 John A. Ligon
John A. Ligon Kristin M. Wessel
Kristin M. Wessel Nirali N. Shah
Nirali N. Shah John Glod
John Glod
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 22 February 2022
Sec. T Cell Biology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.846346
This article is part of the Research TopicNew Frontiers in Gene-Modified T Cell TechnologyView all 16 articles
Advances from novel adoptive cellular therapies have yet to be fully realized for the treatment of children and young adults with solid tumors. This review discusses the strategies and preliminary results, including T-cell, NK-cell and myeloid cell-based therapies. While each of these approaches have shown some early promise, there remain challenges. These include poor trafficking to the tumor as well as a hostile tumor microenvironment with numerous immunosuppressive mechanisms which result in exhaustion of cellular therapies. We then turn our attention to new strategies proposed to address these challenges including novel clinical trials that are ongoing and in development.
Immunologically “hot” solid tumors (e.g. melanoma) (1) with a tumor microenvironment (TME) marked by infiltrating CD8+ T-cells (2, 3), high programmed death ligand 1 (PD-L1) expression (4), or a high tumor mutational burden have shown remarkable responses to immunotherapy including immune checkpoint inhibitors (ICIs) (5). Unfortunately, these benefits have not extended to “cold” tumors (e.g. prostate or pancreatic cancer) (1) where T-cells are either entirely absent (“immune desert”) or sequestered at the periphery (“immune-excluded”) (3, 6). Many pediatric/adolescent and young adult solid tumors are cold tumors (7, 8) and have failed to respond to ICIs (9).
Several approaches have attempted to harness cellular therapy to cure these tumors. Autologous hematopoietic stem cell transplant (HSCT) has enabled maximal chemotherapy dosing in susceptible tumors with varying levels of effectiveness in neuroblastoma (10), Ewing sarcoma (11), breast cancer (12), retinoblastoma (13), hepatoblastoma (14), and other diseases. Recently some groups have piloted allogeneic HSCT to treat solid tumors. Though durable responses are rare, evidence for graft-vs-tumor effect has been observed (15). Finally, as adoptive cellular therapy (ACT) has proven transformative for leukemia and lymphoma, the development of novel ACT for solid tumors has exploded (Figure 1). In this review, we discuss ACT in solid tumors in clinical development, consider challenges plaguing the field, and highlight proposed strategies which will be tested in future clinical trials.
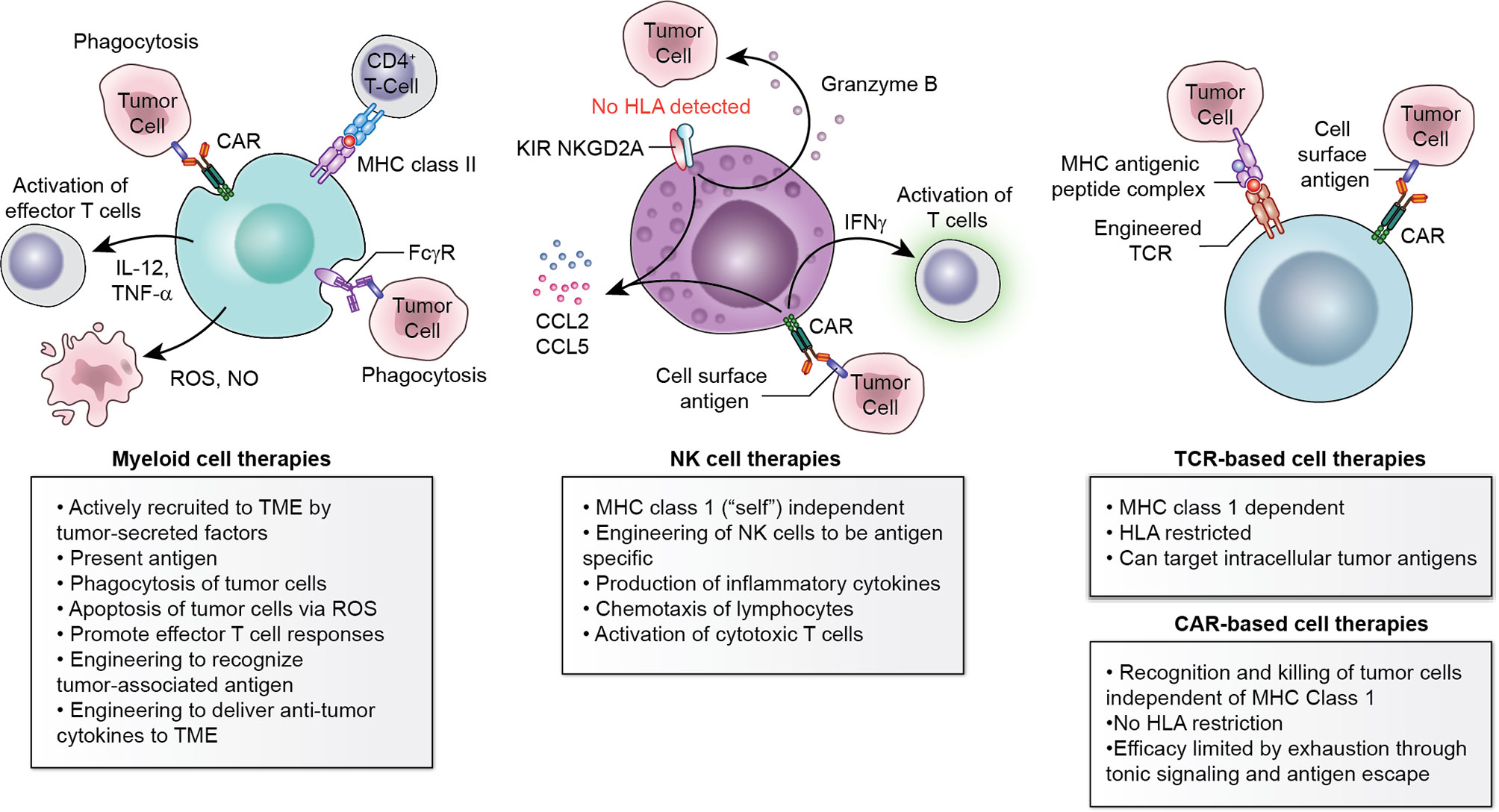
Figure 1 Multiple cell types available to engineer for adoptive cellular therapy. Myeloid cells, NK cells, and T-cell-based therapies each have advantages and disadvantages which should be considered within the context of the histology to be targeted.
T-cells are critical in immune surveillance for cancer. The T-cell receptor (TCR) can recognize cancer-specific antigens processed by major histocompatibility complex (MHC) and presented on the cell surface. TCR engagement by MHC-presented non-self antigens leads to activation and T-cell mediated killing (16). T-cell cytotoxicity in solid tumors has been leveraged using both native T-cells and autologous T-cells genetically engineered to express a specific TCR. Future efforts in allogeneic “off-the-shelf” approaches are being actively studied.
Early studies demonstrated that heterogeneous tumor infiltrating lymphocytes (TILs) collected from a freshly-resected tumor and expanded in vitro were able to specifically lyse autologous tumor (17). Subsequent clinical investigations showed transient responses in patients with metastatic melanoma after TIL infusion, typically under high IL-2 conditions (17). Early TIL trials reported responses in 49-72% of patients with melanoma (18, 19). Pretreatment with lymphodepleting chemotherapy led to improved TIL persistence (18) and recent advances include selection of TILs that recognize patient-specific tumor antigens using single cell sequencing (20). Selected autologous TILs have shown activity in several epithelial malignancies (21, 22).
While advances using TILs continue, the inability to isolate and effectively expand TILs from some solid tumors remains a challenge.
Initial attempts at engineering T-cells for ACT concentrated on genetic engineering of specific TCRs into autologous T-cells collected via peripheral blood apheresis with subsequent reinfusion.
