
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Immunol., 30 March 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.843712
 Hongxia Li1,2,3
Hongxia Li1,2,3 Zhiqiang Cao1,2,3
Zhiqiang Cao1,2,3 Lili Wang1,2,3
Lili Wang1,2,3 Chang Liu1,2,3
Chang Liu1,2,3 Hongkun Lin1,2,3Yuhan Tang1,2,3
Hongkun Lin1,2,3Yuhan Tang1,2,3 Ping Yao1,2,3*
Ping Yao1,2,3*Cardiovascular diseases, the notorious killer, are mainly caused by atherosclerosis (AS) characterized by lipids, cholesterol, and iron overload in plaques. Macrophages are effector cells and accumulate to the damaged and inflamed sites of arteries to internalize native and chemically modified lipoproteins to transform them into cholesterol-loaded foam cells. Foam cell formation is determined by the capacity of phagocytosis, migration, scavenging, and the features of phenotypes. Macrophages are diverse, and the subsets and functions are controlled by their surrounding microenvironment. Generally, macrophages are divided into classically activated (M1) and alternatively activated (M2). Recently, intraplaque macrophage phenotypes are recognized by the stimulation of CXCL4 (M4), oxidized phospholipids (Mox), hemoglobin/haptoglobin complexes [HA-mac/M(Hb)], and heme (Mhem). The pro-atherogenic or anti-atherosclerotic phenotypes of macrophages decide the progression of AS. Besides, apoptosis, necrosis, ferroptosis, autophagy and pyrotopsis determine plaque formation and cardiovascular vulnerability, which may be associated with macrophage polarization phenotypes. In this review, we first summarize the three most popular hypotheses for AS and find the common key factors for further discussion. Secondly, we discuss the factors affecting macrophage polarization and five types of macrophage death in AS progression, especially ferroptosis. A comprehensive understanding of the cellular and molecular mechanisms of plaque formation is conducive to disentangling the candidate targets of macrophage-targeting therapies for clinical intervention at various stages of AS.
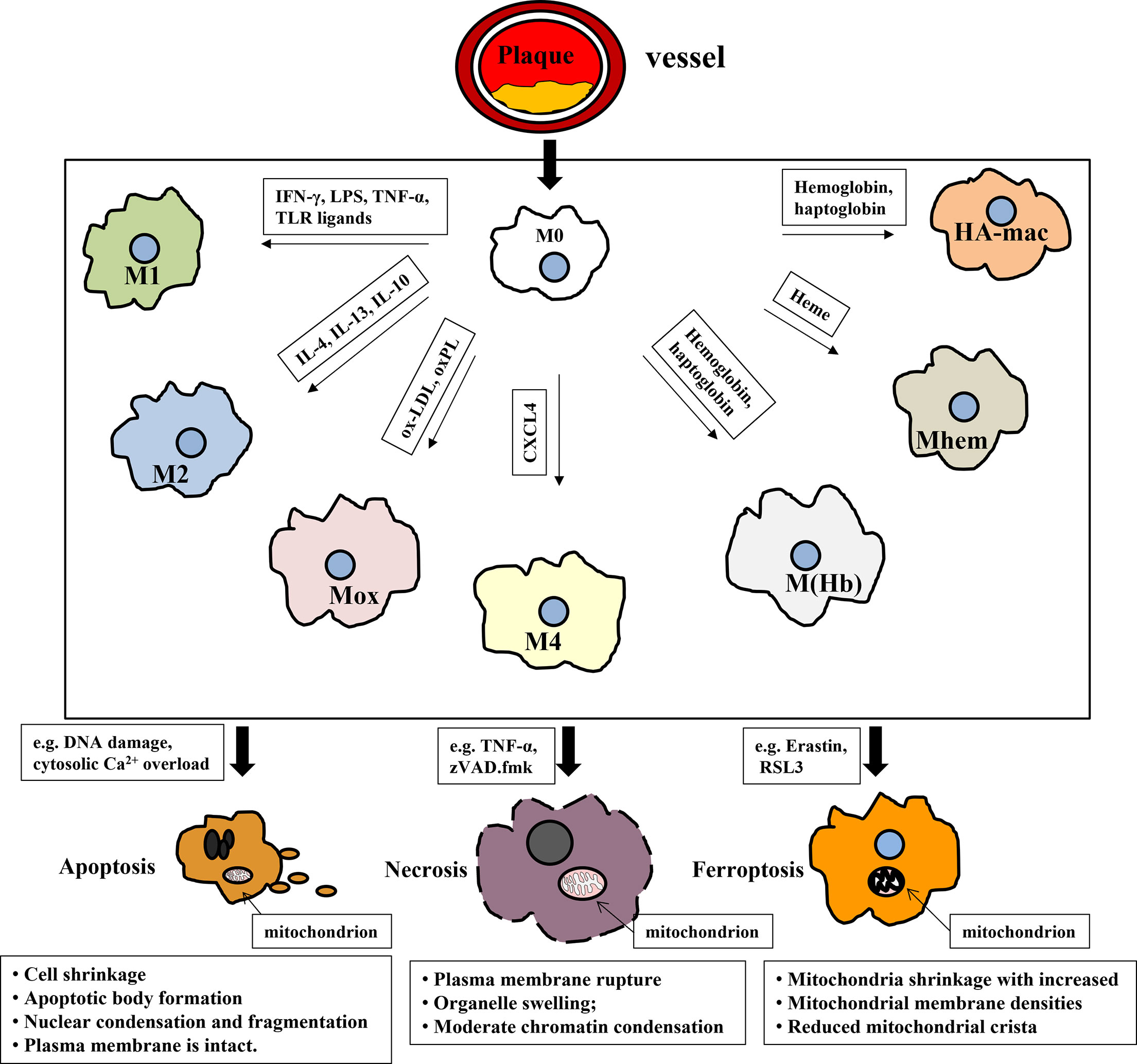
Graphical Abstract In atherosclerotic plaques, macrophages differentiate into different subtypes according to the vascular microenvironment. Subsequently, macrophages die from the inducement (iron or lipids overload, DNA damage) by the way of apoptosis, necrosis and ferroptosis etc., which, in turn, accelerates the atherosclerotic plaque formation.
Cardiovascular disease (CVD) is the leading killer globally and has complex complications, such as stroke, myocardial infarction, heart failure, and hypertension (1). CVD has been a hot topic because of its high incidence and mortality. A report from the Global Burden of Disease Study showed that the total prevalence of CVD and years lived with a disability nearly have doubled from 271 million to 523 million and 17.7 million to 34.4 million in 1999–2019, and the cases of CVD deaths stably increased from 12.1 million to 18.6 million from 1990 to 2019 (2). What is more, the global trends for disability-adjusted life years and years of life lost also increased significantly (2–4). Atherosclerosis (AS), an underlying pathology of CVD, is a chronic, sterilized, inflammatory disease involved in large and medium arteries and accompanied by lipids, cholesterol, iron deposition, and fibrous cap thinning (5, 6). The occurrence and development of AS are strongly linked to local inflammation related to the vascular microenvironment and macrophage death.
Despite the fact that the exploration of cellular and molecular mechanisms has been inspiring over the last decades, the complex mechanisms of plaque formation in AS progression limit us in understanding the AS etiology comprehensively. Since the 19th century, researchers have thought of this disease related to the aging process (7). As the research further developed, three hypotheses have been formed. Firstly, it is the response-to-injury hypothesis (8). According to this hypothesis, various factors, including hemodynamic forces and pro-inflammatory factors, promote the dysfunction of the endothelium (8). Then, platelets aggregate, macrophages and vascular smooth muscle cells (VSMCs) engulf oxidized lipids and eventually form plaques, that is, the injury of vessel is mainly caused by endothelial detachment. However, subsequent studies have shown that the endothelium remains intact during the development of AS, even at the sites of plaque injury (9), which contradicts the core of this hypothesis. Secondly, it is the response-to-retention hypothesis. In the early stage of AS, proteoglycan binds to apolipoprotein B and traps lipids into macrophages (10). In addition, lipolytic enzyme and lysosomal enzyme play an important part in the residence of oxidized low-density lipoprotein (ox-LDL) in the vascular wall. In vitro, the lipoprotein lipase improved the adhesion of ox-LDL, leading to ox-LDL being more easily engulfed by macrophages and VSMCs and increasingly being foam cells (11). Thirdly, that is the oxidative modification hypothesis (12). Circulating LDL is chemically modified to ox-LDL by reactive oxygen species (ROS) and metal ions such as iron or copper, which becomes the pro-inflammatory factor in the vascular microenvironment. Macrophages and VSMCs internalize ox-LDL, an important atherogenic component of the vascular microenvironment via scavenger receptors, such as CD36, causing the transformation of foam cells containing cholesterol esters and subsequently to be the necrotic core.
As shown in Table 1, although the key factors for AS initiating are different among the three hypotheses, they are not mutually repulsive but stress diverse elements as the necessary and sufficient conditions to demonstrate the development of AS. For example, these hypotheses all consider that inflammation, a known feature of AS, is involved in atherosclerotic progression (16), and ox-LDL, an important component of the vascular microenvironment, is the key factor in promoting atherosclerotic lesions. Assuredly, the reduction of ox-LDL cholesterol is effective in AS treatment (7). Furthermore, these hypotheses all highlighted foam cell plays a critical part in every stage of AS, from the initial attack of the arterial lesion and lesion extension to cell death (apoptosis, necrosis, ferroptosis, etc.) causing plaque rupture and the complications of AS.
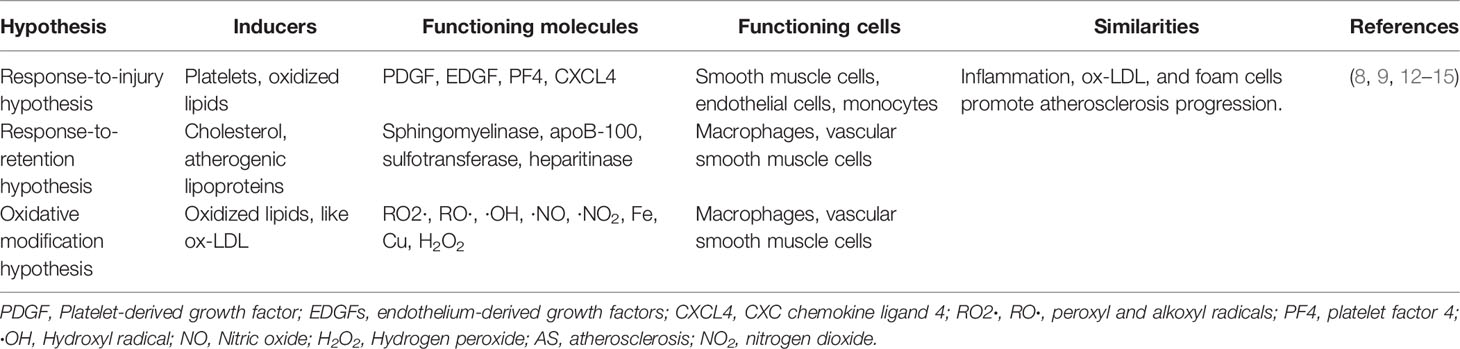
Table 1 Summary of three hypothesis in atherosclerosis progression.
