 Ling-ling Yan1,2†Li-ping Ye1,2,3†Ya-hong Chen4Sai-qin He2Chen-yang Zhang5Xin-li Mao1,2,3*
Ling-ling Yan1,2†Li-ping Ye1,2,3†Ya-hong Chen4Sai-qin He2Chen-yang Zhang5Xin-li Mao1,2,3* Shao-wei Li1,2,3*
Shao-wei Li1,2,3*- 1Key Laboratory of Minimally Invasive Techniques & Rapid Rehabilitation of Digestive System Tumor of Zhejiang Province, Taizhou Hospital Affiliated to Wenzhou Medical University, Linhai, China
- 2Department of Gastroenterology, Taizhou Hospital of Zhejiang Province Affiliated to Wenzhou Medical University, Linhai, China
- 3Institute of Digestive Disease, Taizhou Hospital of Zhejiang Province Affiliated to Wenzhou Medical University, Linhai, China
- 4Health Management Center, Taizhou Hospital of Zhejiang Province Affiliated to Wenzhou Medical University, Linhai, China
- 5Taizhou Hospital of Zhejiang Province Affiliated to Wenzhou Medical University, Linhai, China
Clinical islet transplantation has the potential to cure type 1 diabetes. Despite recent therapeutic success, it is still uncommon because transplanted islets are damaged by multiple challenges, including instant blood mediated inflammatory reaction (IBMIR), inflammatory cytokines, hypoxia/reperfusion injury, and immune rejection. The transplantation microenvironment plays a vital role especially in intraportal islet transplantation. The identification and targeting of pathways that function as “master regulators” during deleterious inflammatory events after transplantation, and the induction of immune tolerance, are necessary to improve the survival of transplanted islets. In this article, we attempt to provide an overview of the influence of microenvironment on the survival of transplanted islets, as well as possible therapeutic targets.
Introduction
Although type 1 diabetes cannot be prevented or reversed, islet transplantation—which restores insulin independence and prevents severe hypoglycemia—is currently proving to be a promising treatment (1, 2). However, the results of multiple clinical trials have shown that most transplant recipients fail to achieve complete insulin independence. Although the immunosuppressive regimen reported from Edmonton, Canada, has achieved unprecedented success in achieving insulin independence in islet transplantation (3), there are still some problems that affect the outcome of islet transplantation. Advancements in the acquisition of pancreatic islets, immunosuppression of islet recipients, and an increase in the number of transplanted islets are required (4). The average human pancreas has 300,000 to 1.5 million pancreatic islets, and only 60% of this islet cell mass is needed to maintain a normal glucose metabolism (5). However, 72% of islet recipients still require more than two successfully processed islet preparations to obtain a sufficient beta cell mass to compensate for islet death in the post-transplantation period (6). However, the source of pancreatic islets is limited.
It was observed that only 10% of recipients remain insulin dependent for more than 5 years and that most recipients re-use insulin because of a decline in the islet function over time (7). During transplantation, islet grafts are damaged by multiple challenges, including enzyme and mechanical damage caused by the isolation process, hypoxia, inflammation, immune rejection and toxicity of immunosuppressive drugs (8). The liver is currently the preferred transplantation site because the procedure is minimally invasive, easy to perform, and has low rates of bleeding and thrombosis (9). Early inflammatory responses strongly influence islet engraftment and survival after intrahepatic transplantation. This early immune response is triggered by immediate blood-mediated inflammatory response (IBMIR) and ischemia-reperfusion injury (10). Elevated inflammatory cytokines interleukin-1β (IL-1β), tumor necrosis factor α (TNF-α), interferon-γ (IFN-γ), were observed during islet transplantation, and macrophages also involved in regulating the cell injury of transplanted pancreatic islets (11). It was observed that concomitant transplantation of islets and vascular endothelial cells in diabetic rats can prolong the survival of islet grafts (12). Previous studies have found that cytokine inhibitors or drugs that inhibit the activation of liver macrophage activation can improve the function of pancreatic islets after transplantation (13, 14). Changes in the microenvironment, mainly involving inflammatory cytokines, endothelial cells, and immune cells, play a key role in the survival of islet grafts.
This review discusses the changes in the microenvironment (inflammatory cytokines, immune cells, endothelial cells, etc.) during clinical intraportal islet transplantation, and their influence on the survival of the islet grafts, with the aim of improving the success of clinical islet transplantation
Status and Recent Progress of Clinical Intraportal Islet Transplantation
In 1990, David Scharp et al. reported the first case of type 1 diabetes allogeneic intraportal islet transplantation resulting in a short-term insulin independence, which opened the prelude to clinical islet transplantation (15). The International Islet Registry collected data from 267 islet allogeneic transplants from multiple centers between 1990 and 2001, only 12.4% of cases achieved insulin independence periods of at least 1 week, and 8.2% maintained insulin independence for more than 1 year. Appropriate immunosuppression remained a critical piece of the unsolved puzzle in order to improve long-term graft function and sustained insulin independence (16). Until 2000, Edmonton’s group reported that insulin independence was achieved in seven patients with type 1 diabetes who underwent islet transplantation. This protocol established the need for sufficient islet β cell mass for transplantation and also proposed a glucocorticoid-free immunosuppressive regimen (3). Since the publication of the Edmonton protocol, many countries around the world have successively carried out allogeneic islet transplantation for the treatment of type 1 diabetes. Extended follow-up of these trials sho wed a gradual loss of insulin independence over time with renewed need for exogenous insulin, with only 10% of patients showing insulin independence at 5 years after transplantation (7). Hering et al. reported in 2016 that the North American Clinical Islet Transplant Consortium conducted a multicenter, single-arm, phase 3 trial to further evaluate the efficiency of allogeneic islet transplantation. The assay, using product purified human pancreatic islets, achieved an HbA1c of 5.6% in the absence of severe hypoglycemia (17). A subsequent multicenter, open-label, randomized controlled trial in Europe reconfirmed the benefit of islet transplantation over intensive insulin therapy in patients with type 1 diabetes with severe hypoglycemia or after kidney transplantation (18). Recently, Marco et al. proposed that vitamin D alone or in combination with other anti-inflammatory agents may serve as a new immunomodulatory strategy to reduce the recurrence of autoimmune/allogeneic transplant rejection in intraportal islet transplantation, but its safety and efficacy further validation in large prospective studies is required (19). In intraportal islet transplantation model, islet pretreatment with mitomycin C prolonged graft survival by suppressing pro-inflammatory events and inducing latent regulatory lymphocytes (20). Xenogeneic and stem cell-derived islet tissues have entered early clinical trials, although much remains to be learned about the in vivo physiology and immunogenicity of various products. These advances provide more options for cellular therapy in diabetes treatment, providing an unlimited source of pancreatic islet tissue for future applications (21).
Intraportal Islet Transplantation Challenges and Strategies
Instant blood mediated inflammatory reaction (IBMIR)
The IBMIR, which is triggered by exposed tissue factor on the islet surface and characterized by platelet activation and aggregation, and activation of the coagulation and complement systems, is a major obstacle after islet transplantation. Moreover, it increases the infiltration of neutrophils, monocytes and macrophages (13). These reactions are thought to cause the immediate loss of most of the transplanted islets (22), and may also increase the risk of islet rejection later through adaptive immune responses (23). Potential means for reducing islet inflammation and protecting islets can be achieved by the addition of anti-inflammatory agents, anticoagulants or coating islets with various protective macromolecules during islet culture (24). In vitro and in vivo experiments demonstrated that low molecular weight dextran sulfate prevented IBMIR, leukocyte infiltration was eliminated at high doses, and transplanted pig islets survived significantly longer in recipients treated with low molecular weight dextran sulfate (25). Another phase II, multicenter, active-controlled, randomized study, the Clinical Islet Transplant Consortium 01 study, revealed that systemic low molecular weight dextran sulfate treatment showed similar efficacy to heparin treatment in preventing IBMIR and promoting islet engraftment (26). However, the reason for the current reluctance to use it is that its target- activated partial thromboplastin time is 3 times longer than heparin’s (27). The use of biotin/avidin technology to bind preformed heparin complexes to the islet surface resulted in protection against IBMIR in vitro loop model and in an allogeneic porcine model of clinical islet transplantation (28). In vitro and in vivo data suggested that activated protein C exerted anti-inflammatory and anti-apoptotic activities by directly acting on cells after exposure of hepatic endothelial cells to pancreatic islets (29). However, the above remain to be verified by clinical studies (30, 31). And it has been confirmed that Cibinetide can reduce IBMIR related platelet consumption in a pro-inflammatory environment and protect isolated human islets (32). The evidence showed that α-1 antitrypsin inhibited IBMIR, which resulted in improved outcome of intraportal islet transplantation in mouse model (33). Currently, the addition of heparin is the standard approach but it is insufficient (27).
Complement system
Complement activation is triggered by natural immunoglobulin (Ig)G or IgM. When isolated islets are exposed to blood, the complement system is rapidly activated and causes lysis of islet cells (34). Complement activation occurs via the classical pathway and an alternative pathway, which leads to the formation of a complex composed of C5b-9, which forms a “channel” through the cell membrane, leading to cell lysis and death. Another major function of the complement system is the production of anaphylatoxins C3a and C5a, which enhance the inflammatory response to islets (23). C3a and C5a produced by complement activation are powerful chemoattractants for macrophages and neutrophils (10). Activation through the C5a receptor can cause granulocytes to release enzymes, such as myeloperoxidase and elastase, and promote monocytes to release cytokines, such as IL-1, IL-6, IL-8, and TNF-a. C5a stimulates endothelial cells to release heparin sulfate, upregulate tissue factor, secrete von Willebrand factor and express P-selectin, which is conducive to fibrin deposition and which enhances thrombin-mediated platelet aggregation and polymorphonuclear leukocyte adhesion (23). Tissue factor and many adhesion molecules can be expressed by endothelial cells triggered by soluble C5b-9 (sC5b-9) (35). Complement activation may therefore induce direct inflammation and indirect effects mediated by endothelial cells.
In allogeneic transplantation, C3 is one of the important factors triggering rejection in mice (36, 37) and humans (38). Numerous studies have shown that the combination of C3 fragment C3dg with an antigen can be used as a strong adjuvant to promote both cellular and humoral responses (39). Therefore, it can be reasonably expected that complement activation and C3dg binding will trigger an immune response, leading to an adaptive immune response to the graft (34). Therefore, small interfering RNA (siRNA) targeting C3 and C5a receptors may increase the viability of transplanted islets (40).