Expression of cancer/testis antigens (CTAs) including melanoma antigen gene (MAGE) family proteins, synovial sarcoma X breakpoint (SSX) family proteins, and New York esophageal squamous cell carcinoma (NY-ESO-1) is normally restricted to the germline. However, solid tumors including melanoma, SS, myxoid/round cell liposarcoma (MRCL), and osteosarcoma express CTAs. Robbins and colleagues targeted NY-ESO-1 using a transduced TCR recognizing the peptide epitope SLLMWITQC in the context of HLA-A*02. Transgenic T-cells combined with IL-2 following lymphodepletion led to responses in 5/11 patients with SS and 2/11 patients with melanoma (23). A subsequent study showed responses in 6/12 patients with NY-ESO-1+ SS in an initial cohort (24), with one complete response (CR) and 14 partial responses (PR) in the first 42 patients (25). This response rate represents a potentially significant improvement over previous therapies for SS (26). These T-cells maintained clonal diversity over time and persisting cells were primarily of central memory and stem cell memory populations (24). Ongoing trials are further investigating genetically engineered NY-ESO-1 targeting autologous T-cells in solid tumors including SS, MRCL and non-small cell lung cancer (NCT02992743, NCT03967223, NCT03709706).
Other successfully targeted CTAs include MAGE-A3 and MAGE-A4. Seventeen patients were treated in a dose escalation study of autologous T-cells genetically modified to express an MHC class II-restricted TCR recognizing MAGE-A3 combined with IL-2 (27). One patient with cervical cancer had a CR and several PRs were observed in patients with esophageal cancer, urothelial cancer, and osteosarcoma. Despite encouraging responses, the significant neurotoxicity observed in this and a subsequent trial targeting MAGE-A3 has hampered development of this strategy (28). In a phase I trial of a TCR developed in a transgenic murine model and recognizing residues 112-120 (KVAELVHFL) of MAGE-A3, 3/9 patients developed significant neurotoxicity (29). Preliminary data for the SURPASS trial utilizing autologous T-cells transduced with a MAGE-A4 TCR and CD8a co-receptor reported 2/5 patients with PRs (30). A MAGE-A4 targeting TCR is also being evaluated in a phase II study for patients with SS and MRCL (NCT04044768). Additional TCR-based strategies targeting CTAs are in development (31).
Viral antigens have also been successfully targeted for treating solid tumors using ACT. TILs targeting human papillomavirus (HPV) antigens E6 and E7 have shown efficacy in early phase clinical trials in HPV-associated carcinomas with responses in 5/18 patients with cervical cancer and 2/11 with head and neck cancer (32). Subsequent work identified TCRs recognizing epitopes of HPV16 E6 and E7 in the context of HLA-A*02:01 and T-cells genetically engineered to express these TCRs led to responses in two early phase studies (33, 34). Epstein-Barr virus (EBV) is associated with several solid tumors (e.g., nasopharyngeal carcinoma [NPC] and post-transplant lymphoproliferative disorder [PTLD]). EBV-specific cytotoxic T-lymphocytes (CTLs) were tested to treat PTLD following HSCT (35). EBV-specific CTLs resulted in PR for 2/10 patients with EBV-associated NPC (36). A phase 3 trial comparing chemotherapy with EBV-specific CTLs combined with chemotherapy for NPC is underway (NCT02578641). TCR-based therapy has also been explored for Merkel cell carcinoma, a skin cancer associated with Merkel cell polyomavirus (37). Autologous T-cells with TCRs recognizing an epitope of Merkel cell polyomavirus, large T antigen and small T antigen, led to durable regression of metastatic lesions in several patients (38).
HLA-restriction (limiting patient access) and reliance on tumor MHC expression have limited TCR-based therapy utility. Chimeric antigen receptor T-cells (CART) are autologous T-cells engineered ex vivo to enable MHC-independent tumor cell killing without HLA restriction. First-generation CAR have 3 components: a specific antibody-derived single chain variable fragment (scFv), a hinge/transmembrane domain, and a T-cell signaling (CD3ζ) domain. Second-generation CAR incorporate one additional co-stimulatory domain, while third-generation CAR incorporate 2 additional co-stimulatory domains. Fourth-generation CAR, also known as TRUCKs (T cells redirected for antigen unrestricted cytokine-initiated killing) include a CAR-inducible transgene product, often pro-inflammatory cytokines which may enhance CART cytotoxicity and activate other immune cells in an immunosuppressive TME (39).
Human epidermal growth factor receptor 2 (HER2) is expressed on several solid tumor types and has attracted interest as a CART target. A clinical trial utilizing 1010 of a third-generation CART incorporating a scFV derived from the humanized monoclonal antibody trasztuzumab following lymphodepletion for HER2+ solid tumors. A patient with metastatic colorectal cancer developed fatal respiratory failure 15 minutes after CART infusion. This was thought to be due to massive cytokine release upon recognition of HER2 at low levels on lung epithelium and prompted concerns about the safety of HER2-CART (40). A subsequent HER2-CART trial in HER2+ sarcomas instead utilized the FRP5 scFV, omitted lymphodepletion, and selected a lower starting dose of 104/m2. There were no dose-limiting toxicities, but also no CART expansion. Doses greater than 106/m2 were associated with greater persistence. 4/17 evaluable patients had stable disease and 1 patient had a PR after a second CART infusion (41). To improve CART expansion and persistence, an ongoing phase I HER2-CART trial (NCT00902044) has incorporated lymphodepletion and HER2-CART doses up to 108/m2. Thus far two CRs have been reported (42, 43).
Clinical experience with CART targeting the diaganglioside GD2, which is highly expressed on osteosarcoma, neuroblastoma, and many central nervous system (CNS) tumors, also suggests that lymphodepletion and adequate cell dose are important for CART expansion and persistence. A phase 1 trial utilizing first-generation GD2-CART without lymphodepletion in neuroblastoma demonstrated safety and clinical activity with 3 CRs, but showed limited expansion and persistence (44, 45). A subsequent trial (NCT02107963) utilized a third-generation GD2-CART with lymphodepletion, and demonstrated good expansion (46). A phase I study of a third-generation GD2-CART with or without lymphodepletion in relapsed/refractory neuroblastoma showed increased CART expansion following lymphodepletion (47). A phase I trial utilizing escalating doses of a second-generation GD2-CART with lymphodepletion of varying intensity in relapsed/refractory neuroblastoma showed regression of soft tissue and bone marrow disease following CART doses of at least 108/m2 (48). GD2-CART have shown promising clinical activity in a phase 1 trial in H3K27M+ diffuse midline gliomas, which are universally fatal malignancies (NCT04196413) (49). Based on preclinical data suggesting that incorporation of IL-15 into CART further enhances persistence and cytotoxicity (50, 51), ongoing trials are utilizing GD2-CART and GD2-CAR-NKT cells engineered to express IL-15 (NCT03721068, NCT03294954).
The checkpoint molecule B7-H3(CD276) is another CART target of interest given its high expression on multiple solid tumor types. Preclinical studies have demonstrated encouraging activity of B7-H3-CART in various xenograft models (52–54). Clinical trials utilizing B7-H3-CART are underway in pediatric and adult solid tumors (NCT04897321, NCT04483778, NCT04432649, NCT05211557, NCT04670068) and CNS tumors (NCT04185038, NCT04385173, NCT04077866).
TCR fusion constructs (TRuCs) also enable HLA-independent cell killing. In contrast to CART, which incorporate only the intracellular signaling domain of the CD3ζ chain, TRuCs involve fusion of the scFv to the N-terminus of any of the other five subunits in the TCR complex. TRuCs are incorporated into the TCR on translation, engage the TCR complex upon activation, and are efficacious in solid tumor xenograft models (55). Anti-mesothelin TRuCs are being studied in a phase 1/2 clinical trial (NCT03907852) with preliminary evidence of activity, with 3/7 patients (2 with mesothelioma, 1 with ovarian cancer) achieving a PR (56).
