 Peter M. Kloetzel
Peter M. Kloetzel
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 21 February 2022
Sec. Antigen Presenting Cell Biology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.849863
This article is part of the Research TopicInsights in Antigen Presenting Cell Biology: 2021View all 9 articles
Proteasome generates spliced peptides by ligating two distant cleavage products in a reverse proteolysis reaction. The observation that CD8+ T cells recognizing a spliced peptide induced T cell rejection in a melanoma patient following adoptive T cell transfer (ATT), raised some hopes with regard to the general therapeutic and immune relevance of spliced peptides. Concomitantly, the identification of spliced peptides was also the start of a controversy with respect to their frequency, abundancy and their therapeutic applicability. Here I review some of the recent evidence favoring or disfavoring an immune relevance of splicetopes and discuss from a theoretical point of view the potential usefulness of tumor specific splicetopes and why against all odds it still may seem worth trying to identify such tumor and patient-specific neosplicetopes for application in ATT.
The majority of defined antitumor T cell responses involves the proteasomal processing of intracellular proteins and their presentation in the context of MHC class I molecules to peptide specific CD8+ T cells. Such antigenic peptides generated by the proteasome are 8-10 amino acids in length and mirror the linear sequence of the parental protein. Work over the past three decades has proven that the vast majority of these peptides are generated by the 26S proteasome, i.e. its 20S catalytic core as part of the ubiquitin proteasome system (UPS) (1–3).
One reason why the proteasome seems to be so ideally suited for the production of antigenic peptides is that its three active site β-subunits (β1s, β2s, β5s) (s-subunits) of the standard proteasome exhibit different cleavage specificities. These can be further modulated by replacing these s-subunits by alternative β1i, β2i and β5i immunosubunits (i-subunits) forming either immunoproteasomes (i-proteasomes) or by pairing with the standard β-subunits to form intermediate type 20S proteasomes. This allows proteasomes to cleave C-terminally of almost any of the 20 amino acids thereby meeting the diverse demands of the more than 10000 HLA class I allele variants for peptide binding (4–7). The combination of different active site β-subunits not only affects the cleavage site usage but also the cleavage strength of the 20S proteasome within a natural protein substrate (8, 9). In consequence, this provides the immune system with antigenic peptides of different linear sequences with different C-terminal anchor residues and also altered relative peptide abundancies that together affect the cellular immune response.
It was undisputed that antigenic peptides of 8-12 amino acid residues in length generated by the 20S proteasome during the degradation of viral, bacterial or human proteins are peptide fragments with a linear sequence identical to that found in the unprocessed parental protein. However, two pioneering reports (10, 11) demonstrated the existence of HLA-1 bound CD8+ T cells reactive peptides which possessed an amino acid sequence that differed from that of the substrate protein and that were the result of a peptide splicing reaction. The spliced epitopes (splicetopes) were identified with the help of tumor patient derived T-cells and shown to be produced by the proteasome via a transpeptidation reaction. This involves the formation of an O-acyl-enzyme intermediate between a N-terminal peptide fragment and the Thr1 residue of one of the β-subunit active sites (Figure 1A). Thus, proteasome catalyzed peptide splicing (PCPS) represented a genuine novel catalytic function of the proteasome (11–15). Peptides can be spliced by PCPS in a cis or inverse order and theoretically even in trans, meaning that peptides derived from two different proteins are ligated and that the substrate proteins have to be present in the catalytic cavity of the 20S proteasome for degradation at the same time (Figure 1B). Importantly, the potential of splicetopes in cancer therapy was suggested by the fact that adoptive transfer of splicetope-specific CD8+ T cells into the autologous melanoma patient was shown to be followed by tumor regression (16). Moreover, CD8+ T cells directed against a spliced peptide expressed by human acute myeloid leukemia cells were shown to inhibit the engraftment of these leukemia cells in nonobese diabetic/severe combined immune deficient (SCID) mice (12). This data highlighted a potential immune relevance of such tumor antigen-derived splicetopes leading to the idea that establishing prediction algorithm aided pipelines for the CD8+ T cell independent identification of new splicetope may be a means to identify new targets for tumor therapy (17).