Recently, with the discovery of various polarization subsets of macrophages, the effect of different populations on the AS condition has been reexamined. Macrophage polarization and death, whose outcomes directly lead to plaque formation, are also considerable causes. Although there are inconsistent viewpoints on the mechanisms of occurrence and development of AS, the common denominator is the recognition of the essential role of inflammation, oxidized lipids, macrophage polarization, and macrophage death events in atherosclerotic vascular disease. This review focuses on inflammation and plaque formation in the initiation and development of AS and teases out the candidate proteins of macrophage-targeting therapies for clinical intervention at various stages of AS.
The formation of atherosclerotic plaque is closely related to macrophages. Firstly, macrophages in plaques are differentiated from monocytes continuously recruited by chemokines, which is the prelude of macrophages transforming to foam cells. Subsequently, macrophages (M0) polarize into different macrophage subsets determined by the different vascular microenvironments. Macrophages with different phenotypes have different phagocytosic abilities to internalize ox-LDL; thus, the outcomes of being foam cells are distinct. Subsequently, foam cells undergo programmed or non-programmed death, causing the formation of plaques and necrotic cores, although different macrophage subsets may display unlike sensitivities for apoptosis, necrosis, ferroptosis, autophagy, and pyroptosis. Polarization and cell death play irreplaceable roles in plaque formation, and they also interplay with each other to control the AS condition. The two main inducements for plaque formation mentioned above are crucial for AS prevention and treatment and will be summarized below.
Macrophages display pro-inflammatory or anti-inflammatory properties due to different phenotypes. In the stimulation of cytokines, macrophages firstly migrate to the inflammatory sites to eliminate inflammation. However, in the process of AS, due to sustaining inflammation response, macrophages are continuously recruited to lesion sites. Monocytes firstly differentiate to macrophages (M0) due to cytokines, such as M-CSF (17). Although the differentiation of monocytes to macrophages is unreversible, macrophages possess eminent plasticity, i.e., switching their phenotypes and functioning according to external signals (18). As shown in Table 2, M0 could mainly polarize to seven types of macrophages according to the vascular microenvironment: classically activated macrophages (M1), alternatively activated macrophages (M2), oxidized phospholipid-induced macrophages (Mox), chemokine (C-X-C motif) ligand 4 or platelet factor 4-induced macrophages (M4), erythrocyte and hemoglobin-induced macrophages [HA-mac, M(Hb), and Mhem].
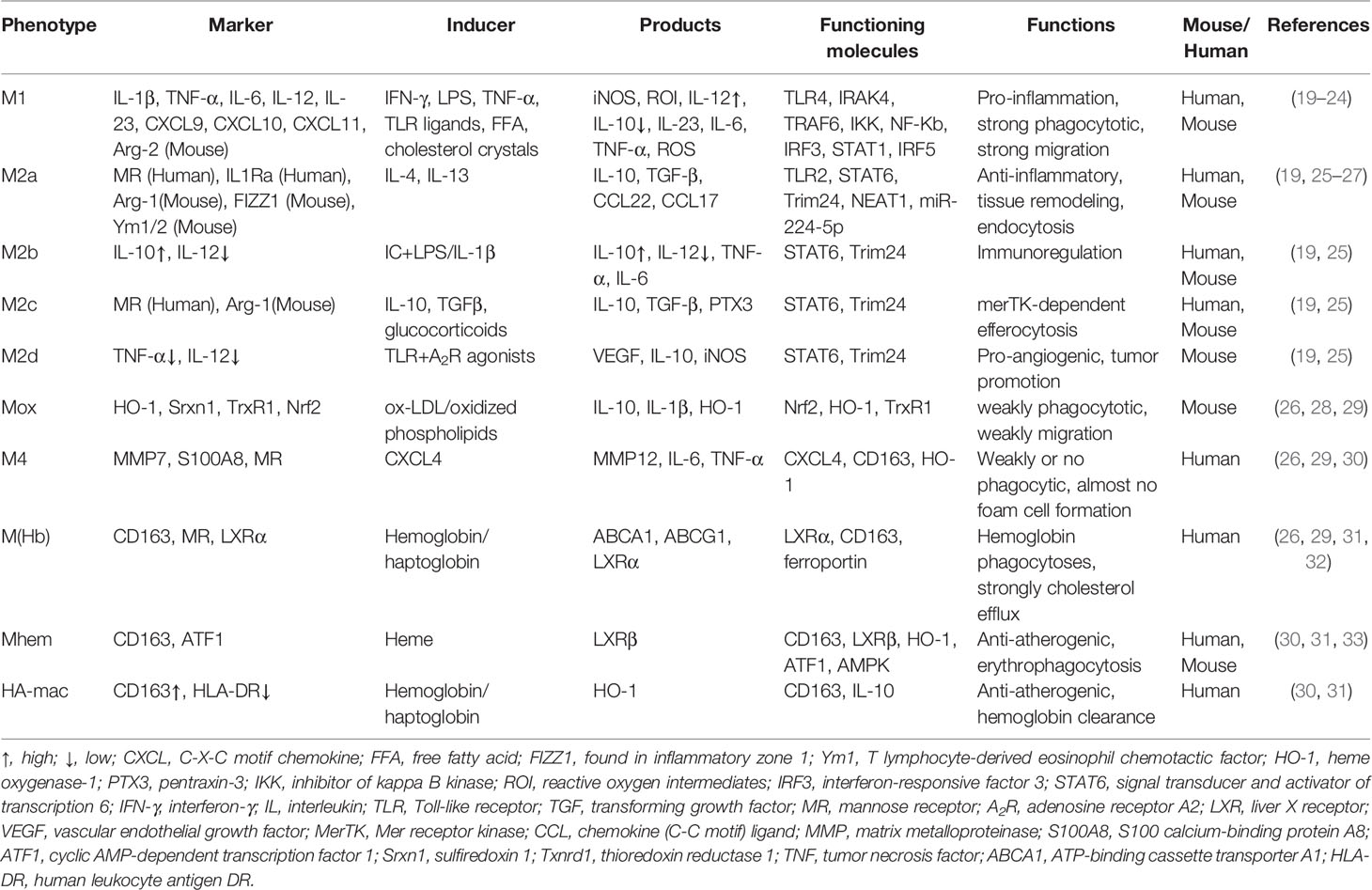
Table 2 Summary of the different macrophage subsets existing in atherosclerotic lesions of humans and mice.
As illustrated in Table 2, M1 macrophages, highly expressed iNOS, are differentiated from the M0 phenotype by lipopolysaccharide (LPS), interferons, pathogen-associated molecular patterns, and lipoproteins via Toll-like receptor signaling, especially TLR4/MyD88/NF-κB (34). Moreover, TIR domain-containing adaptor inducing interferon-beta (TRIF), another downstream protein of TLR4, is also involved in M1 activation except for MyD88. TRIF could activate the transcription factor interferon-responsive factor 3 (IRF3) and then promotes IFNα and IFNβ secretion. Therefore, interferons bind to the interferon receptor (IFNAR) to activate the transcription factor STAT1 to skew macrophages to M1-like or M1 polarization (35) M1 macrophages are pro-inflammatory to destruct tissue and secrete cytokines, for example, IL-1β, IL-6, TNF-α, and IL-12 (36). Pro-inflammatory cytokines sustain to recruit immune cells, causing many macrophages to migrate to lesion sites. Compared with M0, the phagocytosis capacity of M1 is not inferior; even M1 macrophages are more likely to be foam cells, an indispensable part of plaques. If foam cells die and could not be removed, they will become new pro-inflammatory factors, which becomes a vicious circle, that is, recruitment–death–recruitment.
M2 macrophages with high expression of arginase-1 could be polarized by the cytokines like IL-4 and IL-13, and subsequently divided into M2a, M2b, M2c, and M2d, depending on the stimulation (37). As shown in Table 2, IL-4 or IL-13 induces macrophages to the M2 subset through activating STAT6 by the IL-4 receptor α, and IL-10 induces macrophages to M2 polarization via STAT3 through the IL-10 receptor (35). Recently, Cao and colleagues reported that the knockdown of the long noncoding lncRNA-MM2P reduced cytokine-driven M2 polarization and M2-marker genes by decreasing phosphorylation on STAT6 (38). M2 macrophages initiate anti-inflammatory signaling by generating cytokines like the IL-1 receptor agonist, collagen, IL-10, and TGF-β1, making M1 transform into M2 macrophages to strengthen exocytosis (39). M2 macrophages are differentiated by Th2 cytokines and generate IL-10. Due to various scavenger receptors such as CD36, macrophage scavenger receptor 1 (MSR1), macrophage receptor with collagenous structure (MRC1), and mannose receptor (40, 41), the phagocytosis of M2 macrophages is rather strong, causing the phagocytosis capacity of M2 to be superior to M0 (28). It means that if M2 could not well cope with intracellular lipids, they will be a hidden danger for plaque formation due to their “greed.”
Mox is polarized from M0 by oxidized phospholipids and keeps them away from oxidative stress via nuclear factor E2-related factor 2 (Nrf2)-regulated expression of antioxidant enzymes like HO-1, thioredoxin reductase 1 (TrxR1), and sulfiredoxin-1 (Srxn1) (28). In atherosclerotic lesions of ldlr-/- mice, Mox macrophages were extensively distributed in plaque and account for 30% while M1 and M2 macrophages took up 40% and 20% of all plaque macrophages, respectively (28). Notably, the phagocytosis and migration capacities of Mox are inferior to M1 and M2 subsets. What is more, antioxidant proteins were dramatically upregulated by Nrf2 in Mox, suggesting that Mox macrophages may be an anti-atherosclerotic subset. However, it is unclear whether Mox is pro-atherosclerotic or anti-atherosclerotic in AS progression until now. Certainly, M1, M2, and Mox macrophages are all present in atherosclerotic lesions, and the imbalance of the ratio of macrophage subsets may be the cause of plaque formation and the impediment of inflammation alleviation (42).
Iron-loaded M4 macrophages are induced by CXC chemokine ligand 4 (CXCL4) (43). The M4 phenotype is found in human plaque lesions and marked by matrix metalloprotease 7 (MMP-7) and Ca2+-binding protein S100A8 (44). The combination of CD68, S100A8, and MMP7 is a reliable marker to recognize the M4 subset both in vitro and in vivo (43). The M4 subset is dominatingly presented in the adventitia and intima of human arteries to trigger inflammation and promote plaque instability. Moreover, CD163 alleviates AS progression by upregulating the atheroprotective enzyme HO-1 in response to hemoglobin. It was reported that CXCL4 aggravated AS by suppressing CD163 and scavenger receptors CD36 or SR-1, which was consistent with the conclusion in ApeE-/- mice (45, 46), indicating the proatherogenic effects of this population.