NK-cells are innate immune cells and protect against infections and cancer (57, 58). Efforts to harness NK-cell biology for ACT in cancer treatment has gained considerable interest as an alternative to T-cell based immunotherapeutics. NK-cells possess qualities which may allow them to overcome the hostile TME (58–60). While T-cells recognize unique tumor antigens, NK-cell-mediated cytotoxicity depends on the sum of activating and inhibitory signals, including tumor cell lack of MHC class 1 expression or antibody-dependent cell-mediated cytotoxicity (61). Furthermore, NK-cells can produce inflammatory cytokines such as IFNγ and TNFα which can activate CD8+ TILs and enhance their cytotoxicity (62).
These properties allow NK-cells to be engineered or manipulated via different mechanisms from T-cell-centric immunotherapies. Examples include the administration of agonist cytokines or engineering NK-cells which constitutively secrete these cytokines (63). Others have proposed NK-cells which constitutively secrete chemotactic factors to recruit cytotoxic lymphocytes to the TME (64). Tri-specific NK-cell engagers (TriKEs) have been proposed to confer tumor-specificity to NK-cells and enhance NK-cell activation by engaging stimulatory receptors such as the IL-15 receptor (65, 66). Additionally, CAR NK-cells (CAR-NK) designed from stem cell progenitors represent another way to generate tumor-specific NK-cells. Attractively, CAR-NK may be less toxic and could be produced at lower cost than CART (67). Recent experience with CD19-CAR-NK in B-cell malignancies provides proof-of-concept that this strategy can be safely and effectively utilized and with potential for persistence (68). Barriers remain to production and monitoring of persistence of these cells, but additional alterations to the NK-cell product and manufacturing strategies have been proposed to mitigate these issues. Finally, NK-cells also express immune checkpoint molecules such as PD-1, and either combination with ICIs or intrinsic downregulation of these checkpoint molecules have been proposed as mechanisms to further enhance the efficacy of NK-cell-based approaches (69, 70).
Myeloid cells readily infiltrate primary tumors and metastases. Harnessing this property for ACT shows promise in the treatment of solid malignancies (71). Myeloid cells are highly plastic and may acquire a wide spectrum of immune-stimulatory or immune-suppressive phenotypes in response to the local milieu. Tumor associated macrophages (TAMs) are polarized to an anti-tumor M1 phenotype in response to pro-inflammatory factors such as IFNγ, GM-CSF and lipopolysaccharide. M1 TAMs promote Th1 responses, phagocytosis of tumor cells, and antigen presentation. Tumor-associated cytokines such as IL-10, IL-4, IL-13 and TGF-β promote polarization towards an immunosuppressive M2 phenotype. M2 TAMs promote tumor progression through mechanisms including angiogenesis, extracellular matrix (ECM) remodeling and regulatory T-cell recruitment (72). This M1/M2 classification is an oversimplification, however induction of an M1-like, anti-tumor phenotype is important for the success of myeloid-based ACT. The first myeloid-based ACT utilized macrophages polarized to the M1 phenotype ex vivo with IFNγ. Clinical trials showed limited efficacy, but these therapies were generally well-tolerated (73–75).
Subsequent work has focused on engineering myeloid cells towards a more potent and durable anti-tumor phenotype. Anti-HER2 CAR-macrophages (CARM) reduced tumor growth and prolonged survival while reprogramming the immune-suppressive TME in xenograft models (76). A first-in-human trial evaluating CARM is now underway in HER2-overexpressing solid tumors (NCT04660929). Preclinical work has shown that myeloid cells can also be used to deliver cargo to the TME. Administration of myeloid cells genetically engineered to express IL-12, a potent anti-tumor cytokine, resulted in durable cures in a syngeneic model of embryonal rhabdomyosarcoma through activation of T-cell responses in the tumor and metastatic microenvironment (77).
Significant remaining challenges for optimization of solid tumor ACT are outlined in this section. Additionally, we will summarize proposed strategies to overcome these challenges (Figure 2).
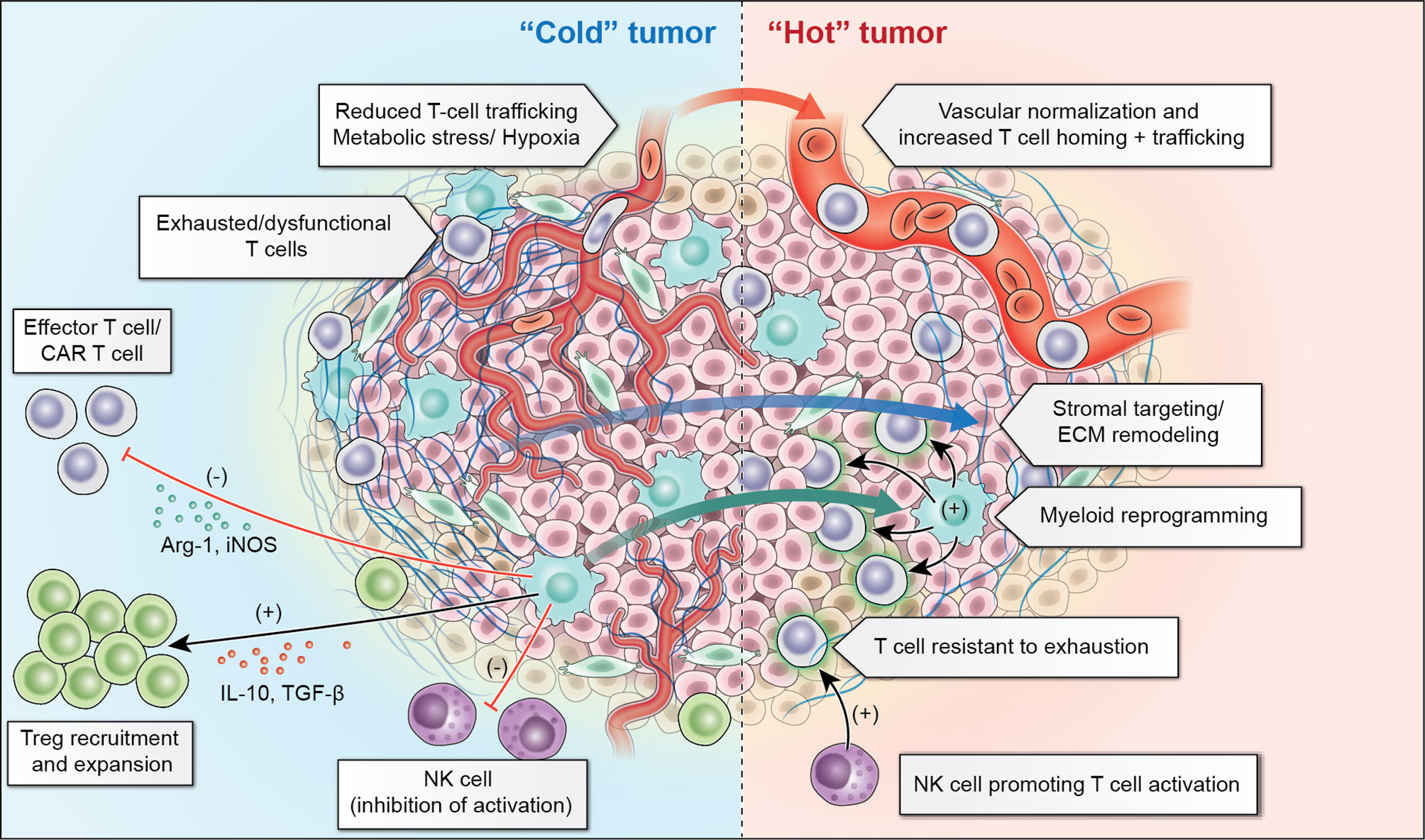
Figure 2 “Cold” solid tumors present a number of challenges within their tumor microenvironment including reduced trafficking related to abnormal tumor vasculature and resident inhibitory myeloid cells which recruit regulatory T cells (Treg) and lead to exhaustion of T-cells and NK-cells. Adoptive cellular therapies aim to overcome these challenges through vascular normalization and extracellular matrix (ECM) remodeling to promote improved trafficking, as well as myeloid cell reprogramming to diminish the inhibitory contribution of these cells. Additionally, T-cells which are resistant to inhibition or “armored” T-cells, or NK-cells which can augment T-cell responses may make it possible to overcome the inhibitory tumor microenvironment.