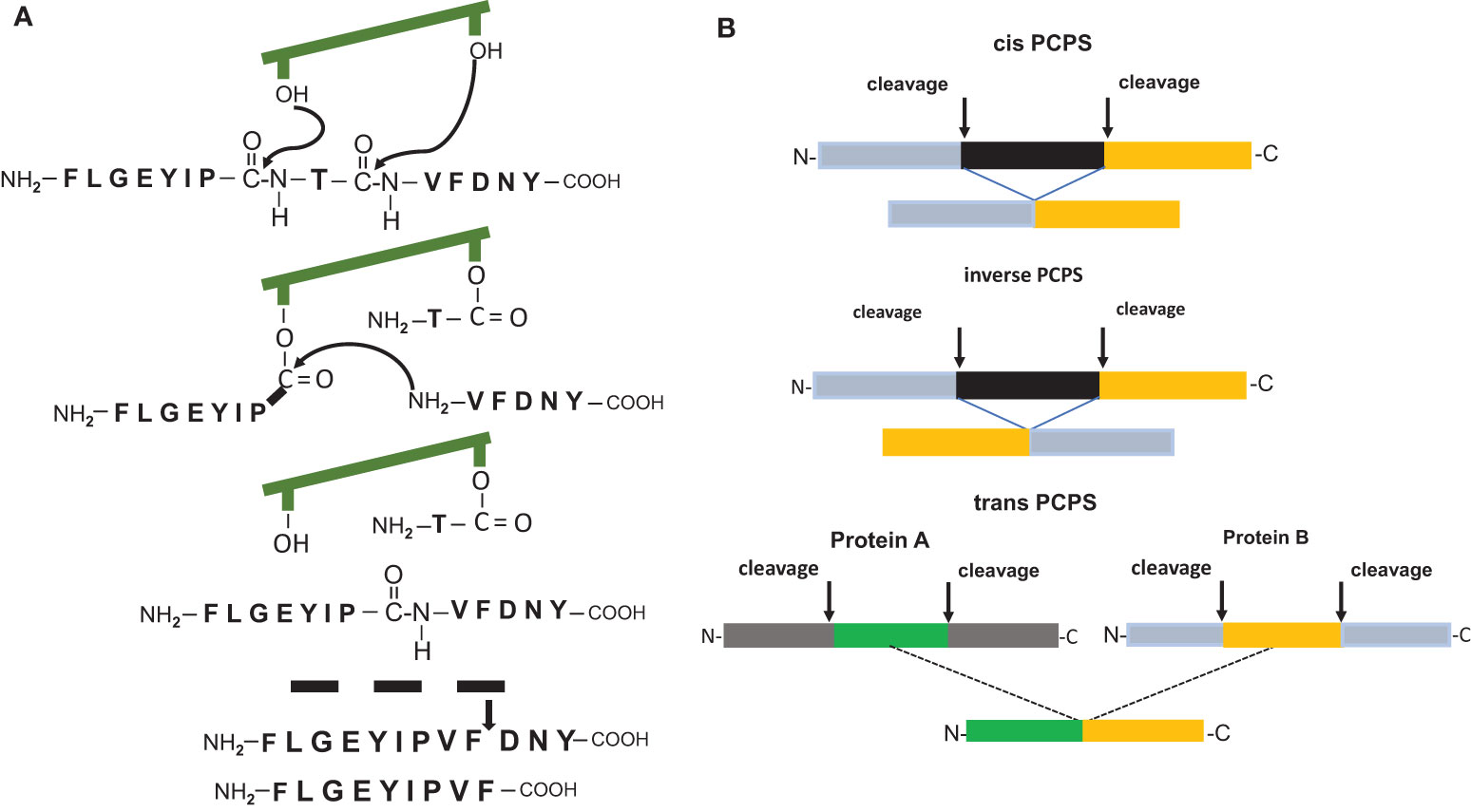
Figure 1 (A) Model of the peptide splicing reaction for the putative RAC2 P29L neosplicetope, adapted from Vigneron et al. (11). The spliced peptide requires an additional proteasomal cleavage for 9mer generation. (B) Proteasome catalyzed peptide splicing in cis, inverse and in trans.
One issue which accompanied the identification T cell reactive splicetopes from the very beginning and which led to controversial discussions was the question on how frequent such splicetope producing splicing events indeed are, how abundant splicetopes are and whether spliced epitopes rather present a rare curiosity than being of genuine importance for the immune system. Initial calculations based on in vitro experiments estimated that epitope splicing efficacy ranged only between 0.0002% and 0.01% of the total proteasome-dependent epitope generation (11, 14, 18, 19) and that epitope production by PCPS was an extremely rare event. Indeed, one has to state that the number of verified proteasome-generated splicetopes derived from human tumor associated antigens that are recognized by CD8+ T cell still doesn´t exceed six and thus is the same number as almost twenty years ago. Aiming at a largescale identification of HLA-1 bound spliced epitopes (20) a complex algorithm-aided approach was developed to analyze HLA-1 immunopeptidome mass spectrometry data derived from the analysis of GR-LCL, CIR cell lines and human primary fibroblast. This analysis came to the exciting conclusion that approximately 25-30% of the HLA-1 immunopeptidome is comprised of spliced peptides, suggesting that spliced epitopes are by far more abundant than was previously estimated. Similarly, the application of a novel integrated bioinformatics workflow to analyze MS data and to discriminate between linear and spliced peptides eluted from HLA precipitates revealed a substantial contribution of both cis and trans spliced peptides to the immunopeptidome (21). Quite in contrast, by de novo sequencing and reanalyzing previously published data (22, 23) it was concluded that the amount of cis spliced peptides in the HLA-1 immunopeptidome ranges at most between 2-6% or only slightly above and are by far less prevalent than proposed. But even this considerably lower number was refuted in two recent reports. From the data it was concluded that peptides generated by PCPS do not play a major role among the HLA-1 presented peptides and amount to rather less than 1% of the HLA-1 immunopeptidome (24) or may not be generated at all (25). These analyses of the HLA-1 immunopeptidome severely questioned the general immune relevance of splicetopes and certainly seed doubts on the applicability of neosplicetopes for targeted TCR generation and adoptive T cell therapy. However, since some spliced peptides appear to be involved in tumor regression after adoptive T cell transfer (12, 16) this does not completely preclude their role in an anti-tumor response.
