Limiting CAR-T Cell Dysfunction and Differentiation. Cancers (2021) 13(14):3534. doi: 10.3390/cancers13143534
127. Klebanoff CA, Finkelstein SE, Surman DR, Lichtman MK, Gattinoni L, Theoret MR, et al. Il-15 Enhances the in Vivo Antitumor Activity of Tumor-Reactive CD8+ T Cells. Proc Natl Acad Sci USA (2004) 101(7):1969–74. doi: 10.1073/pnas.0307298101
128. Hurton LV, Singh H, Najjar AM, Switzer KC, Mi T, Maiti S, et al. Tethered Il-15 Augments Antitumor Activity and Promotes a Stem-Cell Memory Subset in Tumor-Specific T Cells. Proc Natl Acad Sci USA (2016) 113(48):E7788–e97. doi: 10.1073/pnas.1610544113
129. Dubois S, Patel HJ, Zhang M, Waldmann TA, Müller JR. Preassociation of Il-15 With Il-15r Alpha-Igg1-Fc Enhances Its Activity on Proliferation of Nk and CD8+/CD44high T Cells and Its Antitumor Action. J Immunol (2008) 180(4):2099–106. doi: 10.4049/jimmunol.180.4.2099
130. Miller JS, Morishima C, McNeel DG, Patel MR, Kohrt HEK, Thompson JA, et al. A First-In-Human Phase I Study of Subcutaneous Outpatient Recombinant Human Il15 (Rhil15) in Adults With Advanced Solid Tumors. Clin Cancer Res (2018) 24(7):1525–35. doi: 10.1158/1078-0432.Ccr-17-2451
131. Feng J, Xu H, Cinquina A, Wu Z, Chen Q, Zhang P, et al. Treatment of Aggressive T Cell Lymphoblastic Lymphoma/Leukemia Using Anti-CD5 CAR T Cells. Stem Cell Rev Rep (2021) 17(2):652–61. doi: 10.1007/s12015-020-10092-9
132. Waldmann TA, Dubois S, Miljkovic MD, Conlon KC. Il-15 in the Combination Immunotherapy of Cancer. Front Immunol (2020) 11:868. doi: 10.3389/fimmu.2020.00868
133. Li Y, Yee C. Il-21 Mediated Foxp3 Suppression Leads to Enhanced Generation of Antigen-Specific CD8+ Cytotoxic T Lymphocytes. Blood (2008) 111(1):229–35. doi: 10.1182/blood-2007-05-089375
134. Tian Y, Cox MA, Kahan SM, Ingram JT, Bakshi RK, Zajac AJ. A Context-Dependent Role for Il-21 in Modulating the Differentiation, Distribution, and Abundance of Effector and Memory CD8 T Cell Subsets. J Immunol (2016) 196(5):2153–66. doi: 10.4049/jimmunol.1401236
135. Allard EL, Hardy MP, Leignadier J, Marquis M, Rooney J, Lehoux D, et al. Overexpression of Il-21 Promotes Massive CD8+ Memory T Cell Accumulation. Eur J Immunol (2007) 37(11):3069–77. doi: 10.1002/eji.200637017
136. Tian Y, Yuan C, Ma D, Zhang Y, Liu Y, Zhang W, et al. Il-21 and Il-12 Inhibit Differentiation of Treg and Th17 Cells and Enhance Cytotoxicity of Peripheral Blood Mononuclear Cells in Patients With Cervical Cancer. Int J Gynecol Cancer (2011) 21(9):1672–8. doi: 10.1097/IGC.0b013e3182358955
137. Zeng R, Spolski R, Finkelstein SE, Oh S, Kovanen PE, Hinrichs CS, et al. Synergy of Il-21 and Il-15 in Regulating CD8+ T Cell Expansion and Function. J Exp Med (2005) 201(1):139–48. doi: 10.1084/jem.20041057
138. Alves NL, Arosa FA, van Lier RA. Il-21 Sustains CD28 Expression on Il-15-Activated Human Naive CD8+ T Cells. J Immunol (2005) 175(2):755–62. doi: 10.4049/jimmunol.175.2.755
139. Li Y, Bleakley M, Yee C. Il-21 Influences the Frequency, Phenotype, and Affinity of the Antigen-Specific CD8 T Cell Response. J Immunol (2005) 175(4):2261–9. doi: 10.4049/jimmunol.175.4.2261
140. Hinrichs CS, Spolski R, Paulos CM, Gattinoni L, Kerstann KW, Palmer DC, et al. Il-2 and Il-21 Confer Opposing Differentiation Programs to CD8+ T Cells for Adoptive Immunotherapy. Blood (2008) 111(11):5326–33. doi: 10.1182/blood-2007-09-113050
141. Søndergaard H, Galsgaard ED, Bartholomaeussen M, Straten PT, Odum N, Skak K. Intratumoral Interleukin-21 Increases Antitumor Immunity, Tumor-Infiltrating CD8+ T-Cell Density and Activity, and Enlarges Draining Lymph Nodes. J Immunother (2010) 33(3):236–49. doi: 10.1097/CJI.0b013e3181c0c1cb
142. Chapuis AG, Ragnarsson GB, Nguyen HN, Chaney CN, Pufnock JS, Schmitt TM, et al. Transferred WT1-Reactive CD8+ T Cells Can Mediate Antileukemic Activity and Persist in Post-Transplant Patients. Sci Transl Med (2013) 5(174):174ra27. doi: 10.1126/scitranslmed.3004916
143. Deng S, Sun Z, Qiao J, Liang Y, Liu L, Dong C, et al. Targeting Tumors With Il-21 Reshapes the Tumor Microenvironment by Proliferating PD-1intTIM-3-CD8+ T Cells. JCI Insight (2020) 5(7):e132000. doi: 10.1172/jci.insight.132000