Hypoxic damage
Islets are easily damaged under hypoxic conditions prior to transplantation (including pancreas procurement, islet isolation and culture) and exposure to hypoxic environment of the transplant site after transplantation (41). Hypoxia damages islet β cell function, which manifested anaerobic glycolysis, showed elevated lactate and reduced responsiveness to high glucose levels (42). The decrease of blood oxygen partial pressure can lead to irreversible β cell dysfunction, resulting in higher fasting blood-glucose and lower C-peptide levels (43). Calcium influx into islet cells has also been shown to cause cell damage in rat and human islets cultured under hypoxic conditions (44). In order to cope with the impact of hypoxia on the quality and function of islets, the following explorations were attempted: (1) Compared with static cold storage, perfusion effectively reduced anoxic death of islet cells, and islet production was higher after perfusion (45, 46). Perfluorohexyloctan, a semi-fluorinated liquid fluorocarbon, maintained higher intrapancreatic pO2 and improved islet viability and function with porcine pancreas (47, 48). In addition, oxygen supply can be increased and oxygenation of islets can be improved by decreasing the culture density of islets before transplantation (46, 49). (2) Photosynthesis of thermostable microalga (Chlorella sorokiniana) was applied as a method to supply oxygen to cultured islets coencapsulating in alginate gel (50). Microparticle-mediated-oxygenation has been studied to improve islet transplantation.Co-transplantation of oxygen-generating microparticles and minimal islet mass within fibrin-conjugated heparin/VEGF collagen scaffold has enormous potential to enhance islet revascularization, diabetes reversal and oxygenation (51). (3) As mentioned above, the influx of calcium ions into islet cells can also induce islet injury. It was found that potassium channel activator (diazoxide) and calcium channel blocker (nifedipine) were helpful to restore the synthesis of insulin protoplasts and islet cell necrosis caused by hypoxia when used as preconditioning agents (44, 52). (4) Curcumin has the ability to protect β cells from hypoxia damage (53). Puerarin could alleviate β cell apoptosis and malfunction by hypoxic injury of β cells in corpulent mice induced by cobalt chloride induced via PI3K/Akt pathway activation (54). (5) Reconstructing the capillary network in islets is very important to prevent hypoxia and preserve function. Previous studies have shown the effectiveness of prevascularization of the graft bed for subcutaneous islet graft survival (55–58). The use of islet-cell cluster in clinical islet transplantation may be a strategy to prevent islet loss caused by hypoxia after transplantation (59).
Immunosuppressive drug toxicity and immune tolerance induction
During intraportal islet transplantation, isolated islets are exposed to high levels of immunosuppressive drugs, which are detrimental to islet engraftment/survival and long-term function (60). Previously, the standard protocol of immunosuppression for islet transplantation included a combination of corticosteroids, calcineurin inhibitors (tacrolimus and cyclosporine), and purine analogs (mycophenolate mofetil). Many of the above drugs had been shown to be diabetogenic, impairing insulin secretion (61). The Edmonton protocol in 1999 took a major step forward in islet transplantation with the introduction of steroid-free therapy based on low-dose sirolimus, tacrolimus, and daclizumab (3). Although the results of multicenter clinical trials suggested that this protocol could provide short-term insulin independence and reduce the incidence of acute rejection (62), patients receiving long-term immunosuppressive drugs are susceptible to multiple adverse effects, such as infections (63), malignancies (64), de novo diabetes (65) and organ toxicities (66).
Immune tolerance induction is a promising strategy to accept histocompatibility complex (MHC)-mismatched allografts without reducing resistance to infection or increasing other complications (67). For immunological tolerance to allografts, the high proportion of MHC alloreactive T cell is considered as a major barrier to tolerance induction. Central T cell tolerance refers to the deletion of reactive clones in the thymus during negative selection. Peripheral T cell tolerance includes peripheral deletion, anergy/exhaustion, and suppressive function of regulatory T cells (Treg) (68). At present, how to apply the inherent immune tolerance mechanism of the human system to induce donor-specific immune tolerance is the key to solving transplant rejection (69). Singh et al. reported that apoptotic donor lymphocyte infusion prior to transplantation induced long-term tolerance (>1 year) of islet grafts in a non-human primate (NHP) model, which had made a breakthrough in the tolerance induction protocol for allogeneic islet transplantation (70).
Intrathymic inoculation of recipient APCs pulsed with allopeptides can induce intrathymic tolerance, however it is an invasive technique and the thymus regresses with age and has limited potential in adults (71). Another potentially more effective approach to achieving central tolerance is the generation of hematopoietic chimerism or mixed allogeneic chimerism, with lethal total body irradiation or sublethal total body irradiation (assisted by anti-CD4, CD8 monoclonal antibodies or costimulatory blockade) in the prospective transplant recipients, in order to make room for the transplanted bone marrow. Bone marrow cell transplantation can reconstitute the recipient’s hematopoietic compartment with donor hematopoietic stem cells, inducing donor-specific tolerance to islet allografts (62, 72). Another strategy to induce tolerance is to deplete alloreactive T cells prior to transplantation, promoting a hyporesponsive environment that drives tolerance transition (73). T cell depletion can be achieved by total body irradiation, lymphocyte depleting alloantibodies. Among them, antithymocyte globulin (ATG) is a potent inducer of T cell depletion, and ATG alone or in combination with other drugs can prolong the survival of allografts (74, 75). Other pathways for inducing immune tolerance include costimulatory signal blockades, induction and expansion of regulatory T (Treg) cells, etc (62). And in the intrahepatic mouse allogeneic islet transplantation model, Lee et al. demonstrated for the first time that short-term single administration of anti-CD154 monoclonal antibody could induce FoxP3+ Treg cell-mediated immune tolerance (67).
Key Inflammatory Factors and Cells Associated With Islet Cell Dysfunction
Several mediators have been found to cause islet dysfunction and/or cellular death after islet transplantation, including inflammatory cytokines (IL-1β, TNF-α, and IFN-γ), nitric oxide (NO), and nitric oxide synthase (iNOS) (76).
IL-1β
IL-1β is one of the most important mediators of islet injury and plays an important role in the process of pancreatic islets dysfunction, which may represent an early inflammatory marker of graft failure (77). IL-1β is secreted by Kupffer cells, islet resident macrophages, and neutrophils around the transplantation site (78). IL-1β secretion increases during islet acquisition, islet isolation, islet culture, and islet transplantation (77). IL-1β binds to the IL-1β receptor (IL-1βR) on the surface of pancreatic islet cells, causing TNF receptor-related factor 6 (TRAF6) to be activated by IL-1 receptor-related kinase (IRAK), which in turn leads to the phosphorylation and degradation of IκB. Then NF-κB is released from inhibitory IκB, transferred from the cytoplasm to the nucleus, and regulates the transcription of various genes, including IL-1, IL-6, TNF-α, and iNOS (76, 79) (Figure 1). Activation of iNOS results in the production of NO, which is directly related to β-cell apoptosis (80).
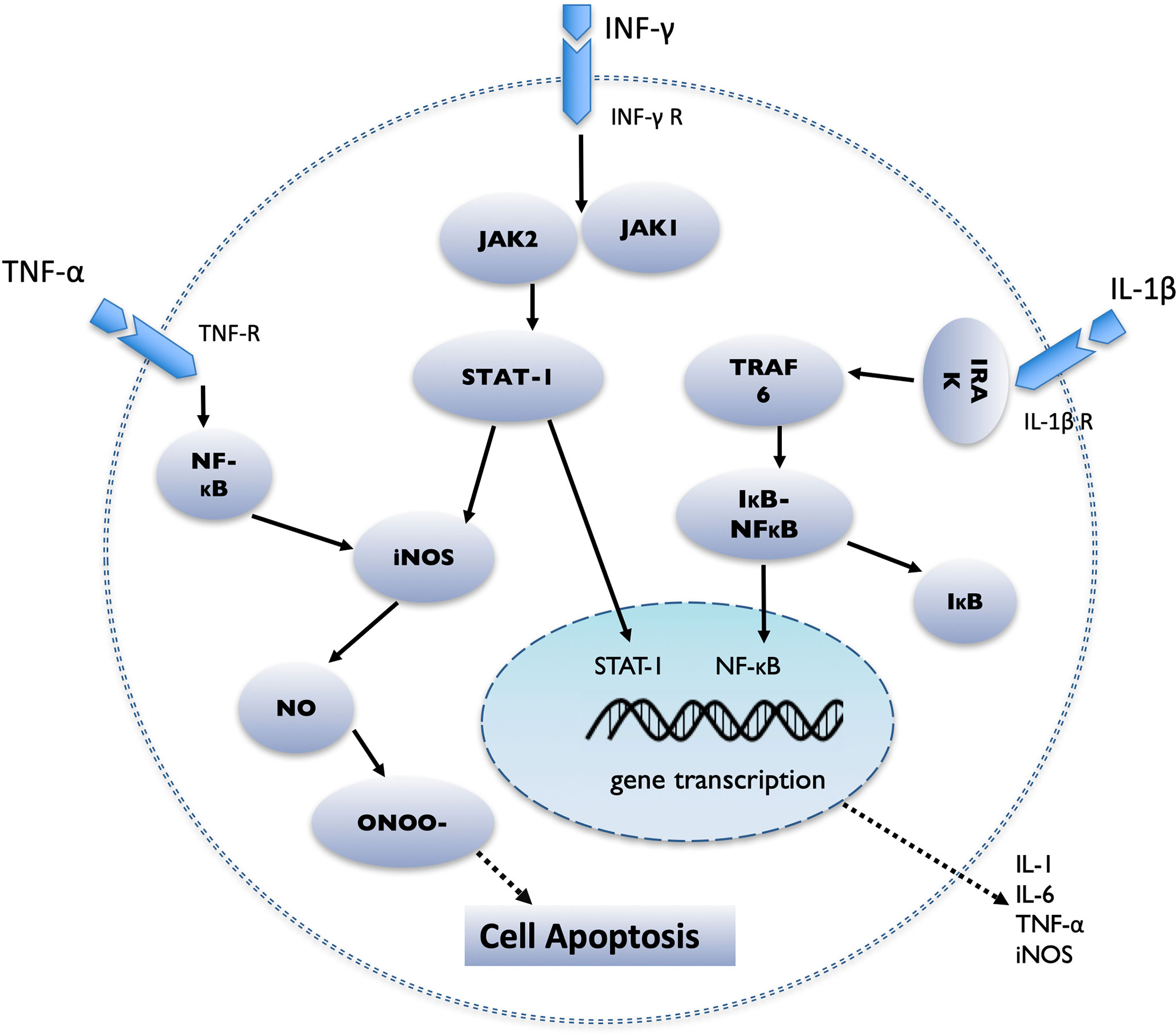
Figure 1 Schematic diagram demonstrating the intracellular cascade of events that occur in the pancreatic islet after stimulation by IL-1β, IFN-γ and TNF-α. IL-1β binds to the IL-1β receptor (IL-1βR), causing TNF receptor-related factor 6 (TRAF6) to be activated by IL-1 receptor-related kinase (IRAK), then NF-κB is released from inhibitory IκB, transferred from the cytoplasm to the nucleus, and regulates the transcription of various genes, including IL-1, IL-6, TNF-α, and iNOS; TNF-α binding to the TNF receptor (TNF-R), initiates the activation of NF-κB and the induction of apoptosis; IFN-γ binds to the IFN-γ receptor (IFN-γR) and causes the activation of JAK1 and JAK2. JAK2 then activates signal transducers and activators of transcription 1 (STAT1), which then transfers to the nucleus, where it performs gene regulation.