Adoptive T-cell therapy (ATT) is today the most effective form of immunotherapy. It involves the use of tumor-infiltrating lymphocytes (TILs), gene-modified T cells expressing a specific T cell receptor (TCR) and chimeric antigen receptor gene-modified T cells (CARs) (17, 26–29). Both, proteins with restricted expression (e.g. cancer/testis antigens (30) that are poorly or not expressed at all in normal adult tissues) and mutant proteins can be recognized by TCRs. When specific TCRs are used to target cancer cells, the quality of the target epitope (quantity and affinity to HLA-1 molecules) and the quality of the T cells (avidity) determine whether or not a tumor is eradicated by T cells expressing tumor-reactive TCRs (31). Until recently, mostly non-mutated shared tumor-associated (self-) antigens (TAAs) have been used as targets in ATT trials. However, having undergone thymic selection many epitopes derived from these self-antigens possess low affinities to the patients’ HLA-molecules and the immune response is not sufficient for long-term eradication of tumors. In addition, many of these antigens are not driving the oncogenic events and their expression is therefore not essential for cancer cell outgrowth. Ideally, many of these problems could be avoided by targeting so called tumor neoantigens that harbor somatic driver mutations whose expression is crucial for the survival of cancer cells and which are not expressed in non-tumor cells. For example, long term responses in melanoma patients correlated with neoantigen specific T cells in the in vitro expanded and infused tumor infiltrating lymphocytes (31). Neoantigens thus represent critical targets for effective anti-tumor T-cell responses and for eradication of advanced cancers with respect to both, efficacy and safety (32–36).
Although targeting cancer-specific neoepitopes by TCR-mediated adoptive T cell transfer (ATT) represents a very promising approach for personalized cancer therapy (26, 33, 36) there are several limitations which may restrict their broad application of cancer immunotherapy. For example, often neoepitopes will not exhibit HLA class I binding affinities sufficient to trigger an efficient T cell response or will not be generated efficiently by the proteasome. In fact, many if not most of the mutations within neoantigens will not be part of or lead to the generation of high-affinity neoepitopes (36). For example, frequent recurrent mutations involving the same amino acid, such as the KRAS G12V mutation do not fit into the most frequent HLA-alleles of the general population, i.e. the HLA-A*02:01 allele which can be found in about 40% of Caucasians. Even if a suitable neoepitope is generated, its haplotype specificity frequently does not match with the patient´s HLA class I allele, consequently excluding these tumor patients from ATT. Another example is the adaptor protein MyD88. A missense mutation almost consistently changing leucine in position 265 to proline (L265P) is one of the most common driver mutations found in about one-fifth of all lymphoid malignancies (37–40). The resulting MyD88 L265P derived neo-epitope however exclusively binds to HLA-B*07:02. Therefore, only patients that carry the HLA-B*07:02 haplotype will potentially benefit from ATT by using a recently published MyD88 L265P specific TCR (41). So far, problems like these prevented the large-scale use of mutation-specific TCRs using T cells directed against mutational epitopes. Theoretically, given that peptide splicing leads to the generation of peptides containing distant fragments of proteins, PCPS harbors the potential to generate mutant spliced peptides with slightly altered sequences that bind efficiently to patient HLA-1 and might therefore represent more suitable TCR targets. This in particular in those cases where the recurrent somatic mutations in a tumor antigen do not lead to a non-spliced unique tumor neoepitope either exhibiting a sufficient HLA class I binding affinity or the appropriate HLA class I haplotype (36). Thus, spliced neoepitopes, i.e. neosplicetopes containing the tumor specific mutation, harbor the theoretical potential to extend the haplotype diversity or HLA-1 binding affinity of tumor specific epitopes for generation of tumor and patient specific TCRs. In theory, PCPS may thus represent an interesting novel approach. However, the major problem connected with it is to develop a method allowing the reliable identification of such spliced mutation harboring neosplicetopes.
In light of the above results and conclusions drawn from the immunopeptidome analyses, the search for splicetopes within the HLA-1 immunopeptidome for application in tumor therapy may seem like looking for a needle in the haystack. Nevertheless, the identification of tumor patient derived CD8+ T-cells recognizing splicetopes and their successful application in tumor therapy by ATT indicates that peptide splicing may not be such an accidental event and that proteasomal generation of tumor cells derived splicetopes represents an ordered, non-random event of sufficient repeatability and frequency. Furthermore, following the outlined theoretical arguments that identification of suitable tumor specific neosplicetopes for ATT may allow to make neoantigens visible for the immune systems which cannot be recognized otherwise alternative approaches allowing a more direct splicetope seemed worthy to consider. With this in mind the spliced peptide predictions algorithms ProteaJ and ProtAG (42, 43) were developed. In combination with mass spectrometric analyses of in vitro PCPS assays these permit the identification spliced peptides that are generated by the 20S proteasome from synthetic polypeptide substrates. It is well established that the in vitro generation of “conventional” non-spliced linear epitopes from polypeptides substrates harboring viral, bacterial or tumor antigen derived epitopes often reflect the in vivo situation of antigen processing with high fidelity both with regard to peptide sequence and amounts (44–51). Alike the fidelity of the generation of linear epitopes, in vitro PCPS experiments showed that published immune reactive spliced epitopes, derived from fibroblast growth factor FGF-5, the melanocyte growth protein gp100mel, tyrosinase and the SP110 nuclear protein were also efficiently generated by in vitro PCPS assays (12, 18, 19, 51–53). In fact, the in vitro PCPS assays suggested that splicetopes that are recognized by CD8+ T cells on the cell surface were produced with amounts reminiscent of what is obtained when virus or tumor derived non-spliced linear epitopes are generated by purified 20S proteasomes in vitro from synthetic peptide substrates. Furthermore, in vitro application of the prediction algorithm ProteaJ allowed the de novo identification of a gp100mel derived spliced epitope eliciting a peptide specific T cell response (53). Interestingly, judging by the number of different spliced peptides generated in an in vitro PCPS reaction the proteasomal splicing reaction as such appears to be a relatively frequent event during proteasomal peptide hydrolysis (15). The relative number of different spliced peptides generated in an in vitro PCPS reaction can however vary considerably between different in vitro substrates indicating that the monitored proteasomal peptide splicing frequency is sequence dependent (15). This conclusion is underlined by a recent study in which a large number of different substrates were studied in vitro in order to determine sequence motives that support proteasomal splicing reactions (54) as well as by the observation that proteasome subtypes with slightly different cleavage site preferences also differ in their peptide splicing efficiencies (51, 54). Underlining the feasibility of an algorithm aided “reverse immunology” approach, two high affinity splicetopes within the secreted bacterial phospholipase PlcB priming antigen-specific CD8+ T cells in L. monocytogenes-infected mice were successfully identified (55). Corroborating experimental data with tumor derived splicetopes both PlcB derived splicetopes were also generated by PCPS in vitro. These experiments were the first to show that PCPS expands the CD8+ T cell response against L. monocytogenes by exposing splicetopes on the cell surface.
