 Julia M. Murphy
Julia M. Murphy Louis Ngai
Louis Ngai Arthur Mortha
Arthur Mortha Sarah Q. Crome
Sarah Q. Crome- 1Department of Immunology, University of Toronto, Toronto, ON, Canada
- 2Toronto General Hospital Research Institute, University Health Network, Toronto, ON, Canada
- 3Ajmera Transplant Centre, University Health Network, Toronto, ON, Canada
Tissue-resident immune cells reside in distinct niches across organs, where they contribute to tissue homeostasis and rapidly respond to perturbations in the local microenvironment. Innate lymphoid cells (ILCs) are a family of innate immune cells that regulate immune and tissue homeostasis. Across anatomical locations throughout the body, ILCs adopt tissue-specific fates, differing from circulating ILC populations. Adaptations of ILCs to microenvironmental changes have been documented in several inflammatory contexts, including obesity, asthma, and inflammatory bowel disease. While our understanding of ILC functions within tissues have predominantly been based on mouse studies, development of advanced single cell platforms to study tissue-resident ILCs in humans and emerging patient-based data is providing new insights into this lymphocyte family. Within this review, we discuss current concepts of ILC fate and function, exploring tissue-specific functions of ILCs and their contribution to health and disease across organ systems.
Introduction
Innate lymphoid cells (ILCs) orchestrate immune responses to signals such as cytokines, alarmins, neuropeptides and hormones, interacting with hematopoietic and non-hematopoietic cells alike. ILCs lack rearranged antigen receptors and while predominantly tissue-resident, are also observed in circulation and secondary lymphoid tissues where they exhibit distinct spatial and temporal functions (1). Outside of roles in immunity, ILCs have key roles in maintaining tissue homeostasis, promoting tissue repair, and regulating inflammation. Via crosstalk with parenchymal cells, ILCs are also involved in processes previously thought to lack immune system influence, such as thermal regulation, neuronal signal transduction, circadian rhythms, and tissue remodeling (2–6). The regulation of both immune functions and tissue-specific processes by ILCs highlights the importance of understanding how they respond and function within tissue niches, and conversely how ILC biology is controlled by the microenvironment in which they reside.
Development of ILCs in non-lymphoid tissues occurs when circulating ILC progenitors seed tissue niches, and requires the expression of local survival factors including IL-7 and thymic stromal lymphopoietin (TSLP) (7, 8). Differentiated ILCs express signature cytokines and transcription factors that parallel CD4+ and CD8+ T cells in both humans and mice (Figure 1) (6, 8), and can be broadly categorized as cytotoxic (NK cells) or non-cytotoxic ‘helper’ ILCs. Human NK cells express TBET and Eomesodermin (EOMES), release IFN-γ and TNF-α and are grouped into CD56dimCD16+ or CD56brightCD16- NK cells. CD56dimCD16+ NK cells express killer cell immunoglobulin-like receptors (KIRs) and exhibit profound cytotoxic potential (6, 8). CD56brightCD16- NK cells lack KIR expression but are superior producers of IFN-γ and TNF-α (9, 10). NK cells discriminate between self and non-self or altered-self and function in anti-viral and anti-tumor immunity similar to CD8+ cytotoxic T cells (6, 8). ‘Helper’ ILC (hILCs) are non-cytotoxic and are classified based on function and development into Group 1 (ILC1s), Group 2 (ILC2s), Group 3 (ILC3s) as well as Lymphoid Tissue inducer LTi cells (6). ILC1s produce IFN-γ in a TBET-dependent but EOMES-independent manner (6). ILC2s express GATA-3 and RORα and secrete interleukin (IL)-4, IL-5, IL-9, IL-13 and Amphiregulin (AREG), aiding in anti-parasite immunity or the promotion of allergic responses (6). ILC3s rely on the transcription factor RORC and produce IL-22, IL-17, and GM-CSF (6). ILC3s include subsets which express natural cytotoxicity receptors (NCRs) NKp44 (human) and NKp46 (mouse and human). LTi cells express ILC3-associated transcription factors and cytokines but also express surface Lymphotoxin (sLT) (11). Further, ILCs with immunosuppressive activity have been identified in cancer, intestinal inflammation, allergy, autoimmunity and ischemia reperfusion injury (12–18). These include both NK-like ILCs, IL-10 producing ILC2s (ILC210) and ID3+ regulatory ILCs [reviewed in Jegatheeswaran et al. (19)]. Despite growing appreciation of ILCs with regulatory functions, their development and function are poorly characterized, particularly in humans.
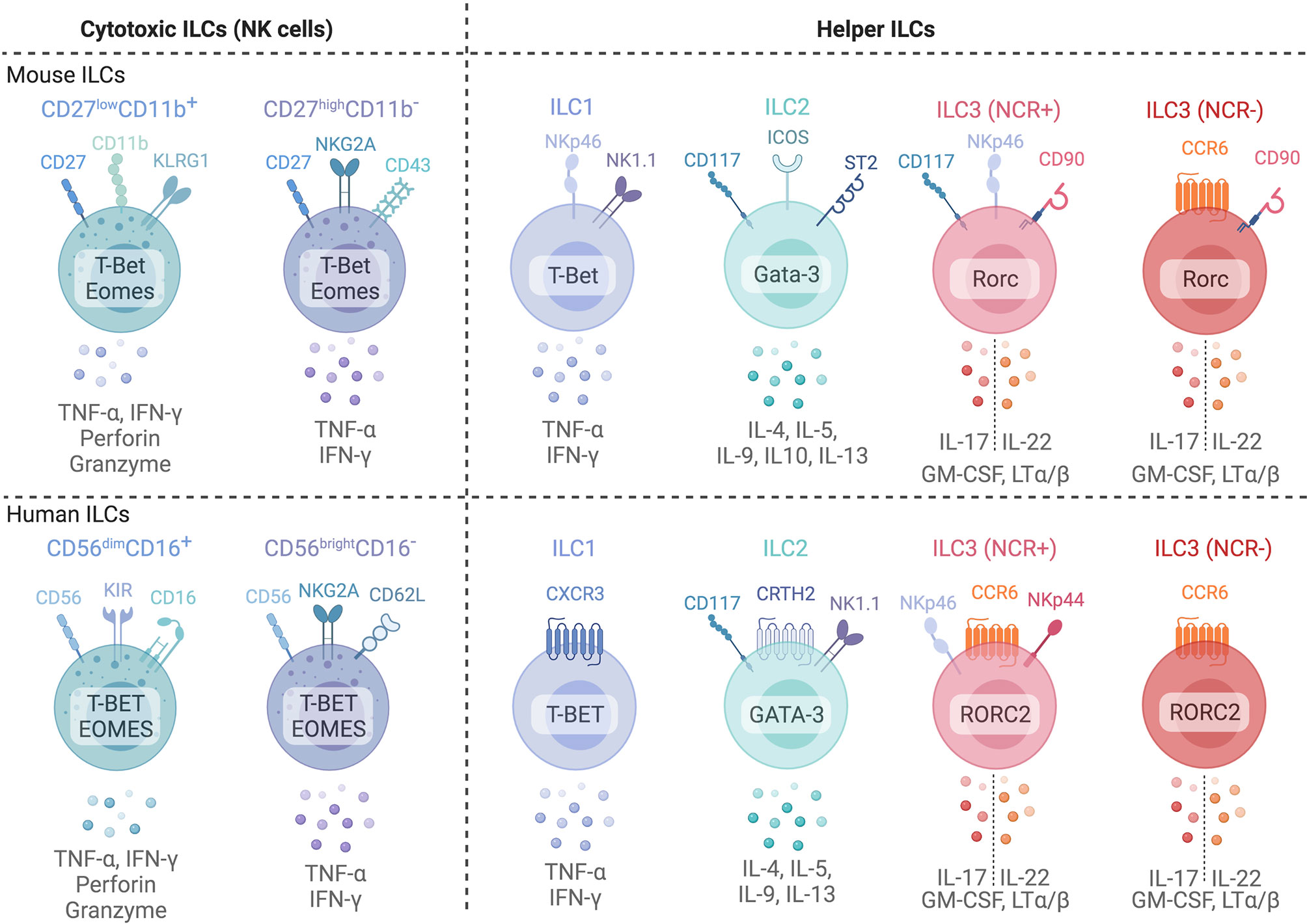
Figure 1 Common phenotypic markers of mice (top) and human (bottom) ILCs and their common cytokine expression profiles. Cytotoxic Natural Killer cells can be subdivided into two major subsets based on surface marker expression in both mice and humans. In mice, NK cells are subdivided into two subsets based on CD27 and CD11b expression: CD27highCD11b- immature NK cells and mature CD27lowCD11b+ NK cells. In humans, CD56brightCD16- and CD56dimCD16+ are generally used to identify immature and mature NK cells in blood. However, tissue NK cells often display a CD56brightCD16- phenotype. ILC1s, ILC2s, and ILC3s are classified based on surface marker and transcription factor expression profiles that parallel CD4+ T helper subsets. ILC3s are further subdivided into natural cytotoxicity receptor (NCR)+ and NCR- subsets. Created with Biorender.org.
Mouse studies identified central roles for ILCs in regulating tissue homeostasis, repair and remodeling, transforming our understanding of cellular interactions between immune cells and the tissues in which they reside. Across tissue microenvironments, ILCs adapt and acquire distinct phenotypes and functional properties (Figure 2). While ILC subsets have important functions within these tissues, dysregulation of ILC numbers and functions is associated with diverse human pathologies including arthritis, diabetes, psoriasis, asthma, and inflammatory bowel disease [reviewed in (20)], highlighting the need to identify how local tissue factors promote or inhibit inflammatory ILC responses. Within this review, we explore NK cell and hILC biology across different tissues in health and disease, highlighting evidence of similarities between human and mouse ILC function where data is available. We summarize current understanding of organ-specific functions of ILCs, focusing on their contributions to tissue homeostasis, host-defense, and inflammatory disease progression across the body.
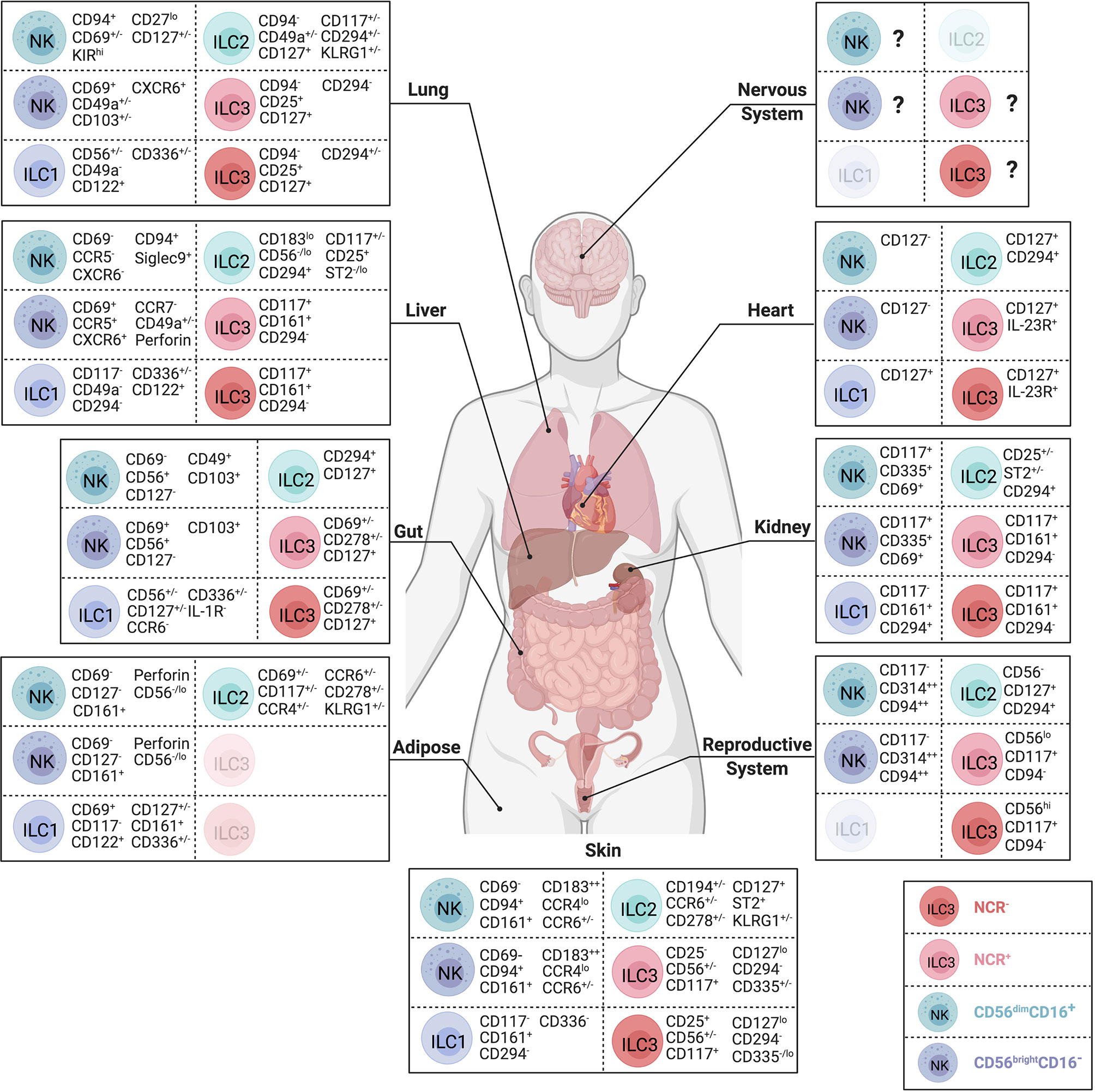
Figure 2 Body-wide distribution and surface phenotypes of human ILCs. Surface marker expression of CD56dim NK cells (teal), CD56brightNK cells (dark blue), ILC1s (purple), ILC2s (green), NCR- ILC3s (red) and NCR+ ILC3s (pink) in nervous system, lung, heart, liver, kidney, gut, reproductive system, adipose tissue, and skin. ILC subsets that have not yet been identified during steady state visualized in lighter color. Represented selection of ILC markers is based on the consistent use of these markers across multiple independent studies. Created with Biorender.org.
ILCs in the Nervous System
While the central nervous system (CNS) is considered an immune-privileged site with minimal immune infiltrate, ILCs have been identified in the CNS of healthy humans and mice, accounting for ~2.5% of leukocytes by sequencing (21–25). CNS-resident NK cells are present in low proportions in the naïve mouse brain and enriched in a IL-2R+ CD27+ CD62Lhigh subset, suggesting a more mature phenotype compared to infiltrating NK cells (22). CNS ILC2s accumulate with age and reside in the healthy murine meninges, localizing within dural sinuses and surrounding blood vessels (23–25). Interestingly, the transcriptional profile of meningeal ILC2s showed downregulation of genes related to metabolism, signal transduction, and inflammation compared to lung-derived ILC2s, suggesting a tissue-specific quiescent adaptation to the CNS environment (23). Upon spinal cord injury in mice, ILC2s migrate to the injured site independently of IL-33 and upregulate Calca (CGRP) and its receptor Ramp3, associated with nerve regeneration (23), yet the regenerative activity of ILC2s in the spinal cord remains to be demonstrated experimentally.
ILCs in Multiple Sclerosis
Multiple sclerosis (MS) is a demyelinating and neurodegenerative autoimmune disease that is one of the most common neurological disabilities in young adults (26). NK cells mediate several treatment-related effects in MS patients (Figure 3A). For example, Daclizumab targets the high affinity IL-2 receptor (CD25), inhibiting activated T cells and resulting in greater availability of IL-2, which expands CD56bright NK cells expressing high levels of the medium-affinity IL-2 receptor chain (CD122) (18, 27). This expansion of CD56bright NK cells or elevated baseline expression of CD122 in patients correlated with lower inflammation and fewer inflammatory lesions (18, 28). While T cells are only modestly depleted by Daclizumab directly, induction of T cell apoptosis by CD56bright NK cells is supported by findings of Granzyme K+ NK cell co-localization with T cells in active MS lesions (18, 29, 30). Takahashi et al. further found that during the remission phase of MS, CD95 expression increased on NK cells alongside decreased response of memory T cells, suggesting that CD95+ NK cells regulate autoimmune memory T cell responses during remission (31). In autologous hematopoietic stem cell transplantation, another MS treatment modality, NK cells reconstitute faster than CD4+ T cells and regulate disease-promoting Th17 cells via NKG2D-mediated cytotoxicity, preventing lesion formation and relapse (32).
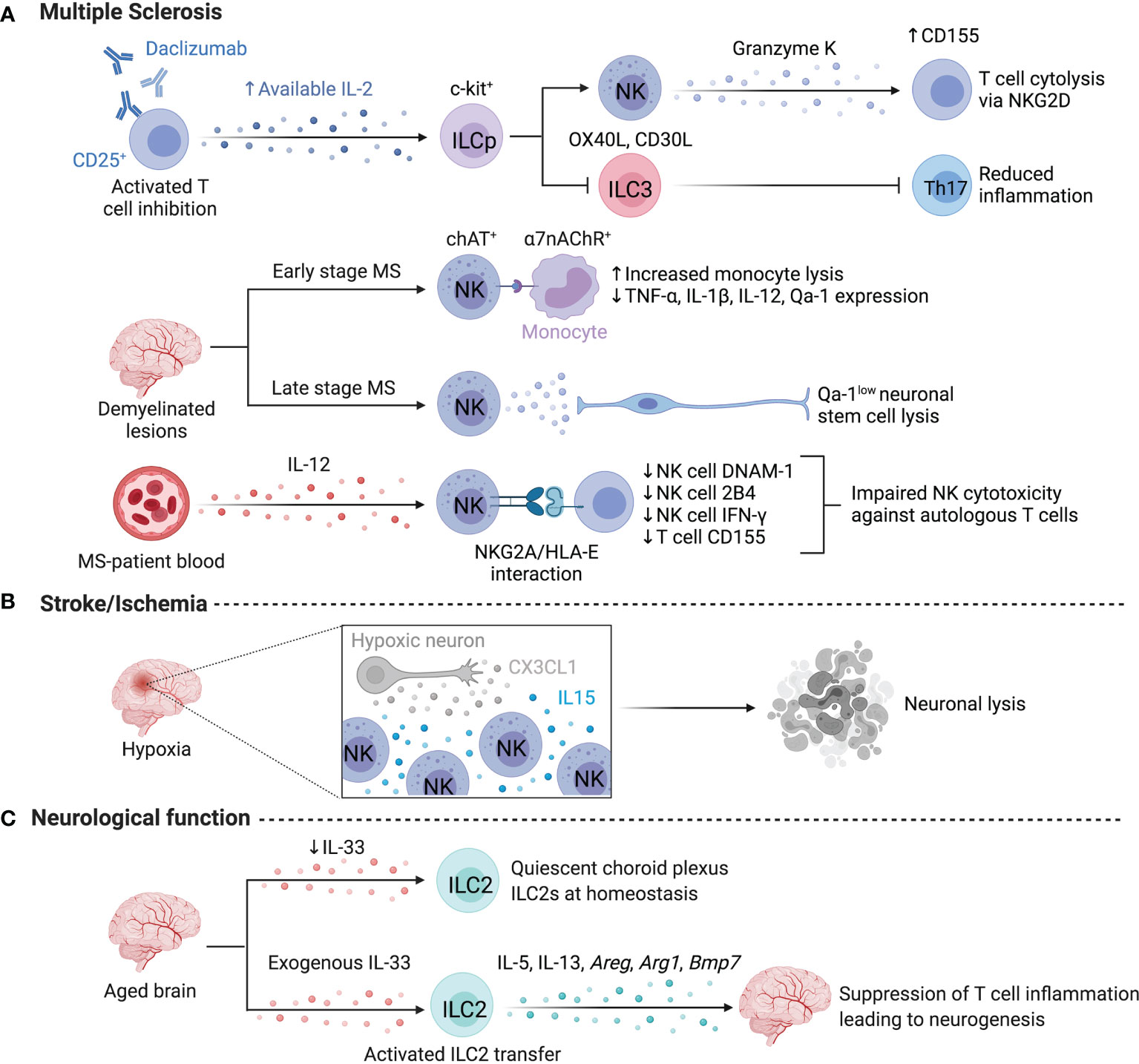
Figure 3 ILCs in the nervous system. Limited information exists on human ILCs in the nervous system at steady state due to challenges in obtaining samples, however, several studies focus on ILC activity in multiple sclerosis or stroke. (A) Daclizumab-driven inhibition of T cells resulted in the expansion of NK cells and the elevated lysis of T cells. Daclizumab treatment lowers the abundance of Lin-c-kit+RORC2+ ILCs and dampens Th17-associated inflammation by lowering IL-17 and GM-CSF. ChAT+ NK cells preferentially localize to demyelinated lesions in the human brain to dampen monocyte-driven inflammation via the a7-nicotinic acetylcholine receptor (a7nAChR), rendering myeloid cells more susceptible to lysis at early stages of MS. Conversely, NK cell-mediated lysis negatively impacts regeneration during later stages of MS by targeting, Qa-1low neuronal stem cells. NK cell activation may be impaired by NKG2A/HLA-E interactions with autologous CD4+ T cells. (B) After a stroke, CX3CL1 from hypoxic neurons recruits NK cells, while local IL-15 levels facilitate NK cell enrichment and promote high NKG2D expression and neuronal lysis. (C) ILC2s impact neurological functions in murine brain and are supported by exogenous IL-33 to suppress T cell inflammation and enhance neurogenesis. Created with Biorender.org.
A higher ratio of CD56bright to CD56dim NK cells is observed in the cerebrospinal fluid of patients with MS compared to those with other inflammatory and non-inflammatory neurological diseases, suggesting an MS-specific alteration in resident NK cells with controversial effects on the abundance of NK cells in circulation (30, 33, 34). Despite conflicting findings regarding abundance, circulating CD56bright NK cells from MS patients have reduced IFN-γ production in response to IL-12 and an impaired ability to regulate autologous CD4+ T cells compared to healthy controls (33, 35). This impaired regulatory capacity was due to HLA-E upregulation on autologous T cells engaging the inhibitory receptor NKG2A on NK cells (35). Further, DNAM-1 and 2B4 were reduced on NK cells alongside reduced expression of the DNAM-1 ligand CD155 on CD4+ T cells, while Daclizumab treatment induced CD155 upregulation on T cells to partially rescue the impaired ability of NK cells to regulate autologous T cells (30). A genome-wide association study of MS patients demonstrated lower expression of TBX21 and EOMES in NK cells, supporting that impairment of NK cells may be a driver of MS (36).
Using the experimental autoimmune encephalomyelitis (EAE) model of MS, Hao et al. demonstrated the importance of CX3CR1-mediated recruitment in generating disease-ameliorating CNS-resident mouse NK cells (37). Transmigration of NK cells into the CNS partially depends on VLA-4 binding to endothelial VCAM-1, as antibody blockade of VLA-4 reduces NK cell recruitment by 40-70% (30, 38). Absence from or blocked transmigration results in excessive proliferation of myelin-reactive CD4+ T helper 17 (Th17) cells, indicating that NK cells must be within the CNS to limit myelin-specific T cell activity and disease progression (37). Mouse NK cell-mediated disease amelioration required an NCR- and perforin-dependent lysis of microglia to abrogate Th17 expansion (37). The tight proximity of microglia and NK cells requires reciprocal chemoattraction through secretion of MIP-1α and MCP-1 by NK cells and microglia, respectively (37, 39). Additionally, NK cells dampen EAE pathogenesis by directly modulating infiltrating CCR2+Ly6Chi monocytes in an acetylcholine-dependent fashion. Adoptive-transfer of choline acetyltransferase (ChAT)-expressing NK cells into the CNS of Cx3cr1-/- mice reduced the abundance of infiltrating monocytes (40). ChAT+ NK cells dampened TNF-α, IL-1β, IL-12 and Qa-1 expression by monocytes through engaging the α7-nicotinic acetylcholine receptor, rendering myeloid cells more susceptible to lysis (40). ChAT+ NK cells preferentially localize to active demyelinated lesions in the human brain, suggesting this mechanism of microglial regulation may translate to human MS as well (40). While dampening myeloid and T cell activity reduces disease severity, murine NK cells negatively impact regeneration through lysis of Qa-1low neuronal stem cells in the sub-ventricular zone, altering neuronal repair and impairing recovery in later disease (41). Of note, NK cell activation via NKG2D triggered motor neuron destruction in models of amyotrophic lateral sclerosis, suggesting pathological NK cell-mediated lysis of neurons is not specific to MS/EAE (42).
Other ILCs have been identified in MS too, although inconsistent phenotyping has hindered identification of these ILCs. A sizable fraction of CD3- IL-17+ RORγt+ cells associate with newly formed meningeal lymphoid follicles of MS patients, suggestive of ILC3 involvement (43). In mice, CD3-RORγt+ populations in the cerebellum after EAE induction were predominantly CD4-, consistent with ILC3 identity (44). Hatfield et al. reported both NCR+ and NCR- ILC3s and CD4+ CD3- LTi-like ILC3s within the meninges of healthy mice which proliferated and accumulated downstream of c-kit signaling during EAE induction (45). Meningeal ILC3s produce IL-17 and GM-CSF, and express co-stimulatory molecules OX40L and CD30L. They accumulated near Th17 cells and antigen presenting cells (APCs) and facilitated T cell activation and entry into the brain parenchyma in a T-bet-dependent fashion, highlighting a role for ILC3s in establishing a microenvironment that sustains Th17 responses in EAE (45, 46).
Helper ILCs (hILCs) are also affected by Daclizumab treatment and appear to play a sex-biased role in MS/EAE. Untreated MS patients presenting with elevated white blood cell counts displayed higher levels of RORγt+ ILCs in their cerebrospinal fluid (47). Daclizumab treatment lowered CXCL13 levels and the abundance of Lin-c-kit+RORγt+ ILCs, suggesting that ILC3 inhibition may be another beneficial effect of Daclizumab treatment (48). In vitro differentiation of c-kit+ ILC precursors and CD34+ hematopoietic progenitor cells under high IL-2 conditions favored the development of CD56bright NK cells and restrained ILC3 differentiation, implying that greater in vivo IL-2 availability affects the development of ILCs by altering subset composition (48). MS has a higher prevalence in females and is correlated with reduced accumulation of ILC2s in EAE models (49). Interestingly, male mice that have reduced c-kit signaling (KitW/Wv) failed to accumulate ILC2s and adopted a female disease phenotype suggesting a sex-dependent role for ILC2s in protection from EAE pathogenesis (49). Il33 expression is only upregulated in male mice after myelin peptide immunization, and IL-33 administration in female mice expands ILC2s and provides protection from EAE, while anti-IL-33 treatment abrogates protection in male mice, further supporting sex effects on ILC2 function, dependent on differential IL-33 availability (50).
ILCs in Cerebral Ischemia (Stroke)
After a stroke, human peripheral blood NK cells are reduced early (< 72h) and the degree of reduction as well as expression of activation markers positively correlates with infarct volume (51, 52). Within 12 hours of intracerebral hemorrhage, CD69+Perforin+ NK cells become the dominant immune cell type in perihematomal regions (21). 24h following a stroke, CD69+NKp46+ cell numbers peaked in the brain and remained elevated (52). In mice, the accumulation of NK cells during the acute phase of stroke is mediated by the release of CX3CL1 by hypoxic neurons (53). Recruited NK cells accumulate in an IL-15-rich environment, adopt an activated phenotype, and mediate neuronal lysis through missing-self activation (Figure 3B) (53). Ischemia-reperfusion injury (IRI) induces IL-15 production by neurons, astrocytes and microglia, blockade of which reduced IFN-γ+ NK cells in the murine brain (54). Liu et al. reported that cholinergic signaling in the brain and catecholaminergic signaling in the periphery suppressed NK cell function after cerebral ischemia, contributing to post-stroke susceptibility to infection (52). While adrenergic activation suppressed NK cell abundance and function in the periphery, cholinergic signaling reduces Runx3 expression in CNS NK cells, leading to a decline in NK cell responsiveness and demonstrating the involvement of distinct neural pathways in regulating the spatial activation of NK cells in mice and humans (52). In humans, the microRNA (miRNA) profile of peripheral NK cells is altered after stroke and inhibition of miRNA-451a and miRNA-122-5p partially restored CD69 and NKG2D expression, suggesting that targeting miRNAs may alleviate immunosuppression observed after a stroke (51). Although data supporting a role for helper ILCs in response to stroke is scarce, early after an acute cerebral infarction circulating ILC1s increased and ILC2s decreased, correlating to serum ox-LDL levels, suggesting lipid-mediated regulation of ILC1 and ILC2 abundance (55).
ILCs in Neurological Function
Murine studies support a role for ILCs in regulating neurological function. Depletion of NK cells using anti-NK1.1 improved cognitive function, enhanced neurogenesis, and reduced microglial inflammation but did not affect β-amyloid concentration in a mouse model of Alzheimer’s disease (56). NK cells exhibited altered expression profiles in the disease model, with higher expression of Icam1, Ctsb, Ctsc, Ccl3 and Ccl4 (56). Following NK cell depletion, microglia exhibited a return to homeostatic morphology, reduced proliferation, and reduced expression of pro-inflammatory mediators including Il18, Il1a, Il1b and Tnf, suggesting that NK cells and type I immunity contribute to cognitive decline by promoting microglial inflammation (56). In line with these findings, choroid plexus ILC2s accumulated and displayed a quiescent state in the aged brain, which was reversed with IL-33 stimulation (57). In comparison to meningeal ILC2s, choroid plexus ILC2s were resistant to senescence and exhibited higher expression of Arg1 and genes associated with glycolysis that may underlie their enhanced proliferative and cytokine-producing capacity, and suggest niche-specific functionality (57). Intriguingly, activation of ILC2s in aged mice or transfer of activated ILC2s to the aged brain increased cognitive function, potentially through IL-5-mediated suppression of T cell inflammation leading to enhanced neurogenesis (Figure 3C) (57). After traumatic brain injury, ILCs are increased in frequency in human meninges and cerebrospinal fluid, and treatment with AMPK-activating metformin in a murine model specifically enhanced IL-10-producing ILC2s and improved neurological outcomes (58). Together, this suggests that ILC2s support neurological function and resolution of inflammation while NK cells exacerbate cognitive decline.
ILCs in Peripheral Nervous System
Nervous system signaling in the periphery is also impacted by ILC activity. Specialized pro-resolving mediators (SPMs) such as PCTR1 are important for resolving inflammation and promoting tissue repair (59). Acetylcholine promotes the enzymatic activity of ILC3-derived 15-LOX-1, the initiating enzyme in PCTR1 biosynthesis (60). Production of SPMs is regulated by the vagus nerve, and loss of vagus nerve signaling reduced peritoneal ILC3s in mice resulting in poor resolution of Escherichia coli infection (60). The circuit between ILC3s, SPMs, and macrophages is key for resolving infection and inflammation in the peritoneum (60).
ILCs in the Lung
NK cells account for 10-20% of all lymphocytes in human and murine lungs (61–63). Lung NK cells are marked by higher CD57 and KIR expression, and lower CD27, indicative of a mature phenotype (64). Despite their high KIR expression, human lung CD56dimCD16+ NK cells are hypofunctional and some CD56bright subsets are characterized by the expression of markers associated with tissue-residency (e.g., CD69, ITGA1 (CD49a), ITGAE (CD103), and CXCR6) (61, 64). A review by Hervier et al. nicely summarizes the development and function of NK cell subsets in the human lung (65). In addition to NK cells, all other helper ILC subsets have been observed in human lung, albeit with conflicting reports on the relative abundance of ILC1s, ILC2s and ILC3s that may reflect small sample sizes, sampling location, or inter-donor heterogeneity (66, 67).
