 Brandie C. Taylor
Brandie C. Taylor Justin M. Balko
Justin M. Balko
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 28 February 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.844866
Immunotherapy has become a key therapeutic strategy in the treatment of many cancers. As a result, research efforts have been aimed at understanding mechanisms of resistance to immunotherapy and how anti-tumor immune response can be therapeutically enhanced. It has been shown that tumor cell recognition by the immune system plays a key role in effective response to T cell targeting therapies in patients. One mechanism by which tumor cells can avoid immunosurveillance is through the downregulation of Major Histocompatibility Complex I (MHC-I). Downregulation of MHC-I has been described as a mechanism of intrinsic and acquired resistance to immunotherapy in patients with cancer. Depending on the mechanism, the downregulation of MHC-I can sometimes be therapeutically restored to aid in anti-tumor immunity. In this article, we will review current research in MHC-I downregulation and its impact on immunotherapy response in patients, as well as possible strategies for therapeutic upregulation of MHC-I.
Recent advances in immuno-oncology have shown the benefit of targeting the immune system for the treatment of cancer. Immunotherapies, such as immune checkpoint inhibitors (ICI), have drastically improved the prognosis and overall survival for some cancer patients. However, only a small percentage of patients respond to ICI treatment due to intrinsic or acquired resistance to therapy. Currently, there are few clinically useful biomarkers of response (e.g., Programmed Death-Ligand 1 [PD-L1], tumor mutational burden, microsatellite instability) and limited approved therapies to augment response to ICI. As immunotherapies become more prominent in the treatment of cancer, there is an immense need for understanding the mechanisms of ICI resistance and for the development of new therapeutic strategies to induce long-term responses.
Tumors often adapt various mechanisms to evade immune destruction (1). Some factors that tumors often utilize include immune suppressive mediators (i.e. immune suppressive cytokines, TGF-β), regulatory and suppressive cell (i.e. regulatory T cells), expression of inhibitory molecules (i.e. PD-L1), and defective antigen presentation (2). CD8+ T cells are one of the primary effector cell types that mediate response to ICI and are activated following the recognition of peptide:major histocompatibility complex class I (MHC-I) complexes (pMHC-I) and co-stimulatory signals. T-cell receptors (TCRs) mediate cytotoxic CD8+ T cell activation by binding to their target through interactions with cognate pMHC-I which are ubiquitously expressed by all nucleated cells (3). The efficacy of ICI depends on T cell recognition of pMHC-I on the tumor surface. Activated CD8+ T cells can induce tumor cell death through injecting granzymes and other cytotoxic molecules through perforin-permeabilized membranes at an immunologic synapse. Thus, one mechanism whereby tumors can avoid T cell cytotoxicity is by downregulation of MHC-I. Loss and reduction of MHC-I expression has been correlated with worse overall prognosis and response to ICI in several cancer patient cohorts and clinical trials (4–8). Aberrant MHC-I expression may be reversible or irreversible because of genetic, epigenetic, transcriptional, or post-transcriptional alterations. The reversibility of some mechanisms of MHC-I downregulation creates a potential avenue for therapeutic strategies to restore MHC-I antigen presentation as a possible combinatorial approach with ICI in cancer. This review will address the current known mechanisms of MHC-I downregulation in cancer, implications in patient immunotherapy efficacy, and potential therapeutic MHC-I upregulation strategies to overcome ICI resistance in patients.
MHC-I is a heterodimer consisting of two domains, a polymorphic human leukocyte antigen (HLA)-encoded heavy α-chain and an invariant light chain termed β2-microglobulin (β2M) (3). There are three classical HLA class I heavy chains, HLA-A, -B, and -C, which are encoded by three separate genes. MHC-I has a high degree of polymorphism derived from each heavy chain’s peptide binding domain. The various HLA alleles have diverse peptide-binding specificities due to the sequence divergence between alleles (9). This diversity of MHC-I allows for a large repertoire of peptides, also known as the immunopeptidome, that can be recognized by CD8+ T cells.
Antigen presentation by MHC-I begins when endogenous proteins are ubiquitinated for proteasomal fragmentation into peptides (10). The peptides are then translocated from the cytosol into the endoplasmic reticulum (ER) through the transporters associated with antigen processing (TAP1/TAP2) and undergo further processing by ER aminopeptidases (ERAP) (11, 12). Processed peptides of 8 to 10 amino acids in length are required for stabilization of the MHC-I complex and are bound by their side chains through molecular interactions (8). The folding and assembly of the α-heavy chain and β2M is promoted by calnexin and this initial complex is stabilized by the chaperone protein tapasin in complex with ERp57 via calreticulin (13). Interaction between TAP1/2 and tapasin permits exchange of peptides into the MHC-I binding groove, release of chaperone proteins, and stabilization of the MHC-I complex. Finally, the peptide:MHC-I complex travels via the Golgi apparatus to the cell surface and antigens are presented to CD8+ T cells (Figure 1). Cross presentation of exogenous antigens, normally presented by MHC-II on the surface of professional antigen presenting cells, can also be presented through the MHC-I pathway through endocytic mechanisms in which antigens are transported from the endosomal compartment into the cytosol and then degraded by the proteasome and transported into the ER (14). MHC-I expression is induced by both type I and type II interferons and this transcriptional activation is regulated by various conserved cis-regulatory elements at their promoters.
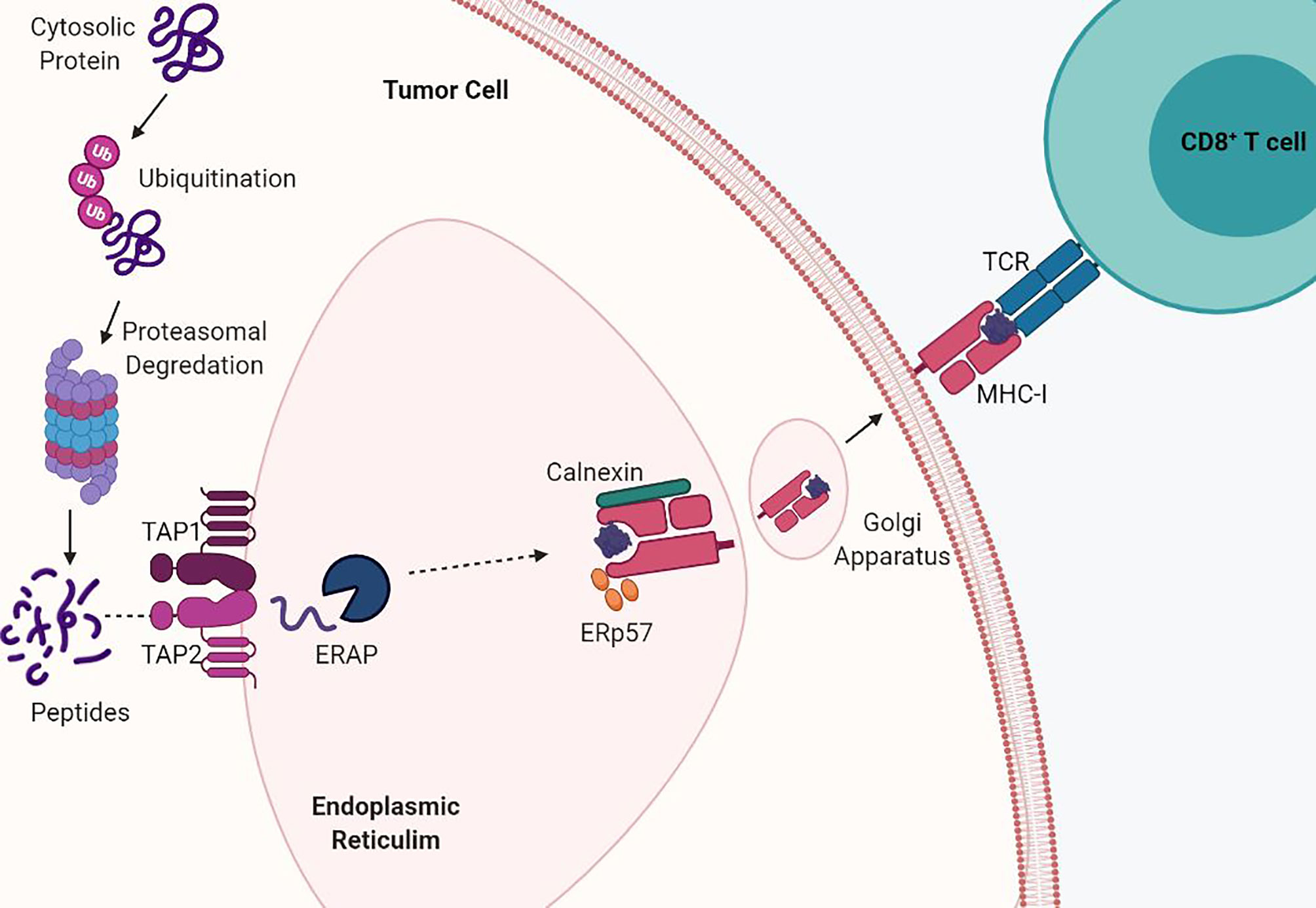
Figure 1 MHC-I antigen processing and presentation pathway. MHC-I presents endogenously derived peptide antigens to CD8+ T cells. Cytosolic proteins are degraded by the proteasome into peptides. These peptides are then shuttled into the endoplasmic reticulum (ER) by TAP transporter proteins. Antigen peptides are then loaded into the assembled MHC-I α-heavy chain and beta-2-microglobulin (β2M) complex and shuttled via the Golgi to the cell surface. Acquired mutations in the antigen presentation pathway drive loss of MHC-I.