The damage of islet cells starts from the donor. Although most pancreatic islet transplants use organs from heart-beating brain-dead (BD) donors, acute physiological changes after brain death of BD donors may still cause significant damage to islets from inflammatory events. Brain death can stimulate various cells to produce pro-inflammatory cytokines, and produce a so-called “cytokine storm”, including IL-1β in BD donors, which greatly reduces the islet yield, functionality, vitality, and engraftment after transplantation (8). One study has shown that the administration of exendin-4 to BD donors can reduce the expression of IL-1β, thereby increasing both the islet viability and insulin secretion in the pancreas after glucose stimulation in a BD rat model (81). In the BD rat model, treatment with a selective neutrophil elastase inhibitor, sivelestat sodium, decreased the expression of IL-1β, significantly improved the islet yield and function in vitro, and suppressed hypercytokinemia-mediated beta-cell death (82).
Then, in the process pancreas digestion and islet purification, enzymatic and mechanical stress can induce inflammatory mediators, such as IL-1β, in the islets (77). The islet basement membrane is lysed during the digestion and separation of the pancreas, which interrupts the communication between islet basement membrane proteins and the integrins expressed by the islets (83). The loss of the interaction between the internal cells and the external microenvironment also interrupts the transmission of pro-survival signals (84). The isolation of pancreatic islets eventually leads to the increased expression of many stress kinases, which subsequently activates pro-inflammatory and pro-apoptotic pathways (43, 85). Furthermore, the functional clustering of differentially expressed genes revealed the upregulation of genes related to cell growth, angiogenesis, inflammation, and apoptosis after isolation and culture (43). A study has shown that islets in early culture (2 days) express more genes, including IL-1β, than islets in long-term culture (7-11 days). It seems that culturing islets before transplantation is beneficial for reducing the expression of inflammatory mediators (86). However, in cultured adult porcine islets, IL-1β mRNA was continuously detected at 1, 4, 8, and 11 days after isolation, and slightly increased over time (87). It is worth noting that a pretreatment culture with anakinra (IL-1 receptor antagonist) prior to human islet transplantation can improve the survival and function of human islets during culture (88).
The early damage of pancreatic islets is mainly manifested by an IBMIR, leading to a cytokine storm, involving IL-1β, TNF-α, and IFN-γ. These cytokine-activated macrophages produce IL-1β, which triggers the cytokine storm at the transplantation site, leading to a negative chain of events (89). Syngeneic transplant models have shown that the nonspecific inflammatory response increases IL-1β at the transplant site and affects early graft failure. In that study, the expression of IL-1β mRNA was maximal on day 1 after transplantation and then declined towards pre-transplantation levels on day 7 (90). Previous animal studies have indicated that approximately 60% of islet grafts lose their function via non-specific inflammation within 3 days after transplantation (91, 92). The overexpression of IL-1β receptor antagonist protein in transplanted islets can improve the outcome of the transplantation (93).
TNF-α
In humans, it appears that IL-1β must act in combination with IFN-γ and/or TNF-α (76). After TNF-α binds to the TNF receptor (TNF-R), it forms a trimer and undergoes conformational changes, which leads to the exposure of the intracellular death domain, and initiates the activation of NF-κB, the activation of the MAPK pathway, and the induction of apoptosis (40). Wen et al. revealed that TNF-α was significantly elevated in patients following allogeneic islet cell infusion compared with patients receiving autologous transplantation (94). Recently, multiple studies have shown that after pancreatic islet allotransplantation, the early use of a combination of anti-IL-1β (anakinra) and TNF-α (etanercept) inflammation blockade is beneficial for reducing islet damage caused by nonspecific inflammation and presumably led to better engraftment (95) (Figure 1).
IFN-γ
IFN-γ binds to the IFN-γ receptor (IFN-γR) and causes the activation of JAK1 and JAK2. JAK2 then activates signal transducers and activators of transcription 1 (STAT1), which then transfers to the nucleus, where it performs gene regulation (96). One study showed that IFN-γ transcripts were found in allografts at 1, 3, 5, and 7 days after transplantation, and peaked on day 5, but there were no such cytokines in syngeneic grafts (97). Some studies have shown that combined cytokines such as IL-1β, TNF-α, and IFN-γ lead to islet cell dysfunction or death (98).
NO and Inducible Nitric Oxide Synthase
Cytokines, such as IL-1β, TNF-α and IFN-γ, mainly stimulate the large expression of iNOS in β cells and macrophages to synthesize excessive NO, thereby causing damage to the pancreatic islets (99); the latter can form a highly active free radical peroxynitrite (ONOO-) by losing an electron and combining with superoxide free radicals, which have strong cytotoxicity and promote apoptosis (100, 101) (Figure 1). NO affects many physiological β-cell processes, including inhibition of oxidative metabolism, changes in the expression of target genes, inhibition of glucose-stimulated insulin secretion, damage to DNA, and induction of endoplasmic reticulum (ER) stress. If exposure to NO is prolonged, it activates various signal cascades and eventually leads to the death of β cells (102). In xenogeneic islet transplantation animal models, selective iNOS inhibitors can suppress the production of induced NO, to prevent early islet graft failure (103).
Immune Cells
Macrophages
Macrophages, which are one of the cellular components of the innate immune system, show great heterogeneity in physiological and pathological conditions. They can be polarized into pro-inflammatory macrophages (M1) or anti-inflammatory macrophages (M2) in different environments (104). Lipopolysaccharide and interferon IFN-γ can activate M1 macrophages to secrete TNF-α, iNOS and superoxide anion to play pro-inflammatory and host defense functions. In contrast, M2 macrophages have a protective role in the immune response and inflammation. Other studies showed that M2 macrophages can be activated by interleukin (IL)-4 and IL-13, and play an immune regulatory and anti-inflammatory role, through the secretion of IL-1Rα, IL-1,TNF-α, IL-10, and other cytokines (104, 105).
After the islets are infused into the portal vein, the activated liver-resident macrophages (Kupffer cells) play a central role in the inflammatory responses within the liver, secreting a series of factors (including arachidonic acid metabolites, TNF-α, IFN-γ, IL-1, IL-6, complement, coagulation factors, reactive oxygen species and nitrogen) to recognize and respond to various signals from the surrounding microenvironment, directly affecting the survival of intrahepatic islets (11, 106, 107). Furthermore, Kupffer cells can induce a response of other non-immune cell subsets, including endothelial cells (108). In addition, it has been suggested that ischemia reperfusion phenomena may directly trigger the activation of sinusoidal endothelial cells and help trigger nonspecific inflammatory responses (109). The transplanted islets, residual endotoxin produced in the process of islet isolation, and liver sinusoidal endothelial damage can all activate Kupffer cells (11, 110). In addition, tissue damage can recruit inflammatory macrophages (M1), which in turn cause islet damage (111). At the same time, dying β cells also produce high mobility group box 1 (HGMB) and iNOS, attracting more macrophages to the liver, and enhancing inflammation and β cell death (14, 112) (Figure 2).