Recruitment as well as local proliferation of ILC precursors in the lung during development shape the pool of tissue-resident ILC subsets (Figure 4A). Oherle et al. identified that murine pulmonary ILC3s develop from a local precursor pool sustained by insulin-like growth factor 1 provided by alveolar fibroblasts (68). Early-life seeding of ILC3s was protective against pneumonia in a CCR4-dependent fashion, driven by a gut commensal microbiota – dendritic cell (DC) axis (69). Similar interactions between adventitial stromal cells and mouse ILC2s were reported to sustain and regulate ILC2s homeostasis and function (24, 70). Adventitial stromal cells release TSLP, promoting basal IL-13 release by ILC2s, which in turn activates adventitial stromal cells to produce IL-33 in a homeostatic circuit (24). Interestingly, ILC2s localize around the peribronchial and perivascular adventitial cuff regions independent of microbial signals, IL-25, IL-33 or TSLP, indicating that additional unknown signals regulate pulmonary ILC2 development and recruitment (24, 70, 71). Whether pulmonary ILC2s in mice and humans originate from other tissues at steady state remains unclear, however mouse intestinal ILC2s were demonstrated to traffic to the lungs in an S1P-dependent manner after intraperitoneal IL-25 administration or helminth infection, demonstrating coordination between tissue sites to resolve multi-organ infections (72). Additional niche-signals may be delivered through the ICOS : ICOSL axis that has been demonstrated to sustain the pool of pulmonary ILC2s by elevating anti-apoptotic genes and IL-2 responsiveness (73). Intriguingly, ILC2s express both ICOS and ICOS-L, suggesting that both self-sustaining and helper cell-dependent interactions promote ILC2 homeostasis (73).
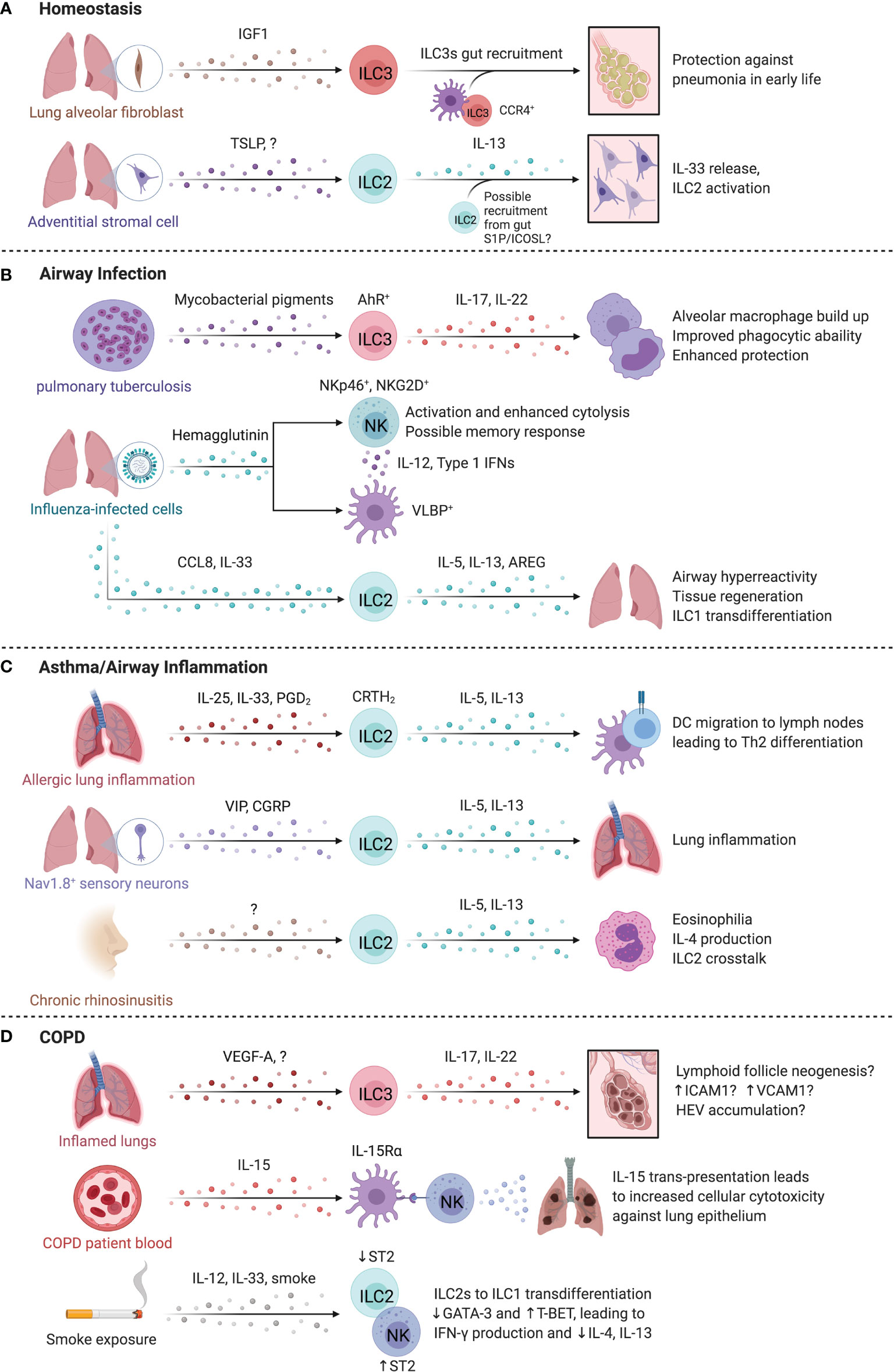
Figure 4 ILCs in the lung. At homeostasis (A), IL-22 producing ILC3s are required for protection against pneumonia and require commensal gut bacteria for their recruitment to the lungs. Once in the lung, ILC3s are sustained locally through insulin-like growth factor 1 (IGF1) from alveolar fibroblasts. In the steady state, Adventitial stromal cell-derived TSLP promotes IL-13 production by ILC2s that drives stromal cells to produce IL-33. (B) During pulmonary tuberculosis (PTB), ILC3 accumulation in the lung is regulated by pathogen-derived AhR-ligands that in turn promote phagocyte function, formation of tertiary lymphoid structures and enhanced protection. Similarly, infection by influenza virus triggers NKp46-dependent activation of NK cells resulting in IL-12 and type 1 interferon secretion by DCs that promote NK cell activation. The release of CCL8 and IL-33 during respiratory viral infection facilitated ILC2 activation and AREG-dependent epithelial repair. (C) In allergic lung inflammation, IL-13 from ILC2s induced Th2 cell differentiation by promoting migration of activated DCs to the draining lymph nodes. In mice, administration of the CRTH2 ligand, prostaglandin D2, promotes ILC2 accumulation. Nociceptor Nav1.8+ sensory neurons activated lung ILC2s through vasoactive intestinal peptide (VIP), while pulmonary neuroendocrine cells produced calcitonin gene related-peptide (CGRP) collectively promoting allergic inflammation in the murine lung. Nasal polyps accumulate ILC2s in chronic rhinosinusitis, which supports eosinophils and promotes chronic airway inflammation. (D) IL-17A+IL-22+ ILCs and NCR- ILC3s are increased in COPD. The lungs of COPD patients and smokers contain Neuropilin 1 (NRP1)-expressing ILC3s surrounding high endothelial venules. NK cells may contribute to COPD, with higher CD57 expression, IL-15-dependent activation, and greater cytotoxicity against lung epithelial cells. Smoke exposure may lead to a sustained loss of ST2 expression on ILC2s, reducing their responsiveness to IL-33 while paradoxically promoting ST2 expression on NK cells that supports a type 1 response. Created with Biorender.org.
ILCs in Airway Infections
Airborne pathogens are a constant challenge within the lung, and ILCs have a key role in anti-bacterial and anti-viral host defense (Figure 4B). Helper ILCs accumulate in the lungs of patients with pulmonary tuberculosis (PTB), while circulating ILCs are reduced (74), suggesting trafficking of ILCs to the lung. ILC3s are critical for host defense in PTB, as specific deletion of ILC3s (Ahrfl/flRorγtCre) increased mycobacterial burden, and impaired the accumulation of alveolar macrophages and formation of protective lymphoid follicles in granulomas (74). Mycobacterial pigments serve as ligands for Aryl hydrocarbon receptor (AhR), a key transcription factor for ILC3 development and function, suggesting an alternative mechanism of ILC3 activation in tuberculosis infection (75). In addition, ILC3s recruited to murine lungs produced IL-17A and IL-22 to enhance protection and support phagocytic functions of inflammatory monocytes to mediate clearance of bacterial infections (76, 77).
NK cells are critical in controlling viral infections in the lung. Indeed, influenza infection is lethal in Ncr1-/- mice (78). However, adoptive transfer and antibody-depletion experiments showed that NK cells exacerbated influenza morbidity and mortality in a manner dependent on virus titer (79). Differences in mouse genetic backgrounds, influenza strains, and infectious dosage complicate the interpretation and translation of these findings. In humans, viral hemagglutinin on infected cells triggered NKp46-dependent activation of NK cells, and upregulation of the NKG2D ligand ULBP on infected DCs and elevated secretion of IL-12 and type 1 interferon facilitated NK cell activation and cytolysis in response to influenza (80, 81). In a human lung tissue explant model, CD56brightCD49a+ NK cells robustly responded to influenza A infection, hinting at an NK cell subset-specific memory response (82). Dou et al. found that seasonal influenza vaccination induced a short-term (6 month) memory response in NK cells, correlating with downregulation of surface NKp46 and a concomitant increase in intracellular NKp46 expression (83). This memory response to re-challenge was not strain-specific, suggesting broader protection to influenza after seasonal strain-specific vaccination (83). While the role of ILC1s separate from NK cells is less clear, murine ILC1s promote antiviral defense and DC maturation, potentially through the glucocorticoid-induced TNFR-related protein (GITR):GITR-L axis (84). GITR upregulation on ILC1s resulted in stronger IFN-γ and TNF-α responses to influenza A, supporting host defense against alveolar viral infections (84).
ILC2s have conflicting roles in influenza infection response, promoting airway hyperreactivity in an IL-13-dependent manner while supporting epithelial cell integrity and tissue repair via the secretion of AREG following viral infections (85, 86). In response to CCL8, IL-33-activated ILC2s produce more IL-5 and IL-13, and exhibit ameboid-like movements to traffic to peribronchial and perivascular sites in mice, particularly at locations of increased collagen-I deposition (71). Human ILC2s also exhibited a chemotactic response to CCL8, suggesting shared lung recruitment responses across species (71). Infections with respiratory syncytial virus (RSV) leads to a viral titer-independent increase in respiratory disease severity in young infants driven by elevated ILC2 cytokine release (87, 88). Interestingly, patients older than 3 months had fewer ILC2s in their lungs, greater IFN-γ levels and experienced less severe disease, suggesting that the immunological changes occurring with age and development confer protection to RSV infections by balancing type 1 and type 2 immunity (88). The plasticity of ILC2s may also play a role in promoting type 1 immunity to viral infections. Silver et al. found that adoptively transferred murine ILC2s trans-differentiate into ILC1s near IL-12- and IL-18-expressing myeloid cells during influenza A infection (89). Overall, this suggests that age and plasticity shape ILC2 responses to viral infections.
ILCs in Asthma and Allergic Airway Inflammation
Asthma is a chronic inflammatory disease of the airways marked by elevated type 2 inflammation (90, 91). ILC2 activity is implicated in airway inflammatory diseases (Figure 4C). ILC2-derived IL-13 is critical for inducing Th2 cell differentiation in response to allergic lung inflammation by promoting the migration of activated DCs to the draining lymph nodes, supporting the development of allergic adaptive immune responses (92). Circulating ILC2s from asthmatic patients produced more IL-5 and IL-13 in response to IL-25 and IL-33 stimulation relative to controls, and administration of prostaglandin D2, the ligand for CRTh2, promoted ILC2 accumulation in murine lungs (93, 94). A single nucleotide polymorphism resulting in elevated CRTh2 expression positively associates with asthma development in humans, although whether this corresponds directly to increased ILC2 presence is unknown (95). Interestingly, the prevalence of asthma is lower in adult males versus females, indicating sex-specific differences in type 2 immunity (96). Several animal studies recapitulated these sex-dependent changes in abundance, phenotype, and responsiveness of ILC2s and implicated the role of sex-hormones in facilitating sex-specific responses to alveolar diseases (96–100). For example, androgen-receptor signaling negatively regulated ILC2 cytokine secretion and differentiation and reduced IL-33-dependent lung inflammation in male mice (97, 100, 101).
Strikingly, neuronal and neuroendocrine-driven stimulation of ILC2s promotes allergic lung inflammation (102, 103). IL-5-stimulated nociceptor Nav1.8+ sensory neurons activated ILC2s through vasoactive intestinal peptide (VIP), while pulmonary neuroendocrine cells trigger ILC2s through the calcitonin gene-related peptide (CGRP) to promote allergic inflammation in the murine lung (102, 103). CGRP-secreting pulmonary neuroendocrine cells were increased in asthmatic patients suggesting that this mechanism could also support ILC2-mediated allergic inflammation in humans, inspiring several pathways of therapeutic interventions (103). Constitutive activation of ILC2s may lead to long-lasting alterations in the lung as found in other pulmonary diseases. For example, ILC2s are enriched in nasal polyps of chronic rhinosinusitis patients along with elevated IL5 and IL13 transcripts, suggesting an ILC2-dependent contribution to the disease-associated eosinophilia and chronic airway inflammation (104, 105). Polyp tissues identified with eosinophilia revealed a co-localization of ILC2s and eosinophils, indicating a possible cross-talk between IL-5-producing ILC2s and IL-4-producing eosinophils to support reciprocal activation and survival (106).
Complicating our understanding of ILC2s in allergic responses are recent findings from Golebsky and colleagues that ILC210s are reduced in abundance in allergic individuals relative to non-allergic controls, while sublingual immunotherapy for grass pollen allergy restores this IL-10-producing subset which may confer protection and restoration of epithelial barrier integrity (16). Interestingly, murine lung ICOS+ST2+ ILC2s exhibit memory in response to allergen challenge dependent on ICOS and IL-33, marked by transcriptional and epigenetic programs involving the scaffold protein Four And A Half LIM Domains 2 (FHL2) (107). Further, adoptive transfer of FHL2+CRTh2+ human ILC2s induced airway hyperreactivity in mice and were partially steroid resistant, suggesting memory ILC2s may be relevant to steroid-resistant asthma (107).
Similar to ILC2s, ILC3s have been linked to asthma pathology. IL-17 levels and IL-17+ ILC3s were elevated in bronchial alveolar lavage fluid of asthmatic patients, especially in patients with severe disease (108, 109). An ILC3 gene signature was upregulated in nasal brushings of adult-onset severe asthma patients, while bronchial brushings revealed elevated type 2 related gene profiles, supporting the idea of an anatomic preference of distinct ILC responses that may selectively contribute to site-specific characteristics of disease (110).
ILCs also contribute to chronic pulmonary inflammation through regulation of adaptive immune cells. CD40L expression by human and murine T helper cells induces an IgE response by B cells, contributing to airway hyper-responsiveness (111, 112). CD40L expression on T cells is induced by cAMP only in the presence of CD56+CD16+ NK cells through a contact-dependent manner to drive asthmatic IgE responses (112). In patients with severe asthma, NK cells expressed higher levels of CD69 and NKG2D in line with an activated phenotype. Despite higher activation status, NK cell ability to induce eosinophil apoptosis was impaired (113). IL-13 production by ILC2s was attenuated and NK cell-induced eosinophil apoptosis was greatly increased by lipoxin A4 (LXA4), a pro-resolving mediator negatively affected during severe allergic asthma (113, 114). Lacking efficiency in resolution of eosinophilic inflammation due to a lack of LXA4 production in severe asthma suggests another axis of interaction promoting pulmonary dysfunction of NK cell and ILC2 responses to inflammation (113). Collectively, multiple layers of regulation affect the localized activity and accumulation of ILCs in asthma, emphasizing the need to understand tissue signals that control ILCs to develop more targeted therapies.
ILCs in COPD
Chronic obstructive pulmonary disease (COPD) is an inflammatory condition characterized by permanent and progressive loss of lung function, associated with smoking and exposure to noxious stimuli (115). ILC1s are increased in abundance in COPD patient lungs, correlating with smoking status and symptom severity (116). All helper ILC subsets localized with lymphoid aggregates in COPD lungs (116). IL-17 upregulation in end-stage COPD is implicated in lymphoid follicle neogenesis, and De Grove et al. found trends of elevated abundance of NCR- ILC3s and IL-17A+ and IL-22+ ILCs in the lungs of COPD patients (66, 117). While this seems to support the involvement of ILC3s in COPD, data supporting a specific role for ILC3-derived IL-17 is lacking. Co-culture of expanded human lung ILC3s with mesenchymal stromal cells induced upregulation of ICAM-1 and VCAM-1, suggestive of LTi activity, contrasting with observations in Rorc-/- and Id2-/- mice that develop lung lymphoid follicles even in the absence of ILC3s/LTis (118, 119). Interestingly, a subset of Neuropilin1+ ILC3s were recruited to high endothelial venules in lung tissues of smokers and COPD patients in a VEGF-A-dependent manner, although the specific role for ILC3s in COPD development and pathogenesis remains unresolved (118).
Circulating NK cells from smokers and COPD patients express higher levels of CD57 and have greater cytotoxicity against autologous lung epithelium than non-smokers or smokers without COPD (120, 121). The increase in cytotoxicity was mirrored in a murine COPD model after cigarette smoke exposure, demonstrating that trans-presentation of IL-15Rα by lung DCs was required to prime high NK cell cytotoxicity against autologous epithelial cells (120). Interestingly, cigarette smoke exposure induces a sustained loss of ST2 expression on ILC2s, dramatically reducing their responsiveness to IL-33, despite increased IL-33 production in severe COPD (122). Conversely, smoke induces an upregulation of ST2 on NK cells, leading to IL-33-mediated activation of NK cells instead of ILC2s, explaining the increase in type 1 immunity despite elevation of the type 2-activating cytokine IL-33 (122). Paralleling this, elevation of circulating ILC1s with a strong inverse correlation to ILC2 abundance was observed in COPD patients, further suggesting a misguided immune activation and cytokine-driven ILC plasticity, similar to mechanisms observed in the response to murine influenza infections (89, 106). Stimulation of human blood-derived ILC2s with IL-12 promoted their trans-differentiation into ILC1-like cells accompanied by the downregulation of GATA3 and an upregulation of T-BET, increasing IFN-γ release while dampening IL-4 and IL-13 production (89, 106). These results collectively demonstrate that persistent lung inflammation and exposure to smoke leads to changes in the local ILC composition and function (Figure 4D).
ILCs in the Skin
The skin is a barrier organ that employs immunological, microbial, and physiochemical mechanisms to protect the body from pathogens and harmful environmental factors. The skin is composed of three distinct layers: the epidermis (mainly comprised of keratinocytes), the underlying dermis, and the innermost subcutis. Tissue-resident and long-lived ILC subsets have been identified in mice and humans with varying proportions identified across studies (123–127). A more granular analysis of skin by layer revealed a predominant accumulation of ILC3s in the epidermis, ILC2s in the subcutis and comparable abundance of both subsets within the dermis in mice (128). This distribution has been attributed to the localized release of IL-7 and TSLP by either hair follicle keratinocytes or epithelial cells (128, 129). Mirroring murine models, human skin ILC2s can be activated by IL-25, IL-33, and TSLP, with high expression of IL-33 and TSLP during chronic skin inflammation (130–132).
ILCs regulate essential homeostatic functions of the skin (Figure 5A). For example, murine ILC3s negatively regulate the size of the lipid-secreting sebaceous glands, while the differentiation, proliferation, and expression of antimicrobial proteins by keratinocytes depends on IL-22 stimulation from ILC3s or epidermal T cells (128, 133). These interactions regulate the skin microbiome which can alter susceptibility to inflammatory disorders, impact repair pathways and influence host defense (128, 133, 134).
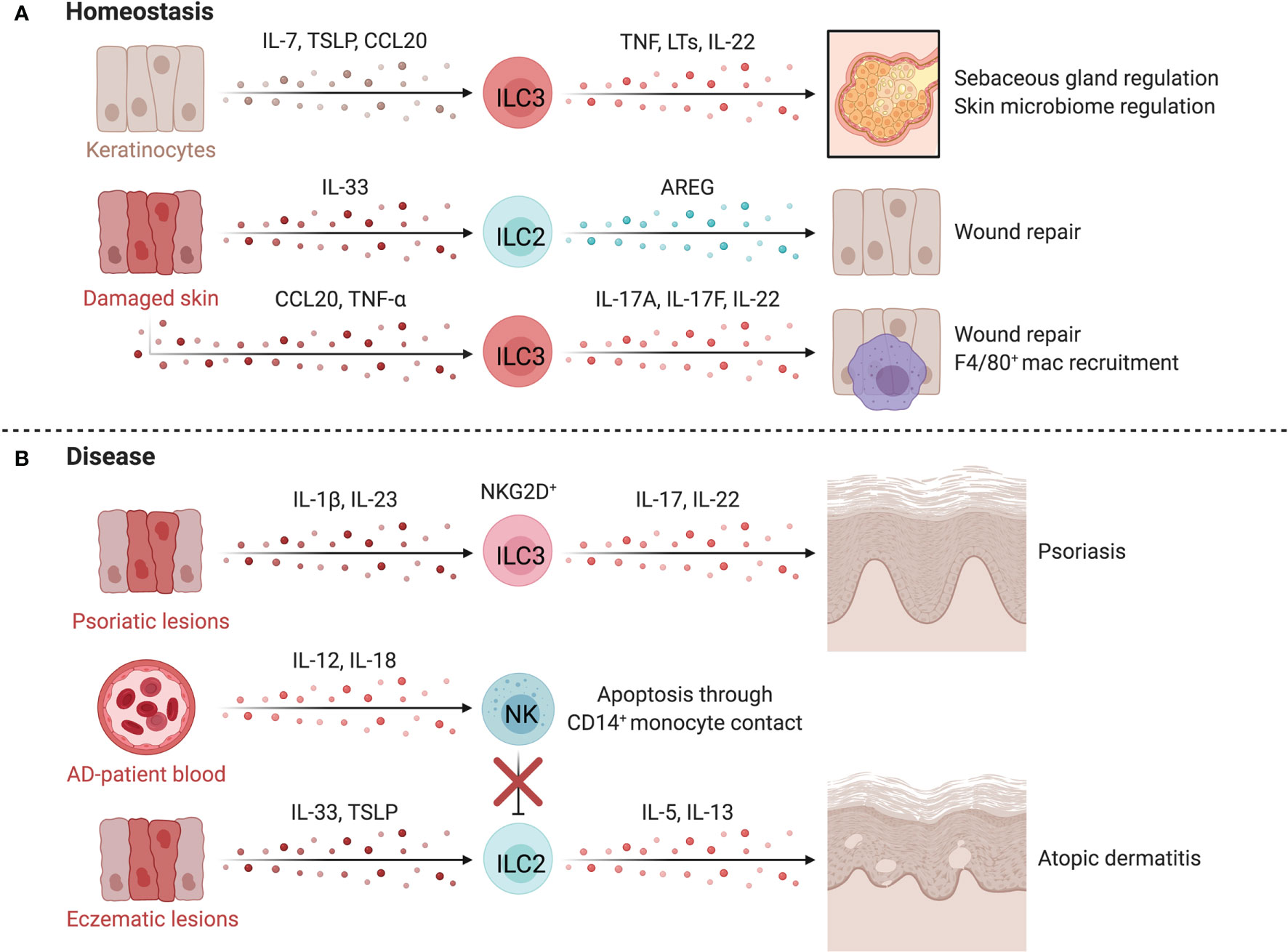
Figure 5 ILCs in the skin. At homeostasis (A), ILCs are retained by IL-7 and TSLP released by hair follicle keratinocytes or epithelial skin cells. TNF-α+LTα1β2+CCR6+ ILC3s negatively regulate lipid-secreting sebaceous glands, regulating the skin microbiome, which can alter susceptibility to inflammatory conditions or affect tissue repair pathways. Upon tissue damage, injured epithelial cells release IL-33, inducing proliferation of skin-resident ILC2s. In mice, anti-CD90 depletion in Rag1-/- mice delays wound healing, suggesting a role for ILC2s in promoting epithelial repair via AREG. The epithelium also produces TNF-α downstream of damage-induced Notch signaling in keratinocytes, recruiting CD4+NKp46low/- ILC3s that participate in wound closure. TNF-α driven release of CCL20 and CXCL13 by keratinocytes recruit ILC3s which facilitates the recruitment of F4/80+ reparative macrophages. In disease (B), psoriatic lesions in mice accumulate IL-17+ IL-22+ NCR+ ILC3s which is mirrored by the increase of CD56+RORγt+ ILC3s in both inflamed and non-inflamed skin of psoriasis patients. ILC2s promote atopic dermatitis (AD) when activated by epithelial-derived TSLP and IL-33 in inflammatory lesions. Dermal NK cells are decreased in AD and prone to apoptosis through contact with CD14+ monocytes. NK cells are proposed to regulate ILC2 abundance in AD, as therapeutic expansion of NK cells lowers ILC2 counts and improves disease scores in an AD mouse model. Created with Biorender.org.
ILCs in Wound Healing
ILCs directly influence skin repair after damage. Murine skin-resident ILC2s activated by IL-33 from injured epithelial cells proliferate at sites of injury while anti-CD90 depletion of ILC2s in Rag1-/- mice delays wound healing (135). CD4+NKp46low/- ILC3s are recruited by damage-induced CXCL13 and CCL20 and promote wound closure via IL-17A, IL-17F and IL-22 and indirectly through CCL3-mediated macrophage recruitment (136). Comparable findings were observed in IL-22-/- mice, where deficiency in IL-22 impaired keratinocyte proliferation, impeding repair (137). These results support a role for ILC2s and ILC3s in regenerative remodeling of the skin, yet research is needed to translate animal findings to humans and to define the differential impacts of ILCs and T helper cells (138).
ILCs in Psoriasis
Psoriasis is a chronic inflammatory skin disease that manifests as red scaly plaques caused by hyperproliferation of keratinocytes downstream of excessive repair pathways (139). Elevated IL-17 levels and Th17-associated gene expression signatures are found in psoriatic lesions and mouse models, implicating IL-17 and IL-22 in pathogenesis (140–142). IL-22- and IL-17-producing NCR+ ILC3s and CD56+RORγt+ ILC3s are enriched in inflamed and non-inflamed skin of psoriasis patients (Figure 5B) (123, 124, 126). ILC3s in inflamed lesions express higher NKG2D, which likely interacts with elevated MICA on keratinocytes (143). Anti-TNF treatment reduced circulating ILC3s in patients, corresponding with a decrease in inflammatory lesions (124). Further, ILC3-derived IL-22 induces an upregulation of MHC-II on keratinocytes, which promotes T cell polarization and skin inflammation, demonstrating a key circuit mediating skin inflammation (144). Skin ILC2s are also capable of driving T cell activation directly by presenting lipid antigens in a CD1a-dependent manner, leading to local activation of T cells in response to dermal bacteria (145).
There is limited and sometimes conflicting evidence for the role of NK cells in psoriasis. Studies have indicated that circulating NK cells are reduced in psoriasis patients (146, 147), or that no change was observed compared to healthy controls (148, 149). Within psoriatic plaques, Ottaviani et al. observed CD56+CD16- NK cells that co-expressed CD161, NKG2A, and CD69 (150). Supernatants from culturing these NK cells activated keratinocytes, increasing MHC-I, ICAM-1 and HLA-DR expression, along with CXCL10 and CCL5 secretion (150). These chemokines induced migration of skin-derived NK cells, supporting NK cell-keratinocyte cross-talk in psoriatic inflammation (150). NK cells appear to be hypofunctional in psoriasis, with reduced degranulation and IFN-γ potential (146, 149). The role of helper type 1 ILCs is even less defined, however expansion of ILC1s was observed in psoriatic lesions (126).
ILCs in Atopic Dermatitis
Atopic dermatitis (AD) is a common inflammatory skin disorder characterized by high levels of IL-4, IL-5 and IL-13 (151, 152). AD skin lesions are enriched for skin-resident ILC2s, which are activated by TSLP or IL-33, promoting type 2 inflammation (132, 153, 154). This is supported by murine models where anti-CD90 and anti-CD25 depletion of skin ILC2s in T and B cell deficient Rag1-/- mice attenuated dermatitis symptoms (153). Interestingly, KLRG1 ligation by E-cadherin reduces IL-5 and IL-13 production by human ILC2s, implicating dysregulation of parenchymal-ILC interactions in AD where E-cadherin levels are canonically downregulated on keratinocytes and ILC2s have elevated KLRG1 expression (132).
ILC3s have also been implicated in the pathogenesis of AD. Circulating ILC2s and ILC3s are elevated in AD patients, and increased IL-17 levels are apparent during acute disease (154, 155). Using several AD models, Kim et al. demonstrated AD lesions had increased numbers of IL17A+ ILC3s, which induced IL-33 release by keratinocytes and fibroblasts, promoting type 2 responses and exacerbating disease in mice (154). Further supporting a role for ILC3s in AD, ILC2s and ILC3s were elevated in AD lesions, with AHR+ ILC3s representing the most abundant subset. ILC3s in AD lesions were frequently surrounded by T cells, suggesting cellular interactions between ILC3s and T cells in AD (126).
NK cells are also altered in AD and are prone to apoptosis via a CD14+ monocyte-driven, contact-dependent mechanism, aligning with observed reductions in peripheral NK cell abundance in AD (146, 152, 156). Mack et al. found particularly reduced levels of circulating mature CD56dimCD16+ NK cells with high expression of KIRs and CD57 in patients with moderate-to-severe AD (152). A regulatory circuit between NK cells and ILC2s is supported by three lines of evidence: NK cell recovery occurring after IL-4 blockade; ILC2 accumulation in AD lesions of NK cell-deficient mice; and NK cell recovery and activation after IL-15 superagonist treatment leading to reduced ILC2 levels and disease scores in an AD model (152). Thus, cross-talk between ILC subsets may underlie the development and severity of AD (Figure 5B).
ILCs in the Intestine
The intestine is the largest mucosal surface in the human body and faces unique challenges. As a barrier surface, immune function in the intestine must balance tolerance and control of commensal microbes with protection from pathogens. Among immune residents of the intestine, ILCs have key roles in sustaining gut barrier integrity, repair, immune homeostasis, and host defense (Figure 6A). ILC distribution along the human intestine was reported by Simoni et al. and Yudanin et al. (67, 127). In line with observations made in mice, both groups demonstrated the presence of NK cells, ILC1s, ILC2s, and ILC3s across the intestinal tract, with predominance of ILC1s and ILC3s (67, 127, 157). NK cells are low in abundance and mainly CD56bright with distinct surface marker expression (64) (Figure 2). Intestinal ILC1s are heterogeneous, including a population of CD103+ ILC1s located in the epithelium, and CD127+ ILC1s residing in the lamina propria (LP) (158, 159). ILC3 subsets also localize within distinct microanatomic compartments of the gut epithelium/isolated lymphoid follicles(ILFs)/LP, but it remains to be shown if a similar distribution applies to humans (160, 161).