Selection of antigens such as GD2 (78) and CTAs (79), which are expressed on numerous solid tumors, leverages the possibility that a single ACT could be active across multiple histologies. However, few antigens are tumor-specific. Thus, identifying a target antigen which will allow tumor clearance without unacceptable normal tissue toxicity (on-target/off-tumor effect) is problematic. In addition to selecting the proper target antigen, low antigen density and antigen downregulation within heterogeneous solid TMEs have emerged as additional barriers to ACT (80, 81).
ACT trafficking is also challenging in solid tumors. Trafficking can be inhibited by physical barriers, loss of MHC class 1 expression, repellent cytokine gradients, expression of inhibitory ligands such as PD-L1, and abnormal tumor vasculature (82). CNS tumors are further shielded by the blood-brain barrier (83). If ACTs cannot traffic to the tumor and engage their target antigen, they fail to be activated and expand, leading to rapid loss of ACT.
Finally, the TME present in many solid tumors is hostile to the ACT. Tumors recruit immunosuppressive TAMs and myeloid-derived suppressor cells (MDSCs) (84) which express inhibitory molecules such as PD-L1 (84), secrete inactivating cytokines such as IL-10 (85), and promote a hypoxic TME (86) which can thwart ACT cytotoxicity. These tumor-sustaining programs promote rapid and irreversible ACT exhaustion, inhibit expansion, and result in failure of tumor clearance. Further ACT engineering or combination with agents to allow ACTs to overcome these challenges, will be necessary for ACT optimization in solid tumors.
Aberrant tumor vasculature and ECM deposition impede ACT trafficking. Regional ACT administration is one strategy to overcome this hurdle. A recent phase I trial demonstrated that intrapleural administration of mesothelin-CART combined with pembrolizumab was safe and feasible, and showed potential efficacy with 2 patients demonstrating metabolic CR on PET scan (87). Intraventricular CART administration for both primary brain tumors and CNS metastases is also under evaluation in early-phase clinical trials and in preclinical models (88, 89) (NCT04196413). Additionally, methods to disrupt the blood-brain barrier to allow trafficking of ACT to CNS tumors, such as focused ultrasound (90) or other mechanical or pharmacological methods (91) have been piloted.
Further genetic modification of ACTs to overcome and leverage features of the hostile TME is currently being explored. Many solid tumor types recruit TAMs by producing chemokines such as CXCL8 and CXCL2. Preclinical data suggest that chemokine secretion can be leveraged to enhance CART trafficking by engineering CART to express chemokine receptors. For example, CXCR2-modified GPC3-CART had improved trafficking in a hepatocellular carcinoma model (92), while a CXCR1/2-modified CD70-CART enhanced CART trafficking and efficacy in murine GBM, ovarian cancer and pancreatic cancer models (93). Many groups have also sought to generate a more “fit” ACT through enhanced cytokine secretion [thoroughly reviewed by Bell and Gottschalk (94)]. Additional modifications include creation of ACT which is resistant to exhaustion [e.g. DNA methyltransferase 3 alpha knock-out (95) or PD-1 deletion (96)] or tuning ACT to be effective despite low antigen density [e.g. c-Jun overexpression (97, 98)]. These modifications of ACTs are now entering clinical trials (e.g. TGF-βR knockout CART NCT04976218).
Tumor-associated vasculature is characterized by pericyte loss, resulting in leakiness and adhesion molecule down-regulation impairing T-cell migration into the tumor (99). VEGF inhibitors, which promote vascular normalization, may enhance CD8+ T-cell infiltration into tumors (100). Anti-VEGF agents have shown synergy with ICIs in select solid malignancies, resulting in FDA approval of these combinations in hepatocellular carcinoma and renal cell carcinoma (101). Preclinical studies suggest that antiangiogenics can also improve ACT trafficking (102, 103). Combining ACTs with antiangiogenics warrants further study in clinical trials.
ECM-remodeling agents may enhance the ability of ACTs to infiltrate tumors. In gastric cancer models, hyaluronic acid reduced mesothelin-CART infiltration, however these CART had superior efficacy when combined with infusion of a secreted form of the human hyaluronidase PH20 (104). CART engineered to express heparinase, which degrades heparan sulfate proteoglycans, showed superior anti-tumor activity and were associated with increased T-cell infiltration in preclinical models (105).
The solid TME contributes to T-cell exhaustion via multiple mechanisms, including repeated TCR stimulation and metabolic stress, thereby reducing the ACT efficacy. Engineering CART to reduce tonic signaling through incorporation of the 4-1BB costimulatory domain vs CD28 costimulatory domain showed reduction in CART exhaustion and enhanced persistence and efficacy in preclinical studies (106). Induction of transient rest periods in CART, such as by dasatinib utilization, has shown exhaustion reversal and improved efficacy (107). A dasatinib-containing culture platform is being used to manufacture GD2-CART in ongoing clinical trials (NCT04539366, NCT04196413). CART combination with ICIs is also under evaluation in clinical trials (108).
The ability of myeloid cells to orchestrate immune responses in the TME makes them an attractive therapeutic target. Low-dose chemotherapy has shown reduction of tumor MDSCs (109–111). MDSC differentiation with ATRA reduced their immune-suppressive function and enhanced efficacy of GD2-CART in preclinical models (112). In a pilot trial studying ipilimumab vs ipilimumab combined with ATRA, patients receiving ATRA had fewer circulating MDSCs (113). Inhibiting myeloid cell trafficking through CSF1R inhibition is another potential avenue to reduce myeloid cell immune-suppression in the TME. CSF1R-targeting agents are generally well-tolerated in the clinic, and the multi-TKI CSF1R inhibitor Pexidartinib is FDA-approved to treat tenosynovial giant-cell tumor (114, 115). Clinical trials studying CSF1R inhibitors with ICIs are underway (NCT02777710, NCT02829723, NCT03502330, NCT04848116, NCT02526017).
While ACT has yet to yield the transformative results in solid tumors that CART have shown for hematologic malignancies, evidence exists that some patients with solid tumors may respond to ACT. T-cells, NK-cells, and myeloid cells have each been engineered to target these tumors, and each have advantages and unique challenges. Further engineering ACTs to overcome tumor immune resistance mechanisms and better understanding how to combine with TME-modifying agents will be critical to expanding the number of patients with solid tumors who may derive therapeutic benefit.
JL, KW, and JG wrote the first version of the manuscript. NS provided critical feedback and additions. All authors contributed to the final version of the manuscript.
The content of this publication does not necessarily reflect the views of policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
This work was supported in part by the Intramural Research Program, Center of Cancer Research, National Cancer Institute. We appreciate the contributions of Erina He and the NIH Medical Arts team in aiding in the production of the figures for this manuscript.