144. White L, Krishnan S, Strbo N, Liu H, Kolber MA, Lichtenheld MG, et al. Differential Effects of Il-21 and Il-15 on Perforin Expression, Lysosomal Degranulation, and Proliferation in CD8 T Cells of Patients With Human Immunodeficiency Virus-1 (Hiv). Blood (2007) 109(9):3873–80. doi: 10.1182/blood-2006-09-045278
145. Darcy PK. Il-21 Priming Enhances T-Cell Immunotherapy. Blood (2008) 111(11):5268–9. doi: 10.1182/blood-2008-03-142505
146. Yi JS, Du M, Zajac AJ. A Vital Role for Interleukin-21 in the Control of a Chronic Viral Infection. Science (New York NY) (2009) 324(5934):1572–6. doi: 10.1126/science.1175194
147. Li Y, Cong Y, Jia M, He Q, Zhong H, Zhao Y, et al. Targeting Il-21 to Tumor-Reactive T Cells Enhances Memory T Cell Responses and Anti-PD-1 Antibody Therapy. Nat Commun (2021) 12(1):951. doi: 10.1038/s41467-021-21241-0
148. Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, Suppressors and Antigen Presenters: How Lymphodepletion Enhances T Cell-Mediated Tumor Immunotherapy. Trends Immunol (2005) 26(2):111–7. doi: 10.1016/j.it.2004.12.003
149. Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, et al. Removal of Homeostatic Cytokine Sinks by Lymphodepletion Enhances the Efficacy of Adoptively Transferred Tumor-Specific CD8+ T Cells. J Exp Med (2005) 202(7):907–12. doi: 10.1084/jem.20050732
150. Hirayama AV, Gauthier J, Hay KA, Voutsinas JM, Wu Q, Gooley T, et al. The Response to Lymphodepletion Impacts Pfs in Patients With Aggressive Non-Hodgkin Lymphoma Treated With CD19 CAR T Cells. Blood (2019) 133(17):1876–87. doi: 10.1182/blood-2018-11-887067
151. Muranski P, Boni A, Wrzesinski C, Citrin DE, Rosenberg SA, Childs R, et al. Increased Intensity Lymphodepletion and Adoptive Immunotherapy–How Far Can We Go? Nat Clin Pract Oncol (2006) 3(12):668–81. doi: 10.1038/ncponc0666
152. Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, et al. Acquisition of Full Effector Function in Vitro Paradoxically Impairs the in Vivo Antitumor Efficacy of Adoptively Transferred CD8+ T Cells. J Clin Invest (2005) 115(6):1616–26. doi: 10.1172/jci24480
153. Sabatino M, Hu J, Sommariva M, Gautam S, Fellowes V, Hocker JD, et al. Generation of Clinical-Grade CD19-Specific CAR-Modified CD8+ Memory Stem Cells for the Treatment of Human B-Cell Malignancies. Blood (2016) 128(4):519–28. doi: 10.1182/blood-2015-11-683847
154. Biasco L, Izotova N, Rivat C, Ghorashian S, Richardson R, Guvenel A, et al. Clonal Expansion of T Memory Stem Cells Determines Early Anti-Leukemic Responses and Long-Term CAR T Cell Persistence in Patients. Nat Cancer (2021) 2(6):629–42. doi: 10.1038/s43018-021-00207-7
155. Alvarez-Fernández C, Escribà-Garcia L, Vidal S, Sierra J, Briones J. A Short CD3/CD28 Costimulation Combined With Il-21 Enhance the Generation of Human Memory Stem T Cells for Adoptive Immunotherapy. J Transl Med (2016) 14(1):214. doi: 10.1186/s12967-016-0973-y
156. Arcangeli S, Falcone L, Camisa B, De Girardi F, Biondi M, Giglio F, et al. Next-Generation Manufacturing Protocols Enriching TScm CAR T Cells Can Overcome Disease-Specific T Cell Defects in Cancer Patients. Front Immunol (2020) 11:1217. doi: 10.3389/fimmu.2020.01217
157. Singh H, Figliola MJ, Dawson MJ, Huls H, Olivares S, Switzer K, et al. Reprogramming CD19-Specific T Cells With Il-21 Signaling Can Improve Adoptive Immunotherapy of B-Lineage Malignancies. Cancer Res (2011) 71(10):3516–27. doi: 10.1158/0008-5472.Can-10-3843
158. Melero I, Rouzaut A, Motz GT, Coukos G. T-Cell and Nk-Cell Infiltration Into Solid Tumors: A Key Limiting Factor for Efficacious Cancer Immunotherapy. Cancer Discov (2014) 4(5):522–6. doi: 10.1158/2159-8290.CD-13-0985
159. Bobanga ID, Petrosiute A, Huang AY. Chemokines as Cancer Vaccine Adjuvants. Vaccines (Basel) (2013) 1(4):444–62. doi: 10.3390/vaccines1040444
160. Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine Expression in Melanoma Metastases Associated With CD8+ T-Cell Recruitment. Cancer Res (2009) 69(7):3077–85. doi: 10.1158/0008-5472.Can-08-2281
161. Mullins IM, Slingluff CL, Lee JK, Garbee CF, Shu J, Anderson SG, et al. Cxc Chemokine Receptor 3 Expression by Activated CD8+ T Cells Is Associated With Survival in Melanoma Patients With Stage Iii Disease. Cancer Res (2004) 64(21):7697–701. doi: 10.1158/0008-5472.Can-04-2059