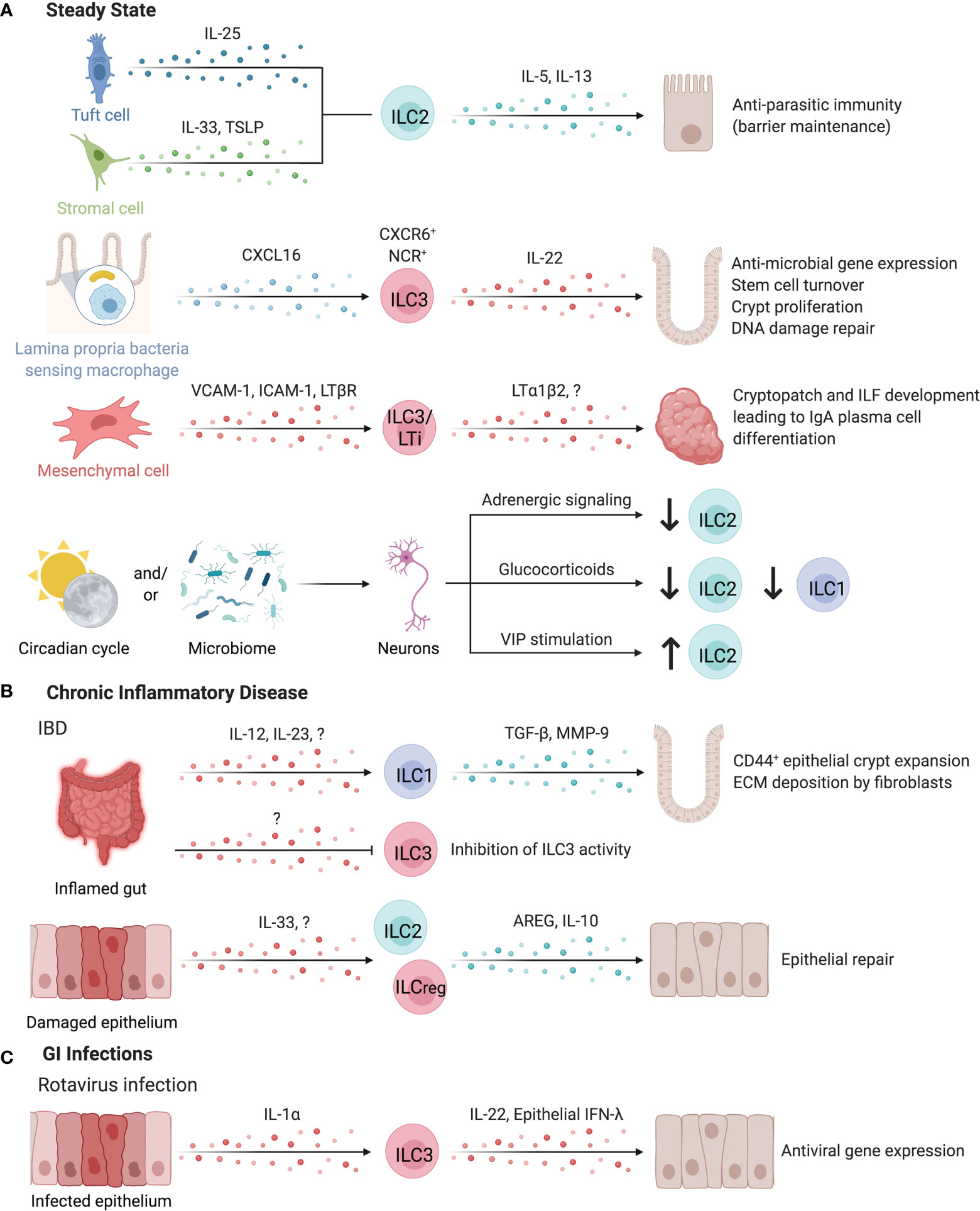
Figure 6 ILCs in the intestines. ILCs have important roles in maintaining intestinal homeostasis (A). ILC2s can be activated by both IL-25-producing tuft cells or IL-33 and TSLP-secreting stromal cells to promote anti-parasitic immunity in the intestines. ILC2s secrete IL-5 and IL-13 to promote host defense through parasite expulsion. Similarly, microbiota-sensing CX3CR1+ macrophages position IL-22-secreting CXCR6+ NCR+ ILC3s in the lamina propria via CXCL16. IL-22 supports anti-microbial gene expression in Paneth cells and promotes stem cell turnover, crypt proliferation, and DNA damage repair. Interaction of ILC3/LTi surface LTα1β2 and LTBR on mesenchymal cells leads to the upregulation of VCAM-1 and ICAM-1, resulting in the formation of cryptopatches and ILF. These tertiary lymphoid structures support the differentiation of IgA-producing plasma cells to promote barrier defense and host-microbiota mutualism. Clock genes and circadian cycles, modulated through feeding and the microbiota drive important homeostatic neuro-immune interactions in the gut. Disruption of circadian regulation alters ILC3 function, abundance, and trafficking into the intestines while negatively regulating ILC2s through adrenergic signaling. Glucocorticoids or vasoactive intestinal peptide further control ILC1 and ILC2 responses. (B) In chronic inflammatory disease such as intestinal bowel disease (IBD), the inflamed gut induced TGF-β and Matrix metalloproteinase 9 production by ILC1s leading to the expansion of epithelial crypt cells and extracellular matrix deposition by fibroblasts, exacerbating fibrosis. In contrast, pro-tolerogenic ILC3 functions including the release of GM-CSF, IL-2 or the expression of MHC-II are impaired in IBD patients, suggesting an anti-inflammatory role for ILC3s. IL-10 producing regulatory ILC2s or ILCregs may also suppress intestinal inflammation. (C) Enteric infection by Rotavirus induces epithelial-derived IL-1α to promote ILC3 production of IL-22, which synergizes with epithelial IFN-λ to promote the induction of antiviral responses in intestinal epithelial cells. Created with Biorender.org.
Intestinal ILCs promote host immunity against pathogenic and commensal microbes through interactions with sentinel immune and tissue cells. For example, murine ILC2s activated by IL-25-producing tuft cells or IL-33- or TSLP-secreting stromal cells promote anti-parasitic immunity, while DC-derived IL-33 promotes regulatory T cell (Treg) responses, suppressing anti-parasitic immunity (4, 24, 162–164). Myeloid cells, especially CXCL16-producing CX3CR1+ macrophages are critical for sustaining lamina propria-resident CXCR6+ NCR+ ILC3s as a major source of IL-22 in the intestinal LP (160). These ILC3s support IL-22-dependent intestinal epithelial anti-microbial gene expression, stem cell turnover, crypt proliferation, and DNA damage repair (160, 165–168).
While ILC3-derived IL-22 protects the intestinal epithelium against genotoxic stress, risk-associated single nucleotide polymorphisms have been identified within Il22 and the IL-23 signaling pathways as a driver of colorectal cancer in patients (165, 169, 170). Nevertheless, ILC3-derived IL-22 and LTα positively alter the glycosylation activity of epithelial cells, supporting glycan-scavenging intestinal commensal microbes and balanced host-microbe interactions and providing protection from infection (171, 172). sLT, expressed by human and mouse LTis, is essential to initiate the development of cryptopatches (CPs) and ILFs in the gut (11, 173). These tertiary lymphoid tissues support the differentiation of IgA-producing plasma cells to promote barrier defense (174, 175). Mouse CP and ILFs contain a unique subset of DCs that require LTβR signaling for their development. These DCs released IL-22 binding protein, which in turn alter intestinal epithelial IL-22R signaling and lipid transport (176).
Tregs have key functions in inducing tolerance to luminal antigens (177). IL-2 and GM-CSF-producing ILC3s directly and indirectly support the generation of Tregs in the healthy murine gastrointestinal tract, upon stimulation by microbiota-sensing IL-1β-producing macrophages. The cooperation and reciprocal crosstalk between macrophages, DCs, and ILC3s supports Treg homeostasis and T cell immunity against orally ingested antigens (177–179). MHC-II expression on murine ILC3s has been demonstrated to regulate T cell responses to microbial antigens via a mechanism analogous to negative selection in the thymus (180, 181). Lehmann et al. reported organ-specific expression levels of MHC-II on murine ILC3s and demonstrate that microbiota-induced IL-23 stimulation of ILC3s reversibly downregulated their MHC-II expression (182). Noteworthy, Rao et al. reported an accumulation of HLA-DR+ ILC3s in T cell-rich areas of colorectal cancers suggesting antigen-presenting capacity of ILC3s in humans as well (183). Together, this suggests that ILC3s both positively and negatively regulate T cell immunity dependent on microenvironmental signals.
Several environmental factors regulate murine intestinal ILC abundance. The metabolite-sensing Ahr is highly expressed by ILCs in the gut, with an important role in sustaining ILC3s and promoting IL-22 production (165, 184, 185). In contrast to ILC3s, gut ILC2 function is suppressed by Ahr signaling, suggesting a role for Ahr ligands in regulating the balance of intestinal ILC subset abundance (186). A similar divergent stimulation between ILC2s and ILC3s has been reported for other dietary components (187, 188). Microbial short chain fatty acids (SCFAs) differentially affect mouse ILCs in a subset- and location-specific manner, generally promoting ILC3 proliferation and IL-22 production while inhibiting ILC2 expansion (189–191). Free Fatty Acid Receptor 2 (Ffar2) acts as a SCFA receptor, and agonism leads to ILC2 proliferation, yet SCFA feeding leads to contraction of ILC2 abundance, suggesting the involvement of several receptors in coordinating the response to microbial fermentation products (191). This along with reports of age and body-mass index-associated alterations in the abundance of ILC subsets suggests age and metabolism-dependent regulation of intestinal ILCs in humans (67).
Cholinergic neurons in the gut and lung of mice produce neuromedin U in response to helminth challenge, which stimulates ILC2 proliferation and production of IL-4 and IL-13 in an IL-33-independent manner (192). The neuromedin U receptor does not appear to be expressed by other hematopoietic cells besides ILC2s at significant levels (192). In humans, the NMUR1 transcript was detected in intestinal ILC2s, yet direct evidence for this ILC2-neuronal interaction in humans is lacking (192). Other modalities where the nervous system regulates ILCs includes negative regulation of ILC2s by adrenergic signaling, glucocorticoid dampening of ILC1 and ILC2 responses, VIP stimulation of ILC2s, and ILC3 co-localization with neurons in enteric CPs, as detailed in a review by Klose and Artis (3). Interestingly, circadian light-dark cycles regulated neuron-immune interactions and intestinal ILC3-specific gene expression through diurnal oscillations of Rorc, Il17a, and Il22, while disruption of ILC3 circadian regulation altered their function, abundance, and trafficking in the murine intestine (2, 193–195). Interestingly, the gut microbiota contributed to control of this circuit, as antibiotic treatment partially restored ILC3 abundance and constrained cytokine production in circadian-disrupted mice (195). In mice, VIP promotes ILC3 intestinal recruitment and maintains expression of gut-homing receptor CCR9 (196). Talbot et al. reported a feeding-induced inhibition of ILC3s by VIPergic neurons, regulating mucosal immunity by dampening IL-22-induced antimicrobial peptide production in exchange for enhanced absorptive capacity of the intestinal epithelium marked by increased fatty acid transporter (Fabp2) expression (197). This contrasts with findings by Seillet et al. that VIP stimulation increased IL-22 production by enteric ILC3s, although the reason for these conflicting results is unclear, suggesting complex signals regulate intestinal ILC3 activity (198). Together, intestinal ILC3s are regulated by a complex circadian network involving light-dark cycles, microbial signals, and nutrient-driven neuronal regulation. Of note, the production of IL-5 by murine ILC2s was also circadian regulated (198).
Chronic Inflammatory Diseases
Chronic inflammation of the intestinal tract is a hallmark of inflammatory bowel disease (IBD) and fosters a local cytokine milieu that promotes differentiation of ILC1s (199). ILC1 expansion in inflamed intestinal tissue is location-specific, with greater expansion of LP-resident CD127+ ILC1s versus intraepithelial ILC1s in Crohn’s disease (CD) patients (158, 159, 200, 201). Specific expansion of CD127+CD94+Granulysin+ ILC1s is observed in the inflamed LP of CD patients (202). With elevated secretion of TGF-β and MMP9, mouse ILC1s facilitate the expansion of CD44+ epithelial crypt cells and extracellular matrix deposition by fibroblasts, collectively supporting matrix remodeling and epithelial proliferation that may exacerbate inflammation-associated fibrosis (Figure 6B) (203). In contrast, ILC3 abundance and homeostatic functions in circadian oscillation, production of IL-2, and expression of MHC-II were critically impaired in IBD patients, supporting anti-inflammatory contribution of ILC3s (178, 180, 193, 195). ILC3 secretion of IL-22 is enhanced by G Protein-Coupled Receptor 34 (GPR34) recognition of lysophosphatidylserine from apoptotic neutrophils, further supporting a role of ILC3s in sensing intestinal injury and initiating repair responses (204). However, ILC3s may contribute to intestinal inflammation under permissive circumstances (205). Further, destabilizing RORγt expression promoted the differentiation of ILC3s into ILC1/ex-ILC3 in mice and humans and correlated with intestinal IBD-like inflammation (206, 207). Interestingly, this differentiation was not static, but was regulated by the myeloid cytokine milieu in the intestinal tract (159). Counterbalancing the elevated type 1 and type 3 immunity reported in IBD, ILC2-derived AREG was sufficient to reduce DSS-induced damage in mice by promoting epithelial integrity and mucus production (162). Bando et al. further identified murine ILC2s as a dominant source of IL-10 in the intestine, while Wang et al. identified a distinct subset of IL-10-producing regulatory ILCs in humans and mice, supporting that IL-10 producing ILC2s or ILCregs may suppress intestinal inflammation (Figure 6B) (13, 17). Targeting ILC3-to-ILC1 plasticity, ILC1 activation, and ILC3 abundance may be a promising approach to restore intestinal immune homeostasis under chronic inflammatory conditions (208–211).
Gastrointestinal Infection
ILCs play a critical role in the response to intestinal pathogens in humans, highlighted by cases of deficiency in RORC resulting in severe mucosal fungal and bacterial infections (212). Along this line, susceptibility to infections by enteric extracellular pathogens are increased in the absence of IL-22 or GM-CSF, highlighting a critical role for ILC3-associated cytokines in barrier defense (213–216). Mouse ILC3s and ILC1s/ex-ILC3s promote antimicrobial responses via surface lymphotoxin-mediated differentiation of goblet cells and IFN-γ-induced production of mucins, further emphasizing the synergistic actions of ILC1s and ILC3s that require underlying microbial recognition and activation by myeloid cells (207, 217–219). Whether this permits discrimination of commensal and pathogenic microbes requires further investigation (219). In response to mouse enteric rotavirus infections, epithelial IL-1α induced ILC3-derived IL-22 which synergized with epithelial IFN-λ, promoting the induction of antiviral gene expression in intestinal epithelial cells, limiting viral replication and tissue damage (Figure 6C) (220). While ILC3s can promote antiviral immunity, they experience cytokine-dependent depletion in the intestinal tract of HIV+ human and SIV+ non-human primates, altering epithelial permeability and homeostasis (221–223). Collectively, ILCs promote intestinal barrier defense against enteric bacterial, fungal, and viral infections by exerting cytokine or cell contact-dependent effects on intestinal epithelial cells.
Enteric parasites and worms constitute a major global health burden. Murine NK cell recruitment to the intestine early after helminth infection does not affect parasite burden but limits the tissue damage induced by infection (224). Experimental models of worm infections revealed the importance of ILC2s and ILC2-derived cytokines in intestinal host defense in mice (225–227). For example, IL-13 from murine ILC2s promoted tuft and goblet cell differentiation from crypt progenitors, contributing to epithelial remodeling and worm expulsion in the characteristic “weep and sweep” response (4, 228). ILC2s actively promoted Th2 cell responses via MHC-II and co-stimulatory molecules, partially acquired through trogocytosis, while T cell-derived IL-2 activated ILC2s for efficient helminth expulsion in mice (229). Further, ILC2s are activated by acetylcholine and upregulate ChAT to produce acetylcholine in response to helminth infection, supporting efficient helminth expulsion through a potential autocrine signaling mechanism (230). The activation of ILC2s following worm infection could be blunted through parasite-derived, bio-active components interfering with the IL-33-ST2 axis (231). Intriguingly, helminth infection changed the global distribution and activation of murine ILC2s through the induction of S1PR1-dependent egress of gut ILC2s and accumulation in the lungs, suggesting a coordinated response to protect distal body sites targeted by helminth infection (72, 232).
While the fetal and adult human intestine hosts a population of ILC2s capable of releasing type 2 cytokines following stimulation with IL-2, IL-25, and IL-33, their role during human parasitic infections has not been well detailed (104). Lack of sample availability has hampered investigation of intestinal ILC abundance and function of worm infected patients (233). To date only two studies analyzed ILCs in worm infected patients. Nausch et al. observed a reduced frequency of ILC2s in children infected with Schistosoma, while Boyd et al. observed an increase in circulating c-kit+ ILCs and elevated IL-13 secretion in adult patients with filarial infections, suggesting heterogeneity in ILC responses dependent on age and/or helminth species (234, 235).
Collectively, intestinal ILCs support host immunity, barrier defense and tissue repair during infection and homeostasis, but also may perpetuate inflammation under permissive microenvironmental conditions.
ILCs in the Liver
The liver is critical for metabolism and blood detoxification. Constant exposure to an array of antigens and microbial products within liver sinusoids promotes tolerance to predominantly harmless antigens (236, 237). The liver contains a large proportion of innate immune cells such as Kupffer cells (specialized macrophages), inflammatory and non-inflammatory macrophages, NKT cells, NK cells and ILCs (238–240). These innate lymphocytes influence the activation and function of the various adaptive immune populations that include αβ T cells, γδ T cells and B cells, as well as parenchymal cells within the liver niche.
In humans, CD56brightCD16- NK cells comprise 50% of all liver NK cells (241). These NK cells express CD69, CCR5 and CXCR6, but not SELL or CCR7, and are localized to sinusoids by CCL3, CCL5, and CXCL16 produced by Kupffer cells, T and NK cells, and endothelial cells, respectively (241, 242). NK cells in healthy liver of deceased donors highly express EOMES, CD7, KLRD1(CD94), GZMK, NCR1(NKp46) and NCAM1(CD56), and lowly express FCGR3A(CD16) and ITGA1(CD49a) (240). Although CD49a+ NK cells akin to murine liver-resident NK cells have been identified in humans, they represent only a small subset of human liver-resident NK cells, while lack of CD49e protein expression differentiated human liver-resident NK cells from conventional (cNK) cells (243, 244). CD49a+CD16− NK cells in liver have a transcriptional program consistent with cytotoxic activity and exhibited antigen-specific killing of autologous targets presenting viral or metal antigens (245). Notably, a donor-derived EOMEShi tissue-resident NK cell population persisted in the liver up to 13 years post-transplant in a study of HLA-mismatched liver transplants (246). This NK cell population had a phenotype consistent with those reported in transcriptomic studies of healthy human liver (240–243).
While group 1 ILCs are the most abundant hILC population in human liver, NCR+ and NCR- ILC3s and ILC2s are also present (247). Liver ILC2s are CRTH2+CD161+CD69+ and highly express fibronectin-binding VLA-5, laminin-binding VLA-6, and the chemokine receptor CCR6 (247). In contrast to mice, only 10% of intrahepatic human ILC2s express the IL-33 receptor ST2, and primarily produce IL-13 and AREG, with very little IL-5 (247).
ILCs in Viral Hepatitis
NK cells are implicated in both Hepatitis C (HCV) and Hepatitis B (HBV) infections, which are major causes of liver inflammation and cirrhosis, leading to development of hepatocellular carcinoma (248) (Figure 7A). Peripheral NK cell abundance is reduced in both HCV- and HBV-infected patients, with reduced IFN-γ and TNF-α potential particularly in HBV, suggesting functional dysregulation (249). Cytotoxic impairment is associated with chronic infection establishment, while acute HCV infection induces NK cell activation, including increased NKG2D expression and greater capacity for cytotoxicity and IFN-γ production (250). Despite shared dysregulation, NK cell phenotype differs between chronic HBV and HCV; an enrichment of NKG2C+ NK cells are observed in HBV, whereas increased CD69 expression and decreased inhibitory KIR expression are observed in HCV (249). Differences in NK cell KIR and HLA allele expression may differentiate infections that are self-limited versus those that become chronic; KIR2DL3 and HLA-C1 expression is reported to be protective in HCV infection (251, 252). Weaker inhibitory signals by HLA-C1 may allow for increased NK cell activation and viral clearance (251, 252). In agreement, degranulation marker CD107a was increased on NK cells with KIR2DL2/3 and was highest in those with self-limiting infections (250). Engagement of HLA-E with elevated NKG2A and CD94 receptors on NK cells of HCV-infected individuals results in TGF-β and IL-10 production and impaired ability to activate DCs for virus-specific T cell responses, in line with findings that hepatocyte and Kupffer cell HLA-E expression correlates with HCV severity (253, 254). Of note, intrahepatic CD56brightCD16- NK cell abundance correlates with better liver function and lower disease scores in HCV-positive patients undergoing liver transplantation (255).
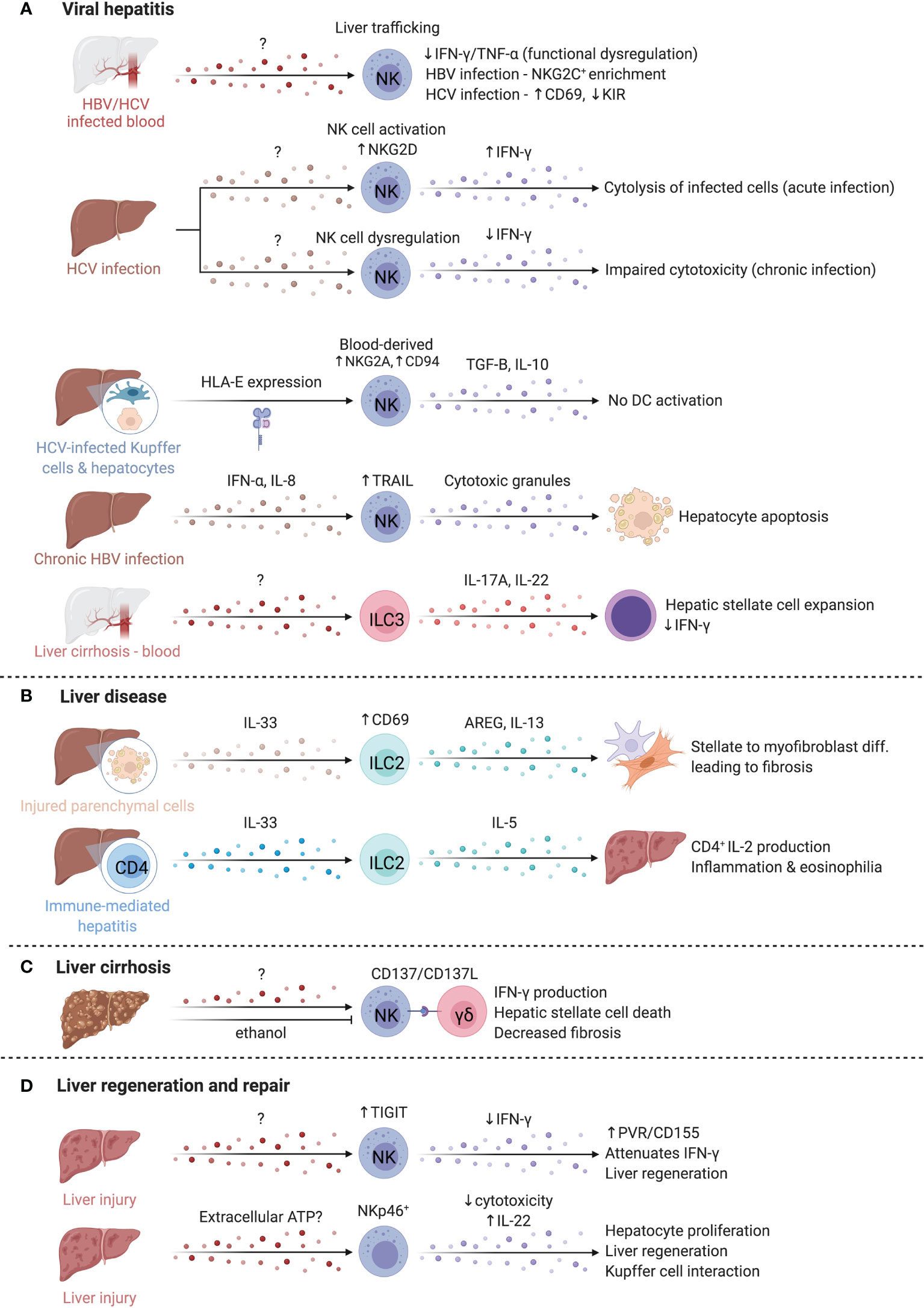
Figure 7 ILCs in the liver. The liver is occupied by a variety of ILCs that play diverse roles in viral hepatitis, liver disease, liver cirrhosis, and liver regeneration and repair. (A) Hepatitis C (HCV) and Hepatitis B (HBV) are key inducers of liver inflammation and cirrhosis and lead to the development of hepatocellular carcinoma. The abundance of NK cells in the blood of both HBV/HCV infected patients is reduced, suggesting elevated homing to the liver where functional deficiencies like impaired IFN-γ and TNF-α production are reported. NK cells in chronic HBV and HCV infected patients adopt distinct phenotypes that manifest in an enrichment of NKG2C-expressing NK cells or altered CD69 and inhibitory KIR expression. In contrast, acute HCV infection promotes elevated NKG2D and IFN-γ expression while IFN-γ is reduced in chronic infections. HCV-infected hepatocytes and Kupffer cells express higher levels of HLA-E that boost TGF-B and IL-10 production by NK cells through NKG2A and CD94. CD56bright NK cells in chronic HBV infection facilitate TRAIL-dependent hepatocyte death. ILC3s separately support hepatic stellate expansion and counteract IFN-γ. (B) In liver disease, ILC2s are increased and activated, driving liver fibrosis via AREG and IL-13. Similarly, in immune-mediated hepatitis, ILC2s were expanded and produced high levels of IL-5, recruiting eosinophils, and driving inflammation. (C) In liver cirrhosis, crosstalk between NK cells and γδ T cells through the CD137-CD137L axis enhanced cytotoxicity of NK cells against HSCs. Alcohol exacerbates fibrosis chronically, but also attenuates NK-mediated cell killing, and reduced NKG2D, TRAIL and IFN-γ expression on NK cells. (D) NK cells upregulate TIGIT while hepatocytes upregulated the ligand PVR/CD155, attenuating IFN-γ production and promoting liver regeneration. Finally, extracellular ATP is elevated after liver injury and regulates regeneration in the liver via NKp46+ NK cells. Created with Biorender.org.
In chronic HBV, circulating and intrahepatic NK cells highly express TRAIL and CD69, especially the CD56bright subset (256). Elevated IFN-α and IL-8 upregulate TRAIL expression on NK cells and TRAILR-2 expression on hepatocytes, respectively, suggesting TRAIL-dependent targeting of hepatocytes by CD56bright NK cells mediates damage during chronic HBV flares (256). Notably, HBV-specific T cells also have high expression of TRAIL-R2 and are susceptible to targeting by NK cells, supporting a role for NK cells in regulating anti-HBV T cell responses (257).
Comparatively little is known about the role of human hILCs in hepatitis infections. Increased hILCs were reported in the circulation of patients with chronic HBV (258, 259). HBV-related cirrhosis progression correlated with IL-17A and IL-22 production by ILC3s, suggesting ILC3 promotion of fibrosis, likely in part due to IL-22-mediated suppression of anti-fibrotic IFN-γ (Figure 7A) (259). While HCV/HBV do not infect mice, other viral hepatitis models provide some context into ILC viral responses in the liver more generally. Hepatic ILC3s produce IL-17A/F alongside γδT cells to promote antiviral T cell responses and inflammation early after infection (260). At later timepoints post-infection, ILC2s induce immunosuppressive neutrophils via IL-13 to limit T cell damage (261). This suggests that hepatic ILCs may have time-dependent roles to balance viral clearance and tissue protection.
ILCs in Liver Fibrosis
Liver disease is characterized by fibrogenesis of the liver, driven by type 2 immunity, with an implication for ILC2 activity (Figure 7B) (262). Hepatic stellate cells become activated and transdifferentiate into myofibroblasts that produce copious extracellular matrix proteins, driving fibrosis and loss of function resulting in cirrhosis (263). Patients with cirrhosis have elevated serum IL-33 and increased intrahepatic ILC2s, correlating with disease severity (247, 264, 265). Expansion of ILC2s and activation by IL-33 from damaged parenchymal cells results in IL-13 production driving fibrotic gene expression in hepatic stellate cells in fibrosis models, or IL-5 production with resultant hepatic inflammation and eosinophilia in immune-mediated hepatitis models (265, 266). While both effects are IL-13-dependent, additional signals which influence IL-5 versus IL-13 dominant responses by ILC2s are unknown. Interestingly, liver ILC2s present antigen to CD4+ T cells which produce IL-2 to sustain ILC2 expansion (267). High levels of IL-6, linked to liver regeneration, were produced by IL-33-activated liver ILC2s, indicating that ILC2s may have a dual roles in immune-mediated liver disease (267).
Intrahepatic human CD49a+ NK cells are expanded in cirrhotic livers (268). CD49a+CD25+ NK cells positively correlate with serum alanine aminotransferase, linking CD49a+CD25+ NK cells to liver inflammation (268). Conversely, liver NK cells dampen fibrosis by killing activated hepatic stellate cells in an NKG2D- and TRAIL-dependent manner, while IFN-γ reduces hepatic stellate cell activation and matrix protein deposition (269, 270). CD137-CD137L crosstalk between NK cells and γδ T cells enhances NK cell cytotoxicity (Figure 7C) (271). Chronic alcohol consumption exacerbates fibrosis, and ethanol attenuates NK cell cytotoxicity towards hepatic stellate cells by reducing NKG2D, TRAIL, and IFN-γ expression, suggesting immunological and environmental mechanisms of NK cell regulation (272).
ILCs in Non-Alcoholic Liver Disease
Non-alcoholic fatty liver disease (NAFLD) is the most common non-infectious chronic liver disease and can develop into non-alcoholic steatohepatitis (NASH) and progress to cirrhosis (273). NK cells are elevated in liver biopsies of NAFLD and NASH patients, with greater than two times increased NK cell abundance in NASH compared to NAFLD (274). NKG2D and TRAIL-DR5 transcript levels also have higher expression in NASH (274). Upregulation of MIC-A/B positively correlates with disease score and degree of fibrosis, suggesting NK cell engagement with MIC-A/B stress ligands could be a key factor in NASH development (274). In agreement, circulating NK cells from NASH patients had higher NKG2D expression (275). Depletion of IFN-γ-producing NKp46+DX5+ NK cells in a NASH mouse model altered macrophage phenotype, suggesting that IFN-γ from NK cells reduces fibrosis by polarizing macrophages away from a TGF-β+ pro-fibrotic phenotype (276). Additional studies are required to delineate the mechanisms that control whether NK cells limit or promote fibrosis.
While fewer studies have focused on hILCs, ILC3s appear to mitigate NAFLD. High fat diet increases ILC3 abundance in mice, while deficiency of ILC3s leads to liver fibrosis and an increase of pro-inflammatory gene expression with concomitant accumulation of saturated fatty acids (277).
ILCs in Liver Regeneration and Repair
The liver is uniquely capable of self-regeneration, including regenerating entire lobes after resection. Group 1 ILCs interact with injured tissue and influence regenerative capacity (Figure 7D). In models where NK cells are pre-activated by viral infection or TLR3 agonism to produce higher levels of IFN-γ, as well as in aged livers that have elevated IFN-γ signaling, regeneration is impaired (278, 279). NK cells upregulate T cell immunoreceptor with Ig and ITIM domains (TIGIT) while hepatocytes upregulate the ligand PVR/CD155, attenuating IFN-γ production and promoting liver regeneration (280). Mouse NKp46+ cells co-localized with F4/80+ cells in liver sinusoids, and NKG2D blockade abrogated regeneration, suggesting NKG2D-mediated crosstalk with Kupffer cells regulates regeneration (281). Extracellular ATP is elevated after injury and regulates liver regeneration (281, 282). ATP limits NK cell cytotoxicity while antagonism of ATP-receptor P2X1 reduces IL-22 production by group 1 ILCs in a murine liver resection model, resulting in dampened hepatocyte proliferation and elevated hepatocellular injury and stress (281, 283). Taken together, extracellular ATP released after resection may dampen NK cell cytotoxicity and promote IL-22 production to modulate time-dependent group 1 ILC functions supporting liver regeneration. Future studies that characterize marker expression in greater detail may clarify whether the cells identified were also inclusive of CD56+ ILC3s or were NK cells or ILC1s that converted to ILC3s.