1. Maleki Vareki S. High and Low Mutational Burden Tumors Versus Immunologically Hot and Cold Tumors and Response to Immune Checkpoint Inhibitors. J Immunother Cancer (2018) 6:157. doi: 10.1186/s40425-018-0479-7
2. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-Gamma-Related mRNA Profile Predicts Clinical Response to PD-1 Blockade. J Clin Invest (2017) 127:2930–40. doi: 10.1172/JCI91190
3. Hegde PS, Karanikas V, Evers S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin Cancer Res (2016) 22:1865–74. doi: 10.1158/1078-0432.CCR-15-1507
4. Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Updated Analysis of KEYNOTE-024: Pembrolizumab Versus Platinum-Based Chemotherapy for Advanced Non-Small-Cell Lung Cancer With PD-L1 Tumor Proportion Score of 50% or Greater. J Clin Oncol (2019) 37:537–46. doi: 10.1200/JCO.18.00149
5. Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor Mutational Load Predicts Survival After Immunotherapy Across Multiple Cancer Types. Nat Genet (2019) 51:202–6. doi: 10.1038/s41588-018-0312-8
6. Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell (2018) 175:984–997.e24. doi: 10.1016/j.cell.2018.09.006
7. Chen L, Oke T, Siegel N, Cojocaru G, Tam AJ, Blosser RL, et al. The Immunosuppressive Niche of Soft-Tissue Sarcomas Is Sustained by Tumor-Associated Macrophages and Characterized by Intratumoral Tertiary Lymphoid Structures. Clin Cancer Res (2020) 26:4018–30. doi: 10.1158/1078-0432.CCR-19-3416
8. Ligon JA, Choi W, Cojocaru G, Fu W, Hsiue EH, Oke TF, et al. Pathways of Immune Exclusion in Metastatic Osteosarcoma Are Associated With Inferior Patient Outcomes. J Immunother Cancer 9 (2021) 9(5):e001772. doi: 10.1136/jitc-2020-001772
9. Davis KL, Fox E, Merchant MS, Reid JM, Kudgus RA, Liu X, et al. Nivolumab in Children and Young Adults With Relapsed or Refractory Solid Tumours or Lymphoma (ADVL1412): A Multicentre, Open-Label, Single-Arm, Phase 1-2 Trial. Lancet Oncol (2020) 21:541–50. doi: 10.1016/S1470-2045(20)30023-1
10. Park JR, Kreissman SG, London WB, Naranjo A, Cohn SL, Hogarty MD, et al. Effect of Tandem Autologous Stem Cell Transplant vs Single Transplant on Event-Free Survival in Patients With High-Risk Neuroblastoma: A Randomized Clinical Trial. JAMA (2019) 322:746–55. doi: 10.1001/jama.2019.11642
11. Ladenstein R, Potschger U, Le Deley MC, Whelan J, Paulussen M, Oberlin O, et al. Primary Disseminated Multifocal Ewing Sarcoma: Results of the Euro-EWING 99 Trial. J Clin Oncol (2010) 28:3284–91. doi: 10.1200/JCO.2009.22.9864
12. Pedrazzoli P, Martino M, Delfanti S, Generali D, Rosti G, Bregni M, et al. High-Dose Chemotherapy With Autologous Hematopoietic Stem Cell Transplantation for High-Risk Primary Breast Cancer. J Natl Cancer Inst Monogr (2015) 2015:70–5. doi: 10.1093/jncimonographs/lgv010
13. de Jong MC, Kors WA, de Graaf P, Castelijns JA, Kivela T, Moll AC. Trilateral Retinoblastoma: A Systematic Review and Meta-Analysis. Lancet Oncol (2014) 15:1157–67. doi: 10.1016/S1470-2045(14)70336-5
14. Haberle B, Maxwell R, Schweinitz DV, Schmid I. High Dose Chemotherapy With Autologous Stem Cell Transplantation in Hepatoblastoma Does Not Improve Outcome. Results of the GPOH Study Hb99. Klin Padiatr (2019) 231:283–90. doi: 10.1055/a-1014-3250
15. Llosa NJ, Cooke KR, Chen AR, Gamper CJ, Klein OR, Zambidis ET, et al. Reduced-Intensity Haploidentical Bone Marrow Transplantation With Post-Transplant Cyclophosphamide for Solid Tumors in Pediatric and Young Adult Patients. Biol Blood Marrow Transplant (2017) 23:2127–36. doi: 10.1016/j.bbmt.2017.08.012
16. Waldman AD, Fritz JM, Lenardo MJ. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat Rev Immunol (2020) 20:651–68. doi: 10.1038/s41577-020-0306-5
17. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of Tumor-Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients With Metastatic Melanoma. A Preliminary Report. N Engl J Med (1988) 319:1676–80. doi: 10.1056/NEJM198812223192527
18. Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive Cell Therapy for Patients With Metastatic Melanoma: Evaluation of Intensive Myeloablative Chemoradiation Preparative Regimens. J Clin Oncol (2008) 26:5233–9. doi: 10.1200/JCO.2008.16.5449
19. Rosenberg SA, Dudley ME. Adoptive Cell Therapy for the Treatment of Patients With Metastatic Melanoma. Curr Opin Immunol (2009) 21:233–40. doi: 10.1016/j.coi.2009.03.002
20. Yamamoto TN, Kishton RJ, Restifo NP. Developing Neoantigen-Targeted T Cell-Based Treatments for Solid Tumors. Nat Med (2019) 25:1488–99. doi: 10.1038/s41591-019-0596-y
21. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer Immunotherapy Based on Mutation-Specific CD4+ T Cells in a Patient With Epithelial Cancer. Science (2014) 344:641–5. doi: 10.1126/science.1251102
22. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
23. Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, Feldman SA, et al. A Pilot Trial Using Lymphocytes Genetically Engineered With an NY-ESO-1-Reactive T-Cell Receptor: Long-Term Follow-Up and Correlates With Response. Clin Cancer Res (2015) 21:1019–27. doi: 10.1158/1078-0432.CCR-14-2708
24. D'Angelo SP, Melchiori L, Merchant MS, Bernstein D, Glod J, Kaplan R, et al. Antitumor Activity Associated With Prolonged Persistence of Adoptively Transferred NY-ESO-1 (C259)T Cells in Synovial Sarcoma. Cancer Discov (2018) 8:944–57. doi: 10.1158/2159-8290.CD-17-1417
25. Ramachandran I, Lowther DE, Dryer-Minnerly R, Wang R, Fayngerts S, Nunez D, et al. Systemic and Local Immunity Following Adoptive Transfer of NY-ESO-1 SPEAR T Cells in Synovial Sarcoma. J Immunother Cancer (2019) 7:276. doi: 10.1186/s40425-019-0762-2
26. Desar IME, Fleuren EDG, van der Graaf WTA. Systemic Treatment for Adults With Synovial Sarcoma. Curr Treat Options Oncol (2018) 19:13. doi: 10.1007/s11864-018-0525-1
27. Lu YC, Parker LL, Lu T, Zheng Z, Toomey MA, White DE, et al. Treatment of Patients With Metastatic Cancer Using a Major Histocompatibility Complex Class II-Restricted T-Cell Receptor Targeting the Cancer Germline Antigen MAGE-A3. J Clin Oncol (2017) 35:3322–9. doi: 10.1200/JCO.2017.74.5463
28. Martin AD, Wang X, Sandberg ML, Negri KR, Wu ML, Toledo Warshaviak D, et al. Re-Examination of MAGE-A3 as a T-Cell Therapeutic Target. J Immunother (2021) 44:95–105. doi: 10.1097/CJI.0000000000000348
29. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer Regression and Neurological Toxicity Following Anti-MAGE-A3 TCR Gene Therapy. J Immunother (2013) 36:133–51. doi: 10.1097/CJI.0b013e3182829903
