 Fang Liang
Fang Liang Weixiao Qin
Weixiao Qin Yilan Zeng2
Yilan Zeng2 Dan Wang
Dan Wang- 1Department of Hematology, The Third Xiangya Hospital, Central South University, Changsha, China
- 2Department of Dermatology, The Third Xiangya Hospital, Central South University, Changsha, China
Autoimmune diseases and autoinflammatory diseases are two types of the immune system disorders. Pyroptosis, a highly inflammatory cell death, plays an important role in diseases of immune system. The gasdermins belong to a pore-forming protein gene family which are mainly expressed in immune cells, gastrointestinal tract, and skin. Gasdermins are regarded as an executor of pyroptosis and have been shown to possess various cellular functions and pathological effects such as pro-inflammatory, immune activation, mediation of tumor, etc. Except for infectious diseases, the vital role of gasdermins in autoimmune diseases, autoinflammatory diseases, and immune-related neoplastic diseases has been proved recently. Therefore, gasdermins have been served as a potential therapeutic target for immune disordered diseases. The review summarizes the basic molecular structure and biological function of gasdermins, mainly discusses their role in autoimmune and autoinflammatory diseases, and highlights the recent research on gasdermin family inhibitors so as to provide potential therapeutic prospects.
Introduction of Autoimmune and Autoinflammatory Diseases
The immune system mainly consists of innate immune system and adaptive immune system. Disorders in our immune system lead to a range of diseases, including autoimmune diseases and autoinflammatory diseases. Deficiency and/or overactivation of multiple factors within innate or adaptive immune systems may participate in the occurrence and development of these diseases, such as experimental autoimmune encephalomyelitis (EAE), Inflammatory bowel disease (IBD), psoriasis, etc. (1). During the occurrence of autoimmune diseases, the immune system is out of balance and immunocytes are overactive, the release of cytokines further lead to abnormal secretion of autoantibodies, resulting in inflammation and damage to tissues and organs (2, 3). While the progression of the diseases can be accelerated because of other key factors, such as gene, environment, hormones, and so on. In 1999, the genetic mechanism of a rare familial disease characterized by recurrent inflammatory episodes which are defined as tumor necrosis factor receptors associated periodic fever syndrome (TRAPS) was discovered. Another Familial Mediterranean Fever (FMF), an autosomal recessive hereditary disease characterized by recurrent peritonitis. TRAPS and FMF are both autoinflammatory diseases (4, 5). Gill et al. envisaged autoinflammatory diseases as counterparts to autoimmune disorders, and proposed that autoinflammatory diseases are related to innate immune system dysfunction and are determined by genes (1). McGonagle and McDermott proposed that dysfunction of the innate immunity or the adaptive immune system is the differentiated element of autoimmune diseases and autoinflammatory diseases (6). Approximately, disorders of innate immune system represent autoinflammatory diseases, while disorders of adaptive immune system reflect autoimmune diseases. However, innate and adaptive immunities are inextricably intertwined and influence each other (7).
Autoimmune diseases afflict nearly 5%-8% of the population worldwide and represent a major global socioeconomic issue (8, 9). So far, there are no fewer than 80 autoimmune diseases have been discovered which are divided into systemic or organ-specific. Systemic Lupus Erythematosus (SLE) and Central Nervous System (CNS) reflect the systemic autoimmune diseases, while type 1 diabetes images the organ-specific autoimmune diseases such as the pancreas (9, 10). During such diseased conditions, the immune system is out of balance, immunocytes are overactive, and there is imbalance of cytokine which further leads to abnormal secretion of autoantibodies, resulting in inflammation and damage to tissues and organs (11). At this stage anti-inflammatory and immunosuppressive therapy are commonly used in the treatment of autoimmune diseases to the symptoms (12–16). Because of the complex mechanisms involved in the pathogenesis of the autoimmune diseases, there is yet no effective treatment available, and therefore, it is important to understand these processes.
Autoinflammatory diseases are characterized by antigen-independent-immune pathways overactivation arising primarily inflammation. Common autoinflammatory diseases include FMF, TRAPS, Cryopyrin-associated periodic syndrome (CAPS) and Mevalonate kinase deficiency (MKD), but not all autoinflammatory diseases manifest in their canonical forms. Hyperactivity of proinflammatory cytokine is found to mediate and participate in autoinflammatory diseases. FMF was identified as the first autoinflammatory disease, which is an inflammasomopathy that arises from the MEFV gene mutations, coding pyrin (expressed predominately by innate lineages) (4, 17). Other autoinflammatory diseases mostly arise out of NF-κB and/or aberrant TNF activity, interferon production and complement activation, and excessive interleukin-1 (IL-1) signaling (7, 18, 19). So far, IL-1 blockers have been approved for a variety of autoinflammatory diseases. New categories of autoinflammatory disease and more treatment will emerge over time.
Autoimmune diseases and autoinflammatory diseases are difficult to cure and seriously affect the quality of life of patients which involve various pathogenic mechanisms such as genes, immunity, inflammation, etc. Therefore, the existing research is summarized to deepen the understanding of the disease and provide help for the diagnosis and treatment of those diseases.
Gasdermin Family, Pyroptosis and Inflammasomes
As early as in 2000s, the gasdermin family was first discovered as a gene family associated with corneal opacity in mice (20, 21). The gasdermin family is predominantly expressed in immune cells, gastrointestinal tract, and skin and linked with many diseases including autoimmune diseases and autoinflammatory diseases (Table 1). This family consists of six genes in humans, GSDMA through GSDMF. In mice, it encodes ten members, which respectively are GSDMA1 to GSDMA3, GSDMC, GSDMC2 to GSDMC4, GSDMD, GSDME, and DFNB59 (22, 23). All of these members possess a semblable molecular structure except DFNB59. A pore-forming N-terminal domain and a regulatory C-terminal domain connected by highly variable and flexible linking regions compose the nuclear structure, which is thought to be necessary for GSDM activation (24). The N-terminal domain (GSDM-NT) is required for forming pores in membranes while the auto-inhibitory C-terminal domain (GSDM-CT) is asked to keep its inactivation (24, 25). Structurally, GSDM-NT predominantly contains loops and β-strands, and sustains drastic conformational changes in pore formation (24). Nevertheless, GSDM-CT is nearly α-helical and remains a compact globular conformation. GSDM-CT forms electrostatic, hydrophobic, and hydrogen bonding interactions after folding back on GSDM-NT (24–26). In vitro, GSDM-NT and GSDM-CT keep bound without lipids, even if the interdomain container is cleaved, this finding suggests that the lipid may play a critical role in the separation of GSDM-NT from GSDM-CT. In simple terms, lipid binding, oligomerization, and membrane insertion make up the pore formation of GSDM (25, 27).