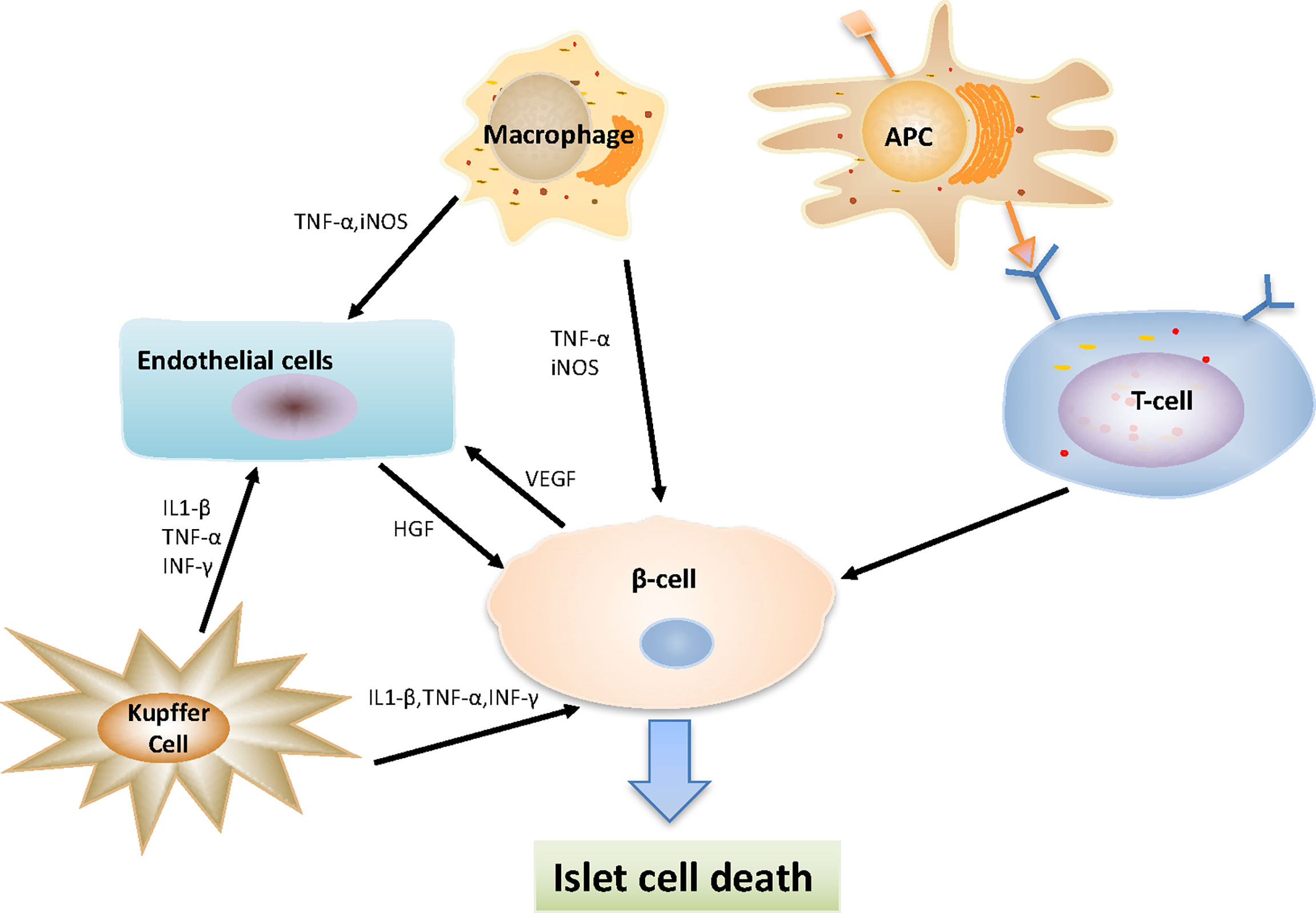
Figure 2 Interaction between various types of cells after islet transplantation. Macrophages secreting TNF-α and iNOS, which act on beta-cells and endothelial cells to play a pro-inflammatory function; Kupffer cells secreting a series of substances (including IL-1β, IFN-γ and TNF-α), directly affecting the survival of intrahepatic islets and endothelial cells; Antigen presenting cells (APCs) take and process antigens from donor and present antigens to host T cells, then reactive CD8+, CD4+ T-cells destroy transplanted islet β cells.
Macrophage depletion can significantly reduce the expression of IL-1β and TNF-α, indicating the role of macrophages in the production of inflammatory cytokines (11). Gou et al. demonstrated that Alpha-1 antitrypsin can protect the survival of islet grafts, in part by inhibiting the polarization of M1 macrophages both in vivo and in vitro (14). One study has mentioned that the local sustained-release of dexamethasone in grafts promotes the survival of mouse pancreatic islet grafts by inducing the differentiation of M2 macrophages in the graft microenvironment as well as the secretion of anti-inflammatory factors (104). Chappell et al. showed that activated M2 macrophages could improve the survival conditions of grafts in a mouse model by improving revascularization or neovascularization, which had a repair function in the graft reaction (113).
Lymphocytes
Type I diabetes is characterized by the autoimmune-mediated damage of islet β-cells, and the transplanted islets will also be attacked by the same stresses that destroy host β-cells (40). CD8+ T cells and CD4+ T cells are the major players in the destruction of β cells. Activated T cells produce cytokines such as IFN-γ, TNF-α, and lymphotoxin to induce β cell apoptosis. T cells also express ligands for the Fas receptor and TNF-related apoptosis-inducing ligands, both of which lead to apoptosis by activating effector caspases. In addition, CD8+ T cells directly contact and promote the release of granzyme B into the cytoplasm of target cells through perforin, thereby activating nucleases and caspases to kill the target cells (40, 98). In some studies, CD8+ T cell infiltration was observed in the pancreas of type 1 diabetic patients and the transplanted pancreas (114, 115). In mouse models, deficiency of CD4+ T cells was observed to stop progression to insulitis (116).
During the rejection of allogeneic transplantation, the host immune response can directly or indirectly be activated by T cells recognizing the donor tissue. Direct graft recognition involves an interaction between the donor tissue resident antigen-presenting cells (APCs) and host T cells via major histocompatibility complex (MHC) (117). Indirect recognition involves the treatment of donor graft peptides by host APCs and corresponding MHC interactions to stimulate host T cells (118). APCs, including macrophages, dendritic cells (DC), passenger leukocytes, from both donors and host are involved in the antigen presentation (40). The activation and maturation of T cells depends on the signals from the above APCs. If these signals are blocked, T cells will undergo apoptosis (Figure 2).
DCs can not only initiate an immune response, but also induce central or peripheral immune tolerance (40). Mature or activated DCs can initiate a positive immune response, while immature DCs or DC precursors show tolerance.  is a transcription factor that is necessary for DC differentiation and maturation. The inhibition of the pathway has been shown to produce tolerogenic DCs (119). RelB is a major protein, which inhibits the expression of MHC-II, CD80, and CD86, and ultimately prevents the maturation of dendritic cells. RelB silenced DCs can inhibit antigen-specific alloreactive immune rejection and reduce the proliferation of antigen-specific T cells (120). In another study, tolerogenic DCs were generated by inhibiting the expression of CD80 or CD86. The administration of modified dendritic cells (DC) can prolong the survival of allografts, thereby inducing T cell hyporesponsiveness and apoptosis (121).
is a transcription factor that is necessary for DC differentiation and maturation. The inhibition of the pathway has been shown to produce tolerogenic DCs (119). RelB is a major protein, which inhibits the expression of MHC-II, CD80, and CD86, and ultimately prevents the maturation of dendritic cells. RelB silenced DCs can inhibit antigen-specific alloreactive immune rejection and reduce the proliferation of antigen-specific T cells (120). In another study, tolerogenic DCs were generated by inhibiting the expression of CD80 or CD86. The administration of modified dendritic cells (DC) can prolong the survival of allografts, thereby inducing T cell hyporesponsiveness and apoptosis (121).
Granulocytes
The poor outcome of intraportal islet transplantation can be explained by IBMIR, one of which is characterized by leucocyte infiltration. The islets were mixed with ABO-compatible blood in a heparinized tube, and the first neutrophilic granulocytes appeared in the islets after 15 minutes, increasing at 1 hour and peaking at 2 hours (122). Neutrophilic granulocytes induce cell damage through cytotoxic attack and phagocytosis. After neutrophil activation, superoxidase is produced to form reactive oxygen species (ROS) and release protease, both of which are involved in killing microbes (123). Neutrophilic granulocytes are also known to contain a large number of cytokines, which are released upon activation, and there is much evidence that cytokines have a damaging effect on pancreatic islets (124). And their infiltration results in the release of chemokines such as TNF-α and macrophage inflammatory protein 1α (MIP-1α) from T cells and macrophages. The mobilization of this immune effector may have effects on specific immune systems, inducing and enhancing cellular rejection (122). The massive infiltration of neutrophilic granulocytes not only causes functional impairment or reduces the mass of the implanted islets, but may also amplify the subsequent immune response, causing direct damage to the islets (125). Therefore, the development of drugs targeting neutrophil toxicity may markedly improve the outcome of intraportal islet transplantation.
Endothelial Cells
Pancreatic islets have an extensive capillary network, which—in addition to providing nutrients and oxygen to the islet endocrine cells and transporting hormones to the peripheral circulation—is an important source of signals for improving the survival rate and function of islet β cells (126). While islets only constitute 1% of the weight of the pancreas, they receive approximately 15% of the blood flow of the pancreas (127). In the process of the isolation, the islets are disrupted from the surrounding tissue and the capillary networks. Therefore, revascularization of the islets after transplantation is important for the function and survival of the islet graft. The endothelial cells retained in the islets after islet isolation are lost after the islets are cultured for 7 days. The isolated islets are considered to be an avascular tissue, and revascularization is carried out through the blood vessels that grow from the host organ to the islets (128). Angiogenesis begins on the first day after transplantation, and vascular remodeling may continue for up to 3 months (129). In comparison to cultured islets, transplantation of freshly isolated islets containing numerous endothelial cells may significantly improve the vascularization of transplanted islets, which in turn leads to an enhanced endocrine function and the survival of islet grafts (130). Olsson et al. observed that islet grafts obtained from freshly isolated islets have higher vascular density and oxygen tension, as well as higher ability to cure chemically induced diabetes, in comparison to islet grafts obtained from cultured islets (131).
Pancreatic islets and vascular endothelial cells secrete high levels of vascular endothelial growth factor (VEGF), which can recruit neovascularization (132). Cheng et al. used adenovirus containing cDNA from human VEGF isoforms to transfected islets and transplanted it into diabetic nude mice. It was found that the blood glucose was normal and that the revascularization of islets was improved (133). Johansson et al. demonstrated that the production of hepatocyte growth factor (HGF) in vitro by endothelial cells increased the proliferation of beta cells, which in turn required VEGF from beta cells (134). VEGF can also stimulate the release of interleukins and increase blood flow to ischemic tissues (135) (Figure 2). However, the supplementation of VEGF in islet grafts may have a negative impact, which recruits and amplifies inflammation, which may destroy islets (133). Previous in vitro and in vivo studies have shown that resident liver macrophages and endothelial cells can mediate early islet dysfunction by secreting cytokines and activating inducible iNOS (103).
Conclusion
Islet transplantation remains a promising treatment to improve the quality of life for many individuals with type 1 diabetes. If the restoration of normal glucose tolerance can be achieved, non-specific inflammation during transplantation can be reduced, and immune tolerance to islet tissue can be induced, it will be an ideal treatment for this disease. However, during the process of islet transplantation—including isolation, culture, and islet implantation—inflammation, ischemia, hypoxia, and immune responses will occur, resulting in the loss of the graft. The transplantation microenvironment plays an important role. Cytokine-mediated non-specific inflammation, immune cell-mediated rejection, and endothelial cells participate in post-transplant vascular remodeling, which directly and indirectly affect graft survival. Some cytokine inhibitors and siRNA targeting complement receptors have been shown to improve the viability of transplanted islets. Understanding the influence of the microenvironment on the survival of transplanted islets, as well as possible therapeutic targets is significant for the future of islet transplantation. The improvement of the microenvironment and the continuous progress of transplantation strategies will eventually improve the prognosis of transplant recipients.
