 Ata Abbasi1,2
Ata Abbasi1,2 Chixiang Chen3†
Chixiang Chen3† Chintan K. Gandhi4
Chintan K. Gandhi4 Rongling Wu3
Rongling Wu3 Annie Pardo5
Annie Pardo5 Moises Selman6
Moises Selman6 Joanna Floros4,7*
Joanna Floros4,7*- 1Cellular and Molecular Research Center, Cellular and Molecular Medicine Institute, Urmia University of Medical Sciences, Urmia, Iran
- 2Department of Pathology, Faculty of Medicine, Urmia University of Medical Sciences, Urmia, Iran
- 3Department of Public Health Science, Pennsylvania State University College of Medicine, Hershey, PA, United States
- 4Department of Pediatrics, Pennsylvania State University College of Medicine, Hershey, PA, United States
- 5Facultad de Ciencias, Universidad Nacional Autónoma de México, Mexico City, Mexico
- 6Unidad de Investigación, Instituto Nacional de Enfermedades Respiratorias “Ismael Cosio Villegas”, Mexico City, Mexico
- 7Department of Obstetrics & Gynecology, Pennsylvania State University College of Medicine, Hershey, PA, United States
Surfactant proteins (SPs) are important for normal lung function and innate immunity of the lungs and their genes have been identified with significant genetic variability. Changes in quantity or quality of SPs due to genetic mutations or natural genetic variability may alter their functions and contribute to the host susceptibility for particular diseases. Alternatively, SP single nucleotide polymorphisms (SNPs) can serve as markers to identify disease risk or response to therapies, as shown for other genes in a number of other studies. In the current study, we evaluated associations of SFTP SNPs with idiopathic pulmonary fibrosis (IPF) by studying novel computational models where the epistatic effects (dominant, additive, recessive) of SNP-SNP interactions could be evaluated, and then compared the results with a previously published hypersensitivity pneumonitis (HP) study where the same novel models were used. Mexican Hispanic patients (IPF=84 & HP=75) and 194 healthy control individuals were evaluated. The goal was to identify SP SNPs and SNP-SNP interactions that associate with IPF as well as SNPs and interactions that may be unique to each of these interstitial diseases or common between them. We observed: 1) in terms of IPF, i) three single SFTPA1 SNPs to associate with decreased IPF risk, ii) three SFTPA1 haplotypes to associate with increased IPF risk, and iii) a number of three-SNP interactions to associate with IPF susceptibility. 2) Comparison of IPF and HP, i) three SFTPA1 and one SFTPB SNP associated with decreased risk in IPF but increased risk in HP, and one SFTPA1 SNP associated with decreased risk in both IPF and HP, ii) a number of three-SNP interactions with the same or different effect pattern associated with IPF and/or HP susceptibility, iii) one of the three-SNP interactions that involved SNPs of SFTPA1, SFTPA2, and SFTPD, with the same effect pattern, was associated with a disease-specific outcome, a decreased and increased risk in HP and IPF, respectively. This is the first study that compares the SP gene variants in these two phenotypically similar diseases. Our findings indicate that SNPs of all SFTPs may play an important role in the genetic susceptibility to IPF and HP. Importantly, IPF and HP share some SP genetic variants, suggesting common pathophysiological mechanisms and pathways regarding surfactant biogenesis, but also some differences, highlighting the diverse underlying pathogenic mechanisms between an inflammatory-driven fibrosis (HP) and an epithelial-driven fibrosis (IPF). Alternatively, the significant SNPs identified here, along with SNPs of other genes, could serve as markers to distinguish these two devastating diseases.
Introduction
Idiopathic pulmonary fibrosis (IPF) is one of the most common interstitial diseases of unknown etiology and poor prognosis (1, 2). It is characterized by aberrant activation of the lung epithelium which provokes the increase and activation of the fibroblasts population that finally leads to the replacement of lung parenchyma with destructive fibrotic bundles leading to respiratory failure and death (3, 4). IPF is a chronic, progressive, irreversible, and usually lethal disease of middle-aged and elderly patients (5, 6). Multiple efforts on different aspects to find out the etiology of this disease have been made but with little progress (3, 7–10). The clinical course of IPF is heterogeneous and considerable overlap exists in presentation of IPF and chronic hypersensitivity pneumonitis (HP) (11).
Hypersensitivity pneumonitis (HP) is another type of interstitial lung disease caused by an exaggerated immune response to environmental antigens, such as fugal, bacterial or bird proteins (12). Although antigens that cause HP have been identified and are distributed worldwide, only a very small percent of the world population gets affected and the distribution among different nations is not similar (12). In Mexico, the pigeon breeder’s disease is the most common type of HP, caused by proteins from avian serum, feces, and feathers (12).
IPF and HP are different diseases with different etiologies. Validated risk factors for IPF include mainly aging and smoking; exposure to metal dust, wood dust, pesticide, and occupational history of farming or agriculture also increased the risk of IPF. Likewise, the risk of HP is associated mainly to the exposure to organic particles and varies with regional disparities in climate, occupational exposures, and environmental exposures, but not ethnicity. Respiratory viral infections, and high pesticide exposure, have been revealed as risk factors. Paradoxically, cigarette smoke reduces the risk of HP, but when smokers develop HP, they often follow a chronic fibro-proliferative course. However, there is a considerable overlap in pathophysiology and clinical presentation (6, 12–14). In fact, a previous study has shown that almost half of IPF patients, were, subsequently, diagnosed with chronic fibrotic HP, and often the clinical, functional and radiological behavior of both diseases are indistinguishable (13, 14). In both diseases, several members of the same family may be affected, indicating the role of genetic factors in the pathogenesis and progression of these diseases (15, 16). However, the interplay of genetics, environmental factors, and perhaps other factors is poorly understood.
Pulmonary surfactant and surfactant proteins (SPs), have been shown to play roles in host-defense functions, i.e. regulation of pro-inflammatory cytokine production, chemotaxis, and tissue repair (17–20), and in surfactant-related functions, i.e. lowering surface tension and stabilizing the alveoli. Hence, derangement in functional ability, structure, and/or levels of SPs (SP-A, SP-B, SP-C, SP-D) may contribute to the development of interstitial lung diseases, such as IPF and HP (21, 22). Alternatively, genetic polymorphisms of these important molecules may serve, along with other gene variants, as markers to distinguish these two diseases that share overlapping pathology and clinical presentation. Being able to distinguish these two diseases early on is of great importance. In this context, it has been recently reported a diagnostic algorithm which allow a better differential diagnosis between fibrotic HP and IPF (23). Accurate diagnosis is essential for appropriate treatment and management of these diseases. Currently, Nintedanib and pirfenidone, inhibitors of fibrosis pathways, are the treatments of choice for IPF (24). In contrast, HP treatment is based on corticosteroids and immunosuppressive therapy and importantly, administration of corticosteroids and immunosuppressants in some of the IPF patients can worsen their clinical condition, primarily those with severe telomere shortening, causing adverse effects and increased mortality (25). A better understanding of the pathobiology of fibrotic HP and of IPF as well as the genetic and molecular differences between them might help to identify biomarkers that allow distinguishing those patients who would benefit from antigen-exposure avoidance and immunosuppression (i.e., HP), from those with IPF in which the use of these drugs is not only unhelpful but increases the risk of hospitalization and death (12, 23, 25–27). There have been numerous examples in the literature where specific SNPs have been associated with disease susceptibility (28) or drug-response in certain disease populations (27, 29–31). For example, as reviewed and analyzed elsewhere (32) genotypes of molecules carrying certain SNPs were associated with response rates after imatinib treatment in patients with chronic myeloid leukemia. Furthermore, surfactant protein A2 genetic variants and genotypes of the donor lung have been associated with post-transplant clinical outcome i.e., survival in lung transplant patients (33).
The human SP-A is encoded by two similar genes, SFTPA1 and SFTPA2, located on chromosome 10 (19, 34–36). Several genetic polymorphisms of each SFTPA gene, have been identified and characterized, and these are found with different frequencies in the general population (18, 19, 37–39). SP-A plays an important role in both innate lung host defense and surfactant-related processes (40) and the human variants may differentially affect these processes (19). SP-B, SP-C, and SP-D are each encoded by a single gene, SFTPB, SFTPC, and SFTPD, respectively (41), and several polymorphisms have been described for each of these genes (42–44). SP-D also plays a role in lung host defense and the primary role of SP-B and SP-C is in surfactant-related functions/processes. Furthermore, multiple studies showed associations of SPs genetic variants with various acute and chronic lung diseases including acute respiratory distress syndrome (ARDS) (43, 44), chronic obstructive pulmonary disease (COPD) (45, 46), cystic fibrosis (47), and other (44).
In a previous study, we showed associations of SP single nucleotide polymorphisms (SNPs) with IPF in a Mexican population (22). In addition, a number of studies identified significant associations between SP genetic variants or mutations and IPF, pointing to a potential role of SPs in the pathogenesis or specific processes of IPF (1, 7, 10, 48–52). Furthermore, to date many gene polymorphisms are known to be present in both IPF and HP interstitial lung disorders. Common genetic variants in TERC, DSP, MUC5B, ATP11A, FAM13A, and IVD described initially as risk gene factors for IPF also represent risk variants for HP, especially for the fibrotic HP (53, 54). Likewise, rare variants in telomere-related genes, such as TERT, TERC, DKC1, RTEL1, and PARN, have been identified in familial and sporadic IPF, and recently the same mutations were identified in patients with HP (55, 56).