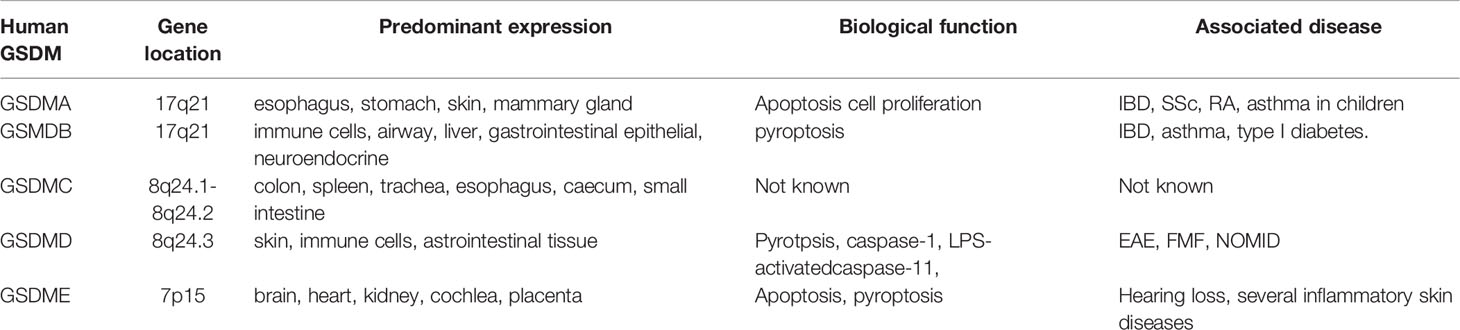
Table 1 Gasdermin family members, functions, and disease correlation.
Pyroptosis is a kind of programmed cell death (PCD) associated with inflammation, which plays an important role in autoimmune and autoinflammatory diseases (28, 29).. Pyroptosis is a protective host defense that protect the cell from intracellular infection by removing the injured cells and concurrently triggering an inflammatory response that executes the following activation of caspase-1 or caspase-11 (21). Activated caspase-1 or caspase-11 cleaves GSDMD independently, releasing the N-terminal fragment (GSDMD-NT) associated with the cytoplasm membrane, which forms the cell membrane pores by oligomerization. Then, the cell swelling causes the membrane lysis and finally leads to pyroptosis (Figure 1) (30–33).
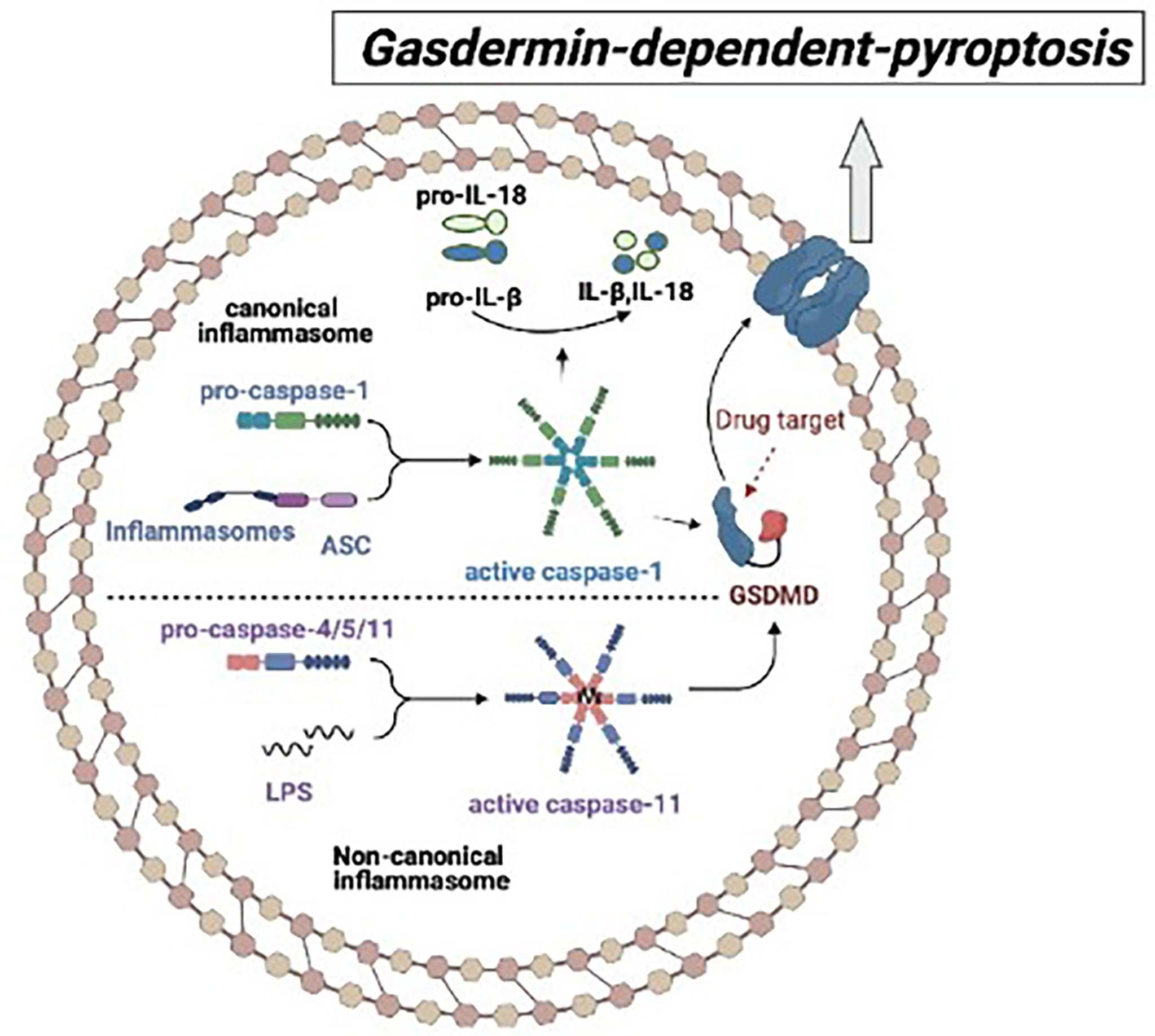
Figure 1 The mechanism of gasdermin-dependent-pyroptosis: In the canonical pyroptosis pathway, ASC recruits intracellular pro-Caspase-1 to bind to the inflammatory complex, and then pro-caspase-1 auto-cleaves into activated Caspase-1, which cleaves GSDMD and promotes IL-1β and IL-18 to maturate. In the non-canonical pyroptosis pathway, inflammatory stimulators such as lipopolysaccharide (LPS) directly bind to Caspase-4/5/11 and cause oligomeric activation of Caspase-4/5/11, activated Caspase-4/5/11 cleaves GSDMD and triggers pyroptosis.
Inflammasome plays a crucial role in the process of pyroptosis. Inflammasomes are cytosolic protein complexes consisting of sensor, adaptor, and effector which are whole protein that is stimulated by pathogen-associated molecular patterns (PAMPs) and endogenous danger-associated molecular patterns (DAMPs) (34). Upon activation by diverse danger signals, members of the NOD-like receptor (NLR) and pyrin and HIN domain-containing (PYHIN) protein families as sensors to combine with apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) adaptor, subsequently recruit effector caspase-1 to form canonical inflammasome so as to initiate a downstream signal. Subsequently, the activated caspase-1 cleaves pro-inflammatory cytokines interleukin 1β (IL-1β) and interleukin 18 (IL-18), allowing them to mature and secret (35, 36). The non-canonical inflammasome is activated by bacterial cell wall component lipopolysaccharide (LPS), and executed by caspase-11 (human caspase-4/5) (30, 31, 37–39). Therefore, both canonical and non-canonical inflammasome participate in the occurrence of pyroptosis by cleaving GSDMD into GSDMD-NT so as to form a pore in the cell membrane (Figure 1).