Author Contributions
All authors contributed to the writing and editing of the manuscript and contributed to the article and approved the submitted version.
Funding
This work was supported in part by Medical Science and Technology Project of Zhejiang Province (2021PY083), Program of Taizhou Science and Technology Grant (20ywb29), Major Research Program of Taizhou Enze Medical Center Grant (19EZZDA2), Open Project Program of Key Laboratory of Minimally Invasive Techniques & Rapid Rehabilitation of Digestive System Tumor of Zhejiang Province (21SZDSYS01, 21SZDSYS09) and Key Technology Research and Development Program of Zhejiang Province (2019C03040).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Min BH, Shin JS, Kim JM, Kang SJ, Kim HJ, Yoon IH, et al. Delayed Revascularization of Islets After Transplantation by IL-6 Blockade in Pig to Non-Human Primate Islet Xenotransplantation Model. Xenotransplantation (2018) 25. doi: 10.1111/xen.12374
2. Shapiro A, Ricordi C, Hering B, Auchincloss H, Lindblad R, Robertson R, et al. International Trial of the Edmonton Protocol for Islet Transplantation. N Engl J Med (2006) 355:1318–30. doi: 10.1056/NEJMoa061267
3. Shapiro A, Lakey J, Ryan E, Korbutt G, Toth E, Warnock G, et al. Islet Transplantation in Seven Patients With Type 1 Diabetes Mellitus Using a Glucocorticoid-Free Immunosuppressive Regimen. N Engl J Med (2000) 343:230–8. doi: 10.1056/NEJM200007273430401
4. Yang Z, Li X, Zhang C, Sun N, Guo T, Lin J, et al. Amniotic Membrane Extract Protects Islets From Serum-Deprivation Induced Impairments and Improves Islet Transplantation Outcome. Front Endocrinol (Lausanne) (2020) 11:587450. doi: 10.3389/fendo.2020.587450
5. London NJ, Robertson DR, Chadwick PR, Johnson RF, James PR, Bell PR. Human Pancreatic Islet Isolation and Transplantation. Clin Transplant (1994) 85:421–59. doi: 10.1016/S0955-470X(10)80005-9
6. Brandhorst D, Brandhorst H, Layland S, Acreman S, Schenke-Layland K, Johnson PRV. Basement Membrane Proteins Improve Human Islet Survival in Hypoxia: Implications for Islet Inflammation. Acta Biomater (2021) 137:92–102. doi: 10.1016/j.actbio.2021.10.013
7. Ryan E, Paty B, Senior P, Bigam D, Alfadhli E, Kneteman N, et al. Five-Year Follow-Up After Clinical Islet Transplantation. Diabetes (2005) 54:2060–9. doi: 10.2337/diabetes.54.7.2060
8. Noguchi H. Regulation of C-Jun NH-Terminal Kinase for Islet Transplantation. J Clin Med (2019) 8:1763. doi: 10.3390/jcm8111763
9. Cayabyab F, Nih LR, Yoshihara E, 732431. Advances in Pancreatic Islet Transplantation Sites for the Treatment of Diabetes. Front Endocrinol (Lausanne) (2021) 12:732431. doi: 10.3389/fendo.2021.732431
10. Citro A, Cantarelli E, Piemonti L. Anti-Inflammatory Strategies to Enhance Islet Engraftment and Survival. Curr Diab Rep (2013) 13:733–44. doi: 10.1007/s11892-013-0401-0
11. Bottino R, Fernandez L, Ricordi C, Lehmann R, Tsan M, Oliver R, et al. Transplantation of Allogeneic Islets of Langerhans in the Rat Liver: Effects of Macrophage Depletion on Graft Survival and Microenvironment Activation. Diabetes (1998) 47:316–23. doi: 10.2337/diabetes.47.3.316
12. Song HJ, Xue WJ, Li Y, Tian XH, Ding XM, Feng XS, et al. Prolongation of Islet Graft Survival Using Concomitant Transplantation of Islets and Vascular Endothelial Cells in Diabetic Rats. Transplant Proc (2010) 42:2662–5. doi: 10.1016/j.transproceed.2010.06.003
13. Szempruch K, Banerjee O, McCall R, Desai CJI. Use of Anti-Inflammatory Agents in Clinical Islet Cell Transplants: A Qualitative Systematic Analysis. Islets (2019) 11:65–75. doi: 10.1080/19382014.2019.1601543
14. Gou W, Wang J, Song L, Kim DS, Cui W, Strange C, et al. Alpha-1 Antitrypsin Suppresses Macrophage Activation and Promotes Islet Graft Survival After Intrahepatic Islet Transplantation. Am J Transplant (2021) 21:1713–24. doi: 10.1111/ajt.16342
15. Scharp D, Lacy P, Santiago J, McCullough C, Weide L, Falqui L, et al. Insulin Independence After Islet Transplantation Into Type I Diabetic Patient. Diabetes (1990) 39:515–8. doi: 10.2337/diabetes.39.4.515
16. Bottino R, Knoll MF, Knoll CA, Bertera S, Trucco MM. The Future of Islet Transplantation Is Now. Front Med (Lausanne) (2018) 5:202. doi: 10.3389/fmed.2018.00202
17. Hering B, Clarke W, Bridges N, Eggerman T, Alejandro R, Bellin M, et al. Phase 3 Trial of Transplantation of Human Islets in Type 1 Diabetes Complicated by Severe Hypoglycemia. Diabetes Care (2016) 39:1230–40. doi: 10.2337/dc15-1988
18. Lablanche S, Vantyghem M, Kessler L, Wojtusciszyn A, Borot S, Thivolet C, et al. Islet Transplantation Versus Insulin Therapy in Patients With Type 1 Diabetes With Severe Hypoglycaemia or Poorly Controlled Glycaemia After Kidney Transplantation (TRIMECO): A Multicentre, Randomised Controlled Trial. Lancet Diabetes Endocrinol (2018) 6:527–37. doi: 10.1016/S2213-8587(18)30078-0
19. Infante M, Ricordi C, Padilla N, Alvarez A, Linetsky E, Lanzoni G, et al. The Role of Vitamin D and Omega-3 PUFAs in Islet Transplantation. Nutrients (2019) 11:2937. doi: 10.3390/nu11122937
20. Yamane K, Anazawa T, Tada S, Fujimoto N, Inoguchi K, Emoto N, et al. AndMitomycin C Treatment Improves Pancreatic Islet Graft Longevity in Intraportal Islet Transplantation by Suppressing Proinflammatory Response. Sci Rep (2020) 10:12086. doi: 10.1038/s41598-020-69009-8
21. Rickels MR, Robertson RP. Pancreatic Islet Transplantation in Humans: Recent Progress and Future Directions. Endocr Rev (2019) 40:631–68. doi: 10.1210/er.2018-00154
22. Razavi M, Zheng F, Telichko A, Wang J, Ren G, Dahl J, et al. Improving the Function and Engraftment of Transplanted Pancreatic Islets Using Pulsed Focused Ultrasound Therapy. Sci Rep (2019) 9. doi: 10.1038/s41598-019-49933-0
23. Bennet W, Groth C, Larsson R, Nilsson B, Korsgren O. Isolated Human Islets Trigger an Instant Blood Mediated Inflammatory Reaction: Implications for Intraportal Islet Transplantation as a Treatment for Patients With Type 1 Diabetes. Ups J Med Sci (2000) 105:125–33. doi: 10.1517/03009734000000059
24. Nilsson B, Ekdahl K, Korsgren O. Control of Instant Blood-Mediated Inflammatory Reaction to Improve Islets of Langerhans Engraftment. Curr Opin Organ Transplant (2011) 16:620–6. doi: 10.1097/MOT.0b013e32834c2393
25. Goto M, Johansson H, Maeda A, Elgue G, Korsgren O, Nilsson BJT. Low Molecular Weight Dextran Sulfate Prevents the Instant Blood-Mediated Inflammatory Reaction Induced by Adult Porcine Islets. Transplantation (2004) 77:741–7. doi: 10.1097/01.TP.0000114872.26990.4F
26. von Zur-Mühlen B, Lundgren T, Bayman L, Berne C, Bridges N, Eggerman T, et al. Open Randomized Multicenter Study to Evaluate Safety and Efficacy of Low Molecular Weight Sulfated Dextran in Islet Transplantation. Transplantation (2019) 103:630–7. doi: 10.1097/TP.0000000000002425
27. Takaki T, Shimoda M. And Medicine, Pancreatic Islet Transplantation: Toward Definitive Treatment for Diabetes Mellitus. Glob Health Med (2020) 2:200–11. doi: 10.35772/ghm.2020.01057
28. Cabric S, Sanchez J, Lundgren T, Foss A, Felldin M, Källen R, et al. Islet Surface Heparinization Prevents the Instant Blood-Mediated Inflammatory Reaction in Islet Transplantation. Diabetes (2007) 56:2008–15. doi: 10.2337/db07-0358
29. Contreras J, Eckstein C, Smyth C, Bilbao G, Vilatoba M, Ringland S, et al. Activated Protein C Preserves Functional Islet Mass After Intraportal Transplantation: A Novel Link Between Endothelial Cell Activation, Thrombosis, Inflammation, and Islet Cell Death. Diabetes (2004) 53:2804–14. doi: 10.2337/diabetes.53.11.2804
30. Koh A, Senior P, Salam A, Kin T, Imes S, Dinyari P, et al. Insulin-Heparin Infusions Peritransplant Substantially Improve Single-Donor Clinical Islet Transplant Success. Transplantation (2010) 89:465–71. doi: 10.1097/TP.0b013e3181c478fd
31. Johansson H, Goto M, Dufrane D, Siegbahn A, Elgue G, Gianello P, et al. Low Molecular Weight Dextran Sulfate: A Strong Candidate Drug to Block IBMIR in Clinical Islet Transplantation. Am J Transplant (2006) 6:305–12. doi: 10.1111/j.1600-6143.2005.01186.x
32. Yao M, Domogatskaya A, Ågren N, Watanabe M, Tokodai K, Brines M, et al. Cibinetide Protects Isolated Human Islets in a Stressful Environment and Improves Engraftment in the Perspective of Intra Portal Islet Transplantation. Cell Transplant (2021) 30. doi: 10.1177/09636897211039739
33. Wang J, Sun Z, Gou W, Adams D, Cui W, Morgan K, et al. α-1 Antitrypsin Enhances Islet Engraftment by Suppression of Instant Blood-Mediated Inflammatory Reaction. Diabetes (2017) 66:970–80. doi: 10.2337/db16-1036