In the present study, we used a statistical approach (57) where novel statistical models were studied to enable investigation of the epistatic effects of SNP-SNP interactions as has been done for other pulmonary diseases (21, 47, 58, 59). Using this novel approach, we reanalyzed IPF data that were previously published. The published study used regression analysis nearly 20 years ago (22). Next we compared these newly analyzed IPF data with our recently published HP data where the same novel statistical models were investigated (21). This comparison study is the first such study where SP variants were compared between IPF and HP. The objectives of the current study are: a) to re-analyze the existing IPF database (22), using the Wang’s newer analytical approach (57), to gain further insight into complex SNP-SNP interactions, and b) to compare the IPF and HP data of SNP-SNP interactions, from the current IPF study and our recently HP published work (21), to identify unique interactions to each disease. The rationale for the latter is that both IPF and HP are members of the ILD family and some unique interactions may help to differentiate these two diseases, while shared interactions may reveal some common pathways.
Methods
Study Population
The IPF and HP study population are the same as the ones described before (21, 22). Briefly, eighty-four and seventy-five unrelated patients were enrolled in the IPF and HP study groups, respectively. The diagnoses of both diseases were supported by clinical observation, pulmonary function, high-resolution computed tomography, and bronchoalveolar lavage findings (4, 12). In some cases, surgical biopsy was done to confirm the diagnosis. Patients with the diagnosis of other interstitial lung disease were excluded. All enrolled HP patients were classified to have a fibrotic HP (4). For both studies, one hundred and ninety-four healthy individuals served as controls. All study members were Hispanic Mexican individuals and enrolled at the National Institute of Respiratory Diseases in Mexico City. The protocol was approved by the Ethics Committee. The demographics and clinical characteristics of the study groups are shown in Table 1. Blood samples from enrolled subjects were collected and the genotyping was done using the PCR-RFLP method as described in the previous studies (21, 22).
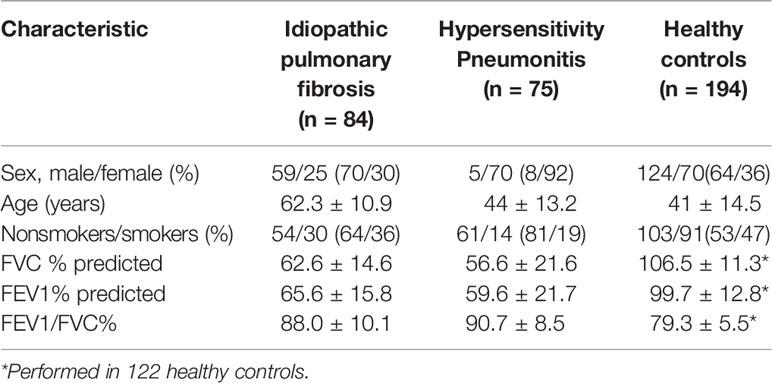
Table 1 Demographics and clinical characteristics of the study groups.
Surfactant Protein Genes and Single Nucleotide Polymorphisms
We evaluated, as shown in Table 2, 17 SNPs of the SP genes using the single SNP model and the two and three-SNP-SNP interaction models described by Wang et al. (57). These included five from SFTPA1, four from SFTPA2, four from SFTPB, two from SFTPC, and two from SFTPD.

Table 2 SNPs of each surfactant protein gene analyzed in this study.
Statistical Analysis
As mentioned previously (57), we used a SNP–SNP interaction method in a case-control setting to study associations of SP genes SNPs with IPF. Wang et al. developed a computational model for detecting additive, dominant and epistatic effects by integrating quantitative genetic theory into a case-control design context (57). This approach integrates the principle of quantitative genetics, and decomposes the overall genetic effect of each SNP into different components: additive (a), dominant (d), and recessive (r) and can characterize high-order epistatic interactions. For example, consider two genes A and B, which may have four types of epistasis, additive x additive, additive x dominant, dominant x additive, dominant x dominant. These four types function differently to affect disease risk. For example, if a x a is important, this means that the two-marker genotype AABB (homozygote AA at gene A and homozygote BB at gene B) performs differently from the two-marker genotype AAbb (homozygote AA at gene A and homozygote bb at gene B). However, if d x d is significant, this means that double heterozygote AaBb performs differently from the other genotypes. Therefore, it is important to distinguish these four types of epistasis.
This SNP-SNP interaction approach (57) has been used and validated in studying associations of high order epistatic interactions with various acute and chronic pulmonary diseases (21, 47, 58, 59). The cases and controls were converted into a 2 × 2 contingency table and various types of epistatic interactions at different orders were tested. The p-value was adjusted for sex and smoking due to their modifying effects on both diseases. To account for multiple testing, false discovery rate (FDR) was set at 5%. The Cochran-Mantel-Haenszel test was used to calculate the Odds ratios (OR) with 95% of confidence interval (95%CI). All possible interactions for two- and three-SNP interaction models were tested and those with p value less than 0.05 are reported.
Results
IPF
Main Effect Analysis of the IPF Group (n = 84) vs the Control Group (n = 194)
In the present study, we observed significant differences in the studied groups with the single- and the three-SNP interaction model. We did not observe any significant SNP-SNP interactions in the two-SNP interaction model.
Single SNP Model
Three SNPs, rs1059047, rs1136450, and rs1059057 of SFTPA1, were each associated with decreased risk of IPF (OR: 0.42 to 0.46, p value: 0.008-0.010) (Table 3).
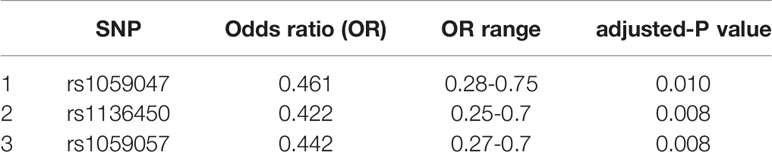
Table 3 IPF association with SFTPA1 single SNPs.
Association of SNP-SNP Interaction with IPF in the Three-SNP Interaction Model
As previously described, each SNP can have additive, dominant or recessive effect on the disease phenotype and these are noted as a, d, and r, respectively. In our analysis, each SNP had dominant or additive effects, and no recessive effect was observed (Supplementary Tables 1–6).
Comparison of the IPF Group (n = 84) vs the Control Group (n = 194)
We found a total of 277 significant SNP-SNP interactions associated with IPF in the three-SNP model. The interactions are shown in detail in Supplementary Tables 1–6. Out of these 277 interactions, we observed the following. First, a) 30 interactions with two additive effects and one dominant effect. Four of these exhibited the axdxa effect pattern and 26 the dxaxa; b) 121 interactions with two dominant and one additive effect, (dxdxa (n=65), axdxd (n=27), dxaxd (n=29)) and c) 126 interactions with three dominant effects (dxdxd). Second, 44 were among SNPs of hydrophilic SPs (SFTPA1, SFTPA2, SFTPD) alone, 14 were among hydrophobic SPs (SFTPB, SFTPC) alone, and the others were among SNPs of both the hydrophilic and hydrophobic SPs. Third, most interactions (n=196) were associated with increased risk of IPF (p=0.001-0.046, OR= 1.48-11.4) and the remaining (n=81) were associated with decreased risk of IPF (p=0.001- 0.049, OR= 0.12-0.69). Among the interactions with increased risk of IPF, 7 were in the same SP gene (intragenic). Of these, 6 were in SFTPA1 (4 with dxdxd, 1 with axdxd and 1 with dxdxa interactions effect) and 1 in SFTPB (with dxaxd interaction effect). Among the interactions with decreased risk of IPF, 2 were intragenic in the SFTPB with dxdxa interactions. No significant interactions were observed with recessive (r x r x r) or additive effects (a x a x a) only.
Overall, out of the 17 SNPs studied, 14 were found in SNP-SNP interactions shown to significantly associate with IPF in the three-SNP model. Of the 14 SNPs, six were SNPs of SFTPA (4 SFTPA1 and 2 SFTPA2), 4 of SFTPB, 2 of SFTPC and 2 of SFTPD. Although all of these SNPs were frequently present in the significant SNP-SNP interactions, the rs1059046 of SFTPA2 encoding amino acid 9 ((AA9), Asn>Thr) was the most frequently present followed by SFTPC SNPs. The frequency of each of the 14 SNPs found in significant SNP-SNP interactions associated IPF are shown in Table 4.
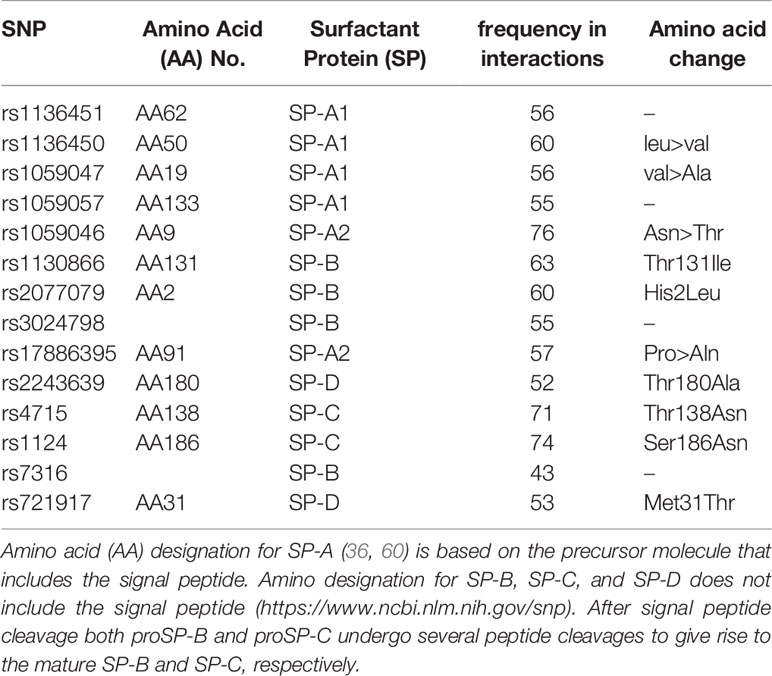
Table 4 Frequency of SNPs in significant SNP-SNP interactions.
