 Bingzhe LV1,2
Bingzhe LV1,2 Yunpeng Wang1,2
Yunpeng Wang1,2 Dongjiang Ma1,2Wei Cheng1,2Jie Liu1,2Tao Yong1,2
Dongjiang Ma1,2Wei Cheng1,2Jie Liu1,2Tao Yong1,2 Hao Chen3,4*†Chen Wang1,3*†
Hao Chen3,4*†Chen Wang1,3*†- 1Department of General Surgery, Lanzhou University Second Hospital, Lanzhou, China
- 2The Second Clinical Medical College, Lanzhou University, Lanzhou, China
- 3Key Laboratory of Digestive System Tumors of Gansu Province, Lanzhou University Second Hospital, Lanzhou, China
- 4Department of Surgical Oncology, Lanzhou University Second Hospital, Lanzhou, China
Tumor immune microenvironment (TIME) include tumor cells, immune cells, cytokines, etc. The interactions between these components, which are divided into anti-tumor and pro-tumor, determine the trend of anti-tumor immunity. Although the immune system can eliminate tumor through the cancer-immune cycle, tumors appear to eventually evade from immune surveillance by shaping an immunosuppressive microenvironment. Immunotherapy reshapes the TIME and restores the tumor killing ability of anti-tumor immune cells. Herein, we review the function of immune cells within the TIME and discuss the contribution of current mainstream immunotherapeutic approaches to remolding the TIME. Changes in the immune microenvironment in different forms under the intervention of immunotherapy can shed light on better combination treatment strategies.
Introduction
The immune system can eliminate tumor cells through the cancer-immune cycle (1). This process is not sustained because tumors can gradually shape the tumor immune microenvironment (TIME) into an immunosuppressive state to combat host immunity, and the balance between pro- and anti-tumor inflammatory mediators may determine tumor progression. Tumors have evolved various mechanisms to evade immune surveillance, such as defecting the antigen presentation machinery, enhancing negative immune regulatory pathways, recruiting tumor-promoting immune cells, and others (2–4). The result is that the function of anti-tumor immune cells is blocked, and it is difficult to maintain anti-tumor immune responses. The tendency for antitumor immunity is determined within the TIME by two immune components, antitumor and pro-tumor (5). Despite heterogeneity across different cancer types and populations, the role of the TIME in tumor progression is similar. The goal of immunotherapy is to restore the killing effect of anti-tumor immune cells on tumors, especially cytotoxic T lymphocytes (CTL). However, pro-tumor immune cells, such as regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAM), and group 2 innate lymphoid cells (ILC2s), play an important role in impairing anti-tumor immune responses and shaping an immunosuppressive microenvironment. Studying the functions and mechanisms of tumor-promoting immune cells will help to improve the response rate of immunotherapy and develop new immunotherapeutic strategies.
Based on the understanding of tumor immune escape, several cancer immunotherapies have been developed to reshape the TIME to subdue tumor cells. Blocking CTLA-4 and PD-1/PD-L1 immune checkpoints can relieve the functional inhibition of T cells (6). Changing the polarization state from M2 to M1 in TAM (dual blocking of PI3K-γ pathway and CSF-1/CSF-1R) can lead to the reduction of immunosuppressive macrophages and the activation of CD8+T cell response (7). DC-based vaccines can activate T cell responses by removing the inhibition of antigen presentation (8). Therapies that reshape TIME could, in theory, remove tumors through the body’s immune system. This mode has higher specificity and lower side effects, and the generation of memory T cells guarantees a sustained response. Understanding the changes in the TIME during tumor development can help to develop targeted therapeutic strategies and improve response rate of immunotherapy. Recently, new advances have been made in the study of the TIME. This review provides a brief overview of the role of tumor-associated immune cells during remodeling of the TIME. In addition, we introduce the contribution of current mainstream immunotherapy approaches to remolding TIME, with a particular focus on immune cell changes.
TIME
Tumor-associated immune cells can be divided into two categories, anti-tumor and tumor-promoting. Antitumor immune cells mainly include effector T cells (including cytotoxic CD8 + T cells and effector CD4 + T cells), natural killer cells (NK), dendritic cells (DC), and M1-polarized macrophages. The tumor-promoting immune cells are mainly Tregs, MDSCs, M2-polarized macrophages, N2-polarized neutrophils, natural killer T Type 2 cells (NKT2) cells, and ILC2s (Figure 1). In addition, metabolic and biochemical components significantly influence immune cell function.
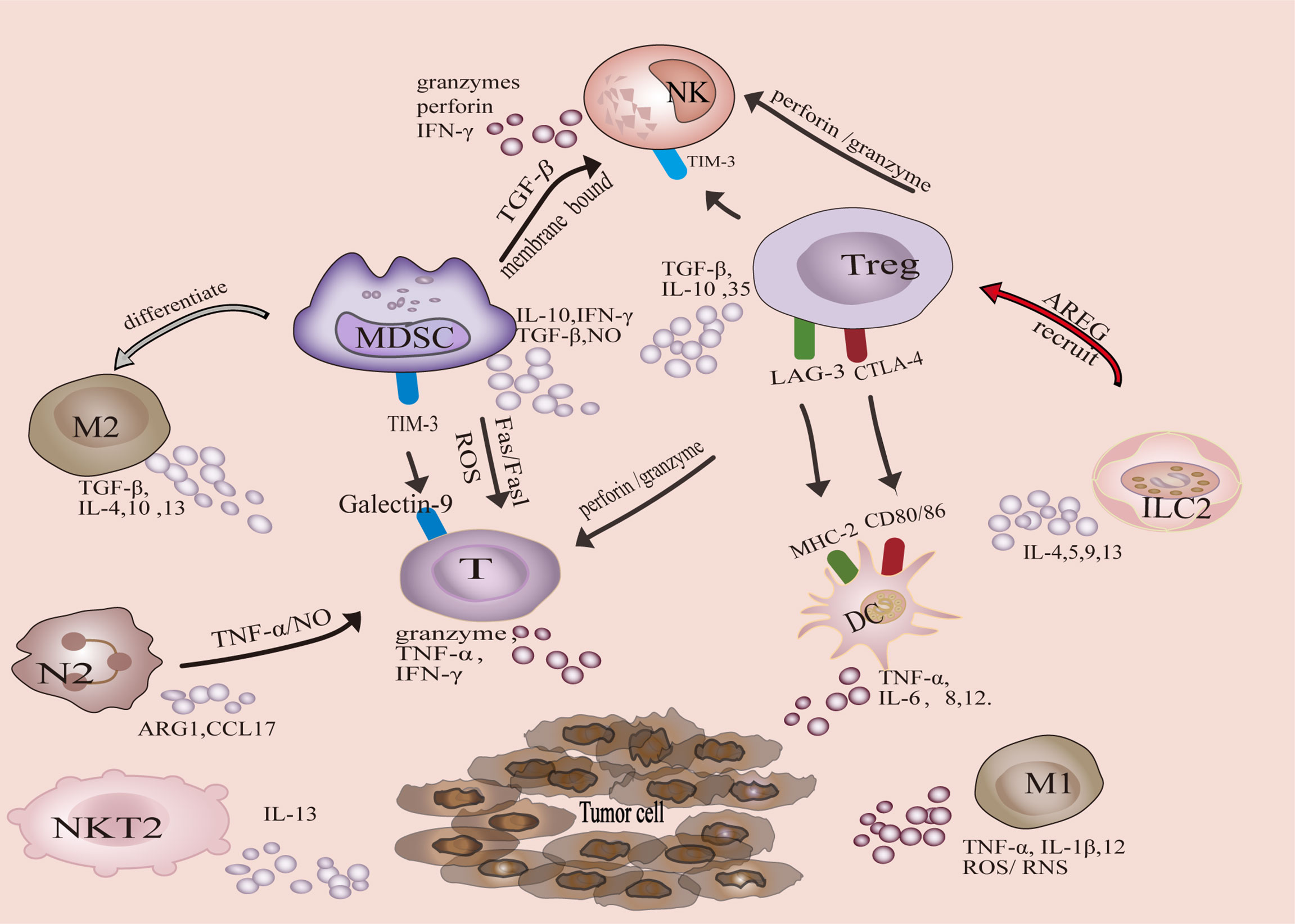
Figure 1 Crosstalk of tumor-associated immune cells in tumor microenvironment.
Anti-Tumor Immune Cells
T cells are the main executor of anti-tumor immune response, including CTL and T helper cells. CTL recognizes MHC-I molecules expressed by tumor cells (9), exerting tumor killing mechanisms through granule exocytosis (granzyme A and B) or death ligand-induced necrosis and apoptosis under the action of chemokines (10). IFN-γ and TNF-α are secreted to induce tumor cells cytotoxicity (10). CD4+T cells can promote CTL proliferation, increase antigen presentation by DCs, promote CTL activation, and promote memory CTL formation (11, 12).
DCs, as the most potent specific antigen-presenting cells (APCs), initiate adaptive immune responses by activating naive T cells (13). DCs can also express CD80/CD86, which interacts with CD28 to generate costimulatory signals that increase T cell activation (14). DCs produce TNF-α, IL-6, IL-8, and IL-12 to participate in anti-tumor immunity. Tumor cells lose MHC-I molecular to evade immune surveillance by T cells (activation of CD8 + T cells requires MHC-I molecule dependent antigen presentation). The activation of NK cells is inhibited by binding of inhibitory receptors to MHC-I molecules, so NK cells can eliminate targets with defective expression of MHC-I molecules through a “missing self” mechanism (15, 16). NK cell lysis of tumor cells is mainly dependent on granzymes and perforin (17).
The polarization of classically activated macrophages (M1) is mainly mediated by GM-CSF, IL-12, IL-18, IFN-γ, and TNF-α (18, 19). M1 promote Th1 response by secreting TNF-α, IL-1β, and IL-12, and promote the recruitment of Th1 cells to inflammatory sites by secreting chemokines CXCL9 and CXCL10 (20). Besides, M1 macrophages exert antitumor effects through the release of reactive oxygen/nitrogen species (ROS/RNS) directly mediated cytotoxicity and antibody-dependent cell-mediated cytotoxicity (21, 22).
Tumor-Promoting Immune Cells
Tregs play a key role in maintaining immune homeostasis and peripheral tolerance (23). The physiological function of Tregs is to prevent the spread of inflammation and limit tissue damage, but it acts as a feedback mechanism to inhibit anti-tumor immune response in the TIME. Tregs inhibit anti-tumor immune response through production of immunosuppressive cytokines, such as TGF-β, IL-10 and IL-35 (24). Furthermore, Tregs can inhibit anti-tumor immune responses in several ways:1) Tregs inhibit CTL-mediated tumor killing via TGF-β -dependent cell contact (25), promoting polarization of M2 macrophages by inhibiting IFN-γ secretion by CTL cells (26), and inhibiting the generation of memory CD8+T cell through CTLA-4 (27). 2) Tregs inhibit NK cell proliferation, IFN-γ production, degranulation and cytotoxicity, which is related to TIM-3 (28). 3) Treg induces DC functional inhibition in these two ways. Treg-expressed CTLA-4 binds to CD80/CD86 on the DC to down-regulate costimulatory signal (29). Furthermore, MHC class II molecules are the major ligands for LAG-3 (30). LAG3 expressed by Tregs can inhibit the expression of MHC II molecules in DCs (31). 4) MDSCs and Tregs reinforce each other to enhance the immunosuppressive microenvironment. Induction of Tregs can be facilitated by TGF-β, IL-10 and IFN-γ secreted by MDSCs. Tregs enhance the function of MDSCs through TGF-β and IL-35 (31). 5) Tregs can induce NK and CD8+T cell death in a granzyme B and perforin dependent manner (32, 33).
MDSCs represent a heterogenous population of immature myeloid cells with different transcriptional activities and differentiation states, characterized by immunosuppressive activity in pathological states (34). MDSCs can be roughly divided into two groups, granulocytic or polymorphonuclear MDSCs (PMN-MDSCs) and monocytic MDSCs (M-MDSCs). PMN-MDSCs can produce ROS and reduce T cells responses to antigens (34). PMN-MDSCs induce CTL apoptosis through the Fas/FasL axis, whereas M-MDSCs produce nitric oxide to inhibit immune activation (35). M-MDSCs can also differentiate into immunosuppressive macrophages and inhibit T cell activation (36). In addition to interacting with Tregs, IL-10, and TGF-β produced by MDSC also impair CTL function (37). MDSCs reduce NK cell numbers and inhibit their function via membrane-bound TGF-β (38). MDSCs express galectin 9, which binds to TIM-3 on lymphocytes and induces T cell apoptosis (39). Inhibition of MDSCs enhances the function of T cells (36, 40).
M2 macrophages are usually the dominant cells in TAM. M2 macrophage polarization is mediated by M-CSF, IL-4, IL-10, IL-13 and TGF-β (41). M2 macrophages secrete immunosuppressive cytokines, TGF-β, IL-4, IL-10 and IL-13 (19, 42–44). M2 macrophages are involved in activating Th2 immune response (45). In addition, M2 macrophages, involved in the recruitment of Tregs cells via M2 derived CCL20/CCL22 (46), as well as by increasing the expression of PD-L1 to attenuate the effects of CTLs and induce MDSC differentiation (47, 48).
Polarization of N2 neutrophils is mainly mediated by TGF-β (49). Recently, IL-6 produced by gastric cancer mesenchymal stem cells was also found to determine N2 polarization (50). N2 neutrophils induce CD8 +T cells apoptosis through TNF-α and NO-dependent mechanism (51). In addition, N2 can also inhibit T cell proliferation by releasing argininase-1 (ARG1) and regulating PD-L1/PD-1 signaling (52), as well as secreting chemokine CCL17 to recruit Tregs (53). Although the exact mechanism remains nonclear, studies have shown that N2 neutrophils inhibit NK cell function (54).