As illustrated in Table 2, HA-mac, M(Hb), and Mhem populations display in the hemorrhagic sites of unstable plaques where they engulf and recycle erythrocyte remnants and hemoglobin, and could be induced by hemoglobin, haptoglobin, and CD163. All of them are regulated by CD163, but HA-mac, M(Hb), and Mhem are also activated by IL-10, LXRα, and ATF1/MAPK, respectively (32). Because of the high expression of LXRa, LXRβ, ABCA1, and ABCG1 which are responsible for the cholesterol efflux, HA-mac, M(Hb), and Mhem subsets are atheroprotective and resist to be foam cells (32, 47), but their effects on AS protection are limited because of the small percentage in plaque lesion.
Cell death and dead cells not being removed effectively are other major causes of plaque and necrotic core formation. The programmed death of macrophages is a complex process involving multiple mechanisms, such as endoplasmic reticulum stress (48), oxidative stress, mitochondrial dysfunction, and lysosome rupture (49). As displayed in Table 3, atherosclerotic plaques contain a large amount of cholesterol as well as necrosis cores comprised of foam cells, collagen, smooth muscle cells, etc. (99), due to the dysfunction of the death-clearance mechanism on macrophage apoptosis, necrosis, and ferroptosis (100, 101). For macrophage death and clearance, the effects of apoptosis–efferocytosis, necrosis, ferroptosis, autophagy, and pyroptosis are highlighted below.
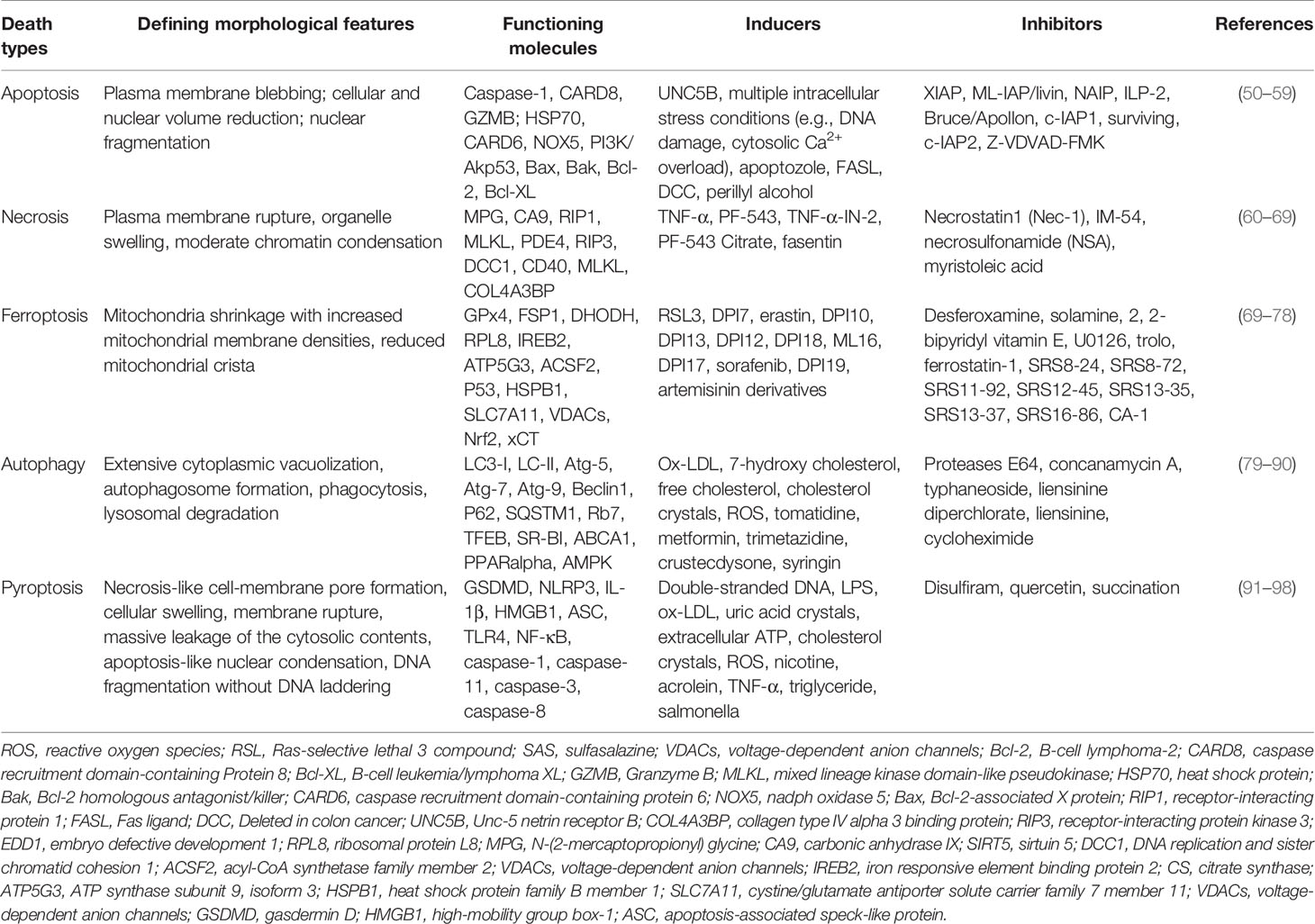
Table 3 Cell death types contributed to plaques and necrosis core in atherosclerosis.
Cell apoptosis has been regarded as a pivotal step for necrotic core formation and unstable plaque rupture. High levels of ox-LDL and cholesterol overload-induced ERs cause macrophage apoptosis (102). Apoptosis executes the programmed progression, which is the activation of caspase-type proteases (103). Macrophage apoptosis is the main cause of necrosis cores and adverse remodeling of the plaque architecture, leading to vulnerable plaques (104). What is more, epidemiological studies showed that AS could be aggravated by hyperhomocysteinemia, which induced inflammation, lipid accumulation, and macrophage apoptosis in arteries (105). However, some cytokines, like colony-stimulating factor 1 (CSF1) derived from VSMCs and endothelial cells promoted macrophage proliferation and reduced macrophage apoptosis in plaques (106), suggesting that vulnerable plaque formation may be effectively prevented by inhibiting macrophage apoptosis. On other hand, because the phagocytic receptors fall off the macrophage cell membrane, in comparison with other human macrophage-rich tissues, efferocytosis is impaired in atherosclerotic plaques (107). This impairment not only decreases the phagocytic capacity but also generates molecules to compete with macrophages for the identification of apoptotic cells, which reduces the clearance of apoptotic cells and subsequently intensifies the inflammatory response (107, 108). Macrophage apoptosis is exacerbating, but efferocytosis is impaired, which enhances atherosclerotic plaque formation under the stimulation of pro-inflammatory factors.
Apoptosis has been concerned with atherosclerotic plaque formation for several decades. This programmed cell death causes the clearance of unhealthy cells but does not produce detrimental substances to the microenvironment. The outcomes of apoptosis closely rely on the stage of the AS (109). It is worth noting that apoptotic macrophages on the early stage of plaques are efficiently cleared by efferocytosis; thereby, secondary necrosis could be prevented. However, efferocytosis is tremendously defective in advanced atherosclerotic plaques; thus, apoptotic macrophages aggregate and secondary necrosis occurs (110). Ox-LDL stimulation, mitochondrial ROS overproduction, ATP depletion, intracellular Ca2+ overload as well as impaired efferocytosis are all inducements to trigger macrophage necrosis. Macrophage necrosis leads to plaque vulnerability and releases massive pro-inflammatory cytokines and DAMPs (damage-associated molecular patterns) like high-mobility group box 1 (HMGB1), heat shock proteins as well as S100 family molecules (109). The result of transmission electron microscopy showed that VSMCs (30 ± 18%) were observed to occur necrosis but not apoptosis (1 ± 2%) in advanced human plaques. Moreover, dying macrophages present the necrotic cell morphology with membrane disruption and swollen, disintegrating organelles (110), implying that macrophage necrosis plays a considerable part in advanced plaques. Recently, GPR32, a receptor for pro-resolving lipid mediators like resolvin D1, was reported to be reduced in human atherosclerotic lesions. Exhilaratingly, the lesion area and necrosis of atherosclerotic plaques were observably decreased when this receptor was overexpressed in mice (111). However, because the applied detection methods for necrosis have not been found, it has not been investigated extensively as apoptosis.
The term ferroptosis was defined to explain the manner of cell death stimulated by the chemical reagent erastin (112). Once the concept of ferroptosis was put forward, researchers paid great attention to it in the field of cancer treatment. Until now, the study of ferroptosis is still limited in AS, especially in macrophages. The differences of ferroptosis in cancer and AS are as follows: firstly, the pivotal challenge is to efficaciously kill cancer cells and keep the healthy cells impervious in cancer research, while macrophage ferroptosis would like to be prevented in AS (113). Secondly, cancer cells are defective in cell death executioner mechanisms including ferroptosis resistance but macrophages are more susceptible to ferroptosis. To enable growth, cancer cells exhibit an increased nutrient demand including iron and lipids compared with macrophages (113); therefore, cancer cells could be more capable of solving crises caused by iron and lipid overload to escape ferroptosis (114). However, due to plaque rupture, iron and lipids are overloaded in AS; macrophages are suffering the challenge from both lipids and iron-containing proteins such as hemoglobin, which could induce macrophage ferroptosis (115).
Ferroptosis, showing the iron-dependent excessive generation of lipid ROS accompanied with the depletion of plasma membrane polyunsaturated fatty acids, has been found to promote the formation and destabilization of plaques (116, 117). Ferroptosis not only controls the death but also the phenotype of macrophages. Macrophages in the vascular microenvironment with excess iron and ox-LDL or LPS/IFN-γ, the proportion of M1 and Mox macrophages increases (115). What is noteworthy is that, compared to M2 macrophages, M1 and Mox macrophages display the ability of ferroptosis resistance (115), implying the interplay between macrophage ferroptosis and polarization. Because lipid peroxidation and iron overload are common and obvious characteristics of plaques, ferroptosis does play a non-negligible role in AS progression (117, 118). Recently, the glutathione (GSH)-dependent antioxidant enzyme glutathione peroxidase 4 (GPx4), ferroptosis suppressor protein 1 (FSP1)-CoQ10, and dihydroorotate dehydrogenase (DHODH) have become a tripod complexion in ferroptosis regulation, which were summarized in Figures 1, 2, and Table 4.