Thirteen interactions had OR greater than 5 indicating a strong association with increased risk of IPF (Table 5). Among them one was an intragenic interaction of SFTPB SNPs (rs2077079, rs3024798, rs7316) and twelve interactions had two-SNPs with additive and one with dominant effect. In all but one, the SFTPA1 SNP (rs1136450, (AA50), leu>val) was the one with the dominant effect.
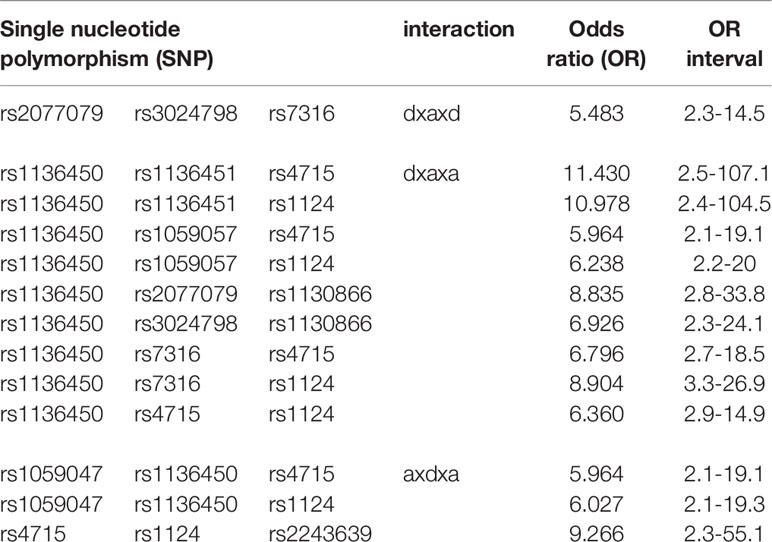
Table 5 SNP interactions associated with IPF with odds ratio higher than 5.
Haplotype Analysis
Haplotype analysis showed that three haplotypes TG (rs1059047 x rs1136450), GA (rs1136450 x rs1136451) and GG (rs1136451 x rs1059057) of the SFTPA1 were associated with increased risk of IPF (OR=2.372, p=0.004, OR=2.368, p=0.004 and OR=2.265, p=0.004, respectively) and all exhibited a dominant effect (Table 6 and Figure 1). The dominant effect of, for example, the TG haplotype displays a higher risk of IPF compared to the combination of non-risk haplotypes (CC, TT, GG). This is also true for haplotypes GA and GG of SFTPA1.
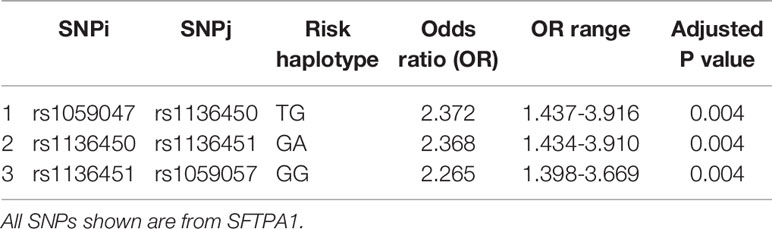
Table 6 Haplotype association with IPF susceptibility.
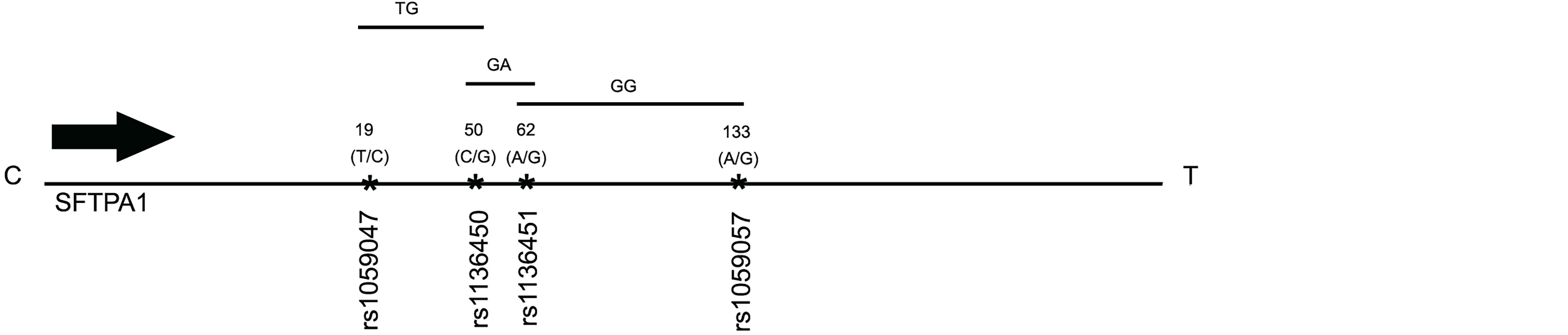
Figure 1 A schematic presentation of the SFTPA1 gene SNPs is shown. The relative location of the gene is shown from centromere (C) to telomere (T). The numbers above the solid black line indicate the amino acid number with the corresponding nucleotide change shown in parenthesis. The SNP id is noted below the black line. The arrow indicates transcriptional orientation. The transmitted haplotypes (TG, GA, and GG) are shown in a two-SNP model and are associated with increased risk of IPF (OR: OR = 2.372, p = 0.004, OR = 2.368, p = 0.004 and OR = 2.265, p = 0.004, respectively). The TG haplotype is constituted by the rs1059047 Val (T) at codon 19 and the rs1136450 Val(G) at codon 50, the GA haplotype by the rs1136450 Val(G) at codon 50 and the rs1136451 Prol(A) at codon 62 and the GG by the rs1136451 Prol(G) at codon 62 and the rs1059057 Thr(G) at codon 133. Of note, the T/C and C/G alleles change the encoded amino acids at codons 19 Val/Ala and 50 Val/Leu, respectively, but the SNPs at codons 62 and 133 do not change the encoding amino acids (19).
Comparison Between IPF (n = 84) and HP (n = 75) Using the Same Control Group (n = 194)
Comparison of SNP Associations in the Single-SNP Model
As shown in Figure 2, the rs1136450 of the SFTPA1 was associated with a decreased risk for both diseases. The rs1059047 and the rs1059057 of the SFTPA1 were associated with decreased risk of IPF, whereas the rs1136451 of the SFTPA1 was associated with increased risk of HP. The rs1130866 of the SFTPB was associated with decreased risk of HP.
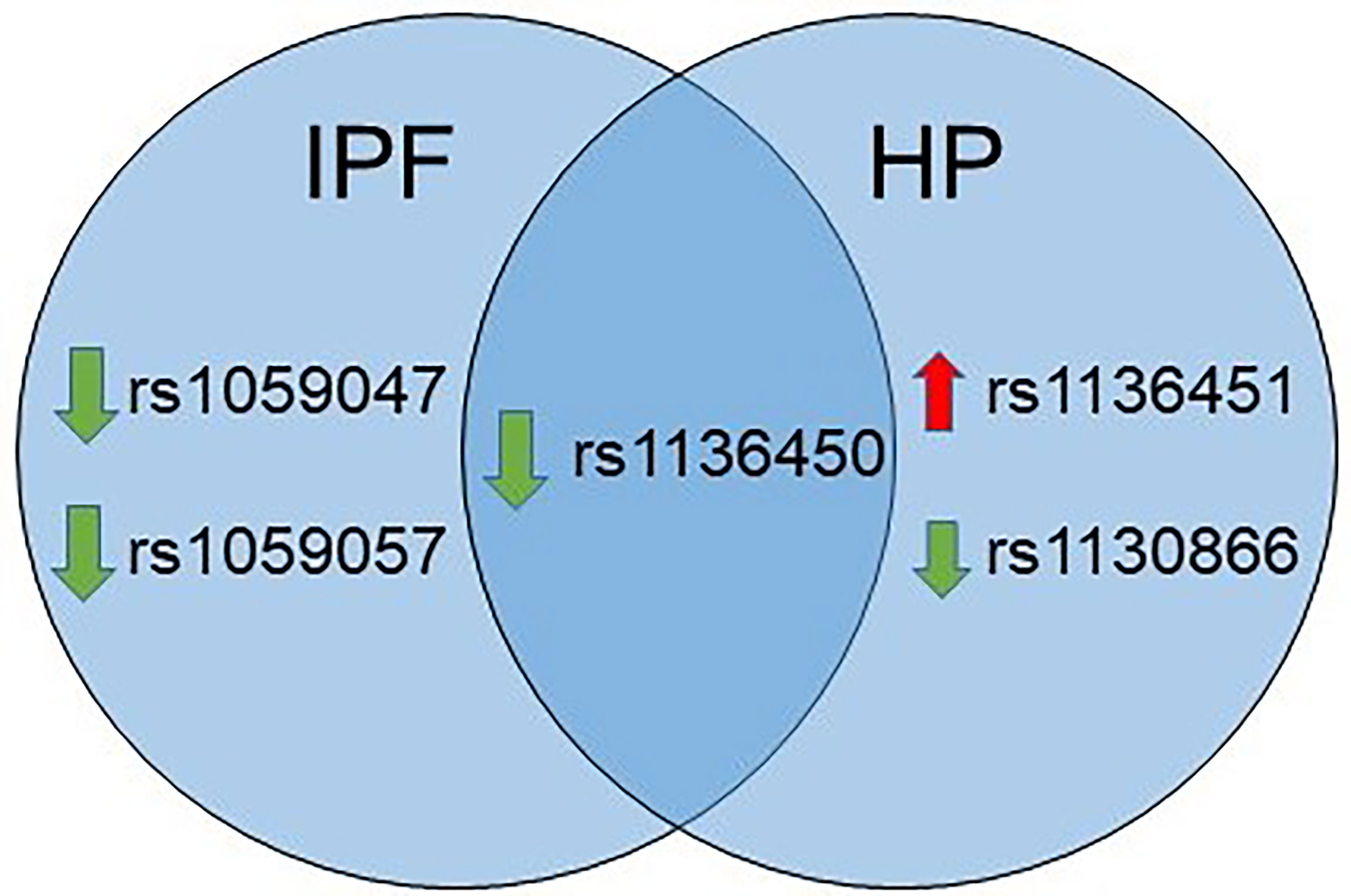
Figure 2 Venn diagram showing similarities and differences in association of SNPs with IPF and HP in a single SNP model. The green and red arrows besides SNPs show association of a given SNP with decreased and increased risk, respectively.