ILC2 secrete cytokines (IL-4, IL-5, IL-9, IL-13) and recruit immunosuppressive cells to shape the TIME (55). ILC2 secrete IL-13, which promotes the aggregation of MDSC and inhibits the anti-tumor response of CTL (56, 57). Besides, ILC2 induce the production of TGF-β from MDSCs, which contributes to the polarization of M2 macrophages (58). LC2s produce the epidermal growth factor (EGF)-like molecule Amphiregulin (AREG), which costimulates ICOSL/ICOS to establish and maintain an immunosuppressive microenvironment, leading to Treg activation and accumulation (59, 60). In addition, ILC2s may inhibit the activity of NK cells (61).
NKT switch between inflammatory and immunosuppressive subsets to respond to the TIME status (62). NKT1 is antitumor, while NKT2 is primarily tumor-promoting.IL-13 produced by NKT2 induces MDSC to produce TGF-β, which inhibits the anti-tumor immune response mediated by CD8+T cells (63). In myeloma, the weakening of NKT2 cell population has the potential to mediate tumor regression (64).
Metabolic: Hypoxia-Adenosinergic Immunosuppression
The numerous and complex cell populations and the limited vasculature within the tumor microenvironment render nutrient and oxygen delivery and waste clearance inefficient. In addition, tumor cells shape the metabolic fitness of tumor infiltrating immune cells by competing for and consuming essential nutrients or otherwise, such as the classical ‘Warburg effect’. Tumors prefer to perform aerobic glycolysis to convert virtually all glucose to lactate even in the presence of oxygen (65). Metabolism in the microenvironment, such as nutrient consumption, increased oxygen consumption, and production of reactive nitrogen and oxygen intermediates, significantly influences antitumor immune responses. As a result, high lactate and low pH, hypoxia, and high levels of ROS are prevalent in the TME. This hostile environment shapes the metabolic adaptation of tumor infiltrating immune cells, and these metabolic changes in immune cells undermine the effectiveness of antitumor immune responses. (A more detailed overview of immunometabolism in review (66, 67)). The main focus here is on the role of the hypoxia adenosine in immunosuppression as well as adenosinergic blockade in reprogramming the TIME. Tumor, especially the solid tumor microenvironment, provides fertile soil for adenosine production. A series of cascades driven by the hypoxia/HIF-1α-CD39/CD73 axis represent major sources of adenosine (68, 69). In addition, some alternative activation modalities, CD38, CD203a, and PAP also contribute to adenosine levels in the TIME (70, 71). There are four receptors for extracellular adenosine, A1, A2a, A2b, and A3. Adenosine is an immunosuppressive metabolite, signaling largely through the A2a receptors on innate and adaptive immune cells (72). A2AR is upregulated due to hypoxia induced HIF-1α transcriptional activity (73). Adenosine accumulation in the TIME can inhibit antitumor immune cell functions by binding to A2AR. For example, T cells and NK cells (68, 74, 75). In addition, adenosine enhances the activity of immunosuppressive cells, such as MDSCs and Tregs, contributing to CAF shaping as well as inducing the formation of new blood vessels (76–80).
Therapy to Reshape TIME
Currently mainstream immunotherapies, Immune checkpoints, CAR-T, DC cell vaccines, contribute to reshaping the TIME, (e.g., changes in immune cells and cytokines after immune checkpoint blockade, changes in T cell function and microenvironmental status after CAR T infusion). This has driven the development of combination therapies. The effects of different immunotherapies on the TIME help us to find effective combination treatment strategies (Figure 2). For example, most classically, CAR T cells provide infiltration while Immune checkpoints inhibitors (ICIs) reverse CAR T cells inhibition and restore functional persistence.
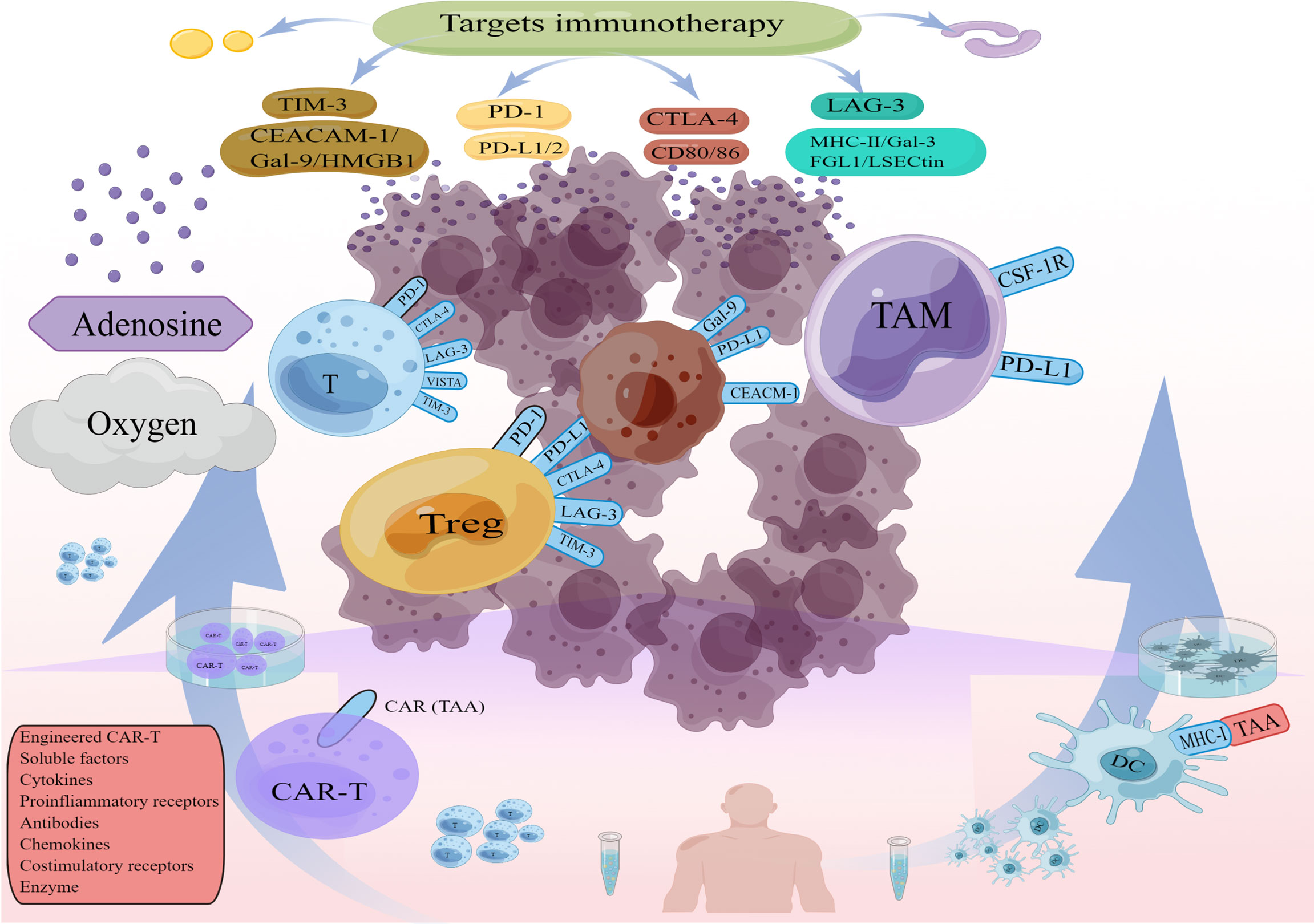
Figure 2 Crosstalk of various treatments in TIME (By Figdraw).
Targets for Antibodies and Small Molecule Inhibitor
At present, the mainstream strategy of immunotherapy is to target some components of the TIME through antibodies or small molecule inhibitors (Table 1). We will summarize the key functions of major immunotherapies in the TIME, with an emphasis on how therapeutically enabling the TIME to generate an anti-tumor immune response. During anti-tumor immunity, negative regulators of T cell activation act as “checkpoint molecules” to modulate the immune response. Depending on checkpoints. This strategy can be implemented by inhibiting inhibitory checkpoints or activating stimulant checkpoints. CTLA-4 and PD-1 are the most effective examples of immune checkpoint therapy.

Table 1 Immunotherapy targets within TIME and the treatment effect.
CTLA-4 is induced on the cell surface of conventional T cells by antigen activation and is constitutively expressed on Treg (85). As having the same B7 ligands as CD28, including B7-1 (CD80) and B7-2 (CD86), CTLA-4 with a higher affinity competes with CD28 expressed on effector T cells for binding to B7-1 and B7-2 which ubiquitously expressed on B lymphocytes, dendritic cells, and other immune cells (112). The result is that costimulatory signals CD80/CD86 which can activate T cells by ligating CD28 are inhibited. As a result, T lymphocyte proliferation and cytokine secretion are hampered (113). CTLA-4 can reduce the activation of T cells by generating inhibitory signals, thereby attenuating the anti-tumor immune response. CTLA-4 can induce indoleamine 2,3-dioxygenase (IDO) and trigger reverse signaling through B7 ligands to inhibit T cell proliferation (114). Inhibition of CTLA-4 enhanced the antitumor activity of effector T cells mainly by inhibiting Treg. Anti-CTLA-4 antibody enhances IL-36 stimulated anti-tumor activity by consuming Tregs, leading to increased CD4+ and CD8+T cells proliferation and IFN-γ levels (115). Anti-CTLA-4 antibody reduces Treg cells in the TIME but do not affect the status of peripheral lymphoid organs, reducing immune-related adverse events (irAEs) (116). Compared with glycoprotein 100 (gp100) peptide vaccine, melanoma patients treated with ipilimumab had improved survival (OS of 10 months/ipilimumab and 6.4 months/GP100, P<0.001) (117). A pooled analysis of 10 prospective studies and 2 retrospective studies, including 1,861 patients with advanced melanoma, found a 22% 3-year survival rate in patients treated with ipilimumab (118).
PD-1 is a transmembrane protein mainly expressed on the surface of activated T cells, B cells and macrophages. PD-L1 and PD-L2 are dual ligands of PD-1, and both have been shown to inhibit T cell activity upon PD-1 engagement (119). PD-L1 over-expressed on cancer cells can interact with PD-1 on activated T cells, inducing T cell inhibition and CTL dysfunction. Correspondingly, blockade of PD-1/PD-L1 promotes pro-inflammatory factors release, T cells proliferation, CTL activation (120). PD-1/PD-L1 is an ideal immunotherapeutic target to restore the effector function of anti-tumor specific T cell. Patients with metastatic melanoma who respond to anti-PD-1 therapy (pembrolizumab) exhibit active CD8+T cell proliferation in the TIME, which is associated with reduced tumor size (121). In addition, it has been observed in melanoma patients that CD4 + T cells expand after PD-1 blockade and that activated CD4 + T cells secrete IFN- γ and chemokines, which contribute to antitumor immunity (86). A trial that enrolled 655 patients with advanced or metastatic melanoma showed long-term antitumor activity and tolerability of pembrolizumab in advanced melanoma, with a mean follow up of 55 mouths. The estimated 5-year overall survival rate was 34% for all patients, 41% for patients receiving initial treatment (122). Pembrolizumab also provided a long-term response and prolonged OS in non-small cell lung cancer, with the combination of pembrolizumab and chemotherapy achieving objective response in 55% of patients compared to 29% of who those treated with chemotherapy alone, with a significantly longer median PFS than chemotherapy (13.0 months vs 8.9 months) (123). Blockade of PD-1/PD-L1 can restore the killing ability of T cells and induce tumor regression, resulting in better clinical outcomes (119).
LAG-3 is expressed on tumor-infiltrating T cells with defective cytokine production and on Tregs (124, 125). Treg cells with high expression of LAG-3 produce immunosuppressive cytokines IL-10 and TGF-β and inhibit effector T cell activity (126). LAG-3 expression levels correlate with tumor progression and poor prognosis (126). Anti-LAG-3 antibodies slow tumor growth in mouse model of fibrosarcoma (127). The combination therapy of anti-LAG-3 antibody and tumor-associated antigen inoculation increases CD8+T cells in the TIME and destroyed tumor parenchyma in prostate cancer tumor models (128). LAG-3 and PD-1 were highly co-expressed in CD4+T cells and CD8+T cells, and the inhibitory effect of the blocking of LAG-3 and PD-1 on tumor progression (129, 130). Moreover, dual blockade of LAG-3 and PD-1 can also increase the number of tumor-infiltrating CD8+ T cells and reduce Treg, thereby synergically enhancing anti-tumor immunity (131). In a Phase I/II study (NCT0198609) evaluating the safety and efficacy of anti-LAG-3 antibody in combination with anti-PD-1 antibody, 61 melanoma patients in a Phase I/II study well tolerated with an ORR of 11.5%. Patients with high LAG-3 expression had a significantly higher objective response rate than those with low expression (132, 133). PD-L1/LAG-3 bispecific antibody induced stronger anti-tumor effect than each parental antibody (134, 135). In addition, PD-1 and LAG-3 blockade improve anti-tumor vaccine efficacy (136). As mentioned above, there is a synergistic effect between anti-LAG-3 and certain immunotherapies, and the combination of anti-LAG3 with more therapies is worth investigating. According to the present findings, LAG-3 is a promising cancer therapeutic target secondary to PD-1/PD-L1 and CTLA-4.