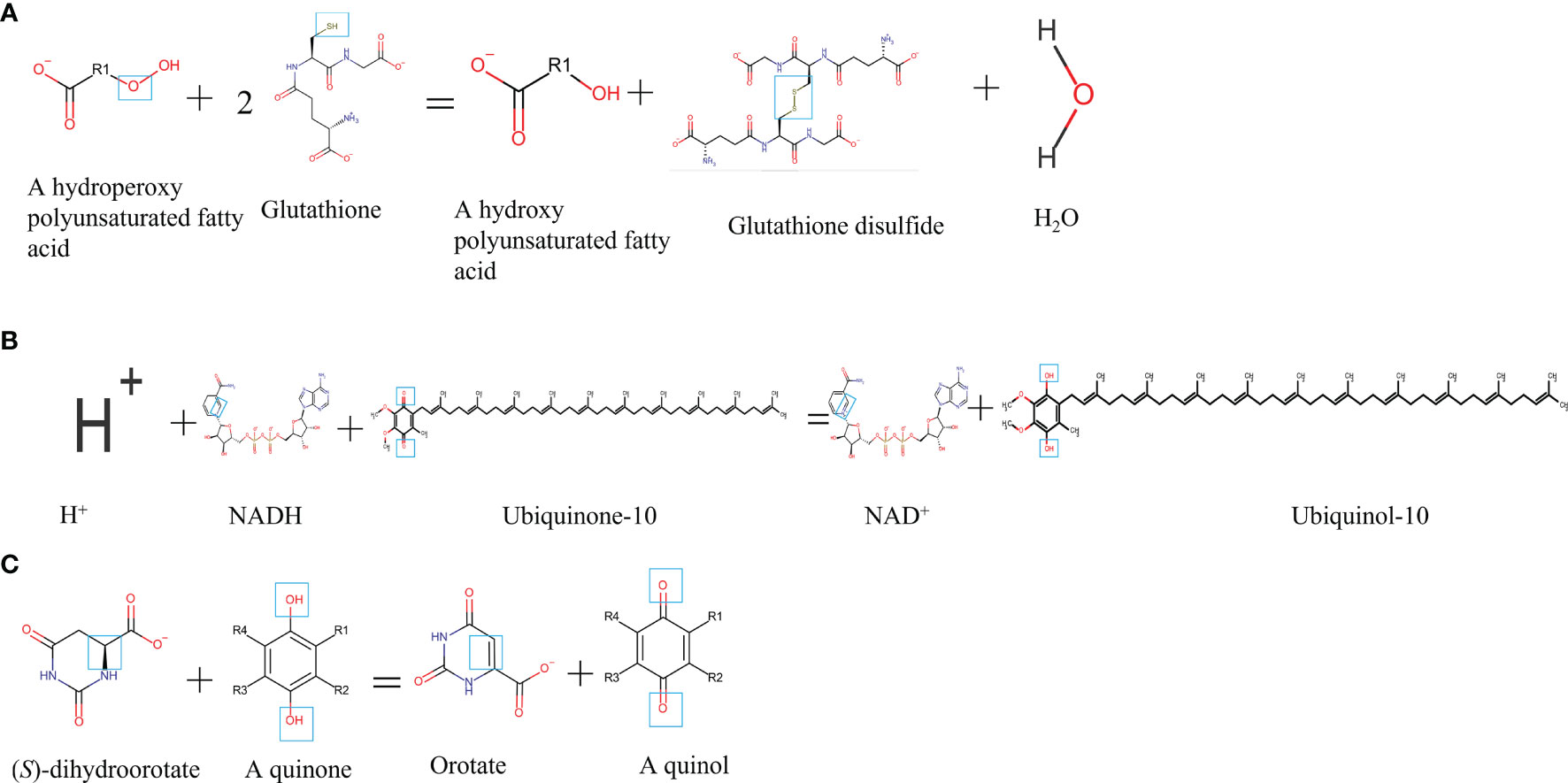
Figure 1 Chemical formula and catalytic activity GPx4, FSP1, and DHODH in ferroptosis regulation. (A) GPx4. (B) FSP1. (C) DHODH. NADH, reduced form of nicotinamide-adenine dinucleotide; NAD+, nicotinamide adenine dinucleotide.
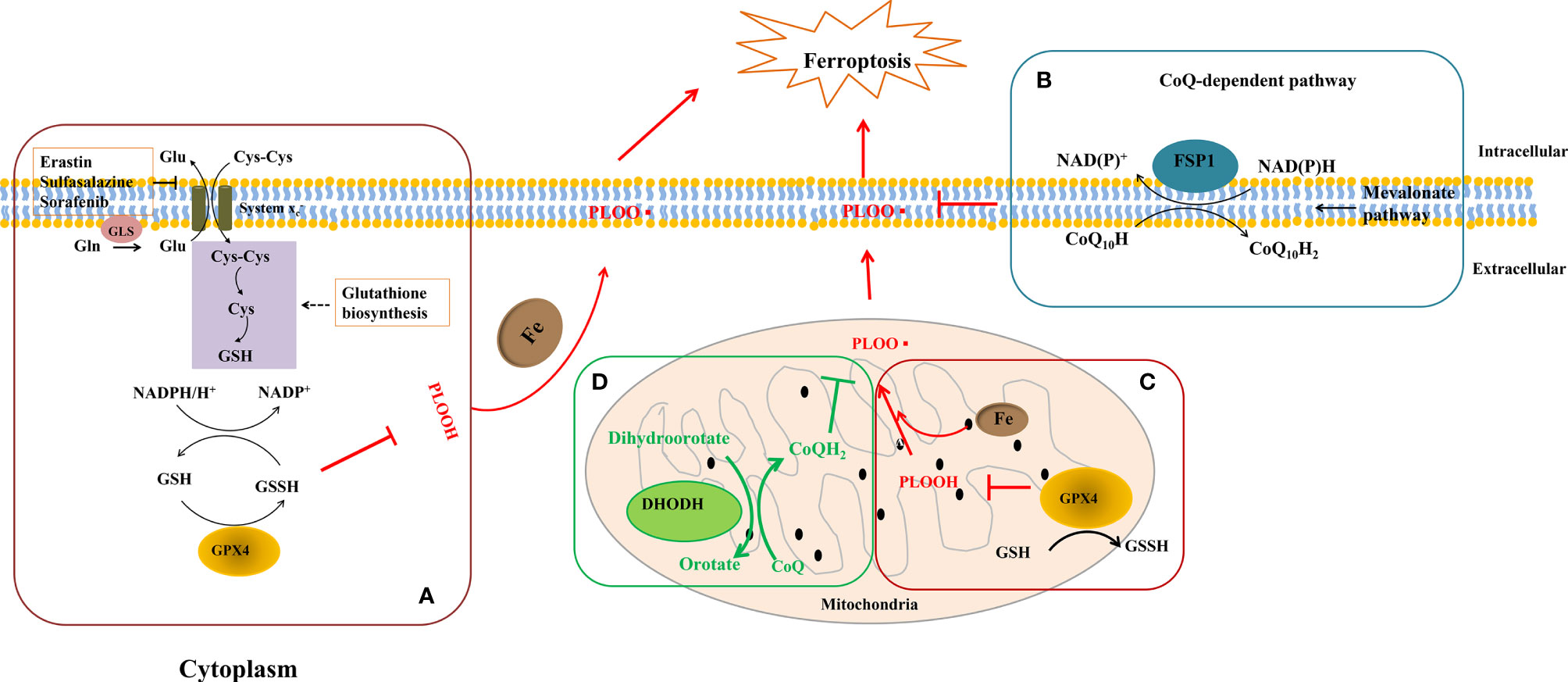
Figure 2 GPx4, FSP1, and DHODH on cell ferroptosis. (A) Firstly, cystine (Cys-Cys) was transported for keeping the homeostasis. Meanwhile, L-glutamine (Gln) was catalyzed by glutaminase (Gls) to become L-glutamate (Glu) and outputted through system Xc- as well. Secondly, GPx4 oxidizes GSH to GSSG to reduce lipid peroxidation in cytoplasm and mitochondria. (B) FSP1 reduces ubiquinol-10 by catalyzing coenzyme Q/ubiquinone-10, which is a process independent of the GPX4 at the plasma membrane. (C, D) In the mitochondrial inner membrane, DHODH operates in parallel to mitochondrial GPx4 to inhibit ferroptosis and catalyzes the conversion of dihydroorotate to orotate via catalyzing the reduction of coenzyme Q/quinone to quinol. NAD(P)H, nicotinamide adenine dinucleotide phosphate; NAD+, nicotinamide adenine dinucleotide; PLOOH, phospholipid hydroperoxide.

Table 4 The action of three main proteins regulating ferroptosis.
GPx4, a GSH-dependent enzyme, is located in the cytoplasm and mitochondria of macrophages and reduces lipid hydroperoxides (L-OOH) to lipid alcohols (L-OH) by catalyzing GSH as shown in Figures 2A, C; therefore, GPx4 generally controls the iron-dependent production of lipid alkoxy radicals (L-O·) from L-OOH, which directly reduces lipid peroxidation that has been produced in the macrophage cell membrane. In our past work, macrophage iron and lipid retention aggravated AS via the autocrine formation of hepcidin in macrophages (121). Notably, the overexpression of GPx4 significantly lessens lipid modifications by the superoxide and impedes the progression of atherosclerotic plaque in ApoE-/- mice (122), indicating that ferroptosis is a risk factor in the progress of CVDs. Breakthroughs have been made in cancer treatment, RSL3, ML162, ML210, 4-hydroxytamoxifen, and the FDA-approved anticancer agent altretamine could suppress GPx4 activity to induce cancer cell ferroptosis (120, 123). Although the evidence has shown that GPx4 participates in the formation of atherosclerotic plaques, the GPx4 knockout animal model, specific inhibitors, and activators of GPx4 may better unveil whether ferroptosis decisively participats in plaque formation in AS.
Recently, Doll et al. and Bersuker et al. simultaneously discovered that FSP1 is a potent protein resistant to ferroptosis independent of GPx4 (71, 120). The great discovery of FSP1 explains the dilemma in the tolerance of anti-cancer treatment by inhibiting GPx4 activation and provides new insights into the ferroptosis inhibition of macrophages in AS. FSP1 contains two mandatory domains for its function in suppressing ferroptosis, i.e., N-myristoylation and a flavoprotein oxidoreductase domain. FSP1 is recruited to the plasma membrane (endoplasmic reticulum, cytomembrane, and Golgi apparatus) by the key site N-myristoylation. Once recruited, FSP1 serves as an NAD(P)H-dependent oxidoreductase to reduce coenzyme Q10 (shown in Figure 2B). Coenzyme Q10 is an active lipophilic electron carrier that is the only one lipid-soluble antioxidant synthesized endogenously (124). Coenzyme Q10 plays a vital role in aerobic respiration to transfer electrons in mitochondria and keeps the lipids of Golgi and cell membranes away from oxidation (125). Although CoQ10 has been reported for decades in mitochondria, it is a novel discovery that CoQ10 is reduced by FSP1 at the cell membrane, which is sufficient to suppress the production of lipid peroxides and ferroptosis. Due to the special pathology of AS, local iron overload and lipid overload provide the necessary conditions for ferroptosis, and the FSP1-CoQ10-NAD(P)H pathway may be a promising strategy for inhibiting atherosclerotic plaque formation.
DHODH is a newly discovered protein operating parallel to GPx4 to inhibit ferroptosis in 2021. In the past decades, DHODH was regarded as an enzyme to catalyze de novo pyrimidine synthesis and produce uridine monophosphate, which would generate pyrimidines to biosynthesize nucleic acids for cell proliferation (125). In metazoans, DHODH is recruited to the outer face of the mitochondrial inner membrane, an environment rich in lipids, indicating that DHODH is a potential enzyme to inhibit ferroptosis by reducing lipid peroxidation (see Figure 2D). Because of the favorable permeability of the mitochondrial outer membrane, DHODH inserted into the outer membrane has access to its substrate dihydroorotate and mitochondrial inner membrane-bound CoQ. Subsequently, the product orotate of DHODH could be utilized in the de novo pyrimidine synthesis pathway in the cytoplasm (126). Therein, DHODH catalyzes dihydroorotate to orotate with quinone as an electron acceptor, which is an essential step for inducing ferroptosis.