162. Lança T, Costa MF, Gonçalves-Sousa N, Rei M, Grosso AR, Penido C, et al. Protective Role of the Inflammatory CCR2/CCL2 Chemokine Pathway Through Recruitment of Type 1 Cytotoxic Γδ T Lymphocytes to Tumor Beds. J Immunol (2013) 190(12):6673–80. doi: 10.4049/jimmunol.1300434
163. Zsiros E, Duttagupta P, Dangaj D, Li H, Frank R, Garrabrant T, et al. The Ovarian Cancer Chemokine Landscape Is Conducive to Homing of Vaccine-Primed and CD3/CD28-Costimulated T Cells Prepared for Adoptive Therapy. Clin Cancer Res (2015) 21(12):2840–50. doi: 10.1158/1078-0432.Ccr-14-2777
164. Korbecki J, Kojder K, Simińska D, Bohatyrewicz R, Gutowska I, Chlubek D, et al. Cc Chemokines in a Tumor: A Review of Pro-Cancer and Anti-Cancer Properties of the Ligands of Receptors CCR1, CCR2, CCR3 and CCR4. Int J Mol Sci (2020) 21(21):8412. doi: 10.3390/ijms21218412
165. Sackstein R, Schatton T, Barthel SR. T-Lymphocyte Homing: An Underappreciated Yet Critical Hurdle for Successful Cancer Immunotherapy. Lab Invest (2017) 97(6):669–97. doi: 10.1038/labinvest.2017.25
166. Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced Tumor Trafficking of Gd2 Chimeric Antigen Receptor T Cells by Expression of the Chemokine Receptor CCR2b. J Immunother (2010) 33(8):780–8. doi: 10.1097/CJI.0b013e3181ee6675
167. Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, et al. Expression of a Functional CCR2 Receptor Enhances Tumor Localization and Tumor Eradication by Retargeted Human T Cells Expressing a Mesothelin-Specific Chimeric Antibody Receptor. Clin Cancer Res (2011) 17(14):4719–30. doi: 10.1158/1078-0432.Ccr-11-0351
168. Asai H, Fujiwara H, An J, Ochi T, Miyazaki Y, Nagai K, et al. Co-Introduced Functional CCR2 Potentiates in Vivo Anti-Lung Cancer Functionality Mediated by T Cells Double Gene-Modified to Express WT1-Specific T-Cell Receptor. PLoS One (2013) 8(2):e56820. doi: 10.1371/journal.pone.0056820
169. Garetto S, Sardi C, Martini E, Roselli G, Morone D, Angioni R, et al. Tailored Chemokine Receptor Modification Improves Homing of Adoptive Therapy T Cells in a Spontaneous Tumor Model. Oncotarget (2016) 7(28):43010–26. doi: 10.18632/oncotarget.9280
170. Hertzer KM, Donald GW, Hines OJ. CXCR2: A Target for Pancreatic Cancer Treatment? Expert Opin Ther Targets (2013) 17(6):667–80. doi: 10.1517/14728222.2013.772137
171. Liu G, Rui W, Zheng H, Huang D, Yu F, Zhang Y, et al. CXCR2-Modified CAR-T Cells Have Enhanced Trafficking Ability That Improves Treatment of Hepatocellular CARcinoma. Eur J Immunol (2020) 50(5):712–24. doi: 10.1002/eji.201948457
172. Whilding LM, Halim L, Draper B, Parente-Pereira AC, Zabinski T, Davies DM, et al. CAR T-Cells Targeting the Integrin Avβ6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy Against Several Solid Malignancies. Cancers (2019) 11(5):674. doi: 10.3390/cancers11050674
173. Peng W, Ye Y, Rabinovich BA, Liu C, Lou Y, Zhang M, et al. Transduction of Tumor-Specific T Cells With CXCR2 Chemokine Receptor Improves Migration to Tumor and Antitumor Immune Responses. Clin Cancer Res (2010) 16(22):5458–68. doi: 10.1158/1078-0432.Ccr-10-0712
174. Idorn M, Skadborg SK, Kellermann L, Halldórsdóttir HR, Holmen Olofsson G, Met Ö, et al. Chemokine Receptor Engineering of T Cells With CXCR2 Improves Homing Towards Subcutaneous Human Melanomas in Xenograft Mouse Model. Oncoimmunology (2018) 7(8):e1450715. doi: 10.1080/2162402x.2018.1450715
175. Kremer V, Ligtenberg MA, Zendehdel R, Seitz C, Duivenvoorden A, Wennerberg E, et al. Genetic Engineering of Human Nk Cells to Express CXCR2 Improves Migration to Renal Cell CARcinoma. J Immunother Cancer (2017) 5(1):73. doi: 10.1186/s40425-017-0275-9
176. Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, et al. CXCR1- or CXCR2-Modified CAR T Cells Co-Opt Il-8 for Maximal Antitumor Efficacy in Solid Tumors. Nat Commun (2019) 10(1):4016. doi: 10.1038/s41467-019-11869-4
177. Hu J, Sun C, Bernatchez C, Xia X, Hwu P, Dotti G, et al. T-Cell Homing Therapy for Reducing Regulatory T Cells and Preserving Effector T-Cell Function in Large Solid Tumors. Clin Cancer Res (2018) 24(12):2920–34. doi: 10.1158/1078-0432.Ccr-17-1365