Transcriptional regulation of MHC-I is mediated by three major promoter elements; enhancer A bound by nuclear factor (NF)-κB, interferon-stimulated response element (ISRE) bound by multiple interferon regulatory factor (IRF) family members, and the SYX-module bound by NOD-like receptor CARD domain containing 5 (NLRC5) (15–17). NF-κB induced class-I expression is largely functional at the HLA-A and HLA-B loci, containing NF-κB binding sites at their Enhancer A regions. In contrast, HLA-C and the non-classical alleles are not transactivated by NF-κB (16, 18).
The IRFs are upregulated in response to both type I interferons (IFNs) (e.g., IFNα and IFNβ) and type II IFNs (e.g., IFNγ) and signal through the JAK/STAT pathway (19). Following phosphorylation by JAK, STAT1/STAT2 dimerizes and translocates to the nucleus, where it binds to Gamma-activated sequence (GAS) elements of IFN-stimulated genes, including IRFs, which can subsequently activate the ISRE in the MHC-I promoter (15). It is important to note that studies show that IRF2 can be constitutively expressed and stable in the absence of IFN stimulation (20, 21). Therefore, not only is the IRSE promoter region implicated under inflammatory conditions, but it can also regulate basal MHC-I expression under non-inflammatory conditions. NLRC5 is a critical transcriptional regulator of MHC-I and its associated genes (e.g. β2M, TAP1/TAP2, and LMP2) (22). NLRC5 lacks a DNA-binding domain and is therefore dependent on the enhanceosome to connect to the promoter region of the SYX-module (23, 24). Activation of each of these three promoter regions facilitates antigen presentation by MHC-I on the tumor cell surface and its accessory molecules β2M, TAP, and LMP2.
Tumor progression is shaped by immunoediting eventually leading to escape from anti-tumor immune responses. Tumor specific MHC-I (tsMHC-I) downregulation has been described in numerous tumor types such as melanoma, breast, colorectal, and cervical cancers (25–28). Loss of tsMHC-I expression is often correlated with shorter overall and progression free survival, and higher instances of metastasis (29–32). The clinical significance of MHC-I is due to the role of antigen presentation and immune control of cancers. Tumors can reduce antigen presentation through several mechanisms including reduced surface expression of tsMHC-I through genetic alterations, antigen depletion, and modulation of transcription. The types of defects that lead to MHC-I disfunction can be divided into two groups: hard lesions (i.e., encoded in the DNA and largely therapeutically intractable) and soft lesions (i.e., those not encoded in the DNA and potentially therapeutically tractable) (Figure 2).
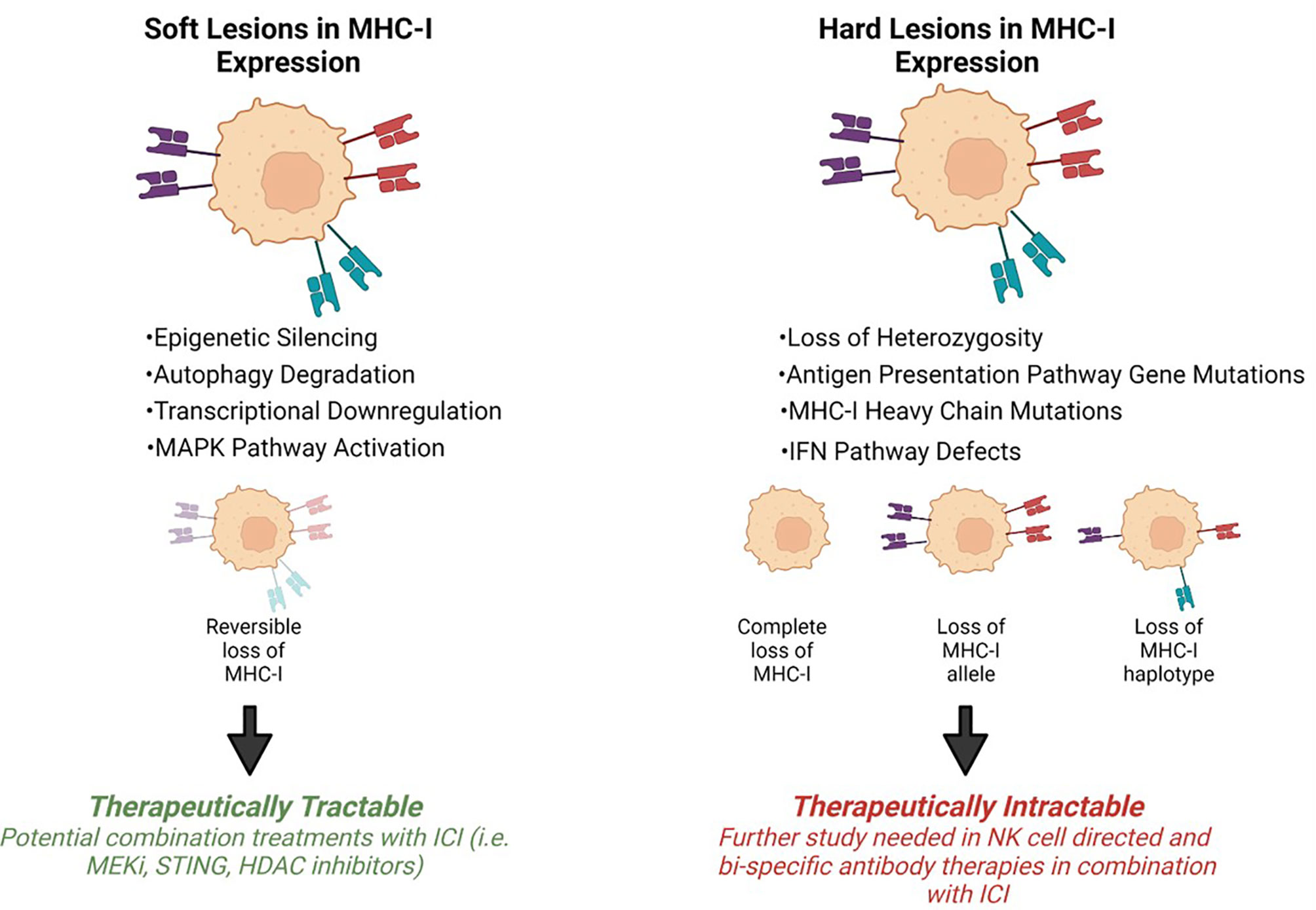
Figure 2 Mechanisms of MHC-I downregulation. Tumor specific MHC-I loss of expression can be classified into soft or hard lesions. MHC-I can be recovered via different treatment approaches depending on the type of mutation. Hard lesions, such as B2m loss, cannot be therapeutically recovered, however, soft lesions, like epigenetic silencing, can be therapeutically reversed.
Defects in the MHC-I antigen presentation pathway are a common mechanism by which tumors can evade the immune response. Reduced levels and complete loss of TAP1/TAP2 expression have been documented in colorectal cancer, renal cell carcinoma, and melanoma resulting in reduced tsMHC-I stability and expression on the tumor surface (33–37). TAP loss has been attributed to a functionally defective allele of Tap1 in numerous solid tumors and cell lines resulting in defective peptide presentation (38). Similarly, studies have shown reduction of tapasin and ERAP have resulted in loss of antigen presentation on the tumor surface (26, 39, 40). Genetic structural alterations of the MHC-I complex are another common finding in tumors that exhibit reduced antigen presentation. Numerous studies document loss of heterozygosity (LOH) associated with chromosome 6p21 resulting in HLA haplotype loss in tumors (41–45). One clinical study designed to test adoptive transfer of T-cells revealed that downregulation of HLA-C was linked with immune evasion and lack of response to treatment (46). Further, a heterozygous deleterious mutation or complete loss of β2M can lead to reduced or absent surface levels of tsMHC-I. Some cancers, such as lymphomas, have high β2M mutation rates in more than 25% of patients (47, 48). Additionally, truncating mutations of β2M were found in metastatic colon and melanoma cancers found to be resistant to the anti-PD-1 therapy, pembrolizumab (25, 49, 50). These observations show that somatic mutations in MHC-I genes are a common mechanism of tumor immune evasion.