ILCs in the Kidney
Kidneys perform essential functions of filtering blood, excreting waste, and regulating the body’s fluid and electrolyte balance. ILCs have been found to contribute to acute and chronic kidney diseases, with protective (Figure 8A) and pathological (Figure 8B) functions in IRI, kidney disease, and lupus nephritis, however, their role in the steady state remains poorly described.
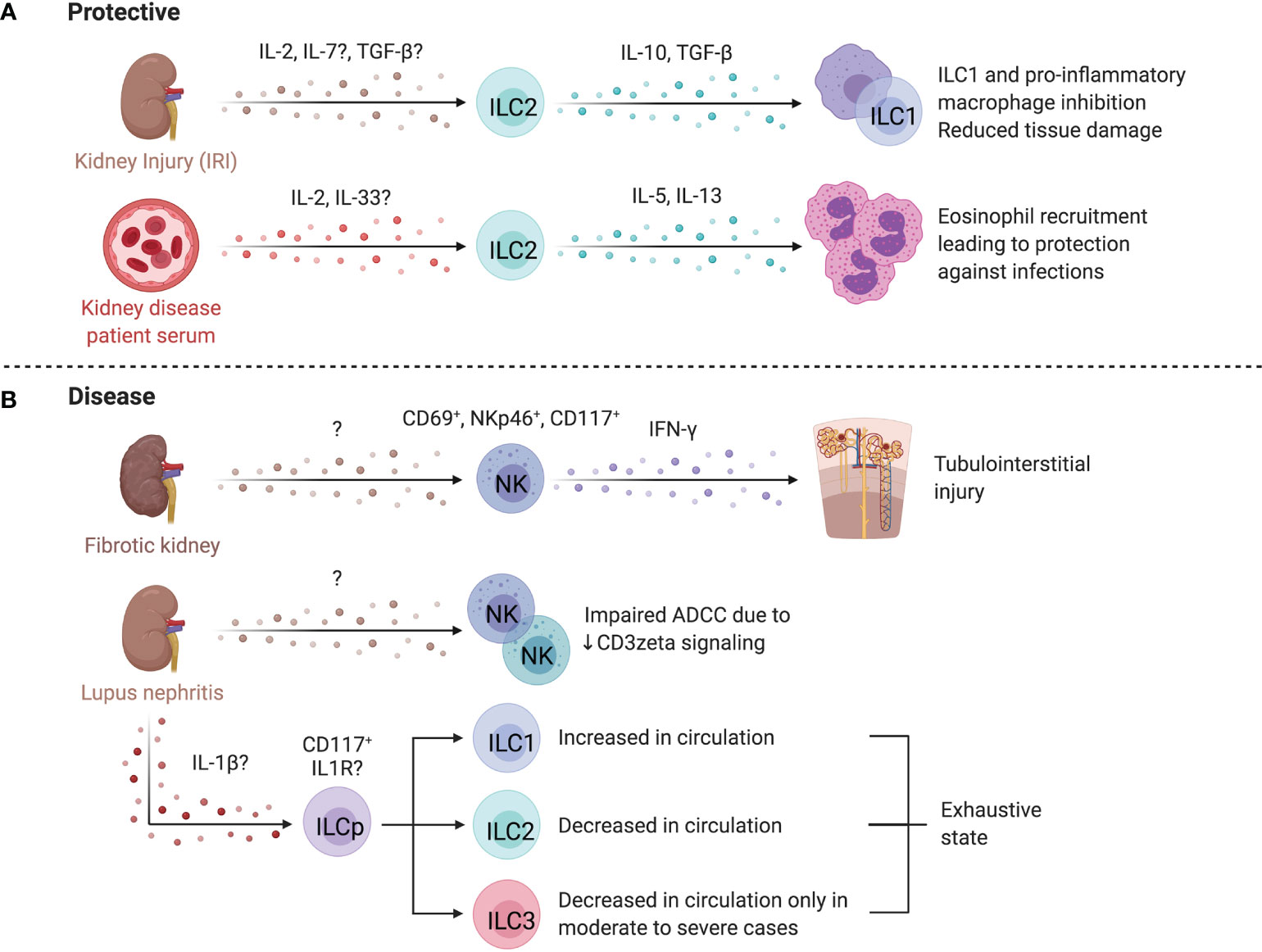
Figure 8 ILCs in the kidney. (A) Mouse and human studies support ILC2s may limit kidney injury. Administration of IL-25 and IL-33 in a humanized mice model attenuates IRI, and in conventional mouse models promotes a Th2 response and M2 macrophage polarization resulting in decreased tissue damage post ischemic injury. ILC2s may also be protective in end-stage renal disease (ESRD), where circulating ILC2 abundance, proliferation, and cytokine release increases. IL-2 is proposed to facilitate this ILC2 expansion via STAT5 and protect against infections through eosinophil support. (B) CD56bright NK cells are more abundant in fibrotic kidney tissue where they upregulate CD69 and co-express NKp46 and CD117, producing the majority of IFN-γ, implying a role in driving inflammation and fibrosis. In lupus nephritis (LN), scRNAseq revealed two distinct NK cell subsets – a CD56dimCD16+ blood-derived and tissue-resident CD56brightCD16- population. Both NK cells showed impaired antibody-dependent cell cytotoxicity because of dampened signaling efficiency by NKp30 and NKp46. LN patients further displayed elevated ILC1s and decreased ILC2s, while patients with moderate to severe disease showed an additional decrease in ILC3s. Created with Biorender.org.
ILCs in Chronic Kidney Disease
End-stage renal disease (ESRD) is associated with high morbidity and mortality, often associated with infections (284). Circulating ILC2 abundance, proliferation, and IL-5/IL-13 production is higher in patients with ESRD versus healthy controls, pointing to ILC responsiveness to the altered environment (285). The IL-2 rich ESRD plasma promotes STAT5 phosphorylation of ILC2s leading to expansion and activation (285). An inverse correlation between circulating ILC2 abundance and infectious complications, as well as elevated IL-33 suggest ILC2 activation as a protective mechanism in ESRD (286). These findings are supported by increased protection from chronic kidney disease by IL-33-induced ILC2 expansion and elevated eosinophil recruitment (287). In contrast, CD56bright NK cells are positively correlated with loss of kidney function in chronic kidney disease and were more abundant in fibrotic biopsies, co-localizing with proximal tubular epithelial cells at sites of tubulointerstitial injury (288). In fibrotic samples, NKp46+CD117+CD56bright NK cells were the dominant source of IFN-γ and upregulated CD69, implying a role in renal injury and fibrosis (288).
ILCs in Ischemia-Reperfusion Injury
IRI occurs when temporary disruptions in blood flow cause hypoxic stress and injury to the kidney. Several lines of evidence suggest that ILCs influence IRI severity. Anti-asialo-GM1 and anti-NK1.1 depletion, or NKG2D blockade ameliorated IRI and prevented killing of Rae-1-expressing tubular epithelial cells by NK cells in mice (289, 290). Interactions between co-stimulatory receptor 4-1BB on NK cells and its activating ligand 4-1BBL on epithelial cells activate NK cells and recruit neutrophils via epithelial cell-derived CXCL1 and CXCL2 (291). Together, these results support a role for NK cell-epithelial cell interactions in aggravating IRI.
IL-25 and IL-33 administration expands ILC2s in mice, attenuating IRI and promoting recovery post ischemic injury (292, 293). ILC2 depletion using anti-CD90 treated Rag1-/- mice abolished the protective effect of IL-33 administration (293). Further, adoptive transfer of either murine (into C57BL/6) or human ILC2s (into NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice) conferred protection from IRI, while AREG-deficient ILC2s failed to abrogate IRI (293). In contrast, Liang et al. reported IL-33 treatment worsened disease scores and fibrosis in mice post-IRI (294). The timing and duration of IL-33 administration and experimental endpoints may account for differences in results, specifically that prolonged IL-33 administration may be detrimental, underscoring the importance of balance between tissue repair and fibrosis processes required to achieve tissue homeostasis.
Despite evidence that expansion or adoptive transfer of ILC2s is beneficial, depletion of ILC2s did not negatively impact IRI, as mice with reduced (Rorafl/+Il7rcre/+), depleted (Icosdtr/+ Cd4cre/+), or deficient for ILC2s (Rorafl/flIl7rcre/+), had no effect on IRI, suggesting ILC2 functions can be compensated for by other cell types (295). A regulatory ILC population (Lin- CD127+ CD25+ IL-10+) was identified in mice that limited ILC1s and pro-inflammatory macrophages in an IL-10- and TGF-β-dependent manner (15). Reduced tissue damage was noted in experimental IRI when these cells were expanded in Rag-/- mice (15). Notably, endogenous ILCregs in IRI produced less IL-10 than expanded ILCregs, suggesting that endogenous ILCreg function is impaired in IRI. While the human counterpart of this regulatory ILC subset was identified, confirmation of their function is needed (15).
ILCs in Lupus Nephritis
ILCs have been linked to kidney autoimmune pathologies such as lupus nephritis (LN), a manifestation of systemic lupus erythematosus (SLE). Arazi et al. identified two distinct NK cell populations within LN kidney tissue by scRNAseq, annotated as CD56dimCD16+ NK cells and tissue-resident CD56brightCD16- NK cells (296). CD56+ NK cells from SLE patients show an impaired antibody-dependent cellular cytotoxicity due to reduced CD3ζ signaling upon NCR engagement (297). Altered abundance of circulating ILCs in LN is associated with disease severity, with increased ILC1s across disease scores and decreased ILC3s in moderate to severe disease scores (298, 299). Further, reduced cytokine production and increased PD-1 expression suggest an exhausted ILC state in active disease (299). Specifically, CD117+ ILCs, likely ILC progenitors, were markedly decreased in LN and preferentially differentiated into ILC1s when cultured in LN plasma. Blockade of IL-1R reversed this effect, suggesting IL-1β-mediated regulation of the ILC progenitor pool (299). In a murine model of LN, renal ILC3s were the dominant source of IL-22 and were increased in abundance, while IL-22 deficiency ameliorated disease, supporting a pathogenic role for ILC3s in LN, yet whether an analogous mechanism applies to humans is unknown (300). These studies collectively support dysregulation of ILCs in SLE and LN.
ILCs in the Female Reproductive System
ILCs have established roles within the reproductive system and influence pregnancy outcomes (Figure 9A). While group 1 and group 3 ILCs are abundant in uterine tissue and participate in the dynamic regulation of reproductive health, little is known about the role of low abundance ILC2s (301).
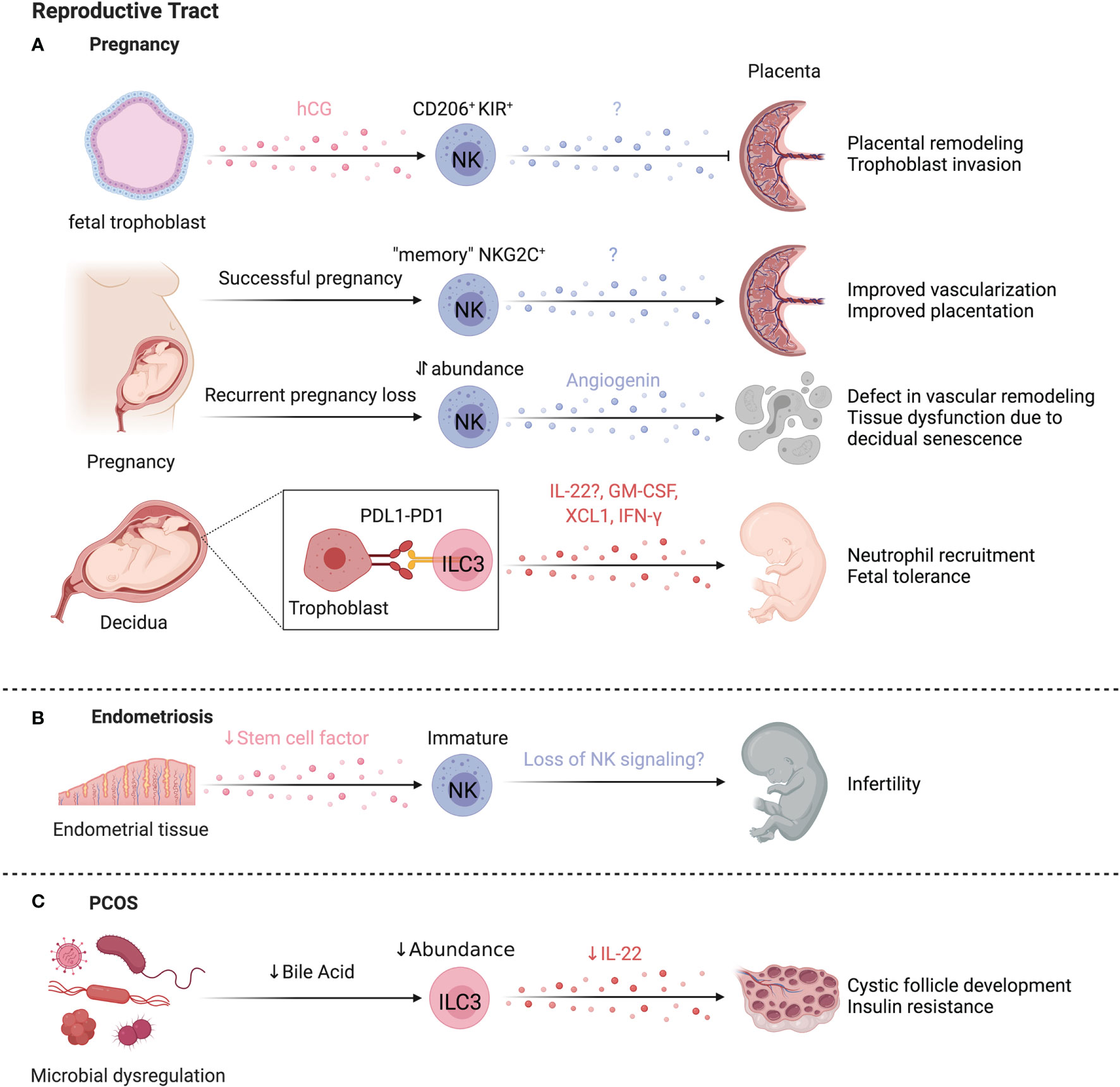
Figure 9 ILCs in the reproductive system. Most ILC data focuses on the female reproductive tract, where NK cells have established roles in pregnancy (A). uNK cells are regulated by human chorionic gonadotropin (hCG) released by fetal trophoblasts, signaling through CD206 (mannose receptor) to facilitate placental remodeling. Women with recurrent pregnancy losses show an increase in uNK cells in the endometrium, coinciding with thickened spiral artery walls and the resulting vascular remodeling affecting the blood flow to the fetus. In support of this, uNK cells from women with recurrent miscarriages also produced more angiogenic factors, fibroblast growth factors, and vascular endothelial growth factors. A special subset of memory uNK cells expressing higher levels of NKG2C contribute to improved reproductive success and lower incidence of pregnancy complications in subsequent pregnancies. In the human decidua, ILC3s express PD-1 and TIM-3, regulating ILC3 cytokine production, in particular IL-33. ILC3-trophoblast interactions may promote fetal tolerance during the first trimester via PD-1:PDL-1 interactions, supported by lower PD-L1 levels in trophoblasts of spontaneous abortions compared to healthy terminated pregnancies. (B) Reduced stem cell factor leads to a higher proportion of immature uNK cells in endometriosis, potentially contributing to infertility associated with endometriosis. (C) In a model of PCOS, microbial dysregulation led to reduced bile acids needed to support ILC3 function. The reduced ILC3 abundance and IL-22 production led to cystic follicle development and insulin resistance. Created with Biorender.org.
ILCs in Pregnancy
Pregnancy is a unique case where non-self is protected from immune-mediated rejection. Dynamic changes occur in both maternal and fetal tissues as pregnancy progresses. The uterine mucosa undergoes cyclic remodeling, termed decidualization, in which the endometrium thickens in preparation for implantation (302). For implantation and placentation to occur, the decidua must be invaded by fetal tissue (trophoblast cells) (303). Together, maternal and fetal tissue interactions promote successful implantation, placentation, and arterial spiralization to facilitate blood and nutrient supply to the developing fetus (303).
Uterine NK (uNK) cells are important cellular regulators of fetal implantation and protect against maternal rejection of the fetus. These uNK cells are the most abundant leukocytes present in the uterine mucosa and exhibit differential functions and limited cytotoxicity compared to cNK cells (304). Despite the discovery of uNK cells in the early 1990’s, the function of uNK cells in healthy and abnormal pregnancy is still the subject of intense research. Uterine NK cells contribute to placental remodeling, striking a balance between excessive trophoblast infiltration and defective placentation, regulated by KIR and MHC-I interactions (305). In general, activating receptor ligation improves reproductive success by promoting trophoblast invasion and vascular transformation (306). Human chorionic gonadotropin (hCG) released by the implanting fetal trophoblast induces uNK cell proliferation through hCG N-linked carbohydrate recognition by CD206 (mannose receptor) on uNK cells, establishing a pathway of uNK cell regulation by the implanting embryo (307).
ScRNAseq defined three distinct subsets of decidual NK cells (dNK cells) in humans. All subsets expressed CD49A and CD9, dNK1 cells expressed CD39, CYP26A1 and B4GALNT1, dNK2 cells expressed ANXA1 and ITGB2, and dNK3 cells expressed ITGB2, CD160, KLRB1 and CD103 (308). In particular, the highly granular and metabolically active dNK1 cells are hypothesized to interact with extravillous trophoblast cells because of high-level expression of KIRs and other HLA-molecule receptors (308). Unlike dNK2 and dNK3 cells, dNK1 cells were mostly IFN-γ- in response to stimulation (309). Computational predictions suggest the mechanism behind the prevention of an inflammatory immune response relies on immune-tissue crosstalk (308). Decidual stromal cells highly express LGALS9 and CLEC2D, pointing to potential NK cell inhibition via interaction with TIM-3 and KLRB1 (308). In line with the scRNAseq findings, Huhn et al. confirmed three subsets of dNK cells by mass cytometry with differential expression of the transcription factors TBET and EOMES (309). NK cells in the uterus or decidua acquire KIRs and CD39 along a developmental trajectory, corresponding to increased immunomodulatory and angiogenic function (310). With KIR acquisition, dNK cell expression of LILRB1, Ki-67, NKp30, and Granzyme B increased while NKG2D, CD161, and TBET decreased, unlike the relatively stable expression of these markers on cNK cells (309). Once acquired, the expression of KIRs remain remarkably stable for successive menstruation cycles (311). In another departure from cNK cells, granules were ~3 times larger in dNK cells and as KIR expression increased, degranulation and cytokine production decreased, supporting that dNK cells are phenotypically and functionally unique (309).
High NKG2C marks uNK cells that have acquired memory, or “trained immunity”, contributing to improved reproductive success in subsequent pregnancies by improving vascularization and placentation (312, 313). Greater abundance of uNK cells and aberrant function resulting in higher expression of angiogenic factors in the endometrium coincides with thickening of the spiral artery walls, suggesting alteration of uNK-mediated vascular remodelling as cause for recurring miscarriage (314, 315). However, lower abundance of uNK cells is also associated with recurrent pregnancy losses through reduced decidual-uNK cell interactions. Chronic senescence of decidual cells leads to tissue dysfunction not conducive to successful pregnancies and uNK cells selectively eliminate senescent decidual cells via NKG2D, while differentiating decidual cells support and recruit uNK cells with CXCL14, IL-15 and TIMP3 (316, 317). Indeed, endometrial biopsies from patients with recurrent pregnancy loss exhibit excessive decidual senescence and reduced uNK cell abundance, indicating that a balanced co-operation between decidual cells and uNK cells promotes healthy pregnancies (316).
dNK cells also protect against pregnancy loss from trophoblast bacterial infection. Here, rather than forming a cytotoxic synapse, dNK cells transfer granulysin to infected trophoblasts using nanotube connections, killing the intracellular bacteria without killing the trophoblast (318). This pathway could account for NK-mediated host defense in a setting that aims to avoid excessive tissue damage.
Other ILCs also have defined roles in pregnancy and reproductive conditions. Vacca et al. reported ILC3s in human decidua express PD-1 and TIM-3, which regulate ILC3 cytokine production, most notably IL-22 (319). Since trophoblast cells are PD-L1high, ILC3-trophoblast interactions may promote fetal tolerance during the first trimester (319). In support of this, PD-L1 levels were much lower or nonexistent in the trophoblast cells of spontaneous abortions compared to healthy terminated pregnancies (319). Subsets of CD127hiCD117hiAhRhiCD94-CD56+/-NKp44+/- decidual ILC3s were capable of producing GM-CSF, XCL1 and low levels of IFN-γ upon stimulation (309). NCR+ILC3s correlate with neutrophil abundance in the human decidua and produce GM-CSF and CXCL8, supporting neutrophil recruitment and survival (320). Based on lower decidual neutrophil numbers in patients with miscarriages, Croxatto et al. hypothesize that the ILC3-neutrophil axis is beneficial, particularly in early stages of pregnancy (320).
ILCs in Endometriosis
Pathological functions of uNK cells are linked to endometriosis, a condition affecting ~10% of women where endometrial tissue grows outside of the uterus, resulting in debilitating pain and infertility. Patients with endometriosis have increased immature uNK cell counts and lower levels of stem cell factor (SCF) in the endometrial tissue, associated with infertility (Figure 9B) (321). Supplementing cultures of immature uNK cells with SCF supports uNK cell maturation (321). Endometrial stromal cells express high levels of SCF, suggesting stromal-uNK cell interactions influence fertility outcomes in endometriosis. SCF receptor expression is also found on helper ILCs, however, their role in endometriosis requires further investigation (322–324). While many questions remain, these reports support a critical role for uNK cell homeostasis in fertility.
Notably, IL-33 elevation is implicated in endometriosis and exogenous IL-33 exacerbates lesion severity and fibrosis dependent on ILC2s in a murine endometriosis model (325). Yet, in endometrial tissue of patients with endometriosis, ILC2s and ILC3s are reduced in abundance relative to non-endometriosis controls (326). Further work will be needed to clarify the role of hILCs in endometriosis.
ILCs in Polycystic Ovary Syndrome
Separate from protective roles in pregnancy, reduced ILC3 activity is associated with polycystic ovary syndrome (PCOS). PCOS encompasses mixed metabolic and reproductive pathologies, such as irregular ovulation, infertility, hyperandrogenism, insulin resistance, and adipose tissue inflammation associated with a complex etiology including hormonal dysregulation and heritability (327). Reductions in bile acids due to microbial dysregulation decreased IL-22 production by ILC3s and promoted insulin resistance and cystic follicle development in a murine PCOS model (328). This was reversed by supplementing missing bile acids or IL-22, supporting a link between the gut and fertility (328). However, hormonal imbalances impact ILC function in PCOS. For example, progesterone driven IL-15, IL-18 and CXCL10 expression promote uNK cell recruitment and proliferation, and are altered in PCOS endometrial tissue (Figure 9C) (329). These findings indicate that tissue-ILC and microbiota-ILC interactions critically regulate reproductive homeostasis on multiple levels.
ILCs in the Heart
In mice, NK cells account for ~3% of cardiac immune cells, ILC2s for ~1.7%, and ILC1s for 0.2%, while ILC3 abundance is negligible (330). Compared to lung ILC2s, murine cardiac ILC2s had lower expression of ICOS, CD25, and Ki-67, and higher expression of Sca-1 and GATA3 (330). Only 2% of cardiac ILC2s in mouse were donor-derived after 2 months of parabiosis, indicating that ILC2s are a stable tissue-resident population in the heart (330). Cardiac-resident ILC2s respond to IL-33 but not IL-25, and a committed cardiac ILC2 precursor (ILC2p) in mice and humans exists in a quiescent state with the capability to differentiate into ILC2s in response to myocardial infarct or myocarditis (331). The existence of undifferentiated ILC2p within tissues has been observed before and suggests a role for this pool of precursors as a reservoir for ILCs to protect from tissue damage (332). Cardiac ILC activity has been implicated in several disease models (Figure 10).
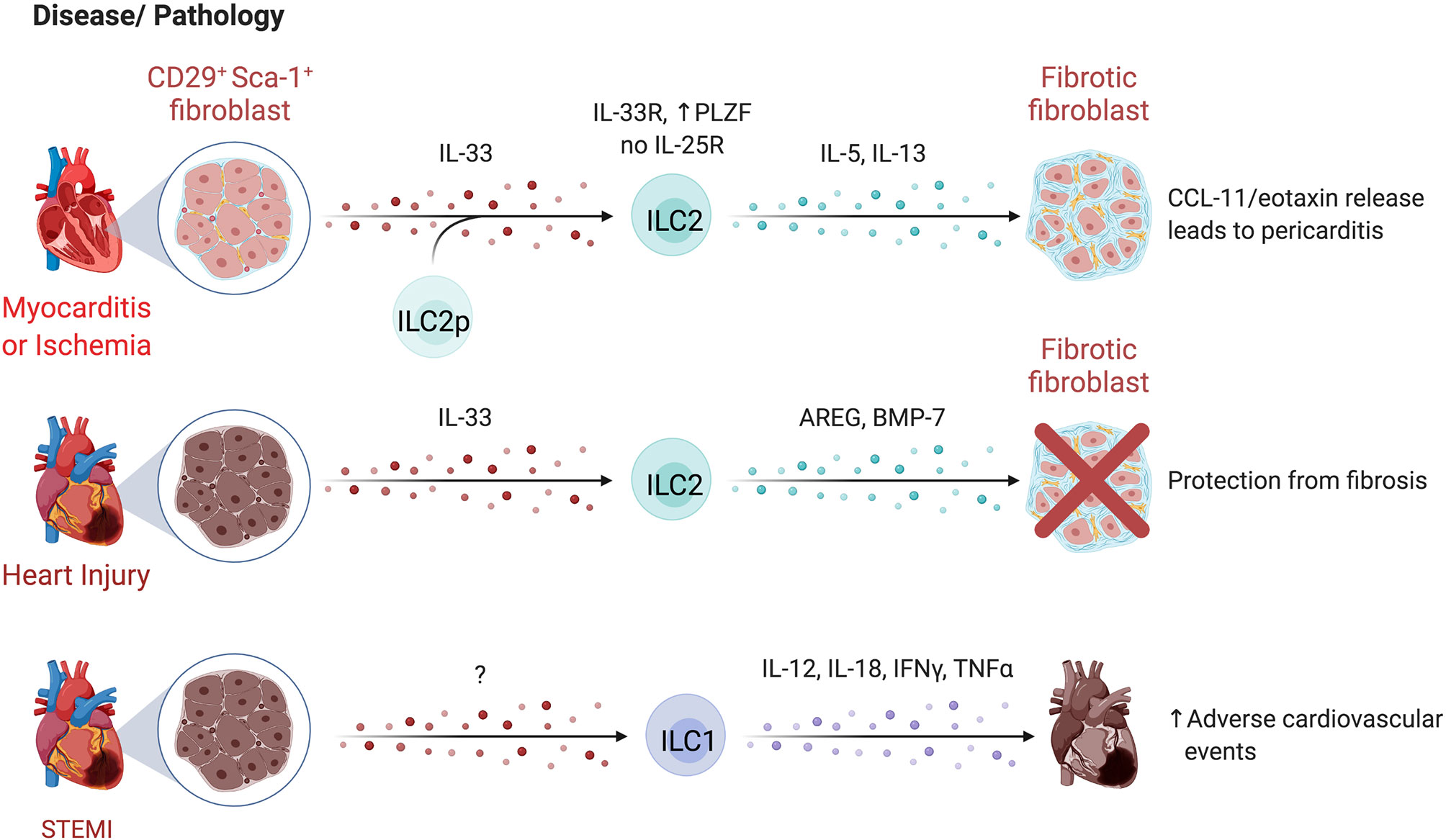
Figure 10 ILCs in the heart. Limited studies have examined ILCs in the heart. In mice, IL-33 administration or CD27+Sca-1+ fibroblast-derived IL-33 expands ILC2s in the pericardium, driving cardiac fibroblasts to secrete CCL-11/eotaxin, leading to the accumulation of eosinophils. In humans, CD127+ ILCs increase in the pericardial fluid during in cardiac disease, particularly during pericarditis. Separately, the expansion of ILC2s by IL-33 was cardioprotective after injury, reducing fibrosis. Within 12 hours of acute ST-segment elevation myocardial infraction (STEMI), ILC1 elevation peaks. Higher ILC1-associated IL-12, IL-18, IFN-γ, and TNF-α levels lead to a higher risk of major adverse cardiovascular events. Created with Biorender.org.
ILCs in Atherosclerosis and Coronary Artery Disease
Conflicting findings on ILC subset functions in atherosclerosis and coronary artery disease (CAD) have been reported. Selathurai et al. found murine NK cells were detrimental to atherosclerotic disease, increasing lesion size in a Perforin- and Granzyme B-dependent manner, while Nour-Eldine et al. found no effect of NK cells on lesion development using a distinct genetic depletion model, likely accounting for divergent findings (333, 334).
In humans, a study of acute ST-segment elevation myocardial infarction (STEMI) found elevated circulating ILC1s within 12 hours of symptom onset which produced more IL-12, IL-18, IFN-γ, and TNF-α, and were associated with a higher risk of major adverse cardiovascular events (335). In contrast, NK cells were reduced and have lower cytotoxicity in CAD patients, both in the case of stable angina and incidence of myocardial infarction or unstable disease (336–338). During follow-up, patients who failed to reconstitute their peripheral NK cells post myocardial infarction had higher levels of serum IL-6 and exhibited characteristics of metabolic syndrome, suggesting poor NK cell recovery corresponds with low-grade inflammation (336). Recovery of NK cells in CAD patients is potentially self-regulated, as apoptotic NK cells both respond to and produce FasL, which is elevated in serum and correlates with NK cell levels and apoptotic susceptibility (339). Increased proportions of CD56bright NK cells were identified in carotid plaques compared to autologous peripheral blood, and greater NK cell infiltration corresponded with symptomatic versus asymptomatic CAD patients (340). Soluble B7-H6 levels of 250 pg/ml were detected in symptomatic patients but not in asymptomatic patients or healthy controls (340). Notably, B7-H6 can interact with NKp30, yet further studies are needed to directly assess B7-H6 and NK cell interactions in this context. Circulating NK cells from atherosclerotic patients had higher TIM-3 expression than healthy controls, with the greatest levels in those with unstable plaques (341). TIM-3 blockade reduced the death of NK cells cultured in TNF-α, suggesting that TIM-3 promotes cytokine-induced NK cell apoptosis in atherosclerosis (341). Whether NK cells are preferentially recruited to unstable carotid plaques, or functionally contribute to plaque destabilization requires additional study (340).
ILC2s appear to have cardioprotective functions based on mouse models. ILC2s protect from cardiac fibrosis and are enhanced by exogenous IL-33, producing AREG and BMP-7 to support cardioprotective responses to injury (342). Expansion of ILC2s reduces atherosclerosis severity and lesion size, while genetic ablation (Staggerer/RoraFlox-CD127Cre) exacerbates disease (343, 344). Notably, protection is IL-5 and IL-13-dependent, recruiting eosinophils and polarizing macrophages towards an anti-inflammatory phenotype (342, 343). Further, pericardial and cardiac ILC2s expand early post-experimental myocardial infarction, peaking at day 3 before returning to homeostatic levels, while the absence of ILC2s impairs cardiac remodeling and results in larger areas of scarring (345). Together, this supports a role for cardiac-resident ILC2s in directing repair pathways in response to injury.
ILCs in Cardiac Inflammation
ILC expansion has been observed in patients with pericarditis (346). In contrast to cardioprotective findings above, ILC2s have been implicated in pericarditis pathology. Exogenous IL-33 expanded murine pericardial ILC2s, driving cardiac fibroblasts to secrete CCL11/eotaxin-1 and recruit eosinophils, initiating pericarditis (346). Pericardial fluid from humans revealed an elevated frequency of CD127+ ILCs in patients with cardiac disease versus controls, indicating that ILCs are also involved in human pericardial pathology (346). In an opposing role, NK cell depletion led to greater inflammation and fibrosis of the heart, dependent on NK cell-mediated prevention of eosinophilic infiltration (347). A possible cross-regulation of NK cells and ILC2s during cardiac inflammation should be investigated.