Even though the analyses of immunopeptidomes failed to identify relevant numbers of spliced epitopes, the observed fidelity of the in vitro splicing reaction in generating known or predicted immune responsive splicetopes suggested that the application of in vitro PCPS assays in combination with prediction algorithms facilitating the identification of splicetopes or neosplicetopes by mass spectrometry may be worthy to test in search for new immune relevant neosplicetopes.
In a proof of principle study Willimsky et al. (43), tested whether an experimental approach solely based on epitope prediction by algorithms or in combination with in vitro PCPS can successfully be applied for the identification of novel neosplicetopes from mutKRASG12V and the mutant RAC2-P29L neoantigens for consecutive generation of splicetope specific TCRs. Both are a so-called driver mutation facilitating tumor growth as well as metastasis and thus present potential targets in ATT. The oncogenic KRASG12V mutant had been chosen because the recurrent G12V mutation does not result in the formation of a high affinity HLA-A*02:01 non-spliced neoepitope. mutRAC2 was selected because the P29L mutation is part of a high affinity HLA-A*02:01 neoepitope and the generation of a putative mutRAC2 derived neosplicetope could therefore be studied in comparison to proteasomal generation of the linear neoepitope. In silico analysis of the mutRAC2 neoantigen predicted a single RAC2-P29L derived putative neosplicetope with high affinity for binding. However, although this putative RAC2-P-29L derived neosplicetope was generated in in vitro PCPS assays, a high affinity TCR generated against this putative mutRAC2 neosplicetope in an established humanized mouse model (56) failed to indicate any immune relevance or to detect any cell surface presentation of this spliced peptide. For comparison the non-spliced mutRAC2 neoepitope was abundantly generated in vitro and efficiently recognized by the corresponding TCR at the cell surface. These combined biochemical/immunological explorative experiments suggest that the predicted mutRAC2 neosplicetope is either not generated in vivo or its amounts are too low to trigger a T-cell response even when the mutRAC2 neoantigen is overexpressed. In silico analysis of the mutKRASG12V neoantigen predicted three putative neosplicetopes with high HLA-A*02:01 binding affinities. However, TCRs generated against these peptides based on in silico predictions failed to demonstrate their immune relevance in T cells assays. Other than reported for a previously proposed in silico-in vitro pipeline for the identification of neosplicetopes (57) also the in vitro generation of the proposed mutKRASG12V derived neosplicetopes could not be validated (43). In addition, applying targeted mass spectrometry and using heavy isotope labeled peptides the identification of the proposed neosplicetopes was proven to be false (58, 59).
Why then did the identification of a mutRAC2 neoantigen derived neosplicetope fail despite its generation by in vitro PCPS? One obvious explanation is that in silico prediction and/or in vitro PCPS are not reliable predictors for the in vivo generation of a splicetope. On the other hand, taken all available experimental evidence (see above) there appears little reason to assume that the 20S proteasome splicing activity as such differs between the in vitro and in vivo situation. What most likely might differ is the efficiency at which a spliced peptide is generated either in vitro or in vivo. One also has to consider that in order to facilitate their mass spectrometric detection in vitro splicing reactions are often driven by increased substrate concentrations (15) potentially leading to a false impression of the actual splicetope generation efficiency. Due to the high substrate density within the catalytic cavity when synthetic polypeptide substrates are used one also cannot entirely exclude that under in vitro conditions splicing events are enforced which in vivo will not occur at all or less frequently than in vivo, when the amino acid sequence motif supporting the generation of the spliced epitope is present within the proteasomal catalytic cavity only once. Furthermore, the proteasomal transpeptidation reaction is most likely less frequent than the normal peptide hydrolysis reaction that results in the generation of non-spliced peptide fragments including linear HLA-1 epitopes. This latter point is nicely reflected by the analysis of mutRAC2P29L where in vitro generation of the linear spliced mutRCA2 derived neoepitope is at least 200fold more efficient than the generation of the putative mutRAC2 neosplicetope (43). From these studies we have to acknowledge that an in silico-in vitro pipeline thought to be a straight forward, easy to use approach to identify spliced peptides for therapeutic use cannot be the method of choice (57) and that in silico predictions of spliced epitopes alone or in combination with algorithms calculating HLA-1 binding affinity (60) and in vitro PCPS reactions seem by no means of sufficient reliability to set out for laborious and time consuming TCR generation.