T cell immunoglobulin and mucin-domain containing-3 (TIM-3), T cell immunoglobulin and ITIM domain (TIGIT), V-domain immunoglobulin suppressor of T cell activation (VISTA) and Siglec-15 are also attractive targets (Table 2). Cancer immune checkpoint therapies are increasingly being developed to restore immune activity against tumor cells. In addition to monotherapy, rational combination of immunotherapy and multi-target combination such as bispecific antibodies, trispecific antibodies have also been considered to achieve synergistic effects to inhibit tumor growth. We anticipate that translating candidate targets into the clinical area may yield better clinical benefits than CTLA-4 or PD-1 inhibitors.

Table 2 Functions of new immune targets.
Small molecule drugs, which are more suitable for oral administration than polymeric antibody drugs, could reduce severe immune-related adverse events (irAEs) resulting from prolonged target occupancy by modulating the half-life of the drug (161, 162). They can cross the cell membrane, penetrate more easily into the tumor tissue and aggregate in a sufficient concentration (161, 162). In addition, they are lower production costs and higher stability (163, 164). We focus here on the role of colony stimulating factor-1 receptor (CSF-1R) in reshaping TIME. CSF-1/CSF-1R signaling is a key activator of the mononuclear phagocyte system, and blockade of CSF-1/CSF-1R creates an environment of reduced immunosuppression and enhanced interferon response that can impede tumor growth (103). Pexidartinib (CSF-1R inhibitor) was demonstrated to alter the distribution of TAMs in TIME and reduce tumor volume in a mouse model of lung adenocarcinoma (165). In a mouse model of BRAF V600E mutant melanoma, Pexidartinib combined with adoptive cell transplantation decreased TAM and increased tumor-infiltrating lymphocyte levels (166). The combination of Pexidartinib and BRAF inhibitors resulted in a significant inhibition of tumor growth by reducing the recruitment of M2 macrophage (104). Moreover, in a pancreatic cancer mouse model, blocking of CSF-1/CSF-1R reduced M2 macrophages within the TIME and polarized the remaining TAMs into an anti-tumor phenotype (103). This study also found that PD-1/PD-L1 expression on TAMs and CTLA-4 expression on CD8+ T cells was increased in the presence of CSF-1/CSF-1R blockade, and the combination of PD-1 or CTLA-4 antagonists resulted in more significant tumor regression (103). A recent clinical study of advanced tenosynovial giant cell tumor found that Pexidartinib significantly reduced tumor size with an overall response rate of 39% (167). To enhance the response of CSF-1/CSF-1R blockade, researchers tend to combine CSF-1/CSF-1R blockade with immunotherapy. The combination of oncolytic virus, CSF-1R inhibition and anti-PD-1 immunotherapy was found to enhances anti-tumor immune response by increasing T cell infiltration and augmenting anti-tumor CD8+ T cell function (168). In addition, the combination of targeted TIME and anti-angiogenesis has been suggested to enhance the antitumor activity of the drug (169). For example, the combination of the tyrosinase inhibitor sorafenib and the immunomodulator lenalidomide has long been found to be more effective than drugs alone (170). Surufatinib is a drug that targets tumor angiogenesis (VEGFR and FGFR1) and tumor immune evasion (CSF-1R). Surufatinib was effective against neuroendocrine tumors in two phase III trials. Whereby it may become a mainstream treatment for neuroendocrine tumors (171). These results suggest that in addition to developing drugs for novel targets, multitarget fusion drugs or combinations of antibodies and small molecule immune modulators also play important roles in reshaping TIME.
Despite the success of anti-PD1/PD-L1 and anti-CTLA4 therapies in advanced cancer. There are a considerable number of patients who remain unresponsive or relapses after initial response. Combination strategy of immunotherapy are used to address these challenges. Furthermore, targeting hypoxic adenosine pathway represents another idea to improve immunotherapy (172, 173). This treatment can be broadly divided into two types.
1. Reduced adenosine production. As mentioned above, hypoxia induces upregulation of CD39 and CD73 and downregulation of adenosine transporters to promote the accumulation of extracellular adenosine. Hyperoxic respiration (60% O2) significantly reduced adenosine levels and gained tumor control and prolong survival. Hyperoxic breathing upregulates antigen-presenting MHC class I molecules on tumor cells, tumor infiltration of CD8 T cells and attenuate immunosuppressive effects of Tregs (69, 174). CD73 can convert AMP produced by catabolism to adenosine. Antagonism of CD73 increases the activity of CD8 + T cells, B cells and related cytokine, and tumor growth and metastatic spread are retarded in CD73 blocked mice (175–177). Compared with anti-PDL1 alone, ORR close to 40% in the dual CD73/PDL1 blockade arm with statistically improved 10-month PFS (64.8 vs 39.2) (NCT03822351). Similar to studies with CD73, tumor growth and metastasis were reduced in CD39-blocked mice (178, 179). CD39 blockade enhance the function of T, NK cells, as well as decreased Treg-mediated immunosuppression (178, 180, 181). Finally, although studies have demonstrated the effectiveness of this approach within tumor-bearing mice. Clinical studies exploring CD39 blockade/inhibition have not yet yielded results.
2. Block the binding of adenosine receptor. A2AR antagonists are a more direct approach to inhibit adenosine induced signal transduction. A2AR-deletion leads to delayed tumor progression and prolonged survival (68, 182). TIME of A2AR antagonist treated mice showed similar changes in immune level as blockade of CD39/73, which was more infiltrated by CD8 T cells and NK cells and contained fewer Tregs (183, 184). Besides, A2AR blockade reduced PD-1 and LAG-3 expression on Tregs and T cells (182). For renal cell cancer, A2AR antagonism (CPI-444) induces durable responses when used as monotherapy as well as in combination with anti-PD-L1. Patients who experienced a positive response included individuals who were resistant or refractory to anti-PD-1/PD-L1 antibodies. Adenosinergic blockade resulted in higher cytotoxic T cell tumor infiltration (172). Besides, A2AR antagonists have shown similar activity in other types of cancer (NCT02403193, NCT03720678, NCT03720678).
Adoptive Cell Therapy
Adoptive cell therapy (ACT) uses autologous immune cells that are isolated, engineered, amplified and injected into a patient to generate durable anti-tumor immune response. T cells genetically modified to express chimeric antigen receptor (CAR), or CAR T, are the most effective cell therapies. CAR T cells specifically recognize tumor-associated antigens (TAA) and kill tumor cells (185). Adoptive transfer of tumor-reactive T cells resulted in persistent clonal repopulation of T cells in patients, with the transferred cells proliferating, displaying functional activity, trafficking to the tumor sites and promoting tumor control (186). Anti-CD19 CAR T cell can produce cytokines that respond specifically to CD19+ target cells and effectively eradicate lymphoma cells (187). In a clinical trial, 82% (89/108) of patients with refractory large B-cell lymphoma (ZUMA-1) achieved an overall response and 58% (63/108) achieved a complete response (188). CAR T cells have achieved remarkable success in treating hematologic malignancies but face unique challenges in solid tumors, such as lack of suitable targets, inefficiency of CAR T cells to infiltrate into tumor sites, and TIME limitations on CAR T efficacy (189). CAR-T holds promise in addressing these issues through diversifying edits and in combination with other therapy approaches. Her-2, a receptor tyrosine kinase overexpressed in many human cancers, is used as a TAA for targeted CAR T in glioblastoma. Although such CAR T cells only expand for a short term, they maintain long-term antitumor activity (190). CAR T cells expressing high levels of the CCR2 receptor can migrate more efficiently to CCL2-secreting tumor sites and exhibit greater antitumor activity (191). In addition, overexpression of heparanase (HPSE), an enzyme that can degrade major components of the extracellular matrix, on CAR T cells effectively promotes tumor T-cell infiltration and antitumor activity (192). CAR-T editing is diverse, for example, PD-1 knockdown can inhibit immune checkpoint signaling (193), LAG-3 knockdown can disrupt negative regulators of T cell activity (194), IL-12-secreting CAR T cells polarized TAMs to M1 phenotype and reduce the levels of MDSCs and Tregs in mouse models (193), IL-18-secreting CAR T cells can increase M1 macrophages, activated DC and activated NK cells while decreasing M2 macrophages and Treg in the TIME (195). CAR T cells co-expressing CCL19 and IL-7 can recruit large numbers of endogenous T cells and DCs to enhance and sustain tumor clearance (196). In addition to the specific operation of CAR T cells, the therapy of reshaping the TIME can theoretically enhance the therapeutic effects of CAR T cells (197).
Cancer Vaccines
Cancer vaccines generate antitumor immune responses against TAAs or tumor-specific antigens (TSAs). DCs are specialized antigen-presenting cells that are key targets for cancer vaccines because of their unique ability to link innate and adaptive immunity (198). The main route of this strategy is to modulate the antigen presenting function of DCs to enhance the antitumor immune response in the TIME. Sipuleucel-T, composed of cultured peripheral blood mononuclear cells containing activated APCs, induces sustained responses of T and B cells to the target antigens prostatic acid phosphatase (PAP) and GM-CSF (199). Patients with localized prostate cancer, when Sipuleucel-T was administered preoperatively, exhibited increased T-cell proliferation and IFN-γ levels. In addition, infiltrating T cells were increased more than threefold in resected tissue specimens after surgery compared with controls (P 0.001) (200). In the early stages of prostate cancer, Sipuleucel-T significantly promotes the activation of APCs and increases the level of antigen presentation (201). In a clinical trial for metastatic prostate cancer (NCT00065442), Sipuleucel-T significantly improved OS compared with placebo (median 25.8 months vs 21.7 months, [HR] 0.78, P =0.03) (202). This suggests that cancer vaccines have considerable potential in tumor immunotherapy. In phase I clinical trial of nine men with metastatic castrate-resistant prostate cancer treated with Sipuleucel-T and escalating doses of ipilimumab showed that IgG and IgM levels against PA2024 and PAP increased significantly after ipilipumab (203). Combination therapy with vaccines and checkpoint inhibitors is effective, although few vaccines are now available for clinical use and multiple clinical studies have been negative (204–207). Vaccines can trigger long-term immunological memory, thus contributing to long-lasting anti-tumor immune response. Antigen and vaccine vectors were developed to achieve optimal antigen presentation by APCs, combined with multiple approaches to overcome immune evasion and immunosuppression by cancer cells. Development of antigens and vaccine vectors to achieve optimal antigen presentation by APCs, as well as combination therapy approaches, hold promise to overcome immune evasion and immunosuppression by cancer cells.
Combination Therapy
Different treatments reshape TIME in different ways. Therefore, we can combine complementary or augmentation strategies to achieve better clinical outcomes. Herein, we discuss the principles and clinical application of combination therapy.
CAR T cells can provide an infiltrate for the TIME, and ICI can reverse CAR T-cell inhibition and restore functional persistence. CAR-T may escalate the expression of PD-1 inhibitory signaling, and interference of PD-1 pathway may restore the effector function of CAR-T cells (208). Combination therapy of oxaliplatin and anti-PD-L1 synergistically improves CAR-T cell-mediated lung tumor control and survival (193, 209, 210). Patients with Malignant Pleural Disease in a Phase I Trial of CAR T-cell combination with Pembrolizumab, have a median overall survival of 23.9 months (211). In another multi-center phase II trial, the combination of anti-PD-1 antibody enhanced CAR-T therapy in lymphoma patients with minimal toxicities (212).
Blocking CSF-1/CSF-1R alone increased PD-1/PD-L1 expression on TAM cells and CTLA-4 expression on CD8+ T cells, whereas combination with PD-1 or CTLA-4 antagonists led to more significant T cell infiltration and tumor regression (213–216). CSF-1R inhibitor overcome the resistance to PD-1/PD-L1 axis blockade in an esophageal adenocarcinoma model, resulting in enhanced T cells infiltration and reduced M2 macrophage polarization in the TIME. It confirms that the direct translation of TAM suppression into clinical benefit (217). Emactuzumab, an anti-CSF-1R antibody, has a manageable safety profile in combination with atezolizumab over atezolizumab monotherapy. The increase in CD8 +TILs after therapy appeared to be associated with persistence of TAM subsets (218). Similarly, combined blockade of different targets is worth exploring its clinical effect. Sabatolimab, an Anti-TIM-3 Antibody, in combination with spartalizumab, an Anti-PD-1 Antibody, shows preliminary signs of antitumor activity (219). The combination of relatlimab, a LAG-3-blocking antibody, and nivolumab has been shown to be safe and to have antitumor activity in patients with previously treated melanoma. The ORR was 33% in some pembrolizumab-refractory patients and 50% in PD-1 naïve patients (220). Afterwards, in patients with previously untreated metastatic or unresectable melanoma, this combination did not show a new safety signal but it provided a greater benefit in progression-free survival than inhibition of PD-1 alone (221).
Vaccines will increase tumor-specific T cells due to intensified immunogenicity. Tumor-specific T cells will still be subject to the immunosuppressive microenvironment, which can be altered by checkpoint inhibitors. Effective vaccines combined with therapies targeting the TIME, such as checkpoint inhibitors, are likely to yield optimal results (222). The personalized neoantigen-based vaccine, NEO-PV-01, combined with nivolumab stimulate durable neoantigen-specific T cell responses in patients with advanced melanoma, non-small cell lung cancer, or bladder cancer (207). GX-188E in combination with pembrolizumab showed preliminary anti-tumor activity in patients with recurrent or advanced cervical cancer (223).