Gasdermin family is well known to play a vital role in pathogen infection, while more and more studies show that both pyroptosis and its related inflammatory cytokines IL-1β and IL-18 are the key factor in the process of non-infection related diseases. IL-1β induces vasodilation, inflammation and immunity extravasation, and also takes part in formation of adaptive immune responses. IL-18 promotes natural killer (NK) cells, T helper 1 (TH1) cells and cytotoxic T cells to produce interferon-γ (IFN-γ) in these cells, and promotes T helper 2 (TH2) cells maturation, and triggers local inflammation. These findings suggest that gasdermins, and autoimmune as well as autoinflammatory disease are inextricably linked (40–43). Therefore, the following section will focus on introducing the gasdermin family and summarizing the role of gasdermins in autoimmune and autoinflammatory diseases.
The Roles of Gasdermins in Autoimmune and Autoinflammatory Diseases
Multiple functions of pyroptosis and inflammasomes are identified in various pathophysiological conditions, and their role in autoimmune and autoinflammatory diseases have been extensively studied recently. Therefore, we will summarize the relation between each subtype of the gasdermin family (GSDMA, GSDMB, GSDMC, GSDMD, GSDME) and autoimmune and autoinflammatory diseases.
GSDMA
The human GSDMA gene is located at 17q21. It is widely expressed in the epithelial cells of the esophagus, stomach, skin and mammary gland, but is often not expressed in primary and the gastric cancer cell lines. It has been suggested that GSDMA suppresses the growth of human gastric epithelial pit cells as it possibly takes part in a regulatory pathway for apoptosis (44). The high expression of GSDMA in non-IBD colonic, noninflamed samples was linked with the IBD susceptibility allele (rs2872507) (45). In childhood asthma, there was a significant correlation of GSDMA (rs7212938) with it as GSDMA may drives the frequency of asthma (46). In addition, GSDMA has been reported to carry rs3894194, which is rich in gene, and regarded as being linked with certain immune diseases such as IBD, asthma and rheumatoid arthritis (RA) (47). Aida et al. reported that the function of r3894194 in systemicsclerosis (SSc) could be mediated by GSDMA expression in macrophages. They found that GSDMA was upregulated in SSc monocyte-derived macrophages, and was an important expression quantitative trait locus (e-QTL) in interferon gamma (IFN-γ) or LPS-triggered monocytes and SSc macrophages (48). Overall, GSMDA is linked with several autoimmune diseases, while the exact pathogenesis still remains blurred, GSDMA may affect those diseases via effects on apoptosis and cell proliferation. It warrants further experimental investigation.
GSDMB
GSMDB is located at 17q12 in the human chromosome (22, 49). It clusters with GSDMA (50). GSDMB is also broadly expressed in the immune cells, airway, gastrointestinal epithelial, liver, and neuroendocrine (51–53). Genome-wide association studies (GWAS) implicated that the polymorphisms in GSDMB were markedly associated with IBD, asthma, and type I diabetes. The minor allele A of rs2872507 in GSDMB was the risk allele for RA and IBD (Crohn’s disease and ulcerative colitis) but was a protective allele for asthma (47). While another study found that the levels of GSDMB were increased remarkably in patients with asthma, and were associated with the severity of asthma (54). A recent study showed that GSDMB was increased in intestinal epithelial cells (IECs) in IBD, and it participated in the regulation of cell proliferation, migration, and adhesion rather than mediating pyroptosis. GSDMB-linked IBD single-nucleotide polymorphisms (SNPs) hinder epithelial recovery/repair (55). Moreover, Luke Jostins et al. reported that numerous IBD loci also contributed to immune diseases (most obviously with ankylosing spondylitis and psoriasis) (56). Those consequences indicate that GSDMB increases the risk for various autoimmune diseases, but the detailed molecular mechanisms remain unclear. There was reported that the GSDMB can bind with caspase-4 to promote non-canonical pyroptosis and facilitate the GSDMD cleavage. This may be associated with the pathogenesis of autoimmune diseases (57).
GSDMC
GSDMC gene has been located at 8q24.1-8q24.2 and is highly expressed in colon, spleen, trachea, esophagus, caecum, and small intestine (38, 49, 58). There are few studies on GSDMC. Lumbar disc degeneration (LDD) mainly manifests as low back pain, disc herniation, spinal stenosis, spinal instability, and radiculopathy. A recent study suggested that autoimmunity may be possible pathogenesis of this disease (59). GWAS in a Chinese population reported that GSDMC was associated with lumbar disc degeneration (LDD). In patients with LDD, the mRNA level of GSDMC was apparently increased and the plasma expression level of GSDMC was also apparently upregulated which were linked with the rs6651255 and rs7833174 (60). GSDMC was also expressed in skin keratinocytes, and the UV radiation increased its expression by triggering NFATc1 signaling (61, 62). Nevertheless, the mechanism of GSDMC in other autoimmune and autoinflammatory diseases is not yet well-understood.
GSDMD
The GSDMD gene is located at 8q24.3 in the human chromosome. It has been extensively studied for decades. As the key substrate of caspase-dependent-pyroptosis, GSDMD was widely expressed in skin, immune cells (especially macrophages and dendritic cells), and gastrointestinal tissue (63). Approximately 480 amino acids constitute GSDMD, which is in two domains: the N-terminal (GSDMD-NT) and the C-terminal (GSDMD-CT). GSDMD is cleaved by activated caspase-1, LPS-activated caspase-11 (human caspase-4/5) to form pore-forming GSDMD-NT (31). The pores of GSDMD-NT can bind with phosphatidylserine and phosphatidylinositol phosphates, causing cell lysis and releasing of IL-1β, IL-18, and HMGB1. GSDMD-NT also combines with cardiolipin in the bacteria membranes, and kills them quickly. In addition, neutrophil elastase (ELANE) and cathepsin G are both able to activate the GSDMD (24, 64–66).