30. Hong D, Clarke J, Johanns T, Kebriaei P, Heymach J, Galal A, et al. Initial Safety, Efficacy, and Product Attributes From the Surpass Trial With ADP-A2M4CD8, A SPEAR T-Cell Therapy Incorporating an Affinity Optimized TCR Targeting MAGE-A4 and a CD8(a) Alpha Co-Receptor. J Immunother Cancer (2020) 8:A231–1. doi: 10.1136/jitc-2020-SITC2020.0379
31. Manfredi F, Cianciotti BC, Potenza A, Tassi E, Noviello M, Biondi A, et al. TCR Redirected T Cells for Cancer Treatment: Achievements, Hurdles, and Goals. Front Immunol (2020) 11:1689. doi: 10.3389/fimmu.2020.01689
32. Stevanovic S, Helman SR, Wunderlich JR, Langhan MM, Doran SL, Kwong MLM, et al. A Phase II Study of Tumor-Infiltrating Lymphocyte Therapy for Human Papillomavirus-Associated Epithelial Cancers. Clin Cancer Res (2019) 25:1486–93. doi: 10.1158/1078-0432.CCR-18-2722
33. Doran SL, Stevanovic S, Adhikary S, Gartner JJ, Jia L, Kwong MLM, et al. T-Cell Receptor Gene Therapy for Human Papillomavirus-Associated Epithelial Cancers: A First-In-Human, Phase I/II Study. J Clin Oncol (2019) 37:2759–68. doi: 10.1200/JCO.18.02424
34. Norberg S, Nagarsheth N, Sinkoe A, Adhikary S, Meyer T, Lack J, et al. Safety and Clinical Activity of Gene-Engineered T-Cell Therapy Targeting HPV-16 E7 for Epithelial Cancers. J Clin Oncol (2020) 38. doi: 10.1200/JCO.2020.38.15_suppl.101
35. Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, et al. Long-Term Outcome of EBV-Specific T-Cell Infusions to Prevent or Treat EBV-Related Lymphoproliferative Disease in Transplant Recipients. Blood (2010) 115:925–35. doi: 10.1182/blood-2009-08-239186
36. Comoli P, Pedrazzoli P, Maccario R, Basso S, Carminati O, Labirio M, et al. Cell Therapy of Stage IV Nasopharyngeal Carcinoma With Autologous Epstein-Barr Virus-Targeted Cytotoxic T Lymphocytes. J Clin Oncol (2005) 23:8942–9. doi: 10.1200/JCO.2005.02.6195
37. Sharma S, Leung WK, Heslop HE. Virus-Specific T Cells for Malignancies - Then, Now and Where to? Curr Stem Cell Rep (2020) 6:17–29. doi: 10.1007/s40778-020-00170-6
38. Chapuis AG, Afanasiev OK, Iyer JG, Paulson KG, Parvathaneni U, Hwang JH, et al. Regression of Metastatic Merkel Cell Carcinoma Following Transfer of Polyomavirus-Specific T Cells and Therapies Capable of Re-Inducing HLA Class-I. Cancer Immunol Res (2014) 2:27–36. doi: 10.1158/2326-6066.CIR-13-0087
39. Thanindratarn P, Dean DC, Nelson SD, Hornicek FJ, Duan Z. Chimeric Antigen Receptor T (CAR-T) Cell Immunotherapy for Sarcomas: From Mechanisms to Potential Clinical Applications. Cancer Treat Rev (2020) 82:101934. doi: 10.1016/j.ctrv.2019.101934
40. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol Ther (2010) 18:843–51. doi: 10.1038/mt.2010.24
41. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol (2015) 33:1688–96. doi: 10.1200/JCO.2014.58.0225
42. Hegde M, Joseph SK, Pashankar F, DeRenzo C, Sanber K, Navai S, et al. Tumor Response and Endogenous Immune Reactivity After Administration of HER2 CAR T Cells in a Child With Metastatic Rhabdomyosarcoma. Nat Commun (2020) 11:3549. doi: 10.1038/s41467-020-17175-8
43. Derenzo C, Navai SA, Joseph S, Sanber K, Byrd T, Zhang H, et al. Administration of HER2-CAR T Cells After Lymphodepletion Safely Improves T Cell Expansion and Induces Clinical Responses in Patients With Advanced Sarcomas. Atlanta, GA: 110th Annual Meeting of the American Association for Cancer Research (2019).
44. Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-Specific T Cells Engineered to Coexpress Tumor-Specific Receptors: Persistence and Antitumor Activity in Individuals With Neuroblastoma. Nat Med (2008) 14:1264–70. doi: 10.1038/nm.1882
45. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor Activity and Long-Term Fate of Chimeric Antigen Receptor-Positive T Cells in Patients With Neuroblastoma. Blood (2011) 118:6050–6. doi: 10.1182/blood-2011-05-354449
46. Stroncek DF, Lee DW, Ren J, Sabatino M, Highfill S, Khuu H, et al. Elutriated Lymphocytes for Manufacturing Chimeric Antigen Receptor T Cells. J Transl Med (2017) 15:59. doi: 10.1186/s12967-017-1160-5
47. Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. CAR T Cells Administered in Combination With Lymphodepletion and PD-1 Inhibition to Patients With Neuroblastoma. Mol Ther (2017) 25:2214–24. doi: 10.1016/j.ymthe.2017.05.012
48. Straathof K, Flutter B, Wallace R, Jain N, Loka T, Depani S, et al. Antitumor Activity Without on-Target Off-Tumor Toxicity of GD2-Chimeric Antigen Receptor T Cells in Patients With Neuroblastoma. Sci Transl Med (2020) 12(571):eabd6169. doi: 10.1126/scitranslmed.abd6169
49. Majzner R, Ramakrishna S, Mochizuki A, Patel S, Chinnasamy H, Yeom K, et al. EPCT-14. GD2 CAR T-Cells Mediate Clinical Activity And Manageable Toxicity In Children And Young Adults With H3K27M-Mutated DIPG And Spinal Cord DMG. Neuro-Oncology (2021) 23:i49–50. doi: 10.1093/neuonc/noab090.200
50. Chen Y, Sun C, Landoni E, Metelitsa L, Dotti G, Savoldo B. Eradication of Neuroblastoma by T Cells Redirected With an Optimized GD2-Specific Chimeric Antigen Receptor and Interleukin-15. Clin Cancer Res (2019) 25:2915–24. doi: 10.1158/1078-0432.CCR-18-1811
51. Xu X, Huang W, Heczey A, Liu D, Guo L, Wood M, et al. NKT Cells Coexpressing a GD2-Specific Chimeric Antigen Receptor and IL15 Show Enhanced In Vivo Persistence and Antitumor Activity Against Neuroblastoma. Clin Cancer Res (2019) 25:7126–38. doi: 10.1158/1078-0432.CCR-19-0421
52. Majzner RG, Theruvath JL, Nellan A, Heitzeneder S, Cui Y, Mount CW, et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin Cancer Res (2019) 25:2560–74. doi: 10.1158/1078-0432.CCR-18-0432
53. Theruvath J, Sotillo E, Mount CW, Graef CM, Delaidelli A, Heitzeneder S, et al. Locoregionally Administered B7-H3-Targeted CAR T Cells for Treatment of Atypical Teratoid/Rhabdoid Tumors. Nat Med (2020) 26:712–9. doi: 10.1038/s41591-020-0821-8
54. Du H, Hirabayashi K, Ahn S, Kren NP, Montgomery SA, Wang X, et al. Antitumor Responses in the Absence of Toxicity in Solid Tumors by Targeting B7-H3 via Chimeric Antigen Receptor T Cells. Cancer Cell (2019) 35:221–237 e8. doi: 10.1016/j.ccell.2019.01.002
55. Baeuerle PA, Ding J, Patel E, Thorausch N, Horton H, Gierut J, et al. Synthetic TRuC Receptors Engaging the Complete T Cell Receptor for Potent Anti-Tumor Response. Nat Commun (2019) 10:2087. doi: 10.1038/s41467-019-10097-0
56. Hong DS, Johnson M, Tanyi JL, MacMullen L, Tighe R, Jalbert L, et al. Abstract CT105: Preliminary Safety and Efficacy of Gavocabtagene Autoleucel (Gavo-Cel, TC-210), a T Cell Receptor Fusion Construct (TRuC™), in Patients With Treatment Refractory Mesothelin Overexpressing Solid Tumors. Cancer Res (2021) 81:CT105–5. doi: 10.1158/1538-7445.AM2021-CT105
57. Mace EM, Orange JS. Emerging Insights Into Human Health and NK Cell Biology From the Study of NK Cell Deficiencies. Immunol Rev (2019) 287:202–25. doi: 10.1111/imr.12725