Multiple non-redundant immunosuppressive mechanisms coexist within the tumour microenvironment. A major immunosuppressive mechanism is the hypoxia adenosinergic immunosuppressive pathway, which now represents an attractive new target for cancer therapy. Several strategies described above can inhibit this mechanism. The ultimate goal of these strategies is to attenuate hypoxia driven and CD39/CD73 mediated accumulation of extracellular adenosine and immunosuppressive signals (174, 224, 225). This liberates the anti-tumor immunity of T and NK cells. In addition to the combination of A2AR inhibitor and anti-PD-1/PD-L1, A2AR inhibitor was also combined with nanovaccine to activate CD8 T and NK cells and inhibit the proliferation of regulatory T cells. Thus, this strategy could trigger a robust systemic antitumor immune response (173, 226). Furthermore, deletion of A2AR enhances the efficacy of CAR T cells (226, 227). Another way to implement this strategy is hyperoxygenation to improve cancer immunotherapies (69, 224).
In addition to the combination of immunotherapy above, the combination of immunotherapy with antiangiogenics, chemotherapy and radiation is under clinical consideration. Nearly every targeted therapy proven to modulate the immune response is currently being tried in combination with immunotherapy (228, 229).
Conclusion
The development of immunotherapy has achieved great clinical results, but the heterogeneity of the TIME makes it difficult to determine the best immunotherapy for individuals. There are still many obstacles in the potential development of immunotherapy. The formation of immunosuppressive microenvironment promotes tumor immune escape and restricts the clinical effect of immunotherapy. The further understanding of the TIME mechanism is conducive to the development of immunotherapy. Combined therapy is more conducive to the remodeling of microenvironment and can bring better clinical benefits. However, this raises the question whether improving anti-tumor immunity will lead to more serious irAEs (More detailed explanations in review (66, 230, 231). On the one hand, the further understanding of microenvironment mechanism is expected to balance the internal environment balance between anti-tumor immunity and Irae. On the other hand, the interpretation of a large number of clinical results, including the detection and summary of adverse immune events, helps to determine the best treatment combination.
Author Contributions
Conceptualization, BL and HC. Writing original manuscript, BL and YW. Visualization, BL. Writing review and editing, BL, YW, DM, WC, JL, TY, CW, and HC. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Key Project of Science and Technology in Gansu province (grant no. 19ZD2WA001).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chen DS, Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
2. Khong HT, Restifo NP. Natural Selection of Tumor Variants in the Generation of “Tumor Escape” Phenotypes. Nat Immunol (2002) 3(11) :999–1005. doi: 10.1038/ni1102-999
3. Thomas DA, Massagué J. TGF-Beta Directly Targets Cytotoxic T Cell Functions During Tumor Evasion of Immune Surveillance. Cancer Cell (2005) 8(5):369–80. doi: 10.1016/j.ccr.2005.10.012
4. Drake CG, Jaffee E, Pardoll DM. Mechanisms of Immune Evasion by Tumors. Adv Immunol (2006) 90:51–81. doi: 10.1016/S0065-2776(06)90002-9
5. Locy H, de Mey S, de Mey W, De Ridder M, Thielemans K, Maenhout SK. Immunomodulation of the Tumor Microenvironment: Turn Foe Into Friend. Front Immunol (2018) 9:2909. doi: 10.3389/fimmu.2018.02909
6. Topalian SL, Drake CG, Pardoll DM. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell (2015) 27(4):450–61. doi: 10.1016/j.ccell.2015.03.001
7. Li M, Li M, Yang Y, Liu Y, Xie H, Yu Q, et al. Remodeling Tumor Immune Microenvironment via Targeted Blockade of PI3K-γ and CSF-1/CSF-1R Pathways in Tumor Associated Macrophages for Pancreatic Cancer Therapy. J Control Release (2020) 321:23–35. doi: 10.1016/j.jconrel.2020.02.011
8. Mastelic-Gavillet B, Balint K, Boudousquie C, Gannon PO, Kandalaft LE. Personalized Dendritic Cell Vaccines-Recent Breakthroughs and Encouraging Clinical Results. Front Immunol (2019) 10:766. doi: 10.3389/fimmu.2019.00766
9. Wherry EJ, Ahmed R. Memory CD8 T-Cell Differentiation During Viral Infection. J Virol (2004) 78(11):5535–45. doi: 10.1128/JVI.78.11.5535-5545.2004
10. Farhood B, Najafi M, Mortezaee K. CD8 Cytotoxic T Lymphocytes in Cancer Immunotherapy: A Review. J Cell Physiol (2019) 234(6):8509–21. doi: 10.1002/jcp.27782
11. Bennett SR, Carbone FR, Karamalis F, Miller JF, Heath WR. Induction of a CD8+ Cytotoxic T Lymphocyte Response by Cross-Priming Requires Cognate CD4+ T Cell Help. J Exp Med (1997) 186(1):65–70. doi: 10.1084/jem.186.1.65
12. Bourgeois C, Rocha B, Tanchot C. A Role for CD40 Expression on CD8+ T Cells in the Generation of CD8+ T Cell Memory. Science (2002) 297(5589):2060–3. doi: 10.1126/science.1072615
13. Fu C, Jiang A. Dendritic Cells and CD8 T Cell Immunity in Tumor Microenvironment. Front Immunol (2018) 9:3059. doi: 10.3389/fimmu.2018.03059
14. Böttcher JP, Reis e Sousa C. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends Cancer (2018) 4(11):784–92. doi: 10.1016/j.trecan.2018.09.001
15. Myers JA, Miller JS. Exploring the NK Cell Platform for Cancer Immunotherapy. Nat Rev Clin Oncol (2021) 18(2):85–100. doi: 10.1038/s41571-020-0426-7
16. Cózar B, Greppi M, Carpentier S, Narni-Mancinelli E, Chiossone L, Vivier E. Tumor-Infiltrating Natural Killer Cells. Cancer Discovery (2021) 11(1):34–44. doi: 10.1158/2159-8290.CD-20-0655
17. Malmberg K-J, Carlsten M, Björklund A, Sohlberg E, Bryceson YT, Ljunggren H-G. Natural Killer Cell-Mediated Immunosurveillance of Human Cancer. Semin Immunol (2017) 31:20–9. doi: 10.1016/j.smim.2017.08.002
18. Zhu J, Zhi Q, Zhou BP, Tao M, Liu J, Li W. The Role of Tumor Associated Macrophages in the Tumor Microenvironment: Mechanism and Functions. Anticancer Agents Med Chem (2016) 16(9):1133–41. doi: 10.2174/1871520616666160520112622
19. Goswami KK, Bose A, Baral R. Macrophages in Tumor: An Inflammatory Perspective. Clin Immunol (2021) 232:108875. doi: 10.1016/j.clim.2021.108875
20. van Dalen FJ, van Stevendaal MHME, Fennemann FL, Verdoes M, Ilina O. Molecular Repolarisation of Tumour-Associated Macrophages. Molecules (2018) 24(1):9. doi: 10.3390/molecules24010009
22. Pan Y, Yu Y, Wang X, Zhang T. Tumor-Associated Macrophages in Tumor Immunity. Front Immunol (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
23. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T Cells and Immune Tolerance. Cell (2008) 133(5):775–87. doi: 10.1016/j.cell.2008.05.009
24. Wei X, Zhang J, Gu Q, Huang M, Zhang W, Guo J, et al. Reciprocal Expression of IL-35 and IL-10 Defines Two Distinct Effector Treg Subsets That Are Required for Maintenance of Immune Tolerance. Cell Rep (2017) 21(7):1853–69. doi: 10.1016/j.celrep.2017.10.090
25. Budhu S, Schaer DA, Li Y, Toledo-Crow R, Panageas K, Yang X, et al. Blockade of Surface-Bound TGF-β on Regulatory T Cells Abrogates Suppression of Effector T Cell Function in the Tumor Microenvironment. Sci Signal (2017) 10(494):eaak9702. doi: 10.1126/scisignal.aak9702
26. Liu C, Chikina M, Deshpande R, Menk AV, Wang T, Tabib T, et al. Treg Cells Promote the SREBP1-Dependent Metabolic Fitness of Tumor-Promoting Macrophages via Repression of CD8 T Cell-Derived Interferon-γ. Immunity (2019) 51(2). doi: 10.1016/j.immuni.2019.06.017
27. Kalia V, Penny LA, Yuzefpolskiy Y, Baumann FM, Sarkar S. Quiescence of Memory CD8(+) T Cells Is Mediated by Regulatory T Cells Through Inhibitory Receptor CTLA-4. Immunity (2015) 42(6):1116–29. doi: 10.1016/j.immuni.2015.05.023
28. Sarhan D, Hippen KL, Lemire A, Hying S, Luo X, Lenvik T, et al. Adaptive NK Cells Resist Regulatory T-Cell Suppression Driven by IL37. Cancer Immunol Res (2018) 6(7):766–75. doi: 10.1158/2326-6066.CIR-17-0498
29. Walker LSK, Sansom DM. The Emerging Role of CTLA4 as a Cell-Extrinsic Regulator of T Cell Responses. Nat Rev Immunol (2011) 11(12):852–63. doi: 10.1038/nri3108
30. Huard B, Mastrangeli R, Prigent P, Bruniquel D, Donini S, El-Tayar N, et al. Characterization of the Major Histocompatibility Complex Class II Binding Site on LAG-3 Protein. Proc Natl Acad Sci U.S.A. (1997) 94(11):5744–9. doi: 10.1073/pnas.94.11.5744
31. Li C, Jiang P, Wei S, Xu X, Wang J. Regulatory T Cells in Tumor Microenvironment: New Mechanisms, Potential Therapeutic Strategies and Future Prospects. Mol Cancer (2020) 19(1):116. doi: 10.1186/s12943-020-01234-1
32. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and Perforin are Important for Regulatory T Cell-Mediated Suppression of Tumor Clearance. Immunity (2007) 27(4):635–46. doi: 10.1016/j.immuni.2007.08.014
33. Ohue Y, Nishikawa H. Regulatory T (Treg) Cells in Cancer: Can Treg Cells be a New Therapeutic Target? Cancer Sci (2019) 110(7):2080–9. doi: 10.1111/cas.14069
34. Gabrilovich DI. Myeloid-Derived Suppressor Cells. Cancer Immunol Res (2017) 5(1):3–8. doi: 10.1158/2326-6066.CIR-16-0297
35. Veglia F, Perego M, Gabrilovich D. Myeloid-Derived Suppressor Cells Coming of Age. Nat Immunol (2018) 19(2):108–19. doi: 10.1038/s41590-017-0022-x
36. Kwak T, Wang F, Deng H, Condamine T, Kumar V, Perego M, et al. Distinct Populations of Immune-Suppressive Macrophages Differentiate From Monocytic Myeloid-Derived Suppressor Cells in Cancer. Cell Rep (2020) 33(13):108571. doi: 10.1016/j.celrep.2020.108571
37. Krishnamoorthy M, Gerhardt L, Maleki Vareki S. Immunosuppressive Effects of Myeloid-Derived Suppressor Cells in Cancer and Immunotherapy. Cells (2021) 10(5):1170. doi: 10.3390/cells10051170
38. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-Expanded Myeloid-Derived Suppressor Cells Induce Anergy of NK Cells Through Membrane-Bound TGF-Beta 1. J Immunol (2009) 182(1):240–9. doi: 10.4049/jimmunol.182.1.240
39. Sakuishi K, Jayaraman P, Behar SM, Anderson AC, Kuchroo VK. Emerging Tim-3 Functions in Antimicrobial and Tumor Immunity. Trends Immunol (2011) 32(8):345–9. doi: 10.1016/j.it.2011.05.003
40. Sun L, Clavijo PE, Robbins Y, Patel P, Friedman J, Greene S, et al. Inhibiting Myeloid-Derived Suppressor Cell Trafficking Enhances T Cell Immunotherapy. JCI Insight (2019) 4(7):e126853. doi: 10.1172/jci.insight.126853
41. Anderson NR, Minutolo NG, Gill S, Klichinsky M. Macrophage-Based Approaches for Cancer Immunotherapy. Cancer Res (2021) 81(5):1201–8. doi: 10.1158/0008-5472.CAN-20-2990