The MHC-I pathway can also be dysregulated through the modulation of various transcription factors. NF-κB, bound to the enhancer A region in the MHC-I promoter, is found to be constitutively active in the majority of cancers (28). However, in some cancer types the loss of NF-κB expression results in MHC-I downregulation (21). This suppression of tsMHC-I expression via NF-κB can be attributed to multiple proteins, specifically Nedd4 Binding Protein 1 (N4BP1) and tumor necrosis factor (TNF)α-induced protein 3 interacting protein 1 (TNIP1) (51). In addition to NF-κB, IFNs dimerize STAT1 resulting in IRF binding to ISRE therefore playing a large role in MHC-I upregulation during inflammation. Downregulation of both type I and II IFN pathways have been identified as mechanisms leading to resistance to ICI and adoptive T-cell therapies (41, 52, 53). Lastly, the expression of NLRC5, a key regulator of MHC-I transcription, has been found to be correlated with MHC-I expression and patient survival in several cancer types including ovarian, breast, and colorectal cancers as a mechanism of immune evasion (24).
The expression of antigen presentation pathway genes can also be downregulated through epigenetic silencing. This can occur as a result of hypermethylation of the NLRC5 and MHC-I promoter regions (24, 38, 54–57). DNA hypermethylation has shown to be reversible with the treatment of DNA methyltransferase inhibitors (DNMTi) leading to MHC-I upregulation in numerous cancer types (58–63). Another common defect resulting in reduced tsMHC-I antigen presentation is deregulation of histone deacetylases (HDACs), specifically classes I, IIA, and IIB, which remove acetyl groups from histones resulting in a more closed chromatin structure preventing the transcription of MHC-I (59, 64, 65). HDAC inhibitors increase the expression of MHC-I both in vitro and in vivo. Many of these drugs are already FDA approved (e.g. panobinostat and vorinostat) while others are in clinical trials to determine their efficacy in various types of cancer (62, 64, 66, 67). Overall, these studies reveal the tumor-intrinsic role of epigenetic silencing to evade immune surveillance and the need for further study to understand the functional effect of these mechanisms in cancer.
In addition, loss of MHC-I in cancer can be due to post-transcriptional regulation mediated by non-coding RNAs. MicroRNAs (miRNA), a class of non-coding RNAs, can bind to the 3’ untranslated region (UTR) of mRNAs. Upregulation of some miRNAs have been reported to repress expression of MHC-I in multiple cancers, including melanoma, esophageal carcinoma, and colorectal cancer (68–70). MiRNAs can affect various parts of the antigen presentation pathway, including TAP1/TAP2 and calreticulin (68, 70, 71). For example, TAP2 and MHC-I expression has been shown to be inhibited in esophageal adenocarcinoma through overexpression of miRNA 125a-5p and 148-3p (69). Furthermore, suppression of chaperone proteins by miRNAs (e.g. calnexin, tapasin, and calreticulin) involved in antigen presentation can downregulate MHC-I expression and CD8+ T cell infiltration (70, 72, 73). Taken together, miRNAs can negatively regulate MHC-I through post-transcriptional modification and further study may potentially lead to new therapeutic targets.
Recent developments in cancer therapy have shown clear advantages in targeting the immune system. Immunotherapies, such as ICI, have revolutionized the treatment for a range of cancers types, such as melanoma and non-small cell lung cancer (74, 75). ICI increases anti-tumor immunity by blocking intrinsic checkpoints that have an inhibitory role in regulating T cell responses, such as cytotoxic T-lymphocyte antigen 4 (CTLA-4) and programmed cell death 1 (PD-1). Compared to previous standards of care (e.g., chemotherapy and surgery), ICI therapy has significantly improved overall and progression free survival in patients (76).
Despite these successes, ICI treatments still have many limitations and challenges. The benefits of ICI are still very low in some common tumor types, including breast and prostate cancer. ICI therapy is further complicated by observations of intertumoral heterogeneity in the degree of response to therapy. Additionally, a large percentage of cancer patients treated with immunotherapy experience intrinsic or acquired resistance, and many experience immune-related adverse events (77). There is currently a limited number of clinically approved multimodal treatments strategies to improve responsiveness to ICI. Furthermore, there are very few biomarkers that can predict immunotherapy benefit.
Acquired resistance to immunotherapy in several forms of cancer is associated with alterations in the MHC-I antigen presentation pathway. In several studies, downregulation of MHC-I is correlated with resistance to ICIs and adoptive T cell therapy (57, 78, 79). This is presumably mediated by defects in the expression of tsMHC-I for tumor antigen presentation to CD8+ T cells. Analysis of somatic genetic alterations through transcriptomic sequencing and IHC in longitudinal samples of melanoma patients revealed LOH of β2M was found to be a common mechanism of acquired resistance to anti-CTLA and anti-PD-1 therapies (25, 50, 80). Mechanisms of transcriptional loss and evolutionary divergence of the class I heavy chain of specific alleles have been reported in many cancers which results in the loss of MHC-I polymorphism leading to ICI resistance (44, 81, 82). Whole exome sequencing and single-cell transcriptome analysis of colorectal patients resistant to ICI therapy have identified β2M biallelic deletions and truncating mutations in both primary and metastatic tumor lesions (49, 83, 84). Furthermore, a recent study in vitro found that β2M suppression in non-small cell lung cancer may be caused by KRAS mutations and STAT5 inhibition (85). Transcriptome and flow cytometric analysis of longitudinal melanoma biopsies collected from patients receiving PD-1 inhibitors identified downregulation of MHC-I to also be associated with TGFβ activity, SNAI1 upregulation, and cancer associated fibroblasts (78). Uncovering additional mechanisms of tsMHC-I and β2M downregulation as potential biomarkers for ICI response and strategies for restoring their expression are areas that merit further study.
Recent studies have demonstrated that in response to anti-PD-1/PD-L1 agents, non-responders have a much higher rate of genomic alterations in the IFNγ pathway as compared to patients that exhibit a clinical benefit. JAK1 or JAK2 loss of function mutations were identified through whole exome sequencing of anti-PD-1 resistant melanoma tumors (25, 86). More specifically, one study determined that JAK1, not JAK2, is the primary mediator of IFNγ induced MHC-I expression and response to anti-PD-L1 therapy (87). Additionally, the transcription factor double homeobox 4 (DUX4) has been shown to block the IFNγ mediated upregulation of tsMHC-I in numerous cancer types in vitro resulting in failure to respond to anti-CTLA-4 and anti-PD-1 therapy and decreased overall survival (88). In addition, Ptpn2, was identified through an in vivo CRISPR screen to negatively regulate the JAK/STAT and antigen presentation pathways leading to ICI resistance (89). Taken together, these studies suggest that negative regulation of the IFNγ pathway is a mechanism of primary resistance to ICI therapy and supports the design of therapeutic strategies to overcome IFNγ downregulation.
TsMHC-I associates with improved response to ICI and increased amounts of tumor infiltrating lymphocytes (44, 81, 83, 87, 90)Loss of MHC-I antigen presentation is common in cancers and allows tumors to evade immune surveillance. Although MHC-I loss due to structural genetic alterations such as β2M loss are irreversible, post-transcriptional downregulation of MHC-I can be reversed (Figure 2). Therefore, novel strategies to upregulate tsMHC-I are anticipated to improve responses to immunotherapies.
One potential target of upregulating tsMHC-I is through the IFN signaling pathway. IFNγ has been shown to increase expression levels of various antigen presentation genes such as MHC-I and TAP (21, 91). Additionally, NLRC5 has been identified as a key transcriptional coactivator of MHC-I and antigen presentation pathway gene expression (23). Genetic alterations, copy number loss, and somatic mutations of NLRC5 results in reduced MHC-I expression in various cancer types (92). In melanomas derived from patients responding to anti-CTLA-4 and anti-PD-1, there was a significantly higher expression of NLRC5 compared to non-responding patients (57). It has been demonstrated in breast cancer in vitro models that NLRC5 was promoted by IFNγ with upregulation of MHC-I suggesting a potential benefit to ICI treatment (93). Although IFN may have a beneficial effect on immunotherapy response, there are limitations to using them in cancer therapy due to potentially life-threatening toxicities seen in patients (94). One possibility of using IFNs for therapeutic upregulation of tsMHC-I without the associated toxicity is through antibody directed, dose-dependent cytokine release at the tumor specific site (95). However, this approach is yet to be tested in large clinical trials.
Additionally, NF-κB pathway activation via immune receptors, such as the TNFα receptor, leads to downstream transcription of MHC-I by binding to the enhancer A regulatory element. Stimulation of NF-κB signaling to induce MHC-I upregulation in cancer is a potential therapeutic target. Retinoids, which are clinically used in neuroblastoma through the induction of apoptosis, have been described to induce MHC-I expression through NF-κB signaling (96, 97). Furthermore, in murine models of melanoma, in tumors with deficient IFN signaling MHC-I expression was restored by intratumoral injection of polyinosinic:polycytidylic acid, BO-112 (98). BO-112 restored MHC-I expression through an NF-κB mediated mechanism and independent of NLRC5. A recent study used a CRISPR screening approach to identify factors that decouple the regulation of MHC-I and PD-L1 (99). It was found that depletion of TRAF3 upregulated MHC-I through the NF-κB pathway and TRAF3 small molecule inhibitors increased immuno-sensitivity of cancer cells specifically through MHC-I expression. Stimulation of the NF-κB and IFN-pathways to induce MHC-I expression is further substantiated by studies of Stimulator of Interferon Genes (STING). STING stimulates both NF-κB and IFN-pathways and several STING agonists are being examined in clinical trials in several solid tumors in combination with anti-PD-1 (ClinicalTrials.gov Identifier: NCT04096638, NCT04144140). These studies demonstrate the potential for increasing tsMHC-I expression through transcriptional regulation and cytotoxic CD8+ T cell infiltration to improve patient responses to immunotherapy.