Recent studies have shown that GSDMD is involved in the process of immune and inflammatory responses. Catherine R et al. reported that AIM2 inflammasome took part in neurodevelopment through regulating GSDMD activation instead of IL-1 and/or IL-18 production. In this study, the DNA damage-induced cell death was significantly reduced in CNS cells lacking GSDMD or caspase-1/11, which means that GSDMD activation may hinder neurodevelopment (67). Sheng Li et al. firstly demonstrated that GSDMD was necessary for EAE and could promote neuroinflammation and demyelination (68). During impaired neurodevelopment, a large amount of DNA damage is produced, which may immunologically activate signals that can cause autoimmune diseases of the nervous system. Another research provided that TPPU (a potent soluble epoxide hydrolase) can attenuate chronic EAE by suppressing caspase-11 and GSDMD in the CNS of EAE mice, a model of multiple sclerosis (MS) (69). Dataset analysis showed a significant elevation of caspase-4/5, pro- IL-1β, and GSDMD in human psoriatic lesions. Besides, caspase-11 contributed to the pathogenesis of imiquimod-induced psoriasis in mice through the mediation of GSDMD activation and cell death (70). The process of GSDMD-dependent pyroptosis also took part in RA through the releasement of inflammatory mediators (71). Another research found that inhibition of the caspase-11 (executor of pyroptosis) could enhance mesenchymal stromal cells (MSCs) efficacy in inflammatory diseases, such as IBD and EAE. MSCs present immunosuppressive properties and were currently being used as promising treatments for various inflammatory diseases, including MS (46, 72, 73). A report demonstrated that the pathogenesis of FMF, an IL-1β-dependent autoinflammatory disease, relies on the GSDMD-dependent-pyroptosis (74). Neonatal-onset multisystem inflammatory disease (NOMID) is the most severe phenotype in cryopyrinopathies (a spectrum of autoinflammatory disorders), and there has been reported that all NOMID-related inflammatory symptoms were prevented in GSDMD-deficient NOMID mice compared to wildtype NOMID mice (75). Overall, these findings suggest that activation of GSDMD aggravates the pathogenesis of various diseases induced by inflammasomes. Targeted GSDMD may provide new perspectives for the treatment of autoimmune and autoinflammatory diseases.
GSDME
GSDME is located at 7p15 in the human chromosome. It was defined as a mutated gene (DFNA5) firstly that was associated with progressive hearing loss (76–83). Subsequent studies found that the DFNA5 might be crucial to chondrocyte development. GSDME is widely expressed in brain, heart, kidney, cochlea, and placenta (76, 84). Similar to GSDMD, GSDME plays a critical role in apoptosis and pyroptosis (85, 86). GSDME releases N-terminal fragment (GSDME-NT) after its cleavage and activation by caspase-3 (27). In addition to forming pores and triggering necrosis or pyroptosis, GSDME-NT permeabilizes the mitochondrial membrane, leading to release cytochrome C, and eventually enhances the activation of caspase-3 during apoptosis (25).
Several studies revealed a link between GSDME and immunity/inflammation. Oncostatin M (OSM), an IL-6 family member, has been found to be overexpressed in several inflammatory skin diseases [including keloid (87), scleroderma (88, 89), and psoriasis (90, 91)]. The level of GSDME in human keratinocytes (HaCaT cells) was up-regulated, while OSMR knockout blocked the upregulation of GSDME. Besides, GSDME knockout inhibited OSM-induced keratinocyte pyroptosis. This study suggested that GSDME played a role in OSM-mediated inflammation (92). Another study found that GSDME-mediate pyroptosis was critical for the trigger of renal tubule injury induced by ureteral obstruction and promoted kidney inflammation and fibrosis (93). Overall, GSDME has pro-inflammatory functions and may be involved in the pathogenesis of more inflammatory diseases.
Therapy Targeting of Gasdermin Family
Based on the important role of gasdermin family in autoimmune and autoinflammatory diseases, this study further summarizes the inhibitors targeting gasdermin family to provide new prospection for disease treatment.
Among all subtypes of the gasdermin family, inhibitors targeting GSDMD have been revealed the most due to its rich biological functions. Necrosulfonamide (NSA) was originally proved to be an inhibitor of necrosis by targeting the Cys86 Residue of MLKL to reduce MLKL-dependent pore formation and death (94). In the work of Joseph K. Rathkey’s group, they found that NSA inhibited pyroptosis and IL-1β release by binding to GSDMD and blocking p30-GSDMD oligomerization in vitro, and NSA did not block GSDME-dependent pyroptosis and the upstream inflammasome activation (95). NSA promoted survival rate and decreased inflammatory cytokine level of sepsis model in vivo (95). Conversely, another study showed that NSA blocked canonical NLRP3 inflammasome priming and activation so as to reduce pyroptosis (96). Both studies showed that NSA inhibited GSDMD-dependent pyroptosis to play a role in treating diseases by two different mechanisms, suggesting that NSA may provide a theoretical basis for the treatment of GSDMD-related inflammatory diseases. Besides, Jun Jacob Hu et al. discovered that disulfiram, a drug used to treat alcohol addiction, inhibited GSDMD-mediated pyroptosis rather than other subtypes of gasdermin family. Disulfiram targeted Cys191/Cys192 in GSDMD so as to inhibit pore formation and IL-1β release, and protected against septic death in mice (97). In another study, LDC7559 from a chemical library was found to inhibit GSDMD-dependent pyroptosis and IL-1β release so as to reduce neutrophil extracellular traps (NETs) formation (98). Activated macrophages present as the Warburg effect, which is characterized as a metabolic change from glycolysis to aerobic glycolysis and the accumulation of Krebs’ cycle intermediates. The alteration of metabolism further impacts the immune state of macrophages (99). Fumarate (DMF), an intermediate of Krebs’ cycle, blocked GSDMD oligomerization and the related pyroptosis by inducing S-(2-succinyl)-cysteine of GSDMD. Dimethyl fumarate with the cell-permeable ability, reduced septic shock and familial Mediterranean fever, and experimental autoimmune encephalitis (100).
Collectively, several studies have found various inhibitors of gasdermin family (mainly GSDMD), and they can improve a variety of diseases. Although these diseases are mainly sepsis, there is also involved to autoimmune and autoinflammation diseases.
Conclusions
Our knowledge of gasdermins function becomes increasingly extensive based on a large of recent studies. Most subtypes of gasdermin play a pro-inflammatory role in the autoimmune and autoinflammation diseases, including IBD, RA, SSc, EAE, psoriasis, etc. Among the whole types of gasdermin, GSDMD is the most studied subtype of gasdermins due to its tight correlation with inflammasomes activation and the related inflammatory cytokines release. Mice GSDMD deficiency presents a protection role in certain autoinflammatory diseases, which suggests that the gasderim family prospects be the key target for the treatment of autoimmune and autoinflammation diseases. Targeted GSDMD has also developed a variety of inhibitors, which are expected to be used for clinical treatment. Besides, GSDME is crucial in several complicated inflammatory skin diseases which are both autoimmune and autoinflammation related, such as psoriasis, keloid, etc.
Although there are more and more researches on gasdermin family in autoimmune and autoinflammation diseases, there are still a lot of unanswered research questions out there: whether GSDMD deficiency or inhibitor play a protective role in various immune-related diseases? Whether targeting GSDME improve the progression of immune disordered diseases? GSDMA and GSDMB have been shown to be important in IBD, whether other family members also play important roles in IBD? At present, the research of GSDMC is relatively lacking, and further discussion is required. To sum up, better understanding of the gasdermin family and exploring targeted inbitors may improve treatment efficiency of autoimmune diseases, and autoinflammatory diseases.
Author Contributions
WQ and FL, writing, reviewing and editing. FL, contributed a lot during the reviewing.