Dihydroorotate and orotate are the substrate and product of DHODH. Mao and his colleagues reported that a separate supplementation with them could attenuate or enhance ferroptosis triggered by the inhibitors of GPx4 (72). When the GPx4 expression is inhibited, DHODH inactivation could induce substantial mitochondrial lipid peroxidation and trigger ferroptosis while cooperating with inducers to initiate ferroptosis in cancer cells with a high expression of GPx4 (72). DHODH inhibited ferroptosis independently of mitochondrial GPx4, cytosolic GPx4, and cytomembrane FSP1 in the mitochondrial inner membrane by reducing ubiquinone to ubiquinol, a radical-trapping antioxidant with anti-ferroptotic activity (see Figure 2D). Meanwhile, brequinar, the inhibitor of DHODH, selectively inhibited cell growth with low-expressed GPx4 by inducing ferroptosis. Furthermore, the authors reported that a combined treatment with brequinar and sulfasalazine synergistically triggered ferroptosis and suppressed tumor growth with a high expression of GPx4 as well (72). Unlike anticancer treatment, iron overload and lipid peroxidation are two key points for macrophage ferroptosis that should be avoided in AS, suggesting that except for the GPx4, FSP1 pathway, DHODH is a promising target for the suppression of macrophage ferroptosis in AS.
Autophagy mediates the degradation and recycling of the damaged organelles and proteins via autophagy-related genes in lysosomes, which is a conserved process for maintaining cellular homeostasis. However, the dysregulation of autophagy has been associated with various metabolic disorders including AS. It was reported that autophagy could be triggered in macrophages, VSMCs, and endothelial cells (127).
In macrophages, autophagy could be induced by oxidized lipids, like ox-LDL and 7-ketocholesterol (128). The activation of autophagy could protect macrophages by digesting the damaged proteins, organelles, or lipids. Notably, lipophagy is selective autophagy that targets lipid droplets to lysosomes for degradation in macrophage foam cells (129). Defective autophagy in macrophages would promote the apoptosis and necrosis of macrophages, causing plaque instability in AS (80). Autophagy is generally regulated by LC3, autophagy-related genes (Atg), p62, AMPK, etc., and it could be regulated by its inhibitors and agonists in CVD, which were summarized in Table 3.
Pyroptosis, a programmed cell death, is closely associated with the activation of NLRP3 inflammasomes and the rapid release of various cytokines, such as IL-1β, IL-18, and HMGB-1 (130). In 2011, Kayagaki and colleagues reported that caspase-11 induces caspase-1-independent pyroptosis in macrophages, which is a noncanonical inflammasome pathway (131). Macrophage pyroptosis induced by the cholesterol crystal or ox-LDL promotes plaque destabilization. Significantly, NLRP3 inflammasome components are highly expressed in macrophages (132); therefore, inhibiting macrophage pyroptosis and reducing inflammation would provide prospective therapeutic strategies for the disease.
The NLRP3 inflammasome consisted of three parts: NLR (NOD-like receptor) families and PYHIN (pyrin and HIN domain-containing protein) families, ASC (apoptosis-associated speck-like protein), and the effector caspase-1. As shown in Table 3, the cholesterol crystal or ox-LDL is phagocytosed by macrophages, which facilitate NLRP3 inflammasome assembly and then activate caspase-1. Subsequently, activated caspase-1 cleaves pro-IL-1β to mature IL-1β, which induces inflammation and competes with cholesterol for access to the ABCA1, causing the retention of cholesterol to form foam cells (97). In addition, gasdermin D (GSDMD) could be cleaved by caspase-1 to N-terminal fragments (GSDMD-NT) and C-terminal fragments (GSDMD-CT) (133). GSDMD-NT translocates to the cytomembrane, self-oligomerizes, and forms the pores to disrupt the homeostasis of intracellular and extracellular osmotic pressure, leading to macrophage pyroptosis to form instability plaques (134).
In the past decades, the hypotheses and mechanisms of AS pathogenesis have always refreshed our perception. The knowledge of innovation on how the stabilization mechanisms that govern cholesterol and lipid transport and stay inside macrophages are operated to be foam cells helps us to tease out the initiating causes and has identified several pathological mechanisms, including inflammation, macrophage polarization, and macrophage death, which can regulate the formation of atherosclerotic plaques. According to the three classical hypotheses above, inflammation, lipid oxidation, and macrophage foam cells display major roles in AS progression, which reminds us to be concerned about the primary cause. Based on the past studies on AS, we summarized two main causes inducing inflammation, promoting foam cell formation and the formation and rupture of plaques, i.e., macrophage polarization and macrophage death.
As the disease progresses, plaques and the necrotic core are built up, although macrophage death is going on, most intense in advanced plaques yet. The recognition of disparate macrophage phenotypes (e.g., M1, M2, and Mox macrophages) has raised the question of who is responsible for foam cell formation the most because of the different abilities for phagocytosis, antioxidation, and death. Notably, it was reported that M1 macrophages could be resistant to ferroptosis compared to the M2 subtype (135). Furthermore, Mox may be an excellent anti-atherosclerotic subtype due to its abilities of weak phagocytosis and strong oxidation resistance. Macrophage death should be prevented, or dead cells should be removed effectively. From the clinical point of view, many therapies, such as statins, whose primary function is lowering serum LDL levels, are not particularly effective (136). It provides new insights that induce macrophages to polarize to anti-atherosclerotic subtypes as well as suppress macrophages’ death by the key molecules of apoptosis, necrosis, ferroptosis, autophagy, and pyroptosis. Taken together, macrophage polarization and death are two main archcriminals that are promising candidates for AS prevention and treatment.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
All authors contributed to the article and approved the submitted version.
This work was supported in part by the National Natural Science Foundation of China (No. 81973044, 82003448).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AS, atherosclerosis; CXCL4, CXC chemokine ligand 4; CVD, cardiovascular disease; DHODH, dihydroorotate dehydrogenase; FSP1, ferroptosis suppressor protein 1; GPx4, glutathione peroxidase 4; LDL, low-density lipoprotein; L-OOH, lipid hydroperoxides; L-OH, lipids alcohols; MMP-7, matrix metalloprotease 7; Nrf2, nuclear factor E2-related factor 2; TrxR1, thioredoxin reductase 1; Srxn1, sulfiredoxin-1; VSMAs, vascular smooth muscle cells.
1. Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation (2019) 139(10):e56–528. doi: 10.1161/CIR.0000000000000659
2. Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990-2019: Update From the GBD 2019 Study. J Am Coll Cardiol (2020) 76(25):2982–3021. doi: 10.1016/j.jacc.2020.11.010
3. Ly KN, Xing J, Klevens RM, Jiles RB, Ward JW, Holmberg SD. The Increasing Burden of Mortality From Viral Hepatitis in the United States Between 1999 and 2007. Ann Intern Med (2012) 156(4):271–8. doi: 10.7326/0003-4819-156-4-201202210-00004
4. Townsend N, Wilson L, Bhatnagar P, Wickramasinghe K, Rayner M, Nichols M. Cardiovascular Disease in Europe: Epidemiological Update 2016. Eur Heart J (2016) 37(42):3232–45. doi: 10.1093/eurheartj/ehw334
5. Barrett TJ, Schlegel M, Zhou F, Gorenchtein M, Bolstorff J, Moore KJ, et al. Platelet Regulation of Myeloid Suppressor of Cytokine Signaling 3 Accelerates Atherosclerosis. Sci Transl Med (2019) 11(517):1–15. doi: 10.1126/scitranslmed.aax0481
6. Grootaert MOJ, Moulis M, Roth L, Martinet W, Vindis C, Bennett MR, et al. Vascular Smooth Muscle Cell Death, Autophagy and Senescence in Atherosclerosis. Cardiovasc Res (2018) 114(4):622–34. doi: 10.1093/cvr/cvy007
7. Steinberg D. Thematic Review Series: The Pathogenesis of Atherosclerosis. An Interpretive History of the Cholesterol Controversy: Part I. J Lipid Res (2004) 45(9):1583–93. doi: 10.1194/jlr.R600009-JLR200
8. Sun P, Dwyer KM, Merz CN, Sun W, Johnson CA, Shircore AM, et al. Blood Pressure, LDL Cholesterol, and Intima-Media Thickness: A Test of the “Response to Injury” Hypothesis of Atherosclerosis. Arterioscler Thromb Vasc Biol (2000) 20(8):2005–10. doi: 10.1161/01.ATV.20.8.2005
9. Ross R. Atherosclerosis–an Inflammatory Disease. N Engl J Med (1999) 340(2):115–26. doi: 10.1056/NEJM199901143400207
10. Manninen S, Lankinen M, Erkkila A, Nguyen SD, Ruuth M, de Mello V, et al. The Effect of Intakes of Fish and Camelina Sativa Oil on Atherogenic and Anti-Atherogenic Functions of LDL and HDL Particles: A Randomized Controlled Trial. Atherosclerosis (2019) 281:56–61. doi: 10.1016/j.atherosclerosis.2018.12.017
11. Ma C, Xia R, Yang S, Liu L, Zhang J, Feng K, et al. Formononetin Attenuates Atherosclerosis via Regulating Interaction Between KLF4 and SRA in Apoe(-/-) Mice. Theranostics (2020) 10(3):1090–106. doi: 10.7150/thno.38115
12. Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. Beyond Cholesterol. Modifications of Low-Density Lipoprotein That Increase Its Atherogenicity. N Engl J Med (1989) 320(14):915–24. doi: 10.1056/NEJM198904063201407
13. Shayo CC, Mladovan AG, Baldi A. Differentiating Agents Modulate Topoisomerase I Activity in U-937 Promonocytic Cells. Eur J Pharmacol (1997) 324(1):129–33. doi: 10.1016/S0014-2999(97)00151-9
14. Wu Q, Sun L, Hu X, Wang X, Xu F, Chen B, et al. Suppressing the Intestinal Farnesoid X Receptor/Sphingomyelin Phosphodiesterase 3 Axis Decreases Atherosclerosis. J Clin Invest (2021) 131(9). doi: 10.1172/JCI142865
15. Ejaz HW, Wang W, Lang M. Copper Toxicity Links to Pathogenesis of Alzheimer’s Disease and Therapeutics Approaches. Int J Mol Sci (2020) 21(20). doi: 10.3390/ijms21207660
16. Soehnlein O, Libby P. Targeting Inflammation in Atherosclerosis - From Experimental Insights to the Clinic. Nat Rev Drug Discov (2021) 20(8):589–610. doi: 10.1038/s41573-021-00198-1
17. Chen YC, Lai YS, Hsuuw YD, Chang KT. Withholding of M-CSF Supplement Reprograms Macrophages to M2-Like via Endogenous CSF-1 Activation. Int J Mol Sci (2021) 22(7):3532. doi: 10.3390/ijms22073532
18. Locati M, Curtale G, Mantovani A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu Rev Pathol (2020) 15:123–47. doi: 10.1146/annurev-pathmechdis-012418-012718
19. Chistiakov DA, Bobryshev YV, Orekhov AN. Changes in Transcriptome of Macrophages in Atherosclerosis. J Cell Mol Med (2015) 19(6):1163–73. doi: 10.1111/jcmm.12591
20. Trombetta AC, Soldano S, Contini P, Tomatis V, Ruaro B, Paolino S, et al. A Circulating Cell Population Showing Both M1 and M2 Monocyte/Macrophage Surface Markers Characterizes Systemic Sclerosis Patients With Lung Involvement. Respir Res (2018) 19(1):186. doi: 10.1186/s12931-018-0891-z
21. Zhao M, Bian YY, Yang LL, Chen YQ, Wang YJ, Ma YT, et al. HuoXueTongFu Formula Alleviates Intraperitoneal Adhesion by Regulating Macrophage Polarization and the SOCS/JAK2/STAT/PPAR-Gamma Signalling Pathway. Mediators Inflamm (2019) 2019:1769374. doi: 10.1155/2019/1769374
22. Yoon J, Um HN, Jang J, Bae YA, Park WJ, Kim HJ, et al. Eosinophil Activation by Toll-Like Receptor 4 Ligands Regulates Macrophage Polarization. Front Cell Dev Biol (2019) 7:329. doi: 10.3389/fcell.2019.00329
23. Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, et al. IRF5 Promotes Inflammatory Macrophage Polarization and TH1-TH17 Responses. Nat Immunol (2011) 12(3):231–8. doi: 10.1038/ni.1990
24. Degboe Y, Rauwel B, Baron M, Boyer JF, Ruyssen-Witrand A, Constantin A, et al. Polarization of Rheumatoid Macrophages by TNF Targeting Through an IL-10/STAT3 Mechanism. Front Immunol (2019) 10:3. doi: 10.3389/fimmu.2019.00003
25. Hu W, Lin J, Lian X, Yu F, Liu W, Wu Y, et al. M2a and M2b Macrophages Predominate in Kidney Tissues and M2 Subpopulations Were Associated With the Severity of Disease of IgAN Patients. Clin Immunol (2019) 205:8–15. doi: 10.1016/j.clim.2019.05.005
26. Jinnouchi H, Guo L, Sakamoto A, Torii S, Sato Y, Cornelissen A, et al. Diversity of Macrophage Phenotypes and Responses in Atherosclerosis. Cell Mol Life Sci (2020) 77(10):1919–32. doi: 10.1007/s00018-019-03371-3
27. Liu D, Wei Y, Liu Y, Wu T, Hu J, Lu H, et al. The Long Non-Coding RNA NEAT1/miR-224-5p/IL-33 Axis Modulates Macrophage M2a Polarization and A1 Astrocyte Activation. Mol Neurobiol (2021) 58(9):4506–19. doi: 10.1007/s12035-021-02405-x
28. Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR, et al. Identification of a Novel Macrophage Phenotype That Develops in Response to Atherogenic Phospholipids via Nrf2. Circ Res (2010) 107(6):737–46. doi: 10.1161/CIRCRESAHA.109.215715
29. Colin S, Chinetti-Gbaguidi G, Staels B. Macrophage Phenotypes in Atherosclerosis. Immunol Rev (2014) 262(1):153–66. doi: 10.1111/imr.12218
30. Skuratovskaia D, Vulf M, Khaziakhmatova O, Malashchenko V, Komar A, Shunkin E, et al. Tissue-Specific Role of Macrophages in Noninfectious Inflammatory Disorders. Biomedicines (2020) 8(10). doi: 10.3390/biomedicines8100400
31. Leitinger N, Schulman IG. Phenotypic Polarization of Macrophages in Atherosclerosis. Arterioscler Thromb Vasc Biol (2013) 33(6):1120–6. doi: 10.1161/ATVBAHA.112.300173
32. Boyle JJ, Johns M, Kampfer T, Nguyen AT, Game L, Schaer DJ, et al. Activating Transcription Factor 1 Directs Mhem Atheroprotective Macrophages Through Coordinated Iron Handling and Foam Cell Protection. Circ Res (2012) 110(1):20–33. doi: 10.1161/CIRCRESAHA.111.247577
33. Wan X, Huo Y, Johns M, Piper E, Mason JC, Carling D, et al. 5’-AMP-Activated Protein Kinase-Activating Transcription Factor 1 Cascade Modulates Human Monocyte-Derived Macrophages to Atheroprotective Functions in Response to Heme or Metformin. Arterioscler Thromb Vasc Biol (2013) 33(11):2470–80. doi: 10.1161/ATVBAHA.113.300986
34. Barrett TJ. Macrophages in Atherosclerosis Regression. Arterioscler Thromb Vasc Biol (2020) 40(1):20–33. doi: 10.1161/ATVBAHA.119.312802
35. Wang N, Liang H, Zen K. Molecular Mechanisms That Influence the Macrophage M1-M2 Polarization Balance. Front Immunol (2014) 5:614. doi: 10.3389/fimmu.2014.00614
36. Luan G, Pan F, Bu L, Wu K, Wang A, Xu X. Butorphanol Promotes Macrophage Phenotypic Transition to Inhibit Inflammatory Lung Injury via Kappa Receptors. Front Immunol (2021) 12:692286. doi: 10.3389/fimmu.2021.692286
37. Yang R, Liao Y, Wang L, He P, Hu Y, Yuan D, et al. Exosomes Derived From M2b Macrophages Attenuate DSS-Induced Colitis. Front Immunol (2019) 10:2346. doi: 10.3389/fimmu.2019.02346
38. Cao J, Dong R, Jiang L, Gong Y, Yuan M, You J, et al. LncRNA-MM2P Identified as a Modulator of Macrophage M2 Polarization. Cancer Immunol Res (2019) 7(2):292–305. doi: 10.1158/2326-6066.CIR-18-0145
39. Martinez FO, Helming L, Gordon S. Alternative Activation of Macrophages: An Immunologic Functional Perspective. Annu Rev Immunol (2009) 27:451–83. doi: 10.1146/annurev.immunol.021908.132532
40. Han IH, Song HO, Ryu JS. IL-6 Produced by Prostate Epithelial Cells Stimulated With Trichomonas Vaginalis Promotes Proliferation of Prostate Cancer Cells by Inducing M2 Polarization of THP-1-Derived Macrophages. PLoS Negl Trop Dis (2020) 14(3):e0008126. doi: 10.1371/journal.pntd.0008126
41. Ono Y, Yoshino O, Hiraoka T, Sato E, Fukui Y, Ushijima A, et al. CD206+ M2-Like Macrophages Are Essential for Successful Implantation. Front Immunol (2020) 11:557184. doi: 10.3389/fimmu.2020.557184
42. Tabas I. Macrophage Death and Defective Inflammation Resolution in Atherosclerosis. Nat Rev Immunol (2010) 10(1):36–46. doi: 10.1038/nri2675
43. Domschke G, Gleissner CA. CXCL4-Induced Macrophages in Human Atherosclerosis. Cytokine (2019) 122:154141. doi: 10.1016/j.cyto.2017.08.021
44. Erbel C, Tyka M, Helmes CM, Akhavanpoor M, Rupp G, Domschke G, et al. CXCL4-Induced Plaque Macrophages can be Specifically Identified by Co-Expression of MMP7+S100A8+ In Vitro and In Vivo. Innate Immun (2015) 21(3):255–65. doi: 10.1177/1753425914526461
45. Gleissner CA. Macrophage Phenotype Modulation by CXCL4 in Atherosclerosis. Front Physiol (2012) 3:1. doi: 10.3389/fphys.2012.00001
46. Gleissner CA, Shaked I, Erbel C, Böckler D, Katus HA, Ley K. CXCL4 Downregulates the Atheroprotective Hemoglobin Receptor CD163 in Human Macrophages. Circ Res (2010) 106(1):203–11. doi: 10.1161/CIRCRESAHA.109.199505
47. Liberale L, Dallegri F, Montecucco F, Carbone F. Pathophysiological Relevance of Macrophage Subsets in Atherogenesis. Thromb Haemost (2017) 117(1):7–18. doi: 10.1160/TH16-08-0593
48. Liang S, Wang F, Bao C, Han J, Guo Y, Liu F, et al. BAG2 Ameliorates Endoplasmic Reticulum Stress-Induced Cell Apoptosis in Mycobacterium Tuberculosis-Infected Macrophages Through Selective Autophagy. Autophagy (2020) 16(8):1453–67. doi: 10.1080/15548627.2019.1687214
49. Hoseini Z, Sepahvand F, Rashidi B, Masoudifar A, Mirzaei H. NLRP3 Inflammasome: Its Regulation and Involvement in Atherosclerosis. J Cell Physiol (2018) 233(3):2116–32. doi: 10.1002/jcp.25930
50. Dull AB, Wilsker D, Hollingshead M, Mazcko C, Annunziata CM, LeBlanc AK, et al. Development of a Quantitative Pharmacodynamic Assay for Apoptosis in Fixed Tumor Tissue and Its Application in Distinguishing Cytotoxic Drug-Induced DNA Double Strand Breaks From DNA Double Strand Breaks Associated With Apoptosis. Oncotarget (2018) 9(24):17104–16. doi: 10.18632/oncotarget.24936
51. Tsuchiya K, Nakajima S, Hosojima S, et al. Caspase-1 Initiates Apoptosis in the Absence of Gasdermin D. Nat Commun (2019) 10(1):2091. doi: 10.1038/s41467-019-09753-2
52. Yu P, Liu B, Hao S, Xing R, Li Y. A New Risk Polymorphism Rs10403848 of CARD8 Significantly Associated With Psoriasis Vulgaris in Northeastern China. BioMed Res Int (2020) 2020:2867505. doi: 10.1155/2020/2867505
53. Zhang H, Guo Z, Guo Y, Wang Z, Tang Y, Song T, et al. Bim Transfer Between Bcl-2-Like Protein and Hsp70 Underlines Bcl-2/Hsp70 Crosstalk to Regulate Apoptosis. Biochem Pharmacol (2021) 190:114660. doi: 10.1016/j.bcp.2021.114660
54. Carnesecchi S, Rougemont AL, Doroshow JH, Nagy M, Mouche S, Gumy-Pause F, et al. The NADPH Oxidase NOX5 Protects Against Apoptosis in ALK-Positive Anaplastic Large-Cell Lymphoma Cell Lines. Free Radic Biol Med (2015) 84:22–9. doi: 10.1016/j.freeradbiomed.2015.02.027
55. Tan S, Liu X, Chen L, Wu X, Tao L, Pan X, et al. Fas/FasL Mediates NF-Kappabp65/PUMA-Modulated Hepatocytes Apoptosis via Autophagy to Drive Liver Fibrosis. Cell Death Dis (2021) 12(5):474.