Numerous oncogenic pathways have been reported to affect the expression of MHC-I and related antigen presentation components, including the MAPK and EGFR pathways (100, 101). The MAPK pathway is thought to negatively influence MHC-I expression through decreased IRF and STAT1 expression resulting in reduced levels of tumor infiltrating lymphocytes (100, 102, 103). MEK inhibitors (MEKi), cobimetinib and trametinib, have been shown to enhance IRF1 expression and increased phosphorylation of STAT1 in human epidermal models (104). MEKi in breast cancer and non-small cell lung cancer models in vivo and in vitro have demonstrated an increase in MHC-I expression and combination of MEKi and ICIs may be a clinically relevant treatment option (79, 103). Overexpression of EGFR has been associated with poor response outcomes to ICI in various cancer models, including non-small cell lung cancer and neuroblastoma (105, 106)Also, the EGFR family member, HER2, was shown to be inversely correlated with MHC-I expression in breast cancer and impairs T cell recognition of peptide: MHC-I complexes (107, 108). Targeting EGFR receptors with antibodies and inhibitors, have resulted in increased expression of MHC-I and antigen presentation components in several cancer cell lines, thus increasing their susceptibility to CD8+ T cell cytotoxicity (109–111). Together, these studies show the importance of targeting oncogenic pathways regulating MHC-I in cancer and the potential for therapeutically restoring anti-tumor immunity and improving immunotherapy response.
A recent study showed that in pancreatic ductal adenocarcinoma (PDAC) MHC-I molecules are selectively targeted for lysosomal degradation through an autophagy-dependent mechanism (112). This study demonstrated that MHC-I is decreased in PDAC through a NBR1-mediated autophagy-lysosomal pathway. It was found that autophagy inhibition using a clinically available antimalarial agent, chloroquine, increased surface levels of MHC-I and anti-tumor T cell responses when combined with ICI therapy (anti-PD-1 and anti-CTLA4). In breast cancer the gene MAL2, which encodes a transmembrane protein associated with protein endocytosis, promotes the turnover of pMHC-I complexes by modulating the interaction between RAB7 and MHC-I (85). MAL2 plays a critical role in the endocytosis-mediated protein degradation of the MHC-I complex and downregulates CD8+ T cell cytotoxicity. Depletion of MAL2 enhanced antigen presentation on tumor cells and activation of CD8+ cytotoxic T cells. Therefore, inhibition of MAL2 is a potentially effective strategy for increasing the efficacy of immunotherapy. Studies have also revealed that high calnexin expression can promote tumor proliferation and growth in lung cancer (113, 114). In both in vitro and an in vivo melanoma models, Scaffold/Matrix- Associated Region-1 (SMAR1) is directly involved in immune evasion mechanisms by positively regulating MHC-I expression through the downregulation of calnexin (115). Calnexin gene expression was found to be suppressed by the formation of a repression complex that binds to the matrix attachment region (MAR) site on the calnexin promoter. This supports a therapeutic need for small molecule compounds for stabilizing SMAR1 expression, increasing CD8+ cytotoxic T cell infiltration to aid ICI treatment.
Other strategies to upregulate tsMHC-I expression are actively being investigated to ameliorate immunotherapy resistance. Current standards of care in oncology like chemotherapy and radiation are known to induce anti-tumor immune responses without stimulating immunogenic cell death. Studies on various tumor cells in vitro have shown that some chemotherapeutic agents augment tsMHC-I expression and in vivo further induce tumor CD8+ T cell infiltration. Docetaxel, a common chemotherapeutic agent, can increase expression of components of the antigen presentation pathway, such as TAP and tapasin, without inducing immunogenic cell death in vitro (116). Topoisomerase inhibitors (e.g. etoposide) and microtubule stabilizers (e.g. paclitaxel) have also been shown to stimulate MHC-I expression and Treg depletion in murine models (8, 117, 118). These pre-clinical studies indicate a therapeutic advantage to increasing tsMHC-I antigen presentation through combination of ICI and chemotherapeutic agents.
As mentioned above, MHC-I downregulation can be categorized into ‘hard’ and ‘soft’ lesions. ‘Hard’ lesions resulting in MHC-I aberrations are irreversible and recovery of these genes is typically only possible by gene editing (5, 119). This has been done through recombinant adenovirus carrying the β2m gene into deficient cells resulting in the induction of MHC-I on the surface (120). Additionally, T-cell bispecific antibodies have been shown to restore cytotoxic T cell activity in tumors that lack MHC-I expression via activating immune synapses in cancer cells without dependency on pMHC-I: cognate TCR interactions (121, 122). Additionally in TAP-deficient tumor cells, it a novel class of antigens presented by these cancers were discovered termed T cell epitopes associated with impaired peptide processing (TEIPP) (123). TEIPP targeting CD8+ T cells could be therapeutically utilized for immunotherapy in TAP-deficient tumor cells with low levels of MHC-I (124).
Although downregulation of tsMHC-I allows tumor evasion from T cell mediated cytotoxicity, Natural Killer (NK) cell activation may hold therapeutic premise in heterogenous MHC-I tumors. NK-cells have inhibitory receptors on their surface (e.g. killer cell immunoglobulin-like receptors (KIRs) and MHC-I can function as a ligand to these receptors. Tumors with absent MHC-I expression can be susceptible to NK-cell mediated cytotoxicity because the KIR-mediated inhibitory signaling is lost, a process known as “missing-self recognition” (125). However, tumor cells can evade immunosurveillance through inhibitory cytokines, such as transforming growth factor-β (TGFβ), that suppress NK-cell activity. The plasticity of tumors evading both T- and NK- cell mediated cytotoxicity resulting in tumor escape substantiates the importance of tsMHC-I expression and the key role it plays in patient overall prognosis.
MHC-I mediated antigen presentation is critical for CD8+ T-cell responses and plays a key role in the adaptive immune response. Although MHC-I is expressed on a wide range of tumor cells, it is often downregulated to evade anti-tumor immunity. There are many mechanisms that can lead to the loss of MHC-I antigen presentation which include defects in the antigen presentation pathway, loss of transcription factors, epigenetic silencing of gene regulatory elements, loss of MHC-I polymorphism, and defects in regulatory signaling pathways. It is important to further understand these mechanisms of MHC-I downregulation and their clinical significance. Furthermore, tsMHC-I has been associated with improved response to anti-PD-1/anti-PD-L1 immunotherapy agents in cancer patients and may be a predictive biomarker in clinical practice. Therefore, identifying novel strategies to upregulate and/or restore tsMHC-I and related pathway components may lead to enhanced anti-tumor response to ICIs.
BT and JB contributed to the writing of the final manuscript. All authors contributed to the article and approved the submitted version.
Funding for this review was provided by NIC/NCI SPORE 2P50CA098131-17 (JB) and Department of Defense Era of Hope Award BC170037 (JB), the Vanderbilt-Ingram Cancer Center Support Grant P30 CA68485, and the NIH T32 CA009592 (BT).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
BioRender.com. was used to make the figures.
1. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
2. Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, et al. Immune Evasion in Cancer: Mechanistic Basis and Therapeutic Strategies. Semin Cancer Biol (2015) 35:S185–98. doi: 10.1016/j.semcancer.2015.03.004
3. Trowsdale J. Genomic Structure and Function in the MHC. Trends Genet (1993) 9:117–22. doi: 10.1016/0168-9525(93)90205-V
4. Anderson P, Aptsiauri N, Ruiz-Cabello F, Garrido F. HLA Class I Loss in Colorectal Cancer: Implications for Immune Escape and Immunotherapy. Cell Mol Immunol (2021) 18:556–65. doi: 10.1038/s41423-021-00634-7
5. Hazini A, Fisher K, Seymour L. Deregulation of HLA-I in Cancer and its Central Importance for Immunotherapy. J Immunother Cancer (2021) 9:e002899. doi: 10.1136/jitc-2021-002899
6. Hurkmans DP, Kuipers ME, Smit J, van Marion R, Mathijssen RHJ, Postmus PE, et al. Tumor Mutational Load, CD8+ T Cells, Expression of PD-L1 and HLA Class I to Guide Immunotherapy Decisions in NSCLC Patients. Cancer Immunol Immunother (2020) 69:771–7. doi: 10.1007/s00262-020-02506-x
7. Montesion M, Murugesan K, Jin DX, Sharaf R, Sanchez N, Guria A, et al. Somatic HLA Class I Loss Is a Widespread Mechanism of Immune Evasion Which Refines the Use of Tumor Mutational Burden as a Biomarker of Checkpoint Inhibitor Response. Cancer Discovery (2021) 11:282–92. doi: 10.1158/2159-8290.CD-20-0672
8. Cornel AM, Mimpen IL, Nierkens S. MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy. Cancers (Basel) (2020) 12. doi: 10.3390/cancers12071760
9. Paul S, Weiskopf D, Angelo MA, Sidney J, Peters B, Sette A. HLA Class I Alleles are Associated With Peptide Binding Repertoires of Different Size, Affinity and Immunogenicity. J Immunol (2013) 191:5831–9. doi: 10.4049/jimmunol.1302101
10. Michalek MT, Grant EP, Gramm C, Goldberg AL, Rock KL. A Role for the Ubiquitin-Dependent Proteolytic Pathway in MHC Class L-Restricted Antigen Presentation. Nature (1993) 363:552–4. doi: 10.1038/363552a0
11. Roelse J, Grommé M, Momburg F, Hämmerling G, Neefjes J. Trimming of TAP-Translocated Peptides in the Endoplasmic Reticulum and in the Cytosol During Recycling. J Exp Med (1994) 180:1591–7. doi: 10.1084/jem.180.5.1591
12. Kaer LV, Ashton-Rickardt PG, Ploegh HL, Tonegawa S. TAP1 Mutant Mice are Deficient in Antigen Presentation, Surface Class I Molecules, and CD4–8+ T Cells. Cell (1992) 71:1205–14. doi: 10.1016/S0092-8674(05)80068-6
13. Chapman DC, Williams DB. ER Quality Control in the Biogenesis of MHC Class I Molecules. Semin Cell Dev Biol (2010) 21:512–9. doi: 10.1016/j.semcdb.2009.12.013
14. Gutiérrez-Martínez E, Planès R, Anselmi G, Reynolds M, Menezes S, Adiko AC, et al. Cross-Presentation of Cell-Associated Antigens by MHC Class I in Dendritic Cell Subsets. Front Immunol (2015) 6:363. doi: 10.3389/fimmu.2015.00363
15. Jongsma MLM, Guarda G, Spaapen RM. The Regulatory Network Behind MHC Class I Expression. Mol Immunol (2019) 113:16–21. doi: 10.1016/j.molimm.2017.12.005
16. Gobin SJP, Keijsers V, van Zutphen M, van den Elsen PJ. The Role of Enhancer A in the Locus-Specific Transactivation of Classical and Nonclassical HLA Class I Genes by Nuclear Factor κb. J Immunol (1998) 161:2276–83.
17. Gobin SJP, Peijnenburg A, Keijsers V, van den Elsen PJ. Site α Is Crucial for Two Routes of Ifnγ-Induced MHC Class I Transactivation: The ISRE-Mediated Route and a Novel Pathway Involving CIITA. Immunity (1997) 6:601–11. doi: 10.1016/S1074-7613(00)80348-9
18. Perkins ND, Edwards NL, Duckett CS, Agranoff AB, Schmid RM, Nabel GJ. A Cooperative Interaction Between NF-Kappa B and Sp1 Is Required for HIV-1 Enhancer Activation. EMBO J (1993) 12:3551–8. doi: 10.1002/j.1460-2075.1993.tb06029.x
19. Chang C-H, Hammer J, Loh JE, Fodor WL, Flavell RA. The Activation of Major Histocompatibility Complex Class I Genes by Interferon Regulatory Factor-1 (IRF-1). Immunogenetics (1992) 35:378–84. doi: 10.1007/BF00179793
20. Watanabe N, Sakakibara J, Hovanessian AG, Taniguchi T, Fujita T. Activation of IFN- β Element by IRF-1 Requires a Post-Translational Event in Addition to IRF-1 Synthesis. Nucleic Acids Res (1991) 19:4421–8. doi: 10.1093/nar/19.16.4421
21. Lorenzi S, Forloni M, Cifaldi L, Antonucci C, Citti A, Boldrini R, et al. IRF1 and NF-kB Restore MHC Class I-Restricted Tumor Antigen Processing and Presentation to Cytotoxic T Cells in Aggressive Neuroblastoma. PloS One (2012) 7:e46928. doi: 10.1371/journal.pone.0046928
22. Downs I, Vijayan S, Sidiq T, Kobayashi KS. CITA/NLRC5: A Critical Transcriptional Regulator of MHC Class I Gene Expression. BioFactors (2016) 42:349–57. doi: 10.1002/biof.1285
23. Meissner TB, Li A, Liu Y-J, Gagnon E, Kobayashi KS. The Nucleotide-Binding Domain of NLRC5 Is Critical for Nuclear Import and Transactivation Activity. Biochem Biophys Res Commun (2012) 418:786–91. doi: 10.1016/j.bbrc.2012.01.104
24. Yoshihama S, Roszik J, Downs I, Meissner TB, Vijayan S, Chapuy B, et al. NLRC5/MHC Class I Transactivator Is a Target for Immune Evasion in Cancer. Proc Natl Acad Sci U.S.A. (2016) 113:5999–6004. doi: 10.1073/pnas.1602069113
25. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated With Acquired Resistance to PD-1 Blockade in Melanoma. New Engl J Med (2016) 375:819–29. doi: 10.1056/NEJMoa1604958
26. Mehta AM, Jordanova ES, Kenter GG, Ferrone S, Fleuren G-J. Association of Antigen Processing Machinery and HLA Class I Defects With Clinicopathological Outcome in Cervical Carcinoma. Cancer Immunol Immunother (2008) 10:197–206. doi: 10.1007/s00262-007-0362-8
27. Cabrera T, Fernandez MA, Sierra A, Garrido A, Herruzo A, Escobedo A, et al. High Frequency of Altered HLA Class I Phenotypes in Invasive Breast Carcinomas. Hum Immunol (1996) 50:127–34. doi: 10.1016/0198-8859(96)00145-0
28. Menon AG, Morreau H, Tollenaar RAEM, Alphenaar E, van Puijenbroek M, Putter H, et al. Down-Regulation of HLA-A Expression Correlates With a Better Prognosis in Colorectal Cancer Patients. Lab Invest (2002) 82:1725–33. doi: 10.1097/01.LAB.0000043124.75633.ED
29. Park HS, Cho U, Im SY, Yoo CY, Jung JH, Suh YJ, et al. Loss of Human Leukocyte Antigen Class I Expression Is Associated With Poor Prognosis in Patients With Advanced Breast Cancer. J Pathol Transl Med (2019) 53:75–85. doi: 10.4132/jptm.2018.10.11
30. Watson NFS, Ramage JM, Madjd Z, Spendlove I, Ellis IO, Scholefield JH, et al. Immunosurveillance Is Active in Colorectal Cancer as Downregulation But Not Complete Loss of MHC Class I Expression Correlates With a Poor Prognosis. Int J Cancer (2006) 118:6–10. doi: 10.1002/ijc.21303
31. Hanagiri T, Shigematsu Y, Kuroda K, Baba T, Shiota H, Ichiki Y, et al. Prognostic Implications of Human Leukocyte Antigen Class I Expression in Patients Who Underwent Surgical Resection for non-Small-Cell Lung Cancer. J Surg Res (2013) 181:e57–63. doi: 10.1016/j.jss.2012.07.029
32. Cordon-Cardo C, Fuks Z, Drobnjak M, Moreno C, Eisenbach L, Feldman M. Expression of HLA-A,B,C Antigens on Primary and Metastatic Tumor Cell Populations of Human Carcinomas. Cancer Res (1991) 51:6372–80.
33. Seliger B, Höhne A, Knuth A, Bernhard H, Ehring B, Tampé R, et al. Reduced Membrane Major Histocompatibility Complex Class I Density and Stability in a Subset of Human Renal Cell Carcinomas With Low TAP and LMP Expression. Clin Cancer Res (1996) 2:1427–33.
34. Kaklamanis L, Townsend A, Doussis-Anagnostopoulou IA, Mortensen N, Harris AL, Gatter KC. Loss of Major Histocompatibility Complex-Encoded Transporter Associated With Antigen Presentation (TAP) in Colorectal Cancer. Am J Pathol (1994) 145:505–9.
35. Seliger B, Maeurer MJ, Ferrone S. TAP Off — Tumors on. Immunol Today (1997) 18:292–9. doi: 10.1016/S0167-5699(97)80026-6
36. Bernal M, García-Alcalde F, Concha A, Cano C, Blanco A, Garrido F, et al. Genome-Wide Differential Genetic Profiling Characterizes Colorectal Cancers With Genetic Instability and Specific Routes to HLA Class I Loss and Immune Escape. Cancer Immunol Immunother (2012) 61:803–16. doi: 10.1007/s00262-011-1147-7
37. Vitale M, Rezzani R, Rodella L, Zauli G, Grigolato P, Cadei M, et al. HLA Class I Antigen and Transporter Associated With Antigen Processing (TAP1 and TAP2) Down-Regulation in High-Grade Primary Breast Carcinoma Lesions. Cancer Res (1998) 58:737–42.