58. Abel AM, Yang C, Thakar MS, Malarkannan S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front Immunol (2018) 9:1869. doi: 10.3389/fimmu.2018.01869
59. Omer N, Nicholls W, Ruegg B, Souza-Fonseca-Guimaraes F, Rossi GR. Enhancing Natural Killer Cell Targeting of Pediatric Sarcoma. Front Immunol (2021) 12:791206. doi: 10.3389/fimmu.2021.791206
60. Rezvani K, Rouce R, Liu E, Shpall E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol Ther (2017) 25:1769–81. doi: 10.1016/j.ymthe.2017.06.012
61. Bryceson YT, March ME, Ljunggren HG, Long EO. Activation, Coactivation, and Costimulation of Resting Human Natural Killer Cells. Immunol Rev (2006) 214:73–91. doi: 10.1111/j.1600-065X.2006.00457.x
62. Castro F, Cardoso AP, Goncalves RM, Serre K, Oliveira MJ. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front Immunol (2018) 9:847. doi: 10.3389/fimmu.2018.00847
63. Jochems C, Hodge JW, Fantini M, Fujii R, M YMo 2nd, Greiner JW, et al. An NK Cell Line (haNK) Expressing High Levels of Granzyme and Engineered to Express the High Affinity CD16 Allele. Oncotarget (2016) 7:86359–73. doi: 10.18632/oncotarget.13411
64. Bernardini G, Antonangeli F, Bonanni V, Santoni A. Dysregulation of Chemokine/Chemokine Receptor Axes and NK Cell Tissue Localization During Diseases. Front Immunol (2016) 7:402. doi: 10.3389/fimmu.2016.00402
65. Felices M, Kodal B, Hinderlie P, Kaminski MF, Cooley S, Weisdorf DJ, et al. Novel CD19-Targeted TriKE Restores NK Cell Function and Proliferative Capacity in CLL. Blood Adv (2019) 3:897–907. doi: 10.1182/bloodadvances.2018029371
66. Felices M, Lenvik TR, Davis ZB, Miller JS, Vallera DA. Generation of BiKEs and TriKEs to Improve NK Cell-Mediated Targeting of Tumor Cells. Methods Mol Biol (2016) 1441:333–46. doi: 10.1007/978-1-4939-3684-7_28
67. Rafei H, Daher M, Rezvani K. Chimeric Antigen Receptor (CAR) Natural Killer (NK)-Cell Therapy: Leveraging the Power of Innate Immunity. Br J Haematol (2021) 193:216–30. doi: 10.1111/bjh.17186
68. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med (2020) 382:545–53. doi: 10.1056/NEJMoa1910607
69. Wang C, Cui A, Bukenya M, Aung A, Pradhan D, Whittaker CA, et al. Reprogramming NK Cells and Macrophages via Combined Antibody and Cytokine Therapy Primes Tumors for Elimination by Checkpoint Blockade. Cell Rep (2021) 37:110021. doi: 10.1016/j.celrep.2021.110021
70. Kim N, Kim HS. Targeting Checkpoint Receptors and Molecules for Therapeutic Modulation of Natural Killer Cells. Front Immunol (2018) 9:2041. doi: 10.3389/fimmu.2018.02041
71. Anderson NR, Minutolo NG, Gill S, Klichinsky M. Macrophage-Based Approaches for Cancer Immunotherapy. Cancer Res (2021) 81:1201–8. doi: 10.1158/0008-5472.CAN-20-2990
72. Koo J, Hayashi M, Verneris MR, Lee-Sherick AB. Targeting Tumor-Associated Macrophages in the Pediatric Sarcoma Tumor Microenvironment. Front Oncol (2020) 10:581107. doi: 10.3389/fonc.2020.581107
73. Ritchie D, Mileshkin L, Wall D, Bartholeyns J, Thompson M, Coverdale J, et al. In Vivo Tracking of Macrophage Activated Killer Cells to Sites of Metastatic Ovarian Carcinoma. Cancer Immunol Immunother (2007) 56:155–63. doi: 10.1007/s00262-006-0181-3
74. Burger M, Thiounn N, Denzinger S, Kondas J, Benoit G, Chapado MS, et al. The Application of Adjuvant Autologous Antravesical Macrophage Cell Therapy vs. BCG in Non-Muscle Invasive Bladder Cancer: A Multicenter, Randomized Trial. J Transl Med (2010) 8:54. doi: 10.1186/1479-5876-8-54
75. Andreesen R, Scheibenbogen C, Brugger W, Krause S, Meerpohl HG, Leser HG, et al. Adoptive Transfer of Tumor Cytotoxic Macrophages Generated In Vitro From Circulating Blood Monocytes: A New Approach to Cancer Immunotherapy. Cancer Res (1990) 50:7450–6.
76. Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human Chimeric Antigen Receptor Macrophages for Cancer Immunotherapy. Nat Biotechnol (2020) 38:947–53. doi: 10.1038/s41587-020-0462-y
77. Kaczanowska S, Beury DW, Gopalan V, Tycko AK, Qin H, Clements ME, et al. Genetically Engineered Myeloid Cells Rebalance the Core Immune Suppression Program in Metastasis. Cell (2021) 184:2033–2052 e21. doi: 10.1016/j.cell.2021.02.048
78. Nazha B, Inal C, Owonikoko TK. Disialoganglioside GD2 Expression in Solid Tumors and Role as a Target for Cancer Therapy. Front Oncol (2020) 10:1000. doi: 10.3389/fonc.2020.01000
79. Iura K, Kohashi K, Ishii T, Maekawa A, Bekki H, Otsuka H, et al. MAGEA4 Expression in Bone and Soft Tissue Tumors: Its Utility as a Target for Immunotherapy and Diagnostic Marker Combined With NY-ESO-1. Virchows Arch (2017) 471:383–92. doi: 10.1007/s00428-017-2206-z
80. Majzner RG, Rietberg SP, Sotillo E, Dong R, Vachharajani VT, Labanieh L, et al. Tuning the Antigen Density Requirement for CAR T-Cell Activity. Cancer Discov (2020) 10:702–23. doi: 10.1158/2159-8290.CD-19-0945
81. Majzner RG, Mackall CL. Tumor Antigen Escape From CAR T-Cell Therapy. Cancer Discov (2018) 8:1219–26. doi: 10.1158/2159-8290.CD-18-0442
82. Wagner J, Wickman E, DeRenzo C, Gottschalk S. CAR T Cell Therapy for Solid Tumors: Bright Future or Dark Reality? Mol Ther (2020) 28:2320–39. doi: 10.1016/j.ymthe.2020.09.015
83. Mo F, Pellerino A, Soffietti R, Ruda R. Blood-Brain Barrier in Brain Tumors: Biology and Clinical Relevance. Int J Mol Sci (2021) 22(23):12654. doi: 10.3390/ijms222312654
84. Loeuillard E, Yang J, Buckarma E, Wang J, Liu Y, Conboy C, et al. Targeting Tumor-Associated Macrophages and Granulocytic Myeloid-Derived Suppressor Cells Augments PD-1 Blockade in Cholangiocarcinoma. J Clin Invest (2020) 130:5380–96. doi: 10.1172/JCI137110
85. Liu Q, Yang C, Wang S, Shi D, Wei C, Song J, et al. Wnt5a-Induced M2 Polarization of Tumor-Associated Macrophages via IL-10 Promotes Colorectal Cancer Progression. Cell Commun Signal (2020) 18:51. doi: 10.1186/s12964-020-00557-2
86. Jeong H, Kim S, Hong BJ, Lee CJ, Kim YE, Bok S, et al. Tumor-Associated Macrophages Enhance Tumor Hypoxia and Aerobic Glycolysis. Cancer Res (2019) 79:795–806. doi: 10.1158/0008-5472.CAN-18-2545
87. Adusumilli PS, Zauderer MG, Riviere I, Solomon SB, Rusch VW, O'Cearbhaill RE, et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-Cell Therapy in Patients With Malignant Pleural Disease, in Combination With the Anti-PD-1 Agent Pembrolizumab. Cancer Discov (2021) 11:2748–63. doi: 10.1158/2159-8290.CD-21-0407
88. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of Glioblastoma After Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
89. Priceman SJ, Tilakawardane D, Jeang B, Aguilar B, Murad JP, Park AK, et al. Regional Delivery of Chimeric Antigen Receptor-Engineered T Cells Effectively Targets HER2(+) Breast Cancer Metastasis to the Brain. Clin Cancer Res (2018) 24:95–105. doi: 10.1158/1078-0432.CCR-17-2041