Taken the available experimental data and in light of the mass spectrometric immunopeptidome analyses (22–25) one could argue that due to the limited cell surface expression of spliced epitopes a search for immune relevant neosplicetope is in vain. On the other hand, considering the therapeutic potential of adoptive T cell transfer for patient specific tumor treatment (35, 36), and the above outlined potential difficulties in finding patient specific appropriate neoepitopes for TCR generation, neosplicetopes generated by PCPS might still carry the potential to overcome some of the restrictions connected with ATT.
Reconsidering the identification of splicetope recognizing T cells derived from melanoma patients (16) one possible way to validate a potential neosplicetope is to isolate tumor patient derived neosplicetope reactive T cell clones in tumor infiltrating lymphocytes for neosplicetope reactivity. However, the number of patients possessing the correct HLA haplotype in combination with a suitable somatic mutation in a tumor specific neoantigen will most likely limit a wide scale application. Alternatively, one may try to generate neosplicetope reactive T cell clones in vitro or in experimental humanized mouse models (56) in order to isolate neosplicetope-specific TCR. However, isolating a T cell clone against a peptide is itself not sufficient to unequivocally demonstrate that this peptide indeed exists and is processed by the tumor. What finally will be needed is experimental evidence that the isolated T cell clone or transduced T cells that express the isolated TCR are able to recognize or to kill tumors expressing the HLA molecules presenting the peptide and the neoantigen from which the putative neosplicetope is derived. One also has to consider that proteasomal generation of neosplicetopes is the result of peptide shuffling bearing the possibility that the generated neosplicetopes are not entirely tumor specific. By setting the search window of the spliced peptide prediction algorithms in way, that neosplicetopes are predicted in which only one or two amino acids are exchanged by the splicing reaction one can significantly minimize the likelihood of cross reactivity. At the end it will depend entirely on the specificity of the generated TCR, which always requires extensive testing because cross reactivity never can be excluded to 100%.
Thus, before starting the screen and despite all caveats one may therefore still want to demonstrate that the in silico predicted neosplicetope is also generated in vitro.
To circumvent the pitfalls connected with the strictly in silico and in vitro based prediction of neosplicetope for TCR generation a á priori experimental proof that in silico predicted neo-splicetopes are generated in vivo and expressed at the cell surface in the context of HLA-1 proteins may seem to be mandatory. However, identification of predicted neosplicetopes among the large population non-spliced peptides eluted from immunoprecipitated HLA-1 molecules will be challenging. It will need the development of a new mass spectrometry compatible algorithm in combination with highly sensitive targeted mass spectrometry as recently described by Beer for KRAS G12V derived peptides (58). For identification by targeted LC-MS/MS predicted spliced peptides have to be synthesized with heavy isotope labeled amino acids and spiked into the eluted peptide preparation before mass spectrometric analysis in order to permit unequivocal validation of the putative neosplicetope. Neither experimental approach represents a straight forward pipeline for spliced epitope identification and may turn out to document that spliced epitopes or neosplicetopes are only of theoretical immune relevance and of theoretical therapeutic potential. However, despite all odds if successful and in light of the expected gain it may still seem worth a try.
The author confirms being the sole contributor of this work and has approved it for publication.
Part of this work was supported by grants from the Berlin Institute of Health (CRG-1), Einstein Stiftung (A- 2013-174) and Berliner Krebsgesellschaft.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Kloetzel PM. Antigen Processing by the Proteasome. Nat Rev Mol Cell Biol (2001) 2:179–87. doi: 10.1038/35056572
2. Strehl B, Seifert U, Krüger E, Heink S, Kuckelkorn U, Kloetzel PM. Interferon Gamma, the Functional Plasticity of the Ubiquitin Proteasome System, and MHC Class I Antigen Processing. Immunol Rev (2005) 207:19–30. doi: 10.1111/j.0105-2896.2005.00308