38. Ye Q, Shen Y, Wang X, Yang J, Miao F, Shen C, et al. Hypermethylation of HLA Class I Gene Is Associated With HLA Class I Down-Regulation in Human Gastric Cancer. Tissue Antigens (2010) 75:30–9. doi: 10.1111/j.1399-0039.2009.01390.x
39. Shionoya Y, Kanaseki T, Miyamoto S, Tokita S, Hongo A, Kikuchi Y, et al. Loss of Tapasin in Human Lung and Colon Cancer Cells and Escape From Tumor-Associated Antigen-Specific CTL Recognition. OncoImmunology (2017) 6:e1274476. doi: 10.1080/2162402X.2016.1274476
40. Stratikos E, Stamogiannos A, Zervoudi E, Fruci D. A Role for Naturally Occurring Alleles of Endoplasmic Reticulum Aminopeptidases in Tumor Immunity and Cancer Pre-Disposition. Front Oncol (2014) 4:363. doi: 10.3389/fonc.2014.00363
41. Maleno I, López-Nevot M, Cabrera T, Salinero J, Garrido F. Multiple Mechanisms Generate HLA Class I Altered Phenotypes in Laryngeal Carcinomas: High Frequency of HLA Haplotype Loss Associated With Loss of Heterozygosity in Chromosome Region 6p21. Cancer Immunol Immunother (2002) 51:389–96. doi: 10.1007/s00262-002-0296-0
42. McGranahan N, Swanton C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell (2017) 168:613–28. doi: 10.1016/j.cell.2017.01.018
43. Yu S, Zhao Z, Chen L, Gu T, Yu H, Tang H, et al. HLA Loss of Heterozygosity-Mediated Discordant Responses to Immune Checkpoint Blockade in Squamous Cell Lung Cancer With Renal Metastasis. Immunotherapy (2021) 13:195–200. doi: 10.2217/imt-2020-0173
44. Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, Makarov V, et al. Patient HLA Class I Genotype Influences Cancer Response to Checkpoint Blockade Immunotherapy. Science (2018) 359:582–7. doi: 10.1126/science.aao4572
45. Lobashevsky AL, Krueger-Sersen M, Britton RM, Littrell CA, Singh S, Cui CP, et al. Pretransplant HLA Typing Revealed Loss of Heterozygosity in the Major Histocompatibility Complex in a Patient With Acute Myeloid Leukemia. Hum Immunol (2019) 80:257–62. doi: 10.1016/j.humimm.2019.02.009
46. Tran E, Robbins PF, Lu Y-C, Prickett TD, Gartner JJ, Jia L, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. New Engl J Med (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
47. Toth DF, Raderer M, Wadsak W, Karanikas G. Beta-2 Microglobulin as a Diagnostic Parameter in Non-Hodgkin Lymphoma: A Comparative Study With FDG-PET. Anticancer Res (2013) 33:3341–5.
48. Castro A, Ozturk K, Pyke RM, Xian S, Zanetti M, Carter H. Elevated Neoantigen Levels in Tumors With Somatic Mutations in the HLA-A, HLA-B, HLA-C and B2M Genes. BMC Med Genomics (2019) 12:107. doi: 10.1186/s12920-019-0544-1
49. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science (2017) 357:409–13. doi: 10.1126/science.aan6733
50. Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane J-P, et al. Resistance to Checkpoint Blockade Therapy Through Inactivation of Antigen Presentation. Nat Commun (2017) 8:1136. doi: 10.1038/s41467-017-01062-w
51. Spel L, Nieuwenhuis J, Haarsma R, Stickel E, Bleijerveld OB, Altelaar M, et al. Nedd4-Binding Protein 1 and TNFAIP3-Interacting Protein 1 Control MHC-1 Display in Neuroblastoma. Cancer Res (2018) 78:6621–31. doi: 10.1158/0008-5472.CAN-18-0545
52. Yan Y, Zheng L, Du Q, Yan B, Geller DA. Interferon Regulatory Factor 1 (IRF-1) and IRF-2 Regulate PD-L1 Expression in Hepatocellular Carcinoma (HCC) Cells. Cancer Immunol Immunother (2020) 69:1891–903. doi: 10.1007/s00262-020-02586-9
53. Kriegsman BA, Vangala P, Chen BJ, Meraner P, Brass AL, Garber M, et al. Frequent Loss of IRF2 in Cancers Leads to Immune Evasion Through Decreased MHC Class I Antigen Presentation and Increased PD-L1 Expression. J Immunol (2019) 203:1999–2010. doi: 10.4049/jimmunol.1900475
54. Burr ML, Sparbier CE, Chan KL, Chan Y-C, Kersbergen A, Lam EYN, et al. An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer Cell (2019) 36:385–401.e8. doi: 10.1016/j.ccell.2019.08.008
55. Nie Y, Yang G, Song Y, Zhao X, So C, Liao J, et al. DNA Hypermethylation Is a Mechanism for Loss of Expression of the HLA Class I Genes in Human Esophageal Squamous Cell Carcinomas. Carcinogenesis (2001) 22:1615–23. doi: 10.1093/carcin/22.10.1615
56. Cho SX, Vijayan S, Yoo J-S, Watanabe T, Ouda R, An N, et al. MHC Class I Transactivator NLRC5 in Host Immunity, Cancer and Beyond. Immunology (2021) 162:252–61. doi: 10.1111/imm.13235
57. Yoshihama S, Cho SX, Yeung J, Pan X, Lizee G, Konganti K, et al. NLRC5/CITA Expression Correlates With Efficient Response to Checkpoint Blockade Immunotherapy. Sci Rep (2021) 11:3258. doi: 10.1038/s41598-021-82729-9
58. Luo N, Nixon MJ, Gonzalez-Ericsson PI, Sanchez V, Opalenik SR, Li H, et al. DNA Methyltransferase Inhibition Upregulates MHC-I to Potentiate Cytotoxic T Lymphocyte Responses in Breast Cancer. Nat Commun (2018) 9:248. doi: 10.1038/s41467-017-02630-w
59. Fonsatti E, Nicolay HJM, Sigalotti L, Calabrò L, Pezzani L, Colizzi F, et al. Functional Up-Regulation of Human Leukocyte Antigen Class I Antigens Expression by 5-Aza-2’-Deoxycytidine in Cutaneous Melanoma: Immunotherapeutic Implications. Clin Cancer Res (2007) 13:3333–8. doi: 10.1158/1078-0432.CCR-06-3091
60. Srivastava P, Paluch BE, Matsuzaki J, James SR, Collamat-Lai G, Taverna P, et al. Immunomodulatory Action of the DNA Methyltransferase Inhibitor SGI-110 in Epithelial Ovarian Cancer Cells and Xenografts. Epigenetics (2015) 10:237–46. doi: 10.1080/15592294.2015.1017198
61. Saleh MH, Wang L, Goldberg MS. Improving Cancer Immunotherapy With DNA Methyltransferase Inhibitors. Cancer Immunol Immunother (2016) 65:787–96. doi: 10.1007/s00262-015-1776-3
62. Moufarrij S, Srivastava A, Gomez S, Hadley M, Palmer E, Austin PT, et al. Combining DNMT and HDAC6 Inhibitors Increases Anti-Tumor Immune Signaling and Decreases Tumor Burden in Ovarian Cancer. Sci Rep (2020) 10:3470. doi: 10.1038/s41598-020-60409-4
63. Siebenkäs C, Chiappinelli KB, Guzzetta AA, Sharma A, Jeschke J, Vatapalli R, et al. Inhibiting DNA Methylation Activates Cancer Testis Antigens and Expression of the Antigen Processing and Presentation Machinery in Colon and Ovarian Cancer Cells. PloS One (2017) 12:e0179501. doi: 10.1371/journal.pone.0179501
64. Glozak MA, Seto E. Histone Deacetylases and Cancer. Oncogene (2007) 26:5420–32. doi: 10.1038/sj.onc.1210610
65. Sun T, Li Y, Yang W, Wu H, Li X, Huang Y, et al. Histone Deacetylase Inhibition Up-Regulates MHC Class I to Facilitate Cytotoxic T Lymphocyte-Mediated Tumor Cell Killing in Glioma Cells. J Cancer (2019) 10:5638–45. doi: 10.7150/jca.34471
66. Mora-García M de L, Duenas-González A, Hernández-Montes J, de la Cruz-Hernández E, Pérez-Cárdenas E, Weiss-Steider B, et al. Up-Regulation of HLA Class-I Antigen Expression and Antigen-Specific CTL Response in Cervical Cancer Cells by the Demethylating Agent Hydralazine and the Histone Deacetylase Inhibitor Valproic Acid. J Transl Med (2006) 4:55. doi: 10.1186/1479-5876-4-55