90. Etame AB, Diaz RJ, Smith CA, Mainprize TG, Hynynen K, Rutka JT. Focused Ultrasound Disruption of the Blood-Brain Barrier: A New Frontier for Therapeutic Delivery in Molecular Neurooncology. Neurosurg Focus (2012) 32:E3. doi: 10.3171/2011.10.FOCUS11252
91. Karmur BS, Philteos J, Abbasian A, Zacharia BE, Lipsman N, Levin V, et al. Blood-Brain Barrier Disruption in Neuro-Oncology: Strategies, Failures, and Challenges to Overcome. Front Oncol (2020) 10:563840. doi: 10.3389/fonc.2020.563840
92. Liu G, Rui W, Zheng H, Huang D, Yu F, Zhang Y, et al. CXCR2-Modified CAR-T Cells Have Enhanced Trafficking Ability That Improves Treatment of Hepatocellular Carcinoma. Eur J Immunol (2020) 50:712–24. doi: 10.1002/eji.201948457
93. Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, et al. CXCR1- or CXCR2-Modified CAR T Cells Co-Opt IL-8 for Maximal Antitumor Efficacy in Solid Tumors. Nat Commun (2019) 10:4016. doi: 10.1038/s41467-019-11869-4
94. Bell M, Gottschalk S. Engineered Cytokine Signaling to Improve CAR T Cell Effector Function. Front Immunol (2021) 12:684642. doi: 10.3389/fimmu.2021.684642
95. Prinzing B, Zebley CC, Petersen CT, Fan Y, Anido AA, Yi Z, et al. Deleting DNMT3A in CAR T Cells Prevents Exhaustion and Enhances Antitumor Activity. Sci Transl Med (2021) 13:eabh0272. doi: 10.1126/scitranslmed.abh0272
96. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-Engineered T Cells in Patients With Refractory Cancer. Science (2020) 367(6481):eaba7365. doi: 10.1126/science.aba7365
97. Heitzeneder S, Bosse KR, Zhu Z, Zhelev D, Majzner RG, Radosevich MT, et al. GPC2-CAR T Cells Tuned for Low Antigen Density Mediate Potent Activity Against Neuroblastoma Without Toxicity. Cancer Cell (2022) 40:53–69 e9. doi: 10.1016/j.ccell.2021.12.005
98. Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, et al. C-Jun Overexpression in CAR T Cells Induces Exhaustion Resistance. Nature (2019) 576:293–300. doi: 10.1038/s41586-019-1805-z
99. Liu G, Rui W, Zhao X, Lin X. Enhancing CAR-T Cell Efficacy in Solid Tumors by Targeting the Tumor Microenvironment. Cell Mol Immunol (2021) 18:1085–95. doi: 10.1038/s41423-021-00655-2
100. Bourhis M, Palle J, Galy-Fauroux I, Terme M. Direct and Indirect Modulation of T Cells by VEGF-A Counteracted by Anti-Angiogenic Treatment. Front Immunol (2021) 12:616837. doi: 10.3389/fimmu.2021.616837
101. Saeed A, Park R, Sun W. The Integration of Immune Checkpoint Inhibitors With VEGF Targeted Agents in Advanced Gastric and Gastroesophageal Adenocarcinoma: A Review on the Rationale and Results of Early Phase Trials. J Hematol Oncol (2021) 14:13. doi: 10.1186/s13045-021-01034-0
102. Bocca P, Di Carlo E, Caruana I, Emionite L, Cilli M, De Angelis B, et al. Bevacizumab-Mediated Tumor Vasculature Remodelling Improves Tumor Infiltration and Antitumor Efficacy of GD2-CAR T Cells in a Human Neuroblastoma Preclinical Model. Oncoimmunology (2017) 7:e1378843. doi: 10.1080/2162402X.2017.1378843
103. Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic Agents can Increase Lymphocyte Infiltration Into Tumor and Enhance the Effectiveness of Adoptive Immunotherapy of Cancer. Cancer Res (2010) 70:6171–80. doi: 10.1158/0008-5472.CAN-10-0153
104. Zhao R, Cui Y, Zheng Y, Li S, Lv J, Wu Q, et al. Human Hyaluronidase PH20 Potentiates the Antitumor Activities of Mesothelin-Specific CAR-T Cells Against Gastric Cancer. Front Immunol (2021) 12:660488. doi: 10.3389/fimmu.2021.660488
105. Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase Promotes Tumor Infiltration and Antitumor Activity of CAR-Redirected T Lymphocytes. Nat Med (2015) 21:524–9. doi: 10.1038/nm.3833
106. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB Costimulation Ameliorates T Cell Exhaustion Induced by Tonic Signaling of Chimeric Antigen Receptors. Nat Med (2015) 21:581–90. doi: 10.1038/nm.3838
107. Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, et al. Transient Rest Restores Functionality in Exhausted CAR-T Cells Through Epigenetic Remodeling. Science (2021) 372(6537):eaba1786. doi: 10.1126/science.aba1786
108. Grosser R, Cherkassky L, Chintala N, Adusumilli PS. Combination Immunotherapy With CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell (2019) 36:471–82. doi: 10.1016/j.ccell.2019.09.006
109. Peereboom DM, Alban TJ, Grabowski MM, Alvarado AG, Otvos B, Bayik D, et al. Metronomic Capecitabine as an Immune Modulator in Glioblastoma Patients Reduces Myeloid-Derived Suppressor Cells. JCI Insight (2019) 4(22):e130748. doi: 10.1172/jci.insight.130748
110. Gebhardt C, Simon SCS, Weber R, Gries M, Mun DH, Reinhard R, et al. Potential Therapeutic Effect of Low-Dose Paclitaxel in Melanoma Patients Resistant to Immune Checkpoint Blockade: A Pilot Study. Cell Immunol (2021) 360:104274. doi: 10.1016/j.cellimm.2020.104274
111. Xu WL, Shi BJ, Li SL, Yu FX, Guo LN, Li M, et al. Targeted Inhibition of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment by Low-Dose Doxorubicin to Improve Immune Efficacy in Murine Neuroblastoma. Chin Med J (Engl) (2020) 134:334–43. doi: 10.1097/CM9.0000000000001234
112. Long AH, Highfill SL, Cui Y, Smith JP, Walker AJ, Ramakrishna S, et al. Reduction of MDSCs With All-Trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas. Cancer Immunol Res (2016) 4:869–80. doi: 10.1158/2326-6066.CIR-15-0230
113. Tobin RP, Jordan KR, Robinson WA, Davis D, Borges VF, Gonzalez R, et al. Targeting Myeloid-Derived Suppressor Cells Using All-Trans Retinoic Acid in Melanoma Patients Treated With Ipilimumab. Int Immunopharmacol (2018) 63:282–91. doi: 10.1016/j.intimp.2018.08.007
114. Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Ruttinger D. Colony-Stimulating Factor 1 Receptor (CSF1R) Inhibitors in Cancer Therapy. J Immunother Cancer (2017) 5:53. doi: 10.1186/s40425-017-0257-y
Keywords: adoptive cell immunotherapy, solid tumor, tumor microenvironment, immune evasion, CAR (chimeric antigen receptor)
Citation: Ligon JA, Wessel KM, Shah NN and Glod J (2022) Adoptive Cell Therapy in Pediatric and Young Adult Solid Tumors: Current Status and Future Directions. Front. Immunol. 13:846346. doi: 10.3389/fimmu.2022.846346
Received: 31 December 2021; Accepted: 03 February 2022;
Published: 22 February 2022.
Edited by:
Shigeki Yagyu, Kyoto Prefectural University of Medicine, JapanReviewed by:
Jorge Ibanez-Vega, St. Jude Children’s Research Hospital, United StatesCopyright © 2022 Ligon, Wessel, Shah and Glod. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John A. Ligon, am9obi5saWdvbkB1ZmwuZWR1; John Glod, am9obi5nbG9kQG5paC5nb3Y=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.