Common SNP-SNP Interactions Between IPF and HP After Comparison to the Same Control Group (n = 194) in the Three SNP Model
We studied the common SNP-SNP interactions in the three SNP model between the 277 interactions resulting from the comparison of the IPF group vs the control group (present study) and the 97 interactions resulting from the comparison of the HP group vs the control group in the three SNP model from our previous study (21). Of the 227 interactions in IPF comparison, as noted above, 81 interactions were associated with decreased risk of IPF, and the remaining 196 with increased risk of the disease. Of the 97 HP interactions, 68 and 29 were associated with decreased and increased risk of HP, respectively. However, in a large number of SNP-SNP interactions, the same SNPs were involved in IPF and HP. These SNP interactions exhibited either the same effect pattern between the two groups, IPF and HP, for example dxdxa (Table 7) or a different effect pattern between the two group comparisons i.e., dxdxa vs dxaxd (Table 8).
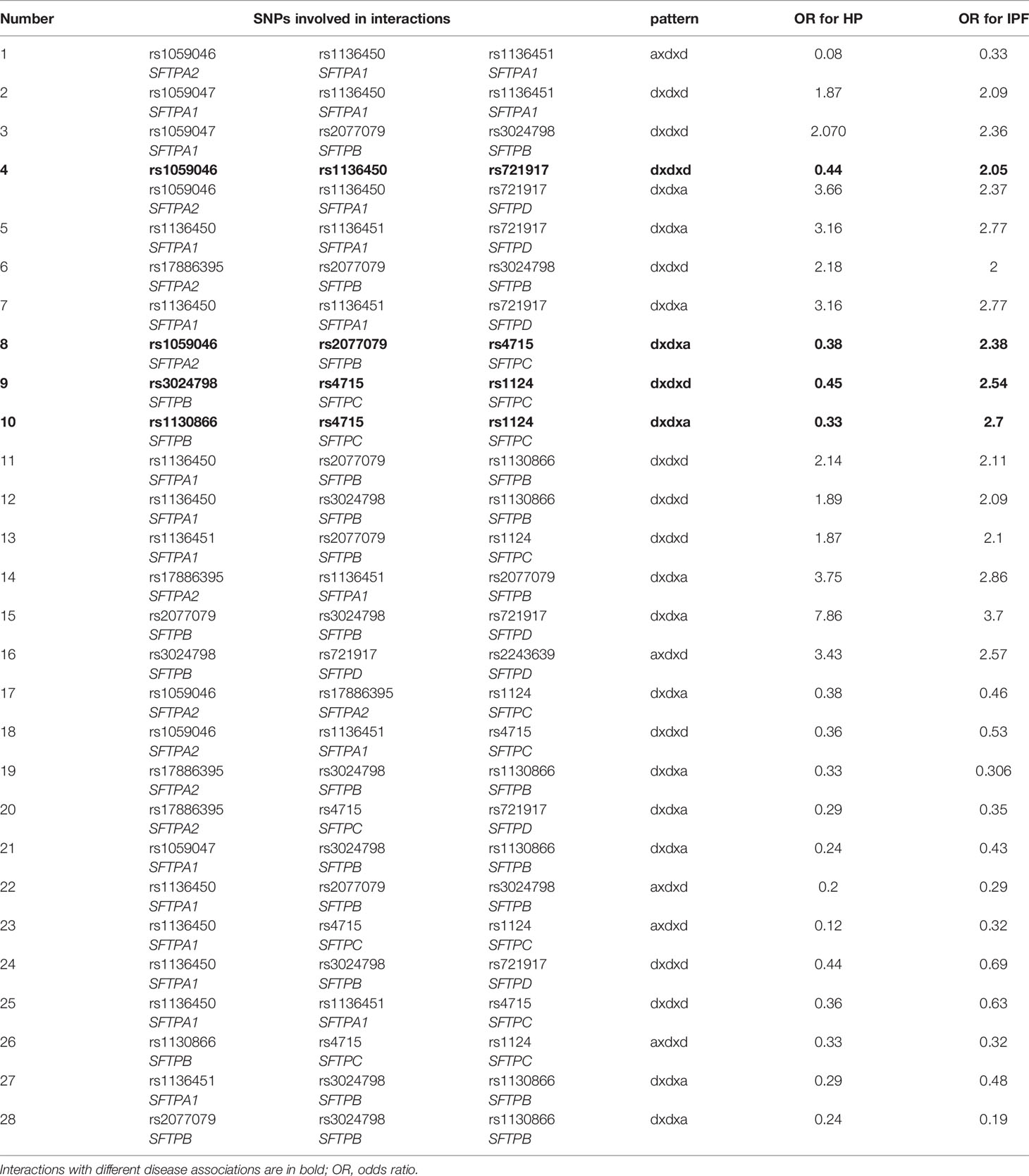
Table 7 Interactions with the same SNPs and the same pattern effect in IPF and HP patients.
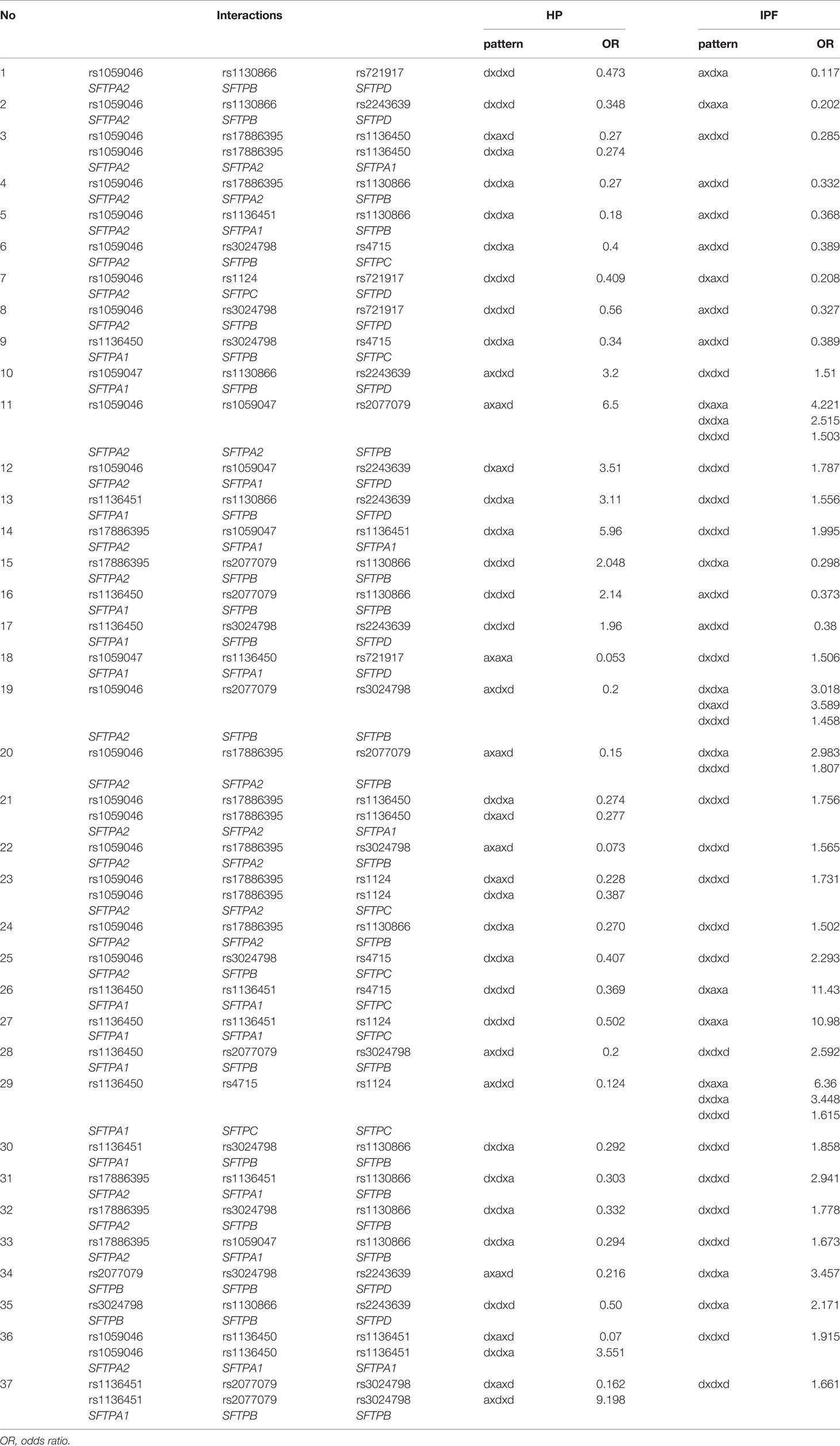
Table 8 Interactions with the same SNPs and different effect pattern in IPF and HP patients.