67. Suraweera A, O’Byrne KJ, Richard DJ. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front Oncol (2018) 8:92. doi: 10.3389/fonc.2018.00092
68. Lazaridou M-F, Gonschorek E, Massa C, Friedrich M, Handke D, Mueller A, et al. Identification of miR-200a-5p Targeting the Peptide Transporter TAP1 and its Association With the Clinical Outcome of Melanoma Patients. OncoImmunology (2020) 9:1774323. doi: 10.1080/2162402X.2020.1774323
69. Mari L, Hoefnagel SJM, Zito D, van de Meent M, van Endert P, Calpe S, et al. microRNA 125a Regulates MHC-I Expression on Esophageal Adenocarcinoma Cells, Associated With Suppression of Antitumor Immune Response and Poor Outcomes of Patients. Gastroenterology (2018) 155:784–98. doi: 10.1053/j.gastro.2018.06.030
70. Colangelo T, Polcaro G, Ziccardi P, Pucci B, Muccillo L, Galgani M, et al. Proteomic Screening Identifies Calreticulin as a miR-27a Direct Target Repressing MHC Class I Cell Surface Exposure in Colorectal Cancer. Cell Death Dis (2016) 7:e2120–0. doi: 10.1038/cddis.2016.28
71. Yi M, Xu L, Jiao Y, Luo S, Li A, Wu K. The Role of Cancer-Derived microRNAs in Cancer Immune Escape. J Hematol Oncol (2020) 13:25. doi: 10.1186/s13045-020-00848-8
72. Hirata T, Yamamoto H, Taniguchi H, Horiuchi S, Oki M, Adachi Y, et al. Characterization of the Immune Escape Phenotype of Human Gastric Cancers With and Without High-Frequency Microsatellite Instability. J Pathol (2007) 211:516–23. doi: 10.1002/path.2142
73. Liu L, Wang Q, Qiu Z, Kang Y, Liu J, Ning S, et al. Noncoding RNAs: The Shot Callers in Tumor Immune Escape. Sig Transduct Target Ther (2020) 5:1–24. doi: 10.1038/s41392-020-0194-y
74. Ribas A, Wolchok JD. Cancer Immunotherapy Using Checkpoint Blockade. Science (2018) 359:1350–5. doi: 10.1126/science.aar4060
75. Leach DR, Krummel MF, Allison JP. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science (1996) 271:1734–6. doi: 10.1126/science.271.5256.1734
76. Bai R, Chen N, Li L, Du N, Bai L, Lv Z, et al. Mechanisms of Cancer Resistance to Immunotherapy. Front Oncol (2020) 10:1290. doi: 10.3389/fonc.2020.01290
77. Postow MA, Sidlow R, Hellmann MD. Immune-Related Adverse Events Associated With Immune Checkpoint Blockade. New Engl J Med (2018) 378:158–68. doi: 10.1056/NEJMra1703481
78. Lee JH, Shklovskaya E, Lim SY, Carlino MS, Menzies AM, Stewart A, et al. Transcriptional Downregulation of MHC Class I and Melanoma De- Differentiation in Resistance to PD-1 Inhibition. Nat Commun (2020) 11:1897. doi: 10.1038/s41467-020-15726-7
79. Lauss M, Donia M, Harbst K, Andersen R, Mitra S, Rosengren F, et al. Mutational and Putative Neoantigen Load Predict Clinical Benefit of Adoptive T Cell Therapy in Melanoma. Nat Commun (2017) 8:1738. doi: 10.1038/s41467-017-01460-0
80. Rodig SJ, Gusenleitner D, Jackson DG, Gjini E, Giobbie-Hurder A, Jin C, et al. MHC Proteins Confer Differential Sensitivity to CTLA-4 and PD-1 Blockade in Untreated Metastatic Melanoma. Sci Trans Med (2018) 10. doi: 10.1126/scitranslmed.aar3342
81. Paulson KG, Voillet V, McAfee MS, Hunter DS, Wagener FD, Perdicchio M, et al. Acquired Cancer Resistance to Combination Immunotherapy From Transcriptional Loss of Class I HLA. Nat Commun (2018) 9:3868. doi: 10.1038/s41467-018-06300-3
82. Roerden M, Nelde A, Heitmann JS, Klein R, Rammensee H-G, Bethge WA, et al. HLA Evolutionary Divergence as a Prognostic Marker for AML Patients Undergoing Allogeneic Stem Cell Transplantation. Cancers (2020) 12:1835. doi: 10.3390/cancers12071835
83. Gurjao C, Liu D, Hofree M, AlDubayan SH, Wakiro I, Su M-J, et al. Intrinsic Resistance to Immune Checkpoint Blockade in a Mismatch Repair–Deficient Colorectal Cancer. Cancer Immunol Res (2019) 7:1230–6. doi: 10.1158/2326-6066.CIR-18-0683
84. Grasso CS, Giannakis M, Wells DK, Hamada T, Mu XJ, Quist M, et al. Genetic Mechanisms of Immune Evasion in Colorectal Cancer. Cancer Discovery (2018) 8:730–49. doi: 10.1158/2159-8290.CD-17-1327
85. Fang Y, Wang L, Wan C, Sun Y, der Jeught KV, Zhou Z, et al. MAL2 Drives Immune Evasion in Breast Cancer by Suppressing Tumor Antigen Presentation. J Clin Invest (2021) 131. doi: 10.1172/JCI140837
86. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discovery (2017) 7:188–201. doi: 10.1158/2159-8290.CD-16-1223
87. Luo N, Formisano L, Gonzalez-Ericsson PI, Sanchez V, Dean PT, Opalenik SR, et al. Melanoma Response to Anti-PD-L1 Immunotherapy Requires JAK1 Signaling, But Not JAK2. Oncoimmunology (2018) 7:e1438106. doi: 10.1080/2162402X.2018.1438106
88. Chew G-L, Campbell AE, De Neef E, Sutliff NA, Shadle SC, Tapscott SJ, et al. DUX4 Suppresses MHC Class I to Promote Cancer Immune Evasion and Resistance to Checkpoint Blockade. Dev Cell (2019) 50:658–71.e7. doi: 10.1016/j.devcel.2019.06.011
89. Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, et al. In Vivo CRISPR Screening Identifies Ptpn2 as a Cancer Immunotherapy Target. Nature (2017) 547:413–8. doi: 10.1038/nature23270
90. Zhao Y, Cao Y, Chen Y, Wu L, Hang H, Jiang C, et al. B2M Gene Expression Shapes the Immune Landscape of Lung Adenocarcinoma and Determines the Response to Immunotherapy. Immunology (2021) 164:507–23. doi: 10.1111/imm.13384
91. Propper DJ, Chao D, Braybrooke JP, Bahl P, Thavasu P, Balkwill F, et al. Low-Dose IFN-γ Induces Tumor MHC Expression in Metastatic Malignant Melanoma. Clin Cancer Res (2003) 9:84–92.
92. Shukla A, Cloutier M, Appiya Santharam M, Ramanathan S, Ilangumaran S. The MHC Class-I Transactivator NLRC5: Implications to Cancer Immunology and Potential Applications to Cancer Immunotherapy. Int J Mol Sci (2021) 22:1964. doi: 10.3390/ijms22041964
93. Zhao M-Z, Sun Y, Jiang X-F, Liu L, Liu L, Sun L-X. Promotion on NLRC5 Upregulating MHC-I Expression by IFN-γ in MHC-I–deficient Breast Cancer Cells. Immunol Res (2019) 67:497–504. doi: 10.1007/s12026-019-09111-w
94. George PM, Badiger R, Alazawi W, Foster GR, Mitchell JA. Pharmacology and Therapeutic Potential of Interferons. Pharmacol Ther (2012) 135:44–53. doi: 10.1016/j.pharmthera.2012.03.006
95. Hemmerle T, Neri D. The Dose-Dependent Tumor Targeting of Antibody–Ifnγ Fusion Proteins Reveals an Unexpected Receptor-Trapping Mechanism. In Vivo Cancer Immunol Res (2014) 2:559–67. doi: 10.1158/2326-6066.CIR-13-0182
96. Segars JH, Nagata T, Bours V, Medin JA, Franzoso G, Blanco JC, et al. Retinoic Acid Induction of Major Histocompatibility Complex Class I Genes in NTera-2 Embryonal Carcinoma Cells Involves Induction of NF-Kappa B (P50-P65) and Retinoic Acid Receptor Beta-Retinoid X Receptor Beta Heterodimers. Mol Cell Biol (1993) 13:6157–69. doi: 10.1128/mcb.13.10.6157
97. Farina AR, Masciulli M-P, Tacconelli A, Cappabianca L, De Santis G, Gulino A, et al. All-Trans-Retinoic Acid Induces Nuclear Factor kappaB Activation and Matrix Metalloproteinase-9 Expression and Enhances Basement Membrane Invasivity of Differentiation-Resistant Human SK-N-BE 9N Neuroblastoma Cells. Cell Growth Differ (2002) 13:343–54.