3. Rock KL, Reits E, Neefjes J. Present Yourself! By MHC Class I and MHC Class II Molecules. Trends Immunol (2016) 37:724–37. doi: 10.1016/j.it.2016.08.010
4. Dahlmann B, Ruppert T, Kuehn L, Merforth S, Kloetzel PM. Different Proteasome Subtypes in a Single Tissue Exhibit Different Enzymatic Properties. J Mol Biol (2000) 303:643–53. doi: 10.1006/jmbi.2000.4185
5. Vigneron N, Van den Eynde BJ. Proteasome Subtypes and the Processing of Tumor Antigens: Increasing Diversity. Curr Opin Immunol (2012) 24:84–91. doi: 10.1016/j.coi.2011.12.002
6. Vigneron N, Van den Eynde BJ. Proteasome Subtypes and Regulators in the Processing of Antigenic Peptides Presented by Class I Molecules of the Major Histocompatibility Complex. Biomolecules (2014) 4:994–1025. doi: 10.3390/biom4040994
7. Mishto M, Liepe J, Textoris-Taube K, Keller C, Henklein P, Weberruß M, et al. Proteasome Isoforms Exhibit Only Quantitative Differences in Cleavage and Epitope Generation. Eur J Immunol (2014) 44:443508–21. doi: 10.1002/eji.201444902
8. Textoris-Taube K, Keller C, Liepe J, Henklein P, Sidney J, Sette A, et al. The T210M Substitution in the HLA-A*02:01 Gp100 Epitope Strongly Affects Overall Proteasomal Cleavage Site Usage and Antigen Processing. J Biol Chem (2015) 290:30417–28. doi: 10.1074/jbc.M115.695189
9. Kisselev AF, Garcia-Calvo M, Overkleeft HS, Peterson E, Pennington MW, Ploegh HL, et al. The Caspase-Like Sites of Proteasomes, Their Substrate Specificity, New Inhibitors and Substrates, and Allosteric Interactions With the Trypsin-Like Sites. J Biol Chem (2003) 278:35869–77. doi: 10.1074/jbc.M303725200
10. Hanada K, Yewdell JW, Yang JC. Immune Recognition of a Human Renal Cancer Antigen Through Post-Translational Protein Splicing. Nature (2004) 427:252–6. doi: 10.1038/nature02240
11. Vigneron N, Stroobant V, Chapiro J, Ooms A, Degiovanni G, Morel S, et al. An Antigenic Peptide Produced by Peptide Splicing in the Proteasome. Science (2004) 304:587–90. doi: 10.1126/science.1095522
12. Warren EH, Vigneron NJ, Gavin MA, Coulie PG, Stroobant V, Dalet A, et al. An Antigen Produced by Splicing of Noncontiguous Peptides in the Reverse Order. Science (2006) 313:1444–7. doi: 10.1126/science.1130660
13. Borissenko L, Groll M. Diversity of Proteasomal Missions: Fine Tuning of the Immune Response. Biol Chem (2007) 388:947–55. doi: 10.1515/BC.2007.109
14. Berkers CR, de Jong A, Ovaa H, Rodenko B. Transpeptidation and Reverse Proteolysis and Their Consequences for Immunity. Int J Biochem Cell Biol (2009) 41:66–71. doi: 10.1016/j.biocel.2008.08.036
15. Mishto M, Goede A, Textoris-Taube KT, Keller C, Janek K, Henklein P, et al. Driving Forces of Proteasome-Catalyzed Peptide Splicing in Yeast and Humans. Mol Cell Proteomics (2012) 11:1008–23. doi: 10.1074/mcp.M112.020164
16. Robbins PF, El-Gamil M, Kawakami Y, Stevens E, Yannelli JR, Rosenberg SA. Recognition of Tyrosinase by Tumor-Infiltrating Lymphocytes From a Patient Responding to Immunotherapy. Cancer Res (1994) 54:3124–6.
17. Mishto M, Liepe J. Post-Translational Peptide Splicing and T Cell Responses. Trends Immunol (2017) 38:904–15. doi: 10.1016/j.it.2017.07.011
18. Dalet A, Robbins PF, Stroobant V, Vigneron N, Li YF, El-Gamil M, et al. An Antigenic Peptide Produced by Reverse Splicing and Double Asparagine Deamidation. Proc Natl Acad Sci USA (2011) 108:E323–31. doi: 10.1073/pnas.1101892108
19. Dalet A, Vigneron N, Stroobant V, Hanada K, Van den Eynde BJ. Splicing of Distant Peptide Fragments Occurs in the Proteasome by Transpeptidation and Produces the Spliced Antigenic Peptide Derived From Fibroblast Growth Factor-5. J Immunol (2010) 184:3016–24. doi: 10.4049/jimmunol.0901277
20. Liepe J, Marino F, Sidney J, Jeko A, Bunting DE, Sette A, et al. A Large Fraction of HLA Class I Ligands Are Proteasome-Generated Spliced Peptides. Science (2016) 354:354–8. doi: 10.1126/science.aaf4384
21. Faridi P, Li C, Ramarathinam SH, Vivian JP, Illing PT, Mifsud NA, et al. A Subset of HLA-I Peptides Are Not Genomically Templated: Evidence for Cis- and Trans-Spliced Peptide Ligands. Sci Immunol (2018) 3:eaar3947. doi: 10.1126/sciimmunol.aar3947
22. Mylonas R, Beer I, Iseli C, Chong C, Pak H-S, Gfeller D, et al. Estimating the Contribution of Proteasomal Spliced Peptides to the HLA-I Ligandome*. Mol Cell Proteomics (2018) 17:2347–57. doi: 10.1074/mcp.RA118.000877