178. Ng YY, Tay JCK, Wang S. CXCR1 Expression to Improve Anti-Cancer Efficacy of Intravenously Injected CAR-Nk Cells in Mice With Peritoneal Xenografts. Mol Ther Oncolytics (2019) 16:75–85. doi: 10.1016/j.omto.2019.12.006
179. Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, et al. T Lymphocytes Coexpressing CCR4 and a Chimeric Antigen Receptor Targeting CD30 Have Improved Homing and Antitumor Activity in a Hodgkin Tumor Model. Blood (2009) 113(25):6392–402. doi: 10.1182/blood-2009-03-209650
180. Khan AB, Carpenter B, Santos ESP, Pospori C, Khorshed R, Griffin J, et al. Redirection to the Bone Marrow Improves T Cell Persistence and Antitumor Functions. J Clin Invest (2018) 128(5):2010–24. doi: 10.1172/jci97454
181. Jamali A, Hadjati J, Madjd Z, Mirzaei HR, Thalheimer FB, Agarwal S, et al. Highly Efficient Generation of Transgenically Augmented CAR Nk Cells Overexpressing CXCR4. Front Immunol (2020) 11:2028. doi: 10.3389/fimmu.2020.02028
182. Müller N, Michen S, Tietze S, Töpfer K, Schulte A, Lamszus K, et al. Engineering Nk Cells Modified With an Egfrviii-Specific Chimeric Antigen Receptor to Overexpress CXCR4 Improves Immunotherapy of Cxcl12/Sdf-1α-Secreting Glioblastoma. J Immunother (2015) 38(5):197–210. doi: 10.1097/cji.0000000000000082
183. Pan YC, Nishikawa T, Chang CY, Tai JA, Kaneda Y. CXCL2 Combined With Hvj-E Suppresses Tumor Growth and Lung Metastasis in Breast Cancer and Enhances Anti-PD-1 Antibody Therapy. Mol Ther Oncolytics (2021) 20:175–86. doi: 10.1016/j.omto.2020.12.011
184. Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, et al. Armed Oncolytic Virus Enhances Immune Functions of Chimeric Antigen Receptor-Modified T Cells in Solid Tumors. Cancer Res (2014) 74(18):5195–205. doi: 10.1158/0008-5472.Can-14-0697
185. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. Il-7 and Ccl19 Expression in CAR-T Cells Improves Immune Cell Infiltration and CAR-T Cell Survival in the Tumor. Nat Biotechnol (2018) 36(4):346–51. doi: 10.1038/nbt.4086
186. Luo H, Su J, Sun R, Sun Y, Wang Y, Dong Y, et al. Coexpression of Il7 and CCL21 Increases Efficacy of CAR-T Cells in Solid Tumors Without Requiring Preconditioned Lymphodepletion. Clin Cancer Res (2020) 26(20):5494–505. doi: 10.1158/1078-0432.Ccr-20-0777
187. Anderson NM, Simon MC. The Tumor Microenvironment. Curr Biol (2020) 30(16):R921–r5. doi: 10.1016/j.cub.2020.06.081
188. Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, et al. Role of Tumor Microenvironment in Tumorigenesis. J Cancer (2017) 8(5):761–73. doi: 10.7150/jca.17648
189. Gunaydin G. Cafs Interacting With Tams in Tumor Microenvironment to Enhance Tumorigenesis and Immune Evasion. Front Oncol (2021) 11:668349. doi: 10.3389/fonc.2021.668349
190. Cha JH, Chan LC, Li CW, Hsu JL, Hung MC. Mechanisms Controlling PD-L1 Expression in Cancer. Mol Cell (2019) 76(3):359–70. doi: 10.1016/j.molcel.2019.09.030
191. Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, et al. Activated Monocytes in Peritumoral Stroma of Hepatocellular CARcinoma Foster Immune Privilege and Disease Progression Through PD-L1. J Exp Med (2009) 206(6):1327–37. doi: 10.1084/jem.20082173
192. Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas Promote Immunosuppression Through Induction of B7-H1 Expression in Tumor-Associated Macrophages. Clin Cancer Res (2013) 19(12):3165–75. doi: 10.1158/1078-0432.Ccr-12-3314
193. Thommen DS, Schumacher TN. T Cell Dysfunction in Cancer. Cancer Cell (2018) 33(4):547–62. doi: 10.1016/j.ccell.2018.03.012
194. Kenderian SS, Ruella M, Shestova O, Klichinsky M, Gill SI. Identification of PD1 and Tim3 as Checkpoints That Limit Chimeric Antigen Receptor T Cell Efficacy in Leukemia. Blood (2015) 126(23):852. doi: 10.1182/blood.V126.23.852.852
195. Abate-Daga D, Hanada K, Davis JL, Yang JC, Rosenberg SA, Morgan RA. Expression Profiling of TCR-Engineered T Cells Demonstrates Overexpression of Multiple Inhibitory Receptors in Persisting Lymphocytes. Blood (2013) 122(8):1399–410. doi: 10.1182/blood-2013-04-495531
196. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T Cells With Cell-Intrinsic PD-1 Checkpoint Blockade Resist Tumor-Mediated Inhibition. J Clin Invest (2016) 126(8):3130–44. doi: 10.1172/jci83092
197. Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual Blockade of PD-1 and Ctla-4 Combined With Tumor Vaccine Effectively Restores T-Cell Rejection Function in Tumors. Cancer Res (2013) 73(12):3591–603. doi: 10.1158/0008-5472.Can-12-4100
198. Kallies A, Zehn D, Utzschneider DT. Precursor Exhausted T Cells: Key to Successful Immunotherapy? Nat Rev Immunol (2020) 20(2):128–36. doi: 10.1038/s41577-019-0223-7
199. Balança CC, Salvioni A, Scarlata CM, Michelas M, Martinez-Gomez C, Gomez-Roca C, et al. PD-1 Blockade Restores Helper Activity of Tumor-Infiltrating, Exhausted PD-1hiCD39+ CD4 T Cells. JCI Insight (2021) 6(2):e142513. doi: 10.1172/jci.insight.142513
200. Pilon-Thomas S, Mackay A, Vohra N, Mulé JJ. Blockade of Programmed Death Ligand 1 Enhances the Therapeutic Efficacy of Combination Immunotherapy Against Melanoma. J Immunol (2010) 184(7):3442–9. doi: 10.4049/jimmunol.0904114
201. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and Activity of Anti-PD-L1 Antibody in Patients With Advanced Cancer. N Engl J Med (2012) 366(26):2455–65. doi: 10.1056/NEJMoa1200694
202. Rowshanravan B, Halliday N, Sansom DM. CTLA-4: A Moving Target in Immunotherapy. Blood (2018) 131(1):58–67. doi: 10.1182/blood-2017-06-741033
203. Tang F, Du X, Liu M, Zheng P, Liu Y. Anti-CTLA-4 Antibodies in Cancer Immunotherapy: Selective Depletion of Intratumoral Regulatory T Cells or Checkpoint Blockade? Cell Biosci (2018) 8:30. doi: 10.1186/s13578-018-0229-z
204. Sharma P, Allison JP. The Future of Immune Checkpoint Therapy. Science (New York NY) (2015) 348(6230):56–61. doi: 10.1126/science.aaa8172
205. Hodi FS, O’Day SJ, MCDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved Survival With Ipilimumab in Patients With Metastatic Melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
206. Carretero-González A, Lora D, Ghanem I, Zugazagoitia J, Castellano D, Sepúlveda JM, et al. Analysis of Response Rate With Anti PD1/PD-L1 Monoclonal Antibodies in Advanced Solid Tumors: A Meta-Analysis of Randomized Clinical Trials. Oncotarget (2018) 9(9):8706–15. doi: 10.18632/oncotarget.24283
207. Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell (2016) 167(6):1540–54. doi: 10.1016/j.cell.2016.11.022
208. Boussiotis VA. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N Engl J Med (2016) 375(18):1767–78. doi: 10.1056/NEJMra1514296
209. Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, et al. Immune-Related Adverse Events With Immune Checkpoint Blockade: A Comprehensive Review. Eur J Cancer (2016) 54:139–48. doi: 10.1016/j.ejca.2015.11.016
210. Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, et al. CRISPR/Cas9-Mediated PD-1 Disruption Enhances Anti-Tumor Efficacy of Human Chimeric Antigen Receptor T Cells. Sci Rep (2017) 7(1):737. doi: 10.1038/s41598-017-00462-8
211. Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res (2016) 76(6):1578–90. doi: 10.1158/0008-5472.Can-15-2524
212. Chen C, Gu YM, Zhang F, Zhang ZC, Zhang YT, He YD, et al. Construction of PD1/CD28 Chimeric-Switch Receptor Enhances Anti-Tumor Ability of C-Met CAR-T in Gastric Cancer. Oncoimmunology (2021) 10(1):1901434. doi: 10.1080/2162402x.2021.1901434
213. Ankri C, Shamalov K, Horovitz-Fried M, Mauer S, Cohen CJ. Human T Cells Engineered to Express a Programmed Death 1/28 Costimulatory Retargeting Molecule Display Enhanced Antitumor Activity. J Immunol (2013) 191(8):4121–9. doi: 10.4049/jimmunol.1203085
214. Schlenker R, Olguín-Contreras LF, Leisegang M, Schnappinger J, Disovic A, Rühland S, et al. Chimeric PD-1:28 Receptor Upgrades Low-Avidity T Cells and Restores Effector Function of Tumor-Infiltrating Lymphocytes for Adoptive Cell Therapy. Cancer Res (2017) 77(13):3577–90. doi: 10.1158/0008-5472.Can-16-1922
215. Liang Y, Liu H, Lu Z, Lei W, Zhang C, Li P, et al. CD19 CAR-T Expressing PD-1/CD28 Chimeric Switch Receptor as a Salvage Therapy for Dlbcl Patients Treated With Different CD19-Directed CAR T-Cell Therapies. J Hematol Oncol (2021) 14(1):26. doi: 10.1186/s13045-021-01044-y