Sex differences in mortality and morbidity of Coxsackievirus B3 (CVB3) viral myocarditis may also reflect sex-based regulation of NK cell function. Male mice experience greater morbidity and mortality from myocarditis following CVB3 infection, with increased IFN-γ+ NK cell infiltration in cardiac tissue (348). Ovariectomized or sexually immature female mice show similar susceptibility to infection-triggered myocarditis when compared to male mice, while estrogen-treated male mice had ameliorated myocarditis (348). CVB3-stimulated NK cells cultured with estrogen down-regulated T-bet expression and consequently had reduced IFN-γ production, indicating that regulation of T-bet expression by estrogen might underlie the decreased IFN-γ+ NK cell infiltration in female mice and contribute to sex differences in myocarditis, in line with prior reports of hormonal regulation of other ILC subsets (100, 348).
Adipose ILCs
Adipocytes are critical regulators of energy and glucose homeostasis. They are a heterogeneous population of cells, comprising energy-storing white adipocytes, thermogenic brown adipocytes that express uncoupling protein-1 (UCP-1) to dissipate energy as heat, as well as beige adipocytes that reside within white adipose tissue (WAT) and upregulate UCP-1 in response to environmental cues (349). Beige adipocyte accumulation protects from insulin resistance, and regulation of the beiging process has become an attractive therapeutic target for metabolic dysregulation and type 2 diabetes (350).
ILC2s are the dominant hILC subtype identified in adipose tissue, with phenotypic variation across different murine adipose compartments: ILC2s in para-aortic adipose tissue have an inflammatory phenotype defined by IL-25 responsiveness and high KLRG1 expression, whereas peri-gonadal adipose ILC2s are IL-33 responsive, expressing ST2 (343, 351). Intriguingly, mouse ILC2s highly express bone morphogenetic protein (Bmp)2 and Bmp7, which promote adipocyte differentiation, while ILC-deficient mice have more CD34+PDGFRα+ precursor adipocytes, supporting a role for ILCs in adipogenesis (352). Under homeostatic conditions, ILC2s orchestrate immune responses in adipose tissues by recruiting eosinophils, promoting alternative activation of macrophages, and regulating beiging, glucose catabolism, and insulin sensitivity in adipose tissues (Figure 11A) (353, 354). In support of this, depletion of IL-5 and IL-13-producing cells (mainly ILC2s) corresponded to a reduction of eosinophils and Arg-1+ adipose macrophages in visceral adipose tissue (355). ILC2s also promote Treg responses through ICOSL and OX40L co-stimulation in adipose tissues, critical for supporting insulin sensitivity (356–358).
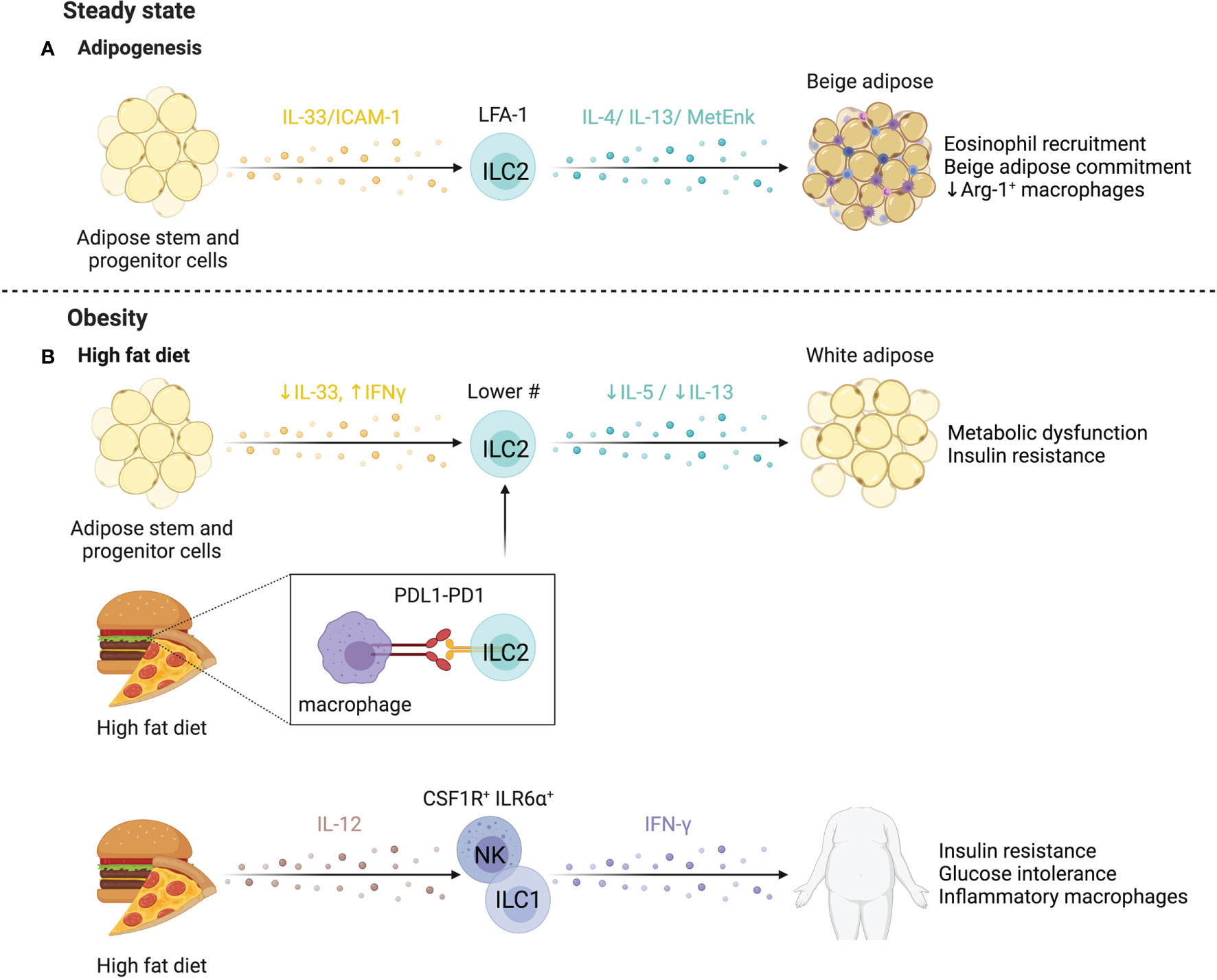
Figure 11 ILCs in adipose tissue. (A) Using mostly mouse models, ILC2s are linked to adipogenesis, where they maintain homeostasis and prevent obesity by promoting adipose beiging. ILC2s are sustained by IL-33 from adipose stem and progenitor cells (ASPCs) and required stromal cell interactions via ICAM-1 and LFA-1. The resulting release of IL-4, IL-13 by ILC2s promotes eosinophil recruitment via stromal cell-derived eotaxin (CCL11). IL-33 stimulates ILC2-produced methionine-enkephalin (MetEnk), an endogenous opioid-like peptide that promotes adipose beiging. (B) Both obese humans and mice, have reduced ILC2s in the white adipose tissue due to diet-driven impairment of IL-33 production by ASPCs. The infiltration of IFN-γ-producing ILCs actively represses cytokine release by ILC2s and propagates ILC2 inhibition through via PD-1 and macrophage-expressed PD-L1. A unique subpopulation of CSF1R+IL16Rα+ NK cells and increased ILC1 abundance positively correlates with glucose intolerance and insulin resistance. Overall, reduction of adipose ILC2s fosters metabolic dysfunction, insulin resistance and obesity. Created with Biorender.org.
Mouse studies support that ILC2s can directly promote adipose beiging, supporting homeostasis and preventing obesity (5, 359, 360). ILC2s are sustained by IL-33 from adipose stem and progenitor cells (ASPCs) (360). Stromal ICAM-1 interactions with LFA-1 on ILC2s promotes ILC2 activation and proliferation in adipose tissue, while IL-4/IL-13 expression by ILC2s induces eotaxin (CCL11) expression in stromal cells, supporting eosinophil recruitment (361). Peritoneal IL-33 administration expands adipocyte precursors and promotes beige lineage commitment in an ILC2-dependent manner, as effects are abolished in the absence of ILC2s and when adipocyte precursors are not receptive to IL-4/IL-13 signaling (Il4rafl/flPdgfraCre) (359). An alternative mechanism of ILC2-dependent beiging of mouse adipose tissue was proposed by Brestoff et al. who found IL-33-stimulated ILC2s produce methionine-enkephalin (MetEnk), an endogenous opioid-like peptide which induces WAT beiging (5). Overall, these studies demonstrate that ILC2-dependent eosinophil-derived IL-4 and ILC2-derived IL-13 and/or MetEnk directly promote murine adipocyte precursor proliferation and beige lineage commitment (5, 359).
Group 1 ILCs are largely resident in murine adipose tissue (362). Although their homeostatic role is poorly understood, ILC1s regulate adipose macrophage homeostasis (363). Alternatively activated macrophages scavenge potentially cytotoxic molecules released during adipose tissue remodeling and upregulate stress ligands (i.e. Rae-1) at steady state, and their selective depletion by adipose type 1 ILCs prevents stress-induced inflammation in macrophages during homeostatic tissue remodeling (363).
ILCs in Obesity
Dysregulation of the immune environment associated with obesity can lead to metabolic dysfunction and insulin resistance, driving type 2 diabetes (T2D) (364). ILC dysregulation has been implicated in obesity (Figure 11B). In obese humans and mice, ILC2s are reduced in WAT, possibly due to reduced IL-33 production by ASPCs in response to high fat diet (5, 360, 365). High PD-1 expression on ILC2s reduced IL-5 and IL-13 production, an effect partially rescued by macrophage depletion, suggesting PD-1/PD-L1 interactions between ILC2s and macrophages dampens ILC2 function in obese conditions in mice (365). Additionally, adipocyte-derived soluble ST2 is induced by obesity and interrupts IL-33 signaling, impairing ILC2 homeostasis (366). Infiltration of IFN-γ-producing cells also contributes to reduced ILC2 abundance and function, as IFN-γ directly represses ILC2s and counteracts IL-33 (357).
Obese mice fed a high fat diet had adipose tissue-specific IL-12-dependent accumulation of ILC1s with elevated IFN-γ production, resulting in insulin resistance and glucose intolerance (362, 367). Interestingly, CD56dim CD16– ILCs accumulating during obesity have reduced cytotoxicity, a potential secondary mechanism contributing to macrophage accumulation and glucose intolerance (363). Wensveen et al. demonstrated NKp46 on adipose-resident mouse NK cells may regulate this effect (368). High fat diet-induced obesity triggered the expression of NCR1 ligands on adipocytes which promoted local NK cell proliferation and production of IFN-γ, inducing the differentiation of pro-inflammatory macrophages and promoting insulin resistance (368). Wang et al. further found that the ILC1 IFN-γ-dependent expansion of pro-inflammatory macrophages exacerbated adipose fibrosis by promoting TGF-β1 and pro-fibrotic programs in macrophages, resulting in higher collagen deposition (367).
In agreement with murine models, circulating and adipose ILC1s are increased in obese patients, especially those with T2D, and the abundance of ILC1s positively correlates with measures of glucose intolerance and insulin resistance (367). A unique subpopulation of CSF1R+IL6Rα+ NK cells is expanded in human and murine obesity (369). Selective depletion of this subset (Csf1r-loxSTOPlox-DTR x NcrCre) resulted in decreased weight gain, better glucose tolerance, and insulin responsiveness in mice fed a high fat diet (369). Further, the expression of RORγt, lymphotoxin and IL-22 all elevated weight gain and adipose tissue size, paralleling findings that IL-22 from Th17 cells exacerbates inflammation in obesity (370, 371). The regulation of metabolism by intestinal ILC3s suggests a gut-adipose axis that remains to be explored. Overall, ILC2s mediate adipose homeostasis and are dysregulated in obesity, while ILC1s and potentially ILC3s have a role in exacerbating inflammation.
Concluding Remarks
Multiple parallels and differences between murine and human ILCs exist. Their evolutionarily conserved transcriptional programs and functional similarity emphasizes their importance across multiple distinct phylogenetic branches. However, a better understanding of homologies and analogies in their surface receptor expression and function are needed to inform conserved mechanisms underlying responses to infections, inflammation and malignancies. This will be of particular interest to enhance our understanding NK cell biology, where receptors regulating NK cells responses differ between mouse and human, but different receptors often perform similar function.
The distinct living conditions of mice and humans require conserved and specified adaptations of organs and tissues to environmental triggers. ILCs as regulators of tissue homeostasis adapt to these species-specific environments. Shared and differing microbiota within humans and mice may explain conserved and distinct functions of ILCs across these organisms. Gnotobiotic technologies, humanized mice, knock-out mouse models or adoptive transfer experiments are suitable to investigate these differences but bring their own pitfalls. Beyond these challenges, a key obstacle for the study of ILCs in situ is the scarcity of ILC-specific models available to tease out cell-specific or even organ-specific functions. Adding to this, ILCs typically function in cellular networks to influence the outcome of a given immune response. To properly understand their function and tease out any redundancy, more systems-based approaches are needed, particularly in humans (295, 372, 373).
Other roadblocks in understanding the role of various ILCs in homeostasis and disease are studies designed only to link the presence or absence of ILCs with disease outcome. This is especially evident in reports of NK cell function. Reporting expansion or reduction of NK cells as having a protective or detrimental effect assumes a homogeneous function of NK cells. While classically, NK cell function has been cytotoxic and inflammatory, NK cells have also been cast in an immunoregulatory role where they dampen an immune response (19). Additionally, identifying NK cells as CD3-CD56+ does not rule out other non-cytotoxic ILC1s and ILC3s that can share CD56 expression, and does not address the large degree of heterogeneity within NK cells (374). Untangling the identity and functional capacity of distinct CD3- CD56+ populations may help to clarify contradictory findings.
The plasticity of ILCs makes definitively assigning them as “good” or “bad” quite problematic. Sometimes ILCs with identical or similar surface phenotype may be functionally distinct (206). While this may be context dependent, ILC plasticity may be partially responsible for conflicting reports of their function in disease. Future studies should consider assessing the functional role of ILC subsets correlated with ILC transcriptional and epigenetic profiles to identify mechanisms underlying distinct ILC functions and whether some level of ‘trained’ immunity contributes to differing findings. Additionally, it remains unclear if the tissue-specific functions of ILCs are due in part to the existence of specific subsets of ILCs that home to their niche, or instead these different functions are a directly due to microenvironment signals leading to niche adaptation. It is also entirely possible that both cases are true and contribute to establishing tissue-specific ILC functions. Moving forward, the characterization of tissue-specific networks and niches for ILCs will transform our understanding of ILC functions and underlying mechanisms controlling their tissue adaptations.
Author Contributions
All authors contributed to conceptualization, topic curation, writing and editing of the manuscript. Figures were designed by LN, with input from JM, AM, and SC. All authors contributed to the article and approved the submitted version.
Funding
JMM was supported by Queen Elizabeth II, Dick and Peggy Sharpe, and Peterborough KM Hunter Scholarships. LN was supported by an Ontario Graduate Scholarship and a NSERC-PGS award. AM's research program is supported by Canadian Institutes for Health Research (CIHR, 388337) and Natural Sciences and Engineering Research Council of Canada (NSERC, RGPIN-2019-04521), and is a Tier 2 Canadian Research Chair in Mucosal Immunology. SQC's research program is supported by funding from CIHR (168960 and 169084) and a NSERC (RGPIN-2021-03672), and is a Medicine By Design Investigator (Canada First Research Excellence Fund).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
All figures were created with BioRender.com.
Glossary

References
1. Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue Residency of Innate Lymphoid Cells in Lymphoid and Nonlymphoid Organs. Science (80-) (2015) 350:981–5. doi: 10.1126/science.aac9593
2. Godinho-Silva C, Domingues RG, Rendas M, Raposo B, Ribeiro H, da Silva JA, et al. Light-Entrained and Brain-Tuned Circadian Circuits Regulate ILC3s and Gut Homeostasis. Nature (2019) 574:254–8. doi: 10.1038/s41586-019-1579-3
3. Klose CS, Artis D. Neuronal Regulation of Innate Lymphoid Cells. Curr Opin Immunol (2019) 56:94–9. doi: 10.1016/j.coi.2018.11.002
4. Von Moltke J, Ji M, Liang HE, Locksley RM. Tuft-Cell-Derived IL-25 Regulates an Intestinal ILC2-Epithelial Response Circuit. Nature (2016) 529:221–5. doi: 10.1038/nature16161
5. Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, et al. Group 2 Innate Lymphoid Cells Promote Beiging of White Adipose Tissue and Limit Obesity. Nature (2015) 519:242–6. doi: 10.1038/nature14115
6. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate Lymphoid Cells: 10 Years on. Cell (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
7. Vonarbourg C, Diefenbach A. Multifaceted Roles of Interleukin-7 Signaling for the Development and Function of Innate Lymphoid Cells. Semin Immunol (2012) 24:165–74. doi: 10.1016/j.smim.2012.03.002
8. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate Lymphoid Cells-a Proposal for Uniform Nomenclature. Nat Rev Immunol (2013) 13:145–9. doi: 10.1038/nri3365
9. Fauriat C, Long EO, Ljunggren H-G, Bryceson YT. Regulation of Human NK-Cell Cytokine and Chemokine Production by Target Cell Recognition. Blood (2016) 115:2167–77. doi: 10.1182/blood-2009-08-238469.A
10. Cooper MA, Fehniger TA, Turner SC, Chen KS, Ghaheri BA, Ghayur T, et al. Human Natural Killer Cells: A Unique Innate Immunoregulatory Role for the CD56bright Subset. Blood (2001) 97:3146–51. doi: 10.1182/blood.V97.10.3146
11. Mebius RE, Rennert P, Weissman IL. Developing Lymph Nodes Collect CD4+CD3- Ltβ+ Cells That Can Differentiate to APC, NK Cells, and Follicular Cells But Not T or B Cells. Immunity (1997) 7:493–504. doi: 10.1016/S1074-7613(00)80371-4
12. Crome SQ, Ohashi PS. Immunoregulatory Functions of Innate Lymphoid Cells. J Immunother Cancer (2018) 6:4–7. doi: 10.1186/s40425-018-0433-8
13. Wang S, Xia P, Chen Y, Qu Y, Xiong Z, Ye B, et al. Regulatory Innate Lymphoid Cells Control Innate Intestinal Inflammation. Cell (2017) 171:201–16. doi: 10.1016/j.cell.2017.07.027
14. Crome SQ, Nguyen LT, Lopez-Verges S, Yang SYC, Martin B, Yam JY, et al. A Distinct Innate Lymphoid Cell Population Regulates Tumor-Associated T Cells. Nat Med (2017) 23:368–75. doi: 10.1038/nm.4278
15. Cao Q, Wang R, Wang Y, Niu Z, Chen T, Wang C, et al. Regulatory Innate Lymphoid Cells Suppress Innate Immunity and Reduce Renal Ischemia/Reperfusion Injury. Kidney Int (2020) 97:130–42. doi: 10.1016/j.kint.2019.07.019
16. Golebski K, Layhadi JA, Sahiner U, Steveling-Klein EH, Lenormand MM, Li RCY, et al. Induction of IL-10-Producing Type 2 Innate Lymphoid Cells by Allergen Immunotherapy Is Associated With Clinical Response. Immunity (2021) 54:291–307.e7. doi: 10.1016/j.immuni.2020.12.013
17. Bando JK, Gilfillan S, Di Luccia B, Fachi JL, Fachi JL, Sécca C, et al. ILC2s are the Predominant Source of Intestinal ILC-Derived IL-10. J Exp Med (2020) 217:e20191520. doi: 10.1084/jem_20191520
18. Bielekova B, Catalfamo M, Reichert-Scrivner S, Packer A, Cerna M, Waldmann TA, et al. Regulatory CD56bright Natural Killer Cells Mediate Immunomodulatory Effects of IL-2rα-Targeted Therapy (Daclizumab) in Multiple Sclerosis. Proc Natl Acad Sci USA (2006) 103:5941–6. doi: 10.1073/pnas.0601335103
19. Jegatheeswaran S, Mathews JA, Crome SQ. Searching for the Elusive Regulatory Innate Lymphoid Cell. J Immunol (2021) 207:1949–57. doi: 10.4049/jimmunol.2100661
20. Shikhagaie MM, Germar K, Bal SM, Ros XR, Spits H. Innate Lymphoid Cells in Autoimmunity: Emerging Regulators in Rheumatic Diseases. Nat Rev Rheumatol (2017) 13:164–73. doi: 10.1038/nrrheum.2016.218
21. Li Z, Li M, Shi SX, Yao N, Cheng X, Guo A, et al. Brain Transforms Natural Killer Cells That Exacerbate Brain Edema After Intracerebral Hemorrhage. J Exp Med (2020) 217:e20200213. doi: 10.1084/jem.20200213
22. Korin B, Ben-Shaanan TL, Schiller M, Dubovik T, Azulay-Debby H, Boshnak NT, et al. High-Dimensional, Single-Cell Characterization of the Brain’s Immune Compartment. Nat Neurosci (2017) 20:1300–9. doi: 10.1038/nn.4610
23. Gadani SP, Smirnov I, Smith AT, Overall CC, Kipnis J. Characterization of Meningeal Type 2 Innate Lymphocytes and Their Response to CNS Injury. J Exp Med (2017) 214:285–96. doi: 10.1084/jem.20161982
24. Dahlgren MW, Jones SW, Cautivo KM, Dubinin A, Ortiz-Carpena JF, Farhat S, et al. Adventitial Stromal Cells Define Group 2 Innate Lymphoid Cell Tissue Niches. Immunity (2019) 50:707–22.e6. doi: 10.1016/j.immuni.2019.02.002
25. Golomb SM, Guldner IH, Zhao A, Wang Q, Palakurthi B, Aleksandrovic EA, et al. Multi-Modal Single-Cell Analysis Reveals Brain Immune Landscape Plasticity During Aging and Gut Microbiota Dysbiosis. Cell Rep (2020) 33:108438. doi: 10.1016/j.celrep.2020.108438
26. Browne P, Chandraratna D, Angood C, Tremlett H, Baker C, Taylor BV, et al. Atlas of Multiple Sclerosis 2013: A Growing Global Problem With Widespread Inequity. Neurology (2014) 83:1022–4. doi: 10.1212/WNL.0000000000000768
27. Martin JF, Perry JSA, Jakhete NR, Wang X, Bielekova B. An IL-2 Paradox: Blocking CD25 on T Cells Induces IL-2–Driven Activation of CD56 Bright NK Cells. J Immunol (2010) 185:1311–20. doi: 10.4049/jimmunol.0902238
28. Sheridan JP, Zhang Y, Riester K, Tang MT, Efros L, Shi J, et al. Intermediate-Affinity Interleukin-2 Receptor Expression Predicts CD56bright Natural Killer Cell Expansion After Daclizumab Treatment in the CHOICE Study of Patients With Multiple Sclerosis. Mult Scler J (2011) 17:1441–8. doi: 10.1177/1352458511414755
29. Jiang W, Chai NR, Maric D, Bielekova B. Unexpected Role for Granzyme K in CD56 Bright NK Cell-Mediated Immunoregulation of Multiple Sclerosis. J Immunol (2011) 187:781–90. doi: 10.4049/jimmunol.1100789
30. Gross CC, Schulte-Mecklenbeck A, Rünzi A, Kuhlmann T, Posevitz-Fejfár A, Schwab N, et al. Impaired NK-Mediated Regulation of T-Cell Activity in Multiple Sclerosis is Reconstituted by IL-2 Receptor Modulation. Proc Natl Acad Sci USA (2016) 113:E2973–82. doi: 10.1073/pnas.1524924113
31. Takahashi K, Aranami T, Endoh M, Miyake S, Yamamura T. The Regulatory Role of Natural Killer Cells in Multiple Sclerosis. Brain (2004) 127:1917–27. doi: 10.1093/brain/awh219
32. Darlington PJ, Stopnicki B, Touil T, Doucet JS, Fawaz L, Roberts ME, et al. Natural Killer Cells Regulate Th17 Cells After Autologous Hematopoietic Stem Cell Transplantation for Relapsing Remitting Multiple Sclerosis. Front Immunol (2018) 9:834. doi: 10.3389/fimmu.2018.00834
33. Lünemann A, Tackenberg B, DeAngelis T, da Silva RB, Messmer B, Vanoaica LD, et al. Impaired IFN-γ Production and Proliferation of NK Cells in Multiple Sclerosis. Int Immunol (2011) 23:139–48. doi: 10.1093/intimm/dxq463
34. Rodríguez-Martín E, Picón C, Costa-Frossard L, Alenda R, Sainz de la Maza S, Roldán E, et al. Natural Killer Cell Subsets in Cerebrospinal Fluid of Patients With Multiple Sclerosis. Clin Exp Immunol (2015) 180:243–9. doi: 10.1111/cei.12580
35. Laroni A, Armentani E, Kerlero de Rosbo N, Ivaldi F, Marcenaro E, Sivori S, et al. Dysregulation of Regulatory CD56bright NK Cells/T Cells Interactions in Multiple Sclerosis. J Autoimmun (2016) 72:8–18. doi: 10.1016/j.jaut.2016.04.003
36. McKay FC, Gatt PN, Fewings N, Parnell GP, Schibeci SD, Basuki MAI, et al. The Low EOMES/TBX21 Molecular Phenotype in Multiple Sclerosis Reflects CD56+ Cell Dysregulation and Is Affected by Immunomodulatory Therapies. Clin Immunol (2016) 163:96–107. doi: 10.1016/j.clim.2015.12.015
37. Hao J, Liu R, Piao W, Zhou Q, Vollmer TL, Campagnolo DI, et al. Central Nervous System (CNS)-Resident Natural Killer Cells Suppress Th17 Responses and CNS Autoimmune Pathology. J Exp Med (2010) 207:1907–21. doi: 10.1084/jem.20092749
38. Gan Y, Liu R, Wu W, Bomprezzi R, Shi FD. Antibody to α4 Integrin Suppresses Natural Killer Cells Infiltration in Central Nervous System in Experimental Autoimmune Encephalomyelitis. J Neuroimmunol (2012) 247:9–15. doi: 10.1016/j.jneuroim.2012.03.011
39. Lünemann A, Lünemann JD, Roberts S, Messmer B, da Silva RB, Raine CS, et al. Human NK Cells Kill Resting But Not Activated Microglia via NKG2D- and NKp46-Mediated Recognition. J Immunol (2008) 181:6170–7. doi: 10.4049/jimmunol.181.9.6170
40. Jiang W, Li D, Han R, Zhang C, Jin WN, Wood K, et al. Acetylcholine-Producing NK Cells Attenuate CNS Inflammation via Modulation of Infiltrating Monocytes/Macrophages. Proc Natl Acad Sci USA (2017) 114:E6202–11. doi: 10.1073/pnas.1705491114
41. Liu Q, Sanai N, Jin WN, La Cava A, Van Kaer L, Shi FD. Neural Stem Cells Sustain Natural Killer Cells That Dictate Recovery From Brain Inflammation. Nat Neurosci (2016) 19:243–52. doi: 10.1038/nn.4211
42. Garofalo S, Cocozza G, Porzia A, Inghilleri M, Raspa M, Scavizzi F, et al. Natural Killer Cells Modulate Motor Neuron-Immune Cell Cross Talk in Models of Amyotrophic Lateral Sclerosis. Nat Commun (2020) 11:1773. doi: 10.1038/s41467-020-15644-8
43. Serafini B, Rosicarelli B, Veroni C, Zhou L, Reali C, Aloisi F. Rorγt Expression and Lymphoid Neogenesis in the Brain of Patients With Secondary Progressive Multiple Sclerosis. J Neuropathol Exp Neurol (2016) 75:877–88. doi: 10.1093/jnen/nlw063
44. Schropp V, Rohde J, Rovituso DM, Jabari S, Bharti R, Kuerten S. Contribution of LTi and TH17 Cells to B Cell Aggregate Formation in the Central Nervous System in a Mouse Model of Multiple Sclerosis. J Neuroinflamm (2019) 16:1–15. doi: 10.1186/s12974-019-1500-x
45. Hatfield JK, Brown MA. Group 3 Innate Lymphoid Cells Accumulate and Exhibit Disease-Induced Activation in the Meninges in EAE. Cell Immunol (2015) 297:69–79. doi: 10.1016/j.cellimm.2015.06.006
46. Kwong B, Rua R, Gao Y, Flickinger J, Wang Y, Kruhlak MJ, et al. T-Bet-Dependent NKp46+ Innate Lymphoid Cells Regulate the Onset of TH17-Induced Neuroinflammation. Nat Immunol (2017) 18:1117–27. doi: 10.1038/ni.3816
47. Degn M, Modvig S, Dyring-Andersen B, Bonefeld CM, Frederiksen JL, Geisler C, et al. Increased Prevalence of Lymphoid Tissue Inducer Cells in the Cerebrospinal Fluid of Patients With Early Multiple Sclerosis. Mult Scler (2016) 22:1013–20. doi: 10.1177/1352458515609795
48. Perry JSA, Han S, Xu Q, Herman ML, Kennedy LB, Csako G, et al. Inhibition of LTi Cell Development by CD25 Blockade is Associated With Decreased Intrathecal Inflammation in Multiple Sclerosis. Sci Transl Med (2012) 4:1–8. doi: 10.1126/scitranslmed.3004140
49. Russi AE, Walker-Caulfield ME, Ebel ME, Brown MA. Cutting Edge: C-Kit Signaling Differentially Regulates Type 2 Innate Lymphoid Cell Accumulation and Susceptibility to Central Nervous System Demyelination in Male and Female SJL Mice. J Immunol (2015) 194:5609–13. doi: 10.4049/jimmunol.1500068
50. Russi AE, Ebel ME, Yang Y, Brown MA. Male-Specific IL-33 Expression Regulates Sex-Dimorphic EAE Susceptibility. Proc Natl Acad Sci USA (2018) 115:E1520–9. doi: 10.1073/pnas.1710401115
51. Kong Y, Li S, Cheng X, Ren H, Zhang B, Ma H, et al. Brain Ischemia Significantly Alters microRNA Expression in Human Peripheral Blood Natural Killer Cells. Front Immunol (2020) 11:759. doi: 10.3389/fimmu.2020.00759
52. Liu Q, Jin WN, Liu Y, Shi K, Sun H, Zhang F, et al. Brain Ischemia Suppresses Immunity in the Periphery and Brain via Different Neurogenic Innervations. Immunity (2017) 46:474–87. doi: 10.1016/j.immuni.2017.02.015
53. Gan Y, Liu Q, Wu W, Yin JX, Bai XF, Shen R, et al. Ischemic Neurons Recruit Natural Killer Cells That Accelerate Brain Infarction. Proc Natl Acad Sci USA (2014) 111:2704–9. doi: 10.1073/pnas.1315943111
54. Lee GA, Lin TN, Chen CY, Mau SY, Huang WZ, Kao YC, et al. Interleukin 15 Blockade Protects the Brain From Cerebral Ischemia-Reperfusion Injury. Brain Behav Immun (2018) 73:562–70. doi: 10.1016/j.bbi.2018.06.021
55. Li Q, Liu M, Fu R, Cao Q, Wang Y, Han S, et al. Alteration of Circulating Innate Lymphoid Cells in Patients With Atherosclerotic Cerebral Infarction. Am J Transl Res (2018) 10:4322–30.