Funding
This study was supported by Longitudinal subject of Municipal Science and Technology Bureau (grant number XY050033).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Grateau G, Hentgen V, Stojanovic KS, Jéru I, Amselem S, Steichen O. How Should We Approach Classification of Autoinflammatory Diseases? Nat Rev Rheumatol (2013) 9(10):624–9. doi: 10.1038/nrrheum.2013.101
2. Wahren-Herlenius M, Dörner T. Immunopathogenic Mechanisms of Systemic Autoimmune Disease. Lancet (2013) 382(9894):819–31. doi: 10.1016/S0140-6736(13)60954-X
3. Ronnblom L, Pascual V. The Innate Immune System in SLE: Type I Interferons and Dendritic Cells. Lupus (2008) 17(5):394–9. doi: 10.1177/0961203308090020
4. Aksentijevich I, Centola M, Deng ZM, Sood R, Doggett N A. Ancient Missense Mutations in a New Member of the RoRet Gene Family are Likely to Cause Familial Mediterranean Fever. Cell (1997) 90(4):797–807. doi: 10.1016/s0092-8674(00)80539-5
5. Bernot A, Clepet C, Dasilva C, Devaud C, Petit J-L, Caloustian C, et al. The French FMF ConsortiumA Candidate Gene for Familial Mediterranean Fever. Nat. Genet. 17: 25-31. Nat Genet (1997) 17(1):25. doi: 10.1038/ng0997-25
6. McGonagle D, McDermott MF. A Proposed Classification of the Immunological Diseases. PloS Med (2006) 3(8):e297. doi: 10.1371/journal.pmed.0030297
7. Nigrovic PA, Lee PY, Hoffman HM. Monogenic Autoinflammatory Disorders: Conceptual Overview, Phenotype, and Clinical Approach. J Allergy Clin Immunol (2020) 146(5):925–37. doi: 10.1016/j.jaci.2020.08.017
8. Cooper GS, Bynum ML, Somers EC. Recent Insights in the Epidemiology of Autoimmune Diseases: Improved Prevalence Estimates and Understanding of Clustering of Diseases. J Autoimmun (2009) 33(3-4):197–207. doi: 10.1016/j.jaut.2009.09.008
9. Davidson A, Diamond B. Autoimmune Diseases. N Engl J Med (2001) 345(5):340–50. doi: 10.1056/NEJM200108023450506
10. Fugger L, Jensen LT, Rossjohn J. Challenges, Progress, and Prospects of Developing Therapies to Treat Autoimmune Diseases. Cell (2020) 181(1):63–80. doi: 10.1016/j.cell.2020.03.007
11. Abramson J, Husebye ES. Autoimmune Regulator and Self-Tolerance - Molecular and Clinical Aspects. Immunol Rev (2016) 271(1):127–40. doi: 10.1111/imr.12419
12. Seibel MJ, Cooper MS, Zhou H. Glucocorticoid-Induced Osteoporosis: Mechanisms, Management, and Future Perspectives. Lancet Diabetes Endocrinol (2013) 1(1):59–70. doi: 10.1016/S2213-8587(13)70045-7
13. Bakshi J, Tejera Segura B, Wincu C, Rahman A. Unmet Needs in the Pathogenesis and Treatment of Systemic Lupus Erythematosus. Clin Rev Allergy Immunol (2018) 55(3):352–67. doi: 10.1007/s12016-017-8640-5
14. Davis LS, Reimold AM. Research and Therapeutics-Traditional and Emerging Therapies in Systemic Lupus Erythematosus. Rheumatology (Oxford) (2017) 56(suppl_1):i100–13. doi: 10.1093/rheumatology/kew417
15. Cheung TT, McInnes IB. Future Therapeutic Targets in Rheumatoid Arthritis? Semin Immunopathol (2017) 39(4):487–500. doi: 10.1007/s00281-017-0623-3
16. Mavragani CP, Moutsopoulos HM. Sjogren's Syndrome: Old and New Therapeutic Targets. J Autoimmun (2020) 110:102364. doi: 10.1016/j.jaut.2019.102364
17. Consortium FF. A Candidate Gene for Familial Mediterranean Fever. Nat Genet (1997) 17(1):25–31. doi: 10.1038/ng0997-25
18. de Jesus AA, Canna SW, Liu Y, Goldbach-Mansky R. Molecular Mechanisms in Genetically Defined Autoinflammatory Diseases: Disorders of Amplified Danger Signaling. Annu Rev Immunol (2015) 33:823–74. doi: 10.1146/annurev-immunol-032414-112227
19. Manthiram K, Zhou Q, Aksentijevich I, Kastner DL. The Monogenic Autoinflammatory Diseases Define New Pathways in Human Innate Immunity and Inflammation. Nat Immunol (2017) 18(8):832–42. doi: 10.1038/ni.3777
20. Saeki N, Kuwahara Y, Sasaki H, Satoh H, Shiroishi T. Gasdermin (Gsdm) Localizing to Mouse Chromosome 11 is Predominantly Expressed in Upper Gastrointestinal Tract But Significantly Suppressed in Human Gastric Cancer Cells. Mamm Genome (2000) 11(9):718–24. doi: 10.1007/s003350010138
21. Sato H, Koide T, Masuya H, Wakana S, Sagai T, Umezawa A, et al. A New Mutation Rim3 Resembling Re Den Is Mapped Close to Retinoic Acid Receptor Alpha ( Rara ) Gene on Mouse Chromosome 11. Mamm Genome (1998) 9(1):20–5. doi: 10.1007/s003359900673
22. Tamura M, Tanaka S, Fujii T, Aoki A, Komiyama H, Ezawa K, et al. Members of a Novel Gene Family, Gsdm, are Expressed Exclusively in the Epithelium of the Skin and Gastrointestinal Tract in a Highly Tissue-Specific Manner. Genomics (2007) 89(5):618–29. doi: 10.1016/j.ygeno.2007.01.003
23. Broz P, Pelegrin P, Shao F. The Gasdermins, a Protein Family Executing Cell Death and Inflammation. Nat Rev Immunol (2020) 20(3):143–57. doi: 10.1038/s41577-019-0228-2
24. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-Forming Activity and Structural Autoinhibition of the Gasdermin Family. Nature (2016) 535(7610):111–6. doi: 10.1038/nature18590
25. Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES, et al. Cleavage of DFNA5 by Caspase-3 During Apoptosis Mediates Progression to Secondary Necrotic/Pyroptotic Cell Death. Nat Commun (2017) 8(1). doi: 10.1038/ncomms14128
26. Liu Z, Wang C, Yang J, Zhou B, Yang R, Ramachandran R, et al. Crystal Structures of the Full-Length Murine and Human Gasdermin D Reveal Mechanisms of Autoinhibition, Lipid Binding, and Oligomerization. Immunity (2019) 51(1):43–49 e4. doi: 10.1016/j.immuni.2019.04.017
27. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy Drugs Induce Pyroptosis Through Caspase-3 Cleavage of a Gasdermin. Nature (2017) 547(7661):99–103. doi: 10.1038/nature22393
28. Kolb JP, Oguin TH 3rd, Oberst A, Martinez J. Programmed Cell Death and Inflammation: Winter Is Coming. Trends Immunol (2017) 38(10):705–18. doi: 10.1016/j.it.2017.06.009
29. Mathur A, Feng S, Hayward JA, Ngo C, Fox D, Atmosukart II, et al. A Multicomponent Toxin From Bacillus Cereus Incites Inflammation and Shapes Host Outcome via the NLRP3 Inflammasome. Nat Microbiol (2019) 4(2):362–74. doi: 10.1038/s41564-018-0318-0
30. Kesavardhana S, Malireddi RKS, Kanneganti TD. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu Rev Immunol (2020) 38:567–95. doi: 10.1146/annurev-immunol-073119-095439
31. Abu Khweek A, Amer AO. Pyroptotic and non-Pyroptotic Effector Functions of Caspase-11. Immunol Rev (2020) 297(1):39–52. doi: 10.1111/imr.12910
32. Kumari P, Russo AJ, Shivcharan S, Rathinam VA. AIM2 in Health and Disease: Inflammasome and Beyond. Immunol Rev (2020) 297(1):83–95. doi: 10.1111/imr.12903
33. Zhang Y, Chen X, Gueydan C, Han J. Plasma Membrane Changes During Programmed Cell Deaths. Cell Res (2018) 28(1):9–21. doi: 10.1038/cr.2017.133
34. Rashidi M, Wicks IP, Vince JE. Inflammasomes and Cell Death: Common Pathways in Microparticle Diseases. Trends Mol Med (2020) 26(11):1003–20. doi: 10.1016/j.molmed.2020.06.005
35. Man SM, Karki R, Kanneganti T-D. Molecular Mechanisms and Functions of Pyroptosis, Inflammatory Caspases and Inflammasomes in Infectious Diseases. Immunol Rev (2017) 277(1):61–75. doi: 10.1111/imr.12534
36. Jorgensen M, Edward A. Pyroptotic Cell Death Defends Against Intracellular Pathogens. Immunol Rev (2015) 265(1):130–42. doi: 10.1111/imr.12287
37. Chen KW, Demarco B, Broz P. Beyond Inflammasomes: Emerging Function of Gasdermins During Apoptosis and NETosis. EMBO J (2020) 39(2):e103397. doi: 10.15252/embj.2019103397
38. Alehashemi S, Goldbach-Mansky R. Human Autoinflammatory Diseases Mediated by NLRP3-, Pyrin-, NLRP1-, and NLRC4-Inflammasome Dysregulation Updates on Diagnosis, Treatment, and the Respective Roles of IL-1 and IL-18. Front Immunol (2020) 11:1840. doi: 10.3389/fimmu.2020.01840
39. Cox JH, Starr AE, Kappelhoff R, Yan R, Roberts CR, Overall CM, et al. Matrix Metalloproteinase 8 Deficiency in Mice Exacerbates Inflammatory Arthritis Through Delayed Neutrophil Apoptosis and Reduced Caspase 11 Expression. Arthritis Rheum (2010) 62(12):3645–55. doi: 10.1002/art.27757
40. De Schutter E, Croes L, Ibrahim J, Pauwels P, Op deBeeck K, Vandenabeele P, et al. GSDME and its Role in Cancer: From Behind the Scenes to the Front of the Stage. Int J Cancer (2021) 148(12):2872–83. doi: 10.1002/ijc.33390
41. Liu X, Xia S, Zhang Z, Wu H, Lieberman J. Channelling Inflammation: Gasdermins in Physiology and Disease. Nat Rev Drug Discov (2021) 20(5):384–405. doi: 10.1038/s41573-021-00154-z
42. Andrade WA, Zamboni DS. NLRC4 Biology in Immunity and Inflammation. J Leukoc Biol (2020) 108(4):1117–27. doi: 10.1002/JLB.3MR0420-573R
43. Bourdonnay E, Henry T. Transcriptional and Epigenetic Regulation of Gasdermins. J Mol Biol (2022) 434(4):167253. doi: 10.1016/j.jmb.2021.167253
44. Saeki N, Kim DH, Usui T, Aoyagi K, Tatsuta T, Aoki K, et al. GASDERMIN, Suppressed Frequently in Gastric Cancer, is a Target of LMO1 in TGF-Beta-Dependent Apoptotic Signalling. Oncogene (2007) 26(45):6488–98. doi: 10.1038/sj.onc.1210475
45. Soderman J, Berglind L, Almer S. Gene Expression-Genotype Analysis Implicates GSDMA, GSDMB, and LRRC3C as Contributors to Inflammatory Bowel Disease Susceptibility. BioMed Res Int (2015) 2015:834805. doi: 10.1155/2015/834805
46. Yu J, Kang M-J, Kim B-J, Kwon J-W, Song Y-H, Choi W-A, et al. Polymorphisms in GSDMA and GSDMB are Associated With Asthma Susceptibility, Atopy and BHR. Pediatr Pulmonol (2011) 46(7):701–8. doi: 10.1002/ppul.21424
47. Li X, Ampleford EJ, Howard TD, Moore WC, Torgerson DG, Li H, et al. Genome-Wide Association Studies of Asthma Indicate Opposite Immunopathogenesis Direction From Autoimmune Diseases. J Allergy Clin Immunol (2012) 130(4):861–8.e7. doi: 10.1016/j.jaci.2012.04.041
48. Moreno-Moral A, Bagnati M, Koturan S, Ko J-H, Fonseca C, Harmston N, et al. Changes in Macrophage Transcriptome Associate With Systemic Sclerosis and Mediate GSDMA Contribution to Disease Risk. Ann Rheum Dis (2018) 77(4):596–601. doi: 10.1136/annrheumdis-2017-212454
49. Saeki N, Usui T, Aoyagi K, Kim DH, Sato M, Mabuchi T, et al. Distinctive Expression and Function of Four GSDM Family Genes (GSDMA-D) in Normal and Malignant Upper Gastrointestinal Epithelium. Genes Chromosomes Cancer (2009) 48(3):261–71. doi: 10.1002/gcc.20636
50. Katoh M, Katoh M. Evolutionary Recombination Hotspot Around GSDML-GSDM Locus is Closely Linked to the Oncogenomic Recombination Hotspot Around the PPP1R1B-ERBB2-GRB7 Amplicon. Int J Oncol (2004) 24(4):757–63. doi: 10.3892/ijo.24.4.757
51. Saeki N, Usui T, Aoyagi K, Kim DH, Sato M, Mabuchi T, et al. Distinctive Expression and Function of Four GSDM Family Genes (GSDMA-D) in Normal and Malignant Upper Gastrointestinal Epithelium. Genes Chromosomes Cancer (2009) 48(3):261–71. doi: 10.1002/gcc.20636.