216. Liu H, Lei W, Zhang C, Yang C, Wei J, Guo Q, et al. CD19-Specific CAR T Cells That Express a PD-1/CD28 Chimeric Switch-Receptor Are Effective in Patients With PD-L1-Positive B-Cell Lymphoma. Clin Cancer Res (2021) 27(2):473–84. doi: 10.1158/1078-0432.Ccr-20-1457
217. Hoogi S, Eisenberg V, Mayer S, Shamul A, Barliya T, Cohen CJ. A Tigit-Based Chimeric Co-Stimulatory Switch Receptor Improves T-Cell Anti-Tumor Function. J Immunother Cancer (2019) 7(1):243. doi: 10.1186/s40425-019-0721-y
218. Shin JH, Park HB, Oh YM, Lim DP, Lee JE, Seo HH, et al. Positive Conversion of Negative Signaling of CTLA4 Potentiates Antitumor Efficacy of Adoptive T-Cell Therapy in Murine Tumor Models. Blood (2012) 119(24):5678–87. doi: 10.1182/blood-2011-09-380519
219. Lin S, Cheng L, Ye W, Li S, Zheng D, Qin L, et al. Chimeric CTLA4-CD28-CD3z T Cells Potentiate Antitumor Activity Against CD80/CD86-Positive B Cell Malignancies. Front Immunol (2021) 12:642528. doi: 10.3389/fimmu.2021.642528
220. Angioni R, Sánchez-Rodríguez R, Viola A, Molon B. TGF-β in Cancer: Metabolic Driver of the Tolerogenic Crosstalk in the Tumor Microenvironment. Cancers (2021) 13(3):401. doi: 10.3390/cancers13030401
221. Turley SJ, Cremasco V, Astarita JL. Immunological Hallmarks of Stromal Cells in the Tumour Microenvironment. Nat Rev Immunol (2015) 15(11):669–82. doi: 10.1038/nri3902
222. Bhowmick NA, Neilson EG, Moses HL. Stromal Fibroblasts in Cancer Initiation and Progression. Nature (2004) 432(7015):332–7. doi: 10.1038/nature03096
223. Thomas DA, Massagué J. TGF-Beta Directly Targets Cytotoxic T Cell Functions During Tumor Evasion of Immune Surveillance. Cancer Cell (2005) 8(5):369–80. doi: 10.1016/j.ccr.2005.10.012
224. Mueller MM, Fusenig NE. Friends or Foes - Bipolar Effects of the Tumour Stroma in Cancer. Nat Rev Cancer (2004) 4(11):839–49. doi: 10.1038/nrc1477
225. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature (2018) 554(7693):544–8. doi: 10.1038/nature25501
226. Ravi R, Noonan KA, Pham V, Bedi R, Zhavoronkov A, Ozerov IV, et al. Bifunctional Immune Checkpoint-Targeted Antibody-Ligand Traps That Simultaneously Disable TGFβ Enhance the Efficacy of Cancer Immunotherapy. Nat Commun (2018) 9(1):741. doi: 10.1038/s41467-017-02696-6
227. Bendle GM, Linnemann C, Bies L, Song JY, Schumacher TN. Blockade of TGF-β Signaling Greatly Enhances the Efficacy of TCR Gene Therapy of Cancer. J Immunol (2013) 191(6):3232–9. doi: 10.4049/jimmunol.1301270
228. Bollard CM, Tripic T, Cruz CR, Dotti G, Gottschalk S, Torrano V, et al. Tumor-Specific T-Cells Engineered to Overcome Tumor Immune Evasion Induce Clinical Responses in Patients With Relapsed Hodgkin Lymphoma. J Clin Oncol (2018) 36(11):1128–39. doi: 10.1200/jco.2017.74.3179
229. Sukumaran S, Watanabe N, Bajgain P, Raja K, Mohammed S, Fisher WE, et al. Enhancing the Potency and Specificity of Engineered T Cells for Cancer Treatment. Cancer Discov (2018) 8(8):972–87. doi: 10.1158/2159-8290.CD-17-1298
230. Roth TL, Li PJ, Blaeschke F, Nies JF, Apathy R, Mowery C, et al. Pooled Knockin Targeting for Genome Engineering of Cellular Immunotherapies. Cell (2020) 181(3):728–44. doi: 10.1016/j.cell.2020.03.039
231. Oda SK, Anderson KG, Ravikumar P, Bonson P, Garcia NM, Jenkins CM, et al. A Fas-4-1bb Fusion Protein Converts a Death to a Pro-Survival Signal and Enhances T Cell Therapy. J Exp Med (2020) 217(12):e20191166. doi: 10.1084/jem.20191166
232. Kageyama S, Ikeda H, Miyahara Y, Imai N, Ishihara M, Saito K, et al. Adoptive Transfer of Mage-A4 T-Cell Receptor Gene-Transduced Lymphocytes in Patients With Recurrent Esophageal Cancer. Clin Cancer Res (2015) 21(10):2268–77. doi: 10.1158/1078-0432.Ccr-14-1559
233. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer Regression in Patients After Transfer of Genetically Engineered Lymphocytes. Science (New York NY) (2006) 314(5796):126–9. doi: 10.1126/science.1129003
234. Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene Therapy With Human and Mouse T-Cell Receptors Mediates Cancer Regression and Targets Normal Tissues Expressing Cognate Antigen. Blood (2009) 114(3):535–46. doi: 10.1182/blood-2009-03-211714