23. Rolfs Z, Muller M, Shortreed MR, Smith LM, Bassani-Sternberg M. Comment on A Subset of HLA-I Peptides Are Not Genomically Templated: Evidence for Cis- and Trans-Spliced Peptide Ligands. Sci Immunol (2019) 4:eaaw1622. doi: 10.1126/sciimmunol.aaw1622
24. Erhard F, Dölken L, Schilling B, Schlosser A. Identification of the Cryptic HLA-I Immunopeptidome. Cancer Immunol Res (2020) 8:1018–26. doi: 10.1158/2326-6066.CIR-19-0886
25. Admon A. Are There Indeed Spliced Peptides in the Immunopeptidome? Mol Cell Proteomics (2021) 20:100099. doi: 10.1016/j.mcpro.2021.100099
26. Hinrichs CS, Rosenberg SA. Exploiting the Curative Potential of Adoptive T-Cell Therapy for Cancer. Immunol Rev (2014) 257:56–71. doi: 10.1111/imr.12132
27. Yee C. Adoptive T Cell Therapy: Points to Consider. Curr Opin Immunol (2018) 51:197–203. doi: 10.1016/j.coi.2018.04.007
28. June CH, Sadelain M. Chimeric Antigen Receptor Therapy. N Engl J Med (2018) 379:64–73. doi: 10.1056/NEJMra1706169
29. Gjerstorff MF, Andersen MH, Ditzel HJ. Oncogenic Cancer/Testis Antigens: Prime Candidates for Immunotherapy. Oncotarget (2015) 6:15772–87. doi: 10.18632/oncotarget.4694
30. Engels B, Engelhard VH, Sidney J, Sette A, Binder DC, Liu RB, et al. Relapse or Eradication of Cancer Is Predicted by Peptide-Major Histocompatibility Complex Affinity. Cancer Cell (2013) 23:516–26. doi: 10.1016/j.ccr.2013.03.018
31. Robbins PF, Lu Y-C, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining Exomic Sequencing Data to Identify Mutated Antigens Recognized by Adoptively Transferred Tumor-Reactive T Cells. Nat Med (2013) 19:747–52. doi: 10.1038/nm.3161
32. Hacohen N, Fritsch EF, Carter TA, Lander ES, Wu CJ. Getting Personal With Neoantigen-Based Therapeutic Cancer Vaccines. Cancer Immunol Res (2013) 1:11–5. doi: 10.1158/2326-6066.CIR-13-0022
33. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer Immunotherapy Based on Mutation Specific CD4+ T Cells in a Patient With Epithelial Cancer. Science (2014) 344:641–5. doi: 10.1126/science.1251102
34. Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of Somatic Mutations in Human Gastrointestinal Cancers. Science (2015) 350:1387–90. doi: 10.1126/science.aad1253
35. Blankenstein T, Leisegang M, Uckert W, Schreiber H. Targeting Cancer-Specific Mutations by T Cell Receptor Gene Therapy. Curr Opin Immunol (2015) 33:112–9. doi: 10.1016/j.coi.2015.02.005
36. Ott PA, Dotti G, Yee C, Goff SL. An Update on Adoptive T-Cell Therapy and Neoantigen Vaccines. Am Soc Clin Oncol Educ Book (2019) 39:e70–8. doi: 10.1200/EDBK_23800
37. Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, et al. Oncogenically Active MYD88 Mutations in Human Lymphoma. Nature (2011) 470:115–21. doi: 10.1038/nature09671
38. Rovira J, Karube K, Valera A, Colomer D, Enjuanes A, Colomo L, et al. MYD88 L265P Mutations, But No Other Variants, Identify a Subpopulation of DLBCL Patients of Activated B-Cell Origin, Extranodal Involvement, and Poor Outcome. Clin Cancer Res (2016) 22:2755–64. doi: 10.1158/1078-0432.CCR-15-1525
39. Lee JH, Jeong H, Choi JW, Oh H, Kim YS. Clinicopathologic Significance of MYD88 L265P Mutation in Diffuse Large B-Cell Lymphoma: A Meta- Analysis. Sci Rep (2017) 7:1–8. doi: 10.1038/s41598-017-01998-5
40. Xu L, Hunter ZR, Yang G, Zhou Y, Cao Y, Liu X, et al. MYD88 L265P in Waldensträm Macroglobulinemia, Immunoglobulin M Monoclonal Gammopathy, and Other B-Cell Lymphoproliferative Disorders Using Conventional and Quantitative Allele-Specific Polymerase Chain Reaction. Blood (2013) 121:2051–8. doi: 10.1182/blood-2012-09-454355
41. Çınar Ö., Brzezicha B, Grunert C, Kloetzel PM, Beier C, Peuker CA, et al. High-Affinity T-Cell Receptor Specific for MyD88 L265P Mutation for Adoptive T-Cell Therapy of B-Cell Malignancies. J Immunother Cancer (2021) 9:e002410. doi: 10.1136/jitc-2021-002410
42. Liepe J, Mishto M, Textoris-Taube K, Janek K, Keller C, Henklein P, et al. The 20S Proteasome Splicing Activity Discovered by Splicemet. PloS Comput Biol (2010) 6:e1000830. doi: 10.1371/journal.pcbi.1000830
43. Willimsky G, Beier C, Immisch L, Papafotiou G, Scheuplein V, Goede A, et al In Vitro Proteasome Processing of Neo-Splicetopes Does Not Predict Their Presentation In Vivo. Elife (2021) 10:e62019. doi: 10.7554/eLife.62019
44. Boes B, Hengel H, Ruppert T, Multhaup G, Koszinowski UH, Kloetzel PM. Interferon Gamma Stimulation Modulates the Proteolytic Activity and Cleavage Site Preference of 20S Mouse Proteasomes. J Exp Med (1994) 179:901–9. doi: 10.1084/jem.179.3.901