52. Carl-McGrath S, Schneider-Stock R, Ebert M, Röcken C. Differential Expression and Localisation of Gasdermin-Like (GSDML), A Novel Member of the Cancer-Associated GSDMDC Protein Family, in Neoplastic and Non-Neoplastic Gastric, Hepatic, and Colon Tissues. Pathology (2008) 40(1):13–24. doi: 10.1080/00313020701716250
53. Hu Y, Jin S, Cheng L, Liu G, Jiang Q. Autoimmune Disease Variants Regulate GSDMB Gene Expression in Human Immune Cells and Whole Blood. Proc Natl Acad Sci (2017) 114(38):E7860. doi: 10.1073/pnas.1712127114
54. Das S, Miller M, Beppu AK, Mueller J, McGeough MD, Vuong C, et al. GSDMB Induces an Asthma Phenotype Characterized by Increased Airway Responsiveness and Remodeling Without Lung Inflammation. Proc Natl Acad Sci U S A (2016) 113(46):13132–7. doi: 10.1073/pnas.1610433113
55. Rana N, Privitera G, Kondolf HC, Bulek K, Lechuga S, De Salvo C, et al. GSDMB is Increased in IBD and Regulates Epithelial Restitution/Repair Independent of Pyroptosis. Cell (2022) 185(2):283–98.e17. doi: 10.1016/j.cell.2021.12.024
56. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-Microbe Interactions Have Shaped the Genetic Architecture of Inflammatory Bowel Disease. Nature (2012) 491(7422):119–24. doi: 10.1038/nature11582
57. Chen Q, Shi P, Wang Y, Zou D, Wu X, Wang D, et al. GSDMB Promotes non-Canonical Pyroptosis by Enhancing Caspase-4 Activity. J Mol Cell Biol (2019) 11(6):496–508. doi: 10.1093/jmcb/mjy056
58. Shi J, Gao W, Feng S. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci (2017) 42(4):245–54. doi: 10.1016/j.tibs.2016.10.004
59. Wang J, Tang T, Yang H, Yao X, Chen L, Liu W, et al. The Expression of Fas Ligand on Normal and Stabbed-Disc Cells in a Rabbit Model of Intervertebral Disc Degeneration: A Possible Pathogenesis. J Neurosurg Spine (2007) 6(5):425–30. doi: 10.3171/spi.2007.6.5.425
60. Jiang H, Moro A, Liu Y, Wang J, Meng D, Zhan X, et al. Two GWAS-Identified Variants are Associated With Lumbar Spinal Stenosis and Gasdermin-C Expression in Chinese Population. Sci Rep (2020) 10(1):21069. doi: 10.1038/s41598-020-78249-7
61. Kusumaningrum N, Hun Lee D, Yoon H-S, Park C-H, Ho Chung J. Ultraviolet Light-Induced Gasdermin C Expression is Mediated via TRPV1/calcium/calcineurin/NFATc1 Signaling. Int J Mol Med (2018) 42(5):2859–66. doi: 10.3892/ijmm.2018.3839
62. Kusumaningrum N, Hun Lee D, Yoon H-S, Yeon K K, Park C-H, Ho Chung J. Gasdermin C is Induced by Ultraviolet Light and Contributes to MMP-1 Expression via Activation of ERK and JNK Pathways. J Dermatol Sci (2018) 90(2):180–9. doi: 10.1016/j.jdermsci.2018.01.015
63. Liu X, Lieberman J. Knocking 'Em Dead: Pore-Forming Proteins in Immune Defense. Annu Rev Immunol (2020) 38:455–85. doi: 10.1146/annurev-immunol-111319-023800
64. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli V G, Wu H, et al. Inflammasome-Activated Gasdermin D Causes Pyroptosis by Forming Membrane Pores. Nature (2016) 535(7610):153–8. doi: 10.1038/nature18629
65. Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD Membrane Pore Formation Constitutes the Mechanism of Pyroptotic Cell Death. EMBO J (2016) 35(16):1766–78. doi: 10.15252/embj.201694696
66. Mulvihill E, Sborgi L, Mari S A, Pfreundschuh M, Hiller S, Müller DJ, et al. Mechanism of Membrane Pore Formation by Human Gasdermin-D. EMBO J (2018) 37(14):e98321. doi: 10.15252/embj.201798321
67. Lammert CR, Frost EL, Bellinger CE, Bolte AC, McKee CA, Hurt ME, et al. AIM2 Inflammasome Surveillance of DNA Damage Shapes Neurodevelopment. Nature (2020) 580(7805):647–52. doi: 10.1038/s41586-020-2174-3
68. Li S, Wu Y, Yang D, Wu C, Ma C, Liu X, et al. Gasdermin D in Peripheral Myeloid Cells Drives Neuroinflammation in Experimental Autoimmune Encephalomyelitis. J Exp Med (2019) 216(11):2562–81. doi: 10.1084/jem.20190377
69. Biliktu M, Senol SP, Temiz-Resitoglu M, Guden DS, Horat MF, Sahan-Firat S, et al. Pharmacological Inhibition of Soluble Epoxide Hydrolase Attenuates Chronic Experimental Autoimmune Encephalomyelitis by Modulating Inflammatory and Anti-Inflammatory Pathways in an Inflammasome-Dependent and -Independent Manner. Inflammopharmacology (2020) 28(6):1509–24. doi: 10.1007/s10787-020-00691-w
70. Kenealy S, Manils J, Raverdeau M, Munoz-Wolf N, Barber G, Liddicoat A, et al. Caspase-11-Mediated Cell Death Contributes to the Pathogenesis of Imiquimod-Induced Psoriasis. J Invest Dermatol (2019) 139(11):2389–2393 e3. doi: 10.1016/j.jid.2019.05.010
71. Chadha S, Behl T, Bungau S, Kumar A, Arora R, Gupta A, et al. Mechanistic Insights Into the Role of Pyroptosis in Rheumatoid Arthritis. Curr Res Transl Med (2020) 68(4):151–8. doi: 10.1016/j.retram.2020.07.003
72. Chen Y, Qin X, An Q, Yi J, Feng F, Yin D, et al. Mesenchymal Stromal Cells Directly Promote Inflammation by Canonical NLRP3 and Non-Canonical Caspase-11 Inflammasomes. EBioMedicine (2018) 32:31–42. doi: 10.1016/j.ebiom.2018.05.023
73. Dang S, Xu H, Xu C, Cai W, Li Q, Cheng Y, et al. Autophagy Regulates the Therapeutic Potential of Mesenchymal Stem Cells in Experimental Autoimmune Encephalomyelitis. Autophagy (2014) 10(7):1301–15. doi: 10.4161/auto.28771
74. Kanneganti A, Subbarao Malireddi RK, Pedro Saavedra HV, Walle LV, Gorp HV, Kambara H, et al. GSDMD is Critical for Autoinflammatory Pathology in a Mouse Model of Familial Mediterranean Fever. J Exp Med (2018) 215(6):1519–29. doi: 10.1084/jem.20172060
75. Xia J, Wang C, Yao J-C, Alippe Y, Xu C, Kress D, et al. Gasdermin D Mediates the Pathogenesis of Neonatal-Onset Multisystem Inflammatory Disease in Mice. PloS Biol (2018) 16(11):e3000047. doi: 10.1371/journal.pbio.3000047
76. Van Laer L, Huizing EH, Verstreken M, van Zuijlen D, Wauters JG, Bossuyt P J, et al. Nonsyndromic Hearing Impairment is Associated With a Mutation in DFNA5. Nat Genet (1998) 20(2):194–7. doi: 10.1038/2503