34. Tjernberg J, Ekdahl KN, Lambris JD, Korsgren O, Nilsson B. Acute Antibody-Mediated Complement Activation Mediates Lysis of Pancreatic Islets Cells and may Cause Tissue Loss in Clinical Islet Transplantation. Transplantation (2008) 85:1193–9. doi: 10.1097/TP.0b013e31816b22f3
35. Tedesco F, Pausa M, Nardon E, Introna M, Mantovani A, Dobrina A. The Cytolytically Inactive Terminal Complement Complex Activates Endothelial Cells to Express Adhesion Molecules and Tissue Factor Procoagulant Activity. J Exp Med (1997) 185:1619–27. doi: 10.1084/jem.185.9.1619
36. Peng Q, Li K, Patel H, Sacks S, Zhou W. Dendritic Cell Synthesis of C3 Is Required for Full T Cell Activation and Development of a Th1 Phenotype. J Immunol (2006) 176:3330–41. doi: 10.4049/jimmunol.176.6.3330
37. Pratt J, Basheer S, Sacks S. Local Synthesis of Complement Component C3 Regulates Acute Renal Transplant Rejection. Nat Med (2002) 8:582–7. doi: 10.1038/nm0602-582
38. Brown K, Kondeatis E, Vaughan R, Kon S, Farmer C, Taylor J, et al. Influence of Donor C3 Allotype on Late Renal-Transplantation Outcome. N Engl J Med (2006) 354:2014–23. doi: 10.1056/NEJMoa052825
39. Barrault D, Steward M, Cox V, Smith R, Knight A. Efficient Production of Complement (C3d)3 Fusion Proteins Using the Baculovirus Expression Vector System. J Immunol Methods (2005) 304:158–73. doi: 10.1016/j.jim.2005.07.013
40. Li F, Mahato RI. RNA Interference for Improving the Outcome of Islet Transplantation. Adv Drug Deliv Rev (2011) 63:47–68. doi: 10.1016/j.addr.2010.11.003
41. Komatsu H, Kandeel F, Mullen YJP. Impact of Oxygen on Pancreatic Islet Survival. Pancreas (2018) 47:533–43. doi: 10.1097/MPA.0000000000001050
42. Garcia-Contreras M, Tamayo-Garcia A, Pappan K, Michelotti G, Stabler C, Ricordi C, et al. Metabolomics Study of the Effects of Inflammation, Hypoxia, and High Glucose on Isolated Human Pancreatic Islets. J Proteome Res (2017) 16:2294–306. doi: 10.1021/acs.jproteome.7b00160
43. Negi S, Jetha A, Aikin R, Hasilo C, Sladek R, Paraskevas S. Analysis of Beta-Cell Gene Expression Reveals Inflammatory Signaling and Evidence of Dedifferentiation Following Human Islet Isolation and Culture. PloS One (2012) 7:e30415. doi: 10.1371/journal.pone.0030415
44. Ma Z, Moruzzi N, Catrina S, Hals I, Oberholzer J, Grill V, et al. Preconditioning With Associated Blocking of Ca2+ Inflow Alleviates Hypoxia-Induced Damage to Pancreatic β-Cells. PLoS ONE (2013) 8:e67498. doi: 10.1371/journal.pone.0067498
45. Scott W, O’Brien T, Ferrer-Fabrega J, Avgoustiniatos E, Weegman B, Anazawa T, et al. Persufflation Improves Pancreas Preservation When Compared With the Two-Layer Method. Transplant Proc (2010) 42:2016–9. doi: 10.1016/j.transproceed.2010.05.092
46. Reddy M, Carter N, Cunningham A, Shaw J, Talbot D. Portal Venous Oxygen Persufflation of the Donation After Cardiac Death Pancreas in a Rat Model Is Superior to Static Cold Storage and Hypothermic Machine Perfusion. Transpl Int (2014) 27:634–9. doi: 10.1111/tri.12313
47. Brandhorst H, Iken M, Scott W, Papas K, Theisinger B, Johnson P, et al. Quality of Isolated Pig Islets Is Improved Using Perfluorohexyloctane for Pancreas Storage in a Split Lobe Model. Cell Transplant (2013) 22:1477–83. doi: 10.3727/096368912X657639
48. Brandhorst H, Asif S, Andersson K, Theisinger B, Andersson H, Felldin M, et al. A New Oxygen Carrier for Improved Long-Term Storage of Human Pancreata Before Islet Isolation. Transplantation (2010) 89:155–60. doi: 10.1097/TP.0b013e3181c9266c
49. Rodriguez-Brotons A, Bietiger W, Peronet C, Magisson J, Sookhareea C, Langlois A, et al. Impact of Pancreatic Rat Islet Density on Cell Survival During Hypoxia. J Diabetes Res (2016) 2016:3615286. doi: 10.1155/2016/3615286
50. Bloch K, Papismedov E, Yavriyants K, Vorobeychik M, Beer S, Vardi P. Photosynthetic Oxygen Generator for Bioartificial Pancreas. Tissue Eng (2006) 12:337–44. doi: 10.1089/ten.2006.12.337
51. Montazeri L, Hojjati-Emami S, Bonakdar S, Tahamtani Y, Hajizadeh-Saffar E, Noori-Keshtkar M, et al. Improvement of Islet Engrafts by Enhanced Angiogenesis and Microparticle-Mediated Oxygenation. Biomaterials (2016) 89:157–65. doi: 10.1016/j.biomaterials.2016.02.043
52. Wang Y, Wang S, Harvat T, Kinzer K, Zhang L, Feng F, et al. Diazoxide, a K(ATP) Channel Opener, Prevents Ischemia-Reperfusion Injury in Rodent Pancreatic Islets. Cell Transplant (2015) 24:25–36. doi: 10.3727/096368913X673441
53. Han J, Oh J, Ihm S, Lee M. Peptide Micelle-Mediated Curcumin Delivery for Protection of Islet β-Cells Under Hypoxia. J Drug Target (2016) 24:618–23. doi: 10.3109/1061186X.2015.1132220
54. Li Z, Shangguan Z, Liu Y, Wang J, Li X, Yang S, et al. Puerarin Protects Pancreatic β-Cell Survival via PI3K/Akt Signaling Pathway. J Mol Endocrinol (2014) 53:71–9. doi: 10.1530/JME-13-0302
55. Kawakami Y, Iwata H, Gu Y, Miyamoto M, Murakami Y, Yamasaki T, et al. Modified Subcutaneous Tissue With Neovascularization Is Useful as the Site for Pancreatic Islet Transplantation. Cell Transplant (2000) 9:729–32. doi: 10.1177/096368970000900523
56. Kawakami Y, Iwata H, Gu Y, Miyamoto M, Murakami Y, Balamurugan A, et al. Successful Subcutaneous Pancreatic Islet Transplantation Using an Angiogenic Growth Factor-Releasing Device. Pancreas (2001) 23:375–81. doi: 10.1097/00006676-200111000-00007
57. Halberstadt C, Williams D, Emerich D, Goddard M, Vasconcellos A, Curry W, et al. Subcutaneous Transplantation of Islets Into Streptozocin-Induced Diabetic Rats. Cell Transplant (2005) 14:595–605. doi: 10.3727/000000005783982792
58. Pileggi A, Molano R, Ricordi C, Zahr E, Collins J, Valdes R, et al. Reversal of Diabetes by Pancreatic Islet Transplantation Into a Subcutaneous, Neovascularized Device. Transplantation (2006) 81:1318–24. doi: 10.1097/01.tp.0000203858.41105.88
59. Pathak S, Regmi S, Gupta B, Pham T, Yong C, Kim J, et al. Engineered Islet Cell Clusters Transplanted Into Subcutaneous Space Are Superior to Pancreatic Islets in Diabetes. FASEB J (2017) 31:5111–21. doi: 10.1096/fj.201700490R
60. Vallabhajosyula P, Hirakata A, Shimizu A, Okumi M, Tchipashvili V, Hong H, et al. Assessing the Effect of Immunosuppression on Engraftment of Pancreatic Islets. Transplantation (2013) 96:372–8. doi: 10.1097/TP.0b013e31829f7515
61. Shapiro A, Geng Hao E, Lakey J, Finegood D, Rajotte R, Kneteman NJT. Defining Optimal Immunosuppression for Islet Transplantation Based on Reduced Diabetogenicity in Canine Islet Autografts. Transplantation (2002) 74:1522–8. doi: 10.1097/00007890-200212150-00008
62. Bhatt S, Fung JJ, Lu L, Qian S. Tolerance-Inducing Strategies in Islet Transplantation. Int J Endocrinol (2012) 2012:396524. doi: 10.1155/2012/396524
63. van Doesum W, Gard L, Bemelman F, de Fijter J, Homan van der Heide J, Niesters H, et al. Incidence and Outcome of BK Polyomavirus Infection in a Multicenter Randomized Controlled Trial With Renal Transplant Patients Receiving Cyclosporine-, Mycophenolate Sodium-, or Everolimus-Based Low-Dose Immunosuppressive Therapy. Transpl Infect Dis (2017) 19:e12687. doi: 10.1111/tid.12687
64. Domhan S, Zeier M, Abdollahi AJN. Dialysis, Transplantation: Official Publication of the European Dialysis and T.A.-E.R. Association, Immunosuppressive Therapy and Post-Transplant Malignancy. Nephrol Dial Transplant (2009) 24:1097–103. doi: 10.1093/ndt/gfn605
65. Mizrahi N, Braun M, Ben Gal T, Rosengarten D, Kramer M, Grossman AJE. Post-Transplant Diabetes Mellitus: Incidence, Predicting Factors and Outcomes. Endocrine (2020) 69:303–9. doi: 10.1007/s12020-020-02339-9
66. Rother K, Harlan D. Challenges Facing Islet Transplantation for the Treatment of Type 1 Diabetes Mellitus. J Clin Invest (2004) 114:877–83. doi: 10.1172/JCI200423235
67. Lee SJ, Kim HJ, Byun NR, Park CG. Donor-Specific Regulatory T Cell-Mediated Immune Tolerance in an Intrahepatic Murine Allogeneic Islet Transplantation Model With Short-Term Anti-CD154 mAb Single Treatment. Cell Transplant (2020) 29:963689720913876. doi: 10.1177/0963689720913876
68. Anderton S, Burkhart C, Metzler B, D.J.I.r W. Mechanisms of Central and Peripheral T-Cell Tolerance: Lessons From Experimental Models of Multiple Sclerosis. Immunol Rev (1999) 169:123–37. doi: 10.1111/j.1600-065X.1999.tb01311.x
69. Sato N, Marubashi S. Induction of Immune Tolerance in Islet Transplantation Using Apoptotic Donor Leukocytes. J Clin Med (2021) 10:5306. doi: 10.3390/jcm10225306
70. Singh A, Ramachandran S, Graham ML, Daneshmandi S, Heller D, Suarez-Pinzon WL, et al. Long-Term Tolerance of Islet Allografts in Nonhuman Primates Induced by Apoptotic Donor Leukocytes. Nat Commun (2019) 10:3495. doi: 10.1038/s41467-019-11338-y