42. Wu K, Lin K, Li X, Yuan X, Xu P, Ni P, et al. Redefining Tumor-Associated Macrophage Subpopulations and Functions in the Tumor Microenvironment. Front Immunol (2020) 11:1731. doi: 10.3389/fimmu.2020.01731
43. Zhu S, Luo Z, Li X, Han X, Shi S, Zhang T. Tumor-Associated Macrophages: Role in Tumorigenesis and Immunotherapy Implications. J Cancer (2021) 12(1):54–64. doi: 10.7150/jca.49692
44. Boutilier AJ, Elsawa SF. Macrophage Polarization States in the Tumor Microenvironment. Int J Mol Sci (2021) 22(13):6995. doi: 10.3390/ijms22136995
45. Griess B, Mir S, Datta K, Teoh-Fitzgerald M. Scavenging Reactive Oxygen Species Selectively Inhibits M2 Macrophage Polarization and Their Pro-Tumorigenic Function in Part, via Stat3 Suppression. Free Radic Biol Med (2020) 147:48–60. doi: 10.1016/j.freeradbiomed.2019.12.018
46. Najafi M, Hashemi Goradel N, Farhood B, Salehi E, Nashtaei MS, Khanlarkhani N, et al. Macrophage Polarity in Cancer: A Review. J Cell Biochem (2019) 120(3):2756–65. doi: 10.1002/jcb.27646
47. Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas Promote Immunosuppression Through Induction of B7-H1 Expression in Tumor-Associated Macrophages. Clin Cancer research: an Off J Am Assoc Cancer Res (2013) 19(12):3165–75. doi: 10.1158/1078-0432.CCR-12-3314
48. Albini A, Bruno A, Noonan DM, Mortara L. Contribution to Tumor Angiogenesis From Innate Immune Cells Within the Tumor Microenvironment: Implications for Immunotherapy. Front Immunol (2018) 9:527. doi: 10.3389/fimmu.2018.00527
49. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-Beta: “N1” Versus “N2” TAN. Cancer Cell (2009) 16(3):183–94. doi: 10.1016/j.ccr.2009.06.017
50. Zhu Q, Zhang X, Zhang L, Li W, Wu H, Yuan X, et al. The IL-6-STAT3 Axis Mediates a Reciprocal Crosstalk Between Cancer-Derived Mesenchymal Stem Cells and Neutrophils to Synergistically Prompt Gastric Cancer Progression. Cell Death Dis (2014) 5:e1295. doi: 10.1038/cddis.2014.263
51. Michaeli J, Shaul ME, Mishalian I, Hovav A-H, Levy L, Zolotriov L, et al. Tumor-Associated Neutrophils Induce Apoptosis of non-Activated CD8 T-Cells in a Tnfα and NO-Dependent Mechanism, Promoting a Tumor-Supportive Environment. Oncoimmunology (2017) 6(11):e1356965. doi: 10.1080/2162402X.2017.1356965
52. Giese MA, Hind LE, Huttenlocher A. Neutrophil Plasticity in the Tumor Microenvironment. Blood (2019) 133(20):2159–67. doi: 10.1182/blood-2018-11-844548
53. Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, et al. Arginase I-Producing Myeloid-Derived Suppressor Cells in Renal Cell Carcinoma are a Subpopulation of Activated Granulocytes. Cancer Res (2009) 69(4):1553–60. doi: 10.1158/0008-5472.CAN-08-1921
54. Spiegel A, Brooks MW, Houshyar S, Reinhardt F, Ardolino M, Fessler E, et al. Neutrophils Suppress Intraluminal NK Cell-Mediated Tumor Cell Clearance and Enhance Extravasation of Disseminated Carcinoma Cells. Cancer Discovery (2016) 6(6):630–49. doi: 10.1158/2159-8290.CD-15-1157
55. Maggi E, Veneziani I, Moretta L, Cosmi L, Annunziato F. Group 2 Innate Lymphoid Cells: A Double-Edged Sword in Cancer? Cancers (2020) 12(11):3452. doi: 10.3390/cancers12113452
56. Chevalier MF, Trabanelli S, Racle J, Salomé B, Cesson V, Gharbi D, et al. ILC2-Modulated T Cell-to-MDSC Balance is Associated With Bladder Cancer Recurrence. J Clin Invest (2017) 127(8):2916–29. doi: 10.1172/JCI89717
57. Trabanelli S, Chevalier MF, Martinez-Usatorre A, Gomez-Cadena A, Salomé B, Lecciso M, et al. Tumour-Derived PGD2 and NKp30-B7H6 Engagement Drives an Immunosuppressive ILC2-MDSC Axis. Nat Commun (2017) 8(1):593. doi: 10.1038/s41467-017-00678-2
58. Gong D, Shi W, S-j Yi, Chen H, Groffen J, Heisterkamp N. Tgfβ Signaling Plays a Critical Role in Promoting Alternative Macrophage Activation. BMC Immunol (2012) 13:31. doi: 10.1186/1471-2172-13-31
59. Mattner J, Wirtz S. Friend or Foe? The Ambiguous Role of Innate Lymphoid Cells in Cancer Development. Trends Immunol (2017) 38(1):29–38. doi: 10.1016/j.it.2016.10.004
60. Molofsky AB, Van Gool F, Liang H-E, Van Dyken SJ, Nussbaum JC, Lee J, et al. Interleukin-33 and Interferon-γ Counter-Regulate Group 2 Innate Lymphoid Cell Activation During Immune Perturbation. Immunity (2015) 43(1):161–74. doi: 10.1016/j.immuni.2015.05.019
61. Long A, Dominguez D, Qin L, Chen S, Fan J, Zhang M, et al. Type 2 Innate Lymphoid Cells Impede IL-33-Mediated Tumor Suppression. J Immunol (2018) 201(11):3456–64. doi: 10.4049/jimmunol.1800173
62. Hinshaw DC, Shevde LA. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res (2019) 79(18):4557–66. doi: 10.1158/0008-5472.CAN-18-3962
63. Terabe M, Berzofsky JA. Tissue-Specific Roles of NKT Cells in Tumor Immunity. Front Immunol (2018) 9:1838. doi: 10.3389/fimmu.2018.01838
64. Dhodapkar MV, Kumar V. Type II NKT Cells and Their Emerging Role in Health and Disease. J Immunol (2017) 198(3):1015–21. doi: 10.4049/jimmunol.1601399
65. Warburg O. On Respiratory Impairment in Cancer Cells. Science (1956) 124(3215):269–70. doi: 10.1126/science.124.3215.269
66. Ferrari SM, Fallahi P, Elia G, Ragusa F, Ruffilli I, Patrizio A, et al. Autoimmune Endocrine Dysfunctions Associated With Cancer Immunotherapies. Int J Mol Sci (2019) 20(10):2560. doi: 10.3390/ijms20102560
67. Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N, et al. The Cancer Metabolic Reprogramming and Immune Response. Mol Cancer (2021) 20(1):28. doi: 10.1186/s12943-021-01316-8
68. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, et al. A2A Adenosine Receptor Protects Tumors From Antitumor T Cells. Proc Natl Acad Sci U.S.A. (2006) 103(35):13132–7. doi: 10.1073/pnas.0605251103
69. Hatfield SM, Kjaergaard J, Lukashev D, Belikoff B, Schreiber TH, Sethumadhavan S, et al. Systemic Oxygenation Weakens the Hypoxia and Hypoxia Inducible Factor 1alpha-Dependent and Extracellular Adenosine-Mediated Tumor Protection. J Mol Med (Berl) (2014) 92(12):1283–92. doi: 10.1007/s00109-014-1189-3
70. Horenstein AL, Bracci C, Morandi F, Malavasi F. CD38 in Adenosinergic Pathways and Metabolic Re-Programming in Human Multiple Myeloma Cells: In-Tandem Insights From Basic Science to Therapy. Front Immunol (2019) 10:760. doi: 10.3389/fimmu.2019.00760
71. Kong HY, Byun J. Emerging Roles of Human Prostatic Acid Phosphatase. Biomol Ther (Seoul) (2013) 21(1):10–20. doi: 10.4062/biomolther.2012.095
72. Cekic C, Linden J. Purinergic Regulation of the Immune System. Nat Rev Immunol (2016) 16(3):177–92. doi: 10.1038/nri.2016.4
73. St Hilaire C, Carroll SH, Chen H, Ravid K. Mechanisms of Induction of Adenosine Receptor Genes and its Functional Significance. J Cell Physiol (2009) 218(1):35–44. doi: 10.1002/jcp.21579
74. Sitkovsky MV, Hatfield S, Abbott R, Belikoff B, Lukashev D, Ohta A. Hostile, Hypoxia-A2-Adenosinergic Tumor Biology as the Next Barrier to Overcome for Tumor Immunologists. Cancer Immunol Res (2014) 2(7):598–605. doi: 10.1158/2326-6066.CIR-14-0075
75. Young A, Ngiow SF, Gao Y, Patch AM, Barkauskas DS, Messaoudene M, et al. A2AR Adenosine Signaling Suppresses Natural Killer Cell Maturation in the Tumor Microenvironment. Cancer Res (2018) 78(4):1003–16. doi: 10.1158/0008-5472.CAN-17-2826
76. Ludwig N, Yerneni SS, Azambuja JH, Gillespie DG, Menshikova EV, Jackson EK, et al. Tumor-Derived Exosomes Promote Angiogenesis via Adenosine A2B Receptor Signaling. Angiogenesis (2020) 23(4):599–610. doi: 10.1007/s10456-020-09728-8
77. Ryzhov S, Novitskiy SV, Goldstein AE, Biktasova A, Blackburn MR, Biaggioni I, et al. Adenosinergic Regulation of the Expansion and Immunosuppressive Activity of CD11b+Gr1+ Cells. J Immunol (2011) 187(11):6120–9. doi: 10.4049/jimmunol.1101225
78. Sorrentino C, Miele L, Porta A, Pinto A, Morello S. Myeloid-Derived Suppressor Cells Contribute to A2B Adenosine Receptor-Induced VEGF Production and Angiogenesis in a Mouse Melanoma Model. Oncotarget (2015) 6(29):27478–89. doi: 10.18632/oncotarget.4393
79. Mediavilla-Varela M, Luddy K, Noyes D, Khalil FK, Neuger AM, Soliman H, et al. Antagonism of Adenosine A2A Receptor Expressed by Lung Adenocarcinoma Tumor Cells and Cancer Associated Fibroblasts Inhibits Their Growth. Cancer Biol Ther (2013) 14(9):860–8. doi: 10.4161/cbt.25643
80. Sorrentino C, Miele L, Porta A, Pinto A, Morello S. Activation of the A2B Adenosine Receptor in B16 Melanomas Induces CXCL12 Expression in FAP-Positive Tumor Stromal Cells, Enhancing Tumor Progression. Oncotarget (2016) 7(39):64274–88. doi: 10.18632/oncotarget.11729
81. Ji D, Song C, Li Y, Xia J, Wu Y, Jia J, et al. Combination of Radiotherapy and Suppression of Tregs Enhances Abscopal Antitumor Effect and Inhibits Metastasis in Rectal Cancer. J Immunother Cancer (2020) 8(2):e000826. doi: 10.1136/jitc-2020-000826
82. Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, et al. Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3 Regulatory T Cells (Tregs) in Human Cancers. Clin Cancer research: an Off J Am Assoc Cancer Res (2019) 25(4):1233–8. doi: 10.1158/1078-0432.CCR-18-0762
83. Boomer JS, Green JM. An Enigmatic Tail of CD28 Signaling. Cold Spring Harb Perspect Biol (2010) 2(8):a002436. doi: 10.1101/cshperspect.a002436
84. Hosseini A, Gharibi T, Marofi F, Babaloo Z, Baradaran B. CTLA-4: From Mechanism to Autoimmune Therapy. Int Immunopharmacol (2020) 80:106221. doi: 10.1016/j.intimp.2020.106221
85. Fritz JM, Lenardo MJ. Development of Immune Checkpoint Therapy for Cancer. J Exp Med (2019) 216(6):1244–54. doi: 10.1084/jem.20182395
86. Takeuchi Y, Tanemura A, Tada Y, Katayama I, Kumanogoh A, Nishikawa H. Clinical Response to PD-1 Blockade Correlates With a Sub-Fraction of Peripheral Central Memory CD4+ T Cells in Patients With Malignant Melanoma. Int Immunol (2018) 30(1):13–22. doi: 10.1093/intimm/dxx073
87. Hartley GP, Chow L, Ammons DT, Wheat WH, Dow SW. Programmed Cell Death Ligand 1 (PD-L1) Signaling Regulates Macrophage Proliferation and Activation. Cancer Immunol Res (2018) 6(10):1260–73. doi: 10.1158/2326-6066.CIR-17-0537
88. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 Expression by Tumour-Associated Macrophages Inhibits Phagocytosis and Tumour Immunity. Nature (2017) 545(7655):495–9. doi: 10.1038/nature22396
89. Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault M-C, Trevino TN, et al. Contribution of NK Cells to Immunotherapy Mediated by PD-1/PD-L1 Blockade. J Clin Invest (2018) 128(10):4654–68. doi: 10.1172/JCI99317
90. Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al. Fibrinogen-Like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell (2019), 176(1–2):334–47.e12. doi: 10.1016/j.cell.2018.11.010
91. Brignone C, Gutierrez M, Mefti F, Brain E, Jarcau R, Cvitkovic F, et al. First-Line Chemoimmunotherapy in Metastatic Breast Carcinoma: Combination of Paclitaxel and IMP321 (LAG-3Ig) Enhances Immune Responses and Antitumor Activity. J Transl Med (2010) 8:71. doi: 10.1186/1479-5876-8-71
92. Acharya N, Sabatos-Peyton C, Anderson AC. Tim-3 Finds its Place in the Cancer Immunotherapy Landscape. J Immunother Cancer (2020) 8(1):e000911. doi: 10.1136/jitc-2020-000911
93. Ma CJ, Ren JP, Li GY, Wu XY, Brockstedt DG, Lauer P, et al. Enhanced Virus-Specific CD8+ T Cell Responses by Listeria Monocytogenes-Infected Dendritic Cells in the Context of Tim-3 Blockade. PloS One (2014) 9(1):e87821. doi: 10.1371/journal.pone.0087821
94. Nagahara K, Arikawa T, Oomizu S, Kontani K, Nobumoto A, Tateno H, et al. Galectin-9 Increases Tim-3+ Dendritic Cells and CD8+ T Cells and Enhances Antitumor Immunity via Galectin-9-Tim-3 Interactions. J Immunol (2008) 181(11):7660–9. doi: 10.4049/jimmunol.181.11.7660
95. Wu W, Shi Y, Li S, Zhang Y, Liu Y, Wu Y, et al. Blockade of Tim-3 Signaling Restores the Virus-Specific CD8+ T-Cell Response in Patients With Chronic Hepatitis B. Eur J Immunol (2012) 42(5):1180–91. doi: 10.1002/eji.201141852
96. Gautron A-S, Dominguez-Villar M, de Marcken M, Hafler DA. Enhanced Suppressor Function of TIM-3+ FoxP3+ Regulatory T Cells. Eur J Immunol (2014) 44(9):2703–11. doi: 10.1002/eji.201344392
97. Ji XJ, Ma CJ, Wang JM, Wu XY, Niki T, Hirashima M, et al. HCV-Infected Hepatocytes Drive CD4+ CD25+ Foxp3+ Regulatory T-Cell Development Through the Tim-3/Gal-9 Pathway. Eur J Immunol (2013) 43(2):458–67. doi: 10.1002/eji.201242768
98. Ge Z, Zhou G, Campos Carrascosa L, Gausvik E, Boor PPC, Noordam L, et al. TIGIT and PD1 Co-Blockade Restores Ex Vivo Functions of Human Tumor-Infiltrating CD8 T Cells in Hepatocellular Carcinoma. Cell Mol Gastroenterol Hepatol (2021) 12(2):443–64. doi: 10.1016/j.jcmgh.2021.03.003
99. Lozano E, Mena M-P, Díaz T, Martin-Antonio B, León S, Rodríguez-Lobato L-G, et al. Nectin-2 Expression on Malignant Plasma Cells Is Associated With Better Response to TIGIT Blockade in Multiple Myeloma. Clin Cancer research: an Off J Am Assoc Cancer Res (2020) 26(17):4688–98. doi: 10.1158/1078-0432.CCR-19-3673
100. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the Checkpoint Receptor TIGIT Prevents NK Cell Exhaustion and Elicits Potent Anti-Tumor Immunity. Nat Immunol (2018) 19(7):723–32. doi: 10.1038/s41590-018-0132-0
101. Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, et al. VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Res (2014) 74(7):1933–44. doi: 10.1158/0008-5472.CAN-13-1506
102. Wang J, Sun J, Liu LN, Flies DB, Nie X, Toki M, et al. Siglec-15 as an Immune Suppressor and Potential Target for Normalization Cancer Immunotherapy. Nat Med (2019) 25(4):656–66. doi: 10.1038/s41591-019-0374-x
103. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R Blockade Reprograms Tumor-Infiltrating Macrophages and Improves Response to T-Cell Checkpoint Immunotherapy in Pancreatic Cancer Models. Cancer Res (2014) 74(18):5057–69. doi: 10.1158/0008-5472.CAN-13-3723
104. Ngiow SF, Meeth KM, Stannard K, Barkauskas DS, Bollag G, Bosenberg M, et al. Co-Inhibition of Colony Stimulating Factor-1 Receptor and BRAF Oncogene in Mouse Models of BRAF Melanoma. Oncoimmunology (2016) 5(3):e1089381. doi: 10.1080/2162402X.2015.1089381
105. Serrels A, Lund T, Serrels B, Byron A, McPherson RC, von Kriegsheim A, et al. Nuclear FAK Controls Chemokine Transcription, Tregs, and Evasion of Anti-Tumor Immunity. Cell (2015) 163(1):160–73. doi: 10.1016/j.cell.2015.09.001
106. Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting Focal Adhesion Kinase Renders Pancreatic Cancers Responsive to Checkpoint Immunotherapy. Nat Med (2016) 22(8):851–60. doi: 10.1038/nm.4123
107. Stokes JB, Adair SJ, Slack-Davis JK, Walters DM, Tilghman RW, Hershey ED, et al. Inhibition of Focal Adhesion Kinase by PF-562,271 Inhibits the Growth and Metastasis of Pancreatic Cancer Concomitant With Altering the Tumor Microenvironment. Mol Cancer Ther (2011) 10(11):2135–45. doi: 10.1158/1535-7163.MCT-11-0261
108. Holmgaard RB, Schaer DA, Li Y, Castaneda SP, Murphy MY, Xu X, et al. Targeting the Tgfβ Pathway With Galunisertib, a Tgfβri Small Molecule Inhibitor, Promotes Anti-Tumor Immunity Leading to Durable, Complete Responses, as Monotherapy and in Combination With Checkpoint Blockade. J Immunother Cancer (2018) 6(1):47. doi: 10.1186/s40425-018-0356-4
109. Martin CJ, Datta A, Littlefield C, Kalra A, Chapron C, Wawersik S, et al. Selective Inhibition of Tgfβ1 Activation Overcomes Primary Resistance to Checkpoint Blockade Therapy by Altering Tumor Immune Landscape. Sci Transl Med (2020) 12(536):eaay8456. doi: 10.1126/scitranslmed.aay8456
110. Kim CG, Jang M, Kim Y, Leem G, Kim KH, Lee H, et al. VEGF-A Drives TOX-Dependent T Cell Exhaustion in Anti-PD-1-Resistant Microsatellite Stable Colorectal Cancers. Sci Immunol (2019) 4(41):eaay0555. doi: 10.1126/sciimmunol.aay0555
111. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet A-L, et al. VEGF-A Modulates Expression of Inhibitory Checkpoints on CD8+ T Cells in Tumors. J Exp Med (2015) 212(2):139–48. doi: 10.1084/jem.20140559
112. Sobhani N, Tardiel-Cyril DR, Davtyan A, Generali D, Roudi R, Li Y. CTLA-4 in Regulatory T Cells for Cancer Immunotherapy. Cancers (2021) 13(6):1440. doi: 10.3390/cancers13061440
113. Chen Q, Wang J, Chen W, Zhang Q, Wei T, Zhou Y, et al. B7-H5/CD28H is a Co-Stimulatory Pathway and Correlates With Improved Prognosis in Pancreatic Ductal Adenocarcinoma. Cancer Sci (2019) 110(2):530–9. doi: 10.1111/cas.13914
114. Rowshanravan B, Halliday N, Sansom DM. CTLA-4: A Moving Target in Immunotherapy. Blood (2018) 131(1):58–67. doi: 10.1182/blood-2017-06-741033
115. Qu Q, Zhai Z, Xu J, Li S, Chen C, Lu B. IL36 Cooperates With Anti-CTLA-4 Mabs to Facilitate Antitumor Immune Responses. Front Immunol (2020) 11:634. doi: 10.3389/fimmu.2020.00634
116. Pai C-CS, Simons DM, Lu X, Evans M, Wei J, Wang Y-H, et al. Tumor-Conditional Anti-CTLA4 Uncouples Antitumor Efficacy From Immunotherapy-Related Toxicity. J Clin Invest (2019) 129(1):349–63. doi: 10.1172/JCI123391
117. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved Survival With Ipilimumab in Patients With Metastatic Melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
118. Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J Clin Oncol (2015) 33(17):1889–94. doi: 10.1200/JCO.2014.56.2736
119. Han Y, Liu D, Li L. PD-1/PD-L1 Pathway: Current Researches in Cancer. Am J Cancer Res (2020) 10(3):727–42.
120. Mehdizadeh S, Bayatipoor H, Pashangzadeh S, Jafarpour R, Shojaei Z, Motallebnezhad M. Immune Checkpoints and Cancer Development: Therapeutic Implications and Future Directions. Pathol Res Pract (2021) 223:153485. doi: 10.1016/j.prp.2021.153485
121. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 Blockade Induces Responses by Inhibiting Adaptive Immune Resistance. Nature (2014) 515(7528):568–71. doi: 10.1038/nature13954
122. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Five-Year Survival Outcomes for Patients With Advanced Melanoma Treated With Pembrolizumab in KEYNOTE-001. Ann Oncol (2019) 30(4):582–8. doi: 10.1093/annonc/mdz011
123. Leighl NB, Hellmann MD, Hui R, Carcereny E, Felip E, Ahn M-J, et al. Pembrolizumab in Patients With Advanced non-Small-Cell Lung Cancer (KEYNOTE-001): 3-Year Results From an Open-Label, Phase 1 Study. Lancet Respir Med (2019) 7(4):347–57. doi: 10.1016/S2213-2600(18)30500-9
124. Gandhi MK, Lambley E, Duraiswamy J, Dua U, Smith C, Elliott S, et al. Expression of LAG-3 by Tumor-Infiltrating Lymphocytes is Coincident With the Suppression of Latent Membrane Antigen-Specific CD8+ T-Cell Function in Hodgkin Lymphoma Patients. Blood (2006) 108(7):2280–9. doi: 10.1182/blood-2006-04-015164
125. Camisaschi C, Casati C, Rini F, Perego M, De Filippo A, Triebel F, et al. LAG-3 Expression Defines a Subset of CD4(+)CD25(high)Foxp3(+) Regulatory T Cells That are Expanded at Tumor Sites. J Immunol (2010) 184(11):6545–51. doi: 10.4049/jimmunol.0903879
126. Chocarro L, Blanco E, Zuazo M, Arasanz H, Bocanegra A, Fernández-Rubio L, et al. Understanding LAG-3 Signaling. Int J Mol Sci (2021) 22(10):5282. doi: 10.3390/ijms22105282
127. Deng W-W, Mao L, Yu G-T, Bu L-L, Ma S-R, Liu B, et al. LAG-3 Confers Poor Prognosis and its Blockade Reshapes Antitumor Response in Head and Neck Squamous Cell Carcinoma. Oncoimmunology (2016) 5(11):e1239005. doi: 10.1080/2162402X.2016.1239005
128. Grosso JF, Kelleher CC, Harris TJ, Maris CH, Hipkiss EL, De Marzo A, et al. LAG-3 Regulates CD8+ T Cell Accumulation and Effector Function in Murine Self- and Tumor-Tolerance Systems. J Clin Invest (2007) 117(11):3383–92. doi: 10.1172/JCI31184
129. Maruhashi T, Sugiura D, Okazaki I-M, Okazaki T. LAG-3: From Molecular Functions to Clinical Applications. J Immunother Cancer (2020) 8(2):e001014. doi: 10.1136/jitc-2020-001014
130. Zelba H, Bedke J, Hennenlotter J, Mostböck S, Zettl M, Zichner T, et al. PD-1 and LAG-3 Dominate Checkpoint Receptor-Mediated T-Cell Inhibition in Renal Cell Carcinoma. Cancer Immunol Res (2019) 7(11):1891–9. doi: 10.1158/2326-6066.CIR-19-0146
131. Qin S, Xu L, Yi M, Yu S, Wu K, Luo S. Novel Immune Checkpoint Targets: Moving Beyond PD-1 and CTLA-4. Mol Cancer (2019) 18(1):155. doi: 10.1186/s12943-019-1091-2
132. Ascierto PA, Bono P, Bhatia S, Melero I, Nyakas MS, Svane IM, et al. Efficacy of BMS-986016, a Monoclonal Antibody That Targets Lymphocyte Activation Gene-3 (LAG-3), in Combination With Nivolumab in Pts With Melanoma Who Progressed During Prior Anti-PD-1/PD-L1 Therapy (Mel Prior IO) in All-Comer and Biomarker-Enriched Populations. Ann Oncol (2017) 28:v611–2. doi: 10.1093/annonc/mdx440.011
133. Sordo-Bahamonde C, Lorenzo-Herrero S, González-Rodríguez AP, Payer ÁR, González-García E, López-Soto A, et al. LAG-3 Blockade With Relatlimab (BMS-986016) Restores Anti-Leukemic Responses in Chronic Lymphocytic Leukemia. Cancers (2021) 13(9):2112. doi: 10.3390/cancers13092112
134. Jiang H, Ni H, Zhang P, Guo X, Wu M, Shen H, et al. PD-L1/LAG-3 Bispecific Antibody Enhances Tumor-Specific Immunity. Oncoimmunology (2021) 10(1):1943180. doi: 10.1080/2162402X.2021.1943180