235. Xue SA, Gao L, Thomas S, Hart DP, Xue JZ, Gillmore R, et al. Development of a Wilms’ Tumor Antigen-Specific T-Cell Receptor for Clinical Trials: Engineered Patient’s T Cells Can Eliminate Autologous Leukemia Blasts in Nod/Scid Mice. Haematologica (2010) 95(1):126–34. doi: 10.3324/haematol.2009.006486
236. Tawara I, Kageyama S, Miyahara Y, Fujiwara H, Nishida T, Akatsuka Y, et al. Safety and Persistence of WT1-Specific T-Cell Receptor Gene-Transduced Lymphocytes in Patients With Aml and Mds. Blood (2017) 130(18):1985–94. doi: 10.1182/blood-2017-06-791202
237. Garg H, Suri P, Gupta JC, Talwar G, Dubey S. Survivin: A Unique Target for Tumor Therapy. Cancer Cell Int (2016) 16(1):1–14. doi: 10.1186/s12935-016-0326-1
238. Echchannaoui H, Petschenka J, Ferreira EA, Hauptrock B, Lotz-Jenne C, Voss RH, et al. A Potent Tumor-Reactive P53-Specific Single-Chain TCR Without on- or Off-Target Autoimmunity In Vivo. Mol Ther (2019) 27(1):261–71. doi: 10.1016/j.ymthe.2018.11.006
239. Kantoff PW, Schuetz TJ, Blumenstein BA, Glode LM, Bilhartz DL, Wyand M, et al. Overall Survival Analysis of a Phase Ii Randomized Controlled Trial of a Poxviral-Based Psa-Targeted Immunotherapy in Metastatic Castration-Resistant Prostate Cancer. J Clin Oncol (2010) 28(7):1099–105. doi: 10.1200/jco.2009.25.0597
240. Ali M, Giannakopoulou E, Li Y, Lehander M, Virding Culleton S, Yang W, et al. T Cells Targeted to Tdt Kill Leukemic Lymphoblasts While Sparing Normal Lymphocytes. Nat Biotechnol (2021) 1–11. doi: 10.1038/s41587-021-01089-x
241. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, et al. T Cells Targeting CARcinoembryonic Antigen Can Mediate Regression of Metastatic Colorectal Cancer But Induce Severe Transient Colitis. Mol Ther (2011) 19(3):620–6. doi: 10.1038/mt.2010.272
242. Palmer DC, Chan CC, Gattinoni L, Wrzesinski C, Paulos CM, Hinrichs CS, et al. Effective Tumor Treatment Targeting a Melanoma/Melanocyte-Associated Antigen Triggers Severe Ocular Autoimmunity. Proc Natl Acad Sci USA (2008) 105(23):8061–6. doi: 10.1073/pnas.0710929105
243. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer Regression and Neurological Toxicity Following Anti-Mage-A3 TCR Gene Therapy. J Immunother (2013) 36(2):133–51. doi: 10.1097/CJI.0b013e3182829903
244. Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular Toxicity and Titin Cross-Reactivity of Affinity-Enhanced T Cells in Myeloma and Melanoma. Blood (2013) 122(6):863–71. doi: 10.1182/blood-2013-03-490565
245. Faridi R, Zahra A, Khan K, Idrees M. Oncogenic Potential of Human Papillomavirus (Hpv) and Its Relation With Cervical Cancer. Virol J (2011) 8:269. doi: 10.1186/1743-422x-8-269
246. Raab-Traub N. Novel Mechanisms of Ebv-Induced Oncogenesis. Curr Opin Virol (2012) 2(4):453–8. doi: 10.1016/j.coviro.2012.07.001
247. Lupberger J, Hildt E. Hepatitis B Virus-Induced Oncogenesis. World J Gastroenterol (2007) 13(1):74–81. doi: 10.3748/wjg.v13.i1.74
248. Wang XG, Revskaya E, Bryan RA, Strickler HD, Burk RD, Casadevall A, et al. Treating Cancer as an Infectious Disease–Viral Antigens as Novel Targets for Treatment and Potential Prevention of Tumors of Viral Etiology. PLoS One (2007) 2(10):e1114. doi: 10.1371/journal.pone.0001114
249. Tashiro H, Brenner MK. Immunotherapy Against Cancer-Related Viruses. Cell Res (2017) 27(1):59–73. doi: 10.1038/cr.2016.153
250. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic Instability–an Evolving Hallmark of Cancer. Nat Rev Mol Cell Biol (2010) 11(3):220–8. doi: 10.1038/nrm2858
251. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic Correlates of Response to CTLA-4 Blockade in Metastatic Melanoma. Science (New York NY) (2015) 350(6257):207–11. doi: 10.1126/science.aad0095
252. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer Immunology. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non-Small Cell Lung Cancer. Science (New York NY) (2015) 348(6230):124–8. doi: 10.1126/science.aaa1348
253. Spranger S, Bao R, Gajewski TF. Melanoma-Intrinsic B-Catenin Signalling Prevents Anti-Tumour Immunity. Nature (2015) 523(7559):231–5. doi: 10.1038/nature14404
254. Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor Mutational Load Predicts Survival After Immunotherapy Across Multiple Cancer Types. Nat Genet (2019) 51(2):202–6. doi: 10.1038/s41588-018-0312-8
255. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint Blockade Cancer Immunotherapy Targets Tumour-Specific Mutant Antigens. Nature (2014) 515(7528):577–81. doi: 10.1038/nature13988
256. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Löwer M, et al. Personalized RNA Mutanome Vaccines Mobilize Poly-Specific Therapeutic Immunity Against Cancer. Nature (2017) 547(7662):222–6. doi: 10.1038/nature23003
257. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An Immunogenic Personal Neoantigen Vaccine for Patients With Melanoma. Nature (2017) 547(7662):217–21. doi: 10.1038/nature22991
258. Ott PA, Hu-Lieskovan S, Chmielowski B, Govindan R, Naing A, Bhardwaj N, et al. A Phase Ib Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients With Advanced Melanoma, Non-Small Cell Lung Cancer, or Bladder Cancer. Cell (2020) 183(2):347–62. doi: 10.1016/j.cell.2020.08.053
259. van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor Exome Analysis Reveals Neoantigen-Specific T-Cell Reactivity in an Ipilimumab-Responsive Melanoma. J Clin Oncol (2013) 31(32):e439–42. doi: 10.1200/jco.2012.47.7521
260. Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining Exomic Sequencing Data to Identify Mutated Antigens Recognized by Adoptively Transferred Tumor-Reactive T Cells. Nat Med (2013) 19(6):747–52. doi: 10.1038/nm.3161
261. Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, et al. Isolation of Neoantigen-Specific T Cells From Tumor and Peripheral Lymphocytes. J Clin Invest (2015) 125(10):3981–91. doi: 10.1172/jci82416
262. Lu YC, Yao X, Li YF, El-Gamil M, Dudley ME, Yang JC, et al. Mutated Ppp1r3b Is Recognized by T Cells Used to Treat a Melanoma Patient Who Experienced a Durable Complete Tumor Regression. J Immunol (2013) 190(12):6034–42. doi: 10.4049/jimmunol.1202830
263. Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of Somatic Mutations in Human Gastrointestinal Cancers. Science (New York NY) (2015) 350(6266):1387–90. doi: 10.1126/science.aad1253
264. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer Immunotherapy Based on Mutation-Specific CD4+ T Cells in a Patient With Epithelial Cancer. Science (New York NY) (2014) 344(6184):641–5. doi: 10.1126/science.1251102
265. Veatch JR, Jesernig BL, Kargl J, Fitzgibbon M, Lee SM, Baik C, et al. Endogenous CD4(+) T Cells Recognize Neoantigens in Lung Cancer Patients, Including Recurrent Oncogenic Kras and Erbb2 (Her2) Driver Mutations. Cancer Immunol Res (2019) 7(6):910–22. doi: 10.1158/2326-6066.Cir-18-0402
266. Blass E, Ott PA. Advances in the Development of Personalized Neoantigen-Based Therapeutic Cancer Vaccines. Nat Rev Clin Oncol (2021) 18(4):215–29. doi: 10.1038/s41571-020-00460-2
267. Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. Cancer Immunotherapy. A Dendritic Cell Vaccine Increases the Breadth and Diversity of Melanoma Neoantigen-Specific T Cells. Science (New York NY) (2015) 348(6236):803–8. doi: 10.1126/science.aaa3828
268. Strønen E, Toebes M, Kelderman S, van Buuren MM, Yang W, van Rooij N, et al. Targeting of Cancer Neoantigens With Donor-Derived T Cell Receptor Repertoires. Science (New York NY) (2016) 352(6291):1337–41. doi: 10.1126/science.aaf2288
269. Ali M, Foldvari Z, Giannakopoulou E, Böschen ML, Strønen E, Yang W, et al. Induction of Neoantigen-Reactive T Cells From Healthy Donors. Nat Protoc (2019) 14(6):1926–43. doi: 10.1038/s41596-019-0170-6
Keywords: T cell receptor, genetic engineering, cancer immunotherapy, TCR-engineering, new strategies
Citation: Wei F, Cheng X-X, Xue JZ and Xue S-A (2022) Emerging Strategies in TCR-Engineered T Cells. Front. Immunol. 13:850358. doi: 10.3389/fimmu.2022.850358
Received: 07 January 2022; Accepted: 03 March 2022;
Published: 30 March 2022.
Edited by:
Ignazio Caruana, University Children’s Hospital Würzburg, GermanyReviewed by:
Jochen Buechner, Oslo University Hospital, NorwayDonovan Flumens, Laboratory of Experimental Hematology, University of Antwerp, Belgium
Sara Mastaglio, San Raffaele Hospital (IRCCS), Italy
Copyright © 2022 Wei, Cheng, Xue and Xue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shao-An Xue, c2hhby1hbi54dWVAdWNsLmFjLnVr