71. Oluwole O, Depaz H, Gopinathan R, Ali A, Garrovillo M, Jin M, et al. Indirect Allorecognition in Acquired Thymic Tolerance: Induction of Donor-Specific Permanent Acceptance of Rat Islets by Adoptive Transfer of Allopeptide-Pulsed Host Myeloid and Thymic Dendritic Cells. Diabetes (2001) 50:1546–52. doi: 10.2337/diabetes.50.7.1546
72. Britt L, Scharp D, Lacy P, Slavin SJD. Transplantation of Islet Cells Across Major Histocompatibility Barriers After Total Lymphoid Irradiation and Infusion of Allogeneic Bone Marrow Cells. Diabetes (1982) 31:63–8. doi: 10.2337/diab.31.4.S63
73. Liang Y, Huang T, Zhang C, Todorov I, Atkinson M, Kandeel F, et al. Donor CD8+ T Cells Facilitate Induction of Chimerism and Tolerance Without GVHD in Autoimmune NOD Mice Conditioned With Anti-CD3 mAb. Blood (2005) 105:2180–8. doi: 10.1182/blood-2004-06-2411
74. Préville X, Flacher M, LeMauff B, Beauchard S, Davelu P, Tiollier J, et al. Mechanisms Involved in Antithymocyte Globulin Immunosuppressive Activity in a Nonhuman Primate Model. Transplantation (2001) 71:460–8. doi: 10.1097/00007890-200102150-00021
75. Friedman E, Beyer MJN. Effect of Antithymocyte Globulin on Islet of Langerhans Transplantation. Nephron (1978) 22:212–6. doi: 10.1159/000181451
76. Barshes NR, Wyllie S, Goss JA. Inflammation-Mediated Dysfunction and Apoptosis in Pancreatic Islet Transplantation: Implications for Intrahepatic Grafts. J Leukoc Biol (2005) 77:587–97. doi: 10.1189/jlb.1104649
77. Chen C, Rong P, Yang M, Ma X, Feng Z, Wang W. The Role of Interleukin-1beta in Destruction of Transplanted Islets. Cell Transplant (2020) 29:963689720934413. doi: 10.1177/0963689720934413
78. Kanak MA, Takita M, Kunnathodi F, Lawrence MC, Levy MF, Naziruddin B. Inflammatory Response in Islet Transplantation. Int J Endocrinol (2014) 2014:451035. doi: 10.1155/2014/451035
79. Thomas H, McKenzie M, Angstetra E, Campbell P, Kay T. Beta Cell Apoptosis in Diabetes. Apoptosis (2009) 14:1389–404. doi: 10.1007/s10495-009-0339-5
80. Nano E, Petropavlovskaia M, Rosenberg L. Islet Neogenesis Associated Protein (INGAP) Protects Pancreatic β Cells From IL-1β and Ifnγ-Induced Apoptosis. Cell Death Discov (2021) 7:56. doi: 10.1038/s41420-021-00441-z
81. Carlessi R, Lemos NE, Dias AL, Oliveira FS, Brondani LA, Canani LH, et al. Exendin-4 Protects Rat Islets Against Loss of Viability and Function Induced by Brain Death. Mol Cell Endocrinol (2015) 412:239–50. doi: 10.1016/j.mce.2015.05.009
82. Shinichiro O, Kuroki T, Adachi T, Kosaka T, Okamoto T, Tanaka T, et al. The Effect of Selective Neutrophil Elastase Inhibitor on Pancreatic Islet Yields and Functions in Rat With Hypercytokinemia. Ann Transplant (2011) 16:99–106. doi: 10.12659/AOT.882225
83. Cross SE, Vaughan RH, Willcox AJ, McBride AJ, Abraham AA, Han B, et al. Key Matrix Proteins Within the Pancreatic Islet Basement Membrane Are Differentially Digested During Human Islet Isolation. Am J Transplant (2017) 17:451–61. doi: 10.1111/ajt.13975
84. Parnaud G, Hammar E, Rouiller DG, Armanet M, Halban PA, Bosco D. Blockade of Beta1 Integrin-Laminin-5 Interaction Affects Spreading and Insulin Secretion of Rat Beta-Cells Attached on Extracellular Matrix. Diabetes (2006) 55:1413–20. doi: 10.2337/db05-1388
85. Cowley M, Walters S, Zammit N, Hawthorne W, Gunton J, Chapman J, et al. Human Islets Express a Marked Pro-Inflammatory Molecular Signature Prior to Transplantation. Transplant J (2012) 94:162. doi: 10.1097/00007890-201211271-00300
86. Johansson U, Olsson A, Gabrielsson S, Nilsson B, Korsgren O. Inflammatory Mediators Expressed in Human Islets of Langerhans: Implications for Islet Transplantation. Biochem Biophys Res Commun (2003) 308:474–9. doi: 10.1016/S0006-291X(03)01392-5
87. Ehrnfelt C, Kumagai-Braesch M, Uzunel M, Holgersson JJX. Adult Porcine Islets Produce MCP-1 and Recruit Human Monocytes In Vitro. Xenotransplantation (2004) 11:184–94. doi: 10.1046/j.1399-3089.2003.00104.x
88. Sahraoui A, Kloster-Jensen K, Ueland T, Korsgren O, Foss A, Scholz H. Anakinra and Tocilizumab Enhance Survival and Function of Human Islets During Culture: Implications for Clinical Islet Transplantation. Cell Transplant (2014) 23:1199–211. doi: 10.3727/096368913X667529
89. Matsuoka T, Yoshimatsu G, Sakata N, Kawakami R, Tanaka T, Yamada T, et al. Inhibition of NLRP3 Inflammasome by MCC950 Improves the Metabolic Outcome of Islet Transplantation by Suppressing IL-1beta and Islet Cellular Death. Sci Rep (2020) 10:17920. doi: 10.1038/s41598-020-74786-3
90. Montolio M, Biarnes M, Tellez N, Escoriza J, Soler J, Montanya E. Interleukin-1beta and Inducible Form of Nitric Oxide Synthase Expression in Early Syngeneic Islet Transplantation. J Endocrinol (2007) 192:169–77. doi: 10.1677/joe.1.06968
91. Arita S, Nagai T, Ochiai M, Sakamoto Y, Shevlin L, Smith C, et al. Prevention of Primary Nonfunction of Canine Islet Autografts by Treatment With Pravastatin. Transplantation (2002) 73:7–12. doi: 10.1097/00007890-200201150-00003
92. Biarnés M, Montolio M, Nacher V, Raurell M, Soler J, Montanya EJD. Beta-Cell Death and Mass in Syngeneically Transplanted Islets Exposed to Short- and Long-Term Hyperglycemia. Diabetes (2002) 51:66–72. doi: 10.2337/diabetes.51.1.66
93. Téllez N, Montolio M, Estil-les E, Escoriza J, Soler J, Montanya EJD. Adenoviral Overproduction of Interleukin-1 Receptor Antagonist Increases Beta Cell Replication and Mass in Syngeneically Transplanted Islets, and Improves Metabolic Outcome. Diabetologia (2007) 50:602–11. doi: 10.1007/s00125-006-0548-1
94. Chung WY, Pollard CA, Kumar R, Drogemuller CJ, Naziruddin B, Stover C, et al. A Comparison of the Inflammatory Response Following Autologous Compared With Allogenic Islet Cell Transplantation. Ann Transl Med (2021) 9:98. doi: 10.21037/atm-20-3519
95. Onaca N, Takita M, Levy MF, Naziruddin B. Anti-Inflammatory Approach With Early Double Cytokine Blockade (IL-1beta and TNF-Alpha) Is Safe and Facilitates Engraftment in Islet Allotransplantation. Transplant Direct (2020) 6:e530. doi: 10.1097/TXD.0000000000000977
96. Tau G, Rothman PJA. Biologic Functions of the IFN-Gamma Receptors. Allergy (1999) 54:1233–51. doi: 10.1034/j.1398-9995.1999.00099.x
97. Ozasa T, Newton M, Dallman M, Shimizu S, Gray D, Morris PJT. Cytokine Gene Expression in Pancreatic Islet Grafts in the Rat. Transplantation (1997) 64:1152–9. doi: 10.1097/00007890-199710270-00013
98. Eizirik D, Mandrup-Poulsen TJD. A Choice of Death–the Signal-Transduction of Immune-Mediated Beta-Cell Apoptosis. Diabetologia (2001) 44:2115–33. doi: 10.1007/s001250100021
99. Saldeen J. Cytokines Induce Both Necrosis and Apoptosis via a Common Bcl-2-Inhibitable Pathway in Rat Insulin-Producing Cells*. Endocrinology (2000) 141:2003–10. doi: 10.1210/endo.141.6.7523
100. Kwon K, Kim E, Jeong E, Lee Y, Lee Y, Park J, et al. Cortex Cinnamomi Extract Prevents Streptozotocin- and Cytokine-Induced Beta-Cell Damage by Inhibiting NF-Kappab. World J Gastroenterol (2006) 12:4331–7. doi: 10.3748/wjg.v12.i27.4331
101. Li B, Liu Y, Cheng Y, Zhang K, Li T, Zhao N. Protective Effect of Inducible Nitric Oxide Synthase Inhibitor on Pancreas Transplantation in Rats. World J Gastroenterol (2007) 13:6066–71. doi: 10.3748/wjg.v13.45.6066
102. Oleson BJ, Corbett JA. Dual Role of Nitric Oxide in Regulating the Response of Beta Cells to DNA Damage. Antioxid Redox Signal (2018) 29:1432–45. doi: 10.1089/ars.2017.7351
103. Brandhorst D, Brandhorst H, Zwolinski A, Nahidi F, Bretzel RJT. Prevention of Early Islet Graft Failure by Selective Inducible Nitric Oxide Synthase Inhibitors After Pig to Nude Rat Intraportal Islet Transplantation. Transplantation (2001) 71:179–84. doi: 10.1097/00007890-200101270-00002
104. Li Y, Ding X, Tian X, Zheng J, Ding C, Li X, et al. Islet Transplantation Modulates Macrophage to Induce Immune Tolerance and Angiogenesis of Islet Tissue in Type I Diabetes Mice Model. Aging (Albany NY) (2020) 12:24023–32. doi: 10.18632/aging.104085
105. Gordon S, Plüddemann A, Martinez Estrada F. Macrophage Heterogeneity in Tissues: Phenotypic Diversity and Functions. Immunol Rev (2014) 262:36–55. doi: 10.1111/imr.12223
106. Decker K. Biologically Active Products of Stimulated Liver Macrophages (Kupffer Cells). Eur J Biochem (1990) 192:245–61. doi: 10.1111/j.1432-1033.1990.tb19222.x
107. Nussler A, Carroll PB, Silvio M, Rilo H, Simmons R, Starzl TE, et al. Hepatic Nitric Oxide Generation as a Putative Mechanism for Failure of Intrahepatic Cell Grafts. Transplant Proc (1992) 24:2297.