56. Naziroglu M. A Novel Antagonist of TRPM2 and TRPV4 Channels: Carvacrol. Metab Brain Dis (2022). doi: 10.1007/s11011-021-00887-1
57. Saraei R, Soleimani M, Movassaghpour Akbari AA, Farshdousti Hagh M, Hassanzadeh A, Solali S. The Role of XIAP in Resistance to TNF-Related Apoptosis-Inducing Ligand (TRAIL) in Leukemia. BioMed Pharmacother (2018) 107:1010–9. doi: 10.1016/j.biopha.2018.08.065
58. Peilin W, Songsong T, Chengyu Z, Zhi C, Chunhui M, Yinxian Y, et al. Directed Elimination of Senescent Cells Attenuates Development of Osteoarthritis by Inhibition of C-IAP and XIAP. Biochim Biophys Acta Mol Basis Dis (2019) 1865(10):2618–32. doi: 10.1016/j.bbadis.2019.05.017
59. Zhang R, Zhou W, Yu Z, Yang L, Liu G, Yu H, et al. miR-1247-3p Mediates Apoptosis of Cerebral Neurons by Targeting Caspase-2 in Stroke. Brain Res (2019) 1714:18–26. doi: 10.1016/j.brainres.2019.02.020
60. Chaudhary GR, Yadav PK, Yadav AK, Tiwari M, Gupta A, Sharma A, et al. Necrosis and Necroptosis in Germ Cell Depletion From Mammalian Ovary. J Cell Physiol (2019) 234(6):8019–27. doi: 10.1002/jcp.27562
61. Peng QL, Zhang YM, Liu YC, Liang L, Li WL, Tian XL, et al. Necroptosis Contributes to Myofiber Death in Idiopathic Inflammatory Myopathies. Arthritis Rheumatol (2022). doi: 10.1002/art.42071
62. Mauney EE, Power-Hays A, Flamand Y, Vrooman L, Silverman LB, Grover AS. Clinical Characteristics and Short-Term Outcomes of Children With Asparaginase-Associated Pancreatitis. J Pediatr Gastroenterol Nutr (2021). doi: 10.1097/MPG.0000000000003334
63. Fritsch M, Gunther SD, Schwarzer R, Albert MC, Schorn F, Werthenbach JP, et al. Caspase-8 is the Molecular Switch for Apoptosis, Necroptosis and Pyroptosis. Nature (2019) 575(7784):683–7. doi: 10.1038/s41586-019-1770-6
64. Szebenyi J, Gede N, Hegyi P, Szakacs Z, Solymar M, Eross B, et al. Efficacy of Biologics Targeting Tumour Necrosis Factor-Alpha, Interleukin-17 -12/23, -23 and Small Molecules Targeting JAK and PDE4 in the Treatment of Nail Psoriasis: A Network Meta-Analysis. Acta Derm Venereol (2020) 100(18):adv00318. doi: 10.2340/00015555-3640
65. Hamada M, Kameyama H, Iwai S, Yura Y. Induction of Autophagy by Sphingosine Kinase 1 Inhibitor PF-543 in Head and Neck Squamous Cell Carcinoma Cells. Cell Death Discov (2017) 3:17047. doi: 10.1038/cddiscovery.2017.47
66. Wood TE, Dalili S, Simpson CD, Hurren R, Mao X, Saiz FS, et al. A Novel Inhibitor of Glucose Uptake Sensitizes Cells to FAS-Induced Cell Death. Mol Cancer Ther (2008) 7(11):3546–55. doi: 10.1158/1535-7163.MCT-08-0569
67. Shen B, Mei M, Pu Y, Zhang H, Liu H, Tang M, et al. Necrostatin-1 Attenuates Renal Ischemia and Reperfusion Injury via Meditation of HIF-1alpha/Mir-26a/TRPC6/PARP1 Signaling. Mol Ther Nucleic Acids (2019) 17:701–13. doi: 10.1016/j.omtn.2019.06.025
68. Menacher G, Balszuweit F, Lang S, Thiermann H, Kehe K, Gudermann T, et al. Necrosulfonamide - Unexpected Effect in the Course of a Sulfur Mustard Intoxication. Chem Biol Interact (2019) 298:80–5. doi: 10.1016/j.cbi.2018.10.030
69. Yao Y, Chen Z, Zhang H, Chen C, Zeng M, Yunis J, et al. Selenium-GPX4 Axis Protects Follicular Helper T Cells From Ferroptosis. Nat Immunol (2021) 22(9):1127–39. doi: 10.1038/s41590-021-00996-0
70. Zhang Y, Wu D, Fan Z, Li J, Gao L, Wang Y, et al. Microcystin-LR Induces Ferroptosis in Intestine of Common Carp (Cyprinus Carpio). Ecotoxicol Environ Saf (2021) 223:112610. doi: 10.1016/j.ecoenv.2021.112610
71. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ Oxidoreductase FSP1 Acts Parallel to GPX4 to Inhibit Ferroptosis. Nature (2019) 575(7784):688–92. doi: 10.1038/s41586-019-1705-2
72. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-Mediated Ferroptosis Defence Is a Targetable Vulnerability in Cancer. Nature (2021) 593(7860):586–90. doi: 10.1038/s41586-021-03539-7
73. Park JM, Mau CZ, Chen YC, Su YH, Chen HA, Huang SY, et al. A Case-Control Study in Taiwanese Cohort and Meta-Analysis of Serum Ferritin in Pancreatic Cancer. Sci Rep (2021) 11(1):21242. doi: 10.1038/s41598-021-00650-7
74. Fei Z, Lijuan Y, Jing Z, Zhao Z, Zhao R, Xu X, et al. Molecular Characteristics Associated With Ferroptosis in Hepatocellular Carcinoma Progression. Hum Cell (2021) 34(1):177–86. doi: 10.1007/s13577-020-00431-w
75. Fang X, Cai Z, Wang H, Han D, Cheng Q, Zhang P, et al. Loss of Cardiac Ferritin H Facilitates Cardiomyopathy via Slc7a11-Mediated Ferroptosis. Circ Res (2020) 127(4):486–501. doi: 10.1161/CIRCRESAHA.120.316509
76. Cui Y, Zhang Z, Zhou X, Zhao Z, Zhao R, Xu X, et al. Microglia and Macrophage Exhibit Attenuated Inflammatory Response and Ferroptosis Resistance After RSL3 Stimulation via Increasing Nrf2 Expression. J Neuroinflammation (2021) 18(1):249. doi: 10.1186/s12974-021-02231-x
77. Yang Y, Luo M, Zhang K, Zhang J, Gao T, Connell DO, et al. Nedd4 Ubiquitylates VDAC2/3 to Suppress Erastin-Induced Ferroptosis in Melanoma. Nat Commun (2020) 11(1):433. doi: 10.1038/s41467-020-14324-x
78. Li Y, Zeng X, Lu D, Yin M, Shan M, Gao Y. Erastin Induces Ferroptosis via Ferroportin-Mediated Iron Accumulation in Endometriosis. Hum Reprod (2021) 36(4):951–64. doi: 10.1093/humrep/deaa363
79. Chen Y, Hua Y, Li X, Arslan IM, Zhang W, Meng G. Distinct Types of Cell Death and the Implication in Diabetic Cardiomyopathy. Front Pharmacol (2020) 11:42. doi: 10.3389/fphar.2020.00042
80. Evans TD, Jeong SJ, Zhang X, Sergin I, Razani B. TFEB and Trehalose Drive the Macrophage Autophagy-Lysosome System to Protect Against Atherosclerosis. Autophagy (2018) 14(4):724–6. doi: 10.1080/15548627.2018.1434373
81. Tao H, Yancey PG, Blakemore JL, Zhang Y, Ding L, Jerome WG, et al. Macrophage SR-BI Modulates Autophagy via VPS34 Complex and PPARalpha Transcription of Tfeb in Atherosclerosis. J Clin Invest (2021) 131(7). doi: 10.1172/JCI94229
82. Tur J, Pereira-Lopes S, Vico T, Marin EA, Munoz JP, Hernandez-Alvarez M, et al. Mitofusin 2 in Macrophages Links Mitochondrial ROS Production, Cytokine Release, Phagocytosis, Autophagy, and Bactericidal Activity. Cell Rep (2020) 32(8):108079. doi: 10.1016/j.celrep.2020.108079
83. Jeong SJ, Zhang X, Rodriguez-Velez A, Evans TD, Razani B. P62/SQSTM1 and Selective Autophagy in Cardiometabolic Diseases. Antioxid Redox Signal (2019) 31(6):458–71. doi: 10.1089/ars.2018.7649
84. Yuan P, Hu Q, He X, Long Y, Song X, Wu F, et al. Laminar Flow Inhibits the Hippo/YAP Pathway via Autophagy and SIRT1-Mediated Deacetylation Against Atherosclerosis. Cell Death Dis (2020) 11(2):141. doi: 10.1038/s41419-020-2343-1
85. Fang S, Wan X, Zou X, Sun S, Hao X, Liang C, et al. Arsenic Trioxide Induces Macrophage Autophagy and Atheroprotection by Regulating ROS-Dependent TFEB Nuclear Translocation and AKT/mTOR Pathway. Cell Death Dis (2021) 12(1):88. doi: 10.1038/s41419-020-03357-1
86. Papini A. Investigation of Morphological Features of Autophagy During Plant Programmed Cell Death. Methods Mol Biol (2018) 1743:9–19. doi: 10.1007/978-1-4939-7668-3_2
87. Liang X, Wang S, Wang L, Ceylan AF, Ren J, Zhang Y. Mitophagy Inhibitor Liensinine Suppresses Doxorubicin-Induced Cardiotoxicity Through Inhibition of Drp1-Mediated Maladaptive Mitochondrial Fission. Pharmacol Res (2020) 157:104846. doi: 10.1016/j.phrs.2020.104846
88. Ahsan A, Zheng Y, Ma S, Liu M, Cao M, Li Y, et al. Tomatidine Protects Against Ischemic Neuronal Injury by Improving Lysosomal Function. Eur J Pharmacol (2020) 882:173280. doi: 10.1016/j.ejphar.2020.173280
89. Bharath LP, Agrawal M, McCambridge G, Nicholas DA, Hasturk H, Liu J, et al. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab (2020) 32(1):44–55.e6. doi: 10.1016/j.cmet.2020.04.015
90. Zhu HY, Huang ZX, Chen GQ, Sheng F, Zheng YS. Typhaneoside Prevents Acute Myeloid Leukemia (AML) Through Suppressing Proliferation and Inducing Ferroptosis Associated With Autophagy. Biochem Biophys Res Commun (2019) 516(4):1265–71. doi: 10.1016/j.bbrc.2019.06.070
91. He Y, Hara H, Nunez G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci (2016) 41(12):1012–21. doi: 10.1016/j.tibs.2016.09.002
92. Tavakoli Dargani Z, Singla DK. Embryonic Stem Cell-Derived Exosomes Inhibit Doxorubicin-Induced TLR4-NLRP3-Mediated Cell Death-Pyroptosis. Am J Physiol Heart Circ Physiol (2019) 317(2):H460–71. doi: 10.1152/ajpheart.00056.2019
93. Volchuk A, Ye A, Chi L, Steinberg BE, Goldenberg NM. Indirect Regulation of HMGB1 Release by Gasdermin D. Nat Commun (2020) 11(1):4561. doi: 10.1038/s41467-020-18443-3
94. He B, Nie Q, Wang F, Han Y, Yang B, Sun M, et al. Role of Pyroptosis in Atherosclerosis and Its Therapeutic Implications. J Cell Physiol (2021) 236(10):7159–75. doi: 10.1002/jcp.30366
95. Jia C, Chen H, Zhang J, Zhou K, Zhuge Y, Niu C, et al. Role of Pyroptosis in Cardiovascular Diseases. Int Immunopharmacol (2019) 67:311–8. doi: 10.1016/j.intimp.2018.12.028
96. Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, et al. FDA-Approved Disulfiram Inhibits Pyroptosis by Blocking Gasdermin D Pore Formation. Nat Immunol (2020) 21(7):736–45. doi: 10.1038/s41590-020-0669-6
97. Li H, Xiao L, He H, Zeng H, Jiang J, Mei C, et al. Quercetin Attenuates Atherosclerotic Inflammation by Inhibiting Galectin-3-NLRP3 Signaling Pathway. Mol Nutr Food Res (2021) 65(15):e2000746. doi: 10.1002/mnfr.202000746
98. Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, et al. Succination Inactivates Gasdermin D and Blocks Pyroptosis. Science (2020) 369(6511):1633–7. doi: 10.1126/science.abb9818
100. Su G, Yang W, Wang S, Geng C, Guan X. SIRT1-Autophagy Axis Inhibits Excess Iron-Induced Ferroptosis of Foam Cells and Subsequently Increases IL-1Beta and IL-18. Biochem Biophys Res Commun (2021) 561:33–9. doi: 10.1016/j.bbrc.2021.05.011
101. Wang L, Li H, Tang Y, Yao P. Potential Mechanisms and Effects of Efferocytosis in Atherosclerosis. Front Endocrinol (Lausanne) (2020) 11:585285. doi: 10.3389/fendo.2020.585285
102. Guo C, Ma R, Liu X, Chen T, Li Y, Yu Y, et al. Silica Nanoparticles Promote oxLDL-Induced Macrophage Lipid Accumulation and Apoptosis via Endoplasmic Reticulum Stress Signaling. Sci Total Environ (2018) 631–2:570–9. doi: 10.1016/j.scitotenv.2018.02.312
103. Yu H, Guo P, Xie X, Wang Y, Chen G. Ferroptosis, a New Form of Cell Death, and Its Relationships With Tumourous Diseases. J Cell Mol Med (2017) 21(4):648–57. doi: 10.1111/jcmm.13008
104. Simion V, Zhou H, Haemmig S, Pierce JB, Mendes S, Tesmenitsky Y, et al. A Macrophage-Specific lncRNA Regulates Apoptosis and Atherosclerosis by Tethering HuR in the Nucleus. Nat Commun (2020) 11(1):6135. doi: 10.1038/s41467-020-19664-2
105. Zhang N, Zhu L, Wu X, Yan R, Yang S, Jiang X, et al. The Regulation of Ero1-Alpha in Homocysteine-Induced Macrophage Apoptosis and Vulnerable Plaque Formation in Atherosclerosis. Atherosclerosis (2021) 334:39–47. doi: 10.1016/j.atherosclerosis.2021.08.015
106. Sinha SK, Miikeda A, Fouladian Z, Mehrabian M, Edillor C, Shih D, et al. Local M-CSF (Macrophage Colony-Stimulating Factor) Expression Regulates Macrophage Proliferation and Apoptosis in Atherosclerosis. Arterioscler Thromb Vasc Biol (2021) 41(1):220–33.