56. Zhang Y, Fung ITH, Sankar P, Chen X, Robison LS, Ye L, et al. Depletion of NK Cells Improves Cognitive Function in the Alzheimer Disease Mouse Model. J Immunol (2020) 205:502–10. doi: 10.4049/jimmunol.2000037
57. Fung ITH, Sankar P, Zhang Y, Robison LS, Zhao X, D’Souza SS, et al. Activation of Group 2 Innate Lymphoid Cells Alleviates Aging-Associated Cognitive Decline. J Exp Med (2020) 217:e20190915. doi: 10.1084/jem.20190915
58. Baban B, Braun M, Khodadadi H, Ward A, Alverson K, Malik A, et al. AMPK Induces Regulatory Innate Lymphoid Cells After Traumatic Brain Injury. JCI Insight (2021) 6:1–13. doi: 10.1172/jci.insight.126766
59. Dalli J, Serhan CN. Immunoresolvents Signaling Molecules at Intersection Between the Brain and Immune System. Curr Opin Immunol (2018) 50:48–54. doi: 10.1016/j.coi.2017.10.007
60. Dalli J, Colas RA, Arnardottir H, Serhan CN. Vagal Regulation of Group 3 Innate Lymphoid Cells and the Immunoresolvent PCTR1 Controls Infection Resolution. Immunity (2017) 46:92–105. doi: 10.1016/j.immuni.2016.12.009
61. Marquardt N, Kekäläinen E, Chen P, Kvedaraite E, Wilson JN, Ivarsson MA, et al. Human Lung Natural Killer Cells are Predominantly Comprised of Highly Differentiated Hypofunctional CD69–CD56dim Cells. J Allergy Clin Immunol (2017) 139:1321–30.e4. doi: 10.1016/j.jaci.2016.07.043
62. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of Natural Killer Cells. Nat Immunol (2008) 9:503–10. doi: 10.1038/ni1582
63. Wang J, Li F, Zheng M, Sun R, Wei H, Tian Z. Lung Natural Killer Cells in Mice: Phenotype and Response to Respiratory Infection. Immunology (2012) 137:37–47. doi: 10.1111/j.1365-2567.2012.03607.x
64. Dogra P, Rancan C, Ma W, Toth M, Senda T, Carpenter DJ, et al. Tissue Determinants of Human NK Cell Development, Function, and Residence. Cell (2020) 180:749–63.e13. doi: 10.1016/j.cell.2020.01.022
65. Hervier B, Russick J, Cremer I, Vieillard V. NK Cells in the Human Lungs. Front Immunol (2019) 10:1263. doi: 10.3389/fimmu.2019.01263
66. De Grove KC, Provoost S, Verhamme FM, Bracke KR, Joos GF, Maes T, et al. Characterization and Quantification of Innate Lymphoid Cell Subsets in Human Lung. PloS One (2016) 11:1–12. doi: 10.1371/journal.pone.0145961
67. Yudanin NA, Schmitz F, Flamar AL, Thome JJC, Tait Wojno E, Moeller JB, et al. Spatial and Temporal Mapping of Human Innate Lymphoid Cells Reveals Elements of Tissue Specificity. Immunity (2019) 50:505–19.e4. doi: 10.1016/j.immuni.2019.01.012
68. Oherle K, Acker E, Bonfield M, Wang T, Gray J, Lang I, et al. Insulin-Like Growth Factor 1 Supports a Pulmonary Niche That Promotes Type 3 Innate Lymphoid Cell Development in Newborn Lungs. Immunity (2020) 52:275–94.e9. doi: 10.1016/j.immuni.2020.01.005
69. Gray J, Oehrle K, Worthen G, Alenghat T, Whitsett J, Deshmukh H. Intestinal Commensal Bacteria Mediate Lung Mucosal Immunity and Promote Resistance of Newborn Mice to Infection. Sci Transl Med (2017) 9:1–14. doi: 10.1126/scitranslmed.aaf9412
70. Ricardo-Gonzalez RR, Van Dyken SJ, Schneider C, Lee J, Nussbaum JC, Liang HE, et al. Tissue Signals Imprint ILC2 Identity With Anticipatory Function. Nat Immunol (2018) 19:1093–9. doi: 10.1038/s41590-018-0201-4
71. Puttur F, Denney L, Gregory LG, Vuononvirta J, Oliver R, Entwistle LJ, et al. Pulmonary Environmental Cues Drive Group 2 Innate Lymphoid Cell Dynamics in Mice and Humans. Sci Immunol (2019) 4:eaav7638. doi: 10.1126/sciimmunol.aav7638
72. Huang Y, Mao K, Chen X, Sun MA, Kawabe T, Li W, et al. S1P-Dependent Interorgan Trafficking of Group 2 Innate Lymphoid Cells Supports Host Defense. Science (80-) (2018) 359:114–9. doi: 10.1126/science.aam5809
73. Maazi H, Patel N, Sankaranarayanan I, Suzuki Y, Rigas D, Soroosh P, et al. ICOS: ICOS-Ligand Interaction Is Required for Type 2 Innate Lymphoid Cell Function, Homeostasis, and Induction of Airway Hyperreactivity. Immunity (2015) 42:538–51. doi: 10.1016/j.immuni.2015.02.007
74. Ardain A, Domingo-Gonzalez R, Das S, Kazer SW, Howard NC, Singh A, et al. Group 3 Innate Lymphoid Cells Mediate Early Protective Immunity Against Tuberculosis. Nature (2019) 570:528–32. doi: 10.1038/s41586-019-1276-2
75. Moura-Alves P, Faé K, Houthuys E, Dorhoi A, Kreuchwig A, Furkert J, et al. AhR Sensing of Bacterial Pigments Regulates Antibacterial Defence. Nature (2014) 512:387–92. doi: 10.1038/nature13684
76. Xiong H, Keith JW, Samilo DW, Carter RA, Leiner IM, Pamer EG. Innate Lymphocyte/Ly6Chi Monocyte Crosstalk Promotes Klebsiella Pneumoniae Clearance. Cell (2016) 165:679–89. doi: 10.1016/j.cell.2016.03.017
77. Van Maele L, Carnoy C, Cayet D, Ivanov S, Porte R, Deruy E, et al. Activation of Type 3 Innate Lymphoid Cells and Interleukin 22 Secretion in the Lungs During Streptococcus Pneumoniae Infection. J Infect Dis (2014) 210:493–503. doi: 10.1093/infdis/jiu106
78. Gazit R, Gruda R, Elboim M, Arnon TI, Katz G, Achdout H, et al. Lethal Influenza Infection in the Absence of the Natural Killer Cell Receptor Gene Ncr1. Nat Immunol (2006) 7:517–23. doi: 10.1038/ni1322
79. Zhou G, Juang SWW, Kane KP. NK Cells Exacerbate the Pathology of Influenza Virus Infection in Mice. Eur J Immunol (2013) 43:929–38. doi: 10.1002/eji.201242620
80. Draghi M, Pashine A, Sanjanwala B, Gendzekhadze K, Cantoni C, Cosman D, et al. NKp46 and NKG2D Recognition of Infected Dendritic Cells Is Necessary for NK Cell Activation in the Human Response to Influenza Infection. J Immunol (2007) 178:2688–98. doi: 10.4049/jimmunol.178.5.2688
81. Mandelboim O, Lieberman N, Lev M, Paul L, Arnon TI, Bushkin Y, et al. Recognition of Haemagglutinins on Virus-Infected Cells by NKp46 Activates Lysis by Human NK Cells. Nature (2001) 409:1055–60. doi: 10.1038/35059110
82. Cooper GE, Ostridge K, Khakoo SI, Wilkinson TMA, Staples KJ. Human CD49a+ Lung Natural Killer Cell Cytotoxicity in Response to Influenza A Virus. Front Immunol (2018) 9:1671. doi: 10.3389/fimmu.2018.01671
83. Dou Y, Fu B, Sun R, Li W, Hu W, Tian Z, et al. Influenza Vaccine Induces Intracellular Immune Memory of Human NK Cells. PloS One (2015) 10:1–17. doi: 10.1371/journal.pone.0121258
84. Vashist N, Trittel S, Ebensen T, Chambers BJ, Guzmán CA, Riese P. Influenza-Activated ILC1s Contribute to Antiviral Immunity Partially Influenced by Differential GITR Expression. Front Immunol (2018) 8:505. doi: 10.3389/fimmu.2018.00505
85. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CGK, Doering TA, et al. Innate Lymphoid Cells Promote Lung-Tissue Homeostasis After Infection With Influenza Virus. Nat Immunol (2011) 12:1045–54. doi: 10.1038/ni.2131
86. Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie ANJ, Smith DE, et al. Innate Lymphoid Cells Mediate Influenza-Induced Airway Hyper-Reactivity Independently of Adaptive Immunity. Nat Immunol (2011) 12:631–8. doi: 10.1038/ni.2045
87. Breese Hall C, Weinberg GA, Iwane MK, Blumkin AK, Edwards KM, Staat MA, et al. Burden of Respiratory Syncytial Virus Infection in Young Children. N Engl J Med (2009) 360:588–98. doi: 10.5409/wjcp.v1.i3.8
88. Vu LD, Siefker D, Jones TL, You D, Taylor R, DeVincenzo J, et al. Elevated Levels of Type 2 Respiratory Innate Lymphoid Cells in Human Infants With Severe Respiratory Syncytial Virus Bronchiolitis. Am J Respir Crit Care Med (2019) 200:1414–23. doi: 10.1164/rccm.201812-2366OC
89. Silver JS, Kearley J, Copenhaver AM, Sanden C, Mori M, Yu L, et al. Inflammatory Triggers Associated With Exacerbations of COPD Orchestrate Plasticity of Group 2 Innate Lymphoid Cells in the Lungs. Nat Immunol (2016) 17:626–35. doi: 10.1038/ni.3443
90. Hamelmann E, Gelfand EW. IL-5-Induced Airway Eosinophilia - The Key to Asthma? Immunol Rev (2001) 179:182–91. doi: 10.1034/j.1600-065X.2001.790118.x
91. van Rijt L, von Richthofen H, van Ree R. Type 2 Innate Lymphoid Cells: At the Cross-Roads in Allergic Asthma. Semin Immunopathol (2016) 38:483–96. doi: 10.1007/s00281-016-0556-2
92. Halim TYF, Steer CA, Mathä L, Gold MJ, Martinez-Gonzalez I, McNagny KM, et al. Group 2 Innate Lymphoid Cells are Critical for the Initiation of Adaptive T Helper 2 Cell-Mediated Allergic Lung Inflammation. Immunity (2014) 40:425–35. doi: 10.1016/j.immuni.2014.01.011
93. Bartemes KR, Kephart GM, Fox SJ, Kita H. Enhanced Innate Type 2 Immune Response in Peripheral Blood From Patients With Asthma. J Allergy Clin Immunol (2014) 134:671–8.e4. doi: 10.1016/j.jaci.2014.06.024
94. Tait Wojno ED, Monticelli LA, Tran SV, Alenghat T, Osborne LC, Thome JJ, et al. The Prostaglandin D2 Receptor CRTH2 Regulates Accumulation of Group 2 Innate Lymphoid Cells in the Inflamed Lung. Mucosal Immunol (2015) 8:1313–23. doi: 10.1038/mi.2015.21
95. Campos Alberto E, MacLean E, Davidson C, Palikhe NS, Storie J, Tse C, et al. The Single Nucleotide Polymorphism CRTh2 Rs533116 Is Associated With Allergic Asthma and Increased Expression of Crth2. Allergy Eur J Allergy Clin Immunol (2012) 67:1357–64. doi: 10.1111/all.12003
96. Laffont S, Blanquart E, Guéry J-C. Sex Differences in Asthma: A Key Role of Androgen-Signaling in Group 2 Innate Lymphoid Cells. Front Immunol (2017) 8:1069. doi: 10.3389/fimmu.2017.01069
97. Kadel S, Ainsua-Enrich E, Hatipoglu I, Turner S, Singh S, Khan S, et al. A Major Population of Functional KLRG1 – ILC2s in Female Lungs Contributes to a Sex Bias in ILC2 Numbers. ImmunoHorizons (2018) 2:74–86. doi: 10.4049/immunohorizons.1800008
98. Mathä L, Shim H, Steer CA, Yin YH, Martinez I, Fumio Takei G. Female and Male Mouse Lung Group 2 Innate Lymphoid Cells Differ in Gene Expression Profiles and Cytokine Production. PloS One (2019) 14:1–13. doi: 10.1371/journal.pone.0214286
99. Warren KJ, Sweeter JM, Pavlik JA, Nelson AJ, Devasure JM, Dickinson JD, et al. Sex Differences in Activation of Lung-Related Type 2 Innate Lymphoid Cells in Experimental Asthma. Ann Allergy Asthma Immunol (2017) 118:233–4. doi: 10.1016/j.anai.2016.11.011
100. Cephus JY, Stier MT, Fuseini H, Yung JA, Toki S, Bloodworth MH, et al. Testosterone Attenuates Group 2 Innate Lymphoid Cell-Mediated Airway Inflammation. Cell Rep (2017) 21:2487–99. doi: 10.1016/j.celrep.2017.10.110
101. Laffont S, Blanquart E, Savignac M, Cénac C, Laverny G, Metzger D, et al. Androgen Signaling Negatively Controls Group 2 Innate Lymphoid Cells. J Exp Med (2017) 214:1581–92. doi: 10.1084/jem.20161807
102. Talbot S, Abdulnour REE, Burkett PR, Lee S, Cronin SJF, Pascal MA, et al. Silencing Nociceptor Neurons Reduces Allergic Airway Inflammation. Neuron (2015) 87:341–54. doi: 10.1016/j.neuron.2015.06.007
103. Sui P, Wiesner DL, Xu J, Zhang Y, Lee J, Van Dyken S, et al. Pulmonary Neuroendocrine Cells Amplify Allergic Asthma Responses. Science (80-) (2018) 360:eaan8546. doi: 10.1126/science.aan8546
104. Mjösberg JM, Trifari S, Crellin NK, Peters CP, Van Drunen CM, Piet B, et al. Human IL-25-and IL-33-Responsive Type 2 Innate Lymphoid Cells Are Defined by Expression of CRTH2 and CD161. Nat Immunol (2011) 12:1055–62. doi: 10.1038/ni.2104
105. Van Bruaene N, Pérez-Novo CA, Basinski TM, Van Zele T, Holtappels G, De Ruyck N, et al. T-Cell Regulation in Chronic Paranasal Sinus Disease. J Allergy Clin Immunol (2008) 121:16–8. doi: 10.1016/j.jaci.2008.02.018
106. Bal SM, Bernink JH, Nagasawa M, Groot J, Shikhagaie MM, Golebski K, et al. IL-1β, IL-4 and IL-12 Control the Fate of Group 2 Innate Lymphoid Cells in Human Airway Inflammation in the Lungs. Nat Immunol (2016) 17:636–45. doi: 10.1038/ni.3444
107. Verma M, Michalec L, Sripada A, McKay J, Sirohi K, Verma D, et al. The Molecular and Epigenetic Mechanisms of Innate Lymphoid Cell (ILC) Memory and its Relevance for Asthma. J Exp Med (2021) 218:e20201354. doi: 10.1084/jem.20201354
108. Molet S, Hamid Q, Davoine F, Nutku E, Taha R, Pagé N, et al. IL-17 Is Increased in Asthmatic Airways and Induces Human Bronchial Fibroblasts to Produce Cytokines. J Allergy Clin Immunol (2001) 108:430–8. doi: 10.1067/mai.2001.117929
109. Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, et al. Interleukin-17-Producing Innate Lymphoid Cells and the NLRP3 Inflammasome Facilitate Obesity-Associated Airway Hyperreactivity. Nat Med (2014) 20:54–61. doi: 10.1038/nm.3423
110. Hekking PP, Loza MJ, Pavlidis S, de Meulder B, Lefaudeux D, Baribaud F, et al. Pathway Discovery Using Transcriptomic Profiles in Adult-Onset Severe Asthma. J Allergy Clin Immunol (2018) 141:1280–90. doi: 10.1016/j.jaci.2017.06.037
111. Mehlhop PD, Van de Rijn M, Brewer JP, Kisselgof AB, Geha RS, Oettgen HC, et al. CD40L, But Not CD40, Is Required for Allergen-Induced Bronchial Hyperresponsiveness in Mice. Am J Respir Cell Mol Biol (2000) 23:646–51. doi: 10.1165/ajrcmb.23.5.3954
112. Wingett D, Nielson CP. Divergence in NK Cell and Cyclic AMP Regulation of T Cell CD40L Expression in Asthmatic Subjects. J Leukoc Biol (2003) 74:531–41. doi: 10.1189/jlb.0303103
113. Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S, et al. Lipoxin A4 Regulates Natural Killer Cell and Type 2 Innate Lymphoid Cell Activation in Asthma. Sci Transl Med (2013) 5:174ra26. doi: 10.1126/scitranslmed.3004812
114. Levy BD, Bonnans C, Silverman ES, Palmer LJ, Marigowda C, Israel E. Diminished Lipoxin Biosynthesis in Severe Asthma. Am J Respir Crit Care Med (2005) 172:824–30. doi: 10.1164/rccm.200410-1413OC
115. Mannino DM, Buist AS. Global Burden of COPD: Risk Factors, Prevalence, and Future Trends. Lancet (2007) 370:765–73. doi: 10.1016/S0140-6736(07)61380-4
116. Blomme EE, Provoost S, De Smet EG, De Grove KC, Van Eeckhoutte HP, De Volder J, et al. Quantification and Role of Innate Lymphoid Cell Subsets in Chronic Obstructive Pulmonary Disease. Clin Transl Immunol (2021) 10:1–16. doi: 10.1002/cti2.1287
117. Roos AB, Sandén C, Mori M, Bjermer L, Stampfli MR, Erjefält JS. IL-17A Is Elevated in End-Stage Chronic Obstructive Pulmonary Disease and Contributes to Cigarette Smoke-Induced Lymphoid Neogenesis. Am J Respir Crit Care Med (2015) 191:1232–41. doi: 10.1164/rccm.201410-1861OC
118. Shikhagaie MM, Björklund ÅK, Mjösberg J, Erjefält JS, Cornelissen AS, Ros XR, et al. Neuropilin-1 Is Expressed on Lymphoid Tissue Residing LTi-Like Group 3 Innate Lymphoid Cells and Associated With Ectopic Lymphoid Aggregates. Cell Rep (2017) 18:1761–73. doi: 10.1016/j.celrep.2017.01.063
119. Rangel-Moreno J, Carragher DM, de la Luz Garcia-Hernandez M, Hwang JY, Kusser K, Hartson L, et al. The Development of Inducible Bronchus-Associated Lymphoid Tissue Depends on IL-17. Nat Immunol (2011) 12:639–46. doi: 10.1038/ni.2053
120. Finch DK, Stolberg VR, Ferguson J, Alikaj H, Kady MR, Richmond BW, et al. Lung Dendritic Cells Drive Natural Killer Cytotoxicity in Chronic Obstructive Pulmonary Disease via IL-15ra. Am J Respir Crit Care Med (2018) 198:1140–50. doi: 10.1164/rccm.201712-2513OC
121. Osterburg AR, Lach L, Panos RJ, Borchers MT. Unique Natural Killer Cell Subpopulations are Associated With Exacerbation Risk in Chronic Obstructive Pulmonary Disease. Sci Rep (2020) 10:1–11. doi: 10.1038/s41598-020-58326-7
122. Kearley J, Silver JS, Sanden C, Liu Z, Berlin AA, White N, et al. Cigarette Smoke Silences Innate Lymphoid Cell Function and Facilitates an Exacerbated Type I Interleukin-33-Dependent Response to Infection. Immunity (2015) 42:566–79. doi: 10.1016/j.immuni.2015.02.011
123. Teunissen MBM, Munneke JM, Bernink JH, Spuls PI, Res PCM, Te Velde A, et al. Composition of Innate Lymphoid Cell Subsets in the Human Skin: Enrichment of NCR + ILC3 in Lesional Skin and Blood of Psoriasis Patients. J Invest Dermatol (2014) 134:2351–60. doi: 10.1038/jid.2014.146
124. Villanova F, Flutter B, Tosi I, Grys K, Sreeneebus H, Perera GK, et al. Characterization of Innate Lymphoid Cells in Human Skin and Blood Demonstrates Increase of NKp44+ ILC3 in Psoriasis. J Invest Dermatol (2014) 134:984–91. doi: 10.1038/jid.2013.477
125. Simoni Y, Newell EW. Dissecting Human ILC Heterogeneity: More Than Just Three Subsets. Immunology (2018) 153:297–303. doi: 10.1111/imm.12862
126. Brüggen MC, Bauer WM, Reininger B, Clim E, Captarencu C, Steiner GE, et al. In Situ Mapping of Innate Lymphoid Cells in Human Skin: Evidence for Remarkable Differences Between Normal and Inflamed Skin. J Invest Dermatol (2016) 136:2396–405. doi: 10.1016/j.jid.2016.07.017
127. Simoni Y, Fehlings M, Kløverpris HN, McGovern N, Koo SL, Loh CY, et al. Human Innate Lymphoid Cell Subsets Possess Tissue-Type Based Heterogeneity in Phenotype and Frequency. Immunity (2017) 46:148–61. doi: 10.1016/j.immuni.2016.11.005
128. Kobayashi T, Voisin B, Kim DY, Kennedy EA, Jo JH, Shih HY, et al. Homeostatic Control of Sebaceous Glands by Innate Lymphoid Cells Regulates Commensal Bacteria Equilibrium. Cell (2019) 176:982–97.e16. doi: 10.1016/j.cell.2018.12.031
129. Adachi T, Kobayashi T, Sugihara E, Yamada T, Ikuta K, Pittaluga S, et al. Hair Follicle-Derived IL-7 and IL-15 Mediate Skin-Resident Memory T Cell Homeostasis and Lymphoma. Nat Med (2015) 21:1272–9. doi: 10.1038/nm.3962
130. Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, et al. Human Epithelial Cells Trigger Dendritic Cell-Mediated Allergic Inflammation by Producing TSLP. Nat Immunol (2002) 3:673–80. doi: 10.1038/ni805
131. Chen YL, Gutowska-Owsiak D, Hardman CS, Westmoreland M, MacKenzie T, Cifuentes L, et al. Proof-Of-Concept Clinical Trial of Etokimab Shows a Key Role for IL-33 in Atopic Dermatitis Pathogenesis. Sci Transl Med (2019) 11:eaax2945. doi: 10.1126/scitranslmed.aax2945
132. Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, et al. A Role for IL-25 and IL-33-Driven Type-2 Innate Lymphoid Cells in Atopic Dermatitis. J Exp Med (2013) 210:2939–50. doi: 10.1084/jem.20130351
133. Wolk K, Witte E, Wallace E, Döcke WD, Kunz S, Asadullah K, et al. IL-22 Regulates the Expression of Genes Responsible for Antimicrobial Defense, Cellular Differentiation, and Mobility in Keratinocytes: A Potential Role in Psoriasis. Eur J Immunol (2006) 36:1309–23. doi: 10.1002/eji.200535503
134. Erin Chen Y, Fischbach MA, Belkaid Y. Skin Microbiota-Host Interactions. Nature (2018) 553:427–36. doi: 10.1038/nature25177
135. Rak GD, Osborne LC, Siracusa MC, Kim BS, Wang K, Bayat A, et al. IL-33-Dependent Group 2 Innate Lymphoid Cells Promote Cutaneous Wound Healing. J Invest Dermatol (2016) 136:487–96. doi: 10.1038/JID.2015.406
136. Li Z, Hodgkinson T, Gothard EJ, Boroumand S, Lamb R, Cummins I, et al. Epidermal Notch1 Recruits Rorγ + Group 3 Innate Lymphoid Cells to Orchestrate Normal Skin Repair. Nat Commun (2016) 7:11394. doi: 10.1038/ncomms11394
137. McGee HM, Schmidt BA, Booth CJ, Yancopoulos GD, Valenzuela DM, Murphy AJ, et al. IL-22 Promotes Fibroblast-Mediated Wound Repair in the Skin. J Invest Dermatol (2013) 133:1321–9. doi: 10.1038/jid.2012.463
138. Eyerich S, Eyerich K, Pennino D, Carbone T, Nasorri F, Pallotta S, et al. Th22 Cells Represent a Distinct Human T Cell Subset Involved in Epidermal Immunity and Remodeling. J Clin Invest (2009) 119:3573–85. doi: 10.1172/JCI40202
139. Kobayashi T, Ricardo-Gonzalez RR, Moro K. Skin-Resident Innate Lymphoid Cells – Cutaneous Innate Guardians and Regulators. Trends Immunol (2020) 41:100–12. doi: 10.1016/j.it.2019.12.004
140. Di Cesare A, Di Meglio P, Nestle FO. The IL-23/Th17 Axis in the Immunopathogenesis of Psoriasis. J Invest Dermatol (2009) 129:1339–50. doi: 10.1038/jid.2009.59
141. Chiricozzi A, Suarez-Farinas M, Fuentes-Duculan J, Cueto I, Li K, Tian S, et al. Increased Expression of Interleukin-17 Pathway Genes in Nonlesional Skin of Moderate-to-Severe Psoriasis Vulgaris. Br J Dermatol (2016) 174:136–45. doi: 10.1111/bjd.14034
142. Muromoto R, Hirao T, Tawa K, Hirashima K, Kon S, Kitai Y, et al. IL-17A Plays a Central Role in the Expression of Psoriasis Signature Genes Through the Induction of Iκb-ζ in Keratinocytes. Int Immunol (2016) 28:443–52. doi: 10.1093/intimm/dxw011
143. Dyring-Andersen B, Geisler C, Agerbeck C, Lauritsen JPH, Gúdjonsdottir SD, Skov L, et al. Increased Number and Frequency of Group 3 Innate Lymphoid Cells in Nonlesional Psoriatic Skin. Br J Dermatol (2014) 170:609–16. doi: 10.1111/bjd.12658
144. Tamoutounour S, Han SJ, Deckers J, Constantinides MG, Hurabielle C, Harrison OJ, et al. Keratinocyte-Intrinsic MHCII Expression Controls Microbiota-Induced Th1 Cell Responses. Proc Natl Acad Sci USA (2019) 116:23643–52. doi: 10.1073/pnas.1912432116
145. Hardman CS, Chen YL, Salimi M, Jarrett R, Johnson D, Järvinen VJ, et al. CD1a Presentation of Endogenous Antigens by Group 2 Innate Lymphoid Cells. Sci Immunol (2017) 2:eaan5918. doi: 10.1126/sciimmunol.aan5918
146. Luci C, Gaudy-Marqueste C, Rouzaire P, Audonnet S, Cognet C, Hennino A, et al. Peripheral Natural Killer Cells Exhibit Qualitative and Quantitative Changes in Patients With Psoriasis and Atopic Dermatitis. Br J Dermatol (2012) 166:789–96. doi: 10.1111/j.1365-2133.2012.10814.x
147. Cameron AL, Kirby B, Griffiths CE. Circulating Natural Killer Cells In Psoriasis. Br J Dermatol (2003) 149:160–4. doi: 10.1159/000328011
148. Skrzeczyńska-Moncznik J, Stefańska A, Zabel BA, Kapińska-Mrowiecka M, Butcher EC, Cichy J. Chemerin and the Recruitment of NK Cells to Diseased Skin. Acta Biochim Pol (2009) 56:355–60. doi: 10.18388/abp.2009_2468
149. Dunphy SE, Sweeney CM, Kelly G, Tobin AM, Kirby B, Gardiner CM. Natural Killer Cells From Psoriasis Vulgaris Patients Have Reduced Levels of Cytotoxicity Associated Degranulation and Cytokine Production. Clin Immunol (2017) 177:43–9. doi: 10.1016/j.clim.2015.10.004
150. Ottaviani C, Nasorri F, Bedini C, de Pità O, Girolomoni G, Cavani A. CD56brightCD16- NK Cells Accumulate in Psoriatic Skin in Response to CXCL10 and CCL5 and Exacerbate Skin Inflammation. Eur J Immunol (2006) 36:118–28. doi: 10.1002/eji.200535243
151. Brandt EB, Sivaprasad U. Th2 Cytokines and Atopic Dermatitis. J Clin Cell Immunol (2011) 2:110. doi: 10.4172/2155-9899.1000110.Th2
152. Mack MR, Brestoff JR, Berrien-Elliott MM, Trier AM, Yang TLB, McCullen M, et al. Blood Natural Killer Cell Deficiency Reveals an Immunotherapy Strategy for Atopic Dermatitis. Sci Transl Med (2020) 12(532):eaay1005 . doi: 10.1126/scitranslmed.aay1005
153. Kim BS, Siracusa MC, Saenz SA, Noti M, Monticelli LA, Sonnenberg GF, et al. TSLP Elicits IL-33-Independent Innate Lymphoid Cell Responses to Promote Skin Inflammation. Sci Transl Med (2013) 5:170ra16. doi: 10.1126/scitranslmed.3005374
154. Kim MH, Jin SP, Jang S, Choi JY, Chung DH, Lee DH, et al. IL-17a–Producing Innate Lymphoid Cells Promote Skin Inflammation by Inducing IL-33–Driven Type 2 Immune Responses. J Invest Dermatol (2020) 140:827–37.e9. doi: 10.1016/j.jid.2019.08.447
155. Gittler JK, Shemer A, Suárez-Fariñas M, Fuentes-Duculan J, Gulewicz KJ, Wang CQF, et al. Progressive Activation of TH2/TH22 Cytokines and Selective Epidermal Proteins Characterizes Acute and Chronic Atopic Dermatitis. J Allergy Clin Immunol (2012) 130:1344–54. doi: 10.1016/j.jaci.2012.07.012
156. Katsuta M, Takigawa Y, Kimishima M, Inaoka M, Takahashi R, Shiohara T. NK Cells and γδ + T Cells Are Phenotypically and Functionally Defective Due to Preferential Apoptosis in Patients With Atopic Dermatitis. J Immunol (2006) 176:7736–44. doi: 10.4049/jimmunol.176.12.7736
157. Bernink JH, Mjösberg J, Spits H. Human ILC1: To Be or Not to be. Immunity (2017) 46:756–7. doi: 10.1016/j.immuni.2017.05.001
158. Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, et al. Intraepithelial Type 1 Innate Lymphoid Cells are a Unique Subset of IL-12- and IL-15-Responsive IFN-γ-Producing Cells. Immunity (2013) 38:769–81. doi: 10.1016/j.immuni.2013.02.010
159. Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M, et al. Interleukin-12 and -23 Control Plasticity Of CD127+ Group 1 And Group 3 Innate Lymphoid Cells In The Intestinal Lamina Propria. Immunity (2015) 43:146–60. doi: 10.1016/j.immuni.2015.06.019
160. Satoh-Takayama N, Serafini N, Verrier T, Rekiki A, Renauld JC, Frankel G, et al. The Chemokine Receptor CXCR6 Controls the Functional Topography of Interleukin-22 Producing Intestinal Innate Lymphoid Cells. Immunity (2014) 41:776–88. doi: 10.1016/j.immuni.2014.10.007
161. Emgård J, Kammoun H, García-Cassani B, Chesné J, Parigi SM, Jacob JM, et al. Oxysterol Sensing Through the Receptor GPR183 Promotes the Lymphoid-Tissue-Inducing Function of Innate Lymphoid Cells and Colonic Inflammation. Immunity (2018) 48:120–32.e8. doi: 10.1016/j.immuni.2017.11.020
162. Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DMW, Artis D. IL-33 Promotes an Innate Immune Pathway of Intestinal Tissue Protection Dependent on Amphiregulin-EGFR Interactions. Proc Natl Acad Sci USA (2015) 112:10762–7. doi: 10.1073/pnas.1509070112
163. Schneider C, O’Leary CE, von Moltke J, Liang HE, Ang QY, Turnbaugh PJ, et al. A Metabolite-Triggered Tuft Cell-ILC2 Circuit Drives Small Intestinal Remodeling. Cell (2018) 174:271–84.e14. doi: 10.1016/j.cell.2018.05.014