135. Kraman M, Faroudi M, Allen NL, Kmiecik K, Gliddon D, Seal C, et al. FS118, a Bispecific Antibody Targeting LAG-3 and PD-L1, Enhances T-Cell Activation Resulting in Potent Antitumor Activity. Clin Cancer research: an Off J Am Assoc Cancer Res (2020) 26(13):3333–44. doi: 10.1158/1078-0432.CCR-19-3548
136. Zahm CD, Moseman JE, Delmastro LE, G Mcneel D. PD-1 and LAG-3 Blockade Improve Anti-Tumor Vaccine Efficacy. Oncoimmunology (2021) 10(1):1912892. doi: 10.1080/2162402X.2021.1912892
137. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-Specific Cell Surface Protein Tim-3 Regulates Macrophage Activation and Severity of an Autoimmune Disease. Nature (2002) 415(6871):536–41. doi: 10.1038/415536a
138. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 Ligand Galectin-9 Negatively Regulates T Helper Type 1 Immunity. Nat Immunol (2005) 6(12):1245–52. doi: 10.1038/ni1271
139. Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, et al. Tim-3 Mediates Phagocytosis of Apoptotic Cells and Cross-Presentation. Blood (2009) 113(16):3821–30. doi: 10.1182/blood-2008-10-185884
140. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-Infiltrating DCs Suppress Nucleic Acid-Mediated Innate Immune Responses Through Interactions Between the Receptor TIM-3 and the Alarmin HMGB1. Nat Immunol (2012) 13(9):832–42. doi: 10.1038/ni.2376
141. Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. CEACAM1 Regulates TIM-3-Mediated Tolerance and Exhaustion. Nature (2015) 517(7534):386–90. doi: 10.1038/nature13848
142. Gao X, Zhu Y, Li G, Huang H, Zhang G, Wang F, et al. TIM-3 Expression Characterizes Regulatory T Cells in Tumor Tissues and is Associated With Lung Cancer Progression. PloS One (2012) 7(2):e30676. doi: 10.1371/journal.pone.0030676
143. Ndhlovu LC, Lopez-Verges S, Barbour JD, Jones RB, Jha AR, Long BR, et al. Tim-3 Marks Human Natural Killer Cell Maturation and Suppresses Cell-Mediated Cytotoxicity. Blood (2012) 119(16):3734–43. doi: 10.1182/blood-2011-11-392951
144. Kang CW, Dutta A, Chang LY, Mahalingam J, Lin YC, Chiang JM, et al. Apoptosis of Tumor Infiltrating Effector TIM-3+CD8+ T Cells in Colon Cancer. Sci Rep (2015) 5:15659. doi: 10.1038/srep15659
145. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 Expression is Associated With Tumor Antigen-Specific CD8+ T Cell Dysfunction in Melanoma Patients. J Exp Med (2010) 207(10):2175–86. doi: 10.1084/jem.20100637
146. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The Surface Protein TIGIT Suppresses T Cell Activation by Promoting the Generation of Mature Immunoregulatory Dendritic Cells. Nat Immunol (2009) 10(1):48–57. doi: 10.1038/ni.1674
147. Boles KS, Vermi W, Facchetti F, Fuchs A, Wilson TJ, Diacovo TG, et al. A Novel Molecular Interaction for the Adhesion of Follicular CD4 T Cells to Follicular DC. Eur J Immunol (2009) 39(3):695–703. doi: 10.1002/eji.200839116
148. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The Interaction of TIGIT With PVR and PVRL2 Inhibits Human NK Cell Cytotoxicity. Proc Natl Acad Sci U.S.A. (2009) 106(42):17858–63. doi: 10.1073/pnas.0903474106
149. Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD, et al. Cutting Edge: TIGIT has T Cell-Intrinsic Inhibitory Functions. J Immunol (2011) 186(3):1338–42. doi: 10.4049/jimmunol.1003081
150. Lozano E, Dominguez-Villar M, Kuchroo V, Hafler DA. The TIGIT/CD226 Axis Regulates Human T Cell Function. J Immunol (2012) 188(8):3869–75. doi: 10.4049/jimmunol.1103627
151. Liu S, Zhang H, Li M, Hu D, Li C, Ge B, et al. Recruitment of Grb2 and SHIP1 by the ITT-Like Motif of TIGIT Suppresses Granule Polarization and Cytotoxicity of NK Cells. Cell Death Differ (2013) 20(3):456–64. doi: 10.1038/cdd.2012.141
152. Fuhrman CA, Yeh WI, Seay HR, Saikumar Lakshmi P, Chopra G, Zhang L, et al. Divergent Phenotypes of Human Regulatory T Cells Expressing the Receptors TIGIT and CD226. J Immunol (2015) 195(1):145–55. doi: 10.4049/jimmunol.1402381
153. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg Cells Expressing the Coinhibitory Molecule TIGIT Selectively Inhibit Proinflammatory Th1 and Th17 Cell Responses. Immunity (2014) 40(4):569–81. doi: 10.1016/j.immuni.2014.02.012
154. Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a Novel Mouse Ig Superfamily Ligand That Negatively Regulates T Cell Responses. J Exp Med (2011) 208(3):577–92. doi: 10.1084/jem.20100619
155. ElTanbouly MA, Croteau W, Noelle RJ, Lines JL. VISTA: A Novel Immunotherapy Target for Normalizing Innate and Adaptive Immunity. Semin Immunol (2019) 42:101308. doi: 10.1016/j.smim.2019.101308
156. Borggrewe M, Grit C, Den Dunnen WFA, Burm SM, Bajramovic JJ, Noelle RJ, et al. VISTA Expression by Microglia Decreases During Inflammation and is Differentially Regulated in CNS Diseases. Glia (2018) 66(12):2645–58. doi: 10.1002/glia.23517
157. Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, et al. VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Res (2014) 74(7):1933–44. doi: 10.1158/0008-5472.CAN-13-1506
158. Mehta N, Maddineni S, Kelly RL, Lee RB, Hunter SA, Silberstein JL, et al. An Engineered Antibody Binds a Distinct Epitope and is a Potent Inhibitor of Murine and Human VISTA. Sci Rep (2020) 10(1):15171. doi: 10.1038/s41598-020-71519-4
159. Sun J, Lu Q, Sanmamed MF, Wang J. Siglec-15 as an Emerging Target for Next-Generation Cancer Immunotherapy. Clin Cancer Res (2021) 27(3):680–8. doi: 10.1158/1078-0432.CCR-19-2925
160. Wang J, Sun J, Liu LN, Flies DB, Nie X, Toki M, et al. Siglec-15 as an Immune Suppressor and Potential Target for Normalization Cancer Immunotherapy. Nat Med (2019) 25(4):656–66. doi: 10.1038/s41591-019-0374-x
161. Han Y, Zhu L, Wu W, Zhang H, Hu W, Dai L, et al. Small Molecular Immune Modulators as Anticancer Agents. Adv Exp Med Biol (2020) 1248:547–618. doi: 10.1007/978-981-15-3266-5_22
162. Zhan MM, Hu XQ, Liu XX, Ruan BF, Xu J, Liao C. From Monoclonal Antibodies to Small Molecules: The Development of Inhibitors Targeting the PD-1/PD-L1 Pathway. Drug Discovery Today (2016) 21(6):1027–36. doi: 10.1016/j.drudis.2016.04.011
163. Lin X, Lu X, Luo G, Xiang H. Progress in PD-1/PD-L1 Pathway Inhibitors: From Biomacromolecules to Small Molecules. Eur J Med Chem (2020) 186:111876. doi: 10.1016/j.ejmech.2019.111876
164. Skalniak L, Zak KM, Guzik K, Magiera K, Musielak B, Pachota M, et al. Small-Molecule Inhibitors of PD-1/PD-L1 Immune Checkpoint Alleviate the PD-L1-Induced Exhaustion of T-Cells. Oncotarget (2017) 8(42):72167–81. doi: 10.18632/oncotarget.20050
165. Cuccarese MF, Dubach JM, Pfirschke C, Engblom C, Garris C, Miller MA, et al. Heterogeneity of Macrophage Infiltration and Therapeutic Response in Lung Carcinoma Revealed by 3D Organ Imaging. Nat Commun (2017) 8:14293. doi: 10.1038/ncomms14293
166. Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, et al. Inhibition of CSF-1 Receptor Improves the Antitumor Efficacy of Adoptive Cell Transfer Immunotherapy. Cancer Res (2014) 74(1):153–61. doi: 10.1158/0008-5472.CAN-13-1816
167. Tap WD, Gelderblom H, Palmerini E, Desai J, Bauer S, Blay J-Y, et al. Pexidartinib Versus Placebo for Advanced Tenosynovial Giant Cell Tumour (ENLIVEN): A Randomised Phase 3 Trial. Lancet (London England) (2019) 394(10197):478–87. doi: 10.1016/S0140-6736(19)30764-0
168. Shi G, Yang Q, Zhang Y, Jiang Q, Lin Y, Yang S, et al. Modulating the Tumor Microenvironment via Oncolytic Viruses and CSF-1r Inhibition Synergistically Enhances Anti-PD-1 Immunotherapy. Mol Ther (2019) 27(1):244–60. doi: 10.1016/j.ymthe.2018.11.010
169. Bayat Mokhtari R, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B, et al. Combination Therapy in Combating Cancer. Oncotarget (2017) 8(23):38022–43. doi: 10.18632/oncotarget.16723
170. Mangiameli DP, Blansfield JA, Kachala S, Lorang D, Schafer PH, Muller GW, et al. Combination Therapy Targeting the Tumor Microenvironment is Effective in a Model of Human Ocular Melanoma. J Transl Med (2007) 5:38. doi: 10.1186/1479-5876-5-38
171. Koumarianou A, Kaltsas G. Surufatinib - a Novel Oral Agent for Neuroendocrine Tumours. Nat Rev Endocrinol (2021) 17(1):9–10. doi: 10.1038/s41574-020-00439-0
172. Fong L, Hotson A, Powderly JD, Sznol M, Heist RS, Choueiri TK, et al. Adenosine 2a Receptor Blockade as an Immunotherapy for Treatment-Refractory Renal Cell Cancer. Cancer Discovery (2020) 10(1):40–53. doi: 10.1158/2159-8290.CD-19-0980
173. Sitkovsky MV. Lessons From the A2A Adenosine Receptor Antagonist-Enabled Tumor Regression and Survival in Patients With Treatment-Refractory Renal Cell Cancer. Cancer Discovery (2020) 10(1):16–9. doi: 10.1158/2159-8290.CD-19-1280
174. Hatfield SM, Kjaergaard J, Lukashev D, Schreiber TH, Belikoff B, Abbott R, et al. Immunological Mechanisms of the Antitumor Effects of Supplemental Oxygenation. Sci Transl Med (2015) 7(277):277ra30. doi: 10.1126/scitranslmed.aaa1260
175. Forte G, Sorrentino R, Montinaro A, Luciano A, Adcock IM, Maiolino P, et al. Inhibition of CD73 Improves B Cell-Mediated Anti-Tumor Immunity in a Mouse Model of Melanoma. J Immunol (2012) 189(5):2226–33. doi: 10.4049/jimmunol.1200744
176. Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, et al. Anti-CD73 Antibody Therapy Inhibits Breast Tumor Growth and Metastasis. Proc Natl Acad Sci U.S.A. (2010) 107(4):1547–52. doi: 10.1073/pnas.0908801107
177. Stagg J, Divisekera U, Duret H, Sparwasser T, Teng MW, Darcy PK, et al. CD73-Deficient Mice Have Increased Antitumor Immunity and are Resistant to Experimental Metastasis. Cancer Res (2011) 71(8):2892–900. doi: 10.1158/0008-5472.CAN-10-4246
178. Sun X, Wu Y, Gao W, Enjyoji K, Csizmadia E, Muller CE, et al. CD39/ENTPD1 Expression by CD4+Foxp3+ Regulatory T Cells Promotes Hepatic Metastatic Tumor Growth in Mice. Gastroenterology (2010) 139(3):1030–40. doi: 10.1053/j.gastro.2010.05.007
179. Hayes GM, Cairns B, Levashova Z, Chinn L, Perez M, Theunissen JW, et al. CD39 is a Promising Therapeutic Antibody Target for the Treatment of Soft Tissue Sarcoma. Am J Transl Res (2015) 7(6):1181–8.
180. Bastid J, Regairaz A, Bonnefoy N, Dejou C, Giustiniani J, Laheurte C, et al. Inhibition of CD39 Enzymatic Function at the Surface of Tumor Cells Alleviates Their Immunosuppressive Activity. Cancer Immunol Res (2015) 3(3):254–65. doi: 10.1158/2326-6066.CIR-14-0018
181. Hausler SF, Del Barrio IM, Diessner J, Stein RG, Strohschein J, Honig A, et al. Anti-CD39 and Anti-CD73 Antibodies A1 and 7G2 Improve Targeted Therapy in Ovarian Cancer by Blocking Adenosine-Dependent Immune Evasion. Am J Transl Res (2014) 6(2):129–39.