77. Yu C, Meng X, Zhang S, Zhao G, Hu L, Kong X, et al. A 3-Nucleotide Deletion in the Polypyrimidine Tract of Intron 7 of the DFNA5 Gene Causes Nonsyndromic Hearing Impairment in a Chinese Family. Genomics (2003) 82(5):575–9. doi: 10.1016/S0888-7543(03)00175-7
78. Bischoff AMLC, Luijendijk MWJ, Huygen PLM, van Duijnhoven G, De Leenheer EMR, Oudesluijs GG, et al. A Novel Mutation Identified in the DFNA5 Gene in a Dutch Family: A Clinical and Genetic Evaluation. Audiol Neurootol (2004) 9(1):34–46. doi: 10.1159/000074185
79. Cheng J, Han D Y, Dai P, Sun H J, Tao R, Sun Q, et al. A Novel DFNA5 Mutation, IVS8+4 A>G, in the Splice Donor Site of Intron 8 Causes Late-Onset non-Syndromic Hearing Loss in a Chinese Family. Clin Genet (2007) 72(5):471–7. doi: 10.1111/j.1399-0004.2007.00889.x
80. Park H-J, Cho H-J, Baek J-I, Ben-Yosef T, Kwon T-J, Griffith AJ, et al. Evidence for a Founder Mutation Causing DFNA5 Hearing Loss in East Asians. J Hum Genet (2010) 55(1):59–62. doi: 10.1038/jhg.2009.114
81. Li-Yang M-N, Shen X-F, Wei Q -J, Yao J, Lu Y-J, Cao X, et al. IVS8+1 DelG, A Novel Splice Site Mutation Causing DFNA5 Deafness in a Chinese Family. 中华医学杂志:英文版 (2015) 18):6. doi: 10.4103/0366-6999.164980
82. Beeck K, Laer LV, Camp GV. DFNA5, a Gene Involved in Hearing Loss and Cancer: A Review. Ann Otol Rhinol Laryngol (2012) 121(3):197–207. doi: 10.1177/000348941212100310
83. Nishio A, Noguchi Y, Sato T, Naruse T K, Kimura A, Takagi A, et al. A DFNA5 Mutation Identified in Japanese Families With Autosomal Dominant Hereditary Hearing Loss. Ann Hum Genet (2014) 78(2):83–91. doi: 10.1111/ahg.12053
84. Thompson DA, Weigel RJ. Characterization of a Gene That is Inversely Correlated With Estrogen Receptor Expression (ICERE-1) in Breast Carcinomas. Eur J Biochem (1998) 252(1):169–77. doi: 10.1046/j.1432-1327.1998.2520169.x
85. Op deBeeck K, Van Camp G, Thys S, Cools N, Callebaut I, Vrijens K, et al. The DFNA5 Gene, Responsible for Hearing Loss and Involved in Cancer, Encodes a Novel Apoptosis-Inducing Protein. Eur J Hum Genet (2011) 19(9):965–73. doi: 10.1038/ejhg.2011.63
86. Yu J, Li S, Qi J, Chen Z, Wu Y, Guo J, et al. Cleavage of GSDME by Caspase-3 Determines Lobaplatin-Induced Pyroptosis in Colon Cancer Cells. Cell Death Dis (2019) 10(3):193. doi: 10.1038/s41419-019-1441-4
87. Lim C P, Phan T T, Lim I J, Cao X. Cytokine Profiling and Stat3 Phosphorylation in Epithelial-Mesenchymal Interactions Between Keloid Keratinocytes and Fibroblasts. J Invest Dermatol (2009) 129(4):851–61. doi: 10.1038/jid.2008.337
88. Atamas SP, White B. Cytokine Regulation of Pulmonary Fibrosis in Scleroderma. Cytokine Growth Factor Rev (2003) 14(6):537–50. doi: 10.1016/S1359-6101(03)00060-1
89. Marden G, Wan Q, Wilks J, Nevin K, Feeney M, Wisniacki N, et al. The Role of the Oncostatin M/OSM Receptor Beta Axis in Activating Dermal Microvascular Endothelial Cells in Systemic Sclerosis. Arthritis Res Ther (2020) 22(1):179. doi: 10.1186/s13075-020-02266-0
90. Boniface K, Diveu C, Morel F, Pedretti N, Froger J, Ravon E, et al. Oncostatin M Secreted by Skin Infiltrating T Lymphocytes Is a Potent Keratinocyte Activator Involved in Skin Inflammation. J Immunol (2007) 178(7):4615–22. doi: 10.4049/jimmunol.178.7.4615
91. Gazel A, Rosdy M, Bertino B, Tornier C, Sahuc F, Blumenberg M. A Characteristic Subset of Psoriasis-Associated Genes is Induced by Oncostatin-M in Reconstituted Epidermis. J Invest Dermatol (2006) 126(12):2647–57. doi: 10.1038/sj.jid.5700461
92. Liu J, Zhong Y, Liu H, Yang H, Lu P, Shi Y, et al. Oncostatin M Sensitizes Keratinocytes to UVB-Induced Inflammation via GSDME-Mediated Pyroptosis. J Dermatol Sci (2021) 104(2):95–103. doi: 10.1016/j.jdermsci.2021.09.004
93. Li Y, Yuan Y, Huang Z-X, Chen H, Lan R, Wang Z, et al. GSDME-Mediated Pyroptosis Promotes Inflammation and Fibrosis in Obstructive Nephropathy. Cell Death Differ (2021) 28(8):2333–50. doi: 10.1038/s41418-021-00755-6
94. Sun L, et al. Mixed Lineage Kinase Domain-Like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell (2012) 148(1-2):213–27. doi: 10.1016/j.cell.2011.11.031
95. Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, et al. Chemical Disruption of the Pyroptotic Pore-Forming Protein Gasdermin D Inhibits Inflammatory Cell Death and Sepsis. Sci Immunol (2018) 3(26):eaat2738. doi: 10.1126/sciimmunol.aat2738
96. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. The Pyroptotic Cell Death Effector Gasdermin D Is Activated by Gout-Associated Uric Acid Crystals But Is Dispensable for Cell Death and IL-1β Release. J Immunol (2019) 203(3):736–48. doi: 10.4049/jimmunol.1900228
97. Hu J J, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, et al. FDA-Approved Disulfiram Inhibits Pyroptosis by Blocking Gasdermin D Pore Formation. Nat Immunol (2020) 21(7):736–45. doi: 10.1038/s41590-020-0669-6
98. Sollberger G, Choidas A, Lawrence Burn G, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin D Plays a Vital Role in the Generation of Neutrophil Extracellular Traps. Sci Immunol (2018) 3(26):eaar6689. doi: 10.1126/sciimmunol.aar6689
99. Galli G, Saleh M. Immunometabolism of Macrophages in Bacterial Infections. Front Cell Infect Microbiol (2020) 10:607650. doi: 10.3389/fcimb.2020.607650
Keywords: gasdermins, autoimmune diseases, autoinflammatory disease, pyroptosis, inflammasome
Citation: Liang F, Qin W, Zeng Y and Wang D (2022) Modulation of Autoimmune and Autoinflammatory Diseases by Gasdermins. Front. Immunol. 13:841729. doi: 10.3389/fimmu.2022.841729
Received: 22 December 2021; Accepted: 05 May 2022;
Published: 01 June 2022.
Edited by:
Zhongde Ye, Stanford University, United StatesReviewed by:
Nan Wang, Stanford University, United StatesRyu Watanabe, Osaka Metropolitan University, Japan
Copyright © 2022 Liang, Qin, Zeng and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dan Wang, RHJkYW53YW5nQDE2My5jb20=
†These authors have contributed equally to this work