107. Proto JD, Doran AC, Gusarova G, Yurdagul A Jr, Sozen E, Subramanian M, et al. Regulatory T Cells Promote Macrophage Efferocytosis During Inflammation Resolution. Immunity (2018) 49(4):666–77.e6. doi: 10.1016/j.immuni.2018.07.015
108. Coornaert I, Hofmans S, Devisscher L, Augustyns K, Van Der Veken P, De Meyer GRY, et al. Novel Drug Discovery Strategies for Atherosclerosis That Target Necrosis and Necroptosis. Expert Opin Drug Discov (2018) 13(6):477–88. doi: 10.1080/17460441.2018.1457644
109. Back M, Yurdagul A Jr, Tabas I, Öörni K, Kovanen PT. Inflammation and Its Resolution in Atherosclerosis: Mediators and Therapeutic Opportunities. Nat Rev Cardiol (2019) 16(7):389–406. doi: 10.1038/s41569-019-0169-2
110. Chen J, Kos R, Garssen J, Redegeld F. Molecular Insights Into the Mechanism of Necroptosis: The Necrosome As a Potential Therapeutic Target. Cells (2019) 8(12). doi: 10.3390/cells8121486
111. Mena HA, Spite M. Proresolving Receptor Tames Inflammation in Atherosclerosis. J Clin Invest (2021) 131(24). doi: 10.1172/JCI155240
112. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
113. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell (2019) 35(6):830–49. doi: 10.1016/j.ccell.2019.04.002
114. Emmons MF, Smalley KSM. Ironing-Out the Details: New Strategies for Combining Ferroptosis Inhibitors With Immunotherapy in Melanoma. J Invest Dermatol (2022) 142(1):18–20. doi: 10.1016/j.jid.2021.06.014
115. Ouyang S, You J, Zhi C, Li P, Lin X, Tan X, et al. Ferroptosis: The Potential Value Target in Atherosclerosis. Cell Death Dis (2021) 12(8):782. doi: 10.1038/s41419-021-04054-3
116. Martinet W, Coornaert I, Puylaert P, De Meyer GRY. Macrophage Death as a Pharmacological Target in Atherosclerosis. Front Pharmacol (2019) 10:306. doi: 10.3389/fphar.2019.00306
117. Mao H, Zhao Y, Li H, Lei L. Ferroptosis as an Emerging Target in Inflammatory Diseases. Prog Biophys Mol Biol (2020) 155:20–8. doi: 10.1016/j.pbiomolbio.2020.04.001
118. Le Y, Zhang Z, Wang C, Lu D. Ferroptotic Cell Death: New Regulatory Mechanisms for Metabolic Diseases. Endocr Metab Immune Disord Drug Targets (2021) 21(5):785–800. doi: 10.2174/1871530320666200731175328
119. Stockwell BR, Jiang X, Gu W. Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biol (2020) 30(6):478–90. doi: 10.1016/j.tcb.2020.02.009
120. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 Is a Glutathione-Independent Ferroptosis Suppressor. Nature (2019) 575(7784):693–8. doi: 10.1038/s41586-019-1707-0
121. Xiao L, Luo G, Guo X, Jiang C, Zeng H, Zhou F, et al. Macrophage Iron Retention Aggravates Atherosclerosis: Evidence for the Role of Autocrine Formation of Hepcidin in Plaque Macrophages. Biochim Biophys Acta Mol Cell Biol Lipids (2020) 1865(2):158531. doi: 10.1016/j.bbalip.2019.158531
122. Guo Z, Ran Q, Roberts LJ 2nd, Zhou L, Richardson A, Sharan C, et al. Suppression of Atherogenesis by Overexpression of Glutathione Peroxidase-4 in Apolipoprotein E-Deficient Mice. Free Radic Biol Med (2008) 44(3):343–52. doi: 10.1016/j.freeradbiomed.2007.09.009
123. Ye L, Jin F, Kumar SK, Dai Y. The Mechanisms and Therapeutic Targets of Ferroptosis in Cancer. Expert Opin Ther Targets (2021) 25(11):965–86. doi: 10.1080/14728222.2021.2011206
124. Santoro MM. The Antioxidant Role of Non-Mitochondrial CoQ10: Mystery Solved! Cell Metab (2020) 31(1):13–5. doi: 10.1016/j.cmet.2019.12.007
125. Orlando P, Silvestri S, Galeazzi R, Antonicelli R, Marcheggiani F, Cirilli I, et al. Effect of Ubiquinol Supplementation on Biochemical and Oxidative Stress Indexes After Intense Exercise in Young Athletes. Redox Rep (2018) 23(1):136–45. doi: 10.1080/13510002.2018.1472924
126. Evans DR, Guy HI. Mammalian Pyrimidine Biosynthesis: Fresh Insights Into an Ancient Pathway. J Biol Chem (2004) 279(32):33035–8. doi: 10.1074/jbc.R400007200
127. Grootaert MOJ, Roth L, Schrijvers DM, De Meyer GRY, Martinet W. Defective Autophagy in Atherosclerosis: To Die or to Senesce? Oxid Med Cell Longev (2018) 2018:7687083. doi: 10.1155/2018/7687083
128. Cao H, Jia Q, Yan L, Chen C, Xing S, Shen D. Quercetin Suppresses the Progression of Atherosclerosis by Regulating MST1-Mediated Autophagy in Ox-LDL-Induced RAW264.7 Macrophage Foam Cells. Int J Mol Sci (2019) 20(23). doi: 10.3390/ijms20236093
129. Robichaud S, Fairman G, Vijithakumar V, Mak E, Cook DP, Pelletier AR, et al. Identification of Novel Lipid Droplet Factors That Regulate Lipophagy and Cholesterol Efflux in Macrophage Foam Cells. Autophagy (2021) 17(11):3671–89. doi: 10.1080/15548627.2021.1886839
130. He X, Fan X, Bai B, Lu N, Zhang S, Zhang L. Pyroptosis is a Critical Immune-Inflammatory Response Involved in Atherosclerosis. Pharmacol Res (2021) 165:105447. doi: 10.1016/j.phrs.2021.105447
131. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-Canonical Inflammasome Activation Targets Caspase-11. Nature (2011) 479(7371):117–21. doi: 10.1038/nature10558
132. Shi X, Xie WL, Kong WW, Chen D, Qu P. Expression of the NLRP3 Inflammasome in Carotid Atherosclerosis. J Stroke Cerebrovasc Dis (2015) 24(11):2455–66. doi: 10.1016/j.jstrokecerebrovasdis.2015.03.024
133. Burdette BE, Esparza AN, Zhu H, Wang S. Gasdermin D in Pyroptosis. Acta Pharm Sin B (2021) 11(9):2768–82. doi: 10.1016/j.apsb.2021.02.006
134. Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, et al. Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe (2018) 24(1):97–108.e4. doi: 10.1016/j.chom.2018.05.009
135. Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, et al. Redox Lipid Reprogramming Commands Susceptibility of Macrophages and Microglia to Ferroptotic Death. Nat Chem Biol (2020) 16(3):278–90. doi: 10.1038/s41589-019-0462-8
Keywords: atherosclerosis prevention, plaque formation, inflammation, macrophage polarization, macrophage death
Citation: Li H, Cao Z, Wang L, Liu C, Lin H, Tang Y and Yao P (2022) Macrophage Subsets and Death Are Responsible for Atherosclerotic Plaque Formation. Front. Immunol. 13:843712. doi: 10.3389/fimmu.2022.843712
Received: 26 December 2021; Accepted: 17 February 2022;
Published: 30 March 2022.
Edited by:
Huaizhu Wu, Baylor College of Medicine, United StatesReviewed by:
Xueying Peng, Hangzhou First People’s Hospital, ChinaCopyright © 2022 Li, Cao, Wang, Liu, Lin, Tang and Yao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping Yao, eWFvcGluZ0BtYWlscy50am11LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.