Same SNPs and Same Pattern Effect in IPF and HP
We observed 28 interactions that involved the same SNPs (n=28) that exhibited the same effect pattern (n=29); Interaction #4 (Table 7) exhibited two different effect patterns. Of the 28 interactions, 13 of them were associated with decreased risk in both diseases and 12 of them with increased risk in both diseases. However, 4 interactions shown in bold in Table 7, involving the same SNPs and exhibiting the same pattern effect were associated with a decreased risk in HP and an increased risk in IPF indicating disease specificity. Moreover, one of these four interactions (rs1059046, rs1136450, rs721917 of SFTPA2, SFTPA1, SFTPD, respectively), noted as #4 in Table 7, exhibited two different patterns (dxdxd, dxdxa) in both IPF and HP, with the dxdxd (shown in bold) pattern being associated with an outcome that seemed to be disease-specific i.e., with a decreased risk in HP and an increased risk in IPF.
Same SNPs and Different Pattern Effect in IPF and HP
The interactions with the same SNPs (n=37) but with different pattern effects exhibited altered susceptibility in the two diseases and are shown in Table 8. For example, interactions #26 and #27, rs1136450 x rs1136451 x rs4715 and rs1136450 x rs1136451 x rs1124 (Table 8) with the dxaxa pattern were strongly associated with IPF (OR=11.43, 10.98, respectively), but the same SNP interactions in the dxdxd pattern were both associated with low risk of HP (OR=0.36 and 0.5, respectively). The rs1136450 x rs4715 x rs1124 (interaction #29, Table 8) is another interaction which was associated with increased risk of IPF in the dxaxa, dxdxa and dxdxd patterns (OR=6.36, 3.44 and 1.61, respectively) but associated with decreased risk of IPF and HP in the axdxd pattern (interaction #23, Table 7).
Of note, rs1059057 and rs7316 were not present in any of the common SNP-SNP interactions.
Discussion
Surfactant proteins (SPs) are important in normal lung function and also in innate immunity of the lungs. Changes in quantity or quality of SPs due to genetic alterations may alter their functions, whether in host defense and/or surfactant-related processes, and contribute to the host susceptibility for particular diseases. SP-A, for example, plays an important role in innate host defense against various pathogens, irritants, and other (40) and its natural genetic variability may differentially affect these processes (19). In the current study, we evaluated associations of SFTP SNPs with IPF using a novel statistical method and compared the results of the current study with a previously published HP study (21). The goal here was to find SNPs and SNP-SNP interactions that are unique to each of these interstitial lung diseases or common between them. Importantly, IPF is an epithelial-driven fibrosis and is typically progressive, while HP is an immune inflammatory-driven disease, often evolving to fibrosis, and may also develop a progressive fibrotic phenotype. We observed, 1) in terms of IPF: i) three single SFTPA1 SNPs to associate with decreased IPF risk, ii) three SFTPA1 haplotypes to associate with increased IPF risk, and iii) a number of three-SNP interactions to associate with IPF susceptibility. 2) After comparison of IPF and HP, i) three SFTPA1 and one SFTPB SNP were found to associate with decreased or increased risk in IPF and HP and one SFTPA1 SNP to associate with decreased risk in both IPF and HP, ii) a number of three-SNP interactions with the same or different effect pattern associated with IPF and/or HP susceptibility; iii) one of the three-SNP interactions that involved SNPs of SFTPA2, SFTPA1, and SFTPD, with the same effect pattern, was associated with a disease-specific outcome, a decreased and increased risk in HP and IPF, respectively. The findings of the current study may provide an example to start differentiating these two diseases based on their genetic background.
For the current study, we took advantage of the same ethnic background of cases for both diseases (IPF and HP) and controls, i.e. All study subjects were Mexican Hispanic patients and all of them were enrolled from the same center. We hoped that this will help identify genetic patterns that in turn may enable distinguishing these two diseases or at the very least provide a paradigm for further work where genetic models could be used to find disease-specific markers.
IPF
The SFTPA1 gene is shown in the present study, via different analyses, including the use of a single SNP model, haplotype analysis and SNP-SNP interactions, to associate with IPF susceptibility. One of its SNPs, rs1136450, was the most frequently observed SFTPA1 SNP in the 3-SNP interactions. This SNP was found to be significant either by itself, as part of significant haplotypes, or part of significant 3-SNP interactions and has been found previously to associate with susceptibility with other pulmonary diseases including community acquired pneumonia (61), cystic fibrosis (47), acute respiratory distress of the newborn (59) and pediatric acute and chronic respiratory failure (58, 62). The rs1136450 SNP encodes (leu/val) amino acid 50 (AA50), and it is located within the collagen-like region of SP-A1. In the absence of the collagen-like region, functional defects of surfactant are observed as well as the absence of an extracellular structural form of surfactant, the tubular myelin (63). This region is also important for the oligomerization and stability of SP-A (64). This SNP change is SP-A1 variant-specific and not gene-specific. All of the most frequently found and studied SP-A1 variants (6A, 6A3, and 6A4) have a leucine at AA50 except the 6A2 variant that has a valine. On the other hand, all frequently found and studied SP-A2 variants have a valine (19, 38, 39). These two amino acids (Leu/Val), although they share a lot of similarities, as they are both essential non-polar amino acids with aliphatic side chains and neutral charge at pH 7.4, they may however provide differential sites for proteases. Proteases are often highly specific in their proteolytic activity, and even a conservative substitution as this one, provided by the rs1136450 SNP, may have a significant differential effect on the proteolylic process, as demonstrated in detailed studies for the matrix metalloproteinases family (65). Whether and how the AA50 change imparted by this SNP differentially affects any of the studied properties affected by the collagen-like region or provides preferential sites for various enzymes remains to be determined.
Of interest, a previous analysis of the same cohort using the traditional logistic regression analysis showed association of three SNPs of the SFTPA1, rs4253527 (AA219_T), rs1136450 (AA50_C), and rs1136451 (AA62_G) with increased risk of IPF (22). In the present study, the “G” allele of the rs1136450 SNP was associated with decreased risk of IPF. Although these findings appear to contradict one another, both studies do identify the same significant SNPs. Also, this comparison provides an opportunity for a cautionary note where one needs to take into consideration the method of assigning, reference and alternate alleles of a given SNP. Wang et al.’s approach employed in the present study uses the dbSNP database of the NCBI (66) to assign the reference and the alternate alleles, whereas, in the logistic regression analysis, alleles were assigned based on the frequencies in the studied group, i.e., the more frequent allele in that study group was the reference allele. Furthermore, in the current study, we did not observe association of the rs4253527 (AA219_T) with IPF either in the single-SNP or in the three-SNP model. This may be due to the difference in statistical approaches used for the two studies. Although we did not observe association of rs1136451 in the single-SNP model either, this SNP was observed in several SNP-SNP interactions (Table 1).
However, what is of considerable interest are the observations made regarding associations with relative risk or protection of IPF. In the single SNP model, the SFTPA1 SNP (rs1136450) and two other SFTPA1 SNPs (rs1059047, rs1059057) were shown to associate with decreased risk but haplotypes that included the rs1136450 and/or the other two SNPs were associated with increased risk. Moreover, a number of 3-SNP interactions, where the rs1136450 had a dominant effect and any other two SNPs had an additive effect, exhibited a high odds ratio indicating risk. However, when the rs1136450 in 3-SNP interactions was found with different effect patterns, the interactions were associated with either increased or decreased risk. An example to demonstrate this is depicted in the 3-SNP interactions, #9 and #28 in Table 8, where in the former the pattern effect is axdxd and in the latter is dxdxd, where the rs1136450 has additive (a) and dominant (d) effect, respectively. These different effect patterns of the rs113650, were associated with decreased (axdxd) and increased (dxdxd) risk of IPF, respectively. A similar effect pattern observation in 3-SNP interactions was made for the SFTPA2 rs1059046 (interactions, 3 & 21, 4 & 24, 6 & 25, Table 8). We postulate that the effect pattern, whether dominant (d) or additive (a) (no recessive effect was observed in this study), plays an important role in disease susceptibility. Although what contributes to a SNP to exhibit a different effect pattern is not entirely clear, we speculate that the overall cellular microenvironment, its interaction with other SNPs, and other unknown factors contribute to the specific effect pattern and its consequences, as it may be assessed by the strength of the given association with disease susceptibility. For example, different effect patterns among the same SNPs were observed, where each was associated with varying degree of IPF susceptibility as assessed by the odds ratio. Interaction #11 (Table 8) exemplifies this point. Three different effect patterns observed and each was associated with increased IPF risk albeit each exhibited a different odds ratio (range 1.5-4.2). These observations collectively indicate that fully understanding SNP-SNP interactions that may alter the risk of an individual to a disease, is a challenging problem (67, 68) due to the phenomenon of epistasis where the combined effect of one or more genes/SNPs on the phenotype could not have been predicted by their separate effects (69). Thus, this type of observations requires further consideration.
The central pathogenesis path of IPF is a progressive deposition of fibrotic tissue in the lungs secondary to dysregulated activity of the alveolar epithelium to repeated injury (7). Actually, mutations in genes encoding surfactant proteins, have been identified in adults with the phenotype of pulmonary fibrosis through a gain-of-toxic function mechanism (70). In the present study, we show that the SFTP genes play a role in IPF as shown by the numerous 3-SNP significant interactions that included SNPs of all the SFTP genes. Although, single SNPs or haplotypes of SFTPA1 exhibited significant association with IPF susceptibility, no SNPs of other SFTP genes showed any significant associations. Considering the vital role of SP-A in innate immunity and host responses of the lung to foreign particles, these findings are not surprising. Of interest, SFTPA1, compared to SFTPA2, has been shown to be more efficient in surfactant lipid reorganization and in preventing surfactant inhibition by serum proteins, indicating that the importance of this gene in IPF may in part be due to its dual role in host defense and surfactant-related activities. Because the 3-SNP interactions involved SNPs from all SFTPs, we postulate that collectively SFTP variants contribute to IPF in ways that we are not able, to yet fully understand. Further research is needed in studying the impact of the actual SNP-SNP interactions on levels and biophysical/biochemical properties of SPs in appropriate biological models. This may shed light into the complexities of their interactions and advance our understanding of these interactions on complex diseases, such as IPF.