164. Hung LY, Tanaka Y, Herbine K, Pastore C, Singh B, Ferguson A, et al. Cellular Context of IL-33 Expression Dictates Impact on Anti-Helminth Immunity. Sci Immunol (2020) 5:eabc6259. doi: 10.1126/sciimmunol.abc6259
165. Gronke K, Hernández PP, Zimmermann J, Klose CSN, Kofoed-Branzk M, Guendel F, et al. Interleukin-22 Protects Intestinal Stem Cells Against Genotoxic Stress. Nature (2019) 566:249–53. doi: 10.1038/s41586-019-0899-7
166. Aparicio-Domingo P, Romera-Hernandez M, Karrich JJ, Cornelissen F, Papazian N, Lindenbergh-Kortleve DJ, et al. Type 3 Innate Lymphoid Cells Maintain Intestinal Epithelial Stem Cells After Tissue Damage. J Exp Med (2015) 212:1783–91. doi: 10.1084/jem.20150318
167. Romera-Hernández M, Aparicio-Domingo P, Papazian N, Karrich JJ, Cornelissen F, Hoogenboezem RM, et al. Yap1-Driven Intestinal Repair Is Controlled by Group 3 Innate Lymphoid Cells. Cell Rep (2020) 30:37–45.e3. doi: 10.1016/j.celrep.2019.11.115
168. Lindemans CA, Calafiore M, Mertelsmann AM, O’Connor MH, Dudakov JA, Jenq RR, et al. Interleukin-22 Promotes Intestinal-Stem-Cell-Mediated Epithelial Regeneration. Nature (2015) 528:560–4. doi: 10.1038/nature16460
169. Thompson CL, Plummer SJ, Tucker TC, Casey G, Li L. Interleukin-22 Genetic Polymorphisms and Risk of Colon Cancer. Cancer Causes Control (2010) 21:1165–70. doi: 10.1007/s10552-010-9542-5
170. Yao J, Liu L, Yang M. Interleukin-23 Receptor Genetic Variants Contribute to Susceptibility of Multiple Cancers. Gene (2014) 533:21–5. doi: 10.1016/j.gene.2013.09.054
171. Pickard JM, Maurice CF, Kinnebrew MA, Abt MC, Schenten D, Golovkina TV, et al. Rapid Fucosylation of Intestinal Epithelium Sustains Host-Commensal Symbiosis in Sickness. Nature (2014) 514:638–41. doi: 10.1038/nature13823
172. Goto Y, Obata T, Kunisawa J, Sato S, Ivanov II, Lamichhane A, et al. Innate Lymphoid Cells Regulate Intestinal Epithelial Cell Glycosylation. Science (80-) (2014) 345:1–14. doi: 10.1126/science.1254009
173. Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, Littmann DR. An Essential Function for the Nuclear Receptor Rorγt in the Generation of Fetal Lymphoid Tissue Inducer Cells. Nat Immunol (2004) 5:64–73. doi: 10.1038/ni1022
174. Fenton TM, Jørgensen PB, Niss K, Rubin SJS, Mörbe UM, Riis LB, et al. Immune Profiling of Human Gut-Associated Lymphoid Tissue Identifies a Role for Isolated Lymphoid Follicles in Priming of Region-Specific Immunity. Immunity (2020) 52:557–70.e6. doi: 10.1016/j.immuni.2020.02.001
175. Lane PJL, McConnell FM, Withers D, Gaspal F, Saini M, Anderson G. Lymphoid Tissue Inducer Cells: Bridges Between the Ancient Innate and the Modern Adaptive Immune Systems. Mucosal Immunol (2009) 2:472–7. doi: 10.1038/mi.2009.111
176. Guendel F, Kofoed-Branzk M, Gronke K, Tizian C, Witkowski M, Cheng HW, et al. Group 3 Innate Lymphoid Cells Program a Distinct Subset of IL-22BP-Producing Dendritic Cells Demarcating Solitary Intestinal Lymphoid Tissues. Immunity (2020) 53:1015–32.e8. doi: 10.1016/j.immuni.2020.10.012
177. Belkaid Y, Tarbell K. Regulatory T Cells in the Control of Host-Microorganism Interactions. Annu Rev Immunol (2009) 27:551–89. doi: 10.1146/annurev.immunol.021908.132723
178. Zhou L, Chu C, Teng F, Bessman NJ, Goc J, Santosa EK, et al. Innate Lymphoid Cells Support Regulatory T Cells in the Intestine Through Interleukin-2. Nature (2019) 568:405–9. doi: 10.1038/s41586-019-1082-x
179. Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, et al. Microbiota-Dependent Crosstalk Between Macrophages and ILC3 Promotes Intestinal Homeostasis. Science (80-) (2014) 343:1249288. doi: 10.1126/science.1249288
180. Hepworth MR, Fung TC, Masur SH, Kelsen JR, McConnell FM, Dubrot J, et al. Group 3 Innate Lymphoid Cells Mediate Intestinal Selection of Commensal Bacteria-Specific CD4+ T Cells. Science (80-) (2015) 348:1031–5. doi: 10.1126/science.aaa4812
181. Hepworth MR, Monticelli LA, Fung TC, Ziegler CGK, Grunberg S, Sinha R, et al. Innate Lymphoid Cells Regulate CD4 + T-Cell Responses to Intestinal Commensal Bacteria. Nature (2013) 498:113–7. doi: 10.1038/nature12240
182. Lehmann FM, von Burg N, Ivanek R, Teufel C, Horvath E, Peter A, et al. Microbiota-Induced Tissue Signals Regulate ILC3-Mediated Antigen Presentation. Nat Commun (2020) 11:1794. doi: 10.1038/s41467-020-15612-2
183. Rao A, Strauss O, Kokkinou E, Bruchard M, Tripathi KP, Schlums H, et al. Cytokines Regulate the Antigen-Presenting Characteristics of Human Circulating and Tissue-Resident Intestinal ILCs. Nat Commun (2020) 11:2049. doi: 10.1038/s41467-020-15695-x
184. Qiu J, Heller JJ, Guo X, Chen ZME, Fish K, Fu YX, et al. The Aryl Hydrocarbon Receptor Regulates Gut Immunity Through Modulation of Innate Lymphoid Cells. Immunity (2012) 36:92–104. doi: 10.1016/j.immuni.2011.11.011
185. Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, et al. AHR Drives the Development of Gut ILC22 Cells and Postnatal Lymphoid Tissues via Pathways Dependent on and Independent of Notch. Nat Immunol (2012) 13:144–52. doi: 10.1038/ni.2187
186. Li S, Bostick JW, Ye J, Qiu J, Zhang B, Urban JF, et al. Aryl Hydrocarbon Receptor Signaling Cell Intrinsically Inhibits Intestinal Group 2 Innate Lymphoid Cell Function. Immunity (2018) 49:915–28.e5. doi: 10.1016/j.immuni.2018.09.015
187. Wilhelm C, Harrison OJ, Schmitt V, Pelletier M, Spencer SP, Urban JF, et al. Critical Role of Fatty Acid Metabolism in ILC2-Mediated Barrier Protection During Malnutrition and Helminth Infection. J Exp Med (2016) 213:1409–18. doi: 10.1084/jem.20151448
188. Spencer SP, Wilhelm C, Yang Q, Hall JA, Bouladoux N, Boyd A, et al. Adaptation of Innate Lymphoid Cells to a Micronutrient Deficiency Promotes Type 2 Barrier Immunity. Science (80-) (2014) 343:432–7. doi: 10.1126/science.1247606
189. Chun E, Lavoie S, Fonseca-Pereira D, Bae S, Michaud M, Hoveyda HR, et al. Metabolite-Sensing Receptor Ffar2 Regulates Colonic Group 3 Innate Lymphoid Cells and Gut Immunity. Immunity (2019) 51:871–84.e6. doi: 10.1016/j.immuni.2019.09.014
190. Yang W, Yu T, Huang X, Bilotta AJ, Xu L, Lu Y, et al. Intestinal Microbiota-Derived Short-Chain Fatty Acids Regulation of Immune Cell IL-22 Production and Gut Immunity. Nat Commun (2020) 11:4457. doi: 10.1038/s41467-020-18262-6
191. Sepahi A, Liu QY, Friesen L, Kim CH. Dietary Fiber Metabolites Regulate Innate Lymphoid Cell Responses. Mucosal Immunol (2021) 14:317–30. doi: 10.1038/s41385-020-0312-8
192. Klose CSN, Mahlakõiv T, Moeller JB, Rankin LC, Flamar AL, Kabata H, et al. The Neuropeptide Neuromedin U Stimulates Innate Lymphoid Cells and Type 2 Inflammation. Nature (2017) 549:282–6. doi: 10.1038/nature23676
193. Burrows K, Mortha A. Going Green With Solar-Powered ILC3 Homeostasis. Sci Immunol (2019) 4:3–5. doi: 10.1126/sciimmunol.aaz0433
194. Wang Q, Robinette ML, Billon C, Collins PL, Bando JK, Fachi JL, et al. Circadian Rhythm-Dependent and Circadian Rhythm-Independent Impacts of the Molecular Clock on Type 3 Innate Lymphoid Cells. Sci Immunol (2019) 4:eaay7501. doi: 10.1126/sciimmunol.aay7501
195. Teng F, Goc J, Zhou L, Chu C, Shah MA, Eberl G, et al. A Circadian Clock is Essential for Homeostasis of Group 3 Innate Lymphoid Cells in the Gut. Sci Immunol (2019) 4:1–12. doi: 10.1126/sciimmunol.aax1215
196. Yu HB, Yang H, Allaire JM, Ma C, Graef FA, Mortha A, et al. Vasoactive Intestinal Peptide Promotes Host Defense Against Enteric Pathogens by Modulating the Recruitment of Group 3 Innate Lymphoid Cells. Proc Natl Acad Sci USA (2021) 118:e2106634118. doi: 10.1073/pnas.2106634118
197. Talbot J, Hahn P, Kroehling L, Nguyen H, Li D, Littman DR. Feeding-Dependent VIP Neuron–ILC3 Circuit Regulates the Intestinal Barrier. Nature (2020) 579:575–80. doi: 10.1038/s41586-020-2039-9
198. Seillet C, Luong K, Tellier J, Jacquelot N, Shen RD, Hickey P, et al. The Neuropeptide VIP Confers Anticipatory Mucosal Immunity by Regulating ILC3 Activity. Nat Immunol (2020) 21:168–77. doi: 10.1038/s41590-019-0567-y
199. Mortha A, Burrows K. Cytokine Networks Between Innate Lymphoid Cells and Myeloid Cells. Front Immunol (2018) 9:191. doi: 10.3389/fimmu.2018.00191
200. Li J, Doty AL, Iqbal A, Glover SC. The Differential Frequency of Lineage-CRTH2-CD45+NKp44-CD117-CD127+ILC Subset in the Inflamed Terminal Ileum of Patients With Crohn’s Disease. Cell Immunol (2016) 304–305:63–8. doi: 10.1016/j.cellimm.2016.05.001
201. Bernink JH, Peters CP, Munneke M, Te Velde AA, Meijer SL, Weijer K, et al. Human Type 1 Innate Lymphoid Cells Accumulate in Inflamed Mucosal Tissues. Nat Immunol (2013) 14:221–9. doi: 10.1038/ni.2534
202. Krabbendam L, Heesters BA, Kradolfer CMA, Haverkate NJE, Becker MAJ, Buskens CJ, et al. CD127+ CD94+ Innate Lymphoid Cells Expressing Granulysin and Perforin Are Expanded in Patients With Crohn’s Disease. Nat Commun (2021) 12:1–11. doi: 10.1038/s41467-021-26187-x
203. Jowett GM, Norman MDA, Yu TTL, Rosell Arévalo P, Hoogland D, Lust ST, et al. ILC1 Drive Intestinal Epithelial and Matrix Remodelling. Nat Mater (2021) 20:250–9. doi: 10.1038/s41563-020-0783-8
204. Wang X, Cai J, Lin B, Ma M, Tao Y, Zhou Y, et al. GPR34-Mediated Sensing of Lysophosphatidylserine Released by Apoptotic Neutrophils Activates Type 3 Innate Lymphoid Cells to Mediate Tissue Repair. Immunity (2021) 54:1123–36.e8. doi: 10.1016/j.immuni.2021.05.007
205. Song C, Lee JS, Gilfillan S, Robinette ML, Newberry RD, Stappenbeck TS, et al. Unique and Redundant Functions of NKp46+ ILC3s in Models of Intestinal Inflammation. J Exp Med (2015) 212:1869–82. doi: 10.1084/jem.20151403
206. Vonarbourg C, Mortha A, Bui VL, Hernandez PP, Kiss EA, Hoyler T, et al. Regulated Expression of Nuclear Receptor Rorγt Confers Distinct Functional Fates to NK Cell Receptor-Expressing Rorγt+ Innate Lymphocytes. Immunity (2010) 33:736–51. doi: 10.1016/j.immuni.2010.10.017
207. Klose CSN, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, D’Hargues Y, et al. A T-Bet Gradient Controls the Fate and Function of CCR6-Rorγt + Innate Lymphoid Cells. Nature (2013) 494:261–5. doi: 10.1038/nature11813
208. Mazzurana L, Forkel M, Rao A, Van Acker A, Kokkinou E, Ichiya T, et al. Suppression of Aiolos and Ikaros Expression by Lenalidomide Reduces Human ILC3–ILC1/NK Cell Transdifferentiation. Eur J Immunol (2019) 49:1344–55. doi: 10.1002/eji.201848075
209. Sandborn WJ, Feagan BG, Fedorak RN, Scherl E, Fleisher MR, Katz S, et al. A Randomized Trial of Ustekinumab, a Human Interleukin-12/23 Monoclonal Antibody, in Patients With Moderate-To-Severe Crohn’s Disease. Gastroenterology (2008) 135:1130–41. doi: 10.1053/j.gastro.2008.07.014
210. Creyns B, Jacobs I, Verstockt B, Cremer J, Ballet V, Vandecasteele R, et al. Biological Therapy in Inflammatory Bowel Disease Patients Partly Restores Intestinal Innate Lymphoid Cell Subtype Equilibrium. Front Immunol (2020) 11:1847. doi: 10.3389/fimmu.2020.01847
211. Chen J, Haller CA, Jernigan FE, Koerner SK, Wong DJ, Wang Y, et al. Modulation of Lymphocyte-Mediated Tissue Repair by Rational Design of Heterocyclic Aryl Hydrocarbon Receptor Agonists. Sci Adv (2020) 6:1–16. doi: 10.1126/sciadv.aay8230
212. Okada S, Markle JG, Deenick EK, Mele F, Averbuch D, Lagos M, et al. Impairment of Immunity to Candida and Mycobacterium in Humans With Bi-Allelic RORC Mutations. Science (80-) (2015) 349:606–13. doi: 10.1126/science.aaa4282
213. Hirata Y, Egea L, Dann SM, Eckmann L, Kagnoff MF. GM-CSF-Facilitated Dendritic Cell Recruitment and Survival Govern the Intestinal Mucosal Response to a Mouse Enteric Bacterial Pathogen. Cell Host Microbe (2010) 7:151–63. doi: 10.1016/j.chom.2010.01.006
214. Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 Mediates Early Host Defense Against Attaching and Effacing Bacterial Pathogens. Nat Med (2008) 14:282–9. doi: 10.1038/nm1720
215. Abt MC, Buffie CG, Sušac B, Becattini S, Carter RA, Leiner I, et al. TLR-7 Activation Enhances IL-22-Mediated Colonization Resistance Against Vancomycin-Resistant Enterococcus. Sci Transl Med (2016) 8:327ra25. doi: 10.1126/scitranslmed.aad6663
216. Zelante T, Iannitti RG, Cunha C, DeLuca A, Giovannini G, Pieraccini G, et al. Tryptophan Catabolites From Microbiota Engage Aryl Hydrocarbon Receptor and Balance Mucosal Reactivity via Interleukin-22. Immunity (2013) 39:372–85. doi: 10.1016/j.immuni.2013.08.003
217. Pian Y, Chai Q, Ren B, Wang Y, Lv M, Qiu J, et al. Type 3 Innate Lymphoid Cells Direct Goblet Cell Differentiation via the LT–Ltβr Pathway During Listeria Infection. J Immunol (2020) 205:853–63. doi: 10.4049/jimmunol.2000197
218. Songhet P, Barthel M, Stecher B, Müller AJ, Kremer M, Hansson GC, et al. Stromal IFN-γr-Signaling Modulates Goblet Cell Function During Salmonella Typhimurium Infection. PloS One (2011) 6:36–9. doi: 10.1371/journal.pone.0022459
219. Castleman MJ, Dillon SM, Purba C, Cogswell AC, McCarter M, Barker E, et al. Enteric Bacteria Induce Ifnγ and Granzyme B From Human Colonic Group 1 Innate Lymphoid Cells. Gut Microbes (2020) 12:1667723. doi: 10.1080/19490976.2019.1667723
220. Hernández PP, Mahlakõiv T, Yang I, Schwierzeck V, Nguyen N, Guendel F, et al. Interferon-γ and Interleukin 22 Act Synergistically for the Induction of Interferon-Stimulated Genes and Control of Rotavirus Infection. Nat Immunol (2015) 16:698–707. doi: 10.1038/ni.3180
221. Wang Y, Lifshitz L, Gellatly K, Vinton CL, Busman-Sahay K, McCauley S, et al. HIV-1-Induced Cytokines Deplete Homeostatic Innate Lymphoid Cells and Expand TCF7-Dependent Memory NK Cells. Nat Immunol (2020) 21:274–86. doi: 10.1038/s41590-020-0593-9
222. Hueber B, Curtis AD, Kroll K, Varner V, Jones R, Pathak S, et al. Functional Perturbation of Mucosal Group 3 Innate Lymphoid and Natural Killer Cells in Simian-Human Immunodeficiency Virus/Simian Immunodeficiency Virus-Infected Infant Rhesus Macaques. J Virol (2020) 94:e01644–19. doi: 10.1128/jvi.01644-19
223. Li H, Richert-Spuhler LE, Evans TI, Gillis J, Connole M, Estes JD, et al. Hypercytotoxicity and Rapid Loss of NKp44+ Innate Lymphoid Cells During Acute SIV Infection. PloS Pathog (2014) 10:e1004551. doi: 10.1371/journal.ppat.1004551
224. Gentile ME, Li Y, Robertson A, Shah K, Fontes G, Kaufmann E, et al. NK Cell Recruitment Limits Tissue Damage During an Enteric Helminth Infection. Mucosal Immunol (2020) 13:357–70. doi: 10.1038/s41385-019-0231-8
225. Fallon PG, Ballantyne SJ, Mangan NE, Barlow JL, Dasvarma A, Hewett DR, et al. Identification of an Interleukin (IL)-25-Dependent Cell Population That Provides IL-4, IL-5, and IL-13 at the Onset of Helminth Expulsion. J Exp Med (2006) 203:1105–16. doi: 10.1084/jem.20051615
226. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TKA, et al. Nuocytes Represent a New Innate Effector Leukocyte That Mediates Type-2 Immunity. Nature (2010) 464:1367–70. doi: 10.1038/nature08900
227. Saenz SA, Siracusa MC, Perrigoue JG, Spencer SP, Urban JF, Tocker JE, et al. IL25 Elicits a Multipotent Progenitor Cell Population That Promotes TH2 Cytokine Responses. Nature (2010) 464:1362–6. doi: 10.1038/nature08901
228. Anthony RM, Rutitzky LI, Urban JF, Stadecker MJ, Gause WC. Protective Immune Mechanisms in Helminth Infection. Nat Rev Immunol (2007) 7:975–87. doi: 10.1038/nri2199
229. Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. MHCII-Mediated Dialog Between Group 2 Innate Lymphoid Cells and CD4+ T Cells Potentiates Type 2 Immunity and Promotes Parasitic Helminth Expulsion. Immunity (2014) 41:283–95. doi: 10.1016/j.immuni.2014.06.016
230. Chu C, Parkhurst CN, Zhang W, Zhou L, Yano H, Arifuzzaman M, et al. The ChAT-Acetylcholine Pathway Promotes Group 2 Innate Lymphoid Cell Responses and Anti-Helminth Immunity. Sci Immunol (2021) 6:eabe3218. doi: 10.1126/sciimmunol.abe3218
231. Osbourn M, Soares DC, Vacca F, Cohen ES, Scott IC, Gregory WF, et al. HpARI Protein Secreted by a Helminth Parasite Suppresses Interleukin-33. Immunity (2017) 47:739–51.e5. doi: 10.1016/j.immuni.2017.09.015
232. Campbell L, Hepworth MR, Whittingham-Dowd J, Thompson S, Bancroft AJ, Hayes KS, et al. ILC2s Mediate Systemic Innate Protection by Priming Mucus Production at Distal Mucosal Sites. J Exp Med (2019) 216:2714–23. doi: 10.1084/jem.20180610
233. Löser S, Smith KA, Maizels RM. Innate Lymphoid Cells in Helminth Infections—Obligatory or Accessory? Front Immunol (2019) 10:620. doi: 10.3389/fimmu.2019.00620
234. Nausch N, Appleby LJ, Sparks AM, Midzi N, Mduluza T, Mutapi F. Group 2 Innate Lymphoid Cell Proportions Are Diminished in Young Helminth Infected Children and Restored by Curative Anti-Helminthic Treatment. PloS Negl Trop Dis (2015) 9:1–16. doi: 10.1371/journal.pntd.0003627
235. Boyd A, Ribeiro JMC, Nutman TB. Human CD117 (Ckit)+ Innate Lymphoid Cells Have a Discrete Transcriptional Profile at Homeostasis and are Expanded During Filarial Infection. PloS One (2014) 9:1–11. doi: 10.1371/journal.pone.0108649
236. Gao B, Jeong W, Tian Z. Liver: An Organ With Predominant Innate Immunity. Hepatology (2008) 47:729–36. doi: 10.1002/hep.22034
237. Protzer U, Maini MK, Knolle PA. Living in the Liver: Hepatic Infections. Nat Rev Immunol (2012) 12:201–13. doi: 10.1038/nri3169
238. Ju C, Tacke F. Hepatic Macrophages in Homeostasis and Liver Diseases: From Pathogenesis to Novel Therapeutic Strategies. Cell Mol Immunol (2016) 13:316–27. doi: 10.1038/cmi.2015.104
239. Peng H, Wisse E, Tian Z. Liver Natural Killer Cells: Subsets and Roles in Liver Immunity. Cell Mol Immunol (2016) 13:328–36. doi: 10.1038/cmi.2015.96
240. MacParland SA, Liu JC, Ma XZ, Innes BT, Bartczak AM, Gage BK, et al. Single Cell RNA Sequencing of Human Liver Reveals Distinct Intrahepatic Macrophage Populations. Nat Commun (2018) 9:4383. doi: 10.1038/s41467-018-06318-7
241. Hudspeth K, Donadon M, Cimino M, Pontarini E, Tentorio P, Preti M, et al. Human Liver-Resident CD56bright/CD16neg NK Cells are Retained Within Hepatic Sinusoids via the Engagement of CCR5 and CXCR6 Pathways. J Autoimmun (2016) 66:40–50. doi: 10.1016/j.jaut.2015.08.011
242. Stegmann KA, Robertson F, Hansi N, Gill U, Pallant C, Christophides T, et al. CXCR6 Marks a Novel Subset of T-Bet Lo Eomes Hi Natural Killer Cells Residing in Human Liver. Sci Rep (2016) 6:26157. doi: 10.1038/srep26157
243. Aw Yeang HX, Piersma SJ, Lin Y, Yang L, Malkova ON, Miner C, et al. Cutting Edge: Human CD49e – NK Cells Are Tissue Resident in the Liver. J Immunol (2017) 198:1417–22. doi: 10.4049/jimmunol.1601818
244. Marquardt N, Béziat V, Nyström S, Hengst J, Ivarsson MA, Kekäläinen E, et al. Cutting Edge: Identification and Characterization of Human Intrahepatic CD49a + NK Cells. J Immunol (2015) 194:2467–71. doi: 10.4049/jimmunol.1402756
245. Stary V, Pandey RV, Strobl J, Kleissl L, Starlinger P, Pereyra D, et al. A Discrete Subset of Epigenetically Primed Human NK Cells Mediates Antigen-Specific Immune Responses. Sci Immunol (2020) 5:eaba6232. doi: 10.1126/sciimmunol.aba6232
246. Cuff AO, Robertson FP, Stegmann KA, Pallett LJ, Maini MK, Davidson BR, et al. Eomes Hi NK Cells in Human Liver Are Long-Lived and Do Not Recirculate But Can Be Replenished From the Circulation. J Immunol (2016) 197:4283–91. doi: 10.4049/jimmunol.1601424
247. Jeffery HC, McDowell P, Lutz P, Wawman RE, Roberts S, Bagnall C, et al. Human Intrahepatic ILC2 are IL-13positive Amphiregulinpositive and Their Frequency Correlates With Model of End Stage Liver Disease Score. PloS One (2017) 12:e0188649. doi: 10.1371/journal.pone.0188649
248. Kanda T, Goto T, Hirotsu Y, Moriyama M, Omata M. Molecular Mechanisms Driving Progression of Liver Cirrhosis Towards Hepatocellular Carcinoma in Chronic Hepatitis B and C Infections: A Review. Int J Mol Sci (2019) 20:1358. doi: 10.3390/ijms20061358
249. Oliviero B, Varchetta S, Paudice E, Michelone G, Zaramella M, Mavilio D, et al. Natural Killer Cell Functional Dichotomy in Chronic Hepatitis B and Chronic Hepatitis C Virus Infections. Gastroenterology (2009) 137:1151–60. doi: 10.1053/j.gastro.2009.05.047
250. Amadei B, Urbani S, Cazaly A, Fisicaro P, Zerbini A, Ahmed P, et al. Activation of Natural Killer Cells During Acute Infection With Hepatitis C Virus. Gastroenterology (2010) 138:1536–45. doi: 10.1053/j.gastro.2010.01.006
251. Romero V, Azocar J, Zúñiga J, Clavijo OP, Terreros D, Gu X, et al. Interaction of NK Inhibitory Receptor Genes With HLA-C and MHC Class II Alleles in Hepatitis C Virus Infection Outcome. Mol Immunol (2008) 45:2429–36. doi: 10.1016/j.molimm.2008.01.002
252. Khakoo SI, Thio CL, Martin MP, Brooks CR, Gao X, Astemborski J, et al. HLA and NK Cell Inhibitory Receptor Genes in Resolving Hepatitis C Virus Infection. Science (80-) (2004) 305:872–4. doi: 10.1126/science.1097670
253. Jinushi M, Takehara T, Tatsumi T, Kanto T, Miyagi T, Suzuki T, et al. Negative Regulation of NK Cell Activities by Inhibitory Receptor CD94/NKG2A Leads to Altered NK Cell-Induced Modulation of Dendritic Cell Functions in Chronic Hepatitis C Virus Infection. J Immunol (2004) 173:6072–81. doi: 10.4049/jimmunol.173.10.6072
254. Araújo RC, Dias FC, Bertol BC, Silva DM, Almeida PH, Teixeira AC, et al. Liver HLA-E Expression is Associated With Severity of Liver Disease in Chronic Hepatitis C. J Immunol Res (2018) 2018:2563563. doi: 10.1155/2018/2563563
255. Doyle EH, Aloman C, El-Shamy A, Eng F, Rahman A, Klepper AL, et al. A Subset of Liver Resident Natural Killer Cells is Expanded in Hepatitis C-Infected Patients With Better Liver Function. Sci Rep (2021) 11:1–13. doi: 10.1038/s41598-020-80819-8
256. Dunn C, Brunetto M, Reynolds G, Christophides T, Kennedy PT, Lampertico P, et al. Cytokines Induced During Chronic Hepatitis B Virus Infection Promote a Pathway for NK Cell-Mediated Liver Damage. J Exp Med (2007) 204:667–80. doi: 10.1084/jem.20061287
257. Peppa D, Gil US, Reynolds G, Easom NJW, Pallett LJ, Schurich A, et al. Up-Regulation of a Death Receptor Renders Antiviral T Cells Susceptible to NK Cell-Mediated Deletion. J Exp Med (2013) 210:99–114. doi: 10.1084/jem.20121172
258. Yang Z, Tang T, Wei X, Yang S, Tian Z. Type 1 Innate Lymphoid Cells Contribute to the Pathogenesis of Chronic Hepatitis B. Innate Immun (2015) 21:665–73. doi: 10.1177/1753425915586074
259. Wang S, Li J, Wu S, Cheng L, Shen Y, Ma W, et al. Type 3 Innate Lymphoid Cell: A New Player in Liver Fibrosis Progression. Clin Sci (2018) 132:2565–82. doi: 10.1042/CS20180482
260. Jie Z, Liang Y, Hou L, Dong C, Iwakura Y, Soong L, et al. Intrahepatic Innate Lymphoid Cells Secrete IL-17A and IL-17f That Are Crucial for T Cell Priming in Viral Infection. J Immunol (2014) 192:3289–300. doi: 10.4049/jimmunol.1303281
261. Liang Y, Yi P, Yuan DMK, Jie Z, Kwota Z, Soong L, et al. IL-33 Induces Immunosuppressive Neutrophils via a Type 2 Innate Lymphoid Cell/IL-13/STAT6 Axis and Protects the Liver Against Injury in LCMV Infection-Induced Viral Hepatitis. Cell Mol Immunol (2019) 16:126–37. doi: 10.1038/cmi.2017.147
262. Hart KM, Fabre T, Sciurba JC, Gieseck RL, Borthwick LA, Vannella KM, et al. Type 2 Immunity Is Protective in Metabolic Disease But Exacerbates NAFLD Collaboratively With TGF-B. Sci Transl Med (2017) 9:eaal3694. doi: 10.1126/scitranslmed.aal3694
263. Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver Fibrosis and Repair: Immune Regulation of Wound Healing in a Solid Organ. Nat Rev Immunol (2014) 14:181–94. doi: 10.1038/nri3623
264. Gonzalez-Polo V, Pucci-Molineris M, Cervera V, Gambaro S, Yantorno SE, Descalzi V, et al. Group 2 Innate Lymphoid Cells Exhibit Progressively Higher Levels of Activation During Worsening of Liver Fibrosis. Ann Hepatol (2019) 18:366–72. doi: 10.1016/j.aohep.2018.12.001
265. Mchedlidze T, Waldner M, Zopf S, Walker J, Rankin AL, Schuchmann M, et al. Interleukin-33-Dependent Innate Lymphoid Cells Mediate Hepatic Fibrosis. Immunity (2013) 39:357–71. doi: 10.1016/j.immuni.2013.07.018
266. Neumann K, Karimi K, Meiners J, Voetlause R, Steinmann S, Dammermann W, et al. A Proinflammatory Role of Type 2 Innate Lymphoid Cells in Murine Immune-Mediated Hepatitis. J Immunol (2017) 198:128–37. doi: 10.4049/jimmunol.1600418
267. Steinmann S, Schoedsack M, Heinrich F, Breda PC, Ochel A, Tiegs G, et al. Hepatic ILC2 Activity Is Regulated by Liver Inflammation-Induced Cytokines and Effector CD4+ T Cells. Sci Rep (2020) 10:1–13. doi: 10.1038/s41598-020-57985-w
268. Martrus G, Kautz T, Lunemann S, Richert L, Glau L, Salzberger W, et al. Proliferative Capacity Exhibited by Human Liver-Resident CD49a+CD25+ NK Cells. PloS One (2017) 12:e0182532. doi: 10.1371/journal.pone.0182532
269. Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural Killer Cells Ameliorate Liver Fibrosis by Killing Activated Stellate Cells in NKG2D-Dependent and Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand-Dependent Manners. Gastroenterology (2006) 130:435–52. doi: 10.1053/j.gastro.2005.10.055
270. Baroni GS, D’Ambrosio L, Curto P, Casini A, Mancini R, Jezequel AM, et al. Interferon Gamma Decreases Hepatic Stellate Cell Activation and Extracellular Matrix Deposition in Rat Liver Fibrosis. Hepatology (1996) 23:1189–99. doi: 10.1053/jhep.1996.v23.pm0008621153