45. Niedermann G, Butz S, Ihlenfeldt HG, Grimm R, Lucchiari M, Hoschützky H, et al. Contribution of Proteasome-Mediated Proteolysis to the Hierarchy of Epitopes Presented by Major Histocompatibility Complex Class I Molecules. Immunity (1995) 2:289–99. doi: 10.1016/1074-7613(95)90053-5
46. Kessler JH, Beekman NJ, Bres-Vloemans SA, Verdijk P, van Veelen PA, Kloosterman-Joosten AM, et al. Efficient Identification of Novel Hla-A*0201–Presented Cytotoxic T Lymphocyte Epitopes in the Widely Expressed Tumor Antigen Prame by Proteasome-Mediated Digestion Analysis. J Exp Med (2001) 193:73–88. doi: 10.1084/jem.193.1.73
47. Sijts A, Zaiss D, Kloetzel PM. The Role of the Ubiquitin-Proteasome Pathway in MHC Class I Antigen Processing: Implications for Vaccine Design. Curr Mol Med (2001) 1:665–76. doi: 10.2174/1566524013363230
48. Guillaume B, Stroobant V, Bousquet-Dubouch MP, Colau D, Chapiro J, Parmentier N, et al. Analysis of the Processing of Seven Human Tumor Antigens by Intermediate Proteasomes. J Immunol (2012) 189:3538–47. doi: 10.4049/jimmunol.1103213
49. Theobald M, Ruppert T, Kuckelkorn U, Hernandez J, Häussler A, Ferreira EA, et al. The Sequence Alteration Associated With a Mutational Hotspot in P53 Protects Cells From Lysis by Cytotoxic T Lymphocytes Specific for a Flanking Peptide Epitope. J Exp Med (1998) 188:1017–28. doi: 10.1084/jem.188.6.1017
50. Zanker D, Waithman J, Yewdell JW, Chen W. Mixed Proteasomes Function to Increase Viral Peptide Diversity and Broaden Antiviral CD8+ T Cell Responses. J Immunol (2013) 191:52–9. doi: 10.4049/jimmunol.130080
51. Dalet A, Stroobant V, Vigneron N, Van den Eynde BJ. Differences in the Production of Spliced Antigenic Peptides by the Standard Proteasome and the Immunoproteasome. Eur J Immunol (2011) 41:39–46. doi: 10.1002/eji.201040750
52. Michaux A, Larrieu P, Stroobant V, Fonteneau JF, Jotereau F, Van den Eynde BJ, et al. A Spliced Antigenic Peptide Comprising a Single Spliced Amino Acid Is Produced in the Proteasome by Reverse Splicing of a Longer Peptide Fragment Followed by Trimming. J Immunol (2014) 192:1962–71. doi: 10.4049/jimmunol.1302032
53. Ebstein F, Textoris-Taube K, Keller C, Golnik R, Vigneron N, Van den Eynde BJ, et al. Proteasomes Generate Spliced Epitopes by Two Different Mechanisms and as Efficiently as Non-Spliced Epitopes. Sci Rep (2016) 6:24032. doi: 10.1038/srep24032
54. Paes W, Leonov G, Partridge T, Nicastri A, Ternette N, Borrow P. Elucidation of the Signatures of Proteasome-Catalyzed Peptide Splicing. Front Immunol (2020) 11:563800. doi: 10.3389/fimmu.2020.563800
55. Platteel ACM, Liepe J, Textoris-Taube K, Keller C, Henklein P, Schalkwijk HH, et al. Multi-Level Strategy for Identifying Proteasome-Catalyzed Spliced Epitopes Targeted by CD8+ T Cells During Bacterial Infection. Cell Rep (2017) 20:1242–53. doi: 10.1016/j.celrep.2017.07.026
56. Obenaus M, Leitao C, Leisegang M, Chen X, Gavvovidis I, van der Bruggen P, et al. Identification of Human T-Cell Receptors With Optimal Affinity to Cancer Antigens Using Antigen-Negative Humanized Mice. Nat Biotechnol (2015) 33(4):402–7. doi: 10.1038/nbt.3147
57. Mishto M, Mansurkhodzhaev A, Ying G, Bitra A, Cordfunke RA, Henze S, et al. An in Silico-In Vitro Pipeline Identifying an HLA-A*02:01+ KRAS G12V+ Spliced Epitope Candidate for a Broad Tumor-Immune Response in Cancer Patients. Front Immunol (2019) 10:2572. doi: 10.3389/fimmu.2019.02572
58. Beer I. Commentary: An In Silico - In Vitro Pipeline Identifying an HLA-A*02:01 + KRAS G12V + Spliced Epitope Candidate for a Broad Tumor-Immune Response in Cancer Patients. Front Immunol (2021) 12:523906. doi: 10.3389/fimmu.2021.523906
59. Mishto M, Rodriguez-Hernandez G, Neefjes J, Urlaub H, Liepe J. Response: Commentary: An In Silico-In Vitro Pipeline Identifying an HLA-A*02:01+ KRAS G12V+ Spliced Epitope Candidate for a Broad Tumor-Immune Response in Cancer Patients. Front Immunol (2021) 12:679836. doi: 10.3389/fimmu.2021.679836
Keywords: proteasome, antigen processing, peptide splicing, adoptive T cell transfer, prediction algorithms, neosplicetopes
Citation: Kloetzel PM (2022) Neo-Splicetopes in Tumor Therapy: A Lost Case? Front. Immunol. 13:849863. doi: 10.3389/fimmu.2022.849863
Received: 06 January 2022; Accepted: 03 February 2022;
Published: 21 February 2022.
Edited by:
Elodie Segura, Institut Curie, FranceReviewed by:
Nathalie Vigneron, Ludwig Cancer Research, BelgiumCopyright © 2022 Kloetzel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter M. Kloetzel, cC1tLmtsb2V0emVsQGNoYXJpdGUuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.