Comparison of IPF and HP
When we compared findings of the current IPF study with a previously published HP study (21), using the same statistical approach, and cohort as control, a number of similarities and differences were identified between these interstitial lung diseases. Although one single SFTPA1 SNP (rs1136450), was found to associate with decreased risk in both diseases, other SFTPA1 SNPs were associated with IPF or HP disease-specific susceptibility. Thus, these point to a potential use of these SNPs as markers to distinguish between these two diseases. The SFTPA1 significant SNPs that change the encoded amino acid may affect functional aspects of the protein variant. The significance of the rs1136450 has been discussed above under IPF. The rs1059047 of SFTPA1 that associates with decreased risk in IPF, changes amino acid 19 (Val/Ala). This amino acid may or may not be part of all the mature SP-A1 molecules (60) and therefore it is unclear whether and how this may affect the processing of the precursor protein and/or the functional capability of the mature molecules that contain this amino acid.
Moreover, one SFTPB SNP (rs1130866) was associated specifically with decreased risk in HP. The SP-B protein plays a crucial role in surfactant function and some mutations are not compatible with life (71). The SFTPB SNP noted above is responsible for a missense codon (ACT/ATT) that changes amino acid 131 from a Threonine (ACT) to Isoleucine (ATT) and eliminates an N-linked glycosylation site (Asp129-Gln-Thr131) (72). Although amino acid 131 is part of the N-terminal peptide of the SP-B preprotein and not the mature SP-B, animal models have shown that the alleles of this SNP differentially affect the number of lamellar bodies, an extracellular structural form of surfactant, surface tension and levels of SP-B (73). Whether this SNP modifies any surfactant properties in humans and whether the consequences of these contribute to HP susceptibility is unknown. Its location is near a mutation hotspot and whether this, under certain circumstances, impacts other events is currently unknown (74).
Furthermore, we observed four 3-SNP interactions to exhibit a disease specific outcome. These interactions involved the same SNPs with similar effect patterns and associated with increased and decreased risk of IPF and HP, respectively (Table 7). These interactions involved SNPs from all SFTPs, with two of these (interactions #9 and 10) containing SNPs only from the hydrophobic surfactant proteins (SFTPB and SFTPC), one (interaction #4) contained SNPs only from the hydrophilic proteins (SFTPA2, SFTPA1, SFTPD) and one (interaction #8) contained SNPs from both groups (SFTPA2, SFTPB, SFTPC). Collectively, these indicate a role of all the surfactant proteins in the susceptibility of these diseases. Furthermore, the disease-specific lung microenvironment may alter the susceptibility of the host (75) and thus SNP interactions of the same SNPs with the same effect pattern may lead to different outcomes, as shown with the 3-SNP interactions in the present study. These interactions indicate that is plausible to distinguish the two similar interstitial lung diseases (IPF and HP), based on genetic interactions. Furthermore, we observed a number of 3-SNP interactions, with the same SNPs but with different effect patterns, to associate with IPF and HP disease susceptibility (Table 8). These observations are difficult to understand at the current time, as there may be a nonlinear relationship (dominant) between the gene products and disease outcomes, due perhaps to the gene dosage/imbalance and other sources of “more than-additive genetic interactions” that may lead to variable phenotypes of either over-, under- or non-function of the gene products in a given disease state (76).
The strengths of the current study are: a) the use of a newer statistical analysis to study SNP-SNP interactions with adjustment of important variables such as age, sex, and smoking status, b) enrollment of a patient population and control study groups with a similar ethnic background from the same center; therefore, the population structure of the studied groups may not be a major issue. Although, the second strength maybe a limitation (i.e., whether it can be generalized to include other groups of a different race or ethnicity), to our current knowledge ethnicity is not a significant risk factor as both diseases occur similarly in different ethnic groups as reported worldwide. However, there is limited information to determine whether differences in the frequency of the surfactant protein genetic variants, epigenetics, or differences in the SNP-SNP effect patterns studied here exist among different groups that could potentially point to different underlying processes in the pathogenesis of these disease. Thus, it is prudent to replicate the present findings in a larger sample size by case control studies with clinically confirmed cases of IPF and HP of other groups of heterogeneous non-Hispanic patients from various ethnicities in order to validate and strengthen the differential diagnostic potential of the identified SNPs and SNP interactions.
In conclusion, SNPs of all SFTPs appear to play an important role in identifying disease susceptibility in IPF and HP interstitial lung diseases. Similarities between these two diseases with regards to the surfactant protein genes variants were observed in haplotypes or in 3-SNP interactions, as well as differences highlighting the different underlying pathogenic mechanisms between an inflammatory-driven fibrosis (HP) and an epithelial-driven fibrosis (IPF). In addition, the disease-specific associations of the SP polymorphisms hold the potential for these SNPs to be used as markers to distinguish between these two diseases. The information obtained in the current study was enabled via the use of newer/novel statistical methods to study models of single SNPs as well as SNP-SNP interactions where epistasis could be addressed. SFTPA1 and SFTPB SNPs might be of particular interest for future studies, where these could be used as markers either individually or together with other biomarkers in an attempt to distinguish between these two similar diseases. This is of great importance as it could help downstream decision-making of diagnosis and disease-specific therapies of these two devastating diseases.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below:
dbSNP, accession number: 1063345.
Ethics Statement
The studies involving human participants were reviewed and approved by National Institute of Respiratory Diseases in Mexico City. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
AA analyzed the data and wrote the original draft of the manuscript. CC performed the statistical analysis. CG contributed to the manuscript writing. MS and AP were responsible for sample collection and patient screening as noted in previous publications and contributed in manuscript writing. RW worked closely with CC and guided the statistical analysis. JF supervised the entire project and worked closely with all co-authors and especially with AA to ensure quality of data analysis and finalize the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH HL34788 to JF.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank the study participants. We thank Susan DiAngelo for her technical assistance in genotyping samples.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.842745/full#supplementary-material
Abbreviations
IPF, Interstitial pulmonary fibrosis; HP, Hypersensitivity Pneumonitis; SP, surfactant protein; SFTPA1, gene encoding SP-A1; SFTPA2, gene encodi ng SP-A2; SFTPB, gene encoding SP-B; SFTPC, gene encoding SP-C; SFTPD, gene encoding SP-C; SNPs, single nucleotide polymorphisms.
References
1. van Moorsel CH, Hoffman TW, van Batenburg AA, Klay D, van der Vis JJ, Grutters JC. Understanding Idiopathic Interstitial Pneumonia: A Gene-Based Review of Stressed Lungs. BioMed Res Int (2015) 2015:304186. doi: 10.1155/2015/304186
2. Selman M, Pardo A. The Leading Role of Epithelial Cells in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Cell Signalling (2020) 66:109482. doi: 10.1016/j.cellsig.2019.109482
3. Ntolios P, Tzilas V, Bouros E, Avdoula E, Karakasiliotis I, Bouros D, et al. The Role of Microbiome and Virome in Idiopathic Pulmonary Fibrosis. Biomedicines (2021) 9(4):442. doi: 10.3390/biomedicines9040442
4. Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med (2018) 198(5):e44–68. doi: 10.1164/rccm.201807-1255ST
5. Pardo A, Selman M. The Interplay of the Genetic Architecture, Aging, and Environmental Factors in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Am J Respir Cell Mol Biol (2021) 64(2):163–72. doi: 10.1165/rcmb.2020-0373PS
6. King TE Jr, Pardo A, Selman M. Idiopathic Pulmonary Fibrosis. Lancet (2011) 378(9807):1949–61. doi: 10.1016/S0140-6736(11)60052-4
7. Taskar VS, Coultas DB. Is Idiopathic Pulmonary Fibrosis an Environmental Disease? Proc Am Thorac Soc (2006) 3(4):293–8. doi: 10.1513/pats.200512-131TK
8. Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, et al. A Common MUC5B Promoter Polymorphism and Pulmonary Fibrosis. New Engl J Med (2011) 364(16):1503–12. doi: 10.1056/NEJMoa1013660
9. Molyneaux PL, Maher TM. The Role of Infection in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Eur Respir Rev (2013) 22(129):376–81. doi: 10.1183/09059180.00000713
10. Doubková M, Kozubík KS, Radová L, Pešová M, Trizuljak J, Pál K, et al. A Novel Germline Mutation of the SFTPA1 Gene in Familial Interstitial Pneumonia. Hum Genome Variation (2019) 6(1):1–6. doi: 10.1038/s41439-019-0044-z
11. Chiba S, Tsuchiya K, Akashi T, Ishizuka M, Okamoto T, Furusawa H, et al. Chronic Hypersensitivity Pneumonitis With a Usual Interstitial Pneumonia-Like Pattern: Correlation Between Histopathologic and Clinical Findings. Chest (2016) 149(6):1473–81. doi: 10.1016/j.chest.2015.12.030
12. Costabel U, Miyazaki Y, Pardo A, Koschel D, Bonella F, Spagnolo P, et al. Hypersensitivity Pneumonitis. Nat Rev Dis Primers (2020) 6(1):1–19. doi: 10.1038/s41572-020-0191-z
13. Morell F, Villar A, Montero M-Á, Muñoz X, Colby TV, Pipvath S, et al. Chronic Hypersensitivity Pneumonitis in Patients Diagnosed With Idiopathic Pulmonary Fibrosis: A Prospective Case-Cohort Study. Lancet Respir Med (2013) 1(9):685–94. doi: 10.1016/S2213-2600(13)70191-7
14. Yang MM, Wolters PJ. Cut From the Same Cloth: Similarities Between Hypersensitivity Pneumonitis and IPF. Am J Respir Crit Care Med (2021) 205(1):4–6. doi: 10.1164/rccm.202109-2211ED
15. Kaur A, Mathai SK, Schwartz DA. Genetics in Idiopathic Pulmonary Fibrosis Pathogenesis, Prognosis, and Treatment. Front Med (2017) 4:154. doi: 10.3389/fmed.2017.00154
16. Falfán-Valencia R, Camarena A, Pineda CL, Montaño M, Juárez A, Buendía-Roldán I, et al. Genetic Susceptibility to Multicase Hypersensitivity Pneumonitis Is Associated With the TNF-238 GG Genotype of the Promoter Region and HLA-DRB1*04 Bearing HLA Haplotypes. Respir Med (2014) 108(1):211–7. doi: 10.1016/j.rmed.2013.11.004
17. Haque R, Umstead TM, Ponnuru P, Guo X, Hawgood S, Phelps DS, et al. Role of Surfactant Protein-A (SP-A) in Lung Injury in Response to Acute Ozone Exposure of SP-A Deficient Mice. Toxicol Appl Pharmacol (2007) 220(1):72–82. doi: 10.1016/j.taap.2006.12.017
18. Floros J, Wang G, Mikerov AN. Genetic Complexity of the Human Innate Host Defense Molecules, Surfactant Protein A1 (SP-A1) and SP-A2—impact on Function. Crit Reviews™ Eukaryotic Gene Expression (2009) 19(2):125–37. doi: 10.1615/CritRevEukarGeneExpr.v19.i2.30
19. Floros J, Thorenoor N, Tsotakos N, Phelps DS. Human Surfactant Protein SP-A1 and SP-A2 Variants Differentially Affect the Alveolar Microenvironment, Surfactant Structure, Regulation and Function of the Alveolar Macrophage, and Animal and Human Survival Under Various Conditions. Front Immunol (2021) 12:2889. doi: 10.3389/fimmu.2021.681639
20. Perez-Gil J, Weaver TE. Pulmonary Surfactant Pathophysiology: Current Models and Open Questions. Physiology (2010) 25(3):132–41. doi: 10.1152/physiol.00006.2010
21. Gandhi CK, Chen C, Amatya S, Yang L, Fu C, Zhou S, et al. SNP and Haplotype Interaction Models Reveal Association of Surfactant Protein Gene Polymorphisms With Hypersensitivity Pneumonitis of Mexican Population. Front Med (2020) 7:1043. doi: 10.3389/fmed.2020.588404
22. Selman M, Lin H-M, Montaño M, Jenkins AL, Estrada A, Lin Z, et al. Surfactant Protein A and B Genetic Variants Predispose to Idiopathic Pulmonary Fibrosis. Hum Genet (2003) 113(6):542–50. doi: 10.1007/s00439-003-1015-4
23. Guler SA, Wohlfarth E, Berezowska S, Geiser TK, Ebner L, Funke-Chambour M. Performance of a Diagnostic Algorithm for Fibrotic Hypersensitivity Pneumonitis. A Case–Control Study. Respir Res (2021) 22(1):1–7. doi: 10.1186/s12931-021-01727-7
24. Pleasants R, Tighe RM. Management of Idiopathic Pulmonary Fibrosis. Ann Pharmacother (2019) 53(12):1238–48. doi: 10.1177/1060028019862497
25. Network IPFCR. Prednisone, Azathioprine, and N-Acetylcysteine for Pulmonary Fibrosis. N Engl J Med (2012) 366(21):1968–77. doi: 10.1056/NEJMoa1113354
26. Newton CA, Zhang D, Oldham JM, Kozlitina J, Ma S-F, Martinez FJ, et al. Telomere Length and Use of Immunosuppressive Medications in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med (2019) 200(3):336–47. doi: 10.1164/rccm.201809-1646OC
27. Oldham JM, Ma S-F, Martinez FJ, Anstrom KJ, Raghu G, Schwartz DA, et al. TOLLIP, MUC5B, and the Response to N-Acetylcysteine Among Individuals With Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med (2015) 192(12):1475–82. doi: 10.1164/rccm.201505-1010OC
28. Chen H, Xu X, Hua C, Zhang H, Jian J, Ge T, et al. Polymorphisms of Matrix Metalloproteinases Affect the Susceptibility of Esophageal Cancer: Evidence From 20412 Subjects, Systematic Review and Updated Meta-Analysis. Medicine (2021) 100(38):e27229. doi: 10.1097/MD.0000000000027229
29. Megías-Vericat J, Herrero M, Rojas L, Montesinos P, Bosó V, Moscardó F, et al. A Systematic Review and Meta-Analysis of the Impact of WT1 Polymorphism Rs16754 in the Effectiveness of Standard Chemotherapy in Patients With Acute Myeloid Leukemia. Pharmacogenomics J (2016) 16(1):30–40. doi: 10.1038/tpj.2015.80
30. Ojha RP, Gurney JG. Methylenetetrahydrofolate Reductase C677T and Overall Survival in Pediatric Acute Lymphoblastic Leukemia: A Systematic Review. Leukemia Lymphoma (2014) 55(1):67–73. doi: 10.3109/10428194.2013.792336
31. Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 Mutations in AML: Review of Current Knowledge and Evidence. Leukemia (2019) 33(2):299–312. doi: 10.1038/s41375-018-0357-9
32. Cargnin S, Ravegnini G, Soverini S, Angelini S, Terrazzino S. Impact of SLC22A1 and CYP3A5 Genotypes on Imatinib Response in Chronic Myeloid Leukemia: A Systematic Review and Meta-Analysis. Pharmacol Res (2018) 131:244–54. doi: 10.1016/j.phrs.2018.02.005
33. D'Ovidio F, Floros J, Aramini B, Lederer D, DiAngelo SL, Arcasoy S, et al. Donor Surfactant Protein A2 Polymorphism and Lung Transplant Survival. Eur Respir J (2020) 1900618. doi: 10.1183/13993003.00618-2019
34. Hoover RR, Floros J. Organization of the Human SP-A and SP-D Loci at 10q22-Q23. Physical and Radiation Hybrid Mapping Reveal Gene Order and Orientation. Am J Respir Cell Mol Biol (1998) 18(3):353–62. doi: 10.1165/ajrcmb.18.3.3035
35. Bruns G, Stroh H, Veldman GM, Latt SA, Floros J. The 35 Kd Pulmonary Surfactant-Associated Protein Is Encoded on Chromosome 10. Hum Genet (1987) 76(1):58–62. doi: 10.1007/BF00283051
36. Floros J, Steinbrink R, Jacobs K, Phelps D, Kriz R, Recny M, et al. Isolation and Characterization of cDNA Clones for the 35-kDa Pulmonary Surfactant-Associated Protein. J Biol Chem (1986) 261(19):9029–33. doi: 10.1016/S0021-9258(19)84483-6
37. DiAngelo S, Lin Z, Wang G, Phillips S, Ramet M, Luo J, et al. Novel, non-Radioactive, Simple and Multiplex PCR-cRFLP Methods for Genotyping Human SP-A and SP-D Marker Alleles. Dis Markers (1999) 15(4):269–81. doi: 10.1155/1999/961430
38. Floros J, Hoover RR. Genetics of the Hydrophilic Surfactant Proteins A and D. Biochim Biophys Acta (1998) 1408(2-3):312–22. doi: 10.1016/S0925-4439(98)00077-5
39. Floros J, Wang G, Lin Z. Genetic Diversity of Human SP-A, A Molecule With Innate Host Defense and Surfactant-Related Functions; Characteristics, Primary Function, and Significance. Curr Pharmacogenomics (2005) 3(2):87–95. doi: 10.2174/1570160054022935
40. Lynnlee Depicolzuane DSP, Floros J. Surfactant Protein-A Function: Knowledge Gained From SP-A Knockout Mice. Front Pediatrics (2021) 9:799693. doi: 10.3389/fped.2021.799693
41. Floros J, Phelps D. Pulmonary Surfactant. In: Yaksh TL, Lynch C III, Zapol WM, Maze M, Biebuyck JF, Saidman LJ, editors. Anesthesia: Biologic Foundations. Philadelphia, PA, USA: Lippincott-Raven, Philadelphia (1997). p. 1259–79.