182. Leone RD, Sun IM, Oh MH, Sun IH, Wen J, Englert J, et al. Inhibition of the Adenosine A2a Receptor Modulates Expression of T Cell Coinhibitory Receptors and Improves Effector Function for Enhanced Checkpoint Blockade and ACT in Murine Cancer Models. Cancer Immunol Immunother (2018) 67(8):1271–84. doi: 10.1007/s00262-018-2186-0
183. Ma SR, Deng WW, Liu JF, Mao L, Yu GT, Bu LL, et al. Blockade of Adenosine A2A Receptor Enhances CD8(+) T Cells Response and Decreases Regulatory T Cells in Head and Neck Squamous Cell Carcinoma. Mol Cancer (2017) 16(1):99. doi: 10.1186/s12943-017-0665-0
184. Young A, Ngiow SF, Madore J, Reinhardt J, Landsberg J, Chitsazan A, et al. Targeting Adenosine in BRAF-Mutant Melanoma Reduces Tumor Growth and Metastasis. Cancer Res (2017) 77(17):4684–96. doi: 10.1158/0008-5472.CAN-17-0393
185. Figueroa JA, Reidy A, Mirandola L, Trotter K, Suvorava N, Figueroa A, et al. Chimeric Antigen Receptor Engineering: A Right Step in the Evolution of Adoptive Cellular Immunotherapy. Int Rev Immunol (2015) 34(2):154–87. doi: 10.3109/08830185.2015.1018419
186. Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer Regression and Autoimmunity in Patients After Clonal Repopulation With Antitumor Lymphocytes. Science (2002) 298(5594):850–4. doi: 10.1126/science.1076514
187. Kochenderfer JN, Yu Z, Frasheri D, Restifo NP, Rosenberg SA. Adoptive Transfer of Syngeneic T Cells Transduced With a Chimeric Antigen Receptor That Recognizes Murine CD19 can Eradicate Lymphoma and Normal B Cells. Blood (2010) 116(19):3875–86. doi: 10.1182/blood-2010-01-265041
188. Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-Term Safety and Activity of Axicabtagene Ciloleucel in Refractory Large B-Cell Lymphoma (ZUMA-1): A Single-Arm, Multicentre, Phase 1-2 Trial. Lancet Oncol (2019) 20(1):31–42. doi: 10.1016/S1470-2045(18)30864-7
189. Martinez M, Moon EK. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front Immunol (2019) 10:128. doi: 10.3389/fimmu.2019.00128
190. Zhang C, Burger MC, Jennewein L, Genßler S, Schönfeld K, Zeiner P, et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J Natl Cancer Inst (2016) 108(5):108(5). doi: 10.1093/jnci/djv375
191. Moon EK, Carpenito C, Sun J, Wang L-CS, Kapoor V, Predina J, et al. Expression of a Functional CCR2 Receptor Enhances Tumor Localization and Tumor Eradication by Retargeted Human T Cells Expressing a Mesothelin-Specific Chimeric Antibody Receptor. Clin Cancer research: an Off J Am Assoc Cancer Res (2011) 17(14):4719–30. doi: 10.1158/1078-0432.CCR-11-0351
192. Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase Promotes Tumor Infiltration and Antitumor Activity of CAR-Redirected T Lymphocytes. Nat Med (2015) 21(5):524–9. doi: 10.1038/nm.3833
193. Grosser R, Cherkassky L, Chintala N, Adusumilli PS. Combination Immunotherapy With CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell (2019) 36(5):471–82. doi: 10.1016/j.ccell.2019.09.006
194. Zhang Y, Zhang X, Cheng C, Mu W, Liu X, Li N, et al. CRISPR-Cas9 Mediated LAG-3 Disruption in CAR-T Cells. Front Med (2017) 11(4):554–62. doi: 10.1007/s11684-017-0543-6
195. Chmielewski M, Abken H. CAR T Cells Releasing IL-18 Convert to T-Bet FoxO1 Effectors That Exhibit Augmented Activity Against Advanced Solid Tumors. Cell Rep (2017) 21(11):3205–19. doi: 10.1016/j.celrep.2017.11.063
196. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 Expression in CAR-T Cells Improves Immune Cell Infiltration and CAR-T Cell Survival in the Tumor. Nat Biotechnol (2018) 36(4):346–51. doi: 10.1038/nbt.4086
197. Baybutt TR, Flickinger JC, Caparosa EM, Snook AE. Advances in Chimeric Antigen Receptor T-Cell Therapies for Solid Tumors. Clin Pharmacol Ther (2019) 105(1):71–8. doi: 10.1002/cpt.1280
198. Igarashi Y, Sasada T. Cancer Vaccines: Toward the Next Breakthrough in Cancer Immunotherapy. J Immunol Res (2020) 2020:5825401. doi: 10.1155/2020/5825401
199. Madan RA, Antonarakis ES, Drake CG, Fong L, Yu EY, McNeel DG, et al. Putting the Pieces Together: Completing the Mechanism of Action Jigsaw for Sipuleucel-T. J Natl Cancer Inst (2020) 112(6):562–73. doi: 10.1093/jnci/djaa021
200. Fong L, Carroll P, Weinberg V, Chan S, Lewis J, Corman J, et al. Activated Lymphocyte Recruitment Into the Tumor Microenvironment Following Preoperative Sipuleucel-T for Localized Prostate Cancer. J Natl Cancer Inst (2014) 106(11):dju268. doi: 10.1093/jnci/dju268
201. Beer TM, Bernstein GT, Corman JM, Glode LM, Hall SJ, Poll WL, et al. Randomized Trial of Autologous Cellular Immunotherapy With Sipuleucel-T in Androgen-Dependent Prostate Cancer. Clin Cancer research: an Off J Am Assoc Cancer Res (2011) 17(13):4558–67. doi: 10.1158/1078-0432.CCR-10-3223
202. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N Engl J Med (2010) 363(5):411–22. doi: 10.1056/NEJMoa1001294
203. Scholz M, Yep S, Chancey M, Kelly C, Chau K, Turner J, et al. Phase I Clinical Trial of Sipuleucel-T Combined With Escalating Doses of Ipilimumab in Progressive Metastatic Castrate-Resistant Prostate Cancer. Immunotargets Ther (2017) 6:11–6. doi: 10.2147/ITT.S122497
204. Ali OA, Lewin SA, Dranoff G, Mooney DJ. Vaccines Combined With Immune Checkpoint Antibodies Promote Cytotoxic T-Cell Activity and Tumor Eradication. Cancer Immunol Res (2016) 4(2):95–100. doi: 10.1158/2326-6066.CIR-14-0126
205. Sarnaik AA, Yu B, Yu D, Morelli D, Hall M, Bogle D, et al. Extended Dose Ipilimumab With a Peptide Vaccine: Immune Correlates Associated With Clinical Benefit in Patients With Resected High-Risk Stage IIIc/IV Melanoma. Clin Cancer Res (2011) 17(4):896–906. doi: 10.1158/1078-0432.CCR-10-2463
206. Yarchoan M, Huang CY, Zhu Q, Ferguson AK, Durham JN, Anders RA, et al. A Phase 2 Study of GVAX Colon Vaccine With Cyclophosphamide and Pembrolizumab in Patients With Mismatch Repair Proficient Advanced Colorectal Cancer. Cancer Med (2020) 9(4):1485–94. doi: 10.1002/cam4.2763
207. Ott PA, Hu-Lieskovan S, Chmielowski B, Govindan R, Naing A, Bhardwaj N, et al. A Phase Ib Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients With Advanced Melanoma, Non-Small Cell Lung Cancer, or Bladder Cancer. Cell (2020) 183(2):347–62.e24. doi: 10.1016/j.cell.2020.08.053
208. Song W, Zhang M. Use of CAR-T Cell Therapy, PD-1 Blockade, and Their Combination for the Treatment of Hematological Malignancies. Clin Immunol (2020) 214:108382. doi: 10.1016/j.clim.2020.108382
209. Srivastava S, Furlan SN, Jaeger-Ruckstuhl CA, Sarvothama M, Berger C, Smythe KS, et al. Immunogenic Chemotherapy Enhances Recruitment of CAR-T Cells to Lung Tumors and Improves Antitumor Efficacy When Combined With Checkpoint Blockade. Cancer Cell (2021) 39(2):193–208.e10. doi: 10.1016/j.ccell.2020.11.005
210. Baybutt TR, Flickinger JC Jr., Caparosa EM, Snook AE. Advances in Chimeric Antigen Receptor T-Cell Therapies for Solid Tumors. Clin Pharmacol Ther (2019) 105(1):71–8. doi: 10.1002/cpt.1280
211. Adusumilli PS, Zauderer MG, Riviere I, Solomon SB, Rusch VW, O’Cearbhaill RE, et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-Cell Therapy in Patients With Malignant Pleural Disease, in Combination With the Anti-PD-1 Agent Pembrolizumab. Cancer Discovery (2021) 11(11):2748–63. doi: 10.1158/2159-8290.CD-21-0407
212. Sang W, Wang X, Geng H, Li T, Li D, Zhang B, et al. Anti-PD-1 Therapy Enhances the Efficacy of CD30-Directed Chimeric Antigen Receptor T Cell Therapy in Patients With Relapsed/Refractory CD30+ Lymphoma. Front Immunol (2022) 13:858021. doi: 10.3389/fimmu.2022.858021
213. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R Blockade Reprograms Tumor-Infiltrating Macrophages and Improves Response to T-Cell Checkpoint Immunotherapy in Pancreatic Cancer Models. Cancer Res (2014) 74(18):5057–69. doi: 10.1158/0008-5472.CAN-13-3723
214. Przystal JM, Becker H, Canjuga D, Tsiami F, Anderle N, Keller AL, et al. Targeting CSF1R Alone or in Combination With PD1 in Experimental Glioma. Cancers (Basel) (2021) 13(10):2400. doi: 10.3390/cancers13102400
215. Antonios JP, Soto H, Everson RG, Moughon D, Orpilla JR, Shin NP, et al. Immunosuppressive Tumor-Infiltrating Myeloid Cells Mediate Adaptive Immune Resistance via a PD-1/PD-L1 Mechanism in Glioblastoma. Neuro Oncol (2017) 19(6):796–807. doi: 10.1093/neuonc/now287
216. Holmgaard RB, Brachfeld A, Gasmi B, Jones DR, Mattar M, Doman T, et al. Timing of CSF-1/CSF-1R Signaling Blockade is Critical to Improving Responses to CTLA-4 Based Immunotherapy. Oncoimmunology (2016) 5(7):e1151595. doi: 10.1080/2162402X.2016.1151595
217. Omstead AN, Paskewicz M, Gorbunova A, Zheng P, Salvitti MS, Mansoor R, et al. CSF-1R Inhibitor, Pexidartinib, Sensitizes Esophageal Adenocarcinoma to PD-1 Immune Checkpoint Blockade in a Rat Model. Carcinogenesis (2022):bgac043. doi: 10.1093/carcin/bgac043
218. Gomez-Roca C, Cassier P, Zamarin D, Machiels JP, Luis Perez Gracia J, Stephen Hodi F, et al. Anti-CSF-1R Emactuzumab in Combination With Anti-PD-L1 Atezolizumab in Advanced Solid Tumor Patients Naive or Experienced for Immune Checkpoint Blockade. J Immunother Cancer (2022) 10(5):e004076. doi: 10.1136/jitc-2021-004076
219. Curigliano G, Gelderblom H, Mach N, Doi T, Tai D, Forde PM, et al. Phase I/Ib Clinical Trial of Sabatolimab, an Anti-TIM-3 Antibody, Alone and in Combination With Spartalizumab, an Anti-PD-1 Antibody, in Advanced Solid Tumors. Clin Cancer Res (2021) 27(13):3620–9. doi: 10.1158/1078-0432.CCR-20-4746
220. Atkinson V, Khattak A, Haydon A, Eastgate M, Roy A, Prithviraj P, et al. Eftilagimod Alpha, a Soluble Lymphocyte Activation Gene-3 (LAG-3) Protein Plus Pembrolizumab in Patients With Metastatic Melanoma. J Immunother Cancer (2020) 8(2):e001681. doi: 10.1136/jitc-2020-001681
221. Tawbi HA, Schadendorf D, Lipson EJ, Ascierto PA, Matamala L, Castillo Gutierrez E, et al. Relatlimab and Nivolumab Versus Nivolumab in Untreated Advanced Melanoma. N Engl J Med (2022) 386(1):24–34. doi: 10.1056/NEJMoa2109970
222. Gide TN, Quek C, Menzies AM, Tasker AT, Shang P, Holst J, et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell (2019) 35(2):238–55.e6. doi: 10.1016/j.ccell.2019.01.003
223. Youn JW, Hur SY, Woo JW, Kim YM, Lim MC, Park SY, et al. Pembrolizumab Plus GX-188E Therapeutic DNA Vaccine in Patients With HPV-16-Positive or HPV-18-Positive Advanced Cervical Cancer: Interim Results of a Single-Arm, Phase 2 Trial. Lancet Oncol (2020) 21(12):1653–60. doi: 10.1016/S1470-2045(20)30486-1
224. Hatfield SM, Sitkovsky M. Oxygenation to Improve Cancer Vaccines, Adoptive Cell Transfer and Blockade of Immunological Negative Regulators. Oncoimmunology (2015) 4(12):e1052934. doi: 10.1080/2162402X.2015.1052934
225. Hatfield SM, Sitkovsky MV. Antihypoxic Oxygenation Agents With Respiratory Hyperoxia to Improve Cancer Immunotherapy. J Clin Invest (2020) 130(11):5629–37. doi: 10.1172/JCI137554
226. Yan P, Luo Y, Li X, Li Y, Wang Y, Wu J, et al. A Redox-Responsive Nanovaccine Combined With A2A Receptor Antagonist for Cancer Immunotherapy. Adv Healthc Mater (2021) 10(21):e2101222. doi: 10.1002/adhm.202101222
227. Giuffrida L, Sek K, Henderson MA, Lai J, Chen AXY, Meyran D, et al. CRISPR/Cas9 Mediated Deletion of the Adenosine A2A Receptor Enhances CAR T Cell Efficacy. Nat Commun (2021) 12(1):3236. doi: 10.1038/s41467-021-23331-5
228. Yap TA, Parkes EE, Peng W, Moyers JT, Curran MA, Tawbi HA. Development of Immunotherapy Combination Strategies in Cancer. Cancer Discovery (2021) 11(6):1368–97. doi: 10.1158/2159-8290.CD-20-1209
229. Zhu S, Zhang T, Zheng L, Liu H, Song W, Liu D, et al. Combination Strategies to Maximize the Benefits of Cancer Immunotherapy. J Hematol Oncol (2021) 14(1):156. doi: 10.1186/s13045-021-01164-5
230. Wright JJ, Powers AC, Johnson DB. Endocrine Toxicities of Immune Checkpoint Inhibitors. Nat Rev Endocrinol (2021) 17(7):389–99. doi: 10.1038/s41574-021-00484-3
Keywords: tumor immune microenvironment, immunotherapy, immune cell, antibody, small molecule inhibitor
Citation: LV B, Wang Y, Ma D, Cheng W, Liu J, Yong T, Chen H and Wang C (2022) Immunotherapy: Reshape the Tumor Immune Microenvironment. Front. Immunol. 13:844142. doi: 10.3389/fimmu.2022.844142
Received: 27 December 2021; Accepted: 13 June 2022;
Published: 06 July 2022.
Edited by:
Fabio Malavasi, University of Turin, ItalyReviewed by:
Sonja I. Buschow, Erasmus Medical Center, NetherlandsMichael Sitkovsky, Northeastern University, United States
Silvia Martina Ferrari, University of Pisa, Italy
Copyright © 2022 LV, Wang, Ma, Cheng, Liu, Yong, Chen and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chen Wang, Y2hlbndhbmdAbHp1LmVkdS5jbg==; Hao Chen, ZXJ5X2NoZW5oQGx6dS5lZHUuY24=
†These authors share senior authorship