271. Liu M, Hu Y, Yuan Y, Tian Z, Zhang C. γδt Cells Suppress Liver Fibrosis via Strong Cytolysis and Enhanced NK Cell-Mediated Cytotoxicity Against Hepatic Stellate Cells. Front Immunol (2019) 10:477. doi: 10.3389/fimmu.2019.00477
272. Jeong W-I, Park O, Gao B. Abrogation of the Antifibrotic Effects of Natural Killer Cells/Interferon-γ Contributes to Alcohol Acceleration of Liver Fibrosis. Gastroenterology (2008) 134:248–58. doi: 10.1053/j.gastro.2007.09.034
273. Bellentani S. The Epidemiology of Non-Alcoholic Fatty Liver Disease. Liver Int (2017) 37:81–4. doi: 10.1111/liv.13299
274. Kahraman A, Schlattjan M, Kocabayoglu P, Yildiz-Meziletoglu S, Schlensak M, Fingas CD, et al. Major Histocompatibility Complex Class I-Related Chains A and B (MIC A/B): A Novel Role in Nonalcoholic Steatohepatitis. Hepatology (2010) 51:92–102. doi: 10.1002/hep.23253
275. Stiglund N, Strand K, Cornillet M, Stål P, Thorell A, Zimmer CL, et al. Retained NK Cell Phenotype and Functionality in Non-Alcoholic Fatty Liver Disease. Front Immunol (2019) 10:1255. doi: 10.3389/fimmu.2019.01255
276. Tosello-Trampont AC, Krueger P, Narayanan S, Landes SG, Leitinger N, Hahn YS. NKp46+ Natural Killer Cells Attenuate Metabolism-Induced Hepatic Fibrosis by Regulating Macrophage Activation in Mice. Hepatology (2016) 63:799–812. doi: 10.1002/hep.28389
277. Hamaguchi M, Okamura T, Fukuda T, Nishida K, Yoshimura Y, Hashimoto Y, et al. Group 3 Innate Lymphoid Cells Protect Steatohepatitis From High-Fat Diet Induced Toxicity. Front Immunol (2021) 12:648754. doi: 10.3389/fimmu.2021.648754
278. Sun R, Gao B. Negative Regulation of Liver Regeneration by Innate Immunity (Natural Killer Cells/Interferon-γ). Gastroenterology (2004) 127:1525–39. doi: 10.1053/j.gastro.2004.08.055
279. Singh P, Goode T, Dean A, Awad SS, Darlington GJ. Elevated Interferon Gamma Signaling Contributes to Impaired Regeneration in the Aged Liver. J Gerontol - Ser A Biol Sci Med Sci (2011) 66:944–56. doi: 10.1093/gerona/glr094
280. Bi J, Zheng X, Chen Y, Wei H, Sun R, Tian Z. TIGIT Safeguards Liver Regeneration Through Regulating Natural Killer Cell-Hepatocyte Crosstalk. Hepatology (2014) 60:1389–98. doi: 10.1002/hep.27245
281. Graubardt N, Fahrner R, Trochsler M, Keogh A, Breu K, Furer C, et al. Promotion of Liver Regeneration by Natural Killer Cells in a Murine Model Is Dependent on Extracellular Adenosine Triphosphate Phosphohydrolysis. Hepatology (2013) 57:1969–79. doi: 10.1002/hep.26008
282. Gonzales E, Julien B, Serrière-Lanneau V, Nicou A, Doignon I, Lagoudakis L, et al. ATP Release After Partial Hepatectomy Regulates Liver Regeneration in the Rat. J Hepatol (2010) 52:54–62. doi: 10.1016/j.jhep.2009.10.005
283. Kudira R, Malinka T, Kohler A, Dosch M, de Agüero MG, Melin N, et al. P2X1-Regulated IL-22 Secretion by Innate Lymphoid Cells Is Required for Efficient Liver Regeneration. Hepatology (2016) 63:2004–17. doi: 10.1002/hep.28492
284. Dalrymple LS, Go AS. Epidemiology of Acute Infections Among Patients With Chronic Kidney Disease. Clin J Am Soc Nephrol (2008) 3:1487–93. doi: 10.2215/CJN.01290308
285. Liu G-Y, Deng X-H, Li X, Cao Y-J, Xing Y-F, Zhou P, et al. Expansion of Group 2 Innate Lymphoid Cells in Patients With End-Stage Renal Disease and Their Clinical Significance. J Immunol (2020) 205:36–44. doi: 10.4049/jimmunol.1901095
286. Gungor O, Unal HU, Guclu A, Gezer M, Eyileten T, Guzel FB, et al. IL-33 and ST2 Levels in Chronic Kidney Disease: Associations With Inflammation, Vascular Abnormalities, Cardiovascular Events, and Survival. PloS One (2017) 12:1–14. doi: 10.1371/journal.pone.0178939
287. Riedel JH, Becker M, Kopp K, Düster M, Brix SR, Meyer-Schwesinger C, et al. IL-33-Mediated Expansion of Type 2 Innate Lymphoid Cells Protects From Progressive Glomerulosclerosis. J Am Soc Nephrol (2017) 28:2068–80. doi: 10.1681/ASN.2016080877
288. Law BMP, Wilkinson R, Wang X, Kildey K, Lindner M, Rist MJ, et al. Interferon-γ Production by Tubulointerstitial Human CD56bright Natural Killer Cells Contributes to Renal Fibrosis and Chronic Kidney Disease Progression. Kidney Int (2017) 92:79–88. doi: 10.1016/j.kint.2017.02.006
289. Victorino F, Sojka DK, Brodsky KS, McNamee EN, Masterson JC, Homann D, et al. Tissue-Resident NK Cells Mediate Ischemic Kidney Injury and Are Not Depleted by Anti–Asialo-GM1 Antibody. J Immunol (2015) 195:4973–85. doi: 10.4049/jimmunol.1500651
290. Zhang Z-X, Wang S, Huang X, Min W-P, Sun H, Liu W, et al. NK Cells Induce Apoptosis in Tubular Epithelial Cells and Contribute to Renal Ischemia-Reperfusion Injury. J Immunol (2008) 181:7489–98. doi: 10.4049/jimmunol.181.11.7489
291. Kim HJ, Lee JS, Kim JD, Cha HJ, Kim A, Lee SK, et al. Reverse Signaling Through the Costimulatory Ligand CD137L in Epithelial Cells is Essential for Natural Killer Cell-Mediated Acute Tissue Inflammation. Proc Natl Acad Sci USA (2012) 109:13–22. doi: 10.1073/pnas.1112256109
292. Huang Q, Niu Z, Tan J, Yang J, Liu Y, Ma H, et al. IL-25 Elicits Innate Lymphoid Cells and Multipotent Progenitor Type 2 Cells That Reduce Renal Ischemic/Reperfusion Injury. J Am Soc Nephrol (2015) 26:2199–211. doi: 10.1681/ASN.2014050479
293. Cao Q, Wang Y, Niu Z, Wang C, Wang R, Zhang Z, et al. Potentiating Tissue-Resident Type 2 Innate Lymphoid Cells by IL-33 to Prevent Renal Ischemia-Reperfusion Injury. J Am Soc Nephrol (2018) 29:961–76. doi: 10.1681/ASN.2017070774
294. Liang H, Xu F, Wen XJ, Liu HZ, Wang HB, Zhong JY, et al. Interleukin-33 Signaling Contributes to Renal Fibrosis Following Ischemia Reperfusion. Eur J Pharmacol (2017) 812:18–27. doi: 10.1016/j.ejphar.2017.06.031
295. Cameron GJ, Cautivo KM, Loering S, Jiang SH, Deshpande AV, Foster PS, et al. Group 2 Innate Lymphoid Cells are Redundant in Experimental Renal Ischemia-Reperfusion Injury. Front Immunol (2019) 10:826. doi: 10.3389/fimmu.2019.00826
296. Arazi A, Rao DA, Berthier CC, Davidson A, Liu Y, Hoover PJ, et al. The Immune Cell Landscape in Kidneys of Patients With Lupus Nephritis. Nat Immunol (2019) 20:902–14. doi: 10.1038/s41590-019-0398-x
297. Suárez-Fueyo A, Bradley SJ, Katsuyama T, Solomon S, Katsuyama E, Kyttaris VC, et al. Downregulation of CD3ζ in NK Cells From Systemic Lupus Erythematosus Patients Confers a Proinflammatory Phenotype. J Immunol (2018) 200:3077–86. doi: 10.4049/jimmunol.1700588
298. Guo C, Zhou M, Zhao S, Huang Y, Wang S, Fu R, et al. Innate Lymphoid Cell Disturbance With Increase in ILC1 in Systemic Lupus Erythematosus. Clin Immunol (2019) 202:49–58. doi: 10.1016/j.clim.2019.03.008
299. Ryu S, Lee EY, Kim DK, Kim YS, Chung DH, Kim JH, et al. Reduction of Circulating Innate Lymphoid Cell Progenitors Results in Impaired Cytokine Production by Innate Lymphoid Cells in Patients With Lupus Nephritis. Arthritis Res Ther (2020) 22:1–13. doi: 10.1186/s13075-020-2114-5
300. Hu L, Hu J, Chen L, Zhang Y, Wang Q, Yang X. Interleukin-22 From Type 3 Innate Lymphoid Cells Aggravates Lupus Nephritis by Promoting Macrophage Infiltration in Lupus-Prone Mice. Front Immunol (2021) 12:584414. doi: 10.3389/fimmu.2021.584414
301. Miller D, Motomura K, Garcia-Flores V, Romero R, Gomez-Lopez N. Innate Lymphoid Cells in the Maternal and Fetal Compartments. Front Immunol (2018) 9:2396. doi: 10.3389/fimmu.2018.02396
302. Gellersen B, Brosens IA, Brosens JJ. Decidualization of the Human Endometrium: Mechanisms, Functions, and Clinical Perspectives. Semin Reprod Med (2007) 25:445–53. doi: 10.1055/s-2007-991042
303. Colucci F. The Immunological Code of Pregnancy. Science (80-) (2019) 365:862–3. doi: 10.1126/science.aaw1300
304. Kopcow HD, Allan DSJ, Chen X, Rybalov B, Andzelm MM, Ge B, et al. Human Decidual NK Cells Form Immature Activating Synapses and Are Not Cytotoxic. Proc Natl Acad Sci USA (2005) 102:15563–8. doi: 10.1073/pnas.0507835102
305. Gaynor LM, Colucci F. Uterine Natural Killer Cells: Functional Distinctions and Influence on Pregnancy in Humans and Mice. Front Immunol (2017) 8:467. doi: 10.3389/fimmu.2017.00467
306. Kennedy PR, Chazara O, Gardner L, Ivarsson MA, Farrell LE, Xiong S, et al. Activating KIR2DS4 Is Expressed by Uterine NK Cells and Contributes to Successful Pregnancy. J Immunol (2016) 197:4292–300. doi: 10.4049/jimmunol.1601279
307. Kane N, Kelly R, Saunders PTK, Critchley HOD. Proliferation of Uterine Natural Killer Cells is Induced by Human Chorionic Gonadotropin and Mediated via the Mannose Receptor. Endocrinology (2009) 150:2882–8. doi: 10.1210/en.2008-1309
308. Vento-Tormo R, Efremova M, Botting RA, Turco MY, Vento-Tormo M, Meyer KB, et al. Single-Cell Reconstruction of the Early Maternal–Fetal Interface in Humans. Nature (2018) 563:347–53. doi: 10.1038/s41586-018-0698-6
309. Huhn O, Ivarsson MA, Gardner L, Hollinshead M, Stinchcombe JC, Chen P, et al. Distinctive Phenotypes and Functions of Innate Lymphoid Cells in Human Decidua During Early Pregnancy. Nat Commun (2020) 11:381. doi: 10.1038/s41467-019-14123-z
310. Strunz B, Bister J, Jönsson H, Filipovic I, Crona-Guterstam Y, Kvedaraite E, et al. Continuous Human Uterine NK Cell Differentiation in Response to Endometrial Regeneration and Pregnancy. Sci Immunol (2021) 6:eabb7800. doi: 10.1126/sciimmunol.abb7800
311. Ivarsson MA, Stiglund N, Marquardt N, Westgren M, Gidlöf S, Björkström NK. Composition and Dynamics of the Uterine NK Cell KIR Repertoire in Menstrual Blood. Mucosal Immunol (2017) 10:322–31. doi: 10.1038/mi.2016.50
312. Netea MG, Joosten LAB, Latz E, Mills KHG, Stunnenberg HG, Neill LAJO, et al. Trained Immunity: A Program of Innate Immune Memory in Health and Disease. Science (80-) (2017) 352:aaf1098. doi: 10.1126/science.aaf1098
313. Gamliel M, Goldman-Wohl D, Isaacson B, Gur C, Stein N, Yamin R, et al. Trained Memory of Human Uterine NK Cells Enhances Their Function in Subsequent Pregnancies. Immunity (2018) 48:951–62.e5. doi: 10.1016/j.immuni.2018.03.030
314. Kuon RJ, Weber M, Heger J, Santillán I, Vomstein K, Bär C, et al. Uterine Natural Killer Cells in Patients With Idiopathic Recurrent Miscarriage. Am J Reprod Immunol (2017) 78:1–4. doi: 10.1111/aji.12721
315. Chen X, Liu Y, Cheung WC, Zhao Y, Huang J, Chung JPW, et al. Increased Expression of Angiogenic Cytokines in CD56+ Uterine Natural Killer Cells From Women With Recurrent Miscarriage. Cytokine (2018) 110:272–6. doi: 10.1016/j.cyto.2018.01.013
316. Lucas ES, Vrljicak P, Muter J, Diniz-da-Costa MM, Brighton PJ, Kong CS, et al. Recurrent Pregnancy Loss Is Associated With a Pro-Senescent Decidual Response During the Peri-Implantation Window. Commun Biol (2020) 3:1–14. doi: 10.1038/s42003-020-0763-1
317. Brighton PJ, Maruyama Y, Fishwick K, Vrljicak P, Tewary S, Fujihara R, et al. Clearance of Senescent Decidual Cells by Uterine Natural Killer Cells in Cycling Human Endometrium. Elife (2017) 6:1–23. doi: 10.7554/eLife.31274.001
318. Crespo ÂC, Mulik S, Dotiwala F, Ansara JA, Sen Santara S, Ingersoll K, et al. Decidual NK Cells Transfer Granulysin to Selectively Kill Bacteria in Trophoblasts. Cell (2020) 182:1125–1139.e18. doi: 10.1016/j.cell.2020.07.019
319. Vacca P, Pesce S, Greppi M, Fulcheri E, Munari E, Olive D, et al. PD-1 Is Expressed by and Regulates Human Group 3 Innate Lymphoid Cells in Human Decidua. Mucosal Immunol (2019) 12:624–31. doi: 10.1038/s41385-019-0141-9
320. Croxatto D, Micheletti A, Montaldo E, Orecchia P, Loiacono F, Canegallo F, et al. Group 3 Innate Lymphoid Cells Regulate Neutrophil Migration and Function in Human Decidua. Mucosal Immunol (2016) 9:1372–83. doi: 10.1038/mi.2016.10
321. Thiruchelvam U, Wingfield M, O’Farrelly C. Increased uNK Progenitor Cells in Women With Endometriosis and Infertility Are Associated With Low Levels of Endometrial Stem Cell Factor. Am J Reprod Immunol (2016) 75:493–502. doi: 10.1111/aji.12486
322. Kanzaki H, Imai K, Hatayama H, Inoue T, Kojima K, Fujimoto M, et al. The Role of Cytokines in Human Endometrium: The Inhibitory Effect of IL-1 and TNFa on in Vitro Decidualization and mRNA Expression of M-CSF, SCF and LIF in the Human Endometrium. Endocr J (1994) 41:S105–15. doi: 10.1507/endocrj.41.Supplement_S105
323. Hochdörfer T, Winkler C, Pardali K, Mjösberg J. Expression of C-Kit Discriminates Between Two Functionally Distinct Subsets of Human Type 2 Innate Lymphoid Cells. Eur J Immunol (2019) 49:884–93. doi: 10.1002/eji.201848006
324. Cupedo T, Crellin NK, Papazian N, Rombouts EJ, Weijer K, Grogan JL, et al. Human Fetal Lymphoid Tissue-Inducer Cells Are Interleukin 17-Producing Precursors to RORC+ CD127+ Natural Killer-Like Cells. Nat Immunol (2009) 10:66–74. doi: 10.1038/ni.1668
325. Miller JE, Lingegowda H, Symons LK, Bougie O, Young SL, Lessey BA, et al. Interleukin-33 Activates Group 2 Innate Lymphoid Cell Expansion and Modulates Endometriosis. JCI Insight (2021) 6:e149699. doi: 10.1172/jci.insight.149699
326. Sugahara T, Tanaka Y, Hamaguchi M, Fujii M, Shimura K, Ogawa K, et al. Reduced Innate Lymphoid Cells in the Endometrium of Women With Endometriosis. Am J Reprod Immunol (2022) 87(1):e13502. doi: 10.1111/aji.13502
327. Azziz R, Carmina E, Chen Z, Dunaif A, Laven JSE, Legro RS, et al. Polycystic Ovary Syndrome. Nat Rev Dis Prim (2016) 2:16057. doi: 10.1038/nrdp.2016.57
328. Qi X, Yun C, Sun L, Xia J, Wu Q, Wang Y, et al. Gut Microbiota–Bile Acid–Interleukin-22 Axis Orchestrates Polycystic Ovary Syndrome. Nat Med (2019) 25:1459–9. doi: 10.1038/s41591-019-0562-8
329. Matteo M, Serviddio G, Massenzio F, Scillitani G, Castellana L, Picca G, et al. Reduced Percentage of Natural Killer Cells Associated With Impaired Cytokine Network in the Secretory Endometrium of Infertile Women With Polycystic Ovary Syndrome. Fertil Steril (2010) 94:2222–2227.e3. doi: 10.1016/j.fertnstert.2010.01.049
330. Deng Y, Wu S, Yang Y, Meng M, Chen X, Chen S, et al. Unique Phenotypes of Heart Resident Type 2 Innate Lymphoid Cells. Front Immunol (2020) 11:802. doi: 10.3389/fimmu.2020.00802
331. Bracamonte-Baran W, Chen G, Hou X, Talor MV, Choi HS, Davogustto G, et al. Non-Cytotoxic Cardiac Innate Lymphoid Cells Are a Resident and Quiescent Type 2-Commited Population. Front Immunol (2019) 10:634. doi: 10.3389/fimmu.2019.00634
332. Ghaedi M, Shen ZY, Orangi M, Martinez-Gonzalez I, Wei L, Lu X, et al. Single-Cell Analysis of Rorα Tracer Mouse Lung Reveals ILC Progenitors and Effector ILC2 Subsets. J Exp Med (2020) 217:jem.20182293. doi: 10.1084/jem.20182293
333. Selathurai A, Deswaerte V, Kanellakis P, Tipping P, Toh BH, Bobik A, et al. Natural Killer (NK) Cells Augment Atherosclerosis by Cytotoxic-Dependent Mechanisms. Cardiovasc Res (2014) 102:128–37. doi: 10.1093/cvr/cvu016
334. Nour-Eldine W, Joffre J, Zibara K, Esposito B, Giraud A, Zeboudj L, et al. Genetic Depletion or Hyperresponsiveness of Natural Killer Cells Do Not Affect Atherosclerosis Development. Circ Res (2018) 122:47–57. doi: 10.1161/CIRCRESAHA.117.311743
335. Li J, Wu J, Zhang M, Zheng Y. Dynamic Changes of Innate Lymphoid Cells in Acute ST-Segment Elevation Myocardial Infarction and its Association With Clinical Outcomes. Sci Rep (2020) 10:5099. doi: 10.1038/s41598-020-61903-5
336. Backteman K, Ernerudh J, Jonasson L. Natural Killer (NK) Cell Deficit in Coronary Artery Disease: No Aberrations in Phenotype But Sustained Reduction of NK Cells Is Associated With Low-Grade Inflammation. Clin Exp Immunol (2014) 175:104–12. doi: 10.1111/cei.12210
337. Jonasson L, Backteman K, Ernerudh J. Loss of Natural Killer Cell Activity in Patients With Coronary Artery Disease. Atherosclerosis (2005) 183:316–21. doi: 10.1016/j.atherosclerosis.2005.03.011
338. Hak Ł, Mysłiwska J, Wieckiewicz J, Szyndler K, Trzonkowski P, Siebert J, et al. NK Cell Compartment in Patients With Coronary Heart Disease. Immun Ageing (2007) 4:3. doi: 10.1186/1742-4933-4-3
339. Szymanowski A, Li W, Lundberg A, Evaldsson C, Nilsson L, Backteman K, et al. Soluble Fas Ligand is Associated With Natural Killer Cell Dynamics in Coronary Artery Disease. Atherosclerosis (2014) 233:616–22. doi: 10.1016/j.atherosclerosis.2014.01.030
340. Bonaccorsi I, Spinelli D, Cantoni C, Barillà C, Pipitò N, De Pasquale C, et al. Symptomatic Carotid Atherosclerotic Plaques Are Associated With Increased Infiltration of Natural Killer (NK) Cells and Higher Serum Levels of NK Activating Receptor Ligands. Front Immunol (2019) 10:1503. doi: 10.3389/fimmu.2019.01503
341. Hou N, Zhao D, Liu Y, Gao L, Liang X, Liu X, et al. Increased Expression of T Cell Immunoglobulin- and Mucin Domain-Containing Molecule-3 on Natural Killer Cells in Atherogenesis. Atherosclerosis (2012) 222:67–73. doi: 10.1016/j.atherosclerosis.2012.02.009
342. Chen WY, Wu YH, Tsai TH, Li RF, Lai ACY, Li LC, et al. Group 2 Innate Lymphoid Cells Contribute to IL-33-Mediated Alleviation of Cardiac Fibrosis. Theranostics (2021) 11:2594–611. doi: 10.7150/THNO.51648
343. Newland SA, Mohanta S, Clément M, Taleb S, Walker JA, Nus M, et al. Type-2 Innate Lymphoid Cells Control the Development of Atherosclerosis in Mice. Nat Commun (2017) 8:15781. doi: 10.1038/ncomms15781
344. Engelbertsen D, Foks AC, Alberts-Grill N, Kuperwaser F, Chen T, Lederer JA, et al. Expansion of CD25+ Innate Lymphoid Cells Reduces Atherosclerosis. Arterioscler Thromb Vasc Biol (2015) 35:2526–35. doi: 10.1161/ATVBAHA.115.306048
345. Yu X, Newland SA, Zhao TX, Lu Y, Sage AS, Sun Y, et al. Innate Lymphoid Cells Promote Recovery of Ventricular Function After Myocardial Infarction. J Am Coll Cardiol (2021) 78:1127–42. doi: 10.1016/j.jacc.2021.07.018
346. Choi HS, Won T, Hou X, Chen G, Bracamonte-Baran W, Talor MV, et al. Innate Lymphoid Cells Play a Pathogenic Role in Pericarditis. Cell Rep (2020) 30:2989–3003.e6. doi: 10.1016/j.celrep.2020.02.040
347. Ong S, Ligons DL, Barin JG, Wu L, Talor MV, Diny N, et al. Natural Killer Cells Limit Cardiac Inflammation and Fibrosis by Halting Eosinophil Infiltration. Am J Pathol (2015) 185:847–61. doi: 10.1016/j.ajpath.2014.11.023
348. Zhou N, Yue Y, Xiong S. Sex Hormone Contributes to Sexually Dimorphic Susceptibility in CVB3-Induced Viral Myocarditis via Modulating IFN-γ+ NK Cell Production. Can J Cardiol (2018) 34:492–501. doi: 10.1016/j.cjca.2018.01.002
349. Sidossis L, Kajimura S. Brown and Beige Fat in Humans: Thermogenic Adipocytes That Control Energy and Glucose Homeostasis. J Clin Invest (2015) 125:478–86. doi: 10.1172/JCI78362
350. Kajimura S, Spiegelman BM, Seale P. Brown and Beige Fat: Physiological Roles Beyond Heat Generation. Cell Metab (2015) 22:546–59. doi: 10.1016/j.cmet.2015.09.007
351. Huang Y, Guo L, Qiu J, Chen X, Hu-Li J, Siebenlist U, et al. IL-25-Responsive, Lineage-Negative KLRG1 Hi Cells Are Multipotential “Inflammatory” Type 2 Innate Lymphoid Cells. Nat Immunol (2015) 16:161–9. doi: 10.1038/ni.3078
352. Miyajima Y, Ealey KN, Motomura Y, Mochizuki M, Takeno N, Yanagita M, et al. Effects of BMP7 Produced by Group 2 Innate Lymphoid Cells on Adipogenesis. Int Immunol (2020) 32:407–19. doi: 10.1093/intimm/dxaa013
353. Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jourihan HA, Bando JK, et al. Eosinophils Sustain Adipose Alternatively Activated Macrophages Associated With Glucose Homeostasis. Science (80-) (2011) 332:243–7. doi: 10.1126/science.1201475
354. Qiu Y, Nguyen KD, Odegaard JI, Cui X, Tian X, Locksley RM, et al. Eosinophils and Type 2 Cytokine Signaling in Macrophages Orchestrate Development of Functional Beige Fat. Cell (2014) 157:1292–308. doi: 10.1016/j.cell.2014.03.066
355. Molofsky AB, Nussbaum JC, Liang HE, Dyken SJV, Cheng LE, Mohapatra A, et al. Innate Lymphoid Type 2 Cells Sustain Visceral Adipose Tissue Eosinophils and Alternatively Activated Macrophages. J Exp Med (2013) 210:535–49. doi: 10.1084/jem.20121964
356. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, But Not Obese, Fat Is Enriched for a Unique Population of Regulatory T Cells That Affect Metabolic Parameters. Nat Med (2009) 15:930–9. doi: 10.1038/nm.2002
357. Molofsky AB, Van Gool F, Liang HE, Van Dyken SJ, Nussbaum JC, Lee J, et al. Interleukin-33 And Interferon-γ Counter-Regulate Group 2 Innate Lymphoid Cell Activation During Immune Perturbation. Immunity (2015) 43:161–74. doi: 10.1016/j.immuni.2015.05.019
358. Halim TYF, Rana BMJ, Walker JA, Kerscher B, Knolle MD, Jolin HE, et al. Tissue-Restricted Adaptive Type 2 Immunity Is Orchestrated by Expression of the Costimulatory Molecule OX40L on Group 2 Innate Lymphoid Cells. Immunity (2018) 48:1195–207.e6. doi: 10.1016/j.immuni.2018.05.003
359. Lee MW, Odegaard JI, Mukundan L, Qiu Y, Molofsky AB, Nussbaum JC, et al. Activated Type 2 Innate Lymphoid Cells Regulate Beige Fat Biogenesis. Cell (2015) 160:74–87. doi: 10.1016/j.cell.2014.12.011
360. Mahlakõiv T, Flamar AL, Johnston LK, Moriyama S, Putzel GG, Bryce PJ, et al. Stromal Cells Maintain Immune Cell Homeostasis in Adipose Tissue via Production of Interleukin-33. Sci Immunol (2019) 4:eaax0416. doi: 10.1126/sciimmunol.aax0416
361. Rana BMJ, Jou E, Barlow JL, Rodriguez-Rodriguez N, Walker JA, Knox C, et al. A Stromal Cell Niche Sustains ILC2-Mediated Type-2 Conditioning in Adipose Tissue. J Exp Med (2019) 216:1999–2009. doi: 10.1084/jem.20190689
362. O’Sullivan TE, Rapp M, Fan X, El WO, Bhardwaj P, NM A, et al. Adipose-Resident Group 1 Innate Lymphoid Cells Promote Obesity-Associated Insulin Resistance. Immunity (2016) 45:428–41. doi: 10.1016/j.immuni.2016.06.016
363. Boulenouar S, Michelet X, Duquette D, Alvarez D, Hogan AE, Dold C, et al. Adipose Type One Innate Lymphoid Cells Regulate Macrophage Homeostasis Through Targeted Cytotoxicity. Immunity (2017) 46:273–86. doi: 10.1016/j.immuni.2017.01.008
364. Hotamisligil GS. Foundations of Immunometabolism and Implications for Metabolic Health and Disease. Immunity (2017) 47:406–20. doi: 10.1016/j.immuni.2017.08.009
365. Oldenhove G, Boucquey E, Taquin A, Acolty V, Bonetti L, Ryffel B, et al. PD-1 Is Involved in the Dysregulation of Type 2 Innate Lymphoid Cells in a Murine Model of Obesity. Cell Rep (2018) 25:2053–60.e4. doi: 10.1016/j.celrep.2018.10.091
366. Zhao XY, Zhou L, Chen Z, Ji Y, Peng X, Qi L, et al. The Obesity-Induced Adipokine Sst2 Exacerbates Adipose Treg and ILC2 Depletion and Promotes Insulin Resistance. Sci Adv (2020) 6:eaay6191. doi: 10.1126/sciadv.aay6191
367. Wang H, Shen L, Sun X, Liu F, Feng W, Jiang C, et al. Adipose Group 1 Innate Lymphoid Cells Promote Adipose Tissue Fibrosis and Diabetes in Obesity. Nat Commun (2019) 10:3254. doi: 10.1038/s41467-019-11270-1
368. Wensveen FM, Jelenčić V, Valentić S, Šestan M, Wensveen TT, Theurich S, et al. NK Cells Link Obesity-Induced Adipose Stress to Inflammation and Insulin Resistance. Nat Immunol (2015) 16:376–85. doi: 10.1038/ni.3120
369. Theurich S, Tsaousidou E, Hanssen R, Lempradl AM, Mauer J, Timper K, et al. IL-6/Stat3-Dependent Induction of a Distinct, Obesity-Associated NK Cell Subpopulation Deteriorates Energy and Glucose Homeostasis. Cell Metab (2017) 26:171–84.e6. doi: 10.1016/j.cmet.2017.05.018
370. Dalmas E, Venteclef N, Caer C, Poitou C, Cremer I, Aron-Wisnewsky J, et al. T Cell+Derived IL-22 Amplifies IL-1β+Driven Inflammation in Human Adipose Tissue: Relevance to Obesity and Type 2 Diabetes. Diabetes (2014) 63:1966–77. doi: 10.2337/db13-1511
371. Upadhyay V, Poroyko V, Kim TJ, Devkota S, Fu S, Liu D, et al. Lymphotoxin Regulates Commensal Responses to Enable Diet-Induced Obesity. Nat Immunol (2012) 13:947–53. doi: 10.1038/ni.2403
372. Vély F, Barlogis V, Vallentin B, Neven B, Piperoglou C, Perchet T, et al. Evidence of Innate Lymphoid Cell Redundancy in Humans. Nat Immunol (2016) 17:1291–9. doi: 10.1038/ni.3553
373. Piperoglou C, Larid G, Vallentin B, Balligand L, Crinier A, Banzet N, et al. Innate Lymphoid Cell Recovery and Occurrence of GvHD After Hematopoietic Stem Cell Transplantation. J Leukoc Biol (2021) 17:1–12. doi: 10.1038/ni.3553
Keywords: innate lymphoid cell (ILC), NK cell, tissue-resident immune cells, tissue homeostasis, autoimmunity, inflammation, immune tolerance
Citation: Murphy JM, Ngai L, Mortha A and Crome SQ (2022) Tissue-Dependent Adaptations and Functions of Innate Lymphoid Cells. Front. Immunol. 13:836999. doi: 10.3389/fimmu.2022.836999
Received: 16 December 2021; Accepted: 11 February 2022;
Published: 10 March 2022.
Edited by:
Cyril Seillet, Walter and Eliza Hall Institute of Medical Research, AustraliaReviewed by:
Hui Peng, University of Science and Technology of China, ChinaElia Tait Wojno, University of Washington, United States
Copyright © 2022 Murphy, Ngai, Mortha and Crome. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sarah Q. Crome, c2FyYWguY3JvbWVAdXRvcm9udG8uY2E=
†ORCID: Julia M. Murphy, orcid.org/0000-0002-0617-1666
Louis Ngai, orcid.org/0000-0002-6835-1961
Arthur Mortha, orcid.org/0000-0003-2673-0485
Sarah Q. Crome, orcid.org/0000-0001-5117-7453
‡These authors have contributed equally to this work