42. Wert SE, Whitsett JA, Nogee LM. Genetic Disorders of Surfactant Dysfunction. Pediatr Dev Pathol (2009) 12(4):253–74. doi: 10.2350/09-01-0586.1
43. Lin Z, Pearson C, Chinchilli V, Pietschmann S, Luo J, Pison U, et al. Polymorphisms of Human SP-A, SP-B, and SP-D Genes: Association of SP-B Thr131Ile With ARDS. Clin Genet (2000) 58(3):181–91. doi: 10.1034/j.1399-0004.2000.580305.x
44. Silveyra P, Floros J. Genetic Variant Associations of Human SP-A and SP-D With Acute and Chronic Lung Injury. Front Biosci: J Virtual library (2012) 17:407. doi: 10.2741/3935
45. Guo X, Lin H, Lin Z, Montano M, Sansores R, Wang G, et al. Surfactant Protein Gene A, B, and D Marker Alleles in Chronic Obstructive Pulmonary Disease of a Mexican Population. Eur Respir J (2001) 18(3):482–90. doi: 10.1183/09031936.01.00043401
46. Seifart C, Plagens A, Brödje D, Müller B, Von Wichert P, Floros J. Surfactant Protein B Intron 4 Variation in German Patients With COPD and Acute Respiratory Failure. Dis Markers (2002) 18(3):129–36. doi: 10.1155/2002/194075
47. Lin Z, Thorenoor N, Wu R, DiAngelo SL, Ye M, Thomas NJ, et al. Genetic Association of Pulmonary Surfactant Protein Genes, SFTPA1, SFTPA2, SFTPB, SFTPC, and SFTPD With Cystic Fibrosis. Front Immunol (2018) 9:2256. doi: 10.3389/fimmu.2018.02256
48. Nathan N, Giraud V, Picard C, Nunes H, Dastot-Le Moal F, Copin B, et al. Germline SFTPA1 Mutation in Familial Idiopathic Interstitial Pneumonia and Lung Cancer. Hum Mol Genet (2016) 25(8):1457–67. doi: 10.1093/hmg/ddw014
49. van Moorsel CH, Ten Klooster L, van Oosterhout MF, de Jong PA, Adams H, Wouter van Es H, et al. SFTPA2 Mutations in Familial and Sporadic Idiopathic Interstitial Pneumonia. Am J Respir Crit Care Med (2015) 192(10):1249–52. doi: 10.1164/rccm.201504-0675LE
50. Liu L, Qin J, Guo T, Chen P, Ouyang R, Peng H, et al. Identification and Functional Characterization of a Novel Surfactant Protein A2 Mutation (P. N207Y) in a Chinese Family With Idiopathic Pulmonary Fibrosis. Mol Genet Genomic Med (2020) 8(9):e1393. doi: 10.1002/mgg3.1393
51. Takezaki A, Tsukumo S-i, Setoguchi Y, Ledford JG, Goto H, Hosomichi K, et al. A Homozygous SFTPA1 Mutation Drives Necroptosis of Type II Alveolar Epithelial Cells in Patients With Idiopathic Pulmonary Fibrosis. J Exp Med (2019) 216(12):2724–35. doi: 10.1084/jem.20182351
52. Nureki S-I, Tomer Y, Venosa A, Katzen J, Russo SJ, Jamil S, et al. Expression of Mutant Sftpc in Murine Alveolar Epithelia Drives Spontaneous Lung Fibrosis. J Clin Invest (2018) 128(9):4008–24. doi: 10.1172/JCI99287
53. Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, Steele MP, et al. Genome-Wide Association Study Identifies Multiple Susceptibility Loci for Pulmonary Fibrosis. Nat Genet (2013) 45(6):613–20. doi: 10.1038/ng.2609
54. Furusawa H, Peljto AL, Walts AD, Cardwell J, Molyneaux PL, Lee JS, et al. Common Idiopathic Pulmonary Fibrosis Risk Variants are Associated With Hypersensitivity Pneumonitis. Thorax (2022) 77(5):508–10. doi: 10.1164/ajrccm-conference.2021.203.1_MeetingAbstracts.A3143
55. Kam ML, Nguyen TT, Ngeow JY. Telomere Biology Disorders. NPJ Genomic Med (2021) 6(1):1–13. doi: 10.1038/s41525-021-00198-5
56. Ley B, Torgerson DG, Oldham JM, Adegunsoye A, Liu S, Li J, et al. Rare Protein-Altering Telomere-Related Gene Variants in Patients With Chronic Hypersensitivity Pneumonitis. Am J Respir Crit Care Med (2019) 200(9):1154–63. doi: 10.1164/rccm.201902-0360OC
57. Wang Z, Liu T, Lin Z, Hegarty J, Koltun WA, Wu R. A General Model for Multilocus Epistatic Interactions in Case-Control Studies. PloS One (2010) 5(8):e11384. doi: 10.1371/journal.pone.0011384
58. Gandhi CK, Chen C, Wu R, Yang L, Thorenoor N, Thomas NJ, et al. Association of SNP–SNP Interactions of Surfactant Protein Genes With Pediatric Acute Respiratory Failure. J Clin Med (2020) 9(4):1183. doi: 10.3390/jcm9041183
59. Amatya S, Ye M, Yang L, Gandhi CK, Wu R, Nagourney B, et al. Single Nucleotide Polymorphisms Interactions of the Surfactant Protein Genes Associated With Respiratory Distress Syndrome Susceptibility in Preterm Infants. Front Pediatr (2021) 9(1065). doi: 10.3389/fped.2021.682160
60. Wang G, Bates-Kenney SR, Tao J-Q, Phelps DS, Floros J. Differences in Biochemical Properties and in Biological Function Between Human SP-A1 and SP-A2 Variants, and the Impact of Ozone-Induced Oxidation. Biochemistry (2004) 43(14):4227–39. doi: 10.1021/bi036023i
61. García-Laorden M, de Castro FR, Solé-Violán J, Rajas O, Blanquer J, Borderías L, et al. Influence of Genetic Variability at the Surfactant Proteins A and D in Community-Acquired Pneumonia: A Prospective, Observational, Genetic Study. Crit Care (2011) 15(1):1–12. doi: 10.1186/cc10030
62. Gandhi CK, Thomas NJ, Meixia Y, Spear D, Fu C, Zhou S, et al. SNP-SNP Interactions of Surfactant Protein Genes in Persistent Respiratory Morbidity Susceptibility in Previously Healthy Children. Front Genet (2022) 13:815727. doi: 10.3389/fgene.2022.815727
63. Ikegami M, Elhalwagi BM, Palaniyar N, Dienger K, Korfhagen T, Whitsett JA, et al. The Collagen-Like Region of Surfactant Protein A (SP-A) Is Required for Correction of Surfactant Structural and Functional Defects in the SP-A Null Mouse. J Biol Chem (2001) 276(42):38542–8. doi: 10.1074/jbc.M102054200
64. Palaniyar N, Ikegami M, Korfhagen T, Whitsett J, McCormack FX. Domains of Surfactant Protein A That Affect Protein Oligomerization, Lipid Structure and Surface Tension. Comp Biochem Physiol Part A: Mol Integr Physiol (2001) 129(1):109–27. doi: 10.1016/S1095-6433(01)00309-9
65. Eckhard U, Huesgen PF, Schilling O, Bellac CL, Butler GS, Cox JH, et al. Active Site Specificity Profiling of the Matrix Metalloproteinase Family: Proteomic Identification of 4300 Cleavage Sites by Nine MMPs Explored With Structural and Synthetic Peptide Cleavage Analyses. Matrix Biol (2016) 49:37–60. doi: 10.1016/j.matbio.2015.09.003
66. Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: The NCBI Database of Genetic Variation. Nucleic Acids Res (2001) 29(1):308–11. doi: 10.1093/nar/29.1.308
67. Cao X, Liu J, Guo M, Wang J. HiSSI: High-Order SNP-SNP Interactions Detection Based on Efficient Significant Pattern and Differential Evolution. BMC Med Genomics (2019) 12(7):139. doi: 10.1186/s12920-019-0584-6
68. Cordell HJ. Detecting Gene–Gene Interactions That Underlie Human Diseases. Nat Rev Genet (2009) 10(6):392–404. doi: 10.1038/nrg2579
69. Frankel WN, Schork NJ. Who's Afraid of Epistasis? Nat Genet (1996) 14(4):371–3. doi: 10.1038/ng1296-371
70. Nogee LM. Genetic Causes of Surfactant Protein Abnormalities. Curr Opin Pediatrics (2019) 31(3):330. doi: 10.1097/MOP.0000000000000751
71. Nogee LM, Garnier G, Dietz HC, Singer L, Murphy AM, DeMello DE, et al. A Mutation in the Surfactant Protein B Gene Responsible for Fatal Neonatal Respiratory Disease in Multiple Kindreds. J Clin Invest (1994) 93(4):1860–3. doi: 10.1172/JCI117173
72. Wang G, Christensen ND, Wigdahl B, Guttentag SH, Floros J. Differences in N-Linked Glycosylation Between Human Surfactant Protein-B Variants of the C or T Allele at the Single-Nucleotide Polymorphism at Position 1580: Implications for Disease. Biochem J (2003) 369(Pt 1):179–84. doi: 10.1042/bj20021376
73. Yang F, Zhang J, Yang Y, Ruan F, Chen X, Guo J, et al. Regulatory Roles of Human Surfactant Protein B Variants on Genetic Susceptibility to Pseudomonas Aeruginosa Pneumonia-Induced Sepsis. Shock (2020) 54(4):507–19. doi: 10.1097/SHK.0000000000001494
74. Lin Z, deMello DE, Wallot M, Floros J. An SP-B Gene Mutation Responsible for SP-B Deficiency in Fatal Congenital Alveolar Proteinosis: Evidence for a Mutation Hotspot in Exon 4. Mol Genet Metab (1998) 64(1):25–35. doi: 10.1006/mgme.1998.2702
75. Gosens R, Hiemstra PS, Adcock IM, Bracke KR, Dickson RP, Hansbro PM, et al. Host–microbe Cross-Talk in the Lung Microenvironment: Implications for Understanding and Treating Chronic Lung Disease. Eur Respir J (2020) 56(2):1902320. doi: 10.1183/13993003.02320-2019
Keywords: surfactant protein, single nucleotide polymorphism, idiopathic pulmonary fibrosis, hypersensitivity pneumonitis, SNP-SNP interactions
Citation: Abbasi A, Chen C, Gandhi CK, Wu R, Pardo A, Selman M and Floros J (2022) Single Nucleotide Polymorphisms (SNP) and SNP-SNP Interactions of the Surfactant Protein Genes Are Associated With Idiopathic Pulmonary Fibrosis in a Mexican Study Group; Comparison With Hypersensitivity Pneumonitis. Front. Immunol. 13:842745. doi: 10.3389/fimmu.2022.842745
Received: 24 December 2021; Accepted: 09 May 2022;
Published: 02 June 2022.
Edited by:
Taruna Madan, National Institute for Research in Reproductive Health (ICMR), IndiaReviewed by:
Bartosz Pilecki, University of Southern Denmark, DenmarkHaruhiko Furusawa, Tokyo Medical and Dental University, Japan
Copyright © 2022 Abbasi, Chen, Gandhi, Wu, Pardo, Selman and Floros. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joanna Floros, amZsb3Jvc0BwZW5uc3RhdGVoZWFsdGgucHN1LmVkdQ==
†Present address: Chixiang Chen, Department of Epidemiology and Public Health, University of Maryland School of Medicine, Baltimore, MD