108. Deaciuc I, Bagby G, Niesman M, Skrepnik N, Spitzer JJH. Modulation of Hepatic Sinusoidal Endothelial Cell Function by Kupffer Cells: An Example of Intercellular Communication in the Liver. Hepatology (1994) 19:464–70. doi: 10.1002/hep.1840190227
109. Colletti L, Remick D, Burtch G, Kunkel S, Strieter R, Campbell D. Role of Tumor Necrosis Factor-Alpha in the Pathophysiologic Alterations After Hepatic Ischemia/Reperfusion Injury in the Rat. J Clin Invest (1990) 85:1936–43. doi: 10.1172/JCI114656
110. Vargas F, Vives-Pi M, Somoza N, Armengol P, Alcalde L, Martí M, et al. Endotoxin Contamination may be Responsible for the Unexplained Failure of Human Pancreatic Islet Transplantation. Transplantation (1998) 65:722–7. doi: 10.1097/00007890-199803150-00020
111. de Groot M, Schuurs T, Keizer P, Fekken S, Leuvenink H, Van Schilfgaarde R. Response of Encapsulated Rat Pancreatic Islets to Hypoxia. Cell Transplant (2003) 12:867–75. doi: 10.3727/000000003771000219
112. Wang J, Gou W, Kim D, Strange C, Wang HJT. Clathrin-Mediated Endocytosis of Alpha-1 Antitrypsin Is Essential for Its Protective Function in Islet Cell Survival. Theranostics (2019) 9:3940–51. doi: 10.7150/thno.31647
113. Chappell A, Lujan-Hernandez J, Perry D, Corvera S, Lalikos JJP. Alternatively Activated M2 Macrophages Improve Autologous Fat Graft Survival in a Mouse Model Through Induction of Angiogenesis. Plas Reconstr Surg (2015) 136:277e. doi: 10.1097/PRS.0000000000001435
114. Sibley R, Sutherland D, Goetz F, Michael A. Recurrent Diabetes Mellitus in the Pancreas Iso- and Allograft. A Light and Electron Microscopic and Immunohistochemical Analysis of Four Cases. Lab Inves (1985) 53:132–44.
115. Santamaria P, Nakhleh R, Sutherland D, Barbosa JJD. Characterization of T Lymphocytes Infiltrating Human Pancreas Allograft Affected by Isletitis and Recurrent Diabetes. Diabetes (1992) 41:53–61. doi: 10.2337/diabetes.41.1.53
116. Mora C, Wong F, Chang C, Flavell R. Pancreatic Infiltration But Not Diabetes Occurs in the Relative Absence of MHC Class II-Restricted CD4 T Cells: Studies Using NOD/CIITA-Deficient Mice. J Immunol (1999) 162:4576–88.
117. Lin C, Gill R. Direct and Indirect Allograft Recognition: Pathways Dictating Graft Rejection Mechanisms. Curr Opin Organ Tranplant (2016) 21:40–4. doi: 10.1097/MOT.0000000000000263
118. Barra JM, Tse HM. Redox-Dependent Inflammation in Islet Transplantation Rejection. Front Endocrinol (Lausanne) (2018) 9:175. doi: 10.3389/fendo.2018.00175
119. Yoshimura S, Bondeson J, Foxwell B, Brennan F, Feldmann M. Effective Antigen Presentation by Dendritic Cells Is NF-KappaB Dependent: Coordinate Regulation of MHC, Co-Stimulatory Molecules and Cytokines. Int Immunol (2001) 13:675–83. doi: 10.1093/intimm/13.5.675
120. Li M, Zhang X, Zheng X, Lian D, Zhang Z, Ge W, et al. Immune Modulation and Tolerance Induction by RelB-Silenced Dendritic Cells Through RNA Interference. J Immunol (2007) 178:5480–7. doi: 10.4049/jimmunol.178.9.5480
121. Liang X, Lu L, Chen Z, Vickers T, Zhang H, Fung J, et al. Administration of Dendritic Cells Transduced With Antisense Oligodeoxyribonucleotides Targeting CD80 or CD86 Prolongs Allograft Survival. Transplantation (2003) 76:721–9. doi: 10.1097/01.TP.0000076470.35404.49
122. Moberg L, Korsgren O, Nilsson BJC. Neutrophilic Granulocytes Are the Predominant Cell Type Infiltrating Pancreatic Islets in Contact With ABO-Compatible Blood. Clin Exp Immunol (2005) 142:125–31. doi: 10.1111/j.1365-2249.2005.02883.x
123. Lipton P. Ischemic Cell Death in Brain Neurons. Physiol Rev (1999) 79:1431–568. doi: 10.1152/physrev.1999.79.4.1431
124. Andersson A, Flodström M, Sandler SJB. Cytokine-Induced Inhibition of Insulin Release From Mouse Pancreatic Beta-Cells Deficient in Inducible Nitric Oxide Synthase. Biochem Biophys Res Commun (2001) 281:396–403. doi: 10.1006/bbrc.2001.4361
125. Badet L, Titus T, Metzen E, Handa A, McShane P, Chang L, et al. The Interaction Between Primate Blood and Mouse Islets Induces Accelerated Clotting With Islet Destruction. Xenotransplantation (2002) 9:91–6. doi: 10.1034/j.1399-3089.2002.1o040.x
126. Hogan MF, Hull RL. The Islet Endothelial Cell: A Novel Contributor to Beta Cell Secretory Dysfunction in Diabetes. Diabetologia (2017) 60:952–9. doi: 10.1007/s00125-017-4272-9
127. Jansson L, Hellerström CJD. Stimulation by Glucose of the Blood Flow to the Pancreatic Islets of the Rat. Diabetologia (1983) 25:45–50. doi: 10.1007/BF00251896
128. Nyqvist D, Köhler M, Wahlstedt H, Berggren PJD. Donor Islet Endothelial Cells Participate in Formation of Functional Vessels Within Pancreatic Islet Grafts. Diabetes (2005) 54:2287–93. doi: 10.2337/diabetes.54.8.2287
129. Emamaullee J, Shapiro A. Factors Influencing the Loss of β-Cell Mass in Islet Transplantation. Cell Transplant (2007) 16:1–8. doi: 10.3727/000000007783464461
130. Brissova M, Fowler M, Wiebe P, Shostak A, Shiota M, Radhika A, et al. Intraislet Endothelial Cells Contribute to Revascularization of Transplanted Pancreatic Islets. Diabetes (2004) 53:1318–25. doi: 10.2337/diabetes.53.5.1318
131. Olsson R, Carlsson PJD. Better Vascular Engraftment and Function in Pancreatic Islets Transplanted Without Prior Culture. Diabetologia (2005) 48:469–76. doi: 10.1007/s00125-004-1650-x
132. Figliuzzi M, Bonandrini B, Silvani S, Remuzzi A. Mesenchymal Stem Cells Help Pancreatic Islet Transplantation to Control Type 1 Diabetes. World J Stem Cells (2014) 6:163–72. doi: 10.4252/wjsc.v6.i2.163
133. Cheng K, Fraga D, Zhang C, Kotb M, Gaber A, Guntaka R, et al. Adenovirus-Based Vascular Endothelial Growth Factor Gene Delivery to Human Pancreatic Islets. Gene Ther (2004) 11:1105–16. doi: 10.1038/sj.gt.3302267
134. Johansson M, Mattsson G, Andersson A, Jansson L, Carlsson PO. Islet Endothelial Cells and Pancreatic Beta-Cell Proliferation: Studies In Vitro and During Pregnancy in Adult Rats. Endocrinology (2006) 147:2315–24. doi: 10.1210/en.2005-0997
Keywords: islet transplantation, microenvironment, instant blood, mediated inflammatory reaction, inflammatory cytokine, therapeutic target
Citation: Yan L-l, Ye L-p, Chen Y-h, He S-q, Zhang C-y, Mao X-l and Li S-w (2022) The Influence of Microenvironment on Survival of Intraportal Transplanted Islets. Front. Immunol. 13:849580. doi: 10.3389/fimmu.2022.849580
Received: 06 January 2022; Accepted: 03 March 2022;
Published: 28 March 2022.
Edited by:
Lisha Mou, Shenzhen Second People’s Hospital, ChinaReviewed by:
Pradeep Shrestha, University of Texas MD Anderson Cancer Center, United StatesShiva Pathak, Stanford University, United States
Copyright © 2022 Yan, Ye, Chen, He, Zhang, Mao and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin-li Mao, bWFveGxAZW56ZW1lZC5jb20=; Shao-wei Li, bGlfc2hhb3dlaTgxQGhvdG1haWwuY29t
†These authors contributed equally to this work