98. Kalbasi A, Tariveranmoshabad M, Hakimi K, Kremer S, Campbell KM, Funes JM, et al. Uncoupling Interferon Signaling and Antigen Presentation to Overcome Immunotherapy Resistance Due to JAK1 Loss in Melanoma. Sci Trans Med (2020) 12(565). doi: 10.1126/scitranslmed.abb0152
99. Gu SS, Zhang W, Wang X, Jiang P, Traugh N, Li Z, et al. Therapeutically Increasing MHC-I Expression Potentiates Immune Checkpoint Blockade. Cancer Discovery (2021) 11:1524–41. doi: 10.1158/2159-8290.CD-20-0812
100. Brea EJ, Oh CY, Manchado E, Budhu S, Gejman RS, Mo G, et al. Kinase Regulation of Human MHC Class I Molecule Expression on Cancer Cells. Cancer Immunol Res (2016) 4:936–47. doi: 10.1158/2326-6066.CIR-16-0177
101. Seliger B, Harders C, Lohmann S, Momburg F, Urlinger S, Tampé R, et al. Down-Regulation of the MHC Class I Antigen-Processing Machinery After Oncogenic Transformation of Murine Fibroblasts. Eur J Immunol (1998) 28:122–33. doi: 10.1002/(SICI)1521-4141(199801)28:01<122::AID-IMMU122>3.0.CO;2-F
102. Loi S, Dushyanthen S, Beavis PA, Salgado R, Denkert C, Savas P, et al. RAS/MAPK Activation Is Associated With Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin Cancer Res (2016) 22:1499–509. doi: 10.1158/1078-0432.CCR-15-1125
103. Franklin DA, James JL, Axelrod ML, Balko JM. MEK Inhibition Activates STAT Signaling to Increase Breast Cancer Immunogenicity via MHC-I Expression. Cancer Drug Resist (2020) 3:603–12. doi: 10.20517/cdr.2019.109
104. Lulli D, Carbone ML, Pastore S. The MEK Inhibitors Trametinib and Cobimetinib Induce a Type I Interferon Response in Human Keratinocytes. Int J Mol Sci (2017) 18:2227. doi: 10.3390/ijms18102227
105. Hastings K, Yu HA, Wei W, Sanchez-Vega F, DeVeaux M, Choi J, et al. EGFR Mutation Subtypes and Response to Immune Checkpoint Blockade Treatment in non-Small-Cell Lung Cancer. Ann Oncol (2019) 30:1311–20. doi: 10.1093/annonc/mdz141
106. Meric-Bernstam F, Sandhu SK, Hamid O, Spreafico A, Kasper S, Dummer R, et al. Phase Ib Study of MIW815 (ADU-S100) in Combination With Spartalizumab (PDR001) in Patients (Pts) With Advanced/Metastatic Solid Tumors or Lymphomas. JCO (2019) 37:2507–7. doi: 10.1200/JCO.2019.37.15_suppl.2507
107. Inoue M, Mimura K, Izawa S, Shiraishi K, Inoue A, Shiba S, et al. Expression of MHC Class I on Breast Cancer Cells Correlates Inversely With HER2 Expression. Oncoimmunology (2012) 1:1104–10. doi: 10.4161/onci.21056
108. Mimura K, Ando T, Poschke I, Mougiakakos D, Johansson CC, Ichikawa J, et al. T Cell Recognition of HLA-A2 Restricted Tumor Antigens is Impaired by the Oncogene HER2. Int J Cancer (2011) 128:390–401. doi: 10.1002/ijc.25613
109. Garrido G, Rabasa A, Garrido C, Chao L, Garrido F, García-Lora ÁM, et al. Upregulation of HLA Class I Expression on Tumor Cells by the Anti-EGFR Antibody Nimotuzumab. Front Pharmacol (2017) 8:595. doi: 10.3389/fphar.2017.00595
110. Srivastava RM, Trivedi S, Concha-Benavente F, Hyun-bae J, Wang L, Seethala RR, et al. STAT1-Induced HLA Class I Upregulation Enhances Immunogenicity and Clinical Response to Anti-EGFR mAb Cetuximab Therapy in HNC Patients. Cancer Immunol Res (2015) 3:936–45. doi: 10.1158/2326-6066.CIR-15-0053
111. Pollack BP, Sapkota B, Cartee TV. Epidermal Growth Factor Receptor Inhibition Augments the Expression of MHC Class I and II Genes. Clin Cancer Res (2011) 17:4400–13. doi: 10.1158/1078-0432.CCR-10-3283
112. Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, et al. Autophagy Promotes Immune Evasion of Pancreatic Cancer by Degrading MHC-I. Nature (2020) 581:100–5. doi: 10.1038/s41586-020-2229-5
113. Kobayashi M, Nagashio R, Jiang S-X, Saito K, Tsuchiya B, Ryuge S, et al. Calnexin Is a Novel Sero-Diagnostic Marker for Lung Cancer. Lung Cancer (2015) 90:342–5. doi: 10.1016/j.lungcan.2015.08.015
114. Okayama A, Miyagi Y, Oshita F, Nishi M, Nakamura Y, Nagashima Y, et al. Proteomic Analysis of Proteins Related to Prognosis of Lung Adenocarcinoma. J Proteome Res (2014) 13:4686–94. doi: 10.1021/pr4012969
115. Alam A, Taye N, Patel S, Thube M, Mullick J, Shah VK, et al. SMAR1 Favors Immunosurveillance of Cancer Cells by Modulating Calnexin and MHC I Expression. Neoplasia (2019) 21:945–62. doi: 10.1016/j.neo.2019.07.002
116. Hodge JW, Garnett CT, Farsaci B, Palena C, Tsang K-Y, Ferrone S, et al. Chemotherapy-Induced Immunogenic Modulation of Tumor Cells Enhances Killing by Cytotoxic T Lymphocytes and Is Distinct From Immunogenic Cell Death. Int J Cancer (2013) 133:624–36. doi: 10.1002/ijc.28070
117. Tseng C-W, Hung C-F, Alvarez RD, Trimble C, Huh WK, Kim D, et al. Pretreatment With Cisplatin Enhances E7-Specific CD8+ T-Cell–Mediated Antitumor Immunity Induced by DNA Vaccination. Clin Cancer Res (2008) 14:3185–92. doi: 10.1158/1078-0432.CCR-08-0037
118. Iwai T, Sugimoto M, Wakita D, Yorozu K, Kurasawa M, Yamamoto K. Topoisomerase I Inhibitor, Irinotecan, Depletes Regulatory T Cells and Up-Regulates MHC Class I and PD-L1 Expression, Resulting in a Supra-Additive Antitumor Effect When Combined With Anti-PD-L1 Antibodies. Oncotarget (2018) 9:31411–21. doi: 10.18632/oncotarget.25830
119. Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, van Hall T. The Urgent Need to Recover MHC Class I in Cancers for Effective Immunotherapy. Curr Opin Immunol (2016) 39:44–51. doi: 10.1016/j.coi.2015.12.007
120. del Campo AB, Carretero J, Muñoz JA, Zinchenko S, Ruiz-Cabello F, González-Aseguinolaza G, et al. Adenovirus Expressing β2-Microglobulin Recovers HLA Class I Expression and Antitumor Immunity by Increasing T-Cell Recognition. Cancer Gene Ther (2014) 21:317–32. doi: 10.1038/cgt.2014.32
121. Messaoudene M, Mourikis TP, Michels J, Fu Y, Bonvalet M, Lacroix-Trikki M, et al. T-Cell Bispecific Antibodies in Node-Positive Breast Cancer: Novel Therapeutic Avenue for MHC Class I Loss Variants. Ann Oncol (2019) 30:934–44. doi: 10.1093/annonc/mdz112
122. Holland CJ, Crean RM, Pentier JM, de Wet B, Lloyd A, Srikannathasan V, et al. Specificity of Bispecific T Cell Receptors and Antibodies Targeting Peptide-HLA. J Clin Invest (2020) 130:2673–88. doi: 10.1172/JCI130562
123. Oliveira CC, Querido B, Sluijter M, Derbinski J, van der Burg SH, van Hall T. Peptide Transporter TAP Mediates Between Competing Antigen Sources Generating Distinct Surface MHC Class I Peptide Repertoires. Eur J Immunol (2011) 41:3114–24. doi: 10.1002/eji.201141836
124. Doorduijn EM, Sluijter M, Querido BJ, Oliveira CC, Achour A, Ossendorp F, et al. TAP-Independent Self-Peptides Enhance T Cell Recognition of Immune-Escaped Tumors. J Clin Invest (2016) 126:784–94. doi: 10.1172/JCI83671
Keywords: major histocompatibility complex class-I, cancer immunotherapy, immune checkpoint inhibition, antigen presentation, immunoncology
Citation: Taylor BC and Balko JM (2022) Mechanisms of MHC-I Downregulation and Role in Immunotherapy Response. Front. Immunol. 13:844866. doi: 10.3389/fimmu.2022.844866
Received: 28 December 2021; Accepted: 09 February 2022;
Published: 28 February 2022.
Edited by:
Varun Sasidharan Nair, Helmholtz Center for Infection Research, Helmholtz Association of German Research Centers (HZ), GermanyReviewed by:
Dhaneshwar Kumar, National Institute of Diabetes and Digestive and Kidney Diseases (NIH), United StatesCopyright © 2022 Taylor and Balko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Justin M. Balko, anVzdGluLmJhbGtvQHZ1bWMub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.